Полиэфир, способ сообщения белизны полиэфиру, тонирующий состав премикса, формованное изделие - RU2142477C1
Код документа: RU2142477C1
Чертежи





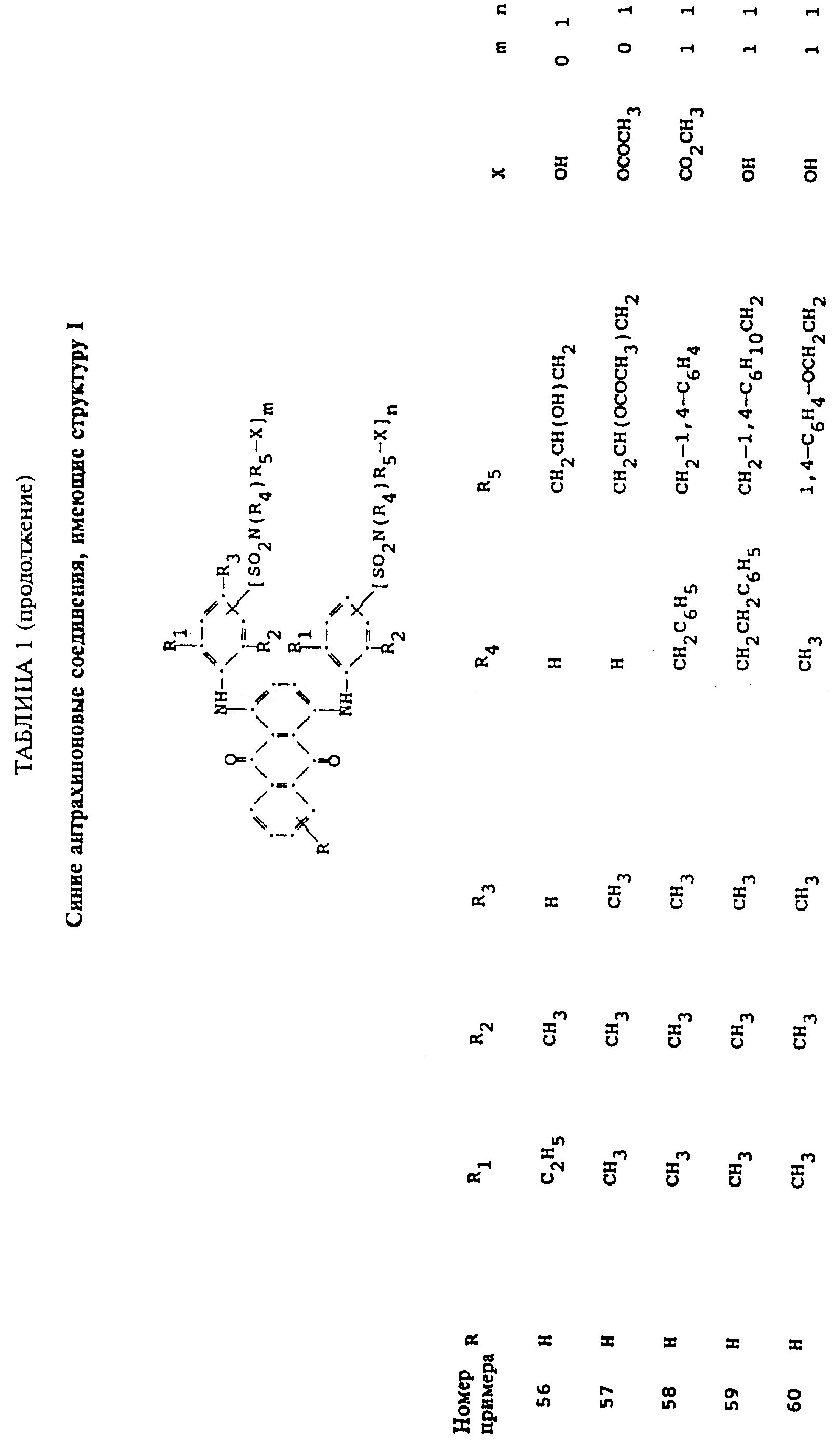
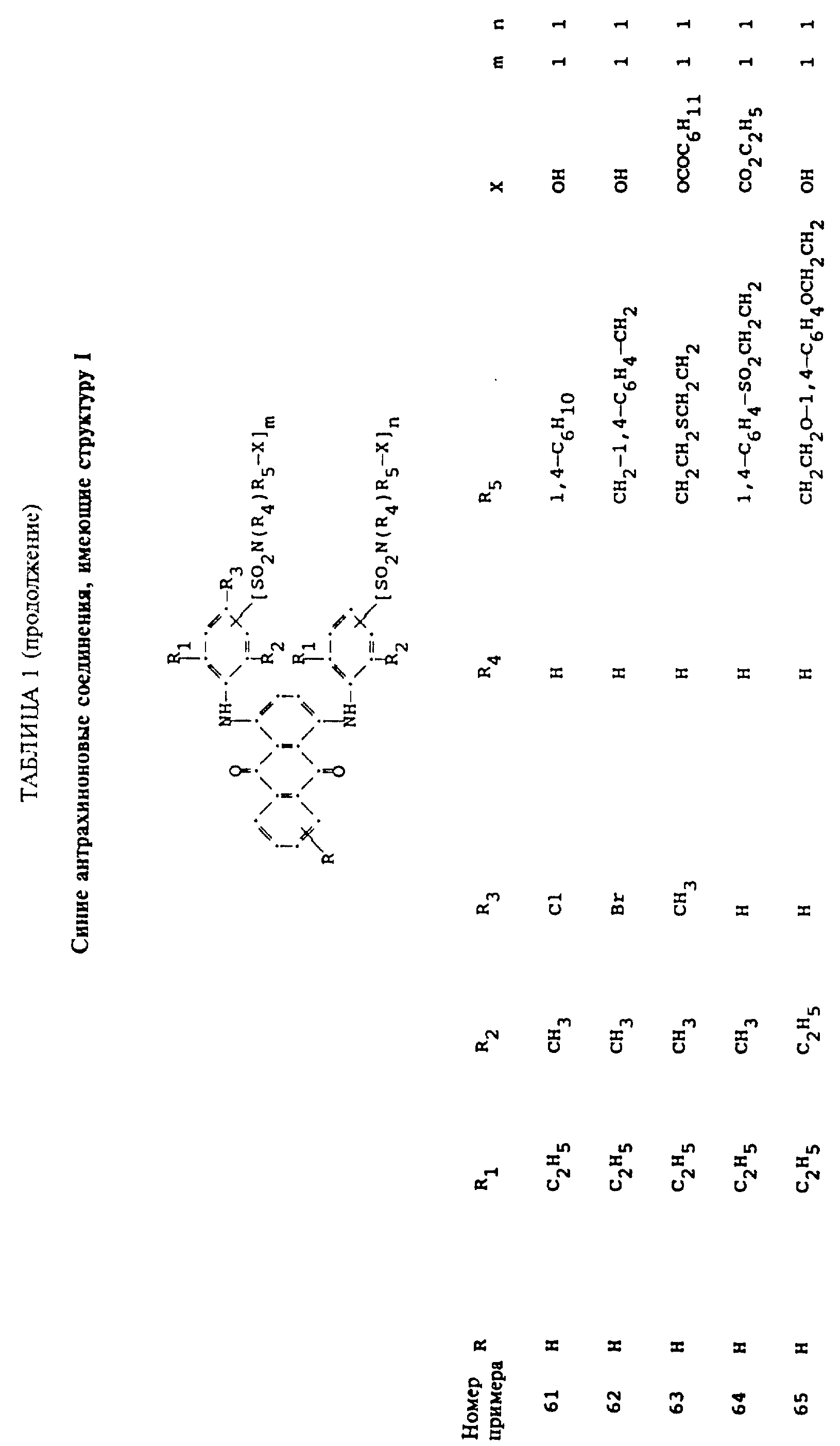
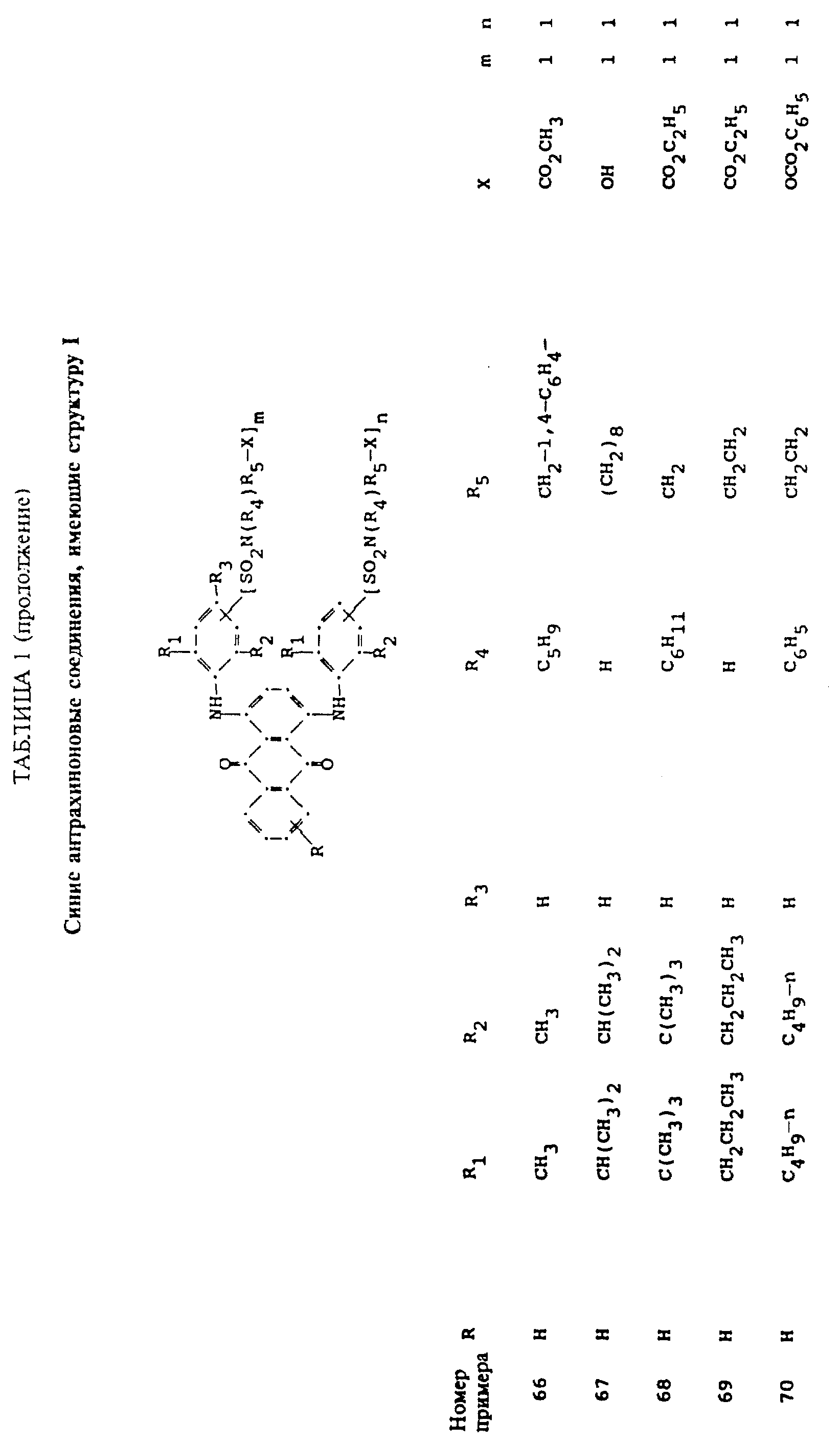
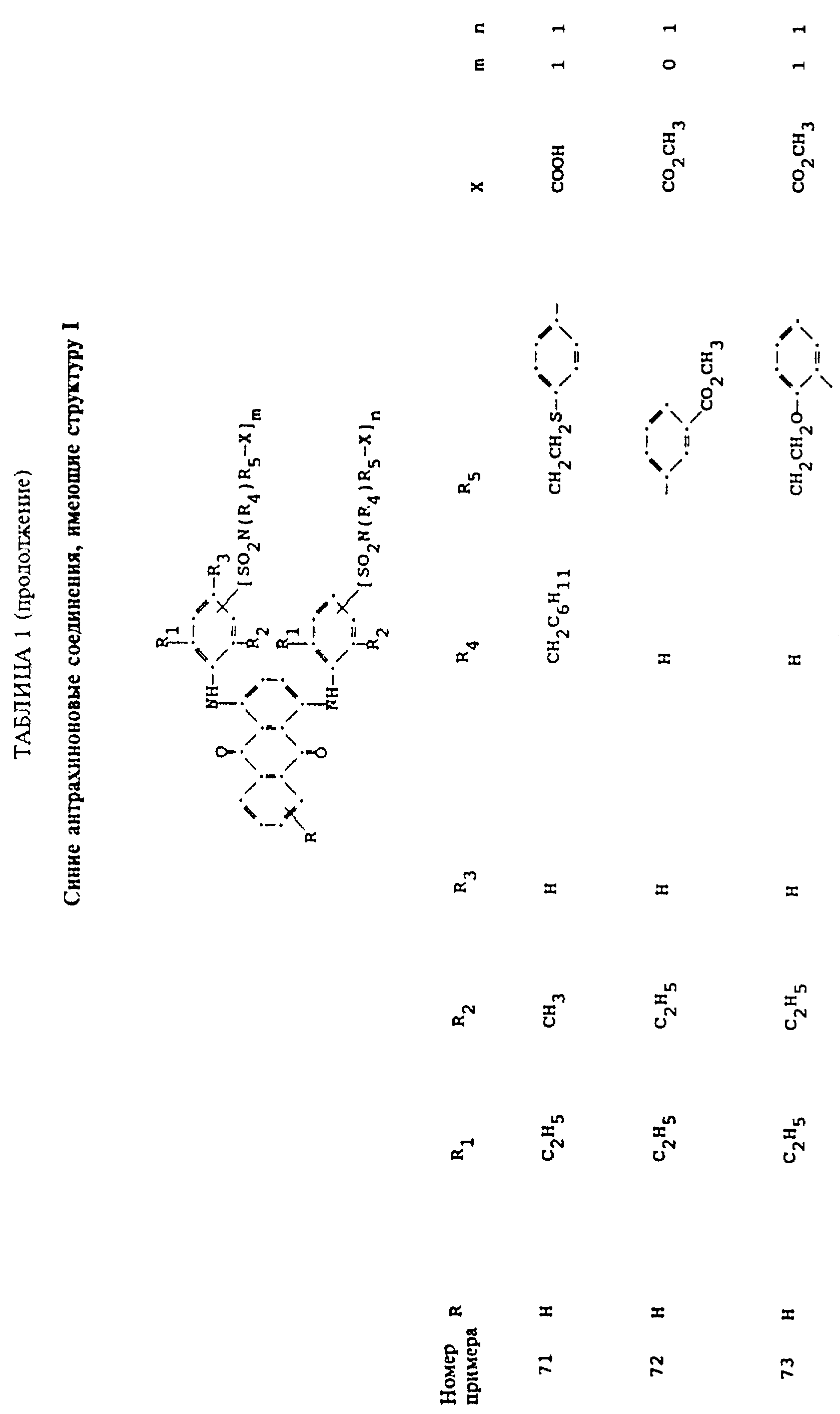




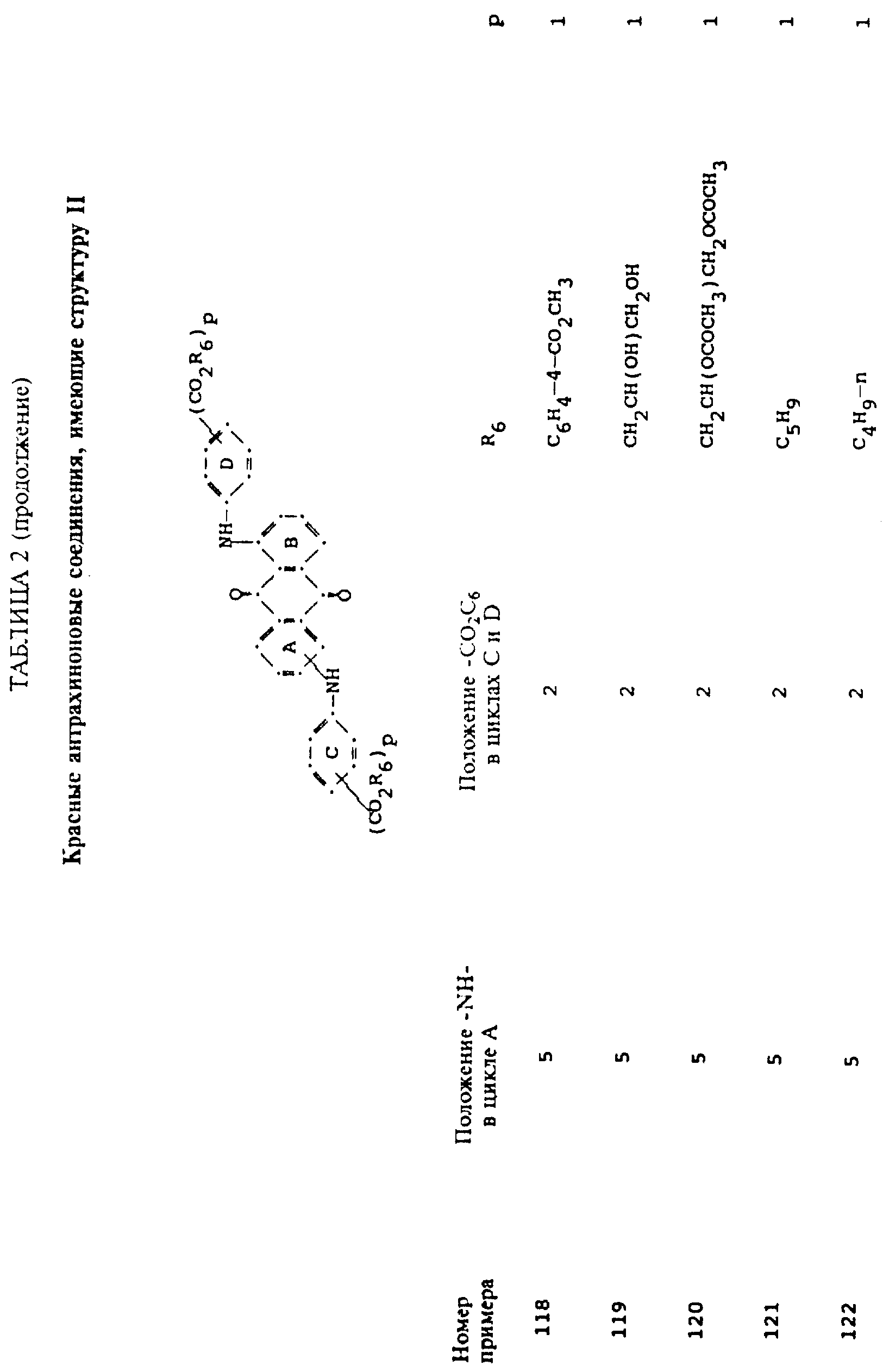

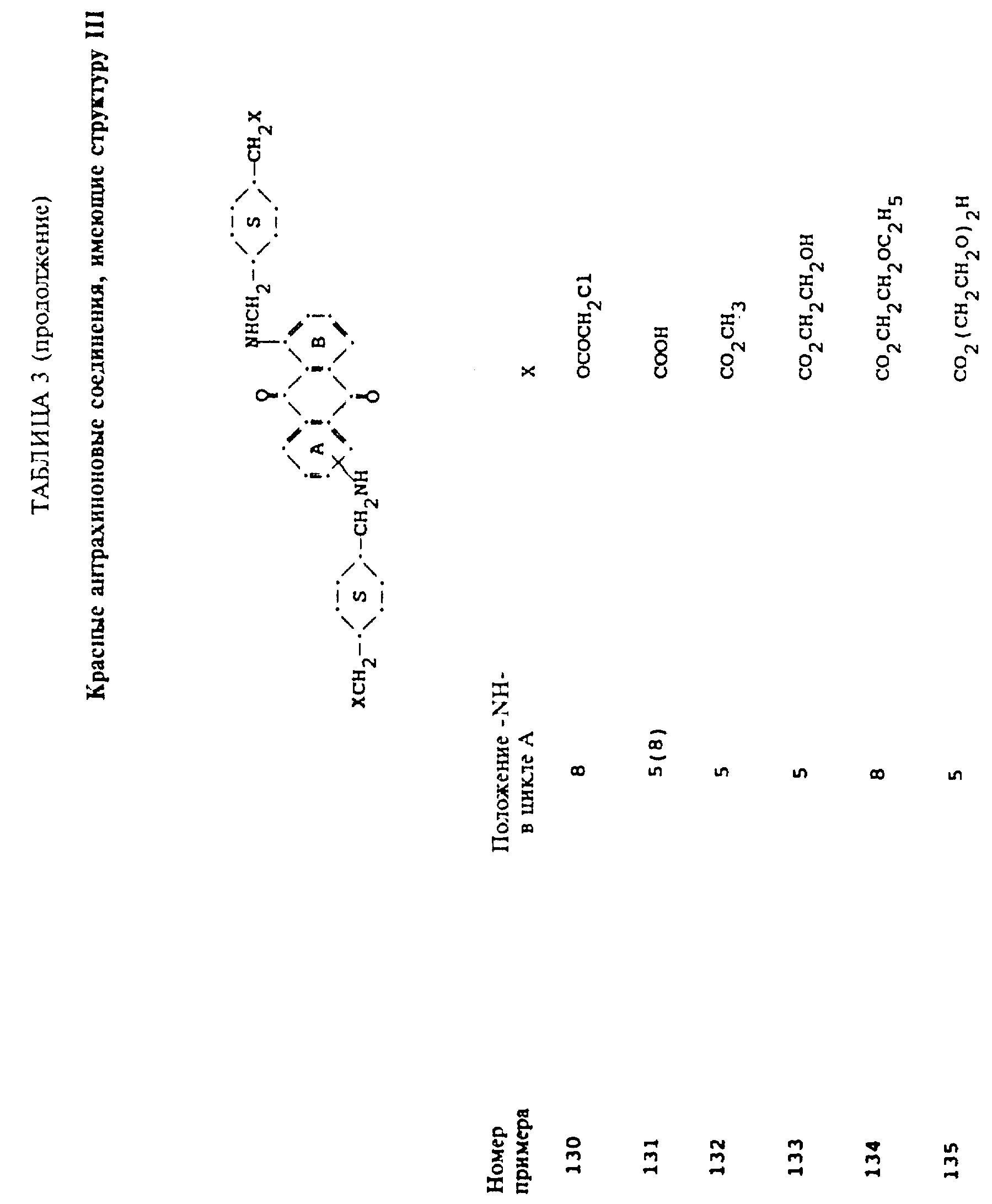

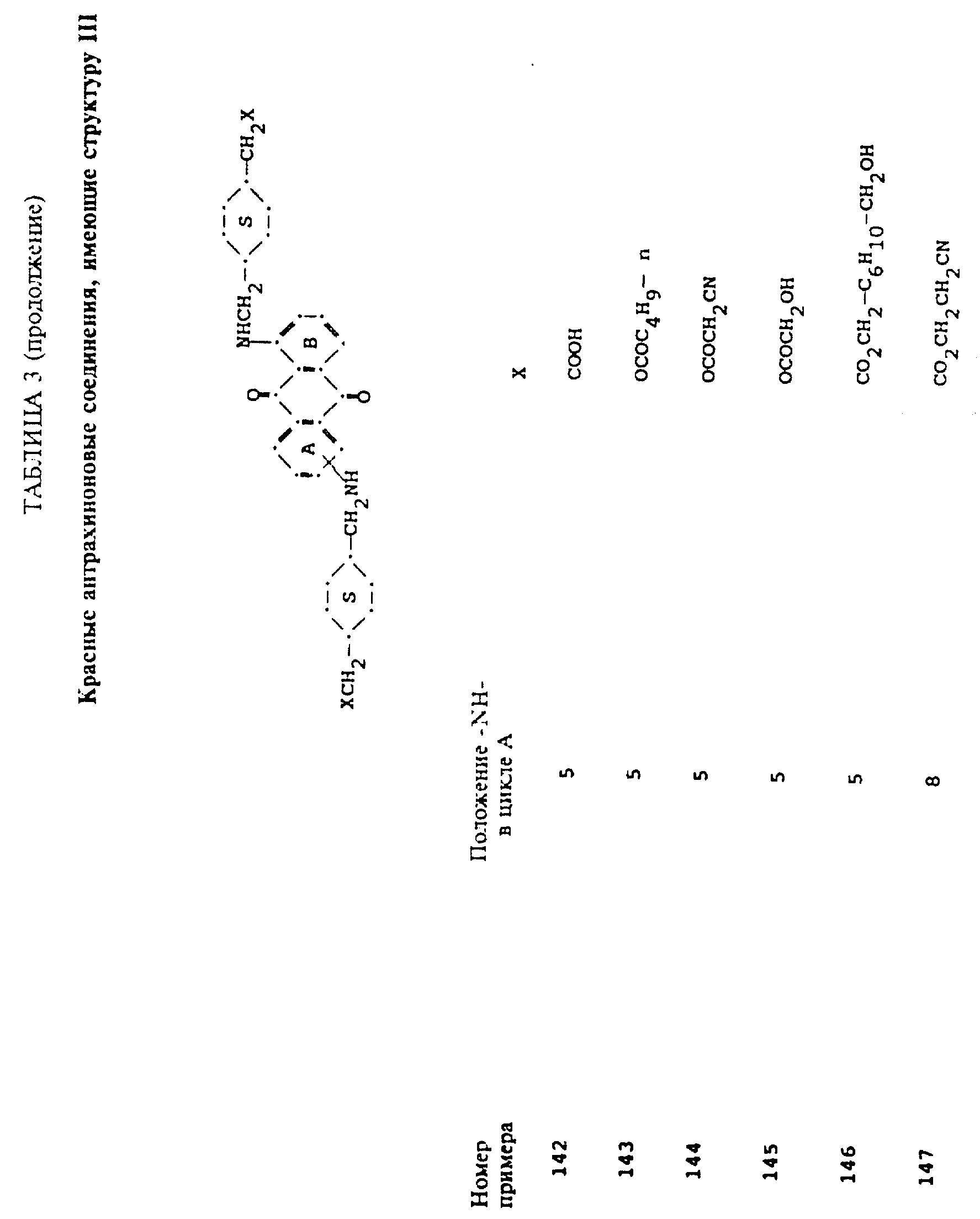
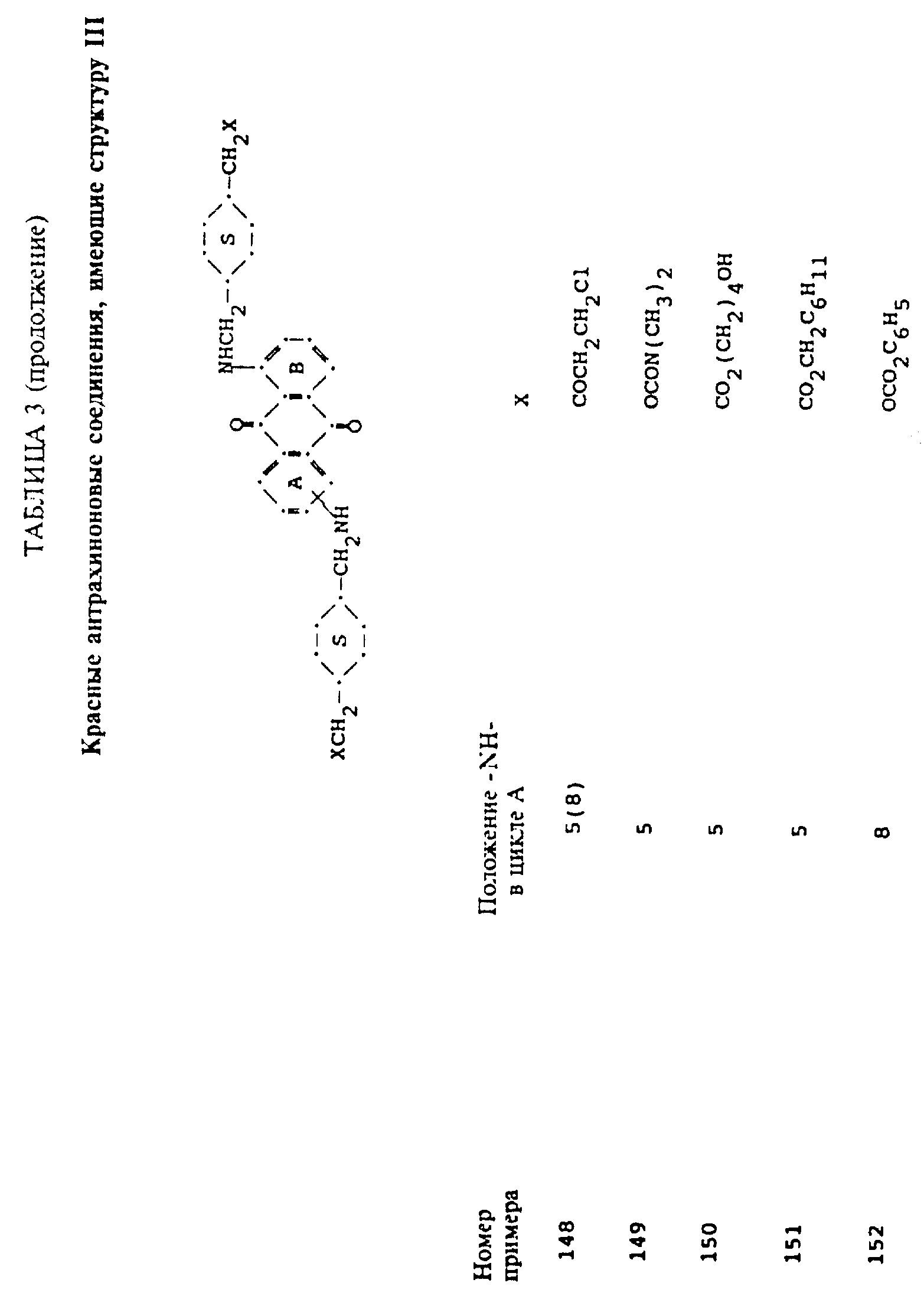











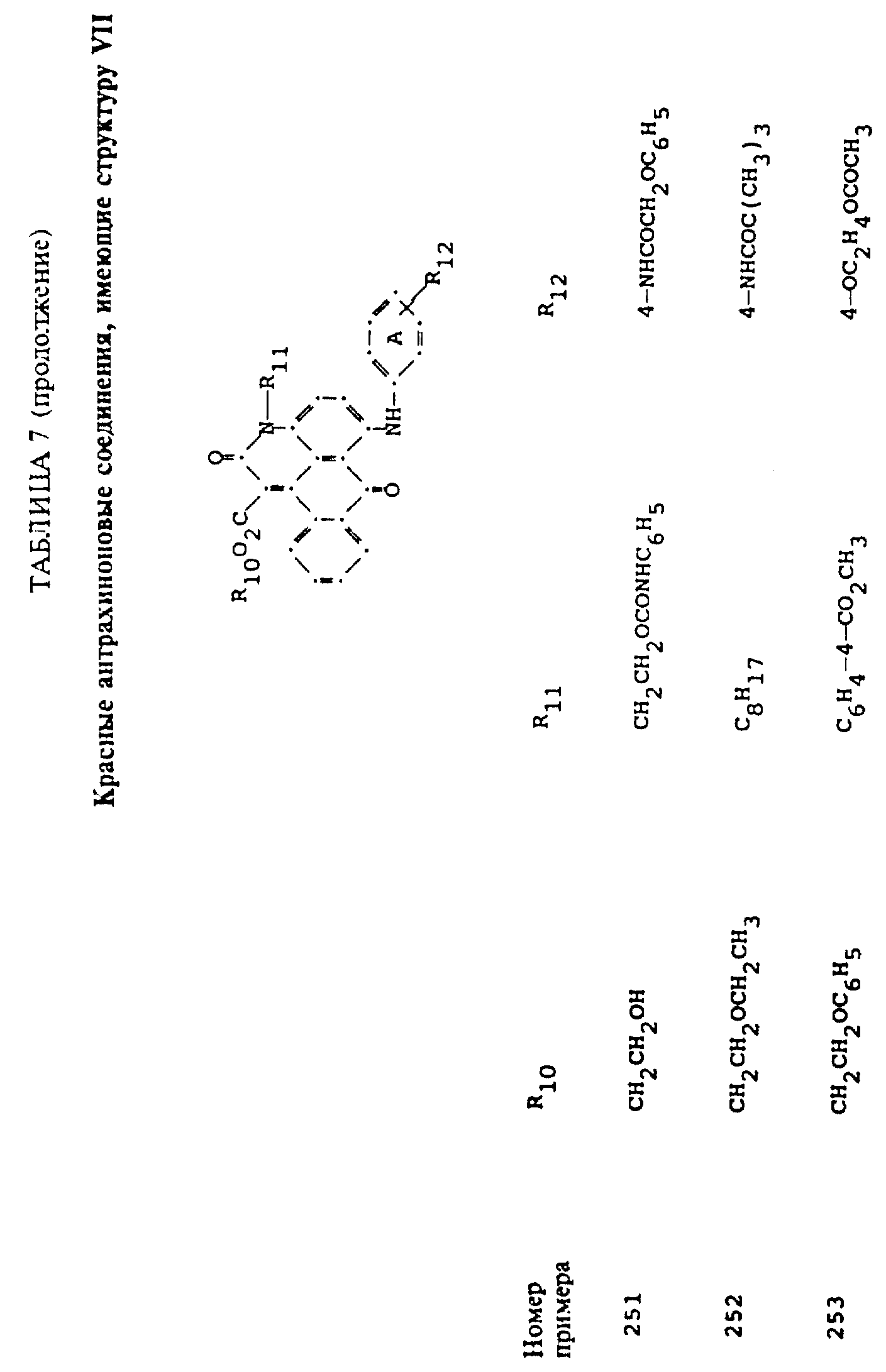

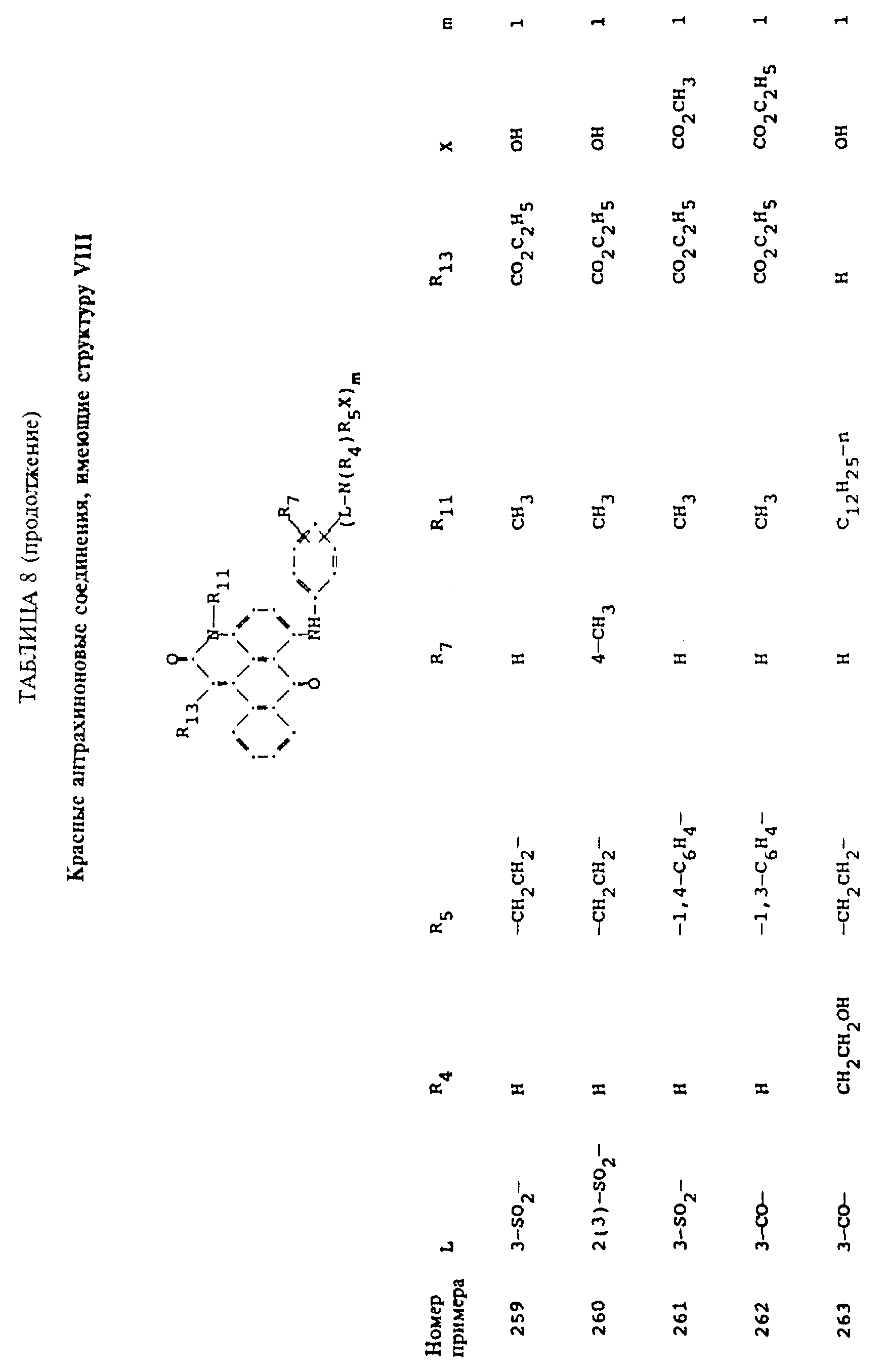
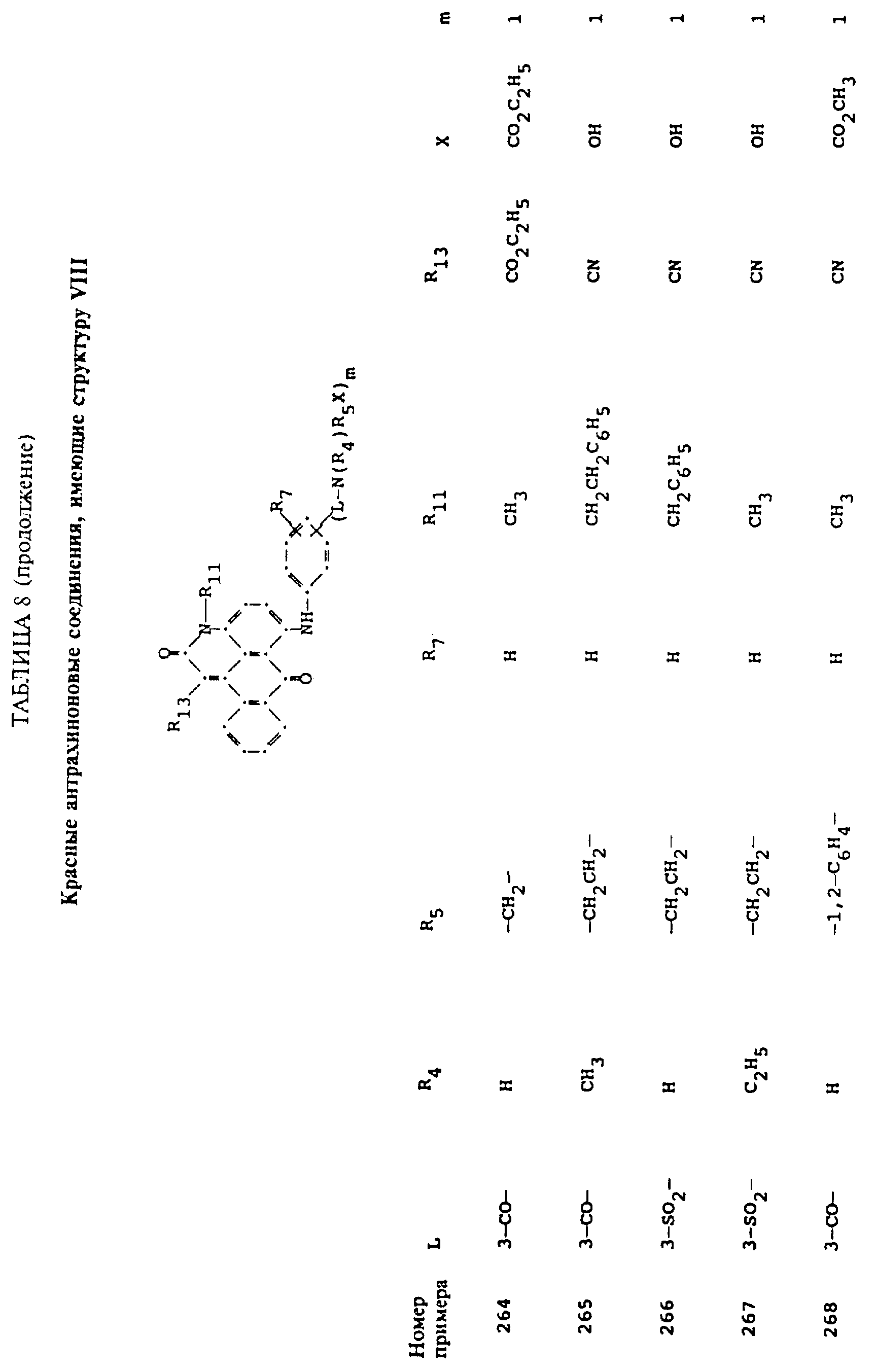
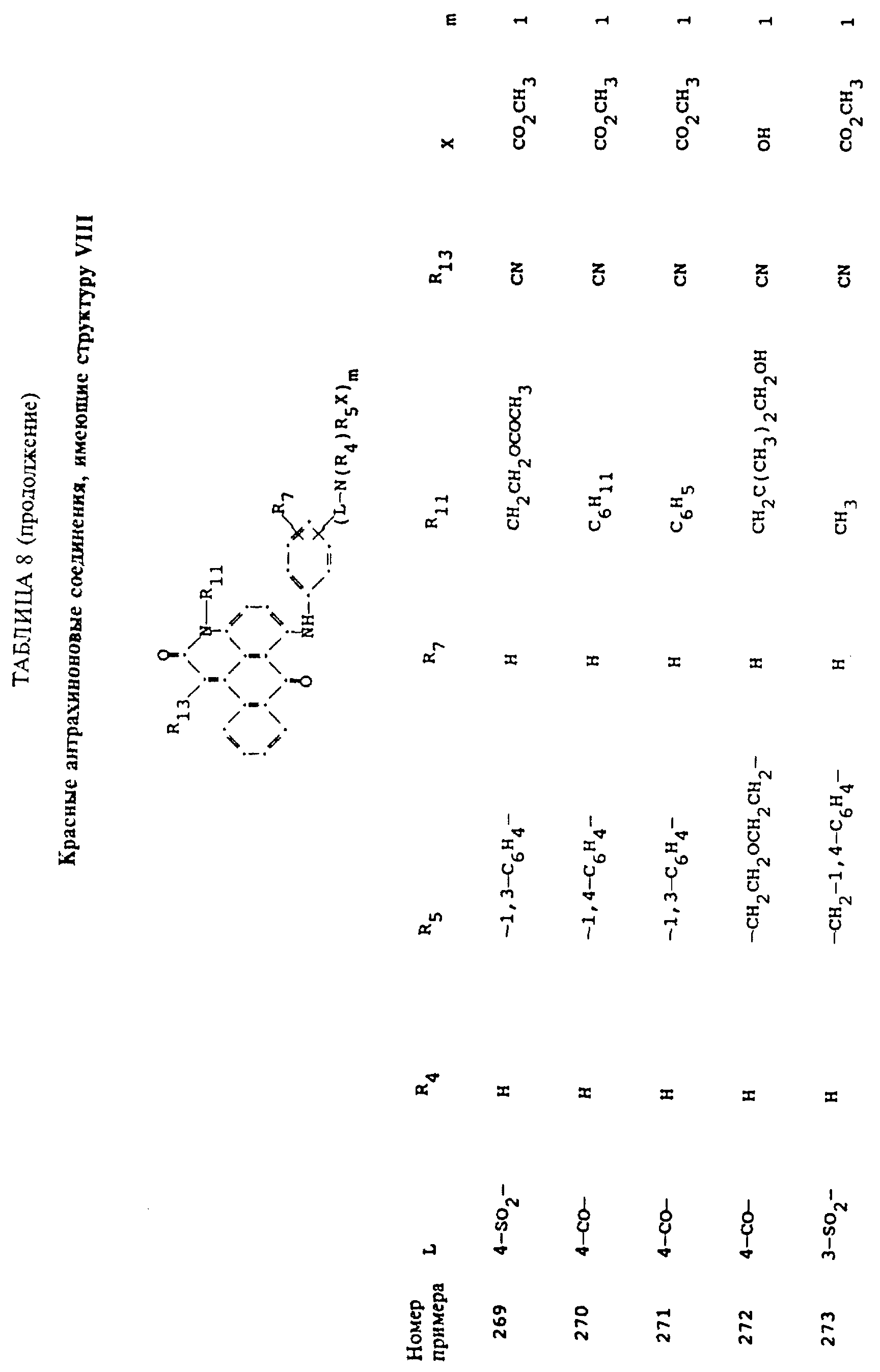




Описание
Настоящее изобретение относится к области органической химии. В частности, это изобретение относится к определенным замещенным 1,4-бис(2,6-диалкиланилино)антрахинонам в сочетании с выбранными соединениями красного антрахинона и антрапиридон(3H- дибенз[f,i,j]изохинолин-2,7-диона), которые используют в качестве тонирующей системы в полиэфирах.
К сожалению, вырабатываемые полиэфирные волокна и пластики обычно имеют нежелательную желтую окраску. В настоящее время для улучшения белизны полиэфирных волокон или нейтральных цветовых характеристик полиэфирных пластиков в полиэфир, чтобы скрыть или нейтрализовать их желтый цвет, вводятся так называемые специальные тонеры.
Наиболее широко используемым промышленным тонером для маскировки желтого цвета полимеров является ацетат кобальта. У последнего имеется, однако, ряд недостатков, которые следует отметить. Например, материалы, тонированные ацетатом кобальта, склонны к нестабильности при хранении, в частности чувствительны к изменениям температуры и влажности; они имеют тенденцию приобретать нежелательный сдвиг окраски в сторону желтого цвета. Далее, если для маскировки желтого цвета некоторых полимеров требуются высокие концентрации кобальта, то при этом возникает тенденция приобретения такими полимерами серого окрашивания.
Другим недостатком применения ацетата кобальта является определенное ограничение уровня кобальта, которое считается допустимым для полиэфирных каталитических систем.
Далее, соли кобальта имеют тенденцию снижать конечную термическую стабильность полимеров и увеличивают образование ацетальдегида в поли(этилентерефталате).
И наконец, для кобальта характерна выраженная тенденция образовывать нерастворимые осадки в промышленном оборудовании, что создает определенные производственные проблемы.
В патенте США N 4745174 раскрываются 1-циано-3H- -дибенз[f, j,i]изохинолин-2,7-дионы, которые используют как тонеры для полиэфирных волокон и пластиков и которые решают многие из упомянутых выше проблем. Однако получение этих соединений дорогостоящее; их производство и использование требует внимания к окружающей среде, к вопросам безопасности и токсикологии.
Для обеспечения наибольшей степени безопасности тонеры, пригодные для полиэфиров, не должны экстрагироваться из этих полимеров. Они должны быть также устойчивы к солнечному свету и быть стабильными в широком диапазоне температур и влажности. Далее, эти тонеры не должны сублимироваться или менять окраску при крайне высоких температурах, имеющих место в процессе производства полиэфиров. В дополнение к сказанному, тонер не должен оказывать вредного воздействия на физические свойства полимерного полиэфира.
В патенте США N 3488195 и 3849139 раскрывается применение определенных 1,4-бис-(ариламино)антрахинонов, включая 1,4-бис-(2, 6-диариламино)антрахиноны, для тонирования фотографической рентгеновской пленки в целях облегчения исследования полученных на ней рентгенограмм для получения точного диагноза. Однако ни в одном из этих патентов не отмечается, что эти соединения синего цвета должны быть особенно эффективны как тонеры в комбинации с определенными красными красителями, чтобы перекрыть свойственную полиэфирам желтую окраску.
