Композиция нуклеированного полимера пропилена с высокой жесткостью - RU2784936C1
Код документа: RU2784936C1
Чертежи
Описание
Настоящее изобретение направлено на новую нуклеированную композицию гомополимера пропилена с улучшенными механическими, в частности, ударными свойствами, а также на формованный продукт, содержащий гомополимер пропилена.
Лишь ограниченное количество документов посвящено влиянию эффектов структуры цепи на экспрессию β-нуклеации. Таким образом, K. Busse et al., Macromolecules 2000, 33, 8775-8780 описывают образование β-фазы в изотактическом полипропилене (iPP), полученном с помощью металлоценового катализатора (концентрация дефектов, определенная13C-ЯМР: 1,1%), при добавлении нуклеирующего агента N,N-дициклогексил-2,6-нафталиндикарбоксамида (0,3 мас. %). В отсутствие нуклеирующего агента β-фаза не наблюдалась, в то время как композиция показывала некоторое содержание β-фазы с нуклеацией. Содержание в диапазоне 30-40 % β-фазы можно измерить по диаграммам широкоугольного рассеяния рентгеновских лучей (WAXS) на Фиг. 2 указанного документа, что явно ниже требований для эффективного улучшения ударных свойств.
M. Varma-Nair и др., J.Therm. J.Therm. Anal. Calorim. 2004, 59, 483-495 раскрывают эффекты нуклеации с α- и β-нуклеирующими агентами для гомополимера пропилена на основе Циглера-Натта и на основе металлоцена с неопределенной структурой цепи. Значительное содержание β-фазы около 83 % для обоих полимеров достигается только после очень медленного охлаждения, то есть не в реальных условиях обработки.
R. Krache и др., Macromolecules 2007, 40, 6871-6878, обнаружили, что добавления 1 мас. % типичного β-нуклеирующего агента недостаточно для развития какого-либо заметного количества β-модификации в гомополимере пропилена на основе металлоцена с неопределенной структурой цепи по крайней мере в используемых условиях кристаллизации, которые охватывают широкий диапазон скоростей охлаждения. Для сравнения, такое же количество β-нуклеирующего агента, добавленного к гомополимеру iPP, полученного с помощью катализатора Циглера-Натта, приводит к образованию почти 100% β-формы при низких скоростях охлаждения.
Создание β-формы полипропилена является эффективным и универсальным способом улучшение ударной вязкости гомополимеров пропилена и статистических сополимеров этилена и пропилена. Для получения достаточно высокого содержания β-формы необходимо не только добавить специальный β-нуклеирующий агент, но также иметь подходящий основной полимер. Общеизвестно, что дефекты в цепи, означающие как стерео-, так и регио-ошибки в случае гомополимеров пропилена, также нарушают образование β-формы. Они могут значительно нарушить образование β-модификации, как обсуждается, например, в EP 2 657 285 A1 и EP 2 657 286 A1. Однако данные изобретения относятся к ограниченному содержанию β-фазы без добавления конкретного нуклеирующего агента. Более того, образование других модификаций полипропилена, таких как значительное количество γ-фазы, пагубно для улучшения ударной вязкости.
Таким образом, целью настоящего изобретения является обеспечение композиции гомополимера пропилена с улучшенными механическими, в частности высокими ударными свойствами, в сочетании с высокой стереорегулярностью и ограниченным количеством региодефектов. Такую композицию гомополимера пропилена следует производить с помощью способа, не требующего или требующего лишь незначительных изменений процесса.
Данные цели могут быть достигнуты путем открытия композиции полимера пропилена согласно настоящему изобретению, которая имеет относительно высокое содержание β-фазы и относительно низкое количество стерео- и региодефектов.
Соответственно, настоящее изобретение направлено на композицию полимера пропилена, содержащую:
(а) не менее 90 мас. % гомополимера пропилена от общей массы композиции гомополимера пропилена, где гомополимер пропилена имеет
(i) изотактичность пентад (mmmm) 98,0 мол. % или более, определенную количественной спектроскопией ЯМР (ядерный магнитный резонанс)13С, как описано в настоящем документе,
(ii) относительное количество 2,1-эритрорегиодефектов не более 2,0 мол. %, определенного количественной спектроскопией ЯМР13С, как описано настоящем документе, и
(b) нуклеирующий агент (B), селективный для бета-модификации изотактического полипропилена в количестве от 0,001 до 1,0 мас. % от общей массы композиции гомополимера пропилена,
где указанная композиция гомополимера пропилена имеет скорость течения расплава MFR2 (230°C, 2,16 кг) согласно ISO 1133 от 0,1 до 10 г/10 мин., содержание бета-фазы не менее 80 %, измеренное методом широкоугольного рассеяния рентгеновских лучей (WAXS), как описано в разделе, посвященному методам, ниже.
Настоящее изобретение дополнительно направлено на способ получения указанной выше композиции гомополимера пропилена, включающий стадии:
(i) полимеризация пропилена в присутствии односайтового катализатора полимеризации в гомополимер пропилена (A), как определено в п. 1,
(ii) смешивание в расплаве подходящего количества нуклеирующего агента (B), селективного для бета-модификации изотактического полипропилена, с указанным гомополимером пропилена (A) в устройстве для смешивания расплава, и
(iii) отверждение полученного расплава в процессе непрерывного или подводного гранулирования.
Более того, настоящее изобретение направлено на формованный продукт из гомополимера пропилена, получаемый прессованием или экструзией указанной выше композиции гомополимера пропилена. Такой формованный продукт предпочтительно может быть частью системы транспортировки жидкости, системы фильтрации или системы хранения жидкости.
Предпочтительно, указанная выше композиция гомополимера пропилена имеет относительное улучшение ударной вязкости по Шарпи образца с надрезом ΔNIS (Дельта NIS) (23°C) не менее 4,0, где ΔNIS (23°C) определяется следующим образом:
ΔNIS=(NISнук-NISPP)/NISPP
где NISнук представляет собой ударную вязкость по Шарпи образца с надрезом при 23°C композиции β-нуклеированного гомополимера пропилена, и NISPP представляет собой ударную вязкость по Шарпи образца с надрезом при 23°C ненуклеированного гомополимера пропилена (a), где ударная вязкость по Шарпи образца с надрезом при 23°C измеряется в соответствии с ISO 179 1eA.
Более предпочтительно композиция гомополимера пропилена согласно настоящему изобретению имеет относительное улучшение ударной вязкости по Шарпи образца с надрезом ΔNIS (23°C) не менее 4,2, даже более предпочтительно не менее 4,5. Верхний предел для ΔNIS (23°C) предпочтительно составляет 20,0.
Соответствующие композиции дополнительно характеризуются низким содержанием фракции растворимых веществ, особенно растворимых в холодном ксилоле (XCS), как описано ниже.
Композиция гомополимера пропилена согласно настоящему изобретению может предпочтительно иметь абсолютную ударную вязкость по Шарпи образца с надрезом при 23°C, измеренную согласно ISO 179 1eA, не менее 30 кДж/м2, более предпочтительно не менее 35 кДж/м2, еще более предпочтительно не менее 40 кДж/м2. Верхний предел указанной абсолютной ударной вязкости по Шарпи образца с надрезом при 23°C предпочтительно составляет 100 кДж/м².
Композиция гомополимера пропилена согласно настоящему изобретению предпочтительно имеет модуль упругости при изгибе, определенный согласно ISO 178, в диапазоне от 1200 до 2000 МПа, более предпочтительно от 1300 до 1900 МПа.
Композиция гомополимера пропилена согласно настоящему изобретению предпочтительно имеет основную температуру плавления (Tm) не менее 140°C, более предпочтительно в диапазоне от 140 до 152°C, еще более предпочтительно в диапазоне от 141 до 150°C и/или температуру кристаллизации (Tc) не менее 114°C, более предпочтительно в диапазоне от 115 до 135°C, еще более предпочтительно в диапазоне от 116 до 127°C, гдеTm иTc измеряются методом дифференциальной сканирующей калориметрии (DSC). Основная температура плавления (Tm) является типичной мерой для определения состава полимера пропилена, представляющего β-фазу.
Композиция гомополимера пропилена согласно настоящему изобретению предпочтительно имеет содержание бета-фазы не менее 85 %, более предпочтительно не менее 88 %, еще более предпочтительно не менее 90 %, определенное методом широкоугольного рассеяния рентгеновских лучей (WAXS), как описано в разделе о методах ниже. Верхний предел содержания бета-фазы предпочтительно составляет 99,99%.
Композиция гомополимера пропилена согласно настоящему изобретению предпочтительно имеет относительное содержание гамма-модификации (содержание гамма-фазы) менее 5,0 %, более предпочтительно менее 4,0 %, даже более предпочтительно менее 3,0 %, определенное методом широкоугольного рассеяние рентгеновских лучей (WAXS), как описано в разделе о методах ниже. Нижний предел указанного содержания гамма-фазы предпочтительно составляет 0,001 %.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
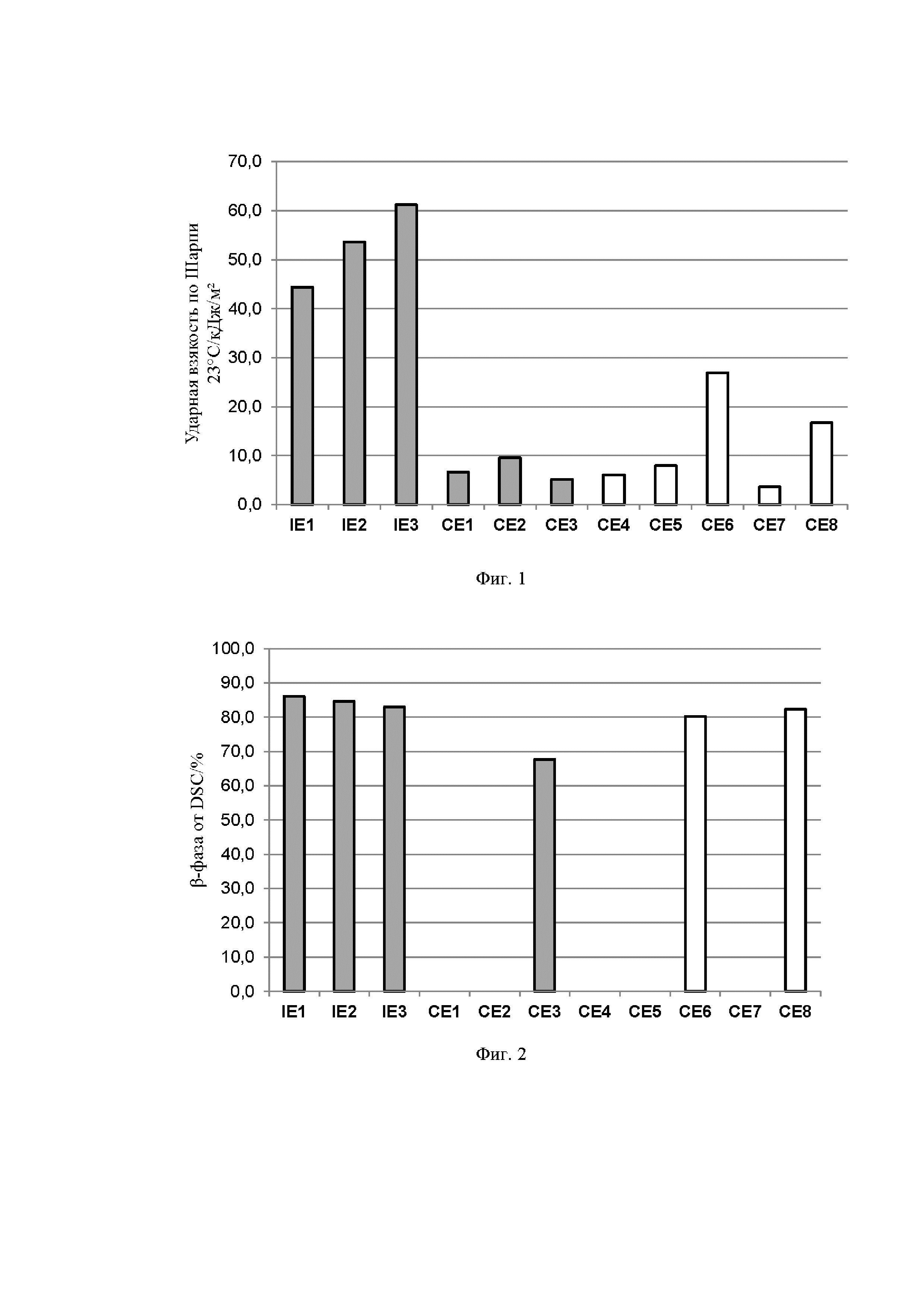
На Фиг. 1 показана диаграмма, сравнивающая значения ударной вязкости по Шарпи (23°C) (NIS) образца с надрезом для композиций согласно изобретению и сравнительных примеров.
