Катализатор на носителе, содержащий связанный активатор, образующий катион - RU2178421C2
Код документа: RU2178421C2
Чертежи


Описание
Изобретение относится к носителям и полученным на их основе катализаторам на носителе, которые используются при полимеризации олефинов. Более конкретно, это изобретение относится к таким носителям, содержащим активирующее соединение, которое химически связано или прикреплено к носителю. Изобретение также относится к получению таких материалов носителя, и катализаторам на носителе, и к их использованию в способе полимеризации олефинов.
В области техники ранее было описано несколько катализаторов на носителе, образующих катион и используемых в способах полимеризации олефинов. В заявке WO-91/09882 описан катализатор на носителе, приготовленный путем сочетания i) бис(циклопентадиенил)металла, содержащего по меньшей мере один лиганд, способный взаимодействовать с протоном, ii) активирующего компонента, содержащего катион, способный отдавать протон, и объемистый, подвижный анион, способный стабилизировать катион металла, образовавшийся в результате взаимодействия между соединением металла и активирующим компонентом, и iii) материала носителя катализатора. Материал носителя может подвергаться термической или химической дегидратации. В некоторых примерах с этой целью добавляют триэтилалюминий. Максимальная насыпная плотность, указанная в примерах заявки WO-91/09882, составляла 0,17 г/см3. Приведенные значения эффективности катализатора были не удовлетворительны.
В заявке WO-94/03506 описан ионный катализатор на носителе, приготовленный путем сочетания i) моно(циклопентадиенил) металла, ii) активирующего компонента, содержащего катион, который может необратимо взаимодействовать по меньшей мере с одним лигандом, содержащимся в этом соединении металла, и анион, который является химически стабильным, не нуклеофильным анионным комплексом, и iii) материала носителя катализатора. Необязательно, ионный катализатор на носителе может проводить предварительную полимеризацию олефинового мономера. Этот материал носителя также может быть обработан гидролизуемой органической добавкой, предпочтительно алкильным соединением металла из Группы 13, таким как триэтилалюминий. В этой ссылке также предложено использование таких ионных катализаторов на носителе в газофазном процессе полимеризации. К сожалению, эффективность катализаторов, полученных в заявке WO-94/03506, вероятно, была недостаточной для промышленного применения.
В патенте США A-5399636 описаны металлоценовые катализаторы на носителе, в которых металлоцен химически присоединен к материалам носителя, которые включают диоксид кремния, оксид алюминия, глину, фосфат алюминия и их смеси. В патенте США A-5427991 описаны некоторые каталитические носители, содержащие полианионные функциональные группы, состоящие из некоординирующих анионных групп, химически связанных со сшитыми остовными полимерными компонентами. В этой ссылке (колонка 19, строки 4-12) указано, что желательно маскировать или защищать гидроксильные группы субстрата посредством стандартных химических обработок. Однако маскировка или защита гидроксильных групп до их взаимодействия с некоординирующим анионным реагентом делает их инертным в дальнейшей реакции, тем самым не выполняется цель изобретения. Проведение маскировки или защиты этих групп после взаимодействия с некоординирующим анионным реагентом отрицательно сказывается на желаемых, химически связанных анионных группах. На фиг. 8 предложена альтернативная схема, включающая функционализацию поверхностных гидроксильных групп посредством взаимодействия с пара-бромфенил(триметокси)силаном. Отсутствуют какие-либо указания о желательности ограничения количества функционализированных поверхностных гидроксильных групп на диоксиде кремния меньше чем 1,0 ммоль/г. Кроме того, в этой ссылке не указывается образование реакционноспособной силановой функциональной группы вместо парабромфенилсилоксановой. По указанным ниже причинам раскрытие этой публикации в отношении исходных материалов на основе диоксида кремния или оксида алюминия считается недейственным или недостаточным.
Катионные гомогенные катализаторы, полученные с использованием образующих катионы активирующих соединений, описаны в многочисленных ссылках уровня техники. В EP-A-277004 и заявке на патент США A-5064802 описано использование солей бренстедовских кислот, способных образовывать катионы за счет переноса водорода. В EP-A-277003 описан аналогичный способ с использованием объемистых анионов, содержащих несколько атомов бора. В патенте WO-93/23412 описаны активаторы, образующие катионы соли карбония. В заявке на патент США A-5321106 рекомендовано использование катионных активаторов - окисляющих солей, а в заявке США 304314, поданной 12 сентября 1994 г. , описано использование катионных активаторов - солей силилия. К сожалению, такие гомогенные катализаторы, нанесенные по обычным методикам физической адсорбции на поверхность материала носителя, могут быть вновь удалены разбавителями, присутствующими в традиционных процессах полимеризации в растворе или суспензии, и разбавителями, которые могут образоваться в традиционных процессах полимеризации в газовой фазе, в таких как с использованием конденсации и рециркуляции либо разбавителей, либо мономеров. Подобная потеря каталитического материала с носителя может отрицательно воздействовать на величину насыпной плотности полученного полимерного продукта.
Было бы желательно разработать катализатор на носителе и способ полимеризации с использованием этого катализатора, в котором было бы возможно получать полимеры при хорошей эффективности катализатора. Кроме того, было бы желательно разработать такой катализатор на носителе, который может использоваться в способе полимеризации в суспензии или газовой фазе и который относительно стабилен в присутствии конденсированных мономеров или разбавителей.
В соответствии с настоящим изобретением предлагается носитель, использующийся для приготовления катализаторов на носителе, содержащих продукт взаимодействия A)
неорганического оксидного материала, включающего твердую матрицу и реакционноспособные гидроксильные группы или производные гидроксильных групп на поверхности матрицы, функционализированные
реакционноспособным силаном, который соответствует формуле -OSiR2H, в которой независимо в каждом случае R является атомом водорода, гидрокарбилом C1-C20 или
гидрокарбилокси-группой C1-C20, причем этот неорганический оксидный материал содержит менее 1,0 ммоль/г реакционноспособных поверхностных гидроксильных групп, и B) активирующего
соединения, включающего:
b1) катион, способный взаимодействовать с соединением переходного металла с образованием каталитически активного комплекса переходного металла, и
b2)
совместимый анион, содержащий по меньшей мере один заместитель, способный взаимодействовать с неорганической оксидной матрицей, с остаточными гидроксильными функциональными группами неорганического
оксида, или с реакционноспособной силановой группой, в результате чего совместимый анион ковалентно связывается с носителем.
Кроме того, изобретение предлагает каталитическую систему на носителе, используемую в аддитивной полимеризации мономеров, способных к такой полимеризации, которая включает указанный выше носитель; и (C) соединение переходного металла, содержащее по меньшей мере одну π-связанную анионную лигандную группу. Причем это соединение переходного металла способно взаимодействовать с указанным выше носителем посредством катиона b1), в результате чего каталитически активный комплекс переходного металла химически связывается с носителем.
Кроме того, изобретение предлагает способ получения носителя, включающий сочетание неорганического оксидного материала, содержащего твердую матрицу и реакционноспособные гидроксильные группы или производные гидроксильных групп на поверхности матрицы, функционализированные реакционноспособным силаном, который соответствует формуле -OSiR2H, в которой независимо в каждом случае R является атомом водорода, гидрокарбилом C1-C20 или гидрокарбилокси-группой C1-C20, причем этот неорганический оксидный материал содержит менее 1,0 ммоль/г реакционноспособных поверхностных гидроксильных групп, с активирующим соединением B) с образованием носителя для катализатора полимеризации олефинов.
В еще одном аспекте настоящее изобретение предлагает способ аддитивной полимеризации, в котором один или несколько полимеризующихся мономеров контактируют с каталитической системой на носителе в соответствии с настоящим изобретением в условиях аддитивной полимеризации.
Носитель и катализаторы на носителе согласно настоящему изобретению легко получают с высоким выходом и эффективностью. Важно, что каталитические системы, приготовленные из указанных выше компонентов катализатора, демонстрируют улучшенные характеристики, измеренные по каталитической активности и/или насыпной плотности продукта, по сравнению с известными ранее каталитическими системами на носителе. Полагают, что это является результатом контроля содержания доступных гидроксильных групп на поверхности неорганического оксида до менее чем 1,0 ммоль/г, до взаимодействия с активирующим соединением В) и/или использования специфических реакционноспособных функциональных силановых групп, которые описаны здесь дополнительно.
Все ссылки в изобретении на элементы или металлы, принадлежащие к определенной Группе, относятся к Периодической таблице элементов, опубликованной издательством Си-Ар-Си Пресс Инк. , 1989. Кроме того, любая ссылка на Группу или Группы будет сделана на Группу или Группы, которые указаны в этой Таблице элементов, используя систему ИЮПАК для нумерации групп.
Неожиданно было найдено, что при использовании указанного в изобретении уникального сочетания активирующих соединений и носителей активирующее соединение может быть присоединено к носителю, причем оно еще способно активировать катализаторы с переходными металлами, которые обычно используются в процессах аддитивной полимеризации. Катализаторы на носителе согласно настоящему изобретению могут использоваться для получения олефиновых полимеров с весьма высокой каталитической эффективностью. Преимущественно эффективность этих катализаторов достигает по меньшей мере 100 кг полимера на 1 г переходного металла, более предпочтительно по меньшей мере 1000 кг полимера на 1 г переходного металла. Сверх того, эти катализаторы на носителе обладают высокой стойкостью к выщелачиванию в типичных условиях, используемых в процессе суспензионной или газофазной полимеризации.
Дополнительные преимущества при использовании катализаторов на носителе согласно изобретения в процессах полимеризации включают то, что исключается образование отложений полимера на стенках реактора и других движущихся частей реактора и что в процессах полимеризации с образованием частиц получаются полимеры, имеющие улучшенную насыпную плотность. В соответствии с настоящим изобретением улучшенной насыпной плотностью для гомополимеров на основе этилена и интерполимеров на основе этилена является насыпная плотность, по меньшей мере равная 0,20 г/см3 и предпочтительно по меньшей мере примерно 0,25 г/см3.
Неорганические оксидные носители, подходящие для использования в настоящем изобретении, включают весьма пористые диоксиды кремния (силикагели), оксиды алюминия, алюмосиликаты, алюмофосфаты, глины, оксиды титана и их смеси. Предпочтительными неорганическими оксидами являются диоксид кремния и оксид алюминия. Наиболее предпочтительным материалом носителя является диоксид кремния. Материал носителя может иметь гранулированную, агломерированную, таблетированную или любую другую физическую форму.
Носители, подходящие для использования в настоящем изобретении, предпочтительно имеют площадь поверхности, измеренную методом азотной порозиметрии с использованием метода БЭТ, от 10 до 1000 м2/г и предпочтительно от 100 до 600 м2/г. Объем пор носителя, измеренный методом адсорбции азота, преимущественно составляет между 0,1 и 3 см2/г, предпочтительно примерно от 0,2 до 2 см2/г. Средний размер частиц не является существенным, но обычно составляет от 0,5 до 500 мкм, предпочтительно от 1 до 150 мкм.
Известно, что неорганические оксиды, особенно диоксид кремния, оксид алюминия и алюмосиликаты, по своей природе содержат небольшие количества гидроксильных групп, связанных с атомной матрицей. При использовании таких оксидов для приготовления компонента A эти материалы предпочтительно сначала подвергают термической и/или химической обработке, чтобы снизить содержание гидроксильных групп до 0,001 - 10 ммоль/г, более предпочтительно 0,01 - 1,0 ммоль/г, наиболее предпочтительно 0,1 - 0,8 ммоль/г. Обычно термическую обработку (прокаливание) проводят при температуре от 150 до 900oC, предпочтительно от 300 до 850oC в течение от 10 минут от 50 часов. Типичная химическая обработка включает контактирование с алкилирующими агентами - кислотами Льюиса, такими как тригидрокарбилалюминиевые соединения, тригидрокарбилхлорсилановые соединения, тригидрокарбилалкоксисилановые соединения или аналогичные агенты. Содержание остаточных гидроксильных групп может быть определено по методике инфракрасной спектроскопии (диффузное отражение) с Фурье-преобразованием (ИКСДОФ), как описано в книге П. Гриффитс и Дж. Де Хасет. "Инфракрасная спектроскопия с Фурье-преобразованием", 83 Химический анализ, издательство Уилей Интерсайенз, 1986, с. 544.
Неорганический оксид может не содержать функциональных групп за исключением гидроксильных групп на поверхности, как описано выше. В этом варианте воплощения изобретения пониженное содержание гидроксильных групп в носителе приводит к улучшению свойств полученного катализатора на носителе, наиболее вероятно вследствие отсутствия взаимодействия между комплексом переходного металла и остаточными гидроксильными группами. Предпочтительное содержание гидроксильных групп в таком носителе составляет менее 0,8 ммоль/г, предпочтительно менее 0,5 ммоль/г.
Неорганический оксид также может быть функционализирован путем обработки силановым, гидрокарбилсилановым или хлорсилановым функционализирующим агентом для того, чтобы присоединить к нему свисающую реакционноспособную силановую группу, как описано ранее. Подходящими функционализирующими агентами являются такие соединения, которые взаимодействуют с поверхностными гидроксильными группами неорганического оксида или реагируют с атомами металла, или металлоида в матрице неорганического оксида. Примеры подходящих функционализирующих агентов включают фенилсилан, дифенилсилан, метилфенилсилан, диметилсилан, диэтилсилан, диэтоксисилан и хлордиметилсилан. Методики получения функционализированных неорганических оксидных соединений были описаны ранее в заявках на патент США A-3687920 и A-3879368.
В предпочтительном варианте воплощения изобретения силан и неорганический оксид контактируют, необязательно в присутствии углеводородного разбавителя, в присутствии основного вспомогательного агента, предпочтительно триалкил C1-C4 амина. Это взаимодействие проводят при температуре от 0 до 110oC, предпочтительно от 20 до 50oC. Обычно используют избыток функционализирующего агента. В расчете на неорганический оксид предпочтительные соотношения функционализирующего агента составляют от 1 до 2500 ммоль/г. В результате указанной реакции функционализации остаточное содержание гидроксильных групп в неорганическом оксиде дополнительно снижается до ранее упомянутой малой величины, менее 1,0 ммоль/г. Предпочтительно содержание остаточных гидроксильных групп на функционализированных носителях составляет 0,8 ммоль/г и наиболее предпочтительно менее 0,5 ммоль/г. Весьма предпочтительно использовать при приготовлении компонента A прокаленный диоксид кремния, имеющий исходное (то есть до функционализации) содержание остаточных гидроксильных групп менее 1,0 ммоль/г, и используют от 1 до 20 ммоль функционализирующего агента на 1 г диоксида кремния. Молярное соотношение используемого основного вспомогательного агента к функционализирующему агенту обычно составляет от 0,7: 1 до 2,0: 1. Непрореагировавший функционализирующий агент предпочтительно удаляют с поверхности неорганического оксида, например, промывкой жидким углеводородом, и носитель тщательно высушивают перед использованием при приготовлении каталитических систем на носителях.
