Днк, повышающая продуцирование белка, и ее применение - RU2375450C2
Код документа: RU2375450C2
Чертежи













































































































































































































































Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к очищенным и выделенным последовательностям ДНК, имеющим активность по повышению продуцирования белка, и, более конкретно, к применению областей прикрепления матрикса (MAR) для повышения активности продуцирования белка в эукариотической клетке. Также описан способ идентификации указанных активных областей, в частности, нуклеотидных последовательностей MAR, и применение этих охарактеризованных активных последовательностей MAR в новом способе множественной трансфекции.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
В настоящее время принятой является модель организации эукариотических хромосом путем петлевых доменов (Boulikas T, "Nature of DNA sequences at the attachment regions of genes to the nuclear matrix", J.Cell Biochem., 52: 14-22,1993). Согласно данной модели хроматин организован в петли, которые охватывают 50-100 т.п.н., присоединенных к ядерному матриксу, белковой сети, состоящей из RNP и других негистоновых белков (Bode J, Stengert-Iber M, Kay V, Schalke T and Dietz-Pfeilstetter A, Crit. Rev. Euk. Gene Exp., 6: 115-138,1996).
Области ДНК, прикрепленные к ядерному матриксу, называются SAR или MAR, соответственно, по областям прикрепления к каркасу (во время метафазы) или матриксу (интерфаза) (Hart C and Laemmli U (1998), "Facilitation of chromatin dynamics by SARs" Curr Opin Genet Dev 8, 519-525).
Данные области как таковые могут определять границы независимых хроматиновых доменов, так что только окружающие цис-регуляторные элементы контролируют экспрессию генов в пределах данного домена.
Однако их способность полностью защищать хромосомный локус от близлежащих хроматиновых элементов и, таким образом, давать независимую от положения генную экспрессию не наблюдалась в стабильно трансфицированных клетках (Poljak L, Seum C, Mattioni T and Laemmli U. (1994) "SARs stimulate but do not confer position independent gene expression", Nucleic Acids Res 22, 4386-4394). С другой стороны, последовательности MAR (или S/MAR), как было показано, взаимодействуют с энхансерами, повышая доступность местного хроматина (Jenuwein T, Forrester W, Fernandez-Herrero L, Laible G, Dull M, and Grosschedl R. (1997) "Extension of chromatin accessibility by nuclear matrix attachment regions" Nature 385, 269-272). Конкретно, элементы MAR могут усиливать экспрессию гетерологичных генов в культивируемых клеточных линиях (Kalos M and Fournier R (1995) "Position-independent transgene expression mediated by boundary elements from the apolipoprotein B chromatin domain" Mol Cell Biol 15, 198-207), в трансгенных мышах (Castilla J, Pintado B, Sola, I, Sanchez-Morgado J, and Enjuanes L (1998) "Engineering passive immunity in transgenic mice secreting virus-neutralizing antibodies in milk" Nat Biotechnol 16, 349-354) и в растениях (Allen G, Hall GJ, Michalowski S, Newman W, Spiker S, Weissinger A, and Thompson W (1996), "High-level transgene expression in plant cells: effects of a strong scaffold attachment region from tobacco" Plant Cell 8, 899-913). Способность последовательностей MAR для разработки улучшенных векторов для генной терапии также выявлена (Agarwal M, Austin T, Morel F, Chen J, Bohnlein E, and Plavec I (1998), "Scaffold attachment region-mediated enhancement of retroviral vector expression in primary T cells" J Virol 72, 3720-3728).
Недавно было показано, что последовательности, модифицирующие структуру хроматина, включающие в себя MAR, на примере 5' MAR куриного лизоцима, способны значительно усиливать репортерную экспрессию в группах стабильных клеток яичника китайского хомячка (CHO) (Zahn-Zabal M, et al., "Development of stable cell lines for production or regulated expression using matrix attachment regions" J Biotechnol, 2001, 87(1): p. 29-42). Указанное свойство применяли для повышения доли высокопродуцирующих клонов, таким образом, снижая число клонов, которые необходимо подвергать скринингу. Указанные преимущества наблюдали в конструкциях с MAR, фланкирующими экспрессирующую трансген кассету, а также при совместной трансфекции конструкций с MAR на отдельной плазмиде. Однако уровни экспрессии после совместной трансфекции с MAR были не так высоки, как те, что наблюдались в конструкции, в которых две MAR ограничивают экспрессирующий трансген блок. Третий и предпочтительный процесс, как показано, представлял собой трансфекцию трансгенов с последовательностями MAR, как связанными с трангеном, так и находящимися на отдельной плазмиде (Girod et al., подано для публикации). Однако одним из сохраняющихся ограничений данного способа является количество ДНК на клетку, которое может трансфицироваться.
Многие протоколы множественной трансфекции разработаны для достижения высокой эффективности трансфекции при характеристике функции представляющих интерес генов. Протокол, применяемый Yamamoto et al, 1999 ("High efficiency gene transfer by multiple transfection protocol", Histochem. J. 31 (4), 241-243), приводит к эффективности трансфекции, примерно составляющей 80%, после 5 событий транфекции, в то время как при обычном протоколе трансфекции достигали уровня <40%. Хотя данный способ может использоваться, когда требуется повысить долю экспрессирующих клеток, он не приводит к образованию клеток с более высокой присущей им продуктивности. Поэтому он не может использоваться для образования моноклональных клеточных линий высокопродуцирующих клеток. Следовательно, ранее описанный способ имеет два основных недостатка: i) указанный способ не образует гомогенной популяции трансфицированных клеток, поскольку он не способствует интеграции дальнейшей генной копии, и не направляет трансгены в предпочтительные локусы хромосомы, ii) применение одного и того же маркера селекции в событиях множественной трансфекции не позволяет осуществлять селекцию дважды или трижды трансфицированных клеток.
В патентной заявке WO 02/074969 также было продемонстрировано применение MAR для разработки стабильных эукариотических клеточных линий. Однако в данной заявке не описано ни консервативной гомологии ДНК-элемента MAR, ни способа предсказания способности последовательности ДНК служить последовательностью MAR.
Фактически не было обнаружено какой-либо четкой консенсусной последовательности MAR (Boulikas T, "Nature of DNA sequences at the attachment regions of genes to the nuclear matrix", J. Cell Biochem., 52: 14-22, 1993), но эволюционно структура данных последовательностей кажется функционально консервативной в эукариотических геномах, поскольку MAR животных могут связываться с ядерными каркасами растений, и наоборот (Mielke C, Kohwi Y, Kohwi-Shigematsu T and Bode J, "Hierarchical binding of DNA fragments derived from scaffold-attached regions: correlation of properties in vitro and function in vivo", Biochemistry, 29: 7475-7485, 1990).
Идентификация MAR в биохимических исследованиях является длительным и непредсказуемым процессом; различные результаты могут быть получены в зависимости от анализа (Razin SV,"Functional architecture of chromosomal DNA domains", Crit Rev Eukaryot Gene Expr., 6: 247-269, 1996). С учетом огромного количества ожидаемых MAR в эукариотическом геноме и количества последовательностей, опубликованных в ходе геномных проектов, очень полезным был бы инструмент, способный отфильтровывать потенциальные MAR для проведения направленных экспериментов.
В настоящее время через Интернет доступны два различных предсказательных инструмента для MAR. Первый из них, MAR-Finder (http://futuresoft.org/MarFinder; Singh GB, Kramer JA and Krawetz SA, "Mathematical model to predict regions of chromatin attachment to the nuclear matrix", Nucleic Acid Research, 25: 1419-1425, 1997), основан на наборе профилей, идентифицированных в пределах различных MAR и на статистическом анализе совместного присутствия данных профилей. Предсказания MAR-Finder зависят от контекста последовательности, и это означает, что предсказанные MAR зависят от поданной на рассмотрение последовательности. Второе предсказательное программное обеспечение, SMARTest (http://www.genomatix.de; Frisch M, Frech K, Klingenhoff A, Cartharius K, Liebich I and Werner T, "In silico prediction of scaffold/matrix attachment regions in large genomic sequences", Genome Research, 12: 349-354,2001), использует весовые матрицы, происходящие из экспериментально идентифицированных MAR.
Сообщается, что SMARTest подходит для проведения широкомасштабных анализов. Но в действительности, из-за его относительно низкой специфичности количество гипотетических MAR при проведении им широкомасштабных анализов быстро становится огромным, и, поскольку SMARTest не имеет пути увеличения специфичности для снижения числа гипотетических MAR, он становится почти бесполезным для скрининга сильных MAR в больших последовательностях ДНК.
Также существуют некоторые другие программы, не доступные через Интернет; они также основаны на частоте мотивов MAR (критерий MRS; Van Drunen CM et al., "A bipartite sequence element associated with matrix/scaffold attachment regions", Nucleic Acids Res, 27: 2924-2930,1999), (ChrClass; Glazko GV et al., "Comparative study and prediction of DNA fragments associated with various elements of the nuclear matrix", Biochim. Biophys. Acta, 1517: 351-356,2001) или основаны на идентификации участков индуцированного стрессом ДНК-дуплекса (SIDD; Benham C and al., "Stress-induced duplex DNA destabilization in scaffold/matrix attachment regions", J. Mol. Biol., 274: 181-196, 1997). Однако остается неизвестным, подходят ли они для анализа геномных последовательностей, и не сообщается, могут ли данные инструменты обеспечить идентификацию повышающих продуцирование белка последовательностей.
Более того, вследствие относительно низкой специфичности данных программ (Frisch M, Frech K, Klingenhoff A, Cartharius K, Liebich I and Werner T, "In silico prediction of scaffold/matrix attachment regions in large genomic sequences", Genome Research, 12: 349-354, 2001) количество гипотетических MAR, идентифицированных в геномах, быстро становится неуправляемым при проведении широкомасштабных анализов, особенно если большинство из них не имеют активности или имеют слабую активность на практике. Таким образом, в отсутствие способа повышения специфичности предсказания для снижения числа гипотетических MAR многие из доступных программ становятся почти бесполезными для идентификации действенных генетических элементов в свете эффективного повышения продуцирования рекомбинантного белка.
Поскольку все указанные выше доступные способы предсказания имеют некоторые недостатки, которые предотвращают широкомасштабный анализ геномов для надежной идентификации новых и потенциальных MAR, целью данного изобретения является 1) понимание функциональных характеристик MAR, которые обеспечат улучшенную экспрессию рекомбинантного белка; 2) получение нового биоинформационного инструмента, компилирующего структурные характеристики MAR в виде предсказания функции, 3) выполнение широкомасштабных анализов геномов с целью идентификации новых и более действенных MAR и, наконец, 4) демонстрация улучшенной эффективности для повышения продуцирования рекомбинантных белков из эукариотических клеток или организмов при использовании новых идентифицированных последовательностей MAR.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данной цели достигали путем предоставления улучшенного и надежного способа идентификации последовательностей ДНК, имеющей активность по повышению продуцирования белка, в частности нуклеотидных последовательностей MAR, и применения данных охарактеризованных активных последовательностей MAR в новом способе множественной трансфекции для повышения продуцирования рекомбинантных белков в эукариотических клетках.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 приведены графики распределения MAR и не относящихся к MAR последовательностей. Гистограммы представляют собой графики плотности (относительная частота, разделенная на ширину элемента) по отношению к значению наблюдаемого параметра. Гистограмма плотности человеческих MAR в базе данных SMARt DB показана в черном цвете, тогда как гистограммы плотности для человеческой хромосомы 22 приведены серым.
На фиг.2 приведены диаграммы рассеяния четырех различных критериев, использованных SMAR Scan®, и содержание AT в человеческих MAR из SMARt DB.
На фиг. 3 приведены графики распределения последовательностей MAR в организме. Последовательности MAR других организмов получены из SMARt и проанализированы. Распределения плотности последовательностей MAR для мыши, курицы, Sorghum bicolor и человека нанесены совместно.
На фиг.4 приведены предсказания SMAR Scan® по человеческой хромосоме 22 и по перетасованной человеческой хромосоме 22. Верхний график: среднее число положительных результатов SMAR Scan®, полученных на пятикратно разрушенной, смешанной перетасованной неперекрывающимися окнами длиной 10 п.н. хромосоме 22 в модели Марковской цепи 1 порядка, и на нативной хромосоме 22. Нижний график: среднее число MAR, предсказанных SMAR Scan®.
На фиг.5 показан анализ способности 5'-MAR гена куриного лизоцима стимулировать экспрессию трансгена в клетках CHO-DG44. Фрагменты B, K и F характеризуются наиболее высокой способностью стимулировать экспрессию трансгена. Указанная относительная сила элементов основана на числе клеток с высоким уровнем экспрессии.
На фиг.6 показан эффект серийных делеций 5'-конца (верхняя часть) и 3'-конца (нижняя часть) 5'-MAR на потерю способности стимулировать экспрессию трансгена. Переход от повышенной к пониженной активности перекрывается с B-, K- и F-фрагментами.
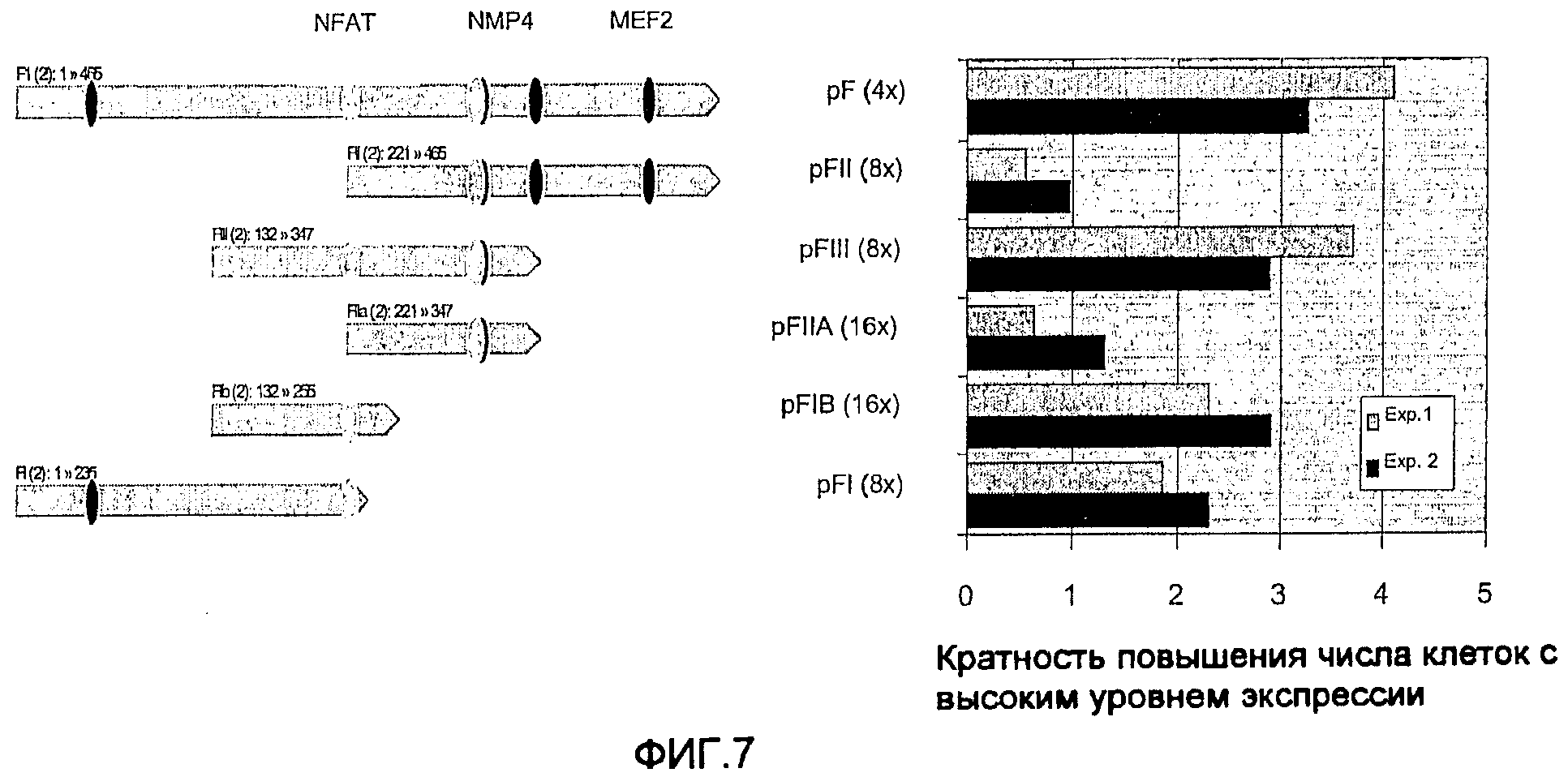
На фиг.7 показано, что части F-фрагмента значимо стимулируют экспрессию трансгена. Области F-фрагмента, обозначенные светло-серой стрелкой, были мультимеризованы, встроены в pGEGFP-контроль и трансфицированы в клетки CHO. Элемент, который характеризуется наивысшей активностью, расположен в центральной части элемента и соответствует фрагменту Fill (помеченная черной полосой минимальная MAR). Кроме того, энхансерная активность расположена в 3'-фланкирующей части фрагмента Fill (помеченный темно-серой полосой энхансер MAR).