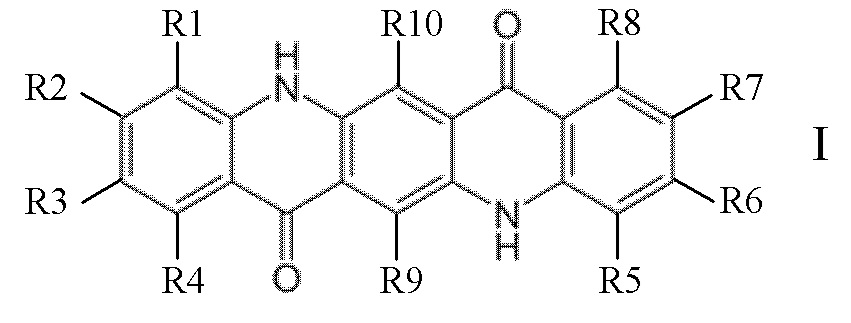
На Фиг. 2 показана диаграмма, сравнивающая содержание β-фазы в композициях согласно изобретению и сравнительных примеров.
Гомополимер пропилена (А)
Композиция гомополимера пропилена согласно изобретению содержит не менее 90 мас. % гомополимера пропилена (A), как описано выше. В дополнительных вариантах осуществления изобретения композиция гомополимера пропилена содержит не менее 95 мас. %, не менее 97 мас. % или не менее 99 мас. % гомополимера пропилена (А), как описано в настоящем документе. В дополнительном варианте осуществления изобретения композиция гомополимера пропилена содержит 100 мас. %, не более 99,9 мас. %, не более 99,7 мас. %, не более 99,5 мас. % или не более 99,2 мас. % гомополимера пропилена (A), как описано в настоящем документе. Следует понимать, что каждая комбинация нижнего и верхнего пределов, как указано выше, рассматривается как раскрытая, например, то, что композиция гомополимера пропилена согласно изобретению содержит гомополимер пропилена (A), как описано в настоящем документе, в диапазоне от 90 до 100 мас. % или, например, в диапазоне от 90 до 99,7 мас. %. В одном варианте осуществления гомополимер пропилена (A) является единственным полимером в композиции гомополимера пропилена по изобретению. В еще одном предпочтительном варианте осуществления композиция гомополимера пропилена состоит из гомополимера полипропилена (А), как описано в настоящем документе.
Существует решающее различие в микроструктуре цепи между полипропиленами, полученными с помощью металлоценового катализатора и катализатора Циглера-Натта. Регулярность цепи полипропилена на основе металлоцена снижается из-за стерео- и региодефектов, тогда как регулярность цепи полипропиленов на основе Циглера-Натта снижается только из-за стереодефектов. Важно отметить, что настоящий гомополимер пропилена (A) может быть получен или получен в присутствии односайтового катализатора полимеризации, предпочтительно катализатора металлоценового типа на носителе, дающего гомополимер пропилена, предпочтительно изотактический гомополимер пропилена с высокой стереорегулярностью и низким количеством региодефектов.
Выражение «гомополимер», используемое в настоящем изобретении, относится к полипропилену, который состоит по существу, т.е. из не менее 99,5 мас. %, более предпочтительно не менее 99,8 мас. %, звеньев пропилена. В предпочтительном варианте осуществления обнаруживаются только звенья пропилена в гомополимере пропилена. Содержание сомономера можно определить с помощью спектроскопии13С ЯМР.
Как очевидно из термина «гомополимер пропилена», настоящее изобретение не определяет композицию различных полимеров. Соответственно, гомополимер пропилена может содержать дополнительные добавки, но не другие полимерные компоненты, кроме гомополимера.
Кроме того, гомополимер пропилена (A) можно определить по основной температуре плавления (Tm). Таким образом, очевидно, что гомополимер пропилена (A) предпочтительно имеет основную температуру плавления (Tm), измеренную с помощью дифференциальной сканирующей калориметрии (DSC), не менее 145°C, более предпочтительно не менее 148°C, наиболее предпочтительно не менее 150°C. Верхний предел основной температуры плавления (Tm) может составлять 170°C, 165°C или 162°C. Таким образом, в частности, следует принимать во внимание, что основная температура плавления (Tm), измеренная с помощью дифференциальной сканирующей калориметрии (DSC) гомополимера пропилена, находится в диапазоне от 145 до 170°C, более предпочтительно в диапазоне от 148 до 165°C, еще более предпочтительно в диапазоне от 150 до 162°C.
Гомополимер пропилена (А) можно определить по его стереорегулярности, то есть по его изотактичности пентад. Таким образом, предпочтительно, чтобы гомополимер пропилена (А) имел довольно высокую изотактичность пентад (mmmm), то есть 98,0 мол. % или более, более предпочтительно 98,5 мол. % или более, еще более предпочтительно 99,0 мол. % или более, еще более предпочтительно в диапазоне от 98,5 или более до менее 100,0 мол. %, например, от 98,9 мол. % или более до 99,8 мол. % или менее. Изотактичность пентад (mmmm) может быть определена с помощью количественной спектроскопии13С ЯМР, как описано в разделе, посвященном методам, ниже.
Гомополимер пропилена (A), например, изотактический гомополимер пропилена на основе металлоцена, содержит обнаруживаемое количество региодефектов, то есть 2,1-эритрорегиодефектов. Соответственно, изотактический гомополимер пропилена, как и изотактический гомополимер пропилена на основе металлоцена, предпочтительно имеет 2,1-эритрорегиодефекты от 0,0 или более до 2,0 мол. % или менее, предпочтительно в диапазоне от 0,1 до 1,5 мол. %, более предпочтительно в диапазоне от 0,2 до 1,2 мол. %. Предпочтительно гомополимер пропилена (A), например, изотактический гомополимер пропилена на основе металлоцена, характеризуется довольно низкой концентрацией региоошибок, таких как 2,1-эритрорегиодефекты, по сравнению с известными полипропиленами на основе металлоцена. Соответственно, предпочтительно, чтобы гомополимер пропилена (А), используемый в настоящем изобретении, имел 2,1-эритрорегиодефекты, предпочтительно от 0,0 или более до 1,8 мол. % или менее, более предпочтительно от 0,05 мол. % или более до 1,4 мол. % или менее, например, от 0,1 мол. % или более до 1,1 мол. % или менее. Следует понимать, что каждая комбинация нижнего и верхнего пределов, как указано выше, считается раскрытой. 2,1-эритрорегиодефекты могут быть определены с помощью количественной спектроскопии13С ЯМР, как описано в разделе, посвященном методам, ниже.
Кроме того, предпочтительно, чтобы гомополимер пропилена (A), используемый в настоящем изобретении, имел скорость течения расплава (MFR), заданную в определенном диапазоне. Соответственно, предпочтительно, чтобы в настоящем изобретении гомополимер пропилена имелMFR2 (230°C) не более 10,0 г/10 мин, более предпочтительно 7,0 г/10 мин или менее, еще более предпочтительно 5,0 г/10 мин или менее. В качестве нижнего предела можно указать значение MFR2 (230°C) 0,1 г/10 мин, предпочтительно 0,15 г/10 мин или более, еще более предпочтительно 0,2 г/10 мин или более. Следует понимать, что каждая комбинация верхнего и нижнего пределов здесь рассматривается как раскрытая.
Кроме того, предпочтительно, чтобы гомополимер пропилена (А), используемый в настоящем изобретении, имел средневесовую молекулярную массу Mw более 300 кг/моль, более предпочтительно, не менее 320 кг/моль, например, не менее 330 кг/моль. Кроме того, предпочтительно, чтобы средневесовая молекулярная масса Mw составляла не более 1200 кг/моль, более предпочтительно не более 1000 кг/моль, например, не более 900 кг/моль. Гомополимер пропилена (А) предпочтительно имеет соотношение между средневесовой и среднечисловой молекулярной массой, Mw/Mn, не более 4,0, более предпочтительно не более 3,5, причем как Mw, так и Mn определяются с помощью эксклюзионной хроматографии (SEC), как описано в разделе о методах ниже. Нижний предел соотношения между средневесовой и среднечисловой молекулярной массой, Mw/Mn, предпочтительно составляет 2,0.
Также предпочтительно, чтобы гомополимер пропилена (A), используемый в настоящем изобретении, имел низкое содержание фракции, растворимой в холодном ксилоле (XCS). Указанная фракция XCS предпочтительно составляет менее 1,5 мас. %, более предпочтительно менее 1,0 мас. %, например менее 0,7 мас. %. Фракция XCS определяется, как описано в разделе о методах ниже. Нижний предел указанного содержания веществ, растворимых в холодном ксилоле (XCS), предпочтительно составляет 0,01 %.
Бета-нуклеирующий агент (B)
Композиция полимера пропилена по настоящему изобретению содержит нуклеирующий агент (B), селективный для бета-модификации полипропилена, предпочтительно изотактического полипропилена, в количестве от 0,001 мас. % до 1,0 мас. % от общей массы композиции гомополимера пропилена. Бета-полипропилен может быть образован добавлением бета-нуклеирующих агентов к расплаву пропилена и последующей кристаллизацией. Бета-кристалличность предпочтительно индуцируется нуклеирующим агентом (В), селективным для бета-модификации полипропилена, выбранным из соединений типа хинакридона и солей дикарбоновых кислот с металлами из группы IIa периодической системы.
Нуклеирующий агент (В), селективный для бета-модификации полипропилена, используемый в настоящем изобретении, может быть предпочтительно выбран из по меньшей мере одного хинакридонового соединения, указанного ниже:
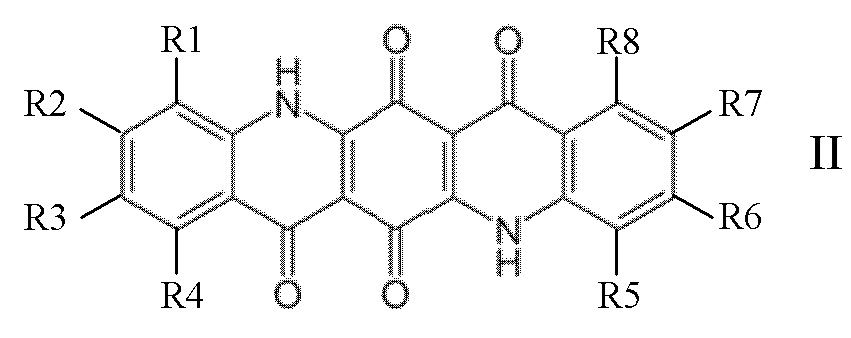

где R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 независимо представляют собой H, алкил, галоген, алкокси или арил;
или по меньшей мере одной соли дикарбоновой кислоты с по меньшей мере одним щелочноземельным металлом или их смеси.
Предпочтительно R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 независимо представляют собой H, CH3, Cl, метокси или фенил.
