Очищенное производное циклодекстрина, клатратный комплекс очищенного производного циклодекстрина с лекарственным средством, фармацевтическая композиция - RU2099354C1
Код документа: RU2099354C1
Чертежи







Описание
Изобретение относится к производным циклодекстрина и к их фармацевтическому применению в качестве клатратобразующих агентов.
Циклодекстрины (ЦД) представляют собой группу циклических гомологичных олигосахаридов, которые получают в результате разложения крахмала путем воздействия ферментом (циклодекстрин- трансглюкозилазой), вырабатываемым бактерией Bacillus macerans. Способы получения циклодекстрин-трансглюкозилазы, а также выделения циклодекстринов описаны в специальной литературе.
Циклодекстрины представляют собой циклические молекулы, содержащие шесть или более единиц α-D-глюкопиранозы, которые в 1,4-положениях связаны так же, как в амилозе. В результате такой циклической упаковки указанная молекула характерна тем, что она не имеет ни восстановительной концевой группы, ни не восстановительной концевой группы.
Ниже представлена схематическая формула указанной молекулы (I), где гидроксильные группы показаны в 2, 3 и 6-положениях единиц глюкопиранозы.

Переменная n может быть числом от 4 до 6, или выше.
Если n 4, то эта молекула известна как α-циклодекстрин или циклогексаамилоза; если n 5, то эта молекула является, как известно, b-циклодекстрином или циклогептаамилозой, а если n 6, то эта молекула известна как g-циклодекстрин или циклоктаамилоза. Под понятием циклодекстрин, используемым в настоящем описании, подразумеваются все указанные выше формы циклодекстрина, а также молекулы, в которых n > 6.
Считается, что вследствие циклической упаковки и конформации единиц a-D-глюкопиранозы, свободное вращение вокруг гликозидных связей ограничено и молекулы циклодекстринов имеют коническую форму, причем на малом конце конуса располагаются первичные гидроксильные группы, а у большого отверстия конуса располагаются вторичные гидроксильные группы. По границе полости располагаются атомы водорода от C3 и C5 вместе с глюкозидными кислородными атомами, в результате чего образуется относительно липофильная полость, но гидрофильная внешняя поверхность.
В результате образования двух отдельных полярных областей и изменений в структуре растворителя, которые имеют место при комплексообразовании, циклодекстрины обладают способностью образовывать комплексы с рядом органических и неорганических молекул. Образование циклодекстриновых комплексов включения с молекулами относится к так называемому феномену "хозяин-гость".
Эти уникальные свойства циклодекстринов позволяют использовать их в сельском хозяйстве, для обработки воды, в качестве поверхностно-активных веществ, а также в системе доставки лекарственного вещества к участку действия. Применение циклодекстринов в фармацевтической промышленности позволяет получить различные лекарственные средства в виде микрокапсул с пролонгированным высвобождением, с повышенной стабильностью и повышенной водной растворимостью.
Известно, что циклодекстрины способствуют увеличению скорости растворения лекарственных средств. Однако, при этом образованные комплексы являются также стабильными в водном растворе, так что повышение скорости растворения сопровождается повышением растворимости с насыщением лекарственного средства. К сожалению, указанный выше b-циклодекстрин, который образует наиболее стабильные комплексы с большинством лекарственных средств, обладает наименьшей водорастворимостью, поэтому лекарственные средства, образующие комплексы с ним, не могут быть введены в раствор в терапевтических концентрациях. Этот факт, очевидно, обусловлен кристаллической структурой самого b-циклодекстрина.
Известно, что химическая модификация циклодекстринов приводит к модулированию их свойств. Электронейтральные циклодекстрины описаны Parmerter и др. (патент США N 3453259) и Gramera и др. (патент США N 3459731). Эти соединения были получены путем реакции конденсации циклодекстринов с различными эпоксидами или органическими галидами.
Были получены и другие производные, например, циклодекстрины с катионными свойствам (Parmerter (I); патент США N 3453257), нерастворимые структурированные циклодекстрины (Solms; патент США N 3420788), и циклодекстрины с анионными свойствами (Parmerter (II); патент США N 3426011). Для получения циклодекстринов с анионными свойствами, к исходным циклодекстринам добавляли карбоновые кислоты, фосфористые кислоты, фосфиновые кислоты, фосфоновые кислоты, фосфорные кислоты, тиофосфиновые кислоты, тиофосфоновые кислоты и сульфоновые кислоты (см. Parmerter (II), см.выше).
Циклодекстрины нашли свое применение в фармацевтическом производстве для обеспечения системы доставки лекарственного средства к участку действия. В качестве "хозяев" для молекул лекарственного средства, называемых "гостями", указанные комплексы включения (или клатраты) способствуют повышению водорастворимости фармацевтических средств, которые по своей природе имеют низкую водорастворимость (Jones, патент США N 4555504).
Указанная солюбилизация некоторых лекарственных средств способствует повышению их биологической доступности. В виде клатратных комплексов некоторые лекарственные средства приобретают повышенную химическую стабильность в водном растворе (Harada и др. патент США N 4497803 и Hayashi и др. патент США N 3816394). Кроме того, циклодекстрины являются эффективными для сообщения высокорастворимым в воде лекарственным средствам способности к пролонгированному высвобождению (Friedman, патент США N 4774329).
Несмотря на все указанные преимущества использования циклодекстринов в фармакологии, однако имеются некоторые факторы, ограничивающие их использование в этой области. Например, использование циклодекстринов в клинических условиях должно быть ограничено пероральными лекарственными формами и лекарственными формами для местного применения, поскольку циклодекстрины при попадании в организм в неметаболизированном состоянии являются нефротоксичными. Так как ферменты млекопитающих являются специфическими к деградации линейных молекул крахмала, то циклодекстрины остаются, в основном, неметаболизированными, и в результате рециркуляции и реадсорбции они аккумулируются в клетках проксимальных канальцев.
Циклодекстрины и их производные являются, в основном, кристаллическими твердыми веществами, и их концентрация в почечных тканях с последующим образованием кристаллов может вызвать некротические поражения клеток. Несмотря на способность циклодекстринов образовывать водорастворимые клатратные комплексы, комплексы кристаллического циклодекстрина с лекарственными средствами могут быть использованы лишь в фармацевтических формах для подъязычного введения.
В соответствии с этим, были предприняты усилия, направленные на разработку способа ингибирования образования кристаллов в комплексах циклодекстрина и лекарственного средства путем дериватизации исходных циклодекстринов неспецифическим образом в целях получения аморфных смесей, содержащих много компонентов в виде циклодекстриновых производных (см. Pitha, патенты США N 4596795 и N 4727064). Эти смеси препятствуют процессу кристаллизации циклодекстринов в комплексах с отдельными соединениями, сообщая тем самым этим комплексам пониженную токсичность.
Настоящее изобретение относится к очищенным производным циклодекстрина, присутствующим как в виде отдельных производных, так и в виде смеси производных. Указанные соединения получают путем нагревания исходного циклодекстринового материала с реагентом (или реагентами), который вводит в циклодекстриновую молекулу специфический заместитель анионного типа, т. е. (C2-6-алкилен)-SO-анионный заместитель. Как было установлено, эти соединения обладают особенно повышенной водорастворимостью и преимущественно низкой токсичностью. Кроме того, было обнаружено, что более высокозамещенные циклодекстриновые производные, преимущественно, не вызывают разрушения мембраны. Такие дериватизированные циклодекстрины могут быть использованы в качестве клатрат-образующих агентов в парентеральных фармацевтических препаратах, и в других целях, аналогичных вышеуказанным.
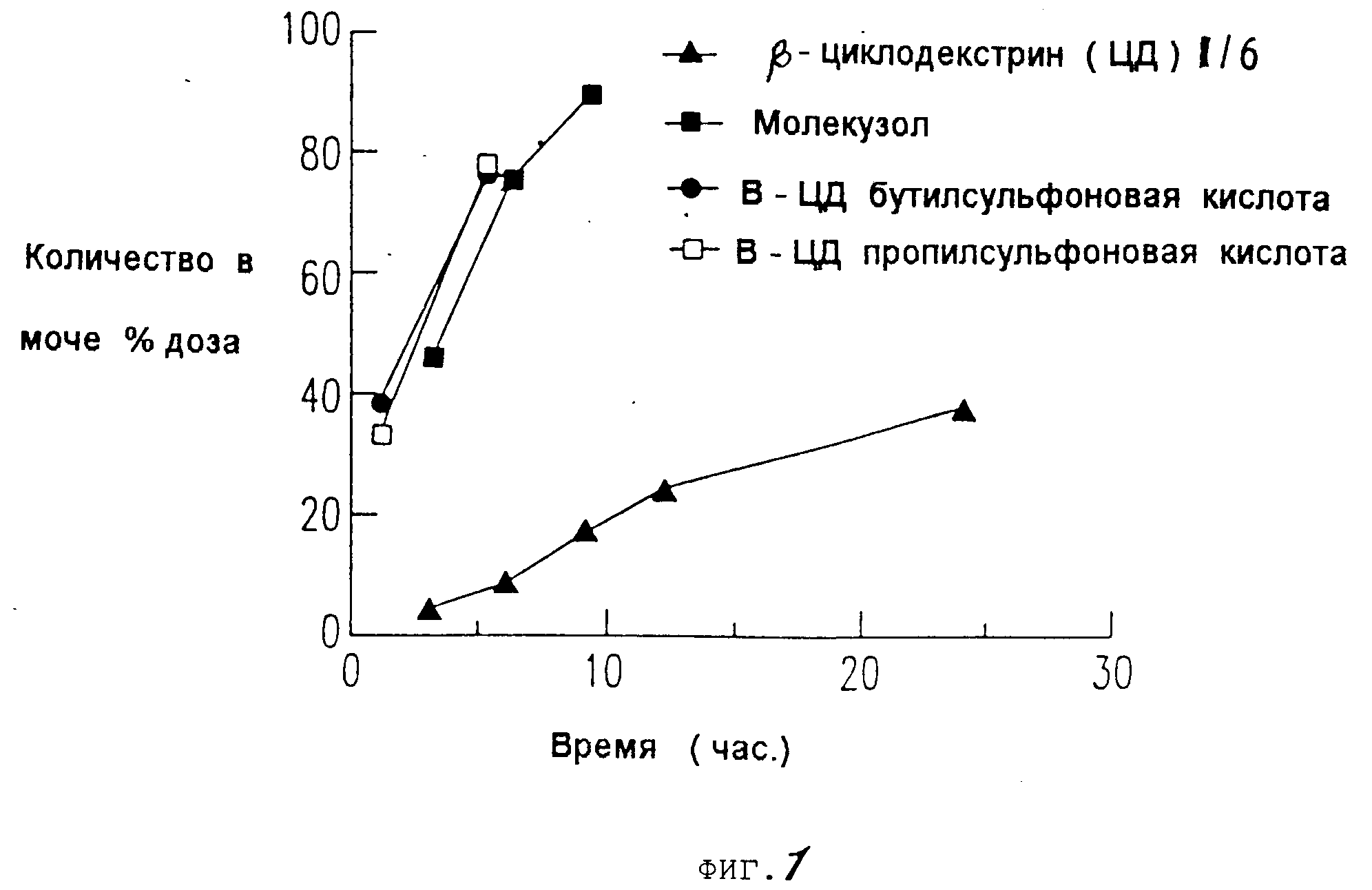
На фиг. 1 представлен график кумулятивного выделения циклодекстрина с мочой у мышей для случаев: недериватизированного циклодекстрина, гидроксипропил-дериватизированного циклодекстрина и двух сульфоалкиловых производных циклодекстрина.
