Состав на основе этиленпропиленовых-1-бутеновых терполимеров - RU2659964C1
Код документа: RU2659964C1
Чертежи
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к составу, содержащему этиленпропиленовый сополимер и этиленпропиленовые-1-бутеновые терполимеры, пригодному для создания пленок, в частности двухосноориентированных пленок, обладающих отличными свойствами в части, касающейся низкого содержания растворимых в ксилоле веществ, температуры плавления, температуры начала сваривания (SIT) и органолептических свойств.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Пленки, изготовленные из пропиленовых сополимеров или терполимеров, известны в отрасли техники, к которой относится данное изобретение.
Пропиленовые сополимеры или терполимеры, по сравнению с пропиленовыми гомополимерами, характеризуются лучшей ударной прочностью, пониженной жесткостью и повышенной прозрачностью. Однако в некоторых случаях трудно найти приемлемый баланс между данными свойствами, особенно когда требуется их противопоставление друг другу. Для получения определенной пластичности, например, требуется увеличение доли фракций растворимых в ксилоле, что недопустимо для изделий, контактирующих с продуктами питания.
В патенте США № 6 221 984 раскрываются статистические сополимеры пропилена с этиленом и, по меньшей мере, одним C4-C10 альфа-олефином и способ получения таких статистических сополимеров, которые могут использоваться в пленках, волокнах или формованных изделиях. В частности, терполимеры, полученные описанным там способом, особенно подходят для производства пленок для упаковки пищевых продуктов, из-за низкого содержания в них растворимых в ксилоле полимерных частиц (Примеры 1-3). С другой стороны, при увеличении фракции, растворимой в ксилоле, значение температуры начала сваривания пленки и ее оптические свойства становятся неудовлетворительными.
В патенте WO 2009/019169 раскрывается этиленпропиленовый-1-бутеновый терполимер полученный в газофазном реакторе, состоящем из двух взаимосвязанных зон полимеризации. Указанный терполимер, помимо других особенностей, обладает: соотношением между концентрацией этилена (вес.%) и концентрацией 1-бутилена (вес.%), составляющим от 0,1 до 0,8 и долей растворимой в ксилоле фракции при 25°C выше 9 вес.%.
В патенте WO 2013/174778 описывается этиленпропиленовый-1-бутеновый терполимер с содержанием этиленовых звеньев, составляющим от 0,5 вес.% до 2,2 вес.% и содержанием 1-бутеновых звеньев, составляющим от 6,0 вес.% до 20,0 вес.%.
В котором, в частности, растворимая в ксилоле фракция при 25°C составляет менее 15,0 вес.%, а минимальное значение составляет 5,0 вес.%. В данном случае возможно улучшение баланса между фракцией растворимой в ксилоле и SIT.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩЕСТВА ИЗОБРЕТЕНИЯ
К удивлению нами было обнаружено, что состав, содержащий этиленпропиленовый сополимер и этиленпропиленовые-1-бутеновые терполимеры, полученные с помощью гетерогенных катализаторов, обладающих особыми признаками, может быть с успехом использован для получения пленок, в частности двухосноориентированных пленок (BOPP), обладающих низкой температурой начала сваривания, лучшей прозрачностью и низкой растворимостью в ксилоле.
Состав полиолефина по настоящему изобретению, содержащий:
A) от 19 вес.% до 50 вес.% этиленпропиленового сополимера с содержанием этиленовых звеньев, составляющим от 1,5 вес.% до 6,0 вес.%;
B) от 50 вес.% до 81 вес.% этиленпропиленового-1-бутенового терполимера с содержанием этиленовых звеньев, составляющим от 1,5 вес.% до 6,0 вес.%, и содержанием 1-бутеновых звеньев, составляющим от 4,8 вес.% до 12,4 вес.%;
где сумма компонента А) и В) составляет 100;