В патенте США N 2731476 описано использование определенных синих сульфонамидных производных 1,4-бис-(ариламино)антрахинона в качестве красящих веществ и пигментов для красящих лаков и пластических масс.
В патентах США N 4359570; 4403092; 4420581; 4790581; 4740581 и 4999418 раскрыты окрашенные полиэфирные композиции, содержащие меняющиеся уровни синего и красного красителей, однако ни в одном из них не упоминается полезность описываемых соединений как компонентов при получении эффективных тонирующих систем для полиэфиров.
Настоящим изобретением обеспечиваются определенные синие замещенные 1,4- бис(2, 6-диалкиланилино)антрахиноны в сочетании с выбранными соединениями красного антрахинона и антрапиридон (3H- дибенз[f,i, j]изохинолин-2,7-диона), которые обеспечивают применимость тонирующей системы к полиэфирам, имеющим желтую окраску для сообщения им требуемого оттенка от нейтрального до светло-голубого. Эти термостойкие окрашенные соединения могут содержать реактивные полиэфирные группы и в процессе полимеризации они предпочтительно включаются в структуры полиэфирного полимера.
Этим изобретением обеспечивается экономичная, безопасная и совместимая с окружающей средой тонирующая система для использования последней в полиэфирных волокнах и пластиках. Эти тонеры имеют адекватные колористические характеристики и стабильны к нагреву, свету и влажности и к разнообразным факторам окружающей среды; они преодолевают многие из проблем, возникающих при использовании описанных выше известных тонирующих систем. Показано, что тонеры настоящего изобретения стабильны к УФ-свету, повышенным температурам, гликолизу и гидролизу. Далее, соотношение красного и синего компонентов можно варьировать, как этого требует получение целевых колористических характеристик тонирующей системы, с тем, чтобы придать надежные тонирующие свойства различным полиэфирам, имеющим меняющуюся интенсивность желтой окраски.
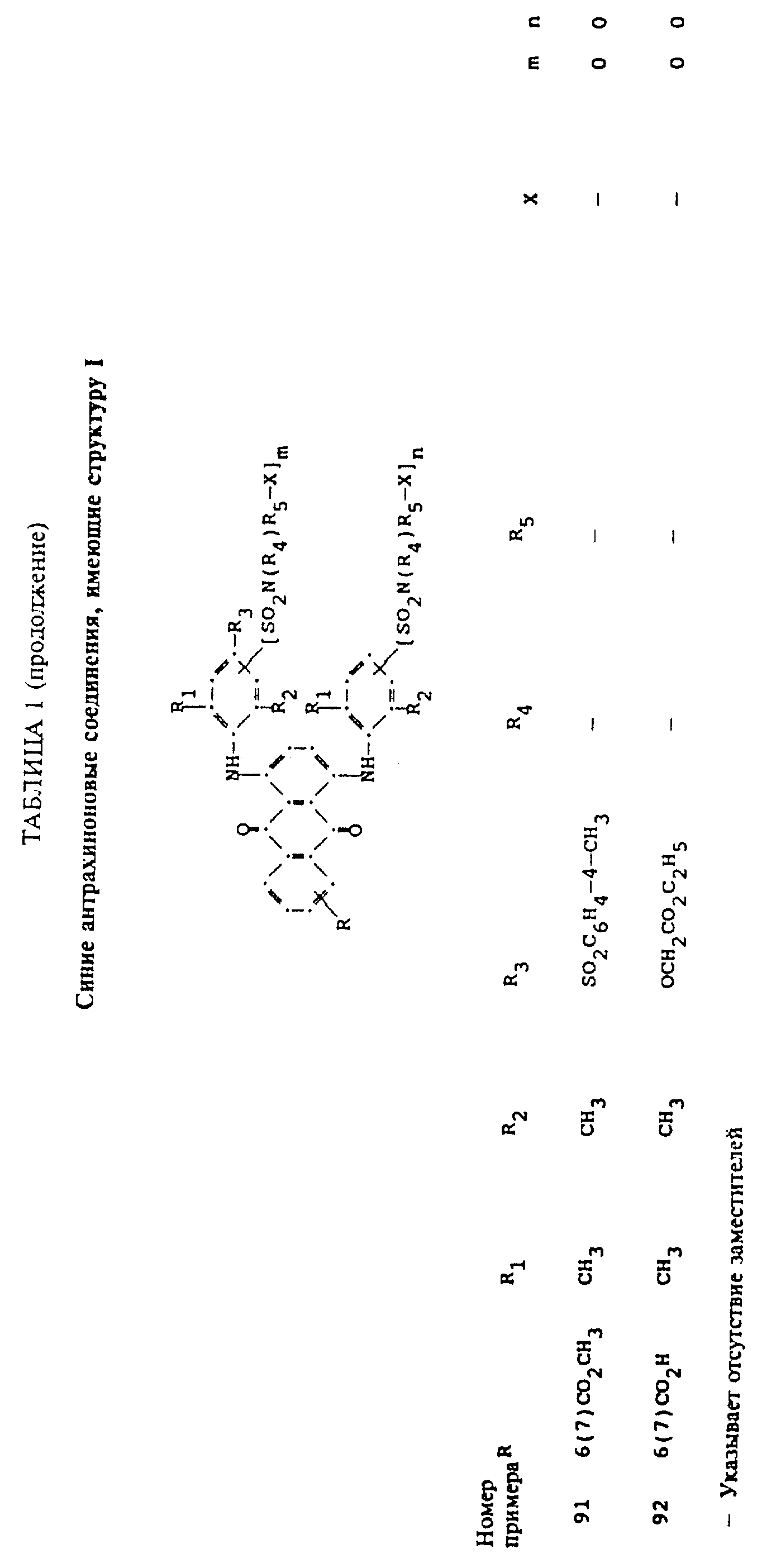
Основная часть тонирующей системы,
используемой в настоящем изобретении, содержит по меньшей мере один синий 1,4-бис(2,6- диалкиланилино)антрахинон(ы)
формулы (I):
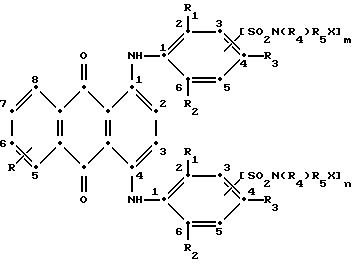
в которой R выбирают из группы, содержащей водород, C1-C6 -алкил, галоген, карбокси и

R1 и R2 независимо друг от друга представляют собой C1-C6 - алкил;
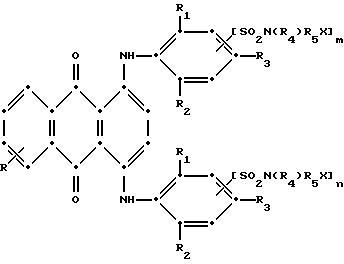
R3 выбирают из группы, содержащей водород, галоген, C1-C6 - алкил, замещенный C1-C6-алкил, гидрокси, C1-C6-алкокси, замещенный C1-C6-алкокси, циано, тиоциано, C1-C6-алкилтио, замещенный C1-C6-алкилтио, C1-C6-алкилсульфонил, замещенный C1-C6-алкилсульфонил, C1-C6-алкоксикарбонил, карбокси, арилокси, арилтио, арилсульфонил и SO2N(R4)R5X, если m и/или n=0;
R4 выбирают из группы, содержащей водород, C1-C6-алкил, замещенный C1-C6-алкил, C3-C8-алкенил, C3-C8-алкинил, C3-C7-циклоалкил и арил;
R5 - связывающая группа, выбранная из группы, содержащей C1-C8-алкилен, C1-C6-алкилен-Z-C1-C6-алкилен, арилен-C1-C6-алкилен. apилeн-Z-C1-C6 -алкилен,
C3-C7-циклоалкилен, C1-C6 -алкилен-циклоалкилен-C1-C6-алкилен, C1-C6-алкилен-арилен-C1-C6-алкилен и C1-C6-алкилен-Z-арилен-Z-C1 -C6-алкилен, где Z выбирают из -O-, -S- или SO2;
X представляет собой галоген или реактивную полиэфирную группу; и
m и n независимо друг от друга представляют собой 0 или 1, при условии, что присутствует по меньшей мере одна полиэфирная реактивная группа.