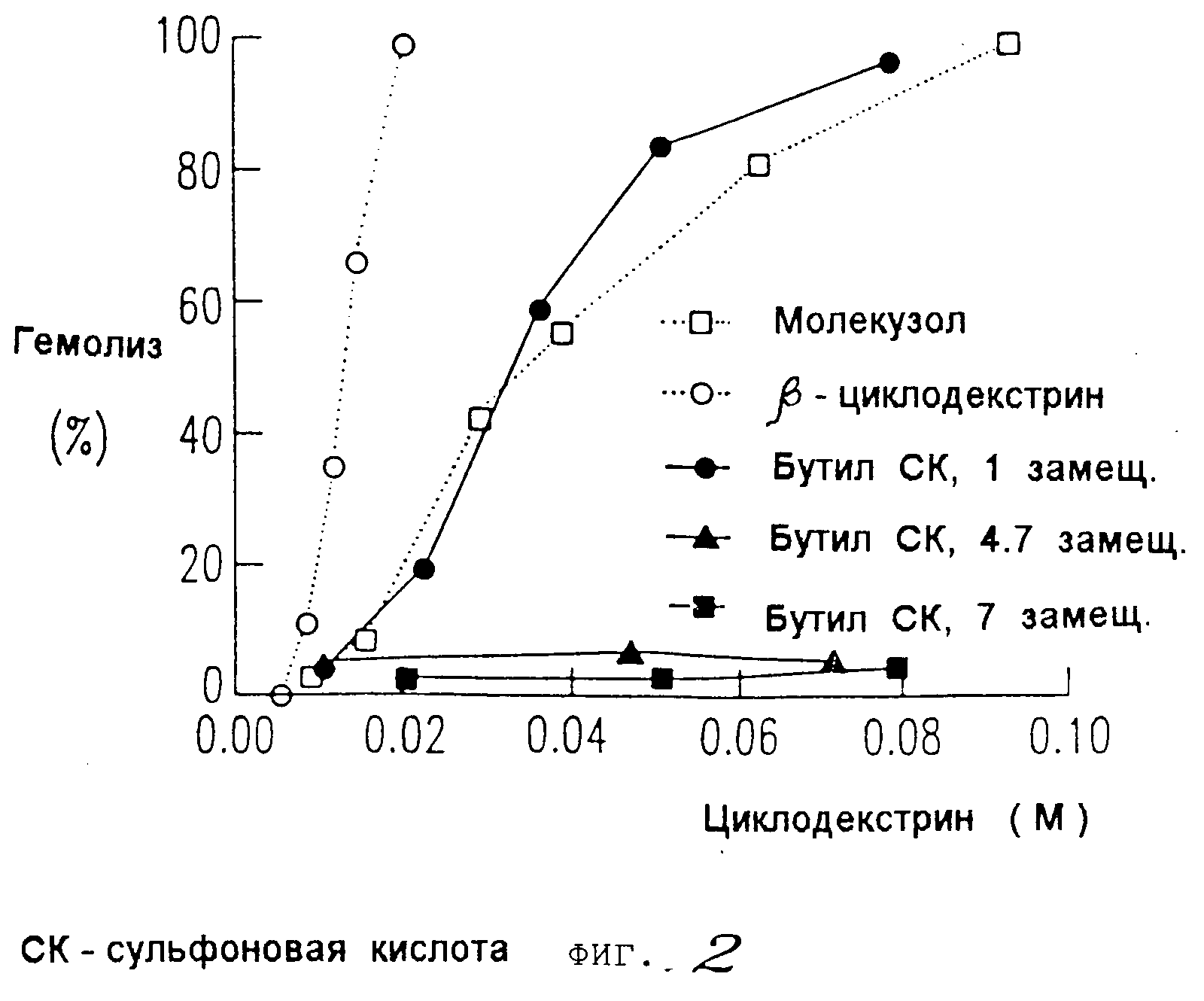
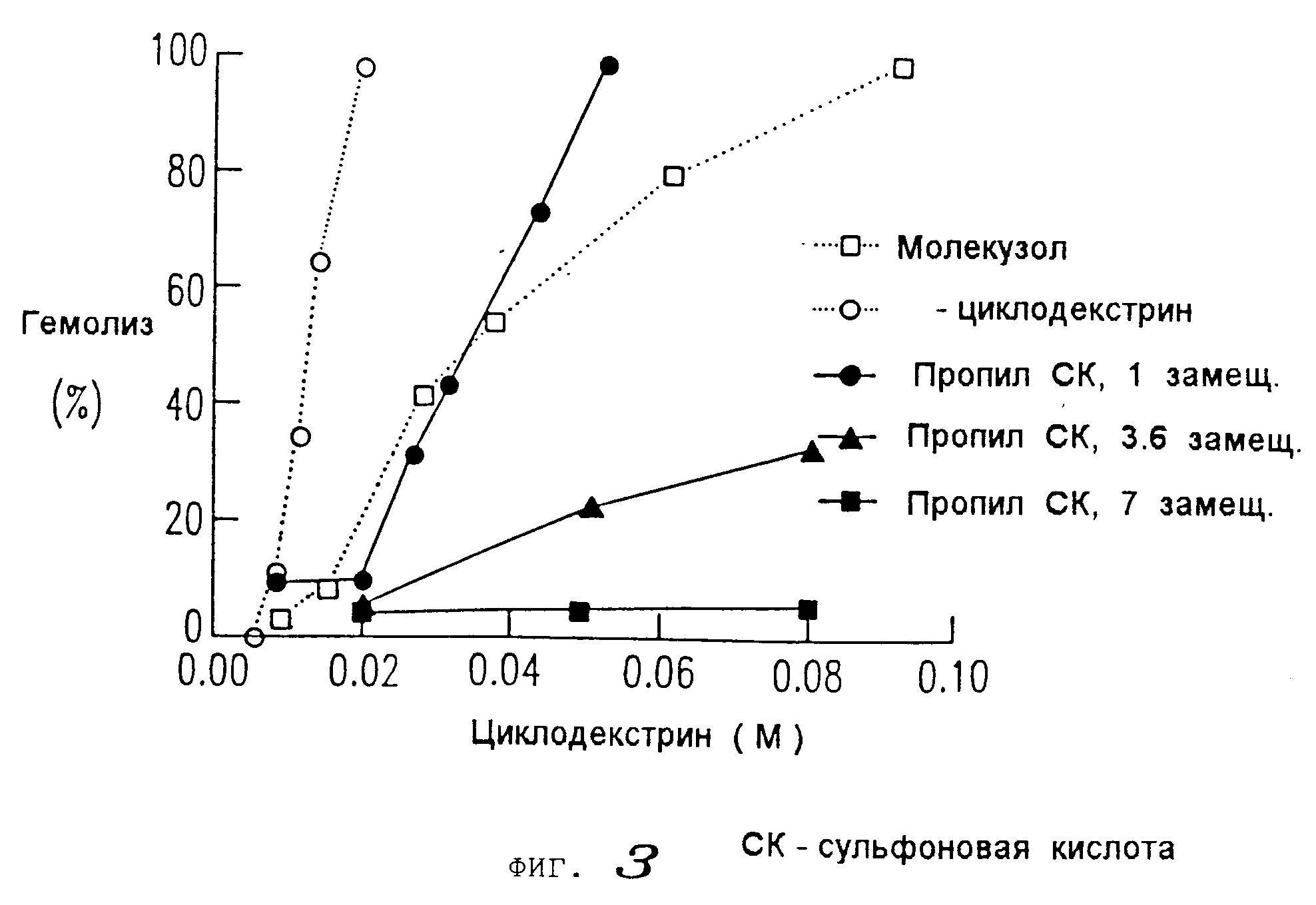
На фиг. 2 и 3 представлены данные, показывающие, что более высокозамещенные алкилсульфоновые кислоты настоящего изобретения вызывают меньшее разрушение мембраны (как было определено путем исследования гемолиза эритроцитов) по сравнению с монозамещенными производными алкилсульфоновой кислоты с недериватизированным циклодекстрином, вызывающим наибольшее разрушение мембраны; и что монозамещенные производные алкилсульфоновой кислоты настоящего изобретения вызывает такую же степень разрушения мембраны, как и гидроксипропиловое производное циклодекстрина (как было также определено путем исследования гемолиза эритроцитов).