состав, характеризуемый следующими свойствами:
- молекулярновесовым распределением (МВР), выраженным как соотношение Mw/Mn, превышающим 4,0;
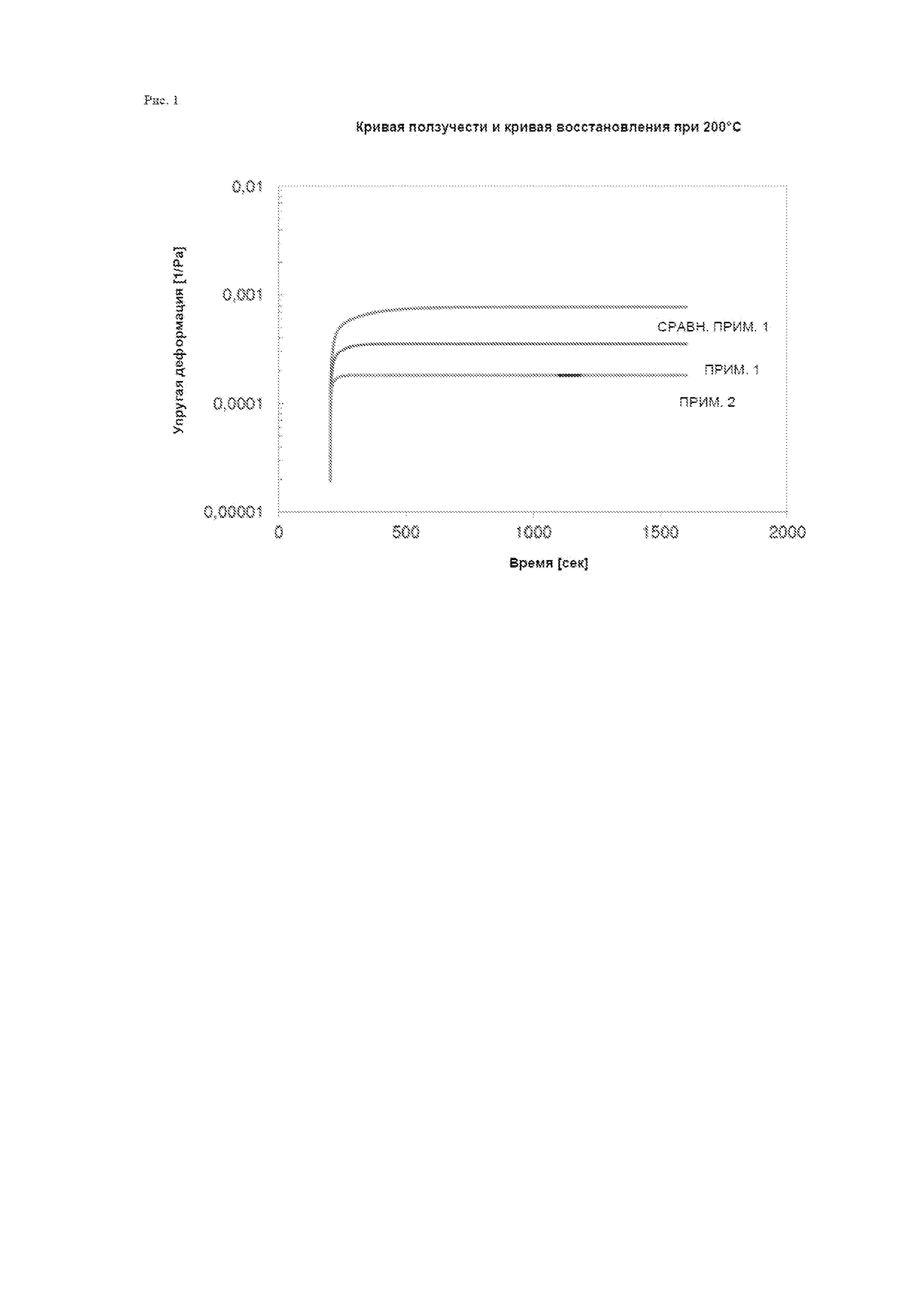
- максимальным значением ниже 53x10-4 1/Па, между 600 и 1200 секундами, кривой ползучести и кривой восстановления, измеренным на расплавленном полимере при 200°C в соответствии с процедурой, приведенной в разделе определения характеристик.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Рис. 1 представлена кривая ползучести и кривая восстановления, измеренные на расплавленном полимере при 200°C в соответствии с процедурой, приведенной в разделе определения характеристик примеров 1 и 2 и сравнительного примера 1.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Состав полиолефина, содержащий:
А) от 19 вес.% до 50 вес.%, предпочтительно от 25 вес.% до 42 вес.%, более предпочтительно от 31 вес.% до 38 вес.%, этиленпропиленового сополимера, с содержанием этиленовых звеньев, составляющим от 1,5 вес.% до 6,0 вес.%, предпочтительно от 2,6 вес.% до 5,2 вес.%, более предпочтительно от 3,1 вес.% до 4,3 вес.%.
В) от 50 вес.% до 81 вес.%, предпочтительно от 58 вес.% до 75 вес.%, более предпочтительно от 62 вес.% до 69 вес.%, этиленпропиленового-1-бутенового терполимера с содержанием этиленовых звеньев, составляющим от 1,5 вес.% до 6,0 вес.%, предпочтительно от 1,9 вес.% до 4,8 вес.%, более предпочтительно от 2,1 вес.% до 3,7 вес.%, и с содержанием 1-бутеновых звеньев, составляющим от 4,8 вес.% до 12,4 вес.%, предпочтительно от 5,1 вес.% до 10,3 вес.%, более предпочтительно от 6,8 вес.% до 9,5 вес.%;
где сумма компонента А) и В) составляет 100;
состав, характеризуемый следующими свойствами:
- молекулярновесовым распределением (МВР), выраженным как соотношение Mw/Mn, превышающим 4,0, предпочтительно превышающим 4,5 и более предпочтительно превышающим 4,7.
- максимальным значением ниже 53x10-5 1/Па, предпочтительно ниже 51x10-5 1/Па, более предпочтительно ниже 45x10-5 1/Па между 600 и 1200 секундами, кривой ползучести и кривой восстановления, измеренным на расплавленном полимере при 200°C в соответствии с процедурой, приведенной в разделе определения характеристик.
Содержание этиленовых звеньев в составе полиолефина составляет от 1,5 вес.% до 6,0 вес.%, предпочтительно от 2,1 вес.% до 5,2 вес.%, более предпочтительно от 2,5 вес.% до 4,3 вес.%, особенно предпочтительно от 2,8 вес.% до 3,9 вес.%.
Содержание 1-бутеновых звеньев в составе полиолефина составляет от 3,2 вес.% до 9,8 вес.%, предпочтительно от 3,6 вес.% до 8,2 вес.%, более предпочтительно от 4,8 вес.% до 7,9 вес.%, особенно предпочтительно от 5,8 вес.% до 7,4 вес.%.
Этиленпропиленовый-1-бутеновый терполимер содержит только пропиленовые, этиленовые и 1-бутеновые сомономеры, а этиленпропиленовый сополимер содержит только пропилен и этилен в качестве сомономера.
Предпочтительный индекс текучести расплава (MFR 230°C, 2,16 кг), относящийся к составу полиолефина реакторной чистоты (т.е. сополимерам, которые не подвергались химическому или физическому легкому крекингу понижения вязкости), составляет от 0,9 до 25 г/10 мин, предпочтительно от 3,0 до 20,0 г/10 мин; более предпочтительно от 4,0 до 18,0 г/10 мин.
Данный состав полиолефина особенно подходит для производства пленок, в частности двухосноориентированных (BOPP) пленок. BOPP-пленка, полученная из данного состава полиолефина, имеет низкую температуру начала сваривания (SIT) и низкое содержание веществ, растворимых в ксилоле при 0/25°C.
В частности, состав полиолефина предпочтительно имеет:
- содержание фракции, растворимой в ксилоле при 0/25°C (XS), составляющее от 1,2 вес.% до 15,1 вес.%;
- температуру начала сваривания (SIT), составляющую от 90 до 110°C.