Неорганический оксид, полученный носитель или каталитическую систему на носителе также можно обрабатывать алюминиевым компонентом, выбранным из алюмоксана или соединения алюминия формулы AlR3, где R такой, как определено выше. Примеры подходящих групп R включают метил, метокси, этил, этокси, пропил (все изомеры), пропокси (все изомеры), бутил (все изомеры), бутокси (все изомеры), фенил и бензил. Предпочтительно, алюминиевый компонент выбирают из группы, состоящей из алюмоксанов и соединений тригидрокарбил (C1-C4)алюминия. Наиболее предпочтительные алюминиевые компоненты представляют собой алюмоксаны, триметилалюминий, триэтилалюминий, триизобутилалюминий и их смеси.
Алюмоксаны (также называемые алюминоксанами) представляют собой олигомерные или полимерные оксиалюминиевые соединения, содержащие цепочки с чередующимися атомами алюминия и кислорода, причем при алюминии имеется заместитель, предпочтительно алкильная группа. Полагают, что структура алюмоксана может быть представлена следующей общей формулой [-Al(R)-O]m для циклических алюмоксанов и R2Al-O[Al(R)-O]m-AlR2 для линейных соединений, в которых R такой, как определено выше, и m является целым числом, изменяющимся от 1 до примерно 50, предпочтительно по меньшей мере 4. Обычно алюмоксаны являются продуктами взаимодействия воды и алюминийалкилов, которые, кроме алкильных групп, могут содержать галогенидные или алкоксидные группы. При взаимодействии нескольких различных алкилалюминиевых соединений, таких как, например, триметилалюминий и триизобутилалюминий, с водой образуются так называемые модифицированные или смешанные алюмоксаны. Предпочтительными алюмоксанами являются метилалюмоксан и метилалюмоксан, модифицированный небольшим количеством алкильных групп C2-C4, особенно таких, как изобутил. Обычно алюмоксаны содержат небольшие или значительные количества исходного алкилалюминиевого соединения.
Конкретные методики приготовления соединений типа алюмоксана путем контактирования алкилалюминиевого соединения с неорганической солью, содержащей кристаллизационную воду, описаны в патенте США 4542199. В особенно предпочтительном варианте осуществления алкилалюминиевое соединение контактирует с регенерируемым содержащим воду веществом, таким как гидратированный оксид алюминия, диоксид кремния или другое вещество. Это описано в заявке на Европатент A-338044. Таким образом, алюмоксан может быть введен в носитель путем взаимодействия гидратированного оксида алюминия или диоксидкремниевого материала, который необязательно функционализирован силаном, силоксаном, гидрокарбилоксисиланом или хлорсилановыми группами, с триалкил(C1-C10)алюминиевым соединением, в соответствии с известными методиками.
Обработка неорганического оксидного материала для того, чтобы также ввести необязательные количества алюмоксана или триалкилалюминия, в дополнение к активирующему соединению, включает его контактирование до, после или одновременно с добавлением активатора, наряду с алюмоксаном или триалкилалюминиевым соединением, особенно триэтилалюминием или триизобутилалюминием. Кроме того, необязательно смесь может быть нагрета в инертной атмосфере в течение периода времени и при температуре, которые достаточны для фиксации алюмоксана или триалкилалюминия на носителе, или компонент носителя, содержащий алюмоксан или триалкилалюминиевое соединение, можно подвергать одной или нескольким стадиям промывки, чтобы удалить алюмоксан или триалкилалюминий, несвязанные с носителем.
Кроме контактирования носителя с алюмоксаном, последний может образовываться in situ, посредством контактирования негидролизованного неорганического оксида или увлажненного неорганического оксида с триалкилалюминиевым соединением, необязательно в присутствии инертного разбавителя. Такой способ хорошо известен из уровня техники и описан в заявках на патенты NN ЕР-A-250600, США 4912075 И США 5008228. Подходящие алифатические углеводородные разбавители включают пентан, изопентан, гексан, гептан, октан, изооктан, нонан, изононан, декан, циклогексан, метилциклогексан и сочетания из двух или нескольких таких разбавителей. Подходящими ароматическими углеводородными разбавителями являются бензол, толуол, ксилол и другие алкил- или галогензамещенные ароматические соединения. Наиболее предпочтительно, разбавитель представляет собой ароматический углеводород, особенно толуол. После приготовления указанным выше образом, по одной из описанных ранее методик, содержание остаточных гидроксильных групп в носителе снижается до желаемого низкого уровня, 1,0 ммоль групп -OH на 1 г носителя.
Анионный
компонент соединения активатора B, используемого в соответствии с настоящим изобретением, соответствует формуле
(DM'Q3)-,
в которой D представляет собой
связывающую группу, содержащую функциональную группу, способную взаимодействовать с неорганической оксидной матрицей, с ее остаточными гидроксильными функциональными группами или с
реакционноспособными силановыми функциональными группами необязательно функционализированного неорганического оксида,
M' является атомом бора или алюминия в степени окисления 3; и
Q
является гидрокарбилом, гидрокарбилокси-группой, фторированным гидрокарбилом, фторированной гидрокарбилокси-группой или фторированной силилгидрокарбильной группой, имеющей до 20 неводородных
атомов.
Наиболее предпочтительно в каждом случае Q является фторированной арильной группой, особенно пентафторфенильной группой.
Предпочтительно, соединения активатора
являются солями формулы
G+e(DM'Q3)e-,
в которой G+e является катионным остатком соли кислоты Бренстеда, окисляющего
катиона, иона карбония или иона силилия; и e представляет собой целое число от 1 до 3, наиболее предпочтительно e равно 1.
Подходящие связывающие заместители D при совместимых анионах, используемых с немодифицированными неорганическими оксидами, или с неорганическими оксидами, содержащими только остаточную гидроксильную функциональную группу, включают группы, содержащие силановые, силоксановые, гидрокарбилоксисилановые, галоидсилановые, амино-, карбоновые кислоты, эфиры карбоновой кислоты, альдегиды, кетоны или эпоксидные функциональные группы, содержащие от 1 до 106 не водородных атомов, более предпочтительно от 2 до 1000 не водородных атомов и наиболее предпочтительно от 4 до 20 не водородных атомов. При практическом использовании силана, содержащего совместимые анионы, может потребоваться использование основного катализатора, такого как три(алкил C1-C4) амин, для осуществления реакции с субстратом, содержащим только остаточную гидроксильную функциональную группу. Предпочтительно, D при использовании с такими немодифицированными неорганическими оксидными соединениями, является силаном или хлорсиланом, замещенным гидрокарбильным радикалом. Предпочтительные, связывающие заместители D включают группы: силилзамещенный арил, силилзамещенный аралкил, силилзамещенный алкарил, силилзамещенный алкил, силилзамещенный галоидарил или силилзамещенные галоидалкильные группы, включающие полимерные связывающие группы, наиболее предпочтительно пара-силилфенил, (-C6H4SiH3), пара-силилтетрафторфенил, (-C6H4SiH3), силилнафтил, (-C10H8SiH3), силилперфторнафтил, (-C10H8SiH3) и 2-силил-1-этил, (-C2H4SiH3).
Подходящие связывающие заместители D при совместимых анионах, используемых с неорганическими оксидами, которые модифицированы реакционноспособной силановой функциональной группой, включают группы, содержащие силановые, силоксановые, гидрокарбилоксисилановые, галоидсилановые, гидроксильные, тиоловые, амино-, карбоновые кислоты, эфиры карбоновой кислоты, альдегидные, кетоновые или эпоксидные функциональные группы, содержащие от 1 до 106 неводородных атомов, более предпочтительно от 2 до 1000 неводородных атомов и наиболее предпочтительно от 4 до 20 неводородных атомов. Предпочтительно, в таких случаях D представляет собой гидроксилзамещенный гидрокарбильный радикал, более предпочтительно гидроксизамещенный арил, гидроксизамещенный аралкил, гидроксизамещенный алкарил, гидроксизамещенный алкил, гидроксизамещенный галоидарил или гидроксизамещенную галоидалкильную группу, включающую полимерные связывающие группы, наиболее предпочтительными являются гидроксифенильная группа, гидрокситолильная, гидроксибензильная, гидроксинафтильная, гидроксибисфенильная, гидроксициклогексильная, гидроксиалкильная C1-C4 и гидроксиполистирильная группы, или их фторированные производные. Наиболее предпочтительными связывающими заместителями D являются п-гидроксифенильная группа, 4-гидроксибензильная, 6-гидрокси-2-нафтильная, 4-(4'-гидроксифенил)фенильная, 4-(4'-гидроксифенил)диметилметилен)фенильная или их фторированные производные. Может также потребоваться использование основного катализатора, такого как три(алкил C1-C4)амин, для осуществления реакции с субстратом.
Наиболее предпочтительно, D является одним из предшествующих гидрозамещенных заместителей, используемых в сочетании с диоксидом кремния, функционализированным реакционноспособным силаном.
Иллюстративные, но не ограничивающие примеры анионных компонентов, (DM'Q3)-, активирующего соединения, которые могут использоваться в настоящем изобретении, включают трис-(пентафторфенил)(4-гидроксифенил)борат, трис-(пентафторфенил)(4-гидрокситетрафторфенил)борат, трис(2, 4-дифторфенил)(4-гидроксифенил)борат, трис(3,5-дифторфенил)(4-гидроксифенил)борат, трис(3,5-ди-трифторметилфенил)(4-гидроксифенил)борат, трис(пентафторфенил)(2-гидроксиэтил)борат, трис(пентафторфенил)(4-гидроксибутил)борат, трис(пентафторфенил)(4-гидроксициклогексил)борат, трис(пентафторфенил)(3,5-диметил-4-гидроксифенил)борат, трис(пентафторфенил)-4-(4'-гидроксифенил)фенилборат и трис(пентафторфенил)-(гидроксинафтил)бораты (все изомеры, особенно, трис(пентафторфенил)(6-гидрокси-2-нафтил)борат.
Катионная часть активирующего соединения может быть любым катионом, который способен взаимодействовать с соединением переходного металла с образованием каталитически активного комплекса переходного металла. Предпочтительно, этот катион выбирают из группы, состоящей из катионов бренстедовских кислот, карбониевых катионов, силильных катионов и катионных окисляющих агентов.
Катионы
бренстедовских кислот могут быть представлены следующей общей формулой:
(L*-H)+,
в которой L* является нейтральным основанием Льюиса, предпочтительно
азот-, фосфор-, кислород или серосодержащим основанием Льюиса; и (L*-H)+ является кислотой Бренстеда.
Иллюстративные, но неограничивающие, примеры бренстедовских кислотных катионов активирующего соединения, которые могут использоваться в настоящем изобретении, являются катионы триалкилзамещенного аммония, такие как триэтиламмоний, трипропиламмоний, три(н-бутил)аммоний, триметиламмоний, три-бутиламмоний. Также являются приемлемыми катионы N, N-диалкиланилина, такие как N, N-диметиланилиний, N, N-диэтиланилиний, N, N-2,4,6-пентаметиланилиний и т. п. ; катионы диалкилзамещенного аммония, такие как ди(изопропил)аммоний, дициклогексиламмоний и т. п. ; и катионы триарилфосфония, такие как трифенилфосфоний, три(метилфенил)фосфоний, три(диметилфенил)фосфоний, диэтилоксоний, диметилсульфоний, диэтилсульфоний и дифенилсульфоний.
Второй тип подходящего катиона (обозначен как ©+ ) является стабильным ионом карбония или силилия, содержащим до 30 неводородных атомов, причем этот катион способен взаимодействовать с заместителем соединения переходного металла и превращать его в каталитически активный комплекс переходного металла. Подходящие примеры карбониевых катионов включают тропилий, трифенилметилий, бензол(диазоний). Соли силилия были обобщенно описаны ранее в J. Chem. Soc. Chem. Comm. , 1993, 383-384, а также Дж. Б. Ламбертом и др. в журнале Organometallics, 1994, 13, 2430-2443. Предпочтительными катионами силилия являются триметилсилилий, триэтилсилилий и их эфирнозамещенные аддукты.
Другой подходящий тип катиона (обозначен как Oxe+) является катионным окисляющим агентом, имеющим заряд e+, причем е - это целое число от 1 до 3.
Примеры катионных окисляющих агентов включают ферроцений, гидрокарбилзамещенный ферроцений, Ag+ и Pb2+.
Активирующие соединения, которые могут использоваться в настоящем изобретении, легко готовятся путем сочетания производной металла Группы 1, Группы 2 или гриньяровской производной металла функционализированного заместителя D, или его "маскированной производной", с нейтральным предшественником аниона, и затем этот продукт реакции контактируют с хлоридной солью катиона, который будет использоваться. Примеры подходящих металлических производных включают соли лития или Гриньяра. Термин "маскированная производная" относится к хорошо известной практике использования инертной функциональной группы при получении и превращении аналогичной желаемой, реакционноспособной функциональной группы в последующей стадии, с использованием методов, которые хорошо известны специалистам в этой области техники. Например, в ходе синтеза может присутствовать триметилсилилокси-группа и в последующем ее превращают в желаемую гидроксильную группу путем гидролиза.
Носитель согласно настоящему изобретению обычно содержит от 0,001 до 10 ммоль активирующего соединения на 1 г неорганического оксида, предпочтительно от 0,01 до 1 ммоль/г. При слишком малых количествах каталитическая эффективность полученного катализатора на носителе становится неприемлемой. Содержание остаточных гидроксильных групп после взаимодействия с активирующим соединением желательно составляет менее 50 молярных % в расчете на содержание желаемого комплекса переходного металла, более предпочтительно, менее 10 молярных % в расчете на содержание желаемого комплекса переходного металла, наиболее предпочтительно, менее 1 молярного % в расчете на содержание желаемого комплекса переходного металла.
Носитель настоящего изобретения может храниться или транспортироваться в инертном состоянии как таковой или может быть суспендирован в инертном разбавителе, таком как алкановый или ароматический углеводород. Он может использоваться для получения катализатора на носителе согласно настоящему изобретению путем контактирования с подходящим соединением переходного металла, необязательно в присутствии жидкого разбавителя.
