Комплексы каскадных полимеров, содержащее их диагностическое контрастное средство и промежуточные соединения - RU2197495C2
Код документа: RU2197495C2
Чертежи

Описание
Изобретение относится к описанному в пунктах формулы предмету изобретения, т. е. к новым комплексам каскадных полимеров, к содержащим эти соединения, средствам, к применению комплексов в диагностике и терапии, а также к способам получения этих соединений и средств.
Используемые в настоящее время в клинике контрастные средства для современных способов формирования изображения ядерно-спиновой томографии (MRI) и компьютерной томографии (СТ) (Magnevist®, Pro Hance®, Ultravist® и Omniscan®) распределяются во всем внеклеточном пространстве тела (внутрисосудистое пространство и внутритканевое пространство). Это пространство для распределения включает примерно 20% объема тела.
Внеклеточные контрастные средства для MRI вначале находили успешное клиническое применение при диагностике церебральных заболеваний и заболеваний спинного мозга, так как здесь складывается совершенно особая ситуация в отношении локального пространства распределения. В мозге и спинном мозге внеклеточные контрастные средства в здоровой ткани не могут покинуть внутрисосудистое пространство из-за барьера между кровью и мозгом. При болезненных процессах, связанных с нарушением барьера между кровью и мозгом (например, злокачественные опухоли, воспаления, демиелизирующие заболевания и т.д.), внутри мозга возникают участки с повышенной проницаемостью кровеносных сосудов для этих внеклеточных контрастных средств (Schmiedl et al., MRI of blood-brain barrier permeability in astrocytic gliomas: application of small and large molecular weight contrast media, Magn. Reson. Med. 22:288, 1991). Используя эти нарушения проницаемости сосудов, можно распознать больную ткань по повышенной по сравнению со здоровой тканью контрастности изображения.
Вне головного мозга и спинного мозга такой проницаемости для указанного выше контрастного средства, правда, не существует (Canty et al., First-pass entry of nonionic contrast agent into the myocardial extravascular space. Effects on radiographic estimate of transit time and blood volume. Circulation 84: 2071, 1991). Таким образом, обогащение контрастным средством не зависит больше от проницаемости сосудов, а лишь от величины внеклеточного пространства в соответствующей ткани. Отграничивание сосудов от окружающего их внутритканевого пространства при применении этого контрастного средства невозможно.
В особенности для изображения сосудов было бы желательно контрастное средство, которое распределяется лишь исключительно в вазальном пространстве (пространство внутри сосудов). Такой кровяной пул-агент должен позволить с помощью ядерно-спиновой томографии разграничить хорошо снабжаемые кровью ткани и плохо снабжаемые и таким образом диагностировать ишемию. Ткани, подвергнутые инфаркту, также можно было бы из-за их анемии отграничить от окружающих здоровых или подвергнутых ишемии тканей, если применить вазальное контрастное средство. Это имеет особенно большое значение, если речь идет, например, о том, чтобы отличить сердечный инфаркт от ишемии.
До настоящего времени большинство пациентов, у которых имеется подозрение на кардиососудистое заболевание (эта болезнь является наиболее частой причиной смерти в западных промышленных странах), подвергаются инвазионным диагностическим исследованиям. В ангиографии в настоящее время применяют рентгенографическую диагностику с помощью контрастных средств, содержащих иод. Эти исследования имеют значительные недостатки: они связаны с риском облучения, а также с неприятными ощущениями и нагрузками, которые происходят, прежде всего, от того, что содержащие иод контрастные средства по сравнению с контрастными средствами для NMR должны применяться в значительно более высоких концентрациях.
Поэтому существует потребность в контрастных средствах для NMR, с помощью которых можно маркировать вазальное пространство (кровяной пул-агент). Эти соединения должны отличаться хорошей переносимостью и высокой эффективностью (повышенное увеличение интенсивности сигнала при ЯМР).
Попытка, по меньшей мере, часть этой проблемы решить путем применения комплексообразователей, которые привязаны к макро- или биомолекулам, имела доныне очень ограниченный успех.
Так, например, количество парамагнитных центров в комплексах, описанных в европейских патентных заявках 0088695 и 0150844, недостаточно для получения удовлетворительного изображения.
Если повысить количество необходимых ионов металла путем многократного введения комплексообразующих мономерных звеньев в макромолекулярную биомолекулу, то это повлечет за собой недопустимое снижение химического сродства и/или специфичности /J. Nucl. Med, 24, 1158 (1983)/.
В общем, макромолекулы могут быть пригодными для применения в ангиографии в качестве контрастного средства. Однако альбумин GdDTPA (Radiology 1987; 162: 205), например, через 24 часа после внутривенной инъекции дает у крысы обогащение в ткани печени, которое составляет почти 30% дозы. Кроме того, через 24 часа устраняется лишь 20% дозы.
Макромолекула полилизина-GdDTPA (Европейская патентная заявка, публикация 0233619) оказалась также пригодной в качестве кровяного пул-агента. Это соединение состоит, однако, из смеси молекул различной величины, что обусловлено способом получения. При опытах с крысой по выделению могло оказаться, что эта макромолекула в неизменном виде выделяется почками с помощью сосудистой фильтрации. Полилизин-GdDTPA, однако, вследствие условий синтеза, может содержать также макромолекулы, которые настолько велики, что они при сосудистой фильтрации не могут пройти через капилляры почек и таким образом остаются в теле.
Также описаны макромолекулярные контрастные средства на основе углеводов, например декстран (Европейская патентная заявка, публикация 0326226). Недостаток этих соединений состоит в том, что они, как правило, содержат лишь примерно 5% парамагнитного катиона, усиливающего сигнал.
Описанные в Европейской патентной заявке 0480863 полимеры представляют собой уже некоторый шаг на пути к кровяному пул-агенту, так как они не обладают больше характерной для упомянутых выше полимеров гетерогенностью в отношении величины и молекулярного веса. Они оставляют, однако, желать лучшего в отношении полноты выделения, переносимости и/или эффективности действия.
Поэтому задача состоит в том, чтобы предоставить в распоряжение новые, не имеющие упомянутых недостатков диагностические средства прежде всего для распознания и локализации заболеваний сосудов. Эта задача решается с помощью данного изобретения.
Было найдено, что комплексы, которые состоят из азотсодержащих каскадных полимеров с комплексообразующими лигандами из, по меньшей мере, 16 ионов элементов с порядковыми номерами 20-29, 39, 42, 44 или 57-83, а также в случае необходимости из катионов неорганических и/или органических оснований, аминокислот или амидов аминокислот, и в случае необходимости, содержат ацилированные аминогруппы, неожиданным образом прекрасно подходят для получения диагностического средства для ЯМР- и рентгеновских исследований, не обнаруживая указанных недостатков.
Комплексообразующие каскадные полимеры согласно изобретению можно описать общей формулой 1:
A-{X-[Y-(Z-
где А означает азотсодержащее каскадное ядро с базисной мультиплетностью а,
Х и Y
независимо друг от друга означают простую связь или репродуктивное каскадное мономерное звено с репродуктивной мультиплетностью x или y,
Z и W независимо друг от друга означают
репродуктивное
каскадное мономерное звено с репродуктивной мультиплетностью z или w,
К означает группу комплексообразователя,
а означает числа от 2 до 12,
x, y, z и w
независимо друг от
друга означают числа от 1 до 4 при условии, что, по меньшей мере, два репродуктивных мономерных звена различны, что для произведения мультиплетностей действительно:
16≤a•
x•y•z•w≤64
и что, по меньшей мере, одно из репродуктивных каскадных мономерных звеньев X, Y, Z, W означает репродуктивное мономерное звено 1,4,
7,
10-тетраазациклододекана или 1,4,8,11-тетраазациклотетрадекана.
В качестве каскадного ядра А пригодны:
атом азота,








где m и n означают числа от 1 до 10,
p означает числа от 0 до 10,
U1 означает Q1 или Е,
U2 означает Q2 или Е, с Е, означающей группу:

причем o означает числа от 1 до 6,
Q1 - атом водорода или Q2,
Q2 - простую связь,
М означает C1-C10-алкиленовую цепь, которая в случае необходимости прервана 1-3 атомами кислорода и/или в случае необходимости замещена 1-2 оксогруппами,
R0 означает разветвленный или линейный C1-С10-алкильный остаток, нитрогруппу, аминогруппу, группу карбоновой кислоты или

причем число Q2 соответствует базисной мультиплетности а.
Простейший случай каскадного ядра представляет собой атом азота, три связи которого (базисная мультиплетность а=3) в первом "внутреннем слое" (генерация 1) заняты тремя репродуктивными мономерными звеньями Х или Y (если Х означает простую связь) или Z (если Х или Y означают, соответственно, простую связь); иначе говоря: три атома водорода, являющегося инициатором каскада аммиака А(Н)a=NH3, замещены тремя репродуктивными мономерными звеньями Х, или Y, или Z. Содержащееся в каскадном ядре А число Q2 представляет собой при этом базисную мультиплетность а.
Репродуктивные мономерные звенья X, Y, Z и W содержат -NQ1 Q2-группы, где Q1 означает атом водорода или Q2, a Q2 означает простую связь. Содержащееся в соответствующем репродуктивном мономерном звене (например, X) число Q2 соответствует репродуктивной мультиплетности этого мономерного звена (например, x в случае X). Произведение всех мультиплетностей a•x• y•z•w дает число связанных в каскадном полимере остатков К комплексообразователя. Полимеры согласно изобретению содержат, по меньшей мере, 16 и максимум 64 остатка К в молекуле, которые могут связать соответственно один до максимум трех (в случае двухвалентного иона), предпочтительно один ион элемента указанного выше порядкового числа.
Последняя генерация, т.е. связанное с остатком К комплексообразователя репродуктивное мономерное звено W, посредством групп NH (-NQ1 Q2 с Q1, означающим атом водорода, и Q2= простая связь) связана с К, в то время как предыдущие репродуктивные мономерные звенья могут быть связаны друг с другом как посредством NHQ2-групп (например, с помощью реакций ацилирования), так и через NQ2Q2-группы (например, с помощью реакций алкилирования).
Комплексы каскадных полимеров согласно изобретению имеют максимум 10 генераций (т. е. в молекуле может быть в наличии также больше, чем одно из репродуктивных мономерных звеньев X, Y и Z), предпочтительно, однако, от 2 до 4 генераций, причем, по меньшей мере, два репродуктивных мономерных звена в молекуле различны.
В качестве предпочтительных каскадных ядер А следовало бы привести такие, которые подпадают под вышеуказанные общие формулы, если
m означает числа 1-3,
особенно предпочтительно число 1,
n означает числа 1-3, особенно предпочтительно число 1,
p означает числа 0-3, особенно предпочтительны числа 1 и 3,
о означает числа 1-2,
особенно предпочтительно число 1,
М означает -СН2-группу, -СО-группу -СН2СО-группу и
R0 означает -CH2NU1U2-, СН3- или NO2-группу.
Предпочтительными являются далее каскадные ядра А, которые подпадают под вторую и четвертую из приведенных восьми общих формул, особенно те,
которые
соответствуют общей формуле:
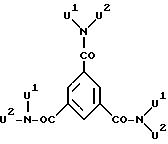
где U1 и U2 означают группу Е и значением "о" 1 или 2.
В качестве предпочтительных инициаторов каскадов А(Н)а следует, например, привести:
(в скобках приводится базисная мультиплетность а,
приведенная для
случая последующего моно- или бизамещения, служащего для построения следующей генерации)
трис(аминоэтил)амин - (а=6 или 3)
трис(аминопропил)амин - (а=6 или 3)
диэтилентриамин - (а=5 или 3)
триэтилентетрамин - (а=6 или 4)
тетраэтиленпентамин - (а=7 или 5)
1,3,5-трис(аминометил)бензол - (а=6 или 3)
триамид
тримезиновой
кислоты - (а=6 или 3)
1,4,7-триазациклононан - (а=3)
1,4,7,10-тетраазациклододекан - (а=4)
1,4,7,10,13-пентаазациклопентадекан - (а=5)
1,4,8,
11-тетраазациклотетрадекан - (а=4)
1,4,7,10,13,16-гексаазациклооктадекан - (а=6)
1,4,7,10,13,16,19,22,25,28-декаазациклотриаконтан - (а=10)
тетракис(аминометил)метан - (а=8
или 4)
1,1,1-трис(аминометил)этан - (а=6 или 3)
трис(аминопропил)-нитрометан - (а=6 или 3)
2,4,6-триамино-1,3,5-триазин - (а=6 или 3)
1,3,5,7-амид
адамантантетракарбоновой кислоты - (а=8 или 4)
3,3',5,5'-дифениловый эфир амида тетракарбоновой кислоты - (а=8 или 4)
амид 1,2-бис(феноксиэтан)3', 3'', 5',5''-тетракарбоновой
кислоты
- (а=8 или 4)
1,4,7,10,13,16,21,24-октаазабицикло (8.8.8)гаксакосан - (а=6)
Следует указать на то, что определение в качестве каскадного ядра А и тем самым разделение
каскадного ядра
и первого репродуктивного мономерного звена можно выбрать чисто формально и, таким образом, независимо от фактического синтетического построения желательных комплексов каскадных
полимеров. Так,
например, использованный в примере 4 трис(аминоэтил)амин можно рассматривать как собственно каскадное ядро А (сравните с первой приведенной для А общей формулой с m=n=p=l, U1=E и "о",
означающим число 1 и U1=U2=Q2), так и в качестве атома азота (=каскадному ядру А), который, как первая генерация, имеет три репродуктивных мономерных
звена;

(сравните с определением Е).
Репродуктивные каскадные мономерные
звенья X, Z, Y и W
независимо друг от друга определяются с помощью E,

предпочтительно


предпочтительно


с t в значении чисел 1 или 2

предпочтительно

где U1 означает Q1 или Е,
U2 означает Q2 Е, с Е, означающим группу

Причем о означает числа 1-6, предпочтительно от 1 до 2,
Q1 означает атом водорода или Q2,
Q2 означает прямую связь,
U3 означает цепь -NHCO- (СН2)0 или C1-C20 -алкиленовую цепь, предпочтительно C1-С10-алкиленовую цепь, которая при необходимости прервана от 1 до 2 атомами кислорода и/или
1-2 остатками -N(CO)q -R2-, 1-2-фениленовыми и/или 1-2 фениленокси-остатками и/или в случае необходимости замещена 1-2 оксо-, тиоксо-, карбокси-, С1-С5-алкилкарбокси-, С1-С5 -алкокси-, гидроксигруппой, С1-С5-алкильными группами, причем
q означает числа 0 или 1 и
R2 означает атом водорода, метиловый или этиловый остаток, который при необходимости замещен 1-2 гидрокси- или 1 карбоксигруппой (группами),
L означает атом водорода или группу

V означает метиновую группу

или V означает группу
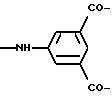
если одновременно U4 и U5 идентичны и означают простую связь или группу М,
U5' означает группу М и
U6 означает группу

или простую связь при условии, что, по меньшей мере, одно из каскадных репродуктивных мономерных звеньев означает приведенное выше репродуктивное мономерное звено 1,4,7,10-тетраазациклододекана или 1,4,8,11-тетраазациклотетрадекана. Предпочтительно при этом репродуктивное мономерное звено 1,4,7,10-тетраазациклододекан.
Предпочтительными каскадными репродуктивными мономерными звеньями X, Y, Z и W являются те, в которых в приведенных выше
общих формулах остаток U3 означает
-CO-, -COCH2OCH2CO-, -COCH2-, -CН2СН2-, -CONHC6H4-,
-NHCOCH2-, -COCH2CH2CO-, -COCH2-CH2CH2CO-,
-COCН2СН2СН2СН2С-, -CONHCH2
CH2NHCOCH2CH2CO-,
-СОСН2СН2NНСОСН2СН2СО-,
остаток U4 означает простую связь, -СН2
СО-,
остаток U5 означает прямую связь, -(СН2)4-, -СН2СО-, -СН(СООН)-, СН2ОСН2СН2-, -СН2С6
Н4-, СН2-С6Н4OСН2СН2-,
остаток Е означает группу