Для данного состава полиолефина фракция, растворимая в ксилоле при 25°C, и температура начала сваривания (SIT) удовлетворяют следующему соотношению
SIT<114-(XSx1.3)
где:
XS = вес.% фракции, растворимой в ксилоле при 0/25°C, определенный способом, приведенным в разделе определения характеристик;
SIT = °C температура начала сваривания (SIT), определенная способом, приведенным в разделе определения характеристик.
Предпочтительное соотношение представляет собой:
SIT<113-(XSx1,3);
В частности, разница между температурой плавления и SIT составляет выше 25°C, предпочтительно выше 26°С.
Состав полиолефина, описанный в настоящем документе, получают способом, включающим полимеризацию пропилена с этиленом на первой стадии, а затем пропилена с этиленом и 1-бутеном на второй стадии в присутствии катализатора, содержащего продукт реакции между:
(i) твердым компонентом катализатора, содержащим Ti, Mg, Cl, и электронодонорным соединением, характеризующимся содержанием от 0,1 до 50 вес.% Bi по отношению к общей массе указанного твердого компонента катализатора;
(ii) соединением алкилалюминия и,
(iii) необязательно, электронодонорным соединением (внешний донор).
Предпочтительное содержание Bi в компоненте катализатора составляет от 0,5 до 40 %, более предпочтительно от 1 до 35 и особенно от 2 до 25 вес.%, а в отдельном варианте осуществления от 2 до 20 вес.%.
Частицы твердого компонента имеют в основном шарообразную форму, а их средний диаметр составляет от 5 до 150 мкм, предпочтительно от 20 до 100 мкм и более предпочтительно от 30 до 90 мкм. Поскольку частицы, имеют в основном шарообразную форму, то соотношение между наибольшей геометрической осью и наименьшей геометрической осью упомянутых частиц, равно или меньше 1,5, а предпочтительно меньше 1,3.
В целом, концентрация Mg составляет от 8 до 30 вес.%, а более предпочтительно от 10 до 25 вес.%.
В целом, концентрация Ti составляет от 0,5 до 5 вес.%, а более предпочтительно от 0,7 до 3 вес.%.
Предпочтительные внутренние электронодонорные соединения выбираются из алкильных и арильных сложных эфиров, необязательно замещенных ароматических многоосновных карбоновых кислот, например, сложных эфиров бензойной и фталевой кислот. Конкретными примерами таких сложных эфиров являются n-бутилфталат, диизобутилфталат, ди-n-октилфталат, этилбензоат и p-этоксиэтилбензоат.
Молярное соотношение Mg/Ti предпочтительно равно или превышает 13, предпочтительно составляет от 14 до 40, более предпочтительно от 15 до 40. Соответственно, молярное соотношение Mg/донор предпочтительно превышает 16, более предпочтительно превышает 17 и, как правило, составляет от 18 до 50.
Атомы Bi предпочтительно получают из одного или нескольких соединений Bi не имеющих Bi-углеродных связей. В частности, соединения Bi выбираются из галогенидов Bi, карбонатов Bi, ацетатов Bi, нитратов Bi, оксидов Bi, сульфатов Bi, сульфидов Bi. Предпочтительными соединениями Bi являются соединения с валентностью 3+. Среди галогенидов Bi предпочтительными являются трихлорид Bi и трибромид Bi. Наиболее предпочтительным соединением Bi является BiCl3.
Получение твердого каталитического компонента может осуществляться различными способами.
В соответствии с одним способом, твердый компонент катализатора получают реакцией титанового соединения формулы Ti(OR)q-yXy, где q представляет собой валентность титана, y представляет собой число от 1 до q, предпочтительно TiCl4, с хлоридом магния, являющегося производным аддукта формулы MgCl2⋅pROH, где р представляет собой число от 0,1 до 6, предпочтительно от 2 до 3,5, а R представляет собой углеводородный радикал, содержащий 1-18 атомов углерода. Аддукты сферической формы получают путем смешивания спирта и хлорида магния в режиме перемешивания при температуре плавления аддукта (100-130°C). Затем аддукт смешивают с инертным углеводородом, неспособным смешиваться с аддуктом, и получают эмульсию, которую быстро охлаждают, в результате чего происходит затвердевание аддукта в форме сферических частиц. Примеры сферических аддуктов, полученных в соответствии с данным способом, описаны в патенте США 4 399 054 и патенте США 4 469 648. Полученный аддукт непосредственно реагирует с соединением титана или предварительно подвергается контролируемой термической обработке для удаления спирта (80-130°C) и получением аддукта, в котором число молей спирта обычно ниже 3, преимущественно составляет от 0,1 до 2,5. Реакция с соединением Ti осуществляется суспендированием аддукта (без спирта или со спиртом) в холодном TiCl4 (обычно 0°C). Смесь нагревают до 80-130°С и выдерживают при этой температуре в течение 0,5-2 часов. Обработка соединением TiCl4 может проводиться один или несколько раз. В процессе обработки TiCl4 может добавляться электронодонорное соединение в требуемых соотношениях.
Существуют несколько способов добавления одного или нескольких соединений Bi в технологический процесс получении катализатора. В соответствии с предпочтительным вариантом осуществления, соединение (я) Bi вводится непосредственно в аддукт MgCl2⋅pROH в процессе его получения. В частности, соединение Bi может добавляться на начальной стадии получения аддукта, путем его смешивания с MgCl2 и спиртом. В альтернативном варианте осуществления, оно может добавляться в расплавленный аддукт перед стадией эмульгирования. Концентрация введенного Bi составляет от 0,1 до 1 моль на моль Mg в аддукте. Предпочтительными соединениями Bi, вводимыми непосредственно в аддукт MgCl2⋅pROH, являются галогениды Bi, в частности BiCl3.
Алкилалюминиевое соединение (ii), предпочтительно, выбирают из группы, включающей триалкилалюминевые соединения, например, триэтилалюминий, триизобутилалюминий, три-n-бутилалюминий, три-n-гексилалюминий, n-гексилалюминий, три-n-октилалюминий. Можно также использовать алкилалюминийгалогениды, алкилалюминийгидриды или алкилалюминийсесквихлориды, такие как AlEt2Cl и Al2Et3Cl3, необязательно, в смеси с указанными триалкилалюминиевыми соединениями. Соотношение Al/Ti выше 1 и обычно составляет от 50 до 2000.