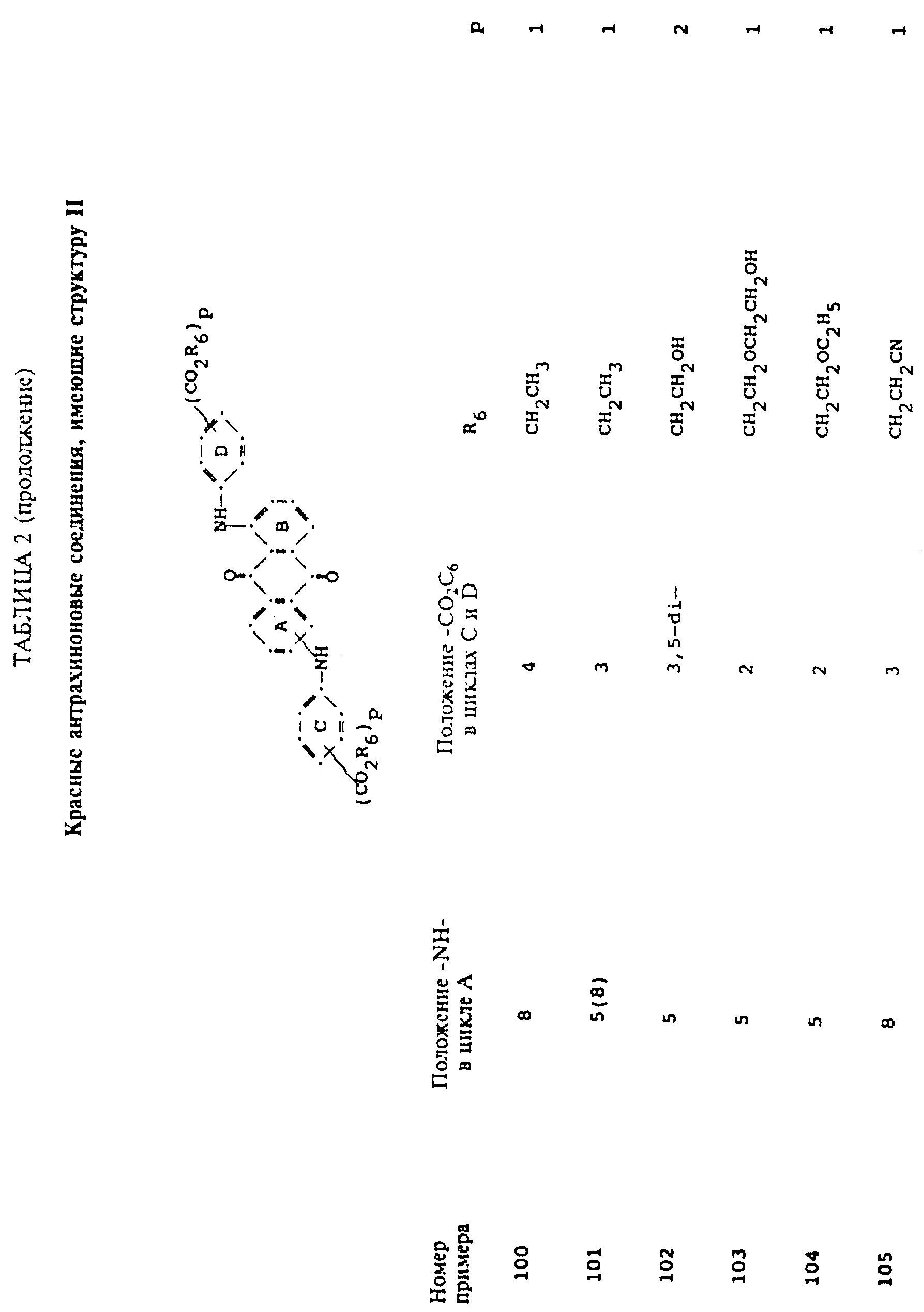
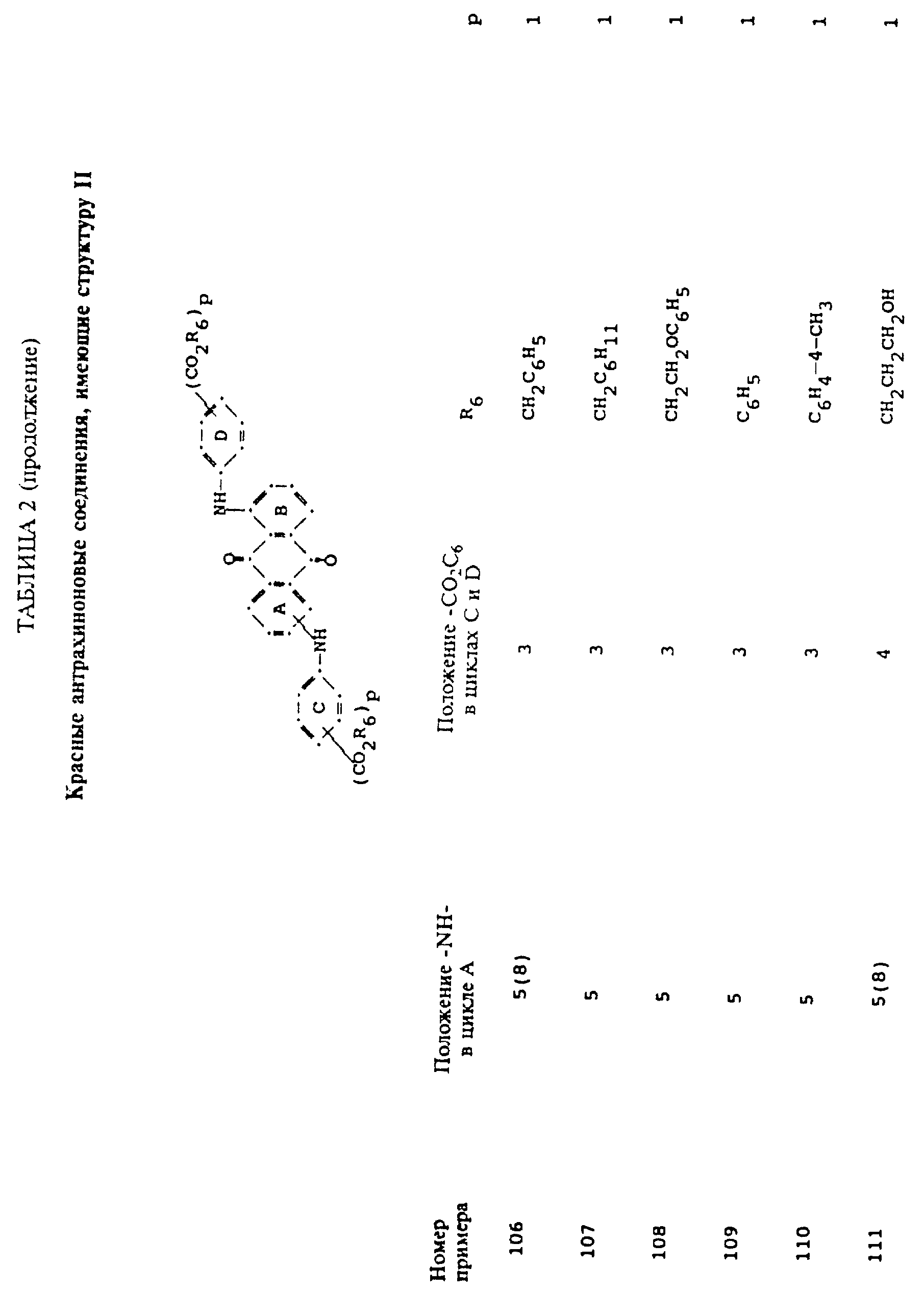
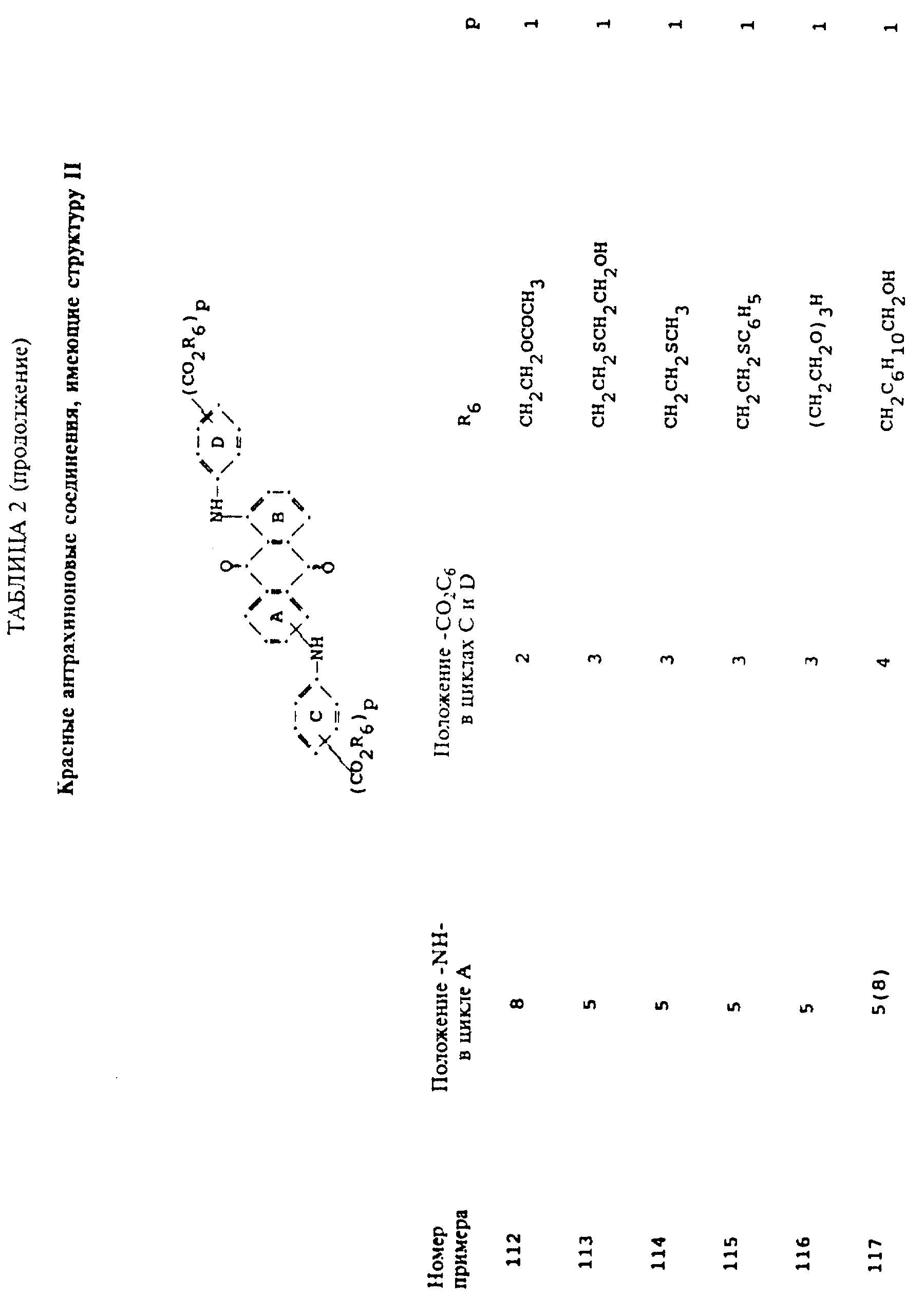
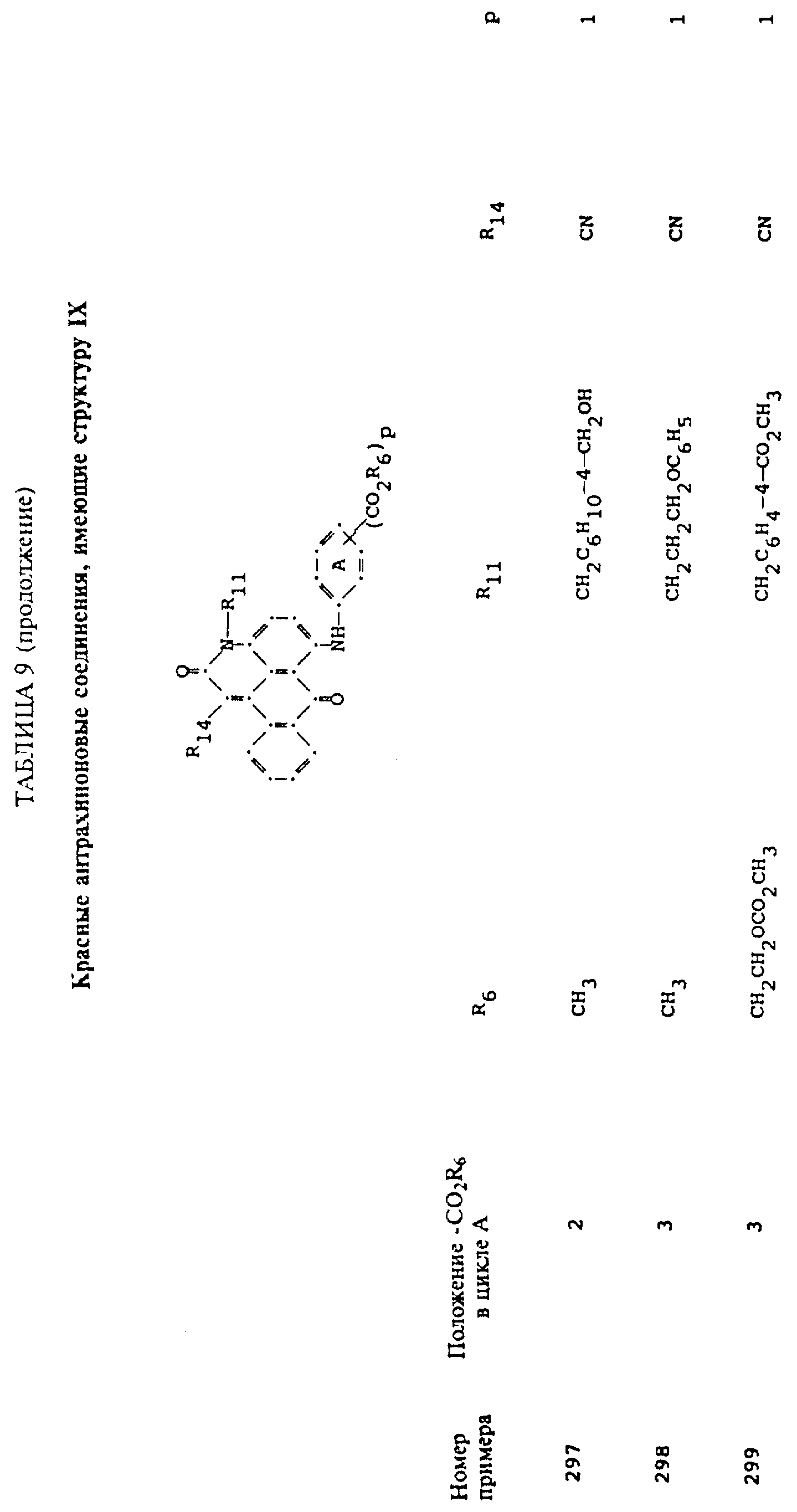
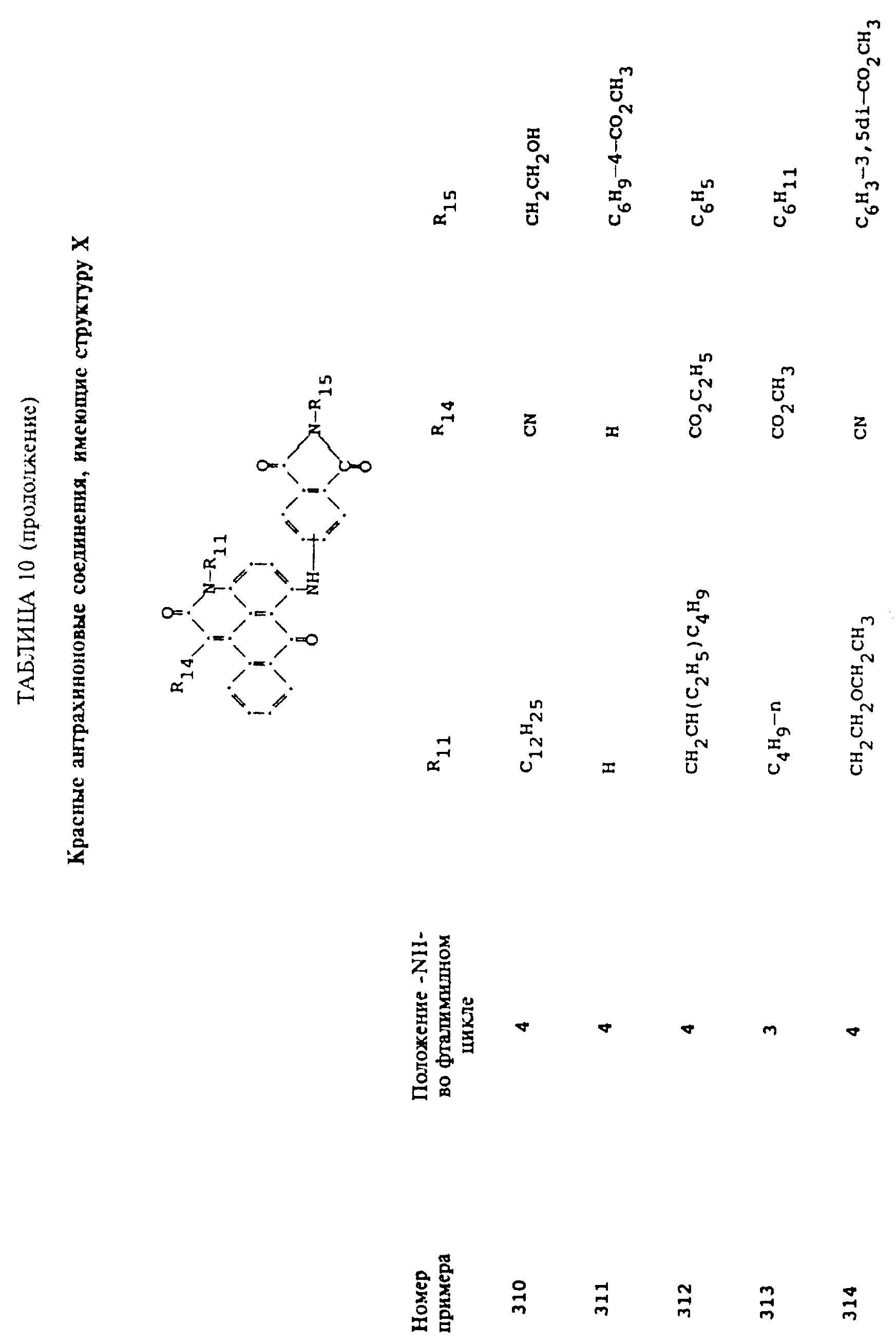
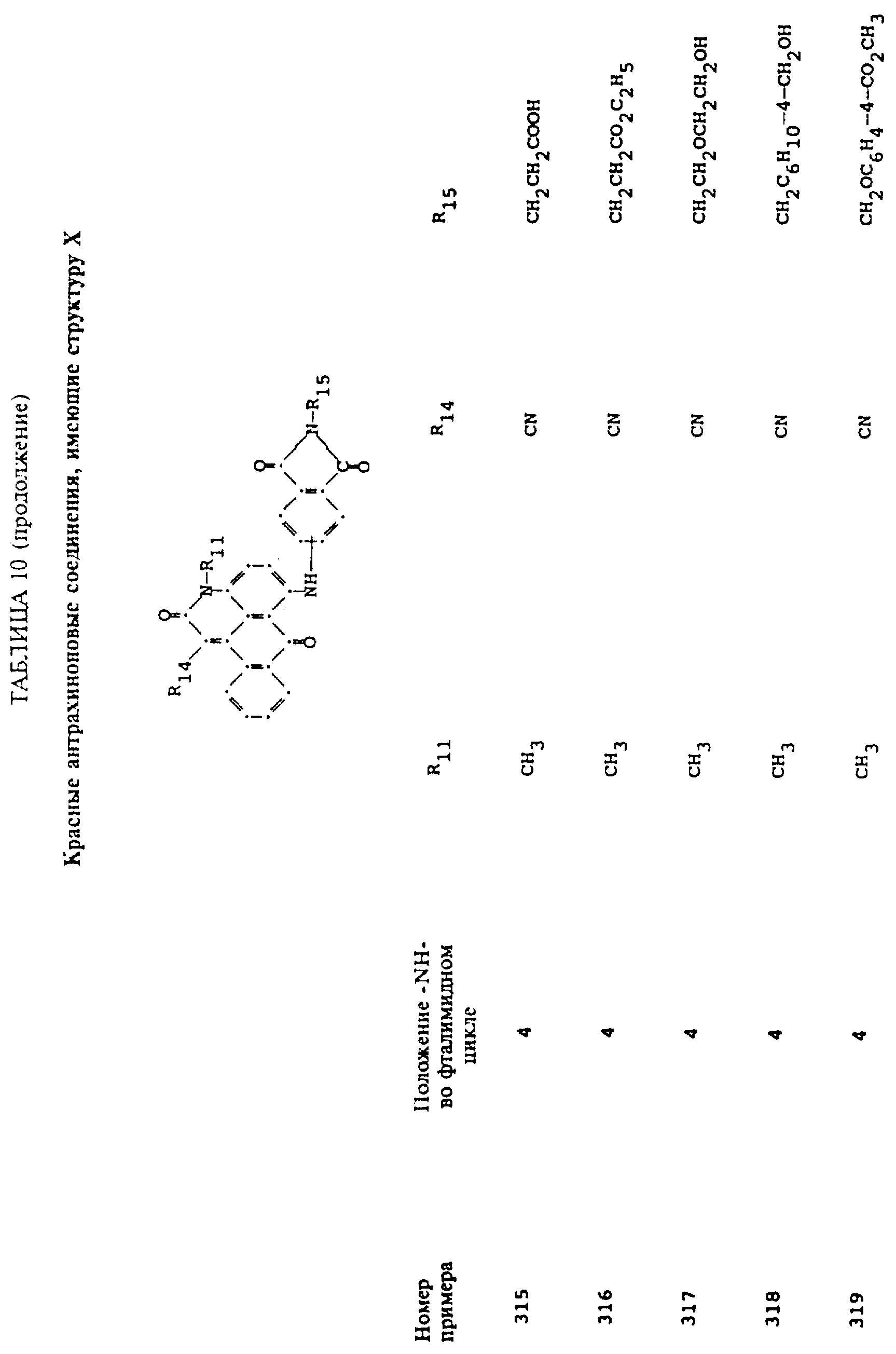
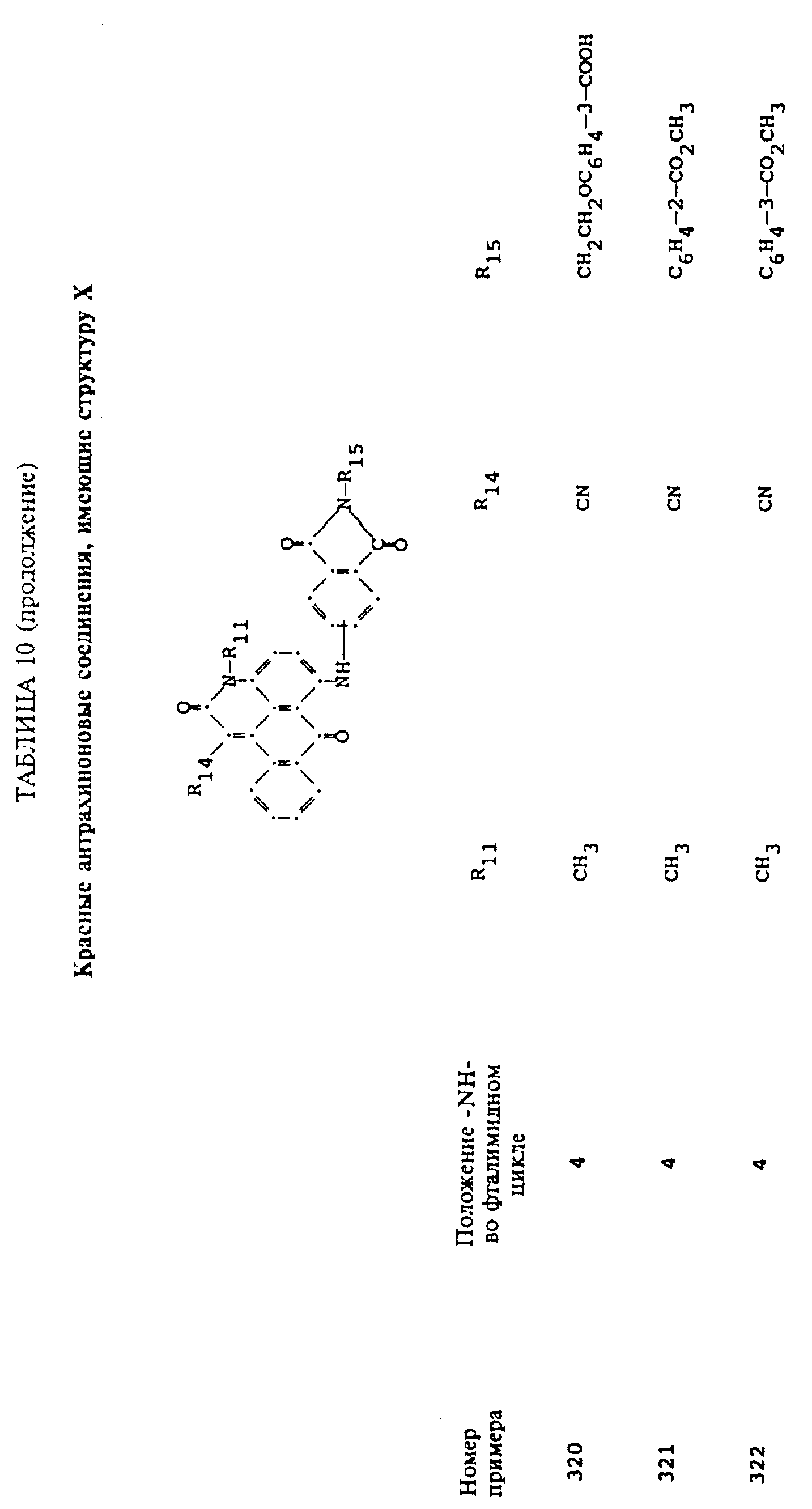
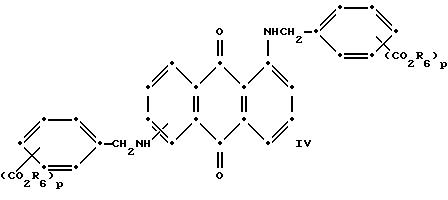
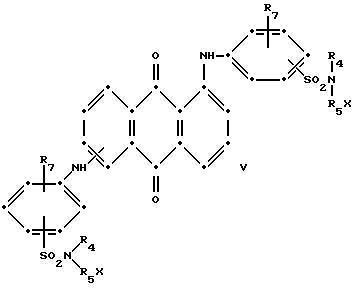
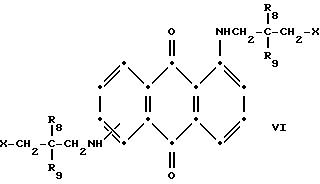
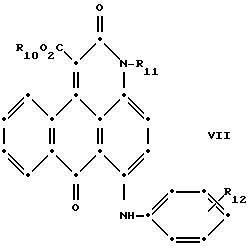
Красные компоненты, которые могут быть смешаны с синими компонентами приведенной выше
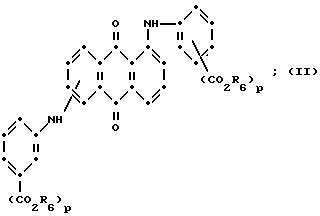
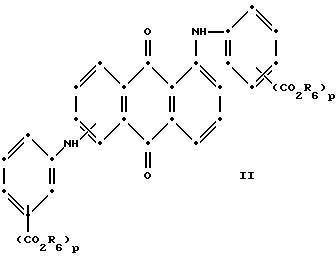
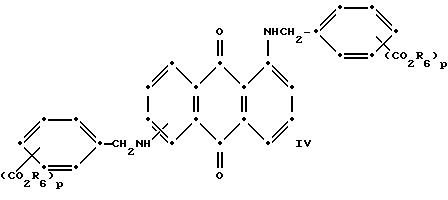
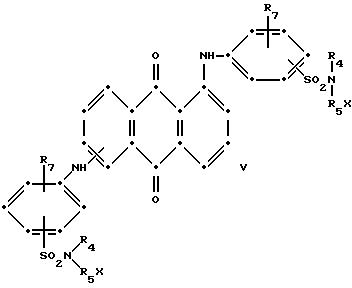
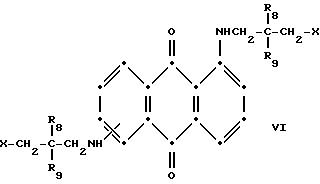
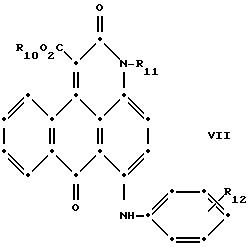
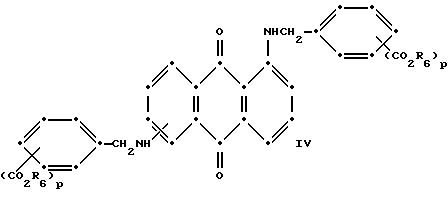
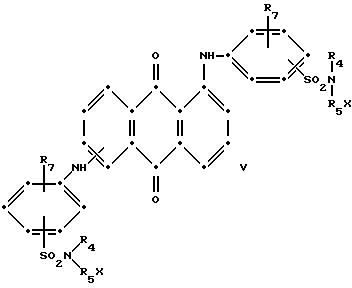
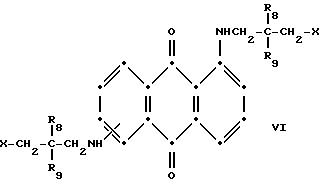
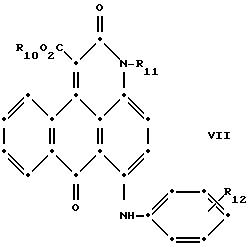
формулы (I), имеют следующие структурные формулы (II)-(X):
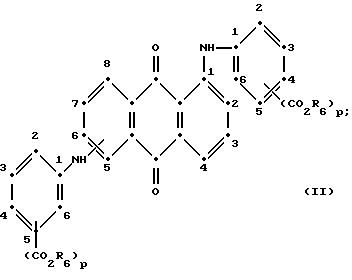
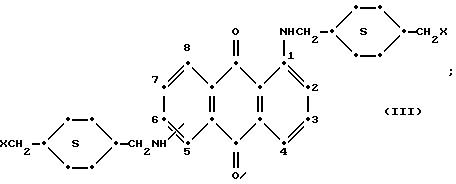
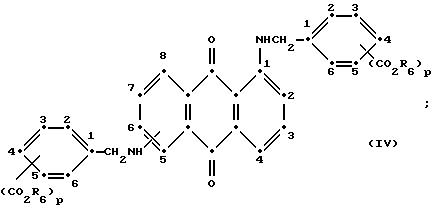
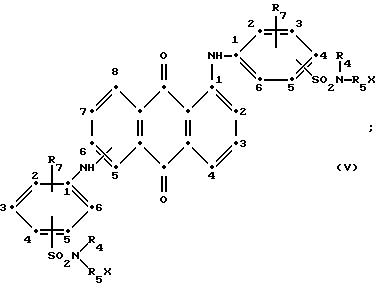
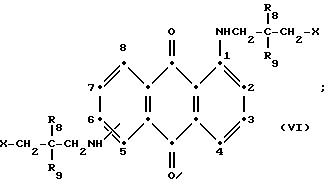
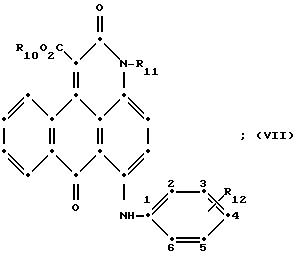
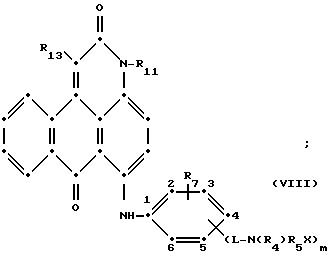
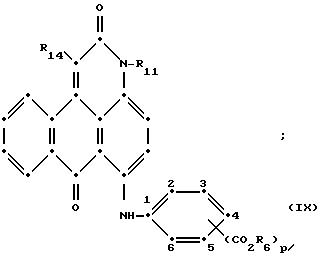
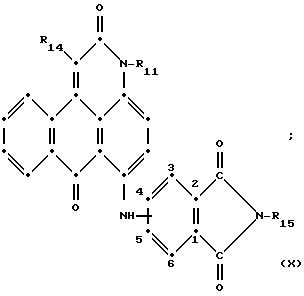
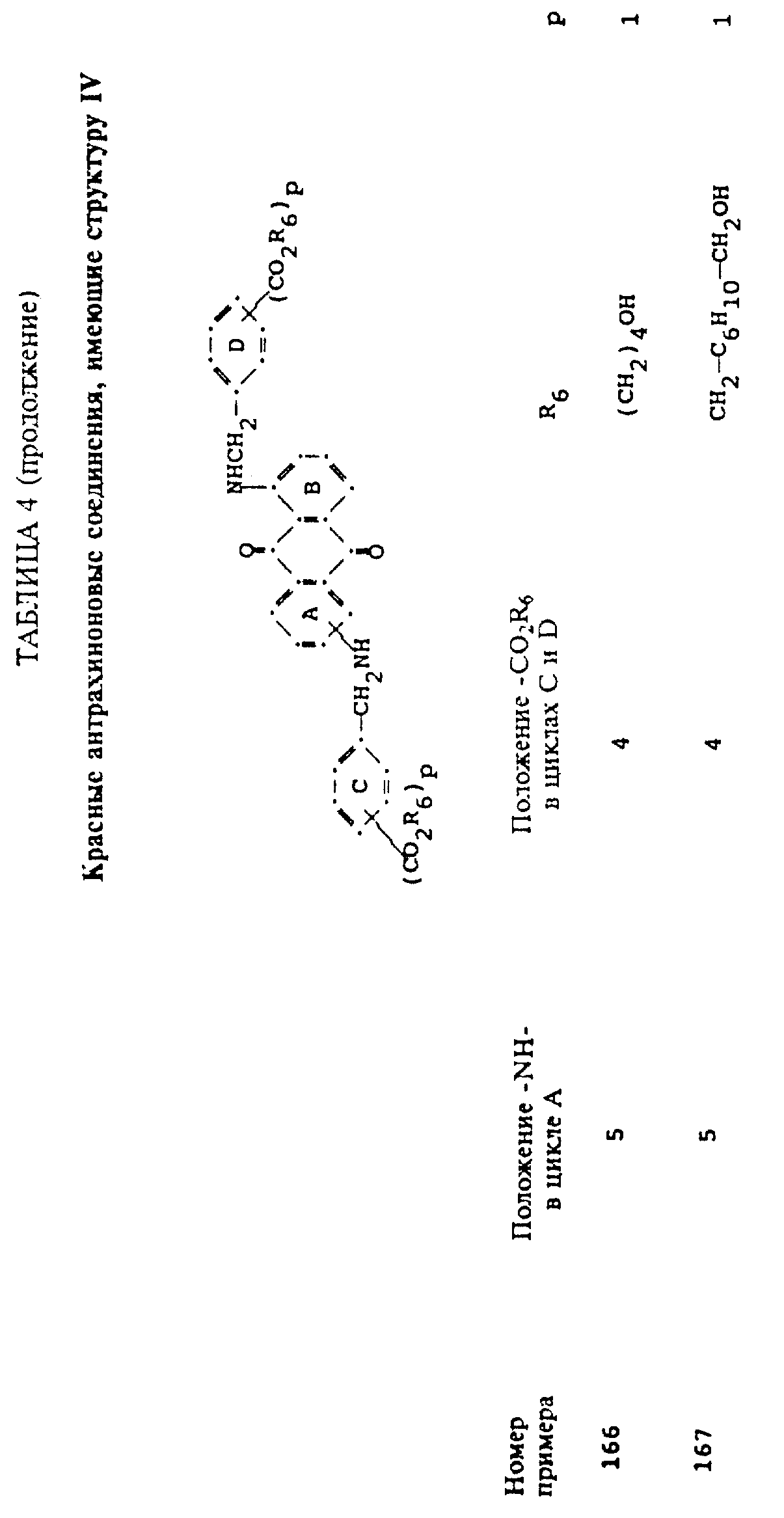
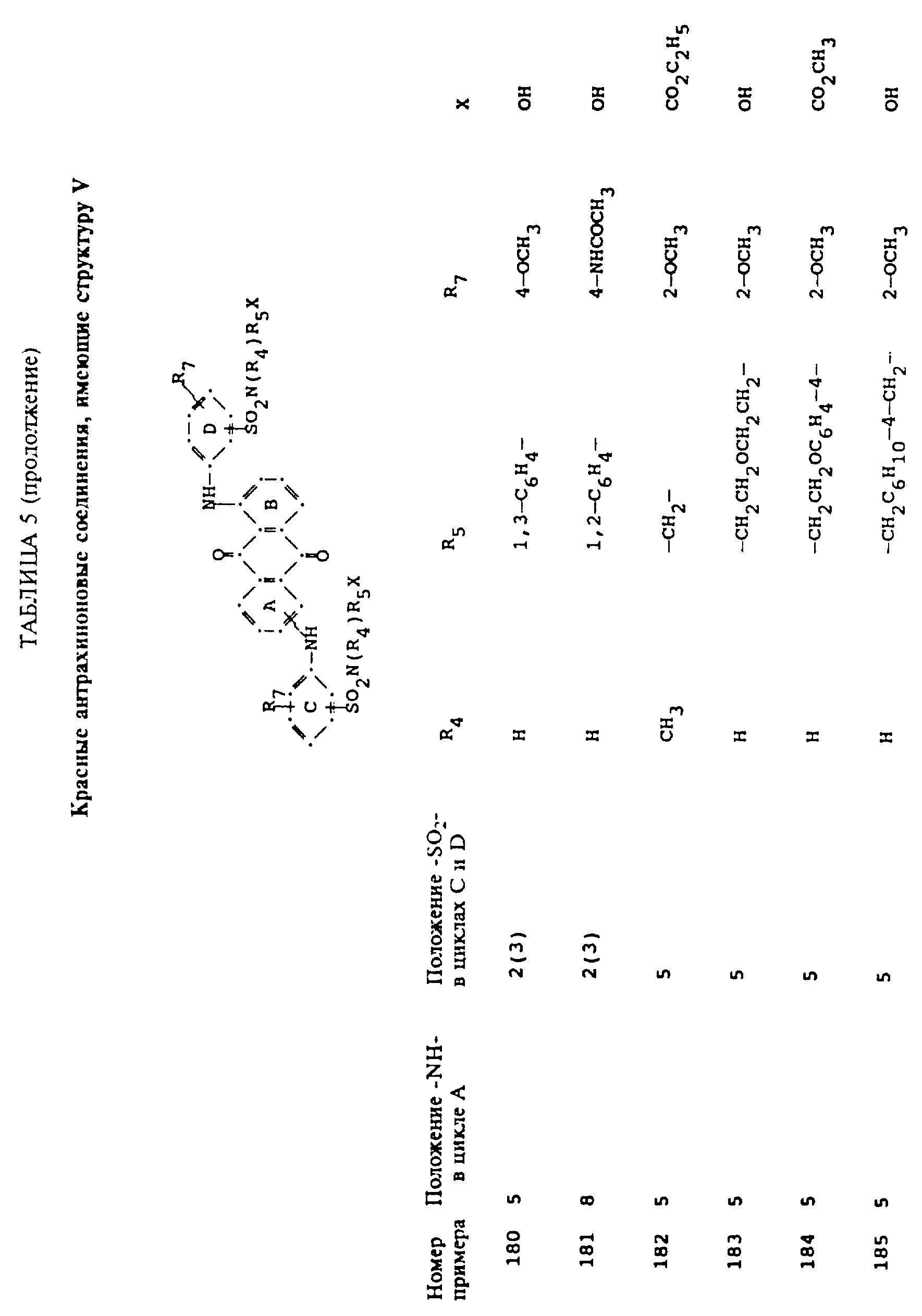