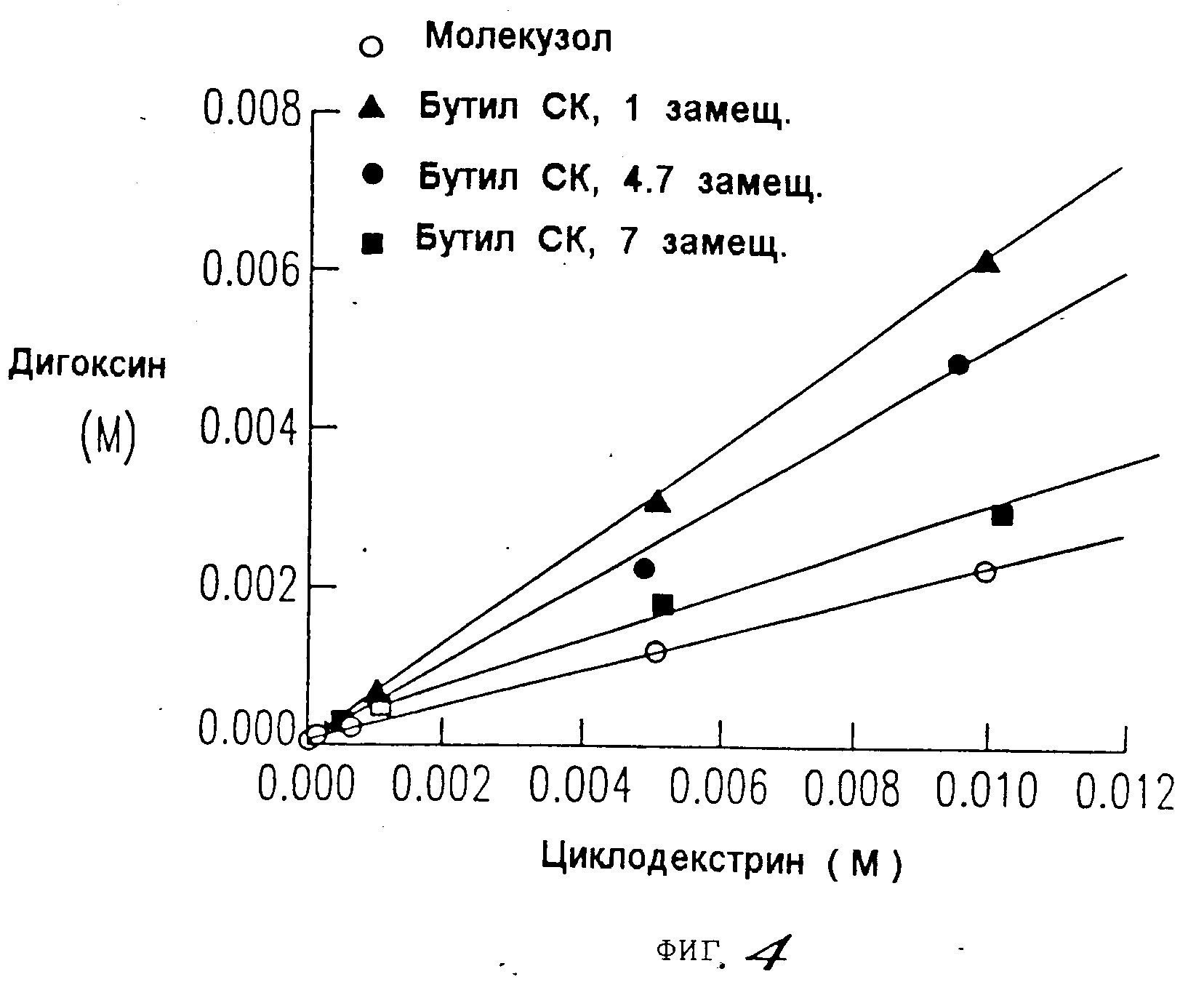
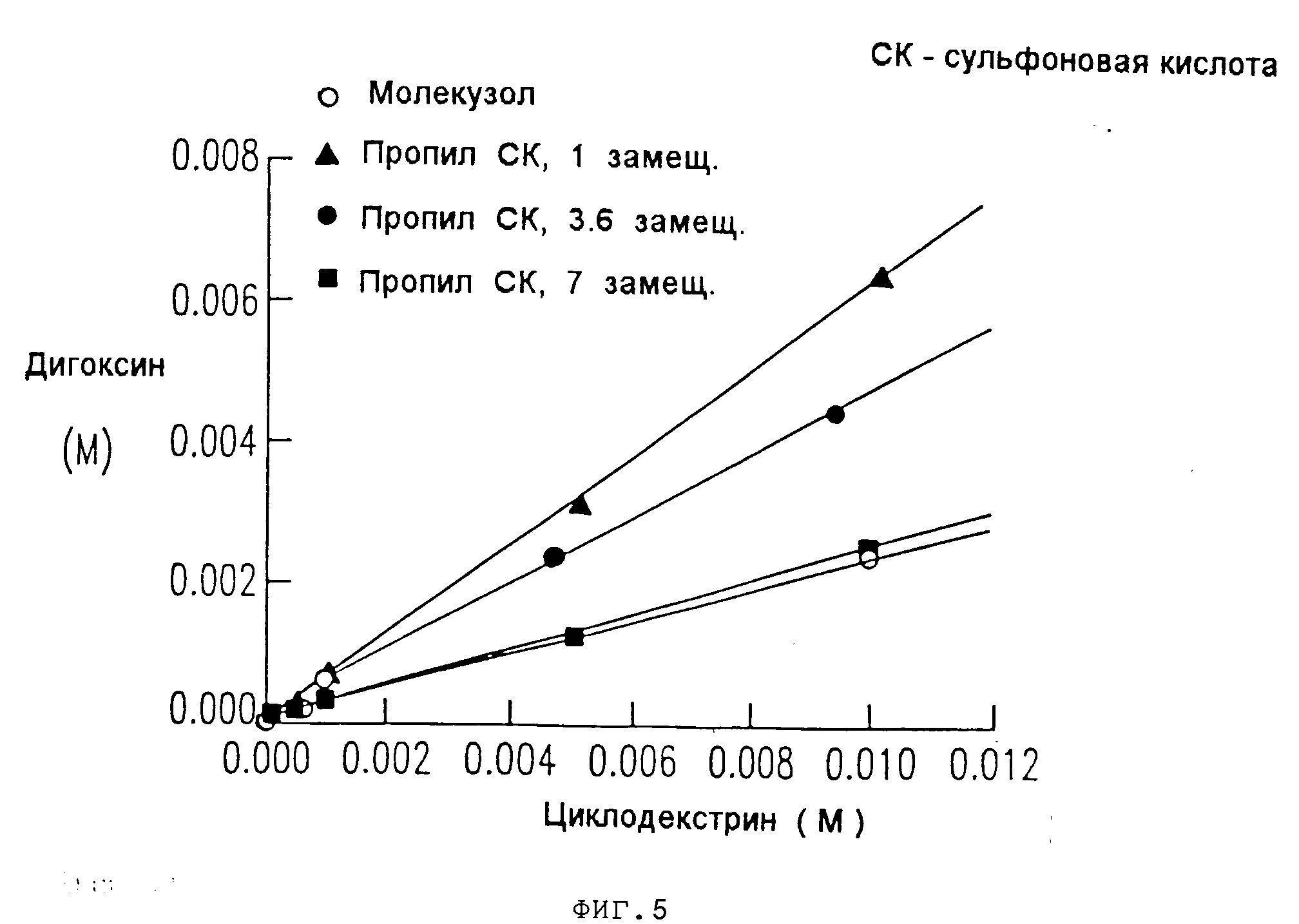
На фиг.4, 5, 6, 7 показано, что константы ассоциации для равновесия между сульфоалкил-циклодекстриновыми производными настоящего изобретения и дигоксином или прогестероном значительно выше, чем константа ассоциации для равновесия между гидроксипропил-циклодекстриновым производным и дигоксином или прогестероном, соответственно.
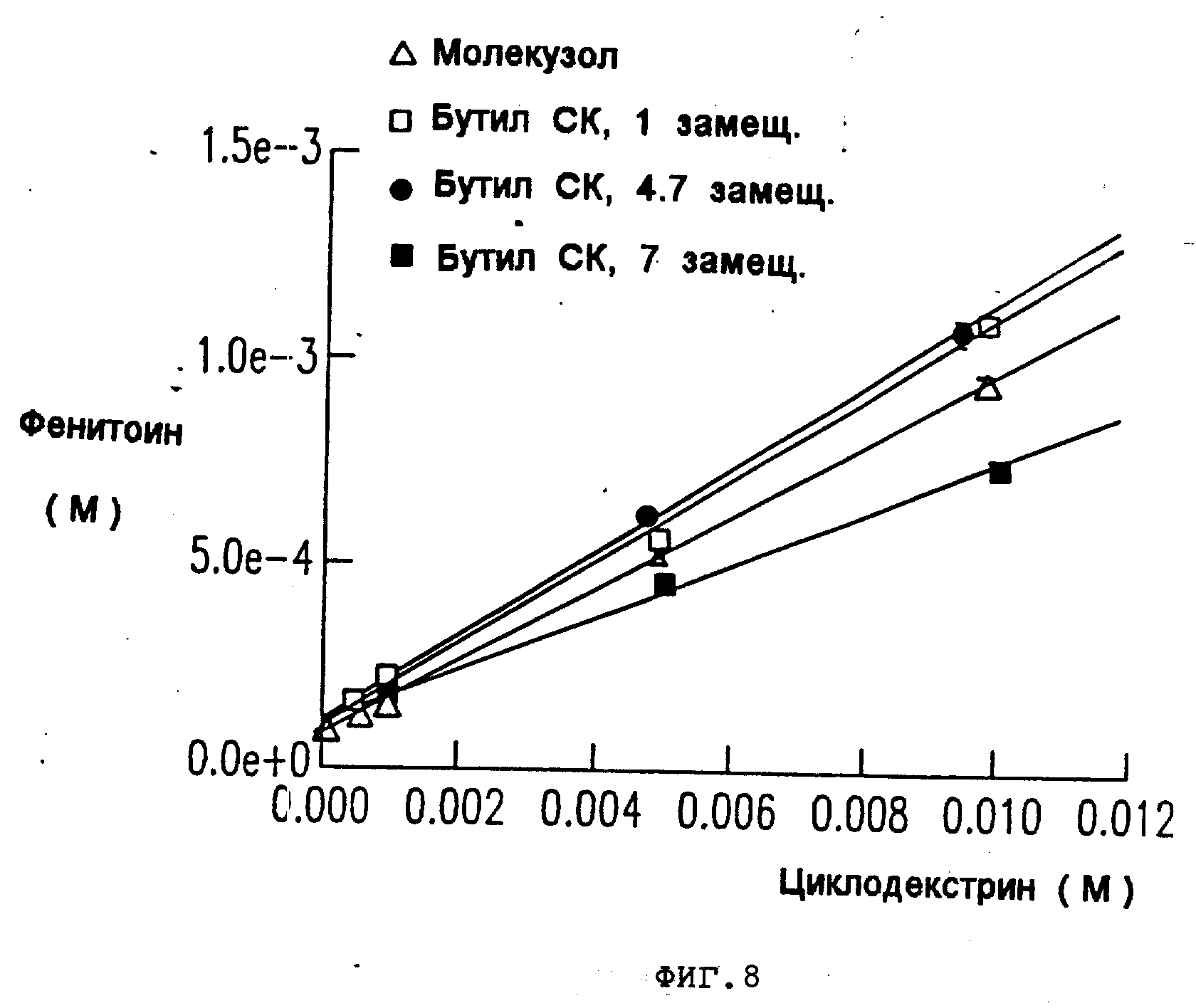
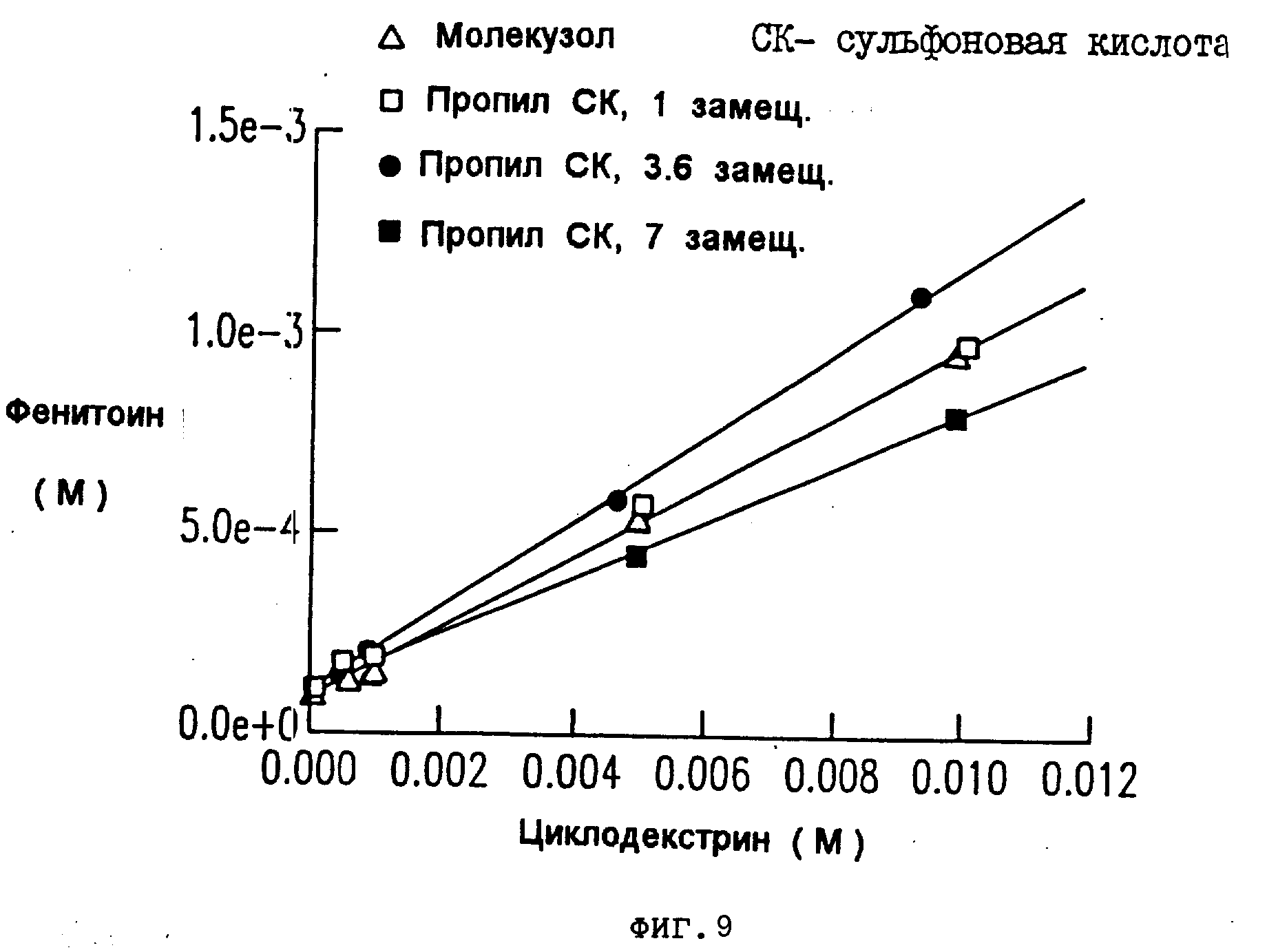
На фиг.8, 9, 10, 11 показано, что в комплексе с фенитоином и тестостероном, сульфоалкил-циклодекстриновые производные настоящего изобретения имеют особенно высокие константы ассоциации по сравнению с гидроксипропил-циклодекстриновым производным.
Настоящее изобретение относится к циклодекстриновым производным, предназначенным для фармацевтического использования. Эти производные могут быть использованы в качестве клатратобразующих агентов в сочетании с лекарственными средствами в целях получения клатратных комплексов, предназначенных для парентеральных и других фармацевтических препаратов. В настоящем изобретении также описаны способы получения и выделения производных циклодекстрина.
Производные циклодекстрина настоящего изобретения имеют функциональные группы (C2-6-алкилен)-SO и поэтому являются заряженными молекулами. Обнаружение того, что указанные соединения обладают очень низким уровнем токсичности, является неожиданным в свете предшествующих работ в данной области, где высказывается мнение, что производные циклодекстрина должны оставаться электронейтральными для сохранения своей нетоксичности (см. Pitha, "Amorphous Water-Soluble" Third Int'l Symposium on Recent Advances in Drug Delivery Systems, Salt Lake City, Utah, Feb. 23-27, 1987).
Тот факт, что производные циклодекстринов настоящего изобретения имеют высокую водорастворимость и низкую нефротоксичность, также являются неожиданным в свете раскрытия патента США N 4727064, где указывается, что для поддержания высокого уровня растворимости для циклодекстриновых производных, необходимо использовать смесь этих производных.
Водорастворимость, которой обладают сульфоалкил-циклодекстриновые производные настоящего изобретения, может быть получена путем сольватации частей сульфоновой кислоты. Таким образом, гетерогенная смесь производных циклодекстринов настоящего изобретения не является необходимым условием для того, чтобы имело место заметное повышение сольватации. Хотя в соответствии с настоящим изобретением может быть использована смесь сульфоалкилэфирных производных, однако для повышения растворимости использование такой смеси не является обязательным.
В
предпочтительном варианте (I) осуществления настоящего изобретения, производные циклодекстрина имеют структуру, которая может быть представлена следующей формулой (2):
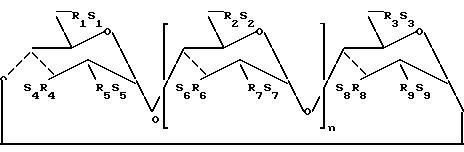
где n 4, 5 или 6;
каждый из R1, R2, R3, R4, R5, R6, R7, R8 и R9 являются независимо 0- или 0-(C2-6-алкилен)-SO-группой, где, по крайней мере, один из R1 и R2 является независимо 0-(C2-6-алкилен)-SO-группой, а предпочтительно 0-(CH2)m-SO -группой, где m 2 6, а предпочтительно 2 4 (например, OCH2CH2CH2SO или OCH2CH2CH2CH2
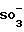
каждый из S1, S2, S3, S4, S5, S6, S7, S8 и S9 являются независимо фармацевтически приемлемым катионом, например, H+, ионом щелочного металла (например, Li+, Na+), щелочноземельного металла (например, Ca+2, Mg+2), аммония, и катионом аминов, например, C1-6-алкиламинов, пиперидина, пиразина, C1-6-алканоламина и C4-8-циклоалканоламина.
В другом предпочтительном варианте (2) осуществления настоящего изобретения:
R1 является 0-(C2-6-алкилен)-
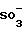

