Способ конструирования рекомбинантной плазмидной днк, способной экспрессировать иммунный интерферон человека - RU2056460C1
Код документа: RU2056460C1
Чертежи


Описание
Изобретение относится к построению вектора экспрессии рекомбинантной ДНК, кодирующей иммунный интерферон человека.
Иммунный интерферон человека (γ-интерферон, 11F или 1FN-γ), который составляет предмет изобретения, в противоположность α и β -интерферонам, рН 2 лабилен его получают главным образом митогенной индукцией лимфоцитов, и он также имеет оптигенные отличия. Иммунный интерферон человека можно было получать лишь на очень незначительных уровнях, что затрудняло его характеризацию.
Известно соединение для частичной очистки иммунного интерферона человека (1), которое получено из культур лимфоцитов, стимулированных сочетанием фитогемагглютина и сложного эфира форбола, и очищено последовательными хроматографическими разделениями. В результате этой процедуры получили продукт с молекулярным весом 58000.
Иммунный интерферон человека получали в очень малых количествах трансляцией ИРНК в ооцитах, причем ДНК иммунного интерферона можно будет синтезировать и клонировать (2).
Полученное количество иммунного интерферона недостаточно для проведения экспериментов по характеризации и определению биологических свойств очищенной компоненты. Однако при исследованиях in vitro, проведенных с неочищенными препаратами, также как и in vivo экспериментах с препаратами γ-интерферона крыс, предполагалось, что основной функцией иммунного интерферона может быть функция иммуннорегулирующего агента (3, 4). Иммунный интерферон обладает не только противовирусной и противоклеточной активностью вообще ко всем интерферонам человека, а потенцирующим действием на эти активности с α и β-интерфероном. Кроме того, in vitro антипролиферативное действие γ-интерферона на опухолевые клетки, приблизительно в 10-100 раз выше, нежели действие других классов интерферонов (3, 6, 7). Этот результат вместе с выраженной иммуннорегуляторной ролью интерферона (3, 4) предполагает гораздо более выраженную противоопухолевую способность для IFN-γ, нежели для IFN-α и IFN-β. Однако в экспериментах in vivo с мышами и крысами IFN-γ препараты демонстрируют заметное превосходство по сравнению с противовирусными стимулированными интерферонами в плане противоопухолевой активности против остесаркомы (8).
Исследования до изобретения приходилось выполнять на загрязненных препаратах из-за их малой доступности. Однако они определенно подтверждали очень важные биологические функции иммунного интерферона. Иммунный интерферон обладает не только способностью, связанной с противовирусной активностью, но также сильной иммунорегуляторной и противоопухолевой активностью, что указывает на него как на потенциально многообещающий клинический объект.
Изобретение основано на использование технологии рекомбинантной ДНК для получения имунного интерферона человека, предпочтительно в зрелой форме и в количествах, достаточных для проведения тестов на животных и для клинических испытаний, что необходимо перед выходом на рынок. Этот продукт пригоден для использования для профилактики и терапевтического лечения людей при вирусных инфекциях, злокачественных новообразованиях и состояний и иммуносупрессии и иммунодефицита.
Первый рекомбинантный иммунный интерферон человека, полученный в соответствии с изобретением, имеет метионин в качестве своей первой аминокислоты (кодон стартового сигнала ATG находится перед структурным геном), или, в том случае, если метионин находится внутри- или внеклеточно отщеплен, имеет в качестве первой аминокислоты цистеин. Зрелый иммунный интерферон человека можно также получить вместе с конъюгированным белком, отличным от обычного сигнального полипептида, причем конъюгат можно специфически отщеплять во внутри- или внеклеточное окружение. Наконец, зрелый иммунный интерферон человека можно получить прямой экспрессией без необходимости отщепления каких-либо сигнальных полипептидов. Это важно в тех случаях, когда данный хозяин может не удалять или удалять не столь эффективно сигнальный пептид, где вектор экспрессии предназначен экспрессировать зрелый интерферон человека вместе с его сигнальным пептидом. Полученный таким образом зрелый иммунный интерферон человека выделяют и очищают до уровня, удовлетворяющего требованиям для применения при лечении вирусных заболеваний, злокачественных новообразований и состояний иммуносупрессии или иммунодефицита.
Иммунный интерферон человека получают следующим образом:
Ткани человека, например, ткань селезенки человека или периферические
лимфоциты крови, культивировали с митогенами для стимулирования продуцирования иммунного интерферона.
Осадок клеток из такой клеточной культуры экстрагировали в присутствии ингибитора рибонуклеазы для выделения всей цитоплазмической РНК.
На олиго-АТ колонке выделили полностью информационную РНК (μ РНК) в полиаденилированной форме. Эту РНК расфракционировали по размерам, используя градиент плотности сахарозы и гель-электрофорез в кислоте-мочевине.
Соответствующую РНК (от 12S до 18S) превратили в соответствующую однотяжевую комплементарную ДНК (кДНК), из которой получили двунитевую ДНК. После поли-dC сшивания ее включили в такой вектор как плазмида, несущая один или более фенотипический маркер.
