Эфиры альдоновой кислоты, способ их получения и способ получения фармацевтических биологически активных веществ, связанных по свободным аминогруппам с полисахаридами или производными полисахаридов - RU2330046C2
Код документа: RU2330046C2
Чертежи
Описание
Настоящее изобретение относится к эфирам альдоновой кислоты, твердым веществам и растворам, которые содержат эти эфиры, а также к способу их получения. Далее, настоящее изобретение относится к способам получения фармацевтических биологически активных веществ, связанных по свободным аминогруппам с полисахаридами или производными полисахаридов, которые осуществляют при использовании эфиров альдоновой кислоты, а также к фармацевтическим биологически активным веществам, которые получают этими способами.
Конъюгация фармацевтических биологически активных веществ, в особенности протеинов, с производными полиэтиленгликоля ("ПЭГилирование") или полисахаридами, как декстраны или, в особенности, гидроксиэтилкрахмал (Hydroxyethylstarke=HES) (HESилирование), в последние годы получила большое значение вследствие увеличения числа фармацевтических протеинов в связи с исследованиями в области генной инженерии.
Такие протеины часто обладают слишком коротким периодом полураспада, который можно целенаправленно удлинять за счет связывания с вышеуказанными полимерными соединениями, как ПЭГ или HES. Благодаря связыванию, однако, также можно положительно влиять на антигенные свойства протеинов. В случае других фармацевтических биологически активных веществ за счет связывания можно значительно повышать растворимость в воде.
В патентах DE 19628705 и DE 10129369 описываются способы, как связывание с гидроксиэтилкрахмалом в безводном диметилсульфоксиде (ДМСО) через соответствующий лактон альдоновой кислоты гидроксиэтилкрахмала, которое может осуществляться по свободным аминогруппам гемоглобина, соответственно амфотерицина В.
Так как в случае протеинов часто нельзя работать непосредственно в безводных апротонных растворителях, либо по причинам растворимости, либо также по причинам денатурации протеинов, существуют также способы связывания с гидроксиэтилкрахмалом в содержащей воду среде. Например, связывание селективно окисленного на восстанавливающемся конце цепи до альдоновой кислоты гидроксиэтилкрахмала осуществляют посредством водорастворимого карбодиимида EDC (1-этил-3-(3-диметиламино-пропил)карбодиимид) (РСТ/ЕР 02/02928). Однако очень часто использование карбодиимидов связано с недостатками, так как карбодиимиды очень часто вызывают внутримолекулярные или межмолекулярные реакции сшивки протеинов в качестве побочных реакций.
В случае содержащих фосфатные группы соединений, как нуклеиновые кислоты, связывание часто совсем невозможно, так как фосфатные группы также могут реагировать с EDC (S.S. Wong, Chemistry of Protein Conjugation and Cross-Linking, CRC-Press, Boca Raton, London, New York, Washington D.C., 1993, c. 199).
Принимая во внимание вышеприведенный уровень техники, задачей изобретения является получение соединений, которые при отсутствии вышеописанных недостатков позволяют целенаправленно осуществлять связывание полисахаридов или их производных с содержащими аминогруппы биологически активными веществами, в особенности с протеинами, в чисто водных системах или также в смесях растворителей с водой.
Далее, такое соединение должно обладать таким свойством, чтобы происходило по возможности количественное присоединение биологически активного вещества через ковалентную связь к полисахариду или производному полисахарида.
Далее, задачей изобретения является получение соединений, которые позволяют осуществлять по возможности мягкое связывание полисахарида или его производного с биологически активным веществом. Так, за счет взаимодействия по возможности незначительно должны изменяться, в особенности, структура, активность и совместимость биологически активного вещества. Например, нужно избегать межмолекулярных и внутримолекулярных реакций сшивки. Сверх того, должна существовать возможность связывания также биологически активных веществ, обладающих фосфатными группами.
Далее, задачей настоящего изобретения, следовательно, является получение соединений, которые обеспечивают по возможности селективное связывание с биологически активным веществом. Так, в особенности должна быть обеспечена целенаправленная стехиометрия конъюгата, причем в особенности должна существовать возможность получения конъюгатов 1:1 за счет использования этих соединений.
Наконец, задачей изобретения является разработка по возможности простого и экономичного способа получения таких соединений и продуктов связывания полисахаридов или производных полисахаридов с биологически активными веществами.
Поставленные задачи, а также другие, которые, правда, дословно не указаны, однако, как само собой разумеется, могут возникать из обсуждаемых в данном контексте зависимостей или неизбежно следуют из них, решаются с помощью описанных в пункте 1 формулы изобретения эфиров альдоновой кислоты. Целесообразные вариации предлагаемых согласно изобретению эфиров альдоновой кислоты, а также устойчивые и используемые в способах получения конъюгатов эфиры альдоновой кислоты приведены в зависимых от пункта 1 пунктах 2-19 формулы изобретения.
В отношении способа получения эфиров альдоновой кислоты поставленная задача решается согласно пунктам 20-28 формулы изобретения.
В пунктах 29-34 формулы изобретения описываются способы получения конъюгатов полисахарид-биологически активное вещество и получаемые этими способами фармацевтические биологически активные вещества.
Благодаря эфирам альдоновой кислоты, которые образуются из селективно окисленных на восстанавливающемся конце цепи до альдоновых кислот полисахаридов или производных полисахаридов, получают соединения, с помощью которых решаются поставленные задачи. Такие сложные эфиры можно рассматривать как активированные кислоты. В водной среде за счет взаимодействия с нуклеофильными NH2-группами они превращаются в (более стабильные) амиды.
Далее, благодаря настоящему изобретению достигают, в частности, следующих преимуществ:
Предлагаемые согласно изобретению эфиры альдоновой кислоты позволяют легко осуществлять присоединение биологически активного вещества за счет ковалентной связи к полисахариду или производному полисахарида.