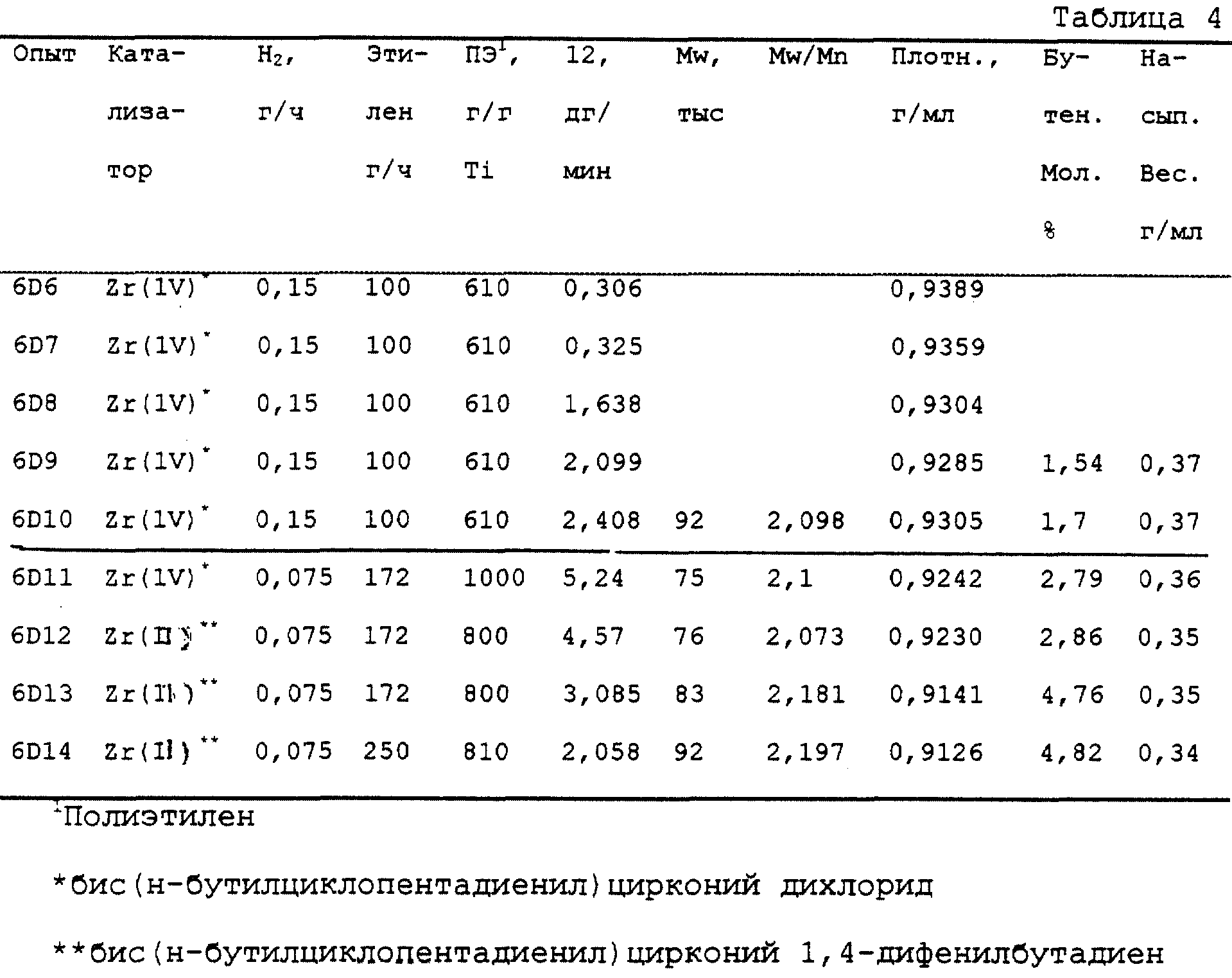
Подходящими соединениями (C) переходного металла для использования в катализаторе на носителе согласно настоящему изобретению могут быть производные любого переходного металла, включая лантаниды, но предпочтительно из Групп 3, 4 или лантанидов, в которых переходный металл имеет формальную степень окисления +2, +3 или +4, соответствующую ранее упомянутым требованиям. Предпочтительные соединения включают комплексы металла, содержащие от 1 до 3 π-связанных групп анионного лиганда, которые могут быть циклической или нециклической, делокализованной π-связанной анионной лигандной группой. Примерами такой π-связанной анионной лигандной группы являются сопряженные или несопряженные, циклические или нециклические диенильные, аллильные и арильные группы. Термин "π-связанные" означает, что лигандная группа связана с переходным металлом посредством π -связи.
Каждый атом в делокализованной π-связанной группе может быть независимо замещен радикалом, выбранным из группы, состоящей из галогена, гидрокарбила, галоидгидрокарбила и гидрокарбилзамещенных металлоидных радикалов, в которых металлоид выбирают из Группы 14 Периодической таблицы элементов. Термин "гидрокарбил" включает неразветвленные, разветвленные и циклические алкильные радикалы C1-C20, ароматические радикалы C6-C20, алкилзамещенные ароматические радикалы C7-C20 и арилзамещенные алкильные радикалы C7-C20. Кроме того, два или несколько таких радикалов могут образовать совместно конденсированную циклическую систему или гидрированную конденсированную циклическую систему. Подходящие гидрокарбилзамещенные металлоидорганические радикалы включают моно-, ди- и тризамещенные металлоидорганические радикалы элементов 14-й Группы, в которых каждая гидрокарбильная группа содержит от 1 до 20 атомов углерода. Примеры подходящих гидрокарбилзамещенных металлоидорганических радикалов включают триметилсилил, триэтилсилил, этилдиметилсилил, метилдиэтилсилил, трифенилгерманил, триметилгерманил.
Примеры подходящих анионных, делокализованных π-связанных групп включают циклопентадиенильную, инденильную, флуоренильную, тетрагидроинденильную, тетрагидрофлуоренильную, октагидрофлуоренильную, пентадиенильную, циклогексадиенильную, дигидроантраценильную, гексагидроантраценильную и декагидроантраценильную группы, а также гидрокарбил(C1-C10)замещенные производные этих групп. Предпочтительными анионными, делокализованными π-связанными группами являются циклопентадиенил, пентаметилциклопентадиенил, тетраметилциклопентадиенил, инденил, 2,3-диметилинденил, флуоренил, 2-метилинденил и 2-метил-4-фенилинденил.
Подходящие соединения (C) переходного металла могут представлять собой любые производные любого переходного металла, включая лантаниды, но предпочтительно из
переходных металлов Групп 3, 4 или лантанидов. Более предпочтительны комплексы металлов, соответствующие формуле
L1MXmX'nX''p,
или их
димеры, в которых L является анионной, делокализованной π-связанной группой, которая присоединена к M и содержит до 50 неводородных атомов, необязательно две группы L могут быть совместно
связаны через один или несколько заместителей, в результате чего образуется мостиковая структура, и, кроме того, необязательно одна группа L может быть связана с X через один или несколько
заместителей L;
M представляет собой металл из Группы 4 Периодической таблицы элементов, имеющий формальную степень окисления +2, +3 или +4;
X является необязательным двухвалентным
заместителем, имеющим до 50 неводородных атомов, который вместе с группой L образует металлоцикл с M;
X' является необязательным нейтральным основанием Льюиса, имеющим до 20 неводородных
атомов;
X'' в каждом случае представляет собой одновалентную анионную группу, имеющую до 40 неводородных атомов, необязательно две группы X'' могут быть ковалентно связаны вместе с
образованием двухвалентной, дианионной группы, имеющей обе валентности, связанные с металлом M, или образуют нейтральный, сопряженный или несопряженный диен, который связан π-связью с M (при
этом металл M имеет степень окисления +2), или, кроме того, необязательно одна или несколько групп X" и одна или несколько групп X'' могут быть связаны вместе с образованием группы, которая ковалентно
связана с M и координирована к нему с помощью функциональной группы основания Льюиса;
l равно 1 или 2;
m равно 0 или 1;
n является числом от 0 до 3;
p - целое число
от 0 до 3; и
сумма l+m+p равна формальной степени окисления металла M.
Предпочтительные комплексы представляют собой те, которые содержат или одну, или две группы L. Последние комплексы включают те, что содержат мостиковую группу, связывающую две группы L. Предпочтительными мостиковыми группами являются те, которые соответствуют формуле (ER2* )x, в которой E является атомом кремния или углерода, R* независимо в каждом случае является атомом водорода или группой, выбранной из силила, гидрокарбила, гидрокарбилокси-группы и их сочетания, причем R* содержит до 30 атомов углерода или кремния и x равно от 1 до 8. Предпочтительно, R* независимо в каждом случае является метилом, бензилом, трет-бутилом или фенилом.
Примерами указанных выше бис (L)-содержащих комплексов являются соединения, которые соответствуют формуле

или

в которых M - это титан, цирконий или гафний, предпочтительно цирконий или гафний, имеющий формальную степень окисления +2 или +4;
R3 в каждом случае независимо выбирают из группы, состоящей из атома водорода, гидрокарбила, силила, германила, циано-, галогенида и их сочетаний, указанный радикал R3 имеет до 20 неводородных атомов, или соседние группы R3 вместе образуют двухвалентную производную (то есть, гидрокарбадиил, силадиил, гермадиил), в результате образуется конденсированная кольцевая система, и
X'' независимо в каждом случае представляет собой анионную лигандную группу, имеющую до 40 неводородных атомов, или две группы X'' вместе образуют двухвалентную, дианионную лигандную группу, имеющую до 40 неводородных атомов, или вместе представляют собой сопряженный диен, имеющий от 4 до 30 неводородных атомов и который образует π-комплекс с металлом M (при этом M имеет степень окисления +2) и
R*, E и x - такие, как определено ранее.
Указанные выше комплексы металлов особенно пригодны для получения полимеров, которые имеют стереорегулярную молекулярную структуру. В таком качестве предпочтительно, чтобы этот комплекс обладал Cs симметрией или обладал хиральной стереожесткой структурой. Примерами первого типа являются соединения, имеющие различные делокализованные π-связанные системы, такие как одна циклопентадиенильная группа и одна флуоренильная группа. Аналогичные системы на основе титана (IV) или циркония (IV) были описаны Эвеном и др. для получения синдиотактических олефиновых полимеров в J. Am. Chem. Soc. 110, 6255-6256 (1980). Примеры хиральной структуры включают бис-инденильные комплексы. Аналогичные системы на основе титана (IV) или циркония (IV) были описаны Уилдом и др. для получения изотактических олефиновых полимеров в J. Organomet. Chem. 232, 233-247 (1982).
Примерами мостиковых лигандов, содержащих две π-связанные группы, являются (диметилсилил-бис-циклопентадиенил), (диметилсилил-бис-метилциклопентадиенил), (диметилсилил-бис-этилциклопентадиенил), (диметилсилил-бис-трет-бутилциклопентадиенил), (диметилсилил-бис-тетраметилциклопентадиенил), (диметилсилил-бис-инденил), (диметилсилил-бис-тетрагидроинденил), (диметилсилил-бис-флуоренил), (диметилсилил-бис-тетрагидрофлуоренил), (диметилсилил-бис-2-метил-4-фенилинденил), (диметилсилил-бис-2-метилинденил), (диметилсилил-циклопентадиенилфлуоренил), (1,1,2,2-тетраметил-1,2-дисилил-бис-циклопентадиенил), 1, 2-бис(циклопентадиенил)этан и (изопропилиден-циклопентадиенилфлуоренил).
Предпочтительные группы X'' выбирают из гидрида, гидрокарбила, силила, германила, галоидгидрокарбила, галоидсилила, силилгидрокарбила и аминогидрокарбильной группы, или две группы X'', взятые вместе образуют двухвалентную производную сопряженного диена, или еще взятые вместе, они образуют нейтральный, π-связанный сопряженный диен. Наиболее предпочтительными группами X'' являются гидрокарбильные группы C1-C20.
Дополнительный класс комплексов металлов, используемых в настоящем
изобретении, соответствует формуле
L1MXmX'nX''p,
или их димеры, в которых L является анионной, делокализованной π-связанной группой,
которая присоединена к металлу M и содержит до 50 неводородных атомов;
M представляет собой металл из Группы 4 Периодической таблицы элементов, имеющий формальную степень окисления +2, +3
или +4;
X является двухвалентным заместителем, имеющим до 50 неводородных атомов, который вместе с группой L образует металлоцикл с M;
X' является необязательным нейтральным
основанием Льюиса, имеющим до 20 неводородных атомов;
X'' в каждом случае представляет собой одновалентную анионную группу, имеющую до 20 неводородных атомов, необязательно две группы X''
вместе могут образовать двухвалентную, анионную группу, имеющей обе валентности, связанные с металлом M, или образуют нейтральный, сопряженный диен C5-C30, и, кроме того, необязательно группы X'' и X'
могут быть связаны вместе с образованием группы, которая ковалентно связана с металлом M и координирована с помощью функциональной группы основания Льюиса:
l равно 1 или 2;
m равно 1;
n является числом от 0 до 3;
p - целое число от 1 до 2; и
сумма l+m+p равна формальной степени окисления металла M.
Предпочтительные двухвалентные заместители X предпочтительно включают группы, которые содержат до 30 неводородных атомов, включающих по меньшей мере один атом, то есть атом кислорода, серы, бора или представитель из Группы 14 Периодической таблицы элементов, непосредственно присоединенный к делокализованной π-связанной группе, и другой атом, выбранный из группы, состоящей из азота, фосфора, кислорода или серы, который ковалентно связан с металлом M.
Предпочтительный класс таких координационных комплексов металлов Группы 4, используемых в настоящем изобретении, соответствует формуле
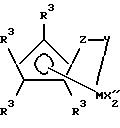
в которой M представляет собой титан или цирконий в формальной степени окисления +2 или +4;
в каждом случае R3 независимо выбирают из группы, состоящей из атома водорода, гидрокарбила, силила, германила, галоида, циано- и их сочетаний, причем эти заместители R3 имеют до 20 неводородных атомов, или соседние группы R3 вместе образуют двухвалентную производную (то есть гидрокарбадиил, силадиил, гермадиил), в результате образуется конденсированная кольцевая система,
каждый X'' является галоидом, гидрокарбилом, гидрокарбилокси- или силильной группой, имеющей до 20 неводородных атомов, или две группы X'' вместе образуют сопряженный диен C5-C30;
группой, которая образует π-комплекс с металлом M;
Y является -O-, -S-, -NR*-, -PR*-; и
Z представляет собой группы SiR2*, CR2*, SiR2* SiR2*, CR2* CR2*, CR*= CR*, CR2* SiR2* или GeR2*,
в которых R* такой, как определено ранее.