На фиг.8 приведена карта положений различных мотивов последовательности ДНК в cLysMAR.
На фиг.8 (B) представлена карта положений различных мотивов последовательности ДНК в cLysMAR. Вертикальные линии представляют собой положение предсказанных на компьютере участков или мотивов последовательности по 3034 парам оснований cLysMAR и его активных областей, как представлено на фиг.5. Предполагаемые участки факторов транскрипции (MEF2 05, Oct-1, USF-02, GATA, NFAT) для активаторов и (CDP, SATB1, CTCF, ARBP/MeCP2) для репрессоров транскрипции идентифицированы с использованием Matinspector (Genomatix), и островки CpG идентифицировали с использованием CPGPLOT. Мотивы, ранее ассоциированные с MAR-элементами, помечены черным и включают в себя CpG-динуклеотиды и CpG-островки, мотивы раскручивания (AATATATT и AATATT), поли-As и Ts, поли-Gs и Cs, участки связывания топоизомеразы II дрозофилы (GTNWAYATTNATTNATNNR), которые идентичны ядру длиной 6 п.н. и участкам связывания группы высокой подвижности I (HMG-I/Y). Другие структурные мотивы включают в себя участки связывания нуклеосомы и участки, неблагоприятные для связывания нуклеосомы, и мотив, который, как полагают, ослабляет сверхспирализованную цепь ДНК. На фиг.8 (A) приведено сравнение способности частей cLysMAR активировать транскрипцию с профилями коэффициента предсказания MAR, полученными посредством MarFinder. На верхней диаграмме показана активность фрагмента MAR, как на фиг.5, в то время как средняя и нижняя кривые показывают предсказанный MARFinder потенциал в плане активности MAR и в плане образования структур изогнутой ДНК соответственно.
На фиг.9 показана корреляция физико-химических свойств ДНК с активностью MAR. На фиг.9(A) представлены профили температуры плавления ДНК, изгибания двойной спирали, глубины большой бороздки и ширины малой бороздки 5'-MAR, и они определены с использованием алгоритмов Levitsky et al (Levitsky VG, Ponomarenko MP, Ponomarenko JV, Frolov AS, Kolchanov NA "Nucleosomal DNA property database", Bioinformatics, 15; 582-592, 1999). Наиболее активные фрагменты B, K и F, обозначенные вверху, соответствуют показанным на фигуре 1. На фиг.9(B) представлено увеличение данных, представленных на панели A для демонстрации карты фрагмента F, выровненной с графиками, соответствующими температуре плавления (верхняя кривая) и изгибанию ДНК (нижняя кривая). Положение наиболее активного фрагмента FIB и участка связывания белков - специфичных факторов транскрипции соответствует указанному.
На фиг.10 показано распределение предполагаемых участков связывания факторов транскрипции в пределах 5'-cLysMAR. Большие стрелки указывают положение элементов CUE, идентифицированных SMAR Scan®.
На фиг.11 показана схема соединения различных частей MAR. Указанные части cLysMAR амплифицировали путем ПЦР, вводили линкерные элементы BglII-BamHI с каждого конца и соединяли для получения обозначенных составных элементов. Например, верхняя конструкция состоит из соединения всех CUE и фланкирующих последовательностей в их исходном расположении, за исключением того, что каждый элемент отделяют линкерные последовательности BglII-BamHI.
На фиг.12 представлены карты плазмид.
На фиг.13 показано действие повторной транфекции первичных трансфектантов на экспрессию GFP. Клетки (CHO-DG44) повторно трансфицировали pSV40EGFP (левая пробирка) или pMAR-SV40EGFP (центральная пробирка) и pSVneo в качестве плазмиды устойчивости. Клетки, трансфицированные pMAR-SV40EGFP, повторно трансфицировали через 24 часа той же плазмидой и другой плазмидой селекции, pSVpuro (правая пробирка). После селекции в течение двух недель фенотип стабильно трансфицированной клеточной популяции анализировали посредством FACS.
На фиг.14 показано действие множественной загрузки содержащей MAR плазмиды. Вторичные трансфектанты pMAR-SV40EGFP/pMAR-SV40EGFP использовались в третьем цикле трансфекции в конце процесса селекции. Третичную трансфекцию проводили pMAR или pMAR-SV40EGFP с получением третичных трансфектантов. Через 24 часа клетки опять трансфицировали каждой плазмидой, происходящей из четвертичных трансфектантов (см. таблицу 4).
На фиг.15 показана сравнительная действенность алгоритмов предсказания SMAR на примере области WP18A10A7.
(A) Анализ путем SMAR Scan® проводили с настройками по умолчанию.
(B) Анализ SIDD (верхняя кривая и шкала слева) и связывание некоторых фрагментов ДНК с ядерным матриксом in vitro (гистограмма, шкала справа) приводили по Goetze et al (Goetze S, Gluch A, Benham C, Bode J, "Computational and in vitro analysis of destabilized DNA regions in the interferon gene cluster: potential of predicting functional gene domains." Biochemistry, 42: 154-166, 2003).
На фиг. 16 представлены результаты протокола, подобного генной терапии, с использованием MAR. Группа мышей, подвергнутая инъекции MAR-системой, индуцированной в начале эксперимента, характеризовалась улучшенной индукцией гематокрита по сравнению с мышами, инъецированными исходной системой без MAR. Через 2 месяца значения гематокрита в «МAR-содержащей группе» оставались по-прежнему на более высоком уровне (65%) по сравнению с нормальным гематокритом (45-55%).
На фиг.17 представлена диаграмма рассеяния процентных долей нуклеотидов AT (верх) и TA (низ) для 1757 последовательностей S/MAR в зависимости от предсказанного изгибания ДНК, вычисленного посредством SMAR Scan®.
На фиг.18 представлены графики распределения процентных долей динуклеотидов по 1757 не относящимся к S/MAR последовательностям.
На фиг.19 показано влияние различных элементов S/MAR на продуцирование рекомбинантного зеленого флуоресцирующего белка (GFP). Популяции клеток CHO, трансфицированных экспрессирующим GFP вектором, содержащим MAR-элемент, как указано, анализировали на сортере активированных флуоресценцией клеток (FACS®), и приведены типичные профили. Профили показывают подсчет числа клеток как функцию уровня флуоресценции GFP.
На фиг.20 обозначено действие индукции гематокрита у мышей, подвергнутых инъекции MAR-системой.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к очищенным и выделенным последовательностям ДНК, имеющим активность по повышению продуцирования белка, характеризующимся тем, что указанная последовательность ДНК включает в себя по меньшей мере один элемент изогнутой ДНК и по меньшей мере один участок связывания ДНК-связывающего белка.
Некоторые последовательности ДНК, как известно, образуют относительно «статичную кривую», где ДНК следует конкретному 3-мерному пути. Таким образом, вместо пребывания в нормальной В-конформации ДНК («прямой»), отрезок ДНК может образовывать плоскую, планарную кривую, также определяемую как изогнутая ДНК (Marini, et al, 1982 "Bent helical structure in kinetoplast DNA", Proc. Natl. Acad. Sci. USA, 79: 7664-7664).
Неожиданно авторами настоящего изобретения было показано, что элемент изогнутой ДНК из очищенной и выделенной последовательности ДНК, имеющей активность по повышению продуцирования белка, согласно изобретению обычно содержит по меньшей мере 10% динуклеотида TA и/или по меньшей мере 12% динуклеотида AT в отрезке из 100 следующих друг за другом пар оснований. Предпочтительно элемент изогнутой ДНК содержит по меньшей мере 33% динуклеотида TA и/или по меньшей мере 33% динуклеотида AT в отрезке из 100 следующих друг за другом пар оснований. Эти данные получены описанным далее способом.
Очищенная и выделенная последовательность ДНК согласно изобретению обычно содержит нуклеотидную последовательность MAR, выбранную из группы, состоящей из последовательностей SEQ ID No: 1-27, или элемент cLysMAR или его фрагмент.
Предпочтительно очищенная и выделенная последовательность ДНК представляет собой нуклеотидную последовательность MAR, выбранную из группы, состоящей из последовательностей SEQ ID No: 1-27, более предпочтительно из последовательностей SEQ ID No: 24-27.
Также к настоящему изобретению относятся комплементарные последовательности указанных выше последовательностей SEQ ID No: 1-27 и элемент или фрагмент cLysMAR, который может быть продуцирован с использованием ПЦР или другими средствами.
«Элемент» представляет собой консервативную нуклеотидную последовательность, которая несет общие функциональные свойства (т.е. участки связывания факторов транскрипции) или структурные черты (т.е. последовательность изогнутой ДНК).
Часть последовательностей SEQ ID No: 1-27 и элемента или фрагмента cLysMAR относится к последовательностям, имеющим по меньшей мере 70% нуклеотидов в длину с последовательностью, соответствующей SEQ ID No: 1-27. Данные последовательности могут использоваться до тех пор, пока они проявляют те же свойства, что и природная последовательность, из которой они происходят. Предпочтительно данные последовательности имеют более 80%, в частности более 90%, нуклеотидов в длину по сравнению с соответствующей последовательностью SEQ ID No: 1-27.
Настоящее изобретение также включает в себя варианты указанных выше последовательностей SEQ ID No: 1-27 и элемента или фрагмента cLysMAR, то есть нуклеотидные последовательности, которые изменяются по сравнению с указанными последовательностями за счет консервативных нуклеотидных замен, при которых один или несколько нуклеотидов замещаются на другие с теми же характеристиками.
Последовательности SEQ ID No: 1-23 идентифицировали путем сканирования человеческой хромосомы 1 и 2 с использованием SMAR Scan®, показав, что идентификации новых последовательностей MAR реальна с использованием описанных здесь и далее инструментов, тогда как SEQ ID No: 24-27 идентифицировали путем сканирования полного генома человека с использованием комбинированного способа SMAR Scan®.
На первой стадии полноразмерную хромосому 1 и 2 подвергали скринингу для идентификации элемента изогнутой ДНК как области, соответствующей наиболее сильному изгибу, глубине большой бороздки, ширине малой бороздки и минимальной температуре плавления, показанным на фигуре 3. На второй стадии данный набор последовательностей сканировали на предмет участков связывания регуляторных белков, таких как SATB1, GATA, и т.д., как показано на фигуре 8B), что давало последовательности SEQ ID No: 1-23.
Более того, последовательности 21-23, как было показано далее, расположены за известным геном из Human Genome Data Base.
Что касается SEQ ID No: 24-27, то данные последовательности были получены путем сканирования человеческого генома комбинированным способом и выбраны в качестве примеров из 1757 MAR-элементов, выявленных таким путем.
Молекулярные химеры последовательностей MAR также относятся к настоящему изобретению. Под молекулярной химерой подразумевается нуклеотидная последовательность, которая может включать в себя функциональную часть элемента MAR и которая может быть получена способами молекулярной биологии, известными специалистам в данной области.
Конкретные комбинации элементов или фрагментов MAR или их подчастей также относятся к настоящему изобретению. Данные фрагменты могут быть получены различными способами, известными в данной области. Данные способы включают в себя в качестве неограничивающих примеров расщепление ферментами рестрикции и получение фрагментов, химический синтез или полимеразные цепные реакции (ПЦР).
Поэтому конкретные комбинации элементов или фрагментов последовательностей SEQ ID No: 1-27 и элементов или фрагментов cLysMAR также относятся к настоящему изобретению, в зависимости от функциональных результатов, которые нужно получить. Элементы cLysMAR, например, представляют собой B-, K- и F-области, описанные в WO 02/074969, описание которых включено сюда полностью в качестве ссылки. Предпочтительные элементы cLysMAR, использованные согласно изобретению, представляют собой B-, K- и F-области. Только один элемент может использоваться, или могут использоваться множественные копии одного и того же или различных элементов (мультимеризованные элементы) (см. фиг.8 A)).
Под фрагментом подразумевается часть соответствующей нуклеотидной последовательности. Фрагменты нуклеотидной последовательности могут сохранять биологическую активность и, следовательно, связываться с очищенными ядерными матриксами и/или изменять профили экспрессии кодирующих последовательностей, функционально связанных с промотором. Фрагменты нуклеотидной последовательности MAR могут изменяться примерно от 100 до 1000 п.н., предпочтительно примерно от 200 до 700 п.н., более предпочтительно примерно от 300 до 500 п.н. Также рассматриваются любые комбинации фрагментов, которые имеют то же число нуклеотидов, что присутствует в синтетической последовательности MAR, состоящей из природного элемента MAR и/или фрагментов. Фрагменты предпочтительно соединены линкерными последовательностями. Предпочтительные линкеры представляют собой линкер BglII-BamHI.
«Активность по повышению продуцирования белка» относится к активности очищенной и выделенной последовательности ДНК, определяемой следующим образом: после введения в подходящих условиях в эукариотическую клетку хозяина последовательность способна повышать уровень продуцирования белка в культуре клеток по сравнению с культурой клеток, трансфицированной без указанной последовательности ДНК. Обычно повышение составляет от 1,5- до 10-кратного, предпочтительно от 4- до 10-кратного. Это соответствует скорости продуцирования или специфичной клеточной продуктивности, составляющей по меньшей мере 10 пг на клетку в сутки (см. пример 11 и фиг.13).
При использовании здесь предложены следующие определения для облегчения понимания данного изобретения.
«Хроматин» представляет собой белковый материал и материал нуклеиновой кислоты, составляющий хромосомы эукариотической клетки, и относится к ДНК, РНК и ассоциированным белкам.
«Элемент хроматина» означает последовательность нуклеиновой кислоты на хромосоме, обладающую свойствами по модификации структуры хроматина при интеграции в данную хромосому.
«Цис» относится к расположению двух или большего числа элементов (таких как элементы хроматина) на одной молекуле нуклеиновой кислоты (такой как один и тот же вектор, плазмида или хромосома).
«Транс» относится к расположению двух или большего числа элементов (таких как элементы хроматина) на двух или большем числе различных молекул нуклеиновой кислоты (например, на двух векторах или двух хромосомах).
Модифицирующие хроматин элементы, которые потенциально способны преодолевать эффекты положения и, следовательно, вызывают интерес в плане разработки стабильных клеточных линий, включают в себя граничные элементы (BE), участки прикрепления матрикса (MAR), области контроля локуса (LCR) и универсальные элементы открытия хроматина (UCOE).
Граничные элементы («BE»), или инсуляторные элементы, во многих случаях определяют границы хроматина (Bell A and Felsenfeld G. 1999; "Stopped at the border: boundaries and insulators, Curr Opin Genet Dev 9, 191-198) и могут играть роль в определении транскрипционного домена in vivo. BE не имеют присущей промоторной/энхансерной активности, но, тем не менее, как полагают, защищают гены от транскрипционного влияния регуляторных элементов в окружающем хроматине. Для идентификации инсуляторных элементов обычно применяют анализ энхансерного блока. В данном анализе элемент хроматина помещают между энхансером и промотором и измеряют активированную энхансером транскрипцию. Граничные элементы, как было показано, могут защищать стабильно трансфицированные репортерные гены от эффектор положения в клетках дрозофилы, дрожжей и млекопитающих. Также было показано, что они повышают долю трансгенных мышей с индуцированной экспрессией трансгена.
Области контроля локуса («LCR») являются цис-регуляторными элементами, требуемыми для исходной активации хроматина локуса и последующей транскрипции генов в их природных участках (Grosveld, F. 1999,"Activation by locus control regions?" Curr Opin Genet Dev 9, 152-157). Активирующая функция LCR также обеспечивает экспрессию связанного трансгена в подходящей ткани у трансгенных мышей, независимо от участка интеграции в геном хозяина. Хотя LCR, в общем, обеспечивают тканеспецифический уровень экспрессии связанных генов, эффективная экспрессия почти во всех тканях трансгенных мышей сообщалась для укороченного LCR человеческого T-клеточного рецептора и LCR крысиного LAP. Наиболее интенсивно охарактеризованным LCR является таковой из локуса глобина. В настоящее время оценивается его применение в векторах для генной терапии серповидноклеточной анемии и β-талассемии.