Полученные таким образом векторы использовали для трансформации бактериальных клеток, получая библиотеку клонов. Меченные радиоизотопами кДНК, полученные как из индуцированных, так и из неиндуцированных РНК, как описано ранее, использовали в качестве проб для анализа дуплицированных клонов библиотеки. Затем избыток ДНК удаляли и клоны экспонировали рентгеновскими лучами для идентификаци индуцированных клонов кДНК.
Из клонов индуцированных кДНК выделили соответствующие плазмидные ДНК и определили последовательность оснований.
В первом варианте воплощения секвенированную ДНК in vitro встроили в соответствующий вектор экспрессии, который использовали для трансформации клеток E.coli, которые, в свою очередь, культивировали с экспрессией целевого иммунного интерферона человека.
Экспрессированный таким образом иммунный интерферон человека содержал 146 аминокислот в зрелой форме, начиная с цистеина, и является основным. Его мономерный молекулярный вес был рассчитан как 17140. Возможно из-за наличия многочисленных основных остатков, гидрофобности, образования солевых мостиков и т.д. молекулы могут ассоциироваться в олигомерные формы, например, в димеры, тримеры или тетрамеры. Высокие значения молекулярных весов, наблюдавшиеся ранее для природных материалов (1), которые нельзя объяснить из основании только одной последовательности аминокислот, могут быть обусловлены такими олигомерными формами, так же, как и углеводами после пострансляционного гликозилирования.
В некоторых системах клеток хозяина, особенно при лигировании в вектор экспрессии так, чтобы экспрессировать интерферон вместе с его сигнальным пептидом, зрелую форму иммунного интерферона человека экспортируют в среду клеточной культуры, что облегчает выделение и очистку.
Технология культивирования. Известны устройства и способы поддержания перманентных линий клеток, полученных последовательной серией пассажей выделенных исходных клеток. Для применения в исследованиях такие клеточные линии поддерживают на твердых подложках в жидкой среде или выращивают в суспензии, содержащей поддерживающие питательные вещества. Более подробное описание предпосылок изобретения можно найти в Microbiology, 2nd Edition, Harper and Row, Publishers, Inc. Hagerstown, Maryland (1973) особенно на с. 1122 и далее Scientific American 245, c. 66 и далее (1981).
А. Культуры клеток (микроорганизмы)
Бактериальные штаммы/промоторы
Описываемую здесь работу осуществляли, используя микроорганизм E.coli К-12 штамм 294 (end A,
thi-, hsr-, khsm+) как описано в патенте Великобритании N 2055382 А. Этот штамм был депонирован в Американской Коллекции Типовых культур, АТСС под номером 31446.
Однако могут быть использованы и различные другие микроорганизмы, такие, как E. coli B, E.coli X 1776 (ATCC N 31537) и E. coli W 3110 (F-, λ- протрофный) (АТСС N 27325)
или другие микробные штаммы, многие из которых депонированы и являются доступными (см. также открытую патентную выкладку Западной Германии 26444432). Эти микроорганизмы включают, например, такие
Bacilli как Bacillus Subtilis и другие энтеробактерии, среди которых в качестве примера можно упомянуть Salmonella typhimurium и Serratia marcesans.
Беталактамозная и лактозная промоторные системы могут быть использованы для экспрессии гетерологичных белков и микроорганизмах. Конструкция этих промоторных систем описана в Change et al. Nature 275, 617 (1978) и Itakura et al. Science 198, 1056 (1977). Позднее была разработана система на основе триптофана, так называемая trp промоторная система (Goeddel et al. Nucleic Acids Research 8, 4057 (1980) и Kleid et al. U.S.S.N. 133, 296, 1980). Было открыто и использовано большое число других микробных промоторов (Siebenlist et al. Cell 20, 269 (1980)).
Дрожжевые штаммы/дрожжевые промоторы
Плазмида
YRp7 (9.10.11) способна реплицироваться как в E.coli так и в дрожжах, Saccharomyces cerevisiae. Для отбора в клетках дрожжей плазмида содержит TRPI ген (9, 10, 11), который комплементарен (позволяет
расти в отсутствии триптофана (мутациям дрожжей в этом гене, обнаруженном на хромосоме IV дрожжей 12). Использовался штамм RH218 (13) депонирован в Американской коллекции типовых культур (АТСС N
44076). Однако любой штамм Saccharomyces cerevisiae, содержащий мутацию, который делает клетку trp 1, эффективен для экспрессии. Примерами других штаммов, которые могут быть использованы, является
рер4-1 (14). Этот триптофановый ауксотропный штамм также имеет точковую мутацию в гене TRP-I. Помещенная на 5' конец недрожжевого гена 5'-фланкирующая последовательность ДНК (промотор) из дрожжевого
гена (для алкогольдегидрогеназы 1) способна обеспечивать экспрессию чужеродного гена в дрожжах в составе плазмиды. Кроме промотора, для экспрессии недрожжевого гена в дрожжах требуется вторая
дрожжевая последовательность, расположенная на 3'-конце недрожжевого гена на плазмиде так, чтобы обеспечить соответствующую терминацию транскрипции и полиаденилирование. Этот промотор можно с успехом
использовать в изобретении наряду с другими. В предпочтительных вариантах, 5'-фланкирующую последовательность дрожжевого гена 3-фосфоглицераткиназы (15) размещали выше структурного гена, после
которого следует ДНК, содержащая сигналы терминации полиаденилирования, например, TRPI (9, 10, 11) ген или PGK ген (15).
