Новые марганецсодержащие наноструктуры - RU2638535C2
Код документа: RU2638535C2
Чертежи



Описание
Область настоящего изобретения
Настоящее изобретение относится к хелатообразующим полимерным наноструктурам, включающим парамагнитные ионы марганца(II), а также способам получения указанных наноструктур, а также использованию наноструктур для визуализации или томографирования биологического материала.
Предпосылки настоящего изобретения
Магнитно-резонансная томография, MRI, является способом медицинской визуализации, при которой мягкие ткани тела визуализируются благодаря использованию намагниченности атомных ядер. Существует множество применений в медицинской практике способа, таких как томографирование нервной системы, сосудистой системы и визуализация опухоли.
Как правило, визуализируются обильные водородные ядра молекул воды тела. Сила MRI сигнала зависит от характера ядра, его относительного содержания и его локальной магнитной среды. Эти факторы влияют на продольные (Т1, постоянная времени перемагничивания) и поперечные (Т2, постоянная времени затухания сигнала) интервалы времени релаксации, которые в свою очередь влияют на силу сигнала. Таким образом, источник контраста в MPI представляет собой комбинацию локальной концентрации ядер и их магнитной среды. Различные морфологические характеристики могут быть усилены увеличением Т1 или Т2 контраста. Локальная магнитная среда может быть модифицирована присутствием контрастных веществ и, в зависимости от их магнитных свойств, сигнал может быть увеличен (положительный контраст) или снижен (отрицательный контраст). Положительные контрастные вещества часто предпочтительны, поскольку интерпретация изображений упрощается, когда больше яркости указывает на наличие дополнительного контрастного вещества. Принцип положительных контрастных веществ представляет собой сокращение продольного времени релаксации, Т1, которая описывает, как быстро молекулы воды перемагничиваются после каждого сканирования. При наличии положительного контрастного вещества дополнительный сигнал можно накопить за определенный период времени.
Кроме того, эффект соединения на Т1 задается как релаксивность, таким образом высокая релаксивность дает более мощное усиление сигнала. Релаксивность (r) зависит от частоты в структурно-зависимом пути, что затрудняет сравнение данных литературы из разных источников. Клинические сканеры обычно имеют магнитное поле 1,5 или 3 тесла, таким образом в качестве разумного компромисса релаксивности измерялись при 1,91 Т, что соответствует протонной резонансной частоте 81,3 МГц. Измерения также могут быть выполнены при других частотах, например, 60 МГц. Релаксивность для коммерчески доступного гадолиния на основе контрастных веществ близка к 4/мМ Gd/с при соответствующих с клинической точки зрения областях.
На рынке в настоящее время превалируют водорастворимые хелаты гадолиния. Из-за их малого физического размера (<1 нм) они быстро распределяются во внеклеточном пространстве (кровь плюс интерстициальное пространство между клетками тканей), что несколько ограничивает эффект контраста. Проблемой с использованием in vivo ионов парамагнитных металлов, таких как гадолиний является их токсичность, и хелаты в представленных на рынке в настоящее время контрастных веществах весьма успешно решают эту проблему. Тем не менее, недавно было обнаружено, что хелаты высвобождают небольшие количества гадолиния, что становится проблематичным у пациентов с практически отсутствующей очень слабой функцией почек, у которых был обнаружен серьезный побочный эффект, называемый нефрогенным системным фиброзом, NSF (Grobner et al. Nephrology, Dialysis and Transplantation 2006, 27, 1104; Sieber et al. Invest. Radiol. 2008, 43, 65).
Проблема с NSF приводит к вопросу об использовании другого вещества взамен гадолиния в качестве Т1 - сокращающего контрастного вещества; Mangafodipir представляет собой хелат марганца(II), который использовали в качестве контрастного вещества (Elizondo, G. et al. Radiology, 178, 73, 1991). Он имеет умеренную стабильностью in vivo и большая часть марганца высвобождается в виде ионов после инъекции и накапливается в печени, обеспечивая хороший контраст между здоровой и опухолевой тканью. Высвобождение умеренных количеств марганца не является большой проблемой, так как он является незаменимым микроэлементом для живых организмов и существуют механизмы для утилизации марганца. Mangafodipir сейчас не представлен на рынке из-за низкого объема продаж. Ионы марганца менее магнитноактивные, чем гадолиний и большинство соединений марганца имеют низкие релаксивности, с несколькими характерными примерами (Pan, D. et al. WIREs Nanomed Nanobiotechnol, 3, 162, 2011), демонстрирующими потенциал контрастных веществ на основе марганца. Если бы полезные свойства могли быть включены в более химически доступную, подходящего размера и более биологически инертную структуру, это явилось бы явным преимуществом. В настоящем изобретении раскрыта группа материалов, в которых это достигается. Есть целый ряд контрастных веществ на основе наночастиц, известных в данной области техники. Некоторые из них, на основе оксида железа, были использованы в качестве специфических контрастных веществ печени, но они уже не представлены на рынке из-за низких объемов продаж. Существует огромное количество литературы об экспериментальном использовании этих частиц (например, Bulte, J. W. M. and Modo, М. М. J. Eds. "Nanoparticles in Biomedical Imaging" Springer, 2008). Хотя настоящее изобретение относится к наноразмерным структурам, они не относятся к типу ядро-оболочка, который обычно подразумевают под термином, а скорее основаны на сшитом полимере.
Существует огромное количество литературы о полимерных материалах, несущих хелатирующие группы и ионы парамагнитных металлов для использования в MRI. В целом достигается устойчивое увеличение в релаксивности, хотя и не такое высокое, как описано в настоящем изобретении. Насколько нам известно, ни один из указанных литературных источников не раскрывает полимерные структуры, включающие бисфосфонаты по настоящему изобретению.
Следующие примеры литературы являются примерами соответствующих справочных изданий, которые никоим образом не должны быть истолкованы как входящие в объем настоящего изобретения.
В Rongved P. Carbohydr Res 214, 315 (1991) описывается ряд полимерных материалов с углеводной основой и присоединенными хелатирующими группами. Веществом, ближайшим к настоящему изобретению является гадолиний(III) - фосфат декстрана, который обладает релаксивностью в 16/мМ Gd/c при частоте 20 МГц. Существенно ниже, чем для материалов, описанных в настоящем изобретении. Данное вещество является веществом с неустановленной стабильностью. Фосфор входит в фосфатные группы, связанные также как и фосфатные эфиры, в отличие от фосфонатов по настоящему изобретению. Вещество, таким образом, выходит за рамки настоящего изобретения.
Rongved также раскрывает конъюгат марганец(II)-EDTA - сахароза-эпихлоргидрин с релаксивностью в 19,2/мМ Mn/c и конъюгат марганец(II)-EDTA - аминоэтилдекстран с релаксивностью в 12,8/мM Mn/с. Оба значения существенно ниже, чем у веществ по настоящему изобретению, тем более, что они были измерены при низкой частоте 20 МГц. Вещества выходят за рамки настоящего изобретения.
В WO 2010135167 описан бисфосфонатный материал на основе полистирола. Имеют место попытки создавать не наноструктуры, а скорее сыпучие материалы.
Литература, описывающая использование дендримеров с определенной химической структурой (в отличие от структур на основе полимера по настоящему изобретению) и молекулярной массой, очень обширна.
Наиболее известным примером дендримера, используемого в качестве контрастного вещества MR является Gadomer 17 (Turetschek, K. et al. J. Magn. Reson. Imaging 20, 138 (2004)), который, однако, так и не попал в клиническую практику. Данный подход к наноразмерным структурам страдает от очень дорогостоящего химического синтеза. Отчасти по причине многочисленных химических стадий и отчасти из-за трудности очистки и идентификации многих возможных примесей. Кроме того, релаксивности настолько высокие, как те, которые обнаружены авторами настоящего изобретения, насколько нам известно, никогда не были зарегистрированы в этой области.
Обоснованием хороших результатов от использования высокомолекулярных веществ в качестве контрастных веществ MRI с высокой чувствительностью и селективностью для диагностики опухоли является эффект повышенной проницаемости и удерживания (ЕРР), называемый также пассивной доставкой в область опухоли. Он основан на том, что капилляры здоровых тканей практически непроницаемы для молекул крупнее 3-4 нм, тогда как капилляры быстро растущей опухолевой ткани значительно более проницаемые. Хотя и без всякой определенности и ограничений, можно предположить, что EPR эффект является основой подходящих свойств для визуализации опухоли по настоящему изобретению.
Существует целый ряд фосфонатсодержащих мономерных хелатов гадолиния, известных в данной области техники, в то время как комбинации марганец-фосфор менее распространены, хотя и не безызвестны (Pan, D. et al. WIREs Nanomed Nanobiotechnol 3, 162, 2011). Они не являются в любом случае примечательными своими MRI свойствами и описанные релаксивности являются умеренными. Пример одного из них, включающего бисфосфонат можно найти в Vitha, T. et al. Dalton Transactions p 3204 (2009).
Краткое описание настоящего изобретения
Первый аспект настоящего изобретения относится к наноструктурам, содержащим наноразмерные структуры на основе полимерной структуры или подложки, включающей или обрамленной по меньшей мере пятью геминальными бисфосфонатными группами -P=O(OR1)(OR2)-, которые в контексте настоящего изобретения эквивалентны -R4R3C(P=O(OR1)(OR2))2-, где R1 и R2 независимо выбраны из отрицательного заряда, Н, алкила и арила.
Второй аспект настоящего изобретения относится к наноструктурам, содержащим парамагнитные ионы марганца, включенные в наноразмерные структуры на основе полимерной структуры или подложки, включающей или обрамленной по меньшей мере пятью геминальными бисфосфонатными группами -P=O(OR1)(OR2)-, которые в контексте настоящего изобретения эквивалентны -R4R3C(P=O(OR1)(OR2))2, как упоминалось выше, где R1 и R2 независимо выбраны из отрицательного заряда, Н, алкила и арила.
Хотя обычно подразумевают, что термин "наноразмерные" охватывает все, что меньше 100 нм, в центре настоящего изобретения объекты с сильно разветвленными или перекрестно сшитыми структурами почти сферической формы и среднего размера (гидродинамический диаметр) 1-100 нм или, в некоторых вариантах осуществления, 2-50 нм, 3-10 нм или 3-7 нм.
Второй аспект настоящего изобретения относится к способам получения таких наноструктур, как содержащих ионы марганца, так и не содержащих такие ионы.
Третий аспект настоящего изобретения относится к композициям, таким как фармацевтические композиции, содержащие такие наноструктуры, в частности такие, которые содержат парамагнитные ионы марганца, а также к использованию таких наноструктур, в частности тех, которые содержат парамагнитные ионы марганца в качестве контрастных веществ для клинического применения, в частности использования в качестве контрастных веществ для MRI.
Некоторыми преимуществами наноструктур, раскрытых в данном документе по сравнению с предшествующим уровнем техники являются сочетание релаксивности на порядок более высокой, чем у материалов, присутствующих в настоящее время на рынке, в сочетании с размером, подходящим для избирательного накопления в опухолевой ткани и хорошей биологической переносимости. Это делает наноструктуры по настоящему изобретению, в частности те наноструктуры, которые содержат парамагнитные ионы марганца, подходящими для использования в качестве контрастного вещества для MRI и, в частности, для визуализации опухоли.
Кроме того, использование марганца вместо гадолиния в качестве парамагнитной составляющей обходит проблемы токсичности, связанные с гадолинием.
Использование обильного марганца вместо относительно разреженного гадолиния также имеет положительные экономические эффекты в производстве материала.
Конкретные варианты осуществления.
1. Наноструктура, содержащая ионы марганца, включенные в полимерную структуру, содержащую по меньшей мере пять геминальных бисфосфонатных групп, где геминальные бисфосфонатные группы независимо друг от друга включены как -R3R4C(P=O(OR1)(OR2))2 (которая эквивалентна -R4R3C(P=O(OR1)(OR2))2), где R1 и R2 независимо выбраны из группы, содержащей отрицательный заряд, Н, алкил и арил, и где по меньшей мере одна из R3 и R4 представляет собой группу, связанную с полимерной структурой при условии, что если одна из R3 и R4представляет собой такую связанную группу, остальные R3 и R4представляют собой либо группу, способную связываться с полимерной структурой, либо остаток такой группы, либо выбраны из группы, состоящей из H, OH, OR5 и R5, где R5 представляет собой низший алкил.
2. Наноструктура согласно варианту осуществления 1, где ионы марганца представляют собой ионы марганца(II).
3. Наноструктура согласно варианту осуществления 1 или 2, где R1и R2 независимо выбраны из группы, состоящей из отрицательного заряда, Н, алкила и метила.
4. Наноструктура по любому из вариантов осуществления 1-3, где группа, связанная с полимерной структурой, и/или группа, способная связываться с полимерной структурой или остаток такой группы выбирают из группы, состоящей из:
(CH2)nSi(Rx)2, где Rx независимо представляет собой низший алкил, ОН, O-, или О-, где - обозначает связь с полимерной структурой, и где n равно 1-5,
(CH2)nCORy, где Ry представляет собой О-, NH2, NHRZ, NRZ2 или связь с полимерной структурой, где Rz представляет собой низший алкил и где n равно 1-5 и - обозначает связь с полимерной структурой, и
(СН2)nSO2Ry, где Ry представляет собой O-, NH2, NHRZ, NRZ2 или связь с полимерной структурой, Rz представляет собой низший алкил, n равно 1-5, и - обозначает связь с полимерной структурой.
5. Наноструктура по любому из вариантов осуществления 1-4, которая содержит атомы кремния.
6. Наноструктура по любому из вариантов осуществления 1-5, где R3 и/или R4 выбрана(ы) из группы, состоящей из -(СН2)n-Si(Rx)3, где Rx независимо представляет собой низший алкил, ОН, O-, или O-, где - обозначает связь с полимерной структурой, и где n равно 1-5.
7. Наноструктура по любому из вариантов осуществления 1-6, где гидродинамический диаметр наноструктуры составляет 2-50 нм.
8. Наноструктура по любому из вариантов осуществления 1-7, где гидродинамический диаметр наноструктуры составляет 3-10 нм.
9. Наноструктура по любому из вариантов осуществления 1-8, где гидродинамический диаметр наноструктуры составляет 3-7 нм.
10. Наноструктура по любому из вариантов осуществления 1-7, где гидродинамический диаметр наноструктуры составляет 10-50 нм.
11. Наноструктура по любому из вариантов осуществления 1-7 или 10, где гидродинамический диаметр наноструктуры составляет 10-20 нм.
12. Наноструктура по любому из вариантов осуществления 1-11, где полимерная структура содержит мономерные остатки, содержащие геминальную бисфосфонатную группу и две органо-оксисилановые группы.
13. Наноструктура по любому из вариантов осуществления 1-11, где полимерная структура создана на основе полиэтиленимина.
14. Наноструктура согласно варианту осуществления 13, где молярное отношение P/N составляет 0,1-3.
15. Наноструктура по любому из вариантов осуществления 1-14, где молярное отношение P/Mn составляет 7-20.
16. Наноструктура по любому из вариантов осуществления 5 или 6-12 или 14-15 в случае зависимости от варианта осуществления 5, где молярное отношение Si/Mn составляет 5-20.
17. Наноструктура по любому из вариантов осуществления 5 или 6-16 в случае зависимости от варианта осуществления 5, где молярное отношение Si/P составляет 0,7-1,3.
18. Наноструктура по любому из пп. 1-17, где ионы марганца координируются с фосфонатными группами.
19. Наноструктура по любому из вариантов осуществления 1-18, где указанная наноструктура дополнительно содержит гидрофильные группы, присоединенные к наружным частям.
20. Наноструктура по варианту осуществления 19, где гидрофильные группы содержат -(CH2CH2O)nCH3 функциональные группы, где n=4-50.
21. Наноструктура по любому из вариантов осуществления 1-20, где полимерная структура содержит мономерные остатки с общей структурой
{(X7aO)(X7bO)PO}2-(C){(CH2)nSi(OX7c)(OX7d)(OX7e)}{(CH2)оSi(OX7c)(OX7d)(OX7e)},
где
X7a, X7b, X7c, X7d, X7e независимо выбраны из H, C1-8алкила и бензила; и n и o независимо выбраны из 1-5.
22. Композиция, содержащая наноструктуру по любому из вариантов осуществления 1-21.
23. Фармацевтическая композиция, содержащая наноструктуру по любому из вариантов осуществления 1-21.
24. Использование наночастицы по любому из вариантов осуществления 1-21 или композиции по варианту осуществления 22 или 23 в качестве контрастного вещества MRI.
25. Способ получения наноструктуры по любому из вариантов осуществления 1-21, включающий:
получение наноструктур с полимерной структурой, содержащей геминальные бисфосфонаты и контактирование указанных наноструктур с ионами марганца.
26. Способ по варианту осуществления 25, дополнительно содержащий этап, где наноструктуры очищают посредством ультрафильтрации.
27. Способ получения наноструктуры по любому из вариантов осуществления 13 или 14-21 в случае зависимости от варианта осуществления 13, где геминальный бифосфонат привит к полимерной структуре, полученной из полиэтиленимина, который впоследствии насыщается ионами марганца.
28. Способ получения наночастиц по любому из вариантов 13-21 или 12, в случае зависимости от варианта осуществления 12, где указанные силаны представлены в смеси растворителей, содержащей воду и один или несколько других растворителей, которые смешиваются с водой.
29. Продукт, получаемый способом по любому из вариантов осуществления 25-28.
30. Наноструктура, содержащая полимерную структуру, содержащую по меньшей мере пять геминальных бисфосфонатных групп, где геминальные бисфосфонатные группы независимо друг от друга включены как
-R3R4C(P=O(OR1)(OR2))2 (которые эквивалентны -R4R3C(P=O(OR1)(OR2))2), где R1 и R2 независимо выбраны из группы, содержащей отрицательный заряд, Н, алкил и арил, и где по меньшей мере одна из R3 и R4 представляет собой группу, связанную с полимерной структурой при условии, что если одна из R3 и R4представляет собой такую связанную группу, остальные R3 и R4представляют собой либо группу, способную связываться с полимерной структурой, либо остаток такой группы, либо выбраны из группы, состоящей из Н, ОН, OR5 и R5, где R5 представляет собой низший алкил.
31. Наноструктура по варианту осуществления 30, где R1 и R2независимо выбраны из группы, состоящей из отрицательного заряда, Н, алкила и метила.
32. Наноструктура по варианту осуществления 30 или 31, где группа, связанная с полимерной структурой и/или группа, способная связываться с полимерной структурой или остаток такой группы выбран из группы, состоящей из:
(СН2)nSi(Rx)3, где Rx независимо представляет собой низший алкил, OH, O-, или O-, где - обозначает связь с полимерной структурой, и где n равно 1-5,
(СН2)nCORy, где Ry представляет собой O-, NH2, NHRZ, NRZ2 или связь с полимерной структурой, Rz представляет собой низший алкил, n равно 1-5, и - обозначает связь с полимерной структурой, и
(СН2)nSO2Ry, где Ry представляет собой O-, NH2, NHRZ, NRZ2, или связь с полимерной структурой, Rz представляет собой низший алкил. и n равно 1-5, а - обозначает связь с полимерной структурой.
33. Наноструктура по любому из вариантов осуществления 30-32, которая содержит атомы кремния.
34. Наноструктура по любому из вариантов осуществления 30-33, где R3 и/или R4 выбраны/выбрана из группы, состоящей из -(CH2)n-Si(Rx)3, где Rx независимо представляет собой низший алкил, ОН, O-, или O-, где - обозначает связь с полимерной структурой, и где n равно 1-5.
35. Наноструктура по любому из вариантов осуществления 30-34, где гидродинамический диаметр наноструктуры составляет 2-50 нм.
36. Наноструктура по любому из вариантов осуществления 30-35, где гидродинамический диаметр наноструктуры составляет 3-10 нм.
37. Наноструктура по любому из вариантов осуществления 30-36, где гидродинамический диаметр наноструктуры составляет 3-7 нм.
38. Наноструктура по любому из вариантов осуществления 30-35, где гидродинамический диаметр наноструктуры составляет 10-50 нм.
39. Наноструктура по любому из вариантов осуществления 30-35 или 38, где гидродинамический диаметр наноструктуры составляет 10-20 нм.
40. Наноструктура по любому из вариантов осуществления 30-39, где полимерная структура содержит мономерные остатки, содержащие геминальную бисфосфонатную группу и две органо-оксисилановые группы.
41. Наноструктура по любому из вариантов осуществления 30-40, где полимерную структуру создают на основе полиэтиленимина.
42. Наноструктура согласно варианту осуществления 41, где молярное отношение P/N составляет 0,1-3.
43. Наноструктура по любому из вариантов осуществления 33 или 34-42 в случае зависимости от варианта осуществления 33, где молярное отношение Si/P составляет 0,7-1,3.
44. Наноструктура по любому из вариантов осуществления 30-43, где указанная наноструктура дополнительно содержит гидрофильные группы, присоединенные к наружным частям.
45. Наноструктура по варианту осуществления 44, где гидрофильные группы содержат компоненты -(CH2CH2O)nCH3, где n=4-50.
46. Наноструктура по любому из вариантов осуществления 30-45, где полимерная структура содержит мономерные остатки с общей структурой
{(X7aO)(X7bO)PO}2-(C){(CH2)nSi(OX7c)(OX7d)(OX7e)}{(CH2)оSi(OX7c)(OX7d)(OX7e)}, где X7a, X7b, X7c, X7d, X7e независимо выбраны из H, C1-8алкила и бензила; а n и o независимо выбраны из 1-5.
Определения терминов
Термин "наноструктура", используемый в данном документе, относится к объекту с общим диаметром от 1-100 нм по сути сферической формы, т.е. исключая хлопья, палочки, трубочки и клочья. Используемый в данном документе, термин не учитывает структуры часто именуемые "наночастицами ядро-оболочка" или просто "наночастицами", которые имеют минеральное или металлическое ядро и органическое покрытие.
Термин "полимерная структура", используемый в данном документе относится к ковалентно связанной группе атомов, образующих либо древовидную структуру с многочисленными ответвлениями либо сетчатую структуру с множественными поперечными сшивками. Полимерные структуры формируются из связывания мономеров и/или олигомеров и/или сшивающих средств посредством ковалентных связей. Типичные мономеры можно найти в учебниках по химии полимеров, таких как J.R. Fried, "Polymer Science and Technology" Prentice Hall 1995. Некоторыми примерами мономеров являются стирол, пропилен, этилен, тетрафторэтилен, трифторэтилен, дифторэтилен, метилакрилат, этилакрилат, гидроксиэтилакрилат, акриламид, метилметакрилат, этилметакрилат, гидроксиэтилметакрилат, H2N-(CH2)n-COOH, где n равно 1-10, 3-аминобензойная кислота, 4-аминобензойная кислота, N-винилпиролидон и предшественники кремния наподобие (CH3COO)2Si(CH3)2. Некоторыми примерами полимеров являются полимеры, полученные из соответствующих пар мономеров, таких как терефталевая кислота + 1,4 диамино бензол, терефталевая кислота + этиленгликоль, и HCOO-(CH2)nCOOH+H2N-(CH2)m-NH2, где n и m независимо равны 1-10. Олигомеры с 2-10 мономерными соединенными звеньями могут быть использованы в качестве предшественников. Некоторыми примерами олигомеров, отличных от связанных групп указанных выше мономеров являются циклические или полициклические силаны, такие как гексаметилциклотрисилоксан, 2,4,6,8-тетраметилциклотетрасилоксан и декаметилциклопентасилоксан. Типичные сшивающие средства могут быть найдены в учебниках химии полимеров, таких как J.R. Fried, "Polymer Science and Technology" Prentice Hall 1995. Некоторыми примерами сшивающих средств являются N,N'-метиленбис(акриламид), эпихлоргидрин, дивинилбензол, 1,3-дивинилтетраметилдисилоксан, 1,3-фениленедиисоцианат, 3,3' диангидрид бифенилтетракарбоновой кислоты, 1,4-бутандиол дивиниловый эфир, тетраэтоксисилан, олигосиликаты, такие как метасиликат, или силсесквиоксаны, органосиланы, такие как бис(триэтоксисилильный)метан, бис(триэтоксисилил)этан, бис(триэтоксисилил)пропан, бис(триэтоксисилил)бутан, метилтриэтоксисилан, этилтриэтоксисилан и пропилтриэтоксисилан.
Данная полимерная структура представляет собой скелет наноструктуры. Специалист понимает, что случайный характер процесса полимеризации приводит к тому, что материалы являются смесью множества подобных, но не идентичных ответвляющихся образцов, позиции сшивки и молекулярной массы.
Термин "геминальная бисфосфонатная группа" относится к двум фосфонатным группам, разделенным одним атомом углерода, то есть фосфонатные группы связаны с одним и тем же углеродом. Соединения, содержащие такую геминальную бисфосфонатную группу, часто называют 1,1-бисфосфонатами (или 1,1-дифосфонатами). Фосфонатные группы в геминальной бисфосфонатной группе могут быть замещены. В некоторых вариантах осуществления каждая из фосфонатных групп имеет формулу -P=O(OR1)(OR2), где R1 и R2 независимо выбраны из группы, содержащей отрицательный заряд, Н, алкил и арил.
Термин "группа, связанная с полимерной структурой" относится к химической группе, где ковалентная связь формально замещает атом водорода в полимерной структуре. Химические группы, подпадающие под это определение, как правило, представляют собой короткие неразветвленные остатки углеводородов, простых эфиров, амидов или сложных эфиров. Некоторыми типичными примерами являются -(CH2)n-, -(CH2)nCO-, -(СН2)nCOO-, -(СН2)nCONH- и -(CH2)nSi(O-)3. В данном контексте слово "короткий" означает, что n равно 1-8.
Термин "группа, образующая часть полимерной структуры" относится к ситуации, когда две фосфонатные группы располагаются на одном и том же атоме углерода полимерной структуры.
Термин "группа, способная связываться с полимерной структурой" относится к предшественникам вышеуказанных групп, рассматриваемым как связанные с полимерной структурой. Некоторыми примерами являются -(СН2)nOH, -(СН2)nBr, -(СН2)nCOCl-, -(СН2)nCOCH3, -(СН2)nCOCH2CH3, -(СН2)nCOO-, -(СН2)nCONH2 и (СН2)nSi(OEt)3.
Термин "биологически инертный", используемый в данном документе, относится к материалу, который является биологически совместимым, то есть безвредным для живого организма и в то же время устойчивым к деструкции in vivo.
Термин "DLS", используемый в данном документе является аббревиатурой для динамического рассеяния света, способа определения размера частиц, а также может упоминаться как фотон-корреляционная спектроскопия или квазиупругое рассеяние света. Размеры DLS, заданные, как указано в тексте и в формуле изобретения, если ничего другого не указано, относятся к положению максимума среднеобъемного пика для образца, измеренного при 25°C в нейтральном водном растворе с ионной силой, соответствующей 150 мМ NaCl.
Термин "сферический", используемый в данном документе, предназначен для описания наноструктур с такой формой, при которой малая ось составляет не более половины главной оси, т.е. самая длинная ось, проходящая через центр (точку веса) структуры не больше двойной длины самой короткой оси, проходящей через ту же точку.
Термин "гидрофильный органический остаток", используемый в данном документе, относится к органическому остатку, который способствуют растворимости в водных растворителях и в настоящем изобретении подразумевается, что они являются биологически инертными, за исключением полипептидов и сложных углеводов. Примерами подходящих гидрофильных органических остатков является любая группа, содержащая углерод с молекулярной композицией (aO+bN)/(cC+dS+eSi+fP)>0,3, где a, b, c, d, e и f представляют собой молекулярный процент кислорода (O), азота (N), углерода (С), серы (S), кремния (Si) и фосфора (Р), соответственно.
Термин "активированный силан", используемый в данном документе, относится к силану следующего типа RnSi(Х)4-n, где X представляет собой алкоксигруппу, арилоксигруппу, атом галогена, диалкиламиногруппу, азотсодержащий гетероцикл или ацилоксигруппу.
Термин "оксисилан", используемый в данном документе относится к любым органическим соединениям с одним или несколькими атомами кислорода, связанными с атомом кремния. Их неограничивающими примерами являются:

Термин "органосилан", используемый в данном документе, относится к органическим соединениям, содержащим одну или несколько углеродно-кремниевых связей.
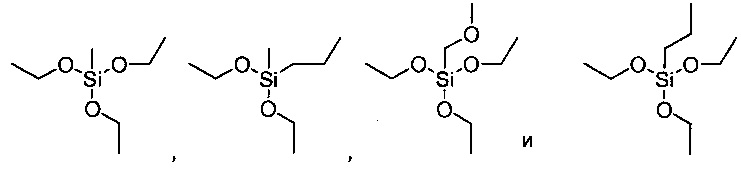
Термин "органо-оксисилан", используемый в данном документе, относится к органическим соединениям, содержащим один или несколько атомов углерода и один или несколько атомов кислорода, присоединенных к атому кремния. Их неограничивающими примерами являются:
Термины "углеводород" и "углеводородная цепь" используются в данном документе для обозначения органического остатка, состоящего из водорода и углерода. Углеводород может быть полностью насыщенным или он может содержать один или несколько ненасыщенных связей. Углеводород может содержать любое количество атомов углерода от 1 до 50.
Термин "алкил", используемый в данном документе, относится к прямой или разветвленной углеводородной цепи, полностью насыщенной (без двойных или тройных связей) углеводородной группой. Алкильная группа может в настоящем тексте иметь от 1 до 15 атомов углерода. Алкильная группа соединений может быть обозначена как "С1-15алкил" или аналогичными обозначениями. Типичные алкильные группы включают, но никоим образом не ограничиваются ими, метил, этил, пропил, изопропил, бутил, изобутил, третичный бутил, пентил, гексил и т.п.
Термин "низший алкил", используемый в данном документе, относится к алкилу, имеющему 1-8 атомов углерода.
Термин "низший спирт", используемый в данном документе, относится к спирту, имеющему 1-8 атомов углерода.
Всякий раз при использовании в данном описании, если не указано иное, числовой диапазон, такой как "от 1 до 8" или "1-8" относится к каждому целому числу в заданном диапазоне; например, "от 1 до 8 атомов углерода" означает, что алкильная группа может состоять из 1 атома углерода, 2 атомов углерода, 3 атомов углерода и т.д., вплоть до 8 атомов углерода. Есть, однако, некоторые исключения, которые являются очевидными для квалифицированных специалистов. В частности, во всех случаях, когда диапазон приводят в данном документе для молярного отношения, такого как молярное отношение P/N или молярное отношение Si/P в наноструктурах, для диаметра или размера, pH, периода времени, концентрации, осмотического давления или температуры, диапазон включает также все десятичные числа, попадающие в указанный диапазон.
Используемый в данном документе, термин "алкокси" относится к формуле -OR, где R представляет собой C1-8алкил, например, метокси, этокси, н-пропокси, 1-метилэтокси (изопропокси), н-бутокси, изобутокси, втор-бутокси, трет-бутокси, амилокси, изоамилокси и т.п. Алкокси может быть необязательно замещен.
Используемый в данном документе термин "арилокси" относится к RO-, в котором R представляет собой арил, где "арил" относится к карбоциклическому (углеродному) кольцу или двум или более конденсированным кольцам (кольцам, которые имеют общие два соседних атома углерода), которые имеют полностью делокализованную пи-электронную систему. Арильное кольцо может быть 4-20-членным кольцом. Примеры арильных групп включают, но не ограничиваются ими, бензол, нафталин и азулен. Арильная группа может быть необязательно замещенной, например, фенокси, нафталенилокси, азуленилокси, антраценилокси, нафталенилтио, фенилтио и т.п. Арилокси могут быть необязательно замещенными.
Используемый в данном документе, термин "ацил" относится к карбонильной группе, т.е. -C(=O)-.
Используемый в данном документе, термин "ацилокси" относится к атому кислорода, связанному посредством карбонильной группы, т.е. -C(=O)-O-.
Используемый в данном документе, термин "гетероцикл" относится к стабильному 3-18-членному кольцу, которое состоит из атомов углерода и одного-пяти гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы. Гетероцикл может быть моноциклическим, бициклическим или трициклическим.
Термин "сильное основание", используемый в данном документе, относится в данном контексте к основаниям, которые сильнее, чем гидроксид и не совместимы с водными средами.
Термин "гидродинамический диаметр", используемый в данном документе, относится к диаметру гипотетической твердой сферы, которая диффундирует с той же скоростью, что и частица. Гидратация и форма входят в поведение сферы. Термин также известен как "диаметр Стокса" или "диаметр Стокса-Эйнштейна".
Термин "конъюгат", используемый в данном документе, относится к молекулярному объекту, который является флюоресцентным маркером, красителем, спин-меткой, радиоактивным маркером, лигандом к биологическому рецептору, хелатом, ингибитором фермента, субстратом фермента, антителом или родственной антителу структурой. См., например, "Bioconjugate Techniques", Greg T. Hermanson second edition, Elsevier 2008, ISBN 978-0-12-370501-3 для справки поданному вопросу.
Термины "обрабатывать для конъюгации" и "точка крепления" оба относятся к бифункциональным молекулам, которые могут связываться с полимерной сеткой или быть включенными в полимерную сетку, но оставляя одну реакционноспособную группу, которая может быть связана с конъюгатом, как определено выше. Типичным, но не исключительным, примером может быть (EtO)3SiCH2CH2CH2NH2.
Аббревиатура TEOS обозначает тетраэтоксисилан.
Аббревиатура DCM обозначает дихлорметан.
Аббревиатура "бисбис" обозначает 1,1-бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан, продукт примера 1b.
Подробное описание настоящего изобретения
В первом аспекте настоящего изобретения рассматривают парамагнитные ионы марганца, введенные в наноразмерные структуры (наноструктуры) на основе полимерной структуры или подложки, включающей или обрамленной множеством фосфонатных групп -P=O(OR1)(OR2), где R1 и R2 независимо выбирают из отрицательного заряда, H, алкила или арила. В случае, если по меньшей мере одна из R1или R2 представляет собой Н, то полученная фосфоновая кислота ионизируется до степени, зависящей от pH.
Как упоминалось выше, термин "наноструктуры" относится к структуре с общим диаметром 1-100 нм.
В некоторых вариантах осуществления настоящего изобретения R1 и R2 независимо выбраны из группы, состоящей из отрицательного заряда, Н и метила.
Для атома углерода, разделяющего бифосфонатные группы, т.е. промежуточного атома углерода, существует/существуют одна или несколько связей с полимерной структурой. Таким образом, промежуточный атом углерода может быть либо частью полимерной структуры, либо может быть прикрепленным к полимерной структуре. Особый интерес представляют структуры типа (R3R4C(P=O(OR1)(OR2))2, где R1 и R2 независимо выбраны из Н или алкила или арила и по меньшей мере одна из R3 и R4 представляет собой группу, способную связываться с полимерной структурой материала. В случае, если одна из R3 и R4является такой группой, то оставшаяся группа выбрана из группы, состоящей из H, OH, OR5 (при этом R5 представляет собой низший алкил) и низших алкилов.
В некоторых вариантах осуществления настоящего изобретения R3и/или R4 выбрана/выбраны из группы, состоящей из -(CH2)n-Si(Rx)3, где Rx независимо представляет собой низший алкил, OH, O- или O-, где - обозначает связь с полимерной структурой, и где n равно 1-5.
В некоторых вариантах осуществления настоящего изобретения R3представляет собой -(CH2)nCO- (с карбонильной группой, формирующей связь с полимерной структурой), и R4 представляет собой H, и n=1-5. В некоторых из этих вариантов осуществления n=1.
В некоторых вариантах осуществления настоящего изобретения R3 и R4 представляют собой независимо -(CH2)n-SiO3, где n=1-5, и силан является частью полимерной структуры по причине образования Si-O-Si связей, как подробно изложено далее.
В некоторых вариантах осуществления настоящего изобретения R3и R4 оба представляют собой -(CH2)n-SiO3, где n=3, и силан является частью указанной полимерной структуры в указанном выше порядке.
Также возможно использование фосфоновых амидов, хлоридов или фторидов вместо фосфоновых эфиров или фосфоновой кислоты в качестве компонентов или исходных материалов соединений, описанных в данном документе. Фосфонаты могут присутствовать в их свободной форме или в виде сложных эфиров или в виде амидов или любой их смеси.
В некоторых вариантах осуществления изобретения фосфонаты представляет собой смесь свободных фосфонатов и сложных метиловых эфиров указанного фосфоната.
Полимерная структура, с которой связаны указанные фосфонатные группы, может быть построена из большого числа известных мономеров, что можно обнаружить в любой книге по химии полимеров (например, J.R. Fried, "Polymer Science and Technology" Prentice Hall 1995). Некоторыми неограничивающими примерами являются полиакрилаты, полиметакрилаты, полиамиды, полистирол, полидиметилсилоксаны-силоксаны (силиконы), полиорганосиланы, полиамины, такие как полиэтиленимин или углеводы, особенно сильно разветвленные или сшитые структуры.
Наноструктуры по настоящему изобретению являются, как упоминалось выше, структурами почти сферической формы и средним размером (гидродинамическим диаметром) 1-100 нм; в некоторых вариантах средний размер может быть 2-50 нм, в других вариантах средний размер может составлять 3-10 нм, или 3-7 нм, или 10-50 нм, или 10-20 нм.
В некоторых вариантах осуществления настоящего изобретения, неограничивающим примером которого является использование в качестве внутривенного контрастного вещества, средний гидродинамический диаметр наноструктур составляет 3-7 нм.
В некоторых вариантах осуществления настоящего изобретения, например, вариантах осуществления, где материал используется для визуализации лимфатических узлов, средний гидродинамический диаметр наноструктур составляет 10-50 нм или 10-20 нм.
Указанный гидродинамический диаметр представляет собой диаметр эквивалентной твердой сферы, вычисленный из коэффициента диффузии согласно уравнению Стокса-Эйнштейна. Коэффициент диффузии в свою очередь рассчитывается по данным рассеяния света, зависящим от времени, полученным с помощью методики динамического рассеяния света (DLS). Для сравнения, бычий сывороточный альбумин согласно измерениям имеющий гидродинамический диаметр 6,5 нм по данным DLS в водном растворе, в высокой степени соответствует кристаллической структуре. В зависимости от того, используется ли среднее количество, средний объем или средняя интенсивность рассеянного излучения, значения могут несколько отличаться. Средний объем, как правило, самый используемый, так как он показывает, какой размер частиц основная часть материала имеет. Средние диаметры, указанные в настоящем тексте, соотносятся со средними объемами.
Целесообразно использовать структуры с разветвленной или сеткообразной структурой для формирования сферических структур, требуемых в настоящем изобретении. Одним установленным способом получения сетчатой структуры является введение поперечных связей с помощью включения фракции бифункциональных мономеров в процессе полимеризации. Хорошо изученным примером является сшивка полистирола с дивинилбензолом.
Разветвленные структуры могут быть сформированы благодаря наличию более одного реакционноспособного положения в мономерах ("The architecture and surface behavior of highly branched molecules" Peleshanko, S., Tsukruk, V.V., Prog. Polym. Sci. 33, 523 (2008)). Хорошо изученным примером является образование высоко разветвленного полиэтиленимина путем полимеризации азиридина. Полиэтиленимин содержит смесь первичных, вторичных и третичных аминогрупп и имеет разветвленные хаотичные структуры, как показано на схеме ниже. Точную структуру следует рассматривать всего лишь как типичную и никоим образом не ограничивающую настоящее изобретение. Бисфосфонаты, необходимые для настоящего изобретения могут быть присоединены к первичным и/или вторичным аминогруппам.
В некоторых вариантах осуществления настоящего изобретения полимерная структура представляет собой полиэтиленимин. Ниже показан типичный структурный фрагмент полиэтиленимина. Пунктирные связи указывают на то, что полимерная сетка продолжается.

При включении бисфосфоната в структуру полиакрилата возможно прикрепить бисфосфонат к амидному азоту через короткий линкер. Типичным, но не ограничивающим примером структурного фрагмента из такого материала будет структура ниже с R1 и R2, как определено ранее в тексте, n от 1-5, и пунктирными связями, указывающими на то, что фрагмент принадлежит полимеру. Также является возможным прикрепить бисфосфонат непосредственно к углеродному скелету.