R2-R9 являются 0-;
S1-S9 являются такими, как они были определены в варианте (1) (см.выше).
В
другом предпочтительном варианте (3) осуществления настоящего изобретения:
каждый из R1, R2 и R3 являются независимо 0-(C2-6-алкилен)- SO группой, а предпочтительно 0-(CH2)m-SO-группой (например, OCH2CH2CH2SO или OCH2CH2CH2CH2SO);
R4-R9 являются 0-; и
S1-S9 являются такими, как они были определены в варианте (1), (см.выше).
В
другом предпочтительном варианте (4) осуществления настоящего изобретения:
R1-R3 являются такими, как они были определены в вариантах (2) и (3), (см.выше);
по
крайней мере, один из R4, R6 и R8 является 0-(C2-6-алкилен)-SO-группой, а предпочтительно 0-(CH2)m-SO-группой (например,
OCH2CH2CH2SO или OCH2CH2CH2
CH2SO).
R5, R7 и R9 являются 0-; и
S1-S9 являются такими, как они были определены в варианте (1) (см.выше).
В другом предпочтительном варианте (6) осуществления настоящего
изобретения:
R1, R2, R3, R4, R5, R6 и R8 независимо являются 0-(C2-6-алкилен)-SO-группой, а предпочтительно 0-(CH2)m-SO-группой (например,


R5, R7 и R9 являются 0-; и
S1-S9 являются такими, как они
были определены в варианте (I) (см.выше).
Термины алкилен и алкил, используемые в настоящем описании (например, в 0-(C2-6-алкилен)-SO-группе или в алкиламинах), означают линейные или разветвленные, насыщенные или ненасыщенные (т.е. содержащие одну двойную связь) бивалентные алкиленовые группы и моновалентные алкильные группы, соответственно. Аналогично, термин алканол, используемый в настоящем описании, означает линейные или разветвленные, насыщенные или ненасыщенные алькильные компоненты алканольных групп, в которых гидроксильные группы могут находиться в любом положении на алкильной части. Термин циклоалканол означает незамещенные или замещенные (например, метиловые или этиловые) циклические спирты.
Настоящее изобретение относится к композициям, содержащим смесь производных циклодекстрина, имеющих структуру, представленную формулой (2), причем указанные композиции, в среднем, содержат, по крайней мере, от 1 до 3n + 6 частей алкилсульфоновой кислоты на одну молекулу циклодекстрина. Настоящее изобретение также относится к композициям, содержащим, в основном, лишь один-единственный тип циклодекстринового производного.
Производные циклодекстрина настоящего изобретения являются либо замещенными, по крайней мере, в одной первичной гидроксильной группе (т.е. по крайней мере, один из R1-R3 является заместителем), либо они являются замещенными как в первичной гидроксильной группе, так и в 3-положении гидроксильной группы (т.е. по крайней мере, один из R1-R3 и, по крайней мере, один из R4, R6 и R8 является заместителем). Замещение во 2-положении гидроксильной группы, хотя теоретически возможно (как показали исследования, проведенные авторами настоящей заявки), однако, фактически, оно, очевидно, не происходит в продуктах настоящего изобретения.
Производные циклодекстринов настоящего изобретения получают (как описывается ниже) в виде очищенных композиций, т.е. композиций, содержащих, по крайней мере, 95 мас. циклодекстринового(ных) производного(ных), имеющего замещение, по крайней мере, на первичной гидроксильной группе молекулы циклодекстрина (т. е. R1, R2 или R3 формулы (2) как было определено с помощью 300 МГц1H-ЯМР). В предпочтительном варианте осуществления настоящего изобретения очищенные композиции содержат, по крайней мере, 98 мас. производного(ных) циклодекстрина.
Указанные соединения значительно отличаются от соединений, раскрытых в патенте США N 3426011, где получают лишь продукты реакции циклодекстрина с сультоновым реагентом. В результате, эти реакционные продукты содержат значительное количество незамещенного циклодекстринового исходного материала.