Примеры комплексов металлов Группы 4 которые могут быть использованы на практике настоящего изобретения, включают: циклопентадиенилтитантриметил, циклопентадиенилтитантриэтил,
циклопентадиенилтитантриизопропил, циклопентадиенилтитантрифенил, циклопентадиенилтитантрибензил, циклопентадиенилтитан-2,4-пентадиенил, циклопентадиенилтитандиметилметоксид,
циклопентадиенилтитандиметилхлорид, пентаметилциклопентадиенилтитантриметил, инденилтитантриметил, инденилтитантриэтил, инденилтитантрипропил, инденилтитантрифенил, тетрагидроинденилтитантрибензил,
пентаметилциклопентадиенилтитантриизопропил, пентаметилциклопентадиенилтитантрибензил, пентаметилциклопентадиенилтитандиметилметоксид, пентаметилциклопентадиенилтитандиметилхлорид, ( η5 -2,4-диметил-1,3-пентадиенил)титантриметил, октагидрофлуоренилтитантриметил, тетрагидроинденилтитантриметил, тетрагидрофлуоренилтитантриметил, (1,1-диметил-2,3,4,9,10- η -1,4,5,6,7,
8-гексагидронафталенил)титантриметил, (1,1,2,3-тетраметил-2,3,4,9,10- η -1,4,5,6,7,8-гексагидронафталенил)титантриметил, (трет-бутиламидо)(тетраметил- η5
-циклопентадиенил)диметилсилантитандихлорид, (трет-бутиламидо)(тетраметил- η5 -циклопентадиенил)диметилсилантитандиметил, (трет-бутиламидо)(тетраметил- η5
-циклопентадиенил)-1,2-этандиилтитандиметил, (трет-бутиламидо)(тетраметил- η5 -инденил)диметилсилантитандиметил, (трет-бутиламидо)(тетраметил- η5
-циклопентадиенил)диметилсилантитан(III)-2-(диметиламино)бензил, (трет-бутиламидо)(тетраметил- η5 -циклопентадиенил)диметилсилантитан(III)аллил,
(трет-бутиламидо)(тетраметил- η5 -циклопентадиенил)диметилсилантитан(II)-1,4-дифенил-1,3-бутадиен, (трет-бутиламидо)(2-метилинденил)диметилсилантитан(II)-1,4-дифенил- 1,3-бутадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан(IV)-1,3-бутадиен, (трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан(II)-1,4- дифенил-1,3-бутадиен, (трет-бутиламидо)(2,
3-диметилинденил)диметилсилантитан(IV)-1,3-бутадиен, (трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан(II)-1,3-пентадиен, (трет-бутиламидо)(2-метилинденил)диметилсилантитан(II)-1,3-пентадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан(IV)диметил, (трет-бутиламидо)(2-метил-4-фенилинденил)диметилсилантитан(II)-1,4- дифенил-1,3-бутадиен, (трет-бутиламидо)(тетраметил- η5 -циклопентадиенил)диметилсилантитан(IV)-1,3-бутадиен, (трет-бутиламидо)(тетраметил- η5 -циклопентадиенил)диметилсилантитан(II)-1,4-дибензил-1,3-бутадиен,
(трет-бутиламидо)(тетраметил- η5 -циклопентадиенил)диметилсилантитан(II)-2,4-гексадиен, (трет-бутиламидо)(тетраметил- η5
-циклопентадиенил)диметилсилантитан(II)-3-метил-1,3-пентадиен, (трет-бутиламидо)(2,4-диметил-1,3-пентадиен-2-ил)диметилсилантитандиметил, (трет-бутиламидо)(1,1-диметил-2,3,4,9,10- η -1,4,5,6,7,
8-гексагидронафталин-4-ил)диметилсилантитандиметил и (трет-бутиламидо)(1,1,2,3-тетраметил-2,3,4,9,10- η -1,4,5,6,7,8-гексагидронафталин-4-ил)диметилсилантитандиметил.
Бис(L)-содержащие комплексы, включающие мостиковые комплексы, пригодные для использования в настоящем изобретении, включают: бисциклопентадиенилцирконийдиметил, бисциклопентадиенилтитандиэтил, бисциклопентадиенилтитандиизопропил, бисциклопентадиенилтитандифенил, бисциклопентадиенилцирконийдибензил, бисциклопентадиенилтитан-2,4-пентадиенил, бисциклопентадиенилтитанметилметоксид, бисциклопентадиенилтитанметилхлорид, биспентаметилциклопентадиенилтитандиметил, бисинденилтитандиметил, инденилфлуоренилтитандиэтил, бисинденилтитанметил-(2-(диметиламино)бензил), бисинденилтитанметилтриметилсилил, бистетрагидроинденилтитанметилтриметилсилил, биспентаметилциклопентадиенилтитандиизопропил, биспентаметилциклопентадиенилтитандибензил, биспентаметилциклопентадиенилтитанметилметоксид, биспентаметилциклопентадиенилтитанметилхлорид, (диметилсилил-бис-циклопентадиенил)цирконийдиметил, (диметилсилил-бис-пентаметилциклопентадиенил)титан-2, 4-пентадиенил, (диметилсилил-бис-трет-бутилциклопентадиенил)цирконийдихлорид, (метилен-бис-пентаметилциклопентадиенил)титан(III)-2-(диметиламино)бензил, (диметилсилил-бис-инденил)цирконийхлорид, (диметилсилил-бис-2-метилинденил)цирконийдиметил, (диметилсилил-бис-2-метил-4-фенилинденил)цирконийдиметил, (диметилсилил-бис-2-метилинденил)цирконий-1,4-дифенил-1,3-бутадиен, (диметилсилил-бис-2-метил-4-фенилинденил)цирконий(II)-1,4-дифенил-1,3- бутадиен, (диметилсилил-бис-тетрагидроинденил)цирконий(II)-1,4-дифенил-1,3-бутадиен, (диметилсилил-бис-флуоренил)цирконийхлорид, (диметилсилил-бис-тетрагидрофлуоренил)цирконийди(триметилсилил), (изопропилиден)(циклопентадиенил)(флуоренил)цирконийдибензил и (диметилсилилпентаметилциклопентадиенил)(флуоренил)цирконийдиметил.
Прочие соединения, которые используются при получении каталитических композиций согласно этому изобретению, особенного соединения, содержащие другие металлы Группы 4, конечно, являются очевидными для специалистов в этой области техники.
Вообще в катализаторе на носителе соотношение числа молей активирующего соединения (B) к числу молей соединения переходного металла (C) составляет от 0,5: 1 до 2: 1, предпочтительно от 0,5: 1 до 1,5: 1, и наиболее предпочтительно составляет от 0,75: 1 до 1,25: 1. При слишком низких соотношениях катализатор на носителе не будет обладать высокой активностью, в то время как при слишком высоких соотношениях стоимость катализатора станет чрезмерной из-за использования относительно большого количества активирующего соединения. Количество комплекса переходного металла, химически связанного с матрицей неорганического оксида в полученном катализаторе на носителе, предпочтительно составляет от 0,0005 до 20 ммоль/г, более предпочтительно от 0,001 до 10 ммоль/г.
Катализатор на носителе настоящего изобретения может быть приготовлен путем сочетания материала носителя, активирующего соединения и комплекса металла в любой последовательности. Предпочтительно, сначала неорганический оксидный материал обрабатывают активирующим соединением, путем сочетания этих обоих компонентов в подходящем жидком разбавителе, таком как алифатический или ароматический углеводород, с образованием суспензии. Температура, давление и время контактирования не являются существенными, но обычно они изменяются в интервалах от -20 до 150oC, от 1 Па до 10000 МПа, более предпочтительно при атмосферном давлении (100 кПа), в течение от 5 минут до 48 часов. Обычно суспензию перемешивают. После этой обработки обычно твердое вещество отделяют от разбавителя.
Перед использованием носителя изобретения разбавитель или растворитель предпочтительно удаляют, чтобы получить легко текучий порошок. Предпочтительно для этого используют методику, при которой удаляют только жидкость и оставляют полученное твердое вещество, такую как использование нагрева, пониженного давления, выпаривания или их сочетания. Альтернативно носитель может дополнительно контактировать с соединением переходного металла (C) до удаления жидкого разбавителя. В случае такого контактирования соединение переходного металла предпочтительно используют в растворе подходящего растворителя, такого как жидкий углеводородный растворитель, преимущественно алифатические или циклоалифатические углеводороды C5-C10 или ароматические углеводороды C6-C10. Альтернативно также можно использовать суспензию или дисперсию соединения переходного металла в нерастворяющем разбавителе. Температура контактирования не является существенной при условии, что она ниже температуры разложения соединения переходного металла и активатора. Хорошие результаты получаются в интервале температур от 0 до 100oC. Это контактирование может представлять собой полное погружение в жидкую среду или контакт с распыленной струей раствора, дисперсии или суспензии. Все стадии этого процесса должны осуществляться в отсутствие кислорода и влаги. Полученный катализатор на носителе может храниться или транспортироваться в виде свободно текучего порошка в инертном состоянии после удаления растворителя.
Катализатор на носителе настоящего изобретения может использоваться в процессах аддитивной полимеризации, в которых один или несколько полимеризующихся мономеров контактируют с нанесенным на носитель катализатором настоящего изобретения в условиях аддитивной полимеризации.
Подходящие мономеры, аддитивно полимеризующиеся, включают этиленненасыщенные мономеры, ацетиленовые соединения, сопряженные или несопряженные диены и полиены. Предпочтительные мономеры включают олефины, например, альфа-олефины, имеющие от 2 до 20000 атомов углерода, предпочтительно от 2 до 20, более предпочтительно от 2 до 8 атомов углерода, и сочетания из двух или более таких альфа-олефинов. Особенно пригодные альфа-олефины включают, например, этилен, пропилен, бутен-1, пентен-1, 4-метилпентен-1, гексен-1, гептен-1, октен-1, нонен-1, децен-1, ундецен-1, додецен-1, тридецен-1, тетрадецен-1, пентадецен-1 или их сочетания, а также длинноцепочечные с винильной терминальной группой олигомерные или полимерные продукты реакции, полученные при полимеризации и альфа-олефины C10-C30, специально добавленные в реакционную смесь для того, чтобы получить в полимерных продуктах относительно длинные цепочки разветвлений. Предпочтительными альфа-олефинами являются этилен, пропилен, бутен-1, 4-метилпентен-1, гексен-1, октен-1 и сочетания этилена и/или пропилена с одним или несколькими такими альфа-олефинами. Другие предпочтительные мономеры включают стирол, галоид- и алкилзамещенные стиролы, тетрафторэтилен, винилциклобутен, 1,4-гексадиен, дициклопентадиен и этилиденнорборнен и 1, 7-октадиен. Также могут использоваться смеси указанных выше мономеров.
Катализатор на носителе может быть получен in situ, в полимеризационной смеси посредством введения в указанную смесь носителя согласно настоящему изобретению или его компонентов, а также подходящего соединения переходного металла (C). Катализатор на носителе предпочтительно может использоваться в процессах полимеризации при высоком давлении, в растворе, в суспензии или в газовой фазе. Процесс полимеризации при высоком давлении обычно проводят при температурах от 100 до 400oC и при давлениях свыше 500 бар. В суспензионном процессе обычно используют инертный углеводородный разбавитель и температуру от 0 до температуры ниже той, при которой образующийся полимер приобретает существенную растворимость в инертной полимеризационной среде. Предпочтительная температура составляет от 40 до 115oC. Процесс полимеризации в растворе проводят при температуре от той, при которой образующийся полимер растворим в инертном растворителе, до 275oC, предпочтительно при температуре от 130 до 260oC, более предпочтительно от 150 до 240oC. Предпочтительными инертными растворителями являются углеводороды C1-C20 и предпочтительно алифатические углеводороды C5-C10, включая их смеси. Процессы полимеризации в растворе или в суспензии обычно проводят при давлении между 100 кПа и 10 МПа. Типичными рабочими условиями для полимеризации в газовой фазе являются температура от 20 до 100oC, более предпочтительно от 40 до 80o . В газофазном процессе типичное давление полимеризации составляет от 10 кПа до 10 МПа. Конденсирующийся мономер или разбавитель могут впрыскиваться в реактор, чтобы способствовать отводу тепла с помощью скрытой теплоты испарения.
Предпочтительно, в газофазном процессе полимеризации используют носитель, который имеет средний диаметр частиц от 20 до 200 мкм, более предпочтительно от 30 до 150 мкм и наиболее предпочтительно от 50 до 100 мкм. Предпочтительно, в суспензионном процессе полимеризации используют носитель, который имеет средний диаметр частиц от 1 до 200 мкм, более предпочтительно от 5 до 100 мкм и наиболее предпочтительно от 20 до 80 мкм. Предпочтительно, в процессе полимеризации в растворе или при высоком давлении используют носитель, который имеет средний диаметр частиц от 1 до 40 мкм, более предпочтительно от 1 до 30 мкм и наиболее предпочтительно от 1 до 20 мкм.
В процессе полимеризации согласно настоящему изобретению можно использовать поглотитель примесей, назначением которых является защита нанесенного на носитель катализатора от каталитических ядов, таких как вода, кислород и полярные соединения. Эти поглотители вообще могут использоваться в количествах, которые зависят от количества примесей. Предпочтительными поглотителями являются упомянутые выше алюминийорганические соединения формулы AlR3 или алюмоксаны.
В процессе полимеризации согласно настоящему изобретению также можно использовать агенты, регулирующие молекулярный вес. Примеры таких агентов, регулирующих молекулярный вес, включают водород, триалкилалюминиевые соединения или другие известные агенты передачи цепи. Особенным преимуществом использования нанесенных на носитель катализаторов изобретения является их способность (в зависимости от условий реакции) получать гомополимеры и сополимеры альфа-олефинов с узким распределением молекулярного веса. Предпочтительные полимеры имеют соотношение Mw/Mn менее 2,5, более предпочтительно, менее 2,3. Такое узкое распределение молекулярного веса полимерных продуктов, особенно полученных в суспензионном процессе, является весьма желательным вследствие улучшенных прочностных свойств.
Описав это изобретение, мы предоставляем следующие примеры, дополнительно иллюстрирующие изобретение, которые не следует рассматривать как ограничивающие. Если не указано другое, все части и проценты выражены в расчете на массу. Насыпная плотность полимеров, полученных в представленных примерах, была определена в соответствии со стандартом ASTM 1895.
ПРИМЕРЫ
A1a. Синтез (4-бромфенокси)триметилсилана, Br(C6H4)-п-OSiMe3
К 40,3 г пара-бромфенола
(116 ммоль) добавляют 100 мл 1,1,1,3,3,3-гексаметидисилазана (чистота 98%, 464 ммоль) и смесь нагревают до кипения с обратным холодильником в течение 2 ч. После охлаждения до 25oC избыток 1,
1,1,3,3,3-гексаметилдисилазана удаляют при перегонке (120oC), и остаток очищают методом флэш-хроматографии, используя силикагель Дэвисон 948 (800oC, пентан). Продукт представляет
собой бесцветную жидкость; выход 50 г (88%).