Эфиры альдоновой кислоты согласно настоящему изобретению могут взаимодействовать с биологически активным веществом в мягких условиях. При этом, в особенности, структура, активность и совместимость биологически активного вещества за счет взаимодействия изменяются только в незначительном объеме. Таким образом, можно избегать, в частности, в особенности межмолекулярных и внутримолекулярных реакций сшивки. Далее, можно связывать фармацевтические биологически активные вещества, которые обладают фосфатными группами, без изменения этих групп.
Предлагаемые согласно изобретению эфиры альдоновой кислоты обеспечивают очень селективное связывание с биологически активным веществом. Далее, можно обеспечивать, например, целенаправленную стехиометрию желательного конъюгата, причем за счет использования этих соединений становится возможным в особенности получение конъюгатов 1:1.
Сверх того, согласно изобретению разработаны простые и экономичные способы получения активированных эфиров альдоновой кислоты и продуктов связывания полисахаридов или производных полисахаридов с биологически активными веществами.
Эфиры альдоновой кислоты согласно настоящему изобретению образуются из полисахаридов или производных полисахаридов, которые могут селективно окисляться на восстанавливающемся конце цепи. Такого рода полисахариды, а также получаемые из них производные, кроме того, известны специалистам и могут быть коммерчески доступны. Полисахариды представляют собой макромолекулярные углеводы, молекулы которых имеют большое число (по меньшей мере >10, обычно, однако, значительно больше) гликозидно связанных друг с другом моносахаридных молекул (глюкоза). Среднемассовая молекулярная масса предпочтительных полисахаридов составляет величину в области предпочтительно 1500-1000000 Дальтонов, особенно предпочтительно 2000-300000 Дальтонов и в высшей степени предпочтительно 2000-50000 Дальтонов. Молекулярную массу Mw определяют обычными способами. К ним относятся, например, водная гель-проникающая хроматография (ГПХ), высокоэффективная жидкостная хроматография (ВЭЖХ), светорассеяние и тому подобное.
За счет молекулярной массы полисахаридного остатка можно изменять, в частности, время пребывания в организме.
К предпочтительным полисахаридам относятся крахмал, а также получаемые путем гидролиза фракции крахмала, которые можно рассматривать как продукты расщепления крахмала. Крахмал обычно подразделяют на амилозу и амилопектин, которые различаются степенью разветвления. Согласно изобретению особенно предпочтителен амилопектин.
Под амилопектином при этом понимают прежде всего вообще разветвленные крахмалы или продукты, получаемые из крахмала, с a-(1-4)- и a-(1-6)-связями между молекулами глюкозы. Разветвление цепи при этом происходит через a-(1-6)-связи. В случае естественно встречающихся амилопектинов они неравномерно расположены примерно в случае каждых 15-30 сегментов глюкозы. Молекулярная масса нативного амилопектина является очень высокой и составляет величину в области от 107 Дальтонов до 2х108 Дальтонов. Исходят из того, что амилопектин в известных пределах также образует спирали.
Для амилопектинов можно определять степень разветвления. Мерой разветвления является соотношение числа молекул ангидроглюкозы, которые содержат точки разветвления (a-(1-6)-связи), к общему числу молекул ангидроглюкозы амилопектина, причем это соотношение выражают в мол.%. Встречающийся в природе амилопектин имеет степени разветвления примерно 4 мол.%. Используемые для получения эфиров альдоновой кислоты амилопектины предпочтительно имеют среднее разветвление в пределах от 5 мол.% до 10 мол.%.
Далее, можно использовать гиперразветвленные амилопектины, которые имеют степень разветвления, значительно превышающую известную естественную степень разветвления амилопектинов. При этом в случае степени разветвления в каждом случае речь идет о среднем значении (средняя степень разветвления), так как амилопектины являются полидисперсными веществами.
Такие гиперразветвленные амилопектины обладают значительно более высокими степенями разветвления, выражаемыми в виде мол.% ангидроглюкоз разветвления, по сравнению с неизмененным амилопектином, соответственно гидроксиэтилкрахмалом, и вследствие этого по своей структуре подобны гликогену.
Средняя степень разветвления гиперразветвленных амилопектинов обычно составляет величину в пределах от >10 мол.% до 25 мол.%. Это означает, что эти амилопектины в среднем примерно на каждые 10-4 глюкозных звеньев имеют одну a-(1-6)-связь и, таким образом, одну точку разветвления.
Предпочтительно используемые в области медицины типы амилопектина характеризуются степенью разветвления от 11 мол.% до 16 мол.%.
Другие предпочтительные гиперразветвленные амилопектины имеют степень разветвления в пределах от 13 мол.% до 16 мол.%.
Используемые согласно изобретению амилопектины имеют величину среднемассовой молекулярной массы Mw в пределах предпочтительно от 2000 Дальтонов до 800000 Дальтонов, в особенности от 2000 Дальтонов до 300000 Дальтонов и особенно предпочтительно от 2000 Дальтонов до 50000 Дальтонов.
Вышеуказанные крахмалы могут быть коммерчески доступны. Далее, их получение известно из литературы. Так, крахмал можно получать, в частности, из картофеля, тапиоки, маниока, риса, пшеницы или кукурузы. Получаемые из этих растений крахмалы прежде всего многократно подвергают гидролитической реакции расщепления. При этом молекулярная масса от примерно 20000000 Дальтонов снижается до нескольких миллионов Дальтонов, причем также известно дальнейшее уменьшение молекулярной массы до вышеуказанных значений. В частности, особенно предпочтительно можно использовать фракции, образующиеся в результате расщепления крахмала восковидной кукурузы, для получения предлагаемых согласно изобретению эфиров альдоновой кислоты.
Вышеуказанные гиперразветвленные фракции крахмала описываются, в частности, в немецкой патентной заявке 10217994.
Далее, для получения предлагаемых согласно изобретению эфиров альдоновой кислоты можно использовать также производные полисахаридов. К ним относятся, в особенности, гидроксиалкилкрахмалы, например гидроксиэтилкрахмал и гидроксипропилкрахмал, которые можно получать путем гидроксиалкилирования из вышеуказанных крахмалов, в особенности из амилопектина. При этом предпочтителен гидроксиэтилкрахмал (HES).