Подходящие внешние электронодонорные соединения включают в себя соединения кремния, простые эфиры, сложные эфиры, амины, гетероциклические соединения и, в частности, 2,2,6,6-тетраметилпиперидины и кетоны.
Другим предпочтительным классом внешних электронодонорных соединений являются соединения кремния формулы (R6)a(R7)bSi(OR8)c, где a и b представляют собой целые числа от 0 до 2, c представляет собой целое число от 1 до 4, а сумма (a+b+c) равна 4; R6, R7 и R8 представляют собой алкильные, циклоалкильные или арильные радикалы, содержащие от 1 до 18 атомов углерода, необязательно содержащие гетероатомы. Особенно предпочтительными являются соединения кремния, в которых a равно 1, b равно 1, c равно 2 и, по меньшей мере, одно из R6 и R7 выбирается из разветвленных алкильных, циклоалкильных или арильных групп с 3-10 атомами углерода, необязательно содержащих гетероатомы, а R8 представляет собой C1-C10 алкильную группу, в частности метил. Примерами таких предпочтительных соединений кремния являются: метилциклогексилдиметоксисилан (С донор), дифенилдиметоксисилан, метил-трет-бутилдиметоксисилан, дициклопентилдиметоксисилан (D донор), диизопропилдиметоксисилан, 2-этилпиперидинил-трет-бутилдиметоксисилан, 2-этилпиперидинилтексилдиметоксисилан, 3,3,3-трифторо-n-пропил-2-этилпиперидинилдиметоксисилан, метил-3,3,3-трифторо-n-пропилдимпетоксисилан. Кроме того, предпочтительными являются соединения кремния, в которых а равно 0, с равно 3, R7 представляет собой разветвленную алкильную или циклоалкильную группу, необязательно содержащую гетероатомы, а R8 представляет собой метил. Примерами таких предпочтительных соединений кремния являются циклогексилтриметоксисилан, трет-бутилтриметоксисилан и тексилтриметоксисилан.
Электронодонорное соединение (iii) используется в таком количестве, чтобы получить молярное соотношение между алюминийорганическим соединением и указанным электронодонорным соединением (iii) в пределах от 0,1 до 500, предпочтительно от 1 до 300 и более предпочтительно от 3 до 100.
Состав полиолефина по настоящему изобретению получают смешиванием компонентов А) и В), причем указанные компоненты получают способом полимеризации, известным в отрасли техники, к которой относится данное изобретение, например, газофазной полимеризацией в одном или нескольких реакторах с псевдоожиженным или механически перемешиваемым слоем, суспензионной полимеризацией с использованием инертного углеводородного растворителя в качестве разбавителя или объемной полимеризацией с использованием жидкого мономера (например, пропилена) в качестве реакционной среды. Состав полиолефина по настоящему изобретению также получают способом полимеризации в двух или более стадиях, в которых компонент А) получают на первых стадиях, а компонент В) получают на вторых стадиях в присутствии компонента А). Каждая стадия может представлять собой газофазную полимеризацию в одном или нескольких реакторах с псевдоожиженным или механически перемешиваемым слоем, суспензионную полимеризацию с использованием инертного углеводородного растворителя в качестве разбавителя или объемную полимеризацию с использованием жидкого мономера (например, пропилена) в качестве реакционной среды.
Полимеризацию обычно проводят при температуре от 20 до 120°С, предпочтительно от 40 до 80°С. При газофазной полимеризации рабочее давление, как правило, составляет от 0,5 до 5 МПа, предпочтительно от 1 до 4 МПа. При объемной полимеризации рабочее давление обычно составляет от 1 до 8 МПа, предпочтительно от 1,5 до 5 МПа. Водород обычно используют в качестве регулятора молекулярного веса.
Следующие примеры приведены для иллюстрации изобретения и не ограничивают его объем.
ПРИМЕРЫ
ОПРЕДЕЛЕНИЕ ХАРАКТЕРИСТИК
Определение содержания Mg, Ti
Определение содержания Mg и Ti в твердом компоненте катализатора, выполнялось способом эмиссионной спектроскопии с индуктивно-связанной плазмой на спектрометре "I.C.P Spectrometer ARL Accuris".
Образец получали путем взвешивания на аналитических весах в платиновом тигле "Fluxy" 0,1÷0,3 грамм катализатора и 2 граммов смеси метабората /тетрабората лития в соотношении 1/1. После добавления нескольких капель раствора KI, тигель вставляли в специальный аппарат "Claisse Fluxy" для полного сжигания. Остаток собирали раствором с 5% объемным содержанием HNO3 и подвергали анализу.
Определение содержания Bi
Определение содержания Bi в твердом компоненте катализатора выполнялось способом эмиссионной спектроскопии с индуктивно-связанной плазмой на спектрометре "I.C.P Spectrometer ARL Accuris".
Образец получали путем взвешивания на аналитических весах 0,1÷0,3 грамм катализатора в мерной колбе емкостью 200 см3. После медленного добавления 10 миллилитров раствора с объемным содержанием 65% HNO3 и 50 см3 дистиллированной воды, образец подвергали дегидрированию в течение 4-6 часов. Затем содержание мерной колбы разбавляли деионизованной водой до отметки. Полученный раствор подвергали анализу с помощью ICP на следующей длине волны: висмут – 223,06 нм.
Определение содержания внутреннего донора
Определение содержания внутреннего донора в твердом каталитическом соединении осуществлялось способом газовой хроматографии. Твердый компонент растворяли в ацетоне, добавляли внутренний стандарт и анализировали образец органической фазы в газовом хроматографе, чтобы определить концентрацию доноров в исходном каталитическом соединении.