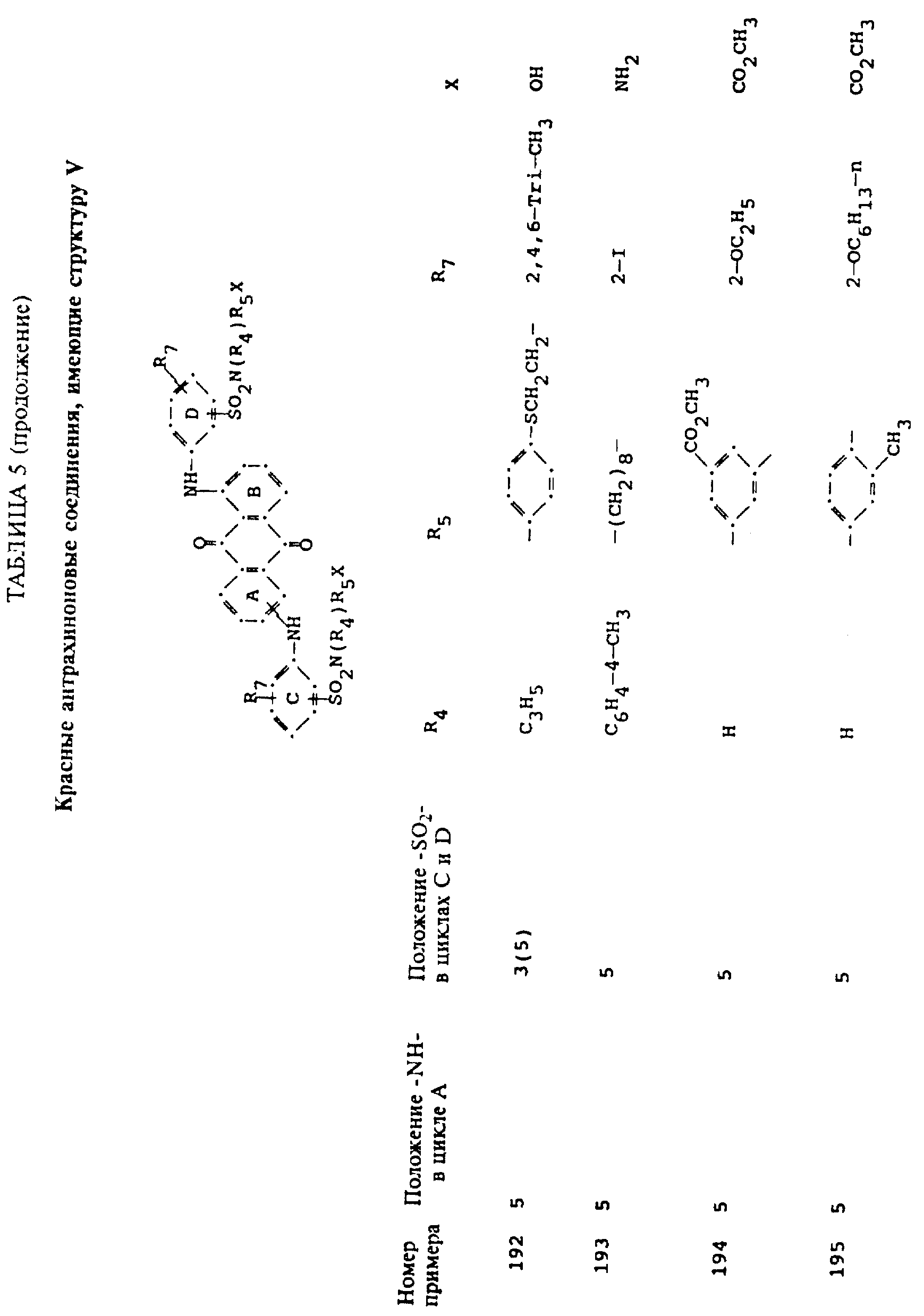
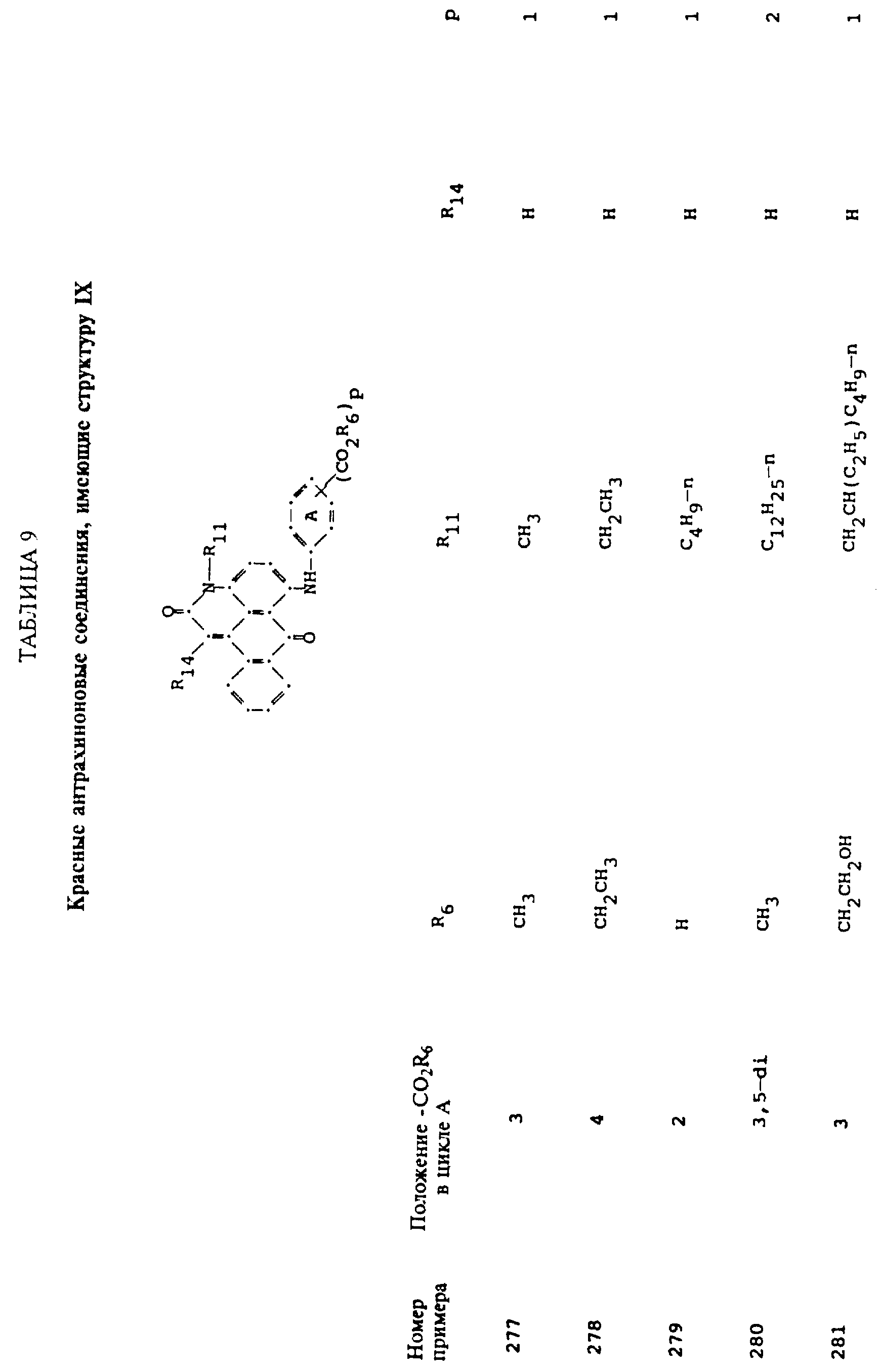
в которых R6 выбирают из группы, содержащей водород, C1-C6-алкил, замещенный C1-C6-алкил, C3-C7-циклоалкил или арил;
R7 представляет собой водород или от одной до трех групп, которые выбирают из C1-C6-алкила, замещенного C1-C6-алкила, C1-C6-алканоиламино, галогена, гидрокси, C1 -C6-алкил-C1-C6-алкокси, C1 -C6-алкилтио;
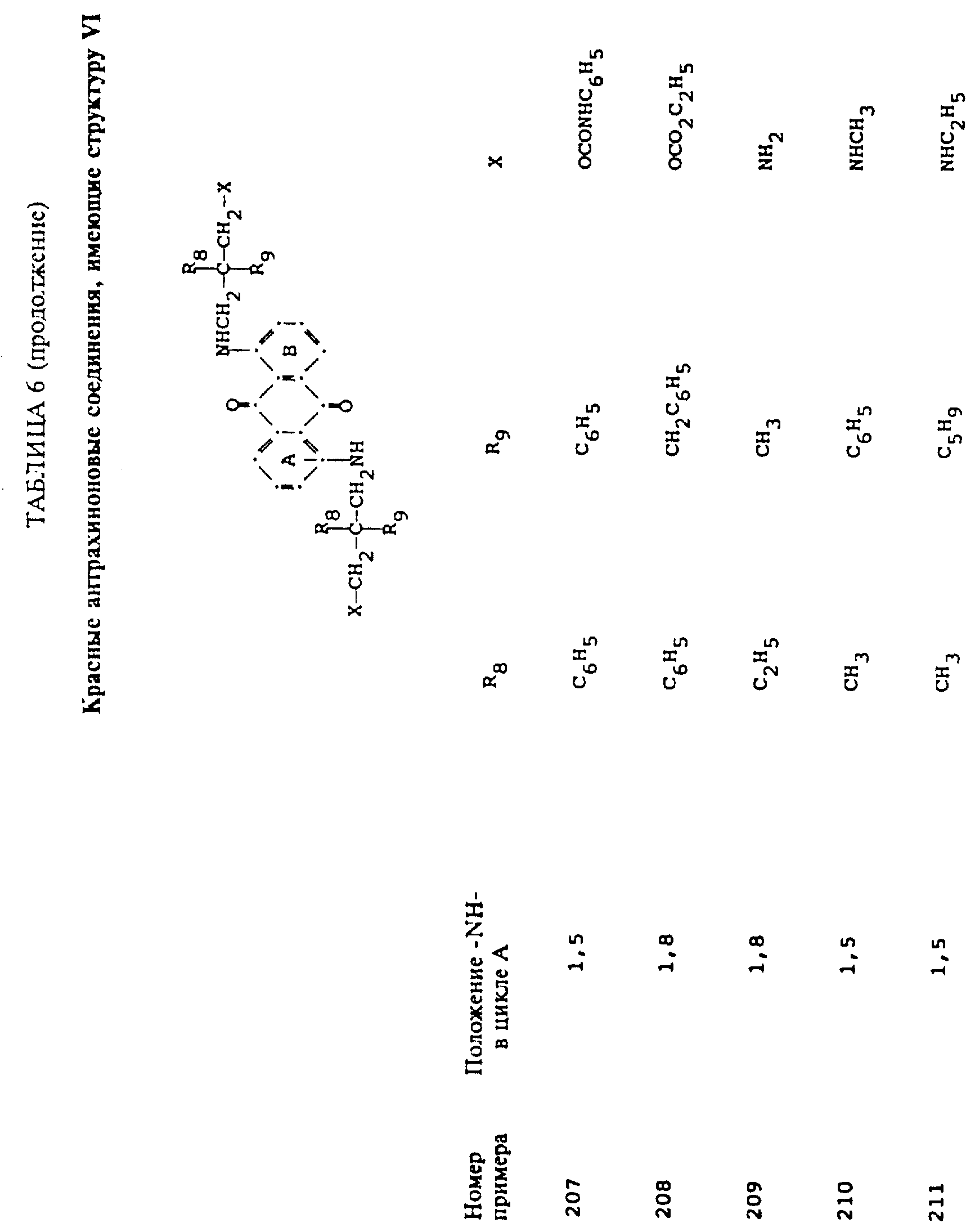
R8 и R9 одинаковые или разные, выбранные из группы, включающей C1-C6-алкил, замещенный C1-C6-алкил, C3-C7-циклоалкил или арил;
R10 выбирают из группы, включающей C1-C6-алкил, замещенный C1-C6-алкил, C3 -C7-циклоалкил или арил;
R11 выбирают из группы, содержащей водород, C1-C12-алкил, замещенный C1-C12-алкил, C3 -C7-циклоалкил или арил;
R12 представляет собой водород или от одной до трех групп, которые выбирают из C1-C6-алкила, замещенного C1 -C6-алкила, C1-C6-алкокси, замещенного C1-C6 -алкокси, C1-C6-алкилтио, замещенного C1-C6-алкилтио, галогена, гидрокси, C1-C6-алканоиламино, ароиламино, C1-C6 -алкил-сульфониламино и арилсульфониламино;
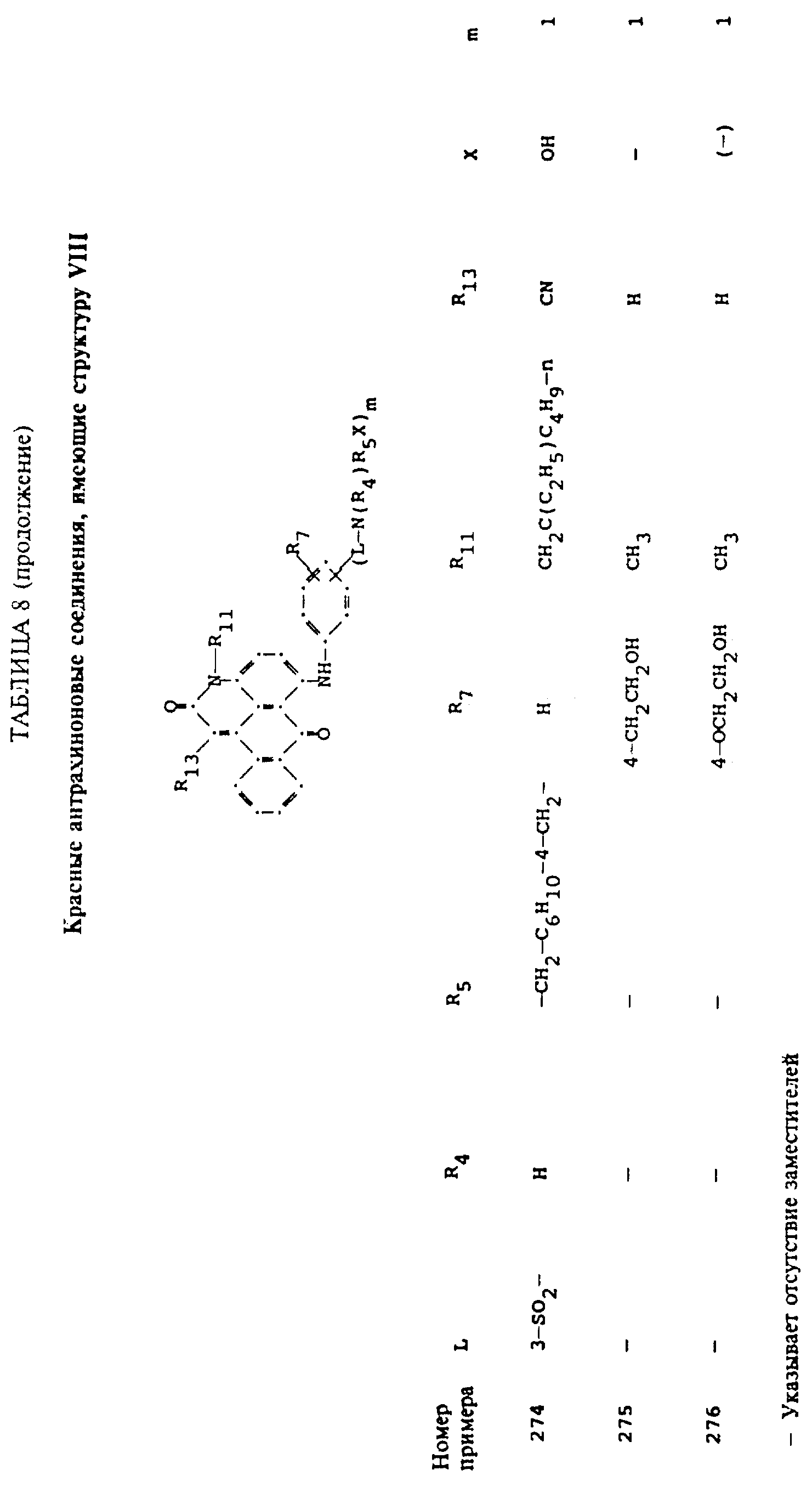
R13 и R14 выбирают из водорода, циано или CO2R10;
R15 представляет собой определенные ранее R4 или R5X;
L представляет собой -CO- или -SO2 -; X имеет значения, определенные выше; m=0 или 1; p=1 или 2 при условии, что R13 представляет собой водород, когда m= 0, и что присутствует по меньшей мере одна реактивная полиэфирная группа.