В качестве примера следует привести названные каскадные репродуктивные мономерные звенья X, Y, Z и W:
-CH2CH2NH-; -CH2 CH2N<;
-COCH(NH-)(CH2)4NH-;
-COCH(N<)(CH2)4N<;
-COCH2OCH2CON(CH2 CH2NH-)2;
-COCH2OCН2CON(CН2CH2N<)2;
-COCH2N(CH2CH2NH-)2 ;
-COCH2N(CH2CH2N<)2;
-COCH2NH-; -COCH2N<;
-COCH2CH2CON(CH2 CH2NH-)2;
-COCH2CH2CON(CH2CH2N<)2;
-COCH2OCH2CONH-C6H4 -CH[CH2CON(CH2CH2NH-)2]2;
-COCH2OCH2CONH-C6H4-CH[CH2CON(CH2 CH2N<)2]2;
-COCH2CH2CO-NH-C6H4-CH[CH2CON(CH2CH2NH-)2]2;
-СОСН2СН2СО-NН-С6Н4-СН[СН2СОN(СН2СН2N<)2]2;
-CONH-C6 H4 -CH[CH2CON(CH2CH2NH-)2]2;
-CONH-C6H4-CH[CH2CON(CH2CH2N<)2]2;
-COCH(NH-)CH(COOH)NH-; -COCH(N<)CH(COOH)N<;




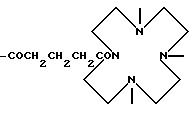
-COCH(CH2CH2CH2-N)2
-COCH(CH2 CH2-N)2

-COCH(CH2-N)2



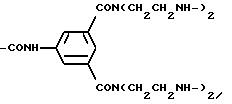



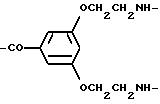





Остатки К комплексообразователей описываются общими формулами IA, IB, IC:



где R1 независимо друг от друга означают атом водорода или эквивалент иона металла порядковох номеров 20-29, 39, 42-44 или 57-83,
R2 означает атом водорода, метиловый или этиловый остаток, который при необходимости замещен 1-2-гидрокси- или одной карбоксигруппой (группами),
R3 означает группу

или группу

R4 означает атом водорода или линейную, разветвленную, насыщенную или ненасыщенную C1-С30-алкильную цепь, которая при необходимости прервана 1-10 атомами кислорода, 1-й фениленовой, 1-й фениленоксигруппами и/или в случае необходимости замещена 1-5 гидроксигруппами, 1-3 карбоксигруппами, 1 фенильной группой,
R5 означает атом водорода или R4,
U7 означает линейную, разветвленную, насыщенную или ненасыщенную C1-C20-алкиленовую группу, содержащую при необходимости 1-5 иминогрупп, 1-3 фениленовые группы, 1-3 фениленоксигруппы, 1-3 фенилениминогруппы, 1-5 амидных групп, 1-2 гидразидных групп, 1-5 карбонильных групп, 1-5 этиленоксигрупп, 1 мочевинную группу, 1 тиомочевинную группу, 1-2 карбоксиалкилиминогруппы, 1-2 сложноэфирные группы, 1-10 атомов кислорода, 1-5 атомов серы, и/или 1-5 атомов азота, и/или в случае необходимости замещенную 1-5 гидрокси-, 1-2 меркапто-, 1-5 оксо-, 1-5 тиоксо-, 1-3 карбокси, 1-5 карбоксиалкильной группой (группами), 1-5 сложноэфирной группой и/или 1-3 аминогруппой (группами), причем содержащиеся в случае необходимости фениленовые группы могут быть замещены 1-2-мя карбокси-, 1-2-мя сульфоновыми группами или 1-2 гидроксигруппами,
Т означает -CO-α-, -NHCO-α- или -NHCS-α-группу и
α означает место связи с концевым атомом азота последней генерации воспроизводимого мономерного звена W.
Предпочтительны остатки К комплексообразователей общей формулы IA.
В качестве предпочтительных остатков К комплексообразователей следует далее назвать те, при которых в приведенной выше формуле IA
означающая U7 C1-C20-алкиленовая цепь, предпочтительно C1-C12-алкиленовая, содержит группы
-СН2-, -CH2NHCO-,
-NHCOCH2O-, -NНСОСН2OС6Н4-, -N(CH2CO2H)-, -NНСОСН2С6Н4-, -NHCSNHC6H4-, -СН2OС6Н4-, -СН2СН2О-,
и/или замещена группами -СООН, -СН2СООН.
В качестве примера для U7 можно привести
следующие группы:
-CH2-, -CH2CH2-, -CH2CH2CH2-, -C6H4-, -C6H10-, -CH2
C6H5-,
-CH2NHCOCH2CH(CH2CO2H)-C6H4-,
-CH2NHCOCH2OCH2-,
-CH2NHCOCH2C6H4-,
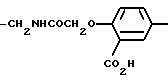
-CH2 NHCSNH-C6H4-CH(CH2COOH)CH2-,
-CH2OC6H4-N(CH2COOH)CH2-,
-СН2NНСОСН2O(СН2СН2O)4-С6Н4-,
-СН2O-С6Н4-,
-СН2СН2-O-СН2СН2-, -СН2СН2-O-СН2СН2-O-СН2СН2-,

Особенно предпочтительно U7 означает группу СН2.
В
качестве примера для R4 можно привести следующие группы:
-СН3, -С6Н5, -СН2-СООН,
-СН2-С6Н5,
-СН2-O-(СН2СН2-O-)6СН3, -СН2-ОН.
Предпочтительны атом водорода и метиловая группа,
Т предпочтительно
означает
группу -СО-α-.
Если средство согласно изоретению предназначено для применения в ЯМР-диагностике, то центральный ион комплексной соли должен быть парамагнитным. Это, в частности, двух- и трехвалентные ионы элементов порядковых номеров 21-29, 42, 44 и 58-70. Пригодными ионами являются, например, ионы хрома (III), железа (II), кобальта (II), никеля (II), меди (II), празеодима (III), неодима (III), самария (III) и иттербия (III). Из-за своего очень сильного магнитного момента особенно предпочтительны ионы гадолиния (III), тербия (III), диспрозия (III), гольмия (III), эрбия (III), марганца (II) и железа (III).
Если средство согласно изобретению предназначено для диагностики с помощью рентгенографии, то центральный ион должен быть элементом с более высоким порядковым номером, чтобы достичь достаточной абсорбции рентгеновских лучей. Было найдено, что для этой цели пригодными являются диагностические средства, которые содержат физиологически переносимую комплексную соль с центральными ионами элементов с порядковыми номерами 21-29, 39, 42, 44, 57-83; это, к примеру, ион лантана (III) и указанные выше ионы ряда лантанидов.
Комплексы каскадных полимеров согласно изобретению содержат, по меньшей мере, 16 ионов элемента указанных выше порядковых номеров.
Остаточные кислотные атомы водорода, т. е. те, которые не замещены центральным ионом, могут быть в случае необходимости полностью или частично замещены катионами неорганических или органических оснований, аминокислот или амидов аминокислот.
Пригодными неорганическими катионами являются, например, ион лития, ион калия, ион кальция, ион магния и особенно ион натрия. Пригодными катионами органических оснований являются, среди прочих, катионы первичных, вторичных или третичных аминов, как, например, этаноламин, диэтаноламин, морфолин, глюкамин, N, N-диметилглюкамин и особенно N-метилглюкамин. Соответствующими катионами аминокислот являются, например, катионы лизина, аргинина и орнитина, а также амиды в ином случае кислых или нейтральных аминокислот.
Соединения согласно изобретению, которые имеют молекулярный вес от 10.000 до 80.000D, предпочтительно 15.000-40.000D, имеют вышеперечисленные желательные свойства. Они содержат необходимое для их применения большое число ионов металлов, стабильно связанных в комплексы.
Они накапливаются в областях с повышенной проницаемостью сосудов, как, например, в опухолях, позволяют создать мнение о перфузии
тканей, дают возможность определить объем
крови в тканях, избирательно сократить время релаксации или плотность крови и получить изображение проницаемости кровеносных сосудов. Такую физиологическую
информацию нельзя получить при применении
внеклеточных контрастных средств, как, например, Gd-DTPA (Magnevist®).
Исходя из этого получаются также в современных способах
получения изображения путем ядерно-спиновой
томографии и компьютерной томографии: более специфические диагнозы злокачественных опухолей, ранний контроль терапии при цитостатической,
антифлогистической или вазодилатативной терапии, раннее
распознание малоперфундированных областей (например, в миокарде), ангиография при заболеваниях сосудов и распознавание и диагноз (стерильных
или инфекционных) воспалительных процессов.
В качестве другого преимущества перед внеклеточными контрастными средствами, как, например, Gd-DTPA (Magnevist®), следует выделить более высокую эффективность контрастного средства в ядерно-спиновой томографии (более высокая релаксивность), что ведет к заметному снижению необходимой для диагностики дозы. Одновременно контрастные средства согласно изобретению можно сформировать в виде растворов, измомолярных по отношению к крови, и благодаря этому снизить осмотическую нагрузку тела, что отражается в снижении токсичности вещества (более высокий порог токсичности). Меньшая доза и более высокий порог токсичности приводят к значительному повышению безопасности применения контрастных средств в современных способах формирования изображения.
По сравнению с макромолекулярными контрастными средствами на основе углеводов, например декстран (Европейская патентная заявка, публикация 0326226), которые, как упоминалось выше, как правило, содержат примерно лишь 5% парамагнитного катиона, усиливающего сигнал, полимерные комплексы согласно изобретению содержат, как правило, примерно 20% парамагнитного катиона. Таким образом, макромолекулы согласно изобретению вызывают значительно более высокое усиление сигнала на одну молекулу, что одновременно приводит к тому, что необходимая для ядерно-спиновой томографии доза значительно меньше по сравнению с макромолекулярным контрастным средством на основе углеводов.
С помощью полимерных комплексов согласно изобретению удалось получить макромолекулы с унифицированно определенным молекулярным весом. Таким образом, неожиданно оказалось возможным так влиять на величину макромолекулы, чтобы она была достаточно большой и медленно покидала вазальное пространство, но достаточно малой, чтобы еще проходить через капилляры почек, которые имеют величину

По сравнению с другими упомянутыми в уровне техники полимерными соединениями комплексы каскадных полимеров согласно изобретению отличаются улучшенным выделением, повышенной эффективностью, более высокой стабильностью и/или более высокой переносимостью.
Другое преимущество данного изобретения состоит в том, что теперь стали доступны комплексы с гидрофильными или липофильными, макроциклическими или открытыми цепями, низкомолекулярными или высокомолекулярными лигандами. Благодаря этому создается возможность управлять переносимостью и фармакокинетикой этих полимерных комплексов с помощью химического замещения.
Получение комплексов каскадных полимеров согласно изобретению осуществляется благодаря
тому, что соединения общей формулы I
A-{X-[Y-(Z-(W-βw)z)y]x}a (I′)
где А означает азот, содержащее каскадное
ядро базисной мультиплетности а,
Х и Y независимо друг от друга
означают простую связь или каскадное репродуктивное мономерное звено с репродуктивной мультиплетностью x или y,
Z и W
независимо друг от друга означают каскадное репродуктивное мономерное
звено с репродуктивной мультиплетностью z или w,
а означает числа от 2 до 12,
x, y, z и w независимо друг от
друга означают числа от 1 до 4,
β означает место связи
конечных групп NH последней генерации, репродуктивного мономерного звена w,
при условии, что, по меньшей мере, два
репродуктивных мономерных звена различны, что для произведения
мультиплетностей действительно:
16≤a•x•y•z•w≤64
и что, по меньшей мере, одно
из каскадных репродуктивных мономерных звеньев X, Y, Z, W
означает репродуктивное мономерное звено 1,4,7,10-тетраазациклододекана или 1,4,8,11-тетраазациклотетрадекана,
подвергают
взаимодействию с комплексом или комплексообразователем К' общей
формулы I'А, I'В или I'С



причем R1' независимо друг от друга означает атом водорода, эквивалент ионов металла порядковых номеров 20-29, 39, 42-44 или 57-83 или кислотозащитную группу,
R2 означает атом водорода, метиловый или этиловый остаток, который в случае необходимости замещен 1-2 гидрокси- или 1 карбоксигруппой (группами),
R3 означает группу
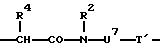
или группу

R4 означает атом водорода или линейную, разветвленную, насыщенную или ненасыщенную C1-С30-алкильную цепь, которая в случае необходимости может быть прервана 1-10 атомами кислорода, 1 фениленовой группой, 1 фениленоксигруппой и/или в случае необходимости замещена 1-5 гидроксигруппами, 1-3 карбоксигруппами, 1 фенильной группой.
R5 означает атом водорода или R4,
U7 означает линейную, разветвленную, насыщенную или ненасыщенную С1-С20-алкиленовую группу, содержащую
при необходимости 1-5 имино-, 1-3 фениленовую, 1-3
фениленокси-, 1-3 фениленимино-, 1-5 амидных, 1-2 гидразидных, 1-5 карбонильных, 1-5 этиленокси-, 1 мочевинную, 1 тиомочевинную, 1-2
карбоксиалкилимино-группы, 1-2 сложноэфирные группы, 1-10 атомов
кислорода, 1-5 атомов серы и/или 1-5 атома (атомов) азота и/или в случае необходимости замещенную 1-5 гидрокси-, 1-2 меркапто-, 1-5
оксо-, 1-5 тиоксо, 1-3 карбокси-, 1-5 карбоксиалкилгруппой
(группами), 1-5 сложноэфирными группами (группой) и/или 1-3 аминогруппами (группой), причем содержащиеся в случае необходимости фениленовые
группы могут быть замещены 1-2 карбокси-, 1-2 сульфоновой
или 1-2 гидроксигруппой (группами),
Т' означает -С*O-, -СООН-, -N= C=O- или -N=С=S-группы и С*O означает активированную
карбоксильную группу при условии, что поскольку К' означает комплекс,
по меньшей мере, два (для двухвалентных металлов) или три (для трехвалентных металлов) из заместителей R1 означают
эквивалент иона металла вышеуказанных элементов и что, по желанию, имеют
место другие карбоксильные группы в виде их солей с неорганическими и/или органическими основаниями, аминокислотами или
амидами аминокислот, отщепляют имеющиеся в случае необходимости защитные группы,
полученные таким образом каскадные полимеры - поскольку К' означает комплексообразователь известным само по себе
способом приводят во взаимодействие с, по меньшей мере, одним окислом металла или солью
металла элемента с порядковыми номерами 20-29, 39, 42, 44 или 57-83 и в случае необходимости затем в полученных
таким образом комплексах каскадных полимеров имеющиеся кислотные атомы водорода
полностью или частично замещают катионами неорганических и/или органических оснований, аминокислотами или амидами
аминокислот и в случае необходимости еще имеющиеся свободные концевые аминогруппы в
случае необходимости перед или после комплексообразования металла подвергают ацилированию.
Другой
аспект данного изобретения представляют новые соединения общей формулы I'A

причем R1' означает атом водорода, эквивалент ионов металлов порядковых номеров 20-29, 39, 42-44 или 57-83 или кислотозащитную группу,
R2 означает атом водорода, метиловый или этиловый остаток, который в случае необходимости замещен 1-2 гидрокси- или 1 карбоксигруппой (группами),
R3' означает группу