Определение фракции растворимой в ксилоле при 0/25°C
В стеклянную колбу, оснащенную обратным холодильником и магнитной мешалкой, вводят 2,5 г полимера и 250 мл o-ксилола. Температуру повышают в течение 30 минут до температуры кипения растворителя. Полученный раствор выдерживают в обратном холодильнике при перемешивании в течение дальнейших 30 минут. Закрытую колбу затем выдерживают в течение 30 минут в ванне со льдом и водой и термостатической водной бане при 25°С также в течение 30 минут. Полученное таким образом твердое вещество отфильтровывают бумагой для быстрого фильтрования, а отфильтрованную жидкость объемом 100 мл выливают в предварительно взвешенный алюминиевый контейнер, который нагревают на обогревающей плите в потоке азота, выпаривая растворитель. Затем контейнер выдерживают в печи при 80°С под вакуумом до получения постоянной массы. Содержание указанной растворимой в ксилоле фракции выражалось как процент от исходных 2,5 г, а затем по разности (дополнительно к 100), как X.I. %.
Нахождение молекулярно-весового распределения (Mw/Mn)
Молекулярные веса и молекулярно-весовое распределение измеряли при 150°C прибором Waters Alliance GPCV/2000, оснащенным четырьмя колонками со смешанным сорбентом PLgel Olexis с размерами частиц 13 мкм. Размеры колонок составляли 300×7,8 мм. В качестве подвижной фазы использовали 1,2,4-трихлорбензол (ТСВ) после перегонки в вакууме, а скорость потока поддерживали на уровне 1,0 мл/мин. Раствор образца получали путем нагревания образца с перемешиванием при 150°С в ТСВ в течение от одного до двух часов. Концентрация составляла 1 мг/мл. Для предотвращения деградации добавляли 0,1 г/л 2,6-ди-трет-бутил-p-крезола. В набор колонок впрыскивали 300 мкл раствора. Калибровочную кривую получали с помощью 10 стандартных образцов полистирола (набор EasiCal компании Agilent) с молекулярными весами в интервале от 580 до 7 500 000. Предполагалось, что значения K уравнения Марка-Хувинка были равны:
K = 1,21 × 10-4 дл/г, а α = 0,706 для стандартных образцов полистирола
K = 1,90 × 10-4 дл/г, а α = 0,725 для экспериментальных образцов.
Для интерполяции экспериментальных данных и получения калибровочной кривой использовалась аппроксимация с помощью полинома третьего порядка. Сбор и обработка данных осуществлялась с помощью программного обеспечения «Waters Empowers 3 Chromatography Data Software» с опцией ГПХ.
Индекс текучести расплава (MIL)
Индекс текучести расплава (MIL) полимера определяли в соответствии с ISO 1133 (230°C, 2,16 кг).
Определение содержания сомономера
Содержание сомономеров определяли способом инфракрасной спектроскопии на инфракрасном спектрометре с преобразованием Фурье (FTIR), путем определения ИК-спектра образца относительно атмосферного фона, причем собираемыми данными являются:
время промывки: минимум 30 секунд
время отбора: минимум 3 минуты
аподизация: Happ-Genzel
разрешение: 2 см-1.
Приготовление образца
Толстый лист получают прессованием на гидравлическом прессе около 1 г образца между двумя слоями алюминиевой фольги. Если речь идет об однородности, то рекомендуется использовать минимум две операции прессования. Из данного листа вырезают небольшую часть для формования пленки. Рекомендуемая толщина пленки составляет от 0,02 до 0,05 см (8 – 20 мил).
Температура прессования составляет 180 ± 10°С (356°F) при давлении прессования около 10 кг/см2 (142,2 фунт/кв. дюйм) в течение приблизительно одной минуты. Затем давление снимается, образец вынимается из пресса и охлаждается до комнатной температуры.
Спектр прессованной пленки полимера регистрируется в показаниях оптической плотности по сравнению с волновыми числами (см-1). Для вычисления содержания этилена и 1-бутена используются следующие измерения.
Площади (At) комбинации полос спектра поглощения между 4482 и 3950 см-1, используемой для спектрометрической нормализации толщины пленки.
Площади (AC2) полосы поглощения между 750-700 см-1 после двух надлежащих последовательных спектроскопических вычитаний: изотактического неаддитивного спектра полипропилена и затем эталонного спектра статистического сополимера 1-бутен-пропилена между 800-690 см-1.
Высоты (DC4) полосы поглощения при 769 см-1 (максимальное значение), после двух надлежащих последовательных спектроскопических вычитаний: изотактического неаддитивного спектра полипропилена и затем эталонного спектра этилен-пропиленового статистического сополимера в диапазоне 800-690 см-1.
Для расчета прямых линий калибровки содержания этилена и 1-бутена для этилена и 1-бутена, полученных с использованием образцов известного количества этилена и 1-бутена, необходима:
Калибровка этилена
Калибровочная прямая получается путем построения графика AC2/At по сравнению с молярным процентом этилена (% C2m). Наклон GC2 рассчитывается линейной регрессией.
Калибровка 1-бутена
Калибровочная прямая получается путем построения графика DC4/At по сравнению с молярным процентом бутена (% C4m). Наклон GC4 рассчитывается линейной регрессией.
