Новый многостадийный способ получения полипропилена - RU2526259C2
Код документа: RU2526259C2
Описание
Настоящее изобретение относится к новому многостадийному способу получения полипропилена путем полимеризации.
При получении полипропилена преимущественно используют катализаторы Циглера-Натта или катализаторы с единым центром полимеризации на металле. В настоящее время при получении полипропилена наблюдается тенденция к использованию катализаторов с единым центром полимеризации на металле. Полипропилены, получаемые при использовании катализаторов с единым центром полимеризации на металле, имеют в высокую степень гомогенности и организованную структуру. Одним из эффектов этого является довольно низкое содержание фракции, растворимой в холодном ксилоле, по сравнению с полипропиленами, полученными в присутствии катализаторов Циглера-Натта. Одной из проблем катализаторов с единым центром полимеризации на металле является то, что они растворимы в пропилене. В связи с этим полученные каталитические системы с единым центром полимеризации на металле, так называемые нанесенные каталитические системы с единым центром полимеризации на металле, позволяют осуществлять контроль гетерогенного процесса. Недостатком таких нанесенных каталитических систем является то, что остатки катализатора попадают в полученный полипропилен. Такие остатки нежелательны для некоторых конечных применений и, следовательно, необходимы стадии трудоемкой промывки. По этой причине была разработана новая каталитическая система с единым центром полимеризации на металле, которая значительно снижает недостатки известных гетерогенных каталитических систем. Такие новые каталитические системы являются самодостаточными, то есть отсутствует необходимость в каталитически инертном внешнем материале-носителе, и дополнительно характеризуются низкой площадью поверхности и низкой пористостью. Такие новые каталитические системы с единым центром полимеризации на металле не загрязняют полученный полипропилен нежелательными остатками, такими как кремний, что позволяет повысить производительность, поскольку может быть повышена объемная плотность полимеризованного полипропилена. Впервые такие новые каталитические системы с единым центром полимеризации на металле описаны, например, в WO 03/051934 или в WO 2006/069733. Также в ЕР 1 847 552 описываются новые каталитические системы с единым центром полимеризации на металле, позволяющие получить мультиразветвленные полипропилены. Но даже несмотря на общее понимание того, что такие новые самодостаточные катализаторы с единым центром полимеризации на металле могут быть полезны для получения полипропилена, все еще продолжает существовать необходимость в оптимизации таких каталитических систем для получения специфических требуемых полипропиленов.
Например, существует необходимость в разработке промышленного способа получения чистого полипропилена с высокой жесткостью и высокой теплостойкостью.
Целью настоящего изобретения является получение полипропилена со скоростью течения расплава MFR2 (230°С) более чем 2,0 г/10 минут при многостадийном способе полимеризации в присутствии самодостаточной каталитической системы с низкой пористостью, включающей цирконийорганическое соединение с формулой (I), приведенной ниже.
Следовательно, настоящее изобретение относится к многостадийному способу получения полипропилена путем полимеризации, включающему использование по меньшей мере двух последовательно соединенных реакторов, где указанный способ включает стадии:
(A) получение в первом реакторе (R-1) первой фракции полипропилена (РР-1),
(B) перемещение указанной первой фракции полипропилена (РР-1) во второй реактор (R-2),
(C) полимеризацию в указанном втором реакторе (R-2) второй фракции полипропилена (РР-2) в присутствии указанной первой фракции полипропилена (РР-1) с получением композиции полипропилена (РР-С),
где
(а) указанная первая фракция полипропилена (РР-1)
(i) имеет скорость течения расплава MFR2 (230°С), измеренную согласно ISO 1133,
(α) не более чем 1,5 г/10 минут,
или
(β) более чем 2,0 г/10 минут,
и
(ii) включает пропиленовые единицы и необязательно по меньшей мере один С2 - С10 α-олефин, отличающийся от пропилена,
(b) композиция полипропилена (РР-С) имеет скорость течения расплава MFR2 (230°С), измеренную согласно ISO 1133, более чем 2,0 г/10 минут,
и
(c) скорость течения расплава MFR2 (230°С) композиции полипропилена (РР-С) отличается от скорости течения расплава MFR2 (230°С) первой фракции полипропилена (РР-1),
где дополнительно
в первом реакторе (R-1) и во втором реакторе (R-2) полимеризация проходит в присутствии твердой каталитической системы (SCS), где указанная твердая каталитическая система (SCS)
(d) имеет пористость менее чем 1,40 мл/г, измеренную согласно ASTM 4641, и/или площадь поверхности менее чем 25 м2/г, измеренную согласно ASTM D 3663,
(e) включает катализатор, представляющий цирконийорганическое соединение с формулой (I):
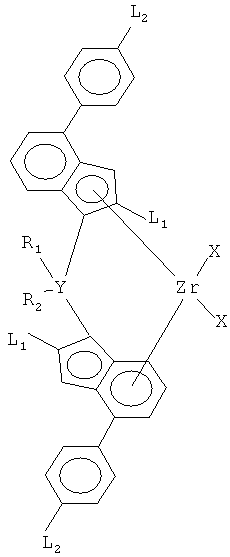
где
X - лиганды, соединенные σ- связью с цирконием (Zr),
L1 - идентичные остатки, выбранные из группы, состоящей из линейного C1 - С20 алкила, разветвленного С3 - С20 алкила, линейного C1 - С20 алкенила, разветвленного С4 - С20 алкенила, С4 - С12 циклоалкила, С1 - С20 алкила, замещенного С5 - С20 циклоалкилом, С6 - С20 арила и С5 - С20 циклоалкила, замещенного C1 - С20 алкилом, где циклоалкильный остаток замещен C1 - С20 алкилом,
L2 - идентичные остатки, выбранные из группы, состоящей из линейного C1 - С20 алкила, разветвленного С3 - С20 алкила, линейного C1 - С20 алкенила, разветвленного С4 - С20 алкенила, С4 - С12 циклоалкила, С1 - С20 алкила, замещенного С5 - С20 циклоалкилом, и С6 - С20 арила,
Y - С, Si или Ge, предпочтительно Si,
R1 - C1 - C20 алкил, C4 - С10 циклоалкил, C6-C12 арил, C7-C12 арилалкил или триметилсилил,
R2 - С1 - С20 алкил, С4 - С10 циклоалкил, С6-С12 арил, С7-С12 арилалкил или триметилсилил и
(f) включает сокатализатор (Со), включающий элемент (Е) из группы 13 периодической таблицы (IUРАС).
Указанный выше способ позволяет получить очень чистый полипропилен, то есть полипропилен, по существу свободный от нежелательных остатков и дополнительно характеризующийся превосходной жесткостью и высокой теплостойкостью.
Далее настоящее изобретение будет описано более детально.
Используемый в описании настоящей патентной заявки термин «многостадийный способ полимеризации» относится к полипропилену, полученному по меньшей мере в двух последовательно соединенных реакторах. Следовательно, способ по настоящему изобретению включает по меньшей мере первый реактор (R-1) и второй реактор (R-2). В одном конкретном варианте воплощения настоящего изобретения способ по настоящему изобретению включает использование двух ректоров полимеризации (R-1) и (R-2). В другом предпочтительном варианте воплощения настоящего изобретения способ по настоящему изобретению включает использование по меньшей мере трех реакторов полимеризации, то есть после второго реактора (R-2) расположен по меньшей мере третий реактор (R-3). Используемый в описании настоящей патентной заявки термин «реактор полимеризации» относится к месту прохождения основной полимеризации. Следовательно, в случае, когда способ включает использование двух реакторов полимеризации, это определение не исключает, что процесс в целом включает, например, стадию предварительной полимеризации в реакторе предварительной полимеризации. Используемый в описании настоящей патентной заявки термин «включает использование» относится к закрытой формулировке только в отношении основных реакторов полимеризации.
Используемый в описании настоящей патентной заявки термин «прямая подача» относится к способу, где содержимое первого реактора (R-1), такое как суспензия реактора (SR), а именно продукт полимеризации и реакционная среда, подается напрямую на следующую стадию, то есть во второй реактор (R-2), такой как первый газофазный реактор (GPR-1).
Используемый в описании настоящей патентной заявки термин «не прямая подача» относится к способу, где содержимое первого реактора (R-1), такое как суспензия реактора (SR), подают во второй реактор (R-2), такой как первый газофазный реактор (GPR-1), а именно продукт полимеризации, через реакционную среду установки для отделения, и реакционную среду в виде газа из установки для отделения. Перед подачей реакционной среды во второй реактор (R-2), такой как первый газофазный реактор (GPR-1), некий компонент, например водород, может быть полностью или частично удален из нее при использовании различных технических средств, известных из предшествующего уровня техники, таких как мембраны.
Используемый в описании настоящей патентной заявки термин «установка для отделения» относится к установке, где некоторые легкие компоненты, например водород и/или необязательно азот, могут быть отделены от мономера(ов) полностью или частично при использовании различных технических средств, таких как мембраны, дистилляция, отгонка или конденсация с отводом паровоздушной смеси.
Используемые в описании настоящей патентной заявки термины «по существу без повторно обрабатываемого мономера» и «с минимальным количеством или без повторно обрабатываемого мономера» являются синонимами, используемыми для указания того, что менее чем 30 масс.%, предпочтительно менее чем 20 масс.%, более предпочтительно менее чем 10 масс.%, в частности 0 масс.% мономеров первого реактора (R-1), такого как суспензионный реактор (SR), подано на повторную обработку в первый реактор (R-1), в отличие от традиционных способов, при которых в норме 50 масс.% или более содержимого первого реактора подается на повторную обработку.
В предпочтительном варианте воплощения настоящего изобретения не проводят повторную обработку мономера(ов) из второго реактора (R-2) подачей обратно в первый реактор (R-1). Следовательно, содержимое первого реактора (R-1) напрямую или не напрямую подают во второй реактор (R-2). Прямая подача - предпочтительна.
В способе с прямой подачей содержимое первого реактора (R-1), продукт полимеризации и реакционную среду, подают напрямую во второй реактор (R-2). Продукт может выходить из первого реактора (R-1) периодически или предпочтительно непрерывно. Все содержимое первого реактора (R-1) подают во второй реактор (R-2) как таковое без отделения какого-либо газа или потоков частиц исходя из различного размера частиц. Частицы не возвращают в первый реактор (R-1).
При непрямой подаче во второй реактор (R-2) содержимое первого реактора (R-1) подают сначала в реакционную среду установки для отделения. Полимер подают во второй реактор (R-2) из указанной установки для отделения. Отходящий газ из установки для отделения подают во второй реактор (R-2) в газообразной форме. Однако перед подачей газовой фазы из нее частично или полностью удаляют, например, водород, при использовании различных технических решений, например, мембран или отгонки. В качестве альтернативы, отходящий газ может быть конденсирован и необязательно водород или другой легкий компонент может быть удален перед подачей жидкого мономера во второй реактор (R-2).
Реакция продолжается во втором реакторе (R-2) и необязательно в дополнительном последующем реакторе(ах). Следовательно, мономер, подаваемый во второй реактор (R-2) из первого реактора (R-1), также может быть превращен в полимер.
Первый реактор (R-1), предпочтительно суспензионный реактор (SR), может представлять любой реактор непрерывного действия или простой реактор с мешалкой периодического действия, или циркуляционный реактор для проведения полимеризации в массе или в суспензии. В массе - означает полимеризацию в реакционной среде, включающей по меньшей мере 60% (масса/масса) мономера. В настоящем изобретении суспензионный реактор (SR) предпочтительно представляет (для полимеризации в массе) циркуляционный реактор (LR).
Второй реактор (R-2) и любой последующий реактор предпочтительно представляет газофазный реактор (GPR). Такие газофазные реакторы (GPR) могут представлять любые реакторы с механическим перемешиванием или реакторы с псевдоожиженным слоем. Предпочтительно газофазные реакторы (GPR) включают реактор с псевдоожиженным слоем с механическим перемешиванием со скоростью потока газа по меньшей мере 0,2 м/секунду. Следовательно, понятно, что газофазный реактор представляет реактор с псевдоожиженным слоем предпочтительно с механической мешалкой.
