Стабильное противоопухолевое лекарственное средство, способ его получения и применения - RU2676279C1
Код документа: RU2676279C1
Описание
Область техники
Изобретение относится к области медицины и фармацевтики, а именно к лекарственному средству для лечения метастатической формы рака молочной железы. Предложен также способ получения стабильного лекарственного средства для лечения рака молочной железы в виде инъекционной формы, включающей труднорастворимый комплекс (5Z,5'Z)-2,2'-(этан-1,2-диилдисульфанилдиил)бис(5-(2-пиридилметилен)-3-аллил-3,5-дигидро-4Н-имидазол-4-она) с хлоридом меди (II). Способ позволяет получить стабильное лекарственное средство, оказывающее эффективное цитостатическое действие на опухолевые клетки при этом обладающее умеренными побочными эффектами.
Уровень техники
Рак молочной железы является второй по распространенности формой рака среди женщин в США, и второй основной причиной смертей от рака среди женщин. Однако при раннем обнаружении шансы на успешное лечение рака молочной железы намного выше.
Рак молочной железы хорошо лечится оперативным путем, радиотерапией, химиотерапией и гормональной терапией. На прогноз и выбор терапии влияют возраст пациента, стадия заболевания, патологические характеристики первичной опухоли, включая некроз опухоли, уровни рецепторов к эстрогену (РЭ) или рецепторов к прогестерону (ПР) в опухолевой ткани, сверхэкспрессия статуса HER2 и показатели пролиферативной способности, наряду с менопаузальным статусом и общим состоянием здоровья. Пациенты с излишним весом могут иметь худший прогноз (Bastarrachea et al., Annals of Internal Medicine, 120: 18 [1994]). Прогноз также может варьироваться в зависимости от расы, при этом афроамериканцы и, в меньшей степени, латиноамериканцы, имеют худший прогноз по сравнению с белой расой (Elledge et al., Journal of the National Cancer Institute 86: 705 [1994]; Edwards et al., Journal of Clinical Oncology 16: 2693 [1998]).
Способы лечения река молочной железы определяются как местные или системные. Операция и облучение считаются местными терапиями, поскольку они напрямую воздействуют на опухоль, молочную железу, лимфатические узлы или другие определенные участки. Лекарственная терапия является системной терапией, потому что ее эффекты распространяются широко. Лекарственные терапии включают лечение классическими химиотерапевтическими препаратами, лечение препаратами, блокирующими гормоны (например, ингибиторы ароматазы, селективные модуляторы рецепторов эстрогена и ингибиторы рецепторов эстрогена) и лечение моноклональными антителами (например, против HER-2). Они могут использоваться по раздельности или, чаще всего, в различных комбинациях.
Многие соединения, особенно сложные комплексы, в частности, противораковые средства, представляют собой труднорастворимые в воде гидрофобные соединения и постоянно увеличивающееся число получаемых фармацевтических лекарственных средств, которые являются слаборастворимыми или нерастворимыми в водных растворах, порождает определенные проблемы при доставке их в инъецируемой форме. Хорошо подобранная композиция должна как минимум обеспечивать терапевтически эффективное количество слаборастворимого лекарственного средства в участке мишени в абсорбируемой форме. Кроме того, известные композиции в форме растворов характеризиются нестабильностью с образованием осадка и/или осаждением активно действующего вещества, до непосредственного введения пациенту препарата. Для обеспечения стабильности и увеличения растворимости активного вещества разрабатываются различные комбинации вспомогательных и действующих веществ.
Из уровня техники известны различные решения, направленные на получение лекарственных средств для лечения рака молочной железы, характеризующихся использованием ингибиторов теломеразы с различными вспомогательными веществами.
В документе WO 2015040622 раскрыты составы с контролируемым высвобождением, включающие ингибиторы теломеразы, а именно металлопорфирины для лечения рака. Описаны композиции металлопорфиринов и сополимеров поли(молочной-со-гликолевой)-кислоты, в различных имплантируемых формах для лечения рака.
В заявке US 2017051287 предложена система доставки, подходящая для контролируемого высвобождения агентов, ингибирующих теломеразу, в течение длительного периода времени в опухолевой ткани. Предложен имплант для введения в опухоль путем имплантации или инъекции, представляющий собой твердое устройство в форме микро- или нанокапсул или микро- или наночастиц, содержащее множество частиц. Как правило, формы и размеры устройства или его частиц выбираются так, чтобы обеспечить прямую доставку в опухоль.
В патенте RU 2541100 раскрыта композиция по меньшей мере одного инкапсулированного химиотерапевтического агента и, по меньшей мере, одного амфифильного блок-сополимера. За счет использования в композиции амфифильного блок-сополимера, которым является поли(этиленоксид)-поли(пропиленоксид)-поли(этиленоксид) триблок-сополимер происходит усиление действия инкапсулированного химиотерапевтического препарата, в частности ингибиторов теломеразы, путем стимулирования выхода активного химиотерапевтического агента из липосом. Предложенная композиция может быть использована для введения путем инъекции, в частности внутривенной инъекции, пациентам с онкологическим заболеванием. При этом, по результатам проведенных доклинических исследований, чувствительность опухолевых клеток к указанному в данной заявке химиотерапевтическом агенту - липосомальному доксорубицину, оказалась ниже по сравнении с комплексом (5Z,5'Z)-2,2'-(этан-1,2- диилдисульфанилдиил)бис(5-(2-пиридилметилен)-3-аллил-3,5-дигидро-4Н-имидазол-4-она) с хлоридом меди (II), описанному в настоящей заявке.
Активная фармацевтическая субстанция - координационное соединение меди с (4Z,4'Z)-2,2'-(этан-1,2-диилбис(сульфадиил))бис(1-аллил-4-(пиридин-2-илметилен)-1Н-имидазол-5(4Н)-она) растворима в метаноле, хлороформе, диметилсульфоксиде. Для внутривенного введения вещество в составе готовой лекарственной формы должно быть в растворенном виде для адекватного распределения в организме, достижения оптимальных фармакокинетических показателей, а также снижения дозы лекарственного средства, необходимой для проявления терапевтического эффекта, и, как следствие, уменьшения побочных эффектов. Ввиду недопустимости использования в терапии метанола и хлороформа, а также больших концентраций ДМСО, стоит задача разработать такой раствор для внутривенного введения, в котором активное вещество находилось бы в растворенном состоянии, а состав и количество вспомогательных веществ было бы допустимо для внутривенного введения с достижением терапевтического эффекта, при этом при добавлении такого раствора в плазму крови не должно происходить выпадение осадка или сворачивания белков плазмы.
В заявке на изобретение РФ №2013147475 разработчиками был предложен препарат, обладающий противоопухолевым действием, включающий раствор координационного соединения меди с (4Z,4'Z)-2,2'-(этан-1,2-диилбис(сульфадиил))бис(1-аллил-4-(пиридин-2-илметилен)-1Н-имидазол-5(4Н)-она) и диметилсульфоксида в дистиллированной воде, при этом объемная концентрация диметилсульфоксида составляет 10%. Данные координационные соединения меди являются малорастворимыми в водных растворах диметилсульфоксида без добавления блок-сополимеров, поэтому для эффективного использования данной композиции потребуется введение большого объема препарата и, как следствие, большого количества диметилсульфоксида, который в больших концентрациях может вызывать побочные эффекты, например, внутрисосудистый гемолиз после внутривенного вливания.
Наиболее близким к заявляемому решению по совокупности существенных признаков является композиция, ингибирующая теломеразу, раскрытая в материалах заявки РФ №20141553863, включающая по крайней мере, один блок-сополимер полиоксиэтилена и полиоксипропилена, а также, по крайней мере, одно координационное соединение производного имидазол-4-она, в частности (4Z,4'Z)-2,2'-(этан-1,2-диилбис(сульфадиил))бис(1-аллил-4-(пиридин-2-илметилен)-1Н-имидазол-5(4Н)-она).
Недостатком указанной композиции является то, что в композиции присутствует только плюроник F-127, который является мицеллообразующим поверхностно-активным веществом, и отсутствуют неионногенные поверхностно-активные вещества для повышения растворимости и стабильности. Таким образом, содержание в композиции координационного соединения меди с (4Z,4'Z)-2,2'-(этан-1,2-диилбис(сульфадиил))бис(1-аллил-4-(пиридин-2-илметилен)-1Н-имидазол-5(4Н)-оном), в концентрации от 3 до 10 мг/л недостаточно для оказания стабильного цитотоксического эффекта, что при применении данного соединения in vivo приводит к необходимости введения больших объемов препарата для достижения действующей концентрации.
Раскрытие изобретения
Задачей заявляемого изобретения являлось получение стабильного лекарственного средства в инъекционной форме для лечения рака молочной железы.
Техническим результатом является получение раствора комплекса (5Z,5'Z)-2,2'-(этан-1,2-диилдисульфанилдиил)бис(5-(2-пиридилметилен)-3-аллил-3,5-дигидро-4Н-имидазол-4-она) с хлоридом меди (II), содержащего в качестве растворителя смесь твин-20, полоксамера 407, диметилсульфоксида, а также гемодеза-Н в качестве разбавителя, обеспечивающие растворимость активного компонента в концентрации до 4 мг/мл, совместимость активного вещества со всеми входящими в состав вспомогательными веществами и стабильность полученного средства в течение 60 минут с момента его приготовления до введения пациенту, что является достаточным для проведения инъекции. Заявляемый раствор является однородным, прозрачным, обладает темно-коричневым цветом.
Поставленная задача решается заявляемым противоопухолевым лекарственным средством для лечения рака молочной железы, представляющим собой раствор для инъекционного введения, включающий в качестве активного вещества координационное соединение меди с (4Z,4'Z)-2,2'-(этан-1,2-диилбис(сульфадиил))бис(1-аллил-4-(пиридин-2-илметилен)-1Н-имидазол-5(4Н)-оном), растворитель, включающий диметилсульфоксид, полисорбат 20 и полоксамер, и разбавитель Гемодез Н, при следующем соотношении компонентов (мас. %):
активное вещество - 1,0-4,0
растворитель:
диметилсульфоксид - 9,4-10,0
полисорбат 20 - 0,1-0,5
полоксамер 407 - 0,08-0,11
разбавитель:
Гемодез Н - остальное.