При преимущественном воплощении соединение(я) синего антрахинона соответствуют приведенной выше структурной формуле (I), в которой R представляет собой водород; R1 и R2 независимо друг от друга выбирают из метила и этила; R3 представляет собой водород, метил или бром; R4 представляет собой водород, C1-C4 -алкил или арил; R5 выбирают из группы, содержащей C1-C6-алкилен, C1-C4-алкилен-O-C1-C4-алкилен, -CH2C6H10CH2-, арилен или -CH2-алкилен-, а красный компонент соответствует формуле (V), в которой R7 представляет собой C1-C6 -алкокси, a R4 и R5 такие, как это определено в пункте 1 патентной формулы.
Предпочтительно, чтобы концентрации синего и красного соединения, взятые вместе, составляли от ~0.5 ppm до ~10 ppm. Более предпочтительно, чтобы общая концентрация синего компонента составляла от 1 до 7 ppm, а общая концентрация красного компонента была от 0.5 до 3 ppm.
Термин "полиэфирная реактивная группа" используется здесь для описания группы,
которая реагирует по меньшей мере с одной из функциональных групп, из которых получают сам полиэфир при
определенных условиях его образования. Примеры таких групп, которые могут представлять собой X,
включают гидрокси, карбокси, эфирную группу, амино, C1-C6-алкиламино и т.д.
Эфирным радикалом может быть любой радикал, имеющий формулы
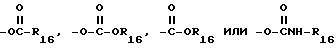
в которых R16 выбирают из групп, содержащих C1-C6-алкил, замещенный C1-C6-алкил, C3-C7-циклоалкил или арил. Реактивная группа X предпочтительно представляет собой гидрокси, карбокси, C1-C2-алкоксикарбонил или ацетокси.
Терминами "замещенный C1-C6-алкил", "замещенный C1-C12 -алкил", "замещенный C1-C6-алкокси", "замещенный C1-C6 -алкилтио", "замещенный C1-C6-алкилсульфонил", "C1-C6 -алкилсульфониламидо", "замещенный алкилен", "C1-C6-алкоксикарбонил" обозначены алкил- и алкилен-группы или части этих групп, они могут содержать в качестве заместителей одну или более групп, предпочтительно от 1 до 3 групп, выбранных из гидрокси, галоген, циано, арил, арилокси, арилтио, C1-C4-алкилтио, C3-C7-циклоалкил, C1-C4-алканоилокси и -(-O-R17-)p-R18, где R17 выбирают из группы, содержащей C1-C6-алкилен, C1-C6-алкиленарилен, циклогексилен, арилен, и C1-C6-алкиленциклогексилен; R18 выбирают из группы, содержащей водород, гидрокси, карбокси, C1-C4 -алканоилокси, C1-C4-алкоксикарбонил, арил, и C3-C7- циклоалкил; а индекс p=1, 2, 3 или 4.
Термином "арил", используемым здесь, предпочтительно обозначают фенил и его замещенные, содержащие 1-3 заместителя, выбранных из группы, которая включает C1-C6-алкил, C1-C6-алкокси, галоген, карбокси, циано, C1-C6-алкилтио, C1-C6-алкилсульфонил, трифторметил, гидрокси, C1-C6-алканоилокси, C1-C6 -алканоиламино и C1-C6-алкоксикарбонил.
Термин "арилен" включает 1,2- 1,3- и 1,4-фенилен и подобные ему радикалы, замещенные 1-3 раза C1-C6 -алкилом, C1-C6-алкокси, C1-C6-алкоксикарбонилом или галогеном.
Термины "C3-C8-алкенил" и "C3-C8 -алкинил" используют для обозначения алифатических углеводородных фрагментов с 3-8 атомами углерода и содержащими соответственно по меньшей мере одну двойную связь углерод-углерод и одну тройную связь углерод-углерод.
Термин "галоген" используют для обозначения брома, хлора, фтора и йода.
Термины "C1-C6-алканоилокси" и "C1-C6-алканоиламино" используют для обозначения радикалов, имеющих формулы
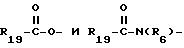
соответственно, где R19 представляет собой прямую или разветвленную цепь радикала C1-C6-алкил, а R6 имеет значения, указанные выше.
Таким образом, настоящее изобретение обеспечивает отформованный полиэфир или фибриллы полиэфира, подвергнутые сополимеризации в таком количестве соединения синего 1,4-бис(2,6-диалкиламино)антрахинона формулы (I) в сочетании с красным антрахиноном или соединением антрапиридона, имеющих приведенные выше формулы (II)-(X), которого достаточно для увеличения белизны, присущей этому полиэфиру. В связи с этим указанные синие и красные соединения не должны присутствовать в количествах, достаточных для приобретения этим полиэфиром интенсивной собственной окраски.
Следующим аспектом настоящего изобретения является обеспечение методом для придания белизны полиэфиру, который в нормальном состоянии имеет желтую окраску; этот метод включает сополимеризацию в указанном полиэфире по меньшей мере одного соединения синего 1,4-бис(2,6-диалкиланилино)антрахинона формулы (I) с красным антрахиноном или соединением антрапиридона, имеющих формулы (II)-(X).
Как отмечено выше, тонирующие смеси настоящего изобретения могут быть добавлены до полимеризации или во время ее. Следовательно, следующим аспектом настоящего изобретения является обеспечение состава премикса, содержащего по меньшей мере одно соединение синего 1,4-бис(2,6-диалкиламино)антрахинона формулы (I) с красным антрахиноном или соединения антрапиридона, имеющих приведенные выше формулы (II)-(X). Этот состав премикса может представлять собой неразбавленную смесь указанных красного и синего соединений, она может быть предварительно растворена в одной из смесей полиэфирных мономеров, например в этиленгликоле.
Очевидно, что общее количество добавляемых тонирующих компонентов зависит от интенсивности желтой окраски, присущей конкретному полиэфиру. Обычно требуемая максимальная концентрация смешанных тонирующих компонентов составляет ~ 10 ppm, а минимальная - ~0.5 ppm; при этом предпочтительно использовать эти компоненты с 1-7 ppm синего компонента формулы (I) в сочетании с ~0.5-3 ppm красного компонента формул (II-X).
В предпочтительном воплощении настоящего изобретения синее антрахиноновое соединение соответствует структуре приведенной выше формулы (I), в которой R представляет собой водород; R1 и R2 независимо друг от друга выбирают из метила и этила; R3 представляет собой водород, метил или бром; R4 представляет собой водород, C1-C4-алкил или арил; R5 выбирают из группы, содержащей C1-C6-алкилен, C1-C4-алкилен-O-C1-C4-алкилен, -CH2C6H10CH2-, арилен, или -CH2 -арилен-. При этом красный компонент соответствует формуле (V), в которой R7 представляет собой C1-C6-алкокси, a R4 и R5 имеют те значения, которые были определены выше дли синего компонента формулы (I).
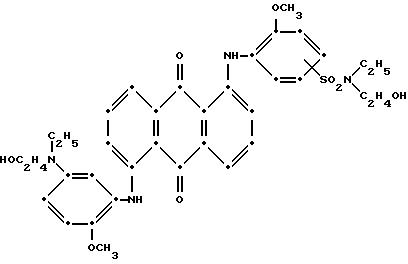
В
особо предпочтительном воплощении настоящего изобретения синий компонент формулы (I) представляет собой
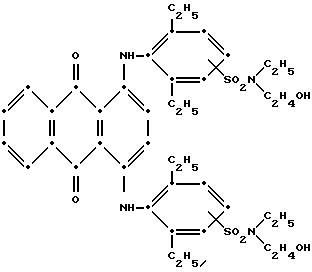
а красное соединение формулы (V) представляет собой
Обычно синие антрахиноны формулы (I) могут быть получены взаимодействием соединения лейкохинизарина (1,4,9,10- тетрагидроксиантрацена) с избытком ароматических аминов, предпочтительно в присутствии кислых катализаторов, таких как борная кислота (как это описано в патенте США N 3918976) согласно приведенной ниже схеме:
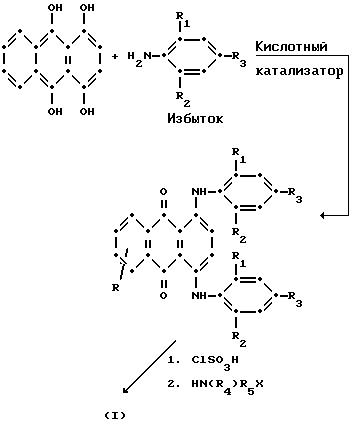
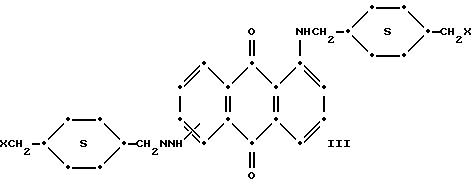
Полученные таким образом 1,4-бис(2,6-диалкиламино)- антрахиноны при необходимости легко взаимодействуют сначала по реакции хлорсульфонирования с хлорсульфоновой кислотой, давая при этом дисульфонил хлориды, которые могут реагировать с аминами, содержащими реактивные полиэфирные группы; основы этого метода раскрываются в патенте США N 2731476, на который ссылаются в этой работе.
Типичные амины, соответствующие формуле HN(R4)R5X, включают 2-аминоэтанол, 2, 2-иминодиэтанол, 1-амино-2,3-пропандиол, 2-метиламиноэтанол, 2-этиламиноэтанол, 2-анилиноэтанол, метилантранилат, метил-м-аминобензоат, п-аминобензойную кислоту, м-аминофенол, 6-амино-гексановую кислоту, β-/ аланин, глицинэтиловый эфир, 2-(п-аминофенил)этанол, 2-(п- аминофенокси)этанол, 4-аминометилциклогексан метанол и 3-амино-2,2-диметил-1-пропанол.
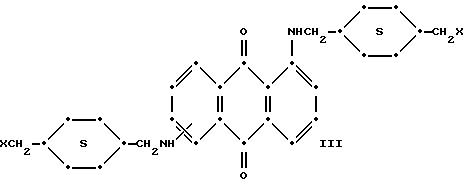
Красные соединения (II) могут быть получены при взаимодействии 1,5-дихлорантрахинона и/или 1,8-дихлорантрахинона или их смеси с о-, м- и п-аминобензойной кислотой (или их сложными эфирами) согласно модифицированной реакции Ульмана, включающей арилирование азота анилина в присутствии медных катализаторов (см. патент США N 4359580, на который в настоящей работе имеется ссылка).
Красные соединения формулы (III) могут быть получены так, как это описано в патенте США N 4420581, а соединения формулы (VI) можно получить, как в патенте США N 4999418, на который ссылаются в этой работе.
Красные антрахиноновые соединения формулы (IV) можно получить путем взаимодействия 1,5-дихлорантрахинона с 1,8-дихлорантрахинона или их смеси с подходящими замещенными бензиламинами по методике, аналогичной той, которая используется при получении соединений формулы (III) и (IV).
Красные антрапиридоновые соединения (VII) могут быть получены так, как это раскрывается в патенте США N 4790581, включенном в ссылки к настоящей работе; методики, пригодные для получения красно- фиолетовых антрапиридоновых соединений (VIII) и (IX) раскрыты в патенте США N 4745174, на который имеется ссылка в настоящей работе.
Полиэфиры, упоминаемые в этом патенте, представляют собой линейные, термопластичные, кристаллические или аморфные полиэфиры, полученные согласно обычной методике полимеризации из одного или более диолов и одной или более дикарбоновых кислот. Нормальные полиэфиры представляют собой литые изделия или фибриллы с вязкостью от ~0.4 до ~1.2 дл/г, измеренной при 25oC в смеси, содержащей фенол и тетрахлорэтан при их весовом соотношении 60/40.
Для описываемых полиэфиров подходящие диолы могут быть выбраны из этиленгликоля, 1,4-циклогександиметанола, 1,2- пропандиола, 1, 3-пропандиола, 1,4-бутандиола, 2,2-диметил-1,3- пропандиола, 1,6-гександиола, 1,2-циклогександиола, 1,4- циклогександиола, 1, 2-циклогександиметанола, 1,3-циклогександиметанола, Z, 8-бис(гидроксиметил)-трицикло-[5.2.1.0]- декана, где Z представляет собой 3, 4, или 5; и диолов, содержащих один или более атомов кислорода в цепи, например, диэтиленгликоль, триэтиленгликоль, дипропиленгликоль, трипропиленгликоль и им подобные. Обычно эти диолы содержат от 2 до 18 атомов углерода, предпочтительно от 2 до 8, могут быть циклоалифатические диолы в цис- или трансконфигурации или в виде смеси обоих этих форм.
Подходящие кислотные компоненты (алифатические, алициклические или ароматические дикарбоновые кислоты) линейных сложных полиэфиров выбирают, например, из терефталевой кислоты, изофталевой кислоты, 1,4-циклогександикарбоновой кислоты, 1,3- циклогександикарбоновой кислоты, янтарной кислоты, глутаровой кислоты, адипиновой кислоты, себациновой кислоты, 1,12-додекановой кислоты, 2,6-нафталиндикарбоновой кислоты и им подобных. При получении полимеров часто предпочтительно использовать функциональные производные этих кислот, такие как диметиловый, диэтиловый или дипропиловый эфир этой дикарбоновой кислоты. Там, где это практически выполнимо, могут быть использованы ангидриды или хлорангидриды этих кислот.
Предпочтительные полиэфиры содержат по меньшей мере ~50 мол.% остатков терефталевой кислоты и по меньшей мере ~50 мол.% этиленгликоля и/или остатков 1, 4- циклогександиметанола и от ~ 2 до ~8 мол.% остатков соединения формулы (I). Особенно предпочтительны полиэфиры, содержащие от ~75 до 100 мол. % остатков терефталевой кислоты и от ~75 до 100 мол.% остатков этиленгликоля.
Линейные полиэфиры могут быть получены согласно хорошо известным условиям образования полиэфира. Например, смесь одной или более дикарбоновых кислот, предпочтительно ароматических дикарбоновых кислот, и одного или более диодов можно нагревать в присутствии катализаторов этерификации или полиэтерификации при температурном диапазоне от ~150 до ~300oC и при давлении от атмосферного до величины ~ 0.2 мм Hg. Обычно дикарбоновая кислота или ее производное этерифицируют или переэтерифицируют диолом (диолами) при атмосферном давлении и при температуре нижней границы особо оговоренного диапазона. Затем проводят поликонденсацию при увеличении температуры и снижении давления, удаляя при этом избыток диола из этой смеси.
Хорошо известны типичные катализаторы или каталитические системы для конденсации полиэфиров. Например, катализаторы, раскрываемые в патентах США N 4025495; 4136089; 4176224; 4238593 и 408527, на которые имеются ссылки в настоящей работе, представляются подходящими для этой задачи. Далее в работе R. E. Wilfong, Journal of Polymer Sciencee, 54, 385 (1961) описаны типичные катализаторы, которые эффективны в реакциях полиэфирной конденсации.
Для полиэфирной конденсации предпочтителен температурный режим от ~260 до ~300oC.
Приведенная далее детальная методика должна проиллюстрировать получение тонирующих компонентов, а примеры из таблиц 1-10 дают дополнительное описание тех синих и красных соединений, которые используются при практическом воплощении настоящего изобретения.
ЭКСПЕРИМЕНТАЛЬНЫЙ
РАЗДЕЛ
Хлорсульфонильное соединение из примера 1 и других примеров может быть также получено согласно процессу, описанному
в совместно поданной заявке США N 08/210785 от 18 марта 1994 г.
Согласно этому процессу реакционную смесь выливают в C3-C6-алифатический кетон или смесь C3-C6-алифатических кетонов при перемешивании и охлаждении;
твердый арилсульфонил галогенид при этом выпадает в осадок. В качестве C3-C6 алифатического кетона предпочтительным
является ацетон. Может быть также использован изопропанол или
смесь изопропанола с ацетоном при их объемном соотношении, предпочтительно составляющем ~80/20. В таком процессе наблюдается меньший
разогрев реакции, что делает выливание реакционной смеси более
безопасным и контролируемым. Более того, при этом из выливаемой смеси выделяется меньше газообразного хлористого водорода, и обычно
получают арилсульфонилхлориды более высокой степени чистоты.
Пример 1
К хлорсульфоновой кислоте (250 мл) порциями при перемешивании добавляли 1,
4-бис(2-этил-6-метиланилино)антрахинон (50.0 г, 0.11 мол) с такой скоростью, чтобы температура
выросла от 25oC до ~45oC. Раствор, полученный при этом, перемешивали в течение 2
часов без нагревания, а затем его добавили тонкой струей в смесь льда с водой (3.0 л) с
энергичным перемешиванием при 0-5oC. Полученный дисульфонил хлорид собрали при
фильтровании, промыли
водой, отжали для удаления большей части воды из него, а затем добавили к
тетрагидрофурану (1.0 л) и перемешивали до растворения. Этаноламин (30.0 г, 0.5 молей) растворили в тетрагидрофуране (50.0 мл),
и раствор, полученный таким образом, добавили по каплям при
перемешивании к раствору дисульфонил хлорида. После перемешивания в течение ~12 часов при комнатной температуре тетрагидрофуран удалили под
давлением ~100 дюймов. Затем остаток растворили в уксусной
кислоте (500 мл), а раствор медленно вылили в воду (3.5 л) при перемешивании. Синий продукт собрали при фильтровании, промыли горячей водой и
высушили (выход составил 77.5 г. 97.7% от теоретически
возможного). Масс-спектрометрией была подтверждена следующая структура:
Максимумы поглощения в видимой области спектра поглощения наблюдались при 578 нм (ε-15,694) и 621 нм (ε-16,817) в N,N-димeтилфopмaмидe, который применяли в качестве растворителя.
Пример 2
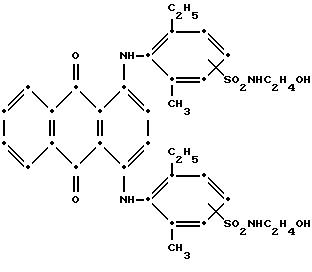
Дисульфонил хлорид антрахинона, полученный при хлорсульфировании 1,4-бис(2-этил-6-метиланилино) антрахинона (0.051 молей) так, как
это описано в примере 1, добавили к суспензии
4- карбометоксибензиламин-HCl (22.3 г, 0.111 молей) в метиленхлориде (250 мл), к которому был прибавлен триэтиламин (26.3 г, 0.260 молей). После того, как
реакционную смесь перемешали при комнатной
температуре, добавили активированный уголь (5.0 г) и сульфат магнезии (5.0 г), полученную смесь отфильтровали для удаления твердых частиц. Растворитель
удаляли из фильтрата под вакуумом, а остаток
суспендировали в ~500 мл воды. Количественно был получен продукт, структура которого, как доказано масс-спектрометрией, следующая:
Максимумы поглощения видимой области спектра поглощения наблюдались при 597 нм (ε-11,971) и 622 нм (ε-15,126) в метиленхлориде.
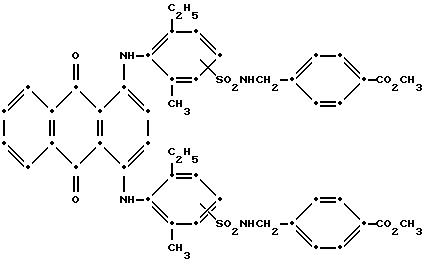
Пример 3
Дисульфонил хлорид антрахинона, полученный при хлорсульфировании 1,4-бис(2-этил-6-метиланилино) антрахинона (0.037 молей) так, как
это описано в примере 1, вступил во взаимодействие
с этилглицинатом•HCl (10.78 г, 0.077 молей), в точности так, как это описано в примере 2, с тем, чтобы получить 29.5 г (99% от теоретического
выхода) приведенного ниже синего продукта:
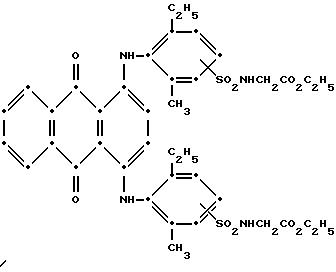
Максимумы поглощения в видимой области спектра поглощения наблюдались при 577 нм (ε-14,713) и 621 нм (ε-15,517) в метиленхлориде.
Пример 4
К о-дихлорбензолу (100 мл) добавили при перемешивании 1,
4- бис(2-этил-6-метиланилино)антрахинон (9.48 г, 0.02 моля). После
полного растворения по каплям при 25-30oC добавляли бром (7.04 г, 0.044 моля), растворенный в о-дихлорбензоле (20 мл).
Когда прикапывание было закончено, реакционную смесь нагревали в
течение 1.0 часа при 25-30oC с последующим охлаждением ее до комнатной температуры, после чего эту смесь вылили в метанол
(500 мл). Полученный продукт перекристаллизовали, собрали при
фильтровании, промыли метанолом и высушили на воздухе. Выход составил 9.44 г (74.7% от теоретического), полученное дибромированное
вещество имело следующую структуру, подтвержденную
масс-спектрометрией:
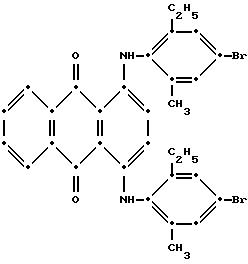
Максимумы поглощения в видимой области спектра поглощения наблюдались при 582 нм (ε-16,953) и 625 нм (ε-16,953) в метиленхлориде.
Пример 5
К хлорсульфоновой кислоте (35.0 мл)
добавляли порциями при энергичном перемешивании при
температуре ниже 35oC 1,4-бис(4- бром-2-этил-6-метиланилино)антрахинон (6.32 г, 0.01 моля), полученный в примере 4. Хлорсульфирование
было завершено при нагревании и перемешивании
реакционного раствора в течение 1.0 часа при температуре ~90-95oC, затем его постепенно при помешивании вылили в ~500 мл смеси льда с водой.
Синий дисульфонил хлорид собрали при
фильтровании и отжали до сухости. Затем полученный продукт добавили к этаноламину (50.0 мл) и реакционную смесь перемешивали в течение 0.5 часа при 90-95oC, а затем вылили в воду (300 мл).