A1b. Синтез MgBr(C6H4)-п-OSiMe3
Смешивают в трехгорлой колбе емкостью 100 мл 1,2 г (49,4 ммоль)
магниевых стружек с 4 мл тетрагидрофурана (ТГФ) и затем добавляют 0,25 мл 1,2-дибромэтана (2,87 ммоль). Смесь доводят до кипения и по каплям добавляют из шприца в течение 15 минут раствор 7,5 мл (38,8
ммоль) 4-бромфенокситриметилсилана в 32 мл тетрагидрофурана. Образовавшуюся реакционную смесь дополнительно кипятят с обратным холодильником в течение 1 часа и затем охлаждают до 25oC.
Темно-серый раствор фильтруют и титруют 2-бутанолом в присутствии 5-метил-1,10-фенантролина. Выход 81% (36 мл 0,87 М).
A1c. Синтез [MgBr•2ТГФ] [(C6F5)3B(C6H4- п-OSiMe3)]
Раствор трис(пентафторфенил)бора (C6F5)3B (15,85 г, 31 ммоль) в 100 мл диэтилового эфира при
комнатной температуре обрабатывают (35,5 мл, 0,87М) свежеприготовленным раствором MgBr(C6H4)-п-OSiMe3 в тетрагидрофуране. Реакционную смесь перемешивают в течение 16 ч,
добавляют 100 мл пентана и смесь дополнительно перемешивают 30 минут, чтобы образовалась двухслойная смесь. Верхний
пентановый слой декантируют, и нижний слой дополнительно промывают двумя порциями
(по 50 мл) пентана. Образовавшийся сироп выпаривают при пониженном давлении, получая белый твердый продукт. Выход 22,1 г (77%), ЯМР-спектроскопия (в пердейтеро-ТГФ) на протонах: б+0,18 (синглет, 9H,
триметилсилан), 6,45 (дублет, 2H, фенилен), 7,06 (дублет, 2H, фенилен); на ядрах19F: -131,1 (дублет), -167,2 (триплет), -169,9 (триплет); на ядрах13C: +0,46 (с, триметилсилан),
117-153 (арильный углерод).
A1d. Синтез диметиланилиний (4-гидроксифенил)трис(пентафторфенил)-бората [PhMe2NH] [(C6F5)3B(C6H4- п-OH)]
Водный 0,312-молярный раствор хлористоводородного диметиланилина (100 мл, 31,2 ммоль) и [MgBr•2 ТГФ] [(C6F5)3B(C6H4- п-OSiMe3)] (22,1 г, 23,9 ммоль) при комнатной температуре перемешивают в течение 16 ч. Образовавшийся водный раствор осторожно декантируют, вязкое твердое вещество промывают
дистиллированной водой (6 раз по 150 мл) и пентаном (3 раза по 100 мл) и сушат при пониженном давлении. Выход 13,2 г (76%), ЯМР-спектроскопия (в пердейтеро-ТГФ) на протонах: б+3,22 (синглет, 6H,
-NH-диметилфенил), 6,40 (дублет, 2H, фенилен), 7,05 (дублет, 2H, фенилен), 7,4-7,7 (мультиплет, 5H, -NH-диметилфенил); на ядрах19F: -131,1 (дублет), -167,8 (триплет), -169,9 (триплет); на
ядрах13C: +46,3 (-NH-диметилфенил), 112-158 (арильный углерод).
A2a. Синтез 4-(4'-бромфенил)фенокси)тримефилсилана, Br(C6H4)-(C6H4)-п-OSiMe3
К 30 г 4-бромфенил-п-фенола (117 ммоль) добавляют 75 мл 1,1,1,3,3,3-гексаметилдисилазана (чистота 98%, 348 ммоль) и нагревают до кипения с обратным холодильником
в течение 4 ч. После охлаждения до 25oC твердый продукт отфильтровывают и промывают 50 мл холодного пентана (0oC). Затем неочищенный продукт растворяют в диэтиловом эфире и
очищают методом флэш-хроматографии, используя силикагель Дэвисон 948 (800oC, пентан). Продукт представляет собой белые кристаллы; выход 33,6 г (89%).
A2b. Синтез MgBr(C6H4)-(C6H4)- п-OSiMe3
В трехгорлой колбе смешивают 0,47 г (19,3 ммоль) магниевых стружек (50 меш) с 5 мл тетрагидрофурана. Затем добавляют в
эту колбу из шприца 0,25 мл 1,2-дибромэтана (2,87 ммоль) и смесь доводят до интенсивного кипения. По каплям добавляют из шприца в течение 20 минут 11 мл раствора 3,0 г 4-бромфенилфенокситриметилсилана
(9,34 ммоль) в условиях кипячения с обратным холодильником. Образовавшуюся горячую реакционную смесь охлаждают до 25oC за 1,5 часа. Темно-серый раствор фильтруют и титруют 2-бутанолом в
присутствии 5-метил-1,10-фенантролина. Выход 87% (10,9 мл, 0,76М).
A2c. Синтез [MgBr•2ТГФ] [(C6F5)3B(C6H4- C6
H4-п-OSiMe3)]
Раствор трис(пентафторфенил)бора (C6F5)3B (3,24 г, 6,33 ммоль) в 50 мл диэтилового эфира при комнатной температуре
обрабатывают 10,4 мл 0,76-молярного (8,13 ммоль) свежеприготовленного раствора MgBrC6H4-C6H4-п-OSiMe3 в тетрагидрофуране. Образовавшуюся
реакционную смесь перемешивают в течение 3 ч, обрабатывают и выделяют, по методике примера Ale. Выход 6,84 г (84%).
A2d. Синтез диметиланилиний
4-(4'-гидроксифенил)фенил)трис(пентафторфенил)бората [PhMe2NH]+[(C6F5)3B(C6H4- C6H4-п-OH)]-
Практически повторяют методику примера A1d. Выход 84%.
A3a. Приготовление 2-бром-6-триметилсилоксинафталина
В атмосфере аргона перемешивают в течение 2 ч
суспензию 10 г 2-бром-6-нафтола в 30 мл бис (триметилсилан) амина (Me3SiNHSiMe3). Через 2 ч удаляют при пониженном давлении избыток силанового реагента. Оставшееся твердое
вещество растворяют в 20 мл пентана и элюируют через слой (5 см) силикагеля. Растворитель удаляют при пониженном давлении, получая 11,5 г 2-бром-6-триметилсилоксинафталина в виде белого
кристаллического вещества. ПМР спектр (в дейтерохлороформе): -0,40 (синглет, 9H, триметилсилан), 6,4-7,3 (мультиплет, 6H, ароматические протоны), ч/млн.
A3b. Приготовление эфирата
диброммагния 6-(триметилсилокси-2-нафтил)-трис(пентафторфенил)бората, MgBr2(Et2O)x[6-Me3SiOC10H6- 2-B(C6F5)3
Суспензию 0,7 г магниевого порошка в 10 мл тетрагидрофурана активируют, добавляя 0,1 мл 1,2-дибромэтана, и осторожно доводят до кипения. Добавляют в течение 30 минут раствор 5,0 г
2-бром-6-триметилсилоксинафталина в 5 мл ТТФ. В это время 0,5 мл охлажденного раствора титруют изопропиловым спиртом. Оставшиеся 13,9 мл 0,758-молярного раствора реактива Гриньяра добавляют к
суспензии 5,39 г трис(пентафторфенил)бора в 30 мл диэтилового эфира. Смесь перемешивают в течение 20 ч, при этом образуется белый осадок. Твердое вещество собирают фильтрацией, промывают диэтиловым
эфиром и пентаном и сушат при пониженном давлении. Выход MgBr2(диэтиловый эфир)x[6-(Me3SiOC10H6- 2-B(C6F5)3
составляет 6,81 г. ПМР спектр (в пердейтеро-ТГФ): 0,28 (синглет, 9H, триметилсилан), 6,8-7,7 (мультиплет, 6H, ароматические протоны), ч/млн. ЯМР на ядрах19F (в пердейтеро-ТГФ): -123,0
(дублет, JF-F = 19,5 Гц, орто-F), -159,4 (мультиплет, мета-фтор), -161,9 (триплет, JF-F = 23 Гц, пара-F).
A3c. Приготовление диметиланилиний
(6-гидрокси-2-нафтил)-трис(пентафторфенил)бората, [PhMe2NH]+[(6-HOC10H6-2- B(C6F5)3]-
Полученный выше
MgBr2 (диэтиловый эфир)x[6-Me3SiOC10H6-2- B(C6F5)3 (6,81 г) суспендируют в дистиллированной воде с избытком
хлористоводородного диметиланилина в течение 4 ч. Водный раствор декантируют и твердое вещество промывают несколькими порциями дистиллированной воды. Полученное твердое вещество растворяют в 10 мл
метанола. Затем метанол удаляют при пониженном давлении, получая [PhMe2NH] [(6-HOC10H6-2- B(C6F5)3] в виде белого кристаллического
вещества. Выход 4,34 г (76%).
ЯМР-спектроскопия (в пердейтеро-ТГФ) на протонах: 3,02 (6H, -N-диметил), 6,6-7,5 (11H, ароматические протоны), ч/млн; на ядрах19F (в пердейтеро-ТГФ): -123,1 (дублет, JF-F = 20,6 Гц, орто-F), -159,4 (мультиплет, мета-фтор), -161,8 (триплет, JF-F = 23 Гц, пара-F).
B. Приготовление силикагеля,
модифицированного фенилсиланом (PhH2Si-O-силикагель)
Суспензию 10 г силикагеля ДэвисонTM 948 (800oC, получен от фирмы Грейс Кемикал Ко. , подразделение
Дэвисон) в 150 мл пентана обрабатывают 2,70 г фенилсилана (Ph SiH3) (25 ммоль) и триэтиламином (2,53 г, 25 ммоль), поданными из шприца в атмосфере аргона при 23oC. Из раствора
интенсивно выделяется газообразный водород. Полученную смесь перемешивают в течение 12 ч. Силикагель, модифицированный фенилсиланом, собирают на пористом стекле в атмосфере аргона, промывают 5 раз
пентаном (по 5 мл) и сушат при пониженном давлении. Выход составляет 10,43 г.
Инфракрасная спектроскопия (диффузное отражение) с Фурье-преобразованием (ИКСДОФ), n (Si-H) 2178 см-1 (оч. сильн. );29Si CPMAS 6 - 23 ч/млн. Содержание гидроксильных групп в функционализированном силикагеле было меньше предела чувствительности (<0,1 ммоль/г).
C. Получение бората анилиния на носителе [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiHPh-O-силикагель)]-
Суспензию 3,0 г силикагеля, модифицированного фенилсиланом, в 30 мл эфира обрабатывают 100 мл эфирного раствора [PhMe2NH]+[(C6F5)3B(C6
H4- п-OH)]- (1,05 г, 1,44 ммоль) при комнатной температуре в атмосфере аргона. Из раствора в течение 10 минут выделяется газообразный водород. Этот раствор перемешивают в
течение 15 ч и полученное твердое вещество фильтруют, промывают 5 порциями (по 20 мл) эфира и 3 порциями (по 20 мл) пентана, сушат при пониженном давлении. Получают 3,71 г продукта. ИКСДОФ, n(Si-H)
2190 см-1 (средн. ); n(N-H) 3239 см-1 (сильн. ).29Si CPMAS: -O-SiHPh-O-силикагель (синглет, 41 ч/млн).13C CPMAS: NHMe2Ph (синглет, 48,5
ч/млн).
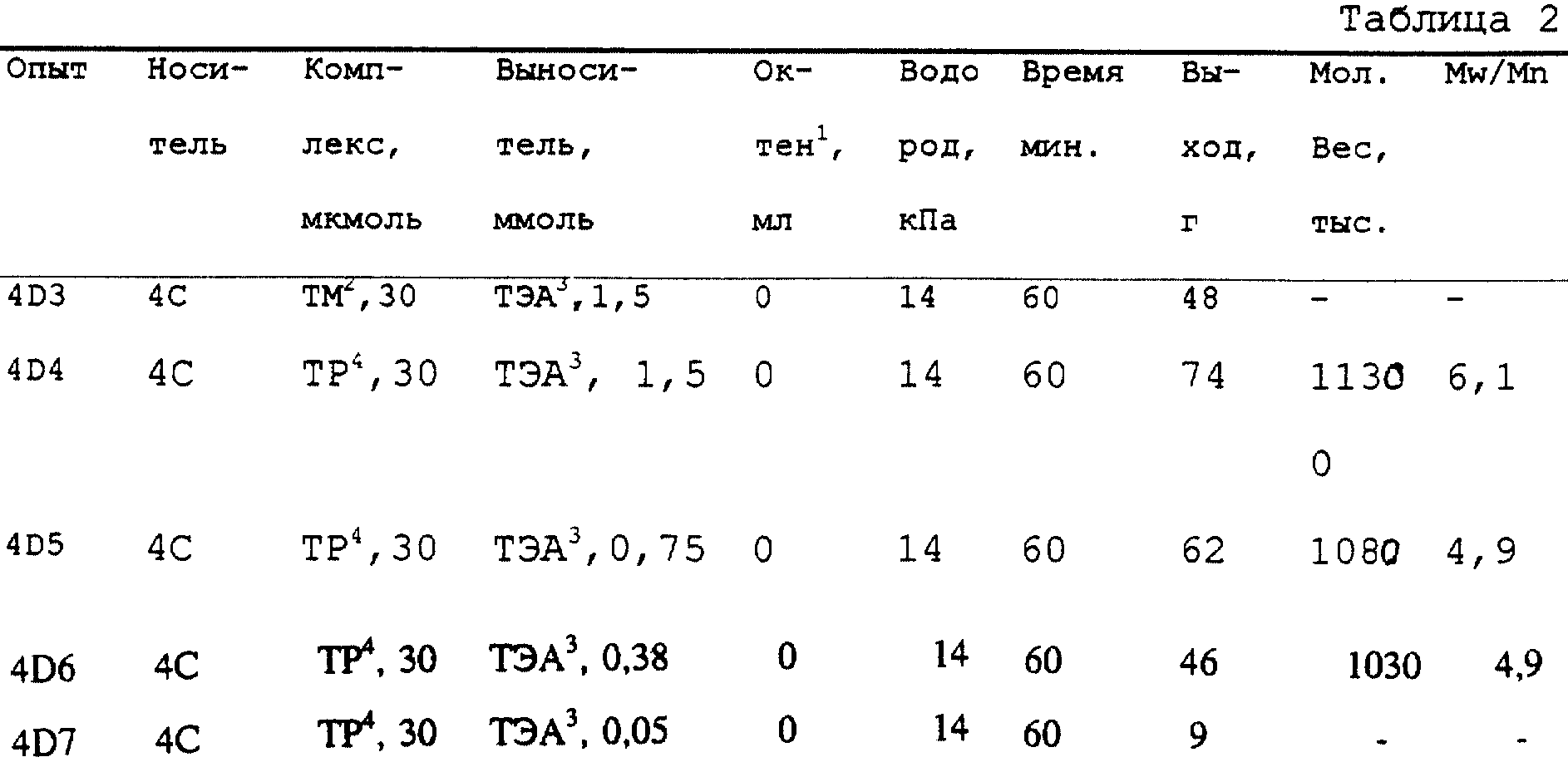
D. Суспензионная полимеризация в реакторе периодического действия
Перед использованием реактор-автоклав емкостью 2 л откачивают при 80oC в течение ночи.