Поставленная задача также решается способом получения указанного лекарственного средства заключающегося в том, что активный компонент - комплекс (5Z,5'Z)-2,2'-(этан-1,2-диилдисульфанилдиил)бис(5-(2-пиридилметилен)-3-аллил-3,5-дигидро-4Н-имидазол-4-она) с хлоридом меди (II) в виде порошка добавляют в предварительно подготовленный растворитель, представляющий смесь компонентов, включающую 94,0-98,2 мас. % ДМСО, 1,0-5,0 мас. % полисорбата 20, 0,8-1,1 мас. % полоксамера 407 из расчета получения раствора активного компонента с концентрацией 10-40 мг/мл, затем полученный раствор обрабатывают ультразвуком до полного растворения активного компоненета, полученный раствор разводят Гемодезом Н до необходимой концентрации активного компонента в лекарственном средстве. При этом ультразвуковую обработку проводят при мощности 60 Вт±10% в течение 3-5 минут.
Также поставленная задача решается способом лечения рака молочной железы, включающим инъекционное ведение указанного лекарственного средства, приготовленного заявялемым способом до истечения 60 минут после приготовления средства в терапевтически эффективном количестве. При этом лекарственное средство вводят внутривенно в количестве из расчета 0,5 мг/кг с периодичностью 1 раз в 3 недели до достижения лечебного эффекта, но не более чем 6 инъекций.
Осуществление изобретения
Вспомогательные вещества в составе лекарственных средств для инъекционного (например, внутривенного) введения выполняют несколько функций: увеличивают биодоступность активного вещества, препятствуют микробной контаминации готового продукта, обеспечивают физическую и химическую стабильность на протяжении всего срока годности.