Структуры на основе полиароматических веществ, подобных полистиролу или поливинилпиридину также могут быть предусмотрены. Бисфосфонат затем присоединяется к ароматической системе. Полиамиды наподобие поливинилпирролидона также являются возможными.
Для формирования наноструктуры по настоящему изобретению требуется подходящая степень сшивки. Включение 1-100% от ди-, три- или тетрафункциональных перекрестно-связывающих веществ является предпочтительным. Не ограничивающий перечень типичных сшивающих веществ представляет собой N,N'-метилен-бис(акриламид), эпихлоргидрин, дивинилбензол, 1,3-дивинилтетраметилдисилоксан, 1,3-фениленедиисоцианат, 3,3'-диангидрид бифенилтетракарбоновой кислоты, 1,4-бутандиолдивиниловый эфир, тетраэтоксисилан, олигосиликаты, такие как метасиликат или силсесквиоксаны, органосиланы, такие как бис(триэтоксисилил)метан, бис(триэтоксисилил)этан, бис(триэтоксисилил)пропан, бис(триэтоксисилил)бутан, метилтриэтоксисилан, этилтриэтоксисилан и пропилтриэтоксисилан.
Степень полимеризации регулируют, чтобы получить продукты необходимого размера путем управления параметрами процесса, как известно в данной области техники. Необходимый размер не может выражаться только гидродинамическим диаметром, но и степенью полимеризации (среднее количество мономеров). Она реже используется, чем гидродинамический диаметр, но это еще один способ концептуализации структуры и включен не в качестве ограничения, а в качестве ссылки. Например, для полимера с плотностью, близкой к 1 г/мл предпочтительные размеры находятся в пределах от 25-3000000 или 25-375000 или 80-3000 или 80-1000 мономеров.
Возможным является смешивание всех указанных полимерных структур в любой химически совместимой комбинации, либо путем смешивания мономеров перед полимеризацией, либо путем прививки второго полимера к первому полимеру.
Особенно предпочтительная структура образуется путем конденсационной полимеризации триалкоксиорганосиланов R5-Si(OR6)3, при этом R5 представляет собой Н или органический остаток, и R6независимо представляет собой низший алкил или арил. Такая структура обладает свойством быть высокополярной, следовательно, совместимой с водой, и степень сшивки можно регулировать с помощью параметров процесса во время производства. Целесообразным является использование мономеров с более, чем одной триалкосилильной группой.
В некоторых вариантах осуществления настоящего изобретения имеются две алкоксисилановые группы, присутствующие в мономере.
В некоторых вариантах осуществления настоящего изобретения указанные алкоксисиланы разделены 1-10 атомами углерода или 3-9 атомами углерода.
В некоторых вариантах осуществления настоящего изобретения указанные алкоксисиланы разделены 7 атомами углерода.
В некоторых вариантах осуществления настоящего изобретения указанные алкоксисиланы разделены тремя или пятью атомами углерода.
В некоторых вариантах осуществления настоящего изобретения две группы фосфоната являются частью группы R5.
В некоторых вариантах осуществления настоящего изобретения указанные два силана разделены 7 атомами углерода и две группы фосфоната являются частью группы R5.
В некоторых вариантах осуществления настоящего изобретения указанные силаны имеют общую структуру:
{(X7aO)(X7bO)PO}2-(C){(CH2)nSi(OX7c)(OX7d)(OX7e)}{(CH2)оSi(OX7c)(OX7d)(OX7e)}, где X7a, X7b, X7c, X7a, X7e независимо выбраны из H, C1-8алкила и бензила; а n и o независимо выбраны из 1-5.
В некоторых вариантах осуществления второй силан, такой как дисилан, прививается к полимерной структуре, образованной первым дисиланом.
Реакционная способность триалкоксисиланов относительно полимеризации изменяется в зависимости от идентичности R6 групп. Было обнаружено, что это является ключевым фактором в контроле размера молекул в процессе производства и обнаружено, что метил и этил, и в особенности последний, является пригодным для получения структуры по настоящему изобретению, хотя является возможным использовать любую другую группу низшего алкила, арил, силил амид, ацил, силилфторид или силилхлорид.
В некоторых вариантах осуществления настоящего изобретения R6 представляет собой этильную группу.
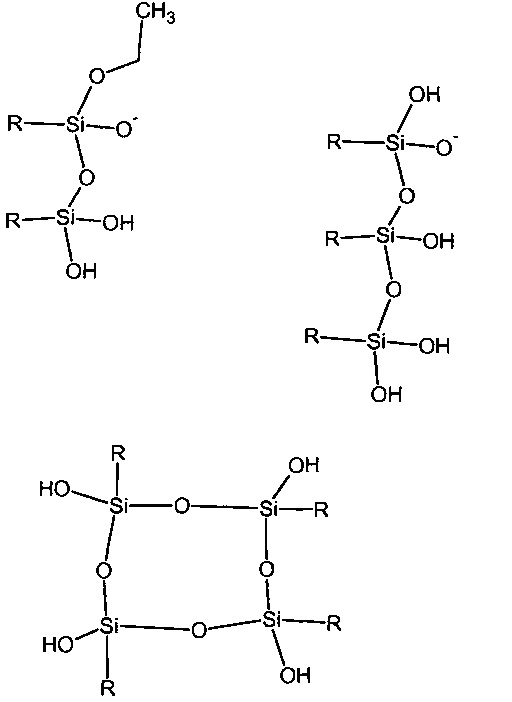
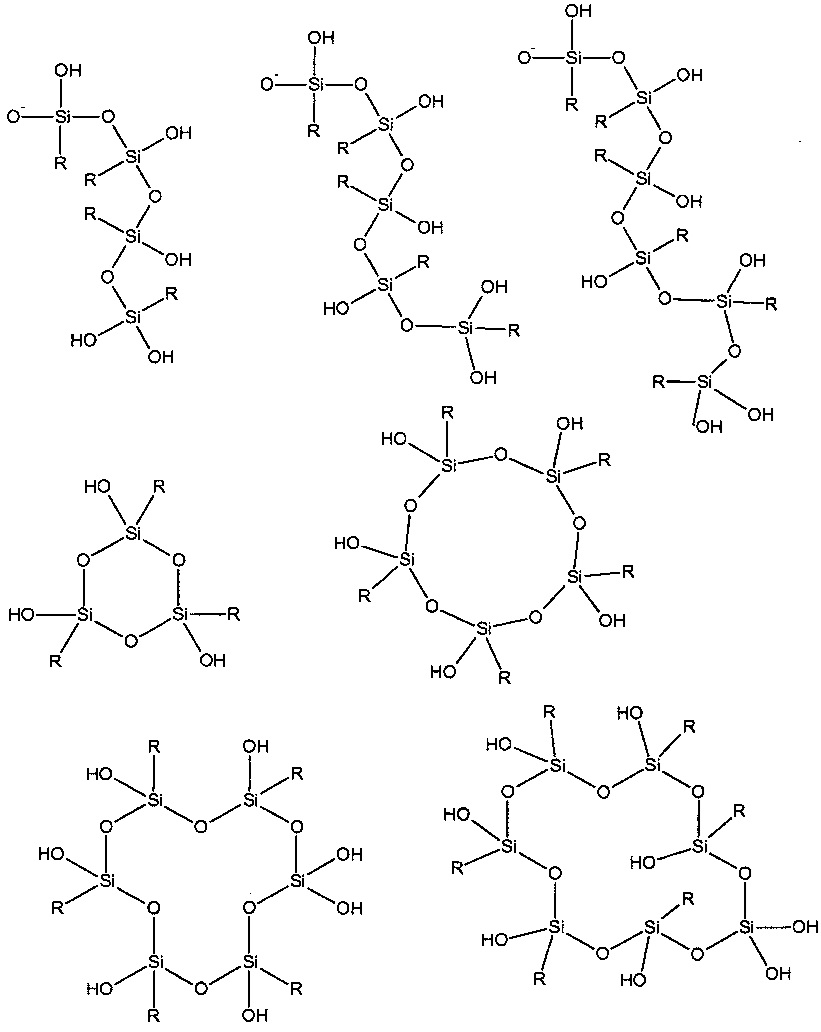
Существует множество различных способов связывания триалкоксисиланов посредством Si-O-Si связей. Известными являются димерные структурные элементы, а также линейные, разветвленные и циклические (R.J. Fessenden, J.S. Fessenden, "Trends in Organosilicon Biological Research" in Advances in Organometallic Chemistry vol. 18 p. 275). Также крем некислородные клеточные структуры различных размеров хорошо известны из литературы (Hanssen R. J. M. et al. Eur. J. Inorg. Chem 675 (2004)) и остаточные группы алкокси или свободных силанольных групп могут также присутствовать в разных пропорциях. Также является возможным, что ионы парамагнитных металлов, необходимые для настоящего изобретения в некоторой степени координируются Si-O-группами. Некоторые структурные элементы, хотя никоим образом не должны быть истолкованы как ограничивающие, которые могут присутствовать в таких структурах, показаны на схеме 1.
Схема 1: Иллюстрация некоторых Si-O-Si структур, которые могут использоваться в настоящем изобретении; R представляет собой любой органический остаток
Хорошо известно, что структура геминальных бифосфонатов R3R4C(P=O(OR1)(OR2))2, которая лежит в основе настоящего изобретения прочно связывает многовалентные катионы, такие как кальций. Преимущество материалов настоящего изобретения состоит в том, что они отдают предпочтение марганцу перед кальцием и магнием при физиологических концентрациях (это дополнительно проиллюстрировано ниже в примере 15).
Фосфонатные группы могут полностью присутствовать в их эфирных формах, полностью или частично гидролизоваться до их кислотной формы, а затем ионизироваться до некоторой степени от частичного до полного соответствия значению pH окружающей среды или любой их смеси. Оптимальным является введение ионов марганца в полимер при нейтральном или щелочном pH. Значения в пределах pH 12-6 или pH 11-8 или предпочтительно от 10,5 до 9,5 являются используемыми. Это указывает на то, что она представляет собой по меньшей мере частично, а иногда даже полностью анионную форму гидрализованного фосфоната, который играет важную роль в связывании ионов металла. Не только фосфонатные эфиры или кислоты, но также фосфоновые амиды могут быть рассмотрены как часть материала или использоваться в качестве исходного материала.
Было обнаружено, что, когда бисфосфонатную структуру вводят в полимерную структуру, предпочтительно гидрофильную, и оставляют связывать марганец(II), достигается заметное увеличение релаксивности. Хелат марганца мономерного бисфосфоната золедроновой кислоты имеет релаксивность 2,3/мМ Mn/c (как показано в примере 17) и марганец с мономерным бисфосфонатом метилендифосфоновой кислоты имеет релаксивность 1/мМ Mn/c. Релаксивности введенных полимерных материалов марганца по настоящему изобретению находятся в пределах от 24-48/мМ Mn/c.
В частности, когда полиэтиленимин образует производное с активированным эфиром геминального бисфосфоната, как описано в примере 9, и впоследствии насыщается ионами марганца(II), получают материал с релаксивностью в 24/мМ Mn/c. Она значительно выше, чем ранее упомянутая релаксивность комплекса золедроновой кислоты, таким образом, демонстрируя преимущества включения бисфосфоната в полимерную структуру. Молярное отношение фосфора к азоту в таких материалах может быть в диапазоне от 0,1 до 3 и предпочтительно от 0,2 до 0,8.
Для того, чтобы дополнительно продемонстрировать, что релаксивность происходит из сочетания геминальных бисфосфонатов с полимером, также были испытаны соответствующие "голые" полимеры. Полиэтиленимин без каких-либо добавок связывает марганец очень слабо и релаксивность того малого количества, которое связано с полимером является очень низким (0,8/мМ/с). Также полиорганосилан, образованный из (EtO)2P=O-CH2-CH2-Si(OEt)3, дает довольно невыразительную релаксивность в 3,0/мМ Mn/c после введения марганца. Все вместе взятое, это доказывает, что удивительно высокая релаксивность, присущая материалам по настоящему изобретению происходит от сочетания всех качеств того, что является полимерным, включающим геминальные бисфосфонаты и включающим парамагнитный металл, такой как марганец.
В примере 22 описано, почему 1,3-бисфосфонат, связанный с полимерной структурой полиэтиленимина, который выходит за рамки настоящего изобретения, имеет более низкую релаксивность (18,5/мМ/с), чем геминальные бисфосфонаты по настоящему изобретению (>=24/мМ/с). Он также имеет низкую стабильность в ионнообменной пробе, которую мы использовали. Таким образом, разумно ожидать, что 1,2-бисфосфонаты, а также любые 1, n-бисфосфонаты, где n>2, являются менее подходящими, чем геминальные бисфосфонаты по настоящему изобретению.
Ионы парамагнитных металлов предположительно, но никоим образом не ограничивая настоящего изобретения, хелатируют с фосфонатными группами, и отношение фосфора к марганцу 10-15 кажется наилучшим компромиссом между релаксивностью и стабильностью, но возможно в пределах от 7 до 20. В таблице 1 примера 11 эффект изменения отношения фосфора к марганцу описан подробно.
Предпочтительное отношение кремния к марганцу будет находиться в пределах от 5 до 20, и отношение фосфора к кремнию должно составлять приблизительно 1, например, от 0,7 до 1,3.
Необязательно, гидрофильный, биологически инертный материал может быть привит на внешние части наноструктур по настоящему изобретению. Указанные внешние части являются теми частями наноструктуры, которые доступны для химических реакций с дериватирующими реагентами. Это может быть целесообразным для биосовместимости материала и многие гидрофильные материалы, такие как углеводы или гидрофильные искусственные полимеры могут быть приняты во внимание. Особый интерес представляют полиэфирные соединения и особенно производные полиэтиленгликоля (PEG). Хотя и без ограничений, производные PEG с метоксильными концевыми группами являются предпочтительными (м-PEG). Они могут быть привиты любым химически приемлемым способом к полимерной структуре, например, к атомам кислорода, азота или углерода, например, к остатку фосфоновой кислоты или силанольных групп после хелатирования металла или, в качестве альтернативы, непосредственно к не содержащему металла полимеру. Подходящими величинами длины цепи линейных производных PEG являются от 4 до 50 единиц -CH2CH2O-. Наиболее предпочтительными являются смеси, содержащие от 5 до 20 единиц и в среднем около 10 или 11. Реагенты, которые наиболее беспрепятственно сочетаются с наноструктурами, представляют собой концевые аминогруппы и могут быть соединены с остаточными фосфоновыми группами. Разветвленные производные PEG также представляют интерес и особенно структуры, подобные тем, что в примере 8, которые являются более компактными и обладают более выраженной способностью к защите поверхности, чем линейные производные PEG с аналогичной молекулярной массой.
Количество биологически инертных полимерных групп на каждой единице наноструктуры может находиться в пределах от 10 до 1000 или от 10 до 100 или от 10 до 50.
В некоторых вариантах осуществления полимерные, насыщенные марганцем бисфосфонатные наноструктуры по настоящему изобретению включают в себя линейные м-PEG группы с длиной цепи от 5 до 20 мономерных остатков, соединенные с фракцией фосфонатных групп посредством амидных связей.
В некоторых вариантах осуществления полимерные, насыщенные марганцем бисфосфонатные наноструктуры по настоящему изобретению включают разветвленные PEG группы структуры X1 (пример 8h), соединенный с фракцией фосфонатных групп посредством амидных связей.
Возможным является введение маркеров для соединения наноструктур по настоящему изобретению с различными активными молекулярными субстанциями, таких как биомаркеры или репортерные элементы. Не ограничивающим перечнем типичных примеров будут пептиды, пептоиды, протеины, антитела, фрагменты ДНК, фрагменты РНК, РНК, фрагменты, флуорофоры, хелаты или низкомолекулярные фармакологические лиганды.
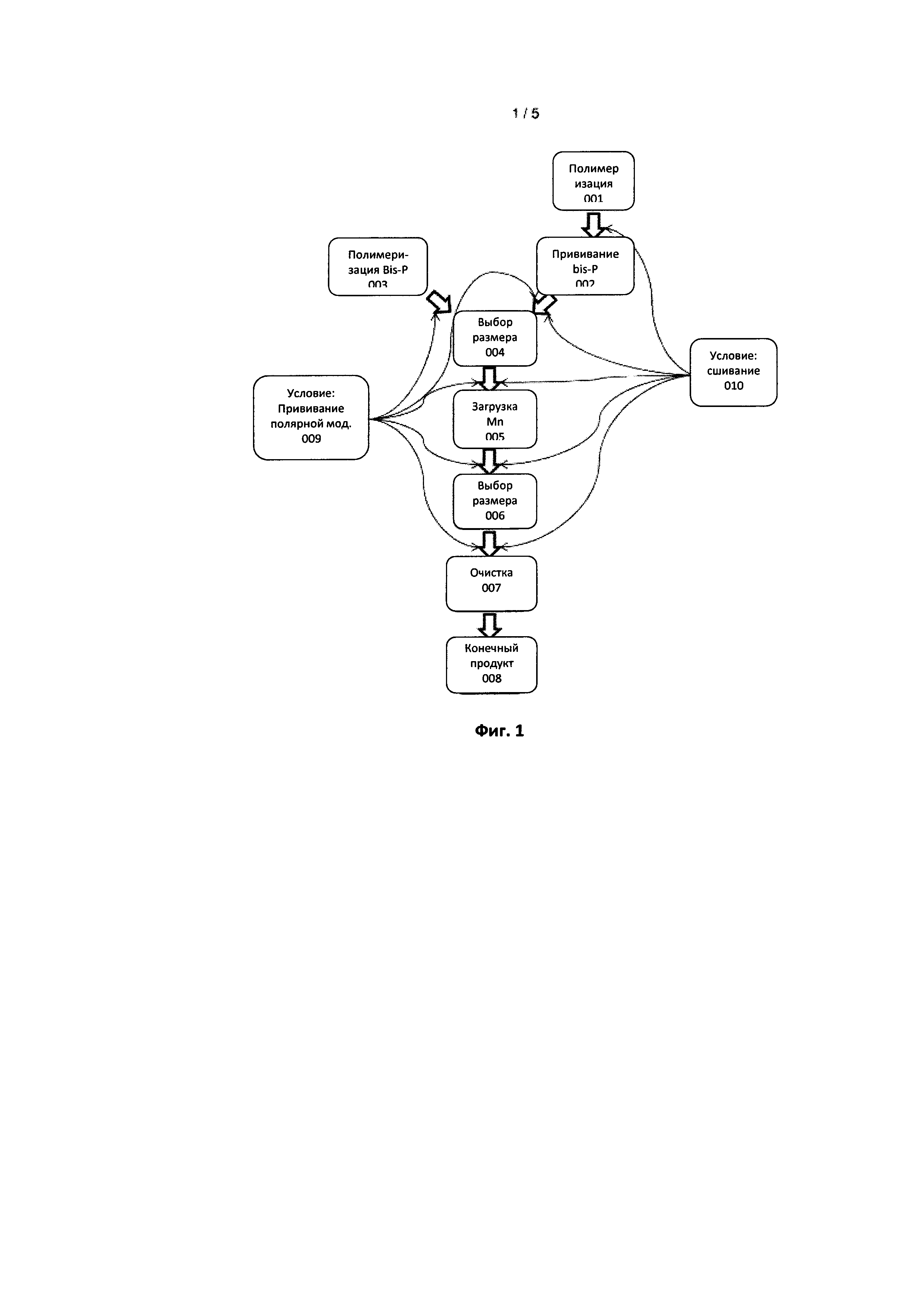
Вторым важным аспектом настоящего изобретения является способ получения указанных наноструктур. В самом широком смысле он, во-первых, включает формирование сферических, наноразмерных полимерных образований, включающих множество бисфосфонатных групп с последующим этапом, где продукт первого этапа контактирует с ионами марганца(II). Необязательно, два этапа, хотя и различных химически, можно проводить одновременно в том же реакционном сосуде. Основные особенности способа отмечены на фигуре 1. В одном или нескольких примерах способа включен выбор размера или этапа очистки путем ультрафильтрации.
Наноразмерную полимерную глобулу, содержащую множество бисфосфонатов получают либо посредством прививки (002) к существующей полимерной глобуле (полученной на этапе полимеризации 001) или путем полимеризации смеси мономеров, включающей бисфосфонаты (003). В зависимости от того, какая полимерная структура необходима, можно рассматривать множество различных инициаторов полимеризации. Для ненасыщенных мономеров, таких как стиролы и акрилаты различные радикальные инициаторы, такие как перекись бензоила или азобисизобутиронитрил, являются предпочтительными. Для мономеров на основе триалкокси силана по одному из предпочтительных вариантов осуществления настоящего изобретения можно использовать спонтанный гидролиз и конденсацию для осуществления полимеризации или использовать кислотный или основный катализ. В некоторых случаях доказано целесообразным использование добавки pH-стабилизирующей соли, например, бикарбоната, в частности бикарбоната натрия или карбоксилата, например, ацетата натрия, формиата натрия или тетраметил формиата аммония к реакционной смеси с целью оптимизации выхода продукта с размерами от 3-10 нм.
Часто растворитель желателен для этапа 003, и, хотя много различных растворителей может быть представлено специалистом в данной области, желательно избегать токсичных растворителей, таким образом, вода и низшие спирты, такие как пропанол, бутанол, этиленгликоль или 1,3-пропандиол являются предпочтительными. Часто желательно оптимизировать выход и качество за счет использования смеси растворителей.
В некоторых вариантах осуществления способа смесь 5-25% воды в низшем спирте используют на этапе 003.
В некоторых вариантах осуществления способа смесь 5-25% воды в этаноле, 1- или 2-пропанол, этиленгликоль или 1,2- или 1,3-пропандиол используют на этапе 003.
Было установлено, что целесообразно применять температуры выше комнатной температуры для этапа 003, например, температуры 40-130°C или 80-120°C или 100-120°C. При использовании низших спиртов необходимо работать с закрытыми, устойчивыми к давлению сосудами для достижения нужной температуры реакции.
Продолжительность этапа 003 зависит от полимерной структуры и способа инициирования и может находиться в диапазоне от нескольких секунд до нескольких дней или недель. Для триалкоксисиланов в некоторых из предпочтительных вариантов осуществления настоящего изобретения оказалось целесообразным использовать промежутки времени 6-200 часов, или 6-48 часов, или 12-36 часов, или приблизительно 24 часа на этапе 003.
В некоторых вариантах осуществления настоящего изобретения условиями этапа 003 являются: температура 105-115°C и продолжительность 20-30 часов.
В некоторых вариантах осуществления настоящего изобретения условиями этапа 003 являются: температура 105-115°C и продолжительность 30-60 часов.
В некоторых вариантах осуществления настоящего изобретения условиями этапа 003 являются: температура сначала 90-100°C в течение 40-50 часов, а затем 105-115°C в течение еще 20-30 часов.Концентрация мономеров на этапе 003 зависит от того, какая полимерная структура требуется, и может варьироваться от молярной концентрации до не содержащих растворителя условий. Однако для триалкоксисиланов в одном из предпочтительных вариантов осуществления настоящего изобретения оказалось целесообразным получать 10-500 мМ или 20-100 мМ и, в частности 40-80 мМ концентрацию мономера.
В некоторых вариантах осуществления настоящего изобретения условиями этапа 003 являются сначала температура 90-100°C в течение 20-50 часов с последующей 105-125°C в течение 20-30 часов и концентрация мономера 40-60 мМ.
На этапе 002, который включает прививки бисфосфонатного реагента к полимерной структуре, условия несколько отличаются. Особенно требования к температуре и концентрации являются более мягкими. Было обнаружено, что начиная с раствора полиэтиленимина в воде необязательно с примесью сорастворителя, при температуре, совместимой с жидкой водой, например, комнатной температуре и приводя его в контакт с бисфосфонатом, способным взаимодействовать с указанным полиэтиленимином, таким как 3,3-бис(диметоксифосфорил)-пропановой кислоты в присутствии соединения, способного образовывать реакционноспособное промежуточное эфирное соединение, такое как N-гидроксисульфосукцинимид натриевой соли в присутствии конденсирующего вещества, такого как N-(диметиламинопропил)-N'-этил карбодиимид при температуре, например комнатной температуре, в течение 1-48 часов, например 20-24 часов, получают материал с бисфосфонатами, привитыми к полимерной структуре.
Этап выбора размера (004) осуществляют на растворе предшественников наноструктуры (X) с целью удаления нежелательных больших или малых элементов. Исходные материалы и остатки растворителя реакционной смеси также удаляют на данном этапе. Ультрафильтрация является предпочтительным способом очистки, особенно при использовании в форме, которую обычно обозначают фильтрацией ламинарного потока или диафильтрацией.Предпочтительно удалять нежелательные большие наноструктуры и/или агрегаты путем пропускания раствора через фильтр с довольно крупными порами, этап 004а. Предпочтительными номинальными пороговыми значениями для таких фильтров являются 300 кДа или 100 кДа или 50 кДа. На этапе 004b требуемый материал собирали на фильтре с порами меньшего размера. Предпочтительные размеры пор для этапа 004b имеют номинальные пороговые значения при 50 кДа, 30 кДа или 10 кДа.
Этап выбора размера (004) может не потребоваться, если исходный материал имеет узкое распределение по размерам.
В некоторых вариантах осуществления настоящего изобретения раствор, полученный на этапе 002 или 003, пропускают сначала через 100 кДа фильтр (этап 004а) и впоследствии собирают на 30 кДа фильтре (этап 004b).
В некоторых вариантах осуществления настоящего изобретения раствор, полученный на этапе 002 или 003, пропускают сначала через 300 кДа фильтр (этап 004а), а затем собирают на 100 кДа фильтре (этап 004b).
В некоторых вариантах осуществления настоящего изобретения раствор, полученный на этапе 002 или 003, пропускают сначала через 50 кДа фильтр (этап 004а) и затем собирают на 30 кДа фильтре (этап 004b).
В некоторых вариантах осуществления настоящего изобретения раствор, полученный на этапе 002 или 003, пропускают сначала через 100 кДа фильтр (этап 004а) и затем собирают на 10 кДа фильтре (этап 004b). Целесообразно промыть материалы несколькими порциями воды после этапа 004b для дальнейшего удаления остатков мономеров или растворителей из этапа 001, 002 или 003.
Другие способы ультрафильтрации, такие как спиновые фильтры или диализ также могут быть использованы, хотя они являются менее масштабируемыми.
Частицы требуемого диапазона размеров могут быть также отобраны с помощью эксклюзионной хроматографии (также называемой гель-фильтрацией).
Этап 005, где указанный полимер насыщается марганцем(II), включает воздействие раствора указанного полимера на ионы марганца(II). Указанные ионы могут быть добавлены к реакционной смеси в виде твердого вещества или в виде раствора. Водорастворимые соли марганца(II), такие как фторид, хлорид, бромид, ацетат, нитрат или сульфат являются предпочтительными. Также является возможным использование менее растворимых источников марганца, например, MnO. Используемые концентрации ионов марганца составляют от 0,1 мМ - 5 М, например, 0,1-600 мМ или 0,1-10 мМ в зависимости от концентрации полимеров. Как обсуждалось ранее, отношение фосфора к марганцу является важным. Оптимально вводить ионы марганца в полимер при нейтральном или щелочном pH. Применяют от pH 12-6 или pH 11-8 или предпочтительно 10,5-9,5. Добавление марганца следует выполнять спустя время, за которое pH стабилизировался на уровне требуемого значения. Было обнаружено, что временные интервалы от 10 минут до 24 часов, например, от одного часа до двух часов, являются достаточными. После введения наноструктур с марганцем, pH доводят до нейтрального (от 8 до 6 или от 7,7 до 7). Температура для загрузки может находиться в пределах от точки замерзания до точки кипения взятого растворителя или смеси растворителей и от комнатной температуры до 60 градусов является предпочтительной.
На необязательном этапе 006 частицы требуемого размера отделяют от неприемлемо больших или малых частиц. Часто это не является необходимым, так как наноструктуры изменяются всего лишь незначительно по размеру в случае добавления марганца. Этап 006 может иметь несколько подэтапов 006а, 006b и т.д. или, для подэтапа в произвольном порядке, 006x.
Ультрафильтрация является предпочтительным способом этапа выбора размеров 006x, особенно при использовании в форме, которую обычно обозначают фильтрацией ламинарного потока или диафильтрацией. Предпочтительно удалять нежелательные большие наноструктуры и/или агрегаты путем пропускания раствора через фильтр с довольно крупными порами, этап 006а. Предпочтительными номинальными пороговыми значениями для таких фильтров являются 300 кДа или 100 кДа или 50 кДа. На этапе 006b требуемый материал собирали на фильтре с меньшего размера порами. Предпочтительные размеры пор для этапа 006b имеют номинальные пороговые значения при 50 кДа, 30 кДа или 10 кДа.
В некоторых вариантах осуществления настоящего изобретения раствор, полученный на этапе процесса 005, пропускают сначала через 100 кДа фильтр (этап 006а) и затем собирают на 30 кДа фильтре (этап 006b).
В некоторых вариантах осуществления настоящего изобретения а раствор, полученный на этапе процесса 005, пропускают сначала через 300 кДа фильтр (этап 006а) и затем собирают на 100 кДа фильтре (этап 006b).
В некоторых вариантах осуществления настоящего изобретения раствор, полученный на этапе процесса 005, пропускают сначала через 50 кДа фильтр (этап 006а) и затем собирают на 30 кДа фильтре (этап 006b).
Целесообразным является промывание материалов несколькими порциями воды после стадии 006b для дополнительного удаления остаточных ионов металлов, мономеров или остатков растворителей с этапа 005.
Другие способы ультрафильтрации, такие как спиновые фильтры или диализ также могут быть применены на этапе 006x.
Частицы требуемого диапазона размеров могут быть отобраны с помощью эксклюзионной хроматографии (также называемой гель-фильтрации) на этапе 006x.
Необязательно указанный насыщенный марганцем полимерный фосфонатный продукт может быть очищен на этапе 007. Этап 007 может иметь несколько подэтапов 007а, 007b и т.д., для подэтапа в произвольном порядке, 007х.
Один предпочтительный способ этапа очистки 007х представляет собой обработку с небольшим количеством катионного обменника, такого как сульфированный полистирол для удаления избытка марганца или слабо связанного марганца. Коммерчески доступные ионообменные смолы часто имеют емкость 1-2 ммоль/г смолы и, как правило, сырой материал из этапа b), содержащий 1 моль марганца, будет обрабатываться с помощью 1-100 г натриевых или калиевых разновидностей ионообменной смолы.
В некоторых вариантах осуществления настоящего изобретения этап 007х включает еще одну диафильтрацию, собирающую материал на 30 кДа фильтре.
В некоторых вариантах осуществления настоящего изобретения этап 007х включает обработку продукта из этапа 006 с натриевой разновидностью ионообменной смолы полистирол-сульфонатного типа.
Последующие этапы очистки для удаления 007х липофильных примесей, таких как следы эндотоксинов (остатки мертвых бактерий), также могут быть добавлены.
В некоторых вариантах осуществления способа продукт этапа 006 обрабатывают активированным углем.
В некоторых вариантах осуществления способа продукт этапа 006 проходит через полиэтилен, или полипропилен, или PVDF фильтр.
В некоторых вариантах осуществления способа продукт этапа 006 обрабатывают иммобилизованным полимиксином В.
Необязательно, этап 009 может быть включен, как показано на фигуре 1, где модификатор биологически инертной поверхности прививают на доступных частях наноструктуры.
Необязательно, сшивка (этап 010) может быть осуществлена путем введения сшивающих веществ во многие места в способе, как показано на фигуре 1. Сшивка путем смешивания в сшивающем веществе на этапе 001 является стандартной процедурой. Мономер может также быть по своей природе склонным к сшивке, например, триалкоксисиланы в одном из предпочтительных вариантов осуществления настоящего изобретения, так что материал, полученный на стадии 002, уже сшит.
В третьем основном аспекте настоящего изобретения материал используют в качестве контрастного вещества для диагностических процедур и, в частности, для магнитно-резонансной томографии (MRI). Материал по настоящему изобретению имеет свойства низкой токсичности и высокой релаксивности, что делает его применимым в качестве контрастного вещества в MRI исследованиях организма, в частности человеческого тела.
Сочетание свойств высокой релаксивности и подходящего размера для тех вариантов осуществления изобретения, где гидродинамический диаметр больше, чем 3 нм или больше, чем 4 нм или больше, чем 5 нм делает композиции, содержащие наноструктуры по настоящему изобретению пригодными для визуализации опухолей, в частности твердых опухолей, с помощью MRI. Также является возможным использование указанных композиций по настоящему изобретению в качестве контрастного вещества для общей анатомической визуализации, например, ангиографии; в частности, ангиография мелких коронарных артерий сердца, или ангиография сонных артерий или почечных артерий или аорты возможна благодаря высокой релаксивности и контрастности настоящего изобретения. Визуализация структур в голове, внутренних органах или конечностях также представляет интерес. Внутренние органы, печень, поджелудочная железа и кишечник представляют особый интерес. Визуализацию толстой кишки можно получить либо путем внутривенного введения, либо введения в виде клизмы. Для визуализации желудка, печени и верхнего отдела кишечника возможно введение контрастного вещества перорально.
Из-за низкой токсичности и высокой релаксивности материалов по настоящему изобретению, они полезны в качестве веществ для мечения клеток. Клетки, например, стволовые клетки или макрофаги для диагностического или терапевтического применения у пациента вводят с наноструктурами по настоящему изобретению ex vivo и затем вводят указанному пациенту, и их распределение в теле может быть визуализировано с помощью MRI.
В некоторых вариантах осуществления настоящего изобретения раствор наноструктур вводят в ткани, часто без ограничения внутрикожно или подкожно, а затем используют для визуализации лимфатических структур пациента с помощью MRI. Особый интерес представляет визуализация лимфатических узлов, которые являются типичным местом локализации метастатических опухолей. Особенно подходящими для этой цели являются наноструктуры с размерами около 10 нм, например, 7-50 нм, или 7-25 нм, или 7-15 нм.
В некоторых вариантах осуществления настоящего изобретения композицию наноструктур по настоящему изобретению со средним гидродинамическим диаметром в диапазоне 8-15 нм вводят пациенту внутрикожно, и лимфатические узлы указанного пациента визуализируют с помощью процедуры MRI. Поскольку наноструктуры по настоящему изобретению имеет свойства высокой релаксивности и низкой токсичности возможно использование материала для мечения клеток. В таком случае клетки, например, стволовые клетки или макрофаги вводят с наноструктурами наружно на тело млекопитающего, например, тело человека, а затем вводят указанному млекопитающему и изображение генерируется сканированием MRI. Это дает возможность наблюдать за клетками по мере того, как они перемещаются в организме.
Использование in vivo наночастиц по настоящему изобретению требует, чтобы они были составлены фармакологически приемлемым способом в соответствии с передовыми наработками, хорошо известными специалистам в данной области. Предпочтительным способом введения является парентеральный, в частности, эффективным является внутривенный способ, но внутриартериальный может иметь преимущества при определенных обстоятельствах. Парентеральное введение часто требует жидкой композиции. Вода является предпочтительным растворителем для введения наноструктур по настоящему изобретению в раствор, но один или несколько сорастворителей или добавки могут быть добавлены в 0,1-10% для улучшения стабильности раствора. Приемлемыми сорастворителями являются спирты, такие как этанол или глицерин, биосовместимые полимеры, такие как этиленгликоль или поливиниловый спирт, диметилсульфоксид или N-метилпирролидинон. Также может быть целесообразным добавить один или несколько осморегуляторов, например, маннит, сорбит, лактозу, глюкозу или другие сахара или сахарные спирты. Желательно, чтобы состав являлся изотоничным жидкостям организма. Предпочтительно раствор для внутривенного применения имеет осмотическое давление от 270-2000 мОсм или 280-1000 мОсм или 280-500 мОсм или, в частности, от 280-300 мОсм. Многие из указанных добавок могут также выполнять функцию криопротекторов, повышающих эффективность восстановления после сублимационной сушки. Также может являться целесообразным добавление электролитов с целью снижения физиологических эффектов введенного раствора. Предпочтительными электролитами будет сочетание нетоксичных солей натрия, кальция или и/или магния. Регулирование pH инъекционного раствора является предпочтительным и любой буфер, подходящий для инъекции может быть рассмотрен, но предпочтительным является трис-HCl. Металло-ионные поглотители могут быть также предусмотрены в качестве добавки. Некоторыми типичными примерами будут EDTA (этилендиаминтетрауксусная кислота), DTPA (диэтилентриамин пентауксусная кислота) и DOTA (1,4,7,10-тетрааза-циклододекана-N,N',N'',N'''-этилендиаминтетрауксусная кислота) или фодипир. Также можно рассматривать использование твердофазных ион-поглощающих смол, добавленных в баллоны для хранения.
Концентрация наноструктур может быть описана множеством разных способов, но двумя наиболее обоснованными являются концентрация по массе, приведенная в г/л растворе и концентрация марганца в ммоль/л раствора. Диапазоны концентрации марганца в композициях, которые пригодны для введения в качестве контрастного вещества, находятся в диапазоне от 1-500 мМ или 10-300 мМ или 10-200 мМ или 50-200 мМ или 100-200 мм. При применении в качестве концентрации по массе и при условии, что отношение марганца к фосфору составляет приблизительно 6, концентрации по массе, которые подходят для композиции контрастного вещества находятся в пределах 0,5-300 г/л, или 5-200 г/л, или 5-250 г/л, или 5-100 г/л, или 100-250 г/л. Концентрации по массе приблизительно соответствуют концентрациям марганца, данным в мм, но соответствие будет изменяться в зависимости от структуры полимера, степени сшивки и наличия биологически инертного покрытия.
Один из вариантов осуществления настоящего изобретения представляет собой фармацевтически приемлемую композицию для внутривенного введения с концентрацией марганца 100-300 мм и отношением фосфора к марганцу 7-20.
Некоторые варианты осуществления настоящего изобретения относятся к фармацевтически приемлемым композициям для внутривенного введения с концентрацией марганца 10-300 мм и отношением фосфора к марганцу 7-20.
Альтернативный вариант осуществления настоящего изобретения представляет собой наноструктуру, содержащую полимерную структуру, содержащую по меньшей мере пять геминальных бисфосфонатных групп, где геминальные бисфосфонатные группы независимо друг от друга включены как
-R3R4C(P=O(OR1)(OR2))2,
что является идентичным -R4R3C(P=O(OR1)(OR2))2,
где R1 и R2 независимо выбраны из группы, содержащей отрицательный заряд, Н, алкил и арил, и где по меньшей мере один из R3 и R4представляет собой группу, связанную с полимерной структурой при условии, что, когда один из R3 и R4 представляет собой такую связанную группу, остальные R3 и R4 представляют собой либо группу, способную соединяться с полимерной структурой, либо остаток такой группы либо выбраны из группы, содержащей Н, ОН, OR5 и R5, где R5 представляет собой низший алкил, т.е. наноструктуру, отмеченную выше, но не содержащую ионов марганца. Такую наноструктуру используют в качестве промежуточного продукта в производстве наноструктур по ранее рассмотренным вариантам осуществления. Такая структура может связывать другие катионы, кроме марганца и использоваться в этом качестве.
Краткое описание графических материалов
В приведенных ниже примерах дается ссылка на прилагаемые графические материалы, на которых:
фиг. 1 представляет собой схематическое изображение способа получения наночастиц по настоящему изобретению, и
на фиг. 2 иллюстрируют повышение контрастности через 5 ч после инъекции у мыши с опухолью;
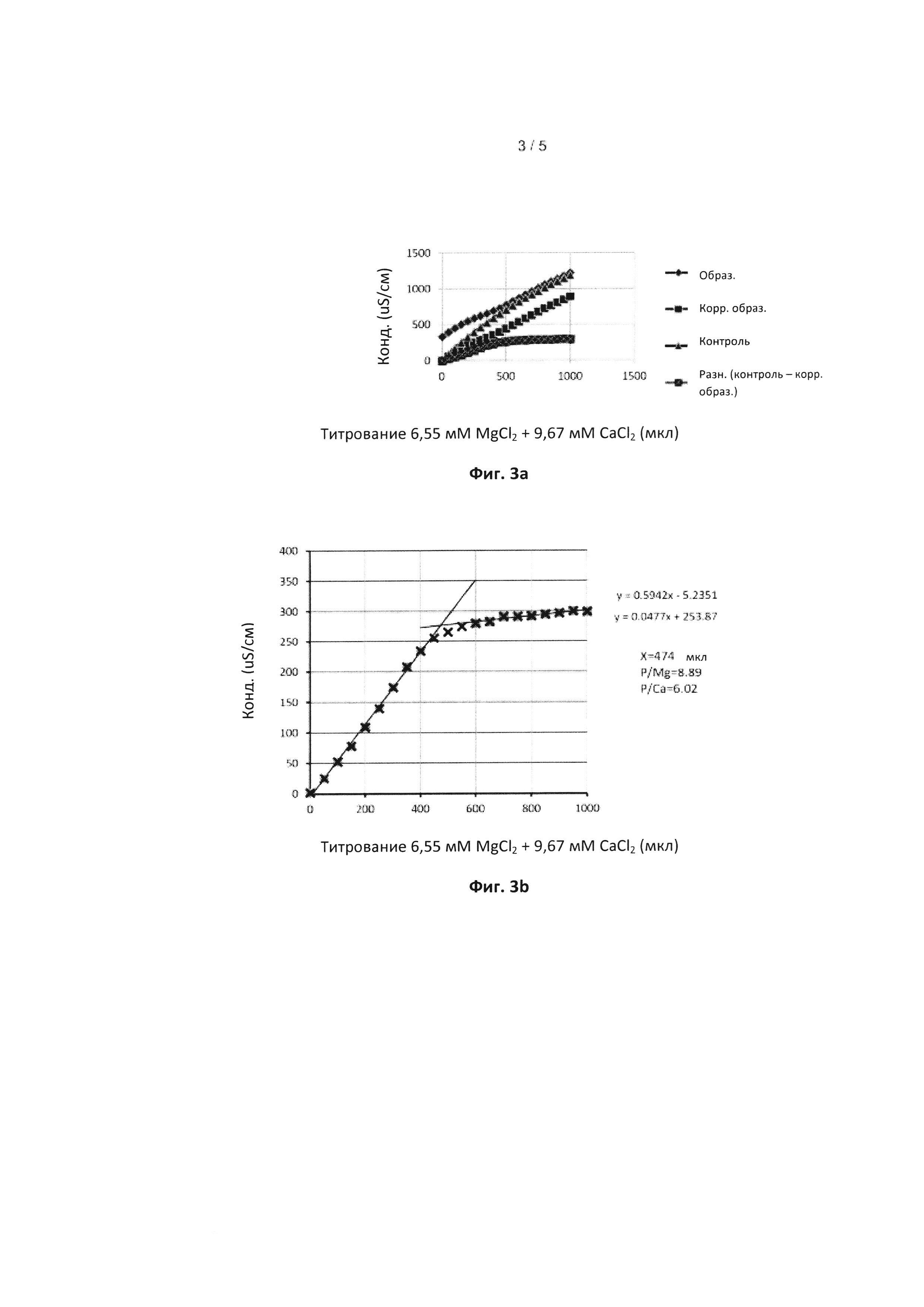
фиг. 3a, 3b и 3c представляют собой кривые, относящиеся к экспериментам по кондуктометрическому титрованию, описанным в примере 23;
на фиг. 4а и 4b иллюстрируют восстановление SI055 во фракциях 1-10 из преиммунной (фиг. 4А) и иммунной колонок (фиг. 4В).
Примеры
Пример 1: Синтез 1,1-бис-(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метана
1а: 1,1-Диаллил-1,1-бисдиметилфосфонат)метан