Следовательно, в предпочтительном варианте воплощения настоящего изобретения первый реактор (R-1) представляет суспензионный реактор (SR), такой как циркуляционный реактор (LR), в то время как второй реактор (R-2) и, если присутствует, любой последующий реактор представляет газофазный реактор (GPR). Следовательно, в одном варианте воплощения настоящего изобретения для способа по настоящему изобретению используют два последовательно соединенных реактора полимеризации, а именно суспензионный реактор (SR), такой как циркуляционный реактор (LR), и первый газофазный реактор (GPR-1). В качестве альтернативы, для способа по настоящему изобретению используют три или четыре последовательно соединенных реактора для полимеризации, а именно суспензионный реактор (SR), такой как циркуляционный реактор (LR), и первый газофазный реактор (GPR-1), второй газофазный реактор (GPR-2) и необязательно четвертый газофазный реактор (GPR-3). Если требуется, перед суспензионным реактором (SR) устанавливают реактор предварительной полимеризации в каждой из указанных выше систем.
Как указанно выше, настоящее изобретение относится к способу получения полипропилена. Полученный полипропилен может представлять гомополимер пропилена, статический сополимер пропилена или гетерофазный сополимер пропилена. Гетерофазный сополимер пропилена по настоящему изобретению включает матрицу и диспергированный в ней эластомерный сополимер. Матрица гетерофазного сополимера пропилена может представлять как гомополимер пропилена, так и статический сополимер пропилена. В любом случае композиция полипропилена (РР-С), полученная при полимеризации во втором реакторе (R-2), представляет статический сополимер пропилена или гомополимер пропилена, последний является предпочтительным. Следовательно, в случае, когда полимеризация по настоящему изобретению продолжается в третьем реакторе (R-3), таком как второй газофазный реактор (GPR-2), и необязательно в четвертом реакторе (R-4), таком как третий газофазный реактор (GPR-3), конечный полипропилен зависит от полимера, полученного в третьем реакторе (R-3) и необязательно в четвертом реакторе (R-4), который может представлять гомополимер пропилена, статический сополимер пропилена или гетерофазный сополимер пропилена. Однако понятно, что конечный полипропилен представляет композицию полипропилена (РР-С) по настоящему изобретению, полученную во втором реакторе (R-2).
Используемый в описании настоящей патентной заявки термин гомополимер относится к полипропилену, по существу состоящему из пропиленовых единиц, то есть по меньшей мере на 99 масс.%, предпочтительно по меньшей мере на 99,5 масс.%, более предпочтительно по меньшей мере на 99,8 масс.%. В предпочтительном варианте воплощения настоящего изобретения гомополимер пропилена состоит только из пропиленовых единиц.
Как указанно выше, сополимер пропилена может представлять как статический сополимер пропилена, так и гетерофазный сополимер пропилена. Используемый в описании настоящей патентной заявки термин «рандом-сополимер» предпочтительно понимается согласно IUPAC, то есть как полимер, в котором можно обнаружить заданную мономерную единицу в любом заданном участке в полимерной цепи.
В отличие от рандом-сополимера пропилена, гетерофазный сополимер пропилена характеризуется фазой матрицы, в которой диспергированна эластомерная фаза. Эластомерная фаза характеризуется довольно низким содержанием фракции, растворимой в холодном ксилоле, и/или аморфностью. Эластомерную фазу получают в третьем реакторе (R-3) (второй газофазный реактор (GPR-2)) и/или четвертом реакторе (R-4) (третий газофазный реактор (GPR-3)). В дополнение матрица представляет композицию пропилена (РР-С) по настоящему изобретению, полученную в первом реакторе (R-1) и втором реакторе (R-2). Следовательно, в предпочтительном варианте воплощения настоящего изобретения содержание фракции, растворимой в холодном ксилоле, в матрице составляет не более чем 15 масс.%, такое как не более чем 8 масс.%, при этом содержание фракции, растворимой в холодном ксилоле, в эластомерной фазе составляет по меньшей мере 20 масс.%, более предпочтительно по меньшей мере 25 масс.%. Дополнительно, матрица может представлять как гомополимер пропилена, так и статический сополимер пропилена. Матрица, то есть композиция пропилена (РР-С), в предпочтительных вариантах воплощения настоящего изобретения детально описана ниже.
Одним из существенных требований настоящего изобретения является то, что композиция полипропилена (РР-С) имеет специфические свойства. Согласно этому требованию композиция полипропилена (РР-С) имеет довольно высокую скорость течения расплава (MFR). Скорость течения расплава, измеренную при нагрузке 2,16 кг при температуре 230°С (ISO 1133), указывают как MFR2 (230°С). Следовательно, в предпочтительном варианте воплощения настоящего изобретения композиция полипропилена (РР-С) имеет MFR2 (230°С) более чем 2,0 г/10 минут, более предпочтительно более чем 3,0 г/10 минут. Следовательно, понятно, что MFR2 (230°С), измеренная согласно ISO 1133, составляет в пределах от 2,0 до 10,0 г/10 минут, более предпочтительно в пределах от 2,0 до 6,0 г/10 минут, такая как в пределах от 2,0 до 4,0 г/10 минут, еще более предпочтительно в пределах от 2,0 до 6,0 г/10 минут.
Как указанно выше, композиция полипропилена (РР-С) может представлять, как сополимер пропилена, так и гомополимер пропилена, последний предпочтителен. Важным аспектом настоящего изобретения является то, что композиция пропилена (РР-С) включает, предпочтительно состоит из двух различных фракций полипропилена, а именно первой фракции полипропилена (РР-1) и второй фракции полипропилена (РР-2). Первую фракцию полипропилена (РР-1) получают в первом реакторе (R-1), предпочтительно суспензионном реакторе (SR), таком как циркуляционный реактор (LR), при этом вторую фракцию полипропилена (РР-2) получают во втором реакторе (R-2), предпочтительно в (первом) газофазном реакторе (GPR-1). Две фракции должны быть получены таким образом, чтобы композиция полипропилена (РР-С) представляла гомогенную смесь первой фракции пропилена (РР-1) и второй фракции полипропилена (РР-2) и имела скорость течения расплава (MFR), как указанно выше. Другим обязательным требованием является то, чтобы первая фракция полипропилена (РР-1) имела скорость течения расплава MFR2 (230°С), измеренную согласно ISO 1133,
(i) не более чем 1,5 г/10 минут, предпочтительно не более чем 1,3 г/10 минут, более предпочтительно в пределах от 0,3 до 1,5 г/10 минут, еще более предпочтительно в пределах от 0,5 до 1,3 г/10 минут,
или
(ii) более чем 2,0 г/10 минут, предпочтительно более чем 3,5 г/10 минут, более предпочтительно более чем в пределах от 2,0 до 50 г/10 минут, еще более предпочтительно более чем в пределах от 3,5 до 35,0 г/10 минут.
В случае, когда первая фракция полипропилена (РР-1) имеет скорость течения расплава MFR2 (230°С) более 2,0 г/10 минут, дополнительным требованием является то, что скорость течения расплава MFR2 (230°С) первой фракции полипропитена (РР-1) отличается от скорости течения расплава MFR2 (230°С) композиции полипропилена (РР-С).
Соответственно, композиция полипропилена (РР-С) по меньшей мере бимодальна по распределению своей молекулярной массы.
Используемый в описании настоящей патентной заявки термин «мультимодальный» или «бимодальный» относится к модальности полимера, то есть
▪ форма кривой распределения молекулярной массы, которая представляет график молекулярной массы фракции, как функцию молекулярной массы,
или
▪ форма кривой распределения содержания сомономера, которая представляет график содержания сомономера, как функцию молекулярной массы фракций полимера.
Следовательно, понятно, что соотношение скорости течения расплава MFR2 (230°С) фракции полипропилена (РР-1) и композиции полипропилена (РР-С) составляет по меньшей мере 1,25 [MFR2 (230°С) РР-1 / MFR2 (230°С) РР-С], более предпочтительно по меньшей мере 1,5, такое как по меньшей мере 2,5, в случае, когда первая фракция полипропилена (РР-1) имеет скорость течения расплава MFR2 (230°С) более 2,0 г/10 минут.
С другой стороны, в предпочтительном случае фракция полипропилена (РР-1) имеет скорость течения расплава MFR2 (230°С), равную или ниже 1,3 г/10 минут, при этом соотношение скорости течения расплава MFR2 (230°С) композиции полипропилена (РР-С) и фракции полипропилена (РР-1) составляет по меньшей мере 1,6 [MFR2 (230°С) РР-С / MFR2 (230°С) РР-1], более предпочтительно составляет по меньшей мере 2,0, такое как по меньшей мере 3,0.
Конечно, скорость течения расплава MFR2 (230°С) композиции полипропилена (РР-С), полученной в первом реакторе (R-1) и втором реакторе (R-2), также очень сильно зависит от разделения по массе полученного в двух реакторах. В частности, установлено, что соотношение по массе между первым ректором (R-1) и вторым реактором (R-2) [кг R-1/ кг R-2] должно быть равно или ниже 1,5 [кг/кг], более предпочтительно равно или ниже 1,2 [кг/кг], еще более предпочтительно в пределах от 0,1 до 1,5 [кг/кг], еще более предпочтительно в пределах от 0,3 до 1,3 [кг/кг], еще более предпочтительно в пределах от 0,4 до 1,1 [кг/кг], такое как в пределах от 0,5 до 1,0 [кг/кг].
В первом реакторе (R-1) в качестве первой фракции пропилена (РР-1) может быть получен статический сополимер пропилена или гомополимер пропилена, последний предпочтителен.
В случае, когда фракция полипропилена (РР-1) представляет статический сополимер пропилена, она включает мономеры, сополимеризуемые с пропиленом, например, сомономеры, такие как этилен и/или С4 - С20 альфа-олефины, в частности, этилен и/или С4 - С10 альфа-олефины, например, 1-бутиен и/или 1-гексен. Предпочтительно статический сополимер пропилена в качестве первой фракции полипропилена (РР-1) включает, по существу состоит из мономеров, сополимеризуемых с пропиленом, из группы, состоящей из этилена, 1-бутиена и 1-гексена. А именно, статический сополимер пропилена включает в отличие от пропилена этиленовые единицы и/или 1-бутен. В предпочтительном варианте воплощения настоящего изобретения статический сополимер пропилена в качестве первой фракции полипропилена (РР-1) включает только этиленовые и пропиленовые единицы. Содержание сомономера в первой фракции полипропилена (РР-1), представляющей статический сополимер пропилена, предпочтительно относительно низкое, то есть вплоть до 6,0 масс.%, более предпочтительно в пределах от 0,5 до 6,0 масс.%, еще более предпочтительно в пределах от 0,5 до 4,0 масс.%, еще более предпочтительно в пределах от 0,5 до 2,0 масс.%.
Также во втором реакторе (R-1) может быть получен статический сополимер пропилена или гомополимер пропилена. Соответственно, полученная композиция полипропилена (РР-С) во втором реакторе (R-2) может представлять гомополимер пропилена или статический сополимер пропилена. Определение термина статический сополимер пропилена (возможные сомономеры и их количества) приведено в предшествующем абзаце.
В случае, когда композиция полипропилена (РР-С) представляет гомополимер полипропилена, две фракции полипропилена РР-1 и РР-2 также обязательно должны представлять гомополимеры пропилена. Соответственно, первая фракция полипропилена (РР-1) и вторая фракция полипропилена (РР-2) отличаются только скоростью течения расплава, а не содержанием сомономера (поскольку сомономер не присутствует).
Для случая, когда композиция полипропилена (РР-С) представляет статический сополимер пропилена, приведены три различных варианта:
(a) В первом реакторе (R-1) и втором реакторе (R-2) получают статический сополимер пропилена. Соответственно, первая фракция полипропилена (РР-1) и вторая фракция полипропилена (РР-2) отличаются скоростью течения расплава и также необязательно содержанием сомономера.
(b) В первом реакторе (R-1) получают статический сополимер пропилена, а во втором реакторе получают (R-2) гомополимер пропилена. Соответственно, первая фракция полипропилена (РР-1) и вторая фракция полипропилена (РР-2) отличаются скоростью течения расплава и также содержанием сомономера.
(c) В первом реакторе (R-1) получают гомополимер пропилена, а во втором реакторе (R-2) получают статический сополимер пропилена. Соответственно, первая фракция полипропилена (РР-1) и вторая фракция полипропилена (РР-2) отличаются скоростью течения расплава и также содержанием сомономера.
В предпочтительном способе получения композиция полипропилена (РР-С) представляет сополимер пропилена, в котором содержание сомономера во втором реакторе (R-2) выше по сравнению с первым реактором (R-1). Соответственно, в первом реакторе (R-1) получают гомополимер пропилена, в то время как во втором реакторе (R-2) получают статический сополимер пропилена (вариант (с)), или в обоих, как в первом реакторе (R-1), так и во втором реакторе (R-2) получают статический сополимер пропилена с отличающимся содержанием сомономера.