При разработке противоопухолевых средств способность вспомогательных веществ оказывать на активную молекулу стабилизирующее действие является очень важной характеристикой, которая зависит от природы активной фармацевтической субстанции и вспомогательных веществ. В настоящее время мицеллообразование является широко распространенной модификацией для лекарственных средств. При этом в процессе гидрофилизации лекарственного средства следует избегать образования слишком стабильных мицелл, поскольку это приведет к снижению биодоступности лекарственного средства, увеличенному времени высвобождения, и, как следствие, снижению его эффективности. Мицеллообразование увеличивает стабильность лекарственного средства в ГЛФ, предотвращает его деструкцию и выпадение, обеспечивают физическую и химическую стабильность на протяжении всего срока годности.
Плюроники, ввиду их хорошей растворимости, поверхностно-активных свойств, и низкой токсичности, широко применяются для формуляции различных терапевтических агентов из-за своей низкой токсичности, биосовместимости с клетками и жидкостями организма, а также слабых иммуногенных свойств. Pluronic F-127 (плюроник F127, полоксамер 407) является широко используемым полимером в системах доставки лекарственных средств. Однако, данный сополимер не является неионногенным поверхностно-активным веществом, способным повышать растворимость. Помимо этого, недостатки плюроника F127 связаны с низкой способностью лекарственной нагрузки и физической стабильностью мицеллярной формулировки. Для повышения стабильности мицеллы обычно используют двойную смешанную систему, состоящую из плюроника F127 и других сополимеров.