После подкисления до pH ниже 4 эту смесь отфильтровали, а полученное твердое вещество промыли водой и высушили на воздухе. Тонкослойная хроматография выявила,
помимо целевого продукта, некоторое
количество сильно полярного вещества, которое, как полагают, представляет собой моно- или дисульфоновую кислоту или их смесь. Очистку проводили растворением части
(1.0 г) сырого продукта в
метиленхлориде (40 мл) с последующей хроматографией в стеклянной воронке через MgO3Si (FLORISILL) с использованием метиленхлорида для удаления небольших количеств
синего материала,
имеющего высокое значение Rf. Затем продукт элюируют, используя тетрагидрофуран. Потом растворитель удаляют из элюата, чтобы получить маслянистый остаток. Последний
растворяют в уксусной
кислоте (10 мл), а полученный раствор затем выливают в воду. Синий продукт собирают при фильтровании, промывают водой и высушивают на воздухе (выход 0.6 г). Масс-спектрометрия и
тонкослойная
хроматография показали, что полученный продукт в основном представляет собой целевое соединение формулы:
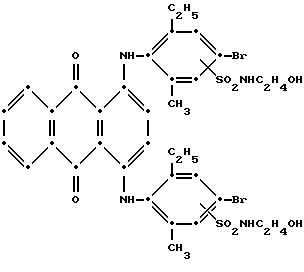
Незначительное количество моносульфонамидного производного также присутствовало. Максимумы поглощения в видимой области спектра поглощения наблюдались при 576 нм (ε-16,519) и 619 нм (ε-17,222)/ в метиленхлориде.
Пример 6
К раствору 1,4-бис(2-этил-6-метиланилино)антрахинона (3.0 r, 0.00632 моля) и п-толуолсульфоновой
кислоты (0.05 г) в N,
N- диметилформамиде (10 мл) по каплям при перемешивании добавили трифторуксусный ангидрид, допуская подъем температуры. Реакционная смесь в течение 15 минут разогревалась до ~
50oC, в
течение этого времени цвет изменился с голубого на красный, что указывает на прошедшее ацилирование. Реакционную смесь вылили в воду (250 мл), а красный продукт собрали при
фильтровании, промыли
водой и высушили на воздухе (выход - 3.50 г, 97.2% от теоретического). Масс-спектрометрией было подтверждено наличие моноацилированного продукта следующей формулы:
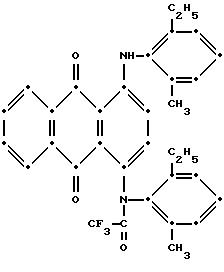
Максимумы поглощения в видимой области спектра поглощения наблюдались при 522 нм (ε-7,079) в метиленхлориде.
Пример 7
Часть продукта (1.5 г, 0.00263 моля), полученного в примере 6, добавляли порциями к хлорсульфоновой кислоте (15 мл) при комнатной температуре. После
перемешивания в
течение ~ 30 минут при комнатной температуре реакционную смесь вылили на смесь льда с водой при помешивании. Полученный желто- красный продукт собрали при фильтровании, промыли водой,
отжали для
удаления большей части воды, а затем добавили к этаноламину (20 мл). После нагревания в течение 1.0 часа до температуры ~90-95oC при перемешивании, для того чтобы получить
сульфонамидный
продукт, удаление трифторуксусных групп с помощью гидролиза было завершено обработкой реакционной смеси 2-этоксиэтанолом (15.0 мл), водой (5.0 мл) и 0.5 мл 50% раствора NaOH, и
нагреванием с
перемешиванием в течение ~0.5 часа. В течение этого времени цвет реакционной смеси сменился с красного до синего. Полученную в результате реакционную смесь вылили на воду (200 мл), а
синий продукт
собрали при фильтровании, промыли водой и высушили на воздухе (выход 1.27 г). Масс-спектрометрией было показано, что целевой моносульфонамидный продукт имеет следующую структуру:
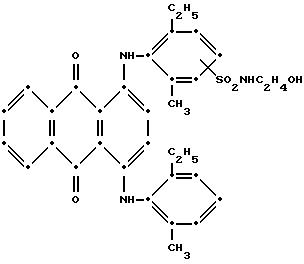
Максимумы поглощения в видимой области спектра поглощения наблюдались при 582 нм (ε-14,706) и 627 нм (ε-15,902) в метиленхлориде.
Пример 8
Дисульфонил хлорид антрахинона, полученный хлорсульфированием 1,4-бис(2-этил-6-метиланилина)антрахинона (0.01 моля)
добавляли
порциями к раствору 2-метиламиноэтанола (1.80 г, 0.024 моля) в метиленхлориде (70.0 мл), содержащему триэтиламин (2.53 г, 0.025 моля), допуская спонтанный подъем температуры до ~27oC.
Тонкослойная хроматография указала на завершение реакции после того, как реакционную смесь перемешивали в течение ~30 минут при комнатной температуре. Органический слой реакционной смеси
дважды
экстрагировали 250 мл воды, а водный слой удалили. Метиленхлорид удалили при пониженном давлении; был получен маслянистый слой, который затвердел при обработке н-гексаном. Собранный при
фильтровании,
промытый н-гексаном и высушенный на воздухе продукт темно-синего цвета весил 6.32 г (84.5% от теоретического выхода). Масс-спектрометрией была подтверждена следующая структура:
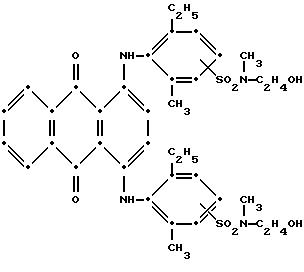
Максимумы поглощения в видимой области спектра поглощения наблюдались при 578 нм (ε-14,511) и 621 нм (ε-15,334)/ в метиленхлориде.
Пример 9
Дисульфонил хлорид антрахинона, полученный хлорсульфированием 1,4-бис(2-этил-6-метиламино)антрахинона (0.0025 моля)
добавили
к перемешиваемому раствору 2-этиламиноэтанола (0.54 г, 0.006 молей) и триэтиламина (0.64 г, 0.0065 моля) в метиленхлориде (15.0 мл) при перемешивании при комнатной температуре. Наблюдался
незначительный разогрев, и реакционную смесь перемешивали в течение ~30 минут. Тонкослойная хроматография (тетрагидрофуран: циклогексан 1: 1) показала завершение реакции. Синий раствор метиленхлорида
промывали 5 раз 200 мл порциями воды, а затем насыщенным раствором хлорида натрия. Метиленхлорид удаляли выпариванием, а остаток обрабатывали в н-гексане для получения твердого вещества. Последнее
собрали при фильтровании, промыли н- гексаном и высушили на воздухе (выход 1.83 г, 94.39% от теоретического). Масс-спектрометрия подтвердила следующую структуру:
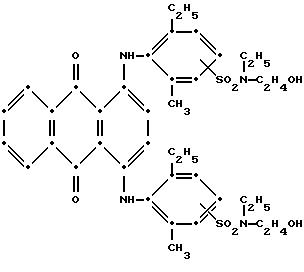
Максимумы поглощения в видимой области спектра поглощения наблюдались при 579 нм (ε-15,054) и 622 нм (ε-15,986) в метиленхлориде.
Пример 10
К хлорсульфоновой кислоте (12.5 мл) порциями добавляли 1,5- бис(2-анизидино)антрахинон (2.25 г, 0.005 молей) при перемешивании при температуре не
более 35oC. Реакционную смесь перемешивали в течение 3.0 часов при комнатной температуре, а затем добавляли по каплям к ацетону (250 мл) при охлаждении. Осадившийся дисульфонил хлорид
собрали при фильтровании, промыли ацетоном и высушили на воздухе (выход 2.85 г). Весь полученный дисульфонил хлорид добавили к 2-аминоэтанолу (35.0 г, 0.57 молей), и реакционную смесь перемешивали в
течение 1.0 часа при температуре ~90-95oC, охладили до комнатной температуры, а затем вылили в ацетон (200 мл). Темно-красный продукт собрали при фильтровании, промыли ацетоном и высушили
на воздухе (выход 2.15 г, 61.7% от теоретического). Предположительная структура, подтвержденная масс-спектрометрией, следующая:
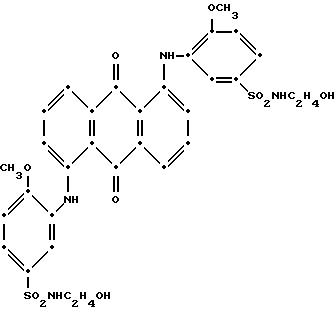
Максимумы поглощения в видимой области УФ-спектра поглощения наблюдались при 333 нм (ε-20,132) и 543 нм (ε-15,508) в метиленхлориде.
Пример 11
Дисульфонил хлорид антрахинона, полученный из 0.005 молей 1,5-бис(2-анизидо)антрахинона, как это описано в примере 10, был добавлен к 2-метиламиноэтанолу (40.0 мл) и
реакционную смесь перемешивали в течение 1.0 часа при 90-95oC; в течение этого времени дисульфониламидный продукт кристаллизовался. Его собрали при фильтровании, промыли ацетоном и
высушили
на воздухе (выход 2.17 г, 59.9% от теоретического выхода, при использовании в качестве исходного 1,4-бис(2-анизидо)антрахинона. Масс- спектрометрией была подтверждена следующая структура:
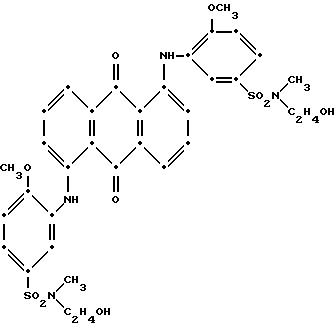
Максимум поглощения в видимой области спектра поглощения наблюдался при 530 нм (ε-15,150) в N, N-диметилформамиде.
Пример 12
Дисульфонил хлорид антрахинона, полученный из 0.01 моля 1,5- бис(2-анизидо)антрахинона, как это описано в примере 10, был добавлен к
раствору
2-(4-аминофенил)этанолу (4.37 г, 0.04 моля) в пиридине (35 мл). Реакционную смесь при перемешивании нагревали до ~55oC, а затем оставили на ночь при комнатной температуре.
Реакционную смесь
при энергичном перемешивании вылили в воду (200 мл), содержащую NaCl (10.0 г) Добавили уксусную кислоту (25.0 мл) и полученную смесь нагрели до ~75oC, а затем оставили
для охлаждения.
Красный осадок был собран при фильтровании, промыт водой и высушен на воздухе (выход 7.14 г). Масс-спектрометрически была подтверждена следующая предположительная структура:
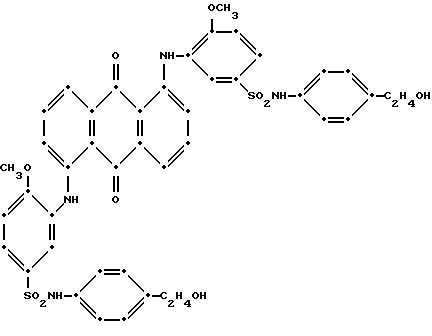
Максимум поглощения в видимой области спектра поглощения наблюдался при 533 нм (ε-14,062) в N, N-диметилформамиде.
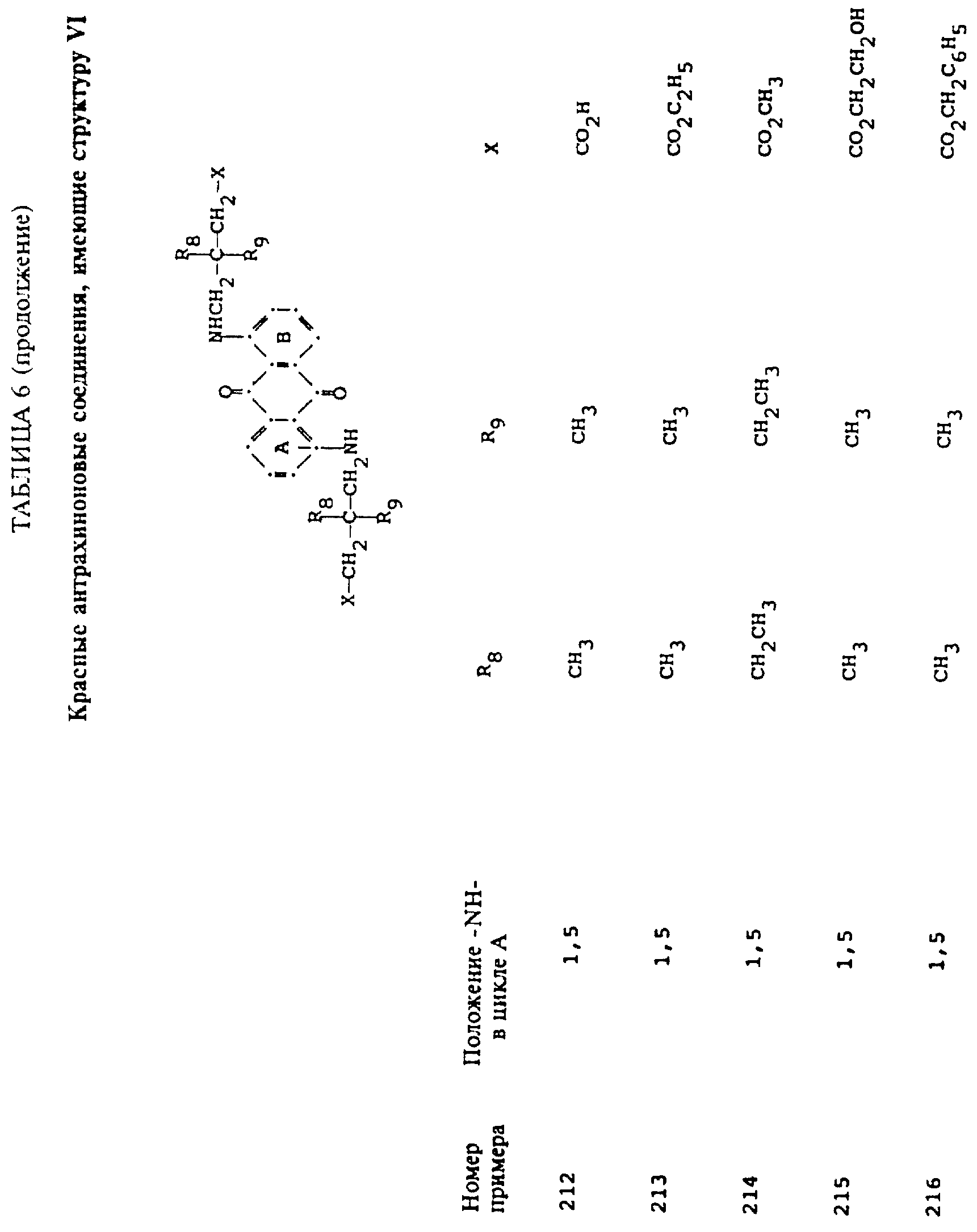
Пример 13
Смесь 6-бром-1-циано-3-метил-3Н-дибенз[f,i,j]изохинолин- 2,7-диона (1.82 г, 0.005 молей), 4-амино-3,5-диметилфенола (2.74 г, 0.02 моля),
карбоната калия (0.35 г), ацетата меди (II) (0.30 г) и N,N-диметилформамида (10.0 мл) нагревали в течение 1 часа при 80-90oC, затем охладили до ~50oC, а затем разбавили
добавлением еще 5.0 мл диметилформамида. Полученный продукт собрали при фильтровании, промыли диметилформамидом, потом метанолом и высушили на воздухе (выход 2.10 г, 48.6% от теоретического).
Масс-спектрометрия подтвердила следующую структуру:
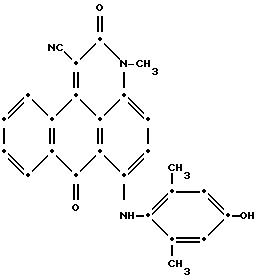
Максимумы поглощения в видимом спектре поглощения наблюдались при 549 нм (ε-9,964) и 585 нм (ε-12,860) в метиленхлориде.
Пример 14
Смесь 6-бром-1-циано-3-метил-3Н-дибенз[f,i,j]изохинолин-2,7-диона
(3.65 г, 0.01 моля), этил-3-аминобензоата (6.60 г, 0.04 моля), карбоната калия (1.00 г), ацетата меди (II) (1.20 г) и N,N-диметилформамид (10.0 мл) нагревали до ~80oC. Реакционную смесь
сконцентрировали, а затем разбавили добавлением 20.0 мл диметилформамида. Постепенно нагрели до ~130oC и добавили еще 10.0 мл диметилформамида для ускорения перемешивания. Реакционную
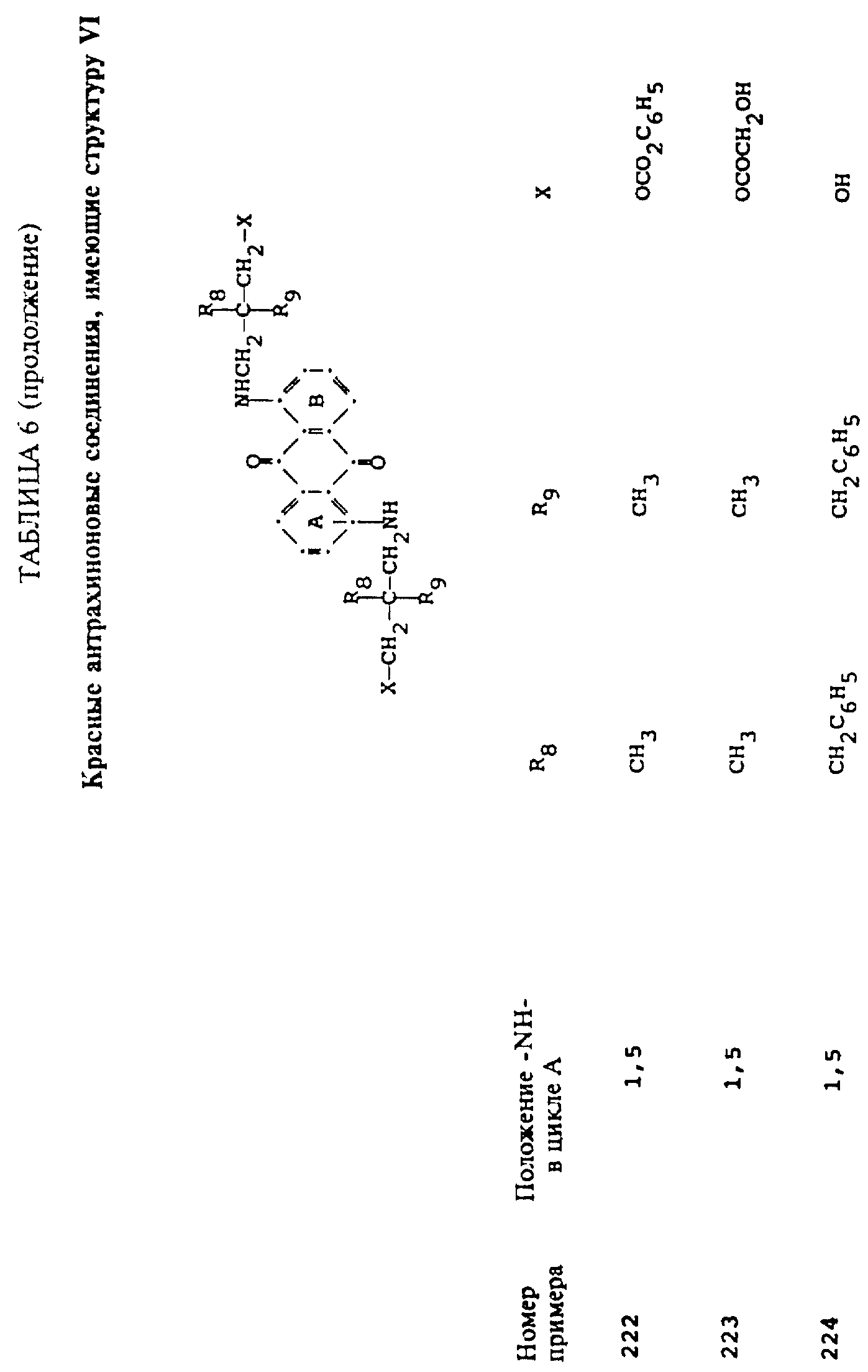
смесь
нагревали в колбе с обратным холодильником в течение 2.0 часов, а затем вылили в химический стакан, пока с помощью горячего диметилформамида вымывали твердое вещество из реакционной склянки.
Полученную смесь затем отфильтровали, а твердое вещество промыли диметилформамидом, потом - этанолом и высушили на воздухе (выход 3.0 г, 66.8% от теоретического). Масс-спектрометрически была
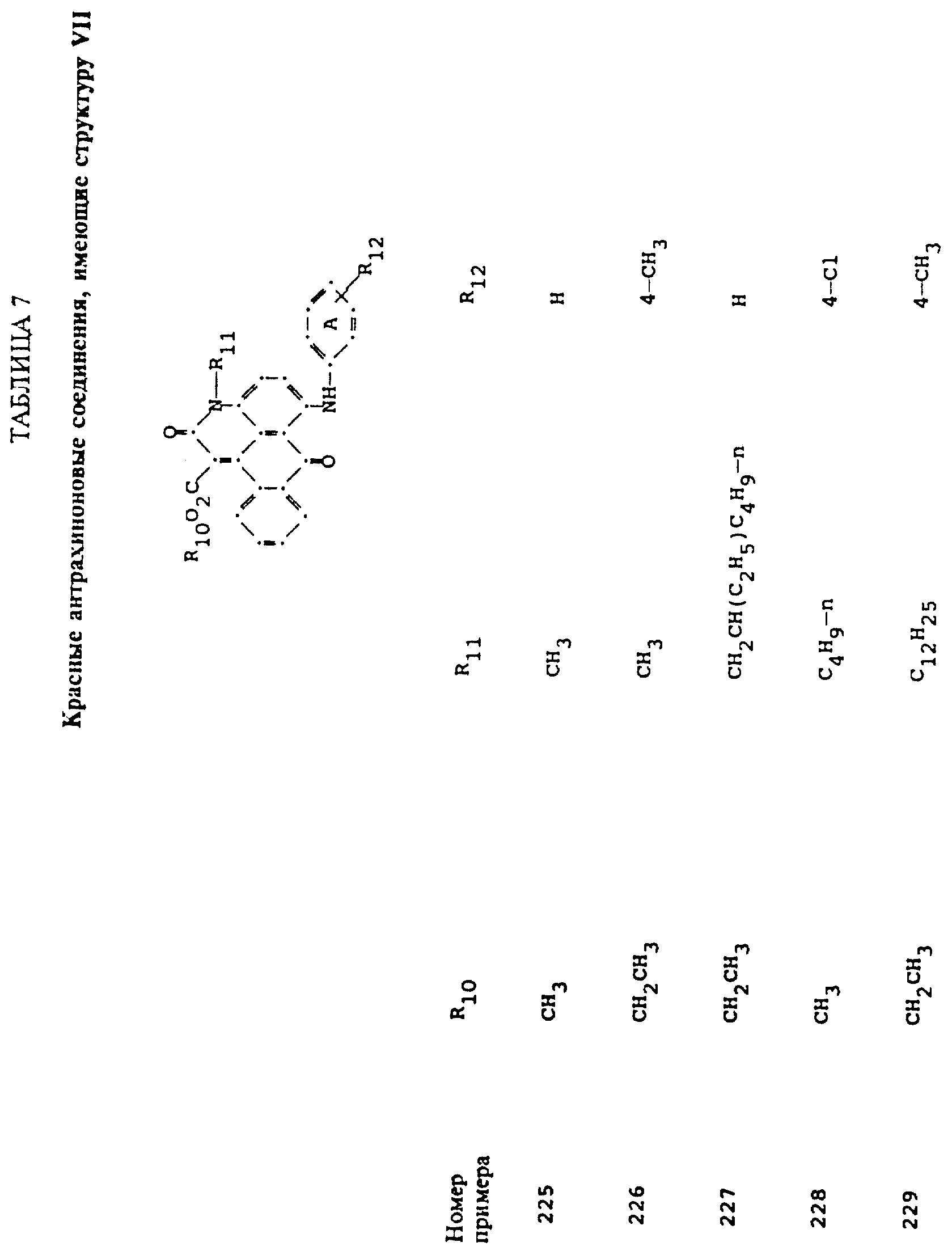
подтверждена следующая структура:
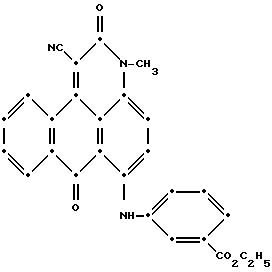
Максимумы поглощения в видимой области УФ-спектра поглощения наблюдались при 359 нм (ε-12,319) и 585 нм (ε-15,313) в метиленхлориде.
Пример 15
Смесь 6-бром-1-циано-3-метил-3Н-дибенз[f,i,j]изохинолин- 2,7-диона
(3.65 г, 0.01 моля), диметил-5-аминоизофталата (8.4.г, 0.04 моля), карбоната калия (1.00 г), ацетата меди (II) (1.20 г) и N,N-диметилформамида (20.0 мл) нагревали до ~ 80oC при
перемешивании. Реакционная смесь стала очень концентрированной, и для ускорения перемешивания было добавлено еще 70.0 мл диметилформамида. После нагревания в течение 15 минут до 140-145oC
реакционную смесь вылили в химический стакан, для промывания склянки при этом использовали диметилформамид. Продукт собрали при фильтровании, промыли диметилформамидом, потом - диметилформамидом и
метанолом 1:1, а в заключении промыли метанолом и высушили на воздухе (выход 3.02 г, 61.1% от теоретического). Масс-спектрометрией была подтверждена следующая предположительная структура:
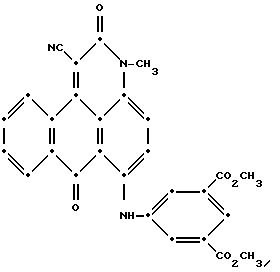
Видимый УФ-спектр показал максимумы поглощения при 353 нм и 584 нм в N, N-диметилформамиде.
