Синтетические мембранные везикулы, содержащие функционально активные слитые пептиды как системы для введения лекарственных средств, способ из получения, применение для изготовления лекарственных средств против спида - RU2125868C1
Код документа: RU2125868C1
Чертежи

Описание
Изобретение относится к синтетическим мембранным везикулам (липосомам), в частности к везикулам, на поверхности которых имеются молекулы слитых пептидов и другие специфические для клеток белки. Молекулы слитых пептидов могут присутствовать в форме синтетического или очищенного пептида либо как часть гликопротеиновых выступов на поверхности оболочки вируса, например, гемагглютининов. У числу таких вирусов относятся вирус гриппа, парагриппа или вирус комариной лихорадки Семлики. Специфические клеточные белки могут быть представлены антителами класса IgG, например антителами СD4.
При приеме больным того или иного лекарственного средства оно сначала должно поступить из места введения в кровь, поэтому способ введения препарата может иметь существенное влияние на его фармакокинетический профиль в кровяном русле. Так, в случае выбора перорального способа введения, действие лекарственного средства развивается медленно, а его оптимальная концентрация в крови достигается не всегда. Напротив, при внутривенном введении концентрация препарата в крови носит предсказуемый характер и достаточно быстро достигает требуемой величины, хотя такой способ связан с известными неудобствами для больного и болезненными ощущениями. Но даже при непосредственном введении медикаментов в системный кровоток, взаимосвязь между введенной дозой, концентрацией препарата в крови и продолжительностью его действия на орган-мишень носит весьма сложный характер. Значения этих параметров определяются сложной системой путем обмена, нередко имеющих конкурентный характер, которые в конечном итоге приводят либо к накоплению активных молекул в ткани-мишени, либо к инактивации и экскреции лекарственного средства. К числу таких путей относится биотрансформация в печени и других органах, выведение через почки или с желчью, связывание препарата с фиксированными или циркулирующими клетками и макромолекулами, пассивное или с помощью других соединений проникновение через мембранные барьеры Creasy W.A, 1979, Drug Disposition in Human, Oxford University Press, New Iork).
В
последние годы заметен большой интерес к перспективам регуляции распределения и обмена лекарственных средства в организме по наиболее благоприятному для больного сценарию посредством применения
различного рода носителей или систем введения медикаментозных препаратов. Такие системы предназначены для контроля одного или нескольких следующих параметров (Juliano R.R., 1975, Can. J. Physiol
Pharmacol., 56, 683-690):
а) скорости включения препарата в определенную ткань организма,
б) распределения и локализации лекарственного средства в организме больного,
в)
устойчивости и скорости превращения препарата.
Важным вкладом в разработку систем направленного введения лекарственных средств явилось конструирование липосом, впервые описанное Bangham
A.D., Standish M. M, Watrins J.C., (1965, J. Mol. Biol. v. 13, 238 - 252). К настоящему времени в литературе имеются многочисленные описания достоинств методики введения лекарственных средств в
составе липосом. Эти способы можно с известной долей условности подразделить на следующие категории:
1. Липосомы могут проникать через биологические мембраны и способствовать транспорту
медицинских препаратов через обычно непроницаемые для последних барьеры. В частности, заключенные в липосомы соединения могут доставляться внутрь клеток.
2. Липосомы могут быть предназначены для взаимодействия с определенными тканями, с целью повышения избирательности действия лекарственных препаратов и снижения их токсического эффекта.
3. Использование липосом позволяет желаемым образом изменять фармакокинетику лекарственных средств, влияя на освобождение, распределение и выведение последних в системном кровотоке.
4. Липосомы могут применяться для предотвращения инактивации химически или метаболически лабильных соединений.
Таким образом можно отбирать соединения, перспективные с точки зрения терапевтического применения, поскольку инкапсулированные препараты обладают, по крайней мере при испытаниях in vitro, измененными, по сравнению с исходными, свойствами. К сожалению, первые надежды на революционизирующие последствия нового подхода к химиотерапии полностью не оправдались, как об этом свидетельствуют результаты экспериментальных исследований (Fildes F.J.T., 1981, Liposomes, From Physical structure to therapeutic applications, Knoght (ed.), Elscvier /North-Holland Biomedical Press/.
Более обнадеживающие результаты при использовании липосом в качестве переносчиков лекарственных средств могут быть получены при нагрузке липосом специфическими белками. Для того чтобы обеспечить более надежную доставку заключенных в липосомы препаратов к нужным органам или тканям, методика нагрузки должна быть направлена на предотвращение задержки липосом в нежелательных местах (таким образом снижается токсичность) и оптимизацию переноса лекарственного средства в определенные типы клеток (для усиления требуемого эффекта).
Некоторые исследователи покрывали липосомы оболочкой из агрегированных иммуноглобулинов для оптимизации их доставки на фагоциты (Jsmail G., Boxer L. A. , Bachner R.L, 1979, Pediatr. Res. v. 13, 769 - 773, Finkelstein M.S., Kuhn S. H. , Schieren H., Weissmann G. and Haffstein S. 1980, Liposomes and Immunobiology (Tam B.H., and Six H.R. eds. 244 - 270, Elsevier /North - Holladn Publishing Co., Amsterdam/.
В дальнейшем методика была усовершенствована Gregoriadis и Neerunjum (1975, Biochem. Biophys. Res. Commun., 65, 537 - 544), которые предложили более эффективный способ нагрузки липосом специфическими для определенного типа клеток антителами (JgG - иммуноглобулинами). Включение липосом, связанных с иммуноглобулинами к определенным клеточным линиям, возрастало при этом в 3-25 раз. Тем не менее, при практическом применении этого метода выяснилось, что нагруженные таким образом липосомы не сливались с клеточными мембранами, что препятствовало эффективной доставке к клеткам заключенных в липосомах лекарственных препаратов (Leserman L., Weinstein J.N., Blumenthal, R., Sharrow S.O. and Texy W.D, (1979, J. Immunol., 122, 585 - 591.
Важным шагом на пути улучшения способов получения систем введения лекарственных средств явилось связывание липосом с вирусными белками. Мембраны липосом, согласно этому способу, подвергают модификации такими белками, как гемагглютинин вируса гриппа (Almeida J.D., Brand C.M., Edwards D.C., Heath T. D., 1975, Lancet, 2, 899 - 901).Эффективность и специфичность взаимодействия вируса с клетками-хозяевами на его ранних стадиях (адсорбция, проникновение в клетки) поддаются регуляции посредством включения соответствующих вирусных белков в мембраны липосомных носителей. Например, включение поверхностных белков вируса Сендай в двуслойную липосому по меньшей мере в 100 раз усиливает включение фрагмента А токсина дифтерии мышиными 1 - клетками по сравнению с наблюдающимся при использовании липосом, содержащих фрагмент А в отсутствие вирусных белков (Uehida T., Kim J., Jamaizumi M., Miyahe Y. and Okada J., 1979, Cell, Bioll., 80, 10 - 20).
Основным недостатком описанной методики являются отсутствие полноценно активных слитых пептидов гемагглютинина вируса гриппа. Вирус гриппа А проникает в клетки
- хозяева в процессе слияния с мембраной. После связывания на поверхности клетки вирусные частицы проникают в нее и переносятся на эндосомы и лизосомы. Кислая среда этих органелл активирует процесс
слияния мембранных компонентов вируса и клетки-хозяина. Преимущество слияния липосомы с клеткой при низких величинах pH состоит в том, что содержимое везикул освобождается после их проникновения в
клеточную цитоплазму (pH среды эндосом приближается к 5,2). Открытие слияния гликопротеинов вируса гриппа при низких значениях pH породило множество попыток получить системы доставки лекарственных
средств с применением липосом, ассоциированных с гемагглютинином вируса гриппа. Kawasaki K. с соавторами (Biochemica et Biophysica Acta, 1983, 733, 286 - 290) использовали везикулы с гликопротеиновыми
геммагглютининами вируса гриппа в системе: желточный фосфатидилхолин /фосфатидилхолин со спиновой меткой/ холестерин в молярном соотношении 1,6: 0,4: 1. При соотношении белка и липидов в препарате
1:44 или 1:105 моль/моль он содержал везикулы диаметром 100 - 300 нм высокой плотности поверхностных выступов. Основные недостатки такого препарата сводились к следующему:
1. Эндоцитоз
фагоцитарными клетками угнетен вследствие высокого содержания холестерина и присутствия остаточных количеств детергента. Активность препарата даже по данным Kawasaki с соавторами не превышала 66%, а
при оценке новейшими, более совершенными методами составляла спустя 7 минут только 1% (!) (см. ниже пример 10). Это может объясняться тем, что использовавшийся авторами тритон X - 100 частично
реагирует с гемагглютинином и полностью не удаляется в процессе диализа.