Так как дрожжевая 5'-фланкирующая последовательность (в сочетании с 3' дрожжевой фланкирующей ДНК) может функционировать для промотирования экспрессии в дрожжах гетерологичных генов, 5' фланкирующие последовательности любого высокоэкспрессирующего дрожжевого гена можно использовать для экспрессии целевых продуктов (16, 17, 18, 19, 20).
Системы клеточные культуры/векторы клеточных культур.
Размножение клеток позвоночных в культуре (культура ткани) стало обычной процедурой за последние годы (см. Tissue Culture, Academic Press, Kruse and Patterson eds, 1973). Использовали COS-7 линию фибробластов почки обезьян в качестве хозяина для получения иммунного интерферона (20а). Однако можно использовать для экспрессии интерферона согласно изобретению любую линию клеток, которая способна к репликации и экспрессии совместимого вектора, например, W138, BHK, 313, CHO, VERO и HeLa. В качестве векторов экспрессии могут быть использованы векторы на основе не только SV40, но и других вирусов, например, Polyoma, Adeno, VSV, BPV.
В. Векторные системы.
Прямая экспрессия зрелого иммунного интерферона в E.coli.
Методика, которую использовали для получения прямой экспрессии IFN-γ (без сигнальной последовательности) в E.coli является вариантом, использованным ранее для гормона роста человека (21) и лейкоцитарного интерферона человека (20б), поскольку включала сочетание синтетической (N-концевой) ДНК и кДНК.
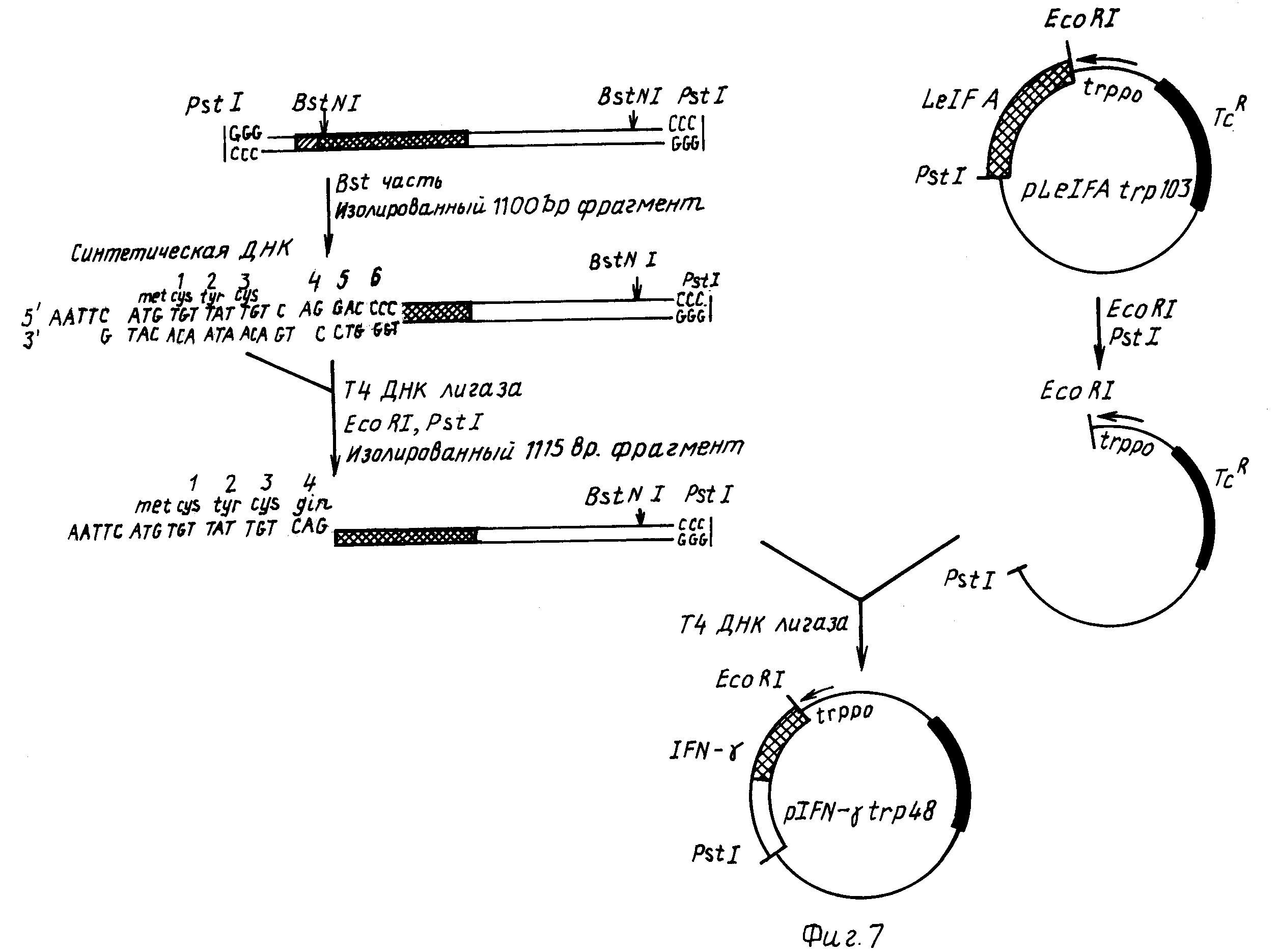
Как следует из нуклеотидной последовательности р69 и из сравнения с известным сайтом расщепления между сигнальным пептидом и зрелым полипептидом для некоторых IFN-γ S (20в), IFN-γ содержит гидрофобный сигнальный пептид из 20 аминокислот, после которого следует 146 аминокислот зрелого IFN-γ (фиг. 5). Как показано на фиг.7 BstNI сайт обычно расположен у аминокислоты 4 зрелого IFN-γ. Два синтетических олигодезоксинуклеотида были сконструированы, которые включили ATG кодон инициации трансляции, кодоны для аминокислот 1, 2 и 3 (цистеин-тирозин-цистеин) и образовывали EcoRI липкий конец. Эти олигонуклеотиды лигировали со 100 п.о. BstNI-PstI фрагментами р69 для создания 1115 п.о. синтетического природного гибридного гена, который кодирует IFN-γ и который связан EcoRI и PstI-сайтами. Этот ген был вставлен в плазмиду pLeIF A trp 103 между EcoRI и PstI участками для получения экспрессионной плазмиды pIFN-γ trp 48. В составе этой плазмиды ген IFN-γ экспрессируется под контролем E.coli trp промотора (pLeIfA trp 103 является производным pLeIF A 25, в котором участок EcoRI, дистальный к гену LeIF A, был удален. Методика удаления этого участка была описана ранее (22)).