2-л Реактор, снабженный механической мешалкой, высушили при 130°C под вакуумом и затем оставили охлаждаться при положительном давлении азота. В реактор загрузили сухой THF (1 л) (сухой Aldrich), 99,9% с 250 частями на миллион ВНТ) посредством передачи по трубке в атмосфере инертного газа. Тетраметилметиленеди(фосфонат) (97,4 г, 420 ммоль) и аллилбромид (183 мл, 255 г, 2,11 моль) (без выделения тепла, кислый газ не определен). Температуру в рубашке установили на 0°C и при внутренней температуре 6°C добавили (разделенный на 6 порций) трет-бутоксид калия (140,3 г в общем, 1,25 моль). Температуру поднимали до 12°C после каждого добавления и позволяли снижаться до 6°C (или ниже) до следующего добавления. Последнее добавление аллилбромида (9,4 мл, 0,11 моль) и т-бутоксида (7,3 г, 65 ммоль) проводили для преобразования нескольких последних процентов моноалкилированного продукта (экзотермический эффект не выявлен).Температуру в рубашке установили на 15°C в течение приблизительно 2 ч и затем густую белую реакционную смесь перемешивали в течение ночи при температуре в рубашке 0°C. Реакцию гасили 50 мл насыщенного NH4Cl (водн.) (температуру повышали от 2 до 5°C), затем добавили толуол (1 л) и 1 л отогнали для удаления THF и избытка аллилбромида. Собирали от 63-73°C с температурой в рубашке от 70-100 градусов более 2 часов. К остатку добавили силикагель (100 г) и активированный уголь (15 г). Реакционную смесь перемешивали в течение нескольких минут и жидкость выкачали фриттованной фильтровальной трубкой (или фильтровали через стекловолоконный фильтр Ватман). Остаточный осадок промыли толуолом (3×100 мл). Объединенный фильтрат фильтровали через стекловолоконный фильтр Ватман с целью удаления нежелательных следов активированного угля (помеченного зеленоватым цветом) и концентрировали на роторном испарителе (температура бани 40°C) с получением указанного в заголовке соединения в виде бледно-желтой жидкости. Сырой продукт повторно ввели в очищенный реактор и растворили в смеси толуола (50 мл) и гептана (380 мл). Кристаллизация может быть индуцирована при внутренней температуре 12-9°C путем посева. Температуру в рубашке снижали до -25°C в течение 2 ч и выдерживали при этой температуре в течение еще двух часов, чтобы завершить кристаллизацию. Маточный раствор удалили из фриттованной фильтровальной трубки и суспензию кристаллов промыли двумя порциями предварительно охлажденного гептана (40 мл). Кристаллы растворили в EtOAc и раствор слили через нижний клапан. Растворитель удалили при пониженном давлении с получением 70,0 г 224 ммоль, 53% продукта в виде твердого вещества с т. пл. чуть выше комнатной температуры.
1Н-ЯМР (CDCl3); 6,35 (м, 2H), 5,20 (м, 4H), 3,68 (д, 12H), 3,00 (abx, 4H).
Также можно было перегонять продукт кратчайшим путем, в тонкопленочном перегонном аппарате.
1b: 1,1-Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан
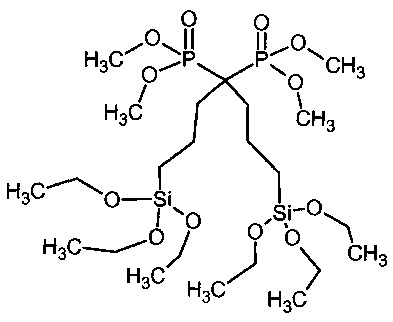
В 2-л реактор с рубашкой контроля температуры, внутренним термометром и механической мешалкой, загрузили толуол (330 мл, сухой Aldrich, герметично закупоренный), тетраметил-1,1-бисаллил-метиленбис(фосфонат) (70 г, 224 ммоль) и триэтоксисилан (123 мл, 655 ммоль). Реакционную смесь обескислородили с помощью трех вакуум-азотных циклов, тщательно поддерживая вакуумные циклы короткими, чтобы избежать потери толуола и силана. Кислород имеет решающее значение для удаления. Температуру в рубашке установили на 30°C. Катализатор Karstedt (4,5 мл, 2% в толуоле 0,053 ммоль) ввели с помощью шприца 0,5 мл порциями с 30 минутным промежутком (в общем 4,5 ч). После завершения добавления катализатора рубашку с контролем температуры установили на 30°C и смесь оставили перемешиваться в течение ночи. На следующее утро температуру в рубашке установили на 40 градусов, добавили дистилляционную головную фракцию и толуол и избыток силана отогнали при давлении от 62 до 13 мбар. Продолжительность 2 ч. Добавили этанол (800 мл) и активированный уголь (15 г) и суспензию перемешивали в течение 10 мин и смесь извлекли через нижний клапан и профильтровали через стекловолоконный фильтр Ватман. Растворитель удаляли при пониженном давлении на роторном испарителе до постоянного веса (температура бани 45°C) с получением 138 г (215 ммоль, 96%) продукта в виде бледно-коричневого масла.
1Н-ЯМР (CDCl3); 3,95 (д, 12H), 3,77 (д, 12H), 2,37 (м, 4H), 2,12 (м, 4H), 1,32 (т, 18H), 0,88(т, 4H).
Также можно было перегонять продукт кратчайшим путем, в тонкопленочном перегонном аппарате при 170°C и 0,3 мбар.
Пример 2: Синтез 1,1-бис(2-триметоксисилилэтил)-1,1-бис(диметилфосфонат)метана
2а: 1,1-Бис(2-трет-бутоксиэтил)-1,1-бис(диметилфосфонат)метан