2. Для изучения роли мембранных компонентов вируса в реакции слияния следует иметь способы манипулирования этими компонентами. Для этой цели необходимо уметь выделять и формировать белки выступов вирусной оболочки, получая таким образом виросомы в полной мере способные вступать в реакцию биологического слияния. Тем не менее, несмотря на все усилия Kawasaki и его соавторов, им не удалось получить вирусы гриппа с оболочками, проявляющими такую биологическую активность.
3. Не удалось доказать связывание нагруженных вирусными белками липосом, несущих фармацевтически активные препараты, со специфическими типами клеток.
Большинство других применявшихся методов получения вирусных оболочек с требуемыми свойствами основаны на солюбилизации вирусных мембран детергентами с последующим удалением детергента из надосадочной жидкости после осаждения внутриклеточных вирусных белков и генетического материала. Детергенты с высокой критической концентрацией мицелл, в частности октилглюкозид, можно достаточно полно удалить с помощью диализа. Методика с использованием октилглюкозида описана при работе с вирусом комариной лихорадки Семлики (ВКЛ) (Helenius A., Sarvas H., and Simons K., 1981, Eur J. Biochem., 116, 27 - 35), вирусом везикулярного стоматита (ВВС) (Eidelman O., Schlegel, R., Tralka T. S. and Blumenthal R. 1984, J. Biol. Chem., 259, 4622 - 4628), вирусом гриппа (Huand R. T.C., Wahu K., Klent H.D. and Rou, R. 1980, Virology 104, 294 - 302) и вирусом Сендай (Harmsen M.D., Wilschut J., Scherphof G., Hulstaert and Hoerstra D., 1985, Sur. J. Biochem. 149, 591 - 599). Однако ни в одном из этих исследований не были получены вирусные оболочки с желаемыми свойствами. Так, виросомы, полученные из ВКЛ, содержали белок и липиды в соотношении, которое отличалось от их соотношения в природной вирусной оболочке, а виросомы из ВВС не обладали способностью к биологическому слиянию.
Еще один описанный в литературе метод состоит в отделении поверхностных выростов от оболочки вируса гриппа бромеленом. Получаемый таким способом гемагглютинин связывался с липосомами (Doms R, Helenius A., and White, 1985, J. Biol. Chem 260, (5), 2973 - 2981). Основным недостатком этого метода является ограниченная биологическая активность таких везикул вследствие расщепления бромеленом.
Таким образом, методический аспект настоящего изобретения состоит в необходимости получения везикул, в которых требуемые лекарственные средства или фармацевтически активные соединения транспортируются на специфические клетки, такие как макрофаги, клетки - хелперы (Т4), клетки мозга и другие клетки того или иного органа, с которыми везикулы прочно связываются, и проникают внутрь этих клеток в процессе фагоцитоза или эндоцитоза, в результате чего требуемое средство поступает сразу после эндоцитоза в цитоплазму нужных клеток.
Решение этой методической проблемы достигается получением везикул вирусного гемагглютинина, характеристика которых приведена в пункте 1 формулы изобретения. Частные пути реализации предлагаемого изобретения и способы улучшения свойств везикул описаны в подпунктах 2 - 9. Способ получения таких везикул описан в пунктах 13 - 18, а способы их применения - в пунктах 9 и 10 формулы изобретения.
В соответствии с этим изобретение позволяет получать виросомы вирусного гемагглютинина, которые содержат идеально подходящие для эндоцитоза липосомы и специфические клеточные маркеры, обладающие полной биологической активностью, предпочтительно гемагглютининовые вирусные гликопротеины или их производные, а также синтетические слитые пептиды, способные индуцировать быстрое слияние указанных везикул с определенным типом клеток сразу после завершения эндоцитоза.
Другой способ реализации изобретения состоит в использовании подходящих агентов, обеспечивающих перекрестную сшивку, которые адсорбируются на специфических липосомах, в смеси со специфическими антителами к рецепторам, для запуска механизма эндоцитоза в клетках данного типа; антитела взаимодействуют со сшивающими агентами таким образом, что полностью сохраняют биологическую активность.
Существенным признаком наиболее предпочтительных везикул является наличие на их поверхности указанных вирусных гликопротеинов или их производных, в полной мере сохраняющих активность, и биологически активных специфических антител, которые обладают способностью прикрепляться к нужным клеткам, подвергаются интернализации в процессе фагоцитоза или эндоцитоза и индицируют быстрое слияние указанных везикул с внутренними цитоплазматическими мембранами и освобождение содержимого виросом в цитоплазму данных клеток. Благодаря использованию активных слитых пептидов согласно настоящему изобретению, освобождение лекарственных средств происходит сразу после фагоцитоза, что позволяет избежать нежелательного длительного пребывания их в эндоцитосомах, которое может приводить к неспецифической деградации фармацевтических продуктов, присутствующих в вирусных гемагглютининовых везикулах согласно настоящему изобретению. При pH 5 слитые пептиды вируса гриппа на поверхности везикул активируются посредством такого же механизма, как в случае живого вируса. Содержимое везикул освобождается в цитоплазму, как то имеет место в случае вируса гриппа от выделяемого нуклеопротеина.
Термином "липосома" обозначаются двуслойные фосфолипиды среднего размера, которые получают в процессе контролируемого удаления детергентов. Первоначальный размер везикул, образующихся сразу после удаления детергента зависит от природы как детергента, так и фосфолипида, а в отдельных случаях - от способа и скорости удаления детергента.
Настоящее изобретение относится также к способу получения везикул, в наибольшей степени пригодных для
фагоцитоза. Способ включает следующие стадии:
1. Разведение одного или двух фосфолипидов в неионном детергенте.
2. Формирование везикул в процессе удаления детергента с использованием микроносителей в форме полистироловых гранул (размер во влажном состоянии 20 - 50 меш).
3. Механическое перемешивание во время удаления детергента.
4. Желательный размер везикул (50 - 100 нм) достигается ультразвуковой обработкой.
Другой способ реализации изобретения сводится к получению везикул, в которых 70 - 95% фосфолипидов (по весу) приходится на долю фосфатидилхолина, а 5 - 30% представлено иным фосфолипидом, например фосфатидилэтаноламином. Содержание холестерина предпочтительно не превышает 1%.
Термин "слитый пептид" относится к гликопротеиновым выростам на поверхности вируса, содержащим слитый пептид. В одном из вариантов настоящее изобретение относится к полному тримеру гемагглютинина из выростов на поверхности вируса или к одному мономеру или к одному или обоим расщепленным субъединицам (гликопротеидам НА1 и НА2), которые содержат функционально активный слитый пептид. В другом варианте изобретение относится к самому слитому пептиду, выделенному из природного материала или полученному синтетическим путем. Предпочтителен такой способ реализации изобретения, при котором эти полипептиды, содержащие слитый пептид, являются гемагглютининами вируса гриппа, особенно гемагглютинином подтипа A-H1N1.