или группу

R4 означает атом водорода или линейную, разветвленную, насыщенную или ненасыщенную C1 -С30-алкильную цепь, которая в случае необходимости прервана 1-10 атомами кислорода, 1 фениленовой-, 1 фениленоксигруппой и/или в случае необходимости замещена 1-5 гидрокси-, 1-3 карбокси-, 1 фенильной группой (группами),
U7 означает линейную, разветвленную, насыщенную или ненасыщенную C1-C20-алкиленовую цепь, содержащую в случае необходимости 1-5 имино-, 1-3 фениленовых, 1-3 фениленокси-, 1-3 фениленимино-, 1-5 амидных, 1-2 гидразидных, 1-5 карбонильных, 1-5 этиленокси-, 1 мочевинную, 1 тиомочевинную, 1-2 карбоксиалкилиминогруппу(группы), 1-2 сложноэфирные группы, 1-10 атомов кислорода, 1-5 атомов серы, и/или 1-5 атомов азота, и/или в случае необходимости замещенную 1-5 гидрокси-, 1-2 меркапто-, 1-5 оксо-, 1-5 тиоксо-, 1-3 карбокси-, 1-5 карбоксиалкильной группой (группами), 1-5 сложноэфирными группами и/или 1-3 аминогруппой (группами), причем содержащиеся в случае необходимости фениленовые группы, могут быть замещены 1-2 карбокси-, 1-2 сульфоновыми или 1-2 гидроксигруппами (группой),
Т' означает -С*О-, -СООН-, -N=C=О- или -N=C=S-группы и
С*О означает активированную карбоксильную группу.
Они пригодны в качестве важных промежуточных продуктов для получения комплексов каскадных полимеров общей формулы I.
В качестве примера активированной карбонильной группы С*О в комплексах или комплексообразователях К' следует назвать ангидрид, n-нитрофениловый сложный эфир, N-гидроксисукцинимидный сложный эфир, пентафторфениловый сложный эфир и хлорангидрид кислоты.
Предпринимаемое для введения мономерных единиц комплексообразователей присоединение или ацилирование проводят с субстратами, которые содержат желательный заместитель К (возможно связанный с летучей группой) или из которых желательный заместитель получают путем реакции.
В качестве примеров реакции присоединения следует назвать взаимодействие изоцианатов и изотиоцианатов, причем взаимодействие изоцианатов проводят предпочтительно в апротонных растворах, как, например, ТГФ, диоксан, ДМФ, ДМСО, метиленхлорид при температурах между 0o-100oC, предпочтительно между 0o и 50oС, в случае необходимости при добавлении органического основания, как, например, триэтиламин, пиридин, лутидин, N-этилдиизопропиламин, N-метилморфолин. Взаимодействие изотиоцианатов проводят, как правило, в растворителях, как, например, в воде или в низших спиртах, как, например, метанол, этанол, изопропанол или их смесях, ДМФ или смесях из ДМФ и воды при температурах между 0o и 10oС, предпочтительно между 0o и 50oС, в случае необходимости при добавлении органического или неорганического основания, как, например, триэтиламин, пиридин, лутидин, N-этилдиизопропиламин, N-метилморфолин или гидроокиси щелочноземельных металлов, гидроокиси щелочных металлов, как, например, лития, натрия, калия, кальция, или их карбонаты, как, например, карбонат магния.
В качестве примера реакций ацилирования следует назвать разложение свободных карбоновых кислот известными специалистам способами (например, J.P. Greenstein, M.Winitz, Chemistry of the Amino Acids, John Wiley & Sons, N.Y, (1961), S. 943-945). Оказалось, однако, предпочтительным карбоксильную группу перед реакцией ацилирования переводить в активную форму, как, например, в ангидрид, активный сложный эфир или хлорангидрид кислоты (например, Е.Gross, J. Meienhofer, The Peptides, Academic Press, N.Y., Vol. 1, S. 65-314; N.F. Albertson, Org. React. 12, 157 (1962).
В случае взаимодействия с активным сложным эфиром следует сослаться на известную специалисту литературу (например, Houben-Weyl, Methoden der organischen Chemie, Georg Thieme Verlag, Stuttgart, Band E5, (1985), 633). Его можно провести в условиях, приведенных выше для реакции с ангидридом. Можно, однако, использовать также апротонные растворители, как, например, метиленхлорид, хлороформ.
В случае взаимодействия с ангидридами кислот следует применять лишь апротонные растворители, как, например, метиленхлорид, хлороформ, толуол или ТГФ при температурах между -20o до 50oС, предпочтительно между 0o до 30oС. Далее следует сослаться на известную специалисту литературу (например, Houben-Weyl, Methoden der Organischen Chemie, Georg Thieme-Verlag, Stuttgart, (1974), Band 15/2, S. 355-364).
В том случае, если R' означает кислотозащитную группу, то речь может идти о низших алкильных, арильных, аралкильных группах, например метильных, этильных, пропильных, бутильных, бензильных, дифенилметильных, трифенилметильных, бис-(п-нитрофенил)-метильных группах, а также триалкилсилильных группах.
В случае необходимости отщепление защитных групп осуществляют известными специалисту способами, например путем гидролиза, гидрогенизации, щелочного омыления сложного эфира щелочью в водно-щелочном растворе при температурах от 0o до 50oС или в случае сложного трет-бутилового эфира с помощью трифторуксусной кислоты.
В случае необходимости не полностью ацилированные лигандом или комплексом концевые аминогруппы можно, если желательно, перевести в амиды или полуамиды. Например, можно назвать взаимодействие с ацетангидридом, ангидридом янтарной кислоты или ангидридом дигликолевой кислоты.
Введение желательных ионов металлов осуществляют способом, который раскрыт, например, в выложенной заявке ФРГ 3401052, растворяя или суспендируя окись металла или соль металла (например, нитрат, ацетат, карбонат, хлорид или сульфат) элемента с порядковыми числами 20-29, 42, 44, 57-83 в воде и/или низшем спирте (как, например, метанол, этанол или изопропанол) и разлагая с помощью раствора или суспензии эквивалентного количества комплексообразующего лиганда, а затем, если желательно, имеющиеся атомы водорода кислотных групп, замещая катионами неорганических или органических оснований, аминокислот или амидов аминокислот.
Введение желательных ионов металлов можно осуществить как на ступени комплексообразователей I'А или I'Б, т.е. перед связыванием с каскадным полимером, так и после связывания с неметаллическими лигандами I'А, I'Б или I'В.
Нейтрализация осуществляется при этом с помощью неорганических оснований (например, гидроокисей, карбонатов или бикарбонатов), например, натрия, калия, лития, магния или кальция и/или органических оснований, как, например, среди прочих первичные, вторичные и третичные амины, например этаноламин, морфолин, глюкамин, N-метил- и N,N-диметилглюкамин, а также основных аминокислот, как, например, лизин, аргинин и орнитин, или амидов первоначально нейтральных или кислых аминокислот, как, например, гиппуровая кислота, глицинацетамид.
Для получения нейтральных комплексных соединений можно, например, кислую комплексную соль в виде водного раствора или суспензии добавить к желательному основанию в таком количестве, чтобы достичь нейтральной точки. Полученный раствор можно затем концентрировать в вакууме до получения твердого вещества. Если образованную нейтральную соль выделить в осадок путем добавки смешиваемых с водой растворителей, как, например, низших спиртов (метанол, этанол, изопропанол и другие), низших кетонов (ацетон и другие), полярных простых эфиров (тетрагидрофуран, диоксан, 1, 2-диметоксиэтан и другие) и таким образом можно получить легко выделяемые и хорошо очищаемые кристаллизаты, что также является преимуществом. Особенно предпочтительно добавлять желательное основание к реакционной смеси уже во время комплексообразования и благодаря этому сэкономить одну перацию способа.
Если кислые комплексные соединения содержат несколько свободных кислотных групп, то часто целесообразно получить нейтральные смешанные соли, которые содержат как неорганические, так и органические катионы в качестве противоиона.
Это может, к примеру, случиться, если комплексообразующий лиганд в водной суспензии или растворе подвергнуть реакции с окисью или солью элемента, представляющего собой центральный ион, и половиной требуемого для нейтрализации количества органического основания полученную комплексную соль выделить, при желании очистить, а затем для полной нейтрализации подвергнуть реакции с необходимым количеством неорганического основания. Последовательность введения оснований может быть обратной.
Очистка полученных таким образом комплексов каскадных полимеров осуществляется при необходимости после установления значения рН до рН 6-8, предпочтительно 7, путем добавления кислоты или основания, предпочтительно путем ультрафильтрации с помощью мембран с соответствующим размером пор (например, Amicon® ХМ30,

В случае нейтральных комплексных соединений часто является преимуществом, если полимерные комплексы для отделения ионных компонентов пропустить через анионообменную установку, например IRA 67 (в форме ОН-), и при необходимости дополнительно пропустить через катионообменную установку, например IRC 50 (форма Н+).
Получение необходимых для связывания с комплексообразователем К' (или также с соответствующими металлосодержащими комплексами) содержащих концевые аминогруппы каскадных полимеров основано, в общем, на имеющихся в продаже или на известных из литературных данных или аналогичных им способах получения инициаторов каскадов A(H)α, содержащих азот. Введение генераций X, Y, Z и W осуществляют известными из литературы методами (например, B.J.March, Advanced Organic Chemistry, 3-rd ed.; John Wiley & Sons, (1985), 364-381) с помощью реакций ацилирования или алкилирования с имеющими желательную структуру защищенными аминами, которые содержат способные к связыванию с каскадным ядром функциональные группы, как, например, карбоновые кислоты, изоцианаты, изотиоцианаты или активированные карбоновые кислоты (как, например, анигидриды, активный сложный эфир, ангидриды кислот) или галогениды (как, например, хлориды, бромиды, иодиды), азиридин, мезилаты, тозилаты или другие, известные специалисту летучие группы.
Следует, однако, еще раз подчеркнуть, что разница между каскадным ядром А и репродуктивным мономерным звеном чисто формальная. С позиций синтеза может являться преимуществом то, что используют не формальный инициатор каскада A(H)α, а вводят относящиеся по определению к каскадному ядру атомы азота лишь вместе с первой генерацией. Так, например, для синтеза описанного в примере 1в) соединения более предпочтительно алкилировать не формальное каскадное ядро амид тримезиновой кислоты с помощью, например, бензилоксикарбонилазиридина (шестикратно), а разложить трихлорид тримезиновой кислоты с помощью бис-[2-(бензилоксикарбониламино)-этил]-амина (трижды).
Таким же образом при встраивании имеющихся при необходимости репродуктивных мономерных звеньев 1,4,7,10-тетраазациклододекана (циклен) или 1,4,8,11-тетраазациклотетрадекана (циклам) может иметь преимущества, если подстроить каскадный полимер неформально, оболочку за оболочкой.
Так, например, можно в процессе предварительной реакции следующее за оболочкой цикла репродуктивное мономерное звено связать с тремя атомами азота циклена. Затем можно таким образом после функционализации четвертого атома азота цикла оба репродуктивных мономерных звена одновременно связать с растущим каскадом.
В качестве аминозащитных групп следует назвать известные специалисту бензилоксикарбонильную группу, группу третичного бутоксикарбонила, трифторацетильную группу, флуоренилметоксикарбонильную группу, бензильную и формильную группы [Th.W.Greene, P.G.M Wuts, Protective Groups in Organic Syntheses, 2nd ed., John Wiley and Sons (1991), S. 309-335]. После отщепления этих защитных групп, которое осуществляется также известными из литературы способами, можно вводить в молекулу следующую желательную генерацию. Наряду с этим состоящим из соответственно двух ступеней реакции (алкилирование или ацилирование и отщепление защитных групп) построением генерации, возможно также осуществляемое с помощью лишь двух ступеней реакции одновременное введение двух, например X-[Y]x, или нескольких генераций, например X-[Y-(Z)y]x. Построение многогенерационных мономерных звеньев осуществляют путем алкилирования или ацилирования незащищенных аминов ("репродуктивный амин"), обладающих структурами желательных репродуктивных мономерных звеньев, вторым репродуктивным амином, аминогруппы которого находятся в защищенной форме.
Необходимые в качестве инициаторов каскадов
соединения общей формулы A(H)α можно
позаимствовать из имеющихся в продаже или получить известными из литературы способами [например, Houben-Weyl, Methoden der Org. Chemie, Georg-Tieme - Verlag,
Stuttgart (1957), Bd. 11/1; M.Micheloni
et al., Inorg. Chemie (1985), 24, 3702; T.J.Atkins et al., Org. Synth., Vol. 58 (1978), 86-98; The Chemistry of Heterocyclic Compounds: J.S.Bradshaw et al.,
Aza-Crown-Macrocycles, John Wiley &
Sons, N.Y. (1993)]. В качестве примеров можно привести:
Трис(аминоэтил)амин (например, Fluka Chemie AG, Швейцария; Aldrich-Chemie, Германия);
Трис(аминопропил) амин [например,
Z.Woerner et al., Angew. Chem. Int. Ed. Engl. (1993), 32, 1306];
Диэтилентриамин [например, Fluka; Aldrich];
Триэтилентетрамин [например,
Fluka; Aldrich];
Триэтиленпентамин
[например, Fluka; Aldrich];
1,3,5-трис(аминометил)бензол [например, T.M.Carrett et al., J. Am. Chem. Soc. (1991), 113, 2965)];
Триамид
тримезиновой кислоты [например, B.H.Kurihara;
Jpn. Kokai Tokkio Koho JP 04077481; CA 117, 162453];
1,4,7-Триазациклононан [например, Fluka; Aldrich];
1,4,7,10,
13-Пентаазациклопентадекан [например, K. W. Aston, Eur. Pat. Appl.
0524161, CA 120, 44580];
1,4,7,10-Тетраазациклододекан [например, Aldrich];
1,4,8,11-Тетраазациклотетрадекан
[например, Fluka, Aldrich];
1,4,7,10,13,16,19,22,25,
28-Декаазациклотриаконтан [например, A.Andres et al., J. Chem. Soc. Dalton Trans. (1993), 3507];
1,1,1-Трис(аминометил)этан
[например, R. J.Geue et al., Aust. J. Chem. (1983), 36, 927];
Трис(аминопропил)-нитрометан [например, G.R.Newkome et al., Angew. Chem. 103, 1205 (1991) аналогично R.C.Larock, Comprehensive
Organic Transformations, VCH Publishers, N.Y. (1989) 419-420];
Амид 1,3,5,7-Адамантантетракарбоновой кислоты [например, H.Stetter et al., Tetr. Lett. 1967, 1841];
1,
2-Бис[феноксиэтан] -амид 3', 3'', 5', 5''-Тетракарбоновой кислоты [например,
J.P.Collman et al.; J. Am. Chem. Soc. (1988), 110, 3477-86, аналогично примеру 1б)];
1,4,7,10,13,16,21,
24-Октаазабицикло[8.8.8] гексакосан (например, Р. Н. Smith et al., J. Org. Chem. (1993),
58, 7939).
Получение репродуктивных аминов, необходимых для построения генераций, содержащих указанные функциональные группы, осуществляют в соответствии с описанными в экспериментальной части предписаниями или способами, известными из литературы.
В качестве примера следует
назвать:
сложный Nα,Nε
- Ди-бензилоксикарбонил-лизин-п-нитрофениловый эфир;
HOOС-СН2OСН2СО-N(СН2СН2
NН-СО-O-СН2С6Н5)2;
НООС-СН2N(СН2СН2NН-СО-O-СН2С6Н5)2;
НООС-СН2СН2СО-N(СН2СН2NН-СОСF3)2
[получают в соответствии с примером 5а, исходя из бис(трифторацетиламиноэтил)амина вместо
бис(бензилоксикарбониламиноэтил)амина];
НООС-СН2OСН2СОNН-С6Н4-СН[СН2СОN(СН2СН2NН-СО-O-СН2С6
Н5)2]2;
O=C=N-C6H4-CH[CH2CON(CH2CH2NH-CO-O-CH2C6H5)2]2

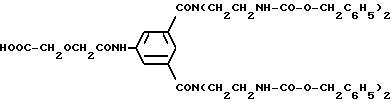

N-Бензилоксикарбонил-азиридин получают в соответствии с M.Zinic et al., J. Chem. Soc., Perkin Trans 1,21-26 (1993).
N-Бензилоксикарбонил-глицин
имеется в продаже. Например, в Bachem, Калифорния

получают согласно C.J.Cavallito et al., J. Amer. Chem. Soc., 1943, 65, 2140, исходя из N-CO-О-СН2С6Н5-(2-брометил)амина вместо хлористого бензила [A.R.Jacobson et al., J. Med. Chem. (1991), 34, 2816].
Получение комплексов и комплексообразователей общей формулы I'А и I'Б осуществляется в соответствии с методиками, приведенными в экспериментальной части, или аналогичным способом, или же известными из литературы способами (см. , например, Европейские патентные заявки 0512661, 0430863, 0255471 и 0565930).
Так, например, получение соединений общей формулы I'А может осуществляться, например, благодаря тому, что в качестве предварительной ступени функциональной группы Т' служит группа Т'' либо в значении защищенной кислотной функции, которую, независимо от кислотозащитных групп R1', приведенным выше способом можно перевести в свободную кислотную функцию, либо в значении защищенной аминной функции, которую можно деблокировать с помощью известных из литературы способов [Th.W.Greene, P.G.M.Wuts, Protective Groups in Organic Synthesis, 2nd edition, John Wiley & Sons (1991), S. 309-385], а затем перевести в изоцианаты или изотиоцианаты [Methoden der Org. Chemie (Houben-Weyl), E4, S. 742-749, 837-843, Georg Thieme Verlag, Stuttgart, New York: (1983)]. Такие соединения можно получить в соответствии с методиками, представленными в экспериментальной части, или аналогичными способами путем моноалкилирования циклов соответствующими амидами α-галогензамещенных карбоновых кислот (в апротонных растворителях, например в хлороформе).
Получить соединения общей формулы I'Б можно, например, благодаря тому, что в
качестве предварительной ступени активированной карбоксильной
группы-С*О- служит защищенная кислотная функция, которую, независимо от защитных кислотных групп R1', описанным выше способом
можно перевести в свободную кислотную функцию и активировать с
помощью также описанных выше, известных из литературы способов. Такие соединения можно получить в соответствии с приведенными в
экспериментальной части методиками, или аналогичными способами, или,
например, благодаря тому, что производную аминокислоты общей формулы II