Регистрируется спектр рядовой пробы, а затем рассчитываются (At), (AC2) и (DC4) рядовой пробы. Содержание этилена (% молярной доли C2m) в образце рассчитывают следующим образом:
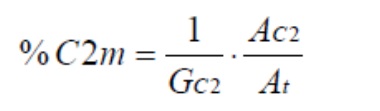
Содержание 1-бутена (% молярной доли C4m) в образце рассчитывают следующим образом:
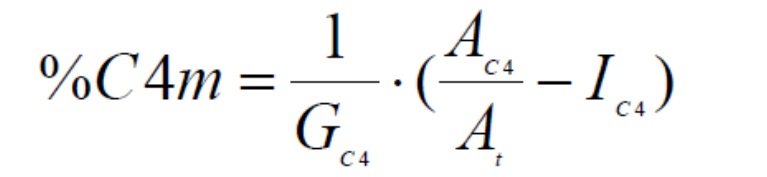
Содержание пропилена (молярной доли C3m) рассчитывают следующим образом:
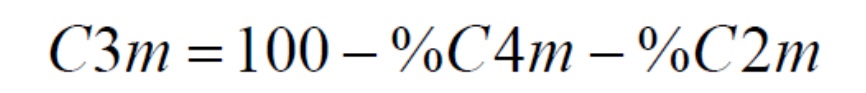
Содержание этилена, 1-бутена по массе рассчитывают следующим образом:
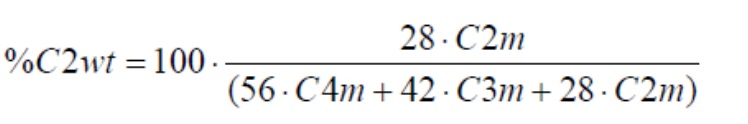
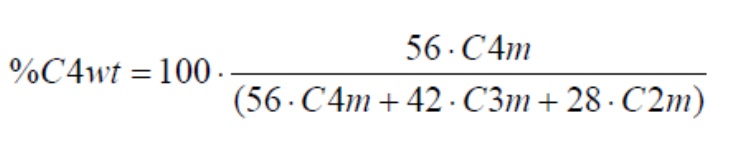
Содержание сомономера в компонентах А) и В) рассчитывалось по формулам:
C2tot=XAC2A+XBC2B и C4tot=XBC4B
где
C2 представляет собой вес.% этиленовых звеньев компонента А, В или общее содержание этилена.
C4 представляет вес.% 1-бутеновых звеньев компонента B или общее содержание 1-бутена.
XA представляет собой величину компонента A, выраженную в вес.%/100
XВ представляет собой величину компонента В, выраженную в вес.%/100
Температура плавления, определенная способом дифференциальной сканирующей калориметрии (ДСК)
Температуры плавления полимеров (Tm) измерялись способом дифференциальной сканирующей калориметрии (ДСК) со скоростью 20°C/мин на калориметре Perkin Elmer DSC-1 ранее откалиброванном по точкам плавления индия, в соответствии со стандартами ISO 11357-1, 2009 и 11357-3, 2011. Вес образцов в каждом тигле ДСК выдерживали на уровне 6,0 ± 0,5 мг.
Для измерения температуры плавления, взвешенный образец запечатывали в алюминиевые лотки и нагревали до 200°С со скоростью 20°С/мин. Образец выдерживали при 200°С в течение 2 минут до полного расплавления всех кристаллитов, а затем охлаждали до 5°С со скоростью 20°С/мин. После выдерживания в течение 2 мин при 5°С, образец нагревали во второй раз до 200°С со скоростью 20°С/мин. При втором нагреве пиковая температура (Tp,m) принималась за температуру плавления.
Определение мутности
Образцы пленки толщиной 50 мкм размером 5х5 см получали экструзией каждой испытываемой композиции в одношнековом экструдере «Collin» (отношение длина/диаметр шнека равно 1:25) со скоростью вытягивания пленки 7 м/мин и температуре расплава 210-250°С. Показатель мутности измеряют с использованием фотометрического устройства «Gardner», подключенного к прибору для определения мутности типа «Hazemeter UX-10» или эквивалентному прибору, снабженному лампой G.E. 1209 с фильтром "C". Эталонные образцы известной мутности используются для калибровки прибора.
Температура начала сваривания (SIT)
Подготовка образцов пленки
Некоторые пленки толщиной 50 мкм получали экструзией каждого испытываемого состава в одношнековом экструдере «Collin» (отношение длина / диаметр шнека равно 1:25) со скоростью вытягивания пленки 7 м/мин и температуре расплава 210-250°С. Каждую полученную пленку накладывают на пленку из гомополимера пропилена толщиной 1000 мкм, растворимая в ксилоле фракция которого составляла 97 вес.%, а MFR L 2 г/10 мин. Наложенные пленки спрессовывали друг с другом прессом «Carver» при 200°С под нагрузкой 9000 кг, которую выдерживали в течение 5 минут. Полученные слоистые материалы растягивали в продольном и в поперечном направлении, то есть подвергают двухосной ориентации с коэффициентом 6 на растяжном устройстве «ТОМ Long» при 150°С, получая, таким образом, пленку толщиной 20 мкм (18 мкм гомополимера + 2 мкм испытуемого материала). Из пленок вырезают образцы размером 2×5 см.
Определение SIT
Для каждого испытания два из указанных выше образцов накладывают друг на друга с выравниванием, причем примыкающие слои представляют собой слои конкретного испытуемого состава. Наложенные образцы сваривают на 2 см вдоль одной из сторон сварочным аппаратом «Brugger Feinmechanik Sealer», модели HSG-ETK 745. Время сваривания составляет 5 секунд при давлении 0,1 Н/мм2. Температура сваривания увеличивалась на 1°С с каждой сваркой, начиная с температуры приблизительно на 30°C меньше, чем температура плавления испытуемого состава. Сваренные образцы оставляли охлаждаться, а затем их несваренные концы крепились к прибору «Instron», где они подвергались испытанию на силу сцепления при скорости 50 мм/мин.
SIT представляет собой минимальную температуру начала сваривания, при которой сварка не ломается при нагрузке не менее 2 ньютонов в указанных условиях проведения испытаний.
Испытание на ползучесть и восстановление
Кривые ползучести и восстановления деформации измерялись реометрам «Physica MCR301» с конической пластиной сверху радиусом 25 мм и углом измерительного конуса, равным 1,992°. Температура испытаний составляет 200°C.
Определение времени ползучести
Комплексная вязкость определялась в режиме качания частоты, при угловой частоте от 100 рад/сек до 0,01 рад/сек и при постоянной амплитуде относительной деформации 5%, а значение на частоте 0,01 рад/сек выбиралось для расчета времени ползучести (без установки времени в профиле ). Время ползучести затем вычислялось по формуле:
Время ползучести=комплексная вязкость@0.01rad/s/100; [1]
Где 100 - приложенное напряжение в Паскалях
Определение времени восстановления
Время восстановления рассчитывалось по следующей формуле:
Время восстановления=время ползучести*7 [2]
Испытание на ползучесть и восстановление
a) ползучесть
ползучесть измерялась один раз в течение времени ползучести, рассчитанным по уравнению [1]. Приложенное сдвиговое напряжение составляет 100 Па.
b) восстановление
восстановление измерялось один раз в течение времени восстановления, рассчитанным по уравнению [2]. Приложенное сдвиговое напряжение составляет 0 Па.