Экспрессия в дрожжах.
Для экспрессии гетерологичного гена, например, кДНК иммунного интерферона, в дрожжах, необходимо сконструировать плазмидный вектор, содержащий четыре компоненты. Первой компонентной является часть, которая позволяет осуществлять трансформацию как E.coli, так и дрожжей и поэтому вектор должен содержать селективный ген из каждого организма. (В этом случае ген является геном устойчивости к ампициллину E.coli и геном TRPI дрожжей). Также требуется источник репликации из обоих организмов. (В этом случае ori E.coli из pBR322 и область ars 1 из хромосомы III дрожжей).
Второй компонентой плазмиды является 5'-фланкирующая последовательность из высоко экспрессивного гена дрожжей. В этом случае используется последовательность дрожжевого гена 3-фосфоглицератиназны (PGK). Этот фрагмент сконструирован таким образом, чтобы удалить ATG структурной последовательности PGK 8 п. о. выше ATG. Эту последовательность заменили последовательностью, содержащей XbaI и EcoRI-сайты для присоединения 5'-фланкирующей последовательности к структурному гену.
Третьей компонентой системы является структурный ген, созданный таким образом, что он содержит как ATG трансляционный стартовый, так и трансляционный стоп сигналы. Выделение и конструирование такого гена описано выше.
Четвертой компонентой является последовательность дрожжевой ДНК, содержащая 3'-фланкирующую последовательность дрожжевого гена, которая содержит соответствующие сигналы терминации и полиаденилирования.
Экспрессия в культуре клеток млекопитающихся.
Репликация вектора экспрессии в культуре ткани осуществлялась за счет обеспечения области на гала репликации ДНК (от вируса SV40) и обеспечения вспомогательной функции (Т антиген) путем введения вектора в линию клеток, эндогенно экспрессирующих этот антиген (23, 24). Поздний промотор вируса SV40 предшествует стрктурному гену интерферона и обеспечивает транскрипцию гена.
Вектор, который использовали для получения экспрессии IFN- γ, состоял из последовательностей pBR322, которые содержат маркер для отбора в E.coli (устойчивость к ампициллину) и ori репликации E.coli. Эти последовательности были получены из плазмиды pML-1 (23) и охватывают область, содержащую EcoRI и Bam HI сайты. Ori SV40 получена из 342 п.о. фрагмента в PvuII-Hind III (25, 26), причем оба конца превращены в концы EcoRI. Эти последовательности дополнительно содержат промотор для ранней и поздней единиц транскрипции. Ориентация ori участка SV40 такова, что промотор для поздней транскрипции расположен проксимально к гену, кодирующему интерферон.