где R5' имеет значение, указанное для R5, причем при необходимости содержащиеся в R5 гидроксильные или карбоксильные группы при необходимости имеются в защищенной форме и
V1 означает линейную или разветвленную C1-C6-алкильную группу, бензильную группу, триметилсилильную, триизопропилсилильную группу, 2,2,2-трифторэтоксигруппу или 2,2,2, -трихлорэтоксигруппу, причем V1 отличается от R1'',
подвергают взаимодействию с алкилирующим агентом общей формулы III:

где R1'' означает защитную группу и
Hal означает атом галогена, как, например, С1, Вr или I, предпочтительным является, однако, С1 (см. также М.А.Williams, H.Rapoport, J. Org. Chem. 58, 1151, (1993)).
Предпочтительными производными аминокислот являются сложные эфиры α-аминокислот естественного происхождения.
Реакция соединения (II) с соединением (III) осуществляется предпочтительно в процессе буферированной реакции алкилирования, причем в качестве буфера служит водный фосфатный буферный раствор. Взаимодействие проходит при значениях рН 7-9, предпочтительно, однако, при рН 8. Концентрация буфера может составлять 0,1-2,5 М, предпочтительно, однако, используют 2М-фосфатный буферный раствор. Температура алкилирования может лежать между 0o и 50oС, предпочтительна комнатная температура.
Реакцию проводят в полярном растворителе, как, например, ацетонитрил, тетрагидрофуран, 1,4-диоксан или 1,2-диметоксиэтан. Предпочтительно применение ацетонитрила.
Фармацевтическое средство согласно изобретению получают также известным способом, суспендируя или растворяя в водной среде комплексные соединения согласно изобретению при необходимости с добавлением обычно используемых в галеновой практике добавок, а затем при необходимости, стерилизуя суспензию или раствор. Пригодными добавками являются, например, физиологически приемлемые буферы (как, например, триметамин), добавки комплексообразователей или слабых комплексов (как, например, диэтилентриаминпентауксусная кислота или соответствующие кальциевые каскадные полимерные комплексы) или, если требуется, электролиты, как, например, хлористый натрий или, если требуется, антиоксиданты, как, например, аскорбиновая кислота.
Если для энтерального накопления или иных целей желательны суспензии или растворы средств согласно изобретению в воде или в физиологическом соляном растворе, то их смешивают с одним или несколькими обычно применяемыми в галеновой практике вспомогательными веществами [к примеру, метилцеллюлоза, лактоза, маннит], и/или с ферментами [например, лецитин, Tween®, Myrj®], и/или с ароматическими веществами для коррекции вкусовых качеств (например, эфирные масла).
В принципе также можно получать фармацевтические средства согласно изобретению даже без выделения комплексных солей. В любом случае особую тщательность следует проявить при образовании хелатов с тем, чтобы соли и растворы солей согласно изобретению были практически свободны от незакомплексированных ионов металлов, оказывающих токсическое действие.
Это можно обеспечить, например, с помощью цветных индикаторов, как, например, ксиленоловый оранжевый, путем контрольного титрования во время процесса получения. Изобретение относится поэтому также к способу получения комплексных соединений и их солей. В качестве последней гарантии безопасности остается очистка комплексной соли.
Фармацевтические средства согласно
изобретению содержат предпочтительно 1 мкмоль 1 моль/л комплексной соли и, как правило, добавляются в количествах
0,0001-5 ммольей/кг. Они предназначены для энтерального и парентерального применения.
Комплексные соединения согласно изобретению используют:
1) для ЯМР- и рентгенодиагностики - в виде их
комплексов с ионами элементов с порядковыми номерами 21-29, 39, 42, 44 и 57-83;
2) для радиодиагностики и радиотерапии - в виде их комплексов с радиоизотопами элементов порядковых номеров 27,
29, 31, 32, 37-39, 43, 49, 62, 64, 70 и 77.
Средства согласно изобретению удовлетворяют многосторонним требованиям в отношении пригодности в качестве контрастных средств для ядерно-спиновой томографии. Так, например, они особенно пригодны для того, чтобы после орального или парентерального применения улучшать выразительную способность полученного с помощью ядерно-спиновой томографии снимка путем повышения интенсивности сигнала. Далее они проявляют высокую эффективность действия, которая необходима, чтобы тело подвергать воздействию, по возможности, меньшего количества посторонних веществ, и хорошую переносимость, которая необходима для того, чтобы поддерживать неагрессивный характер исследований.
Хорошая растворимость в воде и незначительная осмолярность средства согласно изобретению позволяют получать высококонцентрированные растворы, позволяющие поддерживать в соответствующих границах объемную нагрузку на кровообращение и сбалансировать разбавление жидкостью организма, т.е. диагностическое средство для ЯМР должно быть в 100-1000 раз более водорастворимым, чем применяемое для ЯМР-спектроскопии. Далее средство согласно изобретению не только обладает высокой стабильностью in vitro, но также неожиданно имеет высокую стабильность in vivo, так что освобождение или обмен не связанных в комплексе с помощью ковалентной связи, самих по себе ядовитых ионов в промежуток времени, за который новые контрастные средства снова полностью выделяются, идет чрезвычайно медленно.
В общем, средства согласно изобретению в качестве диагностического средства для ЯМР добавляют в количестве 0,0001-5 ммольь/кг, предпочтительно 0,005-0,5 ммольь/кг. Подробности описаны, например, в [H.-J.Weinmann et al., Am. J. of Roentgenology 142, 619 (1984)].
Особенно низкие дозировки (менее 1 мг/кг веса тела) органоспецифического диагностического средства для ЯМР применяют, например, для обнаружения опухолей или сердечных инфарктов.
Далее комплексные соединения согласно изобретению предпочтительно используют в качестве реактивов на восприимчивость и в качестве реактива на изменения для in vivo-спектроскопии.
Средства согласно изобретению, благодаря благоприятным радиоактивным свойствам и хорошей стабильности содержащихся в них комплексных соединений, пригодны также в качестве диагностических средств в радиодиагностике. Подробности их применения и дозирования описывают в "Radiotracers for Medical Applications", CRC-Press, Boca Raton, Florida.
Другим способом, формирующим изображение, основанным на применении радиоизотопов, является позитронно-эмиссионная томография, в которой используют эмиттирующие позитроны - изотопы, как, например,43Sc,44Sc,52Fe,55Co и68Ga (Heiss, W. D. ; Phels, M.E.; Positron Emission Tomography or Brain, Springer Verlag Berlin, Heidelberg, New York, 1983).
Соединения согласно изобретению неожиданным образом оказываются пригодными также для дифференцирования злокачественных и доброкачественных опухолей в области барьера между кровью и мозгом. Они отличаются тем, что они полностью удаляются из тела и таким образом хорошо переносимы.
Так как вещества согласно изобретению накапливаются в злокачественных опухолях (никакой диффузии в здоровые ткани, но высокая проницаемость для емкостей с опухолями), то они могут оказать помощь также при лучевой терапии злокачественных опухолей. Они отличаются от соответствующих диагностических средств лишь количеством и типом применяемого изотопа. Целью при этом является разрушение клеток опухоли с помощью богатого энергией коротковолнового облучения при возможно меньшей ширине распространения. Для этого используют переменное воздействие содержащихся в комплексах металлов (как, например, железо или гадолиний) ионизированными облучениями (например, рентгеновскими лучами) или с помощью нейтронных лучей. Благодаря этому эффекту заметно повышается локальная лучевая доза в том месте, где находится комплекс металла (например, в опухолях). Для того чтобы генерировать одинаковую дозу облучения в злокачественной опухоли, можно при применении таких металлокомплексов значительно снизить лучевую нагрузку на здоровые ткани и таким образом избежать вредных побочных воздействий на пациента. Сопряженные металлокомплексы согласно изобретению пригодны поэтому также для применения в качестве радиосенсибилизирующего вещества при лучевой терапии злокачественных опухолей (например, использование эффекта Мессбауера при нейтроноулавливающей терапии). Соответствующими эмиттирующими β-лучи ионами являются, например,46sc,47Sc,48Sc,72Ga,73Ga и90Y. Пригодными, обладающими небольшим временем полураспада эмиттирующими β-лучи ионами являются предпочтительно, например,211Bi,212Bi,213Bi и214Bi, причем предпочтителен2l2Bi. Пригодным ионом, эмиттирующим протоны и электроны, является158Gd, который можно получить из157Gd путем захвата нейтронов.
Если средство согласно изобретению предназначено для применения в предложенном R.L.Mills et al. [Nature Vol. 336, (1988), S. 787] варианте лучевой терапии, то центральный ион является производным изотопа Мессбауера, как, например57Fe или151Еu.
При применении in vivo терапевтических средств согласно изобретению их можно использовать вместе с соответствующим носителем, как, например, сыворотка или физиологический соляной раствор, и вместе с другим протеином, как, например, человеческий сывороточный альбумин (Human Serum Albumin). Дозирование зависит при этом от вида клеточного повреждения, примененного иона металла и вида способа, формирующего изображение.
Терапевтические средства согласно изобретению принимают парентерально, например i.v.
Подробно применение радиотерапии описано, например, в R.W.Kozak et al. TIBTEC, Oktober, 1986, 262.
Средства согласно изобретению прекрасно подходят для применения в качестве контрастных средств в рентгенографии, причем особенно следует отметить, что с ними не наблюдают при биофармакологических исследованиях признаков анафилактических реакций известных для содержащих иод контрастных средств. Особенно ценны они из-за благоприятных абсорбционных свойств в области повышенных напряжений на трубке в вычислительной субстракционной технологии.
В общем средства согласно изобретению для применения в качестве контрастного средства для рентгенографии дозируются по аналогии с примером диатризоата меглимина в количествах 0,1-5 ммоль/кг, предпочтительно 0,25-1 ммоль/кг.
Подробно применение контрастных средств для рентгенографии обсуждается, например, у Barke, "Einfuehrung in die Roentgenodiagnostik", G.Thieme, Stuttgart, New York (1977).
В целом удалось синтезировать новые комплексообразователи, металлокомплексы и комплексные соли металлов, которые раскрывают новые возможности в диагностической и терапевтической медицине.
Нижеследующие примеры служат для более подробного пояснения предмета изобретения.
Пример 1
а) 1,4,7-Трис(N, N'-дибензилоксикарбонил-лизил)-1,4,7,10-тетраазациклододекан
49,07 г (95,9 ммоль) сложного эфира
ди-Z-лизин-N-гидроксисукцинимида и 5 г (29
ммоль) циклена (1,4,7,10-тетраазациклододекан) растворяют в смеси, состоящей из 200 мл толуола и 100 мл диоксана. Добавляют 9,7 г (95,9 ммоль) триэтиламина и
нагревают в течение 12 часов до 70oС. Подвергают испарению до получения твердого вещества, осадок растворяют в 600 мл дихлорметана и экстрагируют 3 раза с помощью соответственно 200 мл
5%-ного водного раствора карбоната калия.
Органическую фазу высушивают над сульфатом магния и подвергают выпариванию в вакууме до получения твердого продукта. Остаток хроматографируют на силикагеле
(растворитель:этилацетат/этанол = 15:1).
Выход: 29,61 г (75% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 65,28 Н: 6,81 N: 10,29;
получили: С: 65,41 Н: 6,97 N: 10,
10.
б) 1-(Карбоксиметоксиацетил)-4,7,10-трис(N, N'-дибензилоксикарбонил-лизил)-1,4,7,10-татраазациклододекан
К 28 г (20,56 ммоль)
соединения из заглавия примера 1а
(растворенного в 200 мл тетрагидрофурана) добавляют 3,58 г (30,86 ммоль) ангидрида дигликолевой кислоты и 6,24 г (61,72 ммоль) триэтиламина. Подогревают в течение 6
часов до 50oС. Раствор
выпаривают в вакууме до получения твердого вещества, заливают 300 мл дихлорметана и дважды экстрагируют соответственно 150-ю мл 5%-ного водного раствора соляной
кислоты. Органическую фазу высушивают
над сульфатом магния, выпаривают в вакууме до твердого состояния, и остаток хроматографируют на силикагеле (растворитель: дихлорметан/метанол = 20:1).
Выход: 27,65 г (91%) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 63,40 Н: 6,55 N: 9,48;
получили: С: 63,21 Н: 6,70 N: 9,27.
в)
1-[5-(4-Нитрофенокси)-3-оксаглутарил]-4,7,10-трис(N,N'-дибензилоксикарбонил-лизил)-1,4,7,10-тетраазациклододекан
14,78 г (10 ммоль) описанной в примере 1б) карбоновой кислоты,
растворенной в
150 мл дихлорметана, вначале смешивают с 1,53 г (11 ммоль) 4-нитрофенолом, а затем при 0oС с 2,27 г (11 ммоль) дициклогексилкарбодиимида. После перемешивания в течение ночи
при комнатной
температуре отсасывают от дициклогексилмочевины, фильтрат сгущают и подвергают вторничному осаждению из изопропанола. Из получившегося маслянистого продукта удаляют маточный раствор
путем декантации,
масло поглощается дихлорметаном, и продукт сгущают в вакууме. Получают 15,4 г (96,3%) застывшего в виде пены твердого вещества.
Элементный анализ:
расчетн.:
С: 63,11 Н: 6,24
N: 9,64;
получили: С: 62,98 Н: 6,31 N: 9,80.
г) Бис [2-(бензилоксикарбониламино)-этил]-амин
51,5 г (500 ммоль) диэтилентриамина и 139 мл (1 ммоль)
триэтиламина
растворяют в дихлорметане и при -20oС смешивают с 161 г бензилцианформиата в дихлорметане, а затем перемешивают в течение ночи при комнатной температуре. По окончании реакции
упаривают в
вытяжном шкафу, осадок погружают в диэтилэфир, органическую фазу промывают раствором карбоната натрия и высушивают над сульфатом натрия. Фильтрат смешивают с гексаном, осадок
отфильтровывают и
высушивают.
Выход: 163,4 г (88% от теор.).
Элементный анализ:
расчетн.: С: 64,67 Н: 6,78 N: 11,31;
получили: С: 64,58 Н: 6,83 N:
11,28.
д) триамид-N,N,N',N',N'',N''-гексакис[2- (бензилоксикарбониламино)-этил] тримезиновой кислоты
13,27 г (50 ммоль) треххлористой тримезиновой кислоты (Aldrich) и 34,7 мл
(250 ммоль)
триэтиламина растворяют в диметилформамиде и при 0oС смешивают с 65,0 г (175 ммоль) описанного в примере 1 г) амина, а затем перемешивают при комнатной температуре в течение
ночи. Раствор
выпаривают в вакууме, а осадок хроматографируют с этилацетатом на силикагеле.
Выход: 39,4 г (62% от теор.).
Элементный анализ:
расчетн.: С: 65,
24 Н: 5,95 N:
9,92;
получили: С: 65,54 Н: 5,95 N: 9,87.
е) Полностью защищенный 36-мерный бензилоксикарбонилполиамин, построенный из ядра-триамида N,N,N',N',N'',
N''-гексакис(2-аминоэтил)-тримезиновой кислоты и шести описанных в примере 1б аминозащищенных гексааминмонокарбоновых кислот
1,27 г (1 ммоль) описанного в примере 1д)
гексабензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным раствором бромистого водорода в ледяной уксусной кислоте. По истечении 60 мин с помощью
диэтилового эфира заканчивают начавшееся образование осадка, полученный гексаамингидробромид промывают (простым) эфиром, высушивают в вакууме и без дополнительной очистки подвергают дальнейшему
разложению.
Выход: 0,95 г (количественно).
Затем гексаамингидробромид растворяют в 150 мл ДМФ, смешивают с 15,99 г (10 ммоль) описанного в примере 1в) активного
сложного 4-нитрофенильного эфира и с 4,05 г (40 ммоль) триэтиламина, перемешивают в течение ночи при комнатной температуре, а затем выпаривают в вакууме до твердого состояния. Остаток растворяют в
этилацетате, затем последовательно промывают водой, разбавленной натриевой
щелочью и насыщенным раствором NaCl. Органическую фазу высушивают над сульфатом натрия, и фильтрат выпаривают до
получения твердого вещества, и остаток хроматографируют на силикагеле (растворитель: дихлорметан/метанол= 18:2).
Выход: 6,55 г (71% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 63,63 Н: 6,59 N: 10,48;
получено: С: 53,83 Н: 6,70 N: 10,29.
MALDI-TOF-масс-спектр: Molpeak около 9246(M+Na+
)
ж) 1-(Бензилоксикарбонилметил)-4,7,10-трис(трет-бутоксикарбонилметил)-1,4,7,10-тетраазациклододекан (в качестве натрийбромидного комплекса)
20 г (38,87 ммоль) 1,4,
7-трис(трет-бутоксикарбонилметил)-1,4,7,10-тетраазациклододекана (DO3А-трис-трет-бутилового сложного эфира, полученного по ЕР 0299795, пример 22а, растворяют в 100 мл ацетонитрила. Затем добавляют 11,
45 г (50 ммоль) бензилового сложного эфира бромуксусной кислоты и 10,6 г (100 ммоль) карбоната натрия и перемешивают в течение 12 часов. Отфильтровывают соль, выпаривают фильтрат в вакууме до
получения твердого вещества и хроматографируют остаток на силикагеле (растворитель: метиленхлорид/метанол= 20:1).
Выход: 21,72 г (73% от теор.).
Элементный
анализ:
расчетн.: С: 54,90 Н: 7,63 N: 7,32 Na: 3,00 Br: 10,44;
получили: С: 54,80 Н: 7,72 N: 7,21 Na: 2,89 Br: 10,27.
з) 1-(Карбоксиметил)-4,7,
10-трис(трет-бутоксикарбонилметил)-1,4,7,10-тетраазациклододекан (в качестве натрийбромидного комплекса)
20 г (26,12 ммоль) соединения из заглавия примера 1ж) растворяют в 300 мл
изопропанола