Пример 16
К хлорсульфоновой кислоте (50.0 мл) порциями добавили 6- анилино-1-циано-3-метил-3Н-дибенз[f,i,j]изохинолин-2,7-дион (7.54 г) при энергичном перемешивании и допускаемом
повышении
температуры. Затем реакционную смесь вылили в раствор лед/насыщенный NaCl. Твердое сульфонилхлоридное соединение собрали при фильтровании, промыли насыщенным раствором NaCl, а затем
добавили при
энергичном перемешивании к диэтаноламину (50.0 мл). Далее реакционную смесь нагревали в течение 0.5 часа при 50oC, а затем при помешивании вылили в воду. Полученный твердый
продукт собрали
при фильтровании, промыли водой и высушили на воздухе (выход 9.9 г). Масс-спектрометрией была подтверждена следующая предположительная структура:
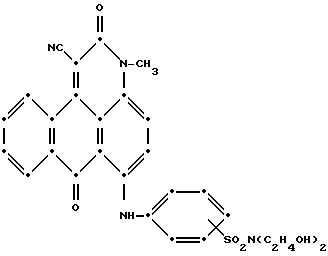
Пример 17
Смесь 6-бром-1-циано-3-метил-3Н-дибенз[f,i,j]изохинолин- 2,7-диона (36.5 г), метил-4-аминобензоата (75.0 г), карбоната калия (10.0 г), ацетата меди (II) (12.0 г) и N, N-диметилформамида (120.0 мл) нагревали в течение 3.0 часов при температуре ~ 110oC. Добавили еще дополнительно диметилформамид (140 мл) и полученную смесь оставили до охлаждения до температуры ~30oC. Полученный продукт собрали при фильтровании, промыли диметилформамидом, потом промыли водой и в заключении изопропанолом, а затем высушили на воздухе (выход 34.0 г). Очистку завершили перекристаллизацией из диметилформамида (620 мл) путем нагревания смеси в колбе с обратным холодильником, а затем охлаждением до 100oC. Полученный продукт собрали при фильтровании, промыли диметилформамидом, изопропанолом, а затем высушили на воздухе (выход 18.8 г). Предположительная структура соединения такова:
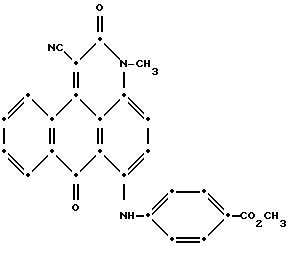
Пример 18
Смесь 6-бром-1-циано-3-метил-3Н-дибенз[f,i, j]изохинолин- 2,7-диона (3.65 г, 0.01 моля), 4-амино-N-(2-гидроксиэтил)фталимида (8.24 г, 0.04 моля), карбоната калия (1.0 г), ацетата меди (II) (1.0 г) и N,N-диметилформамида (20.0 мл) нагревали при перемешивании до 135oC, а затем в течение 1.0 часа выдерживали при 135-140oC. Потом горячую реакционную смесь постепенно выливали в 150 мл ацетона. Твердое вещество собрали при фильтровании, промыли ацетоном, потом диметилформамидом и высушили на воздухе (выход 2.85 г, 58.2% от теоретического). Масс-спектрометрией была подтверждена следующая структура:
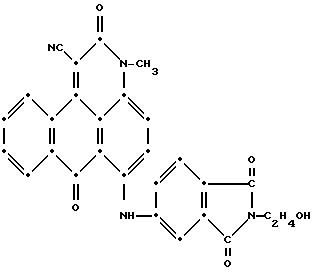
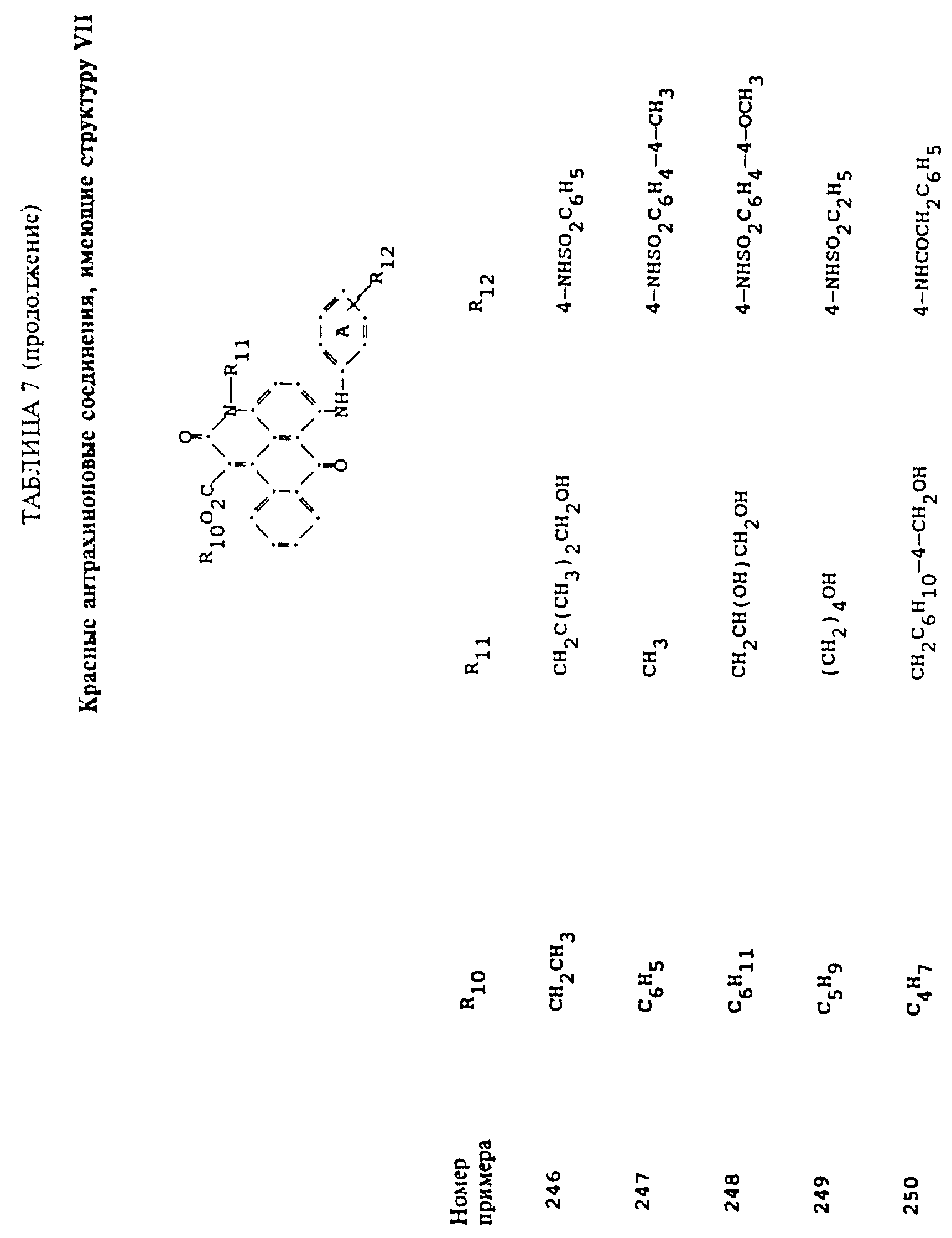
Максимумы поглощения в видимой области УФ-спектра поглощения наблюдались при 367 нм (ε-13,542) и 570 нм
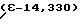
Пример 19
Смесь
6-бром-1-циано-3-метил-3Н-дибенз[f,i,j]изохинолин- 2,
7-диона (1.00 г, 0.003 моля), этил-4-[3-аминобензамидо)] бензоата (3.41 г, 0.012 моля), карбоната калия (0.30 г), ацетата меди (II) (0.36 г) и N,
N-диметилформамида (10.0 мл) нагревали при 120-125oC в течение 6.0 часов с перемешиванием. Реакционная смесь охлаждалась, а затем выливалась при перемешивании в разбавленную водную
уксусную кислоту. Твердый продукт собрали при фильтровании,
промыли водой, повторно суспендировали в метаноле (200 мл), еще раз отфильтровали, промыли метанолом и высушили на воздухе (выход 1.63 г).
Очистку завершили на хроматографической колонке, используя
FLORISIL; образец, растворенный в метиленхлориде, поместили в колонку и элюировали метиленхлоридом, а затем смесью
метиленхлорид:тетрагидрофуран 50:50. После выпаривания растворителя получили твердый
продукт (0.59 г). Масс-спектрометрией была подтверждена следующая структура:
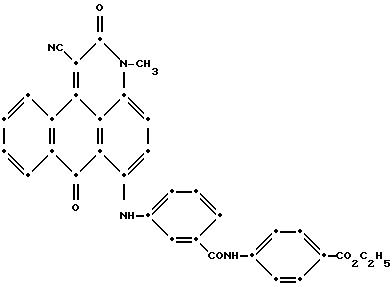
Максимум поглощения в видимом спектре поглощения наблюдался при 584 нм в метиленхлориде.
Пример 20
Смесь 6-бром-1-циано-3-метил-3Н-дибенз[f,i,j]изохинолин- 2,7-диона (1.00 г, 0.003
моля), этил-α-(3- аминобензамидо)ацетата (2.66 г, 0.012 моля), карбоната калия (0.30 г), ацетата меди (II)
(0.36 г) и N,N-диметилформамида (10.0 мл) прореагировали, и был выделен сырой продукт.
Последний очистили с помощью колоночной хроматографии, чтобы получить 0.24 г очищенного продукта, в точности так,
как это описано в примере 19. Масс-спектрометрией была подтверждена следующая
структура:
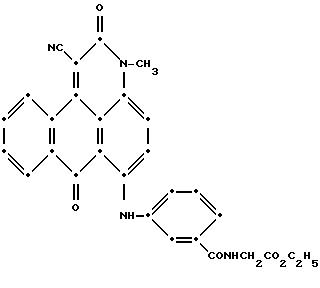
Максимум поглощения в видимом спектре поглощения наблюдался при 585 нм (ε -16,431) в метиленхлориде.
Пример 21
Дисульфонилхлорид антрахинона, полученный из 0.005 молей
1,5-бис(2-анизидино)антрахинона (который был синтезирован так, как в примере 10),
добавляли порциями при комнатной температуре к раствору 2-этиламиноэтанола (1.07 г, 0.012 моля) и триэтиламина (1.27
г, 0.013 моля), растворенного в метиленхлориде (50.0 мл). После перемешивания в
течение 3.0 часов при комнатной температуре реакционную смесь отфильтровали, а полученный темно-красный твердый продукт
промыли метиленхлоридом, потом - изопропанолом, а затем высушили на воздухе
(выход 2.24 г, 59.6% от теоретического). Масс-спектрометрия подтвердила следующую структуру:
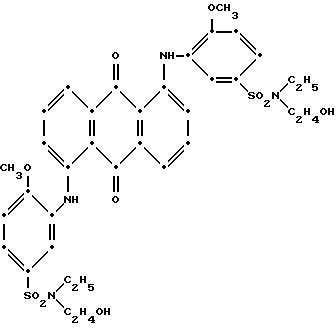
Максимум поглощения в видимом спектре поглощения наблюдался при 529 нм (ε-15,040) в метиленхлориде.
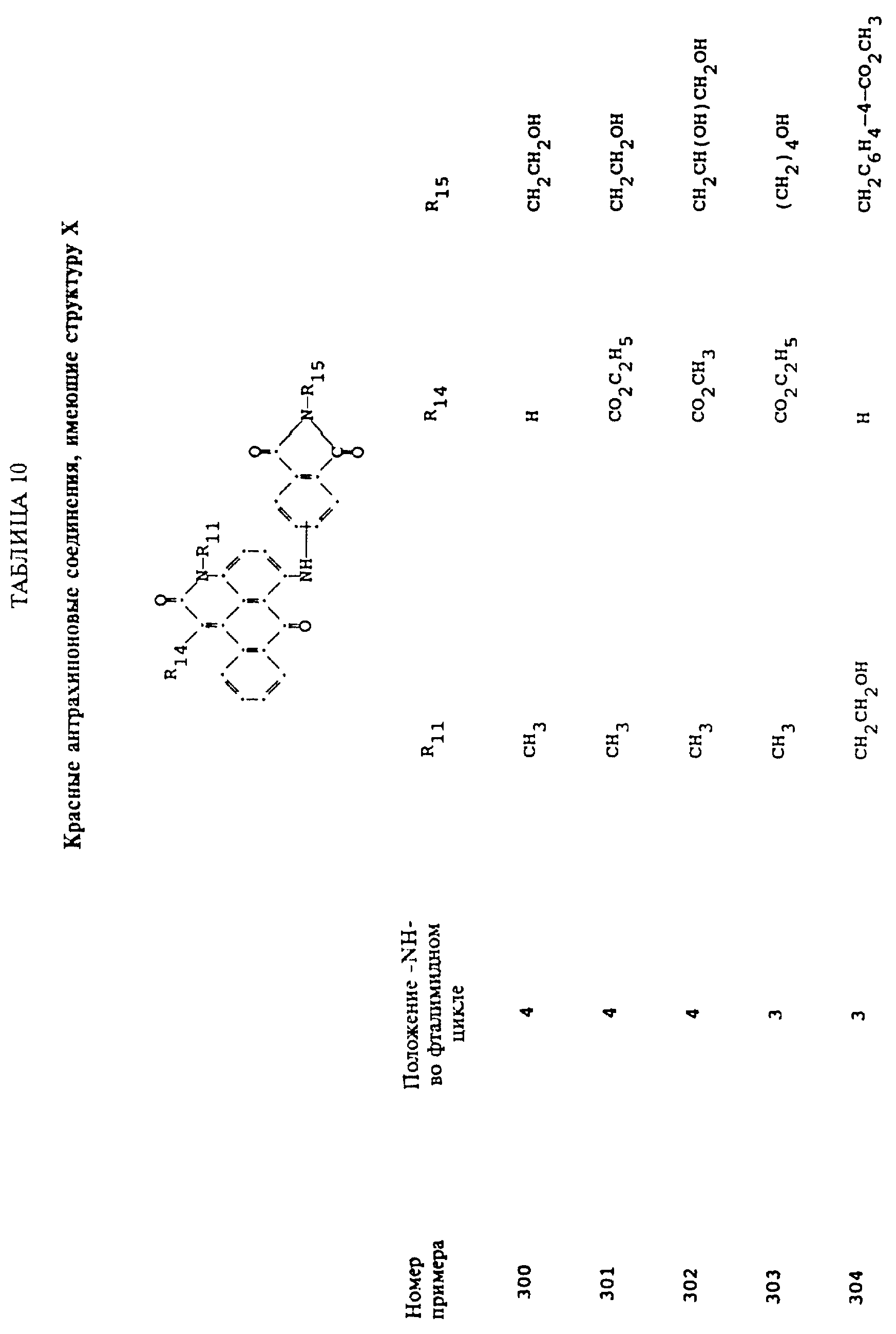
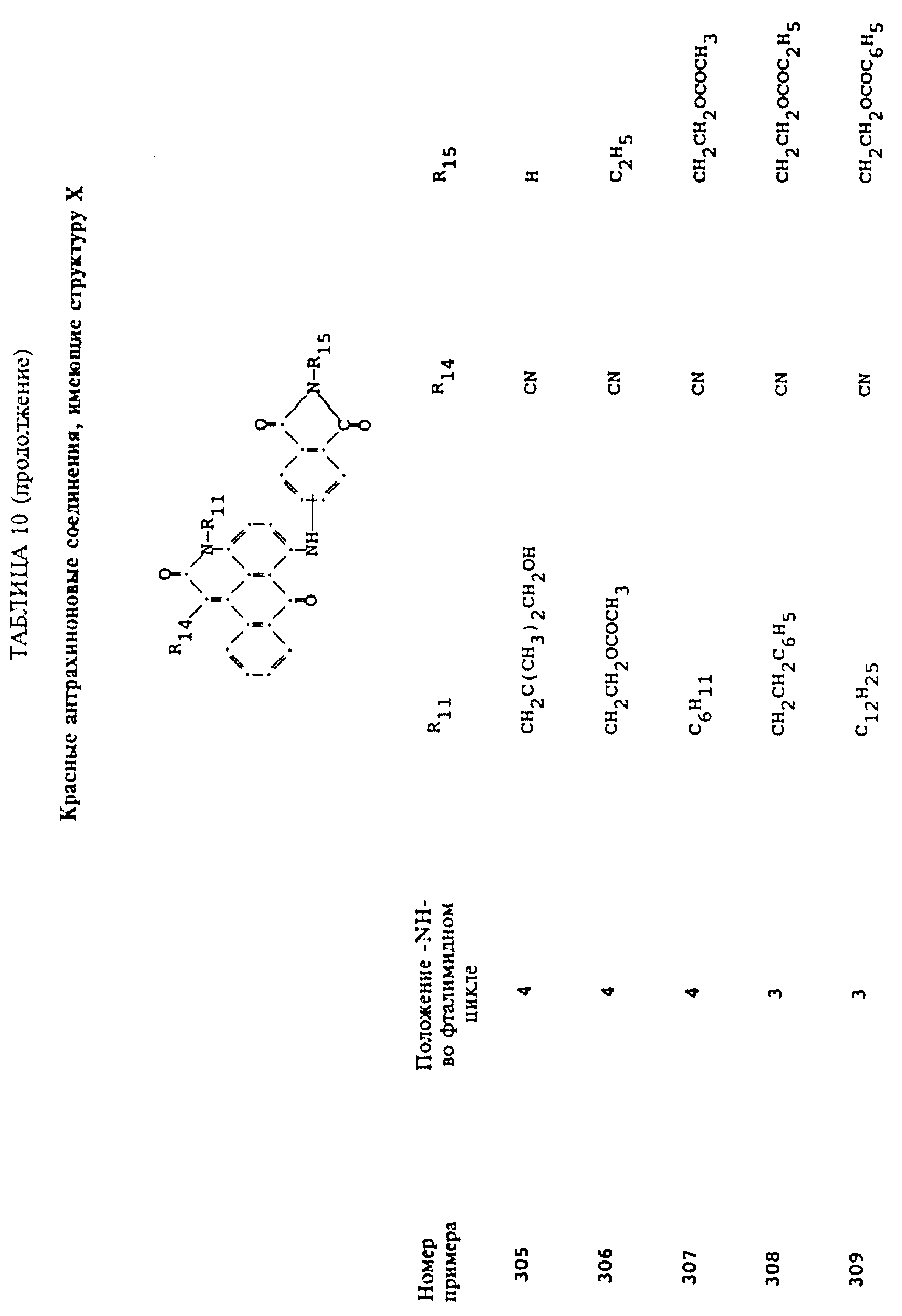
Примерами 22-322, помещенными в таблицах 1-10 продолжена иллюстрация границ настоящего изобретения, что касается структур тонирующих компонентов.
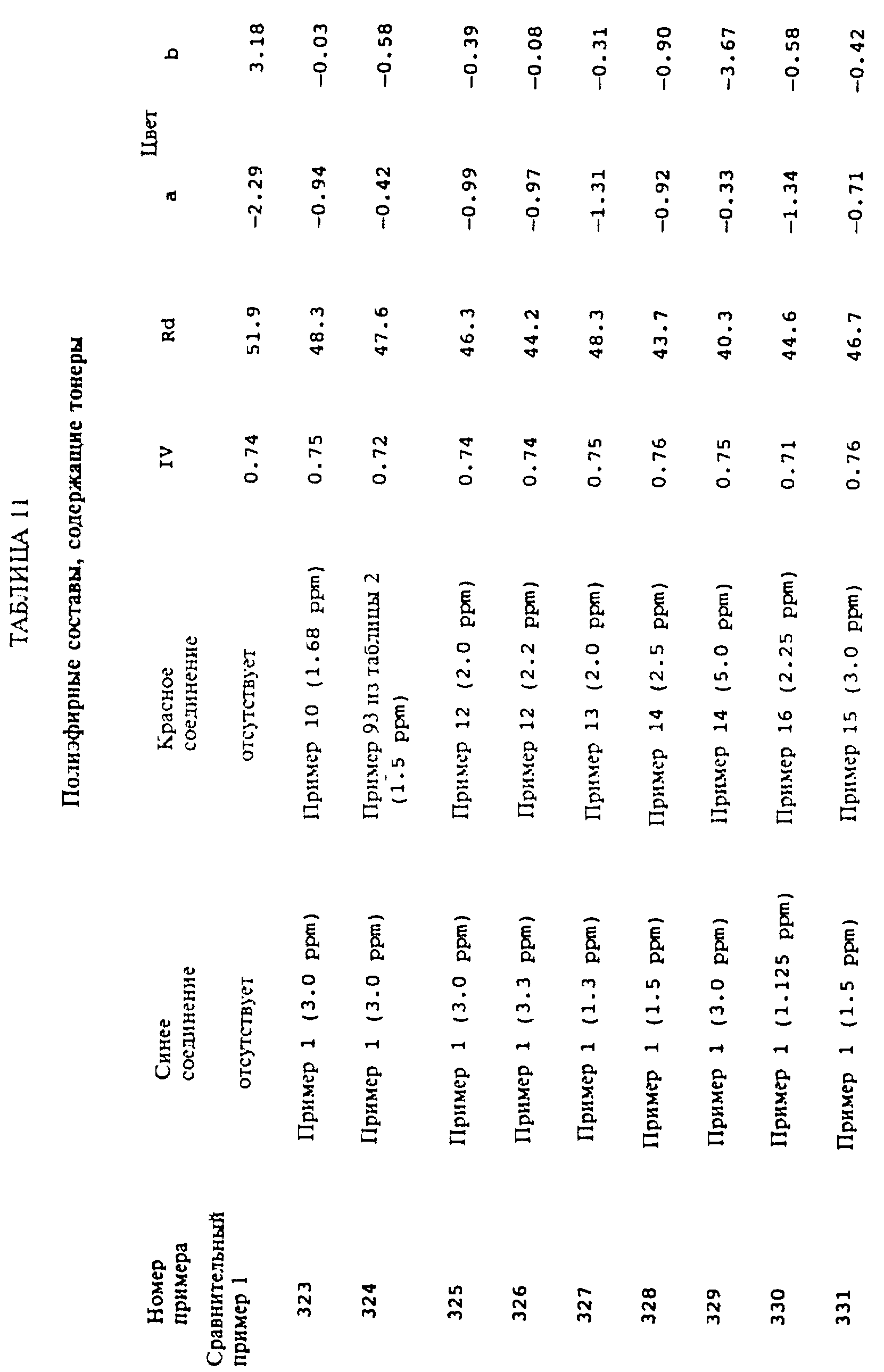
Примеры 323-343 иллюстрируют получение модифицированных поли(этилентерефталатных) полиэфирных полимеров, которые содержат характерные количества подходящих сополимеризующихся синего и красного компонентов. Цветовые характеристики измерялись с использованием спектроколориметра HUNTER LAB ULT- RASCAN (Hunter Associates Laboratory, Inc); были определены величины Hunter Rd, a и b. Измерение цветности
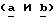
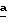
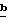
Пример 323 (получение полиэфирного полимера)
В 500 мл
одногорлую круглодонную колбу были загружены следующие вещества:
- 97 г (0.5 молей) диметилфталата;
- 42.8 г (0.69 молей) этиленгликоля;
- 22.3 г (0.16 моля) 1,
4-циклогександиметанола;
- 0.00609 г Ti из н-бутанольного раствора
ацетил-триизопропил титаната;
- 0.0050 г Mn из этиленгликольного раствора ацетата магния;
- 0.0003261 г
(3.0 ppm) синего соединения из примера 1 (из этиленгликольного раствора);
- 0.000168 г (1.68 ppm) красного соединения из примера 10 (из этиленгликольного раствора).
Эта колба была снабжена азотным вводом, мешалкой, вакуумным выводом и конденсационной емкостью. Колбу и ее содержимое нагревали до 200oC на металлической бане Belmont в течение 80 минут при притоке азота и с перемешиванием (скорость мешалки в первые 15 минут доходила до 25 об/минуту, за последующие 5 минут ее увеличили с 25 до 200 об/минуту и это конечное значение скорости мешалки поддерживали вплоть до последней стадии поликонденсации). Температура бани была поднята до 225oC за 5 минут. Колбы и их содержимые нагревались при 225oC в течение 60 минут. Затем добавили 1.05 мл раствора смешанных фосфорных эфиров (состав Merpol A) в этиленгликоле/н-бутаноле, содержащего 0.0087 г фосфора. В течение 20 минут температуру бани увеличили до 282oC. В течение 5 минут проводили вакуумирование до тех пор, пока давление не уменьшилось до 400 мм Hg. Затем в течение 5 минут давление снизили с 400 до 100 мм Hg. В течение следующих 5 минут давление снижали со 100 до 10 мм Hg. Затем в течение 5 минут уменьшили давление с 10 до 0.2 мм Hg. Колбу и ее содержимое нагревали еще 45 минут при 282oC и давлении 0.2 мм Hg, скорость мешалки при этом составляла от 200 об/минуту в начале до 25 об/минуту в конце процесса. Колбу сняли с металлической бани, а полученный полимер оставили охлаждаться в атмосфере азота. Вязкость полученного полимера составила 0.76, ее измеряли в смеси, содержащей по весу фенол/тетрахлорэтан в соотношении 60/40 при концентрации 0.5 г в 100 мл. Полимер охладили в жидком азоте (~ -195oC), затем разбили на куски, которые измельчили на мельнице Wiley так, чтобы измельченный продукт проходил сквозь 3 мм сито. Почти 8.0 г полученных гранул сплавили в лист с примерным диаметром 1.5 дюймов и 125 мм толщиной; процесс проводили на гидравлическом прессе Wabash (Wabash Metal Products, Wabash, IN) при усилии 20.000 фунтов в течение 1.0 минуты. Цветовые характеристики определяли на листе так, как это описано выше, эти характеристики приведены в таблице 11.
Сравнительный пример 1
Был повторен пример 323 за исключением того, что пренебрегли указанными синим и красным тонирующими компонентами,
обеспечивая тем самым контрольный
образец для сравнения. Цветовые величины, приведенные в таблице 11, показывают, что этот образец сам по себе был желтым.