Во всех композициях настоящего изобретения, непрореагировавший циклодекстрин, в основном, удаляется с остаточными примесями, составляющими ≅ 5 мас. от композиции и не оказывающими неблагоприятного воздействия на качество композиции, содержащей производное циклодекстрина.
Было обнаружено, что более высокозамещенные алкилсульфоновокислотные производные циклодекстрина настоящего изобретения обладают, помимо особенно повышенной растворимости и низкой токсичности, преимущественным свойством, способствующим меньшему разрушению мембраны. В результате проведенных исследований по гемолизу эритроцитов было показано, что более высокозамещенные производные циклодекстрины вызывают ничтожно малую степень разрушения мембраны. Монозамещенные производные циклодекстрина вызывают примерно такой же уровень разрушения мембраны, что и гидроксипропиловое производное.
Рассматриваемые производные циклодекстрина могут быть получены, в основном, путем растворения циклодекстрина в водном основании при соответствующей температуре, например при 70-80oC, при возможно наиболее высокой концентрации. Например, для получения производных циклодекстрина варианта (4) (см. выше), к циклодекстрину добавляют некоторое количество подходящего алкилсультона, соответствующее числу молей первичной гидроксильной группе, присутствующей в ЦД, энергично размешивая при этом для обеспечения максимального контакта гетерогенной фазы.
Для получения производных циклодекстрина варианта (2) (см.выше) используют молярное количество алкилсультона, соответствующее числу молей ЦД. Любой специалист может легко определить, что для получения производных циклодекстрина варианта (1), который включает в себя как вариант (4), так и вариант (2), количество алкилсультона должно находиться в пределах между установленными выше значениями. Другие ЦД, используемые в настоящем изобретении, получены Mutatis Mutandis.
Смеси подвергают реакции до тех пор, пока не получат одну фазу, что указывает на истощение алкилсультона. Затем реакционную смесь разбавляют равным объемом воды и нейтрализуют кислотой, такой, как соляная кислота. После этого раствор диализуют для удаления примесей, а затем концентрируют путем ультрафильтрации.
Концентрированный раствор подвергают ионнообменной хроматографии для удаления непрореагировавшего циклодекстрина, и после осушки вымораживанием получают целевой продукт.
ЦД, используемые в настоящем изобретение, могут быть любыми ЦД, полученными известными способами, например путем обработки крахмала циклодекстрин-глюканотрансферазой (CGTase, E.C. 2.4.1.19). Таким образом, под понятием "ЦД" в настоящем описании подразумеваются α-ЦД, в которых шесть единиц глюкозы связаны вместе с помощью a-1,4-связи; b-ЦД, в которых семь единиц глюкозы связаны вместе; или g-ЦД, в которых восемь единиц глюкозы связаны вместе; или их смеси. ИЗ указанных ЦД для получения частично дериватизированных продуктов широкого применения наиболее предпочтительными являются b-ЦД.
Как было указано выше и в зависимости от типа целевого производного циклодекстрина (ЦД), количество алкилсультона, используемого в качестве дериватизирующего агента, не должно превышать приблизительно 1 молярный эквивалент в отношении числа первичных гидроксильных групп, присутствующих в ЦД, хотя его оптимальное количество до некоторой степени зависит от концентрации реагентов. В качестве ускорителя могут быть использованы гидроксид лития, гидроксид натрия и гидроксид калия. Из экономических соображений, наиболее предпочтительным является гидроксид натрия. Его количество должно быть более 30 молярных эквивалентов, а предпочтительно от 80-200 молярных эквивалентов, при уровне концентрации реагента, превышающем 10% (по массе), а предпочтительно от 40-60% (по массе).
В качестве реакционной среды может быть использован любой растворитель, который является инертным в отношении частичного алкилирования. Примерами таких растворителей являются вода, ДМФ, ДМСО и их смеси, однако использование только одной воды наиболее предпочтительно для облегчения последующей обработки.
Тип и концентрация алкилсультона и щелочи не являются критическими для данной реакции. Однако, в основном, реакцию осуществляют при размешивании в течение 1 ч при температуре 10-80oC, а предпочтительно, в течение 5-20 ч при температуре 20-50oC.
Для выделения и очистки целевых соединений из реакционной смеси может быть использована стандартная техника, обычно применяемая в таких случаях. В качестве примеров могут служить экстракция органическими растворителями, диализ, адсорбционная хроматография с использованием активированного угля, силикагеля, окиси алюминия и других адсорбентов; хроматография с использованием в качестве носителя структурированный декстрин, сополимеры стирола и дивинилбензола, также другие структурированные полимеры и их комбинации.
Клатратные комплексы настоящего изобретения могут быть получены любыми известными способами, обычно используемыми для получения комплексов циклодекстрина.
Например, для получения клатратных комплексов, производное циклодекстрина, растворенное в воде или в смешивающемся с водой органическом растворителе, может быть добавлено к физиологически активному соединению (лекарственному средству), растворенному в смешивающемся с водой органическом растворителе. Затем смесь нагревают, а нужный продукт получают путем концентрирования смеси при пониженном давлении или путем его осаждения после охлаждения. В этом случае отношение органического растворителя и воды в смеси может соответствующим образом варьироваться в зависимости от растворимостей исходных материалов и продуктов.
Примерами лекарственных средств, которые могут образовывать комплексы с производными циклодекстрина, являются: дифенилгидантоин, адифенин, аллобарбитал, аминобензойная кислота, аминобарбитал, ампициллин, анетол, аспирин, азопропазон, азуленбарбитуратовая кислота, беклометазон, беклометазона дипропионат, бенциклан, бензальдегид, бензокаин, бензодиазепины, бензотиазид, бетаметазон, бетаметазон 17-валерат, бромбензойная кислота, бромоизовалеримочевина, бутил-п-аминобензоат, хлоралгидрат, хлорамбуцил, хлорамфеникол, хлоробензойная кислота, хлорпромазин, коричная кислота, клофибрат, кофермент А, кортизон, кортизонацетат, циклобарбитал, циклогексилантранилат, деоксихолевая кислота, дексаметазон, дексаметазонацетат, диазепам, дигитоксон, дигоксин, экстрадиол, флуфенаминовая кислота, флуоцинолонацетонид, 5-фторурацил, флурбипрофен, гризеофульвин, гвайазулен, гидрокортизон, гидрокортизона ацетат, ибупрофен, индикан, индометацин, йод, кетопрофен, ланкацидиновая группа антибиотиков, мефанамовая кислота, менадион, мефорбарбитал, метбарбитал, метициллин, метронидазол, метомицин, нитразепам, нитроглицерин, нитромочевины, параметазон, пенециллин, пентобарбитал, фенобарбитал, фенобарбитон, фенил-бутировая кислота, фенил-валериановая кислота, фенитоин, преднизолон, преднизолона ацетат, прогестерон, пропилпарабен, просцилларидин, серии простагландина A, серии простагландина B, серии простагландина E, серии простагландина F, хинолон противомикробное средство, резерпин, спиронолактон, сульфацетамид натрия, сульфонамид, тестостерон, талидомид, тиамин, дилаурилсульфат, тиамфениколпалмитат, тиопентал, триамцинолон, витамин A, витамин D3, витамин E, витамин K3 и варфарин.
Указанные лекарственные средства могут быть растворены в воде или органическом растворителе (либо смешиваемом, либо несмешиваемом с водой). Примерами подходящих растворителей могут служить: диэтилэфир, тетрагидрофуран, диоксан, ацетон, диметилсульфоксид, диметилформамид и низшие алифатические спирты. В предпочтительном варианте осуществления настоящего изобретения лекарственное средство растворяют либо в воде, либо в смешивающемся с водой растворителе, таком, как метанол или этанол. Лекарственное средство может быть также суспендировано в воде.
После установления равновесия полученный комплекс может быть выделен с помощью стандартной техники, такой, как лиофилизация, выпаривание растворителя, преципитация, кристаллизация при низкой температуре или распылительная сушка. Циклодекстриновые комплексы включения могут быть также получены путем физического измельчения или размешивания циклодекстрина и молекул-гостей с небольшим количеством растворителя или без растворителя.
Для получения клатратных комплексов настоящего изобретения могут быть использованы любые подходящие соотношения производного циклодекстрина и лекарственного средства, но предпочтительно, если производное циклодекстрина используется в молярном избытке.
В соответствии с настоящим изобретением преимущественный результат может быть получен при молярном соотношении производного циклодекстрина и лекарственного средства в пределах 10: 1 1:10, а предпочтительно 2:1 5:1 (например, 3: 1) и при использовании способов и отношений, описанных выше. Полученные комплексы обычно содержат до 20 мас. лекарственного средства. Однако, ввиду использования обычно низких доз лекарственного средства и трудности получения гомогенных смесей активного ингредиента и наполнителей, предпочтительно изготавливать комплексы с избыточным содержанием производного циклодекстрина, т.е. комплексы, содержащие около 0,1-10 мас. лекарственного средства, а предпочтительно 0,5-0,2 мас. лекарственного средства.
Получение клатратных комплексов настоящего изобретения позволяет избрать более предпочтительный путь введения лекарственного средства, причем в указанных комплексах циклодекстрин действует лишь в качестве солюбилизирующего агента, не влияя при этом на терапевтическое действие лекарственного средства в любом случае.
Настоящее изобретение также относится к комплексам включения, определенным в настоящей заявке и предназначенным для использования в медицине или ветеринарии. Комплекс для использования в качестве фармацевтического средства может быть представлен в виде фармацевтического препарата.
Таким образом, в одном из своих вариантов настоящее изобретение относится к фармацевтической композиции, содержащей клатратный комплекс, включающий в себя лекарственное средство и производное циклодекстрина в сочетании с фармацевтически приемлемым носителем и необязательно с другими терапевтическими и/или профилактическими ингредиентами. Под термином "приемлемый" по отношению к носителю подразумевается, что указанный носитель является совместимым с другими ингредиентами препарата и не оказывает вредного воздействия на реципиента. Предпочтительно, чтобы фармацевтический препарат был изготовлен в виде разовых лекарственных форм. При этом, желательно, чтобы каждая разовая форма содержала такое количество лекарственного средства, которое обычно вводится в организм в одноразовой дозе указанного средства при отсутствии циклодекстрина.
Фармацевтические композиции могут быть изготовлены в виде любого препарата, содержащего комплексы включения, предназначенные для перорального, интранасального, внутриглазного или парентерального (например, внутримышечного и внутривенного) введения. По необходимости, указанные препараты могут быть изготовлены в виде дробных лекарственных форм стандартными способами, обычно применяемыми в фармации. Эти способы предусматривают стадию введения активного соединения в сочетании с жидкими носителями или с тонко измельченными твердыми носителями, или с теми и другими, а затем, если это необходимо, стадию формования продукта в соответствующий препарат.