К охлажденному льдом раствору 1,1-бис(диметил)фосфонат)метана (50 г, 215 ммоль) в сухом THF (500 мл) в атмосфере азота добавляют гидрид натрия (18,9 г, 60% в минеральном масле, 474 ммоль) тремя порциями в течение 30 мин. Смесь перемешивают в течение 3 часов, а затем добавляют 1-бром-2-трет-бутоксиэтан (90,5 г, 500 ммоль) 5 мл порциями. Ледяную баню удаляют через три часа и перемешивание продолжают при комнатной температуре в течение ночи. Реакционную смесь снова охлаждают на бане со льдом и гасят добавлением 50 мл насыщенного водного раствора хлорида аммония. Летучие вещества удаляют in vacuo и органические вещества растворяют в дихлорметане (300 мл). Кремний (100 г) перемешивают и суспензию фильтруют и осадок на фильтре промывают 3×200 мл дихлорметана. Продукт получают после удаления растворителей.
2b: 1,1-Бис(2-гидроксиэтил)-1,1-бис(диметилфосфонат)метан
Трифторуксусную кислоту (TFA, 50 мл) и дихлорметан (DCM, 50 мл) добавляют к 2 г 1,1-бис(2-трет-бутоксиэтил)-1,1-бис(диметилфосфонат)метану (пример 2а). Смесь перемешивают при комнатной температуре в течение 1 ч и летучие вещества удаляют при пониженном давлении с получением продукта.
2с: Синтез 1,1-бис(2-мезилоксиэтил)-1,1-бис(диметилфосфонат)метана

Продукт из примера 2b (1,1-бис(2-гидроксиэтил)-1,1-бис(диметилфосфонат)метан) (10 ммоль) растворяют в охлажденном льдом дихлорметане (10 мл). Добавляют пиридин (40 ммоль, 3,24 мл) и метансульфонилхлорид (3,44 г, 30 ммоль), поддерживая внутреннюю температуру <5°C. Через три часа добавляют эфир (30 мл) и воду (7 мл). После разделения фаз органический слой промывают 2 М HCl, 5% водным раствором гидрокарбоната натрия и водой. После сушки над сульфатом магния выпаривают летучие вещества с получением продукта.
2d: 1,1-Дивинил-1,1-бисдиметилфосфонат)метан

К охлажденному льдом раствору 1,1-бис(2-мезилоксиэтил)-1,1-бис(диметилфосфонат)метана (50 г, 104 ммоль) в сухом THF (500 мл) в атмосфере азота добавляют диэтилизопропиламин (300 ммоль). Ледяную баню удаляют через 30 минут и перемешивание продолжают при комнатной температуре в течение ночи. Летучие вещества удаляют in vacuo и органические вещества растворяют эфиром (300 мл). Кремний (100 г) и активированный уголь (15) перемешивают и суспензию фильтруют и осадок на фильтре промывают 3×200 мл эфира. Продукт получают после удаления растворителя.
2е: 1,1-Бис(2-триметоксисилилэтил)-1,1-бис(диметилфосфонат)-метан
К раствору 1,1-дивинил-1,1-бис(диметилфосфонат)метана (пример 2d) (2,0 ммоль) в сухом толуоле (20 мл) добавляют 80 мкл раствора катализатора Karstedt в толуоле (2% Pt) и токсисилане (6,0 ммоль, 4,1 мл). Раствор выдерживают при комнатной температуре в течение 2 дней. Летучие вещества удаляют in vacuo и остаточный силан удаляют посредством еще двух вакуумный циклов с добавлением толуола. Остаток растворяют в толуоле, обрабатывают небольшим количеством активированного угля, пропускают через 5 мкм PTFE фильтр и очищают с помощью флэш-хроматографии на колонке с силикагелем с использованием смеси дихлорметана с + 0-10% метанолом в качестве элюента с получением желаемого продукта.
Пример 3: Синтез 1,1-бис(триметоксисилилметил)-1,1-бис(диметилфосфонат)метана

К охлажденному льдом раствору 1,1-бис(диметил)фосфонат)метана (50 г, 215 ммоль) в сухом THF (500 мл) в атмосфере азота добавляют гидрид натрия (18,9 г, 60% в минеральном масле, 474 ммоль) тремя порциями в течение 30 минут. Смесь перемешивают в течение 3 часов и затем хлорметилтриэтоксисилан (500 ммоль) добавляют порциями, поддерживая внутреннюю температуру ниже 5°C. Ледяную баню убирают через три часа и перемешивание продолжают при комнатной температуре в течение ночи. Реакционную смесь охлаждают на бане со льдом и еще раз гасят добавлением 50 мл насыщенного водного раствора хлорида аммония. Летучие вещества удаляют in vacuo и органические вещества растворяют в дихлорметане (300 мл). Кремний (100 г) перемешивают и суспензию фильтруют и осадок на фильтре промывают 3×200 мл дихлорметана. Продукт получают после удаления растворителей.
Пример 4: Синтез 1,1-бис(3-триметоксисилилпропил)-1,1-бис(диэтилфосфонат)метана
4а: 1,1-Диаллил-1,1-бис(диэтилфосфонат)метан
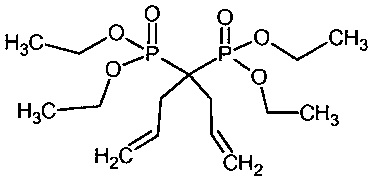
К охлажденному льдом раствору 1,1-бис(диэтил)фосфонат)метана (4,97 мл, 20 ммоль) в сухом THF (50 мл) в атмосфере азота добавили аллилбромид (8,7 мл, 100 ммоль). В течение двух часов добавляли трет-бутоксид калия (6,8 г, 60 ммоль). Раствор перемешивали при комнатной температуре в течение ночи и затем гасили добавлением 50 мл насыщенного водного раствора хлорида аммония. Летучие вещества удалили in vacuo и органические вещества растворили в дихлорметане. С помощью флэш-хроматографии на силикагеле в дихлорметане с метанолом (0-10% градиент) получили, по сути, чистый продукт (ЯМР).
4b: 1,1-Бис(3-триметоксисилилпропил)-1,1-бис(диэтилфосфонат)метан
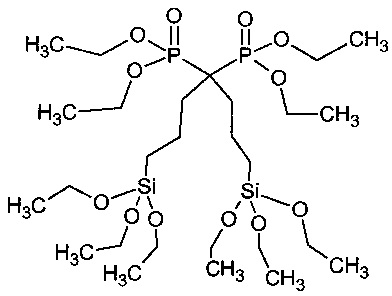
К раствору 1,1-диаллил-1,1-бис(диэтилфосфонат)метана (пример 2а) (4,4 г, 14,2 ммоль) в сухом толуоле (25 мл) добавили 212 мкл раствора катализатора Karstedt в толуоле (2% Pt) и триэтоксисилан (42,7 ммоль, 7,8 мл). Раствор выдерживали при комнатной температуре в течение ночи. Летучие вещества удалили in vacuo и остаточный силан удалили посредством еще двух вакуумных циклов с добавлением толуола. Остаток растворили в толуоле, обработали небольшим количеством активированного угля, пропустили через 5 мкм PTFE фильтр и очистили с помощью флэш-хроматографии на колонке с силикагелем с использованием смеси дихлорметана + 0-10% метанола в качестве элюента. Выход: 6,9 г вещества с 90% чистотой ЯМР.
Пример 5: Синтез 1,1-бис(3-триметоксисилилпропил)-1,1-бис(диизопропилфосфонат)метана
5а: 1,1-Диаллил-1,1-бис(диизопропилфосфонат)метан
К охлажденному льдом раствору 1,1-бис(диизопропил)фосфонат)метана (6,44 мл, 20 ммоль) в сухом THF (50 мл) в атмосфере азота добавили аллилбромид (8,7 мл, 100 ммоль). В течение двух часов добавили трет-бутоксид калия (6,8 г, 60 ммоль). Раствор перемешивали при комнатной температуре в течение ночи и затем гасили добавлением 50 мл насыщенного водного раствора хлорида аммония. Летучие вещества удаляли in vacuo и органические вещества растворяли в дихлорметане. С помощью флэш-хроматографии на силикагеле в дихлорметане с метанолом (0-10% градиент) получили 6,5 г по сути чистого продукта (ЯМР).
5b: 1,1-Бис(3-триметоксисилилпропил)-1,1-бис(диизопропилфосфонат)-метан

К раствору 1,1-диаллил-1,1-бис(диизопропилфосфонат)метана (пример 2а) (0,736 г, 2,0 ммоль) в сухом толуоле (20 мл) добавили 80 мкл раствора катализатора Karstedt в толуоле (2% Pt) и триэтоксисилан (6,0 ммоль, 4,1 мл). Раствор выдерживали при комнатной температуре в течение 2 дней. Летучие вещества удалили in vacuo и остаточный силан удалили посредством еще двух вакуумный циклов с добавлением толуола. Остаток растворили в толуоле, обработали небольшим количеством активированного угля, пропустили через 5 мкм PTFE фильтр и очистили с помощью флэш-хроматографии на колонке с силикагелем с использованием смеси дихлорметана с 0-10% метанолом в качестве элюента. Выход: 1,0 г вещества с 90% чистотой ЯМР.
Пример 6: Синтез 1,1-бис(3-триметоксисилилпропил)-1,1-бис(ди-(3-метоксифенилил)фосфонат)метана
6а: 1,1-Бис(ди-(3-метоксифенилил)фосфонат)метан
К охлажденному льдом раствору бис(дихлорфосфонат)метана в сухом дихлорметане (50 мл) добавили 3-метоксифенол (1,76 мл, 16 ммоль). Раствор триэтиламина (4,91 мл, 32 ммоль) добавляли в течение 1 ч. Реакционную смесь затем перемешивали при комнатной температуре в течение четырех часов, после чего ее вылили на воду со льдом (150 мл). Добавили дихлорметан и разделили фазы (медленно!). Водную фазу еще раз экстрагировали дихлорметаном и объединенные органические фазы высушили над сульфатом натрия. После выпаривания неочищенный продукт очистили с помощью флэш-хроматографии на силикагеле (колонка высотой 10 см, диаметр 3 см). Продукт получили в виде бледно-коричневого масла, и ЯМР-спектроскопия показала высокую чистоту. Выход 0,93 г.
6b: 1,1-Диаллил-1,1-бис(ди-(3-метоксифенилилметоксифенилил)фосфонат)метан
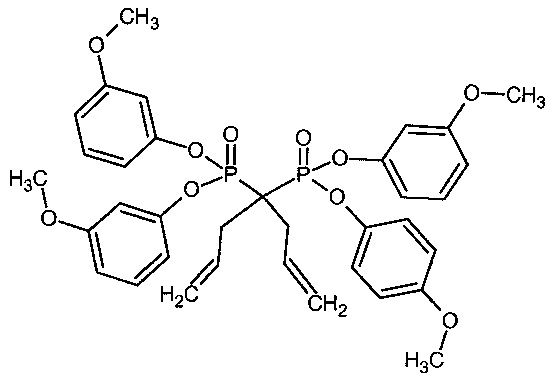
Гидрид натрия (0,683 г 60% в минеральном масле, 17,1 ммоль) суспендировали в сухом THF (150 мл) и охладили до -30°C. Раствор 1,1-бис(ди-(3-метоксифенилил)фосфонат)метан (пример 4а, 2,92 г, 4,87 ммоль) в сухом THF добавляли в течение 30 минут, поддерживая температуру при -30°C. Добавили аллилбромид (48,8 ммоль, 4,21 мл) и реакционную смесь выдерживали при -15°C в течение одного часа, затем нагревали до 40°C в течение 5 дней. Содержимое добавили к 75 мл насыщенного водного раствора хлорида аммония. Летучие вещества выпарили при пониженном давлении и твердые вещества растерли с дихлорметаном для извлечения органических веществ. После сушки над сульфатом натрия и выпаривания растворителя неочищенный продукт очистили с помощью флэш-хроматографии на силикагеле с гептаном: этилацетат 6:4 в качестве элюента. Выход 1,28 г.
6с: 1,1-Бис(3-триметоксисилилпропил)-1,1-бис(ди-(3-метоксиФенилил)-фосфонат)метан

К раствору 1,1-диаллил-1,1-бис(ди-(3-метоксифенилил)фосфонат)метана (пример 4b) (0,794 г, 1,16 ммоль) в сухом толуоле (20 мл) добавили 50 мкл раствора катализатора Karstedt в толуоле (2% Pt) и триэтоксисилан (1,16 ммоль, 0,459 мл). Раствор выдерживали при комнатной температуре в течение 4 дней и каждый день добавляли еще 0,7 г силана и 25 мкл катализатора. Летучие вещества удалили in vacuo и остаточный силан удалили путем еще двух добавлений толуол-вакуумных циклов. Остаток растворили в толуоле, обработали небольшим количеством активированного угля, пропустили через 5 мкм PTFE фильтр и очистили с помощью флэш-хроматографии на колонке с силикагелем с использованием смеси этилацетат: толуол 1:1 в качестве элюента. Выход 150 мг.
Пример 7: Синтез 1,1-бис(3-триметоксисилилпропил)-1,1-бис(ди-циклопропилметил)фосфонат)метана
7а: 1,1-Бис(ди-циклопропилметил)фосфонат)метан