Понятие "сшивающий агент" применяется для обозначения органической гетерофункциональной молекулы, которая связывается с поверхностью везикул, полученных согласно настоящему изобретению, и присоединяет полипептиды. Предпочтительно, чтобы такая молекула представляла собой органическую бифункциональную молекулу, содержащую карбоксильную группу и тиоловую группу, в частности сульфосукцинимидил-(S)- производное, например S-4-(р-малеимидо-фенил)-бутират, S-ацетат, S-2-(m-азидо-о- нитробензамидо) этил-1,3'-дитиопропионат, S-6-(4'-азидо-2'-нитрофениламино)гексаноат. S-(4-азидофенилдитио) пропионат, S-2-(m-азидосалициламидо) этил-1,3-дитиопропионат, S-2-(биотинамидо)-этил-1,3-дитиопропионат, S-6-(биотинамидо) гексаноат, S-3-(4-гидроксифенил) пропионат, S-(4-йодоацетил) аминобензоат, S-4-(II-малеимидометил) циклогексан-1-карбоксилат, S-2,2, 5, 5-тетраметилпирролин-1-оксил-НСl.
Термин "специфичный для клетки" белок или маркер используется для обозначения белка, способного присоединиться к сшивающему агенту на поверхности везикула и связываться с клеточными рецепторами, что приводит к индукции механизма эндоцитоза. В предпочтительном способе реализации изобретения это понятие относится к специфичным для клеточных рецепторов антителам, особенно к моноклональным антителам.
Пример 1.
Получение синтетических мембранных везикул фосфатидилхолина с полностью функционально активными слитыми пептидами в тримере гемагглютинина из вируса гриппа, содержащих в качестве антивирусного препарата декстран-сульфат.
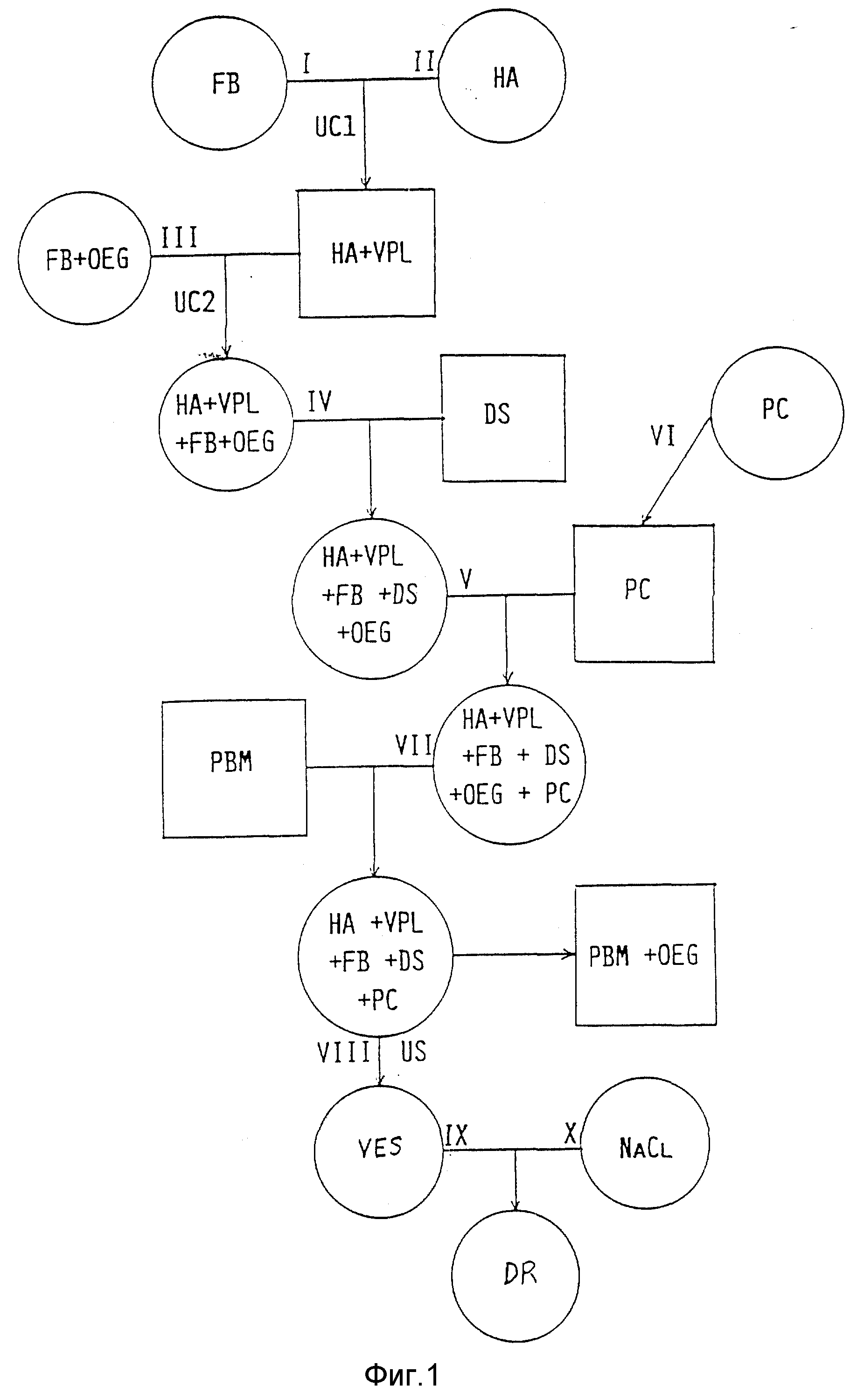
Фиг. 1 иллюстрирует принцип предлагаемой процедуры. Круги обозначают жидкий раствор или суспензию, квадраты - твердый осадок или преципитат.
В общем виде процедура состоит в следующем.
Слитой буферный раствор (FB), содержащий 2700 мг твердого материала и суспензию гемагглютинина (HA), содержащую 46 мг агглютинина и 31,1 мкмоль вирусного фосфолипида (YPL), перемешивают и центрифугируют (UCI). Образующий осадок разводят в растворе III, содержащем слитой буфер (FB) и монододециловый эфир октаэтиленгликоля (OEG), служащего слабым детергентом. После повторного ценрифугирования (UC2) получают надосадочную жидкость (IV), содержащую FB, HA, YPL и OEG и добавляют к ней противовирусный препарат декстран-сульфат (DS). Конечный раствор обозначается символом (V). Раствор VI фосфатидилхолина (PC) упаривают под вакуумом во вращающейся колбе с округлым дном для получения тонкого слоя РС на ее внутренней стенке. К этому слою добавляют раствор V и разводят его в растворе VII, в который трижды последовательно помещают микроноситель в форме полистироловых бусин (PBM) для удаления OEG. В результате получают раствор VIII, в котором с помощью ультразвуковой обработки (US) формируют везикулы (VES) требуемого размера. Конечную суспензию IX разбавляют раствором NaCl(X), получая суспензию лекарственного средства (DR).
Подробное описание.
Слитой буферный раствор (I) содержит 7,9 мг NaCl/мл, 4,4 мг/мл дигидрата тринатрата - цитрата, 2,1 мг/мл 2-морфолиноэтанового моногидрата сульфоновой кислоты (MES) и 1,2 мг/мл N - оксиэтилпиперазин-N'-2-этан-сульфоновой кислоты в H2O, доводя pH до 7,3 добавлением 1 h.NaOH.