Согласно изобретению предпочтительно используют гидроксиэтилкрахмал, который представляет собой гидроксиэтилированное производное полимера глюкозы амилопектина, содержащегося в крахмале восковидной кукурузы в количестве более 95%. Амилопектин состоит из глюкозных звеньев, которые содержат α-1,4-гликозидные связи и имеют α-1, 6-гликозидные разветвления.
Гидроксиэтилкрахмал обладает предпочтительными реологическими свойствами и в настоящее время клинически используется в качестве кровезаменителя и в гемодилюционной терапии (Sommermeyer и др., Krankenhauspharmazie, 8, 271-278 (1987), и Weidler и др., Arzneimittelforschung/Drug Res., 41, 494-498 (1991)).
Гидроксиэтилкрахмал по существу отличается среднемассовой молекулярной массой Mw, среднечисловой молекулярной массой Mn, молекулярно-массовым распределением и степенью замещения. Замещение гидроксиэтильными группами по простой эфирной связи при этом возможно у атомов углерода 2,3 и 6 ангидроглюкозных звеньев. Степень замещения при этом можно указывать как DS ("степень замещения"), которая относится к доле замещенных молекул глюкозы по отношению ко всем звеньям глюкозы, или как MS ("молярное замещение"), которое обозначает среднее число гидроксиэтильных групп на глюкозное звено.
Степень замещения MS (молярное замещение) определяют как среднее число гидроксиэтильных групп на ангидроглюкозное звено. Ее определяют из общего числа гидроксиэтильных групп в образце, например, согласно Morgan, путем расщепления простого эфира и последующего количественного определения образующихся при этом этилиодида и этилена.
Напротив, степень замещения DS (степень замещения) определяют как долю замещенных ангидроглюкозных звеньев по отношению ко всем ангидроглюкозным звеньям. Ее можно определять из измеренного количества незамещенной глюкозы после гидролиза образца. Из этих определений получается, что MS > DS. В случае, когда имеется только монозамещение, следовательно, каждое замещенное ангидроглюкозное звено содержит только одну гидроксиэтильную группу, MS=DS.
Остаток гидроксиэтилкрахмала предпочтительно имеет степень замещения MS от 0,1 до 0,8. Особенно предпочтительно остаток гидроксиэтилкрахмала имеет степень замещения MS от 0,4 до 0,7.
Реакционная способность отдельных гидроксильных групп в незамещенном ангидроглюкозном звене по отношению к гидроксиэтилированию различна в зависимости от условий реакции. В известных пределах благодаря этому оказывают влияние на тип замещения, следовательно, на отдельные, различно замещенные ангидроглюкозные звенья, которые статистически распределены в отдельных полимерных молекулах. Предпочтительно преобладающе гидроксиэтилируются положение С2 и положение С6, причем положение С6 из-за своей более легкой доступности замещается чаще.
В рамках настоящего изобретения предпочтительно используют замещенные преобладающе в положении С2 гидроксиэтилкрахмалы (HES), которые замещены по возможности однородно. Получение таких гидроксиэтилкрахмалов описывается в патенте ЕР-0402724-В2. Они в течение физиологически приемлемого времени расщепляются без остатка и, с другой стороны, все-таки обладают регулируемым поведением при элиминировании. Преобладающее замещение в положении С2 делает гидроксиэтилкрахмал относительно трудно расщепляемым α-амилазой. Преимуществом является то, что по возможности не образуются замещенные последовательно друг за другом внутри полимерной молекулы ангидроглюкозные звенья, чтобы обеспечивать безостаточную расщепляемость. Далее, такие гидроксиэтилкрахмалы, несмотря на низкую степень замещения, обладают достаточно высокой растворимостью в водной среде, так что растворы также в течение более продолжительных промежутков времени являются стабильными и не образуются никакие агломераты, соответственно гели.
В пересчете на гидроксиэтильные группы ангидроглюкозных звеньев остаток гидроксиэтилкрахмала имеет предпочтительно соотношение замещения С2:С6 в пределах от 2 до 15. Особенно предпочтительно соотношение замещения С2:С6 составляет от 3 до 11.
Селективное окисление альдегидной группы вышеуказанных полисахаридов, соответственно производных полисахаридов, до альдоновой кислоты само по себе известно. Его можно осуществлять с помощью мягкого окислителя, например смеси иод/гидроксид калия согласно заявке DE 19628705-А1, или с помощью ферментов.
Для взаимодействия можно использовать свободную альдоновую кислоту. Далее, можно использовать также соли. К ним относятся в особенности соли щелочных металлов, как, например, натриевая и/или калиевая соль альдоновой кислоты.
Для получения предлагаемых согласно изобретению эфиров альдоновой кислоты используют спирты. Понятие спирт включает соединения, содержащие ОН-группы. Эти ОН-группы, в частности, могут быть связаны с атомом азота или с фенильным остатком.
Предпочтительно используют кислые спирты, которые известны специалисту. К ним относятся, в частности, N-гидроксиимиды, например N-гидроксисукцинимид и сульфо-N-гидроксисукцинимид, замещенные фенолы и гидроксиазолы, например гидроксибензтриазол, причем особенно предпочтительны N-гидроксисукцинимиды и сульфо-N-гидроксисукцинимид.
Другие пригодные кислые спирты для получения предлагаемых согласно изобретению эфиров альдоновой кислоты указаны в литературе (V.H.L. Lee (Ed.), Peptide and Protein Drug Delivery, Marcel Dekker, 1991, c. 65).
Согласно особому аспекту настоящего изобретения используют спирты, ОН-группа которых имеет значение pKs в области от 6 до 12, предпочтительно в области от 7 до 11. Это значение относится к определяемой при температуре 25°С константе диссоциации кислоты, причем это значение многократно указано в литературе.