Обрабатывают суспендированный в 300 мл гептана модифицированный фенилсиланом силикагель с нанесенным на него анилиний (4-гидроксифенил) трис (пентафторфенил) боратом
([NHMe2Ph]+[(C6F5)3B(C6H4- п-O-SiHPh-O-силикагель)] (200 мг) 10 мг (31 ммоль) (трет-бутиламидо)диметил(тетраметил- η5
-циклопенадиенил)силантитандиметила. Каталитическую смесь переносят в вакууме в предварительно нагретый реактор. Быстро напускают этилен до давления 1,4 МПа, причем температуру реактора поддерживают
при 75oC с помощью водяной бани с циркулирующей водой. Этилен подают в случае необходимости через регулятор потока массы. Выход полиэтилена после 10 минут реакции составляет 7,14 г;
молекулярный вес равен 745600, отношение Mw/Mn = 2,465.
Пример 2
A. Используют активатор примера 1A1d.
B. Приготовление силикагеля, модифицированного
дифенилсиланом (Ph2HSi-O-силикагель)
Суспензию 20 г силикагеля ДэвисонTM948 (800oC) в 200 мл пентана обрабатывают 8,67 г (Ph2SiH2) (47
ммоль) и триэтиламином (5,08 г, 50 ммоль), поданными из шприца в атмосфере аргона при комнатной температуре. Из раствора интенсивно выделяется газообразный водород. Полученную смесь перемешивают в
течение 12 ч. Силикагель, модифицированный дифенилсиланом, собирают на пористом стекле в атмосфере аргона, промывают 5 раз пентаном (по 10 мл) и сушат при пониженном давлении. Выход составляет 20,87
г. ИКСДОФ: n(Si-H) 2169 см-1 (средн. ). Содержание гидроксильных групп в функционализированном силикагеле было меньше предела чувствительности (<0,1 ммоль/силикагеля).
C. Получение модифицированного дифенилсиланом силикагеля с нанесенным анилиний (4-фенил)трис(пентафторфенил)боратом [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiPh2-O-силикагель)]-
Суспензию 3,0 г силикагеля, модифицированного дифенилсиланом, в 30 мл эфира обрабатывают 100 мл эфирного раствора
[PhMe2NH]+[(C6F5)3B(C6H4- п-OH]- (1,0 г, 1,38 ммоль) при комнатной температуре в атмосфере аргона. Раствор
перемешивают в течение 15 ч, полученное белое твердое вещество фильтруют, промывают 5 порциями (по 20 мл) эфира и 3 порциями (по 20 мл) пентана и сушат при пониженном давлении. Получают 3,11 г
продукта.
D1. Суспензионная полимеризация в реакторе периодического действия
В автоклав Хоппса емкостью 3,78 л, закупленный на фирме Аутоклав Инжинирз Инк. , сначала загружают
1850 г безводного гексана. Затем газовое пространство реактора продувают 2 раза смесью 5 мол. % водорода/этилен, которую сбрасывают после каждой продувки. Затем реактор нагревают до температуры 80oC и сбрасывают газ до давления паров растворителя (190 кПа). Затем добавляют в реактор смесь водород/этилен до давления 470 кПа. Этилен подают через регулятор расхода при установленном давлении
1,3 МПа. Суспендированный катализатор готовят, смешивая 70 мг модифицированного дифенилсиланом силикагеля с нанесенным диметиланилиний боратом, [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiPh2-силикагель)]-, 20 мл смешанного алканового растворителя (ИзопарTME, поставлен фирмой Эксон Кемикалз Инк. ),
и 0,21 мл 0,0717-молярного раствора (15 ммоль) (трет-бутиламидо)диметил(тетраметил- η5 -циклопентадиенил)силантитандиметила, и перемешивают 15 минут. Затем суспендированный
катализатор переносят в реактор через опрессованную емкость из нержавеющей стали. Через 60 минут из реактора удаляют образец полимера, отфильтровывают, порошок помещают примерно на 30 минут на тарелку
в вакуумном сушильном шкафу при 80oC. Выделяют 20,4 г полиэтилена (эффективность катализатора составляет 29800 г полиэтилена на 1 г титана).
D2. Полимеризация в растворе в
реакторе периодического действия
В автоклав емкостью 3,78 л при перемешивании загружают 1445 г ИзопараTME и 126 г октена-1 и нагревают до температуры 130oC. Затем в
реактор загружают водород до давления 360 кПа и после этого этилен, доводя суммарное давление до 3,1 МПа. Катализатор готовят, смешивая 150 мг модифицированного дифенилсиланом силикагеля с нанесенным
диметиланилиний боратом, [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiPh2-O-силикагель)]-, 20 мл растворителя
ИзопарTM E, и 0,42 мл 0,0717-молярного раствора (30 ммоль) (трет-бутиламидо)диметил(тетраметил- η5 -циклопентадиенил)силантитандиметила в течение 15 минут. Затем
суспендированный катализатор переносят в реактор и температуру, и давление в реакторе поддерживают, непрерывно подавая в ходе полимеризации этилен и охлаждая реактор в случае необходимости. Через 10
минут содержимое реактора переносят в продутую азотом пластиковую емкость, содержащую 0,2 г антиокислителя Ирганокс 1010 (поставляется фирмой Циба Гейги Ко. ), образец сушат в течение 15 ч в вакуумном
сушильном шкафу, получая 93,4 г сополимера (эффективность катализатора 65000 г полиэтилена на 1 г титана).
Пример 3
B1. Приготовление силикагеля, модифицированного
фенилсиланом (PhH2Si-O-силикагель)
Силикагели, имеющие остаточное содержание гидроксильных групп 0,5 ммоль/г, получают прокаливанием различных силикагелей при 800oC
(Дэвисон 948, Дэвисон 952 и СилополTM-2212 (поставляется Грейс Дэвисон Корпорейшн). Прокаленный силикагель (20 г) суспендируют в 150 мл пентана и обрабатывают 6 мл фенилсилана (PhSiH3) (48 ммоль) и 6 мл триэтилалюминия (43 ммоль), поданными из шприца в атмосфере аргона при комнатной температуре. Из раствора интенсивно выделяется газообразный водород. Полученную смесь
перемешивают встряхиванием в течение 12 ч. Силикагели, модифицированные фенилсиланом, собирают на пористом стекле в атмосфере аргона, промывают 5 раз пентаном (по 20 мл) и сушат при пониженном
давлении. Выходы составляют примерно 21,0 г.
ИКСДОФ: v(Si-H) 2178 см-1 (оч. сильн. );29Si CPMAS: б -23 ч/млн. Содержание остаточных гидроксильных групп было меньше предела чувствительности (<0,1 ммоль/г силикагеля).
B2. Приготовление силикагеля, модифицированного диметилсиланом (Me2HSi-O-силикагель)
Суспензию 30
г силикагеля Грейс Дэвисон 948 (800oC, 0,5 ммоль - OH/г) в 200 мл пентана обрабатывают 3,0 г (Me2SiH)2NH (22,5 ммоль). Полученную смесь перемешивают встряхиванием в
течение 12 ч. Силикагель, модифицированный диметилсиланом, собирают на пористом стекле в атмосфере аргона, промывают 5 раз пентаном (по 20 мл) и сушат при пониженном давлении. Выход составляет 30,95
г.
ИКСДОФ: v(Si-H) 2158 см-1 (сильн. );29Si CPMAS: б -1,3 ч/млн.
B3. Приготовление силикагеля, модифицированного диэтилсиланом Et2
HSi-O-силикагель
Суспензию 2,5 г силикагеля Грейс Дэвисон 948 (800oC, 0,5 ммоль - ОН/г) в 50 мл толуола обрабатывают 0,9 г Et2SiH2 (97%, 10,2 ммоль) и 1,05 мл
триэтиламина (7,5 ммоль). Полученную смесь кипятят с обратным холодильником в течение 12 ч. Полученный раствор охлаждают до 25oC и силикагель, модифицированный диэтилсиланом, собирают на
пористом стекле в атмосфере аргона, промывают 5 раз пентаном (по 20 мл) и сушат при пониженном давлении. Выход составляет 2,7 г.
ИКСДОФ: v(Si-H) 2141 см-1 (сильн. );29Si CPMAS: б +5 ч/млн.
B4. Приготовление силикагеля, модифицированного фенилметилсиланом (PhMeHSi-O-силикагель)
Суспензию 30 г силикагеля Грейс Дэвисон 948 (800oC, 0,5 ммоль - OH/г) в 350 мл гептана обрабатывают 9,15 г PhMeSiH2 (97%, 72,8 ммоль) и 10,5 мл триэтиламина (75 ммоль). Полученную смесь кипятят с обратным холодильником в течение 12
ч с верхней мешалкой. Полученный раствор охлаждают до 25oC и силикагель, модифицированный фенилметилсиланом, собирают на пористом стекле в атмосфере аргона, промывают 5 раз пентаном (по 30
мл) и сушат при пониженном давлении. Выход составляют 31,73 г.
ИКСДОФ: v(Si-H) 2160 см-1 (сильн. );29Si CPMAS: б -6 ч/млн.
BB5. Приготовление
силикагеля, модифицированного дифенилсиланом Ph2HSi-O-силикагель
Суспензию 20 г силикагеля Грейс Дэвисон 948 (800oC, 0,5 ммоль - OH/г) в 150 мл пентана обрабатывают из
шприца 9 миллилитрами Ph2SiH2 (98%, 44,7 ммоль) и 6,2 мл триэтиламина (44,5 ммоль) в атмосфере аргона при комнатной температуре. Из раствора интенсивно выделяется газообразный
водород. Полученную смесь перемешивают встряхиванием в течение 12 ч. Силикагель, модифицированный дифенилсиланом, собирают на пористом стекле в атмосфере аргона, промывают 5 раз пентаном (по 30 мл) и
сушат при пониженном давлении. Выход составляет 21,6 г.
ИКСДОФ: v(Si-H) 2169 см-1 (сильн. ).
C1. Получение нанесенного на силикагель бората анилиния,
функционализированного w/фенилсиланом [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiHPh-O-силикагель)]-
Суспензию
10,0 г силикагеля, модифицированного фенилсиланом (получен, как в примере 3B1), в 100 мл эфира обрабатывают 100 мл эфирного раствора диметиланилиний(4-гидроксифенил)трис(пентафторфенил)бората
[PhMe2NH] [(C6F5)3B(C6H4-п-OH)] (получен как в примере Al(a-d) (2,94 г, 4,03 ммоль) при комнатной температуре в атмосфере аргона. Этот
раствор перемешивают в сухом боксе в течение 1,5 суток, и полученное твердое вещество фильтруют, промывают 5 порциями (по 20 мл) эфира и 3 порциями (по 20 мл) пентана, и сушат при пониженном давлении.
Выход 11,99 г продукта.
ИКСДОФ: v(Si-H) 2190 см-1 (средн. ); v(N-H) 3239 см-1 (слаб. )29Si CPMAS: -O-SiHPh-O-силикагель (синглет, -41 ч/млн).13C CPMAS: NHMe2Ph (синглет, 48,5 ч/млн). Содержание бора ICP: 0,231%.
C2. Получение нанесенного на силикагель бората анилиния, функционализированного
w/диметилсиланом [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiMe2-O-силикагель)]-
Суспензию 10,0 г
силикагеля, модифицированного диметилсиланом (получен, как в примере 3B2), в 100 мл эфира обрабатывают 100 мл эфирного раствора диметиланилиний (4-гидроксифенил)трис(пентафторфенил)бората [PhMe2NH] [(C6F5)3B(C6H4-п-OH)] (2,9 г, 4,02 ммоль) при 25oC в атмосфере аргона. Этот раствор перемешивают в сухом боксе в течение 2,
5 суток, полученное твердое вещество фильтруют, промывают 5 порциями (по 20 мл) эфира и 3 порциями (по 20 мл) пентана и сушат при пониженном давлении. Выход 12,21 г продукта.
ИКСДОФ: v(N-H) 3240 см-1 (слаб. );29Si CPMAS: -OSiHPh-O-силикагель (синглет, 7,7 ч/млн).
C3. Получение нанесенного на силикагель бората анилиния, функционализированного
w/диэтилсиланом [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiEt2-O-силикагель)]-
Суспензию 10,0 г
силикагеля, модифицированного диэтилсиланом (получен, как в примере 3B3), в 100 мл эфира обрабатывают 100 мл эфирного раствора диметиланилиний (4-гидроксифенил) трис (пентафторфенил) - бората
[PhMe2NH] [(C6F5)3B(C6H4-п-OH)] (2,9 г, 4,02 ммоль) при 25oC в атмосфере аргона. Этот раствор перемешивают в сухом боксе в
течение 2,5 суток, полученное твердое вещество фильтруют, промывают 5 порциями (по 20 мл) эфира и 3 порциями (по 20 мл) пентана и сушат при пониженном давлении.
C4. Получение
нанесенного на силикагель бората анилиния, функционализированного w/фенилметилсиланом [PhMe2NH]+[(C6F5)3B(C6H4
- п-O-SiMePh-O-силикагель)]-
Суспензию 10,0 г силикагеля, модифицированного фенилметилсиланом (получен, как в примере 3B4), в 100 мл эфира обрабатывают 100 мл эфирного раствора
диметиланилиний (4-гидроксифенил)трис(пентафторфенил)бората [PhMe2NH] [(C6F5)3B(C6H4-п-OH] (2,90 г, 4,02 ммол) при 25oC в
атмосфере аргона. Этот раствор перемешивают в сухом боксе в течение 2,5 суток, полученное твердое вещество фильтруют, промывают 5 порциями (по 20 мл) эфира и 3 порциями (по 20 мл) пентана и сушат при
пониженном давлении.
C5. Получение нанесенного на силикагель диметиланилиний (4-гидроксифенил)трис(пентафторфенил)бората, функционализированного w/дифенилсиланом [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiPh2-O-силикагель)]-
Суспензию 10,0 г силикагеля, модифицированного
дифенилсиланом (получен, как в примере 3B5), в 100 мл эфира обрабатывают 100 мл эфирного раствора диметиланилиний(4-гидроксифенил)трис(пентафторфенил)бората [PhMe2NH] [(C6F5)3B(C6H4)-п-OH)] (2,9 г, 4,02 ммоль) при 25oC в атмосфере аргона. Этот раствор перемешивают в сухом боксе в течение 2,5 суток, полученное твердое
вещество фильтруют, промывают 5 порциями (по 20 мл) эфира и 3 порциями (по 20 мл) пентана, и сушат при пониженном давлении.