К охлажденному льдом раствору бис(дихлорфосфонат)метана (1,00 г) в сухом дихлорметане (50 мл) добавили циклопропилметанол (1,15 г, 16 ммоль). Раствор триэтиламина (4,91 мл, 32 ммоль) добавляли в течение 1 ч. Реакционную смесь затем перемешивали при комнатной температуре в течение четырех часов, после чего ее вылили на воду со льдом (150 мл). Добавили дихлорметан и разделили фазы (медленно!). Водную фазу еще раз экстрагировали дихлорметаном и объединенные органические фазы высушили над сульфатом натрия. После выпаривания неочищенный продукт очистили с помощью флэш-хроматографии на силикагеле (колонка высотой 10 см, диаметром 3 см). Продукт получили в виде бесцветного масла, и ЯМР-спектроскопия показала высокую чистоту. Выход 1,04 г, 66%.
7b: 1,1-Диаллил-1,1-бис(ди-(цикпопропилметил)фосфонат)метан
К охлажденному льдом раствору 1,1-бис(ди-циклопропилметил)-фосфонат)метана (пример 5а) (0,794 г, 1,16 ммоль) в сухом THF (20 мл) в атмосфере азота добавили аллилбромид (0,864 мл, 10 ммоль). В течение двух часов добавляли трет-бутоксид калия (0,66 г). Раствор перемешивали при комнатной температуре в течение 4 ч и затем гасили добавлением 3 мл насыщенного водного раствора хлорида аммония. Летучие вещества удалили in vacuo и органические вещества растворили в дихлорметане. С помощью флэш-хроматографии на силикагеле в смеси гептан: этилацетат 3:07 получили 0,4 чистого продукта. 64%.
7с: 1,1-Бис(3-триэтоксисилилпропил)-1,1-бис(ди-циклопропилметил)-фосфонат)метан

К раствору 1,1-диаллил-1,1-бис(ди-(циклопропилметил)фосфонат)-метан (пример 5b) (0,373 г, 0,76 ммоль) в сухом толуоле (20 мл) добавили 30 мкл раствора катализатора Karstedt в толуоле (2% Pt) и триэтоксисилан (1,59 ммоль, 0,299 мл). Раствор выдерживали при комнатной температуре в течение 4 дней и каждый день добавляли еще 0,15 мл силана и 15 мкл катализатора. Летучие вещества удалили in vacuo и остаточный силан удалили путем еще двух добавлений толуол-вакуумных циклов. Остаток растворили в толуоле, обработали небольшим количеством активированного угля, пропустили через 5 мкм PTFE фильтр и очистили с помощью флэш-хроматографии на колонке с силикагелем с использованием смеси дихлорметана + 5% метанола в качестве элюента. Выход 376 мг.
Пример 8: Синтез наноструктуры Y1, конъюгированной с N-(2-аминоэтил)-16,16-ди-2-,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18-гексаоксаикосан-20-ным амидом
Пример 8а: Амид-3-(3-бром-2,2-бис(бромметил)пропокси)проп-1-ен
Гидрид натрия (1,67 г, 42 ммоль) осторожно добавили к 3-бром-2,2-бис(бромметил)пропанолу (9,75 г, 30 ммоль) и аллилбромиду (12,9 мл, 150 ммоль) в сухом и дегазированном DMF (40 мл) в атмосфере азота при 0°C. Температуру затем увеличили до комнатной температуры (22°C) и реакционную смесь перемешивали в течение еще 3 часов. Реакционную смесь затем осторожно добавили к водному насыщенному NH4Cl (50 мл). H2O-фазу затем экстрагировали диэтиловым эфиром (2×50 мл), объединенные органические фазы промыли H2O (5×50 мл) и затем насыщенным раствором соли (50 мл). Органическую фазу высушили над Na2SO4 с последующим фильтрованием. Летучие вещества удалили при пониженном давлении с получением бледно-желтого масла (9,7 г). С помощью колоночной хроматографии на силикагеле (гептан: EtOAc 9:1) получили 6,6 г (62%) продукта в виде прозрачного масла.1Н-ЯМР (CDCl3); 5,93 (м, 1H), 5,28 (м, 2H), 4,05 (д, 2H), 3,58 (с, 6H), 3,52 (с, 2H).
Пример 8b: 16-(Аллилоксиметил)-16-2,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18,21,24,27,30-денаоксагентриаконтан
Тетраэтиленгликоля монометиловый эфир (1,91 мл, 9 ммоль), растворенный в сухом и дегазированном DMF (3,5 мл, высушенный в течение 24 ч, 4Á MS) осторожно добавили к гидриду натрия (365 мг, 9 ммоль) в сухом и дегазированном DMF (15 мл, высушенный в течение 24 ч, 4Á MS) в атмосфере азота при 0°C с помощью шприца. Температуру затем повысили до комнатной температуры и реакционную смесь перемешивали в течение еще 30 мин 3-(3-Бром-2,2-бис(бромметил)пропокси)проп-1-ен (730 мг, 2,0 ммоль) затем добавили и температуру повысили до 100°C. Через 14 ч после завершения реакции (HPLC-ELSD-C18, от 95:5 до 5:95 H2O/ACN в течение 25 мин, Rt продукта = 19,5 мин) температуру понизили до комнатной температуры и реакционную смесь осторожно добавили на H2O (150 мл) и H2O-фазу промывают диэтиловым эфиром (2×50 мл). Хлорид натрия затем добавляли к H2O-фазе до насыщения. H2O-фазу экстрагировали EtOAc (4×50 мл) и объединенные органические фазы промыли насыщенным раствором соли (2×30 мл). Сульфат натрия и уголь добавили к органической фазе. Очищенную органическую фазу профильтровали и летучие вещества удалили при пониженном давлении (8 мм рт. ст., 40°C, затем 0,1 мм рт. ст. (масляный насос) и 40°C с целью удаления остаточного DMF). С помощью колоночной хроматографии (EtOAc:MeOH 9:1) получили 1,05 г (70%) продукта.1Н-ЯМР (CDCl3); 5,90 (м, 1H), 5,20 (м, 2H), 3,94 (дт, 2H), 3,70-3,55 (м, 48H), 3,45 (с, 6H), 3,43 (с, 2H), 3,40 (с, 9H).
Пример 8с: 16,16-Ди-2,5,8,11,14-пентаоксапентадецил-2,5,8,11,14-пентаоксагептадекан-17-ол (4)
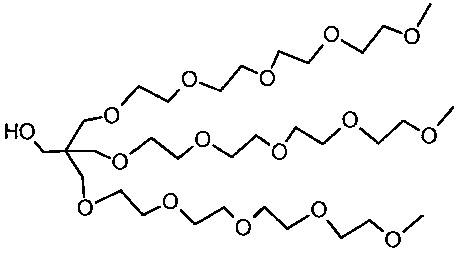
Трет-бутоксид калия (74 мг, 0,66 ммоль) добавляли к 2 (500 мг, 0,66 ммоль) в DMSO (3 мл). Реакционную смесь встряхивали при 100°C в течение 15 мин. Анализ HPLC (HPLC-ELSD-C18, от 95:5 до 5:95 H2O/ACN в 25 мин) показал полное превращение в продукт. Рассол (20 мл) добавили при комнатной температуре и водную фазу экстрагировали этилацетатом (3×20 мл). Объединенные органические фазы промыли рассолом (3×20 мл) и высушили над сульфатом натрия. Фильтрование и удаление летучих веществ при пониженном давлении дает 16-2,5,8,11,14-пентаоксапентадецил-16-((проп-1-энилокси)метил)-2,5,8,11,14,18,21,24,27,30-декаоксагентриаконтан в виде прозрачного масла. HCl (0,1 М) затем добавили в масло, растворенное в ацетоне (4 мл) и смесь взбалтывали при 55°C в течение 30 мин. Летучий материал затем удалили при пониженном давлении, что дало 420 мг (89%) 4 в виде прозрачного масла.1Н-ЯМР (CDCl3); 3,66-3,52 (м, 48H), 3,47 (с, 6H), 3,37 (с, 9H).
Пример 8d: Ттерт-бутил-16,16-ди-2,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18-гексаоксаикосан-20-оат (5)
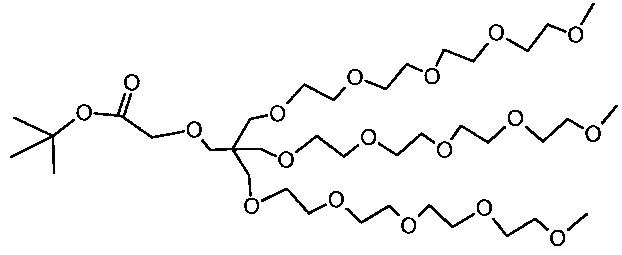
Трет-бутоксид калия (32 мг, 0,28 ммоль) добавили к 4 (100 мг, 0,14 ммоль) и трет-бутил-2-бромацетат (105 мг, 0,54 ммоль) в сухом THF (3 мл). Реакционную смесь взбалтывали в течение 30 мин. Добавили диэтиловый эфир (10 мл) и рассол (5 мл) и водную фазу экстрагировали этиловым ацетатом (3×20 мл). Объединенные органические фазы промыли рассолом, а затем высушили над сульфатом натрия. Летучие вещества удалили при пониженном давлении и сырой продукт очистили с помощью колоночной хроматографии (этилацетат/метанол 9:1), что дало 60 мг (52%) 5.1Н-ЯМР (CDCl3); 3,91 (с, 2H), 3,66-3,52 (м, 48H), 3,51 (с, 2H), 3,45 (с, 6H), 3,37 (с, 9H), 1,46 (с, 9H).
Пример 8е: 16,16-Ди-2,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18-гексаоксаикосан-20-овая кислота (6)

Трифторуксусную кислоту (TFA, 0,5 мл) и дихлорметан (0,5 мл) добавили к 20 мг 5. Смесь взбалтывали при комнатной температуре в течение 1 ч и летучие вещества затем удаляли при пониженном давлении с получением 18 мг 6 в виде желтого масла.
Пример 8f: N-(2-трет-Бутоксикарбониламидоэтил)-16,16-ди-2,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18-гексаоксаикосан-20-овый амид
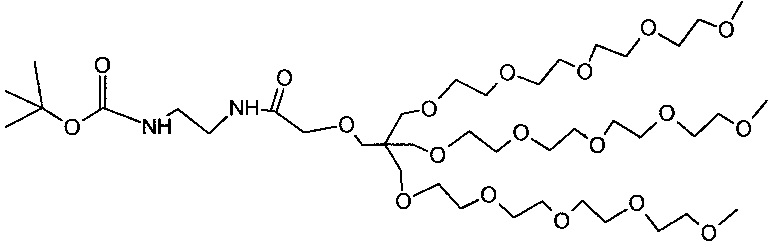
2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (95 мг, 0,25 ммоль) добавили к 16,16-ди-2-,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18-гексаоксаикосан-20-вой кислоте (Пример 8е) (153 мг, 0,2 ммоль), N-BOC-этилендиамин (40 мг, 0,25 ммоль) и диизопропилэтиламин (87 мкл, 0,5 ммоль) в DMF (1 мл, высушенный 4 МС и дегазированный) при комнатной температуре в атмосфере азота. Реакционную смесь взбалтывали в течение 20 часов. К реакционной смеси добавили диметил эфир и смесь экстрагировали 3 раза H2O. Объединенные водные фазы насытили NaCl (с) и затем экстрагировали 4 раза этилацетатом. Объединенные органические фазы высушили над NaSO4, профильтровали и растворитель удалили при пониженном давлении с получением 190 мг (выход) продукта в виде бледно-желтого масла. HPLC анализ (HPLC-ELSD-C18, 90:10-5:95 TFA 0,1% в H2O/ACN в течение 20 мин) показал отдельный пик на 14,5 мин.
Пример 8g: N-(2-Аминоэтил)-16,16-ди-2,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18-гексаоксаикосан-20-вый амид
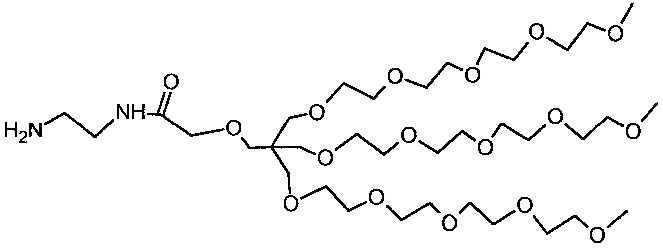
TFA (2 мл) добавили к N-(2-т-бутоксикарбониламидоэтил)-16,16-ди-2,5,8,11,14-пентаоксапентадецил-2,5,8,11,14,18-гексаоксаикосан-20-вому амиду (пример 8f, 160 мг, 0,18 ммоль) в дихлорметане (2 мл). Смесь перемешивали в течение 1 ч при комнатной температуре. Летучие компоненты удалили при пониженном давлении и остаток совыпаривали дважды с сухим толуолом (Al2O3) и затем высушили с помощью масляного насоса. Это дало 130 мг продукта. Анализ HPLC (HPLC-ELSD-C18, 90:10 до 5:95 TFA 0,1% в H2O/MeCN в течение 20 мин) показал отдельный пик на 10,7 мин. МС (ESP+) [М]:807,5.
Пример 8h: Сопряжение связей (конъюгация)
Наноструктуру Х1 (пример 10 с, 100 мг, 0,4 ммоль Р экв.) растворили в H2O (2 мл) путем обработки ультразвуком. PH доводили от 1,9 до 10,4, используя 6 и 1 М NaOH (водн.). Затем добавили хлорид марганца (12,5 мг, 0,065 ммоль), растворенный в H2O (2 мл). Смесь взбалтывали в течение 30 мин при 30°C, pH доводили от 8,5 до 7,1, используя 0,1 HCl (водн.) и материал из примера 8g (37 мг, 0,04 ммоль) и N-гидроксисульфосукцинимида натриевую соль (9 мг, 0,04 ммоль) растворили в H2O (2 мл). N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (24 мг, 0,12 ммоль) затем добавили. Реакционную смесь взбалтывали при комнатной температуре в течение 21 ч и затем профильтровали (5 мкм трубчатый фильтр). Фильтрат профильтровали спиновой фильтрацией (10k отсечка, 3000 G в течение 30 мин) и концентрат (4 мл) разбавили до 15 мл с помощью H2O. Эту процедуру повторили 4 раза. РН концентрата (4 мл) доводили от 4,7 до 7,1 с помощью 0,1 М NaOH (водн.). Фильтрат профильтровали спиновой фильтрацией (10k отсечка, 3000 G в течение 15 мин) и концентрат (0,5 мл) развели в 4 мл с помощью H2O. Эту процедуру повторяли 4 раза. Итоговый концентрат профильтровали (шприцевой фильтр 0,2 мкм) и развели до 2 мл с помощью H2O. Объемное распределения частиц по размерам = 4-5 нм. GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH 7,4, поток 1 мл/мин) Rt=10,3 мин.
Пример 9: Синтез полиэтиленимин-бисфосфонатной наноструктуры Z
Пример 9а: Трет-бутиловый эфир 3,3-бис(диметоксифосфорил)пропановой кислоты

К охлажденному льдом раствору бис(диметоксифосфорил)метана (4,64 г, 20 ммоль) и трет-бутилового эфира бромуксусной кислоты (7,35 мл, 50 ммоль) в сухом THF (40 мл) в атмосфере азота добавили трет-бутоксид калия (5,8 г, 43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем гасили с помощью 4 мл насыщенного раствора хлорида аммония. Летучие вещества удалили in vacuo и с помощью двух циклов с добавлением толуола и выпаривания. С помощью флэш-хроматографии в дихлорметане: метанол, 95:5 получили продукт в виде масла. Выход 4,0 г.
Пример 9b: 3,3-Бис(диметоксифосфорил)пропановая кислота