Предпочтительными хинакридоновыми соединениями формулы I являются хинакридон, диметилхинакридон и диметоксихинакридон. Предпочтительными хинакридоновыми соединениями формулы II являются хинакридонхинон, смешанный кристалл 5,12-дигидро-хино(2,3b)акридин-7,14-диона и хино(2,3b)акридин-6,7,13,14-(5H,12H)-тетрона, как описано в EP-B0177961, и диметоксихинакридонхинон. Предпочтительными хинакридоновыми соединениями формулы III являются дигидрохинакридон, диметоксидигидрохинакридон и дибензодигидрохинакридон.
Альтернативно, нуклеирующий агент (В), селективный для бета-модификации полипропилена, используемый в настоящем изобретении, может быть предпочтительно выбран из по меньшей мере одной соли дикарбоновой кислоты с металлами из группы IIa периодической системы элементов. Соли дикарбоновых кислот с щелочноземельными металлами являются особенно предпочтительными. Еще более предпочтительно использовать кальциевую соль пимелиновой кислоты и кальциевую соль субериновой кислоты и их смеси.
Также можно использовать смеси таких нуклеирующих агентов.
Наиболее предпочтительно бета-нуклеирующий агент (В), селективный для бета-модификации полипропилена, используемый в настоящем изобретении, представляет собой Cinquasia Gold или кальциевую соль пимелиновой кислоты, особенно предпочтительно Cinquasia Gold.
Другими подходящими типами бета-нуклеирующих агентов являются:
диамидные соединения типа производных дикарбоновой кислоты из C5-C8-циклоалкил моноаминов или C6-C12-ароматическихмоноаминов и C5-C8-алифатических, C5-C8-циклоалифатических или C6-C12-ароматических дикарбоновых кислот,
диамидные соединения типа производного диамина из C5-C8-циклоалкил монокарбоновых кислот или C6-C12-ароматических монокарбоновых кислот и C5-C8-циклоалифатических или C6-C12-ароматических диаминов,
диамидные соединения типа производных аминокислот, полученных в результате реакции амидирования C5-C8-алкил-, C5-C8-циклоалкил- илиC6-C12-ариламиновых кислот, C5-C8-алкил-, C5-C8-циклоалкил- или хлоридов C6-C12-ароматических монокарбоновых кислот и C5-C8-алкил-, C5-C8-циклоалкил- или C6-C12-ароматических моноаминов,
соли щелочноземельных металлов и имидокислот формулы IV

где х представляет собой 1-4; R представляет собой H, -COOH, C1-C12-алкил, C5-C8-циклоалкил или C6-C12-арил, и Y представляет собой C1-C12-алкил, C5-C8-циклоалкил или C6-C12-арил-замещенный двухвалентные C6-C12-ароматические остатки, предпочтительно кальциевые соли фталоилглицина, гексагидрофталоилглицина, N-фталоилаланина и/или N-4-метилфталоилглицина.
Бета-нуклеированный полипропилен с бета-кристалличностью по меньшей мере 85 % может быть получен, например, с использованием бета-нуклеирующего агента в количестве от 0,005 мас. % до 0,2 мас. % от композиции полипропилена.
Бета-нуклеирующий агент может быть предоставлен в виде маточной смеси. Маточная смесь предпочтительно состоит из бета-нуклеирующего агента, полипропиленовой смолы и/или полиолефинового воска.
Каталитическая система
Композиция гомополимера пропилена может быть предпочтительно получена в присутствии металлоценового катализатора на носителе, более предпочтительно в присутствии каталитической системы, содержащей
(i) комплекс формулы (I)
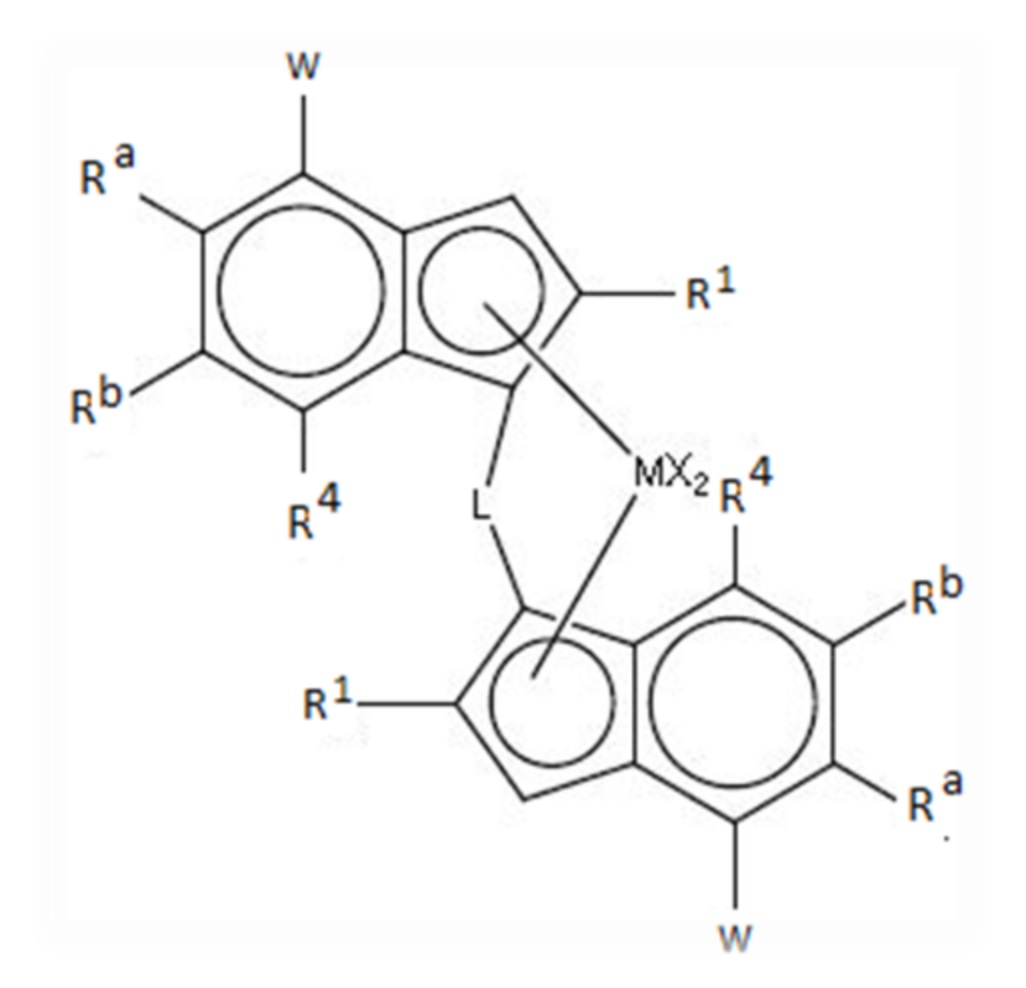
где
М представляет собой цирконий или гафний;
каждый X представляет собой сигма-лиганд;
L представляет собой двухвалентный мостик, выбранный из -R'2C-, -R'2C-CR'2-, -R'2Si-, -R'2Si-SiR'2-, -R'2Ge-, где каждый R' независимо представляет собой атом водорода, C1-C20-гидрокарбил, три(C1-C20-алкил)силил, C6-C20-арил, C7-C20-арилалкил или C7-C20-алкиларил;
каждый R1 независимо представляет собой C1-C20гидрокарбильный радикал, необязательно содержащий один или более гетероатомов из групп 14-16 периодической системы элементов, или C4-C20 гидрокарбильный радикал, разветвленный по β-атому относительно циклопентадиенильного кольца, необязательно содержащий один или более гетероатомов, принадлежащих к группам 14-16 периодической системы элементов, или представляет собой C3-C20гидрокарбильный радикал, разветвленный по β-атому относительно циклопентадиенильного кольца, где β-атом представляет собой атом Si;
каждый Ra независимо представляет собой водород или алифатическую C1-C20 гидрокарбильную группу, необязательно содержащую один или более гетероатомов из групп 14-16 периодической системы элементов;
каждыйRb независимо представляет собой водород или алифатическую C1-C20 гидрокарбильную группу, необязательно содержащую один или более гетероатомов из групп 14-16 периодической системы элементов; или
группы Ra иRb на соседних атомах углерода могут быть взяты вместе с образованием 5-членного насыщенного углеродного кольца, которое необязательно замещено n группами R2, где n составляет от 0 до 3;
каждая R2 является одинаковой или отличной и может быть C1-C20 гидрокарбильной группой или C1-C20гидрокарбильным радикалом, необязательно содержащим один или более гетероатомов, принадлежащих к группам 14-16 периодической системы элементов;
каждая R4 независимо представляет собой алифатическую C1-C20 гидрокарбильную группу, необязательно содержащую один или более гетероатомов из групп 14-16 периодической системы элементов, или атом водорода;
каждая группа W независимо представляет собой арильную или гетероарильную группу, содержащую вплоть до 20 атомов углерода, необязательно замещенную одной или более группами R3;
каждая R3 является одинаковой или отличной и представляет собой C1-C20гидрокарбильную группу, необязательно содержащую один или более гетероатомов, принадлежащих к группам 14-16 периодической системы элементов; или две группы R3 на соседних атомах углерода, взятые вместе, могут образовывать конденсированное 5- или 6-членное неароматическое кольцо с группой W, где само указанное кольцо необязательно замещено одной или более C1-C20 гидрокарбильными группами, или две соседние группы R3, взятые вместе, могут образовывать дополнительное моно- или полициклическое кольцо, конденсированное с W, необязательно замещенное одной или двумя группами R3;
и
(ii) необязательно сокатализатор, содержащий соединение металла группы 13 периодической системы элементов.
Термин «C1-C20 гидрокарбильная группа», следовательно, включает C1-C20-алкил, C2-C20-алкенил, C2-C20-алкинил, C3-C20-циклоалкил, C3-C20-циклоалкенил, C6-C20-арильные группы, C7-C20-алкиларильные группы или C7-C20-арилалкильные группы или смеси указанных групп, такие как циклоалкил, замещенный алкилом.
Если не указано иное, предпочтительными C1-C20-гидрокарбильными группами являются C1-C20-алкил, C4-C20-циклоалкил, C5-C20-циклоалкилалкильные группы, C7-C20-алкиларильные группы, C7-C20-арилалкильные группы или C6-C20-арильные группы, особенно C1-C10-алкильные группы, C6-C10-арильные группы или C7-C12-арилалкильные группы, например, C1-C8-алкильные группы. Наиболее предпочтительными гидрокарбильными группами являются метил, этил, пропил, изопропил, трет-бутил, изобутил, C5-C6-циклоалкил, циклогексилметил, фенил или бензил.
Термин «галоген» включает фтор-, хлор-, бром- и йод-группы, особенно хлор-группы, когда речь идет о комплексном определении.
Термин «гетероциклическая группа» означает предпочтительно моноциклическую неароматическую кольцевую структуру, содержащую по меньшей мере один гетероатом, например, пиперидинил или пиперазинил.
Термин «гетероарил» означает предпочтительно моноциклическую ароматическую кольцевую структуру, содержащую по меньшей мере один гетероатом. Предпочтительные гетероарильные группы содержат от 1 до 4 гетероатомов, выбранных из O, S и N. Предпочтительные гетероарильные группы включают фуранил, тиофенил, оксазол, тиазол, изотиазол, изооксазол, триазол и пиридил.
Любая группа, включающая «один или более гетероатомов, принадлежащих к группам 14-16 периодической системы элементов», предпочтительно означает O, S или N. N-группы могут присутствовать как -NH- или -NR"-, где R" представляет собой C1-C10-алкил. Например, может быть от 1 до 4 гетероатомов. Группа, включающая один или более гетероатомов, принадлежащих к группам 14-16 периодической системы элементов, также может быть алкоксигруппой, например, C1-C10-алкокси группой.
Степень окисления иона металла определяется в первую очередь природой рассматриваемого иона металла и стабильностью отдельных степеней окисления каждого иона металла.
Следует понимать, что в комплексах по изобретению ион металла М координирован лигандами X, чтобы удовлетворить валентность иона металла и заполнить его доступные координационные центры. Природа этих σ-лигандов может сильно различаться.