Готовят физиологический солевой раствор /0,9 % NaCl/, содержащий живой вирус гриппа с титром 109 яичных инфекционных доз 50 /EID50/ на 1 мл, 0,1 мл/яйцо этой суспензии инокулируют в аллантоиновую полость куриного яйца. После 48 часов инкубационного периода при 35oC жидкость из аллантоиновой полости отдувают и собирают. Эту жидкость дважды пропускают через градиент сахарозы при ультрацентрифугировании, как описано Skehel and Schild/ 1971, Virdogy, 44, 396 - 408). Вкратце: Три различных раствора сахарозы /10%, 30% и 50% сахарозы/ и 0,9% раствор NaCl /500 мл каждого раствора/ подают в непрерывный поток ротора KII ультрацентрифуги /Electronucleo nics, Heidelberg, Германия/: Суспензию вируса гриппа пропускают через ротор при скорости потока 5 л/час и ультрацентрифугируют при 100 000 g. Загруженный градиент затем собирают и вирусную фракцию выделяют из фракции сахарозы с плотностью 32-37% сахарозы. Эту концентрированную фракцию вируса (500 мл) разбавляют физиологическим солевым раствором /0,9% NaCl/ до объема 50 л и снова пропускают через градиент плотности сахарозы, как описано выше. Очищенная суспензия вируса гриппа II содержит 266 мкг гемагглютинина вируса гриппа в мл и суммарно 31,1 мкмоль вирусных фосфолипидов, которые были определены следующим образом. Вирусные фосфолипиды экстрагируют по методу Folch с соавторами (Folch L., Lees M., and Sloane S.G.H. (1957); J.Biol. Chem., 266, 497-509). Для анализа фосфолипидов нижнюю органическую фазу упаривают, а остаток либо используют для определения фосфата (Chen P.S., Toribara T.J. and Warner H., 1956, Anal. Chem. 2, 1756 - 1758), либо разводят в небольшом объеме смеси хлороформа и метанола для последующего анализа с помощью тонкослойной хроматографии на фосфолипиды. Используют пластинки из силикагеля (Merck), которые проявляют в системе хлороформ-метанол-уксусная кислота-вода (25:15:4: 2, объемное соотношение). Индивидуальные фосфолипиды выявляют обработкой йодом и идентифицируют на основании значений их RF в сопоставлении с контрольными образцами. Белок определяют с помощью модифицированной методики Лоури (Peterson G.L. 1971, Anal.Biochem 83, 346 - 356).
Раствор (II) в количестве 173 мл смешивают с равным объемом указанного объединенного буфера (I). Этот раствор, содержащую вирус группа, подвергают центрифугированию при 100000 x g в течение 10 мин до получения осадка.
15,3 мл раствора детергента, содержащего 54 мг/мл (= 100 мкмоль/мл) монододецилового эфира октаэтиленгликоля (OEG Sigma) в описанном слитом буферном растворе добавляют к осадку, содержащему вирус гриппа, позволяя выделить слитые пептиды группа из оболочки вируса гриппа. Это соответствует 18 мкг = 33,3 нмоля OEG на 1 мкг гемагглютинина. Полное растворение осадка происходит через 10 мин. Полученный раствор в течение часа подвергают ультрацентрифугированию при 100000 g. Надосадочный слой IV содержит солюбилизированный гемагглютиан и фосфолипиды оболочки вируса гриппа (Luscher M., nd Glück Antiviral Res., 14, 39 - 50) добавляют к оставшейся надосадочной жидкости (IV), которая содержит растворенный тример гемагглютинина вируса гриппа, в комбинации с остатком заменимой нейраминидазы и другими компонентами, получая таким образом раствор V. Эта процедура позволяет спонтанного ввести декстрансульфат в везикулы.
В смеси хлороформа и метанола (2:1) разводят фосфатидилхолин (Sigma) до конечной концентрации 10 мг/мл, 28 мл этого раствора (VI) тщательно упаривают под вакуумом в роторном испарителе, формируя на внутренней поверхности круглодонной колбы тонкий слой фосфолипидов, содержащий их в количестве 280 мг.
К фосфолипидному слою в круглоднной колбе добавляют раствор V. После встряхивания колбы на протяжении по меньшей мере 15 минут и полного растворения фосфатидилхолина, переносят образующий раствор VII в стеклянный сосуд вместе с 4,8 г микроносителя в форме полистироловых гранул (РВМ) размером 25 - 50 меш (во влажном состоянии).
Сосуд встряхивают таким образом, чтобы его содержимое перемещалось из одного конца в другой со скоростью два раза в секунду. Спустя час суспензию отсасывают через тонкую пипетку и переносят в другой сосуд, содержащий 2,4 г полистироловых гранул такого же размера, которые служат микроносителями. Колонку встряхивают на протяжении 30 минут. Процедуру повторяют дважды. По завершении ее образовавшийся раствор (VIII) снимают с гранул, помещают на водяную баню и подвергают ультразвуковой обработке с помощью аппарата Bransonie (Branson Europe BV) при частоте 50 кГц ± 10%. Продолжительность каждой обработки 10 с, ее повторяют дважды с интервалом 10 с. В результате получают среднего размера везикулы диаметром приблизительно 50 - 100 нм. Конечный раствор (IX) разбавляют физиологическим раствором NaCl (X) в соотношении 1:100.
Пример 2.
Получение синтетических мембранных везикул, содержащий противовирусные антитела, адсорбированные на тримерах гемагглютинина вируса гриппа, и моноклональные антитела СД 4.
Везикулы получают, как описано в примере 1, со следующими модификациями: вместо 10 мг фосфатидилхолина используют только 9 мг, в сочетании с 1 мг фосфатидилэтаноламина (кефалина) на 1 мл; указанные соединения разводят в смеси хлороформа с метанолом в соотношении 2:1.
После получения гемагглютининовых везикул согласно процедуре, описанной в примере 1, к раствору добавляют 20 мг сульфосукцинимидил-4-(N-малеимидофенил)-бутирата (СМФБ) в 2 мл воды, который служит сшивающим агентом. Смесь выдерживают в течение 1 часа при комнатной температуре, слегка встряхивая ее, после чего подвергают ультрацентрифугированию при 100000g в течение 10 мин для осаждения везикул.
К осадку добавляют 8 мл (CD4 моноклональных антител антилейцина 3A1 мг/мл (Becton и Diekinson). Ресуспендированный материал осторожно встряхивают в течение нескольких секунд каждые 5 минут на протяжении часа, при комнатной температуре. На последней стадии полученный материал снова осаждают ультрацентрифугированием при 100000g в течение 10 мин (для удаления несвязанных антител). Осадок ресуспендируют в 1500 мл физиологического раствора NaCl.
Пример 3.
Повторяют процедуру, описанную в примере 2, со следующими модификациями.
К раствору (IV) (см. пример 1) вместо декстран-сульфата добавляют по 1000 мг имидазол-карбоксамида и оксимочевины, которая по данным Brunner и Nagel обладает фармацевтической активностью в отношении меланом (Springer Verlag 2-е издание, Mtesnistische Krebstherapie стр. 93, 1979).