Молекулярная масса спирта составляет величину предпочтительно в пределах от 80 г/моль до 500 г/моль, в особенности от 100 г/моль до 200 г/моль.
Спирт можно добавлять к реакционной смеси в свободной форме. Далее, для реакции можно использовать также соединения, которые при добавке воды в случае необходимости при кислотном катализе высвобождают спирт.
Согласно особому аспекту настоящего изобретения для взаимодействия с альдоновой кислотой или солью альдоновой кислоты используют диэфиры угольной кислоты. Эти соединения обеспечивают протекание особенно быстрой и мягкой реакции, причем образуются только угольная кислота, соответственно карбонаты, спирты и желательные эфиры альдоновой кислоты.
Предпочтительными диэфирами угольной кислоты являются, среди прочих, N'N-сукцинимидилкарбонат и сульфо-N'N-сукцинимидил-карбонат.
Эти диэфиры угольной кислоты можно использовать в относительно небольших количествах. Так, диэфир угольной кислоты можно использовать в 1-3-кратном молярном избытке, предпочтительно в 1-1,5-кратном избытке, в пересчете на альдоновую кислоту и/или соль альдоновой кислоты. Продолжительность реакции при использовании диэфиров угольной кислоты является относительно незначительной. Так, реакция часто может заканчиваться спустя 2 часа, предпочтительно спустя 1 час.
Превращение в эфир альдоновой кислоты предпочтительно осуществляют в безводном апротонном растворителе. Содержание воды должно составлять предпочтительно самое большее 0,5 мас.%, особенно предпочтительно самое большее 0,1 мас.%. Пригодными растворителями являются, в частности, диметилсульфоксид (ДМСО), N-метилпирролидон, диметилацетамид (ДМА) и/или диметилформамид (ДМФА).
Реакция этерификации сама по себе известна, причем можно использовать любой способ. Превращение в эфир альдоновой кислоты можно осуществлять, в частности, при применении активаторов. При использовании свободного спирта рекомендуется такого рода образ действий. К активаторам относятся в особенности карбодиимид, как, например, дициклогексилкарбодиимид (DCC) и 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC).
При применении свободного спирта его можно использовать в молярном избытке. Согласно особому аспекту настоящего изобретения спиртовой компонент используют предпочтительно в 5-50-кратном молярном избытке, особенно предпочтительно в 8-20-кратном избытке в пересчете на альдоновую кислоту и/или производное альдоновой кислоты.
Превращение в эфир альдоновой кислоты протекает в мягких условиях. Так, вышеописанные реакции можно проводить при температурах предпочтительно в диапазоне от 0°С до 40°С, особенно предпочтительно в диапазоне от 10°С до 30°С.
Согласно особому аспекту настоящего изобретения превращение осуществляют при незначительной основности. Незначительной основности можно достигать путем добавления реакционной смеси в 10-кратный избыток воды. При этом вода перед добавкой имеет значение рН=7,0 при температуре 25°С, причем вода по существу не содержит никакого буфера. Путем измерения значения рН при температуре 25°С после добавки реакционной смеси получают основность реакционной смеси. Эта смесь после добавки предпочтительно имеет значение рН самое большее 9,0, особенно предпочтительно самое большее 8,0 и особенно предпочтительно самое большее 7,5.
Взаимодействие с HES-альдоновыми кислотами, например с N-гидроксисукцинимидом, протекает в безводном ДМА при исключении воды с помощью EDC путем реакции при комнатной температуре с образованием N-гидроксисукцинимидного эфира HES-кислоты. При этом в особенности является неожиданным то, что не происходит никакой побочной реакции молекул HES за счет взаимодействия имеющихся в большом избытке ОН-групп ангидроглюкоз с EDC, а также предотвращается перегруппировка первично образующейся О-ацилизомочевины из EDC и альдоновой кислоты до соответствующей N-ацилмочевины.
Полученные путем вышеописанного взаимодействия растворы можно использовать без выделения эфиров альдоновой кислоты в реакциях связывания. Поскольку, как правило, объем предварительно активированной альдоновой кислоты в апротонном растворителе является небольшим по сравнению с растворенным в объеме буфера протеином-мишенью, количества апротонного растворителя чаще всего не оказывают мешающего влияния. Предпочтительные растворы включают по меньшей мере 10 мас.% эфира альдоновой кислоты, предпочтительно по меньшей мере 30 мас.% эфира альдоновой кислоты и особенно предпочтительно по меньшей мере 50 мас.% эфира альдоновой кислоты.
Эфиры альдоновой кислоты можно осаждать из раствора в апротонном растворителе, например в DMA, с помощью известных осадителей, как, например, безводный этанол, изопропанол или ацетон, и очищать путем многократного повторения процесса. Предпочтительные твердые вещества включают по меньшей мере 10 мас.% эфира альдоновой кислоты, предпочтительно по меньшей мере 30 мас.% эфира альдоновой кислоты и особенно предпочтительно по меньшей мере 50 мас.% эфира альдоновой кислоты.
Такие эфиры альдоновой кислоты, выделенные в виде вещества, затем можно использовать для связывания, например, для HESилирования. При этом тогда не возникают никакие побочные реакции, как описанные выше в случае активированной с помощью EDC кислоты.
Далее, для связывания раствор активированной альдоновой кислоты можно добавлять к водному раствору фармацевтического биологически активного вещества, который предпочтительно забуферен, при пригодном значении рН. Фармацевтические биологически активные вещества включают по меньшей мере одну аминогруппу, которая может превращаться в амид альдоновой кислоты. К предпочтительным биологически активным веществам относятся протеины и пептиды.
Значение рН при взаимодействии зависит от свойств биологически активного вещества. Предпочтительно, если это возможно, значение рН находится в пределах от 7 до 9, особенно предпочтительно в пределах от 7,5 до 8,5.