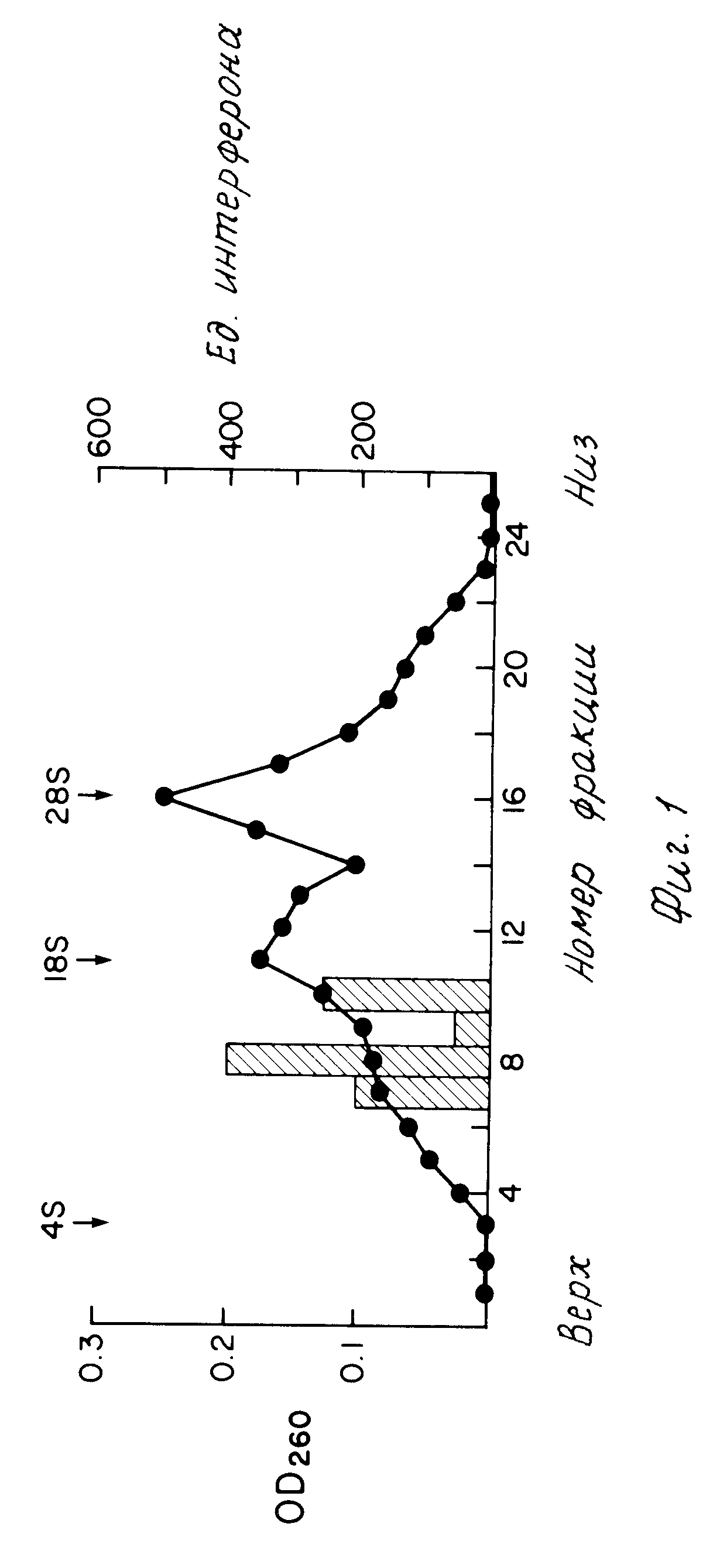
На фиг. 1 изображено центрифугирование в градиенте сахарозы стимулированных лимфоцитов периферической крови (PBL(поли)А) + РНК. Наблюдается два максимума активности интерферона, соответствующих размерам 12S и 16S. Расположение рибосомных маркеров РНК (центрифугированных независимо) помечено над контуром поглощения.