К раствору 3,3-бис(диметоксифосфорил)пропановой кислоты трет-бутилового эфира (2,5 г) в дихлорметане (10 мл) добавили трифторуксусную кислоту. После перемешивания при комнатной температуре выпарили летучие вещества in vacuo. В результате трех цикла вакуумного испарения 5 мл порциями толуола получили 2,2 г продукта.
Пример 9с: Ссинтез полиэтиленимин-бисфосфонатной наноструктуры Z
Разветвленный полиэтиленимин со средней молекулярной массой 25 кДа (100 мг, 2,5 ммоль простейших аминогрупп), 3,3-бис(диметоксифосфорил)пропановой кислоты (1,1 г, 3,87 ммоль) и N-гидроксисульфосукцинимида натриевой соли (100 мг, 0,46 ммоль) растворили в H2O (10 мл) путем обработки ультразвуком (10 мин). РН доводили от 1,8 до 6,5 с помощью 1М NaOH, после чего добавили N-(3-диметиламинопропил)-N'-этилкарбодиимид (1,0 г, 5,2 ммоль). Реакционную смесь взбалтывали при комнатной температуре в течение 23 ч и затем профильтровали (5 мкм шприцевой фильтр). Фильтрат профильтровали спиновой фильтрацией (10k отсечка, 3000 G в течение 30 мин) и ретентат (2 мл) разбавили до 10 мл с помощью H2O. Эту процедуру повторили 4 раза. Конечный ретентат (2 мл) разбавили до 6 мл с использованием H2O. Средний объем распределения частиц по размерам согласно DLS: 4-5 нм. GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=9,1 мин.
Пример 10: Полимеризация 1,1-бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метана с получением наноструктур X
1,1-Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан (XG, у ммоль, см. таблицу 1 ниже) растворили в 200 мл водного 80%-1-пропанола в сосуде высокого давления. Реакционную смесь перемешивали в течение 48 ч при 95°C и затем в течение 24 ч при 110°C. После охлаждения очищенного раствора до комнатной температуры его разбавили MilliQ H2O (800 мл) и затем профильтровали диафильтрованием, используя 300k номинальную границу отсечки по молекулярному весу задерживаемых компонентов (NMWC) колонки распределения пор (ультрафильтрующий картридж Midgee от Heathcare, модель: UFP-300-С-3МА). Собранные фильтраты (~980 мл) затем собирали на диафильтрационной колонке с размером пор 100k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-100-С-3МА) для концентрации наноструктурного раствора. Кроме того, фильтры от Pall Life Sciences также использовали, в частности кассеты Centramate Т-серии OS0100T02 (размер пор кассеты 100k NMWC). Выполнили повторное добавление воды MilliQ и фильтрацию собранного концентрата. Конечный объем собранного концентрата (X1) составил приблизительно 50 мл.
Кроме того, собрали фильтрат, который прошел через диафильтрационную колонку 100k и затем профильтровали с использованием NMWC фильтрующего картриджа с размером пор 30k (GE ультрафильтрующий картридж Midgee от Heathcare, модель: UFP-30-С-3МА). Выполнили повторное добавление (2х) MilliQ воды и фильтрацию собранного концентрата. Итоговый объем собранного концентрата (X2) составил приблизительно 50 мл.
Пример 10а: Полимеризация 100 мМ 1,1-бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метана
X1a. Количества: x=12,8 г, y=20 ммоль. Восстановление после диафильтрации = 26% (на основе восстановления Р); конечное значение pH ~2; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH 7,4, поток 1 мл/мин) Rt=9,2 мин; Состав (ICP, мольное отношение): P/Si=0,9.
X2a. Количества: x=12,8 г, y=20 ммоль. Восстановление после диафильтрации = 17% (на основе восстановления Р); конечное значение pH ~2; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=10,3 мин; композиция (ICP, молярное отношение): P/Si=0,9.
Пример 10b: Полимеризация 80 мМ 1,1-бис(триэтоисисилилпропил)-1,1-бис(диметилфосфонат)метана
X1b. Количества: x=10,4 г, y=16 ммоль. Восстановление после диафильтрации = 31% (на основе восстановления Р); конечное значение pH ~2; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=9,2 мин; композиция (ICP, молярное отношение): P/Si=1,1.
X2b. Количества: x=10,4 г, y=16 ммоль. Восстановление после диафильтрации = 19% (на основе восстановления Р); конечное значение pH ~2; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=10,4 мин; композиция (ICP, молярное отношение): P/Si=1,1.
Пример 10с: Полимеризация 50 мМ 1,1-бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метана
X1c. Количества: x=6,4 г, y=10 ммоль. Восстановление после диафильтрации = 21% (на основе восстановления Р); Конечное значение pH ~2; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=9,7 мин; композиция (ICP, молярное отношение): P/Si=0,9.
X2c. Количества: x=6,4 г, y=10 ммоль. Восстановление после диафильтрации = 25% (на основе восстановления Р); Конечное значение pH ~2; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=10,5 мин; композиция (ICP, молярное отношение): P/Si=0,9.
Пример 10d: Полимеризация 1,1-бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метана с получением наноструктур X в различных растворителях
1,1-Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан (3,2 г, 5 ммоль) растворили в водном 80% этиленгликоле (100 мл). Реакционную смесь перемешивали в течение 21 ч при 116°C. Полимеризацию можно также проводить, как описано выше, но в водном 80% 1,2-пропандиоле, перемешанном в течение 24 ч при 111°C и затем в течение 4 ч при 114°C или в водном 80% диэтиленгликоле, перемешанном в течение 20 ч при 108°C и затем в течение 2 ч при 114°C или в водном 80% триэтиленгликоле, перемешанном в течение 22 ч при 115°C или в водном 80%-ди (этиленгликоль) метиловом эфире, перемешанном в течение 18 ч при 106°C и затем в течение 18 ч при 111°C или в водном 80%-моноэтиловом эфире диэтиленгликоля, перемешанном в течение 35 ч при 107°C или в водном 80% глицерине, перемешанном в течение 19 ч при 114°C.
Пример 10е: Pt-поглощение 1,1-бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метана
SIR-200 (100 г, хелатирующие смолы, тиол, Н формы), предоставленный Resintech взбалтывали два раза с 5% водным раствором бикарбоната натрия (500 мл) и затем взбалтывали два раза с MilliQ водой. Воду отфильтровали и сухой толуол (100 мл) добавили к SIR-200. Летучие вещества удалили in vacuo и остаточную воду удалили посредством еще двух вакуумных циклов с добавлением толуола. 1.1 Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат) метан (30 г, содержание платины: 39 частей на миллион) растворили в сухом толуоле (300 мл) в сосуде. Добавили SIR-200 (10 г, обработанный, как описано выше) и затем взбалтывали в течение ночи. SIR-200 отфильтровали и летучие вещества удалили in vacuo с получением материала с содержанием платины 0,38 частей на миллион.
Пример 11. Введение марганца в наноструктуру X и очистка посредством тангенциальной проточной фильтрации с получением наноструктур Y
PH раствора наноструктур X (пример 13) доводили от pH 2 до pH 10,4, используя 6 М и 1 М NaOH (водн.) и оставили настаиваться в течение 2 ч. Марганца(II) хлорид тетрагидрат (xx мг, yy ммоль) затем добавили и растворили. Смесь взбалтывали в течение 1 ч при 30°C. PH смеси после реакции составил приблизительно pH 7,6, и его доводили до pH 7,4 с использованием 1 М HCl (водн.). Реакционную смесь разбавили до 50 мл MilliQ H2O и затем подвергли диафильтрации с использованием колонки распределения пор 10k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-10-C-3МА) с целью удаления свободного иона Mn. В качестве альтернативы, фильтр от Pall Life Sciences также используют, в частности кассеты Centramate серии T OS010T02 (размер пор кассеты 10k NMWC). Концентрат собрали и процедуру диафильтрации повторили трижды.
Y1a. В наноструктуре X использованы: пример X1a, 15 мл, 3,2 ммоль P. MnCl2 4H2O использованы: xx=106,7 мг, yy=0,54 ммоль. Конечное значение pH 7,4; максимальный объемный размер частиц (DLS в 150 мМ NaCl)=5,6 нм; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=9,5 мин; композиция (ICP, молярное отношение): P/Mn=5,7, P/Si=0,9, SI/Mn=6,2; ионнообменная стабильность при pH 5,5=45% и при pH 7=62%.
Y1b. В наноструктуре X использованы: пример X1a, 15 мл, 3,2 ммоль P. MnCl2 4H2O использованы: xx=107 мг, yy=0,54 ммоль. Конечное значение pH 7,4; объемный размер частиц (DLS в 150 мМ NaCl) максимум = 6,5 нм; GPC анализ (супероза 12 10/300 GL 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=10,1 мин & плечо при 9 мин; композиция (ICP, молярное отношение): P/Mn=5,4, P/Si=0,9, Si/Mn=6; ионнообменная стабильность при pH 5,5=47% и при pH 7=66%.
Y2a. В наноструктуре X использованы: пример X2a, 15 мл, 2 ммоль P. MnCl24H2O использованы: xx=71 мг, yy=0,36 ммоль. Конечное значение pH 7,4; объемный размер частиц (DLS в 150 мМ NaCl) максимум = 4,1 нм; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=10,5 мин; композиция (ICP, молярное отношение): P/Mn=6,6, P/Si=0,9, Si/Mn=7,3; ионнообменная стабильность pH 5,5=43% и при pH 7=60%; n при 81,33 МГц, 25°C=41 мМ-1 Mn с-1.
Y2b. В наноструктуре X использованы: пример X2a, 15 мл, 2 ммоль P. MnCl24H2O использованы: xx=71 мг, yy=0,36 ммоль. Конечное значение pH 7,4; объемный размер частиц (DLS в 150 мМ NaCl) максимум = 5,6 нм; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=10,1 мин; композиция (ICP, молярное отношение): P/Mn=5,6, P/Si=0,9, Si/Mn=6,2; ионнообменная стабильность при pH 5,5=44% и при pH 7=63%.
Пример 11а: Полимеризация 1,1-бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метана с получением наноструктур X11a'
1,1-Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан (0,64 г, 1 ммоль) растворили в водном 80% этиленгликоле (12 мл).Формиат натрия в диапазоне от 28 мг (0,42 ммоль) или 140 мг (2,1 ммоль) и марганца(II) тетрагидрат хлорид (33 мг, 0,17 ммоль) растворили в водном 80% этиленгликоле (4 мл) каждый и последовательно добавили к реакционной смеси, которую перемешивали в течение 22 ч при 114°C.
11а'':
1,1-Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан (0,64 г, 1 ммоль) растворили в водном 80% этиленгликоле (12 мл). Формиат калия (52 мг, 0,62 ммоль) и марганца(II) тетрагидрат хлорид (33 мг, 0,17 ммоль) растворили в водном 80% этиленгликоле (4 мл) каждый и последовательно добавили к реакционной смеси, которую перемешивали в течение 21 ч при 116°C.
11а''':
1,1-Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан (0,64 г, 1 ммоль) растворили в водном 80% этиленгликоле (16 мл). Тетраметиламмония формиат (30 мас.% раствор в воде, 0,245 мл, 0,62 ммоль) и марганца(II) тетрагидрат хлорид (33 мг, 0,17 ммоль) растворили в водном растворе 80% этиленгликоля (4 мл) последовательно добавили к реакционной смеси, которую перемешивали в течение 21 ч при 116°C.
Пример 11b: Введение марганца в наноструктуру X, "отвержденную" добавлением силанов и очистка диафильтрацией с получением наноструктур Z
К 2 мл раствора наноструктуры X (пример 10f) добавили × мл дегазированного марганец(II) тетрагидрат хлорида (растворенного в 80% водном этиленгликоле, 100 мМ), соблюдая молярное отношение фосфор-марганец 12. У мл дегазированного формиата натрия (100 мМ), растворенного в 80% водном этиленгликоле добавляют в насыщенный Mn наноструктурный раствор, удовлетворяющий молярному отношению формиата натрия к марганцу 5 или 3. Конечное значение pH проверяют и устанавливают приблизительно на pH 5 или 3, при необходимости добавляя NaOH (водн.) или HCl (водн.). Смесь взбалтывали в течение 12 или 18 ч при 100°C. Z мл тетраэтилортосиликата (TEOS), растворенного в этаноле (120 мМ), добавляют к смеси и взбалтывают еще в течение 18 или 24 ч при 100°C. (альтернативой добавлению TEOS после первого этапа нагревания является введение раствора TEOS непосредственно в насыщенный Mn наноструктурный раствор, содержащий Na-формиат и взбалтывание в течение 12 или 18 ч при 100°C).
После нагрева и взбалтывания pH доводили до pH 7,0±0,5 путем добавления NaOH (водн.) с последующей ультрафильтрацией (UF) с использованием 4-мл 100 кДа центробежного фильтра (Centriprep ® от Millipore). Раствор сначала разбавили Milli-Q до приблизительно 4-мл и центрифугировали в течение 10-15 мин (3000 × г). Фильтрат собрали и переместили к 4-мл 10 кДа центробежному фильтру (Centriprep ® от Millipore), разбавленный до 4-мл Milli-Q водой, тщательно перемешали и профильтровали спиновой фильтрацией 3000 × г, 10 мин), после чего собрали приблизительно 500 мкл концентрата. Процедуру разбавления и диафильтрации повторили три раза. Итоговые собранные 500 мкл концентрата разбавили до 1 мл MilliQ водой. После чего последовали определение концентрации Mn, комплексометрическое испытание на стабильность, оцененное с помощью релаксометрии (пример 14b), GPC анализ и анализ состава (ICP измерение).
Z1a. 3% TEOS относительно бисбис. В наноструктуре X использованы: 2 мл, 0,2 ммоль P; x=167 мкл; отношение Na-формат-Mn: 5; y=835 мкл; z=25 мкл конечное значение pH до нагрева: pH 5; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=12,6 мин; композиция (ICP, молярное отношение): P/Mn=11,61, P/Si=1,01, Si/Mn=10,51; комплексонометрическая стабильность при pH 7=24%.
Z1b. 5% TEOS относительно бисбис] В наноструктуре X использованы: 2 мл, 0,2 ммоль Р; x=167 мкл; отношение Na-формат-Mn: 5; y=835 мкл; z=41,7 мкл конечное значение pH перед нагревом: pH 5; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=12,6 мин; композиция (ICP, молярное отношение): P/Mn=12,05, P/Si=1,02, Si/Mn=11,77; Комплексонометрическая стабильность при pH 7=23%.
Z1c. 3% TEPS относительно бисбис. В наноструктуре X использованы: 2 мл, 0,2 ммоль P; x=167 мкл; отношение Na-формат-Mn: 5; y=835 мкл; z=25 мкл конечное значение pH перед нагревом: pH 3,5; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=12,6 мин; композиция (ICP, молярное отношение): P/Mn=13,03, P/Si=0,93, Si/Mn=13,96; Комплексонометрическая стабильность при pH 7=27%.
Z1d. 5% TEOS относительно бисбис. В наноструктуре X использованы: PL04064, 2 мл, 0,2 ммоль Р; x=167 мкл; отношение Na-формат-Mn: 5; y=835 мкл; z=41,7 мкл конечное значение pH перед нагревом: pH 3,5; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин) Rt=12,6 мин; композиция (ICP, молярное отношение): P/Mn=13,31, P/Si=0,92, Si/Mn=14,53; Комплексонометрическая стабильность при pH 7=26%.

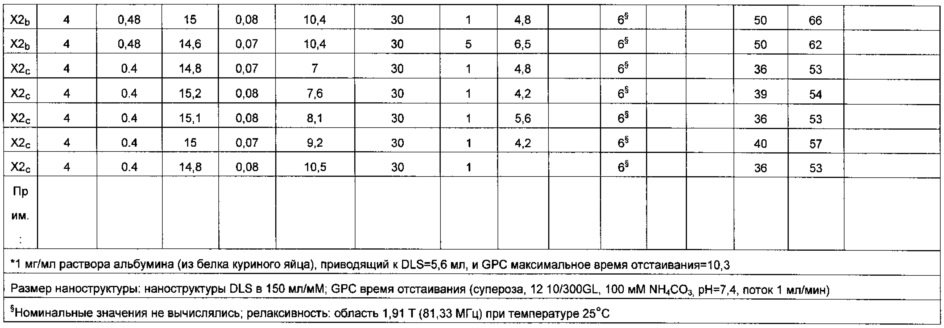
Пример 12. Высушенная замораживанием, составленная, насыщенная Mn наноструктура
1,1-Бис(триэтоксисилилпропил)-1,1-бис(диметилфосфонат)метан (6,4 г, 0,01 ммоль) растворили в 200 мл водного 80% 1-пропанола в сосуде высокого давления. Реакционную смесь перемешивали в течение 48 ч при 95°C и затем 24 ч при 110°C. Температуру понизили до комнатной температуры и прозрачный, бесцветный раствор собрали. Собранный раствор разбавили MilliQ H2O (800 мл) и затем профильтровали с помощью колонки распределения пор 300k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-300-C-3МА). Собранный фильтрат (~980 мл) затем профильтровали с использованием диафильтрационной колонки с размером пор 100k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-100-С-3МА) для концентрирования раствора полимера. Выполнили повторное добавление MilliQ воды и фильтрацию собранного концентрата. Окончательный объем собранного концентрат составил приблизительно 50 мл. Композиция (ICP, молярное отношение): P/Si=0,84.
Кроме того, фильтрат, который проходит через 100k диафильтрационную колонку собрали и затем фильтровали с использованием диафильтрационной колонки с размером пор 30k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-30-С-3МА). Выполнили повторное добавление (2 x) MillIQ воды и фильтрацию собранного концентрата. Окончательный объем собранного концентрата составил приблизительно 50 мл. Композиция (ICP, молярное отношение): P/Si=0,88.
pH наноструктуры, которая проходит через 100k диафильтрацию (25 мл, 2,1 ммоль) установили на pH 2,2-10,5 с помощью 6 М и 1 М NaOH (водн.) и выдерживали в течение 2 часов. Затем добавили марганец(II) хлорид тетрагидрат (45,4 мг, 0,23 ммоль). Смесь взбалтывали в течение 16 ч при 30°C. PH смеси после реакции составила 9,3 и затем ее установили на pH 7,4 с помощью 1 М HCl (водн.). Смесь разбавили до 50 мл MilliQ H2O и затем подвергли диафильтрации с использованием колонки распределения пор 10k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-10-С-3МА). Концентрат собрали и процедуру разбавления и диафильтрации повторили три раза. Итоговый объем собранного раствора составил 10 мл.
К 8,1 мл собранного раствора добавили маннит (0,36 г, 2,0 ммоль) для достижения концентрации 250 мМ. После чего последовала сушка вымораживанием в течение 16 ч, сбор 0,5 г белого рассыпчатого порошка. 20 мг/мл водного раствора лиофилизированного продукта получали и проанализировали. Размер частиц, средневзвешенный (DLS в 150 мМ NaCl) максимум = 4,8 нм; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, рН=7,4, расход 1 мл/мин) Rt=10,3 мин; композиция (ICP, молярное отношение): P/Mn=9,8, P/Si=0,9, Si/Mn=10,7; ионнообменная стабильность при pH 5,5=72% и при pH 7=89%; n при 60 МГц, 37°C=39 мМ-1 Mn с-1.
Пример 13. Дополнительная очистка наноструктуры Y с помощью ионообменной смолы
Для дальнейшего удаления избытка или слабо связанных ионов Mn, образец Y1 обработали катионным обменником (сульфонированный полистирол): ~10 мл насыщенной Mn наноструктуры (~10 мМ Mn) смешали с 1 г Dowex 50WX4 (№форма, предварительно промытая водой) и pH установили на 7,0 с помощью 0,1 М NaOH. Смесь осторожно вращали в течение 16 ч и затем центрифугировали при 3000 оборотах в минуту.
Пример 14. Измерение стабильности для марганецсодержащей наноструктуры (также называемой "ионообменной стабильности")
Сперва определили концентрацию марганца в наноструктурном растворе и затем разбавили водой до концентрации марганца 1,5 мМ и конечного объема 2,2 мл. К 2×1,000 мл разбавленного раствора образца добавили 2×100 мг Dowex 50WX4 (форма Na, промытая водой). PH установили на 7,0 и 5,5 в двух растворах соответственно 0,1 М NaOH или 0,1 М HCl (обычно всего лишь несколько микролитров требуется). Смесь хорошо перемешивали в течение 16 ч при медленном вращении сосуда. IEX частицам дали осесть и 100 мкл аликвоту, взятую из надосадочной жидкости, проанализировали ([Mn]iex). Для определения начальной концентрации марганца в образце оставшийся вышеуказанный раствор использовали для определения [Mn]нач. Стабильность рассчитывали как: [Mn]iEx/[Mn]нач*100 (%)
Пример 14b. Комплексонометрическое испытание на стабильность для марганецсодержащих наноструктур, определены с помощью релаксометрии
Измеряли продольную релаксивность наноструктурного раствора (r1(ns)) с концентрацией 1 мМ Mn. Готовили другой наноструктурный раствор, содержащий 1 мМ Mn и добавьте эквивалентное молярное количество EDTA. Ph данного раствора должен быть отрегулирован до pH 7±0,5, если это необходимо. Измеряли продольную релаксацию данного наноструктурного раствора с добавленным EDTA (r1(NS+EDTA)). В качестве эталонной среды приготовили 1 мМ раствор, используя стандарт марганца для AAS (Fluka 77036), добавили эквимолярное количество EDTA и pH довели до pH 7±0,5. Продольную релаксацию измеряли (r1(MN+EDTA)) и получали в результате значение 1,6 мМ-1 с-1. Для расчета % Mn, высвобожденного из наноструктуры после добавления эквивалентной ЕРТА:

Примечание: релаксацию (T1, в секундах) измеряют с помощью ЯМР анализатора Minispec mq60 (60 МГц) при 37°C, и релаксивность r1 рассчитывается по формуле:

где T1 H2O=0,32 с; C1=1 мМ.
Пример 15. Трансметаллирование с кальцием и марганцем
Буфер, приблизительно имитирующий неорганические компоненты крови, но без кальция смешали с NaCl, (7,14 г), NaHCO3 (1,4 г), KHCO3 (0,43 г), NaH2PO4 (0,165 г), Mr(OAc)2 (0,17 г), разбавленных до объема в 1,00 л. Далее именуемый "буфер крови". С помощью двух образцов наноструктур анализировали А, (Y1, вырезанный между 300 кДа и 100 кДа фильтрами) и В (Y2, вырезанный между 100 кДа и 30 кДа фильтрами). Пробирки подготовили соотвественно:
Образцы смешали с анализируемым А до общей концентрации Mn 1,3 мМ и отнесенными к В до 1,5 мМ. Растворы инкубировали при комнатной температуре в течение одного часа и затем пропускали через отсекающий спиновой фильтр 10 кДа. Фильтраты проанализировали на марганец. Результаты показаны в таблице 2.

Пример 16. Насыщенный Mn диметил-2-(триэтоксисилил) этилфосфонат полимер
Диметил-2-(триэтоксисилил) этилфосфонат (DTEP, 0,9 г, 3,0 ммоль) растворили в 30 мл водного 80%-1-пропанола в сосуде высокого давления. Реакционную смесь перемешивали в течение 48 ч при 95°C и затем в течение 24 ч при 120°C Температуру понизили до комнатной температуры и собрали прозрачный, бесцветный раствор. Собранный раствор разбавили MilliQ H2O (470 мл) и затем профильтровали с использованием колонки распределения пор 100k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-100-С-3МА). Собранный фильтрат (480 мл) затем профильтровали с использованием диафильтрационной колонки с размером пор 30k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-30-С-3МА), чтобы концентрировать раствор полимера. Выполнили повторное добавление MilliQ воды и фильтрацию собранного концентрата. Итоговый объем собранного концентрата составил приблизительно 7,5 мл. Композиция (ICP, молярное отношение): P/Si=0,5.
РН 7,5 мл полимерного раствора DTEP (0,4 ммоль P) отрегулировали от pH 2,6 до pH 10,4, используя 6 М и 1 М NaOH (водн.) и оставили отстаиваться в течение 2 часов. Затем добавили марганец(II) хлорид тетрагидрат (5,9 мг, 0,3 ммоль). Смесь взбалтывали в течение 1 ч при 30°C. PH смеси после реакции составил приблизительно pH 9,4 и затем его доводили до pH 7,4, используя 1 М HCl (водн.). Реакционную смесь разбавили до 50 мл MilliQ H2O и затем подвергли диафильтрации с использованием колонки распределения пор 10k NMWC (ультрафильтрующий картридж Midgee от GE Heathcare, модель: UFP-10-С-3МА) с целью удаления свободных ионов Mn. Концентрат собрали и процедуру разбавления и диафильтрации повторили три раза. Максимальный средневзвешенный размер частиц (DLS в 150 мМ NaCl)=5,6 нм; GPC анализ (супероза 12 10/300 GL, 100 мМ NH4CO3, pH=7,4, поток 1 мл/мин); композиция (ICP, молярное отношение): P/Mn=2,7, P/Si=0,5, Si/Mn=5,7; ионнообменная стабильность pH 5,5=21% и при pH 7=24%; r1 при 81,3 МГц, 25°C=3 мМ-1 Mn с-1.
Пример 17. Насыщенная Mn золедроновая кислота
Золедроновую кислоту (27 мг, 0,01 ммоль) растворили в 10 мл MilliQ H2O. Приготовили 100 мМ водного раствора марганца(II) хлорид тетрагидрата. 10 мкл Mn-раствора смешали с 408 мкл раствора золедроновой кислоты и 582 мкл MilliQ H2O. РН доводили до 7,4 с помощью 6 М NaOH (водн.). Композиция (ICP, молярное отношение): P/Mn=5,26. r1 при 81,3 МГц, 25°C=2,3 мМ-1 Mn с-1.
Пример 18. Насыщенная Mn метилендифосфоновая кислота
Метилендифосфоновую кислоту (9,2 мг, 0,05 ммоль) растворили в 5 мл MilliQ H2O. Приготовили 28 мМ водный раствор марганец(II) хлорид тетрагидрата. 35 мкл Mn-раствора смешивают с 286 мкл раствора метилендифосфоновой кислоты и 679 мкл MilliQ H2O. pH доводили до 7,1 с помощью 6 М NaOH (водн.). Композиция (ICP, молярное отношение): P/Mn=4,6. r1 при 81,3 МГц, 25°C=1 мМ-1 Mn с-1.
Пример 19. Другие ионны металлов, введенные в наноструктуру X
pH наноструктуры Х2а доводили от 2 до 10,4 с помощью 6 М и 1 М NaOH (водн.) и выдерживали в течение 2 часов. Затем добавили соли металлов (xx мг, yy ммоль, см. таблицу 3 ниже), такие как Fe(II) хлорид гидрат, Fe(III) хлорид гидрат, Er(III) хлорид гидрат или Dy(III) хлорид гидрат. Смесь взбалтывали в течение 1 ч при 30°C. PH смеси после реакции изменяли от 4,7 до 7,2 для различных образцов и затем доводили до pH 7,4, используя 1 М HCl (водн.). Смесь профильтровали спиновой фильтрацией (10k MWCO, 3000 × g в течение 15 мин) и концентрат (0,5 мл) разбавили до 4 мл с использованием MilliQ H2O. Эту процедуру повторили 4 раза. Итоговый концентрат разбавили до 4 мл с использованием MilliQ H2O.
а: Использованный предшественник Х2а, 4 мл, 0,11 ммоль Р. FeCl24H2O: xx=7,2 мг, yy=0,04 ммоль. Конечное значение pH 7,4; максимальный средневзвешенный размер частиц (DLS в 150 мМ NaCl)=8,7 нм; композиция (ICP, молярное отношение): P/Fe=4,2, P/Si=0,9, Si/Fe=4,5; r1 при 81,33 МГц, 25°C=3,1 мМ-1 Fe с-1.
b: Использованный предшественник Х2а, 4 мл, 0,11 ммоль Р. FeCl3 6H2O: xx=9 мг, yy=0,03 ммоль. Конечное значение pH 7,4; максимальный средневзвешенный размер частиц (DLS в 150 мМ NaCl)=8,7 нм; композиция (ICP, молярное отношение): P/Fe=6,5, P/Si=0,9, Si/Fe=7,2; r1 при 81,33 МГц, 25°C=8,5 мМ-1 Fe с-1.
c: Использованный предшественник Х2а, 4 мл, 0,11 ммоль Р. ErCl3 6H2O: xx=12,7 мг, yy=0,03 ммоль. Конечное значение pH 7,4; максимальный средневзвешенный размер частиц (DLS в 150 мМ NaCl)=50 нм; композиция (ICP, молярное отношение): P/Er=5,3, P/Si=0,9, Si/Er=5,7; r1 при 81,33 МГц, 25°C=0,4 мМ-1 Er с-1.
d: Использованный предшественник Х2а, 4 мл, 0,11 ммоль Р. DyCl3 6H2O: xx=13,6 мг, yy=0,04 ммоль. Конечное значение pH 7,4; максимальный средневзвешенный размер частиц (DLS в 150 мМ NaCl)=10,1 нм; композиция (ICP, молярное отношение): P/Dy=4,4, P/Si=0,9, Si/Dy=4,7; r1 при 81,33 МГц, 25°C=0,6 мМ-1 Dy с-1.
Пример 20. Релаксивность для насыщенных Mn материалов
Релаксивность некоторых насыщенных Mn материалов показана в таблице 3