Фармацевтические композиции, предназначенные для перорального введения и содержащие твердый носитель, предпочтительно изготавливать в виде разовых лекарственных форм, таких, как болюсы, капсулы, облатки или таблетки, каждая из которых содержит заранее определенное количество активного ингредиента. Таблетки могут быть изготовлены путем прессования или формования, при этом, по желанию, они могут содержать один или несколько дополнительных ингредиентов. Такие таблетки могут быть получены путем прессования в соответствующем устройстве активного соединения, находящегося в свободно текущем состоянии, например в виде порошка или гранул, необязательно смешанных со связующим, замасливателем, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки могут быть получены путем формования из инертного жидкого разбавителя. По желанию, таблетки могут быть покрыты оболочкой или, если они остаются непокрытыми, на их поверхности могут быть сделаны (но необязательно) линии-бороздки. Капсулы могут быть получены путем наполнения оболочки капсулы активным соединением, взятым отдельно или в смеси с одним или несколькими дополнительными ингредиентами, с последующей герметизацией указанной капсулы стандартным способом. Облатки, которые представляют собой лекарственную форму, аналогичную капсулам, содержат активный ингредиент в сочетании с добавкой (или добавками), упакованные в оболочку из рисовой "бумаги".
Указанные таблетки содержат активный ингредиент в сочетании с нетоксичными фармацевтически приемлемыми наполнителями, пригодными для изготовления таблеток. Такими наполнителями могут быть, например, инертные разбавители, лактоза, фосфат кальция или натрия, гранулирующие или дезинтегрирующие агенты, например кукурузный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин или аравийская камедь; и замасливающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки могут оставаться непокрытыми или могут быть покрыты оболочкой с помощью известной техники для замедления их дезинтеграции и адсорбции в желудочно-кишечном тракте, тем самым сообщая этим лекарственным препаратам пролонгированное действие. В этих целях могут быть использованы такие материалы, как моностеарат или дистеарат глицерина, взятые отдельно или в сочетании с воском.
Настоящее изобретение также относится к комплексам, содержащимся в фармацевтических препаратах с пролонгированным высвобождением лекарственного средства. Указанные фармацевтические препараты, т.е. препараты с пролонгированным действием, в основном, известны специалистам. Таким препараты представляют собой системы, изготовленные из инертных полимеров или поликислот, в которых активный ингредиент (комплекс настоящего изобретения) является либо диспергированным, т. е. ковалентно связанным посредством лабильных связей, либо сохраняется между полимерными мембранами как в резервуаре. Пролонгированное высвобождение достигается посредством диффузии активного ингредиента через полимерную матрицу или посредством гидролиза любых присутствующих ковалентных связей.
Пролонгированное действие может быть также обеспечено путем доставки активного ингредиента к месту действия посредством осмотической накачки. Осмотические насосы состоят из резервуара из раствора или суспензии активного ингредиента (т.е. комплекса настоящего изобретения), окруженного полупроницаемой мембраной, содержащей вход для лекарственного средства. Когда вода проникает через полупроницаемую мембрану в резервуар комплекса, раствор комплекса выталкивается через вышеуказанный вход и высвобождается.
В этих системах производные циклодекстрина настоящего изобретения действуют как агенты для стабилизации лекарственного средства. Указанные производные могут также действовать как осмос-регулирующие агенты для стабилизации лекарственного средства. Указанные производные могут также действовать как осмос-регулирующие агенты, обеспечивающие потенциал для притока воды в такие системы.
Фармацевтические препараты, предназначенные для перорального введения и содержащие жидкий носитель, могут быть изготовлены в виде водных или безводных жидких растворов, или в виде жидких эмульсий типа "масло в воде" или "вода в масле". Фармацевтические препараты для парентерального введения обычно изготавливают в виде одноразовых лекарственных форм, или в виде флаконов для многоразового использования, которые могут плотно закрываться после введения очередной требуемой разовой дозы.
Препараты для перорального введения могут быть также изготовлены в виде жестких желатиновых капсул, которые содержат активный ингредиент, смешанный с инертным твердым наполнителем, например, карбонатом кальция, фосфатом кальция или каолином; или в виде мягких желатиновых капсул, которые содержат активный ингредиент, смешанный с водой или масляной средой, такой, как арахисовое масло, жидкое парафиновое масло или оливковое масло.
При этом следует отметить, что помимо вышеуказанных ингредиентов-носителей, описанные выше фармацевтические препараты могут также включать в себя, если это необходимо, один или несколько дополнительных ингредиентов, таких, как разбавители, буферы, ароматизирующие агенты, связующие, консерванты (включая антиоксиданты) и т.п. а также вещества, добавляемые в целях придания лекарственному препарату изотоничности в отношении крови пациента.
Соединения настоящего изобретения могут быть введены перорально, местно, интранасально, путем закапывания в глаза, парентерально, путем ингаляции раствора, или ректально, в виде разовых лекарственных форм, содержащих обычно используемые в этих целях нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. Термин "парентеральный", используемый в настоящем описании, относится к способу введения лекарственного средства путем подкожных, внутривенных, внутримышечных, внутригрудинных инъекций и путем вливания. Соединения настоящего изобретения могут быть использованы для эффективного лечения как теплокровных животных, таких, как мыши, крысы, лошади, собаки, кошки и т.п. так и человека.
Водные суспензии обычно содержат активные ингредиенты в смеси с наполнителями, пригодными для изготовления таких водных суспензий. Такими наполнителями являются суспендирующие агенты, например натрий-карбоксиметилцеллюлоза, метилцеллюлоза, альгинат натрий-гидроксипропилметилцеллюлозы, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующие или смачивающие агенты, например натуральные фосфатиды, такие, как лецитин; или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, происходящие от жирных кислот и гексита, например полиэтиленсорбитмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, происходящими от жирных кислот и гекситангидридов, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например, этил- или н-пропил-р-гидроксибензоат, один или несколько окрашивающих агентов, один или несколько ароматизирующих агентов и один или несколько подслащивающих агентов, например сахарозу или сахарин.
Масляные суспензии могут быть получены путем суспендирования активного ингредиента в растительном масле, например в арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком, как жидкий парафин. Масляные суспензии могут содержать загущающий агент, например пчелиный воск, твердый парафин или цетиловый спирт. Для придания пероральным препаратам приятных вкусовых качеств в эти препараты могут быть добавлены подслащивающие агенты, указанные выше, и ароматизирующие агенты. Указанные композиции будут лучше сохраняться, если в них добавить антиоксидант, например, такой, как аскорбиновая кислота.
Диспергируемые порошки, пригодные для получения водных суспензий путем добавления воды, содержат активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантами. Подходящими диспергирующими или смачивающими и суспендирующими агентами могут быть вещества уже упомянутые выше. При этом могут быть также добавлены подслащивающий, ароматизирующий и окрашивающий агенты.
Фармацевтические композиции настоящего изобретения могут быть также получены в виде эмульсий типа "масло в воде". В качестве масляной фазы, может быть использовано растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смеси. Подходящими эмульгирующими агентами могут быть натуральные камеди, например аравийская или трагакантовая камедь; натуральные фосфатиды, например соевый лецитин; и сложные эфиры или неполные сложные эфиры, происходящие от жирных кислот и гекситангидридов, например сорбитанмоноолеат, и продукты конденсации указанных неполных эфиров с этиленоксидами, например полиоксиэтиленсорбитанмоноолеат. Указанные эмульсии могут также содержать подслащивающие и ароматизирующие агенты.
Сиропы и эликсиры могут быть изготовлены с использованием подслащивающих агентов, например глицерина, сорбита или сахарозы. Такие препараты могут также содержать смягчающие агенты, консерванты, ароматизирующие агенты и красители. Фармацевтические композиции могут быть также получены в виде стерильных препаратов для инъекций, например в виде стерильных инъецируемых водных или масляных суспензий. Такие суспензии получают стандартными способами с использованием соответствующих диспергирующих или смачивающих агентов и суспендирующих агентов, примеры которых приведены выше. Стерильные препараты для инъекций могут быть также получены в виде инъецируемых стерильных растворов или суспензий в нетоксичном парентерально приемлемом разбавителе или растворителе. Такими приемлемыми разбавителями и растворителями могут быть вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды часто используют стерильные жирные масла. В этих целях может быть использовано любое мягкое жирное масло, например синтетические моно- или диглицериды. Помимо этого, в препаратах для инъекций могут быть использованы жирные кислоты, например олеиновая кислота.
Соединения настоящего изобретения могут быть также введены ректально в виде суппозиториев. Указанные композиции получают путем смешивания лекарственного средства с подходящим не раздражающим наполнителем, который при обычной температуре является твердым, а при температуре прямой кишки становится жидким, и поэтому, попадая в прямую кишку, он расплавляется с высвобождением лекарственного средства. Примерами указанных наполнителей могут служить какао-масло и полиэтиленгликоли.
Для местного применения могут быть использованы кремы, мази, желе, растворы или суспензии, содержащие ингредиент.
Количество активного ингредиента, смешиваемого с носителями для изготовления одноразовых лекарственных форм, зависит от состояния пациента и способа введения лекарственного средства. Например, препарат для перорального введения человеку может содержать от 1,0 до 750 мг активного ингредиента в сочетании с соответствующим количеством материала-носителя, которое может варьироваться от около 5 до около 95% по массе полной композиции. Одноразовые лекарственные формы, в основном, содержат от около 1 до около 500 мг активного ингредиента.
Следует отметить, что конкретная доза для любого конкретного пациента зависит от ряда факторов, включая, активность конкретно используемого соединения, возраст, вес тела, пол, общее состояние здоровья пациента, диету, время и способ введения лекарственного средства, скорость его выведения и состав, а также тяжесть конкретного заболевания.
Фармацевтические композиции, содержащие комплексы включения настоящего изобретения, могут быть введены дозами, а также с интервалами между приемами, необходимыми для достижения желаемого фармакологического ответа, обычно ассоциированного с данным лекарственным средством и в данной фазе заболевания при отсутствии циклодекстрина.
Другие преимущества настоящего изобретения будут очевидны из представленных ниже примеров, иллюстрирующих, но ни в коем случае не ограничивающих настоящее изобретение.
Примеры
Гидроксипропил-циклодекстриновое производное, используемое в приведенных ниже
экспериментах, было закуплено у фирмы Pharmatec. Inc. Alachug, F1.