Первоначальное образование мицелл плюроника F127 с гидрофобным активным веществом - комплексом (5Z,5'Z)-2,2'-(этан-1,2-диилдисульфанилдиил)бис(5-(2-пиридилметилен)-3-аллил-3,5-дигидро-4Н-имидазол-4-она) с хлоридом меди (II), формирование первоначальной гидрофильной оболочки мицеллы обеспечивается за счет входящих в состав плюроника гидроксильных групп. Затем, происходит образование «клубков» поливинилпиррилидона с встраиванием мицелл за счет образования водородных связей между гидроксильными группами плюроника F127 и карбонильными фрагментами поливинилпирролидона (в составе Гемодез Н), а, также, встраивание в клубки поливинилпирролидона фрагментов солюбилизатора полисорбата 20 (Tween20). Полученная формуляция повышает растворимость гидрофобного активного действующего вещества, однако, не препятствует высвобождению лекарственного средства в организме.
При разработке состава учитывалась совместимость активного и вспомогательных веществ, чтобы избежать нежелательных физико-химических явлений, результатом которых могут стать образование токсических веществ и потеря биологической активности в процессе хранения и использования лекарственного средства.
Совместимость субстанции со всеми входящими в состав вспомогательными веществами была доказана в процессе изучения стабильности субстанции, а также готовой лекарственной формы.