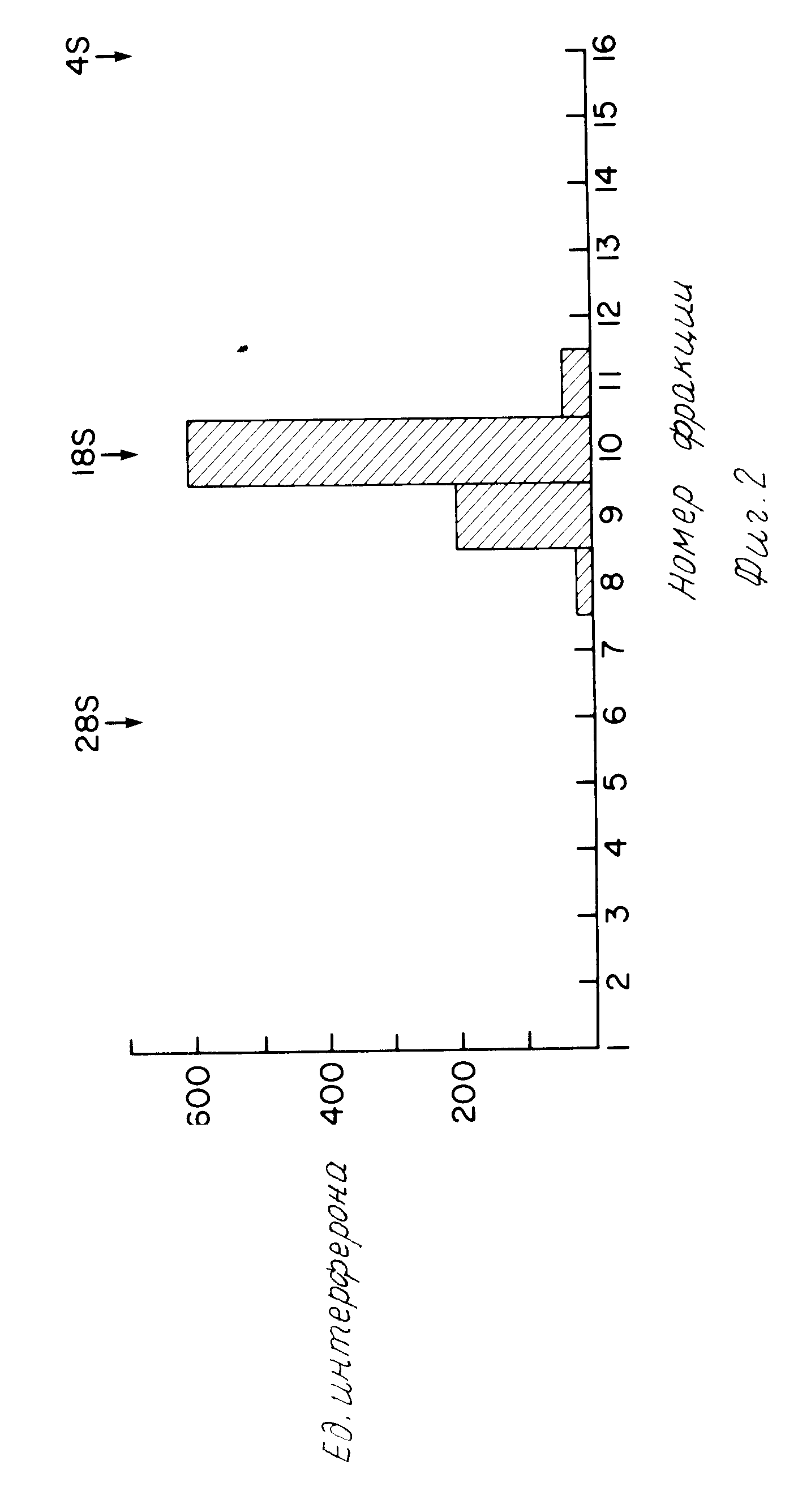
На фиг. 2 изображен электрофорез стимулированных PBL Poly/A + РНК через смесь кислота-мочевина-агароза. Наблюдается только один максимум активности, который мигрирует совместно с 18S РНК. Положения рибосомных маркеров РНК помечены выше контура активности.
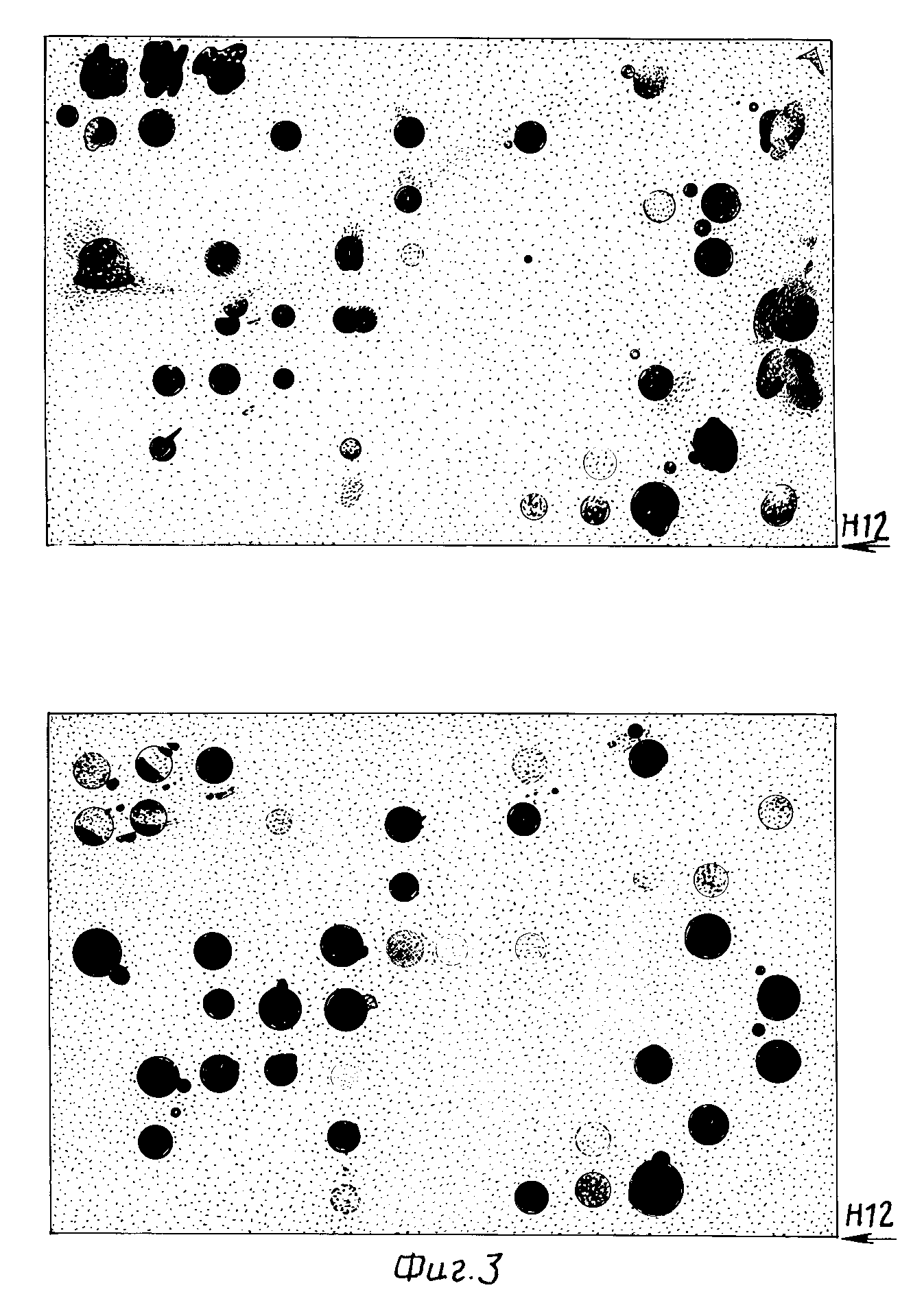
На фиг.3 изображены картины гибридизации 96 колоний с индуцированными и неиндуцированными зондами кДНК, меченными 32Р. 96 индивидуальных трансформантов выращивали на микротитровальной пластине, реплика была помещена на две нитроцеллюлозные мембраны, а затем фильтры гибридизовали с 32р-кДНК образцами, полученными из всех индуцированных иРНК (выше) или иРНК, выделенных из неиндуцированных культур PBL (неиндуцированные, ниже). Фильтры промывали для удаления негибридизованных РНК, а