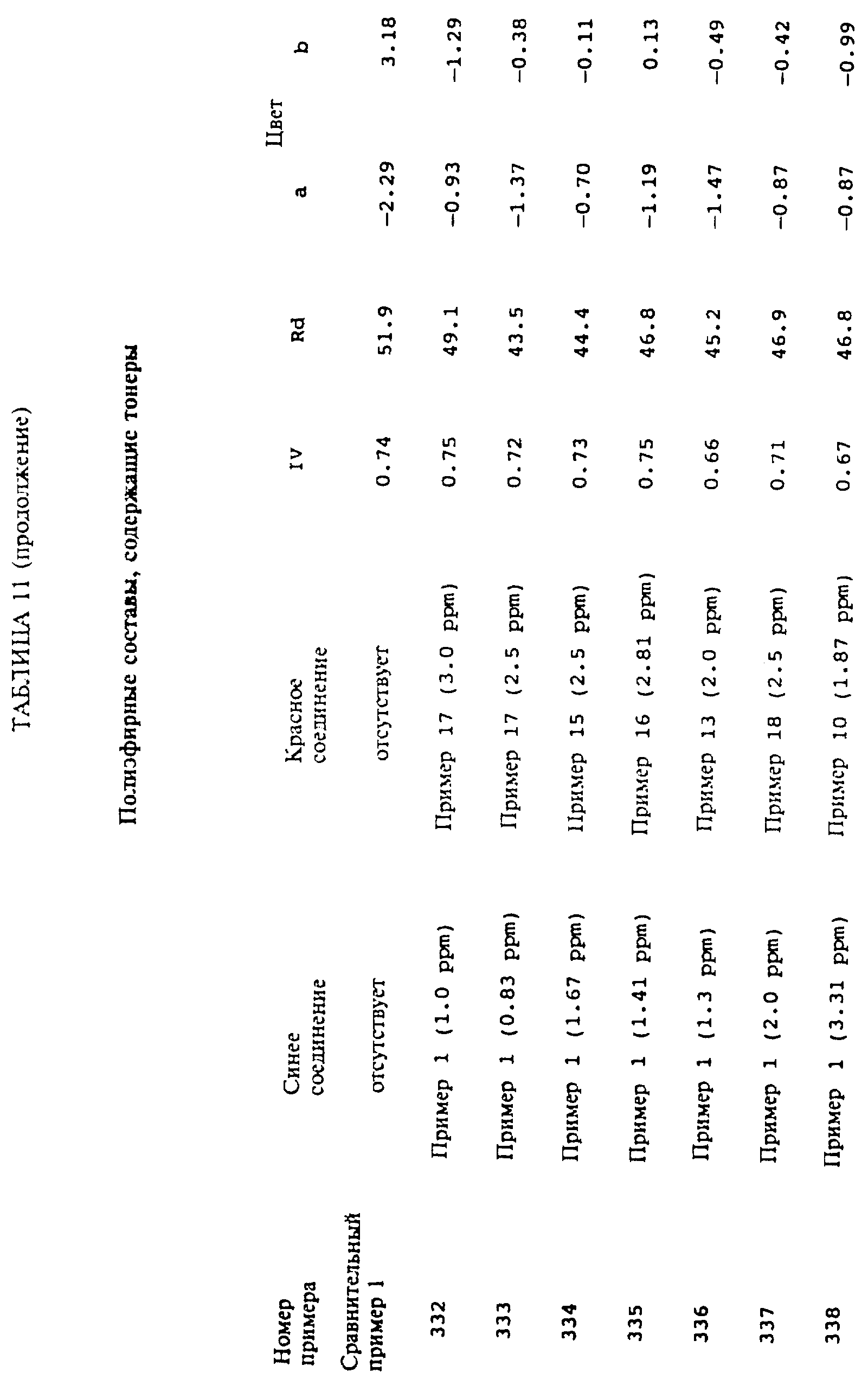
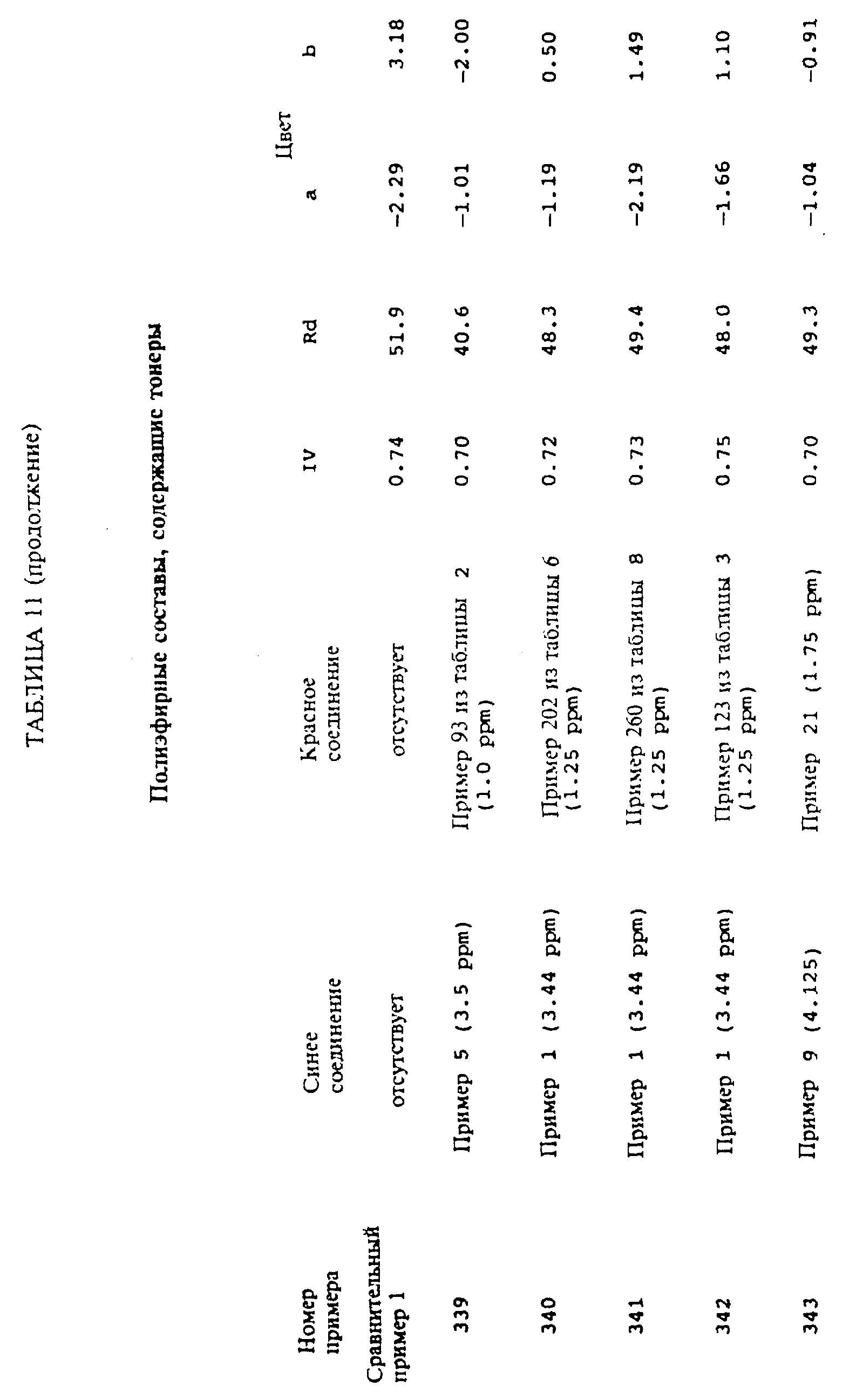
Примеры 324-343
Была
в точности повторена методика,
изложенная в примере 323, за исключением того, что тонирующие синий и красные компоненты варьировались так, как это показано в таблице 11. Цветовые величины определяли
так, как это описано выше и как
это приведено в таблице 11. Эффективность тонирующей системы по удалению желтого цвета полимера продемонстрирована тем фактом, что многие системы демонстрируют величину
"b" в окрестности нуля и что у
всех них величина "b" меньше, чем полимера из сравнительного примера 1, который не содержит никакого тонера.
В следующих примерах (примеры 344-346) приведено получение (поли)этилентерефталата, содержащего разные количества синего и красного тонирующих компонентов. Все цветовые характеристики измерялись на цветовом устройстве HUNTER LAB (Hunter Associates Laboratory, Inc) и CIELAB-L*, были определены величины a* и b*. Как указывалось выше, a* измеряет красноту при знаке + и зеленость при знаке -; b* измеряет желтизну при знаке + и синесть при знаке -.
Пример 344 (Получение (поли)этилентерефталата)
В трехгорлую круглодонную 500 мл колбу были загружены
следующие материалы:
- 97 г (0.5 молей) диметилфталата;
- 62 г (1.0 моль) этиленгликоля;
- 0.00192 г Ti из н-бутанольного раствора ацетил-триизопропил титаната;
- 0.0053 г Mn из
этиленгликольного раствора ацетата магния;
- 0.0345 г трехокиси сурьмы;
- 3.0 ppm синего соединения из примера 1 (из этиленгликольного раствора);
- 1.86 ppm
красного
соединения из примера 10 (из этиленгликольного раствора).
Эта колба была снабжена азотным вводом, мешалкой, вакуумным выводом и конденсационной емкостью. Колбу и ее содержимое нагревали до 200oC на металлической бане Belmont в течение 60 минут при притоке азота и с перемешиванием (скорость мешалки в первые 15 минут была доведена до 165 оборотов/минуту и затем это значение скорости поддерживалось в течение остального времени реакции. Температура бани была поднята до 210oC за 10 минут. Колбы и их содержимые нагревались при указанной температуре в течение 65 минут. Затем добавили 1.57 мл раствора смешанных фосфорных эфиров (состав Merpol A) в этиленгликоле/н-бутаноле, содержащего 0.012 г фосфора. В течение 10 минут температуру бани увеличили до 230oC. В течение 5 минут проводили вакуумирование до тех пор, пока давление не уменьшилось до 200 мм Hg. Колбу и ее содержимое нагревали 25 минут при 230oC и давлении 200 мм Hg. Температура металлической бани была поднята в течение 15 минут до 270oC. Давление снизили до 100 мм Hg в течение 10 минут. Колбу и ее содержимое нагревали в течение 30 минут при 270oC и давлении 100 мм Hg. Температуру металлической бани увеличили до 286oC, а давление снизили до 6 мм Hg в течение 10 минут. Колбу и ее содержимое нагревали при 285oC и 6 мм Hg в течение 25 минут. Давление в течение 3 минут снизили до 0.3 мм Hg, а поликонденсация продолжалась еще 37 минут. Колбу сняли с металлической бани и оставили охлаждаться в атмосфере азота, пока проходила кристаллизация полимера. Вязкость полученного полимера составила 0.65, ее измеряли в 60/40 по весу смеси фенол/тетрахлорэтан и концентрации 0.5 г в 100 мл. При выпаривании не наблюдалось никакой потери цветности в дистиллате, собранном во время поликонденсации. Полученный полимер измельчили на мельнице Wiley так, чтобы он проходил сквозь 3 мм сито; цвет гранул определяли так, как это было описано выше. Характеристики продукта приведены в таблице 12.
Пример 345
Образец (поли)этилентерефталата был получен так, как это описано
в примере 344, за исключением
того, что в качестве тонирующей системы использовали 4.0 ppm синего соединения из примера 8 и 2.0 ppm красного соединения из примера 11. Все свойства приведены в таблице
12.
Пример
346
Образец (поли)этилентерефталата был получен так, как это описано в примере 344, за исключением того, что в качестве тонирующей системы использовали 4.5 ppm
синего соединения из примера 9
и 2.0 ppm красного соединения из примера 168 таблицы 5. Все свойства приведены в таблице 12.
Реферат
Описывается новый литой полиэфир или полиэфир в виде фибрилл, полученный сополимеризацией полиэфира с достаточным для усиления свойственной этому полиэфиру белизны количеством по меньшей мере одного синего соединения 1,4-бис(2,6-диалкиланилино)-антрахинона общей формулы I, где значения R, R1-R5, Х, m, n указаны в п. 1 формулы изобретения вместе с по крайней мере одним красным соединением антрахинона или антрапиридона, выбранным из группы соединений, имеющих приведенные ниже формулы II, где значения R6, p указаны в п. 1 формулы, которые обеспечивают применимость тонирующей системы к полиэфирам, имеющим желтую окраску для сообщения им требуемого оттенка от нейтрального до светло-голубого. Эти термостойкие окрашенные соединения могут содержать реактивные полиэфирные группы и в процессе полимеризации они предпочтительно включаются в структуры полиэфирного полимера. Предлагается также тонирующий состав премикса и формованное изделие. 4 с. и 9 з.п. ф-лы, 12 табл.
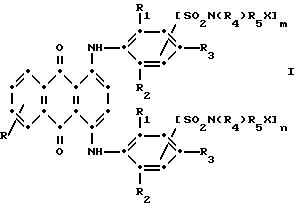
Формула
в которой R выбирают из группы, содержащей водород, С1-С6-алкил, галоген карбокси и

R1 и R2 независимо друг от друга представляют собой С1-С6-алкил;
R3 выбирают из группы, содержащей водород, галоген, С1-С6-алкил, замещенный С1-С6-алкил, гидрокси, С1-С6-алкокси, замещенный С1-С6-алкокси, циано, тиоциано, С1-С6 -алкилтио, замещенный С1-С6-алкилтио, С1-С6-алкилсульфонил, замещенный С1-С6-алкилсульфонил, С1-С6 -алкоксикарбонил, карбокси, арилокси, арилтио, арилсульфонил и SO2N(R4)R5X, если m и/или n = 0;
R4 выбирают из группы, содержащей водород, С1-С6-алкил, замещенный С1-С6-алкил, С3-С8-алкенил, С3-С8алкинил, С3-С7-циклоалкил и арил;
R5 связывающая группа, выбранная из группы, содержащей С1-С8-алкилен, С1-С6-алкилен-Z-С1-С6-алкилен, арилен-С1-С6-алкилен, арилен-Z-С1-С6-алкилен,
С3-С7-циклоалкилен, С1-С6-алкилен-циклоалкилен-С1-С6-алкилен, С1 -С6-алкилен-арилен-С1-С6-алкилен и С1-С6-алкилен-Z-арилен-Z-С1-С6-алкилен, где Z выбирают из -О-, -S- или SO2;
Х представляет собой галоген или реактивную полиэфирную группу;
m и n независимо друг от друга представляют собой 0 или 1; при условии, что присутствует по меньшей мере одна полиэфирная реактивная группа; вместе с по крайней мере одним красным соединением антрахинона или антрапиридона, выбранным из группы соединений, имеющих приведенные ниже формулы II - X:
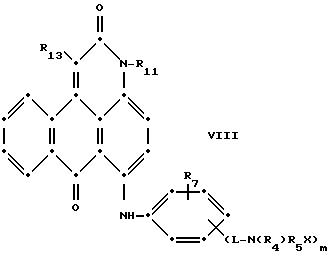
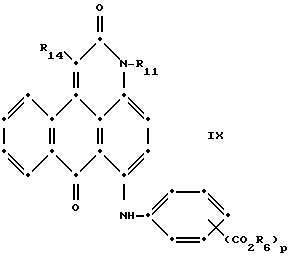
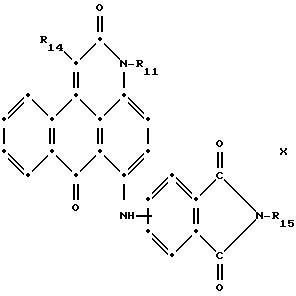
в которых R6 выбирают из группы, содержащей водород, С1-С6-алкил, замещенный С1-С6-алкил, С3-С7-циклоалкил или арил;
R7 представляет собой водород или от одной до трех групп, которые выбирают из С1-С6-алкила, замещенного С1-С6-алкила, С1-С6-алканоиламино, галогена, С1-С6-алкил-С1 -С6-алкокси, С1-С6-алкилтио;
R8 и R9 одинаковые или разные, выбранные из группы, включающей С1-С6-алкил, замещенный С1-С6-алкил, С3-С7-циклоалкил или арил;
R10 выбирают из группы, включающей С1-С6-алкил, замещенный С1-С6-алкил, С3-С7-циклоалкил или арил;
R11 выбирают из группы, содержащей водород, С1-С12-алкил, замещенный С1-С12-алкил, С3-С7-циклоалкил и арил;
R12 представляет собой водород или от одной до трех групп, которые выбирают из С1 -С6-алкила, замещенного С1-С6-алкила, С1-С6-алкокси, замещенного С1-С6-алкокси, С1-С6-алкилтио, замещенного С1-С6 -алкилтио, галогена, гидрокси, С1-С6-алканоиламино, ароиламино, С1-С6-алкилсульфониламино и арилсульфониламино;
R13 и R14 выбирают из водорода, циано или СО2R10;
R15 представляет собой определенные ранее R4 или R5Х;
L представляет собой -СО- или -SO2-; Х имеет значения, определенные выше; m = 0 или 1; р = 1 или 2, при условии, что R13 представляет собой водород, когда m = 0, и что присутствует по меньшей мере одна реактивная полиэфирная группа.
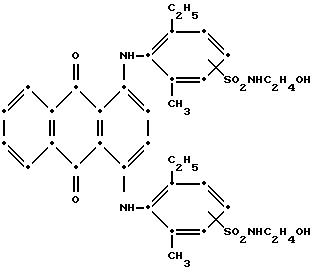
или
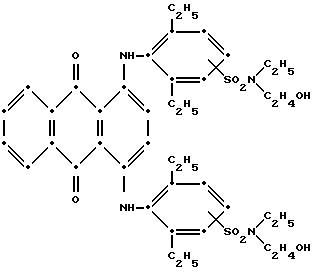
6. Полиэфир по любому из пп.1 - 5, отличающийся тем, что красное соединение формулы V представляет собой
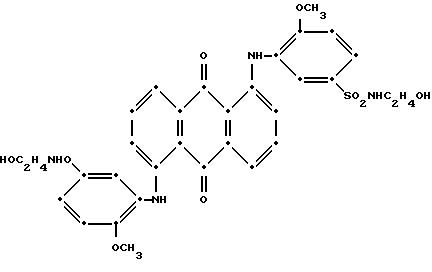
или
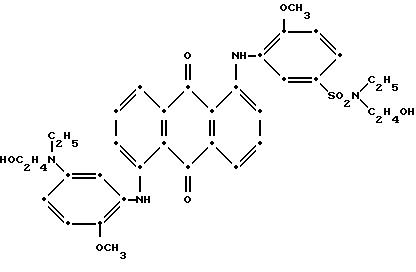
7. Полиэфир по любому из пп.1 - 6, отличающийся тем, что синее соединение формулы I представляет собой
и тем, что красное соединение формулы V представляет собой
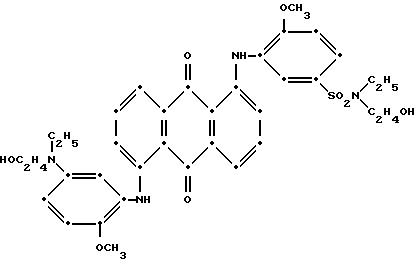
8. Способ сообщения белизны обычно воспринимаемому желтым полиэфиру сополимеризацией или смешением с соединением, усиливающим белизну, отличающийся тем, что в качестве соединения, усиливающего белизну, используют достаточное для усиления белизны количество по меньшей мере одного синего 1,4-бис(2,6-диалкиланилино)антрахинона формулы I
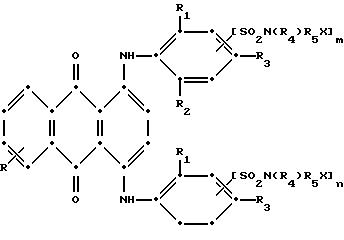
в которой R выбирают из группы, содержащей водород, С1-С6, галоген, карбокси и

R1 и R2 независимо друг от друга представляют собой С1-С6 -алкил;
R3 выбирают из группы, содержащей водород, галоген, С1-С6-алкил, замещенный С1-С6-алкил, гидрокси, С1-С6 -алкокси, замещенный С1-С6-алкокси, циано, тиоциано, С1-С6 -алкилтио, замещенный С1-С6-алкилтио, С1-С6 -алкилсульфонил, замещенный С1-С6-алкилсульфонил, С1-С6 -алкоксикарбонил, карбокси, арилокси, арилтио, арилсульфонил и SO2N(R4)R5X, если m и/или n = 0;
R4 выбирают из группы, содержащей водород, С1-С6-алкил, замещенный С1-С6-алкил, С3-С8-алкенил, С3-С8-алкинил, С3-С7-циклоалкил и арил;
R5 - связывающая группа, выбранная из группы, содержащей С1-С8-алкилен, С1-С6-алкилен-Z-С1-С6-алкилен, арилен-С1-С6-алкилен, арилен-Z-С1-С6-алкилен,
С3-С7-циклоалкилен, С1-С6-алкилен-циклоалкилен-С1-С6-алкилен, С1-С6алкилен-арилен-С1-С6-алкилен и С1-С6-алкилен-Z-арилен- Z-С1-С6-алкилен, где Z выбирают из -О-, -S- или SO2;
Х представляет собой галоген или реактивную полиэфирную группу;
m и n независимо друг от друга представляют собой 0 или 1; при условии, что присутствует по меньшей мере одна полиэфирная реактивная группа; и по меньшей мере одного красного соединения антрахинона или антрапиридона, выбранных из группы соединений, имеющих приведенные ниже формулы II - X:
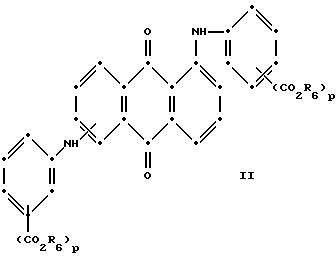
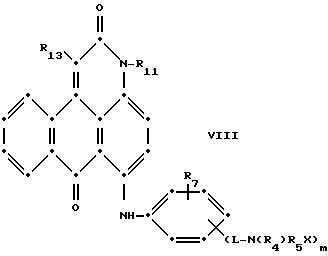
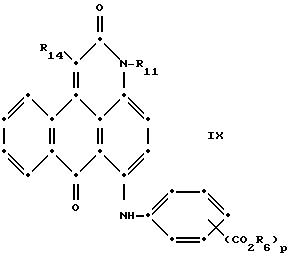
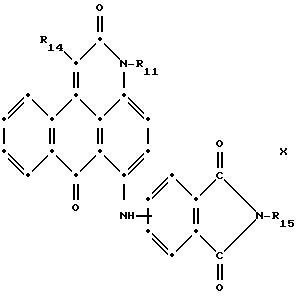
в которых R6 выбирают из группы, содержащей водород, С1-С6-алкил, замещенный С1-С6-алкил, С3 -С7-циклоалкил или арил;
R7 представляет собой водород или от одной до трех групп, которые выбирают из С1-С6-алкила, замещенного С1 -С6-алкила, С1-С6-алканоиламино, галогена, С1-С6-алкил-С1-С6-алкокси, С1-С6-алкилтио;
R8 и R9 одинаковые или разные, выбранные из группы, включающей С1-С6-алкил, замещенный С1-С6-алкил, С3-С7-циклоалкил или арил;
R10 выбирают из группы, включающей С1-С6-алкил, С3-С7-циклоалкил или арил;
R11 выбирают из группы, содержащей водород, С1-С12-алкил, замещенный C1-C12-алкил, С3-С7-циклоалкил или арил;
R12 представляет собой водород или от одной до трех групп, которые выбирают из С1-С6-алкила, замещенного С1-С6-алкила, С1 -С6-алкокси, замещенного С1-С6-алкокси, С1-С6-алкилтио, замещенного С1-С6-алкилтио, С1-С6 -алканоиламино, галогена, ароиламино, С1 -С6-алкилсульфониламино и арилсульфониламино;
R13 и R14 выбирают из водорода, циано или СО2R10;
R15 представляет собой определенные ранее R4 или R5Х;
L представляет собой -СО- или -SO2-, Х имеет значения, определенные выше, m = 0 или 1, р = 1 или 2, при условии что R13 представляет собой водород когда m = 0 и что при сополимеризации соединений формул II - Х присутствует по меньшей мере одна реактивная полиэфирная группа.
-СН2С6Н10СН2-, арилен или СН2-арилен-, а красный компонент соответствует формуле V, в которой R7 представляет собой С1-С6-алкокси, отличающийся тем, что суммарная концентрация синего и красного компонентов составляет от ~ 0,5 ppm до ~ 10 ppm.
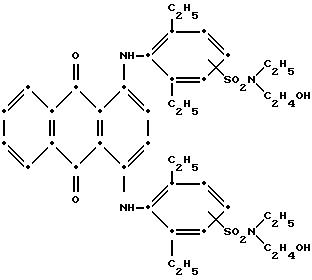
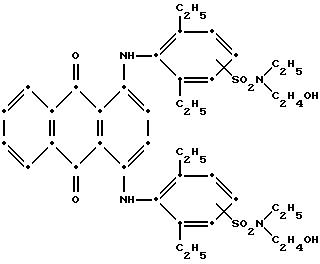
а красное соединение формулы V представляет собой
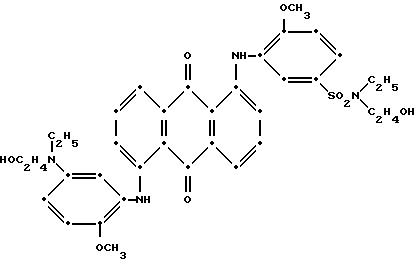
11. Тонирующий состав премикса, содержащий смесь по меньшей мере одного синего соединения 1,4-бис(2, 6-диалкиланилино)антрахинона, имеющего формулу I
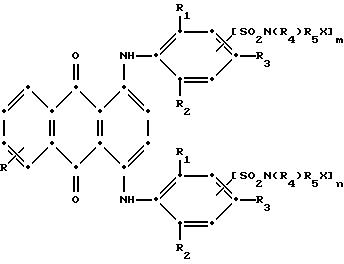
в которой R выбирают из группы, содержащей водород, С1-С6 -алкил, галоген, карбокси и

R1 и R2 независимо друг от друга представляют собой С1-С6-алкил;
R3 выбирают из группы, содержащей водород, галоген, С1-С6-алкил, замещенный С1-С6-алкил, гидрокси, С1-С6-алкокси, замещенный С1-С6-алкокси, циано, тиоциано, С1-С6-алкилтио, замещенный С1-С6-алкилтио, С1-С6-алкилсульфонил, замещенный С1-С6-алкилсульфонил, С1-С6-алкоксикарбонил, карбокси, арилокси, арилтио, арилсульфонил и SO2N(R4)R5X, если m и/или n = 0;
R4 выбирают из группы, содержащей водород, С1-С6-алкил, замещенный С1-С6 -алкил, С3-С8-алкенил, С3-С8-алкинил, С3-С7-циклоалкил и арил;
R5 связывающая группа, выбранная из группы, содержащей С1-С8-алкилен, С1-С6-алкилен-Z-С1-С6-алкилен, арилен-С1-С6-алкилен, арилен-Z-С1-С6-алкилен,
С3-С7-циклоалкилен, С1-С6-алкилен-циклоалкилен-С1-С6-алкилен, С1 -С6 -алкилен-арилен-С1-С6-алкилен и С1-С6-алкилен-Z-арилен-Z-С1-С6-алкилен, где Z выбирают из -О-, -S- или SO2;
X представляет собой галоген или реактивную полиэфирную группу;
m и n независимо друг от друга представляют собой 0 или 1; при условии, что присутствует по меньшей мере одна полиэфирная реактивная группа: вместе с по крайней мере одним красным соединением антрахинона или антрапиридона, выбранным из группы соединений, имеющих приведенные ниже формулы II - X:
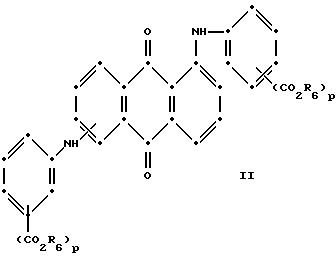
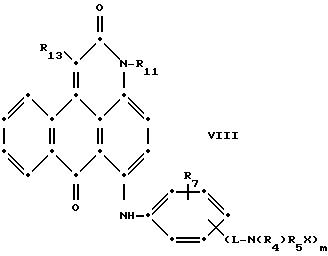
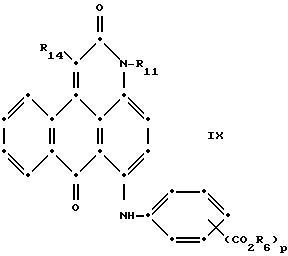
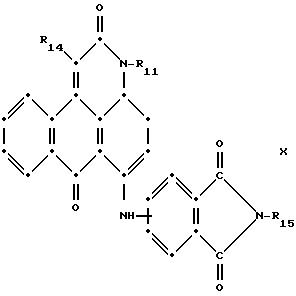
в которых R6 выбирают из группы, содержащей водород, С1-С6-алкил, замещенный С1-С6-алкил, С3-С7-циклоалкил или арил;
R7 представляет собой водород или от одной до трех групп, которые выбирают из С1-С6-алкила, замещенного С1-С6-алкила, С1-С6-алканоиламино, галогена, гидрокси, С1-С6-алкил-С1-С6-алкокси, С1-С6-алкилтио;
R8 и R9 одинаковые или разные, выбранные из группы, включающей С1-С6-алкил, замещенный С1-С6-алкил, С3-С7-циклоалкил или арил;
R10 выбирают из группы, включающей С1-С6-алкил, замещенный С1 -С6-алкил, С3-С7-циклоалкил или арил;
R11 выбирают из группы, содержащей водород, С1-С12-алкил, замещенный С1 -С12-алкил, С3-С7-циклоалкил или арил;
R12 представляет собой водород или от одной до трех групп, которые выбирают из С1-С6 -алкила, замещенного С1-С6-алкила, С1-С6-алкокси, замещенного С1-С6-алкокси, С1-С6-алкилтио, замещенного С1 -С6-алкилтио, С1-С6-алканоиламино, галогена, гидрокси, ароиламино, С1-С6-алкилсульфониламино и арилсульфониламино;
R13 и R14 выбирают из водорода, циано или CO2R10;
R15 представляет собой определенные ранее R4 или R5Х;
L представляет собой -СО- или -SO2-; Х имеет значения, определенные выше; m = 0 или 1; р = 1 или 2, при условии, что R13 представляет собой водород, когда m = 0.
Комментарии