Связывание происходит, в общем, при температурах в диапазоне от 0°С до 40°С, предпочтительно от 10°С до 30°С, причем это не должно рассматриваться как ограничение. Продолжительность реакции можно легко определять пригодным способом. В общем, время реакции составляет величину в области от 1 часа до 100 часов, предпочтительно от 20 часов до 48 часов.
Эфир альдоновой кислоты можно использовать в избытке, в пересчете на фармацевтическое биологически активное вещество. Предпочтительно используют эфир альдоновой кислоты в 1-5-кратном молярном избытке, особенно предпочтительно в 1,5-2-кратном избытке, в пересчете на фармацевтическое биологически активное вещество.
В качестве побочного продукта в случае вышеуказанного взаимодействия осаждается главным образом только спирт, например N-гидроксисукцинимид, который можно легко отделять от продукта связывания, например, путем ультрафильтрации. В качестве побочной реакции может происходить омыление водой до образования свободной кислоты и свободного спирта. Поэтому особенно неожиданным является то, что предлагаемые согласно изобретению эфиры альдоновой кислоты большей частью вступают в реакцию связывания с фармацевтическим биологически активным веществом. Это следует из примеров, в особенности из представленных на фигурах хроматограмм.
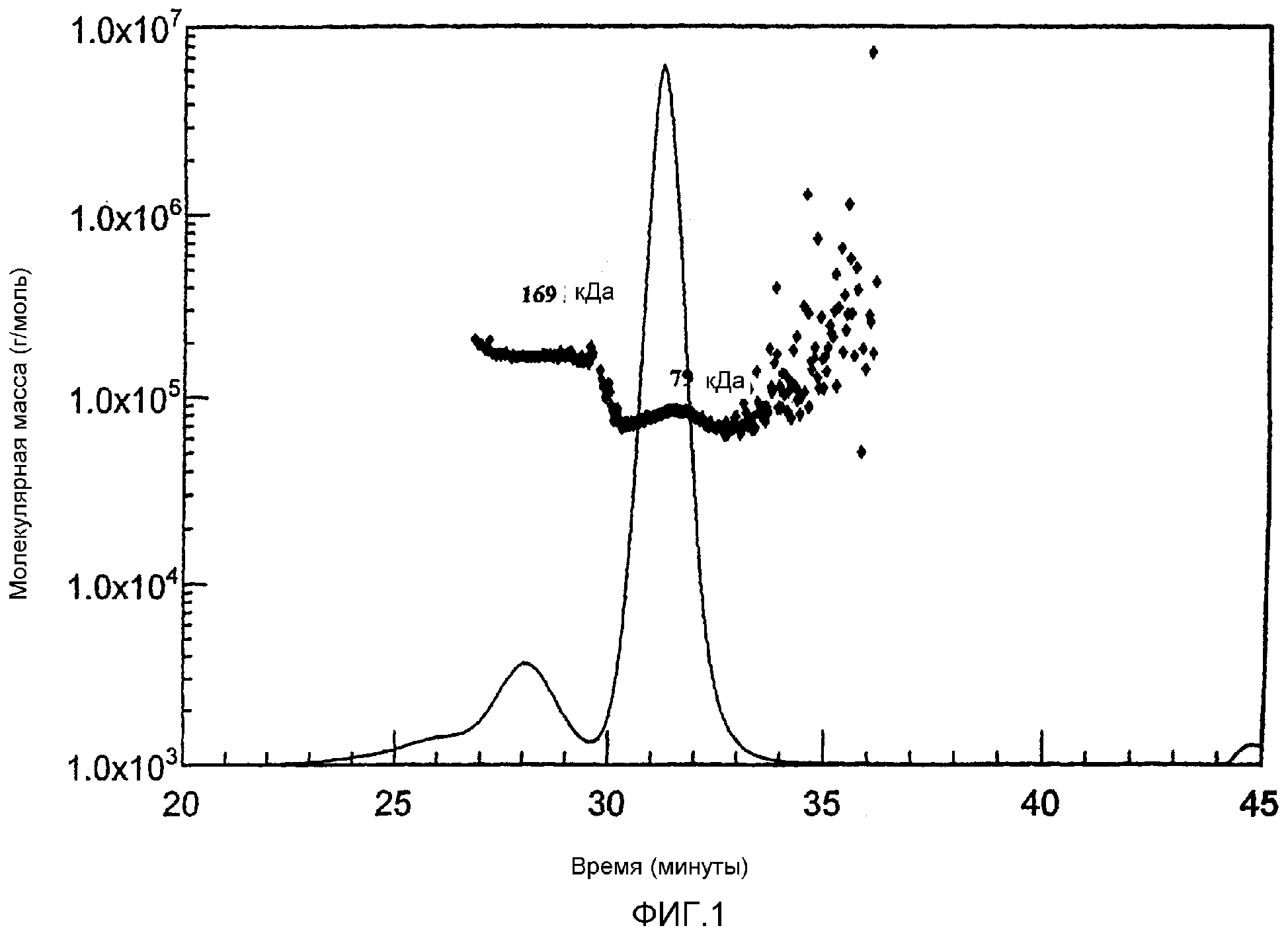
Фиг. 1 MALLS-ГПХ-Хроматограмма непрореагировавшего бычьего сывороточного альбумина (BSA). Мономерный и димерный альбумины четко разделены.