затем экспонировали на пленке рентгеновскими лучами. Эта серия фильтров представлена 86 такими наборами (8300 независимых колоний). Примером "индуцированного" клона является клон, помеченный Н12.
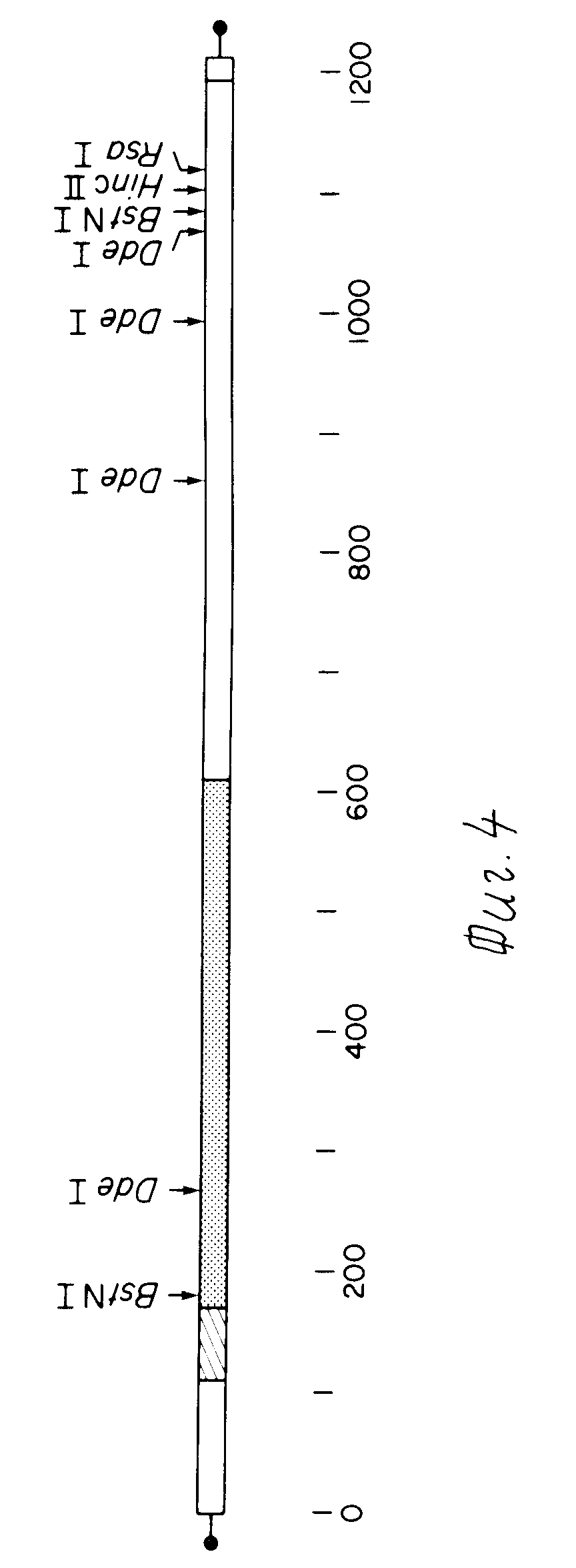
На фиг. 4 представлена карта рестрикции вставки кДНК клона 69. Вставка кДНК связана участками PstI (точки на обоих концов) и олиго-dC-dG хвостами (одна линия). Номер и размер рестрикционных фрагментов были установлены с помощью электрофореза в 6%-ном акриламидном геле. Положения участков были подтверждены секвенированием (на фиг.5). Кодирующая область самой большой открытой рамки считывания обведена в рамку, а заштрихованный участок представляет 20 остатков последовательности сигнального пептида, точечным пунктиром обозначена последовательность зрелого IIF (146 аминокислот).