C6. Получение нанесенного на силикагель бората
диметиланилиний (4-(4'-гидроксифенил)фенил)трис(пентафторфенил)бората, функционализированного w/фенилсиланом [PhMe2NH]+[(C6F5)3B(C6
H4- п-O-SiHPh-O-силикагель)]-
Суспензию 4,0 г силикагеля, модифицированного фенилсиланом, в 80 мл эфира обрабатывают диметиланилиний
(4-(4'-гидроксифенил)фенил)трис(пентафторфенил)бората [PhMe2NH]+[(C6F5)3B(C6H4- п-OH)]- (получен, как в
примере A2(a-d)) (1,2 г, 1,49 ммоль) при 25oC в атмосфере аргона. Этот раствор перемешивают в сухом боксе в течение 2 суток и полученное твердое вещество фильтруют, промывают 5 порциями (по
20 мл) эфира и 3 порциями (по 20 мл) пентана и сушат при пониженном давлении. Выход 5,04 г продукта.
ИКСДОФ: v(Si-H) 2191 см-1 (средн. ); v(N-H) 3244 см-1 (слаб. ).29Si CPMAS: -OSiHPh-O-силикагель (синглет, -41 ч/млн).13C CPMAS: NHMe2Ph (синглет, 47,7 ч/млн). Содержание бора ICP: 0,225%.
C7. Получение
нанесенного на силикагель диметиланилиний (4-(4'-гидроксифенил)фенил)трис(пентафторфенил)бората, функционализированного диметилсиланом [PhMe2NH]+[(C6F5
)3B(C6H4- п-O-SiMe2-O-силикагель)]-
Суспензию 1,0 г силикагеля, модифицированного диметилсиланом, в 60 мл эфира обрабатывают
диметиланилиний (4-(4'-гидроксифенил)фенил)трис(пентафторфенил)боратом [PhMe2NH]+[(C6F5)3B(C6H4)- п-OH)]- (0,
39 г, 0,49 ммоль) при комнатной температуре в атмосфере аргона. Этот раствор перемешивают в сухом боксе в течение 2 суток, полученное твердое вещество фильтруют, промывают 3 порциями (по 20 мл) эфира
и 3 порциями (по 10 мл) пентана и сушат при пониженном давлении. Выход 1,20 г продукта.
ИКСДОФ: v(N-H) 3142 см-1 (слаб. ).29Si CPMAS: -OSiHPh-O-силикагель (синглет, -7,6 ч/млн). Содержание бора ICP: 0,232%.
D. Суспензионная полимеризация в реакторе периодического действия
1) Сополимеризация
Перед использованием
реактор-автоклав емкостью 2 л откачивают при 70oC в течение 90 мин. Гептан (550 мл), содержащий 13 мкмоль триизобутилалюминия (13 мкл 1-молярного раствора в толуоле), разделяют примерно на
две равные части, которые помещают в две емкости высокого давления объемом 600 мл. В одну емкость добавляют 35 мл гексана-1. Во вторую емкость добавляют 0,2 мл толуольного раствора
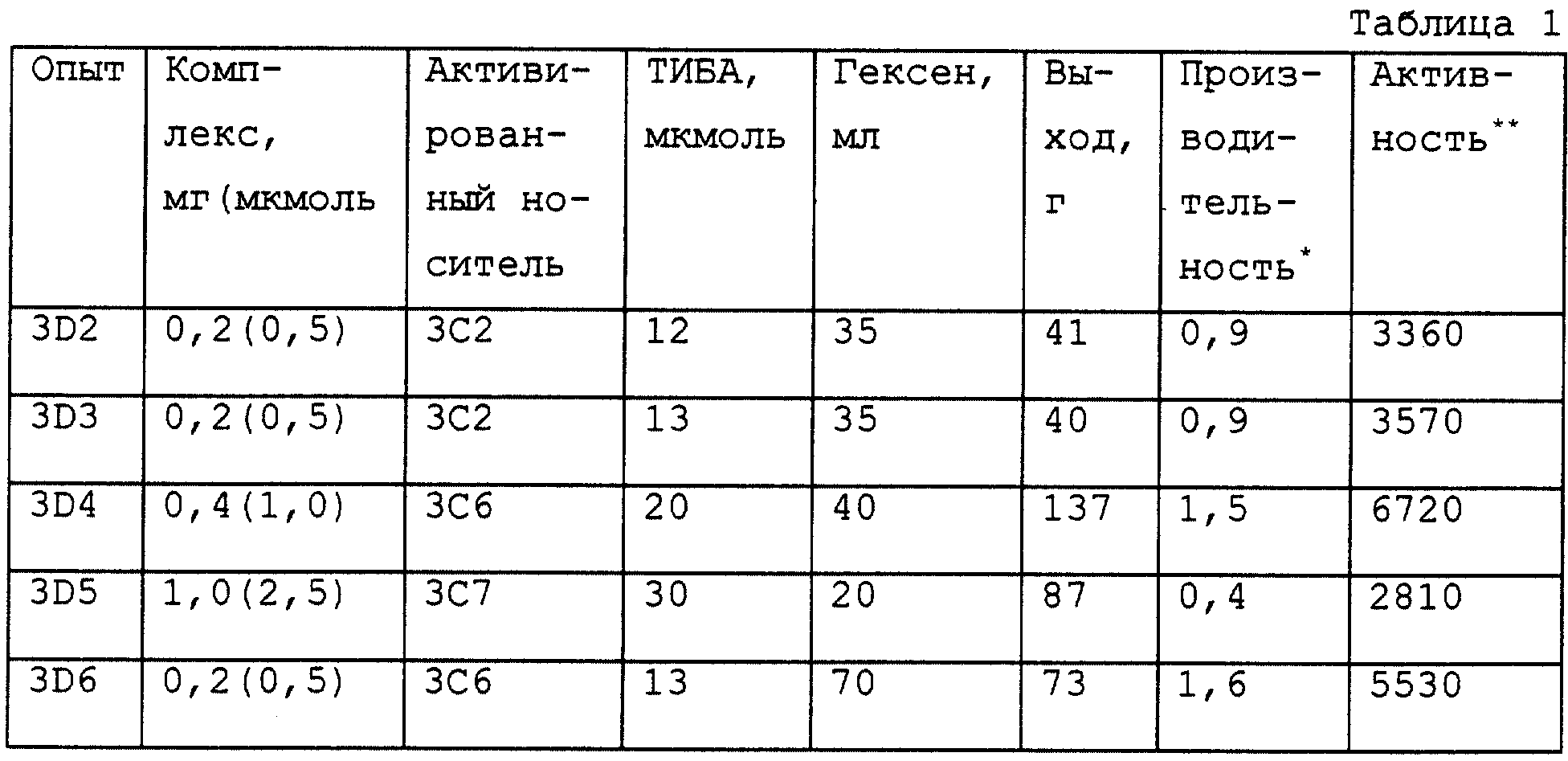
бис(н-бутилциклопентадиенил)цирконий дихлорида (H-BuCp)2ZrCl2 (0,2 мг, 0,494 мкмоль) и 12 мг активированного носителя 3C1 (2,4 мкмоль в расчете на бор). Раствор, содержащий
гексен, переносят в предварительно нагретый реактор и затем добавляют каталитическую смесь. Этилен быстро добавляют в реактор до давления 860 кПа. И реактор выдерживают при 70oC с помощью
водяной бани с циркулирующей водой. Этилен подают в случае необходимости через регулятор потока массы. В этих условиях реакцию проводят в течение 1 часа. Выход полиэтилена составляет 97 г, что
обеспечивает производительность 2,15 • 106 г полимера на 1 кг циркония в час; активность катализатора равна 7950 г полимера на 1 г катализатора в час.
D2-6.
Суспензионная аддитивная полимеризация в реакторе периодического действия
Условия реакции Примера 3D1 практически повторяют, используя различные количества
бис(н-бутилциклопентадиенил)цирконий дихлорида, триизобутилалюм-ния (ТИБА) и гексена, а также различные количества активированных носителей различных типов. Результаты приведены в таблице 1 (см. в
конце описания).
D7. Гомополимеризация этилена
a) Перед использованием реактор-автоклав емкостью 2 л откачивают при 70oC в течение 90 мин. Гептан (350 мл),
содержащий 25 мкмоль триизобутилалюминия (25 мкл 1-молярного раствора в толуоле), и 1,0 мл толуольного раствора бис(н-бутилциклопентадиенил)цирконий диметила Cp2ZrMe2 (0,9 мг, 3,
7 мкмоль) и 30 мг активированного носителя 3C6 (5,0 мкмоль в расчете на бор) переносят в предварительно нагретый реактор. Этилен быстро добавляют в реактор до давления 1200 кПа, и реактор выдерживают
при 80oC с помощью водяной бани с циркулирующей водой. Этилен подают в случае необходимости через регулятор потока массы. В этих условиях реакцию проводят в течение 1 часа. Выход полимера
составляет 160 г, молекулярный вес равен 155000, отношение Mw/Mn = 2,05. Температура плавления (Tпл) по данным дифференциальной сканирующей калориметрии (ДСК) равна 138oC.
b) Практически повторяют условия реакции Примера 3D3a) за исключением того, что количество триизобутилалюминия составляет 50 мкмоль, соединением переходного металла является бис(циклопентадиенил)цирконий дихлорид (2 мг, 6,8 мкмоль), активированным носителем является 3С6 (60 мг, 12 мкмоль в расчете на бор) и поддерживают температуру 75oC. Выход полимера составляет 139 г, молекулярный вес равен 144000, отношение Mw/Mn = 2,41. Tпл по данным ДСК равна 135oC.
c) Практически повторяют условия реакции Примера 3D23) за исключением того, что количество триизобутилалюминия составляет 100 мкмоль, соединением переходного металла является (трет-бутиламидо)диметил(тетраметил-η5 -циклопентадиенил)силанитандихлорид (10 мг, 27,2 мкмоль), активированным носителем является 3C1 (200 мг, 40 мкмоль в расчете на бор) и давление этилена равно 1400 кПа. Выход полимера составляет 94,2 г, молекулярный вес равен 961000, отношение Mw/Mn = 2,00, Tпл по данным ДСК равна 135oC.
d) Практически повторяют условия реакции Примера 3D3a) за исключением того, что количество триизобутилалюминия составляет 50 мкмоль, соединением переходного металла является бис(инденил)цирконий дихлорид (2 мг, 4,8 мкмоль) и активированным носителем является 3C1 (80 мг, 13 мкмоль в расчете на бор). Выход полимера составляет 140 г.
Пример 4
C. Приготовление функционализированного фенилсиланом силикагеля с нанесенным диметиланилиний
(6-гидрокси-2-нафтил)трис(пентафторфенил)боратом, [PhMe2NH]+[силикагель-OSiPhH-O-6-C10H6-2- B(C6F5)3]-
Смесь 1,67 г [PhMe2NH]+[(6-HOC10H6-2- B(C6F5)3]- (получен, как в Примере 1A3) и 5 г
функционализированного фенилсиланом силикагеля (получен, как в Примере 1B) осторожно кипятят с обратным холодильником при перемешивании механической мешалкой в течение 24 ч. Твердое вещество собирают
фильтрацией, промывают диэтиловым эфиром и пентаном и сушат при пониженном давлении.29Si CPMAS ЯМР: -43 ч/млн. Содержание бора в твердом веществе равно 0,154 мас. %.
D.
Суспензионная полимеризация в реакторе периодического действия
D1. Перед использованием реактор-автоклав емкостью 2 л откачивают при 80oC в течение ночи. Суспензию
функционализированного фенилсиланом силикагеля с нанесенным диметиланилиний(6-гидрокси-2-нафтил)трис(пентафторфенил)боратом [PhMe2NH]+[силикагель-OSiPhH-O-C10H6-2- B(C6F5)3]- (100 мг) в гептане (300 мл), 5 мг (трет-бутиламидо)диметил(тетраметил- η5 -циклопентадиенил)силантитандиметила и 0,
1 мл 25%-ного раствора триэтилалюминия в гептане переносят в предварительно нагретый реактор. Этилен быстро добавляют в реактор до давления 1200 кПа, и реактор выдерживают при 80oC с
помощью водяной бани с циркулирующей водой. Этилен подают в случае необходимости через регулятор потока массы. Выход полиэтилена через 60 минут составляет 50 г.
D2. Повторяют методику Примера 4D1) со следующими изменениями: [PhMe2NH]+[силикагель-OSiPhH-OC10H6-2- B(C6F5)3]- (50 мг), 0,05 мл раствора триэтилалюминия, 1,5 мг Cp2ZrMe2 и 20 кПа водорода. Выход полиэтилена через 60 минут составляет 144 г.
D3-22. Практически повторяют условия реакции, указанные выше в D2, используя комплексы переходного металла, активированные носители и другие условия реакции, указанные в таблице 2. Результаты приведены в таблице 2. Значения молекулярного веса определяют методом гель-проникающей хроматографии (ГПС).
Пример 5. Приготовление оксида алюминия, модифицированного диметилсиланом
Суспендируют в 25 мл пентана 5 г диоксида
алюминия ПьюрэлTM (поставляется фирмой Кондеа Хеми АГ, прокален при 600oC в вакууме) и добавляют 4,5 мл (5 ммоль/г) бис(диметилсилан)амина, (HMe2Si)NH (SiMe2
H). Смесь встряхивают в течение 15 ч, твердое вещество собирают на воронке с пористым стеклом, промывают пентаном и сушат при пониженном давлении.
ИКСДОФ: 2958, 2904 (C-H); 2102 см-1 (шир. Si-H).
Получение активированного носителя взаимодействием оксида алюминия, модифицированного диметилсиланом, с диметиланилиний
(4-гидроксифенил)трис(пентафторфенил)боратом [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiMe2-O-Al2O3
)]-
Суспендируют 1,0 г диоксида алюминия, модифицированного диметилсиланом, с диметиланилиний (4-гидроксифенил)трис(пента-фторфенил)боратом [PhMe2NH]+
[(C6F5)3B(C6H4- п-OH)] (0,35 г) в 10 мл эфира в течение 12 ч. Затем твердое вещество собирают фильтрацией, промывают эфиром и пентаном, сушат при
пониженном давлении.
ИКСДОФ: 2960, 2908 (C-H); 2131 (Si-H), 1641, 1623, 1591, 1514, 1461, 1261 (колебания ароматического кольца) см-1.