Получение производного циклодекстрина настоящего изобретения
Пример 1. Моно-сульфобутиловый эфир
b-циклодекстрина
В 100-мл колбу с круглым дном, к водному раствору, состоящему из 30 мл воды и 5,0 гидроксида натрия при температуре 70oC, добавляли 10 г b-циклодекстрина (8,
81•10-3 М). К этому раствору медленно добавляли 1,8 мл (2,40 г, 1,76•10-2 М) бутансультона, энергично размешивая при этом для обеспечения максимального контакта
гетерогенных фаз.
После достижения единой фазы, что указывало на истощение алкилсультона, раствор охлаждали до комнатной температуры и разбавляли 20 мл воды. Полученный раствор нейтрализовали 1н. соляной кислотой и диализовали против 3 х 700 мл воды для удаления солей и гидроксиалкилсульфоновых кислот, образовавшихся в качестве побочных продуктов.
Диализат концентрировали путем ультрафильтрации и помещали на ионнообменную стеклянную колонку (внутр. диам. 1,25" 31,75 мм), упакованную 50 г DEAE-Сефадексом A-25. Непрореагировавший b-циклодекстрин удаляли путем элюирования дистиллированной водой. Монозамещенный сульфобутиловый эфир b-циклодекстрина выделяли путем элюирования 1 н. гидроксидом натрия. Выходящую фракцию, содержащую монозамещенное производное, подвергали ультрафильтрации для удаления остаточных солей. Удерживаемую фракцию доводили до нейтрального значения pH и лиофилизовали, в результате чего получали 2,17 г монозамещенного сульфобутилового эфира b-циклодекстрина в виде белого аморфного твердого вещества. Элементный анализ продукта показал, что отношение углерода к сере составляло 13,7, что соответствует приблизительно 1, 2 замещениям на молекулу.
Пример 2. Моно-сульфопропиловый эфир b-циклодекстрина
Повторяли процедуру, описанную в примере 1, за исключением того, что вместо бутансультона
использовали 1,54 мл (2,15 г 1,76•10-2 М), пропансультона, в результате чего получали 1,97 г моносульфобутилового эфира b-циклодекстрина в виде белого аморфного твердого вещества.
Элементный анализ продукта показал, что отношение углерода к сере составляло 12,1, что соответствует прибл. 1,4 замещениям на молекулу.
Пример 3. Сульфобутиловые эфиры
b-циклодекстрина
В 50-мл колбу с круглым дном, содержащую водный раствор из 10 мл воды и 2,0 г гидроксида натрия при 70oC, добавляли, размешивая, 5 г b-циклодекстрина (4,41•
10-3 М). К этому раствору медленно добавляли 4,5 мл (6,0 г, 4,41•10-2 М) бутансультона, энергично размешивая при этом в целях обеспечения максимального контакта
гетерогенных фаз. После достижения единой фазы, что указывало на истощение алкилсультона, раствор охлаждали до комнатной температуры и разбавляли 20 мл воды. Полученный раствор нейтрализовали 1 н.
соляной кислотой и диализовали против 3 х 700 мл воды для удаления солей и гидроксиалкилсульфоновых кислот, образовавшихся как побочные продукты. Диализат концентрировали путем ультрафильтрации, а
удержанную фракцию доводили до нейтрального значения pH и лиофилизовали, в результате чего получали сульфобутиловый эфир b-циклодекстрина в виде белого аморфного вещества. Элементный анализ продукта
показал, что отношение углерода к сере составляло 3,73, что соответствует приблизительно 7 замещениям на молекулу. Тонкослойная хроматография продукта (2-бутанон:метанол:вода, 4:4:2) показала
отсутствие непрореагировавшего b-циклодекстрина.
Пример 4. Дополнительные сульфоалкилэфиры циклодекстрина
Повторяли процедуры, описанные в примере 3, изменяя при этом реагенты
и их молярные соотношения, в результате чего получали производные циклодекстрина с различными степенями замещения. В табл.1 приводятся типичные результаты.
Кумулятивное мочевыделение
циклодекстрина:
Данные, представленные на фиг.12, показывают, что хотя сульфоалкиловые производные циклодекстрина настоящего изобретения, а также гидроксипропиловые производные выводятся
быстрее и в большей степени у мышей, чем родственное соединение, однако производные настоящего изобретения выводятся быстрее всего. На фиг.12 представлены данные для недериватизированного
циклодекстрина, гидроксипропилового производного, сульфобутилового производного настоящего изобретения и сульфопропилового производного настоящего изобретения.
Острая парентеральная
токсичность
Сульфоалкиловые производные циклодекстрина настоящего изобретения не показали заметного токсического действия при испытаниях на мышах в течение 30-дневного периода после
внутрибрюшинного введения 5,49•10-3 моль/кг указанного производного.
Эта доза эквивалентна 7,1 г/кг для моносульфоалкиловых производных, 12,3 г/кг для сульфобутилового производного с W/7 степенью замещения, и 11,8 г/кг для сульфопропилового производного с W/7 степенью замещения.
Азот мочевины в плазме
Уровень азота мочевины
в плазме являтся показателем функции почек и его повышение свидетельствует о поражении почек. Данные, приведенные в табл.2, показывают, что сульфоалкиловые производные циклодекстрина настоящего
изобретения не вызывают повышения уровней азота мочевины в плазме мышей по сравнению с недериватизированным родственным соединением (контрольное соединение). При этом, однако, статистической разницы
между производными настоящего изобретения и гидроксипропиловым производным не наблюдалось.
Гемолиз эритроцитов
Как видно из данных, представленных на фиг.13 и 14, наиболее
высокозамещенные алкилсульфонокислотные производные настоящего изобретения вызывают меньшее разрушение мембраны, чем монозамещенные производные, о чем свидетельствуют данные по проценту гемолиза.
Монозамещенные производные вызывают примерно такую же степень разрушения мембран, как и гидроксипропиловое производное.
Свойства фазовой растворимости
Как видно из табл.3,
приведенной ниже, и на основании данных, представленных на фиг.4 и 5, константы ассоциации для равновесия между сульфобутиловым производным настоящего изобретения и дигоксином в 5 раз выше, чем
константа для гидроксипропилового производного.
Следует заметить, что максимум по оси X на фиг.4 и 5 соответствует 1,8 мас. циклодекстрина. Из сравнения солюбилизирующих способностей производных настоящего изобретения и гидроксипропилового производного (при 50% растворе, как указано в патенте США N 4727064), можно видеть, что кажущаяся растворимость дигоксина для сульфобутиловых производных настоящего изобретения составляла 216 мг/мл, а для гидроксипропилового производного она составляла 80 мг/мл. Величина 45,0 мг/мл, указанная в патенте США N 4727064, была получена для гидроксипропилового производного с другой степенью замещения, чем то гидроксипропиловое производное, которое было использовано в настоящей заявке для сравнения.
Аналогичные результаты были получены для прогестерона (см. табл.4 и фиг. 6 и 7), фенитоина (см. табл.5 и фиг. 8 и 9) и тестостерона (см. табл.6 и фиг. 10 и 11).
В свете представленного выше описания очевидно, что настоящее изобретение допускает некоторые изменения и модификации, однако при этом следует иметь в виду, что указанные возможные модификации не должны выходить за рамки объема формулы изобретения.
Приложение
Синтез этил и гексилпроизводных циклодекстрина
Получение сульфоэтил-b-циклодекстрина
Информация о получении сульфоэтильного
производного была представлена ранее (пример 5), однако, чтобы защитить полный интервал заявленных алкильных производных, заявитель представляет дополнительную информацию об этом получении.
В круглодонную колбу емкостью 100 мл загружают 10,03 г/8,81• 10-3 моля, 1,0 экв.) при перемешивании к 14 мл 2,5 М водного раствора NaOH при 75oC. К этому раствору в одну порцию прибавляют 9,48 г (4,41•10-3 моля, 5 экв.) натриевой соли 2-бром-1-этансульфоновой кислоты. Реакционную смесь нагревают до 90oC и выдерживают pH около 10 с помощью 9,0 мл 2,5 М NaOH, добавленных в течение первых 16 ч. После этого времени добавляют еще 5,0 мл 2,5 М NaOH для завершения реакции. Реакционная смесь реагирует в этих условиях в течение 72 ч. Наконец, смеси дают остыть до комнатной температуры и промывают ее 2 х 20 мл воды. Объединенные водные растворы нейтрализуют 3н. HCl и диализуют против 2,2 л воды, чтобы удалить соли, непрореагировавшие материалы и побочные продукты. Удержанная часть затем была сконцентрирована и лиофилизована для получения сульфоэтиловых простых эфирных производных b-циклодекстрина в виде аморфного белого твердого продукта.
Капиллярный электрофорез образца (фиг.12) показал пятно анионозамещенного циклодекстрина. Степень замещения была оценена равной 3 по отношению площадей пиков КЭ-анализа и примерно 2,8 по отношению серы к углероду, полученному с помощью элементного анализа.