«MAR» по принятой модели могут опосредовать заякоривание специфичной последовательности ДНК в ядерном матриксе с образованием петлевых доменов хроматина, которые простираются в стороны от ядер гетерохроматина. Хотя MAR не содержат какой-либо очевидной консенсусной или распознаваемой последовательности, их наиболее присущей чертой оказывается общее высокое содержание A/T и преобладание оснований C на одной цепи (Bode J, Schlake T, Rios Ramirez M, Mielke C, Stengart M, Kay V and Klehr Wirth D, "Scaffold/matrix-attached regions: structural propreties creating transcriptionally active loci", Structural and Functional Organization of the Nuclear Matrix: International Review of Citology, 162A: 389-453,1995). Данные области имеют склонность образовывать изогнутые вторичные структуры, которые склонны к разделению цепей. Они часто обозначаются как области неспаренных оснований (BUR), и они содержат элемент-ядро раскручивания (CUE), который может представлять собой точку образования разделения цепей (Benham C and al., Stress induced duplex DNA destabilization in scaffold/matrix attachment regions, J. Mol Biol, 274: 181-196, 1997). Некоторые простые AT-богатые мотивы последовательности часто находятся в последовательностях MAR, но, по большей части, их функциональная значимость и потенциальный способ действия остается неясным. Данные мотивы включают в себя A-бокс (AATAAAYAAA), T-бокс (TTWTWTTWTT), мотивы раскручивания ДНК (AATATATT, AATATT), участки связывания SATB1 (H-бокс, A/T/C25) и консенсусные участки топоизомеразы II позвоночных (RNYNNCNNGYNGKTNYNY) или дрозофилы (GTNWAYATTNATNNR).
Универсальные элементы открытия хроматина («UCOE», также известные как «повсеместно действующие элементы открытия хроматина») описаны в WO 00/05393.
«Энхансер» представляет собой нуклеотидную последовательность, которая действует, усиливая транскрипцию генов независимо от сущности гена, положения данной последовательности по отношению к гену или ориентации данной последовательности. Векторы согласно изобретению необязательно включают в себя энхансеры.
«Ген» представляет собой дезоксирибонуклеотидную (ДНК) последовательность, кодирующую данный зрелый белок.
Используемый здесь термин «ген» не будет включать в себя нетранслируемые фланкирующие области, такие как сигналы инициации транскрипции РНК, участки полиаденилирования, промоторы или энхансеры.
«Ген продукта» представляет собой ген, который кодирует белковый продукт, имеющий требуемые характеристики, такие как диагностическое или терапевтическое применение. Ген продукта включает в себя, например, структурные гены и регуляторные гены.
«Структурный ген» относится к гену, который кодирует структурный белок. Неограничивающие примеры структурных генов включают в себя белки цитоскелета, белки внеклеточного матрикса, ферменты, белки ядерных пор и белки ядерного каркаса, ионные каналы и транспортеры, контрактильные белки и шапероны. Предпочтительные структурные гены кодируют антитела или фрагменты антител.
«Регуляторный ген» относится к гену, который кодирует регуляторный белок. Неогранивающие примеры регуляторных белков включают факторы транскрипции, гормоны, факторы роста, цитокины, молекулы трансдукции сигнала, онкогены, протоонкогены, трансмембранные рецепторы и протеинкиназы.
«Ориентация» относится к порядку нуклеотидов в данной последовательности ДНК. Например, обращенная ориентация последовательности ДНК представляет собой ту, в которой порядок 5'-3' последовательности по отношению к другой последовательности обратен по отношению к точке сравнения в ДНК, из которой получали данную последовательность. Такие точки сравнения могут включать в себя направление транскрипции других определенных последовательностей ДНК в исходной ДНК и/или точки начала репликации реплицируемых векторов, содержащих последовательность.
«Эукариотическая клетка» относится к любой клетке млекопитающего и к не относящейся к млекопитающему клетке из эукариотического организма. В качестве неограничивающего примера к изобретению относится любая эукариотическая клетка, способная поддерживаться в условиях культивирования клеток и впоследствии трансфицироваться. Особо предпочтительные клеточные типы включают в себя, например, стволовые клетки, эмбриональные стволовые клетки, клетки яичника китайского хомячка (CHO), COS, BHK21, NIH3T3, HeLa, C2C12, раковые клетки и первичные дифференцированные или недифференцированные клетки. Другие подходящие клетки хозяина известны специалистам в данной области.
Термины «клетка хозяина» и «рекомбинантная клетка хозяина» используются здесь взаимозаменяемо и обозначают эукариотическую клетку, в которую введены один или несколько векторов по изобретению. Понятно, что такие термины относятся не только к конкретной указанной клетке, но также и к потомству и потенциальному потомству такой клетки. Поскольку в последовательных поколениях могут происходить некоторые модификации вследствие мутации или влияний окружающей среды, такое потомство, фактически, может не быть идентично родительской клетке, но все равно входит в объем применяемого здесь термина.
Термины «введение очищенной ДНК в эукариотические клетки хозяина» или «трансфекция» обозначают любой способ, в котором внеклеточная ДНК, с сопровождающим материалом или без него, входит в клетку хозяина. Термин «клетка трансфицирована» или «трансфицированная клетка» означает клетку, в которую введена внеклеточная ДНК, и она, таким образом, несет внеклеточную ДНК. ДНК может вводиться в клетку так, что нуклеиновая кислота может реплицироваться в качестве интегрированного в хромосому или экстрахромосомного элемента.
Используемый здесь термин «промотор» относится к последовательности нуклеиновой кислоты, которая регулирует экспрессию гена.
«Совместная трансфекция» означает процесс трансфицирования эукариотической клетки более чем одним экзогенным геном, или вектором, или плазмидой, чужеродной в отношении данной клетки, один или несколько из которых придает клетке подлежащий селекции фенотип.
Очищенная и выделенная последовательность ДНК, имеющая активность по повышению продуцирования белка, также включает в себя, кроме одного или нескольких элементов изогнутой ДНК по меньшей мере один участок связывания ДНК-связывающего белка.
Обычно ДНК-связывающий белок представляет собой фактор транскрипции. Примерами факторов транскрипции служит группа, содержащая белки с доменом polyQpolyP. Другим примером фактора транскрипции является фактор транскрипции, выбранный из группы, включающей в себя SATB1, NMP4, MEF2, S8, DLX1, FREAC7, BRN2, GATA 1/3, TATA, Bright, MSX, AP1, C/EBP, CREBP1, FOX, Freac7, HFH1, HNF3-альфа, Nkx25, POU3F2, Pit1, TTF1, XFD1, AR, C/EBP-гамма, Cdc5, FOXD3, HFH3, HNF3-бета, MRF2, Oct1, POU6F1, SRF, V$MTATA_B, XFD2, Bach2, CDP CR3, Cdx2, FOXJ2, HFL, HP1, Myc, PBX, Pax3, TEF, VBP, XFD3, Brn2, COMP1, Evil, FOXP3, GATA4, HFN1, Lhx3, NKX3A, POU1F1, Pax6, TFIIA, или предпочтительна комбинация одного или нескольких факторов транскрипции. Наиболее предпочтительными являются SATB1, NMP4, MEF2 и белки с доменом polyQpolyP.
SATB1, NMP4 и MEF2, например, как известно, регулируют развитие и/или тканеспецифическую генную экспрессию у млекопитающих. Данные факторы транскрипции способны изменять геометрию ДНК, и, наоборот, связывание с ДНК, как с аллостерическим лигандом, модифицирует их структуру. Недавно было обнаружено, что SATB1 образует подобную клетке структуру, ограничивающую хроматин (Cai S, Han HJ, and Kohwi-Shigematsu T, "Tissue-specific nuclear architecture and gene expression regulated by SATB1" Nat Genet, 2003, 34(1): p. 42-51).
Еще одной целью настоящего изобретения является предоставление очищенного и выделенного элемента cLysMAR и/или его фрагмента, комплементарной ему последовательности, его части, захватывающей по меньшей мере 70% нуклеотидов в длину, его молекулярной химеры, их комбинаций и вариантов.
Наиболее предпочтительно элемент cLysMAR и/или его фрагмент состоят по меньшей мере из одной последовательности нуклеиновой кислоты, выбранной из B-, K- и F-областей.
Дальнейшей целью настоящего изобретения является предоставление синтетической последовательности MAR, содержащей природный элемент MAR и/или фрагменты, соединенные друг с другом линкерными последовательностями.
Предпочтительно синтетическая последовательность MAR включает в себя элемент cLysMAR, и/или его фрагмент, комплементарную ему последовательность, его часть, захватывающую по меньшей мере 70% нуклеотидов в длину, его молекулярную химеру, их комбинации и варианты.
Также предпочтительно, что линкерные последовательности представляют собой линкер BglII-BamHI.
Другим аспектом изобретения является предоставление способа идентификации последовательности MAR с использованием биоинформационного инструмента, включающего в себя вычисление значений одной или нескольких характеристик последовательности ДНК, соответствующих изгибанию ДНК, потенциалам глубины большой бороздки и ширины малой бороздки и температуре плавления. Предпочтительно идентификация одной или нескольких характеристик последовательности ДНК далее включает в себя выявление дальнейших характеристик последовательности ДНК, соответствующих участка связывания ДНК-связывающих белков, что также вычисляется данным способом.
Предпочтительно профили или весовые матрицы указанного биоинформационного инструмента основаны на распознавании динуклеотидов.
Биоинформационный инструмент, используемый для настоящего способа, предпочтительно представляет собой SMAR Scan®, который содержит алгоритмы, разработанные Gene Express (http://srs6.bionet.nsc.ru/srs6bin/cgi-bin/wgetz?-e+[FEATURES-SiteID:'nR']) и основанные на Levitsky et al., 1999. Данные алгоритмы распознают профили, основанные на динуклеотидных весовых матрицах, для вычисления теоретических значений конформационных и физико-химических свойств ДНК.
Предпочтительно в SMAR Scan® используются четыре теоретических критерия, также обозначенные как характеристики последовательности ДНК, соответствующие изгибанию ДНК, потенциалам глубины большой бороздки и ширины малой бороздки, температуре плавления, во всех возможных комбинациях, с использованием сканирующих окон различного размера (см. фиг.3). Для каждой применяемой функции следует устанавливать уровень отсечения. Программа возвращает соответствующую критерию область каждый раз, когда вычисленный коэффициент для данной области превышает установленный уровень отсечения для всех выбранных критериев. Два способа выходных данных доступны для управления данными соответствующими критерию областями, первый (называемый «подобный профилю») просто возвращает все положения последовательности запроса и соответствующие им значения для различных выбранных критериев. Второй вариант (называемый «прилегающие соответствующие критерию области») возвращает только положения нескольких прилегающих друг к другу соответствующих критерию областей и их соответствующую последовательность. Для данного варианта минимальное число прилегающих друг к другу соответствующих критерию областей представляет собой еще один уровень отсечения, который опять может быть установлен с применением настраиваемого размера окна. Данный второй вариант является вариантом по умолчанию SMAR Scan®. Действительно, с семантической точки зрения, соответствующая критерию область рассматривается как элемент-ядро раскручивания (CUE), и кластер CUE, сопровождаемый кластерами участков связывания для релевантных белков, считается MAR. Таким образом, SMAR Scan® рассматривает в качестве потенциальной MAR только несколько прилегающих соответствующих критерию областей.
Для настройки установленных по умолчанию уровней отсечения четырех теоретических структурных критериев использовали экспериментально подтвержденные MAR из SMARt DB (http://transfac.gbf.de/-SMARt DB). Получали все человеческие последовательности MAR из этой базы данных и анализировали их посредством SMAR Scan® с использованием «подобного профилю» варианта с четырьмя критериями и без установленного уровня отсечения. Это позволяло установить каждую функцию для каждого положения данных последовательностей. Распределение каждого критерия вычисляли по этим данным (см. фиг.1 и 3).
Установленные уровни отсечения SMAR Scan® для изгиба, глубины большой бороздки и ширины малой бороздки установлены на среднем значении 75-го квантиля и медианы.
Для температуры плавления уровень отсечения по умолчанию следует установить на 75-м квантиле.
Минимальная длина для варианта «прилегающих соответствующих критерию областей» следует установить на уровне 300, поскольку это предполагаемая минимальная длина MAR (см. фиг. 8 и 9). Однако специалист в данной области сможет определить уровни отсечения указанных выше критериев для данного организма с минимальным уровнем экспериментирования.
Предпочтительно уровни изгибания ДНК включают в себя от 3 до 5° (радикальных градусов). Наиболее предпочтительно они располагаются от 3,8 до 4,4, что соответствует наименьшему пику на фиг.1.
Предпочтительно значения глубины большой бороздки составляют от 8,9 до 9,3 Å (Ангстрем), и значения ширины малой бороздки от 5,2 до 5,8 Å. Наиболее предпочтительно значения глубины большой бороздки составляют от 9,0 до 9,2 Å, и значения ширины малой бороздки от 5,4 до 5,7 Å.
Предпочтительно температура плавления составляет от 55 до 75°C (градусов Цельсия). Наиболее предпочтительно температура плавления составляет от 55 до 62°C.
ДНК-связывающий белок, значения, соответствующие которому, могут быть вычислены данным способом, обычно представляет собой фактор транскрипции, предпочтительно с доменом polyQpolyP, или фактор транскрипции, выбранный из группы, включающей в себя SATB1, NMP4, MEF2, S8, DLX1, FREAC7, BRN2, GATA 1/3, TATA, Bright, MSX, AP1, C/EBP, CREBP1, FOX, Freac7, HFH1, HNF3-альфа, Nkx25, POU3F2, Pit1, TTF1, XFD1, AR, C/EBP-гамма, Cdc5, FOXD3, HFH3, HNF3-бета, MRF2, Oct1, POU6F1, SRF, V$MTATA_B, XFD2, Bach2, CDP CR3, Cdx2, FOXJ2, HFL, HP1, Myc, PBX, Pax3, TEF, VBP, XFD3, Brn2, COMP1, Evil, FOXP3, GATA4, HFN1, Lhx3, NKX3A, POU1F1, Pax6, TFIIA, или из комбинации одного или нескольких факторов транскрипции.
Однако специалист в данной области сможет определить другие типы факторов транскрипции для осуществления способа согласно изобретению.
Если SMAR Scan® рассматривается, например, для проведения широкомасштабного анализа, то предпочтительно указанный выше способ далее включает в себя по меньшей мере один фильтр, предсказывающий ДНК-связывающие участки для факторов транскрипции ДНК для снижения объема вычислений.
Принцип данного способа комбинирует SMAR Scan® для сравнения структурных характеристик, описанных выше, и фильтр, такой как, например, pfsearch (из пакета pftools, описанного Bucher P, Karplus K, Moeri N, and Hofmann K, "A flexible search technique based on generalized profiles", Computers and Chemistry, 20: 324, 1996) для предсказывания связывания некоторых факторов транскрипции.
Неограничивающие примеры фильтров включают в себя pfsearch, Matinspector, RMatch Professional и TRANSFAC Professional.
В данном комбинированном способе используются структурные характеристики SMAR Scan® и предсказанное связывание специфичных факторов транскрипции из фильтра, которые могут применяться последовательно в любом порядке для выбора MAR, поэтому в зависимости от фильтра он применяется в начале или в конце осуществления способа.
На первом уровне выбираются последовательности из первичной последовательности ввода, а второй уровень, заключающийся в фильтре, может использоваться для ограничения выбранных последовательностей теми, которые удовлетворяют критериям, использованным фильтром.
В данном комбинированном способе фильтр выявляет кластеры ДНК-связывающих участков с использованием профилей весовых матриц, например, из Matlnspector (Quandt K, Frech K, Karas H, Wingender E, Werner T, "Matlnd and Matlnspector New fast and versatile tools for detection of consensus matches in nucleotide sequence data", Nucleic Acids Research, 23, 4878-4884, 1995). Фильтр также может выявлять плотность кластеров ДНК-связывающих участков.
Комбинированный способ в действительности представляет собой «оболочку», написанную для SMAR Scan® на Perl, и когда в качестве фильтра используется pfsearch, из pftools. Комбинированный способ осуществляет двухуровневую обработку с использованием на каждом уровне одного из данных инструментов (SMAR Scan® или фильтр) в качестве потенциального «фильтра», причем каждый фильтр является необязательным и может использоваться для вычисления предсказанных характеристик без выполнения какого-либо фильтрования.
Если на первом уровне для фильтрации подпоследовательностей используется SMAR Scan®, его требуется использовать для возвращения последовательностей в варианте «прилегающие соответствующие критерию области». Если на первом уровне в качестве первого фильтра используют pfsearch, его следует использовать только с одним профилем и требуется обеспечить расстояние между нуклеотидами. Данное расстояние используют для группировки соответствующих критерию pfsearch областей, которые расположены на расстоянии, меньшем расстояния, предоставляемого для возвращения последовательностей. Комбинированный способ запускает pfsearch, анализирует его выходные данные и возвращает последовательности, соответствующие соответствующим критерию pfsearch областям, которые сгруппированы в соответствии с предоставляемым расстоянием. Какой бы инструмент ни использовали на первом уровне, длина подпоследовательностей, выбранных таким образом, может системно продлеваться с обоих концов согласно параметру, называемому «продление соответствующих критерию областей».