Однако, в частности, предпочтительно, чтобы композиция полипропилена (РР-С) представляла гомополимер пропилена.
Дополнительно, понятно, что композиция полипропилена (РР-С) (следовательно, также его фракции полипропилена РР-1 и РР-2) имеет линейную структуру и, следовательно, не имеет (или нет поблизости) разветвления. Соответственно, понятно, что композиция полипропилена (РР-С) и/или фракция полипропилена (РР-1) по настоящему изобретению предпочтительно имеет индекс разветвления g′ не менее чем 0,90, предпочтительно более чем 0,90, такое как по меньшей мере 0,95. Другими словами, если композиция полипропилена (РР-С) и/или фракция полипропилена (РР-1) имеет некоторое разветвление, то оно довольно умеренное. Соответственно, индекс разветвления g′ композиции полипропилена (РР-С) и/или фракции полипропилена (РР-1) предпочтительно составляет в пределах от 0,90 до 1,00, более предпочтительно в пределах от более чем 0,90 до менее 1,00, такое как в пределах от 0,96 до менее 1,00. Индекс разветвления g′ определяют как g′=[IV]br/[IV]|lin, где g′ представляет индекс разветвления, [IVbr] представляет истинную вязкость разветвленного полипропилена, и [IV]lin представляет истинную вязкость линейного полипропилена с такой же среднемассовой молекулярной массой (в пределах ± 3%), как у разветвленного полипропилена. Таким образом, низкий показатель g′ является индикатором сильно разветвленного полимера. Другими словами, если показатель g′ снижается, то разветвленность полипропилена повышается. Точное определение показателя g′ приведено в Примерах.
Как будет описано детально ниже, в способе по настоящему изобретению используют специфическую каталитическую систему с единым центром полимеризации на металле. Соответственно, фракция полипропилена (РР-1), полученная в первом реакторе (R-1), и/или композиция полипропилена (РР-С), полученная во втором реакторе (R-2), получены без использования катализатора Циглера-Натта. Следовательно, дополнительно предпочтительно, чтобы содержание остатков титана (Ti) в композиции полипропилена (РР-С) предпочтительно составляло менее 10 частей на миллион, даже более предпочтительно титан (Ti) не определяется или даже составляет 0 частей на миллион.
Дополнительно, поскольку катализатор, используемый в способе по настоящему изобретению, не наносится на какой-либо кремниевый носитель, предпочтительно содержание остатков кремния (Si), измеренное согласно ISO 3451-1 (1997), во фракции полипропилена (РР-1) и/или в композиции полипропилена (РР-С) составляет менее 10 частей на миллион, более предпочтительно менее 5 частей на миллион, еще более предпочтительно менее 1,2 частей на миллион, такое как менее 1 часть на миллион. В конкретном варианте воплощения настоящего изобретения кремний (Si) не определяется во фракции полипропилена (РР-1) и/или в композиции полипропилена (РР-С).
Дополнительно, понятно, что содержание остатков магния (Mg), измеренное согласно ISO 3451-1 (1997), во фракции полипропилена (РР-1) и/или в композиции полипропилена (РР-С) составляет не более чем 5 частей на миллион, предпочтительно менее 1,0 частей на миллион, еще более предпочтительно менее 0,5 частей на миллион, еще более предпочтительно 0 частей на миллион или не определяется согласно используемому методу измерения, и/или содержание остатков хлора (Сl), измеренное согласно ISO 3451-1 (1997), составляет менее 1,0 части на миллион, еще более предпочтительно менее 0,5 частей на миллион.
Ввиду специфики используемой каталитической системы полученная фракция полипропилена (РР-1) и/или полученная композиция полипропилена (РР-С) предпочтительно дополнительно характеризуется довольно низким содержанием фракции, растворимой в холодном ксилоле (XCS), то есть, содержание фракции, растворимой в холодном ксилоле (XCS), составляет не более чем 1,5 масс.%, более предпочтительно менее 1,3 масс.%, еще более предпочтительно менее 1,0 масс.%, такое как менее 0,8 масс.%. Следовательно, в частности, понятно, что фракция полипропилена (РР-1) и/или композиция пропилена (РР-С) по настоящему изобретению имеет содержание фракции, растворимой в холодном ксилоле (XCS), составляющее в пределах от 0,3 до 1,5 масс.%, более предпочтительно в пределах от 0,3 до 1,3 масс.%, еще более предпочтительно в пределах от 0,4 до 1,0 масс.%, такое как в пределах от 0,4 до 0,8 масс.%.
Далее будет детально описана композиция полипропилена (РР-С) по настоящему изобретению, полученная при использовании специфической каталитической системы. Соответственно, композиция полипропилена (РР-С) получена не в присутствии катализатора Циглера-Натта. Как правило, полипропилены получают при использовании катализаторов различных типов, также по существу отличающихся по <2,1> региодефектам. Следовательно, понятно, что композиция р-полипропилена (РР-С) имеет <2,1> региодефекты, определяемые при использовании13С-спектроскопии, равные или более чем 0,4 мол.%, более предпочтительно равные или более чем 0,6 мол.%, такие как в пределах от 0,7 до 0,9 мол.%.
Дополнительно, полученная фракция полипропилена (РР-1) и/или полученная композиция полипропилена (РР-С) может быть охарактеризована довольно высокой точкой плавления. Следовательно, понятно, что фракция полипропилена (РР-1) и/или композиция полипропилена (РР-С) имеет температуру плавления (Тm), измеренную согласно ISO 11357-3, по меньшей мере 151,0°С, более предпочтительно по меньшей мере 152°С. Следовательно, в частности понятно, что температура плавления (Тm), измеренная согласно ISO 11357-3, фракции полипропилена (РР-1) и/или композиции полипропилена (РР-С) составляет в пределах от 151 до 160°С, более предпочтительно в пределах от 151 до 155°С, еще более предпочтительно в пределах от 151 до 159°С и еще более предпочтительно в пределах от 151 до 155°С.
Дополнительно, фракция полипропилена (РР-1) и/или композиция полипропилена (РР-С) имеет достаточно высокую температуру кристаллизации (Тс). Следовательно, предпочтительно, чтобы фракция полипропилена (РР-1) и/или композиция полипропилена (РР-С) имела температуру кристаллизации (Тс), измеренную согласно ISO 11357-3, по меньшей мере 110°С, более предпочтительно по меньшей мере 111°С. Соответственно, фракция полипропилена (РР-1) и/или композиция полипропилена (РР-С) предпочтительно имеет температуру кристаллизации (Тс), измеренную согласно ISO 11357-3, в пределах от 110 до 120°С, более предпочтительно в пределах от 111 до 117°С.
Технология ступенчатого изотермического расслоения (stepwise isothermal segregation technique) (SIST) обеспечивает возможность определения распределения толщины ламеллы. Метод точного измерения приведен далее в Примерах (в частности, определение толщины ламеллы каждой фракции и ее энтальпии плавления). Таким образом, довольно большое количество (с довольно высокой энтальпией плавления [Дж/г]) полимерных фракций, кристаллизующихся при высоких температурах, указывает на довольно высокое количество толстых ламелл. Следовательно, понятно, что композиция полипропилена (РР-С) включает по меньшей мере 18,0 масс.%, более предпочтительно по меньшей мере 18,3 масс.%, еще более предпочтительно по меньшей мере 18,6 масс.%, еще более предпочтительно по меньшей мере 19,0 масс.% кристаллической фракции, имеющей толщину ламелл более чем 14,09 нм, где указанную фракцию определяют при использовании технологии ступенчатого изотермического расслоения (SIST). В частности, предпочтительно, чтобы композиция полипропилена (РР-С) включала в пределах от 18,0 до 50,0 масс.%, более предпочтительно в пределах от 18,0 до 45,0 масс.%, еще более предпочтительно в пределах от 18,3 до 40,0 масс.%, еще более предпочтительно в пределах от 19,0 до 35,0 масс.% кристаллической фракции, имеющей толщину ламелл более чем 14,09 нм, где указанную фракцию определяют при использовании технологии ступенчатого изотермического расслоения (SIST).
Дополнительно, понятно, что композиция полипропилена (РР-С) включает не более чем 67,0 масс.%, более предпочтительно не более чем 66,0 масс.%, такое как не более чем 65,0 масс.% кристаллической фракции, имеющей толщину ламелл в пределах от 7,70 до 14,09 нм. С другой стороны, кристаллической фракции, имеющей толщину ламелл в пределах от 7,70 до 14,09 нм, не должно быть слишком мало. Следовательно, дополнительно или в качестве альтернативы, предпочтительно, чтобы верхний предел композиции полипропилена (РР-С) включал более чем 45 масс.%, более предпочтительно более чем 50 масс.%, еще более предпочтительно более чем 55 масс.%, еще более предпочтительно более чем 60 масс.% кристаллической фракции, имеющей толщину ламелл в пределах от 7,70 до 14,09 нм. Следовательно, понятно, что композиция полипропилена (РР-С), полученная способом по настоящему изобретению, включает кристаллическую фракцию, имеющую толщину ламелл в пределах от 7,70 до 14,09 нм, в количестве в пределах от 45,0 до 67,0 масс.%, более предпочтительно в пределах от 55,0 до 67,0 масс.%, еще более предпочтительно в пределах от 60,0 до 66,0 масс.%, еще более предпочтительно в пределах от 61,0 до 65,00 масс.%, такое как в пределах от 63,0 до 65,0 масс.%.
Дополнительно, желательно, чтобы композиция полипропилена (РР-С) включала более чем 12,0 масс.%, более предпочтительно более чем 14,0 масс.%, такое как более чем 15,0 масс.% кристаллической фракции, имеющей толщину ламелл в пределах от 2,52 до 7,69 нм. Следовательно, понятно, что композиция полипропилена (РР-С), полученная способом по настоящему изобретению во втором реакторе (R-2), включает кристаллическую фракцию, имеющую толщину ламелл в пределах от 2,52 до 7,69 нм, в количестве в пределах от 12,0 до 22,0 масс.%, более предпочтительно в пределах от 14,0 до 20,0 масс.%, такое как в пределах от 15,0 до 19,0 масс.%.
Распределение молекулярной массы (MWD) представляет отношение числа молекул в полимере к индивидуальной длине цепи. Распределение молекулярной массы (MWD) выражается как соотношение среднемассовой молекулярной массы (Mw) и среднечисловой молекулярной массы (Мn). Среднечисловая молекулярная масса (Мn) представляет среднюю молекулярную массу полимера, выраженную как число молекул в статический момент графика в каждом пределе молекулярной массы по сравнению с молекулярной массой. По сути это общая молекулярная масса всех молекул, деленная на количество молекул. В свою очередь среднемассовая молекулярная масса (Mw) это статический момент графика массы полимера в каждом пределе молекулярной массы по сравнению с молекулярной массой.
Среднечисловая молекулярная масса (Мn) и среднемассовая молекулярная масса (Mw) наряду с распределением молекулярной массы (MWD) определяют при использовании эксклюзионной хроматографии размеров (SEC) при использовании прибора Waters Alliance GPCV 2000 с он-лайн вискозиметром. Температура термостата 140°С. В качестве растворителя используют трихлорбензол (ISO 16014).
Соответственно, предпочтительно, чтобы первая фракция полипропилена (РР-1) с MFR2 (230°С) с менее 1,5 г/10 минут имела среднемассовую молекулярную массу (Mw) более 300000 г/моль, более предпочтительно более 350000, еще более предпочтительно в пределах от 350000 до 1200000 г/моль, более предпочтительно в пределах от 400000 до 1000000 г/моль. С другой стороны, первая фракция полипропилена (РР-1) с MFR2 (230°С) более чем 2,0 г/10 минут предпочтительно имеет среднемассовую молекулярную массу (Mw) менее 300000 г/моль, более предпочтительно менее 280000, еще более предпочтительно в пределах от 150000 до менее 300000 г/моль, более предпочтительно в пределах от 180000 до 280000 г/моль.
Композиция полипропилена (РР-С) предпочтительно характеризуется среднемассовой молекулярной массой (Mw) менее 400000 г/моль, более предпочтительно менее 350000, еще более предпочтительно в пределах от 150000 до 400000 г/моль, более предпочтительно в пределах от 180000 до 320000 г/моль.
Среднечисловая молекулярная масса (Мn) композиции полипропилена (РР-С) предпочтительно составляет в пределах от 5000 до 400000 г/моль, более предпочтительно в пределах от 10000 до 300000 г/моль.