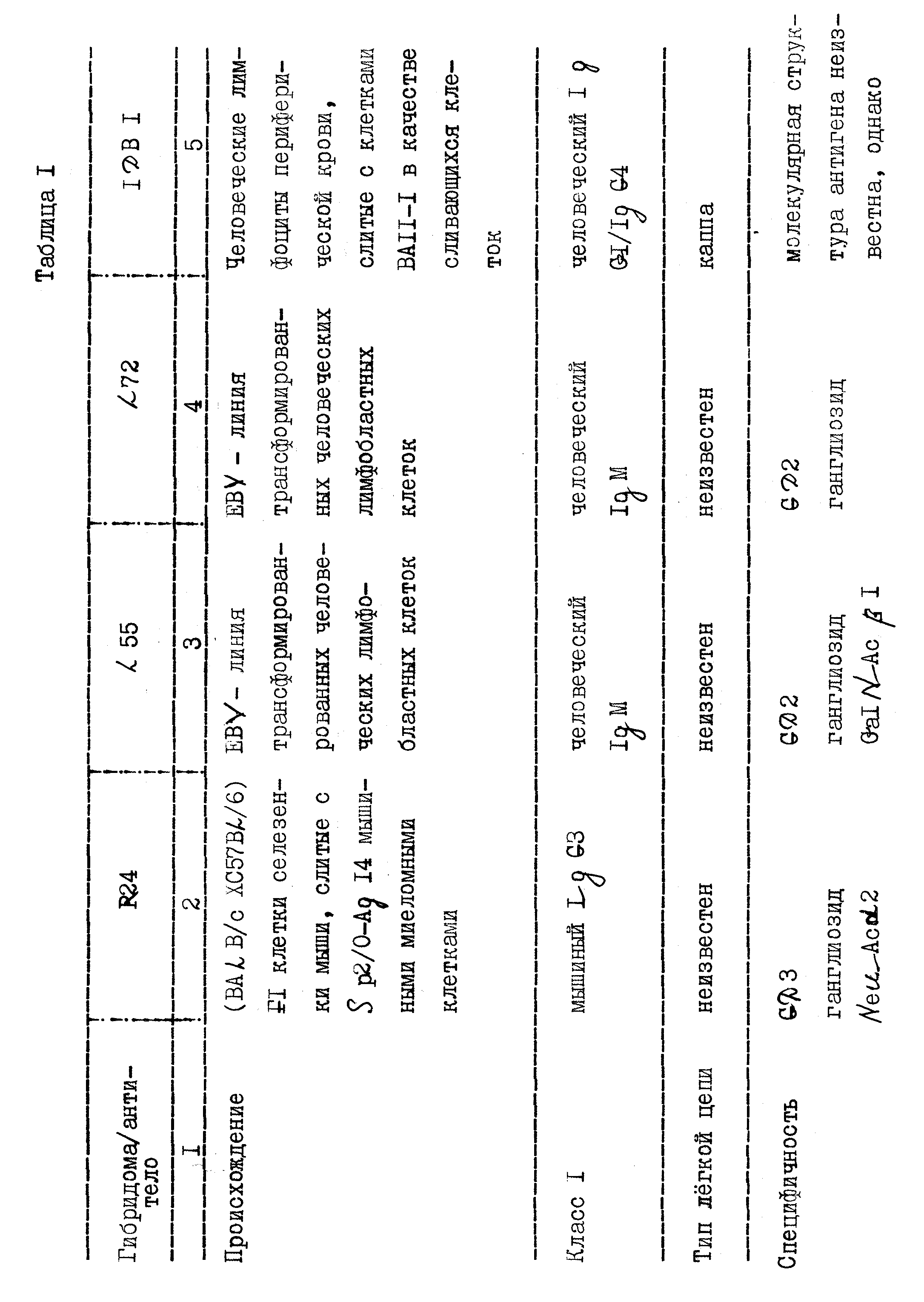
После добавления сшивающего агента и продолжения процедуры в соответствии с описанием примера 2, к раствору везикул (DES) добавляют 1 мг моноклональных антител (либо R 24, как описано Houghton A.N. и др. в Proc, Nat. Ac. Sc. , 82, 1985, стр. 1242, либо 0,5 мг L 55 + 0,5 мг L 72, как описано Lancet 1989, стр. 786 - 787). В результате получают композицию следующего состава, содержащую 1000 доз для введения людям: Получают моноклональные антитела R24 от А.Н.Хугтона в виде водного раствора с концентрацией 3 мг/мл; моноклональные антитела L72 были получены от Р.С.Ирика в виде водного раствора с концентрацией 1 мг/мл. Моноклональные антитела добавляют к везикулам, содержащим сульфо-SMPB-сшивающие молекулы, как описано в примере 2, 46 мг гемагглютинина, 250 мг фосфатидилхолина, 30 мг фосфатидилэтеноламина (кефалина), 20 мг сшивающего агента (сульфо-СМФБ), 1 мг моноклонального антитела, 1000 мг имидазол-карбоксамида, 1000 мг оксимочевины.
Поскольку указанные средства до сих пор использовались примерно в пятикратных количествах, приведенная пропись на самом деле обеспечивает получение лишь 200 доз.
Пример 4.
Повторяют процедуру, описанную в примере 2, со следующими модификациями.
К раствору (IV) (см. пример 1) вместо декстран-сульфата добавляют по 1000 мг по крайней мере одного из нижеупомянутых препаратов: адрипластина, эндоксана, фторурацила, (как описано Brunner и Nagel, Internistische Krebsherapie Springer-Ver lag 2-е издание, стр. 309, 1979) и колхицина (Sigma). Все фармацевтические препараты были куплены на фирме Sigma. Введение одного или нескольких соединений в комбинации было проведено, как описано в Примере 1.
После добавления сшивающего агента и продолжения процедуры в соответствии с описанием примера 2 к раствору везикул (VES) добавляют 1 мг моноклональных антител JDBI, как описано Stelkauskas A.J., Cancer-Immonol. Vol., 23, p. 31, 1986) для ковалентного связывания. В результате получают композицию следующего состава, содержащую 1000 доз фармацевтического средства для лечения больных с карциономой грудной железы: 46 мг гемагглютинина, 250 мг фосфатидилхолина, 30 мг фосфатидилэтаноламина, 20 мг сшивающего агента (сульфо-СМФБ), 1 мг моноклональных антител, 1000 мг адрипластина, 1000 мг эндоксана, 1000 мг фторурацила.
Пример 5.
Повторяют процедуры, описанные в примерах 1 и 2, со следующими модификациями.
Вместо тримеров гемагглютинина вируса гриппа используют мономеры, содержащие слитый пептид. Тримеры гемагглютинина выделяют обработкой целых вирусов гриппа раствором OEG, как описано в примере 1. Мономер гемагглютинина-2, содержащий слитый пептид, получают расщеплением S-H мостиков посредством химического восстановления Д4-дитиотреитолом (ДТТ, Sigma), с последующим отделением от пептида гемагглютинина-1 с помощью гелевой хроматографии на сефадексе G-50 при pH 6. Суспензия очищенного мономера гемагглютинина-2 содержит 180 мкг мономеров, в том числе слитый пептид. Фракцию, содержащую гемегглютинина-1-мономер отбрасывают. Фракцию, содержащую гемаггютин-2-мономер, разбавляют раствором детергента /1000 мкмоль/мл/ до конечного объема 15,3 мл. Этот раствор прибавляют к высушенному противовирусному лекарству декстрансульфату /согласно примеру 1/. Этот новый раствор прибавляют к фосфолипидному слою в круглодонную колбу, как описано в примере 1. Везикулы, в которые был введен НА1 мономер на их поверхности и которые содержат противовирусные лекарство, спонтанно формируются при удалении детергента, как описано в примере 1 /на колонке с микроносителями из шариков полистирола и путем ультрацентрифугирования/.
Пример 6.
Повторяют процедуру, описанную в примере 2, со следующими модификациями.