Таким образом, было разработано лекарственное средство в виде раствора для инъекционного введения, которое включает (мас. %): активное вещество - 1-4 и растворитель, представляющий собой смесь диметилсульфоксида - 9,4-10,0, полисорбата 20 - 0,1-0,5 и полоксамера 407 - 0,08-0,11 и растворитель - гемодез Н - остальное. При этом способ получения средства заключается в следующем: комплекс (5Z,5'Z)-2,2'-(этан-1,2-диилдисульфанилдиил)бис(5-(2-пиридилметилен)-3-аллил-3,5-дигидро-4Н-имидазол-4-она) с хлоридом меди (II) в виде порошка добавляют в растворитель, содержащий 94,0-98,2 мас. % ДМСО, 1,0-5,0 мас. % полисорбата 20, 0,8-1,1 мас. % полоксамера 407, до концентрации комплекса (активного компонента) 10,0-40,0 мг/мл. Полученный раствор обрабатывают в ультразвуковой ванне при мощности 60 Вт±10% до полного растворения активного вещества, приблизительно 3-5 минут. Получают прозрачный раствор темно-коричневого цвета, который затем разводят до необходимой концентрации Гемодезом Н.
Способ получения лекарственного средства заключаются в предварительной подготовке состава растворителя, заключающегося в смешениии ДМСО, полисорбата 20 и полоксамера 407 в необходимых концентрациях, в любой последовательности смешения компонентов. Полученный растворитель хранят при температуре от 2°C до 25°C в защищенном от света месте в течение 24 месяцев. Затем активное вещество в виде порошка добавляют в растворитель, аккуратно перемешивают (без встряхивания) и обрабатывают ультразвуком до полного растворения активного вещества. Полученный концентрированный раствор можно хранить в течение 24 часов. Для введения пациенту концентрированный