и добавляют 3 г палладиевого катализатора (10% Pd/C). Проводят гидрирование в течение ночи при комнатной температуре. Отфильтровывают катализатор и упаривают фильтрат до получения
твердого
вещества.
Выход: 17,47 г (99% от теор.) бесцветного аморфного порошка.
Элементный анализ:
расчета.: С: 49,78 Н: 7,76 N: 8,29 Na: 4,44 Br: 11,83;
получили: С: 49,59 Н: 7,59 N: 8,17 Na: 4,40 Br: 11,70.
и) (4-Карбокси-2-оксо-3-азабутил)-4,7,10-трис(трет-бутоксикарбонилметил)-1,4,7,10-тетраазациклододекан
К 10 г
(14,80
ммоль) соединения из заглавия примера 1з) (растворенного) в диметилформамиде добавляют 1,73 г (15 ммоль) N-гидроксисукцинимида и охлаждают до 0oС, затем добавляют 4,13 г (20 ммоль)
дициклогексилкарбодиимида и перемешивают 1 час при комнатной температуре. Охлаждают до 0oС и затем добавляют 5,1 г (50 ммоль) триэтиламина и 2,25 г (30 ммоль) глицина. Перемешивают в
течение ночи при комнатной температуре. Отфильтровывают выпавшую в осадок мочевину и упаривают фильтрат в вакууме до получения твердого вещества. Остаток заливают водой и дважды экстрагируют с
помощью
метиленхлорида. Органическую фазу высушивают над сульфатом магния и упаривают в вакууме. Остаток хроматографируют на силикагеле (растворитель: метиленхлорид/метанол= 15:1).
Выход: 8, 20 г (88% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 57,21 Н: 8,80 N: 11,12;
получили: С: 57,10 Н: 8,91 N: 11,03.
к)
36-мерный каскадный N-(5-DO3А-ил-4- оксоазапентаноил)полиамид на основе описанного в примере 1е) 36-мерного полиамина
[D03A=1,4,7-Трис(карбоксиметил)-1,4,7,
10-тетраазациклододекан]
1,
84 г (0,2 ммоль) описанного в примере 1е) 36-мерного бензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным
раствором бромистого водорода в ледяной
уксусной кислоте. По истечении 5 часов с помощью диэтилэфира завершают начавшееся выпадение в осадок, получившийся 36-мерный амин-гидробромид промывают простым
эфиром, высушивают в вакууме и без
дополнительной очистки вводят в описанную ниже реакцию.
Выход (количественно): 1,5 г.
14,7 г (20 ммоль) описанной в примере 1и) карбоновой кислоты, 3,0 г (20 ммоль) 1-гидроксибензотриазола и 6,4 г (10 ммоль) 2-(1Н-бензотриазол-1-ил)-тетрафторбората 1,1,3,3-тетраметилурония (TBTU; Peboc Limited, UK) растворяют в ДМФ и перемешивают в течение 15 мин. Раствор затем смешивают с 10,3 мл (60 ммоль) N-этилдиизопропиламина и с 1,5 г (0,2 ммоль) описанного выше 36-мерного амин-гидробромида и в течение 4 дней перемешивают при комнатной температуре. По окончании реакции упаривают в вакууме, остаток растворяют в трифторуксусной кислоте при 0oС, в течение ночи перемешивают при комнатной температуре, упаривают в вакууме и остаток смешивают с простым эфиром. Вещество отсасывают, промывают простым эфиром, высушивают в вакууме, затем промывают водой, натриевой щелочью с рН 7, и раствор подвергают очистке через ультрафильтрационную мембрану YM3, Amicon (cut off:3000D). Фильтрат затем фильтруют и высушивают замораживанием.
Выход: 3,61 г (72% от теор.) хлопьевидного порошка.
Содержание Н2О (Карл-Фишер): 8,9%.
Элементный анализ:
расчетн.: С: 44,86 Н: 5,87 N: 15,34 Na: 10,92;
получили; С: 45,09 Н: 5,80 N: 15,44 Na: 10,
51.
1) 36-мерный Gd-комппекс описанного в
приведенном выше примере лиганда
2,5 г (0,1 ммоль) описанной в предыдущем примере 1к) натриевой соли кислоты комплексообразователя
подкисляют в воде 5 мл ледяной уксусной кислоты, смешивают с
725 мг (2 ммоль) Cd203 и осуществляют комплексообразование в течение 2-х часов при 80oС. После охлаждения
раствор фильтруют, фильтрат подвергают ультраочистке с
помощью устройства YM3 (AMICON®) и с помощью добавки (попеременно) катионообменной смолы IR 120 (Н+-форма) и
анионообменной смолы IRA 410 (ОН--форма)
ультрафильтрату придают минимальную проводимость. Отфильтровывают ионообменные смолы, и фильтрат подвергают сушке замораживанием.
Выход: 1,96 г (70% от теор.) бесцветного хлопьевидного порошка.
Содержание Н2О (Карл-Фишер): 7,4%
Определение Gd (ААС): 19,9%
MALDI-TOP-Macc-спектр:
Molpeak около 25.905 (расч.: 25.911Da)
Элементный анализ (в пересчете на безводное вещество):
расчетн.: С: 39,35 Н: 5,15 Cd: 21,85 N: 13,46;
получили: С: 39,08 Н: 5,29 Cd:
21,03 N: 13,68.
Т-релаксация
(Н2О): 18,0±0,2 (л/ммоль•с)
(плазма): 21,5±0,5 (л/ммоль•с).
Общее остаточное количество в организме после внутривенного ввода (0,1 ммоль гадолиния/кг веса тела) через 14 дней; крыса): 1,09±0,17% дозы.
Соответствующий комплекс европия показывает следующие
значения:
кролик: 0,23±0,12% дозы
мышь: 0,46±0,1% дозы
Пример 2
а) Сложный бензиловый эфир бромпропионилглицина
К 100 г (296,4 ммоль)
сложного глицинбензилового эфира соли п-толуолсульфокислоты
и 33,0 г (326,1 ммоль) триэтиламина, растворенным в 400 мл метиленхлорида, добавляют по капле 55,9 г (326,1 ммоль) хлорида
2-бромпропионовой кислоты. Температуру поддерживают не выше 5oС.
По окончании добавления производят перемешивание в течение 1 часа при 0oС, затем еще 2 часа при комнатной
температуре. Добавляют 500 мл ледяной воды и с помощью 10%-ного водного раствора
соляной кислоты устанавливают значение рН водной фазы, равное 2. Отделяют органическую фазу, по одному разу промывают
300 мл 5%-ного водного раствора соды и 400 мл воды. Высушивают органическую фазу
над сульфатом магния и упаривают в вакууме до получения твердого вещества. Остаток подвергают перекристаллизации из
диизопропилового эфира.
Выход: 68,51 г (75% от теор.) бесцветного кристаллического порошка.
Элементный анализ:
расчетн.: С: 46,76 Н: 7,19 N: 4,54 Br: 25,99;
получили: С: 46,91 Н: 7,28 N: 4,45 Br: 25,81.
б)
1-[4-(Бензилоксикарбонил)-1-метил-2-оксо-3-азабутил] -1,4,7,10-тетраазациклододекан
К 55,8 г (324,4 ммоль) 1,4,7,
10-тетраазациклододекана, растворенного в 600 мл хлороформа, добавляют 50 г
(162,2 ммоль) соединения из заглавия примера 2а) и перемешивают в течение ночи при комнатной температуре. Добавляют 500 мл
воды, отделяют органическую фазу и промывают еще дважды соответственно 400 мл
воды. Высушивают органическую фазу над сульфатом магния и упаривают в вакууме до получения твердого вещества. Остаток
хроматографируют на силикагеле (растворитель: хлороформ/метанол/вода=10:5:1).
Выход: 40,0 г (63% от теор.) слегка желтоватого вязкого масла.
Элементный анализ:
расчетн.: С: 61,36 Н: 8,50 N: 17,89;
получили: С: 61,54 Н: 8,68 N: 17,
68.
в) 1-[4-(Бензилоксикарбонил)-1-метил-2-оксо-3-азабутил] -4,7,
10-трис(трет-бутоксикарбонилметил)-1,4,7,10-тетраазациклододекан натрийбромидный комплекс)
К 20 г (51,08
ммоль) соединения из заглавия примера 2б) и 17,91 (169 ммоль) карбоната натрия в 300
мл ацетонитрила добавляют 33 г (169 ммоль) сложного трет-бутилового эфира бромуксусной кислоты и перемешивают 24
часа при 60oС. Охлаждают до 0oС, отфильтровывают от солей и
упаривают фильтрат до получения твердого вещества. Остаток хроматографируют на силикагеле (растворитель:
этилацетат/этанол: 15: 1). Содержащие продукт фракции упаривают, и остаток подвергают
перекристаллизации из диизопропилэфира.
Выход: 34,62 г (81% от теор.) бесцветного кристаллического порошка.
Элементный анализ:
расчетн.: С: 54,54 Н: 7,59 N:
8,37 Na: 2,74 Br: 9,56;
получено: С: 54,70 Н: 7,65 N: 8,24 Na: 2,60 Br: 9,37.
г) 1-(4-Карбокси-1-метил-2-оксо-3-азабутил)-4,7,10-трис(трет-бутоксикарбонилметил)-1,4,7,
10-тетраазациклододекан натрийбромидный комплекс)
30 г (35,85 ммоль) соединения из заглавия
примера 2в) растворяют в 500 мл изопропанола и добавляют 3 г палладиевого катализатора (10% Pd/C).
Проводят гидрирование в течение ночи при комнатной температуре. Катализатор отфильтровывают, фильтрат
упаривают в вакууме до получения твердого вещества и подвергают перекристаллизации из ацетона.
Выход: 22,75 г (85% от теор.) бесцветного кристаллического порошка.
Элементный анализ:
расчетн.: С: 49,86 Н: 7,69 N: 9,38 Na: 3,07 Br: 10,71;
получили: С: 49,75 Н: 7,81 N: 9,25 Na: 2,94 Br: 10,58.
д)
1-[4-(4-Нитрофеноксикарбонил)-1-метил-2-оксо-3-азабутил]-4,7,10-трис(трет-бутоксикарбонилметил)-1,4,7,
10-тетраазациклододекан натрийбромидный комплекс)
37,3 г (50 ммоль) описанной в
предыдущем примере 2г) карбоновой кислоты в 500 мл дихлорметана смешивают с 7,6 г (55 ммоль) 4-нитрофенола и
охлаждают до 0oС. После добавки 10,8 г (52,5 ммоль) дициклогексилкарбодиимида
перемешивают в течение ночи при комнатной температуре, затем при повторном охлаждении отсасывают от выпавшей в
осадок дициклогексилмочевины, и фильтрат упаривают в вакууме до получения твердого
вещества. Остаток подвергают перекристаллизации из этилацетата.
Выход: 40,3 г (92% от теор.) слегка желтоватого порошка.
Элементный анализ:
расчетн.: С: 51,
21 Н: 6,97 N: 9,68 Na: 2,65 Br: 9,21;
получили: С: 51,06 Н: 7,07 N: 9,82 Na: 2,40 Br: 8,77.
e) 36-мерный каскадный -N-(5-DO3А-ил-4-оксо-3-азагексаноил)полиамид на основе
описанного в примере 1е) 36-мерного полиамина
1,84 г (0,2 ммоль) описанного в примере 1е 36-мерного
бензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают
с 33%-ным бромистым водородом в ледяной уксусной кислоте. По истечении 5 часов с помощью диэтилэфира
завершается начавшееся осаждение, полученный 36-мерный амин-гидробромид промывают простым эфиром,
высушивают в вакууме и подвергают разложению без дополнительной промывки.
Выход: 1,5 г (количественно).
1,5 г описанного 36-мерного амин-гидробромида в 100 мл ДМФ смешивают с 17,4 г (20 ммоль) описанного в примере 2д) активного сложного п-нитрофенилового эфира. Затем в течение одного часа добавляют каплями раствор 5,05 г (50 ммоль) триэтиламина в 20 мл ДМФ настолько медленно, чтобы образующийся вначале осадок снова мог раствориться. Перемешивание проводят в течение ночи при 45oС, затем раствор сгущают в вакууме, остаток растворяют при 0oС в трифторуксусной кислоте и перемешивают в течение ночи при комнатной температуре. Проводят упаривание в вакууме, остаток перемешивают с диэтилэфиром, осадок отсасывают и высушивают в вакууме. Кислый сырой продукт растворяют затем в воде, с помощью разбавленной натриевой щелочи устанавливают рН 7 и производят ультрафильтрование через мембрану (AMICON®) YM3. Das Retentat (ультрафильтрат) высушивают замораживанием.
Выход: 4,0 г (78% от теор.).
Содержание Н2O (Карл-Фишер): 9,3%.
Элементный анализ (в
пересчете на безводное вещество):
расчетн.: С: 45,74 Н: 6,05 N: 15,01 Na: 10,68;
получили: С: 45,84 Н: 5,93 N: 15,22 Na: 10,20.
ж) 36-мерный-Gd-комплекс описанного в
предыдущем примере лиганда
2,5 г (0,1 ммоль) описанной в предыдущем примере
2е) натриевой соли кислоты комплексообразователя подкисляют в воде 5 мл ледяной уксусной кислоты, смешивают с 725
мг (2 ммоль) Gd203 и в течение 2 часов проводят
комплексообразование при 80oС. После охлаждения раствор фильтруют, фильтрат пропускают через устройство YM3 (AMICON®) с целью ультрафильтрации и фильтрат обрабатывают
попеременно добавками катионообменной смолы IR 120 (в форме Н+) и анионообменной смолы IRA 410 (в форме ОН-) с
целью установления минимальной проводимости. Отфильтровывают от
(ионо)обменных смол, и фильтрат высушивают замораживанием.
Выход: 2,14 г (74% от теор.) бесцветного хлопьевидного порошка.
Содержание H2O (Карл-Фишер): 8,
7%
Определение Gd (ААС): 19,4%
MALDI-TOF-масс-спектр: Molpeak около 26.426 Da (расч.: 26/416 Da)
Элементный анализ (в пересчете на безводное вещество):
расчетн.:
С: 40,24 Н: 5,32 Gd: 21,43 N: 13,20;
получили: С: 39,97 Н: 5,50 Gd: 21,19 N: 13,32.
T1
-релаксация (Н2О): 17,5±0,1 (л/ммоль.с)
(плазма):
18,2±0,2 (л/ммоль•с)
Общее остаточное количество в организме после внутривенного введения (0,1 ммоль
гадолиния/кг
веса тела; по истечении 14 дней; крыса): 1,74±0,22%
дозы.
Соответствующий комплекс европия показывает следующие значения:
кролик: 0,32±0,16%
дозы
мышь: 1,0±0,1% дозы
Пример 3
а) 1,4,
7-трис(N-бензилоксикарбонилглицил)-1,4,7,10-тетраазациклододекан
29,37 г (95,9 ммоль) сложного эфира
Z-глицин-N-гидроксисукцинимида и 5 г (29 ммоль) циклена (1,4,7,10-тетраазациклододекана)
растворяют в смеси, состоящей из 100 мл толуола и 50 мл диоксана. Добавляют 9,7 г (95,9 ммоль) триэтиламина и
нагревают 12 часов до 70oС. Упаривают до получения твердого вещества,
погружают остаток в 400 мл дихлорметана и три раза экстрагируют с помощью соответственно в каждом случае 200 мл 5%-ного
водного раствора карбоната калия. Органическую фазу высушивают над сульфатом
магния и упаривают в вакууме до получения твердого вещества. Остаток хроматографируют на силикагеле (растворитель:
этилацетат/этанол=15:1).
Выход: 17,52 г (81% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 61,20 Н: 6,35 N: 13,15;
получили: С: 61,07 Н: 6,45 N: 13,01.
б) 1-(Карбоксиметоксиацетил)-4,7,
10-трис(N-бензилокси-карбонилглицил)-1,4,7,10-тетраазациклододекан
К 17 г (22,79 ммоль) соединения из
заглавия примера 3а) (растворенного в 100 мл тетрагидрофурана) добавляют 3,97 г (34,19
ммоль) ангидрида дигликолевой кислоты и 6,92 г (68,38 ммоль) триэтиламина. Нагревают 6 часов до 50oС.
Раствор упаривают в вакууме до получения твердого вещества, загружают в 250 мл
дихлорметана и дважды экстрагируют с помощью в каждом случае 100 мл 5%-ного водного раствора соляной кислоты.
Органическую фазу высушивают над сульфатом магния, упаривают в вакууме до получения
твердого вещества и остаток хроматографируют на силикагеле (растворитель: дихлорметан/метанол=20:1).
Выход: 17,48 г (89% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 58,53 Н: 5,96 N: 11,38;
получили: С: 58,37 Н: 5,81 N: 11,45.
в) Бис[2-(Nα,Nε
-дибензилоксикарбонил-лизиламино)-этил]-амин
1,03 г (10 ммоль) диэтилентриамина растворяют в 100 мл THF, смешивают с 2,02 г (2,77 мл, 20 ммоль)
триэтиламина и 11,25 г (21 ммоль) сложного
п-нитрофенилового эфира N,N'-ди-бензилоксикарбонил-лизина (О.W.Lever et al., J. Heterocyclic Chem. , 23, 900-903 (1986)) и перемешивают в течение 3-х часов
при комнатной температуре. Полученную
густую суспензию дополняют простым эфиром до 250 мл, перемешивают в течение ночи при комнатной температуре, объемный осадок отсасывают и промывают смесью
ТГФ/эфир=1:1 в количестве 100 мл, а затем еще
раз эфиром. После сушки в вакууме при 40oС получают 8,7 г (97,1% от теор.) бесцветного порошка.
Элементный анализ:
расчетн.: С: 64,34 Н: 6,86 N: 10,94;
получили: С: 64,20 Н: 6,97 N: 10,81.
г) триамид N,N,N',N',N'',N''-гексакис[2-((Nα,Nε
- дибензилоксикарбонил-лизиламино)-этил] тримезиновой кислоты
1,43 г (1,6
ммоль) бис[2-(N,N-дибензилоксикарбонил-лизиламино)-этил]-амина в 20 мл ДМФ смешивают с 1,39 мл (1,01 г, 10 ммоль)
триэтиламина и 0,11 г (0,4 ммоль) трихлорида тримезиновой кислоты (Aldrich),
перемешивают в течение 2-х часов во льду и в течение ночи при комнатной температуре. Затем сгущают в вакууме, погружают в
этилацетат и промывают разбавленной натриевой щелочью, 1М соляной кислотой и
полунасыщенным раствором NaCl и высушивают над сульфатом натрия. После добавки активированного угля фильтруют через
тефлоновый мембранный фильтр, фильтрат сгущают (1,5 г), снова растворяют в примерно
5 мл этилацетата и хроматографируют на силикагеле со смесью этилацетат/метанол (18:2).
Выход: 0,9 г (79,1%) бесцветного порошка.
Элементный анализ:
расчетн.: С: 64,61 Н: 6,48 N: 10,38;
получили: С: 64,45 Н: 6,60 N: 10,28.
д) Полностью защищенный
36-мерный бензилоксикарбонилполиамин, построенный из ядра триамида N, N, N',
N',N'',N''-гексакис[2-(лизиламино)-этил] тримезиновой кислоты и двенадцати описанных в примере 3б) аминозащищенных
триаминмонокарбоновых кислот
2,84 г (1 ммоль) описанного в предыдущем
примере 3г) 12-мерного бензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с
33%-ным раствором бромистого водорода в ледяной уксусной кислоте. По истечении 3-х
часов с помощью диэтилового эфира завершают начавшееся выпадение осадка, полученный 12-мерный амин-гидробромид
промывают эфиром, высушивают в вакууме и без дополнительной очистки вводят в описываемую
ниже реакцию.
Выход: 2,2 г (количественно).
17,2 г (20 ммоль) описанной в примере 3б) цикленкарбоновой кислоты, 3 г (20 ммоль) 1-гидроксибензотриазола и 6,4 г (20 ммоль) 2-(1Н-бензотриазол-1-ил)-тетрафторбората 1,1,3,3-тетраметилурония (TBTU; Peboc Limited, UK) растворяют в ДМФ и перемешивают в течение 15 мин. Затем раствор смешивают с 10,3 мл (60 ммоль) N-этилдиизопропиламина и 2,2 г (1 ммоль) описанного выше 12-мерного амин-гидробромида и перемешивают в течение ночи при комнатной температуре. По окончании реакции упаривают в вакууме, и остаток хроматографируют на силикагеле со смесью дихлорметан/метанол (17:3).
Выход: 9,6 г (84,5% от теор.) бесцветного порошка.
Элементный анализ:
расчетн.: С: 59,
31 Н: 6,20 N: 12,94;
получили: С: 59,20 Н: 6,03 N: 13,19.
MALDI-TOF-масс-спектр: Molpeak
около 11.384 (M+Na+)
е) 36-мерный каскадный
N-(5-DO3А-ил-4-оксо-3-азапентаноил)полиамид на основе описанного в примере 3е) 36-мерного полиамина
2,27 г (0,2 ммоль)
описанного в примере 3д) 36-мерного бензилоксикарбониламина растворяют
в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным бромистым водородом, растворенным в ледяной уксусной кислоте.
Через 5 часов с помощью диэтилэфира завершают начавшееся выпадение