Второй и необязательный уровень может использоваться для отфильтровки последовательностей (уже отфильтрованных последовательностей или нефильтрованных входящих последовательностей) или для получения результатов SMAR Scan® и/или pfsearch без выполнения какой-либо фильтрации данных последовательностей. Если второй уровень комбинированного способа используют для фильтрации, для каждого критерия следует предоставлять рассмотренный уровень отсечения (соответствие каждого нуклеотида) для отфильтровки данных последовательностей (см. фиг. 20).
Другим аспектом настоящего изобретения является предоставление способа идентификации последовательности MAR, включающей в себя по меньшей мере один фильтр, выявляющий кластеры ДНК-связывающих участков с использованием профилей или весовых матриц. Предпочтительно данный способ включает в себя два уровня фильтров, и в данном случае SMAR Scan® полностью отсутствует в указанном способе. Обычно данные два уровня заключаются в pfsearch.
Также к настоящему изобретению относятся очищенные и выделенные последовательности ДНК MAR, которые могут идентифицироваться способом для идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированным способом или способом, включающим в себя по меньшей мере один фильтр.
Анализ комбинированным способом целого генома человека дал всего 1757 предполагаемых MAR, представляющих всего 1 065 305 пар оснований. Для снижения количества результатов на данных 1757 MAR проводили динуклеотидный анализ, вычисляя процентную долю каждого из 16 возможных динуклеотидов для каждой последовательности, рассматривая обе цепи в направлении от 5' к 3'.
Неожиданно настоящими заявителями было показано, что все «супер»-MAR, выявленные комбинированным способом, содержали по меньшей мере 10% динуклеотида TA на протяжении 100 смежных пар оснований. Предпочтительно данные последовательности содержат по меньшей мере 33% динуклеотида TA на протяжении 100 смежных пар оснований.
Настоящими заявителями также было показано, что те же самые последовательности далее содержат по меньшей мере 12% динуклеотида AT на протяжении 100 смежных пар оснований. Предпочтительно они содержат по меньшей мере 33% олигонуклеотида AT смежных пар оснований.
Другим аспектом изобретения является предоставление очищенной и выделенной последовательности ДНК MAR любой из предшествующих описанных MAR, включающей в себя последовательность, выбранную из последовательностей SEQ ID No: 1-27, комплементарную ей последовательность, ее часть, содержащую по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты.
Предпочтительно указанная очищенная и выделенная последовательность ДНК MAR включает в себя последовательность, выбранную из последовательностей SEQ ID No: 24-27, комплементарную ей последовательность, ее часть, содержащую по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты. Данные последовательности 24-27 соответствуют тем, что выявлены комбинированным способом и характеризуются более высокой активностью по повышению продуцирования белка по сравнению с последовательностями 1-23.
Настоящее изобретение также относится к применению очищенной и выделенной последовательности ДНК, содержащей первую выделенную нуклеотидную последовательность области прикрепления матрикса (MAR), которая представляет собой нуклеотидную последовательность MAR, выбранную из группы, включающей в себя
- очищенную и выделенную последовательность ДНК, имеющую активность по повышению продуцирования белка,
- очищенную и выделенную последовательность ДНК MAR, которую можно идентифицировать способом идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированного способа или способа, включающего в себя по меньшей мере один фильтр,
- последовательности SEQ ID No: 1-27,
- очищенный и выделенный элемент и/или фрагмент cLysMAR,
- синтетическую последовательность MAR, содержащую природный элемент MAR и/или фрагменты, соединенные линкерными последовательностями, комплементарную ей последовательность, ее часть, содержащую по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты, или нуклеотидную последовательность MAR элемента и/или фрагмента cLysMAR, комплементарную ей последовательность, его часть, содержащую по меньшей мере 70% нуклеотидов в длину, его молекулярную химеру, их комбинации и варианты для активности по повышению продуцирования белка в эукариотической клетке хозяина.
Указанная очищенная и выделенная последовательность ДНК обычно далее включает в себя одну или несколько регуляторных последовательностей, известных в данной области, например промотор и/или энхансер, участки полиаденилирования и участки соединения после сплайсинга, обычно используемые для экспрессии белка или могут необязательно кодировать селективный маркер. Предпочтительно указанная очищенная и выделенная последовательность ДНК включает в себя промотор, который функционально связан с представляющим интерес геном.
Последовательности ДНК по данному изобретению могут выделяться стандартными протоколами ПЦР и способами, хорошо известными в данной области.
Промоторы, которые могут использоваться при обеспечении того, что такие промоторы совместимы с данной клеткой хозяина, представляют собой, например, промоторы, полученные из геномов вирусов, таких как вирус полиомы, аденовирус (например, аденовирус 2), вирус папилломы (например, вирус папилломы крупного рогатого скота), вирус саркомы птиц, цитомегаловирус (например, немедленный ранний промотор мышиного или человеческого цитомегаловируса), ретровирус, вирус гепатита В и вирус обезьян 40 (например, ранний и поздний промотор SV 40) или промоторы, полученные из гетерологичных промоторов млекопитающих, например промотор актина или иммуноглобулиновый промотор, или промоторы теплового шока. Такие регуляторные последовательности направляют конститутивную экспрессию.
Более того, очищенная и выделенная последовательность ДНК может содержать регуляторные последовательности, которые способны направлять экспрессию нуклеиновой кислоты преимущественно в конкретном клеточном типе (например, тканеспецифические регуляторные элементы используют для экспрессии нуклеиновой кислоты). Тканеспецифические регуляторные элементы известны в данной области. Неограничивающие примеры подходящих тканеспецифических промоторов включают в себя промотор альбумина (специфичный для печени; Pinkert, et al., 1987. Genes Dev. 1: 268-277), специфичные для лимфоидной ткани промоторы (Calame and Eaton, 1988. Adv. Immunol. 43: 235-275), в частности промоторы Т-клеточных рецепторов (Winoto and Baltimore, 1989. EMBOJ. 8: 729-733) и иммуноглобулинов (Banerji, et al., 1983. Cell 33: 729-740; Queen and Baltimore, 1983. Cell 33: 741-748), специфичные для нейронов промоторы (например, промотор нейрофиламентов; Byrne and Ruddle, 1989. Proc. Natl. Acad. Sci. USA 86: 5473-5477), специфичные для поджелудочной железы промоторы (Edlund, et al., 1985. Science 230: 912-916), и промоторы, специфичные для поджелудочной железы (например, промотор сыворотки молока; патент США № 4873316 и заявка на выдачу Европейского патента № 264166).
Регулируемые во время развития промоторы также рассматриваются. Примеры таких промоторов включают в себя, например, мышиные промоторы hox (Kessel and Gruss, 1990. Science 249: 374-379) и промоторы фетопротеина камелии (Campes and Tilghman, 1989. Genes Dev. 3: 537-546).
Регулируемые промоторы генной экспрессии хорошо известны в данной области и включают в качестве неограничивающих примеров любой промотор, который модулирует экспрессию требуемого белка путем связывания экзогенной молекулы, например система CRE/LOX, система TET, доксициклиновая система, система NF-каппа B/УФ свет, система Leu3p/изопропилмалат и система GLVPc/GAL4 (см., например, Sauer, 1998, Methods 14 (4): 381-92; Lewandoski, 2001, Nat. Rev. Genet 2 (10): 743-55; Legrand-Poels etal., 1998, J. Photochem. Photobiol. B. 45: 18; Guo et al., 1996, FEBS Lett. 390 (2): 191-5; Wang et al., PNAS USA, 1999, 96 (15): 84838).
Однако специалист в данной области сможет определить другие типы промоторов, которые могут использоваться при осуществлении настоящего изобретения.
Энхансеры могут необязательно включаться в очищенную последовательность ДНК по изобретению, принадлежа, таким образом, к регуляторной последовательности, например к промотору.
«Представляющий интерес ген» или «трансген» предпочтительно кодирует белок (структурный или регуляторный белок). Используемый здесь термин «белок» в общем относится к пептидам и полипептидам, имеющим более десяти аминокислот. Белки могут быть «гомологичны» в отношении хозяина (т.е. эндогенными в отношении использующейся клетки хозяина), или «гетерологичными» (т.е. чужеродными в отношении используемой клетки хозяина), например белок человека, продуцируемый дрожжами. Белок может продуцироваться в качестве нерастворимого агрегата или в качестве растворимого белка в периплазматическом пространстве или цитоплазме клетки, или во внеклеточной среде. Примеры белков включают в себя гормоны, например гормон роста или эритропоэтин (EPO), факторы роста, например эпидермальный фактор роста, анальгезирующие вещества, например энкефалин, ферменты, такие как химотрипсин, рецепторы гормонов или факторов роста, антитела, и включают в себя также белки, обычно используемые в качестве маркера для визуализации, например зеленый флуоресцентный белок.
Предпочтительно очищенная последовательность ДНК далее включает в себя по меньшей мере вторую выделенную нуклеотидную последовательность области прикрепления матрикса (MAR), выбранную из группы, включающей в себя
- очищенную и выделенную последовательность ДНК, имеющую активность по повышению продуцирования белка,
- очищенную и выделенную последовательность ДНК MAR, которую можно идентифицировать способом идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированного способа или способа, включающего в себя по меньшей мере один фильтр,
- последовательности SEQ ID No: 1-27,
- очищенный и выделенный элемент и/или фрагмент cLysMAR,
- синтетическую последовательность MAR, содержащую природный элемент MAR и/или фрагменты, соединенные линкерными последовательностями, комплементарную ей последовательность, ее часть, содержащую по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты. Выделенная нуклеотидная последовательность области прикрепления матрикса (MAR) может быть идентичной или отличающейся.
Альтернативно используют первую и вторую идентичную нуклеотидную последовательность MAR.
Предпочтительно нуклеотидные последовательности MAR расположены с 5'- и 3'-концов последовательности, содержащей представляющий интерес промотор и ген. Но изобретение также относится к тому факту, что указанные первая и по меньшей мере вторая нуклеотидные последовательности MAR расположены на последовательности, отличной от таковой, содержащей представляющий интерес промотор и ген.
В объем настоящего изобретения также входит выделенная и очищенная последовательность ДНК, включающая первую выделенную нуклеотидную последовательность области прикрепления матрикса (MAR), которая представляет собой нуклеотидную последовательность MAR, выбранную из группы, включающей в себя
- очищенную и выделенную последовательность ДНК, имеющую активность по повышению продуцирования белка,
- очищенную и выделенную последовательность ДНК MAR, которую можно идентифицировать способом идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированного способа или способа, включающего в себя по меньшей мере один фильтр,
- последовательности SEQ ID No: 1-27,
- очищенный и выделенный элемент и/или фрагмент cLysMAR,
- синтетическую последовательность MAR, содержащую природный элемент MAR и/или фрагменты, соединенные линкерными последовательностями, комплементарную ей последовательность, ее часть, содержащую по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты, которые могут использоваться для повышения активности для продуцирования белка в эукариотической клетке хозяина путем введения очищенной и выделенной последовательности ДНК в эукариотическую клетку хозяина хорошо известными протоколами. Обычно применяемые способы введения ДНК в эукариотические клетки хозяина представляют собой, например, прямое введение клонированной ДНК путем микроинъекции или бомбардировки микрочастицами; электропереноса; применения вирусных векторов; инкапсуляции в систему носителя; и применения реагентов для трансфекции, таких как фосфат кальция, диэтиламиноэтил-(DEAE)-декстран или коммерческие системы трансфекции, например, Lipofect-AMINE 2000 (Invitrogen). Предпочтительно способ трансфекции, используемый для введения очищенной последовательности ДНК, в эукариотическую клетку хозяина представляет собой способ трансфекции эукариотической клетки, как описано ниже.
Очищенная и выделенная последовательность ДНК может использоваться в виде кольцевого вектора.
Предпочтительно очищенная и выделенная последовательность ДНК используется в виде линейной последовательности ДНК в виде вектора.
Используемый здесь термин «плазмида» и «вектор» применяются взаимозаменяемо, поскольку плазмида является наиболее часто применяемой формой вектора. Однако изобретение, как подразумевается, относится к таким другим формам экспрессирующих векторах, включая в качестве неограничивающих примеров вирусные векторы (например, дефектные по репликации ретровирусы, аденовирусы и аденоассоциированные вирусы), которые служат в качестве эквивалентных функций.
Настоящее изобретение далее относится к способу трансфекции эукариотической клетки, причем указанный способ включает в себя a) введение в указанную эукариотическую клетку хозяина по меньшей мере одной очищенной последовательности ДНК, включающей в себя по меньшей мере одну представляющую интерес последовательность ДНК, и/или по меньшей мере одной очищенной и выделенной последовательности ДНК, включающей в себя нуклеотидную последовательность MAR или другие модифицирующие хроматин элементы, b) обработку указанной трансфицированной эукариотической клетки хозяина в течение определенного времени по меньшей мере в одной дополнительной стадии трансфекции по меньшей мере одной очищенной последовательностью ДНК, включающей в себя по меньшей мере одну представляющую интерес последовательность ДНК, и/или по меньшей мере одной очищенной и выделенной последовательностью ДНК, включающей в себя нуклеотидную последовательность MAR или другие модифицирующие хроматин элементы, c) селекцию указанной трансфицированной эукариотической клетки хозяина.
Предпочтительно на стадии b) применяют по меньшей мере от двух до четырех стадий трансфекции.
Для выбора успешно трансфицированных клеток ген, который кодирует маркер селекции (например, дающий устойчивость к антибиотикам), в общем, вводят в клетки хозяина вместе с представляющим интерес геном. Ген, который кодирует маркер селекции, может располагаться на очищенной последовательности ДНК, включающей в себя по меньшей мере одну представляющую интерес последовательность ДНК и/или по меньшей мере одну очищенную и выделенную последовательность ДНК, состоящую из нуклеотидной последовательности MAR или других модифицирующих хроматин элементов, или может необязательно быть совместно введен в отдельной форме на плазмиде. Различные маркеры селекции включают в себя те, которые дают устойчивость к лекарственным средствам, таким как G418, гидромицин и метотрексат. Количество лекарственного средства может адаптироваться по требованию для повышения продуктивности.
Обычно используют один или несколько маркеров селекции. Предпочтительно маркеры селекции, используемые на разных стадиях трансфекции, различны. Это позволяет осуществлять селекцию трансформированных клеток, которые являются «мультитрансформированными» с использованием, например, двух различных стадий селекции антибиотиками.
Любая эукариотическая клетка, способная к продуцированию белка и не имеющая клеточной стенки, может использоваться в способах изобретения. Примеры подходящих клеточных линий млекопитающих включают в себя человеческие клетки, такие как линия эмбриональной почки (293 или клетки 293, субклонированные для роста в суспензионной культуре, Graham et al., J. Gen Virol 36,59 (1977)), человеческие клетки карциномы шейки матки (HELA, ATCC CCL 2), человеческие клетки легкого (W138, ATCC CCL 75), человеческие клетки печени (Hep G2, HB 8065); клетки грызунов, например клетки почки карликового хомячка (BHK, ATCC CCL 10), клетки яичника китайского хомячка/-DHFR (CHO, Urlaub and Chasin, Proc. Natl. Acad. Sci. USA, 77, 4216 (1980)), мышиные клетки Сертоли (TM4, Mather, Biol. Reprod 23, 243-251 (1980)), опухоль мышиной молочной железы (MMT 060562, ATCC CCL51); и клетки из других млекопитающих, например линия почки обезьяны CV1, трансформированная SV40 (COS-7, ATCC CRL 1651); клетки почки обезьяны (CV1 ATCC CCL 70); клетки почки африканской зеленой макаки (VERO-76, ATCC CRL-1587); клетки собачьей почки (MDCK, ATCC CCL 34); клетки печени буйволовой крысы (BRL 3A, ATCC CRL 1442); миеломные (например, NS0)/гибридомные клетки.
Предпочтительно выбранные трансфицированные эукариотические клетки хозяина являются клетками-продуцентами высокого уровня белка со скоростью продуцирования по меньшей мере 10 пг на клетку в сутки.
Наиболее предпочтительными для применения здесь являются клетки млекопитающих, более предпочтительными являются клетки CHO.
Представляющая интерес последовательность ДНК из очищенной и выделенной последовательности ДНК обычно представляет собой представляющий интерес ген, предпочтительно кодирующий белок, функционально связанный с описанным выше промотором. Очищенная и выделенная последовательность ДНК, включающая в себя по меньшей мере одну представляющую интерес последовательность ДНК, может включать в себя в дополнение к представляющей интерес последовательности ДНК нуклеотидную последовательность MAR или другие модифицирующие хроматин элементы.
Очищенная и выделенная последовательность ДНК, включающая в себя нуклеотидную последовательность MAR, например, выбрана из группы, включающей в себя последовательности SEQ ID No: 1-27 и/или конкретные элементы cLysMAR, например, области B, K и F, а также фрагменты и элементы, и их комбинации, описанные выше. Другие модифицирующие хроматин элементы представляют собой, например, граничные элементы (BE), области контроля локуса (LCR) и универсальные элементы открытия хроматина (UCOE) (см. Zahn-Zabal et al., уже цитировано). Пример множественных трансфекций клеток хозяина показан в примере 12 (таблица 3). Первая стадия трансфекции (первичная трансфекция) проводится только на представляющем интерес гене (SV40EGFP) одной нуклеотидной последовательностью MAR (MAR) или представляющим интерес геном и нуклеотидной последовательностью MAR (MAR-SV40EGFP). Вторая стадия трансфекции (вторичная трансфекция) проводится только представляющим интерес геном (SV40EGFP), только нуклеотидной последовательностью MAR (MAR) или представляющим интерес геном и нуклеотидной последовательностью MAR (MAR-SV40EGFP), во всех возможных комбинациях, являющихся результатом первой стадии трансфекции.