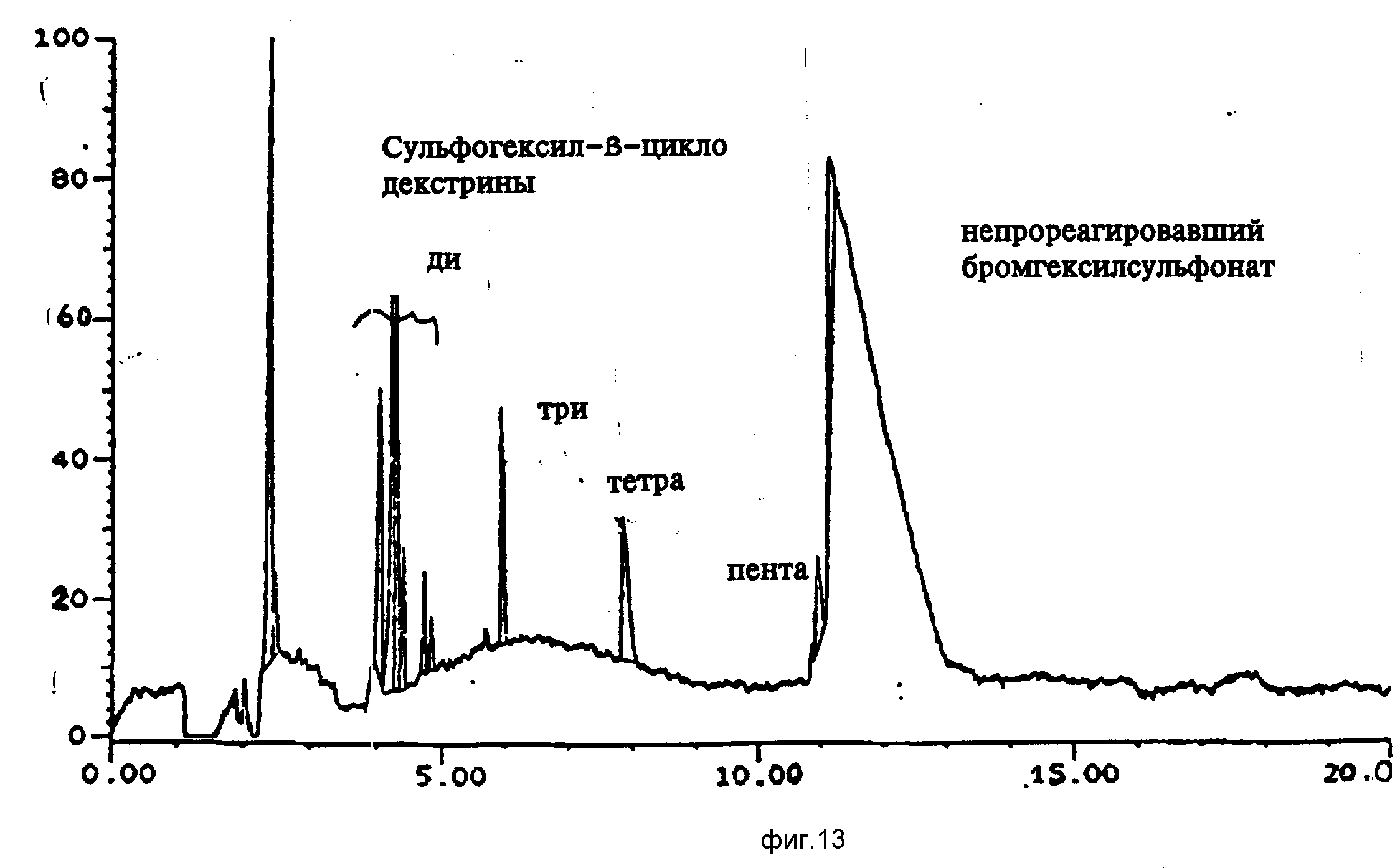
Получение сульфогексил-b- циклодекстрина
В круглодонной
колбе растворяют 10,1 г (8,8•10-3 моля, 1,0 экв.) b-циклодекстрина в 300 мл ДМСО, затем добавляют NaOH (10 экв. в виде 60%-ной дисперсии в минеральном масле) и перемешивают до тех
пор, пока не прекратится выделение H2 (примерно 16-18 ч). Растворяют 4,41•10-2 моля (5,0 экв.) натриевой соли 6-бром-1-гексансульфоновой кислоты в 50,0 мл ДМСО и
прибавляют по каплям в течение 30-45 мин. Затем проводят реакцию в течение 36 ч. После удаления растворителя твердый остаток растворяют в 200 мл воды, устанавливают pH около 7 3н. HCl и диализуют
нейтральный раствор против 2,0 л воды для удаления солей, непрореагировавших материалов и остаточного растворителя. Некоторое количество оставшегося непрореагировавшего b-циклодекстрина удаляют
осаждением. Оставшийся раствор сушат вымораживанием для получения замещенного материала в виде беловатого аморфного твердого продукта.
Капиллярный электрофорез (фиг. 13) показывает наличие полос замещения, соответствующих анионозаряженным сульфогексил-b-циклодекстринам. КЭ-анализ показывает наличие непрореагировавших исходных материалов, b-ЦД при времени удерживания 2,46 мин и бромгексилсульфоната при 11,15 мин. Пики между 4 и 9 мин соответствуют от ди- до пентазамещенным сульфогексилпроизводным b-ЦД.
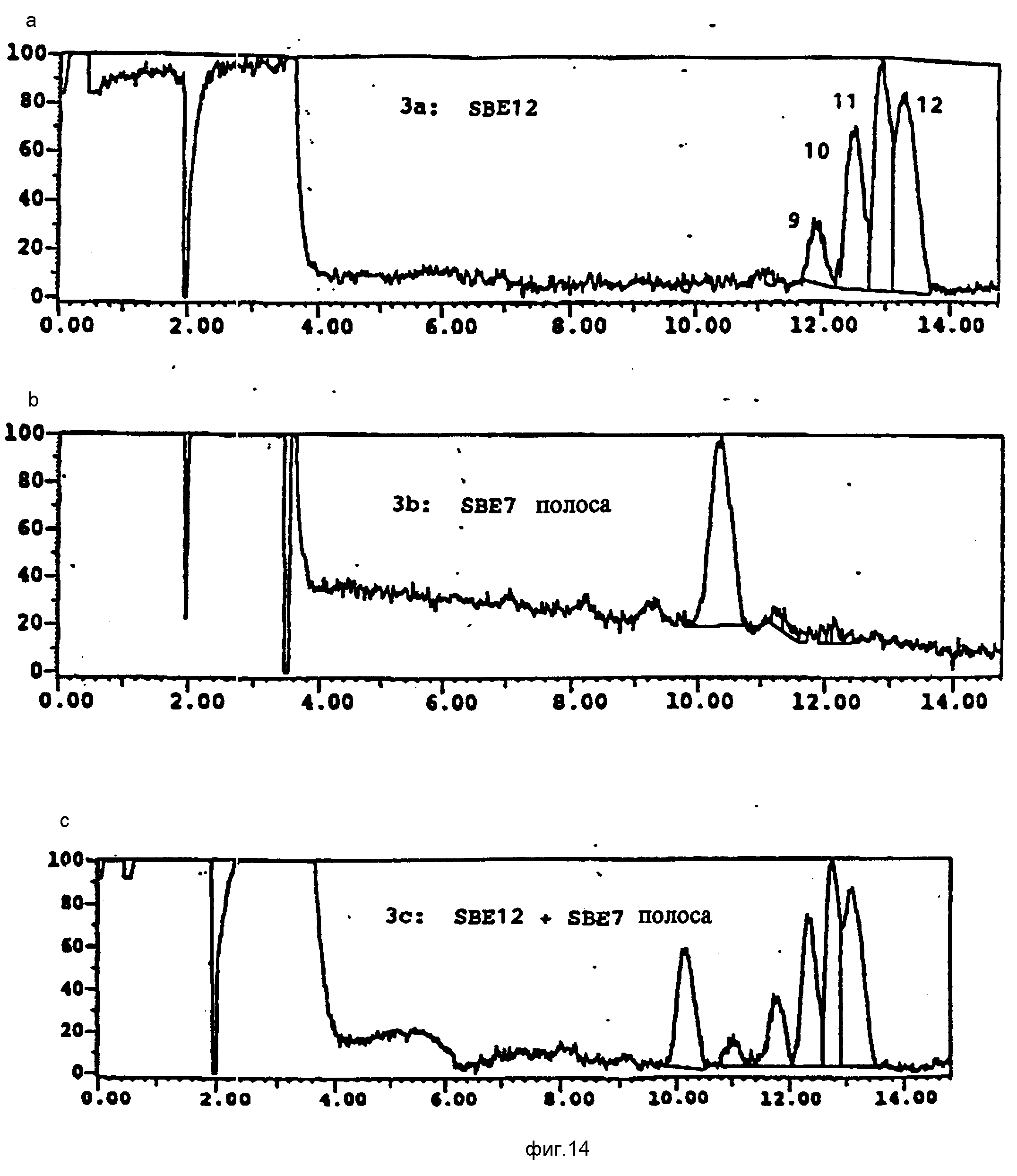
Синтез SBE12-b-CD
В круглодонной колбе
растворяют 10,1 г (8,8•10-3 моля, 1,0 экв.) b-циклодекстрина в 15 мл 4М NaOH. Затем реакционную смесь нагревают до 70oC, прибавляют в одну порцию 6,0 г (4,4•10-2 моля, 5,0 экв.) 1,4-бутансультона и проводят реакцию в течение 2 ч. После этого прибавляют 2,5 г (6,2•10-2 моля, 7,0 экв.) NaOH и 6,0 г (4,4•10-2 моля, 5,0
экв) 1,4-бутансультона и проводят реакцию еще 2 ч. После этого осуществляют третье прибавление 2,5 г (6,2•10-2 моля, 7,0 экв. ) NaOH и 6,0 г (4,4•10-2 моля, 5,0
экв.) 1,4-бутансультона. После этого проводят реакцию в течение 18 ч. Наконец, смесь охлаждают до комнатной температуры и промывают 2 х 20 мл воды; объединенные водные растворы затем нейтрализуют 3н.
HCl и диализуют против 2,5-3,0 л воды для удаления солей, непрореагировавших материалов и побочных продуктов. Оставшуюся часть концентрируют и лиофилизируют, получая сульфобутильное эфирное
производное с двенадцатью в среднем заместителями. Капиллярный электрофорез (КЭ) образца показал получение высокопроизводных со степенью замещения от девяти до двенадцати. Средняя степень замещения
была оценена равной одиннадцати по отношению площадей пиков из КЭ-анализа и примерно 11,5 по отношению серы к углероду, полученному из элементного анализа.
Анализ соотношения углерод-сера показал, что в этом продукте средняя степень замещения составляет 11,5. Капиллярный электрофорез также показал наличие полос в положении 9, 10, 11 и 12 замещения (фиг.14,a); идентичность полос была определена путем сопоставления образца (фиг.14,c) с чистым образцом гепта-замещенного продукта (фиг.14,b).
Реферат
Использование: получение фармацевтических композиций на основе производных циклодекстрина. Сущность изобретения: очищенное производное циклодекстрина - сульфоалкильный простой эфир циклодекстрина; клатратный комплекс указанного производного циклодекстрина с лекарственным средством, характеризующийся молярным соотношением от 10:1 до 1:10; фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и вышеуказанный клатратный комплекс в количестве 1-750 мг на дозу. 2 с.и 1 з.п.ф-лы, 6 табл., 14 ил.
Формула
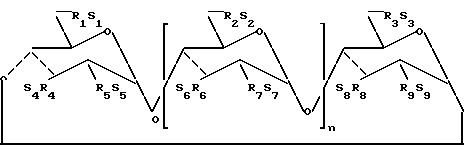
где n 4, 5 или 6;
R1, R2, R3, R4, R5, R6, R7, R8, R9 каждый является независимо O- или O-(C2-6 алкилен)-SO3-группой и по крайней мере один из R1 или R2 является O-(C2-6-алкилен)-SO3--группой и S1, S2, S3, S4, S5, S6, S7, S8 и S9 каждый независимо являются фармацевтически приемлемым катионом, среднее число C2-6-сульфоалкильных групп на молекулу циклодекстрина составляет от 1 до 3n + 6,
и содержание примеси исходного немодифицированного циклодекстрина в смеси сульфоалкильных простых эфиров циклодекстрина приведенной выше структуры не превышает 5 мас.
Комментарии