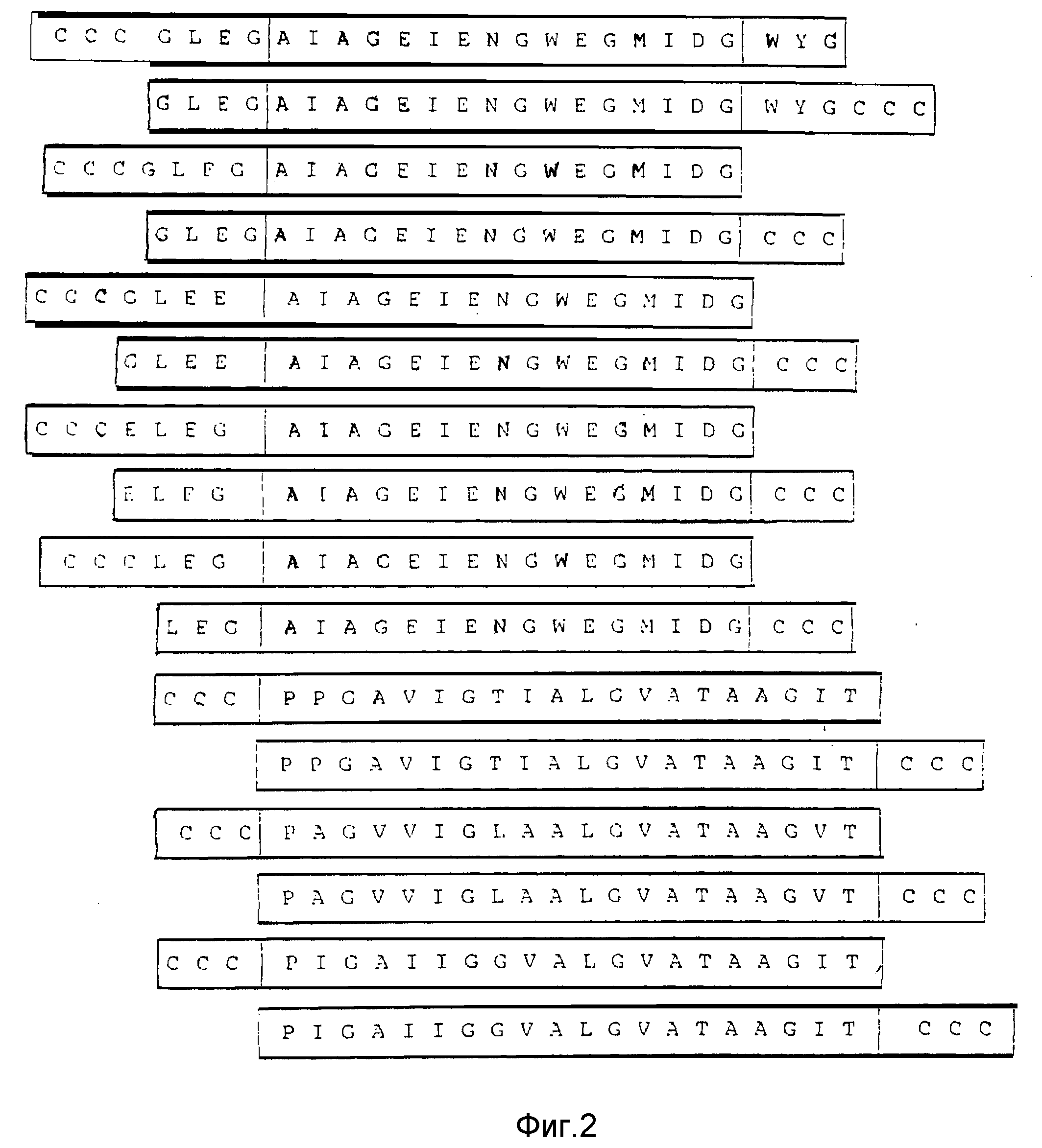
Вместо тримеров гемагглютинина вируса гриппа используют неочищенный слитый пептид. Слитый пептид вируса гриппа, используемый при синтезе мембранных везикул, получают методом химического синтеза. Для этой цели пригодна любая из приведенных на фиг. 2 аминокислотных последовательностей. Для обеспечения слияния концевой фрагмент соответствующей аминокислотной последовательности должен содержать по крайней мере один, предпочтительно три цистеиновых остатка.
Раствор с одним из таких синтетических слитых пептидов содержит его в концентрации 4 мкг/мл.
Везикулы с одним из вышеуказанных слитых пептидов получают следующим образом.
Раствор 9 мг фосфатидилхолина и 1 мг фосфатидилэтаноламина на 1 мл помещали в смесь хлороформа с метанолом в соотношении 2:1. 28 мл конечного раствора осторожно упаривали в вакуумном роторном испарителе, получая таким образом тонкий слой фосфолипидов на внутренних стенках круглодонной колбы.
3 г декстран-сульфата разводили в 15,3 мл раствор детергента, содержащего 100 мкмоль монододецилого эфира октаэтиленгликоля в 1 мл объединенного буферного раствора (композицию см. в примере 1). Конечный раствор переносили на фосфолипидный слой на стенках круглодонной колбы, последнюю встряхивали на протяжении 15 минут после полного растворения фосфолипидного слоя раствор переносили в стеклянный сосуд вместе с 4,8 г полистироловых гранул, служивших микроносителем, размером 25 - 50 меш (во влажном состоянии).
После этого сосуд встряхивали таким образом, чтобы его содержимое перемещалось из одного конца в другой с частотой два раза в секунду. Спустя час суспензию отсасывали тонкой пипеткой и переносили в другой сосуд, содержащий 2,4 г полистироловых гранул вышеуказанного размера. Сосуд встряхивали в течение 30 минут. Процедуру повторяли дважды. По завершении ее образовавшийся раствор снимали с гранул, помещали на водяную баню и подвергали ультразвуковой обработке с помощью аппарата Bransonic (Branson Europe) при частоте 50 кГц ± 10%. Продолжительность каждой обработки составляла 10 с, ее повторяли дважды с интервалом 10 с. В результате получали среднего размера везикулы диаметром приблизительно 50 - 100 нм.
К полученной таким образом суспензии добавляли 2 мл воды содержащей 20 мг сульфо-СМФБ (Pierce). Смесь выдерживали в течение 1 часа при комнатной температуре, слегка встряхивая, после чего подвергали ее ультрацентрифугированию при 100000g в течение 15 мин для осаждения везикул.
Получают синтетические слитые пептиды твердофазным пептидным синтезом. Твердофазный пептидный синтез осуществляют на полуавтоматическом синтезаторе периодического действия /SP G50; Labortec A.G. Budendorf, Швейцария/ с использованием флоренилметоксикарбонил - (Fмок ) - трет-бутильной стратегии. 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийтетрафторборат /ПВТИ/ и 1-гидроксибензотриазол /НОВТ/ используют для активации Fмок-аминокислот. Диметилформамид /ДМФ/ используют в качестве растворителя, а пиперидин /20% в ДМФ/ используют для удаления Fмок - защищающей группы. Каждую реакцию сочетания анализируют на полноту с помощью качественного нингидринового теста /Кайзер-тест/ (Kaiser E.R.L., Colescolt D.D., Bosinger, Coch, P.1, 1970, Anal. Biochem. , 34, 595 - 598/. Отщепление пептидов от смолы и групп, защищающих боковую цепь, осуществляют с помощью трифторуксусной кислоты /ТФУК/, включая подходящие акцепторы /дитиоэритрит и т.п./. Затем осаждают сырые незащищенные пептиды и промывают трет-бутилметиловым эфиром. Очистку пептидов проводят с помощью ВЭЖХ с обращенной фазой в 0,1% ТФУК и элюированием с градиентом ацетонитрила. Используют реагенты Sigma. Пептиды лиофилизируют и повторно суспендируют в слитом буферном растворе (композиция по примеру 1). В концентрации 4 мкг/мл. Ресуспендированный осадок осторожно встряхивали каждые 5 мин (по нескольку секунд на протяжении часа при комнатной температуре. На последней стадии суспензию снова центрифугировали при 100000g в течение 10 мин для удаления несвязанного слитого пептида. Осадок ресуспензировали в 200 мл физиологического раствора NaCl.
Пример 7.
Повторяли процедуру, описанную в примере 6, со следующими модификациями.
2 мл раствора, содержавшего 2 мкг синтетического слитого пептида, и 2 мл, содержащих 100 мкг антилейцина 3А, добавляли к присутствовавшим в осадке везикулам. Ресуспендированный материал осторожно встряхивали в течение нескольких секунд каждые 5 мин на протяжении часа при комнатной температуре. Затем суспензию снова подвергали ультрацентрифугированию при 100000g в течение 10 мин для удаления несвязанных слитых и специфичных для клеток пептидов. Полученный осадок снова ресуспендировали в 200 мл физиологического раствора NaCl.
Пример 8.
В соответствии с процедурами, описанными в примерах 1 - 4 получали везикулы, содержавшие слитый пептид или гемагглютинин рабдовирусов, вирусов парагриппа или тогавирусов.
а) Рабровирус бешенства культивировали в человеческих диплоидных клетках следующим образом. Сливающийся монослой МСК-5 человеческих диплоидных клеток из банка клеток Швейцарского института сывороток и вакцин в Берне инокулируют 1 мл посева Pitman Moore вируса бешенства /из Швейцарского института сывороток и вакцин/. После 1 часа инкубирования при 35oC прибавляют 0,5 мл/см2 тканевой культуры ВМЕ-среды /Гибко, Швейцария/, содержащей 10% фетальной бычьей сыворотки /Гибко/. После 1 недели инкубирования при 35oC собирают среду, содержащую большое количество вируса бешенства.
Полученный материал (надосадочную жидкость), содержащую 107 вирусных частиц в 1 мл, подвергали очистке и концентрировали посредством ультрацентрифугирования в градиенте плотности сахарозы. Ультрацентрифугирование проводят с использованием KII ультрацентрифуги (Electronucleonics, Heidelberg, Германия).
Суспензия очищенного вируса бешенства содержала 210 мкг вирусного гемагглютинина в 1 мл. Ее дальнейшую обработку проводили, как описано в примере 1.