Предпочтительно эукариотическую клетку хозяина трансфицируют путем: a) введения очищенной последовательности ДНК, включающей в себя одну представляющую интерес последовательность ДНК и, дополнительно, нуклеотидную последовательность MAR, b) воздействие на указанную трансфицированную эукариотическую клетку хозяина в течение определенного времени по меньшей мере одной дополнительной стадии трансфекции той же очищенной последовательностью ДНК, включающей в себя одну представляющую интерес последовательность ДНК и, дополнительно, нуклеотидную последовательность MAR со стадии a).
Также предпочтительно нуклеотидная последовательность MAR из очищенной и выделенной последовательности ДНК выбрана из группы, включающей в себя
- очищенную и выделенную последовательность ДНК, имеющую активность по повышению продуцирования белка,
- очищенную и выделенную последовательность ДНК MAR, которую можно идентифицировать способом идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированного способа или способа, включающего в себя по меньшей мере один фильтр,
- последовательности SEQ ID No: 1-27,
- очищенный и выделенный элемент и/или фрагмент cLysMAR,
- синтетическую последовательность MAR, содержащую природный элемент MAR и/или фрагменты, соединенные линкерными последовательностями, комплементарную ей последовательность, ее часть, содержащую по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты.
Неожиданно наблюдали синергию между первой и второй трансфекцией. Особенная синергия наблюдается, когда элементы MAR присутствуют на одной или обеих стадиях трансфекции. Проводили множественные трансфекции клеток одной pMAR или в комбинации с различными экспрессирующими плазмидами, с использованием способа, описанного выше. Например, в таблице 3 показано, что трансфекция клеток дважды плазмидой pMAR-SV40EGFP приводила к наиболее высокой экспрессии GFP и наиболее высокой степени усиления всех условий (4,3-кратному). Наоборот, двукратная трансфекция вектором без MAR приводила к минимальному усилению или его отсутствию, 2,8-кратному вместо ожидаемого двукратного повышения. Это доказывает то, что присутствие элементов MAR на каждой стадии трансфекции вызывает особый интерес в плане достижения максимального синтеза белка.
В качестве конкретного примера способа трансфекции указанная очищенная последовательность ДНК, включающая в себя по меньшей мере одну представляющую интерес последовательность ДНК, может вводиться в форме множественных несвязанных плазмид, содержащих представляющий интерес ген, включающий в себя представляющий интерес ген, функционально связанный с промотором, ген селективного маркера, и/или элементы, повышающие продуцирование белка, такие как последовательности MAR.
Отношения первой и последующих последовательностей ДНК могут адаптироваться, как требуется, для применения специфичных клеточных типов, и это представляет собой рутинное экспериментирование обычных специалистов в данной области.
Определенное время для дополнительных трансформаций первичных клеток непосредственно зависит от клеточного цикла и от его длительности. Обычно определенное время соответствует интервалам, связанным с циклом клеточного деления.
Поэтому данный точный временной режим может адаптировать по требованию для применения конкретных клеточных типов, и представляет собой рутинное экспериментирование обычного специалиста в данной области.
Предпочтительно определенное время представляет собой момент, в который клетка хозяина только что вошла в ту же фазу второго или дальнейшего цикла клеточного деления, предпочтительно второго цикла.
Данное время обычно составляет от 6 ч до 48 ч, предпочтительно от 20 ч до 24 ч после предшествующего события трансфекции.
Также к настоящему изобретению относится способ трансфекции эукариотической клетки хозяина, причем указанный способ включает в себя совместную трансфекцию в указанную клетку хозяина по меньшей мере одной первой очищенной и выделенной последовательности ДНК, включающей в себя по меньшей мере одну представляющую интерес последовательность ДНК, и второй очищенной ДНК, включающей в себя по меньшей мере одну нуклеотидную последовательность MAR, выбранную из группы, включающей в себя
- очищенную и выделенную последовательность ДНК, имеющую активность по повышению продуцирования белка,
- очищенную и выделенную последовательность ДНК MAR, которую можно идентифицировать способом идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированного способа или способа, включающего в себя по меньшей мере один фильтр,
- последовательности SEQ ID No: 1-27,
- очищенный и выделенный элемент и/или фрагмент cLysMAR,
- синтетическую последовательность MAR, содержащую природный элемент MAR и/или фрагменты, соединенные линкерными последовательностями, комплементарную ей последовательность, ее часть, содержащую по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты.
Указанная первая очищенная и выделенная последовательность ДНК также может включать в себя по меньшей мере один нуклеотид MAR, описанный выше.
Также рассматривается способ продуцирования белка, в котором эукариотическую клетку хозяина трансфицируют способами трансфекции согласно изобретению и культивируют в культуральной среде в условиях, подходящих для экспрессии белка. Указанный белок в конце получают любым способом получения, известным специалистам в данной области.
В качестве примера может использоваться следующий способ продуцирования белка.
Эукариотическую клетку хозяина, трансфицированную способом трансфекции согласно изобретению, применяют в способе продуцирования белка путем культивирования указанной клетки в условиях, подходящих для экспрессии указанного белка и получения указанного белка. Подходящие условия культивирования представляют собой те, которые обычно используются для культивирования эукариотических клеток in vitro, как описано в WO 96/39488. Белок может выделяться из культуры клеток общепринятыми способами разделения, такими как, например, фракционирование на иммуноаффинных или ионообменных колонках; преципитация; обращенно-фазовая ВЭЖХ; хроматография; хроматофокусирование; SDS-PAGE; гель-фильтрация. Специалисту в данной области понятно, что способы очистки, подходящие для представляющего интерес полипептида, могут требовать модификации с учетом изменений характера полипептида после экспрессии в рекомбинантной клеточной культуре.
Белки, которые продуцируются по изобретению, могут тестироваться на предмет функциональности различными способами. Например, присутствие антигенных эпитопов и способность белков связывать лиганды может определяться путем анализов вестерн-блота, анализов по флуоресцирующей сортировке клеток, иммунопреципитации, иммунохимических анализов и/или анализов конкурентного связывания, а также в любых других анализах, в которых измеряется специфичная связывающая активность.
Белки по данному изобретению могут использоваться в некотором количестве практических применений, которые включают в себя в качестве неограничивающих примеров следующие.
1. Иммунизация рекомбинантным белковым антигеном хозяина в качестве антагониста инфекции вируса/патогенного микроорганизма.
2. Продуцирование мембранных белков для диагностических и скрининговых анализов.
3. Продуцирование мембранных белков для биохимических анализов.
4. Продуцирование мембранного белка для структурных анализов.
5. Продуцирование антигена для получения антител для иммуногистохимического картирования, включая картирование одиночных рецепторов и ионных каналов.
Также настоящее изобретение относится к эукариотической клетке хозяина, трансфицированной любым из предшествующих способов трансфекции. Предпочтительно эукариотическая клетках хозяина представляет собой клеточную линию млекопитающих.
Как уже описано, примеры подходящих клеток хозяина-млекопитающего включают в себя человеческие клетки, например линия эмбриональной почки (293 или клетки 293, субклонированные для роста в суспензионной культуре, Graham et al., J. Gen Virol 36, 59 (1977)), человеческие клетки карциномы шейки матки (HELA, ATCC CCL 2), человеческие клетки легкого (W138, ATCC CCL 75), человеческие клетки печени (Hep G2, HB 8065); клетки грызунов, например клетки почки карликового хомячка (BHK, ATCC CCL 10), клетки яичника китайского хомячка/-DHFR (CHO, Urlaub and Chasin, Proc. Natl. Acad. Sci. USA, 77,4216 (1980)), мышиные клетки Сертоли (TM4, Mather, Biol. Reprod 23, 243-251 (1980)), опухоль мышиной молочной железы (MMT 060562, ATCC CCL51); и клетки из других млекопитающих, например линия почки обезьяны CV1, трансформированная SV40 (COS-7, ATCC CRL 1651); клетки почки обезьяны (CV1 ATCC CCL 70); клетки почки африканской зеленой макаки (VERO-76, ATCC CRL-1587); клетки собачьей почки (MDCK, ATCC CCL 34); клетки печени буйволовой крысы (BRL 3A, ATCC CRL 1442); миеломные (например, NS0)/гибридомные клетки.
Наиболее предпочтительны для применения здесь клетки CHO.
Настоящее изобретение также относится к смеси или набору для трансфекции клеток, содержащему по меньшей мере одну очищенную и выделенную последовательность ДНК по изобретению.
Изобретение далее относится к трансгенному организму, в котором по меньшей мере некоторое количество из его клеток стабильно включают в себя по меньшей мере одну последовательность ДНК из следующих:
- очищенная и выделенная последовательность ДНК, имеющая активность по повышению продуцирования белка,
- очищенная и выделенная последовательность ДНК MAR, которую можно идентифицировать способом идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированного способа или способа, включающего в себя по меньшей мере один фильтр,
- последовательности SEQ ID No: 1-27,
- очищенный и выделенный элемент и/или фрагмент cLysMAR,
- синтетическая последовательность MAR, содержащая природный элемент MAR и/или фрагменты, соединенные линкерными последовательностями, комплементарная ей последовательность, ее часть, содержащая по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты.
Предпочтительно некоторые из клеток трансгенного организма трансфицированы описанными здесь способами.
Также согласно изобретению рассматривается трансгенный организм, в геном которого стабильно введена по меньшей мере одна последовательность ДНК из следующих:
- очищенная и выделенная последовательность ДНК, имеющая активность по повышению продуцирования белка,
- очищенная и выделенная последовательность ДНК MAR, которую можно идентифицировать способом идентификации последовательности MAR с использованием описанного биоинформационного инструмента, комбинированного способа или способа, включающего в себя по меньшей мере один фильтр,
- последовательности SEQ ID No: 1-27,
- очищенный и выделенный элемент и/или фрагмент cLysMAR,
- синтетическая последовательность MAR, содержащая природный элемент MAR и/или фрагменты, соединенные линкерными последовательностями, комплементарная ей последовательность, ее часть, содержащая по меньшей мере 70% нуклеотидов в длину, ее молекулярную химеру, их комбинации и варианты.
Трансгенные эукариотические организмы, которые могут использоваться согласно изобретению, например, выбраны из группы, состоящей из млекопитающих (мышей, людей, обезьян, и т.д.) и, в частности, лабораторных животных, таких как грызуны в общем смысле, насекомые (дрозофила и т.д.), рыбы (данио и т.д.), амфибии (лягушки, тритоны и т.д.) и другие более простые организмы, такие как С.elegans, дрожжи и т.д.
Еще одной целью настоящего изобретения является предоставление считываемой на компьютере среды, включающей в себя исполняемые компьютером инструкции по выполнению способа идентификации последовательности MAR, описанной согласно изобретению.
Следующее описание будет более полно понятно со ссылкой на следующие примеры. Такие примеры, однако, представляют типовые способы воплощения настоящего изобретения и не предназначены для ограничения объема изобретения.
ПРИМЕРЫ
Пример 1. SMAR Scan® и последовательности MAR
Первую приблизительную оценку SMAR Scan® проводили путем анализа экспериментально определенных человеческих последовательностей MAR и не относящихся к последовательностей MAR. В качестве последовательностей MAR предшествующие результаты анализа человеческих MAR из SMARt Db использовали для построения гистограмм плотности для каждого критерия, показанного на фиг.1. Сходным образом, не относящиеся к MAR последовательности также анализировали и строили. В качестве последовательности MAR использовали все Ref-Seq-контиги из хромосомы 22, принимая во внимание то, что последний достаточно велик, чтобы содержать незначительную часть последовательностей MAR по сравнению с частью не относящихся к MAR последовательностей.
Распределения плотности, показанные на фиг.1, все были скошены с образованием длинного хвоста. Для наибольшего изгибания, наибольшей глубины большой бороздки и наибольшей ширины малой бороздки распределения были скошены вправо. Для минимальной температуры плавления распределения скошены влево, что естественно из-за обратного соответствия данного критерия по отношению к трем другим. Для последовательностей MAR в действительности очевидны были двухфазные распределения со слабым вторым пиком. И также на каждом графике был виден явный сдвиг между распределениями, относящимися к MAR и не к MAR.
Среди всех последовательностей MAR человека в среднем только около 70% из них характеризуются значением, превышающим 75-й квантиль в распределении человека MAR, и это для четырех различных критериев. Что касается второго слабого пика каждого распределения человеческих MAR, то только 15% человеческих последовательностей MAR ответственны за данные отклоняющиеся значения. Среди указанных 15% человеческих последовательностей MAR большинство является хорошо документированными MAR, использованными для отделения трансгена от эффектов положения, например MAR локуса интерферона, MAR локуса бета-глобина (Ramezani A, Hawley TS, Hawley RG, "Performance and safety-enhanced lentiviral vectors containing the human interferon-beta scaffold attachment region and the chicken beta-globin insulator", Blood, 101: 4717-4724, 2003), или MAR аполипопротеина (Namciu, S, Blochinger KB, Fournier REK, "Human matrix attachment regions in-sulate transgene expression from chromosomal position effects in Drosophila melanogaster", Mol. Cell. Biol., 18: 2382-2391, 1998).
Всегда с теми же самыми данными человеческие последовательности MAR также использовали для определения ассоциации между четырьмя вычисленными теоретическими структурными характеристиками и содержанием AT. На фиг.2 представлена диаграмма рассеяния и соответствующий коэффициент корреляции r для каждый пары критериев.
Пример 2. Графики распределения последовательности MAR по организмам
Также получали последовательности MAR из SMARt DB других организмов и анализировали их сходным образом, как объяснялось ранее. Распределение плотностей последовательностей MAR для мыши, курицы, Sorghum bicolor и человека построены вместе на фиг.3.
Пример 3. Предсказание MAR для всей хромосомы 22
Все контиги RefSeq из хромосомы 22 анализировали посредством SMAR Scan® с использованием в данный раз установок по умолчанию. Результат состоит в том, что SMAR Scan предсказал всего 803 MAR, причем их средняя длина составляла 446 п.н., что означает, что в среднем одна MAR составляет 42 777 п.н. Общая длина предсказанных MAR соответствует 1% длины 22 хромосомы. Содержание АT в предсказанных областях изменялось от 65,1% до 93,3%; среднее содержание AT всех данных областей составляет 73,5%. Таким образом, предсказанные MAR были богаты AT, в то время как хромосома 22 не является AT-богатой (52,1% AT).
SMARTest также использовали для анализа целой хромосомы 22 и получали 1387 MAR-кандидатов, причем их средняя длина составляла 494 п.н., что означает, что в среднем одна MAR составляет 24765 п.н. Общая длина предсказанных MAR соответствует 2% длины 22 хромосомы. Из всех MAR, предсказанных двумя программами, 154 предсказанных MAR найдено обеими программами, что представляет соответственно 19% и 11% предсказанных SMAR Scan® и SMARTest MAR. Исходя из предсказанной средней длины MAR SMAR Scan® и SMARTest, вероятность получить случайно перекрывание между предсказаниями SMAR Scan® и SMARTest составляет 0,0027% на одно предсказание.
Для оценки специфичности предсказаний SMAR Scan® анализы SMAR Scan® проводили на случайным образом перетасованных последовательностях хромосомы 22 (фиг. 4). Перетасованные последовательности генерировали с использованием 4 различных способов: путем сегментации хромосомы 22 на неперекрывающияся окна размером 10 п.н. и путем отдельного перетасовывания нуклеотидов в каждом окне; путем «взбалтывания», что означает перестановку всех нуклеотидов хромосомы; путем «разрушения», что означает сегментацию хромосомы на фрагменты размером 10 п.н. и случайное соединение данных фрагментов, и, наконец, посредством Марковских цепей 1 порядка, причем различные состояния представляли все различные динуклеотиды ДНК, и вероятности перехода между данными состояниями основаны на скане хромосомы 22. Для каждого способа перетасовки генерировали пять перетасованных хромосом 22 и анализировали их SMAR Scan® с использованием установок по умолчанию. Что касается числа соответствующих критерию областей, то для пермутированной хромосомы 22 было найдено в среднем 3519170 соответствующих критерию областей (ст.откл.: 18353) в неперекрывающихся окнах по 10 п.н., 1719364 соответствующих критерию областей (ст.откл.: 285904) для полученных путем взбалтывания последовательностей и 247082 соответствующих критерию областей (ст.откл.: 119159) для разрушенной хромосомы 22 и 2282 соответствующих критерию областей в среднем (ст.откл.: 3347) для хромосом, образованных согласно моделям Марковских цепей 1 порядка хромосомы 22, что соответственно представляет собой 185% (ст. откл.: 0,5% от среднего), 9% (ст.откл.: 1,5%), 1% (ст.откл.: 5%) и 0,1% (ст.откл.: 15%) от числа соответствующих критерию областей, обнаруженных в нативной хромосоме 22. Что касается предсказанного количества MAR, которое, таким образом, означает смежные соответствующие критерию области, по длине превышающие 300, то 1 997 MAR было предсказано в перетасованной хромосоме 22 в пределах окон длиной 10 п.н. (ст.откл.: 31,2), только 2,4 кандидатов в MAR было обнаружено в полученных путем взбалтывания последовательностях (ст.откл.: 0,96), и ни одной для разрушенных последовательностей и для последовательностей, генерированных моделью Марковских цепей, что соответственно представляет собой 249% и менее 0,3% от числа предсказанных MAR, обнаруженных в нативной хромосоме 22. Эти данные предоставляют указания на то, что SMAR Scan® выявляет специфичные элементы ДНК, организация которых теряется при перетасовке последовательностей ДНК.