Предпочтительная подгруппа металлоценов принадлежит к группе формулы (I),
где
М представляет собой цирконий или гафний;
каждый X представляет собой сигма-лиганд;
L представляет собой двухвалентный мостик, выбранный из -R'2C-, -R'2C-CR'2-, -R'2Si-, -R'2Si-SiR'2-, -R'2Ge-, где каждый R' независимо представляет собой атом водорода, C1-C20-гидрокарбил, три(C1-C20-алкил)силил, C6-C20-арил, C7-C20-арилалкил или C7-C20-алкиларил;
каждый R1 независимо представляет собой C1-C20 гидрокарбильный радикал, необязательно содержащий один или более гетероатомов из групп 14-16 периодической системы элементов;
каждый Ra независимо представляет собой водород или алифатическую C1-C20 гидрокарбильную группу, необязательно содержащую один или более гетероатомов из групп 14-16 периодической системы элементов;
каждый Rb независимо представляет собой водород или алифатическую C1-C20 гидрокарбильную группу, необязательно содержащую один или более гетероатомов из групп 14-16 периодической системы элементов; или
группы Ra иRb на соседних атомах углерода могут быть взяты вместе с образованием 5-членного насыщенного углеродного кольца, которое необязательно замещено группой R2;
каждая R2 представляет собой C1-C20 гидрокарбильную группу;
каждый R4 независимо представляет собой водород или алифатическую C1-C20 гидрокарбильную группу, необязательно содержащую один или более гетероатомов из групп 14-16 периодической системы элементов;
каждая группа W независимо представляет собой арильную или гетероарильную группу, содержащую вплоть до 20 атомов углерода, необязательно замещенную одной или более группами R3;
каждая R3 представляет собой C1-C20 гидрокарбильную группу, или две группы R3 на соседних атомах углерода, взятые вместе, могут образовывать конденсированное 5- или 6-членное неароматическое кольцо с группой W, где указанное кольцо само необязательно замещено одной или более C1-C20-гидрокарбильными группами;
и (ii) сокатализатор, содержащий соединение металла группы 13 периодической системы элементов, например, бора.
Предпочтительными вариантами каталитической системы согласно формуле (I) являются:
рац-диметилсиландиил-бис[2-метил-4-(3',5'-ди-трет-бутилфенил)-7-метоксиинденил]гафния дихлорид,
рац-диметилсиландиил-бис[2-изо-бутил-4-фенил-5-метокси-6-трет-бутилинден-1-ил]циркония дихлорид,
рац-диметилсиландиил-бис[2-метил-4-(3',5'-ди-трет-бутилфенил)-7-метоксиинденил]циркония дихлорид,
рац-μ-{бис-[η5-2-метил-4-(4'-трет-бутилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]диметилсиландиил}дихлорцирконий),
(рац-анти-диметилсиландиил[2,6-метил-4-(3',5'-диметилфенил)инден-1-ил][2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил]циркония дихлорид), и
(рац-анти-диметилсиландиил[2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил][2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил]гафния дихлорид).
Катализатор по изобретению предпочтительно можно использовать в форме на носителе.
Катализатор, используемый в настоящем изобретении в твердой форме, предпочтительно в форме твердых частиц, может быть нанесен на внешний носитель, такой как диоксид кремния или оксид алюминия.
Два полициклических лиганда, составляющие комплекс формулы (I), предпочтительно идентичны, и, следовательно, комплекс формулы (I) может быть симметричным. Комплексы по изобретению могут быть в мезо- или рацемических формах (или в их смеси). Предпочтительно используется рацемическая (рац) форма.
Для образования активных каталитических частиц обычно необходимо использовать сокатализатор, хорошо известный в данной области. Сокатализаторы, содержащие одно или более соединений металлов группы 13, такие как алюминийорганические соединения или бораты, используемые для активации металлоценовых катализаторов, подходят для использования в этом изобретении.
Система односайтового катализатора полимеризации, используемая в изобретении, включает (i) комплекс, как определено в настоящем документе; и обычно (ii) алюминийалкильное соединение (или другой подходящий сокатализатор) или его продукт реакции. Таким образом, сокатализатор предпочтительно представляет собой алюмоксан, такой как МАО (метилалюмоксан), или алюмоксан, отличный от МАО. Предпочтительным алюмоксаном является метилалюмоксан (МАО). Другими предпочтительными алюминийалкильными соединениями являются триэтилалюминий, триизобутилалюминий, триизогексилалюминий, три-н-октилалюминий и триизооктилалюминий.
Поскольку алюмоксаны, используемые согласно настоящему изобретению в качестве сокатализаторов, не являются чистыми соединениями из-за способа их получения, молярность растворов алюмоксана здесь и далее основывается на содержании в них алюминия. Настоящая твердая каталитическая система включает (I) комплекс, в котором ион металла координирован лигандом, как определено здесь; и обычно (ii) алюминийалкильное соединение (или другой подходящий сокатализатор) или его продукт реакции. Таким образом, сокатализатор предпочтительно представляет собой алюмоксан, такой как МАО (метилалюмоксан), или алюмоксан, отличный от МАО.
Также можно использовать боратные сокатализаторы. Специалисту будет понятно, что при использовании сокатализаторов на основе бора обычно предварительно активируют комплекс путем его реакции с алюминийалкильным соединением, таким как ТИБА (триизобутилалюминий). Данный способ хорошо известен, и можно использовать любой подходящий алюминийалкил, например, Al(C1-6-алкил)3.
Также можно использовать смесь сокатализаторов на основе Al и B. Алюмоксаны образуются при частичном гидролизе алюминийорганических соединений, например, соединений формулы AIR3, AIR2Y и Al2R3Y3, где R может представлять собой, например, C1-C10 алкил, предпочтительно C1-C5 алкил, или C3-10-циклоалкил, C7-C12-арилалкил или алкиларил, и/или фенил, или нафтил, и где Y может быть водородом, галогеном, предпочтительно хлором или бромом, или C1-C10 алкокси, предпочтительно метокси или этокси. Полученные кислородсодержащие алюмоксаны в общем случае не являются чистыми соединениями, а представляют собой смеси олигомеров.
Однако в качестве альтернативы катализаторы изобретения могут использоваться с другими сокатализаторами, например соединениями бора, такими как B(C6F5)3, C6H5N(CH3)2H:B(C6F5)4, (C6H5)3C:B(C6F5)4 или Ni(CN)4[B(C6F5)3]42-.
Подходящие количества сокатализатора хорошо известны специалисту в области техники. Обычно молярные соотношения Al к M составляют от 1:1 до 1000:1 моль/моль. Предпочтительно, когда алюминийалкил используется в качестве сокатализатора, молярное соотношение алюминия в активаторе к переходному металлу в комплексе составляет от 1 до 500 моль/моль, предпочтительно от 10 до 400 моль/моль и, в частности, от 50 до 400 моль/моль.
Обычно каталитическую систему, используемую в настоящем изобретении, можно получить, как описано в WO 2018/122134 A1. Катализатор можно использовать в форме на носителе или без носителя, предпочтительно в форме на носителе. Используемый материал носителя в виде частиц предпочтительно представляет собой органический или неорганический материал, такой как диоксид кремния, оксид алюминия или диоксид циркония, или смешанный оксид, такой как диоксид кремния-оксид алюминия, в частности диоксид кремния, оксид алюминия или диоксид кремния-оксид алюминия. Предпочтительно использование подложки из диоксида кремния. Квалифицированному специалисту известны процедуры, необходимые для нанесения на носитель металлоценового катализатора.
Особенно предпочтительно носитель представляет собой пористый материал, так что комплекс может быть загружен в поры носителя, например, с использованием процесса, аналогичного тем, которые описаны в WO94/14856 (Mobil), WO95/12622 (Borealis) и WO2006/097497. Размер частиц не имеет решающего значения, но предпочтительно находится в диапазоне от 5 до 200 мкм, более предпочтительно от 20 до 80 мкм. Использование этих носителей известно в данной области техники.
Процесс полимеризации
Композиция гомополимера пропилена согласно настоящему изобретению может быть получена способом, включающим стадии:
(i) полимеризация пропилена в присутствии односайтового катализатора полимеризации в гомополимер пропилена (A), как определено выше,
(ii) смешивание в расплаве подходящего количества нуклеирующего агента (B), селективного для бета-модификации изотактического полипропилена, с указанным гомополимером пропилена (A) в устройстве для смешивания расплава, и
(iii) отверждение полученного расплава в процессе непрерывного или подводного гранулирования.
Полимеризация пропилена с использованием указанной выше твердой каталитической системы может быть осуществлена в одном или более, например, одном, двух или трех реакторах полимеризации, с использованием обычных методов полимеризации, например, газофазной полимеризации, полимеризации в жидкой фазе, суспензионной или объемной полимеризации.
Процессы полимеризации, которые подходят для получения статистического сополимера пропилена и этилена, обычно включают одну или две стадии полимеризации, и каждую стадию можно проводить в растворе, суспензии, псевдоожиженном слое, объемной или газовой фазе.
Термин «реактор полимеризации» означает, что происходит основная полимеризация. Таким образом, в случае, если процесс состоит из одного или более реакторов полимеризации, это определение не исключает вариант, когда вся система включает, например, стадию предварительной полимеризации в реакторе предварительной полимеризации. Термин «состоит из» является лишь завершающей формулировкой с точки зрения основных реакторов полимеризации.
Термин «процесс последовательной полимеризации» указывает на то, что гомополимер пропилена получают по меньшей мере в двух реакторах, соединенных последовательно. Соответственно, такая полимеризационная система включает по меньшей мере первый реактор полимеризации (R1) и второй реактор полимеризации (R2) и, необязательно, третий реактор полимеризации (R3). В случае применения «процесса последовательной полимеризации» второй реактор полимеризации (R2) и необязательный третий реактор полимеризации (R3) являются газофазными реакторами (GPR), то есть первым газофазным реактором (GPR1) и вторым газофазным реактором. (GPR2). Газофазный реактор (GPR) предпочтительно представляет собой реактор с псевдоожиженным слоем, реактор с быстрым псевдоожиженным слоем или реактор с неподвижным слоем или любую их комбинацию.
Однако, как правило, предпочтителен одностадийный процесс полимеризации. Первый, соответственно, единственный реактор полимеризации (R1) предпочтительно представляет собой суспензионный реактор и может быть любым непрерывным или простым реактором периодического действия с перемешиванием или петлевым реактором, работающим в объеме или суспензии. Объемная полимеризация означает полимеризацию в реакционной среде, которая содержит по меньшей мере 60 % (мас./мас.) мономера. Согласно настоящему изобретению суспензионный реактор предпочтительно представляет собой (объемный) петлевой реактор.
Для суспензионных реакторов температура реакции обычно находится в диапазоне от 60 до 110°C (например, от 60 до 90°C), давление в реакторе обычно находится в диапазоне от 5 до 80 бар (например, от 20 до 60 бар), а время выдерживания обычно находится в диапазоне от 0,1 до 5 часов (например, от 0,3 до 2 часов). В качестве реакционной среды обычно используется пропилен.
Для газофазных реакторов температура реакции обычно находится в диапазоне от 60 до 115°C (например, от 70 до 110°C), давление в реакторе обычно находится в диапазоне от 10 до 25 бар, а время пребывания обычно составляет от 0,5 до 8 часов (например, от 0,5 до 4 часов). Используемый газ может быть пропиленом, необязательно в виде смеси с инертным газом, таким как азот или пропан. В дополнение к собственно стадиям полимеризации и реакторам, способ может содержать любые дополнительные стадии полимеризации, такие как стадия предварительной полимеризации, и любые дальнейшие стадии обработки после реактора, известные в данной области техники.
Обычно количество используемой твердой каталитической системы будет зависеть от природы катализатора, типов и условий реактора и желаемых свойств полимерного продукта. Как хорошо известно в данной области техники, водород можно использовать для регулирования молекулярной массы полимера.