осадка, полученный 36-мерный амин-гидробромид промывают простым эфиром, высушивают в вакууме и без дополнительной очистки вводят в
описываемую ниже реакцию.
Выход: 1,9 г (количественно).
14,7 г (20 ммоль) описанной в примере 1и) карбоновой кислоты, 3 г (20 ммоль) 1-гидроксибензотриазола и 6,4 г (20 ммоль) 2-(1H-бензотриазол-1-ил)-тетрафторбората 1,1, 3,3-тетраметилурония (TBTU; Peboc Limited, UK) растворяют в ДМФ и перемешивают в течение 15 мин. Затем раствор смешивают с 10,3 мл (60 ммоль) N-этилдиизопропиламина и с 1,9 г (0,2 ммоль) описанного выше 36-мерного амин-гидробромида и в течение 4 дней перемешивают при комнатной температуре. По окончании реакции проводят упаривание в вакууме, остаток растворяют при 0oС в трифторуксусной кислоте, перемешивают в течение ночи при комнатной температуре, упаривают в вакууме, и остаток перемешивают с простым эфиром. Вещество отсасывают, промывают простым эфиром, высушивают в вакууме, с помощью 2Н натриевой щелочи устанавливают рН 7, и раствор очищают через ультрафильтрационную мембрану YM3, Amicon® (cut. off: 3000Da). Фильтрат окончательно фильтруют и высушивают замораживанием.
Выход: 3,93 г (75% от теор.) хлопьевидного порошка.
Содержание Н2О (Карл-Фишер):
5%
Элементный анализ (в пересчете на
безводное вещество):
расчетн.: С: 44,48 Н: 5,75 N: 16,05 Na: 9,98;
получили: С: 44,77 Н: 5,91 N: 15,96 Na: 9,50.
ж)
36-мерный Gd-комплекс описанного в примере 3е
лиганда
2,6 г (0,1 ммоль) описанной в примере 3е) натриевой соли кислоты комплексообразователя подкисляют в воде с помощью 5 мл ледяной уксусной
кислоты, смешивают с 725 мг (2 ммоль) Gd2O3 и проводят комплексообразование в течение 2-х часов при 80oС. После охлаждения раствор фильтруют, фильтрат подвергают
ультрафильтрации через YM3 (AMICON®), и фильтрат подвергают попеременной обработке катионообменной смолой IR 120 (форма Н+) и анионообменной смолой IRA 410 (форма ОН-) с целью обеспечения минимальной
проводимости. Ионообменные смолы отфильтровывают, фильтрат высушивают замораживанием.
Выход: 2,22 г (72% от теор.) бесцветного, хлопьеобразного порошка.
Содержание
Н2О (Карл-Фишер): 8,9%
Определение гадолиния (ААС): 18,5%
MALDI-TOF-масс-спектр: Molpeak около 28.058 Da (расч.:
28.049Da)
Элементный анализ (в пересчете на
безводное вещество):
расчетн.: С: 39,44, Н: 5,10 Gd: 20,18 N: 14,23;
получили: С: 39,56 Н: 5,26 Gd: 19,88 N: 14,09.
Пример 4
а) 36-мерный каскадный
N-(5-DO3А-ил-4-оксо-3-азагексаноил)полиамид на основе описанного в примере 3е 36-мерного полиамина
2,27 г (0,2 ммоль) описанного в примере
3д) 36-мерного бензилоксикарбониламина растворяют в
ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным раствором бромистого водорода в ледяной уксусной кислоте. Через 5 часов с помощью
диметилэфира завершают начавшееся выпадение осадка,
полученный 36-мерный амин-гидробромид промывают простым эфиром, высушивают в вакууме и без дополнительной очистки подвергают дальнейшему
разложению.
Выход: 1,9 г (количественно).
1,9 г описанного выше 36-мерного амин-гидробромида в 100 мл ДМФ смешивают с 17,4 г (20 ммоль) описанного в примере 2д) активного сложного п-нитрофенильного эфира. В течение 1 часа добавляют затем по капле раствор 5,05 г (50 ммоль) триэтиламина в 20 мл ДМФ настолько медленно, чтобы образующийся поначалу осадок мог снова перейти в раствор. Перемешивают в течение ночи при 45oС, затем раствор сгущают в вакууме, остаток растворяют в трифторуксусной кислоте при 0oС и в течение ночи перемешивают при комнатной температуре. Упаривают в вакууме, остаток смешивают с диэтилэфиром, осадок отсасывают и высушивают в вакууме. Кислый сырой продукт растворяют затем в воде, с помощью разбавленной натриевой щелочи устанавливают рН 7 и подвергают ультрафильтрации через мембрану YM3 (AMICON®). Фильтрат высушивают замораживанием.
Выход: 4,0 г (72,9 от теор.).
Содержание Н2О
(Карл-Фишер): 7,5%
Элементный анализ (в пересчете на безводное вещество):
расчетн.: С: 45,30 Н: 5,92 N: 15,73 Na: 9,78;
получили: С:
45,56 Н: 6,10 N: 15,65 Na: 9,47.
б) 36-мерный Gd-комплекс описанного в предыдущем примере 4а лиганда
2,74 г (0,1 ммоль) описанной в предыдущем примере 4а натриевой соли
кислоты комплексообразователя подкисляют в
воде с помощью 5 мл ледяной уксусной кислоты, смешивают с 725 мг (2 ммоль) Gd2O3 и в течение 2-х часов подвергают комплексообразованию
при 80oС. После охлаждения
раствор фильтруют, фильтрат подвергают ультрафильтрации через YM3 (AMICON®), и фильтрат далее подвергают попеременной обработке катионообменнной
смолой IR 120 (Н+-форма)
и анионообменной смолой IRA 410 (ОН--форма) с целью обеспечения минимальной проводимости. Ионообменные смолы отфильтровывают, и фильтрат высушивают
замораживанием.
Выход: 2,46 г (77,8% от теор.) бесцветного, хлопьевидного порошка.
Содержание H2O (Карл-Фишер): 9,7%
Определение Gd (AAC): 18,
1%
MALDI-TOF-Macc-спектр: Molpeak
около 28.563Da (расч.: 28.554Da)
Элементный анализ (в пересчете на безводное вещество)
расчетн.: С: 40,26 Н: 5,26 Gd: 19,83 N: 13,98;
получили: С: 40,01 Н: 5,40 Gd: 19,68 N:
14,11.
Пример 5
а) 1,7-Бис(бензилоксикарбонил)-4-гидроксисукцинил-1,4,7-триазагептан
К 50 г (134,6 ммоль) 1,
7-бис(бензилоксикарбонил)-1,4,7-триазагептана (пример 1
г) в 500 мл тетрагидрофурана добавляют 20,20 г (201,9 ммоль) ангидрида янтарной кислоты и 40,86 г (403,8 ммоль) триэтиламина и перемешивают в
течение ночи при 40oС. Упаривают до получения
твердого вещества, погружают остаток в 1000 мл дихлорметана и дважды промывают соответственно 500 мл 5%-ной соляной кислоты. Органическую фазу
высушивают над сульфатом магния и упаривают до получения
твердого вещества. Остаток хроматографируют на силикагеле (растворитель: дихлорметан/метанол=20:1).
Выход: 56,0 г (93% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 59,05 Н: 6,53 N: 9,39;
получили: С: 59,17 Н: 6,69 N: 9,27.
б) сложный эфир
N-гидроксисукцинимида 1,
7-бис(бензилоксикарбонил)-4-гидроксисукцинил-1,4,7-триазагептана
К 56 г (125,14 ммоль) соединения из заглавия примера 5а), (растворенного) в 300 мл дихлорметана,
добавляют 14,4 г (125,14
ммоль) N-гидроксисукцинимида. Охлаждают до 0oС и добавляют 28,4 г (137,66 ммоль) дициклогексилкарбодиимида. Затем перемешивают в течение 6 часов при комнатной
температуре. Выпавшее в
осадок твердое вещество отфильтровывают и фильтрат упаривают в вакууме до получения твердого вещества. Остаток подвергают перекристаллизации из смеси простой
эфир/2-пропанол.
Выход: 62,01 г (91% от теор.) кристаллического бесцветного вещества.
Элементный анализ:
расчетн.: С: 57,35 Н: 5,92 N: 10,29;
получили: С: 57,24 Н: 5,99 N: 10,12.
в) 1,4,7-трис{ 7-бензилоксикарбониламино-5-[2-бензил-оксикарбониламино)-этил]-4-оксо-5-азагептаноил} 1,4,7,10-тетраазациклододекан
52,22
г (95,9 ммоль) соединения из заглавия
примера 5б) и 5 г (29 ммоль) циклена (1,4,7,10-тетраазациклододекан) растворяют в смеси из 200 мл толуола и 100 мл диоксана. Добавляют 9,7 г (95,9 ммоль)
триэтиламина и нагревают в течение 12 часов
до 70oС. Упаривают до получения твердого вещества, погружают остаток в 600 мл дихлорметана и трижды экстрагируют соответственно 300 мл 5%-ного
водного раствора карбоната калия.
Органическую фазу высушивают над сульфатом магния и упаривают до получения твердого вещества. Остаток хроматографируют на силикагеле (растворитель:
этилацетат/этанол=15:1).
Выход: 28, 95 г (69% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 61,44 Н: 7,04 N: 11,62;
получили: С: 61,57 Н: 6,91 N: 11,69.
г)
1,4,7-Трис{7-бензилоксикарбониламино-5-[2-бензилоксикарбониламино)-этил] -4-оксо-5-азагептаноил} -10-гидроксисукцинил-1,4,7,
10-тетраазациклододекан
К 28 г (19,35 ммоль) соединения из
заголовка примера 5в, растворенного в тетрагидрофуране, добавляют 2,90 г (29 ммоль) ангидрида янтарной кислоты и 5,87 г (58 ммоль)
триэтиламина. Нагревают в течение 6 часов до 50oС.
Раствор упаривают в вакууме до получения твердого вещества, погружают в 200 мл дихлорметана и дважды экстрагируют соответственно 10 мл
5%-ного водного раствора соляной кислоты. Органическую фазу
высушивают над сульфатом магния, упаривают в вакууме до получения твердого вещества и остаток хроматографируют на силикагеле (растворитель:
дихлорметан/метанол=20:1).
Выход: 26,94 г (90% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 60,57 Н: 6,84 N: 10,87;
получили: С: 60,41 Н: 6,95 N: 10,75.
д) 1,4,7,
10,13,16-Гексакис[N-бензилоксикарбонил-β-аланил]-1,4,7,10,13,16-гексаазациклооктадекан
516 мг (2 ммоль) 1,4,7,10,13,
16-гексаазациклооктадекана (гексациклен; Fluka) обезвоживают
азеотропно с помощью толуола. К охлажденному раствору гексациклена в толуоле при комнатной температуре добавляют раствор 3,35 г (15 ммоль)
бензилоксикарбонил-β-аланина (сигма) в
тетрагидрофуране (ТГФ), а также 3,71 г (15 ммоль) 2-этокси-1-этоксикарбонил-1,2-дигидрохинолина (EEDQ; Fluka) и перемешивают в течение ночи. По окончании
реакции продукт, благодаря добавке гексана,
выпадает в осадок, и осадок хроматографируют на силикагеле со смесью дихлорметан/гексан/изопропанол (20:10:1).
Выход: 2,06 г (69% от теор.).
Элементный анализ:
расчетн.: С: 62,89 Н: 6,50 N: 11,28;
получили: С: 62,74 Н: 6,32 N: 11,50.
е) Полностью защищенный 36-мерный
бензилоксикарбонил полиамин, построенный из ядра 1,4,7,10,
13,16-гексакис(β-аланил)-1,4,7,10,13,16-гексаазациклооктадекана из шести описанных в примере 5г) аминозащищенных
гексааминмонокарбоновых кислот
1,49 г (1 ммоль) описанного в
предыдущем примере 5д) гексабензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным
раствором бромистого водорода в ледяной уксусной кислоте. По
истечении 60 мин с помощью диэтилового эфира завершают начавшееся выпадение в осадок, полученный гексаамин-гидробромид промывают простым
эфиром, высушивают в вакууме и без дополнительной очистки
вводят в описываемую ниже реакцию.
Выход: 1,2 г (количественно).
7,0 г (7,5 ммоль) описанной в примере 5г) аминозащищенной гексаамин-монокарбоновой кислоты, 1,2 г (7, 5 ммоль) 1-гидроксибензотриазола и 2,4 г (7,5 ммоль) 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетрафторбората тетраметилурония (TBTU; Peboc Limited, UK) растворяют в ДМФ и перемешивают в течение 15 мин. Этот раствор смешивают затем с 5,16 мл (30 ммоль) N-этилдиизопропиламина и с 1,2 г (1 ммоль) описанного выше гексаамингидробромида и перемешивают в течение ночи при комнатной температуре. По окончании реакции упаривают в вакууме, и остаток хроматографируют на силикагеле в смеси дихлорметан/метанол (17:3).
Выход: 8,5 г (82% от теор.) бесцветного порошка.
Элементный
анализ:
расчетн.: С: 61,83 Н: 6,59 N: 12,15;
получили: С: 61,59 H: 6,71 N: 12,02.
MALDI-TOF-масс-спектр:
Molpeak около 10.397 (M+Na+)
ж)
36-мерный Каскадный N-(5-DO3А-ил-4-оксо-3-азагексаноил)полиамид на основе описанного в примере 5д полиамина
2,07 г (0,2 ммоль) описанного в
примере 5е) 36-мерного бензилоксикрабониламина
растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным раствором бромистого водорода в ледяной уксусной кислоте. По истечении 5
часов с помощью диэтилэфира завершают начавшееся
выпадение осадка, полученный 36-мерный амин-гидробромид промывают простым эфиром и высушивают в вакууме и без дополнительной очистки подвергают
разложению.
Выход: 1,7 г (количественно).
1,7 г описанного выше 36-мерного амин-гидробромида в 100 мл ДМФ смешивают с 217,4 г (20 ммоль) описанного в примере 2д)
активированного сложного п-нитрофенилового
эфира. В течение часа затем добавляют по капле раствор 5,05 г (50 ммоль) триэтиламина в 20 мл ДМФ настолько медленно, чтобы образующийся поначалу осадок смог
снова перейти в раствор. Перемешивают в
течение ночи при 45oС, затем раствор сгущают в вакууме, остаток при 0oС растворяют в трифторуксусной кислоте и перемешивают в течение ночи
при комнатной температуре. Упаривают в
вакууме, остаток перемешивают с диэтилэфиром, осадок отсасывают и высушивают в вакууме. Кислый сырой продукт растворяют затем в воде, с помощью натриевой щелочи
устанавливают рН 7 и подвергают
уьтрафильтрации через мембрану YM3, Amicon®. Retentat высушивают замораживанием
Выход: 4,4 г (83% от теор.).
Содержание
Н2О (Карл-Фишер): 7,
8%
Элементный анализ (в пересчете на безводное вещество):
расчетн.: С: 45,80 Н: 6,08 N: 15,51 Na: 10,18;
получили: С: 45,88 Н: 6,23 N: 15,66
Na: 9,70.
з)
36-мерный Gd-комплекс описанного в предыдущем примере 5ж) лиганда
2,65 г (0,1 ммоль) описанной в предыдущем примере 5ж натриевой соли кислоты
комплексообразователя подкисляют в воде с
помощью 5 мл ледяной уксусной кислоты, смешивают с 725 мг (2 ммоль) Gd2O3 и проводят комплексообразование в течение 2-х часов при 80oС. После охлаждения раствор
фильтруют, фильтрат подвергают ультрафильтрованию через YM3 (Amicon®) и Retentat обрабатывают попеременно добавками катионообменной смолы IR 120
(Н+-форма) и
анионообменной смолы IRА 410 (ОН--форма) с целью установления минимальной проводимости. Ионообменные смолы отфильтровывают, и фильтрат высушивают замораживанием.
Выход: 2, 41 г (81% от теор.) бесцветного, хлопьевидного порошка.
Содержание H2O (Карл-Фишер): 7,5%
Определение Gd (AAC) 18,7%.
MALDI-TOF-Macc-спектр:
Molpeak около 27.580 Da (расч.: 27.566)
Элементный анализ (в пересчете на безводное вещество):
расчетн.: С: 40,52 Н: 5,37 Gd: 20,54 N: 13,72;
получили: С: 40,30 Н: 5,50 Gd:
20,11 N: 13,56.
Пример 6
36-мерный Gd-DTPA Моноамид на основе описанного в примере 5е) 36-мерного полиамина
1,04 г (0,2 ммоль)
описанного в примере 5е) 36-мерного
полибензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным бромистым водородом в ледяной уксусной кислоте. По
истечении 3-х часов с помощью диэтилового
эфира завершается начавшееся выпадение в осадок, полученный 36-мерный амин-гидробромид промывают простым эфиром и высушивают в вакууме. Остаток погружают в
воду и с помощью добавки 1Н натриевой
щелочи доводят до рН 9,5. В этот раствор добавляют 4,35 (10,8 ммоль) N3-(2,6-диоксоморфолиноэтил)-N6-(этоксикарбонилметил)- 3,
6-диазаоктандикислоты (пример 13а из ЕР 0331616) в
твердой форме, при этом рН с помощью дальнейшей добавки натриевой щелочи поддерживается постоянным около 9,5. По окончании добавления для омыления
сложного этилового эфира DTPA с помощью 5Н натриевой
щелочи устанавливают рН 13 и перемешивают в течение ночи при комнатной температуре. Затем с помощью концентрированной соляной кислоты устанавливают
рН 5, смешивают с 1,96 г (5,4 ммоль) Gd2
O3, в течение 30 мин перемешивают при температуре 80oС, после охлаждения устанавливают рН 7 и обессоливают через
ультрафильтрационную мембрану YM3 AMICON®.
Фильтрат затем фильтруют через мембранный фильтр и высушивают замораживанием.
Выход: 2,58 г (92,4% от теор.).
Содержание Н20 (Карл-Фишер): 9,0%
Определение Gd (AAC): 20,3%
Элементный анализ (в пересчете на безводное вещество):
расчетн.: С: 35,46 Н: 4,26 Gd: 22,
29 N: 10,92 Na: 3,26;
получили: С: 35,18 Н: 4,44 Gd: 21,
75 N: 10,83 Na: 3,59.
Пример 7
а)
5-Бензилоксикарбониламино-2-[3-(бензилоксикарбониламино)-пропил]-валериановая кислота
К 10 г (57,39 ммоль) 4-карбокси-1,
7-диаминогептана (получено по A.Reissert, Chem. Ber. 26, 2137 (1893);
27, 979 (1894) в 150 мл воды добавляют по капле при 0oС одновременно 24,48 г (143,5 ммоль) сложного бензилового эфира
хлормуравьиной кислоты и 5Н водного раствора натриевой щелочи и
поддерживают значение рН примерно рН 10. Перемешивают в течение ночи при комнатной температуре. Дважды экстрагируют с помощью
соответственно 150 мл сложного этилового эфира уксусной кислоты. Водную
фазу осторожно подкисляют 4Н водным раствором соляной кислоты (рН 2) и трижды экстрагируют с помощью соответственно 200 мл
этилацетата. Органическую фазу высушивают над сульфатом магния и упаривают в
вакууме до получения твердого вещества.
Выход: 24,13 г (95% от теор.) стекловидного твердого вещества.
Элементный анализ:
расчетн.: С: 59,05 Н: 6,53 N: 9,39;
получили: С: 59,19 Н: 6,71 N: 9,18.
б) сложный эфир N-гидроксисукцинимида
5-бензил-оксикарбониламино-2-[3-(бензилоксикарбониламино)пропил] валериановой кислоты
К 24
г (54,24 ммоль) соединения из заглавия примера 7а), растворенного в 100 мл дихлорметана, добавляют
6,24 г (54,24 ммоль) N-гидроксисукцинимида. Охлаждают до 0oС и добавляют 12,31 г (59,66
ммоль) дициклогексилкарбодиимида. Затем перемешивают 6 часов при комнатной температуре.
Отфильтровывают выпавшее в осадок твердое вещество, и упаривают фильтрат в вакууме до получения твердого
вещества. Остаток подвергают перекристаллизации из (смеси) простой эфир/2-пропанол.
Выход: 27,51 г (94% от теор.) кристаллического бесцветного вещества.
Элементный
анализ:
расчетн.: С: 62,33 Н: 6,16 N: 7,79;
получили: С: 62,17 Н: 6,03 N:
7,85.
в) 1,4,
7-трис{5-Бензилоксикарбониламино-2-[3-бензилоксикарбониламино)-пропил]-валерил}-1,4,7,10-тетраазациклододекан
27 г (50,04 ммоль) соединения из заглавия примера
7б) и 2,61 г (15,16 ммоль)
циклена (1,4,7,10-тетраазациклододекан) растворяют в смеси из 100 мл толуола и 50 мл диоксана. Добавляют 3,07 г (30,32 ммоль) триэтиламина и нагревают в течение 12 часов до