Пример 4. Анализ известных областей прикрепления матрикса в локусе интерферона посредством SMAR Scan®
Релевантность предсказания MAR посредством SMAR Scan® исследовали путем анализа недавно опубликованных областей MAR генного кластера человеческого интерферона короткого плеча хромосомы 9 (9p22). Goetze et al. (уже цитировано) сообщают об исчерпывающем анализе локуса WP18A10A7 с целью выявления предполагаемой корреляции между BUR (названными в данном случае индуцированной стрессом дестабилизацией дуплекса или SIDD) и связыванием ядерного матрикса in vitro (фиг.9, нижняя часть). Три пика SIDD согласовывались с анализом связывания in vitro, в то время как другие не совпадали с участками связывания матрикса. Исследование интерферонового локуса SMAR Scan® (фиг.9, верхняя часть) указывало на то, что три главных пика, сопровождающие кластеры участков связывания регуляторов SATB1, NMP4 и MEF2, хорошо коррелировали с активными MAR. Поэтому авторы заключают, что встречаемость предсказанных CUE и участков связывания данных факторов транскрипции не ограничено cLysMAR, но может быть общим свойством всех MAR. Данные результаты также подразумевают, что программа SMAR Scan® эффективно выявляет элементы MAR из геномных последовательностей.
Пример 5. Точность предсказания SMAR Scan® и сравнение с другими предсказательными инструментами
Точность SMAR Scan® оценивали с использованием шести геномных последовательностей, для которых были картированы экспериментально определенные MAR. Для проведения сравнения с другими предсказательными инструментами проанализированные последовательности совпадали с последовательностями, ранее использованными для сравнения MAR-Finder и SMARTest. Данные геномные последовательности представляют собой три растительные и три человеческие последовательности (таблица 1), в сумме составляющие 310151 п.н. и 37 экспериментально определенные MAR. Результаты SMARTest и MAR-Finder в таблице 1 приходят из предварительного сравнения (Frisch M, Frech K, Klingenhoff A, Cartharius K, Liebich I and Werner T, In silico pre-diction of scaffold/matrix attachment regions in large genomic sequences, Genome Research, 12: 349-354, 2001.).
MAR-Finder использовали с параметрами по умолчанию, за исключением того, что барьер был установлен на 0,4 и для анализа локуса протамина исключали правила AT-богатства (для выявления не богатых AT MAR, как было сделано для локуса протамина).
Шесть разных геномных последовательностей, три растительные и три человеческие последовательности, для которых известны экспериментально определенные MAR, анализировали MAR-Finder, SMARTest и SMAR Scan®. Три положительных совпадения напечатаны жирным, минус (-) указывает на ложноположительные совпадения. Некоторые из более длинных экспериментально определенных MAR содержали более одного предсказания in silico, каждое из которых насчитывало истинно положительного совпадения. Поэтому число истинных предсказаний in silico выше, чем число найденных экспериментально определенных MAR. Специфичность определяется отношением истинно положительных предсказаний, в то время как чувствительность определяется как число экспериментально определенных найденных MAR. *Правило AT-богатства исключали при использовании MAR-Finder.
SMARTest предсказывал как MAR 28 областей, 19 (истинно положительных) из них коррелировали с экспериментально определенными MAR (специфичность: 68%), в то время как 9 (32%) располагались в не относящихся к MAR участкам (ложноположительные). Поскольку некоторые из наиболее длинных экспериментально определенных MAR содержат более одного предсказания in silico, 19 истинно положительных в действительности соответствовали 14 различным экспериментально определенным MAR (чувствительность: 38%). MARFinder предсказывал как MAR 25 областей, 20 (специфичность: 80%) из них коррелировали с экспериментально определенными MAR, соответствующими 12 различных экспериментально определенных MAR (чувствительность: 32%). SMAR Scan® предсказывал 22 области, причем 17 из них были истинно положительными (специфичность: 77%) и совпадали с 14 различными экспериментально определенными MAR (чувствительность: 38%).
В качестве другого примера тот же анализ применяли к человеческим хромосомам 1 и 2, и это приводило к определению 23 последовательностей MAR (SEQ ID Nо: 1-23). Данные последовательности перечислены в Приложении 1 в формате ST25.
Пример 6. Анализ целого генома с использованием комбинированного способа (SMAR Scan®-pfsearch)
Для тестирования потенциальной корреляции между структурными характеристиками, вычисленными SMAR Scan®, и функциональной активностью S/MAR целый геном человека анализировали комбинированными способами с очень жесткими условиями для получения последовательностей с наиболее высокими значениями вычисленных терапевтических характеристик, которые ниже называются «супер»-S/MAR. Этот анализ выполняли с надеждой получения предсказанных элементов MAR с очень высоким потенциалом в плане повышения трансгенной экспрессией и продуцированием рекомбинантного белка. Следовательно, предполагаемые полученные S/MAR вначале анализировали из биоинформационной перспективы в попытках их охарактеризовать и классифицировать.
6.1 S/MAR, предсказанные из анализа всего генома человека
В качестве последовательности всего генома человека использовали все человеческие контиги RefSeq (National Center for Biotechnology Information, The NCBI handbook [Internet]. Bethesda (MD): National Library of Medicine (US), Oct. Chapter 17, The Reference Sequence (RefSeq) Project, 2002 (доступны из http://www.ncbi.nih.gov/entrez/query.fcgi?db=Books) (выпуск 5) и анализировали их комбинированным способом с использованием SMAR Scan в качестве фильтра процессинга первого уровня, с использованием установок по умолчанию, за исключением уровня отсечения наибольшего изгиба, поскольку для критерия изгибания ДНК применяли барьер жесткости, равный 4,0 градусам (вместо 3,202 градусов).
На втором уровне процессинга искали предсказанные участки связывания факторов транскрипции в последовательностях, выбранных на предшествующей стадии, без выполнения фильтрования данных последовательностей.
Анализ комбинированным способом всего генома человека дал всего 1757 предполагаемых «супер»-S/MAR, представляющих всего 1065305 п.н. (0,35% всего генома человека). В таблице 2 для каждой хромосомы показан: ее размер, число генов в ней, число предсказанных в ней S/MAR, плотность генов на S/MAR и количество т.п.н. на S/MAR. В данной таблице показано, что имеет место очень различная плотность генов на S/MAR, предсказанные для различных хромосом (стандартное отклонение представляет более 50% от средней плотности генов на предсказанные S/MAR, и порядок различий между высшей и низшей плотностью генов на S/MAR составляет 6,5). В таблице 2 также показано, что количество т.п.н. на S/MAR изменяется меньше, чем плотность генов на S/MAR (стандартное отклонение представляет собой 25% от среднего, и порядок различий между высшим и низшим количеством т.п.н. на S/MAR составляет 3,2).
Число генов на хромосому соответствует статистике по геному человека NCBI (Build 34 Version 3) (National Center for Biotechnology Information, The NCBI handbook [Internet]. Bethesda (MD): National Library of Medicine (US), Oct. Chapter 17, The Reference Sequence (RefSeq) Project, 2002 (доступно из http://www.ncbi.nih.gov/entrez/query.fcgi?db=Books) на основе аннотаций GenBank. Размеры хромосом представляют собой сумму соответствующих длин человеческих контигов RefSeq (National Center for Biotechnology Information, The NCBI handbook [Internet]. Bethesda (MD): National Library of Medicine (US), Oct. Chapter 17, The Reference Sequence (RefSeq) Project, 2002 (доступно из http://www.ncbi.nih.gov/entrez/query.fcgi?db=Books) (выпуск 5).
6.2 Биоинформационный анализ «супер»-MARS на предмет участков связывания факторов транскрипции
1757 предсказанных последовательностей «супер»-S/MAR, полученных ранее посредством SMAR Scan®, затем анализировали на предмет потенциальных участков связывания факторов транскрипции. Это достигали с использованием RMatch™ Professional (Kel AE, Gossling E, Reuter I, Cheremushkin E, KelMargoulis OV, Wingender E, MATCH: A tool for searching transcription factor binding sites in DNA sequences, Nucleic Acids Res. 31 (13): 35769, 2003), основанного на весовой матрице инструмента на основе TRANSFAC (Wingender E, Chen X, Fricke E, Geffers R, Hehl R, Liebich I, Krull M, Matys V, Michael H, Ohnhauser R, Pruss M, Schacherer F, Thiele S, Urbach S, The TRANSFAC system on gene expression regulation, Nucleic Acids Research, 29(1): 2813, 2001). Match™ 2.0 Professional использовали с большинством настроек Match™ по умолчанию TRANSFAC Professional, выпуск 8.2 (20040630). Суммы всех предсказаний участков связывания на последовательностях 1757, проанализированных по Match™, приведены в таблице 3. Основываясь на данной таблице, для последующего анализа выбирали только те факторы транскрипции, которые насчитывали по меньшей мере 20 отвечающих критерию областей в пределах 1757 проанализированных последовательностей.
Здесь и далее приведены некоторые из факторов транскрипции, которые наиболее часто предсказывались как связывающиеся с 1757 предполагаемыми S/MAR-последовательностями, и их описание по Match™: Cdc5 (белок контроля клеточного деления 5), транскрипционный регулятор/репрессор, Nkx3A, белок с гомеодоменомa, регулируемый андрогеном, POU1F1 (гипофизоспецифичный позитивный фактор транскрипции 1), который специфичен для гипофиза и стимулирует пролиферацию клеток. Таким образом, в дополнение к SATB1, NMP4 и MEF2 в активности MAR могут участвовать другие транскрипционные факторы.
Таблица 3 представляет собой итог всех предсказаний связывания факторов транскрипции (насчитывающих 20 или более соответствующих критерию областей) в 1757 проанализированных последовательностей.
6.3 Биоинформационный анализ предсказанных «супер»-MAR в плане частот динуклеотидов
Разичный компьютерный анализ проводили для упрощенной идентификации «супер»-S/MAR-последовательностей с использованием определенного критерия, который может идентифицироваться без вычислений. Среди них проводили динуклеотидный анализ 1757 супер-MAR, вычисляя процентную долю каждого из 16 возможных динуклеотидов для каждой последовательности при рассмотрении обеих цепей в направлении 5'>3'.
Итог (мин., макс., медиана, среднее значение, 25-й процентиль и 75-й процентиль), также гистограммы для процентной доли каждого динуклеотида в 1757 последовательностях S/MAR, соответственно, представлены в таблице 4. Сходный анализ проводили на случайно выбранных последовательностях из генома человека, представляющих собой случайно выбранные не относящиеся к S/MAR последовательности (которые, однако, могут содержать некоторые MAR). В таблице 5 представлен, соответственно, итог динуклеотидного анализа данных последовательностей.
При рассмотрении результатов по предсказанным элементам S/MAR и не относящимся к S/MAR последовательностям в итоговых таблицах могут отмечаться заметные различия в содержании динуклеотидов AT и TA между данными двумя группами последовательностей. AT и TA составляют соответственно по меньшей мере 18,5 % и 28,6% содержания динуклеотидов в предсказанных последовательностях S/MAR, в то время как минимальные процентные доли тех же динуклеотидов в не относящихся к S/MAR последовательностях составляют соответственно 0,3% и 0%. Сходным образом, максимальное содержание CC и GG в последовательностях S/MAR составляет 4,2%, в то время как в не относящихся к S/MAR последовательностях процентные доли данных двух динуклеотидов могут насчитывать до 20,8%.
Корреляция между процентными долями динуклеотидов AT и TA и максимальным изгибом ДНК, рассчитанная SMAR Scan®, показана на фиг.17 для предсказанных последовательностей S/MAR и на фиг.18 для не относящихся к S/MAR последовательностей. Различные диаграммы рассеяния данных фигур показывают, что процентная доля TA хорошо коррелирует с изгибом, как предсказано SMAR Scan®.
Четыре новых супер-MAR были изъяты случайным образом, и их анализировали на предмет содержания динуклеотидов AT и TA, и сравнивали с ранее известной куриной lysMAR, рассматривания окна по 100 пар оснований (таблица 6).
Неожиданно настоящими заявителями было показано, что все супер-MAR имели частоты динуклеотида AT, более 12%, и динуклеотида TA более 10% от общего числа динуклеотидов при анализе окон по 100 пар оснований ДНК. Наиболее эффективные MAR характнеризуются значениями около 34% этих двух динуклеотидных пар.
6.4 Анализ ортологичных межгенных областей человеческого и мышиного геномов
Для получения представления об эволюции S/MAR ортологичные межгенные области человеческого и мышиного геномов анализировали посредством SMAR Scan®. Использованный набор данных составлен из 87 пар целых ортологичных межгенных областей из человеческого и мышиного геномов (Shabalina SA, Ogurtsov AY, Kondrashov VA, Kondrashov AS, Selective constraint in intergenic regions of human and mouse genomes, Trends Genet, 17 (7): 3736, 2001) (средняя длины ~12000 п.н.), расположенных на 12 человеческих и 12 мышиных хромосомах, синтению данных последовательностей подтверждали парным выравниванием последовательности и за счет рассмотрения аннотации фланкирующих генов (экспериментальной или предсказанной).
Анализ 87 человеческих и мышиных ортологичных межгенных последовательностей проводили посредством SMAR Scan® с применением его установок по умолчанию. Анализ человеческих последовательностей всего давал 12 предсказанных S/MAR (что представляет общую длину 4750 п.н.), расположенных на 5 различных межгенных последовательностях.
Из трех человеческих межгенных последовательностей, в которых, как было предсказано посредством жестких установок SMAR Scan®, содержатся «супер»-S/MAR, одна имела соответствующую ей мышиную ортологичную межгенную последовательность, в которой также предсказывалось содержание одной S/MAR (человеческая EMBL ID: Z96050, положение 28010-76951, ортологична мышиной EMBL ID: AC015932, положения 59884-89963). При проведении локального выравнивания данных двух ортологичных межгенных последовательностей наилучшее локальное выравнивание данных двух больших областей соответствовало областям, предсказанных посредством SMAR Scan® как S/MAR-элементы. Поиск вручную мышиных ортологов двум другим человеческим межгенным последовательностям, которые, как предсказано, содержат «супер»-S/MAR, проводили с использованием Ensembl Genome Browser (http://ensembl.org). Мышиные ортологичные межгенные последовательности данных двух человеческих последовательностей получали с использованием предсказаний ортологов Ensembl (на основе названий генов), с поиском ортологичных мышиных генов для пар человеческих генов, фланкирующих данные межгенные области.
Поскольку SMAR Scan® был настроен на человеческие последовательности и, следовательно, давал мало «супер»-MAR с мышиными геномными последовательностями, его уровень отсечения, принятый по умолчанию, был несколько снижен в плане минимального размера смежных отвечающих критериям областей, рассматриваемых как S/MAR (с использованием 200 п.н. вместо 300 п.н.). Анализ SMAR Scan® данных мышиных последовательностей предсказывал несколько S/MAR, имеющих высокие значения для различных вычисленных структурных характеристик. Это открытие указывает на то, что человеческие элементы MAR консервативны среди видов.
Пример 7. Разделение на фрагменты 5'-MAR гена куриного лизоцима
5'-MAR длиной 3000 пар оснований разделяли на меньшие фрагменты, мониторинг которых проводили в плане воздействия на экспрессию трансгена в клетках яичника китайского хомячка (CHO). Для выполнения этого семь фрагментов длиной ~400 п.н. генерировали в полимеразной цепной реакции (ПЦР).
Данные амплифицированные путем ПЦР фрагменты были следующими друг за другом и покрывали целую последовательность MAR при ориентации конец в конец. Четыре копии каждого из данных фрагментов лигировали в ориентации начало-конец с получением длины, соответствующей примерно половине природной MAR. Тетрамеры встраивали выше промотора SV40 в pGEGFPControl, модифицированную версию вектора pGL3Control (Promega). Плазмиду pGEGFPControl создавали путем обмена гена люциферазы в pGL3Control на ген EGFP из pEGFP-N1 (Clontech). Созданные таким путем плазмиды, содержащие 5'-MAR, трансфицировали совместно с плазмидой устойчивости pSVneo в клетки CHO-DG44 с использованием LipofectAmine 2000 (Invitrogen) в качестве реагента трансфекции, как проводилось ранее (Zahn-Zabal, M., et al., "Development of stable cell lines for production or regulated expression using matrix attachment regions" J Biotechnol, 2001, 87(1): p. 29-42). После селекции устойчивых к антибиотику (G-418) клеток поликлональную клеточную популяцию анализировали посредством FACS на предмет флуоресценции EGFP.