Композицию гомополимера пропилена согласно настоящему изобретению получают, подвергая гомополимер пропилена, как описано в данном документе, стадии формования. Предпочтительно, такая стадия формования представляет собой стадию прессования или стадию экструзии.
Композиция полимера пропилена согласно настоящему изобретению неожиданно имеет улучшенные ударные свойства в сочетании с высокой стереорегулярностью и ограниченным количеством региодефектов. Было обнаружено, что в композиции полимера пропилена согласно настоящему изобретению достигается особенно высокое количество β-фазы с чрезвычайно низкими количествами других модификаций, таких как γ-фаза. Данная микроструктура полимера неожиданно существенно повысила ударную вязкость.
Формованный продукт из гомополимера пропилена
Формованный продукт из гомополимера пропилена по настоящему изобретению предпочтительно представляет собой формованный (прессованием) гомополимер пропилена. Термины «формование» или «формованный» в соответствии с данным изобретением широко понимаются и, таким образом, охватывают изделия, полученные любым способом придания формы посредством формования. Термины «формование» или «формованный», в частности, охватывают, например, изделия, полученные литьем под давлением, прессованием или экструдированием. Также рассматриваются комбинации данных процессов формования, таких как экструзия с раздувом, литье под давлением с раздувом, литье под давлением с вытяжкой и раздувом. В процессе прессования формовочный материал помещается в полость формы и нагревается до температуры выше основной температуры плавления (Tm). Полость формы закрывается, и прикладывается давление сверху, превышающее атмосферное. После этого материал охлаждается, возможно, давление поддерживается во время фазы охлаждения. В процессе литья под давлением формовочный материал подается в нагретый цилиндр (где он нагревается и формуется) и помещается в полость формы, где он охлаждается под давлением. В процессе экструзии материал нагревается, формуется в экструдере и проталкивается через фильеру. В отношении определений экструзии и формования см. "Polypropylene Handbook", Nello Pasquini, 2nd Edition, Hanser.
Согласно настоящему изобретению формованный гомополимер пропилена (продукт) расплавляли либо под давлением, либо без него. В свою очередь, формованный прессованием гомополимер пропилена (продукт) был расплавлен под давлением, то есть под давлением, превышающим атмосферное давление.
В частности, формованный продукт из гомополимера пропилена может представлять собой изделие, образующее часть системы транспортировки жидкости, системы фильтрации или системы хранения жидкости. Более предпочтительно, изделие может представлять собой трубу, фильтрующую пластину, фитинг или любую другую часть системы транспортировки жидкости, системы фильтрации или системы хранения жидкости.
Способы измерения
(а) Скорость течения расплава (MFR).
Скорость течения расплава измеряется как MFR2 в соответствии с ISO 1133 15 (230°C, нагрузка 2,16 кг) для полипропилена. MFR является показателем текучести и, следовательно, технологичности полимера. Чем выше показатель текучести расплава, тем ниже вязкость полимера.
(b) Среднечисловая молекулярная масса (Mn), средневесовая молекулярная масса (Mw) и полидисперсность (Mw/Mn).
Измерения проводили с помощью гель-проникающей хроматографии (GPC). Средневесовую молекулярную массу Mw и полидисперсность (Mw/Mn), где Mn представляет собой среднечисловую молекулярную массу, а Mw представляет собой средневесовую молекулярную массу) измеряли методом на основе ISO 16014-1:2003 и ISO 16014-4:2003.
Инструмент Waters Alliance GPCV 2000, оснащенный рефрактометрическим детектором и онлайн-вискозиметром, использовали с 3xTSK-гелевыми колонками (GMHXL-HT) от TosoHaas и 1,2,4-трихлорбензолом (TCB, стабилизированный 200 мг/л 2,6-ди-трет-бутил-4-метилфенол) в качестве растворителя при 145°С и постоянной скорости потока 1 мл/мин. Для анализа вводили 216,5 мкл раствора образца. Набор колонок был откалиброван с использованием относительной калибровки с 19 узкими стандартами MWD полистирола (PS) в диапазоне от 0,5 кг/моль до 11 500 кг/моль и набором хорошо охарактеризованных широких стандартов полипропилена.
Все образцы приготовили путем растворения 5-10 мг полимера в 10 мл (при 160°С) стабилизированного TCB (того же, что и подвижная фаза) и выдерживания в течение 3 часов при непрерывном встряхивании перед отбором проб в аппарате GPC.
(c) Количественная оценка микроструктуры с помощью ЯМР-спектроскопии.
Количественная спектроскопия ядерного магнитного резонанса (ЯМР) была использована для количественной оценки стереорегулярности (тактичности) и региорегулярности полимеров.
Количественные13C {1H} ЯМР-спектры регистрировали в растворе с помощью спектрометра Bruker Advance III 400 NMR, работающего при 400,15 и 100,62 МГц для1H и13C соответственно. Все спектры регистрировали при использовании13С оптимизированного 10-мм датчика измерения линейных величин при расширенном диапазоне температур при 125°С при использовании во всей пневматике газообразного азота.
Для гомополимеров полипропилена приблизительно 200 мг материала растворяли в 1,2-тетрахлорэтане-d2 (TCE-d2). Для обеспечения гомогенного раствора, после первоначальной подготовки образца в термостате трубку для ЯМР дополнительно нагревали в ротационной печи в течение по меньшей мере 1 часа. При введении в магнит трубка вращалась с частотой 10 Гц. Эта установка была выбрана в первую очередь из-за высокого разрешения, необходимого для количественной оценки распределения тактичности (Busico, V., Cipullo, R., Prog. Polym. Sci. 26 (2001) 443; Busico, V.; Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251). Стандартное одноимпульсное возбуждение использовалось с использованием схемы NOE и двухуровневой развязки WALTZ16 (Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, B., J. Mag. Reson. 187 (2007) 225; Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol. Rapid Commun. 2007, 28, 11289). Всего было получено 8192 (8k) переходных процессов на каждый спектр.
Количественные13C{1H} ЯМР спектры были обработаны, интегрированы, и определены соответствующие количественные свойства из интегралов с использованием специальных компьютерных программ.
Для гомополимеров полипропилена все химические сдвиги внутренне связаны с метилизотактической пентадой (mmmm) при 21,85 млн-1.
Распределение тактичности количественно оценивали путем интеграции метильной области между 23,6-19,7 млн-1 с поправкой на любые сайты, не связанные с интересующими стереопоследовательностями (Busico, V., Cipullo, R., Prog. Polym. Sci. 26 (2001) 443; Busico, V.; Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromolecules 30 (1997) 6251).
В частности, влияние региодефектов и сомономера на количественную оценку распределения тактичности корректировали путем вычитания репрезентативных региодефектов и интегралов сомономера из конкретных интегральных областей стереопоследовательностей.
Изотактичность определялась на уровне пентад и выражалась как процент изотактических последовательностей пентад (mmmm) по отношению ко всем последовательностям пентад:
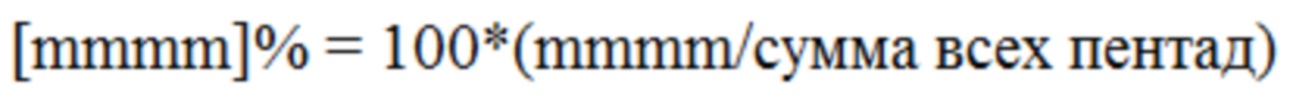
На присутствие 2,1-эритрорегиодефектов указывало присутствие двух метильных сайтов при 17,7 и 17,2 млн-1 и подтверждалось другими характерными сайтами.
Характерные сигналы, соответствующие другим типам региодефектов, не наблюдались (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev.2000, 100, 1253).
Количество 2,1 эритрорегиодефектов было определено количественно с использованием среднего интеграла двух характерных метильных сайтов при 17,7 и 17,2 млн-1.
P21e=(Ie6+Ie8)/2
Количество 1,2-первичного вставленного пропена определяли количественно на основе метильной области с корректировкой, предпринятой для сайтов, включенных в эту область, не связанных с первичной вставкой, и для сайтов первичной вставки, исключенных из этой области:
P12=ICH3+P12e
Общее количество пропена определяли как сумму первичного вставленного пропена и всех других присутствующих региодефектов:
Pобщ=P12+P21e
Молярный процент 2,1-эритрорегиодефекта был определен количественно по отношению ко всему пропену:
[21e]мол. %=100*(P21e/Pобщ)
(d) Измерение широкоугольного рассеяния рентгеновских лучей (WAXS).
Измерение широкоугольного рассеяния рентгеновских лучей (WAXS) образцов проводилось с помощью прибора Bruker D8 Discover. Дифрактометр был снабжен рентгеновской трубкой с медной мишенью, работающей при 30 кВ и 20 мА, и детектором GADDS 2-D. Для направления луча на поверхность использовалась точечная коллимация (0,5 мм). Измерения проводились в геометрии отражения, и были измерены углы 28 в диапазоне от 10° до 32,5°. Данные собирались в течение 300 с. Кривая зависимости интенсивности от 2-тета была получена с теми же параметрами измерения на образце аморфного полипропилена, который был получен экстракцией растворителем. Аморфное гало получено сглаживанием кривой. Аморфное гало был вычтен из измеренной зависимости интенсивности от 2-тета-кривая для получения кристаллической кривой.
Индекс кристалличности Xc может быть определен с помощью площади под кристаллической кривой и исходного спектра с использованием метода Чаллы, Херманса и Виденегра [ChallaG, Hermans PH, Weidinger A, Makromol. Chem. 56, 169 (1962)] следующим образом:

Количество β-формы полипропилена в кристаллической фазе Kβ рассчитывают с использованием метода Джонса [Turner-Jones A, Aizlewood JM, Beckett DR, Makromol. Chem. 75, 134 (1974)] согласно следующему уравнению:

где Iβ (300) представляет собой интенсивность пика β (300), Iα (110) представляет собой интенсивность пика α (110), Iα(040) представляет собой интенсивность пика α (040), и Iα(130) представляет собой интенсивность пика α (130), полученную после вычитания аморфного гало.
Количество γ-формы изотактического полипропилена (iPP) в кристаллической фазе Kγ рассчитывается с использованием метода, разработанного Пэй [Pae KD, J. Polym. Sci., Part A, 6, 657 (1968)] следующим образом:
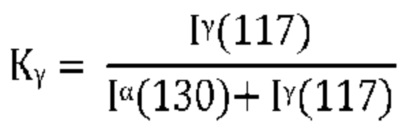
где Iα (130) представляет собой интенсивность пика α (130), и Iγ (117) представляет собой интенсивность пика γ (117), полученную после вычитания базовой линии, соединяющей основание этих пиков.
Количественное определение трехфазной кристаллической системы проводили в соответствии с процедурой, описанной в Obadal M, Cermak R, Stoklasa K, Macromol. Rapid Commun. 26, 1253 (2005). Для трехфазных кристаллических систем следующие уравнения были использованы для определения Kα (количество α-фазы), Kβ (количество β-фазы) и Kγ (количество γ-фазы):



и

(e) Анализ DSC, основная температура плавления (Tm), теплота плавления (Hm) и температура кристаллизации (Tc).
Анализ DSC проводили с помощью дифференциальной сканирующей калориметрии (DSC) Mettler TA Instrument Q2000 на образцах от 5 до 7 мг. DSC выполняется в соответствии с ISO 11357/часть 3/метод C2 в цикле нагрев/охлаждение/нагрев со скоростью сканирования 10°C/мин в диапазоне температур от минус 30 до 225°C. Температура кристаллизации (Tc) определяется на этапе охлаждения, в то время как температура основного плавления (Tm) и теплота плавления (Hm) определяется на втором этапе нагрева.
(f) Ударная вязкость по Шарпи образца с надрезом и дельта NIS.