70oC. Упаривают
до получения твердого вещества, погружают остаток в 300 мл дихлорметана и трижды экстрагируют три раза с помощью соответственно 150 мл 5%-ного водного раствора карбоната
калия. Органическую фазу
высушивают над сульфатом магния и упаривают в вакууме до получения сухого вещества. Остаток хроматографируют на силикагеле (растворитель: этиловый сложный эфир уксусной
кислоты/этанол=15:1).
Выход: 13,81 г (63% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 66,46 Н: 7,26 N: 9,69;
получили:
С: 66,28 Н: 7,39 N: 9,51.
г) 1-[Карбокси-метоксиацетил)-4,7,10-трис{ 5-бензил-оксикарбониламино-2-[3-бензил-оксикарбониламино)-пропил] -валерил} -1,4,7,10-тетрааэациклододекан
К 13 г (9 ммоль) соединения
из заглавия примера 7в) в 80 мл тетрагидрофурана добавляют 1,57 г (13,5 ммоль) ангидрида дигликолевой кислоты и 2,73 г (27 ммоль) триэтиламина. Нагревают 6 часов до
50oС. Раствор упаривают
в вакууме до получения твердого вещества. Погружают в 150 мл дихлорметана и дважды экстрагируют с помощью соответственно 100 мл 5%-ного водного раствора соляной
кислоты. Органическую фазу высушивают
над сульфатом магния, упаривают в вакууме до
получения твердого веществ а, и остаток хроматографируют на силикагеле (растворитель:
дихлорметан/метанол=20:1).
Выход: 12,5 г (89% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 64,60 Н: 6,97 N: 8,97;
получили: С: 64,41 Н: 6,85 N: 8,90.
д) Полностью защищенный 36-мерный бензилоксикарбонил полиамин, построенный из ядра триамида N,N,N',N',N'',N''-гексакис(2-аминоэтил) тримезиновой кислоты и шести описанных в примере 7г) аминозащищенных гексааминмонокарбоновых кислот, 1,27 г (1 ммоль) описанного в предыдущем примере 1д) гексабензилоксикарбонил-амина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным раствором бромистого водорода в ледяной уксусной кислоте. По истечении 60 мин с помощью диметилэфира завершается начавшееся выпадение осадка, полученный гексаамингидробромид промывают эфиром, высушивают в вакууме и без дополнительной очистки вводят в описываемую ниже реакцию.
Выход: 0,95 г (количественно).
11,7 г (7,5 ммоль) описанной в примере 7г) цикленкарбоновой кислоты, 1,2 г (7,5 ммоль) 1-гидроксибензотриазола и 2,4 г (7,5 ммоль) 2-(1Н-бензотриазол-1-ил) тетрафторбората 1,1,3,3-тетраметилурония (TBTU; Peboc Limited, UK) растворяют в ДМФ и перемешивают в течение 15 мин. Этот раствор смешивают затем с 5,16 мл (30 ммоль) N-этилдиизопропиламина и с 0,95 г (1 ммоль) описанного выше гекса-амин-гидробромида и перемешивают в течение ночи при комнатной температуре. По окончании реакции производят упаривание в вакууме и остаток хроматографируют на силикагеле со смесью дихлорметан/метанол (17: 3).
Выход: 7,40 г (76% от теор.) бесцветного порошка.
Элементный анализ:
расчетн.: С: 64,82 Н: 6,99 N: 9,93;
получили: С:
64,58 Н: 7,11 N: 10,04.
MALDI-TOF-масс-спектр: около 9751 (M+Na+).
е) 36-мерный Каскадный N-(5-DO3А-ил-4-оксо-3-азагексаноил)полиамид на основе описанного
в примере 7д полиамина
1,95 г (0,2 ммоль) описанного в
примере 7д 36-мерного бензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным раствором
бромистого водорода в ледяной уксусной кислоте. По истечении 5
часов с помощью диэтилового эфира завершают начавшееся выпадение осадка, полученный 36-мерный амин-гидробромид промывают простым эфиром
и, высушив в вакууме, без дополнительной очистки подвергают
разложению.
Выход: 1,6 г (количественно).
1,6 г Описанного выше 36-мерного амин-гидробромида смешивают с 17,4 г (20 ммоль) описанного в примере 2д) активного сложного п-нитрофенилового эфира. В течение 1 часа затем добавляют по капле раствор 5,05 г (50 ммоль) триэтиламина в 20 мл ДМФ настолько медленно, чтобы образующийся поначалу осадок снова мог перейти в раствор. Перемешивают в течение ночи при 45oС, затем раствор сгущают в вакууме, остаток растворяют в трифторуксусной кислоте при 0oС и в течение ночи перемешивают при комнатной температуре. Упаривают в вакууме, остаток перемешивают с диэтилэфиром, осадок отсасывают и высушивают в вакууме. Кислый сырой продукт растворяют теперь в вакууме, с помощью разбавленной натриевой щелочи устанавливают рН 7 и подвергают ультрафильтрации через мембрану YM3, AMICON®. Фильтрат высушивают замораживанием.
Выход: 3,9 г (76% от теор.).
Содержание Н2О (Карл-Фишер): 8,0%
Элементный анализ (в пересчете на безводное вещество):
расчетн.: С: 46,59 Н: 6,23 N: 14,69 Ма: 10,46;
получили: С: 46,82 Н: 6,
47 N: 14,55 Na: 10,19.
ж) 36-мерный Gd-комплекс описанного в предыдущем примере лиганда
2,28
г (0,1 ммоль) описанной в предыдущем примере 7е натриевой соли кислоты
комплексообразователя в воде подкисляют 5 мл ледяной уксусной кислоты, смешивают с 725 мг (2 ммоль) Gd2О3,
и смесь в течение 2-х часов подвергают комплексообразованию при 80oС. После охлаждения раствор фильтруют, фильтрат подвергают ультрафильтрации через мембрану YM3, AMICON®
. Фильтрат подвергают попеременной обработке катионообменной смолой IR
120 (Н+-форма) и анионообменной смолой IRA 410 (ОН--форма) с целью придания минимальной проводимости.
Ионообменные смолы отфильтровывают, и фильтрат высушивают замораживанием.
Выход: 2,08 г (72% от теор.) бесцветного, хлопьевидного порошка.
Содержание
Н2О (Карл-Фишер): 7,0%
Определение Gd (AAC): 19,3%
MALDI-TOF-масс-спектр: Molpeak около 26.915 Da (расч.: 26.921 Da)
Элементный анализ (в пересчете на безводное
вещество):
расчетн.: С: 41,09 Н: 5,49 Gd: 21,03 N: 12,96;
получили: С: 41,20 Н: 5,60 Gd: 20,66 N: 13,19.
Пример 8
а) 3,
5-Бис[4-(бензилоксикарбонил)-2-оксо-1,4-диазабутил]бензойная кислота
К 30 г (197,17 ммоль) 3,
5-диаминобензойной кислоты в 600 мл дихлорметана добавляют 123,8 г (404,2 ммоль) сложного эфира
N-Z-глицин-N-гидроксисукцинимида. При 0oС в течение 5 мин добавляют по капле 60,7 г (800
ммоль) триэтиламина, растворенного в 100 мл дихлорметана, и перемешивают в течение ночи при
комнатной температуре. Экстрагируют трижды с помощью соответственно 500 мл 10%-ной соляной кислоты,
высушивают органическую фазу над сульфатом магния и упаривают в вакууме до получения твердого
вещества. Остаток подвергают перекристаллизации из ацетона.
Выход: 97,87 г (95% от теор.) бесцветного, кристаллического твердого вещества.
Элементный анализ:
расчетн.: С: 59,77 Н: 5,02 N: 10,72;
получили: С: 59,65 Н: 5,17 N: 10,59.
б)
Сложный эфир N-гидроксисукцинимида 3,5-бис[4-(бензилоксикарбонил-2-оксо-1,
4-диазабутил]-бензойной кислоты
К 60 г (114,8 ммоль) соединения из заглавия примера 8а, растворенного в 300 мл
дихлорметана, добавляют 13,21 г (114,8 ммоль) N-гидроксисукцинимида. Охлаждают
до 0oС и добавляют 26,06 г (126,3 ммоль) дициклогексилкарбодиимида. После этого перемешивают в течение 6 часов
при комнатной температуре. Отфильтровывают выпавшее в осадок твердое вещество,
и упаривают фильтрат в вакууме до получения твердого вещества. Остаток подвергают перекристаллизации из (смеси) простой
эфир/2-пропанол.
Выход: 65,44 г (92% от теор.) кристаллического бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 58,16 Н: 4,72 N: 11,30;
получили: С: 58,31 Н: 4,90 N: 11,15.
в) 1,4,
7-Трис{3,5-бис-[4-бензилоксикарбонил-2-оксо-1,4-диазабутил]-бензоил}-1,4,7,10-тетраазациклододекан
60 г (96,84 ммоль)
соединения из заглавия примера 8б) и 5,05 г (29,34 ммоль) циклена (1,4,
7,10-тетраазациклододекана) растворяют в смеси, состоящей из 200 мл толуола и 100 мл диоксана. Добавляют 5,94 г (58,68 ммоль)
триэтиламина и нагревают в течение 12 часов до 70oС. Упаривают
до получения сухого вещества, остаток погружают в 600 мл дихлорметана и экстрагируют три раза с помощью соответственно 300 мл
5%-ного водного раствора карбоната калия. Органическую фазу высушивают
над сульфатом магния и упаривают в вакууме до получения твердого вещества. Остаток хроматографируют на силикагеле (растворитель:
этилацетат/этанол=15:1).
Выход: 31,65 г (64% от теор.) бесцветного твердого вещества.
Элементный анализ:
расчетн.: С: 61,27 Н: 5,50 N: 13,29;
получили: С: 61,15 Н: 5,61 N: 13,10.
г)
1-(Карбоксиметоксиацетил)-4,7,10-трис{ 3,5-бис-[4-бензилоксикарбонил-2-оксо-1,4-диазабутил]-бензоил}-1,4,7,10-тетраазациклододекан
К
30 г (17,8 ммоль) соединения из заглавия примера 8в,
растворенного в 150 мл тетрагидрофурана, добавляют 3,1 г (26,7 ммоль) ангидрида дигликолевой кислоты и 5,4 г (53,4 ммоль) триэтиламина. Нагревают в
течение 6 часов до 50oС. Раствор
упаривают в вакууме до получения твердого продукта, погружают в 250 мл дихлорметана и дважды экстрагируют соответственно 150 мл 5%-ного водного раствора
соляной кислоты. Органическую фазу высушивают
над сульфатом магния, упаривают в вакууме до получения твердого продукта, и остаток хроматографируют на силикагеле (растворитель:
дихлорметан/метанол=20:1).
Выход: 29,83 г (93% от теор.) бесцветного твердого вещества.
Элементный анализ:
рассчетн.: С: 59,99 Н: 5,37 N: 12,44;
получили: С: 59,81 Н: 5,45 N: 12,29.
д) Полностью
защищенный 36-мерный бензилоксикарбонилполиамин, построенный из ядра-триамида N,N,N',N',N'',N''-гексакис(2-аминоэтил)тримезиновой
кислоты и шести описанных в примере 8г) аминозащищенных
гексаамин-монокарбоновых кислот
1,27 г (1 ммоль) описанного в предыдущем примере 1г) гекса-бензилоксикарбониламина растворяют в
уксусной кислоте и при перемешивании смешивают с 33%-ным
раствором бромистого водорода в ледяной уксусной кислоте. По истечении 60 мин с помощью диэтилэфира завершают начавшееся выпадение в осадок,
полученный гексаамин-гидробромид промывают простым эфиром,
высушивают в вакууме и подвергают описанной ниже реакции.
Выход: 0,95 г (количественно).
13,5 г (7,5 ммоль) описанной в предыдущем примере 8г) циклен-карбоновой кислоты, 1,2 г (7,5 ммоль) 1-гидроксибензотриазола и 2,4 г (7,5 ммоль) 2-(lH-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний-тетрабората TBTU; Peboc Limited, UK) растворяют в ДМФ и перемешивают в течение 15 мин. Этот раствор затем смешивают с 5,16 мл (30 ммоль) N-этилдиизопропиламина и с 0,95 г (1 ммоль) описанного выше гекса-амин-гидробромида и перемешивают в течение ночи при комнатной температуре. По окончании реакции упаривают в вакууме, и остаток хроматографируют на силикагеле с дихлорметан/метанолом в соотношении 8: 1.
Выход: 8,75 г (81% от теор.) бесцветного порошка.
Элементный анализ:
расчетн.: С: 64,34 Н: 5,62 N: 13,61;
получили: С: 64,22 Н: 5,86 N: 13,51.
MALDI-TOF-масс-спектр: Molpeak около 10.832 (M+Na+
)
е) 36-мерный Каскадный N-(5-DO3А-ил-4-оксо-3-азагексаноил)полиамид на основе описанного в примере 8д) полиамина
2,16 г
(0,2 ммоль) описанного в примере 8д 36-мерного
бензилоксикарбониламина растворяют в ледяной уксусной кислоте и при перемешивании смешивают с 33%-ным раствором бромистого водорода в ледяной уксусной
кислоте. Через 5 часов с помощью диэтилового
эфира завершается образование осадка, полученный 36-мерный амин-гидробромид промывают эфиром, высушивают в вакууме и без дополнительной очистки подвергают
разложению.
Выход: 1,8 г (количественно).
1,8 г описанного выше 36-мерного амин-гидробромида в 100 мл ДМФ смешивают с 17,4 г (20 ммоль) описанного в примере 2д) активного сложного п-нитрофенильного эфира. В течение 1 часа добавляют затем по капле раствор 5,05 г (50 ммоль) триэтиламина в 20 мл ДМФ настолько медленно, чтобы образующийся к началу осадок снова мог перейти в раствор. Перемешивают при 45oС в течение ночи, затем раствор сгущают в вакууме, остаток растворяют в трифторуксусной кислоте при 0oС и в течение ночи перемешивают при комнатной температуре. Сгущают в вакууме, остаток перемешивают с диэтиловым эфиром, осадок отсасывают и высушивают в вакууме. Кислый сырой продукт, наконец, растворяют в воде, с помощью разбавленной натриевой щелочи устанавливают рН 7 и подвергают ультрафильтрации через мембрану YM3, АМИКОН. Фильтрат высушивают замораживанием.
Выход: 4,6 г (84% от теор.).
Содержание воды (Карл-Фишер): 9,5%.
Элементный анализ (в пересчете на безводное вещество):
расчетн. С: 47,18 Н: 5,66 N: 16,08 Na: 10,00;
получили: С: 47,
31 Н: 5,52 N: 16,30 Na: 9,57.
ж)
36-мерный Gd-Комплекс описанного в предыдущем примере лиганда
2,74 г (0,1 ммоль) описанной в предыдущем примере 8е) натриевой соли кислоты
комплексообразователя подкисляют в воде с помощью 5
мл ледяной уксусной кислоты, смешивают с 725 мг (2 ммоль) Gd2О3 и подвергают комплексованию в течение 2-х часов при 80oС. После охлаждения раствор фильтруют, фильтрат
подвергают ультрафильтрованию через YM3 (АМИКОН), и полученный фильтрат с помощью попеременной добавки катионообменной смолы IR 129 (Н+
-форма) и анионообменной смолы IRA 410 (ОН-
-форма) устанавливают минимальную проводимость. Ионообменные смолы отфильтровывают, и фильтрат высушивают замораживанием.
Выход: 2,27 г (74% от теор.) бесцветного хлопьевидного порошка.
Содержание воды (Карл-Фишер): 8,6%.
Определение Gd (AAC): 18,2%
MALDI-TOF-масс-спектр: Molpeak около
27.992Da (расч.: 28.001Da)
Элементный
анализ (в пересчете на безводное вещество):
расчетн.: С: 41,82 Н: 5,02 Gd: 20,22 N: 14,26;
получили: С: 41,99 Н: 4,96 Gd: 19,87 N: 14,
40.
ПРИМЕР СРАВНЕНИЯ IN
VIVO
С ВНЕКЛЕТОЧНЫМ КОНТРАСТНЫМ СРЕДСТВОМ
Пригодность описанного в примере 1 л соединения в качестве агента показывают на следующем
эксперименте.
В качестве подопытного животного служат три крысы весом 200-250 г мужского рода (Шеринг-SPF-). Каждому животному вводят по 0,2 мл (соответственно 25 ммоль/л) внутривенно следующего раствора контрастного средства: смесь из соответственно 1 части соединения 1 л, далее называемого соединение 1, и диспрозиевого комплекса 3,6,9-три-(карбоксиметил)-ундекандикислоты (Dy-DTPA), далее называемого соединение 2. Через катетер из сонной артерии берут пробы крови через следующие промежутки времени: 1, 3, 5, 10, 15, 20, 30, 45, 60, 90, 120 мин p.i. В полученных пробах крови замеряют соответственно параллельно концентрации гадолиния (Gd) и диспрозия (Dy) с помощью атомно-эмиссионной спектрометрии (ICP-AES). Оставшуюся в крови часть введенного контрастного средства соединения 1 (Gd) и соединения 2 (Dy, вещество сравнения) можно сравнить с помощью различной маркировки на одинаковых животных. По концентрациям в крови с помощью специальных средств математического обеспечения (программа Topfit) можно рассчитать время полуобмена а и b, объемы распределения, а также полную очистку. Таким образом, эти характеристики представляют данные о пребывании соединений в интравазальном пространстве, соотношении распределения в организме и об удалении их.
Результаты: Прежде всего в первые моменты времени получаются явно более высокие концентрации соединения 16 по сравнению с внеклеточным контрастным средством (соединение 2) (см. чертеж).
Явно более высокие концентрации в крови соединения 1 в первые моменты времени (по сравнению с соединением 2) указывают на заметно меньшие объемы распределения (см. также Vd ss), т.е. соединение 1 распределяется не как соединение 2 в интравазальном пространстве (сосуды) и во внеклеточном пространстве, а большей частью лишь в интравазальном пространстве. В дальнейшем, однако, уровень в крови быстро снижается и время очистки или время β-полуобмена соединения 1 заметно меньше, чем при других кровяных пул-агентах. Полная очистка крови от соединения 1 лишь незначительно меньше по сравнению с соединением 2, что дает возможность сделать заключение об одинаково хорошей очистке почками.
Таким образом, описанное в примере 1 л соединение соответствует требованиям к кровяному пул-агенту: эффективное удаление из крови (через почки), но явно меньший объем распределения, чем при внеклеточном контрастном средстве.
Реферат
Описываются комплексы каскадных полимеров, содержащие а) комплексообразующие лиганды общей формулы I: A-[X-{Y-[Z-(W-KW)z]y }x]a, где A, Z, W, X, Y, a, z, w, x, y принимают значения, указанные в формуле изобретения, К означает остаток комплексообразователя формулы IA, где R1, R2 означает водород, R3 означает -CH(R4)-CO-N(R2 )-U7-T-, R4 означает водород или C1-С30алкильную группу, U7 означает насыщенную C1-C20 алкиленовую группу, Т означает -СО-α, где α означает место соединения с концевыми атомами азота последней генерации репродуктивного мономерного звена W или К означает остаток комплексообразователя формулы IB: (R1O2CCH2)2N-(CH2)2-N(CH2CO2R1)-(CH2)2-N(CH2CO2R1 )(CH2-СО-α), где R1 означает водород, α имеет вышеуказанное значение при условии, что, по меньшей мере, два репродуктивных мономерных звена различны, и для произведения мультиплетностей действительно: а•x•y•z•w равно 24 или 36 и что б) по меньшей мере, 16 ионов элементов, выбранных из лантаноидов, в) при необходимости ацилированные концевые аминогруппы. Также описывается диагностическое контрастное средство, содержащее комплекс каскадного полимера по п.1, производные 1,4,7,10-тетрациклотетрадекана, являющиеся промежуточными соединениями для получения комплексов каскадных полимеров по п.1. 3 с. и 2 з.п.ф-лы, 1 ил.
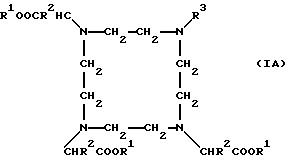
Формула
а) комплексообразующие лиганды общей формулы I
A-[X-{ Y-[Z-(W-Kw)z]y}x]a (I)
где А означает