Экспрессию трансгена выражали в виде процентиля клеток с высокой экспрессией, определенных как клетки, уровни флуоресценции которых по меньшей мере на 4 порядка величины выше, чем средняя флуоресценция клеток, трансфицированных вектором pGEGFPControl без MAR.
На фиг.5 показано, что мультимеризованные фрагменты B, K и F усиливают экспрессию трансгенов, несмотря на их более короткий размер по сравнению с исходной последовательностью MAR. Другие фрагменты, наоборот, слабо активны или полностью неактивны.
Пример 8. Специфичность областей B, K и F в контексте MAR
5'-MAR подвергали серийным делециям от 5'-конца (фиг.6, верхняя часть) или 3'-конца (фиг.6, нижняя часть) соответственно. Эффект укороченных элементов подвергали мониторингу сходным образом, как описано в предыдущем разделе. На фигуре 6 показано, что потеря способности стимулировать экспрессию трансгена в клетках CHO не распределена равномерно.
В данном исследовании делеций потеря активности MAR перекрывалась с дискретными областями перехода, которые перекрываются с B-, K- и F-фрагментом 5'-MAR соответственно. В 5'-делециях активность по большей части терялась при удалении фрагмента K и F. 3'-делеции, которые удаляли F и B-элементы, имеют наиболее выраженный эффект. Наоборот, фланкирующие области A, D, E и G, которые, соответственно, имеют малую способность или отсутствие способности стимулировать экспрессию трансгена сами по себе (фиг.5), не вносят вклад в активность MAR при исследованиях делеции с 5'- и 3'-конца (фиг.6).
Пример 9. Структура F-элемента
F-фрагмент длиной 465 п.н. далее разделяли на меньшие субфрагменты длиной 234, 243, 213 п.н. и 122, 125 и 121 п.н. соответственно. Фрагменты первой группы получали в виде октамеров (8 копий) в ориентации начало-конец, в то время как фрагменты второй группы сходным образом гексадекамеров (16 копий) для поддержания постоянной длины последовательности MAR. Данные элементы клонировали в вектор pGEGFPControl, и их эффекты оценивали в клетках CHO, как описано выше. Интересно, что FIII сохранял большую часть активности полноразмерного фрагмента F, в то время как фрагмент FII, содержащий правостороннюю часть фрагмента FIII, терял всю способность стимулировать экспрессию трансгена (фиг.7). Это указывает на то, что активная область содержалась между нт. 132 и нт. 221 в фрагменте FIB. В соответствии с этим множественные копии фрагментов FI и FIB, которые охватывают данную область, характеризуются сходной активностью. FIIA сам по себе не имеет активности. Однако при добавлении к FIB, результатом чего является FIII, он усиливает активность первого. Поэтому оказывается, что FIIA содержит вспомогательную последовательность, которая имеет малую активность сама по себе, но которая усиливает активность минимального домена, расположенного в FIB.
Анализ распределения индивидуальных мотивов в пределах 5'-MAR гена лизоцима показан на фиг.8A вместе с некоторыми дополнительными мотивами, которые добавлены к анализу авторами изобретения. Было обнаружено, что большинство данных мотивов распределены по элементу MAR и не ассоциированы с активной частью специфично. Например, участки связывания факторов транскрипции и другие мотивы, которые были ассоциированы с MAR, не локализованы преимущественно в активных областях. Также предполагалось, что активные последовательности MAR могут состоять из комбинации различных мотивов. Описаны некоторые компьютерные программы (MAR Finder, SMARTest, стабильность дуплекса SIDD) по идентификации MAR как областей ДНК, которые ассоциированы с ДНК-матриксом. Они обычно основаны на алгоритмах, которые используют предварительно определенную серию специфичных для последовательности профилей, которые, как предполагали ранее, содержат активность MAR, примером чего является MAR Finder, сейчас известный как MAR Wiz. Выходные данные данных программ не обладают высокой корреляцией с транскрипционно активными частями cLysMAR. Например, пики активности, полученные MAR Finder, не имеют четкого соответствия с активной частью MAR, тогда как, например, B-фрагмент довольно активен in vivo, но имеет отрицательный коэффициент при исследовании MAR Finder (фиг.8B, ср. верхнюю и среднюю панели). Изогнутые структуры ДНК, предсказанные данной программой, не имеют высокой корреляции с активностью (фиг. 8B, ср. верхнюю и нижнюю панели). Сходные результаты получали с другими доступными программами (данные не приведены).
Мотивы, идентифицированные доступными компьютерными методами предсказания MAR, таким образом, по всей вероятности, не являются основными детерминантами способности cLysMAR повышать генную экспрессию. Поэтому тестировали некоторое количество других компьютерных инструментов. Неожиданно оказалось, что предсказанные последовательности связывания нуклеосомы и последовательности, неблагоприятные для связывания нуклеосомы, как было обнаружено, расположены в виде повторяемо распределенных по MAR кластеров, причем благоприятные для связывания нуклеосомы участки перекрываются с активными B-, K- и F-областями. Последовательности расположения нуклеосомы, как предполагается, состоят из растяжек ДНК, которые могут легко оборачиваться вокруг гистонов нуклеосомы, и они не были ранее ассоциированы с последовательностями MAR.
Благоприятные для связывания нуклеосомы последовательности могут моделироваться набором характеристик ДНК, которые включают в себя умеренно повторяемые последовательности и другие физико-химические параметры, которые могут обеспечивать правильное фазирование и ориентацию ДНК по искривленной поверхности гистона. Идентификация многих из этих характеристик ДНК может быть компьютеризованна, и до 38 таких различных характеристик применяли для предсказания потенциальных положений нуклеосомы. Поэтому авторы изобретения запланировали определить, могут ли конкретные компоненты программ предсказания участков нуклеосомы коррелировать с активностью MAR, с целью конструирования инструмента, позволяющего идентифицировать новые и возможно более мощные MAR в геномных последовательностях.
Для определения того, могут ли какие-либо аспекты первичной структуры ДНК отличить активные B-, K- и F-области от окружающей последовательности, авторы изобретения анализировали 5'-MAR посредством SMAR Scan®. Из 38 инструментов предсказания нуклеосомных массивов три, как было обнаружено, давали корреляцию с расположением активных субдоменов MAR (фиг. 9A). Расположение B-, K- и F-областей MAR перекрывалось с максимумами изгибания ДНК, глубины большой бороздки и ширины малой бороздки. Более слабую корреляцию также отмечали с минимумом температуры плавления ДНК, определяемого содержанием GC content. Улучшенное картирование по F-фрагменту указывало на то, что падение температуры плавления и подъем изгибания ДНК действительно соответствовали субфрагменту FIB, который содержал минимальный домен MAR (фиг.9B). Таким образом, активные части MAR могут соответствовать областям, предсказанным данной программой как изогнутые области ДНК, и авторы изобретения ссылаются на данные области в тексте, следующем ниже, как на CUE-B, CUE-K и CUE-F. Тем не менее, соответствуют ли данные области действительно изогнутой ДНК и области разматывания пар оснований, неизвестно, поскольку они не соответствуют изогнутой ДНК по предсказаниям MAR Wiz (фиг.9B).
Пример 10. Отпечатки регуляторных элементов в F-фрагменте
Характеристики расположения нуклеосомы могут считаться наиболее специфичными хроматиновыми кодами, содержащимися в геномной ДНК. Хотя данный конкретный код может вносить вклад в активность F-области, маловероятно, что он определяет активность MAR один, так как 3'-часть F-области усиливала активность минимального домена MAR, содержащегося в FIB-части. С использованием программы Matlnspector (Genomatix) авторы изобретения искали участки связывания факторов транскрипции с коэффициентами выше 0,92 и нашли последовательности ДНК для связывания белков NMP4 и MEF2 в 3'-части F-фрагмента (фиг.8B).
Для определения того, могут ли какие-либо из данных участков связывания факторов транскрипции локализоваться вблизи B- и K-активных областей, целую последовательность 5'-MAR анализировали путем связывания NMP4 и MEF2 и белков, о которых сообщается, что они связываются с одноцепочечной или двухцепочечной формой BUR. Среди них SATB1 (специальный связывающий AT-богатые участки белок 1) принадлежит к классу ДНК-связывающих факторов транскрипции, которые могут активировать или подавлять экспрессию окружающих генов. Данное исследование указывает на то, что специфичные белки, такие как SATB1, NMP4 (белок ядерного матрикса 4) и MEF2 (фактор миогенного энхансера 2), имеют специфичное распределение и образуют каркас вокруг минимальных MAR-доменов cLysMAR (фиг. 10). Присутствие некоторых из данных участков связывания NMP4 и SATB1 подтверждали экспериментально посредством EMSA-анализа очищенных рекомбинантных белков (данные не приведены).
Пример 11. Конструирование искусственных MAR путем комбинации определенных генетических элементов
Для дальнейшей оценки относительных функций различных компонентов MAR cLysMAR подвергали делеции всех трех областей CUE (фиг.11, средняя часть), что приводило к потере части ее активности по сравнению с полноразмерной последовательностью MAR, сходным образом собранной из всех ее компонентов в качестве контроля (фиг.11, верхняя часть). Соответственно, одна копия каждой CUE или копии каждой из CUE, соединенные начало к концу, имели малую активность в отсутствие фланкирующих последовательностей. Данные результаты усиливали вывод о том, что оптимальная транскрипционная активность требует комбинации CUE с фланкирующими последовательностями. Интересно, что полноразмерная последовательность MAR, генерированная из каждого из ее компонентов, но содержащая также линкерные последовательности BglII-BamHl (AGATCC), использованные для соединения каждого фрагмента ДНК, характеризовалась высокой транскрипционной активностью (6-кратная активация) по сравнению с 4,8-кратной, отмеченной для исходного элемента MAR в данной серии анализов (см. фиг.5).
Далее авторы изобретения исследовали вопрос, могут ли потенциально изогнутые области ДНК также быть активными в окружении, отличном от того, что находится в их природном MAR-контексте. Поэтому авторы запланировали поменять между собой элементы CUE-F, CUE-B и CUE-K, оставив неизмененными фланкирующие последовательности. Последовательности, фланкирующие CUE-F-элемент, амплифицировали посредством PCR и объединяли так, чтобы они скрепляли различные CUE с поддержанием их исходной ориентации и протяженности, или объединяли без CUE. Данные сконструированные MAR длиной ~1,8 т.н.п затем анализировали на их способность усиливать экспрессию трансгена, как это делали выше. Все три CUE были активны в данном контексте, и поэтому их действие не ограничивается одним данным набором фланкирующих последовательностей. Интересно, что элемент CUE-K был даже более активен, чем CUE-F при встраивании между фланкирующими CUE-F последовательностями, и данная комбинированная конструкция проявляла такую же высокую активность, как наблюдаемая для целой природной MAR (4,8-кратная активация). То, что отличает элемент CUE-K от CUE-F и CUE-B, представляет собой наличие перекрывающихся участков связывания белков MEF-2 и SatB1, в дополнение к его чертам CUE. Поэтому слияние CUE-B с фланкирующим доменом CUE-F приводит к более высокой плотности всех трех участков связывания, что, вероятно, объясняет повышенную активность. Эти результаты указывают на то, что комбинации CUE с последовательностями, содержащими участки связывания таких белков, как NMP4, MEF-2, SatB1, и/или белки polyPpolyQ, составляют мощные искусственные последовательности MAR.
Пример 12. Экспрессирующие векторы
Три экспрессирующих вектора согласно изобретению представлены на фиг.12.
Плазмида pPAG01 представляет собой производное рUC19 длиной 5640 т.п.н. Оно содержит фрагмент куриной ДНК длиной 2960 т.п.н., клонированный в сайты рестрикции BamHI и XbaI. Вставка происходит с края 5'-конца локуса куриного лизоцима и имеет высокое содержание A/T.
Контрольная плазмида pGEGFP (также называемая pSV40EGFP) представляет собой производное вектора pGL3-контроль (Promega), в котором последовательность гена люциферазы замещена последовательностью гена EGFP из вектора pEGFP-N1 (Clontech). Размер плазмиды pGEGFP составляет 4334 т.п.н.
Контрольная плазмида pUbCEGFP является производным pGL3 с промотором убиквитина.
Плазмида pPAG01GFP (также называемая pMAR-SV40EGFP) представляет собой производное pGEGFP с 5'-Lys-MAR-элементом, клонированным в MCS, расположенный непосредственно выше промотора SV40. Размер плазмиды pPAG01 EGF составляет 7285 п.н.
Пример 13. Влияние дополнительной трансфекции первично трансфицированных клеток на экспрессию трансгена
За сутки до трансфекции клетки высевали на 24-луночный планшет в среду роста с плотностью 1,35×105 клеток/лунку для клеток CHO-DG44. Через 16 часов после инокуляции клетки трансфицировали при достижении ими 30-40% сомкнутости с использованием Lipofect-AMINE 2000 (здесь и далее LF2000) согласно инструкциям производителя (Invitrogen). Двадцать семь микролитров бессывороточной среды (Opti-MEM, Invitrogen), содержащих 1,4 мкл LF2000, смешивали с 27 мкл Opti-MEM, содержащими 830 нг линейной плазмидной ДНК. Количество плазмиды селекции антибиотиком (pSVneo) доходило до одной десятой количества репортерной плазмиды, несущей трансген GFP. Смесь инкубировали при комнатной температуре в течение 20 мин для обеспечения образования комплексов ДНК-LF2000. Смесь разбавляли 300 мкл Opti-MEM и выливали в опустошенные перед этим содержащие клетки лунки. Через 3 часа инкубации клеток со смесью ДНК при 37°C в CО2-инкубаторе один мл основанной на DMEM среды добавляли к каждой лунке. Клетки далее инкубировали в течение 24 часов в CО2-инкубаторе при 37°C. Затем клетки трансфицировали второй раз способом, описанным выше, за исключением того, что плазмида устойчивости несла другой ген устойчивости (pSVpuro). Через двадцать четыре часа после второй трансфекции клетки пассировали и размножали во флаконе T-75, содержащем среду селекции, дополненную 500 мкг/мл G-418 и 5 мкг/мл пуромицина. После двухнедельного периода селекции стабильно трансфицированные клетки культивировали в 6-луночных планшетах. Альтернативно популяцию клеток опять трансфицировали с использованием такого же способа, но с pTKhygro (Clontech) и pSVdhfr в качестве плазмид устойчивости. Экспрессию GFP анализировали на сортере активированных флуоресценцией клеток (FACS) и с использованием Fluoroscan.
На фиг.13 показано, что фенотип дважды трансфицированных клеток (здесь и далее называемых вторично трансфицированными) не только характеризовался сильным окрашиванием, так что для визуализации зеленого света от белка GFP не требовалось специальной лампы и фильтра, но также он характеризовался большинством продуцирующих клеток (нижняя правая гистограмма FACS) по сравнению с исходной популяцией (центральная гистограмма). Данный уровень флуоресценции соответствует специфичной клеточной продуктивности, равной по меньшей мере 10 пг на клетку в сутки. Действительно, клетки, трансфицированные только один раз (первично трансфицированные), которые не экспрессировали маркерные белок, почти полностью исчезали из клеточной популяции после повторной трансфекции. Столбцы с флуоресценцией GFP ниже 101 единиц достигали 30% на центральной гистограмме и менее 5% на правой гистограмме. Это указывает на то, что дополнительные клетки были трансфицированы и успешно экспрессировали GFP.
Поразительно, что количество флуоресценции, проявляемой повторно трансфицированными клетками, указывало на то, что субпопуляция клеток, куда была дважды включена ДНК, экспрессировала гораздо больше GFP, чем ожидаемое двукратное повышение. Действительно, результаты, показанные в таблице 2, указывают на то, что вторично трансфицированные клетки проявляют в среднем более двукратного повышения GFP, которое ожидается, если два набора последовательностей, один при каждой из последующих трансфекций, интегрировался независимо со сходной эффективностью. Интересно, что это не зависело от промоторной последовательности, направляющей репортерный ген, поскольку векторы, содержащие как вирусный, так и клеточный промотор, давали сходное усиление GFP (ср. линии 1 и 2). Однако данный эффект был особенно заметным для MAR-содержащего вектора по сравнению с плазмидами без MAR (линия 3), где две последовательные трансфекции приводили к 5,3- и 4,6-кратному повышению экспрессии в двух различных экспериментах.