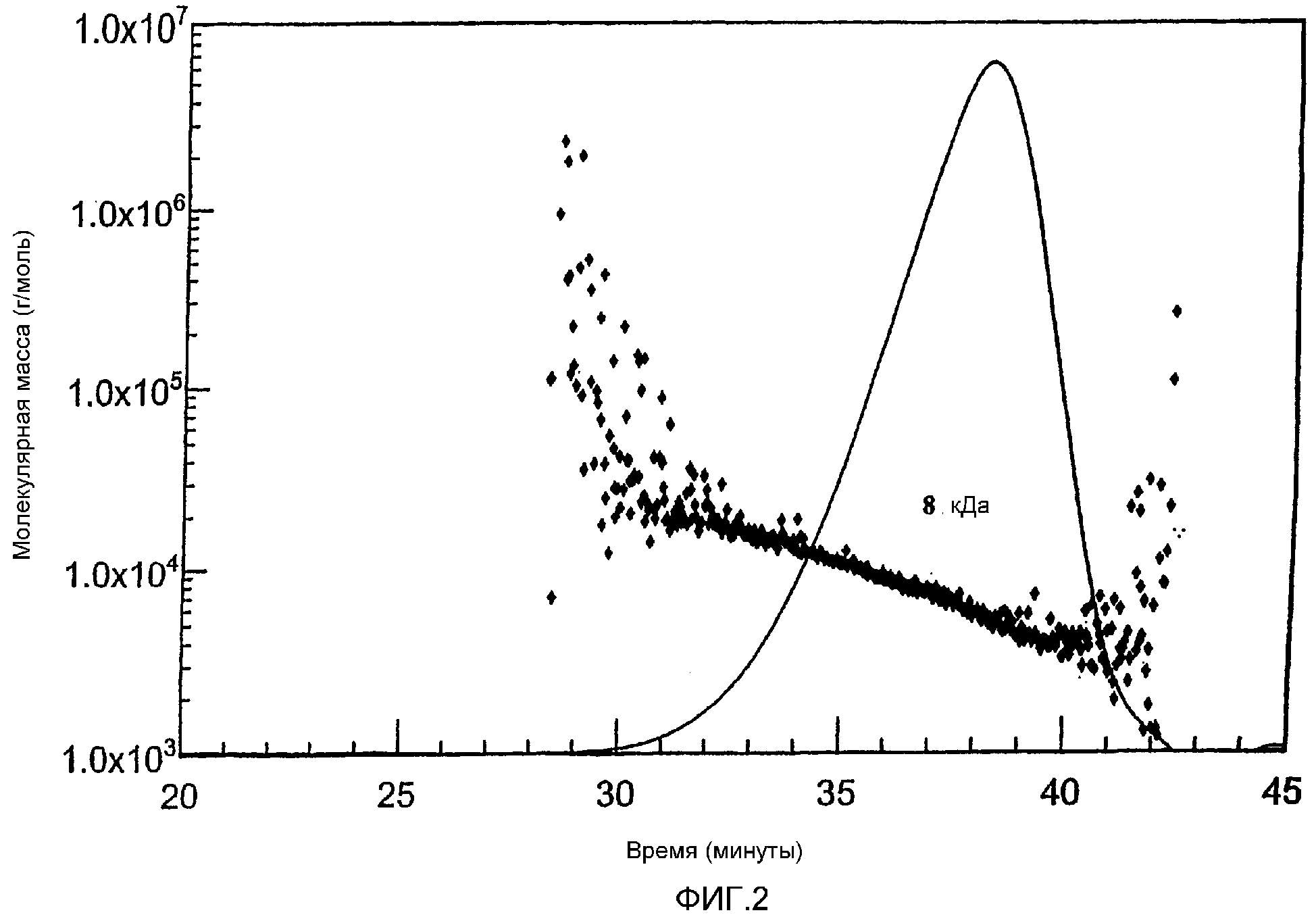
Фиг. 2 MALLS-ГПХ-Хроматограмма непрореагировавшего сложного HES-10/0,4-сукцинимидилового эфира.
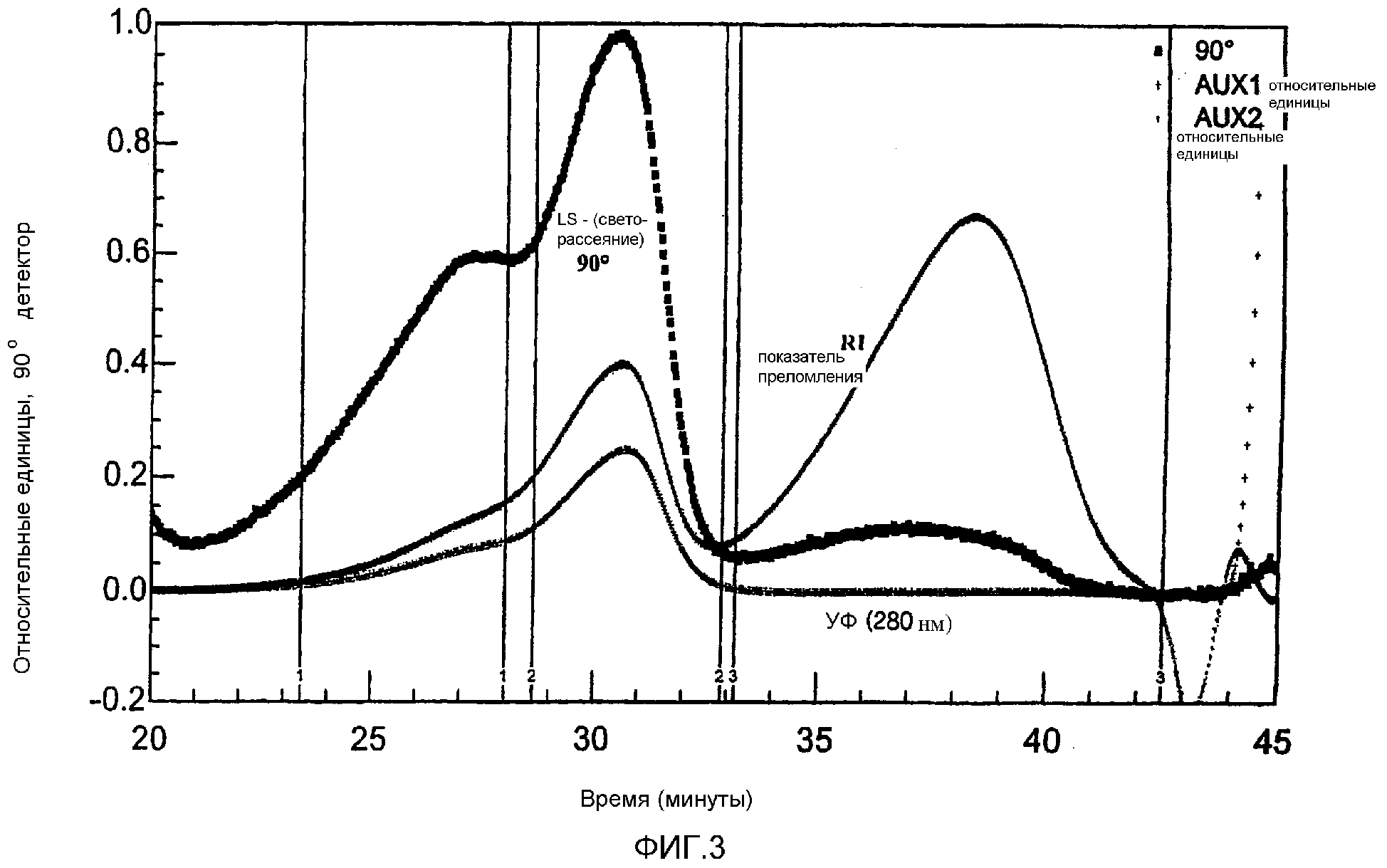
Фиг.3 MALLS-ГПХ-Хроматограмма продукта реакции сложного HES-10/0,4-сукцинимидилового эфира и BSA. Представлены сигналы трехкратного определения показателя преломления (RI), УФ-детектора, а также сигнал светорассеяния при 90°.