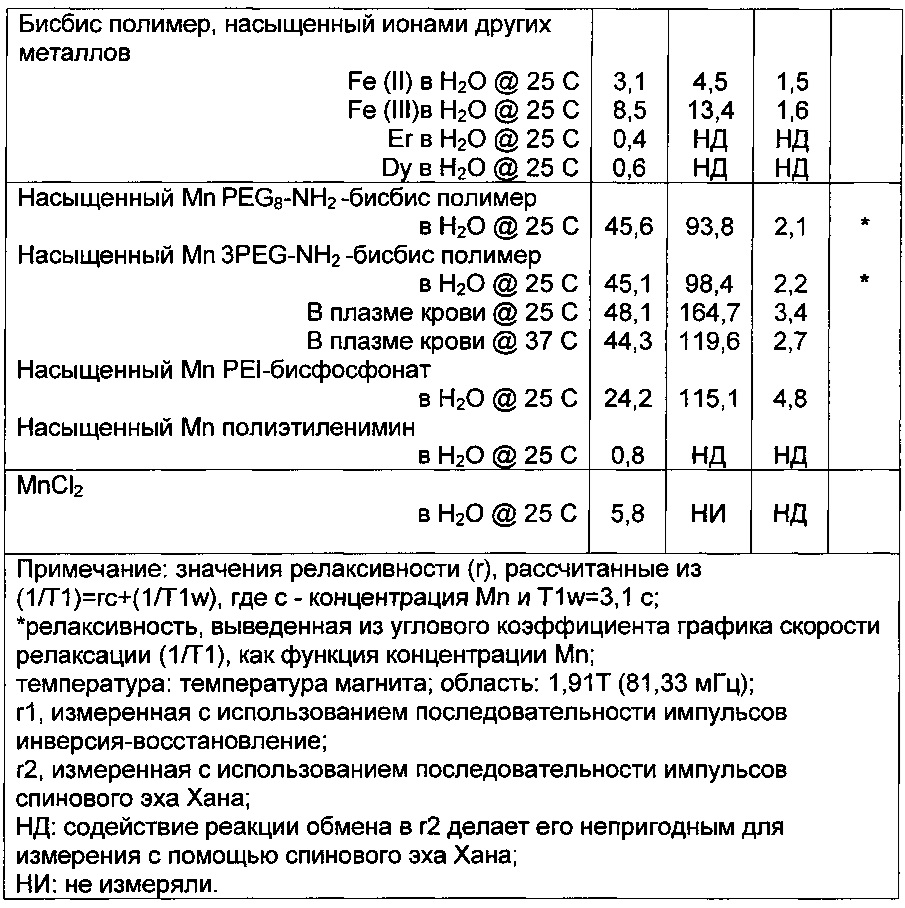
Пример 21: Визуализация in vivo
Качество MR-изображения и контрастность исследовали in vivo у мышей, несущих агрессивно растущую мышиную лимфому EL-4. Клеточную линию мышиной лимфомы EL-4 создали из лимфомы, индуцированной у C57BL/6 мыши. Лимфобластные клетки быстро растут в суспензии в пробирке и в качестве аллотрансплантатов у C57BL/6 мышей.
EL-4 клетки (ECACC 85023105) использовали для развития привитых опухолей у мыши C57BL/6. Суспензию клеток ввели подкожно и опухоли развивались в течение нескольких дней. Через 6-10 дней после инъекции опухоли использовали для визуализации.
MR протокол оптимизировали и испытали на двух животных. МР изображения получили у 7 животных с опухолью EL-4, 4 мыши получили 3 мМ/175 мкл Y2, 3 мыши получили 17 мМ/175 мкл Magnevist, оба вводились за 6 секунд.
Получили Т1-взвешенное GE изображение. Мышь удалили из магнита и подсоединили катетер, содержащий контрастное вещество. Критическое значение имело последующее введение контрастного вещества с целью избегания проникновения в тело животного через катетер. Получили Т1-взвешенные изображения до введения контрастного вещества и непосредственно после этого ввели контрастное вещество, в то время как динамические флеш-изображения получали непрерывно (2 до инъекции, 14 после инъекции, 8 с на изображение).
В экспериментах, помеченных А, В, Е (см. ниже), 10 срезов с областью сканирования 50×50 мМ получили после инъекции либо Y2 либо Magnevist. Изображения, полученные с 8 срезами, область сканирования 50×50 мМ, размер матрицы 256×256 и общее время сканирования немногим больше 10 минут на набор данных. Изображения после введения контрастного вещества получали каждые 15 минут. Из всех наборов данных, полученных после инъекции контрастного вещества, выбрали 1 срез из 10, как образец повышения, см. фигуру 2.
Пример 22: Синтез наноструктур на основе полиэтиленимина с насыщенным марганцем 1,3-бисфосфонатом
22а: Пропилен-1,3-дифосфоновая кислот
Триметил силил бромид (16,84 мл, 25,3 ммоль) и тетераэтилпропилен-1,3-дифосфонат (10,08 г, 31,6 ммоль) растворили в 5 мл охлажденного на льду дихлорметана. Через 10 мин охлаждающую баню удалили и смесь перемешивали при комнатной температуре в течение 16 часов. Летучие вещества удалили на роторном испарителе. К остатку добавили 50 мл воды при перемешивании и охлаждении льдом.Через 20 минут воду удалили на роторном испарителе и остаточную влажность удалили с помощью первых двух циклов дополнительного выпаривания толуола и затем с помощью масляного вакуумного насоса в течение ночи.1Н-ЯМР показал, что этильные группы исчезли, и метиленовые группы все еще присутствовали.
22b: Тетраметилпропилен-1,3-дифосфонат
Пропилен-1,3-дифосфоновую кислоту (600 мг) поместили во взвешенном состоянии в триметил орто формиат (20 мл) и кипятили в течение 6 ч, после чего 10 мл жидкости отогнали, а остальное оставили на ночь. Летучие вещества удалили в вакууме с получением продукта в виде масла.
22с: 1-Трет-бутоксикарбонилметил- O,O,O,O -тетраметилпропилен-1,3-дифосфонат
Тетраметилпропилен-1,3-дифосфонат (206 мг, 0,79 ммоль) растворили в сухом THF (5 мл) в атмосфере азота и раствор охладили в ацетоновой бане с сухим льдом. Трет-BuLi (2,17 М в гептане, 1,66 ммоль) ввели посредством шприца и через 10 минут добавили трет-бутиловый эфир бромуксусной кислоты (0,23 мл, 1,66 ммоль). Спустя 30 минут температуру повышали до -15°C в течение 30 минут. Реакционную смесь гасили добавлением к насыщенному водному раствору хлорида аммония. Путем экстракции эфиром (2×25 мл), высушенным над MgSO4и выпаривания получили маслянистый остаток. С помощью флэш-хроматографии на силикагеле с дихлорметаном + 3% метанолом получили 134 мг требуемого продукта.
22d. 1-Карбоксилметил-O,O,O,O-тетраметилпропилен-1,3-дифосфонат
1-Трет-бутоксикарбонил-метил-O,O,O,O-тетраметилпропилен-1,3-дифосфонат (150 мг) растворили в дихлорметане (10 мл) и добавили трифторуксусную кислоту (0,5 мл). Реакционную смесь оставили на ночь и удалили летучие вещества. Остаточную трифторуксусную кислоту удалили посредством трех циклов дополнительного выпаривания толуола. Выход 132 мг.
22е: Конъюгация 1-карбоксилметил-O,O,O,O-тетраметилпропилен-1,3-дифосфоната с полиэтиленимином и насыщение марганцем
1-Карбоксилметил-O,O,O,O-тетраметилпропилен-1,3-дифосфонат (100 мг), полиэтиленимин (15 мг, средн. Mw 30000) и сульфо-N-гидроксисукцинимид добавили к воде (5 мл) и обрабатывали ультразвуком в течение 10 мин. РН доводили до 6,6 добавлением 0,1 М NaOH. Добавили EDC и раствор поместили в шейкер на 19 часов. Низкомолекулярный материал переместили на 20 кДа отсекающий спиновый фильтр. Остаток промыли 4 раза на том же фильтре. Измеренный диаметр наноструктуры составил 7,5 нм при растворении в 150 мМ NaCl. К 1 мл указанного выше раствора добавили 21 мг в MnCl24 H2O. РН доводили до 7,3 добавлением водного NaOH. Образец промывали водой трижды на 10 кДа номинальном отсекающем спиновом фильтре. Образец анализировали на содержание Mn и установили, что он составляет 2,4 мМ. Установили, что релаксивность составляет 18,5/мМ/с при 60 МГц. Стабильность измерили в соответствии с примером 14: 0,2%.
Пример 23. Кондуктометрическое титрование наноструктур
Кондуктометрическое титрование может быть использовано для определения количества Ca2+ и Mg2+, которые могут адсорбироваться на наноструктурах. Проводимость водного раствора наноструктур измеряют после добавления приращений раствора, содержащего смесь CaCl2 и MgCl2. Проводимость (мс/см) раствора будет увеличиваться с определенной скоростью (наклон кривой титрования) до тех пор, пока Ca2+ и Mg2+ адсорбируют на частицах. Когда наноструктуры насыщаются Ca2+ и Mg2+, проводимость будет увеличиваться с другой скоростью. Для более четкой визуализации конечной точки (где наноструктуры насыщаются Ca и Mg), проводимости в присутствии наноструктур вычитали из проводимостей, полученных путем добавления одинаковых приращений раствора Ca/Mg к воде.
200 мкл образца материала из примера 10 с ([Mn=0; [P]=138 мМ) смешали с 2300 мкл воды. Проводимость измерили после добавления 50 мкл приращений водного раствора, содержащего 6,55 мМ MgCl2 и 9,67 мМ CaCl2. (Данное отношение [Ca]/[Mg] является приблизительно таким же, как то, что обнаружено в крови. См. "обр." на фиг. 3а. Начальное значение проводимости (без добавления раствора Mg/Ca) вычли из "обр." с получением "соотв. обр." на фиг. 3а. Проводимость также измерили после добавления таких же приращений Mg/Ca в воде с получением "слепого обр. "на фиг. 3а. Наконец, проводимости из "соотв. обр." вычли из "слепого обр." с получением "разн. (слепой обр. - соотв. обр.)" на фиг. 3а.
"Разн. (слепой обр. - соотв. обр)" кривую увеличили на фиг. 3b и две прямые подвели к различным частям кривой. Две линии, пересеклись на 474 мкл титранта, что дает [P]/[Mg]=8,89, [P]/[Ca]=6,02 и [P]/[Me]общ.=3,59.
Одну и ту же партию наноструктур, как описано выше, дополнительно концентрировали на фильтре 10 кДа (объем уменьшили приблизительно до одной пятой). Титрование проводили с теми же поправками, что и выше с 40 мкл раствора, смешанного с 2460 воды. Конечную точку определили в 422 мкл. При условии, что отношение адсорбции [P]/[Me] такое же, как и предыдущее титрование, это даст [P]=422/474×138×0,200/0,040=613 мМ. См. фиг. 3с.
Основной целью титрования являлась оценка количества раствора Ca/Mg, которое необходимо добавить для 90% насыщения наноструктур.
Поэтому 506 мкл 400 мМ раствора MgCl2 и 600 мМ раствора CaCl2 добавили к 14,5 мл раствору образца ([P]=138 мМ).
Данный раствор затем дополнительно концентрировали с помощью фильтра 10 кДа, как указано выше, и приблизительно такое же количество титровали, но на этот раз большинство из этих участков должны быть заняты металлами. Результаты показаны на фиг. 3d.
Пример 24. Композиция: насыщение ионами Ca и Mg с последующим добавлением маннита (партия SI055C-PE120208)
2,5 мл образца раствора материала из примера 10с ([Mn]=38 мМ, [P]=508 мМ и Осм ~200 мОсм/кг; партия РЕ 120130) сначала доводили до физиологического уровня pH 7,4. За насыщением (90%) Ca и Mg последовало выполнение следующих этапов. 100 мкл раствора титровали кондуктометрическим титрованием с Ca и Mg ([Ca]/[Mg]=1,48), согласно способу, описанному в примере 23 ("кондуктометрическое титрование наноструктур") и наличие конечной точки определили, когда [Ca]/[Mn]=1,59 или [Mg]/[Mn]=1,08.
Имела место некоторая обеспокоенность, что некоторые из Mn2+ могут быть замещены Ca2+ и Mg2+, тем более, что pH в конце титрования составлял 5,5. Поэтому раствор после титрования профильтровали с помощью 10 кД фильтра и определили, что количество Mn в фильтрате содержит ~15% от общего Mn в образце. Потерю Mn из наноструктуры можно по меньшей мере частично объяснить низким pH в конце титрования.
Для получения соответствующих насыщенных Ca и Mg частиц 500 мкл раствор маннита (Os=280 мОсм/кг) сначала смешали с 21,5 мкл концентрированного раствора Ca/Mg ([Ca]=600 мМ, [Mg]=400 мМ). Данный раствор затем смешали с 580 мкл партии РЕ 120130, pH регулировали от 6,01 до 7,40 с ~8 мкл 1 М NaOH и осмоляльность определили в 270 мОсм/кг.
Пример 25. Испытание на иммуногенность
Способы
Кролику вводили 5-кратно 2×0,5 мл наноструктур в соответствии с примером 24 (объединенных с магнием и кальцием) при концентрации наноструктур, соответствующей 10 мг Mn/мл. Инъекции вводили подкожно, по одной в каждую заднюю ногу. Наноструктуры были смешаны с адъювантом следующим образом:
1) 1:1 об./об. с полным адъювантом Фрейнда (Sigma-Aldrich) (в общей сложности введен 1 мл) для первичной иммунизации и с неполным адъювантом Фрейнда для бустер-инъекций;
2) 1:1 (об./об.) с 40 мг/мл гидроксида алюминия (Pierce) (в общей сложности введен 1 мл) как для первичной иммунизации, так и для бустер-инъекций.
Протокол для инъекций и сбора сыворотки
- День 0: сбор преиммунной сыворотки (20 мл)
- День 0: первичная иммунизация
- День 14: первая бустер-инъекция
- День 28: вторая бустер-инъекция
- День 49: третья бустер-инъекция
- День 70: четвертая бустер-инъекция
- День 84: сбор иммунной сыворотки (60 мл)
Анализ
1. Фракции иммуноглобулина выделили из преиммунной и иммунной сыворотки с помощью хроматографии на протеине G (GE Healthcare).
2. 2 мг фракции IgG конъюгировали с 2 мл агарозной колонкой (Pierce) путем восстановительного амидирования между альдегидными группами на смоле и первичными аминными группами, присутствующими в IgG молекулах. IgG-конъюгированные колонки промыли и уравновесили 0,9% NaCl.
3. 100 мкл раствора наноструктур по примеру 24, соответствующие 1,35 мМ марганца (содержащего приблизительно 21 мкг Si и 7 мкг Mn) внесли в каждую из колонок.
4. Непрерывный поток: колонки промыли 0,9% NaCl, во фракциях 4×1 мл, после чего 2×2 мл (фракции 1-6).
5. Элюат: Связанный SI055 элюировали 4×1 мл 1 М NaCl (фракции 7-10).
6. Непрерывный поток и элюатные фракции проанализировали на содержание Mn и Si с помощью ICP-AES.
Результаты
Как в преиммунной, та и иммунной колонках, практически весь SI055 был обнаружен в непрерывном потоке. В элюате не было никакой разницы между колонками в количестве обнаруженных наноструктур.
Не было ни малейших признаков раздражения или других проблем у кролика за время протокола иммунизации.
Восстановление наноструктур в непрерывном потоке и элюате из преиммунной и иммунной колонок приведены в таблице 4.
Восстановление SI055 во фракциях 1-10 из преиммунной и иммунной колонок показаны на фигуре 4а и 4b, соответственно.
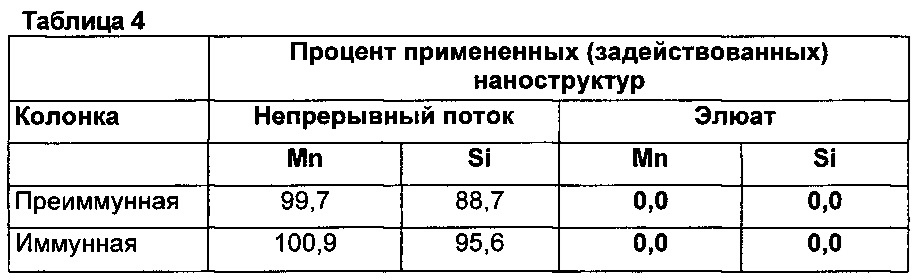
Выводы
Данный пример показал, что не было никакой иммунной реакции на наноструктуры у кроликов, несмотря на достаточно усиленный протокол иммунизации. Этот результат подтверждает тот факт, что наноструктуры являются биологически инертными.
Реферат
Настоящее изобретение относится к пригодным для применения в качестве контрастного вещества для магнитно-резонансной томографии наноструктурам, содержащим парамагнитные ионы марганца (II), введенные в хелатообразующую полимерную структуру, где наноструктура имеет почти сферическую форму и средний размер 3-7 нм; где молярное отношение Р/Mn составляет 7-20; где полимерная структура образована путем полимеризации мономера, представляющего собойс использованием спонтанного гидролиза и конденсации, где степень полимеризации составляет от 25 до 3000000 мономеров; где ионы марганца (II) введены в полимерную структуру путем контактирования полимера с раствором солей марганца (II); где указанная наноструктура необязательно содержит биологически инертные группы -(CHCHO)CH, где n=4, которые прививают к остаточным фосфоновым или силанольным группам полимера после хелатирования марганца путем взаимодействия с,причем количество биологически инертных групп на каждой единице наноструктуры от 10 до 1000. Предложены новые эффективные контрастные вещества для магнитно-резонансной томографии. 3 н. и 2 з.п. ф-лы, 25 пр., 4 табл., 5 ил.
Формула


Документы, цитированные в отчёте о поиске
Лекарственные формы на основе бисфосфонатов
Комментарии