б) Вирус парагриппа типа III, полученный из департамента диагностики вирусов Базельского детского госпиталя Швейцарии, культивировали в аллантонсе куриного яйца и дважды подвергали очистке
посредством ультрацентрифугирования в градиенте плотности сахарозы, как описано в примере 1. Суспензия очищенного вириона парагриппа содержала 245 мг гемагглютинина вируса парагриппа в 1 мл. Ее
дальнейшую обработку проводили, как описано в примере 1;
в) Тогавирус краснухи штамм RA/27, из посева для получения вируса из Швейцарского института сывороток и вакцин в Берне культивировали
в человеческих диплоидных клетках и подвергали очистке, как описано в пункте а). Суспензия очищенного вириона краснухи содержала 205 мкг гемагглютинина в 1 мл. Ее дальнейшую обработку производили, как
описано в примере 1.
Пример 9.
Активирусные средства получали, как описано в примере 1. Мышам линии hu-PBL-SCJD на протяжении 14 дней вводили димер рибонуклеазы, чередуя его введение с введением на протяжении 10 дней только фосфатного буферного раствора (контроль) с декстрансульфатом (500 К Дальтон, Sigma или димера рибонуклеазы (заявка России N 4743095/14) в трех разных дозах, декстран-сульфат в липосомах или димер рибонуклеазы в липосомах. Два последние препарата получали, как описано в примере 1 настоящего изобретения. Введение указанных препаратов производили одновременно с введением вируса гриппа (100 TCID50HIV-IIIIB) Признаки вирусной инфекции (выделение вируса, определение (СНП) провирусных последовательностей) у мышей линии hu-PBL-SC1D проводили спустя 2, 4 и 6 недель после ее начала.
Результаты исследования показывают, что в зависимости от характера вводившихся препаратов частота выделения
возбудителя у мышей различных групп составляла, %:
После введения фосфатного буферного раствора (контроль) - 80
После введения димера рибонуклеазы в дозе 0,001 мг/кг - 92
После введение димера рибонуклеазы в дозе 0,1 мг/кг - 30
При введении димера рибонуклеазы в дозе 10,0 мг/кг - 63
После введения декстран-сульфата в дозе 10 мг/кг - 33
После
введения декстран-сульфата в дозе 10 мг/кг в липосомах - 14
После введения димера рибонуклеазы в липосомах - 25
Результаты исследования свидетельствуют в том, что инъекция каждые 12
часов димера рибонуклеазы в дозе 0,1 мг/кг веса тела, декстран-сульфата в дозе 10 мг/кг липосом, содержащих димер рибонуклеазы, и липосом, содержащих декстран-сульфат, защищают мышей линии hu-PBI-SCID
от инфекции вирусом гриппа. Защита была мене эффективной при использовании препарата димера рибонуклеазы в дозе 10 мг/кг, что может быть связано с подавлением иммунной реакции при высокой дозе димера.
Его защитное действие в дозе 0,001 мг/кг отсутствовало. В то же время, введение димера рибонуклеазы в составе липосом согласно настоящему изобретению повышало эффективность его защитного действия с 63
до 25%, несмотря на высокую дозу. Ожидается, что подбор оптимальных дозировок этого соединения позволит еще больше усилить фармакологическую активность липосомных препаратов.
Этот вывод справедлив и для случая исключения из анализа мышей с неудовлетворительно функционирующими имплантатами человеческих ПЛ (PBL) в конце экспериментального периода, хотя из оценки уровня человеческих иммуноглобулинов и СПП (PCR) β- глобина следует, что введение липосом, содержащих димер рибонуклеазы или декстран-сульфат, снижает выживаемость человеческих клеток. Исключение из анализа мышей линии hu-PBL-S C1D на основании результатов оценки функциональной активности человеческих клеток позволяет дифференцировать непосредственное противовирусное действие липосомных препаратов от их имуномодуляторной активности.
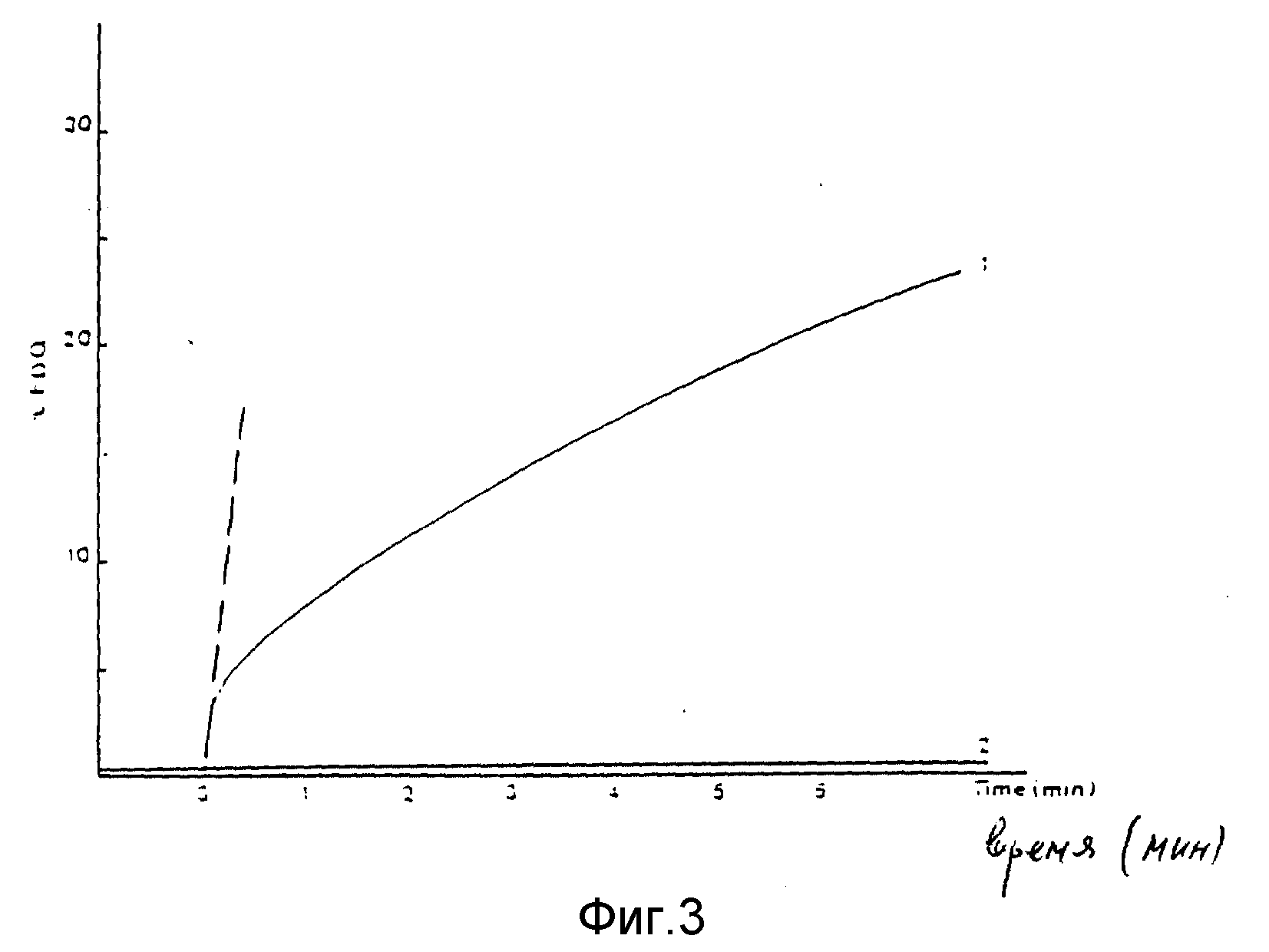
Пример 10.
Везикулы, полученные, как описано в примере 1, сравнивали в тесте Luseher и Gluck, Antiviral Research, 1990, 14, 39 - 50) с рекомбенированными везикулами вируса гриппа, полученными по методу Kawasaku соавторами. Оценивали характер их слияния с модельными мембранами.