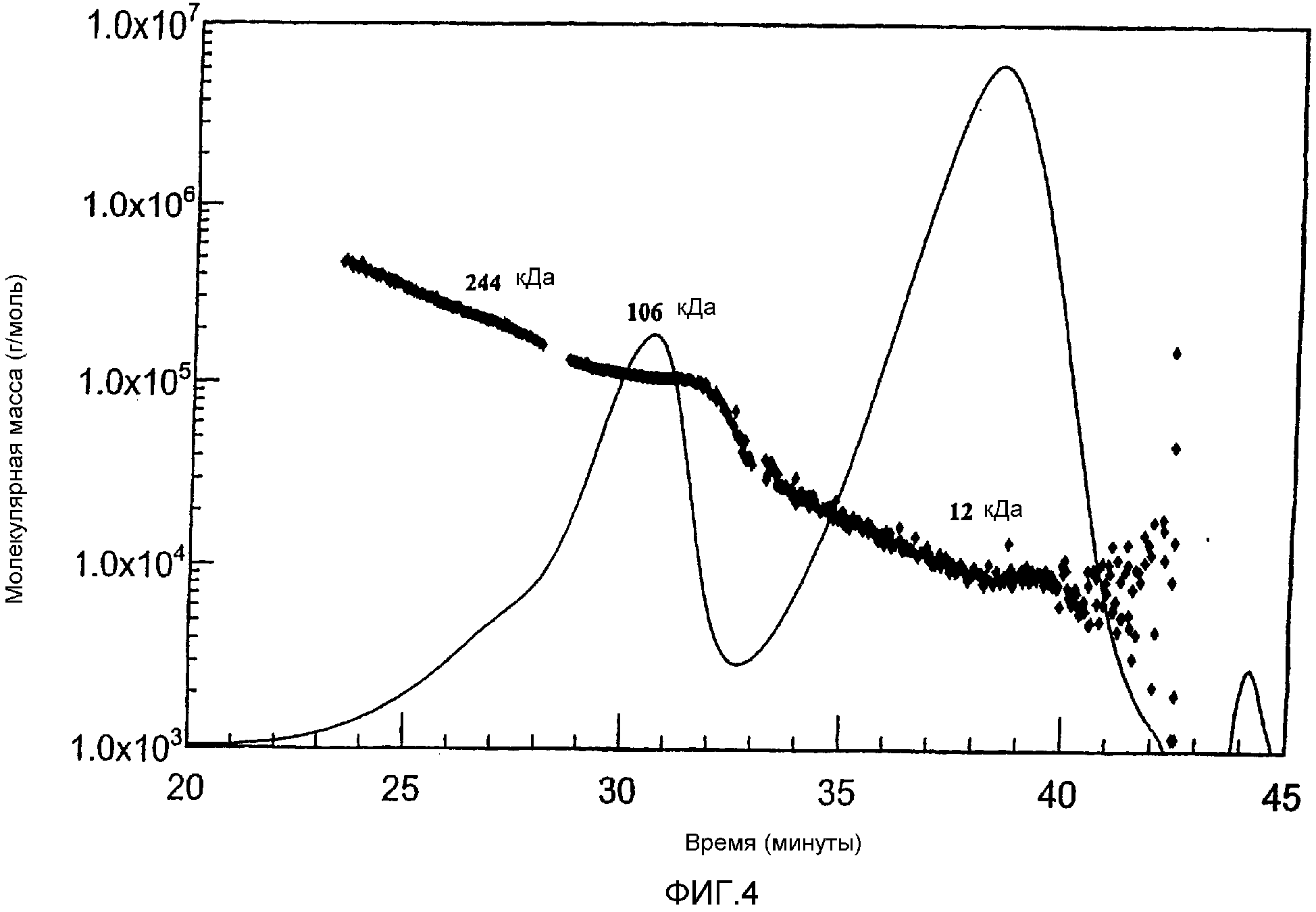
Фиг. 4 MALLS-ГПХ-Хроматограмма продукта реакции сложного HES-10/0,4-сукцинимидилового эфира и BSA с изображением молекулярной массы в зависимости от времени.
Ниже изобретение поясняется подробнее с помощью примеров и сравнительных примеров, причем изобретение не должно ограничиваться этими примерами.