В конце испытания программное обеспечение вычисляет кривую s относительно Па.
Примеры 1 и 2 проведения полимеризации
Процедура получения сферических аддуктов
Микросфероидальный аддукт MgCl2·pC2H5OH получали в соответствии со способом, описанным в сравнительном примере 5 патента WO98/44009, с той разницей, что порошок BiCl3 в количестве 3 моль% по отношению к магнию, добавляли перед подачей масла. Аддукт содержит 11,2 вес. % Mg.
Процедура получения твердого компонента катализатора
В реактор с рубашкой охлаждения объемом 300 л, оснащенный механической мешалкой, холодильником и термопарой, загружали 200 л TiCl4 при комнатной температуре в атмосфере азота. После охлаждения до 0°C и при перемешивании последовательно добавляли диизобутилфталат и 8,0 кг сферического аддукта (полученного согласно вышеприведенному описанию). Концентрацию добавляемого внутреннего донора выдерживали, чтобы получить молярное соотношение Mg/донор равное 8. Температуру повышали до 100°С и поддерживали в течение 1 часов. Затем перемешивание прекращали, твердому продукту давали возможность осесть, а надосадочную жидкость сливали через сифон при 100°С. После удаления надосадочной жидкости дополнительно добавляли первичный TiCl4 для достижения начального объема жидкости. Затем смесь нагревали до 120°С и выдерживали при этой температуре в течение 1/2 часа. После этого перемешивание вновь прекращали, твердому продукту давали возможность осесть, а надосадочную жидкость сливали через сифон при 120°С. Обработку TiCl4 при 120°C повторяли вновь в том же порядке, но снизив время до 15 минут. Твердое вещество промывали безводным гексаном шесть раз с перепадом температур до 60°C и один раз при комнатной температуре. Полученное твердое вещество затем сушили в вакууме.
Осуществление предварительной полимеризации
Перед введением в реакторы полимеризации, твердый компонент катализатора, полученный описанным выше способом, вступал в реакцию с триэтилалюминием (TEAL) и дициклопентилдиметоксисиланом (D донор) в соотношении, указанном в Таблице 1. Затем полученную смесь, перед введением ее в реактор полимеризации, подвергали предварительной полимеризации, выдерживая ее в течение приблизительно 5 минут в виде суспензии в жидком пропилене при 20°С.
Полимеризация
В первом реакторе газофазной полимеризации этиленпропиленовый сополимер получают путем подачи в газообразном состоянии непрерывного и постоянного потока форполимеризованной каталитической системы, водорода (используемого в качестве регулятора молекулярного веса), пропилена и этилена.