Широкое распределение молекулярной массы (MWD) повышает технологические характеристики полипропилена. Следовательно, понятно, что распределение молекулярной массы (MWD), измеренное согласно ISO 16014, композиции полипропилена (РР-С) составляет по меньшей мере 2,8, более предпочтительно по меньшей мере 3,0, такое как 3,3. С другой стороны, широкое распределение молекулярной массы (MWD) указывает на довольно высокое количество фракций с низкой молекулярной массой, которые вносят свой вклад в содержание фракции, растворимой в холодном ксилоле, без улучшения каких-либо диэлектрических характеристик. Таким образом, в качестве альтернативы, в одном варианте воплощения настоящего изобретения распределение молекулярной массы (MWD) композиции полипропилена (РР-С) предпочтительно составляет в пределах от 2,8 до 8,0, более предпочтительно в пределах от 3,0 до 5,0, такое как в пределах от 3,0 до 3,4, еще более предпочтительно в пределах от 3,3 до 3,5.
Соответственно, решающим аспектом способа по настоящему изобретению является получение композиции пропилена (РР-С) в первых двух реакторах. Композиция полипропилена (РР-С) характеризуется низким содержанием остатков и хорошими механическими свойствами. Соответственно, композиция полипропилена (РР-С) предпочтительно представляет конечный продукт способа по настоящему изобретению. Однако композиция полипропилена (РР-С) может быть дополнительно перемещена в дополнительный третий реактор (R-3) и необязательно в четвертый реактор (R-4). Третий реактор (R-3) и четвертый реактор (R-4) предпочтительно представляют газофазные реакторы (GPR). В одном варианте воплощения настоящего изобретения в этих реакторах может быть получен эластомерный сополимер пропилена, который диспергирован в композиции полипропилена (РР-С). Следовательно, способ по настоящему изобретению обеспечивает возможность получения гетерофазного сополимера пропилена, где композиция полипропилена (РР-С) составляет матрицу, а материал полипропилена, полученного в третьем реакторе (R-3) и необязательно в четвертом реакторе (R-4), составляет эластомерную фазу. В качестве альтернативы, в третьем реакторе (R-3) и необязательно в четвертом реакторе (R-4), где оба предпочтительно представляющие газофазные реакторы (GPRs), может быть получен рандом-сополимер пропилена или гомополимеры пропилена, последний по существу предпочтителен. Следовательно, в одном варианте воплощения способ по настоящему изобретению включает три реактора, суспензионный реактор (SR), такой как циркуляционный реактор (LR), и два газофазных реактора, в которых получают гомополимеры.
Условия (температура, давление, время реакции, подача мономера) в каждом реакторе зависят от заданного продукта и известны специалисту в области техники, к которой относится настоящее изобретение. Как указанно выше, первый реактор (R-1) предпочтительно представляет суспензионный реактор (SR), такой как циркуляционный реактор (LR), в то время как второй реактор (R-2) предпочтительно представляет газофазный реактор (GPR-1). Последующие реакторы, если присутствуют, также представляют газофазные реакторы (GPR).
Предпочтительный многостадийный способ представляет способ «циркуляционно-газофазный», такой как предложенный Borealis A/S, Denmark (известный как BORSTAR® technology), описанный, например, в патентной литературе, такой как ЕР 0 887 379 или в WO 92/12182.
Мультимодальные полимеры могут быть получены согласно нескольким способам, описанным, например, в WO 92/12182, ЕР 0 887 379 и WO 98/58976. Содержание этих документов введено здесь ссылкой.
Предпочтительно в способе получения полипропиленового полимера по настоящему изобретению, как было указанно выше, условия для первого реактора (R-1), то есть суспензионного реактора (SR), такого как циркуляционный реактор (LR), на стадии (А) могут быть следующими:
- температура составляет в пределах от 40°С до 110°С, предпочтительно составляет в пределах от 60°С до 100°С, в пределах от 70°С до 90°С,
- давление составляет в пределах от 20 бар до 80 бар, предпочтительно в пределах от 40 бар до 70 бар,
- для контроля молярной массы может быть добавлен водород при использовании известного способа.
Затем реакционная смесь со стадии (А) перемещается во второй реактор (R-2), то есть газофазный реактор (GPR-1), то есть на стадию (С), при этом условия стадии (С) предпочтительно следующие:
- температура составляет в пределах от 50°С до 130°С, предпочтительно в пределах от 60°С до 100°С,
- давление составляет в пределах от 5 бар до 50 бар, предпочтительно в пределах от 15 бар до 40 бар,
- для контроля молярной массы может быть добавлен водород при использовании известного способа.
Время выдержки может варьировать в обеих зонах реактора. В одном варианте воплощения способа получения полипропилена по настоящему изобретению время выдержки в реакторе полимеризации в массе, например в циркуляционном, составляет в пределах от 0,2 до 4 часов, например, в пределах от 0,3 до 1,5 часов, а время выдержки в газофазном реакторе, как правило, составляет в пределах от 0,2 до 6,0 часов, такое как в пределах от 0,5 до 4,0 часов.
Если требуется, полимеризация может быть проведена известным способом при сверхкритических условиях в первом реакторе (R-1), то есть суспензионном реакторе (SR), таком как циркуляционный реактор (LR), и/или конденсацией в газофазном реакторе (GPR-1).
Условия в других газофазных реакторах (GPR), если присутствуют, аналогичны таковым во втором реакторе (R-2).
В частности, хорошие результаты достигаются при использовании способа по настоящему изобретению в случае, когда специфическую предварительную полимеризацию проводят перед полимеризацией в первом реакторе (R-1). Предварительная полимеризация может быть проведена в первом реакторе (R-1), однако предпочтительно проводить предварительную полимеризацию в отдельном реакторе, так называемом реакторе предварительной полимеризации.
Как указанно выше, при использовании способа по настоящему изобретению получают композицию полипропилена (РР-С). Эта композиция пропилена (РР-С) очень подходит для пленочных кондесаторов в виду низкого содержания остатков.
Дополнительно, следует понимать, что температура предварительной полимеризации значительно ниже, то есть составляет в пределах от 10 до 50°С, более предпочтительно в пределах от 12 до 45°С, такая как в пределах от 15 до 42°С.
Давление составляет в пределах от 40 до 80 бар, предпочтительно в пределах от 45 до 75 бар, такое как в пределах от 48 до 70 бар. Время выдержки может варьировать в пределах от 0,1 до 1,0 часов, такое как в пределах от 0,2 до 0,6 часов. При получении для конденсаторов весь водород может быть подан на стадии предварительной полимеризации.
Как указано выше, дополнительным важным аспектом в способе по настоящему изобретению является использование твердой каталитической системы (SCS). Соответственно, настоящее изобретение требует твердой каталитической системы (SCS) с пористостью, измеренной согласно ASTM 4641, менее чем 1,40 мл/г и/или площадью поверхности, измеренной согласно ASTM D 3663, менее чем 25 м2/г, указанная твердая каталитическая система (SCS) включает:
(а) катализатор, представляющий цирконийорганическое соединение с формулой (I):
где
X - лиганды, соединенные σ- связью с цирконием (Zr),
L1 - идентичные остатки, выбранные из группы, состоящей из линейного C1 -С20 алкила, разветвленного С3 - С20 алкила, линейного С1 - С20 алкенила, разветвленного С4 - С20 алкенила, С4 - С12 циклоалкила, C1 - С20 алкила, замещенного С5 - С20 циклоалкилом, С6 - С20 арила и С5 - С20 циклоалкила, замещенного С1 - С20 алкилом, где циклоалкильный остаток замещен C1 - С20 алкилом,
L2 - идентичные остатки, выбранные из группы, состоящей из линейного С1 - С20 алкила, разветвленного С3 - С20 алкила, линейного С1 - С20 алкенила, разветвленного С4 - С20 алкенила, С4 - С12 циклоалкила, C1 - С20 алкила, замещенного С5 - С20 циклоалкилом, и С6 - С20 арила,
Y - С, Si или Ge, предпочтительно Si,
R1 - C1 - С20 алкил, С4 - С10 циклоалкил, С6-С12 арил, С7-С12 арилалкил или триметилсилил,
R2 - C1 - С20 алкил, С4 - С10 циклоалкил, С6-С12 арил, С7-С12 арилалкил или триметилсилил
и
(b) сокатализатор (Со), включающий элемент (Е) из группы 13 периодической таблицы (IUРАС).
Предпочтительно остатки «R1» и «R2» химически различны.
Дополнительно, предпочтительно, чтобы твердая каталитическая система (SCS) имела площадь поверхности менее чем 15 м2/г, более предпочтительно менее чем 10 м2/г и наиболее предпочтительно менее чем 5 м2/г, что является наименьшим, измеряемым пределом. Площадь поверхности в настоящем изобретении измеряют согласно ASTM D 3663 (N2).
Дополнительно, понятно, что твердая каталитическая система (SCS) имеет пористость менее чем 1,30 мл/г и более предпочтительно менее чем 1,00 мл/г. Пористость измеряют согласно ASTM 4641 (N2). В другом предпочтительном варианте воплощения настоящего изобретения пористость не определима при использовании метода, применяемого согласно ASTM 4641 (N2).
В конкретном предпочтительном варианте воплощения настоящего изобретения пористость не определяется при использовании метода согласно ASTM 4641 (N2) и имеет площадь поверхности, измеренную согласно ASTM D 3663 (N2), менее чем 5 м2/г (то есть ниже определяемого предела).
Дополнительно, твердая каталитическая система (SCS), как правило, имеет средний размер частиц более чем 500 µм, то есть предпочтительно в пределах от 2 до 500 µм, более предпочтительно в пределах от 5 до 200 µм. Предпочтительно, чтобы средний размер частиц составлял менее 80 µм, еще более предпочтительно менее 70 µм, предпочтительные пределы среднего размера частиц составляют от 5 до 70 µм или даже от 10 до 60 µм.
Используемый в описании настоящей патентной заявки термин ″δ-лиганд″ имеет общепринятое значение, то есть, группа, связанная с металлом в одном или более положении сигма-связью. Следовательно, лиганды «X» могут быть независимо выбраны из группы, состоящей из водорода, галогена, C1 - С20 алкила, C1 - С20 алкокси, С2 - С20 алкенила, С2 - С20 алкинила, С3 - С12 циклоалкила, С6 - С20 арила, С6 - С20 арилокси, С7 -С20 арилалкила, С7 - С20 арилалкенила, -SR″, -PR″3, -SiR″3, -OSiR″3 и -NR″2,
где каждый R″ представляет независимо водород, C1 - С20 алкил, С2 - С20 алкенил, С2 - С20 алкинил, С3 - С12 циклоалкил или С6 - С20 арил. В предпочтительных вариантах воплощения настоящего изобретения лиганды «X» идентичны и представляют галоген, такой как Сl, или бензил.
В предпочтительных вариантах воплощения настоящего изобретения лиганды «X» представляют таковые, приведенные в пункте 2 формулы изобретения.
Предпочтительный моновалентный анионный лиганд представляет галоген, в частности хлор (Сl).
Остатки «L1» и «L2» должны быть одинаковыми на обоих остатках инденила.
Предпочтительно остатки «L1» выбирают из группы, состоящей из линейного С1 - С20 алкила, разветвленного С3 - С20 алкила, С4 - С12 циклоалкила, C1 - С20 алкила, замещенного С5 - С20 циклоалкилом, и С5 - С20 циклоалкила, замещенного C1 - С20 алкилом, где остаток циклоалкила замещен C1 - С20 алкилом. Более предпочтительно остатки «L1» выбирают из группы, состоящей из метила, этила, n-пропила, изопропила, n-бутила, трет-бутила, циклогексила, метил-(метил-циклогексила).
Предпочтительно остатки «L2» выбирают из группы, состоящей из линейного C1 - С20 алкила, разветвленного С3 - С20 алкила, С4 - С12 циклоалкила, C1 - С20 алкила, замещенного С5 - С20 циклоалкилом, и С6 - С20 арила. Более предпочтительно остатки «L2» выбирают из группы, состоящей из метила, этила, n-пропила, изопропила, n-бутила, трет-бутила, циклогексила и фенила. Наиболее предпочтительно остатки «L2» представляют линейный C1 - С20 алкил или разветвленный С3 - С20 алкил, такой как - С(СН3)3.
Для связывающей группы «-Y(R1)(R2)-» формулы (I)
предпочтительно
Y представляет Si и
R1 и R2 независимо друг от друга выбирают из группы, состоящей из C1 - С10 алкила, С4 - С10 циклоалкила и С6-С12 арила.
Еще более предпочтительно
Y представляет Si и
R1 и R2 независимо друг от друга выбирают из группы, состоящей из C1 - С10 алкила, С4 - С10 циклоалкила и С6-С12 арила,
при условии, что R1 и R2 химически различны.