Примеры и способы получения
Пример 1
Получение эфира HES-10/0,4-кислоты с N-гидроксисукцинимидом
5 г высушенного, селективно окисленного на восстанавливающемся конце цепи, согласно DE 19628705, гидроксиэтилкрахмала со средней молекулярной массой Mw=10000 Дальтонов и степенью замещения MS=0,4 растворяют в 30 мл безводного диметилацетамида при температуре 40°С и после охлаждения раствора в отсутствие влажности смешивают с 10-кратным молярным количеством N-гидроксисукцинимида. Затем порциями добавляют эквимолярное по отношению к HES-кислоте количество EDC и реакционную смесь оставляют реагировать в течение 24 часов. Продукт реакции затем осаждают безводным ацетоном и для очистки многократно переосаждают.
Пример 2
Получение связанного с HES-10/0,4-кислотой миоглобина
15 мг Миоглобина растворяют в 20 мл дистиллированной воды и с помощью раствора гидроксида натрия устанавливают значение рН=7,5. К раствору в течение 1 часа порциями добавляют 1,5 г продукта реакции HES-10/0,4-кислоты и N-гидроксисукцинимида, полученного согласно примеру 1, и значение рН поддерживают постоянным при 7,5 за счет добавки раствора гидроксида натрия. Смесь выдерживают при перемешивании в течение ночи. Образование HESилированного миоглобина определяют путем гель-проникающей хроматографии с выходом 70%, в пересчете на используемый миоглобин.
Пример 3
Получение эфира HES-10/0,4-кислоты с N'N-дисукцинимидилкарбонатом
0,02 ммоль (соответственно, 0,14 г) высушенной HES-10/0,4-кислоты в отсутствие влажности растворяют в 2 мл безводного диметилформамида. К раствору добавляют 0,02 ммоль N'N-дисукцинимидилкарбоната и оставляют реагировать в течение 1 часа при комнатной температуре и при перемешивании.
Пример 4
Получение продукта связывания HES-10/0,4-кислоты с бычьим сывороточным альбумином
50 мг бычьего сывороточного альбумина (BSA; соответственно, 0,7 мкмоль) растворяют в 6 мл 0,3 М раствора гидрокарбоната с рН=8,4. К раствору добавляют смесь, полученную согласно примеру 3, и оставляют реагировать в течение 2 часов при перемешивании и при комнатной температуре.
Доказательство того, что реакция произошла, получают с помощью высокоэффективной гель-проникающей хроматографии низкого давления с многократным детектированием (УФ-детектирование при 280 нм, MALLS-детектор светорассеяния (MALLS=многоугловое лазерное светорассеяние), детектор показателя преломления (RI)).
На фигурах 1-4 в сравнении представлены хроматограммы непрореагировавшего сложного HES-10/0,4-сукцинимидилового эфира, исходного продукта BSA, а также реакционной смеси.
Факт, что произошла реакция, устанавливают по значительному уменьшению пика BSA и появлению пика более высокомолекулярного соединения, который определяют при 280 нм.
Пример 5
Получение эфира HES-50/0,7-кислоты с N'N-дисукцинимидилкарбонатом
0,02 ммоль (0,5 г) высушенной HES-50/0,7-кислоты в отсутствие влажности растворяют в 2 мл безводного диметилформамида. К раствору добавляют 0,02 ммоль N'N-дисукцинимидилкарбоната и оставляют реагировать в течение 1 часа при комнатной температуре и при перемешивании.
Пример 6
Получение продукта связывания HES-50/0,7-кислоты с BSA
50 мг бычьго сывороточного альбумина (BSA) (0,7 мкмоль) растворяют в 6 мл 0,3 М раствора бикарбоната с рН=8,4. К раствору добавляют раствор активированной HES-50/0,7-кислоты согласно примеру 5 и оставляют реагировать в течение 2 часов при перемешивании и при комнатной температуре.
Аналитический контроль в отношении реакционной смеси осуществляют путем высокоэффективной гель-проникающей хроматографии низкого давления с трехкратным детектированием, как описывается в примере 4.
Факт, что произошла реакция, устанавливают по уменьшению сигнала при 280 нм непрореагировавшего BSA и соответствующему появлению отвечающего более высоким молекулярным массам сигнала продукта связывания. Сдвиг соответственно более высокой молекулярной массе HES-кислоты увеличен по сравнению с указанным в примере 4.
Реферат
Изобретение относится к эфирам альдоновой кислоты полисахаридов или производных полисахаридов, селективно окисленных на восстанавливающемся конце цепи до альдоновых кислот, полученные взаимодействием соответствующей альдоновой кислоты или ее соли со спиртом, имеющим молекулярную массу в области от 80 г/моль до 500 г/моль. Настоящее изобретение также относится к одному из охарактеризованных выше эфиров альдоновой кислоты полисахаридов или производных полисахаридов, выделенному в виде твердого вещества, к эфиру, полученному в виде раствора, и к способу получения указанных выше эфиров. В том числе настоящее изобретение относится к способу получения конъюгата фармацевтического биологически активного вещества, связанного по свободным аминогруппам с указанными выше эфирами, и к конъюгату фармацевтического биологически активного вещества. Заявленные эфиры альдоновой кислоты полисахаридов или производных полисахаридов могут взаимодействовать с биологически активным веществом в мягких условиях, что позволяет избегать межмолекулярных и внутримолекулярных реакций сшивки. Также заявленные эфиры не только обеспечивают очень селективное связывание с биологически активным веществом и позволяют легко осуществлять присоединение биологически активного вещества за счет ковалентной связи к полисахариду или производному полисахарида, но и могут связываться с фармацевтическими биологически активными веществами, которые обладают фосфатными группами, без изменения этих групп. Кроме того, согласно настоящему изобретению разработаны простые и экономичные способы получения активированных эфиров альдоновой кислоты и продуктов связывания полисахаридов или производных полисахаридов с биологически активными веществами. 6 н. и 27 з.п. ф-лы, 4 ил.
Формула
Документы, цитированные в отчёте о поиске
Способ получения водорастворимых, физиологически совместимых сложных эфиров крахмала, сложный эфир крахмала
Комментарии