раствор разбавляют до необходимой (терапевтически эффективной) концентрации Гемодезом Н и не позднее, чем через 60 минут после приготовления лекарственного средства вводят пациенту. Предпочтительно вводить средство внутривенно капельно в течение 30 мин.-1 часа 1 раз в 3 недели и не более 6 инъекций.
Выбранные вспомогательные ингредиенты являются фармакопейными, то есть, в ведущих зарубежных фармакопеях (USP, Ph. Eur.) и фармакопее РФ представлены монографии, устанавливающие требования к качеству данных веществ.
Проведенные исследования свойств (стабильности) создаваемых составов лекарственного средства показал, что именно состав, включающий в качестве растворителя - ДМСО, полисорбат 20, полоксамер 407, а в качестве разбавителя гемодез Н позволяет добиться стабильности приготовленного раствора в течение 60 минут. Ниже приведены примеры составов лекарственного средства и данные о стабильности полученных растворов (табл. 1).
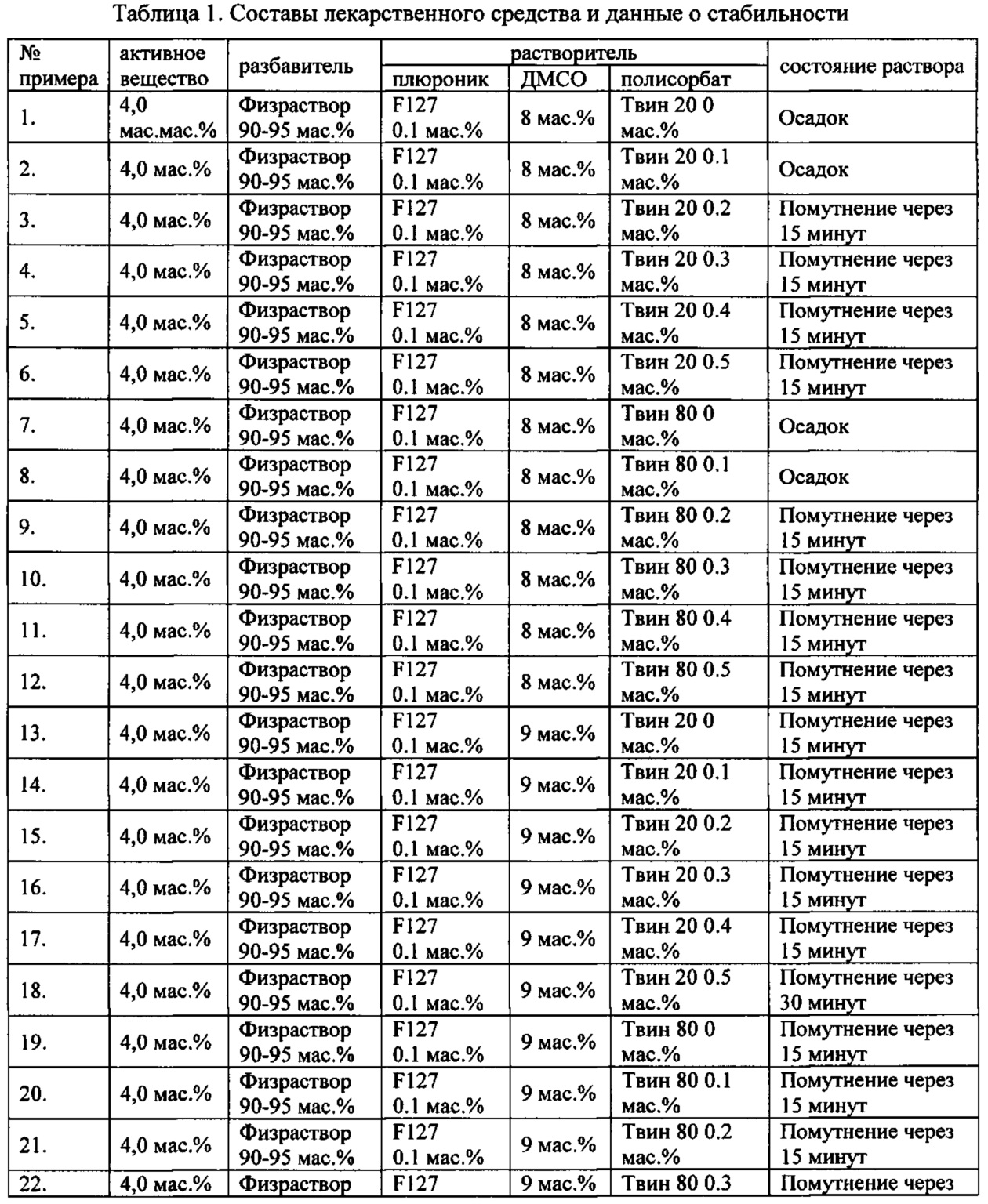
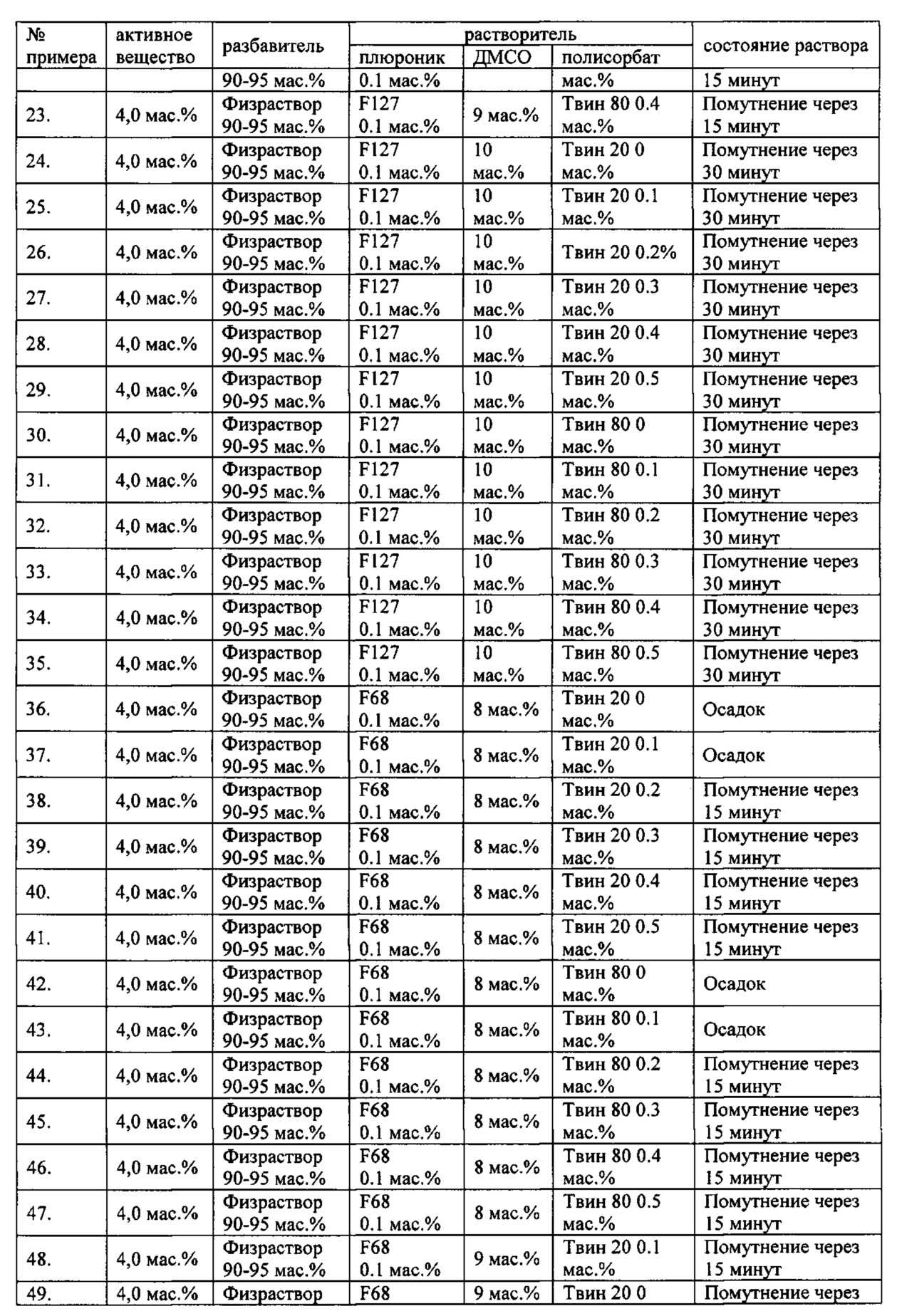
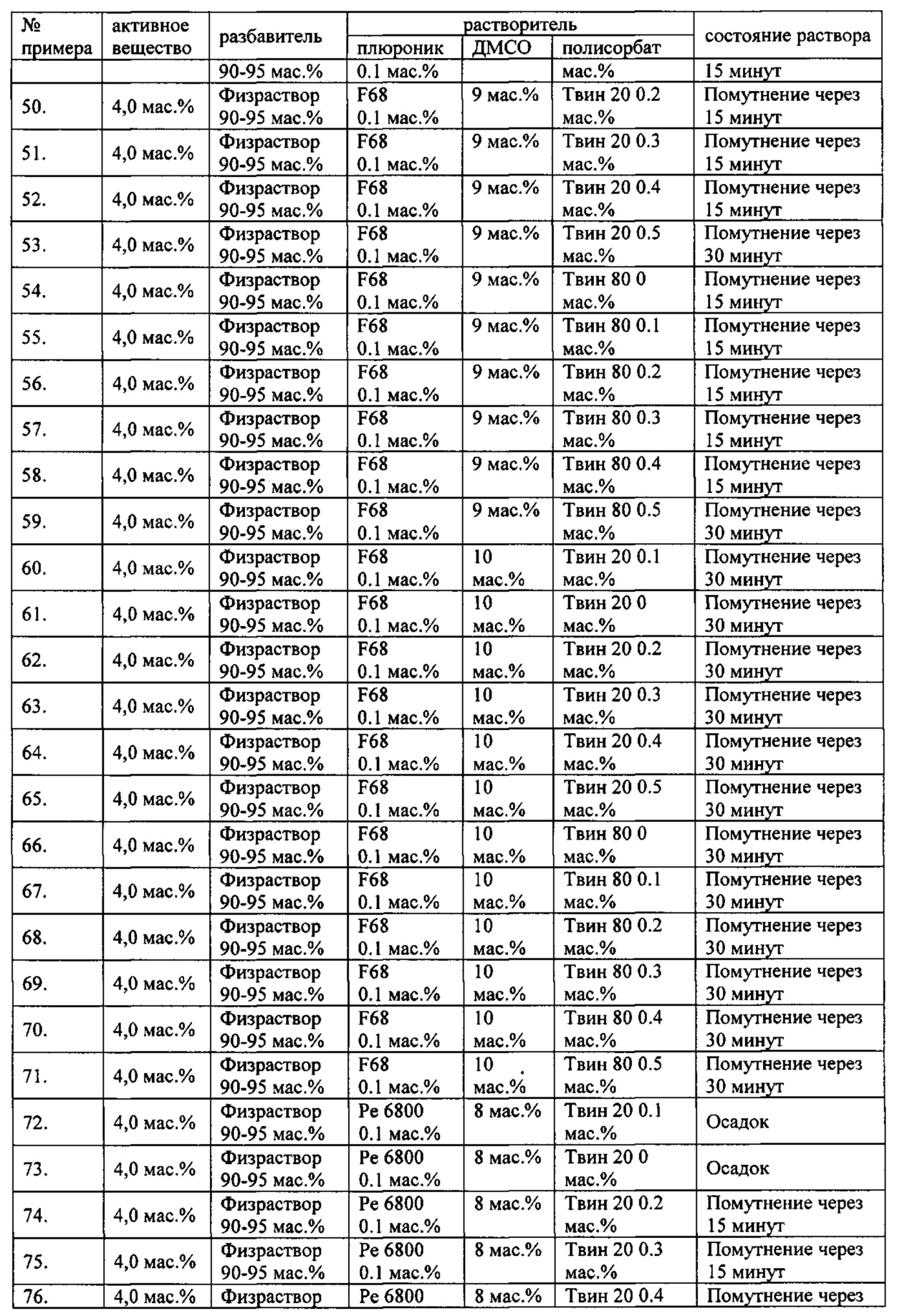
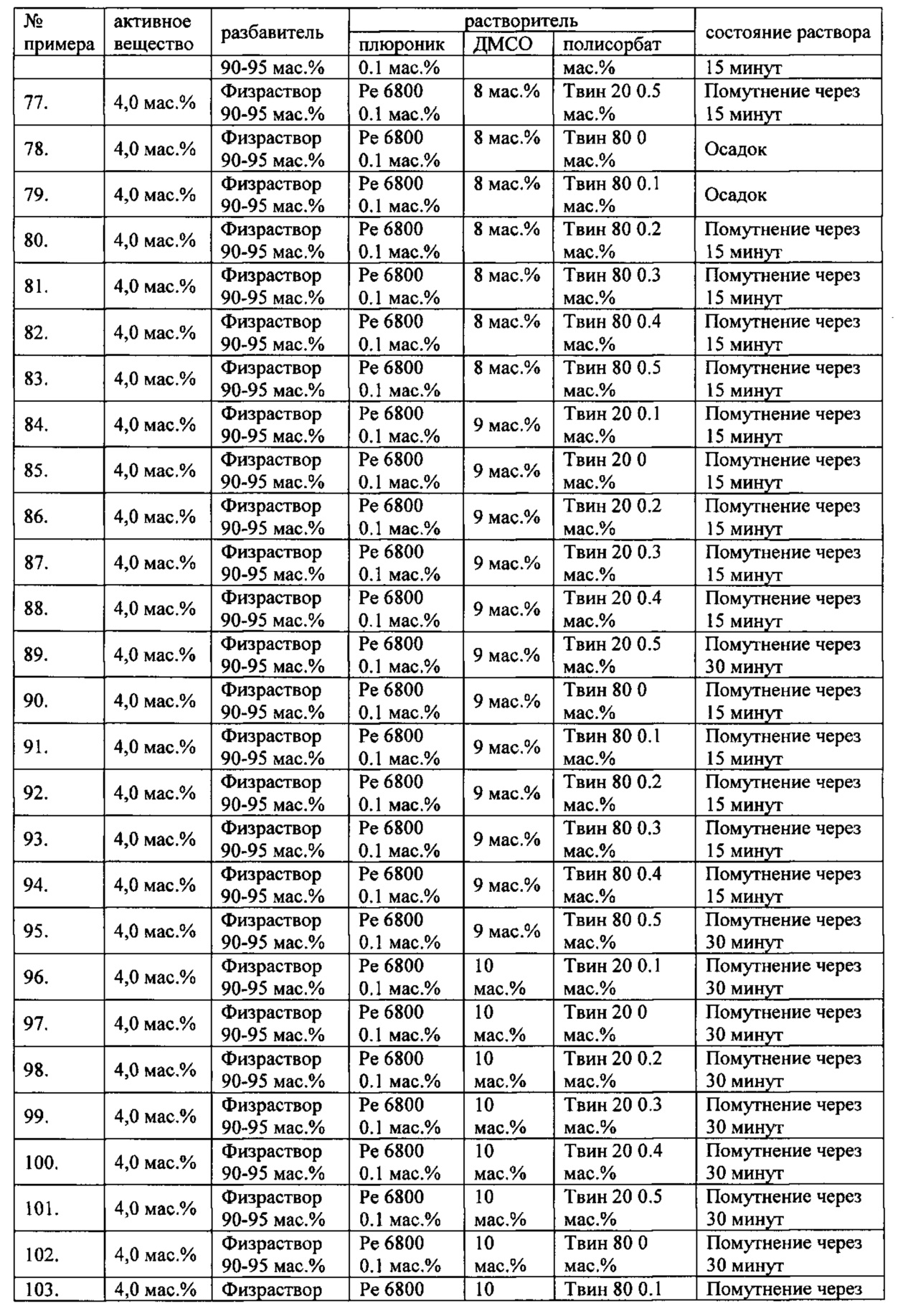
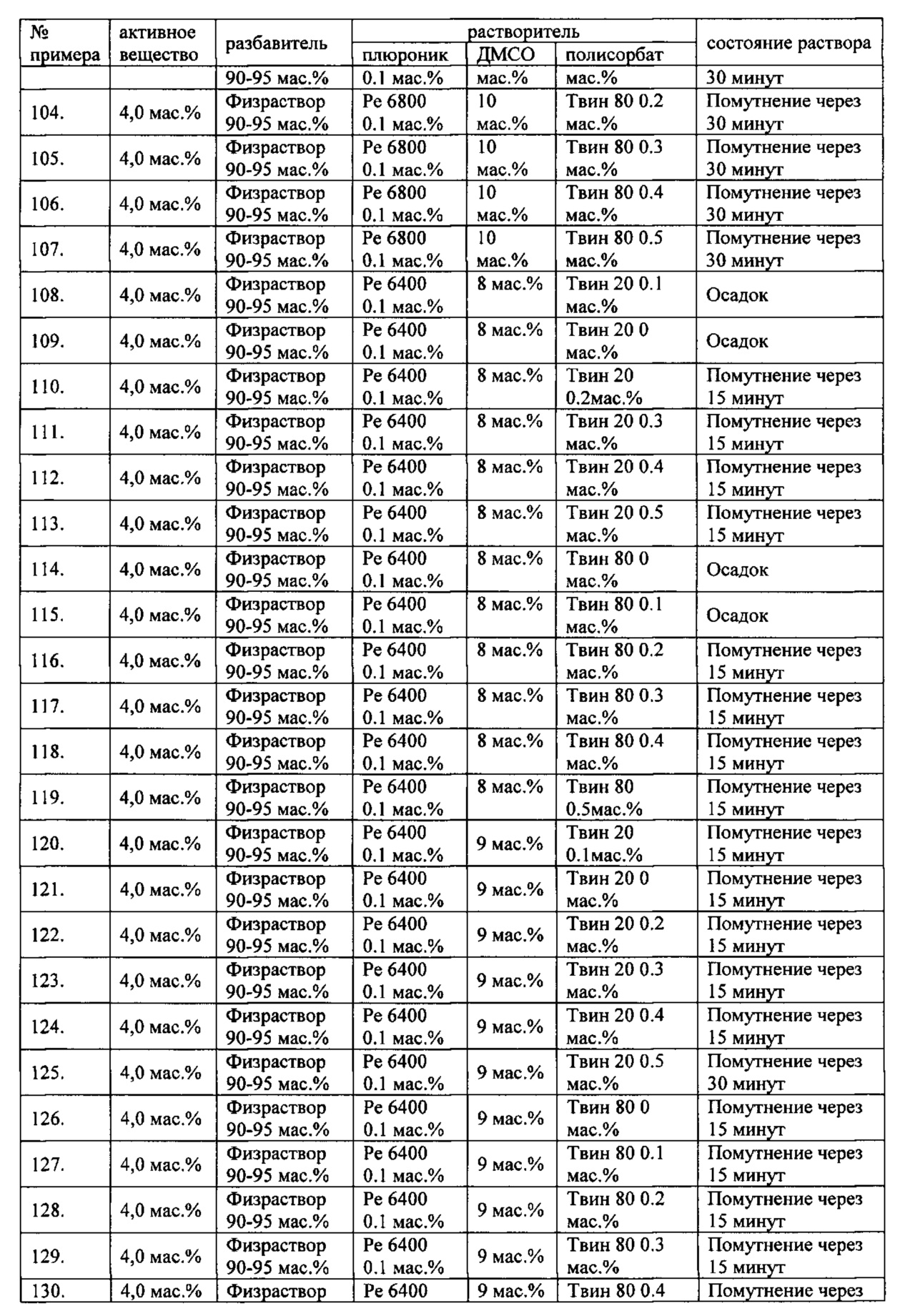
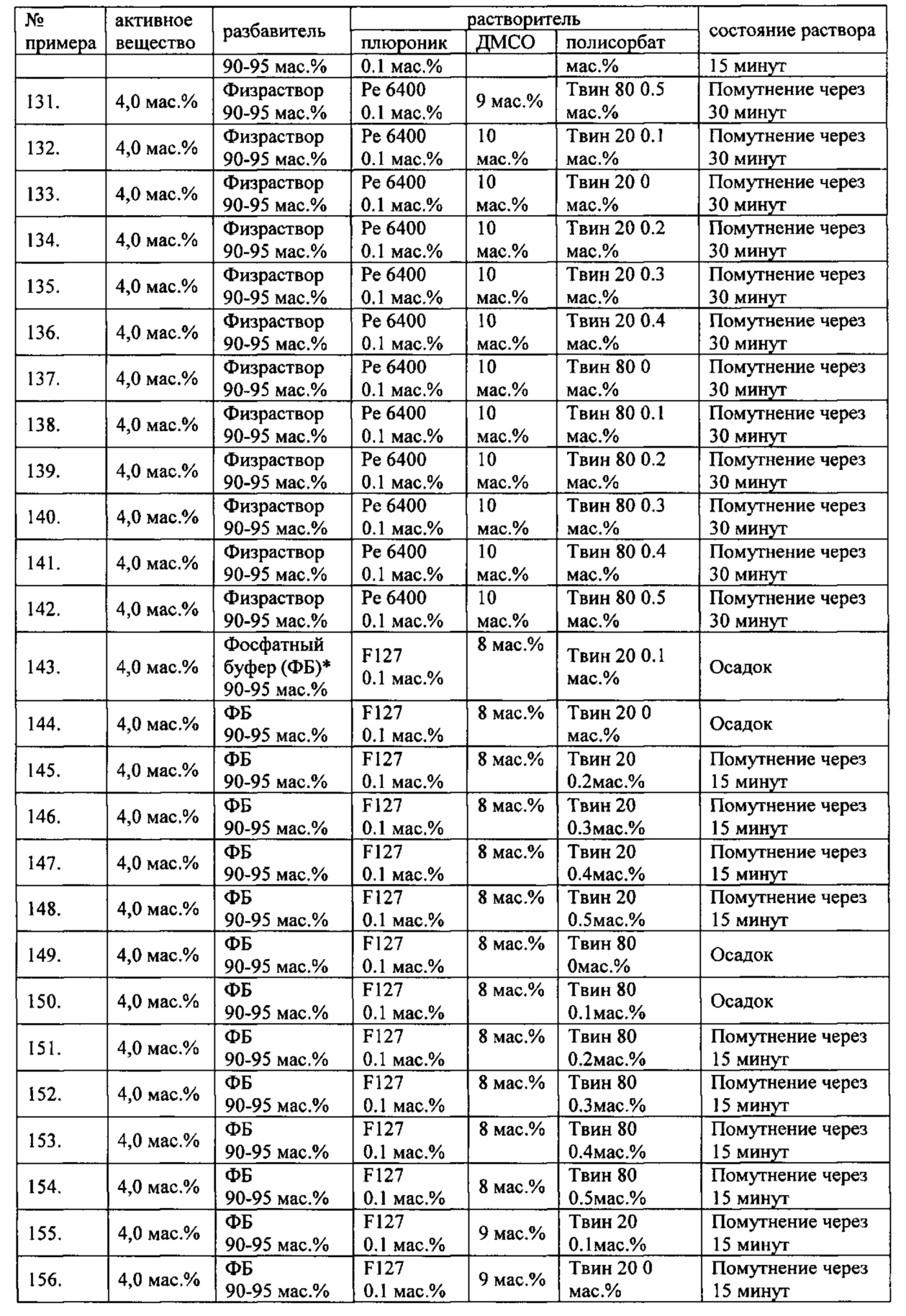
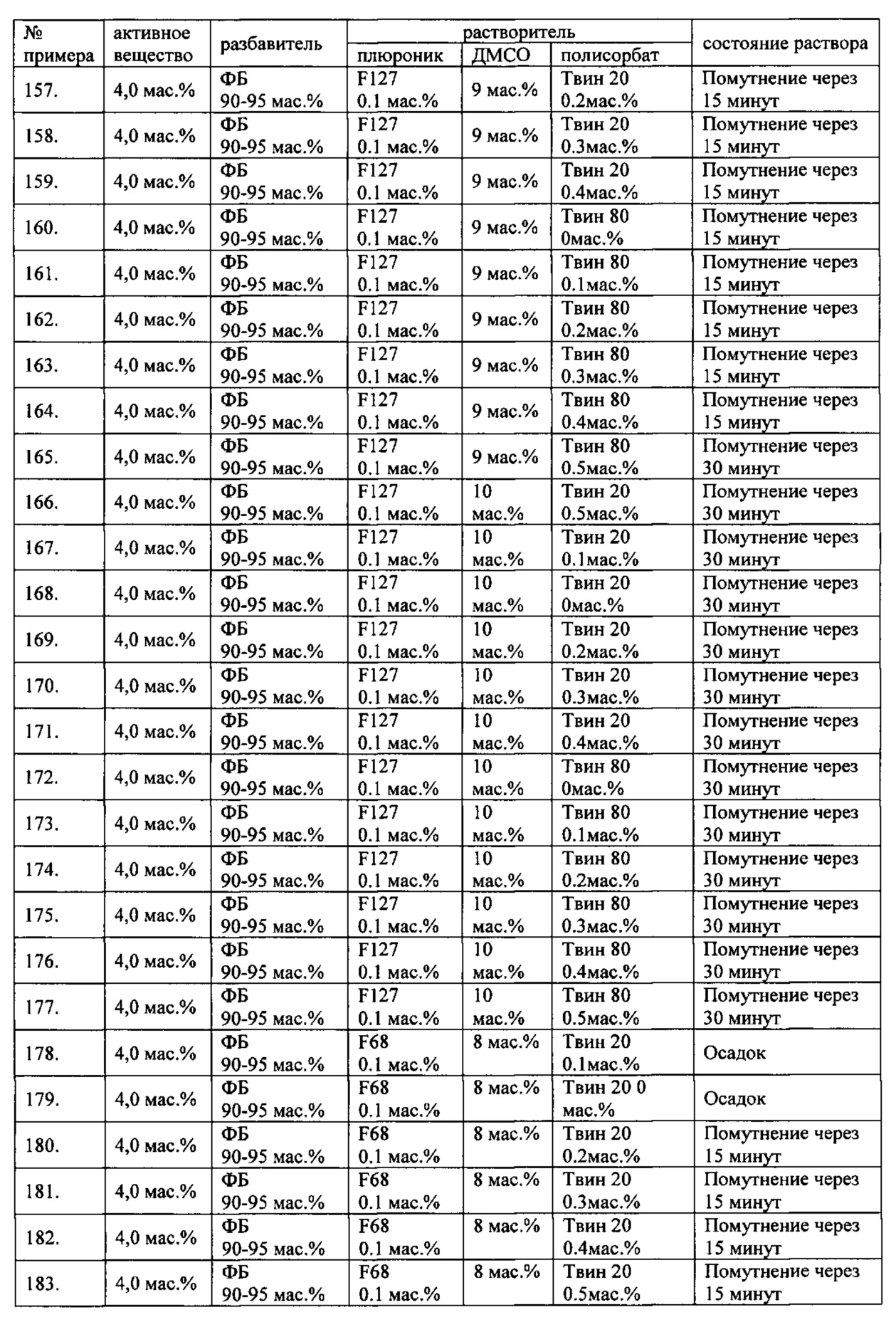
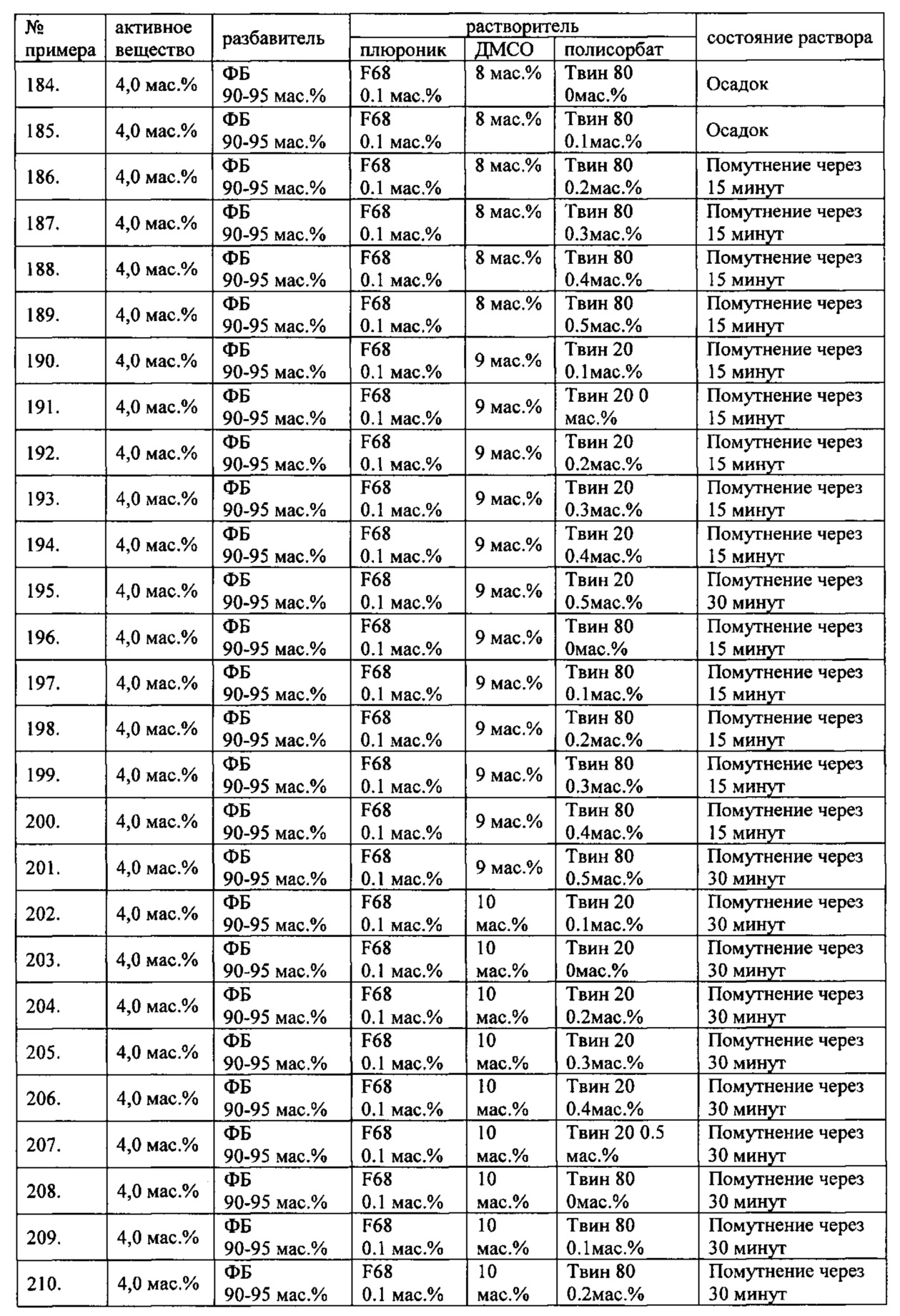
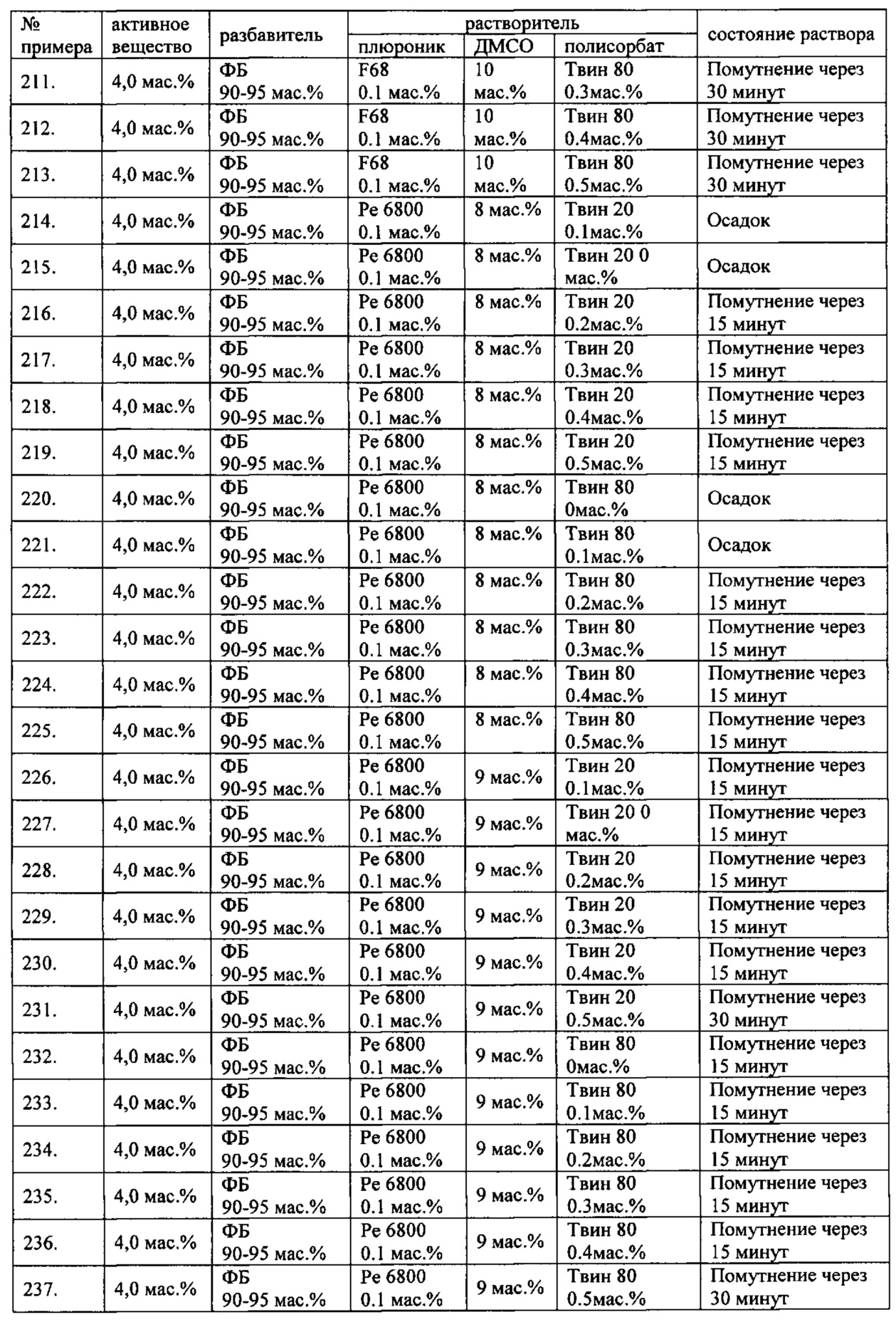
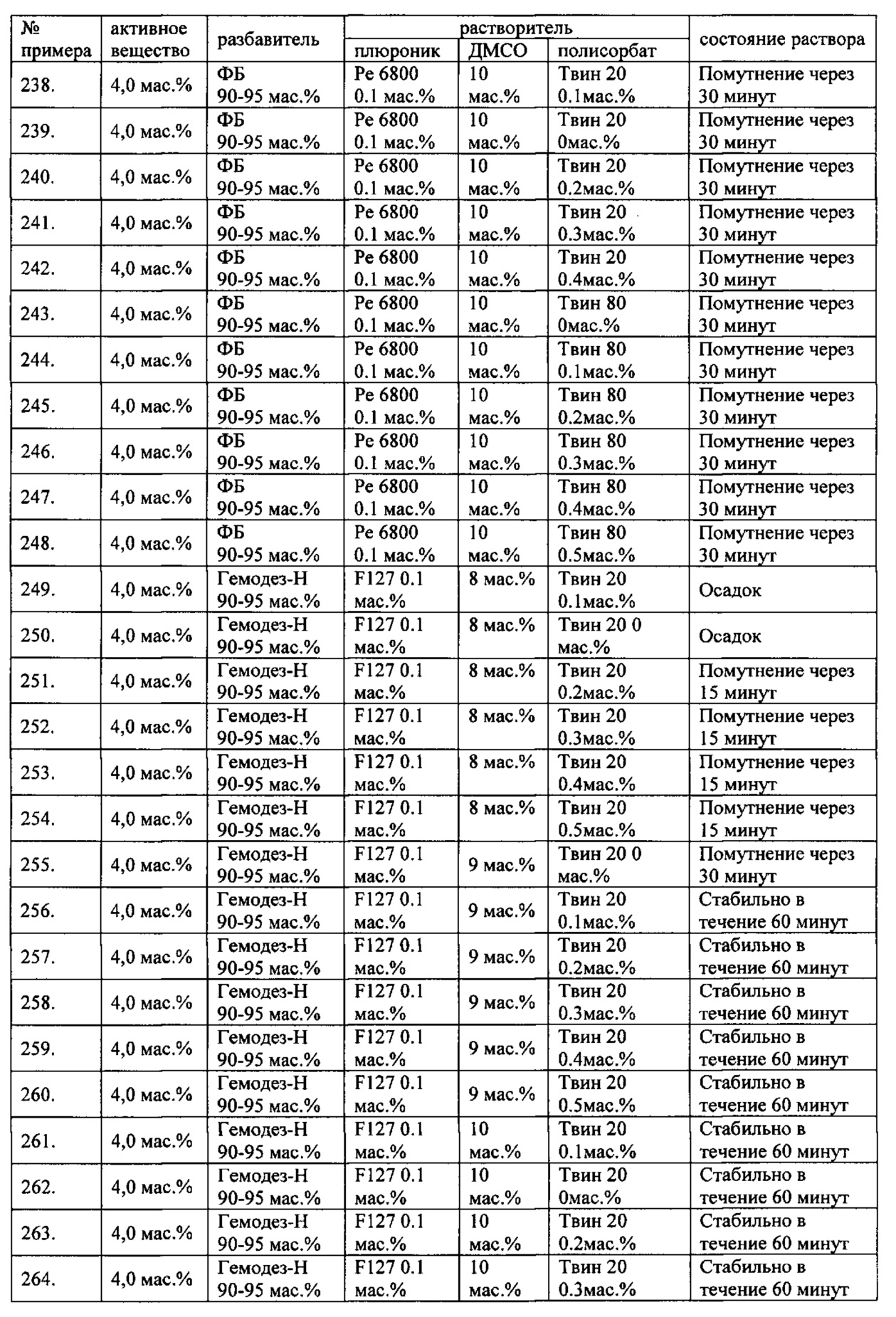
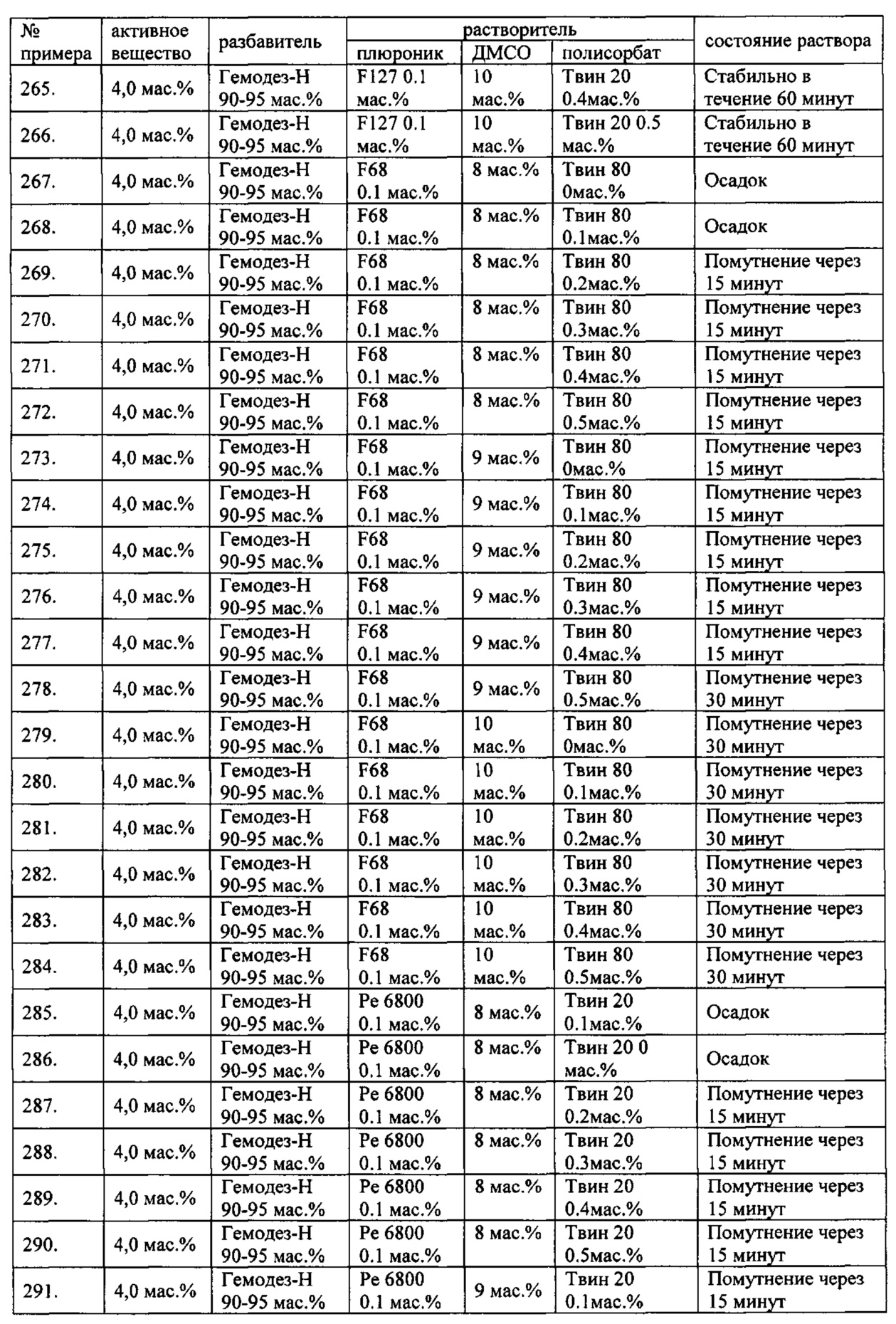
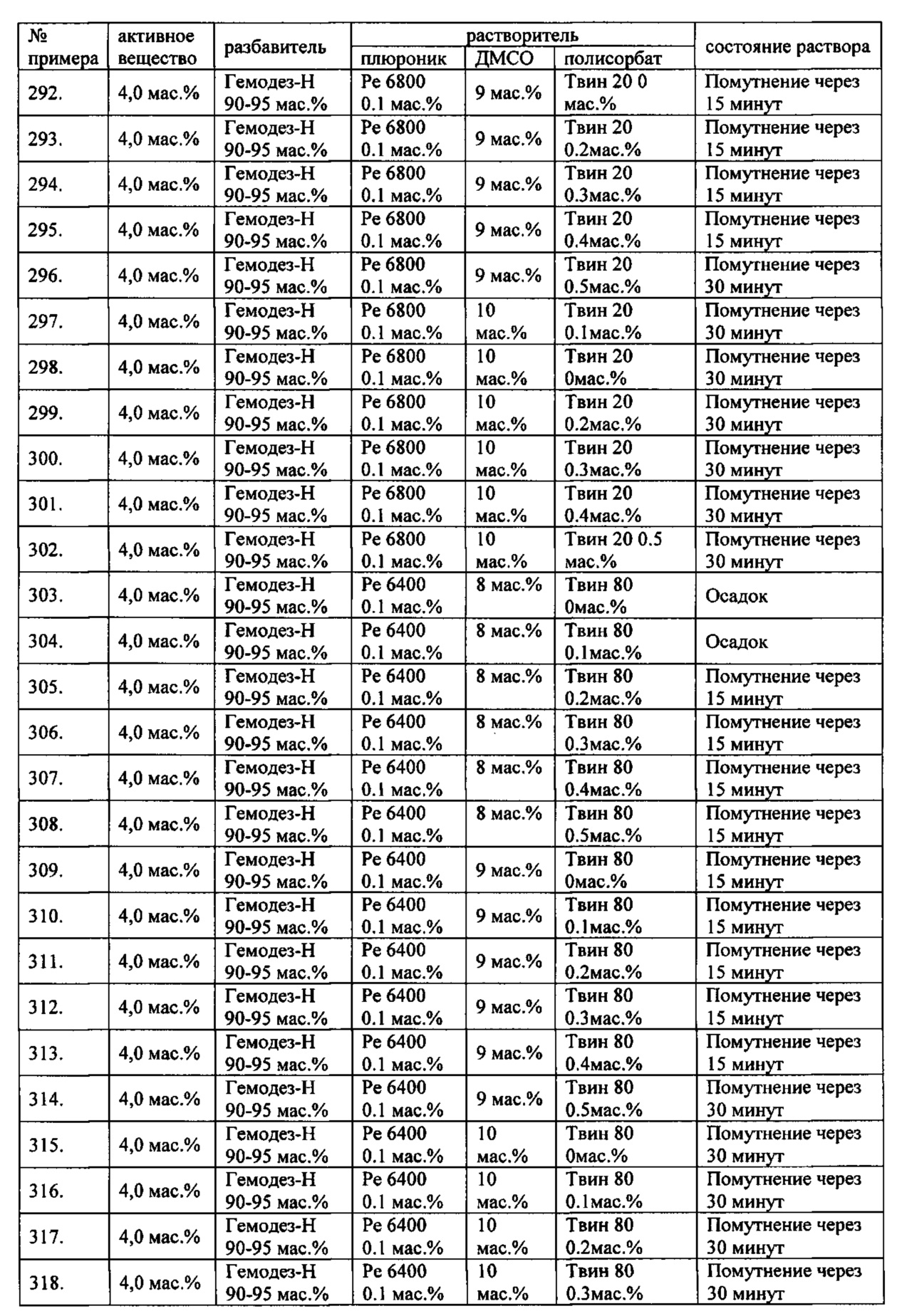

* - фосфатный буфер (ФБ) используемый для приготовления лекарственного средства представляет собой 50 мМ Na фосфатный буфер, рН 7,4.