В предпочтительном варианте воплощения настоящего изобретения используют циркойниорганическое соединение, как приведено в пункте 3 формулы изобретения.
Соответственно, в конкретном варианте воплощения настоящего изобретения катализатор представляет рац-метил(циклогексил)силанедил бис(2-метил-4-(4-трет-бутилфенил)инденил)цирконий дихлорид.
Более предпочтительно катализатор по настоящему изобретению не наносят на какой-либо внешний неорганический или органический носитель, такой как кремний, алюминий или пористый полимерный материал-носитель.
Учитывая дополнительное требование, твердая каталитическая система (SCS) по настоящему изобретению должна включать сокатализатор (Со), включающий элемент (Е) группы 13 периодической таблицы (IUРАС), например, сокатализатор (Со) включает соединение алюминия Аl.
Примерами таких сокатализаторов (Со) являются циркойниорганические соединения, такие как соединения триалкилалюминия и/или соединения алюмоксана или их смеси.
Такие соединения Аl, предпочтительно алюмоксаны, могут быть использованы только в качестве соединений в сокатализаторе (Со) или вместе с другим сокаталитическим(ими) соединением(ами). Таким образом, помимо или дополнительно к соединениям Аl, то есть алюмоксанам, может быть использован другой катионный комплекс, образующий сокаталитические соединения, такие как соединения бора. Указанные сокатализаторы коммерчески доступны или могут быть получены согласно предшествующему уровню техники. Однако предпочтительно использовать в качестве сокатализатора (Со) при получении твердой каталитической системы только соединения Аl.
Предпочтительными сокатализаторами (Со) являются алюмоксаны, в частности C1 - С10-алкилалюмоксаны, наиболее предпочтительно метилалюмоксаны (МАО).
Очень хорошие результаты достигаются в случае, когда молярное соотношение элемента (Е) сокатализатора (Со) и циркония, предпочтительно молярное соотношение Аl сокатализатора (Со) и циркония, находятся в определенном соотношении. Соответственно, понятно, что молярное соотношение элемента (Е) сокатализатора (Со) и циркония [E/Zr], предпочтительно молярное соотношение Аl сокатализатора (Со) (таких как алюмоксаны, такие как метилалюмоксаны (МАО)) и циркония [Al/Zr], составляет в пределах от 100 до 800 моль/моль, более предпочтительно в пределах от 150 до 600 моль/моль, еще более предпочтительно в пределах от 200 до 400 моль/моль, такое как в пределах от 200 до 350 моль/моль.
Предпочтительно циркойниорганическое соединение формулы (I) и сокатализатор (Со) твердой каталитической системы (SCS) составляет по меньшей мере 70 масс.%, более предпочтительно по меньшей мере 80 масс.%, еще более предпочтительно по меньшей мере 90 масс.% и еще более предпочтительно по меньшей мере 95 масс.% твердой каталитической системы. Следовательно, понятно, что твердая каталитическая система характеризуется тем, что является самодостаточной, то есть не включает какой-либо каталитически инертный материал-носитель, такой как, например, кремний, алюминий или MgCl2 или пористый полимерный материал, которые в иных случаях традиционно используют в гетерогенных каталитических системах, то есть катализатор не наносят на внешний носитель или материал-носитель. Это является следствием того, что твердая каталитическая система (SCS) является самодостаточной и имеет достаточно низкую площадь поверхности.
Твердую металлоценовую каталитическую систему (SCS) предпочтительно получают при использовании технологии отверждения эмульсии, основные принципы которой описаны в WO 03/051934. Документ введен здесь ссылкой в полном объеме.
Следовательно, твердая каталитическая система (SCS) предпочтительно имеет форму твердых каталитических частиц, полученных способом, включающим стадии:
a) получение раствора одного или более каталитического компонента;
b) диспергирование указанного раствора во втором растворителе с получением эмульсии, в которой указанный один или более каталитический компонент присутствует в виде капель диспергированной фазы,
c) отверждение указанной диспергированной фазы с превращением указанных капель в твердые частицы и необязательно с извлечением указанных частиц с получением указанного катализатора.
Предпочтительно для получения раствора используют первый растворитель, более предпочтительно первый органический растворитель. Еще более предпочтительно органический растворитель выбирают из группы, состоящей из линейного алкена, циклического алкена, ароматического углеводорода и галогенсодержащего углеводорода.
Дополнительно, второй растворитель, образующий непрерывную фазу, представляет растворитель, инертный в отношении каталитических компонентов. Второй растворитель может не смешиваться с раствором каталитических компонентов по меньшей мере при условиях (таких как температура) стадии диспергирования. Используемый в описании настоящей патентной заявки термин «не смешиваемый с раствором катализатора» означает, что второй растворитель (непрерывная фаза) полностью не смешивается или частично не смешивается, то есть неполностью смешивается с диспергированной фазой раствора.
Предпочтительно не смешиваемый растворитель включает фторированный органический растворитель и/или их функционализированные производные, более предпочтительно не смешивающийся растворитель включает полу-, сильно- или перфторированный углерод и/или их функционализированное производное. Наиболее предпочтительно указанный не смешивающийся растворитель включает перфторуглерод или его функционализированное производное, предпочтительно С3-С30 перфторалканы, -алкены или -циклоалканы, еще более предпочтительно С4-С10 перфторалканы, -алкены или -циклоалкены, по существу предпочтительно перфторгексан, перфторгептан, перфтороктан или перфтор(метилциклогексан) или перфтор (1,3- диметилциклогексан) или их смесь.
Дополнительно, предпочтительно, чтобы эмульсия, включающая указанную непрерывную фазу и указанную диспергированную фазу, представляла би- или мультифазную систему, известную из предшествующего уровня техники. Для образования и стабилизации эмульсии может быть использован эмульгатор. После образования эмульсионной системы в указанном растворе in situ из компонентов катализатора образуется указанный катализатор.
В принципе эмульгирующий агент может представлять любой подходящий агент, участвующий в образовании и/или стабилизации эмульсии и не оказывающий какого-либо негативного воздействия на каталитическую активность катализатора. Эмульгирующий агент может представлять, например, поверхностно-активное вещество на основе углеводородов, необязательно разомкнутых гетероатомом(ами), предпочтительно галогенированные углеводороды, необязательно имеющие функциональную группу, предпочтительно полу-, сильно- или перфторированный углерод и/или их функционализированное производное, известные из предшествующего уровня техники. В качестве альтернативы, эмульгирующий агент может быть получен в процессе получения эмульсии, например, за счет прохождения реакции предшественника поверхностно-активного вещества с соединением раствора катализатора. Указанный предшественник поверхностно-активного вещества может представлять галогенированный углеводород по меньшей мере с одной функциональной группой, например, сильно фторированный C1-n (подходят С4-30- или С5-15) спирт (например, сильно фторированный гептанол, октанол или нонанол), оксид (например, пропеноксид) или эфир акрилата, который реагирует, например, с сокаталитичеким компонентом, таким как алюмоксан, с получением «фактически» поверхностно-активного вещества.
В принципе для получения из диспергированных капель твердых частиц может быть использован любой способ отверждения. В одном предпочтительном варианте воплощения настоящего изобретения отверждение проводят изменением температуры. Эмульсию подвергают постепенному изменению температуры вплоть до 10°С/минуту, предпочтительно в пределах от 0,5 до 6°С/минуту и более предпочтительно в пределах от 1 до 5°С/минуту. Еще более предпочтительно эмульсию подвергают постепенному изменению температуры на более чем 40°С, предпочтительно на более чем 50°С за менее чем 10 секунд, предпочтительно за менее чем 6 секунд.
Предпочтительно извлеченные частицы имеют средний размер в пределах от 5 до 200 µм, более предпочтительно в пределах от 10 до 100 µм.
Дополнительно, предпочтительно отвержденные частицы имеют сферическую форму, заранее определенное распределение размера частиц и площадь поверхности, как указанно выше, предпочтительно менее чем 25 м2/г, более предпочтительно менее чем 20 м2/г, еще более предпочтительно менее чем 15 м2/г, еще более предпочтительно менее чем 10 м2/г и наиболее предпочтительно менее чем 5 м2/г, где указанные частицы получают способом, указанным выше.
Более детальные варианты воплощения и примеры системы с непрерывной и диспергированной фазой, способа получения эмульсии, эмульгирующего агента и способов отверждения приведены, например, в приведенной ссылкой в настоящей патентной заявке международной патентной заявке WO 03/051934.
Все или часть стадий получения может быть проведена непрерывно. В приведенной ссылке WO 2006/069733 описываются принципы таких непрерывных или полунепрерывных способов получения твердого катализатора, полученного способом эмульсия/отверждение.
Указанные выше компоненты катализатора получены согласно способам, описанным в WO 01/48034.
Способ по настоящему изобретению позволяет получить легко осуществимыми средствами в реакторе композиции полипропилена (РР-С), как указанно выше. Эта композиция полипропилена (РР-С) очень подходит для кондесаторов. Соответственно, пленочный конденсатор может быть получен при использовании традиционных вытяжных способов получения, известных из предшествующего уровня техники, при использовании композиции полипропилена (РР-С). Способ получения пленочного конденсатора по настоящему изобретению включает применение композиции полипропилена (РР-С), описанной в настоящей патентной заявке, и получение ее предпочтительно в виде пленки при использовании способа с применением рамы для вытяжки и ориентирования пленки, известного из предшествующего уровня техники.
Способ с применением рамы для вытяжки и ориентирования пленки представляет, в частности, способ, в котором композиция полипропилена (РР-С) расплавлена и экструдируется через щель, такую как Т-образная экструзионная головка, на охлаждающий барабан с получением не вытянутого листа. Указанный лист подвергают предварительному нагреванию при использовании, например, нагретого металлического вала и затем вытягивают в продольном направлении между множеством валов с различной окружной скоростью и затем обе кромки листа захватываются захватами, вытягивают лист в поперечном направлении в термошкафу при использовании рамы для вытягивания и ориентирования пленки с получением в результате двуосноориентированной пленки. Температуру указанного прошедшего вытяжку листа во время вытяжки в продольном направлении предпочтительно контролируют, таким образом, чтобы она составляла в пределах точки плавления полипропилена, как указанно в описании настоящей патентной заявки (- 15 или + 5°С). Равномерность толщины пленки при вытяжке в поперечном направлении оценивают при использовании способа, в котором область фиксации пленки после вытяжки в продольном направлении маскируют и измеряют фактор фактической вытяжки путем измерения размера указанной маскировки после вытяжки в поперечном направлении.
Затем пленку обрабатывают коронным разрядом в воздухе, азоте, газообразном диоксиде углерода или любой их смеси для последующей металлизации поверхности, для улучшения силы адгезии с наносимым металлом и наматывают пленку устройством для намотки.
Полученную пленку помещают в устройство для вакуумной металлизации и на пленку предпочтительно наносят масляный узор для образования изоляционной выемки для заданных целей при использовании устройства для нанесения масляных узоров при помощи специальных аппликационных сеток и тому подобного. Затем металл, подходящий для заданных целей, наносят для достижения заранее заданного слоя сопротивления. Дополнительно, если требуется, металлизацию проводят при использовании предохранительной гребнеообразной пластины для непрерывного изменения показателя сопротивления в поперечном направлении пленки. Металлизированную пленку разрезают с получением двух лент в качестве пары для получения конденсатора. Затем ленты наматывают с получением устройства и устройству придают плоскую форму при использовании термопресса, затем на концы напыляют металл, прикрепляя выводы, пропитывают изоляционным маслом и компонуют с получением конденсатора.
Далее настоящее изобретение будет описано со ссылкой на следующие Примеры.
ПРИМЕРЫ
А. Методы измерения
Для приведенного выше описания настоящего изобретения, если ясно не указанно иное, наряду с приведенными ниже Примерами применяют следующие определения терминов и методы определения.
Количественный анализ микроструктуры при использовании ЯМР спектроскопии.
Количественную спектроскопию ядерно-магнитного резонанса (ЯМР) используют для оценки изотактичности, региорегулярности и содержания сомономера в полимерах.
Количественный анализ13С{1Н} ЯМР спектра записывают в состоянии раствора при использовании ЯМР спектрометра Bruker Advance III 400, работающего на частотах в пределах от 400,15 до 100,62 МГц для1Н и13С, соответственно. Весь спектр записывают при использовании13С оптимизированного 10-мм датчика измерения линейных величин при расширенном диапазоне температур при 125°С при использовании во всей пневматике газообразного азота.