Приведены два независимых эксперимента. Плазмиду устойчивости pSVneo повторно трансфицировали различными экспрессирующими векторами GFP. Через сутки после трансфекции клетки повторно трансфицировали теми же плазмидами с той разницей, что плазмиды устойчивости изменяли на pSVpuro. Клетки, несущие оба гена устойчивости, подвергали селекции на 500 мкг/мл G-418 и 5 мкг/мл пуромицина, и экспрессию маркерного репортерного гена подвергали количественному анализу Fluoroscan. Кратность повышения соответствует отношению флуоресценции, полученной от двух последовательных трансфекций по сравнению с суммарной флуоресценцией, полученной от соответствующих независимых трансфекций. Кратности повышения, которые считали существенно более высокими, приведены жирным шрифтом, и они отвечают уровням флуоресценции, которые, соответственно, превышают в 2 раза сумму значений флуоресценции, полученных от независимых трансфекций.
Повышение уровня экспрессии GFP в множественно трансфицированных клетках не ожидалось исходя из существующего уровня знаний, и данный эффект не наблюдали ранее.
Все вместе представленные здесь данные поддерживают идею о том, что плазмидные последовательности, первично интегрированные в геном хозяина, облегчают интеграцию других плазмид за счет гомологичной рекомбинации со вторым входящим набором плазмидных молекул. События рекомбинации плазмиды происходят в пределах 1-часового интервала после достижения плазмидной ДНК ядра, и частота гомологичной рекомбинации между совместно инъецированными плазмидными молекулами в культивируемых клетках млекопитающих, как было показано, исключительно высока, рекомбинация характеризуется синхронностью (Folger, K. R., K. Thomas, and M.R. Capecchi, Nonreciprocal exchanges of information between DNA duplexes coinjected into mammalian cell nuclei. Mol Cell Biol, 1985, 5(1): p.59-69], что объясняет интеграцию множественных копий плазмид. Однако гомологичная рекомбинация между недавно введенной ДНК и ее хромосомным гомологом обычно происходит очень редко, с максимальной частотой 1 на 103 получающие ДНК клетки [Thomas, K.R., K.R. Folger, and M.R. Capecchi, High frequency targeting of genes to specific sites in the mammalian genome. Cell,1986. 44 (3): p.419-28.]. Таким образом, результаты могут указывать на то, что MAR-элемент неожиданно действует, способствуя таким событиям рекомбинации. MAR не только модифицируют организацию генов in vivo и возможно также обеспечивают репликацию ДНК в связи с вирусными последовательностями ДНК, но также они могут действовать как сигналы рекомбинации ДНК.
Пример 14. MAR опосредуют неожиданно высокие уровни экспрессии в множественно трансфицированных клетках
Если направленные MAR события рекомбинации должны происходить в процессе множественной трансфекции, авторы изобретения ожидают, что синергия между первичной и вторичной плазмидной ДНК подлежит воздействию присутствия элементов MAR на одной или обеих стадиях трансфекции. Авторы изобретения оценивали данную возможность путем множественных трансфекций клеток одной pMAR или в комбинации с различными экспрессирующими плазмидами с использованием описанного ранее способа. В таблице 3 показано, что двукратная трансфекция клеток плазмидой pMAR-SV40EGFP давала наивысшую экспрессию GFP и наивысшую степень усиления всех состояний (в 4,3 раза). Двукратная трансфекция вектором без MAR, наоборот, не давала усиления, или приводила к малому усилению, в 2,8 раза, вместо ожидаемого двукратного усиления. Авторы изобретения заключили, что присутствие элементов MAR на каждой стадии транфекции необходимо для достижения максимального синтеза белка.
Интересно, что когда клетки вначале трансфицировали одним pMAR и затем повторно трансфицировали pSV40EGFP или pMAR-SV40EGFP, уровни GFP более чем удваивались по сравнению с теми, которые получались в результате единичной трансфекции последними плазмидами (в 2,5 и 2,7 раз соответственно вместо ожидаемого 1-кратного). Это указывает на то, что предварительная трансфекция MAR может повышать экспрессию плазмиды, использованной во второй процедуре трансфекции. Поскольку MAR действуют на структуру хроматина и экспрессию генов только местно, это подразумевает, что два типа ДНК могли интегрироваться в один и тот же хромосомный локус. Наоборот, трансфекция одних экспрессирующего GFP векторов с последующей экспрессией элемента MAR на второй стадии приводила к малому увеличению уровня GFP или к отсутствию такого увеличения. Это указывает на то, что важен порядок транфекции плазмид, и что первое событие трансфекции для обеспечения значимо более высокого уровня генной экспрессии должно включать в себя элемент MAR.
Если элементы MAR способствовали бы гомологичной рекомбинации плазмид, остававшихся в форме эписом от первой и второй процедуры трансфекции, с их последующей совместной интеграцией в один хромосомный локус, следовало ожидать, что порядок трансфекции плазмид не повлияет на уровни GFP. Однако приведенные выше открытия указывают на то, что более выгодно трансфицировать элемент MAR в первое, а не во второе событие трансфекции. Это указывает на следующий молекулярный механизм: во время первой процедуры трансфекции элементы MAR могут образовывать конкатамеры и интегрироваться по меньшей мере частично в клеточную хромосому. Данная интегрированная ДНК MAR может, в свою очередь, способствовать дальнейшей интеграции большего количества плазмид во время второй процедуры транфекции в тот же самый или близлежащий хромосомный локус.
Пример 15. MAR как долгосрочные усилители переноса ДНК
Если интегрированные MAR опосредовали образование постоянных склонных к рекомбинации хромосомных структур, следует ожидать высокого уровня экспрессии, даже если вторая трансфекция проводится через много времени после первой, в момент, когда большая часть временно введенной эписомной ДНК элиминировалась. Для проверки данной возможности клетки по таблице 3, подвергнутые селекции на устойчивость к антибиотикам в течение трех недель, опять трансфицировали один раз или дважды и отбирали на включение дополнительных маркеров устойчивости ДНК. Третичный или третичный и четвертичный циклы трансфекции проводили комбинациями pMAR или pMAR-SV40EGFP и анализировали на предмет экспрессии GFP, как делали до этого.
Вторично трансфицированные pMAR-SV40EGFP/pMAR-SV40EGFP клетки использовали в третьем цикле трансфекции в конце процесса селекции. Третичную трансфекцию выполняли с применением pMAR или pMAR-SV40EGFP, и pTKhygro в качестве плазмиды селекции с получением третично трансфицированных клеток. Через 24 часа клетки опять трансфицировали каждой плазмидой и pSVdhfr, результатом чего были четвертично трансфицированные клетки, которые отбирали в ростовой среде, содержащей 500 мкг/мл G-418 и 51 мкг/мл пуромицина, 300 мкг/мл гидромицина B и 5 мкМ метотрексата. Вторично трансфицированные клетки вначале характеризовались флуоресценцией GFP, равной 8300. Кратность повышения соответствует отношению флуоресценции, полученной от двух последовательных трансфекций по сравнению с суммарной флуоресценцией, полученной от соответствующих независимых трансфекций. Кратности повышения, которые считали существенно более высокими, приведены жирным шрифтом, и они отвечают уровням флуоресценции, которые, соответственно, превышают в 2 раза сумму значений флуоресценции, полученных от независимых трансфекций.
Эти результаты показывают, что загрузка большего количества копий pMAR или pMAR-SV40EGFP приводила к сходным двукратным повышениям общей флуоресценции клеток. Загрузка даже большим количеством MAR в четвертичной трансфекции далее повышала данную активность еще в 2,4 раза. Это согласуется с гипотезой авторов изобретения о том, что нововведенные последовательности MAR могут интегрироваться в хромосомный трансгенный локус путем гомологичной рекомбинации и таким образом еще повышать экспрессию трансгена.
Когда клетки трансфицировали третий и четвертый раз плазмидой pMAR-SV40EGFP, активность GFP повышалась далее, и опять до уровней, которые не предсказывались путем сложения уровней флуоресценции, полученных при независимых трансфекциях. Экспрессия GFP достигала уровня, который приводил к тому, что клетки видимо сияли зеленым в дневном свете (фиг.14). Эти результаты далее указывают на то, что эффективность четвертичной трансфекции была намного большей, чем та, что ожидалась исходя из эффективности третьего переноса ДНК, и это означало, что надлежащее время между трансфекциями является критическим для получения оптимального повышения генной экспрессии, причем одни сутки являются предпочтительными по сравнению с периодом, равным трем неделям. Авторы изобретения полагают, что элементы MAR способствуют вторичным событиям интеграции с повышением частоты рекомбинации в участке их хромосомной интеграции посредством расслабления близких хроматиновых структур, поскольку они опосредуют местное повышение ацетилирования гистонов (Yasui, D., et al., SATB1 targets chromatin remodelling to regulate genes over long distances. Nature, 2002.419 (6907): p. 641-5.]. Альтернативно или одновременно с этим MAR потенциально перемещают близлежащие гены в субъядерное пространство, которое, как полагают, обогащено действующими в транс-положении факторами, включая белки, которые могут участвовать в событиях рекомбинации, такие как топоизомеразы. Это может приводить к образованию локуса, в котором последовательности MAR могут окружать повторы pSV40EGFP, эффективно скрывая трансгены от опосредованных хроматином сайленсерных эффектов.
Пример 16. Применение MAR, идентифицированных посредством SMAR Scan®, для повышения экспрессии рекомбинантного белка
Четыре элемента MAR случайным образом выбирали из последовательностей, полученных из анализа всей последовательности генома человека посредством SMAR Scan® или комбинированным способом. Их обозначали 1_6, 1_42, 1_68 (где первое число представляет собой хромосому, из которой происходит последовательность, а второе число специфично для предсказанной MAR на данной хромосоме) и X_S29, «супер»-MAR, идентифицированная на X-хромосоме. Указанные предсказанные MAR встраивали в вектор pGEGFPControl выше промотора SV40 и энхансера, направляющего экспрессию зеленого флуоресцирующего белка, и данные плазмиды трансфицировали в культивируемые клетки CHO, как описано ранее (Zahn-Zabal, M., et al., Development of stable cell lines for production or regulated expression using matrix attachment regions. J. Biotechnol, 2001. 87(1): p. 29-42). Затем экспрессию трансгена анализировали во всей популяции стабильно трансфицированных клеток с использованием устройства-сортера флуоресцентных клеток (FACS). Как можно видеть на фиг.19, все данные новые идентифицированные MAR повышали экспрессию трансгена значимо по сравнению с экспрессией, направляемой MAR куриного лизоцима, причем «супер»-MAR X_S29 была наиболее действенной из всех новых идентифицированных MAR.
Пример 17. Влияние на гематокрит экспрессии mEpo in vivo посредством электропереноса сетевой системы в присутствии человеческой MAR (1-68) и в ее отсутствие
Терапевтический ген кодирует EPO (эритропоэтин), гормон, применяемый для лечения анемии. Ген EPO помещают под контроль индуцируемого доксициклином промотора, в систему переключения генов, описанную ранее и называемую ниже сетевой системой (Imhof, M.O., Chatellard, P., and Mermod, N. (2000). A regulatory network for efficient control of transgene expression. J. Gene. Med. 2, 107-116.). Затем EPO и регуляторные гены вводят инъекцией в мышцы мышей с использованием процедуры электропорации in vivo, называемой электропереносом, так что гены переносятся в ядра мышечных волокон. Когда к питьевой воде мышей добавляют антибиотик доксициклин, данное соединение, как ожидается, индуцирует экспрессию EPO, что приводит к повышению уровня гематокрита вследствие повышения числа красных кровяных клеток, опосредованное высокими уровнями циркулирующих EPO. Таким образом, если MAR повышает экспрессию EPO, будет ожидаться более высокий уровень гематокрита.
Эксперименты in vivo проводили на самках мышей C57BL6 возрастом 5 недель (Iffa Credo-Charles River, Франция). 30 мкг плазмидной ДНК в физиологическом солевом растворе доставляли путем чрезкожных инъекций в переднюю большеберцовую мышцу. Все инъекции проводили под наркозом с использованием кетаминола (75 мг/кг) и наркоксила (10 мг/кг). После внутримышечной инъекции ДНК к мышце прикладывали электрическое поле. Применяли вольтаж 200 В/см при пульсах длительностью 8 мс с частотой 1 Гц (Bettan M, Darteil R, Caillaud JM, Soubrier F, Delaere P, Branelec D, Mahfoudi A, Duverger N, Scherman D. 2000. "High-level protein secretion into blood circulation after electric pulse-mediated gene transfer into skeletal muscle". Mol Ther. 2: 204-10).
16 мышей подвергали инъекции сетевой системой, экспрессирующей EPO без MAR 1_68, и 16 других мышей подвергали инъекции сетевой системой, включающей в себя данную MAR в 5'-направлении от промоторных/энхансерных последовательностей, направляющих экспрессию генов активатора и EPO. В каждой группе половине мышей давали доксициклин в питьевой воде с начала эксперимента (сутки 0 - сутки электропереноса), а второй половине доксициклин помещали в питьевую воду, начиная с 21 суток.
Образцы крови собирали с использованием гепаринизированных капилляров путем ретро-орбитальной пункции в различные моменты времени после инъекции плазмид. Капилляры центрифугировали 10 минут при 5000 об/мин при комнатной температуре, и оценивали объем фракции кровяных клеток по сравнению с общим объемом крови и выражали его виде процентиля, определяя уровень гематокрита.
Как можно заключить из фиг.16, группа мышей, подвергавшаяся инъекции сетевой системой с MAR, индуцированная с начала эксперимента, характеризовалась более высокой индукцией гематокрита по сравнению с мышами, подвергавшимися инъекции исходной сетевой системой без MAR. Через 2 месяца уровни гематокрита в «содержащей MAR группе» еще находились на уровне, более высоком (65%), чем нормальные значения гематокрита (45-55%).
Более важно то, что поздняя индукция (21 сутки) возможна только в присутствии MAR, но не у мышей, в которых сетевую систему вводили без MAR. Таким образом, MAR, вероятно, защищает трансгены от сайленсерных эффектов и обеспечивает индукцию их экспрессии даже после длительного периода в неиндуцирующих условиях.
В общем, элемент MAR способен повышать экспрессию терапевтического гена, что выявляется его повышающим физиологическим действием на гематокрит.
Реферат
Настоящее изобретение относится к области биотехнологии и может быть использовано при производстве различных белковых продуктов с помощью технологии рекомбинантных ДНК. Определены и получены новые последовательности ДНК, относящиеся к областям прикрепления матрикса (MAR), которые характеризуются способностью повышать продуцирование белка в эукариотических клетках. Предложены способы трансфекции эукариотических клеток-хозяев, в том числе новый способ множественной трансфекции, основанные на применении активных последовательностей ДНК MAR по изобретению и обеспечивающие существенное повышение уровня экспрессии рекомбинантного белка по сравнению с аналогичными клетками, трансфицированными традиционными методами. 8 н. и 3 з.п.ф-лы, 20 ил., 9 табл.
Формула
a) последовательность ДНК, характеризующуюся нуклеотидной последовательностью MAR, выбранной из группы, включающей SEQ ID NO: 24-27, комплементарной ей последовательностью, ее частью, содержащей, по меньшей мере, 70% нуклеотидов в длину; или
b) последовательность ДНК, характеризующуюся тем, что она по существу соответствует элементу или фрагменту cLysMAR, включающему, по меньшей мере, одну область, выбранную из В-, K- и F-областей.
очищенную и выделенную последовательность ДНК MAR по п.1,
очищенный и выделенный фрагмент или элемент cLysMAR по п.2 и
искусственную последовательность ДНК MAR по п.3 или 4,
для повышения активности продуцирования рекомбинантного белка в эукариотической клетке-хозяине.
a) введение в указанную эукариотическую клетку-хозяина, по меньшей мере, одной очищенной последовательности ДНК, включающей в себя, по меньшей мере, один функционально связанный с промотором ген, кодирующий представляющий интерес белок, и, по меньшей мере, одной очищенной и выделенной последовательности ДНК MAR, выбранной из группы, включающей
очищенную и выделенную последовательность ДНК MAR по п.1,
очищенный и выделенный фрагмент или элемент cLysMAR по п.2 и
искусственную последовательность ДНК MAR по п.3 или 4;
b) обработку указанной трансфицированной эукариотической клетки-хозяина, по меньшей мере, в одной дополнительной стадии трансфекции, осуществляемой в момент, когда клетка-хозяин только вошла во второй цикл клеточного деления, по меньшей мере, одной очищенной последовательностью ДНК, включающей в себя, по меньшей мере, один функционально связанный с промотором ген, кодирующий представляющий интерес белок, и, по меньшей мере, одной очищенной и выделенной последовательностью ДНК MAR, выбранной из группы, включающей
очищенную и выделенную последовательность ДНК MAR по п.1,
очищенный и выделенный фрагмент или элемент cLysMAR по п.2 и
искусственную последовательность ДНК MAR по п.3 или 4;
c) селекцию указанной трансфицированной эукариотической клетки-хозяина.
очищенную и выделенную последовательность ДНК MAR по п.1,
очищенный и выделенный фрагмент или элемент cLysMAR по п.2 и
искусственную последовательность ДНК MAR по п.З или 4, которая включена во второй вектор.
a) эукариотические клетки-хозяева трансфицируют способом по любому из пп.6-8 или по п.9,
b) трансфицированные клетки культивируют в культуральной среде в условиях, подходящих для экспрессии данного белка, и
c) получают указанный белок из клеток.
Приоритет по пунктам:
Комментарии