Дополнительно была проведена проверка стабильности растворов во всем заявляемом диапазоне содержания компонентов. Результаты представлены в табл. 2.
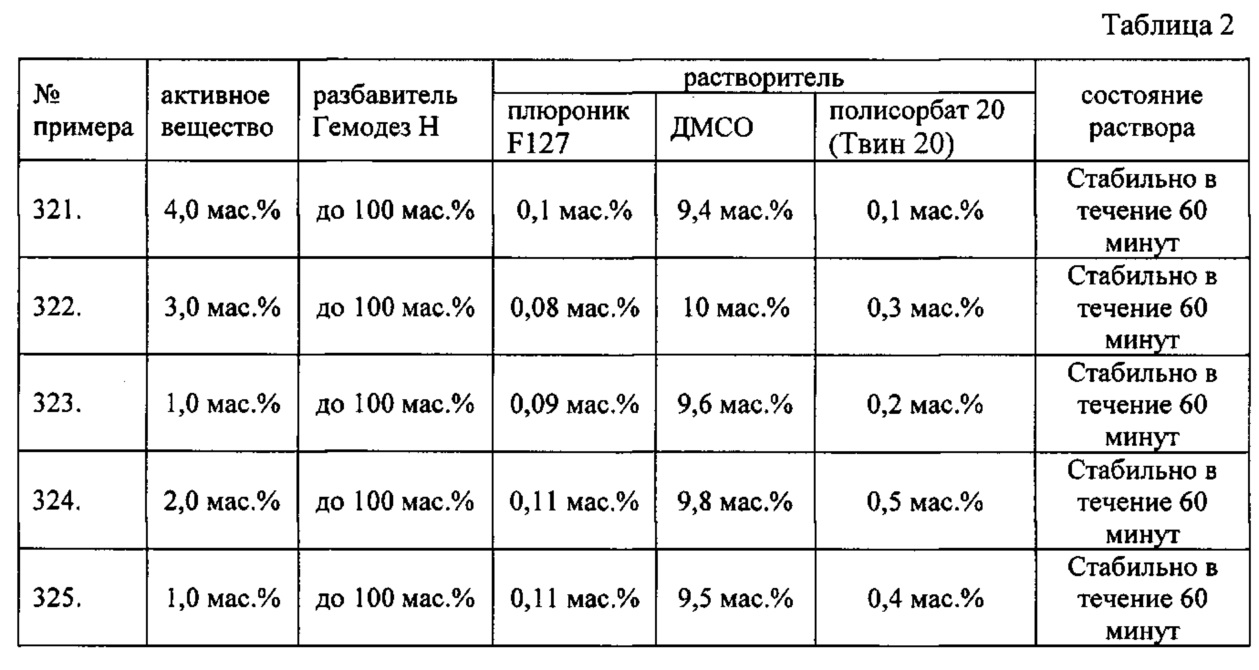
Введение пациенту заявляемого лекарственного средства проводится только под наблюдением врача, имеющего опыт проведения противоопухолевой химиотерапии в условиях специализированного стационара. Заявляемое средство вводится 1 раз в 3 недели. Рекомендуемая доза заявляемого средства составляет 0,5 мг/кг каждые три недели, не более 6 инъекций.
При этом необходимо учитывать, что заявляемое средство должно вводиться при количестве нейтрофилов в периферической крови не менее 1500/мкл. В случае развития фебрильной нейтропении, числа нейтрофилов менее 500/мкл длительностью более одной недели, выраженных или кумулятивных (усиливающихся при повторных введениях) кожных реакций, его доза при следующих введениях должна быть снижена.
Приготовление инфузионного раствора
Если флаконы с порошком и растворителем хранились в холодильнике, то перед разведением их необходимо в течение 5-10 минут выдержать при комнатной температуре (ниже 25°C). Затем в асептических условиях полученный концентрат разводят до концентрации необходимой для введения разбавителем, представляющим собой Гемодез Н. Содержимое с готовым раствором заявляемого средства следует перемешать с помощью вращательных движений. Полученный раствор необходимо использовать в течение 60 минут путем инъекции при комнатной температуре и обычных условиях освещенности.
Проведено исследование острой токсичности заявляемого средства на мышах и крысах при однократном внутривенном введении. Установлено, что мыши несколько менее чувствительны к заявляемому средству, чем крысы. У мышей и крыс выражены половые различия чувствительности к заявляемому средству - самки крыс и мышей несколько менее устойчивы к действию препарата, чем самцы. Средние значения LD50 заявляемого средства для мышей 32,75 мг/кг и для крыс 22,5 мг/кг. Изменений в макроструктуре органов не обнаружено. При введении высоких доз заявляемого средства отмечалось изменение цвета поверхности печени (на 14 день неоднородная) и почек (при гибели животных в первые дни почки были темного цвета). Результаты токсикометрии, данные наблюдений за экспериментальными животными в постинтоксикационном периоде острого отравления, а также данные некропсии позволяют отнести заявялемое средство к III классу умеренно токсичных лекарственных веществ.
Было проведено исследование острой токсичности in vitro. При оценке влияния заявялемого средства на жизнеспособность дифференцированных клеток печени и кишечника человека, культивируемых в условиях микроциркуляции питательной среды в системе Homunculus, установлены значения IC50. IC50 для клеток кишечника составила 38,5 мкМ, для клеток печени - 37,7 мкМ.
Проведено сравнение полученных результатов в исследованиях острой токсичности заявляемого средства in vitro и in vivo. В исследованиях in vivo при внутривенном введении препарата было показано, что среднелетальная доза LD50 для мышей составляет 29,2 мг/кг (или 38,9 мкмоль/кг), для крыс - 23,2 мг/кг (30,9 мкмоль/кг). В исследованиях in vitro IC50 составила 37,7 мкмоль/л для клеточной модели печени, состоящей из дифференцированных клеток HepaRG, и 38,5 мкмоль/л для клеточной модели кишечного эпителия, сформированного из дифференцированных клеток Сасо2. Результаты in vitro и in vivo хорошо коррелируют друг с другом.
Затем было проведено изучение хронической токсичности заявляемого средства на крысах. Проведенные экспериментальные исследования хронической токсичности лекарственного средства показали, что заявялемое средство в условиях хронического (30 дневного) внутривенного введения в дозах 2, 4 и 8 мг/кг оказывает ряд токсических эффектов на организм беспородных белых крыс. Введение заявляемого средства в дозах 4 и 8 мг/кг вызывало лимфопению периферической крови, снижало количество эритроцитов и гемоглобина, снижало костномозговое кроветворение (в особенности эритроидный и лимфоидный ростки), подавляло развитие Т-лимфоцитов в тимусе, угнетало сперматогенез, нарушало морфологическое и функциональное состояние печени и почек; введение заявляемого средства в дозе 2 мг/кг оказывало значительно менее выраженный токсический эффект и преимущественно угнетало костномозговое кроветворение и вызывало лимфопению периферической крови. Все изменения носили транзиторный характер и через 30 дней после отмены препарата практически отсутствовали. Исключение составила группа самцов, получавших заявляемое средство в дозе 8 мг/кг, у которых спустя 4 недели после окончания введения препарата сперматогенный эпителий восстановился не полностью. На основании результатов хронического эксперимента на крысах рассчитан индекс безопасности (ИБ) заявляемого средства при применении в клинике: крысы - доза 8 мг/кг; длительность введения - 30 дней. Гибели нет. Расчетный безопасный курс (РБК) заявляемого средства в днях равен 49 дней. Индекс безопасности (ИБ) составляет 4,1 при продолжительности курса лечения 12 дней. В соответствии с классификацией опасности лекарственных препаратов для клинического применения заявляемого средства относится к II классу умереннотоксичных лекарственных препаратов.
Также проведено изучение хронической токсичности заявляемого средства на кроликах. Проведенные экспериментальные исследования хронической токсичности лекарственного средства показали, что заявялемое средство в условиях хронического (30 дневного) внутривенного введения в дозах 1, 2 и 4 мг/кг оказывает ряд токсических эффектов на организм кроликов. Введение заявляемого средства в дозах 2 и 4 мг/кг вызывало лимфопению периферической крови, снижало количество эритроцитов и содержание в них гемоглобина, снижало костномозговое кроветворение, подавляло развитие лимфоцитов в тимусе, нарушало морфологическое и функциональное состояние печени и почек. Введение заявляемого средства в дозе 1 мг/кг практически не оказывало токсического эффекта и преимущественно выражалось в умеренном местно-раздражающем действии. Местно-раздражающее действие препарата в дозах 2 и 4 мг/кг было ярко выраженным, приводило к тромбозу вен и некрозу окружающих тканей. Нейтрофилия и тромбоцитоз, наблюдаемые в периферической крови животных, получавших препарат в дозах 2 и 4 мг/кг, по всей видимости, являются ответом организма на воспалительную реакцию в месте введения заявляемого средства. Изучение отставленных групп животных, проведенное спустя 30 дней после прекращения введения изучаемого лекарственного средства и контрольных растворов, показало, что практически все вышеперечисленные изменения возвращаются к параметрам физиологической нормы и значительные различия между группами отсутствуют. Исключение составила группа животных, получавших заявляемое средство в дозе 4 мг/кг, у которых, спустя 4 недели после окончания введения препарата, эпителий почечных канальцев восстановился не полностью. На основании результатов хронического эксперимента рассчитан индекс безопасности (ИБ) заявляемого средства при применении в клинике в соответствии с инструкцией (Т.А. Гуськова, 2003 г.): кролики - доза 4 мг/кг; длительность введения - 30 дней. Гибели нет. Расчетный безопасный курс (РБК) препарата равен 45 дням, а индекс безопасности (ИБ) составляет 3,8 при продолжительности курса лечения 12 дней. В соответствии с классификацией опасности лекарственных препаратов для клинического применения заявляемое средство относится к II классу умереннотоксичных лекарственных препаратов.