Для гомополимеров полипропилена около 200 мг материла растворяют в 1,2-тетрахлорэтане-d2 (TCE-d2). Для обеспечения однородности раствора после получения начального образца в термоблоке ампулу для ЯМР-спектроскопии дополнительно нагревают в печи с круглым вращающимся подом в течение по меньшей мере 1 часа. При установке в магнит ампулу подвергают воздействию 10 Гц. Такая схема была выбрана в первую очередь ввиду необходимости высокого разрешения количественного анализа регулярности распределения молекулярной структуры (Busico, V., Cipullo, R., Prog. Polym. Sci. 26 (2001) 443; Busico, V.; Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromoleucles 30 (1997) 6251). Создают стандартное одноимпульсное возбуждение при использовании NOE и двухуровневой WALTZ 16 схемы развязки (Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, В., J. Mag. Reson. 187 (2007) 225; Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol. Rapid Commun. 2007, 28,11289). Всего для спектра потребовалось 8192 (8k) импульсов.
Для сополимеров этиленпропилена около 200 мг материала растворяют в 3 мл 1,2-тетрахлорэтана-d2 (TCE-d2) с хром-(III)-ацетилацетонатом (Сr(асас)3) с получением в результате 65 мМ раствора релаксационного агента в растворителе (Singh, G., Kothari, А., Gupta, V., Polymer Testing 28 5 (2009), 475). Для обеспечения однородности раствора после получения начального образца в термоблоке ампулу для ЯМР-спектроскопии дополнительно нагревают в печи с круглым вращающимся подом в течение по меньшей мере 1 часа. При установке в магнит ампулу подвергают воздействию 10 Гц. Такая схема была выбрана в первую очередь ввиду необходимости высокого разрешения количественного анализа для точного количественного определения содержания этилена. Создают стандартное одноимпульсное возбуждение без использования NOE при оптимизированном угле наклона с 1-секундной задержкой повтора цикла и двухуровневой WALTZ16 схемой развязки (Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, В., J. Mag. Reson. 187 (2007) 225; Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macromol. Rapid Commun. 2007, 28, 11289). Всего для спектра потребовалось 6144 (6k) импульсов.
Проводят количественный анализ на основе13С{1Н} ЯМР спектра с определенным средним значением и определяют соответствующие количественные значения при использовании интеграла с использованием специальных компьютерных программ.
Для сополимеров этиленпропилена все химические сдвиги косвенно указывают на центральную метиленовую группу этиленового блока (ЕЕЕ) при 30,00 частей на миллион при использовании химического сдвига в растворителе. Этот подход позволяет провести сравнение с эталоном даже при отсутствии структурной единицы.
Для гомополимеров полипропилена все химические сдвиги внутренне привязаны к метиловой изотактической пентаде (mmmm) при 21,85 частей на миллион.
Характерные сигналы, соответствующие региодефектам или сомономеру, приведены (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000,100, 1253;; Wang, W-J., Zhu, S., Macromolecules 33 (2000), 1157; Cheng, H. N., Macromolecules 17 (1984), 1950).
Регулярность распределения молекулярной структуры количественно определяют через интеграцию метальной области в пределах 23,6-19,7 частей на миллион с поправкой для любых участков, не связанных с интересующей стереопоследовательностью (Busico, V., Cipullo, R., Prog. Polym. Sci. 26 (2001) 443; Busico, V., Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macromoleucles 30 (1997) 6251).
В частности, воздействие региодефектов и сомономера на количественный анализ регулярности распределения молекулярной структуры корректируют, вычитая интегралы репрезентативного региодефекта и сомономера из конкретной области интеграла стереопоследовательностей.
Изотактичность определяют по уровню пентад и указывают как процент последовательностей изотактических пентад (mmmm) от последовательностей всех пентад:
[mmmm]%=100*(mmmm/сумма всех пентад)
На присутствие 2,1 эритрорегиодефектов указывают два метальных участка 17,7 и 17,2 частей на миллион, что подтверждается другими характерными участками.
Не приведены характерные сигналы, соответствующие другим типам региодефектов (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253).
Количественный анализ 2,1 эритрорегиодефектов проводят при использовании среднего интеграла двух характерных метиловых участков в 17,7 и 17,2 частей на миллион:
Р21е=(Ie6+Iе8)/2
Количественный анализ 1,2 первично вставленного пропена проводят исходя из метиловой области с поправкой на не принятые во внимание участки, включенные в эту область, не связанные с первичной вставкой, и участки первичной вставки, исключенные из этой области:
P12=ICH3+P12e
Общее количество пропена количественно оценивают как сумму первично вставленного пропена и всех других присутствующих региодефектов:
Робщее=Р12+Р21е
Молярный процент 2,1 эритриорегиодефектов количественно оценивают от общего содержания пропена:
[21е] мол.%=100*(Р21е/Робщее)
Для сополимеров приведены характерные сигналы, соответствующие вставке этилена (Cheng, Н. N.. Macromolecules 17 (1984), 1950).
Также сообщается, что (Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000,100,1253; Wang, W-J., Zhu, S., Macromolecules 33 (2000), 1157; Cheng, H. N.. Macromolecules 17 (1984), 1950) для региодефектов требуется поправка за счет воздействия таких дефектов на содержание сомономера.
Молярную фракцию этилена в полимере количественно оценивают при использовании способа Wang et. Al. (Wang, W-J., Zhu, S., Macromolecules 33 (2000), 1157) путем интеграции множества сигналов всей спектральной области13С{1Н} спектра, полученного при заданных условиях. Этот способ был выбран за его точность, надежность и возможность объяснить присутствие региодефектов при необходимости. Интегральные области незначительно регулируют для повышения применяемости к широким пределам содержания сомономеров.
Молярный процент сомономера, введенного в полимер, рассчитывают по молярной фракции согласно:
Е [мол.%]=100*fE
Массовый процент сомономера, введенного в полимер, рассчитывают по молярной фракции согласно:
Е [масс.%]=100*(fE*28,05)/((fE*28,05)+((1-fE)*42,08))
Распределение последовательности сомономера в триадах определяют при использовании метода Kakugo et al. (Kakugo, M., Naito, Y., Mizunuma, K., Miyatake, T. Macromolecules 15 (1982) 1150) путем интеграции множества сигналов всей спектральной области13С{1Н} спектра, полученного при заданных условиях. Этот способ был выбран за его надежность. Интегральные области незначительно регулируют для повышения применяемости к широким пределам содержания сомономеров.
Молярный процент указанной последовательности сомономера в триадах полимера рассчитывают по молярной фракции, определенной методом Kakugo et at. (Kakugo, M., Naito, Y., Mizunuma, K., Miyatake, T. Macromolecules 15 (1982) 1150) согласно:
XXX [мол.%]=100*fXXX
Молярную фракцию сомономера, встроенного в полимер, определенную по распределению последовательности сомономера в триадах, рассчитывают по распределению триад при использовании известных необходимых уравнений (Randall, J. Macromol. Sci., Rev. Macromol. Chem. Phys. 1989, C29,201):
fXEX=fEEE+fPEE+fPEP
fXPX=fPPP+fEPP+fEPE,
где PEE и EPP представляет сумму обратимых последовательностей РЕЕ/ЕЕР и ЕРР/РРЕ соответственно.
Случайность распределения сомономера количественно оценивают как относительное количество изолированных последовательностей этилена по сравнению со всем введенным этиленом. Случайность рассчитывают по распределению последовательностей в триадах при использовании уравнения:
R(E)[%]=100*(fPEP/fXEX)
Наблюдаются характерные сигналы, соответствующие введению 1-гексена, и содержание 1-гексена рассчитывают как молярный процент 1-гексена в полимере Н(мол.%), согласно:
[Н]=Htot/(Ptot+Htot),
где
Htot=I(αB4)/2+I(ααB4)×2,
где I(αВ4) представляет интеграл αВ4 участков при 44,1 частей на миллион, который определяет изолированный 1-гексен, введенный в РРНРР последовательности, и I(ααВ4) представляет интеграл ααВ4 участков при 41,6 частей на миллион, который определяет последовательно введенный 1-гексен в РРННРР последовательности.
Ptot=интеграл всех СН3 площадей метиловой области с поправкой, применяемой при недооценке других пропеновых единиц, не учтенных в этой области, и переоценке из-за других участков, обнаруженных в этой области.
и Н(мол.%)=100×[Н],
которые затем переводят в масс.% при использовании корреляции
Н(масс.%)=(100×Нмол.%×84,16)/(Нмол.%×84,16+(100-Нмол.%)×42,08)
Статистическое распределение рассчитывают исходя из соотношения содержанием гексена, присутствующего в изолированной (РРНРР) и консекутивной (РРННРР) последовательностях введенного сомономера А:
[НН]<[Н]2
Mw, Мn, MWD.
Mw/Mn/MWD измеряют при использовании гельпроникающей хроматографии (GPC) согласно следующему методу:
Среднемассовую молекулярную массу (Mw), среднечисловую молекулярную массу (Мn) и распределение молекулярной массы (MWD=Mw/Mn) измеряют при использовании способа, основанного на ISO 16014-1:2003 и ISO 16014-4:2003. Используют устройство Waters Alliance GPCV 2000 с рефрактометрическим детектором и он-лайн вискозиметром при использовании колонок 3×TSK-gel (GMHXL-HT) от TosoHaas и 1,2,4-трихлорбензола (ТСВ, стабилизированный 200 мг/л 2,6-ди третбутил-4-метил-фенолом) в качестве растворителя при температуре 145°С и постоянной скорости потока 1 мл/минуту. Для анализа инжектируют 216,5 µл образца раствора. Колонку калибруют при использовании относительной калибровки по узким 19 МWD стандартам полистирола (PS) в пределах от 0,5 кг/моль до 11,500 кг/моль и хорошо изученным широким стандартам полипропилена. Все образцы получают, растворяя в пределах от 5 до 10 мг полимера в 10 мл (при 160°С) стабилизированного ТСВ (такой же, как мобильная фаза) и выдерживают в течение 3 часов с непрерывным перемешиванием перед забором образцов в устройство для GPC.
Средние молекулярные массы, распределение молекулярной массы, разветвления g′ (Mn, Mw, MWD, g′) определяют при использовании SEC/VISC-LS.
Средние молекулярные массы (Mw, Mn), распределение молекулярной массы (MWD) и их пределы, описанные через индекс полидисперстности, PDI=Mw/Mn (где Mn представляет среднечисловую молекулярную массу, a Mw представляет среднемассовую молекулярную массу) определяют при использовании гельпроникающей хроматографии (GPC) согласно ISO 16014-4 2003. A PL 220 (Polymer Laboratories) GPC с рефракционным индексом (RI), с он-лайн четырех капиллярным вискозиметром (PL-BV 400-НТ) и детектором двойного светорассеяния (PL-LS 15/90 детектор светорассеяния) под углом 15° и 90°. В качестве неподвижной фазы используют колонки 3× Olexis и 1× Olexis Guard от Polymer Laboratories, а в качестве подвижной фазы 1,2,4-трихлорбензол (ТСВ, стабилизированный 250 мг/л 2,6-ди третбутил-4-метил-фенолом) в качестве растворителя при температуре 160°С и постоянной скорости потока 1 мл/минуту. Для анализа инжектируют 200 µл образца раствора. Соответствующие константы детектора наряду с объемами задержки детектора определяют при использовании узкого стандарта PS (MWD = 1,01) с молярной массой 132900 г/моль и вязкостью 0,4789 дл/г.
Соотвествующее dn/dc для используемого станадарта PS в ТСВ составляет 0,053 см3/г.
Молярную массу каждого элюирования определяют при использовании рассеянного света, используя комбинацию двух углов 15° и 90°. Все данные обрабатывают и проводят расчет при использовании программного обеспечения Cirrus Multi-Offline SEC-Software Version 3.2 (Polymer Laboratories a Varian inc. Company). Молекулярную массу рассчитывают при использовании опции в Cirrus software «use combination of LS angles» в поле «sample calculation options subfield slice MW data from».
Обработка данных детально описана в G. Saunders, P. A. G: Cormack, S. Graham; D. C. Sherrington, Macromolecules, 2005, 38, 6418-6422. Mwi каждого элюирования определяют под углом 90° при использовании следующего уравнения:
Коэффициент Релея R(θ)90° под углом 90° измеряют при использовании LS детектора, и R представляет ответ RI-детектора. Функцию рассеяния частиц Р(θ) определяют при использовании обоих углов (15° и 90°), как описано в С. Jackson and Н. G. Barth(C. Jackson and H. G. Barth, «Molecular Weight Sensitive Detectors» in Handbook of Size Exclusion Chromatography and related techniques, C.-S. Wu, 2nd ed., Marcel Dekker, New York, 2004, стр.103). Для низко- и высокомолекулярной области, в которой слабее сигнал LS детектора или RI детектора соответственно, используют линейный подход для корреляции соотношения объема элюирования к соответствующей молекулярной массе.