Ударная вязкость по Шарпи образца с надрезом (NIS) измеряется в соответствии с ISO 179 1eA при 23°C и 0°C с использованием литых под давлением образцов для испытаний стержней размером 80×10×4 мм3, подготовленных в соответствии с EN ISO 1873-2.
Относительное влияние β-нуклеации на ударную вязкость (дельта NIS) рассчитывали по следующей формуле:
Дельта NIS=(NISнук-NISбаз)/NISбаз
гдеNISнук обозначает значение β-нуклеированного полимера, а NISбаз обозначает значение ненуклеированного базового полимера. Измерение дельта NIS проводилось при 23°C.
(g) Модуль упругости при изгибе.
Модуль упругости при изгибе был определен при трехточечном изгибе при 23°C в соответствии с ISO 178 на испытательных стержнях размером 80×10×4 мм3, отлитых под давлением в соответствии с EN ISO 1873-2.
(h) Прессование
Образцы для измерений WAXS были подготовлены в соответствии с ISO 173-2 на рамной форме:
Условия прессования образцов для испытаний:
(i) Растворимая в холодном ксилоле фракция.
Холодные растворимые вещества ксилола (XCS, мас. %) определяли при 25°C согласно ISO 16152; первое издание; 2005-07-01.
Настоящее изобретение теперь будет проиллюстрировано примерами.
Примеры
Катализаторы, использованные в процессе полимеризации сополимера пропилена в примерах изобретения, были получены следующим образом:
Катализаторы
Катализатор А
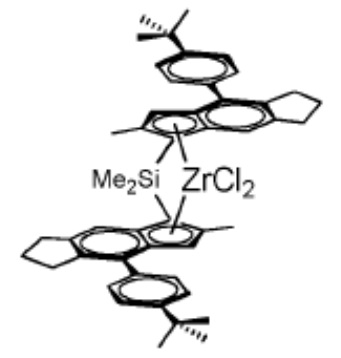
(рац-μ-диметилсиландиил-бис-[η5-2-метил-4-(4'-трет-бутилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]цирконидихлорид) получали как описано в WO 2006/097497 A1. Его спектр1H-ЯМР соответствует спектру, приведенному в упомянутой заявке на патент. Соответствующий металлоцен идентичен MC-CE5 в WO 2018/122134 A1.
Катализаторы диоксид кремния-МАО были получены на диоксиде кремния SUNSPERA DM-L-303 толщиной 30 мкм, произведенном AGC Si-Tech Co, предварительно прокаленном при 600°C в течение 2 часов в электрической муфельной печи в потоке сухого воздуха.
Получение металлоценового катализатора на носителе из диоксида кремния описано в WO 2018/122134.
Катализатор А был получен аналогично с использованием металлоцена MC-CE5.
Катализатор B
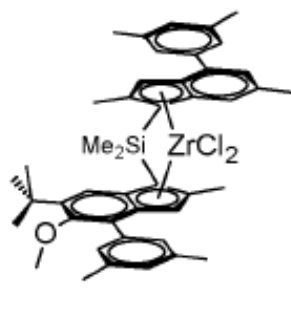
(рац-антидиметилсиландиил[2,6-метил-4-(3',5'-диметилфенил)инден-1-ил][2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутил-инден-1-ил]циркония дихлорид) получали аналогично катализатору A, что также описано в WO 2006/097497 A1. Соответствующий металлоцен идентичен MC-CE5 в WO 2018/122134 A1.
Катализаторы диоксид кремния-МАО были получены на диоксиде кремния SUNSPERA DM-L-303 толщиной 30 мкм, произведенном AGC Si-Tech Co, предварительно прокаленном при 600°C в течение 2 часов в электрической муфельной печи в потоке сухого воздуха.
Получение металлоценового катализатора на носителе из диоксида кремния описано в WO 2018/122134.
Катализатор B был получен аналогично с использованием металлоцена MC-CE7.
Катализатор C

рац-анти-диметилсиландиил[2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил][2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил]-гафния дихлорид.
Катализатор C был получен по следующей методике:
(3,5-диметилфенил)бороновая кислота
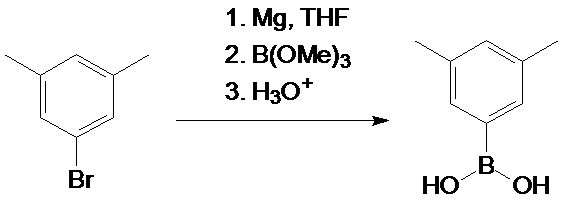
Раствор 3,5-диметилфенилмагния бромида, полученный из раствора 190,3 г (1,03 моль) 1-бром-3,5-диметилбензола в 1000 мл ТГФ (тетрагидрофуран) и 32 г (1,32 моль, 28% избыток) магниевой стружки охлаждали до минус 78°C и добавляли 104 г (1,0 моль) триметилбората одной порцией. Полученную смесь перемешивали в течение ночи при комнатной температуре. Сложный эфир бора гидролизовали осторожным добавлением 1200 мл 2 М HCl. Добавляли 500 мл простого диэтилового эфира, органический слой отделяли и водный слой дополнительно экстрагировали 2×500 мл диэтилового эфира. Объединенный органический экстракт сушили над Na2SO4, а затем упаривали досуха, получая белую массу. Последний растирали с 200 мл н-гексана, фильтровали через стеклянную фритту (G3) и осадок сушили в вакууме. Эта процедура дает 114,6 г (74%) (3,5-диметилфенил) бороновой кислоты.
Результат расчета на C8H11BO2: C, 64.06; H, 7.39. Обнаружено: C, 64.38; H, 7.72.
1H ЯМР (ДМСО-d6): δ 7,38 (s, 2H), 7,00 (s, 1H), 3,44 (very br.s (очень широкий s. 2H), 2,24 (s, 6H).
6-трет-Бутил-4-(3',5'-диметилфенил)-5-метокси-2-метилиндан-1-он

Смесь 49,14 г (157,9 ммоль) 4-бром-6-трет-бутил-5-метокси-2-метилиндан-1-она, 29,6 г (197,4 ммоль, 1,25 экв.) (3,5-диметилфенил) бороновая кислота, 45,2 г (427 ммоль) Na2CO3, 1,87 г (8,3 ммоль, 5 мол. %) Pd (OAc)2, 4,36 г (16,6 ммоль, 10 мол. %) PPh3, 200 мл воды и 500 мл 1,2-диметоксиэтана кипятили с обратным холодильником в течение 6,5 часов. ДМЭ (диметоксиэтан) упаривали на роторном испарителе, к остатку добавляли 600 мл воды и 700 мл дихлорметана. Органический слой отделяли, и водный дополнительно экстрагировали 200 мл дихлорметана. Объединенный экстракт сушили над K2CO3, и затем упаривали досуха, получая черное масло. Неочищенный продукт очищали флэш-хроматографией на силикагеле 60 (40-63 мкм, соотношение гексан:дихлорметан представляет собой 1:1, об., затем 1:3, об.) с получением 48,43 г (91%) 6-трет-бутил-4-(3',5'-диметилфенил)-5-метокси-2-метилиндан-1-она в виде коричневатого масла.
Результат расчета для C23H28O2: C 82,10; H 8,39. Обнаружено: C 82,39; H 8,52.
1H ЯМР (CDCl3): δ 7,73 (s, 1H), 7,02 (s, 1H), 7,01 (s, 2H), 3,32 (s, 3H), 3,13 (dd, J = 17,5 Гц, J = 7,8 Гц, 1H), 2,68-2,57 (m, 1H), 2,44 (dd, J = 17,5 Гц, J = 3,9 Гц), 2,36 (s, 6H), 1,42 (s, 9H), 1,25 (d, J = 7,5 Гц, 3H).
13C{1H} ЯМР (CDCl3): δ 208,90, 163,50, 152,90, 143,32, 138,08, 136,26, 132,68, 130,84, 129,08, 127,18, 121,30, 60,52, 42,17, 35,37, 34,34, 30,52, 21,38, 16,40.
5-трет-Бутил-7-(3',5'-диметилфенил)-6-метокси-2-метил-1H-инден

8,2 г (217 ммоль) NaBH4 добавляли к раствору 48,43 г (143,9 ммоль) 6-трет-бутил-4-(3',5'-диметилфенил)-5-метокси-2-метилиндан-1-она в 300 мл ТГФ, охлажденного до 5°C. Затем к этой смеси по каплям добавляли 150 мл метанола при интенсивном перемешивании в течение прибл. 7 часов при 5°C. Полученную смесь упаривали досуха и остаток распределяли между 500 мл дихлорметана и 500 мл 2 М HCl. Органический слой отделяли, водный слой дополнительно экстрагировали 100 мл дихлорметана. Объединенный органический экстракт упаривали досуха, получая слегка желтоватое масло. К раствору этого масла в 600 мл толуола добавляли 400 мг TsOH, эту смесь кипятили с обратным холодильником с головкой Дина-Старка в течение 10 минут и затем охлаждали до комнатной температуры с использованием водяной бани. Образовавшийся раствор промывали 10% Na2CO3, органический слой отделяли, водный слой экстрагировали 150 мл дихлорметана. Объединенный органический экстракт сушили над K2CO3 и затем пропускали через короткий слой силикагеля 60 (40-63 мкм). Слой силикагеля дополнительно промывали 100 мл дихлорметана. Объединенный органический элюат упаривали досуха, и полученное масло сушили в вакууме при повышенной температуре. Данная процедура дает 45,34 г (98%) 5-трет-бутил-7-(3',5'-диметилфенил)-6-метокси-2-метил-1H-индена, который далее используют без дополнительной очистки.
Результат расчета для C23H28O: C 86,20; H 8,81. Обнаружено: C 86,29; H 9,07.
1H ЯМР(CDCl3): δ 7,20 (s, 1H), 7,08 (br.s, 2H), 6,98 (br.s, 1H), 6,42 (m, 1H), 3,25 (s, 3H), 3,11 (s, 2H), 2,36 (s, 6H), 2,06 (s, 3H), 1,43 (s, 9H).
13C{1H} ЯМР (CDCl3): δ 154,20, 145,22, 141,78, 140,82, 140,64, 138,30, 137,64, 131,80, 128,44, 127,18, 126,85, 116,98, 60,65, 42,80, 35,12, 31,01, 21,41, 16,65.
[2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил]хлордиметилсилан
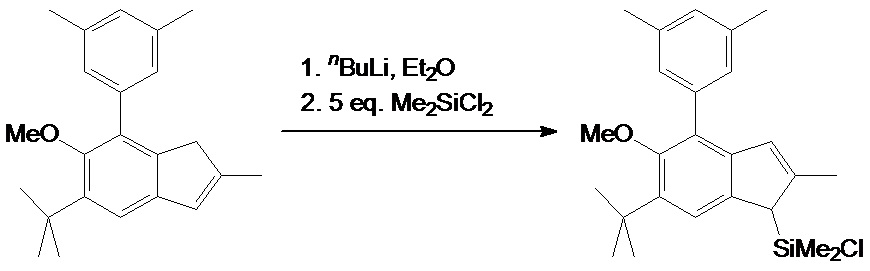
nBuLi (н-Бутиллитий) в гексане (2,43 M, 25,2 мл, 61,24 ммоль) добавляли одной порцией к раствору 19,66 г (61,35 ммоль) 2-метил-5-трет-бутил-6-метокси-7-(3',5'-диметилфенил)-1H-инден в 300 мл простого эфира охлаждают до минус 50°C. Полученную смесь перемешивали в течение 4 часов при комнатной температуре, затем полученную желтую суспензию охлаждали до минус 60°C и добавляли 40,0 мл (42,8 г, 331,6 ммоль, 5,4 экв.) дихлордиметилсилана одной порцией. Полученный раствор перемешивали в течение ночи при комнатной температуре, и затем фильтровали через стеклянную фритту (G3). Фильтрат упаривали досуха, получая [2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил]хлордиметилсилан в виде слегка желтоватого масла, которое в дальнейшем использовали без дополнительной очистки.