Фиг. 3 иллюстрирует кинетику ингибирования затухания флюоресцении гриппа A, меченного P18, под воздействием липосом, содержащих ДОФХ/холестерин. Усиление флюоресценции выражали как степень подавления ее затухания в процентах, рассчитанную по методу Luscher и Gluick (см. выше).
Начальную скорость слияния выражали через значения тангенсов кривых слияния в нулевой точке, соответствовавшей моменту инициирования этого процесса (пунктирная линия на фиг. 3). Кривая 2 характеризует активность слияния везикул, полученных по методике Kawasaki с соавторами.
Пример 11 (новый).
Продукты, описанные в примерах 1 - 8, испытывают на безвредность и нетоксичны на животных следующим образом.
Одну человеческую дозу каждого продукта, описанного в примерах, дают дважды в день /утром и вечером/ взрослым самкам мышей линии Balb/с - в течение 30 дней. Каждая группа состоит из 20 мышей. В конце испытания у всех мышей контролируют состояние здоровья, массу и, после умервщления, аномальности в основных органах. Доказано, что все препараты являются безопасными и нетоксичными.
В другом тесте вещества испытывали на пирогенность на кроликах /2,5 кг/. Вводили 3 человеческие дозы в.в. Каждое вещество испытывали на 10 кроликах. Непрерывно контролировали температуру тела животных. Не было отмечено изменений температуры тела.
Пример 12 (новый).
Противораковые продукты, описанные в примерах 3, 4, 5, 6, 7 и 8, были испытаны на эффективность на моделях голых мышей. Иммуносупрессированным голым мышам инокулировали подкожно опухолевые клетки от человеческих пациентов /меланома карцинома или мамма карцинома; 107 клеток/животное/. Через 7 дней у каждой мыши развивалась видимая опухоль. Мышей обрабатывали внутривенно препаратами, описанными в примерах 3, 4, 5, 6 и 7. Каждая группа состояла из 10 животных. Одной мыши давали ежедневно инъекции одной человеческой дозы в течение 19 дней. Одну группу обрабатывали только физиологическим раствором NaCl. Три группы обрабатывали только фармакологическими веществами /без введения везикул/.
Результаты показали, что обработка физиологическим NaCl дает самые большие опухоли /4 г/мышь/. Обработка только веществом дает опухоли в интервале 2 - 3 г/мышь. Самые хорошие результаты получены с препаратом, описанным в примерах 3 - 7. Масса опухоли составляла 0 - 1 г/мышь, что было значительно лучше, чем в других группах.
В табл. 1 представлены свойства и характеристики заявляемых антител.
Лечение СПИДа и карциномы.
Для дополнительного подтверждения заявляемого применения против СПДа представляется следующая информация, полученная при экспериментах на трансгенных мышах SCID (мыши с тяжелым комбинированным иммунодефицитом, воспроизведенным с помощью ксенопересадки человеческих лейкоцитов периферической крови). Эта модель дает возможность оценки кандидатов в соединения с активностью против ВИЧ.
"НЕР 613" относится к рибонуклеазным димерам.
Мышам hu-PBL-SCID вводили HEP613 в течение 14 дней при трех уровнях дозы (10, 0,1 и 0,001 кг/кг) или в течение 10 дней с сульфатом декстрана, HEP613 в липосомах или сульфат декстрана в липосомах. Лечение начиналось с момента заражения вирусом в дозе 100 TCID50 ВИЧ-1IIIB. Оценку проявления вирусной инфекции у мышей hu-PBL-SCID (выделение вируса, PCR определение провирусных последовательностей проводили через 2, 4 и 6 недель после заражения вирусом. Все результаты суммированы на чертеже ниже, который показывает процент животных в каждой группе, получавшей препараты, от которых выделен вирус. Результаты исследования показывают защиту большинства мышей hu-PBL-SCID от инфицирования ВИЧ после введения через 12 часов 0,1 мг/кг HEP613, 10 мг/кг сульфата декстрана, липосом, содержащих сульфат декстрэна. Защита была минимальной при введении 10 мг/кг HEP613, и никакого защитного эффекта не наблюдалось при дозе HEP613 в 0,001 мг/кг. Те же самые выводы получаются, когда из анализа исключаются мыши с плохо функционирующими трансплантатами человеческих лейкоцитов периферической крови в конце эксперимента (как показано на чертеже ниже), хотя из анализа уровней человеческого иммуноглобулина и результатов PCP β- глобина видно, что лечение или липосомами HEP613 или липосомами с сульфатом декстрана препятствует выживанию человеческих клеток. Исключение мышей hu-PBL-SCID на основе функционирования человеческих клеток дает возможность некоторого разграничения между прямыми противовирусными эффектами и иммуномодулирующей активностью.
HEP613 сам по себе или в липосомной лекарственной форме обладает противовирусной активностью in vivo против ВИЧ-1, и LIDAK рекомендует его последующую оценку.
HEP613 - димер рибонуклеазы
Заявитель предоставляет краткое изложение
результатов клинического изучения на пациентах, инфицированных ВИЧ, применения димера рибонуклеазы (HEP613), включая также применение HEP613 в липосомах.
Целью двойного слепого, переходного, с контролем с плацебо и случайной выборкой исследования была первоначальная оценка переносимости HEP613 человеческим организмом, инфицированным ВИЧ, и определение влияния композиции на течение инфекции ВИЧ у носителей ВИЧ.
Краткое изложение.
Испытания проводились на 20 инфицированных ВИЧ пациентах (16 мужчин, 4 женщины) в возрасте между 18 и 40 лет. Инфицированные пациенты не имели какого-либо другого заболевания и не проявляли симптомов АРС или СПИД. Пациентов лечили HEP613 в липосомах (10 пациентов) или без липосом (10 пациентов) при внутримышечном введении соответствующих препаратов дважды в день.
Истинное/плацебо лечение проводилось в течение 12 недель при дозировке 0,1 мг/кг HEP613 на инъекцию, или начиная с 6 недель введения плацебо и затем 6 недель введения HEP613 или наоборот (двойное слепое переходное исследование). После периода лечения посттерапевтический период продолжался в течение 18 недель, включавших продолжительное медицинское наблюдение и анализ иммунологических характеристик.
Результаты показывают, то липосомы, несущие димер рибонуклеазы (HEP613) в качестве желаемого лекарственного средства, могут безопасно вводиться внутримышечно. Хотя сероположительные носители ВИЧ не могли стать серонегативными с помощью применения препаратов, в соответствии с этим исследованием, могло наблюдаться значительное влияние на виремию и на ряд других иммунологических показателей. Не наблюдалось перехода из состояния переноса к АРС или СПИД, и отмечалось среднее повышение числа T-хелперных лимфоцитов, снижение числа лимфоцитов и повышение отношения хелперов/супрессоров, что подтверждает благоприятное действие противовирусного препарата (HEP613) при лечении пациентов, инфицированных ВИЧ.
Реферат
Изобретение относится к медицине, а именно к синтетическим мембранным везикулам (липосомам), на поверхности которых имеются молекулы слитых пептидов и специфические клеточные белки, способу их получения, а также их применению для изготовления лекарственных средств против СПИДа. Задача изобретения заключается в создании эффективного способа доставки лекарственных веществ к специфическим клеткам организма. Сущность изобретения состоит в создании двуслойных фосфолипидных везикул, содержащих по крайней мере одно фармацевтически активное вещество и имеющих на мембране специфические клеточные маркеры. Предпочтительный диаметр везикул 80 нм, мембрана везикул содержит невирусные фосфолипиды, фосфатидилэтанолами и фосфатидилхолин, с мембранами связан сшивающий агент, желательно в форме сульфосукцинимидильного производного и не менее одного специфического для клеток слитого пептида. Указанные везикулы используют для изготовления лекарственных средств с целью лечения СПИДа и карцином. Преимущество изобретения заключается в повышении специфичности доставки лекарственных веществ к специфическим клеткам. 2 с. и 16 з.п. ф-лы, 1 табл., 3 ил.
Комментарии