Соотношение dn/dc, используемое в уравнении, рассчитывают по константе RI детектора, концентрации с образца и площади пика показаний детектора анализируемого образца.
Относительное количество разветвления определяют при использовании индекса g′ образца разветвленного полимера. Индекс разветвления длинной цепи (LCB) определяют как g′=[η]br/[η]lin. Известно, что если показатель g′ повышается, содержание разветвлений снижается. [η] представляет истинную вязкость при температуре 160°С в трихлорбензоле образца полимера при определенной молекулярной массе, и измеряют при использовании он-лайн вискозиметра и детектора концентрации. Истинную вязкость измеряют, как описано в справочнике Cirrus Multi-Offline SEC-Software Version 3.2 при использовании уравнения Solomon-Gatesman.
Необходимую концентрацию каждого элюирования определяют при использовании RI детектора.
[η]lin представляет истинную вязкость линейного образца и [η]br представляет вязкость разветвленного образца при одной и той же молекулярной массе и химической композиции. Среднечисловое g′n и среднемассовое g′w определяют как следующее:
где аi представляет dW/dlogM фракции I, Аi представляет совокупный dW/dlogM полимера вплоть до фракции i. Показатель [η]]lin линейного контроля (линейный изотактический РР) в зависимости от молекулярной массы измеряют при использовании он-лайн детектора вязкости. Следующие показатели К и α получают (К = 30,68*10-3 и α = 0,681) по линейному контролю с молекулярной массой в пределах logM=4,5-6,1.
Показатель [η]lin молекулярной массы для расчетов g' рассчитывают по следующему уравнению [η]lin,I=К*Мiα. [η]br,I, измеряя для каждого конкретного образца при использовании он-лайн детектора вязкости и детектора концентрации.
Скорость течения расплава (MFR)
Скорость течения расплава измеряют при нагрузке 2,16 кг (MFR2) при 230°С. Скорость течения расплава представляет то количество полимера в граммах, которое устройство, стандартизованное для тестирования согласно ISO 1133, экструдирует в течение 10 минут при температуре 230°С при нагрузке 2,16 кг.
Фракция, растворимая в холодном ксилоле (XCS масс.%)
Фракцию, растворимую в холодном ксилоле (XCS), определяют при температуре 23°С согласно ISO 6427.
Содержание геля считается идентичным фракции, нерастворимой в горячем ксилоле (XHI), которое определяют экстрагированием 1 г тонко нарезанного полимера 350 мл ксилола в аппарате Сокслета в течение 48 часов при температуре кипения. Оставшуюся твердую фракцию сушат при температуре 90°С и взвешивают для определения нерастворимой фракции.
Температуру плавления Тm, температуру кристаллизации Тс определяют при использовании калориметра Mettler ТА820 с проведением дифференциальной сканирующей калориметрии (DSC) 5-10 мг образцов. Обе кривые и кристаллизации, и плавления получают при показателе 10°С/минуту на сканограммах нагревания и охлаждения в пределах от 30°С до 225°С. Температуры кристаллизации и плавления берут как пики эндотерм и экзотерм.
Также при использовании способа DSC согласно ISO 11357-3 измеряют энтальпию плавления и кристаллизации (Нm и Нс).
Элементарный анализ
Ниже приведен элементарный анализ, используемый для определения содержания элементарных остатков, которые главным образом остаются от катализатора, в частности остатки Аl, В и Si в полимере. Указанные остатки Аl, В и Si могут быть в любой форме, например элементарной или ионной форме, и могут быть удалены и определены в полипропилене при использовании описанного ниже ICP способа. Способ также используют для определения содержания в полимере Ti. Понятно, что могут быть использованы другие известные методы, приводящие к получению аналогичных результатов.
ICP спектрометрия (индуктивно соединенная плазменная эмиссионная спектрометрия)
Устройство ICP: Устройство для определения содержания Аl, В и Si представляет ICP Optima 2000 DV, PSN 620785 (поставщик Perkin Elmer Instruments, Belgium) с программным обеспечением.
Пределы определения 0,10 частей на миллион (Аl), 0,10 частей на миллион (В), 0,10 частей на миллион (Si).
Образец полимера сначала сжигают при использовании известного способа, затем растворяют в подходящем кислотном растворителе. Разведения стандартов для калибровочной кривой растворяют в том же растворителе, что и образец, и концентрации выбирают таким образом, что концентрация образца совпадает со стандартом калибровочной кривой.
Частей на миллион по массе:
Содержание золы: содержание золы измеряют согласно стандарту ISO 3451-1 (1997).
Рассчитанная зола, содержание Al, Si и В:
Зола и указанные выше элементы, Аl и/или Si, и/или В также могут быть рассчитаны в полипропилене исходя из полимеризационной активности приведенного в качестве примера катализатора. Эти показатели дают верхний предел присутствия указанных остатков катализатора.
Следовательно, оценку содержания остатков катализатора проводят на основе композиции катализатора и эффективности полимеризации, остатков катализатора в полимере:
Общие остатки катализатора [частей на миллион] = 1/эффективность [кгpp/gкатализатора] × 100
Все остатки [частей на миллион] = wAl, катализатор [%] × общие остататки катализатора [частей на миллион] / 100
Остатки Zr [частей на миллион] = wzr, катализатор [%] × общие остатки катализатора [частей на миллион] / 100
(Аналогичные расчеты также используют для остатков В, Сl и Si.)
Содержание остатков хлора: Содержание остатков Сl в образцах измеряют при использовании известного способа, такого как рентгенофлуоресцентная спектрометрия (XRF). Используют рентгенофлуоресцентный спектрометр Philips PW2400, PSN 620487, (Поставщик: Philips, Belgium) software X47. Пределы определения С1 составляют 1 часть на миллион.
Сила электрического пробоя (ЕВ63%)
Соответствует стандарту IEC 60243-1, Second edition (1998-01).
Метод описывает способ измерения силы электрического пробоя для изоляционных материалов пластин компрессионного прессования.
Определение:
Eb:Eb=Ub/d
Сила электрического поля в тестируемом образце, в котором происходит пробой. В однородных пластинах и пленках это соответствует силе электрического пробоя, разделенной на толщину пластины/пленки (d), единица: кВ/мм. Для каждой БОПП (ВОРР) пленки проводят 10 индивидуальных измерений пробоя. Результаты 10 индивидуальных измерений пробоя БОПП пленки оценивают при использовании графика распределения Вейбулла, где 63 процентиля соответствует силе пробоя (Еb63%). Параметр β представляет наклон кривой линейной регрессии через эти 10 точек (смотрите, также CEI 727-2; First Edition (1993-02)).
Силу электрического пробоя определяют при 50 Гц в высоковольтной камере при использовании металлических стержней в качестве электродов, как указано в IEC 60243-1, Second edition (1998-01) (4.1.2). Напряжение на пленке/пластине повышают со скоростью 2 кВ/с до пробоя.
Пористость: BET с газообразным N2, ASTM 4641, устройство Micromeritics Tristar 3000; подготовка образцов: при температуре 50°С, 6 часов под вакуумом.
Площадь поверхности: BET с газообразным N2 ASTM D 3663, устройство Micromeritics Tristar 3000: подготовку образца проводят при температуре 50°С, 6 часов под вакуумом.
Средний размер частиц (d50) измеряют при использовании Coulter Counter LS200 при комнатной температуре в среде n-гептана.
Технология ступенчатого изотермического расслоения (SIST)
Для SIST анализа проводят изотермическую кристаллизацию в плавителе Mettler ТА820 DSC при использовании 3±0,5 мг образцов при понижающейся температуре в пределах от 200°С до 105°С.
(i) образцы расплавляют при температуре 225°С в течение 5 минут,
(ii) затем охлаждают при 80°С/минуту до 145°С,
(iii) выдерживают в течение 2 часов при температуре 145°С,
(iv) затем охлаждают при 80°С/минуту до 135°С,
(v) выдерживают в течение 2 часов при температуре 135°С,
(vi) затем охлаждают при 80°С/минуту до 125°С,
(vii) выдерживают в течение 2 часов при температуре 125°С,
(viii) затем охлаждают при 80°С/минуту до 115°С,
(ix) выдерживают в течение 2 часов при температуре 115°С,
(x) затем охлаждают при 80°С/минуту до 105°С,
(xi) выдерживают в течение 2 часов при температуре 105°С.
После последней стадии образец охлаждают при 80°С/минуту до -10°С и получают кривую плавления, нагревая охлажденный образец при 10°С/минуту вплоть до 200°С. Все измерения проводят в атмосфере азота. Энтальпию плавления записывают как функцию температуры и оценивают, измеряя энтальпию плавления фракций при плавлении в интервалах температуры
от 50 до 60°С; от 60 до 70°С; от 70 до 80°С; от 80 до 90°С; от 90 до 100°С; от 100 до 110°С; от 110 до 120°С; от 120 до 130°С; от 130 до 140°С; от 140 до 150°С; от 150 до 160°С; от 160 до 170°С; от 170 до 180°С; от 180 до 190°С; от 190 до 200°С.
Таким образом, кривая плавления кристаллизованного материала может быть использована для расчета распределения толщины ламеллы согласно уравнению Thomson-Gibbs (Ур. 1.).
где Т0=457К, ΔН0=134×106 Дж/м3, σ=0,049.6 Дж/м2 и L представляет толщину ламеллы.
Модуль упругости при растяжении измеряют согласно ISO 527-2 (скорость крейцкопфа = 50 мм/минуту; 23°С) при использовании полученных литьем под давлением образцов, как описано в EN ISO 1873-2 (форма кости для собак, толщина 4 мм).
В. Примеры
Используемый катализатор получают по Примеру 5 WO 03/051934, где используемый в нем катализатор был заменен на рац-метил(циклогексил)силанедил бис(2-метил-4-(4-трет-бутилфенил)инденил)циркония дихлорид. Рац-метил(циклогексил)силанедил бис(2-метил-4-(4-трет-бутилфенил)инденил)циркония дихлорид получают согласно WO 2005 105863 А2, Примеры 17-18.
Получение катализатора
В стеклоэмалированном из нержавеющей стали реакторе 90 дм3 с рубашкой получают комплексный раствор при температуре -5°С, добавляя 0,85 кг 24,5 масс.% раствора ((2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,9-гептадекафторнонил)оксиран)/толуол очень медленно (3,4 мл/минуту) в 13,5 кг 30 масс.% раствора МАО(метилалюмоксан)/толуол.
Температуру повышают до 25°С и перемешивают раствор в течение 60 минут. После добавления 210 г комплекса раствор перемешивают в течение дополнительных двух часов. Эту смесь перекачивают при скорости 5 л/час в паре ротор-статор 4М. В роторе-статоре с окружной скоростью 4 м/с смесь перемешивают со скоростью потока 32 л/час с гексадекафтор-1,3-диметилциклогексаном с получением, таким образом, эмульсии. Капли в эмульсии отверждают избыточным потоком 450 л/час гексадекафтор-1,3-диметилциклогексана при температуре 76°С в тефлоновом шланге. Шланг соединен с 160 дм3 реактором с рубашкой из нержавеющей стали, снабженным спиральным смешивающим элементом. В этом реакторе частицы катализатора отделяют от гексадекафтор-1,3-диметилциклогексана за счет разности плотности. После использования комплексного раствора частицы катализатора сушат в 160 дм3 реакторе при температуре 70°С в потоке азота 5 кг/час в течение 7 часов.
Ниже приведено определение пределов пористости и площади поверхности.
Используемые в настоящем изобретении полимеры (СЕ1, E1, Е2, за исключением СЕ2 и СЕ3) получают при использовании указанного выше катализатора непрерывным двухстадийным способом полимеризации, состоящим из процесса полимеризации в массе (циркуляционный реактор) и стадии газофазной полимеризации. Перед подачей на стадию полимеризации в массе катализатор проходит предварительную полимеризацию в циркуляционном реакторе предварительной полимеризации. Уровни водорода в реакторе предварительной полимеризации, в циркуляционном реакторе и газофазном реакторе отличаются, соответственно, и отрегулированы таким образом, чтобы молекулярная масса (MFR) полимера с первой стадии полимеризации отличалась от молекулярной массы (MFR) полимера со второй стадии полимеризации. Масса фракции продукта циркуляционного реактора (разделение) может варьировать в пределах от 30 до 70 масс.%. Температура в реакторе предварительной полимеризации составляет 35°С, в циркуляционном реакторе 70°С (СЕ1, Е1) или 75°С (Е2), а в газофазных реакторах 85°С: давление при полимеризации в циркуляционном реакторе составляет 53 бар, а в газофазном реакторе 30 бар (СЕ1, Е1) или 25 бар (Е2). Разделение и другие параметры процесса, используемые для получения СЕ1, Е1 и Е2, приведены в Таблице 1.