p= 3, а= 6, m= 1 или p= 1, m= 1, а= 4;
U2 означает Q2, a Q2 означает простую связь; или А означает
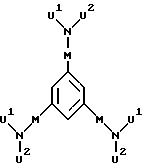
с базисной мультиплетностью а= 6;
где М означает группу СО, U1 и U2 означают Е в значении группы

где о= 1;
Q1 означает атом водорода;
Q2 означает простую связь;
Х и Y, независимо друг от друга, означают простую связь, в этом случае x, y= 1 или репродуктивное мономерное звено формулы -CO-CH(NH-)(CH2)4NH с репродуктивной мультиплетностью x, y= 2; -COCH2CH2NH- с репродуктивной мультиплетностью x, y= 1
Z означает

z= 3;
Z означает

z= 3;
W означает -COCH(NH-)(CH2)4NH с репродуктивной мультиплетностью w= 2, -COCH2NH- с репродуктивной мультиплетностью w= 1, -COCH((CH2 )3-NH-)2 с репродуктивной мультиплетностью w= 2,

с репродуктивной мультиплетностью w= 2;

с репродуктивной мультиплетностью w= 2;
К означает остаток комплексообразователя формулы IA
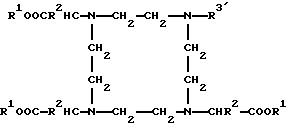
где R1 означает водород;
R2 означает водород;
R3 означает

R4 означает водород или С1-С30алкильную группу,
U7 означает насыщенную С1-С20 алкиленовую группу,
Т означает -СО-α,
α означает место соединения с концевыми атомами азота последней генерации репродуктивного мономерного звена W,
или К означает остаток комплексообразователя формулы IB

где R1 означает водород;
α имеет вышеуказанное значение, при условии, что, по меньшей мере, два репродуктивных мономерных звена различны, и для произведения мультиплетностей действительно:
a• x•y•z•w равно 24 или 36 и что
б) по меньшей мере, 16 ионов элементов, выбранных из лантаноидов,
в) при необходимости, ацилированные концевые аминогруппы.
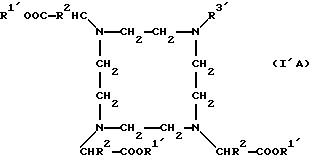
где R1', независимо друг от друга, означают атом водорода или кислотозащитную группу;
R2 означает атом водорода, метильный остаток;
R3' означает группу

где R4 означает атом водорода или линейную, разветвленную насыщенную C1-С30-алкильную группу;
U7 означает линейную насыщенную С1-С20-алкиленовую группу;
Т' означает группу -С*O-, -СООН-, где С*O означает активированную карбонильную группу.
Комментарии