1H ЯМР(CDCl3): δ 7,38 (s, 1H), 7,08 (s, 2H), 6,98 (s, 1H), 6,43 (s, 1H), 3,53 (s, 1H), 3,25 (s, 3H), 2,37 (s, 6H), 2,19 (s, 3H), 1,43 (s, 9H), 0,43 (s, 3H), 0,17 (s, 3H).13C{1H} ЯМР (CDCl3): δ 155,78, 145,88, 143,73, 137,98, 137,56, 137,49, 136,74, 128,32, 127,86, 127,55, 126,64, 120,86, 60,46, 49,99, 35,15, 31,16, 21,41, 17,55, 1,11, минус 0,58.
1-Метокси-2-метил-4-(3',5'-диметилфенил)-1,2,3,5,6,7-гексагидро-s-индацен
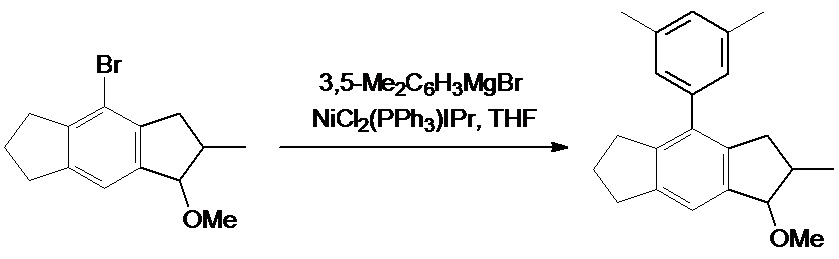
Предшественник 4-бром-1-метокси-2-метил-1,2,3,5,6,7-гексагидро-s-индацен был получен в соответствии с процедурой, описанной в WO 2015/158790 A2 (стр. 26-29).
К смеси 2,0 г (2,56 ммоль, 1,8 мол. %) NiCl2(PPh3)IPr и 40,0 г (142,3 ммоль) 4-бром-1-метокси-2-метил-1,2,3,5,6,7-гексагидро-s-индацен добавляли 200 мл (200 ммоль, 1,4 экв.) 1,0 М 3,5-диметилфенилмагнийбромида в ТГФ. Полученный раствор кипятили с обратным холодильником в течение 3 ч, затем охлаждали до комнатной температуры и добавляли 400 мл воды, а затем 500 мл 1,0 М раствора HCl. Далее эту смесь экстрагировали 600 мл дихлорметана, органический слой отделяли, а водный слой экстрагировали 2х100 мл дихлорметана. Объединенный органический экстракт упаривали досуха, получая слегка зеленоватое масло. Продукт выделяли флэш-хроматографией на силикагеле 60 (40-63 мкм; элюент: гексаны-дихлорметан 2:1 по объему, затем 1:2 по объему). Эта процедура дала 43,02 г (99%) 1-метокси-2-метил-4-(3',5'-диметилфенил)-1,2,3,5,6,7-гексагидро-s-индацена в виде бесцветного густого масла в виде смеси двух диастереоизомеров.
Результат расчета для C22H26O:C 86,23; H 8,55. Обнаружено: C 86,07; H 8,82.
1H ЯМР (CDCl3), Син-изомер: δ 7,21 (s, 1H), 6,94 (br.s, 1H), 6,90 (br.s, 2H), 4,48 (d, J = 5,5 Гц, 1H), 3,43 (s, 3H), 2,94 (t, J = 7,5 Гц, 2H), 2,87-2,65 (m, 3H), 2,63-2,48 (m, 2H), 2,33 (s, 6H), 2,02 (quin, J = 7,5 Гц, 2H), 1,07 (d, J = 6,7 Гц, 3H); Анти-изомер: δ 7,22 (s, 1H), 6,94 (br.s, 1H), 6,89 (br.s, 2H), 4,38 (d, J = 4,0 Гц, 1H), 3,48 (s, 3H), 3,06. (dd, J = 16,0 Гц, J = 7,5 Гц, 1H), 2,93 (t, J = 7,3 Гц, 2H), 2,75 (td, J = 7,3 Гц, J = 3,2 Гц, 2H), 2,51-2,40 (m, 1H), 2,34 (s, 6H), 2,25 (dd, J = 16,0 Гц, J = 5,0 Гц, 1H), 2,01 (quin, J = 7,3 Гц, 2H), 1,11 (d, J = 7,1 Гц, 3H).13C{1H} ЯМР (CDCl3), Син-изомер: δ 142,69, 142,49, 141,43, 139,97, 139,80, 137,40, 135,46, 128,34, 126,73, 120,09, 86,29, 56,76, 39,43, 37,59, 33,11, 32,37, 25,92, 21,41, 13,73; Анти-изомер: δ 143,11, 142,72, 140,76, 139,72, 139,16, 137,37, 135,43, 128,29, 126,60, 119,98, 91,53, 56,45, 40,06, 37,65, 33,03, 32,24, 25,88, 21,36, 19,36.
4-(3',5'-диметилфенил)-6-метил-1,2,3,5-тетрагидро-s-индацен

К раствору 43,02 г (140,4 ммоль) 1-метокси-2-метил-4-(3',5'-диметилфенил)-1,2,3,5,6,7-гексагидро-s-индацена в 600 мл толуола добавляли 200 мг TsOH и полученный раствор кипятили с обратным холодильником с использованием головки Дина-Старка в течение 15 мин. После охлаждения до комнатной температуры реакционную смесь промывали 200 мл 10% NaHCO3. Органический слой отделяли, водный слой дополнительно экстрагировали 100 мл дихлорметана. Объединенный органический экстракт упаривали досуха, получая масло светло-оранжевого цвета. Продукт выделяли флэш-хроматографией на силикагеле 60 (40-63 мкм; элюент: гексаны, затем нексаны:дихлорметан = 10:1 по объему). Эта процедура дает 35,66 г (93%) 4-(3',5'-диметилфенил)-6-метил-1,2,3,5-тетрагидро-s-индацена в виде слегка желтоватого масла, которое самопроизвольно затвердевает с образованием белой массы.
Результат расчета для C21H22: C 91,92; H 8,08. Обнаружено: C 91,78; H 8,25.
1H ЯМР(CDCl3): δ 7,09 (s, 1H), 6,98 (br.s, 2H), 6,96 (br.s, 1H), 6,44 (m, 1H), 3,14 (s, 2H), 2,95 (t, J = 7,3 Гц, 2H), 2,76 (t, J = 7,3 Гц, 2H), 2,35 (s, 6H), 2,07 (s, 3H), 2,02 (quin, J = 7,3 Гц, 2H).13C{1H} ЯМР (CDCl3): δ 145,46, 144,71, 142,81, 140,17, 139,80, 137,81, 137,50, 134,33, 128,35, 127,03, 126,48, 114,83, 42,00, 33,23, 32,00, 25,87, 21,38, 16,74.
[2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил][2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил] диметилсилан

nBuLi в гексане (2,43 M, 25,2 мл, 61,24 ммоль) добавляли одной порцией к раствору 16,83 г (61,33 ммоль) 4-(3',5'-диметилфенил)-6-метил-1,2,3,5-тетрагидро-s-индацен в смеси 300 мл эфира и 40 мл THF, охлаждают до минус 50°C. Эту смесь перемешивали в течение ночи при комнатной температуре, затем полученный красноватый раствор охлаждали до минус 50°C и добавляли 300 мг CuCN. Полученную смесь перемешивали 0,5 ч при минус 25°C, затем раствор [2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил]хлордиметилсилан (полученный выше, прибл. 61,24 ммоль) в 150 мл эфира одной порцией. Данную смесь перемешивали в течение ночи при комнатной температуре, затем фильтровали через слой силикагеля 60 (40-63 мкм), который дополнительно промывали 2х50 мл дихлорметана. Объединенный фильтрат упаривали при пониженном давлении, и остаток сушили в вакууме при повышенной температуре. Эта процедура дает 39,22 г (98%) [2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил][2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил] диметилсилан (прибл. Смесь стереоизомеров 3:2) в виде красноватого стекла.
1H ЯМР (CDCl3): δ 7,48 и 7,33 (2s, сумма 1H), 7,26-7,18 (m, 1H), 7,16-7,07 (m, 2H), 7,04-6,95 (m, 4H), 6,51 и 6,45 (2s, сумма 2H), 3,69 и 3,65 (2s, сумма 2H), 3,28 и 3,26 (2s, сумма 3H), 3,01–2,74 (m, 4H), 2,38 и 2,37 (2s, сумма 12H), 2,20 и 2,15 (2s, сумма 6H), 2,09-1,97 (m, 2H), 1,43 и 1,42 (2s, сумма 9H), минус 0,17, минус 0,18, минус 0,19 и минус 0,24 (4s, сумма 6H).13C{1H} ЯМР (CDCl3): δ 155,29, 147,45, 147,39, 145,99, 145,75, 143,93, 143,90, 143,72, 143,69, 142,06, 142,01, 140,08, 140,06, 139,46, 139,37, 139,26, 139,03, 139,00, 138,24, 137,50, 137,34, 137,07, 136,99, 130,39, 128,23, 128,14, 127,92, 127,50, 127,46, 127,26, 126,12, 126,05, 125,99, 125,94, 120,55, 120,51, 118,46, 118,27, 60,49, 47,33, 46,86, 46,76, 35,14, 33,33, 33,28, 32,18, 31,26, 31,21, 25,95, 25,91, 21,44, 17,96, 17,88, минус 5,27, минус 5,39, минус 5,50, минус 5,82.
Анти-диметилсиландиил[2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил][2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]гафния дихлорид

nBuLi в гексане (2,43 M, 49,6 мл, 120,5 ммоль) добавляли одной порцией к раствору 39,22 г (60,25 ммоль) [2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил][2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]диметилсилан (полученный выше) в 400 мл простого эфира, охлажденный до минус 50°C. Данную смесь перемешивали в течение ночи при комнатной температуре. Затем полученный красный раствор охлаждали до минус 78°C и добавляли 19,3 г (60,26 ммоль) HfCl4. Реакционную смесь перемешивали в течение 24 ч при комнатной температуре, получая оранжевую суспензию. Осадок отфильтровывали (G4), затем промывали 30 мл холодного простого эфира. По данным ЯМР-спектроскопии, данный осадок представлял собой чистый дихлоридсин-гафноцена (с LiCl), тогда как фильтрат содержал прибл. Смесь 4/1 анти- и син-гафноцендихлоридов (в пользу анти-), загрязненную некоторыми другими примесями. Осадок растворяли в 150 мл горячего толуола, и образовавшуюся суспензию фильтровали для удаления LiCl через стеклянную фритту (G4). Фильтрат упаривали прибл. до 45 мл. Оранжевый продаваемый материал, осажденный в течение ночи при комнатной температуре, отфильтровывали (G3) и затем сушили в вакууме. Эта процедура дает 8,1 г (15%) чистого син-комплекса. Маточный раствор упаривали почти досуха, и остаток растирали с 20 мл н-гексана, получая 2,6 г (4,8%) дихлорида син-гафноценав виде оранжевого порошка. Простоэфирный маточный раствор упаривали прибл. до 60 мл, выпавший в осадок желтый порошок отфильтровывали (G4), промывали 20 мл холодного (0°C) эфира, а затем сушили в вакууме. Данная процедура дает 10,2 г (19%) чистого дихлорида антигафноцена. Таким образом, общий выход дихлоридов анти- и син-гафноцена, выделенных в этом синтезе, составил 20,9 г (39%).
Анти-диметилсиландиил[2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил][2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]гафния дихлорид.
Результат расчета для C46H52Cl2OSiHf: C 61,50; H 5,83. Обнаружено: C 61,38; Н 6,15.