Суспензионная
полимеризация в реакторе периодического действия
Перед использованием автоклав емкостью 2 л откачивают при 80oC в течение ночи. Гептановую (300 мл) суспензию 200 мг диоксида
алюминия, модифицированного диметилсиланом, с нанесенным диметиланилиний (4-гидроксифенил)трис(пентафторфенил)боратом [PhMe2NH]+[(C6F5)3B(C6H4- п-O-SiMe2-O-Al2O3)]-, 10 мг бис(циклопентадиенил) цирконий диметила Cp2ZrMe2 и 0,2 мл триэтилалюминия (25%-ный
раствор в гептане), переносят в предварительно нагретый реактор. Этилен быстро добавляют в реактор до давления 1200 кПа, реактор выдерживают при 80oC с помощью водяной бани с циркулирующей
водой. Этилен подают в случае необходимости через регулятор потока массы. Выход полиэтилена спустя 60 минут составляет 2,16 г.
Пример 6
D1-5. Непрерывная суспензионная
полимеризация
Непрерывную суспензионную полимеризацию осуществляют, используя 10-литровый суспензионный реактор, управляемый компьютером и оборудованный внешней водяной рубашкой, мешалкой,
термопарой, капельницей для добавления катализатора, капельницей для добавления разбавителя и датчиком уровня с переменной емкостью. Устанавливают непрерывный поток очищенного разбавителя (изопентан)
на уровне 4000 г/ч, при этом содержание твердого вещества в реакторе приблизительно поддерживается равным 60 мас. %. Уровень в реакторе составляет приблизительно 60% от объема реактора за счет
периодического удаления содержимого реактора. Реактор нагревают до температуры 55oC. Затем в реактор начинают подавать потоки водорода 0,15 л/ч, этилена 650 г/ч и бутена-1 75 г/ч, при этом
давление поддерживают равным 1,5 МПа. Смешанный катализатор готовят сочетанием 81 мг (0,2 ммоль) бис(н-бутилциклопентадиенил)цирконий дихлорида (н-BuCp)2ZrCl2, 4,9 г
модифицированного фенилсиланом силикагеля с нанесенным диметиланилиний 4-(4'-гидроксифенил)фенил)трис(пентафторфенил)боратом, полученным в соответствии с методикой приготовления 3C6, и 2 ммоль
триизобутилалюминия (ТИБА) в 800 мл гексана в сухом боксе и помещают смесь в 1-литровый автоклав. Затем содержимое переносят в атмосфере азота в емкость с перемешиваемым катализатором и разбавляют
изопентаном до объема 8 л. Смешанный катализатор медленно добавляют в реактор из емкости с непрерывно перемешиваемым катализатором. Затем постепенно увеличивают потоки этилена и бутена-1. Из реактора
периодически отбирают пробы смеси, которые подают через выходящие клапаны в нагретую емкость для быстрого испарения разбавителя. Свойства полимера, не содержащего летучих примесей, измеряют и
записывают. Реактор непрерывно работает в течение 8 ч. Приведенные в таблице 3 данные были измерены на образцах, полученных после возвращения в состояние равновесия после изменения условий
процесса.
D6-14. Непрерывная суспензионная полимеризация
Получение бис(н-бутилциклопентадиенил)цирконий 1,4-дифенилбутадиена
В атмосфере азота суспендируют в гексане
2,02 г (4,99 ммоль) перекристаллизованного бис(н-бутилциклопентадиенил)цирконий дихлорида с 1,4-дифенилбутадиеном. Добавляют 2,05 мл 2,5-молярного раствора бутиллития (5,13 ммоль) в гексане, при этом
сразу же появляется красное окрашивание смеси. После перемешивания в течение 30 минут при комнатной температуре смесь кипятят 2 ч с обратным холодильником. Растворитель удаляют в вакууме, а твердое
вещество вновь растворяют примерно в 20 мл горячего гексана. Образуются красные кристаллы. Добавляют небольшое количество гексана, реакционную колбу охлаждают в холодильнике и затем фильтруют через
фильтрующее средство. Выделяют красные кристаллы путем фильтрации через среднюю фильтрующую воронку, промывают 1 раз холодным гексаном и сушат в вакууме. Выход составляет 2,04 г (76,5%).
Опыты полимеризации
Практически повторяют условия реакции Примера 6D1-5 за исключением того, что устанавливают непрерывный поток очищенного изопентана на уровне 2500 г/ч,
температура реактора 65oC и начальная подача потоков этилена и бутена-1 составляет 1000 и 50 г/ч соответственно. Исходный смешанный катализатор готовят сочетанием 40,5 мг (100 мкмоль)
бис(н-бутилциклопентадиенил)цирконий дихлорида, 2,45 г модифицированного фенилсиланом силикагеля с нанесенным диметиланилиний 4-(4'-гидроксифенил)фенил)трис(пентафторфенил)боратом, полученным в
соответствии с методикой приготовления 3C6, и 1 ммоль ТИБА в 400 мл гексана в сухом боксе, и помещают смесь в 1-литровый автоклав. Затем содержимое переносят в атмосфере азота в емкость с
перемешиваемым катализатором и разбавляют изопентаном до объема 8 л. Смешанный катализатор медленно добавляют в реактор из емкости с непрерывно перемешиваемым катализатором. Затем постепенно
увеличивают потоки этилена и бутена-1. Вторую емкость с перемешиваемым катализатором готовят таким же образом, как описано выше, и обе емкости с катализатором переключают на полимеризацию через каждые
3-5 ч в течение 34-часовой полимеризации. В ходе полимеризации предшественник металлоценового катализатора заменяют на бис(н-бутилциклопентадиенил)цирконий(II) 1,4-дифенилбутадиен (н-BuCp)2
Zr(PhCH= CHCH= CHPh). Результаты приведены в таблице 4.
Пример 7
B. Приготовление силикагеля, модифицированного фенилсиланом и обработанного триизобутилалюминием
B1.
Модифицированный фенилсиланом силикагель получают практически по методике Примера 1B, за исключением того, что в качестве силикагеля используют СилополTM 2212, который прокаливают при
температуре 400oC. Суспензию 10 г этого силикагеля, модифицированного фенилсиланом, в 200 мл гептана обрабатывают 20 мл 1-молярного раствора триизобутилалюминия в толуоле при комнатной
температуре. Полученную смесь перемешивают встряхиванием в течение 12 ч, затем фильтруют, промывают 3 раза пентаном (по 50 мл) и сушат в вакууме. Выход составляет 11,3 г.
ИКСДОФ: v(Si-H) 2170 см-1 (средн. );29Si CPMAS: б -24 ч/млн.
B2. Практически повторяют указанные выше условия за исключением того, что силикагель СилополTM 2212 прокаливают при 800oC.
B3. Практически повторяют указанные выше условия за исключением того, что силикагель СилополTM 2212 прокаливают при 500o C.
B4. Практически повторяют указанные выше условия за исключением того, что силикагель СилополTM 2212 прокаливают при 300oC.
C. Приготовление силикагеля, модифицированного фенилсиланом и обработанного триизобутилалюминием, с нанесенным N, N-диметиланилиний (4-(4'-гидроксифенил)фенил)трис(пентафторфенил)боратом [PhMe2NH]+[(C6F5)3B(C6H4)- C6H4-п-O-SiPhH-OSi)]-.
C1. Суспензию силикагеля, модифицированного фенилсиланом, в 200 мл диэтилового эфира обрабатывают [PhMe2NH] [(C6F5)3B(C6H4)- C6H4-п-OH)] (3,01 г, 3,75 ммоль) при комнатной температуре в атмосфере аргона. Этот раствор перемешивают в сухом боксе в течение 2 суток, полученное твердое белое вещество фильтруют, промывают 3 раза (по 50 мл) эфиром и 3 раза (по 10 мл) пентаном и сушат в вакууме. Выход продукта 11,25 г. Содержание бора ICP: 0,244 мас. %.
C2. Время приготовления C1 существенно сокращают (до 2-3 часов), используя толуол вместо диэтилового эфира.
C3. Практически повторяют методику получения C1, используя модифицированный фенилсиланом силикагель 7B4, обработанный триизобутилалюминием.
D. Гомополимеризация этилена
D1. Перед использованием реактор-автоклав емкостью 2 л откачивают при 75oC в течение 90 мин. Нанесенный связанный катализатор готовят сочетанием 0,1 мл
1-молярного раствора триизобутилалюминия (100 ммоль) в толуоле, 500 мл гептана и 0,1 мг бис(н-бутилциклопентадиенил)цирконий дихлорида (н-BuCp)2ZrCl2 (0,247 ммоль) с последующим
добавлением 7 мг силикагеля 7C1 (1,58 ммоль), модифицированного фенилсиланом, с нанесенным диметиланилиний (4-(4'-гидроксифенил)фенил)трис(пентафторфенил)боратом. Смешанный катализатор по вакуумной
линии переносят в предварительно нагретый реактор. Этилен быстро добавляют в реактор до давления 1200 кПа и реактор выдерживают при 75oC с помощью водяной бани с циркулирующей водой. Этилен
подают в случае необходимости через регулятор потока массы. Выход полимера за 1 час составляет 180 г, молекулярный вес равен 169000, отношение Mw/Mn = 2,11, производительность равна 8,0 •
106 г полиэтилена на 1 г циркония в час.
D2. Практически повторяют условия реакции Примера 7D1 за исключением того, что связанный боратный носитель представляет собой силикагель, модифицированный фенилсиланом, с нанесенным диметиланилиний (4-(4'-гидроксифенил)фенил)трис(пентафторфенил)боратом (силикагель СилополTM 2212, прокален при температуре 500oC, 7B3). Количества используемых реагентов: (н-BuCp)2ZrCl2 - 0,2 мг; 0,494 ммоль, триизобутилалюминий - 0,1 мл 1-молярного раствора (100 ммоль) в толуоле, 500 мл гептана и 12 мг (1,97 ммоль) связанного бората. Выход полимера за 1 ч составляет 204 г, молекулярный вес равен 184000, отношение Mw/Mn = 2,38. Производительность 4,5 • 106 г полиэтилена на 1 г Zr в час.
D3. Практически повторяют условия реакции Примера 7D1 за исключением того, что связанный боратный носитель представляет собой силикагель, модифицированный фенилсиланом, с нанесенным диметиланилиний (4-гидроксифенил)трис(пентафторфенил)боратом (силикагель СилополTM 2212, прокален при температуре 800oC, 7B2). Количества используемых реагентов: (н-BuCp)2ZrCl2 - 0,2 мг; 0,494 ммоль, триизобутилалюминий - 0,1 мл 1-молярного раствора (100 ммоль) в толуоле, 500 мл гептана и 13,5 мг (2,16 ммоль) связанного бората. Выход полимера за 1 ч составляет 208 г, молекулярный вес равен 156000, отношение Mw/Mn = 2,09. Производительность 4,6 • 106 г полиэтилена на 1 г Zr в час.
D4. Перед использованием реактор-автоклав емкостью 2 л откачивают при 80oC в течение 90 мин, добавляют 0,35 мл 1-молярного раствора триизобутилалюминия (350 ммоль) в толуоле к 500 мл гептана и раствор обрабатывают (трет-бутиламидо)диметил(тетраметил- η5 -циклопентадиенил)силантитандихлоридом (0,5 мг, 1,36 ммоль) с последующим добавлением 40 мг связанного бората 7C1 (8,5 ммоль). Смешанный катализатор по вакуумной линии переносят в предварительно нагретый реактор. Этилен быстро добавляют в реактор до давления 1400 кПа и реактор выдерживают при 80oC с помощью водяной бани с циркулирующей водой. Этилен подают в случае необходимости через регулятор потока массы. Выход полимера за 1 час составляет 67,7 г, молекулярный вес равен 3220000, отношение Mw/Mn = 2,96, производительность равна 1,0•106 г полиэтилена на 1 г титана в час.
D5. Практически повторяют условия реакции Примера 7D4, используя обработанный металлоценом - этиленбис(инденил)цирконий дихлоридом - и триизобутилалюминием, модифицированный фенилсиланом силикагель с нанесенным боратом 7C3. Количества используемых реагентов: (EBI)2ZrCl2 - 0,13 мг; 0,319 ммоль, триизобутилалюминий - 0,1 мл 1-молярного раствора (100 ммоль) в толуоле и 9,8 мг (1,54 ммоль) связанного бората, нанесенного на силикагель, модифицированный фенилсиланом и обработанный ТИБА. Выход полимера за 1 ч составляет 170 г, молекулярный вес равен 130000, отношение Mw/Mn = 2,63. Производительность 5,9 • 106 г полиэтилена на 1 г Zr в час.
Реферат
Изобретение относится к носителям и полученным на их основе катализаторам на носителе, которые используются при полимеризации олефинов. Описывается носитель, содержащий продукт взаимодействия (А) твердого неорганического оксида, содержащего матрицу диоксида кремния или оксида алюминия и производные гидроксильных групп на поверхности матрицы, функционализированные реакционноспособным силаном, причем указанный реакционноспособный силан соответствует формуле -OSiR2Н, в которой R независимо в каждом случае является водородом или С1-20 углеводородом, причем неорганический оксид содержит менее 1,0 ммол/г гидроксильных групп на поверхности, и (В) активатора, являющегося аммонийной солью трис- (пентафторфенил)-4-гидроксифенилбората, трис(пентафторфенил)-4-(4'-гидроксифенил)фенилбората или трис(пентафторфенил)-(6-гидрокси-2-нафтил) бората. Описываются также катализатор для полимеризации олефинов, содержащий вышеуказанный носитель, и (С) металл IV группы, содержащий, по крайней мере, π-связанную циклопентадиенильную или замещенную циклопентадиенильную анионную лигандную группу и заместитель, способный взаимодействовать с активатором с образованием каталитически активного комплекса переходного металла, и способ полимеризации олефинов с его использованием. Технический эффект - высокая эффективность катализатора, стойкость к выщелачиванию, исключение образования отложений полимера на стенках реактора. 3 с. и 5 з. п. ф-лы, 4 табл.
Формула
-OSiR2Н,
в которой R независимо в каждом случае является водородом или С1-20 углеводородом,
причем неорганический оксид содержит менее 1,0 ммол/г гидроксильных групп на поверхности, и активатор является аммонийной солью трис-(пентафторфенил)-4-гидроксифенилбората, трис(пентафторфенил)-4-(4'-гидроксифенил)фенилбората или трис(пентафторфенил)- (6-гидрокси-2-нафтил)бората.
АlR3,
где R является С1-20 углеводородом.
Документы, цитированные в отчёте о поиске
Способ получения катализатора полимеризации альфа-олефинов, состав для получения катализатора полимеризации альфа-олефинов и способ получения полиолефинов
Комментарии