Полученный в первом реакторе полиэтилен выгружают непрерывным потоком и после продувки потоком непрореагировавших мономеров, подают непрерывным потоком во второй газофазный реактор вместе с количественно постоянными потоками в газообразном состоянии водорода (при использовании), 1-бутена, этилена и пропилена.
Полимерные частицы, покидающие второй реактор, подвергаются обработке паром с целью удаления реакционно-способных мономеров и летучих веществ, а затем сушке.
Основные условия полимеризации приведены в Таблице 1. Свойства полимера приведены в Таблице 2.
Таблица 1
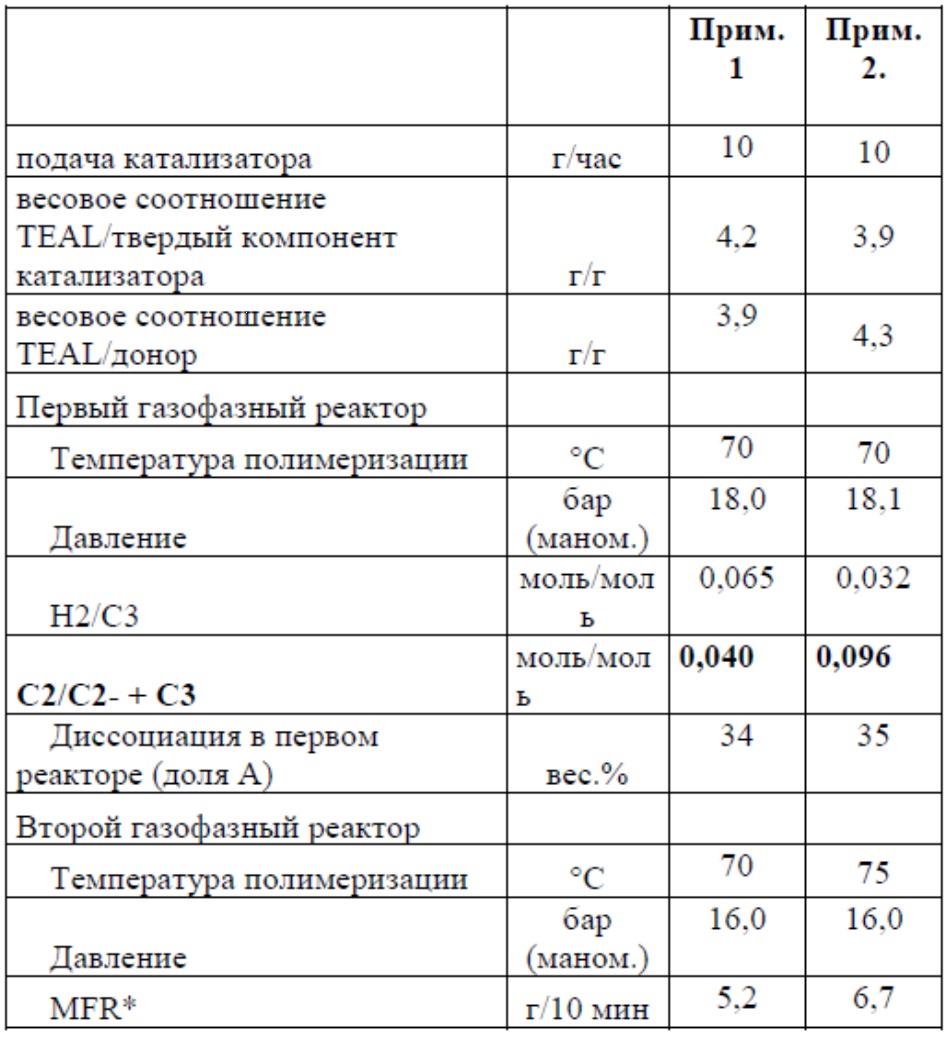
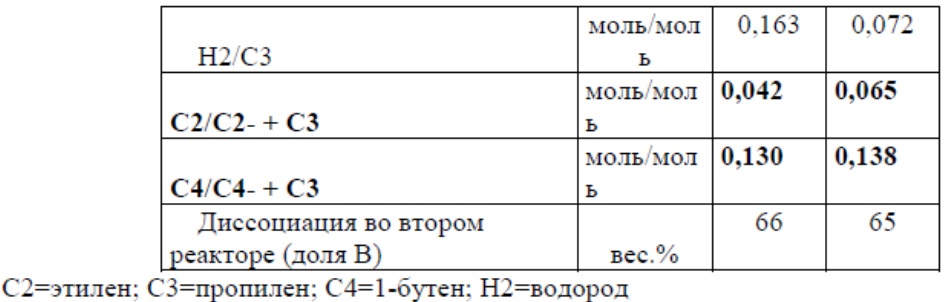
* общий MFR компонента A)+компонента B)
Таблица 2
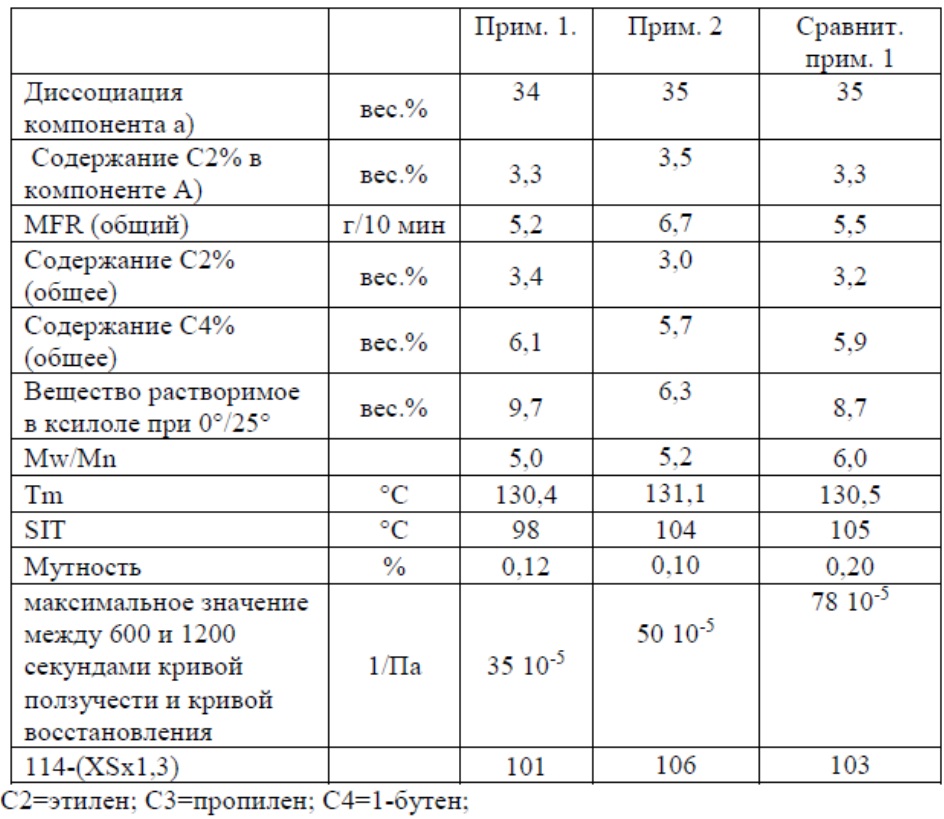
Сравнительный пример 1 представляет собой промышленное изделие, продаваемое компанией «Lyondellbasell» со свойствами, представленными в Таблице 2, и получаемое с использованием катализатора из Примеров 1 и 2, но не содержащее висмута.
Из Таблицы 2 следует, что терполимер, в соответствии с изобретением, показывает улучшенный баланс между веществами, растворимыми в ксилоле и SIT, имея при этом очень низкое значение кривой ползучести и кривой восстановления.
Реферат
Изобретение относится к составу полиолефина, пленке, изготовленной с использованием его, в том числе и изготовлению двухосноориентированной пленке. Состав полиолефина содержит: A) от 19 вес.% до 50 вес.% этиленпропиленового сополимера с содержанием этиленовых звеньев, составляющим от 1,5 вес.% до 6,0 вес.%; B) от 50 вес.% до 81 вес.% этиленпропиленового-1-бутенового терполимера с содержанием этиленовых звеньев, составляющим от 1,5 вес.% до 6,0 вес.% и содержанием 1-бутеновых звеньев, составляющим от 4,8 вес.% до 12,4 вес.%; где сумма компонента А) и В) составляет 100; состав, характеризуемый следующими свойствами: молекулярновесовым распределением (МВР), выраженным как соотношение Mw/Mn, превышающим 4,0; максимальным значением ниже 53x101/Па, между 600 и 1200 секундами, кривой ползучести и кривой восстановления, измеренным на расплавленном полимере при 200°C в соответствии с процедурой, приведенной в разделе определения характеристик. Изобретение позволяет получить полиолефин с необходимым молекулярно-массовым распределением и пленки на его основе с высокими эксплуатационными свойствами. 3 н. и 8 з.п. ф-лы, 2 табл., 2 пр., 1 ил.
Комментарии