1H ЯМР(CDCl3): δ 7,51 (s, 1H), 7,43 (s, 1H), 7,34-7,02 (br.m, 4H), 6,94 (s, 2H), 6,61 (s, 1H), 6,46 (s, 1H), 3,42 (s, 3H), 3,11-2,79 (m, 4H), 2,33 (s, 6H), 2,32 (s, 6H), 2,27 (s, 6H), 2,07-1,92 (m, 2H), 1,38 (s, 9H), 1,27 (s, 3H), 1,26 (s, 3H).13C{1H} ЯМР (CDCl3,): δ 159,55, 144,17, 143,58, 142,84, 138,38, 137,82, 137,57, 136,94, 133,09, 132,67, 132,40, 132,11, 131,23, 128,84, 128,76, 127,40, 126,88, 126,53, 124,97, 121,28, 120,84, 119,76, 119,71, 117,90, 82,92, 82,40, 62,62, 35,68, 33,11, 32,07, 30,43, 26,56, 21,46, 21,38, 18,26, 18,12, 2,63, 2,53.
Анти-диметилсиландиил[2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил][2-метил-4-(3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]гафния дихлорид:
Результат расчета для C46H52Cl2OSiHf: C 61,50; H 5,83. Обнаружено: C 61,59; H 6,06.
1H ЯМР (CDCl3): δ 7,53 (s, 1H), 7,41 (s, 1H), 7,29-7,06 (m, 4H), 6,94 (s, 2H), 6,50 (s, 1H), 6,35 (s, 1H). , 3,26 (s, 3H), 2,95-2,77 (m, 4H), 2,49 (s, 3H), 2,46 (s, 3H), 2,33 (2s, сумма 12H), 1,99-1,86 (m, 1H), 1,86- 1,73 (m, 1H), 1,40 (s, 3H), 1,37 (s, 9H), 1,18 (s, 3H).13C{1H} ЯМР (CDCl3,): δ 158,61, 143,03, 142,46, 142,16, 138,42, 137,73, 137,52, 136,98, 135,33, 134,60, 133,69, 132,53, 131,19, 128,79, 128,71, 127,34, 126,85, 126,00, 125,76, 121,95, 121,45, 119,12, 118,91, 118,55, 84,66, 84,26, 62,31, 35,48, 33,25, 31,94, 30,40, 26,60, 21,44, 18,44, 18,31, 2,93, 2,61.
Катализаторы диоксид кремния-МАО были получены на диоксиде кремния SUNSPERA DM-L-303 толщиной 30 мкм, произведенном AGC Si-Tech Co, предварительно прокаленном при 600°C в течение 2 часов в электрической муфельной печи в потоке сухого воздуха.
Получение металлоценового катализатора на носителе из диоксида кремния описано в WO 2018/122134.
Катализатор C был получен аналогично с использованием металлоценового рац-анти-диметилсиландиил[2-метил-4-(3',5'-диметилфенил)-5-метокси-6-трет-бутилинден-1-ил][2-метил-4- (3',5'-диметилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]гафния дихлорид, полученный выше.
Гомополимеры пропилена от PP-1 до PP-4 были получены в присутствии вышеуказанных катализаторов в лабораторном процессе. Сначала катализаторы, как определено выше (катализатор A, B и C), предварительно полимеризовали в соответствии со следующей процедурой. Автономную предварительную полимеризацию проводили в реакторе под давлением 125 мл, оборудованном линиями подачи газа и верхней мешалкой. Сухой и дегазированный перфтор-1,3-диметилциклогексан (PFC) и желаемое количество катализатора, подлежащего предварительной полимеризации, загружали в реактор внутри бокса с перчатками, и реактор герметично закрывали. Затем реактор вынимали из бокса с перчатками и помещали в водяную баню, поддерживающую температуру 25°C. Затем были подключены верхняя мешалка и линии подачи. Эксперимент начинали с открытия подачи пропилена в реактор и установки скорости мешалки на 450 об/мин. Подачу пропилена оставляли открытой, а расход мономера компенсировали, поддерживая постоянное общее давление в реакторе (около 5 бар изб.). Эксперимент продолжали в течение времени полимеризации, достаточного для обеспечения желаемой степени полимеризации (DP) примерно 3,8 г полимера на грамм катализатора. Затем реактор возвращали внутрь бокса с перчатками перед открытием, и содержимое выливали в стеклянный сосуд. PFC выпаривали до получения постоянного веса предварительно полимеризованного катализатора. Степень полимеризации (DP) определяли гравиметрически и/или анализом золы. Соответствующие гомополимеры PP-1, PP-2, PP-3 и PP-4 затем полимеризовали в настольном реакторе объемом 20 л при 75°C в основной фазе суспензии, используя водород в подходящей концентрации для достижения желаемой скорости течения расплава MFR2 (230 °С). Полимеризацию останавливали через 60 минут путем вентиляции реактора и продувки азотом перед сбором полимера.
Все полимерные порошки были стабилизированы расплавом 0,05 мас. % Стеарата кальция (номер CAS 1592-23-0) и 0,15 мас. % Irganox B215 (распространяется BASF AG, Германия), смесь пентаэритритил-тетракис в соотношении 1:2 (3-(3',5'-ди-трет-Бутил-4-гидроксифенил) пропионат (номер CAS 6683-19-8) и трис-(2,4-ди-трет-бутилфенил) фосфит (номер CAS 31570-04-4).
В качестве β-нуклеирующего агента использовали следующие продукты:
Смесь 5,12-дигидро-хино[2,3-b]акридин-7,14-диона (номер CAS 1047-16-1), хино[2,3-b]акридин-6,7,13,14(5H,12H)-тетрона (номер CAS 1503-48-6) и 5,6,12,13-тетрагидрохино[2,3-b] акридин-7,14-диона (номер CAS 5862-38-4), который коммерчески доступен как Cinquasia Gold YT-923-D (распространяется BASF AG, Германия), разбавленный в порошке гомополимера PP (HC001A-B1) в концентрации 0,25 мас. %, был использован β-нуклеирующий агент «N17000-CM1» .
Хинакридонехинон (номер CAS 1503-48-6; распространяется BASF AG, Германия) использовался в качестве β-нуклеирующего агента «CGNA-7588».
Гетероядерный диметаллический комплекс лантана и кальция (продаваемый Guangdong Winner Functional Materials Co., Китай) использовали в качестве β-нуклеирующего агента «WBG».
Обозначения «SSC» и «ZN» означают, что использовался односайтовый катализатор или катализатор Циглера-Натта соответственно.
AO501 представляет собой антиоксидант Irganox B215, описанный выше.
AS110 представляет собой стеарат кальция, описанный выше.
N17000-CM1, CGNA-7588 и WBG являются β-нуклеирующими агентами, как описано выше (из них было обнаружено, что WBG не дает желаемой степени содержания β-фазы и соответствующих механических характеристик в случае SSC-PP)
PP-1 представляет собой экспериментальный гомополимер пропилена, имеющий скорость течения расплава MFR2 (230°C) 1,05 г/10 мин и Tm 156°C, полученный в присутствии односайтового катализатора A в ходе одностадийной лабораторной полимеризации, имеющий изотактичность пентад (mmmm) 99,4% и относительное количество 2,1-эритрорегиодефектов 0,7 мол. %.
PP-2 представляет собой экспериментальный гомополимер пропилена, имеющий скорость течения расплава MFR2 (230°C) 0,34 г/10 мин и Tm 152°C, полученный в присутствии односайтового катализатора B в одностадийной лабораторной полимеризации, имеющий изотактичность пентад (mmmm) 99,5% и относительное количество 2,1-эритрорегиодефектов 1,0 мол. %.
PP-1 представляет собой экспериментальный гомополимер пропилена, имеющий скорость течения расплава MFR2 (230°C) 0,60 г/10 мин и Tm 152°C, полученный в присутствии односайтового катализатора B в ходе одностадийной лабораторной полимеризации, имеющий изотактичность пентад (mmmm) 99,5% и относительное количество 2,1-эритрорегиодефектов 1,0 мол. %.
PP-4 представляет собой экспериментальный гомополимер пропилена, имеющий скорость течения расплава MFR2 (230°C) 0,24 г/10 мин и Tm 160°C, полученный в присутствии односайтового катализатора C в одностадийной лабораторной полимеризации, имеющий изотактичность пентад (mmmm) 99,7% и относительное количество 2,1-эритрорегиодефектов 0,9 мол. %.
PP-5 представляет собой коммерческий унимодальный гомополимер пропилена B-Powder-10 от Borealis AG, имеющий скорость течения расплава MFR2 (230°C) около 0,3 г/10 мин, Tm 163°C, Mw 1007 кг/моль и Mw/Mn 4,9, полученный в присутствии обычного катализатора Циглера-Натта4-го поколения (катализатор D), имеющий изотактичность пентад (mmmm) 95,2% и не содержащий 2,1-эритрорегиодефектов.
PP-6 представляет собой коммерческий унимодальный гомополимер пропилена HC001A-B1 от Borealis AG, имеющий скорость течения расплава MFR2 (230°C) около 2,5 г/10 мин, Tm 163°C, Mw 565 кг/моль и Mw/Mn 6,2, полученный в присутствии катализатора Циглера-Натта 3-го поколения (катализатор E), имеющий изотактичность пентад (mmmm) 93,1% и не содержащий 2,1-эритрорегиодефектов.
Свойства полученных композиций показаны в Таблице 2 ниже.
Hm2/(Hm1+Hm2) означает количество β-фазы (k(β)), определенное с помощью DSC, как описано выше.
Приведенные выше результаты показывают, что композиции полимера пропилена по настоящему изобретению сочетают в себе превосходные механические свойства, такие как высокая ударная вязкость, исключительное улучшение ударной вязкости по сравнению с ударной вязкостью основного ненуклеированного полимера, выраженное как Дельта NIS (23°C), с по-прежнему достаточно высокой жесткостью. Улучшение ударной вязкости (23°C) также показано на Фиг. 1. Сравнение содержания β-фазы в композициях показано на Фиг. 2.
Это неожиданное сочетание полезных свойств не могло быть достигнуто при полимеризации гомополимеров пропилена с помощью обычного катализатора Циглера-Натта с или без β-нуклеирующего агента (CE5-CE8). Даже при использовании односайтового (металлоценового) катализатора, но в отсутствие эффективного β-нуклеирующего агента, преимущества изобретения не могут быть достигнуты (от CE1 до CE4). CE4 дополнительно показывает, что высокое содержание β-фазы не обеспечивает высокой ударной вязкости.
Реферат
Настоящее изобретение относится к композиции гомополимера пропилена для получения формованных продуктов, а также к способу ее получения и к формованному продукту. Композиция гомополимера пропилена содержит (а) не менее 90 мас. % гомополимера пропилена (А) от общей массы композиции и (b) нуклеирующий агент (B), селективный для бета-модификации изотактического полипропилена, в количестве от 0,001 до 1,0 мас. % от массы общей композиции гомополимера пропилена. Гомополимер пропилена (А) имеет (i) изотактичность пентад (mmmm) 98,0 мол. % или более, определенную количественной спектроскопией ЯМР (ядерный магнитный резонанс)13С, (ii) относительное количество 2,1-эритрорегиодефектов не более 2,0 мол. %, определенное количественной спектроскопией ЯМР13С. Указанная композиция гомополимера пропилена имеет скорость течения расплава MFR2 (230°C, 2,16 кг) согласно ISO 1133 от 0,1 до 10 г/10 мин и содержание бета-фазы не менее 80%, измеренное методом широкоугольного рассеяния рентгеновских лучей (WAXS). Формованный продукт из гомополимера пропилена может быть получен прессованием или экструзией указанной композиции гомополимера пропилена. Полученная композиция гомополимера пропилена обладает улучшенными механическими свойствами, в частности высокими ударными свойствами, в сочетании с высокой стереорегулярностью и ограниченным количеством региодефектов. 3 н. и 12 з.п. ф-лы, 2 ил., 2 табл., 3 пр.
Формула

Документы, цитированные в отчёте о поиске
Полипропилен с чрезвычайно широким распределением молекулярной массы
Гетерофазный сополимер пропилена с низким количеством экстрагируемых веществ
Комментарии