Было проведено исследование хронической токсичности in vitro. Использованы дифференцированные клетки печени и кишечника человека, культивируемых в условиях микроциркуляции питательной среды в системе Homunculus в течение 30 дней. После инкубации клеточных моделей печени и кишечника с заявляемым средством в концентрациях 11 мкмоль/л, 5,5 мкмоль/л и 2,75 мкмоль/л в течение месяца наблюдалось незначительное снижение жизнеспособности клеток в составе клеточной модели печени.
Разработаны модели in vitro и in vivo для оценки эффективности лекарственного средства. В качестве in vitro модели были выбраны опухолевые клетки линии MDA-MB-231 (клетки опухоли молочной железы человека). Эти же клетки были использованы для ксенотрансплантации иммунодефицитным мышам в исследовании специфической активности препарата in vivo. MDA-MB-231 - трижды негативные клетки, в которых отсутствует экспрессия рецепторов эстрогена и прогестерона, а также снижена экспрессия Нег2 рецептора, что делает эти клетки устойчивыми к различным противоопухолевым препаратам, таким как доксорубицин, цисплатин, этопозид и др. Кроме того, в качестве in vitro моделей для анализа противоопухолевого эффекта заявляемого средства были использованы недифференцированные клетки кишечника (Сасо-2) и печени (HepaRG). Эти клетки в недифференцированном состоянии имеют опухолевый фенотип и теряют его при дифференцировке, становясь наиболее приближенными к первичным клеткам в организме человека. В качестве in vivo модели использованы ксенографты - иммунодефицитные мыши линии SCID, которым в область абдоминальной молочной железы (ортотопно) привиты клетки линии MDA-MB-231. Преимущества ортотопного введения клеток заключается в следующем: степень васкуляризации и скорость роста, а также приживаемость ортотопно введенной опухоли выше, чем подкожно введенной. Ортотопная модель несколько ближе к реальной, несмотря на отличия в строении молочных желез человека и мыши. Кроме того, адекватная васкуляризация опухоли должна обеспечить оптимальное распределение внутривенно введенного препарата и попадание в клетки опухоли.
Исследования эффективности заявляемого средства in vitro показали, что спустя 24 часа инкубации клеток с различными концентрациями препарата IC50 для MDA-MB-231 -29,9 мкМ, IC50 для недифференцированных Сасо2 - 18,5 мкМ, IC50 для недифференцированных HepaRG - 12,2 мкМ. Как и ожидалось, наиболее устойчивыми к воздействию препарата оказались трижды негативные клетки рака молочной железы MDA-MB-231, а самыми чувствительными - клетки линии HepaRG, что, вероятно, связано с их происхождением и высоким уровнем транспортеров меди в этих клетках.
При исследовании эффективности заявляемого средства in vivo удалось показать, что однократное введение в дозе 10 мг/кг и пятикратное введение в дозе 6 мг/кг приводит к значительному торможению роста опухоли, при этом от 10 мг/кг заявляемого средства происходит уменьшение размеров опухоли практически вдвое. Дозы 7,5 мг/кг и 5 мг/кг при однократном введении приводят к замедлению роста опухоли, дозы 4 мг/кг и 2 мг/кг при пятикратном введении также приводят к замедлению роста опухоли. Как однократное, так и пятикратное введение растворителя не влияет на рост опухолей по сравнению с контролем.
Спустя 3 недели (21 день) после однократного введения 10 мг/кг заявялемого средства показатель Т/С составляет 11,5%, что говорит о высокой активности препарата в отношении трижды негативной опухоли MDA-MB-231 (Т/С=6-20% +++); показатель ТРО - 88,5%.
Спустя 3 недели (21 день) после пятикратного введения 6 мг/кг заявляемого средства Т/С составляет 29,7%, что также говорит об активности препарата в отношении трижды негативной опухоли MDA-MB-231 (Т/С=21-35% ++); показатель ТРО - 70,3%.
При однократном введении заявляемого средства расчетная доза, которая приводит к торможению роста опухоли на 90% (ЕД90), составляет 10,5-11,8 мг/кг и близка к максимально переносимой дозе (МПД) (при этом ЛД10 в исследовании острой токсичности заявляемого средства на нелинейных мышах-самках составляет примерно 15 мг/кг, на нелинейных крысах-самках примерно 13,5 мг/кг; в данном случае межвидовые отличия практически отсутствуют). Терапевтический индекс (ТИ50) для заявляемого средства при однократном введении равен 4-4,6.
При пятикратном введении заявляемого средства доза, которая приводит к торможению роста опухоли на 90% (ЕД90), составляет примерно 8,3 мг/кг. В исследовании хронической токсичности крысам-самкам в течение месяца внутривенно вводили дозу заявляемого средства 8 мг/кг (дозы больше приводили к гибели или появлению необратимых токсических эффектов). Все показатели возвращались к норме спустя месяц после последнего введения препарата. Для многократного применения доза 8 мг/кг близка к МПД или, точнее, к дозе, приводящей к незначительным обратимым токсическим эффектам. Терапевтический индекс (ТИ50) для заявляемого средства при пятикратном введении равен 2,9-2,6.
Критерием противоопухолевой активности является увеличение lg погибших клеток не менее чем в 2-4 раза. В соответствии с этим критерием эффективным является однократное введение 10 мг/кг заявляемого средства (увеличение lg погибших клеток более чем в 13 раз) и пятикратное введение 6 мг/кг заявляемого средства (увеличение lg погибших клеток примерно в 5 раз).
Согласно статистическому анализу полученных результатов, достоверное уменьшение размеров опухолей по сравнению с контрольной группой происходит в группе мышей после однократного внутривенного введения 10 мг/кг заявляемого средства (p-value <0,005).
Таким образом, если учесть, что в исследовании специфической активности использована развившаяся (более 500 мм3), резистентная к препаратам, трижды негативная опухоль молочной железы человека, то показатели ТРО 88,5% и Т/С 11,5% при однократном внутривенном введении 10 мг/кг заявляемого средства указывают на очень высокую противоопухолевую активность препарата, что удовлетворяет критериям и требованиям Руководства к разрабатываемым противоопухолевым лекарственным средствам, в связи с чем препарат может быть рекомендован для лечения трижды негативных опухолей молочной железы.
Для детекции лекарственного средства в биологических образцах в исследованиях фармакокинетики in vitro и in vivo разработаны и использованы следующие методы: спектроскопия в УФ и видимой областях спектра; LCMS-SIM (масс-спектрометрия в режиме селективного поиска ионов); ААС (атомно-адсорбционная спектроскопия); ICP-MS (масс-спектрометрия с индуктивно связанной плазмой).
Проведено исследование фармакокинетики заявляемого средства in vitro и in vivo.
In vitro. Координационный комплекс меди связывается с альбумином, основным белком плазмы крови. Константа связывания заявляемого средства с альбумином человека Kb=7,65⋅109, число сайтов связывания заявляемого средства с альбумином человека n=2. Анализ стабильности комплекса в плазме крови человека по сравнению со стабильностью в буфере не показал существенных отличий. Кроме того, основной белок плазмы крови, альбумин, не влияет на функциональную активность заявляемого средства: присутствие альбумина не приводит к снижению цитотоксической активности в отношении опухолевых клеток человека. Таким образом, взаимодействие комплекса с альбумином и белками плазмы крови не влияет на его стабильность и функциональную активность, а также фармакокинетику. Кроме того, заявляемое средство не метаболизируется микросомами печени человека и мыши. Это указывает на то, что после попадания в организм заявляемое средство не подвергается метаболизму в печени и попадает в клетки тканей и опухоли в своем исходном неизменном виде. Комплекс активно накапливается в клетках, а точнее в ядрах клеток. При этом комплекс интеркалирует ДНК, приводя к ее разрывам. Это указывает на то, что после введения в организм комплекс накапливается в активно пролиферирующих и обладающих повышенной метаболической активностью клетках тканей, что в свою очередь влияет на его эффективность и фармакокинетику (например, биодоступность, перераспределение, скорость и характер выведения из организма).
In vivo. При анализе результатов, полученных в ходе предварительного эксперимента на мышах, было обнаружено, что через 5 минут после внутривенного введения заявляеомго средства мышам концентрация меди в цельной крови достигает 11 мг/л (расчетная максимальная концентрация в крови после введения 18 мг/л), которая через 4 часа снижается практически до базального уровня, что указывает на то, что заявляемое средство к этому времени выводится и/или перераспределяется, накапливаясь в клетках тканей. Анализ уровня меди в печени мышей, показал, что в течение 4 часов после введения заявляемого средства уровень меди заметно растет. Это указывает на то, что часть лекарственного средства накапливается в печени и, вероятно, других тканях в организме мышей.
Реферат
Группа изобретений относится к химико-фармацевтической промышленности и представляет собой противоопухолевое лекарственное средство для лечения рака молочной железы, представляющее собой раствор для инъекционного введения, включающий в качестве активного вещества комплекс (5Z,5'Z)-2,2'-(этан-1,2-диилдисульфанилдиил)бис(5-(2-пиридилметилен)-3-аллил-3,5-дигидро-4Н-имидазол-4-она) с хлоридом меди (II), растворитель, включающий диметилсульфоксид, полисорбат 20 и полоксамер 407, и разбавитель Гемодез Н. Также изобретения представлены способом получения данного лекарственного средства, включающим добавление активного вещества в виде порошка в предварительно подготовленный растворитель, затем обработку раствора ультразвуком до полного растворения активного компонента и разведение полученного раствора Гемодезом Н до терапевтически эффективной концентрации активного компонента в лекарственном средстве. Изобретения включают способ лечения рака молочной железы, включающий инъекционное введение указанного лекарственного средства, приготовленного названным способом до истечения 60 мин после приготовления средства в терапевтически эффективном количестве. Группа изобретений обеспечивает стабильность полученного средства в течение 60 минут с момента его приготовления до введения пациенту. 3 н. и 2 з.п. ф-лы, 2 табл., 325 пр.
Комментарии