* LG(1/MFR1)*wf1+LG(1/MFR2)*wf-2=LG(1/MFRобщая),
где
MFR1 представляет MFR2 полипропилена, полученного в циркуляционном реакторе,
MFR2 представляет рассчитанную MFR2 полипропилена, полученного в 1GPR, и
MFRoбщая представляет MFR2, измеренную для композиции полипропилена, полученной в 1GPR,
** LG(1/MFR1)*wf1+LG(1/MFR2)*wf-2=LG(1/MFRобщая),
где
MFR1 представляет MFR2 композиции полипропилена, полученной в 1GPR,
MFR2 представляет рассчитанную MFR2 полипропилена, полученного в 2GPR, и
MFRoбщая представляет MFR2, измеренную для композиции полипропилена, полученной в 2GPR.
СЕ1, Е1 и Е2 получают в порошкообразном виде и подвергают гранулированию и стабилизации с Irganox 1010 (4500 частей на миллион).
Сравнительный пример 2 (СЕ 2) представляет коммерческий продукт HB311BF от Borealis AG.
Сравнительный пример 3 (СЕ 3) представляет коммерческий продукт HC300BF от Borealis AG.
СЕ1, СЕ2, СЕ3, E1 и Е2 подвергают последующей обработке для получения БОПП пленок: материалы экструдируют и поливают на охлаждающий вал с получением листов закаленной пленки. Использованные параметры приведены в Таблице 2.
От каждой наливной пленки берут образцы размером 8,5 на 8,5 см, отрезая от центральной части пленки. Эти образцы подвергают двухосной ориентации при использовании лабораторной БОПП установки. Квадратные образцы фиксируют на раме Karo IV для вытяжки и ориентирования пленки пятью захватами с каждой стороны. При использовании рамы для вытяжки и ориентирования пленки поливную пленку растягивают с фактором пять для каждой стороны, таким образом соотношение вытяжки 5×5. После растяжки при этих параметрах БОПП пленку удаляют с рамы для вытяжки и ориентирования пленки и подвергают тестированию на разрыв/пробой, как указанно выше. При использовании описанного выше процесса наливную пленку подвергают двухосной ориентации и проводят тестированиекаждого материала, включая пробой. Указанные конечные результаты представляют среднее двух измерений.
Реферат
Изобретение относится к многостадийному способу получения полипропилена путем полимеризации. Способ включает использование по меньшей мере двух последовательно соединенных реакторов. На стадии (A) в первом реакторе получают первую фракцию полипропилена. На стадии (B) перемещают первую фракцию полипропилена во второй реактор. На стадии (C) проводят полимеризацию во втором реакторе второй фракции полипропилена в присутствии первой фракции полипропилена с получением композиции полипропилена. Первая фракция полипропилена имеет скорость течения расплава MFR(230°С) не более чем 1,5 г/10 минут или более чем 2,0 г/10 минут, включает пропиленовые единицы и необязательно по меньшей мере один С- Сα-олефин, отличающийся от пропилена. Композиция полипропилена имеет скорость течения расплава MFR(230°С) более чем 2,0 г/10 минут и скорость течения расплава MFR(230°С) композиции полипропилена отличается от скорости течения расплава MFR(230°С) первой фракции полипропилена. В первом и во втором реакторе полимеризация проходит в присутствии твердой каталитической системы. Каталитическая система имеет пористость менее чем 1,40 мл/г и/или площадь поверхности менее чем 25 м/г и включает катализатор, представляющий цирконийорганическое соединение с формулой (I). Технически результат - получение чистого полипропилена, по существу свободного от нежелательных остатков и характеризующегося превосходной жесткостью и высокой теплостойкостью. 13 з.п. ф-лы, 9 табл., 5 пр.
Формула
(A) получение в первом реакторе (R-1) первой фракции полипропилена (PP-1),
(B) перемещение указанной первой фракции полипропилена (PP-1) во второй реактор (R-2),
(C) полимеризацию в указанном втором реакторе (R-2) в присутствии указанной первой фракции полипропилена (РР-1) второй фракции полипропилена (PP-2) с получением композиции полипропилена (PP-C),
где
(a) указанная первая фракция полипропилена (PP-1)
(i) имеет скорость течения расплава MFR2 (230°C), измеренную согласно ISO 1133,
(α) не более чем 1,5 г/10 минут,
или
(β) более чем 2,0 г/10 минут,
и
(ii) включает пропиленовые единицы и необязательно по меньшей мере один C2-C10 α-олефин, отличающийся от пропилена,
(b) композиция полипропилена (PP-C) имеет скорость течения расплава MFR2 (230°C), измеренную согласно ISO 1133, более чем 2,0 г/10 минут,
и
(c) скорость течения расплава MFR2 (230°C) композиции полипропилена (PP-C) отличается от скорости течения расплава MFR2 (230°C) первой фракции полипропилена (PP-1),
где дополнительно
в первом реакторе (R-1) и во втором реакторе (R-2) полимеризация проходит в присутствии твердой каталитической системы (SCS), где указанная твердая каталитическая система (SCS)
(d) имеет пористость менее чем 1,40 мл/г, измеренную согласно ASTM 4641, и/или площадь поверхности менее чем 25 м2/г, измеренную согласно ASTM D 3663,
(e) включает катализатор, представляющий цирконийорганическое соединение с формулой (I):
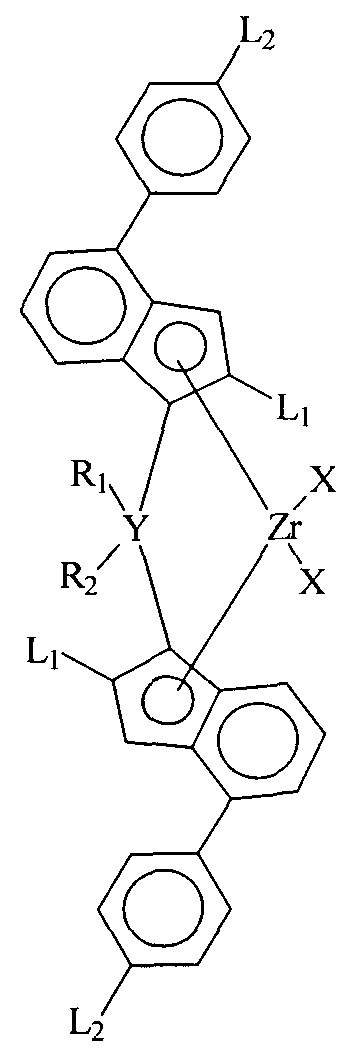
где
X - лиганды, соединенные σ- связью с цирконием (Zr),
L1 - идентичные остатки, выбранные из группы, состоящей из линейного C1-C20 алкила, разветвленного C3-C20 алкила, линейного C1-C20 алкенила, разветвленного C4-C20 алкенила, C4-C12 циклоалкила, C1-C20 алкила, замещенного C5-C20 циклоалкилом, C6-C20 арила и C5-C20 циклоалкила, замещенного C1-C20 алкилом, где циклоалкильный остаток замещен C1-C20 алкилом,
L2 - идентичные остатки, выбранные из группы, состоящей из линейного C1-C20 алкила, разветвленного C3-C20 алкила, линейного C1-C20 алкенила, разветвленного C4-C20 алкенила, C4-C12 циклоалкила, C1-C20 алкила, замещенного C5-C20 циклоалкилом, и C6-C20 арила,
Y - C, Si или Ge, предпочтительно Si,
R1 - C1-C20 алкил, C4-C10 циклоалкил, C6-C12 арил, C7-C12 арилалкил или триметилсилил,
R2 - C1-C20 алкил, C4-C10 циклоалкил, C6-C12 арил, C7-C12 арилалкил или триметилсилил
и
(f) включает сокатализатор (Co), включающий элемент (E) из группы 13 периодической таблицы (IUPAC).
(a) остатки «L2» представляют линейный C1-C20 алкил или разветвленный C3-C20 алкил, такой как -C(CH3)3
и/или
(b) остатки R1 и R2 химически различны
и/или
(c) лиганды «X» могут быть независимо выбраны из группы, состоящей из водорода, галогена, C1-C20 алкила, C1-C20 алкокси, C2-C20 алкенила, C2-C20 алкинила, C3-C12 циклоалкила, C6-C20 арила, C6-C20 арилокси, C7-C20 арилалкила, C7-C20 арилалкенила, -SR″, -PR″3, -SiR″3, -OSiR″3 и -NR″2,
где каждый R″ представляет независимо водород, C1-C20 алкил, C2-C20 алкенил, C2-C20 алкинил, C3-C12 циклоалкил или C6-C20 арил.
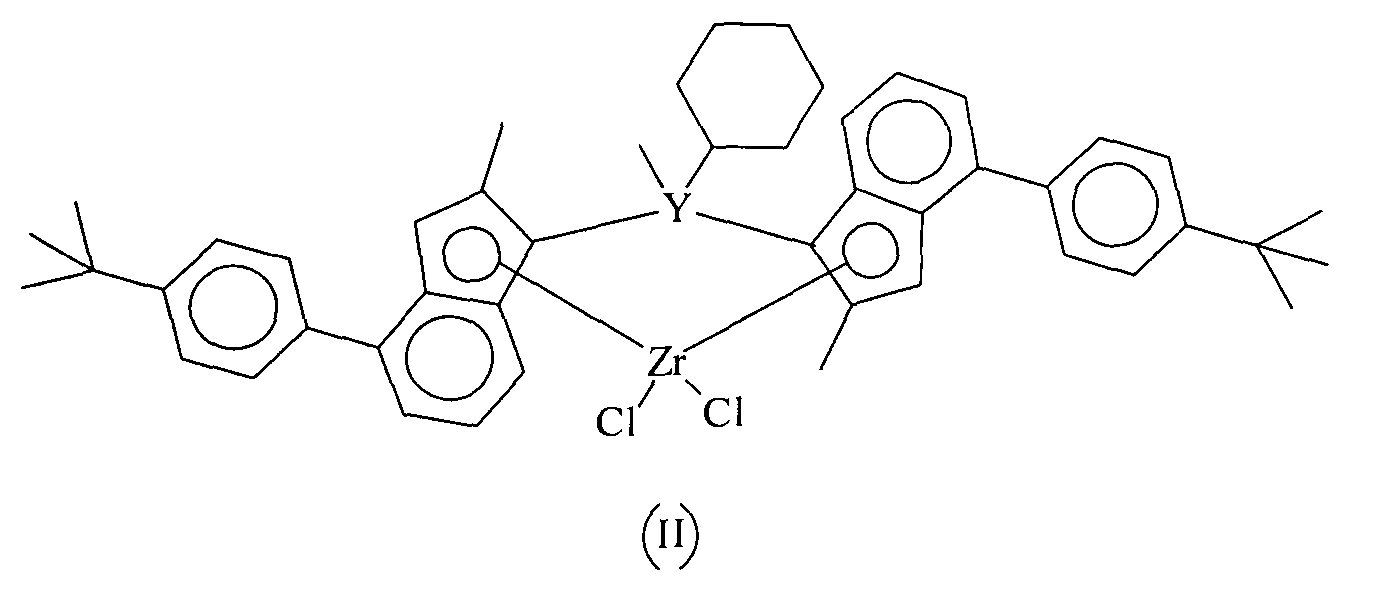
где «Y» представляет C или Si, предпочтительно Si.
(a) первая фракция полипропилена (PP-1) представляет гомополимер пропилена
и/или
(b) композиция полипропилена (PP-C) имеет индекс разветвления g′ по меньшей мере 0,90.
(a) содержание компонентов, растворимых в холодном ксилоле, не более чем 1,5 масс.%,
и/или
(b) содержание <2,1> региодефектов равно или более чем 0,4 мол.%, определенное при использовании13C-спектроскопии, и/или
(c) содержание остатков кремния менее 0,5 частей на миллион.
(a) температуре в пределах от 10 до 50°C
и/или
(b) давлении в пределах от 40 до 80 бар.
Комментарии