На фиг.5 представлена нуклеотидная последовательность вставки плазмидной р69 кДНК, а также выведенная последовательность аминокислот наиболее длинной открытой рамки считывания. Предполагаемая сигнальная последовательность представлена остатками от S1 до S20.
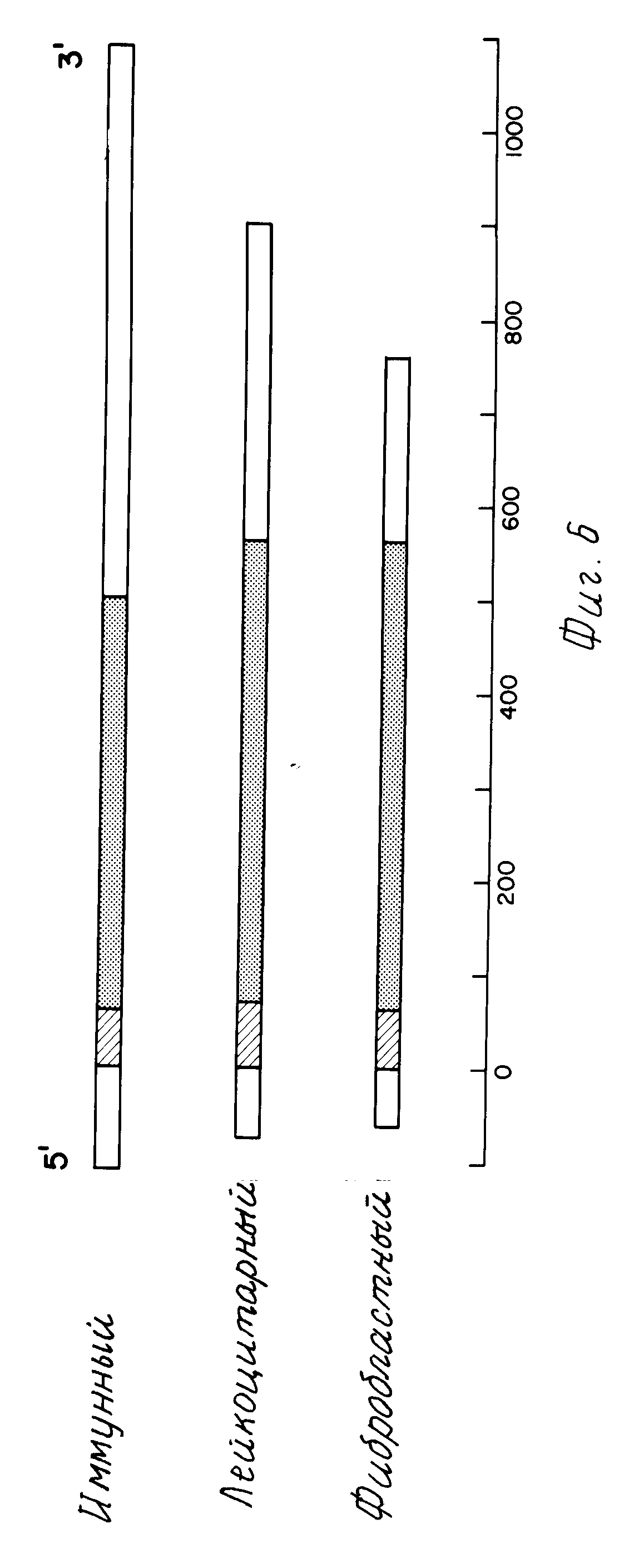
На фиг.6 приводится сравнение структуры IFN-γ иРНК и иРНК IFN-α и IFN-β. Клон 69 иРНК содержит значительно большее количество нетранслированных последовательностей.
На фиг.7 представлена схема конструирования IFN-γ экспрессионной плазмиды pIFN-γ trp 48. Исходным материалом является 1250 п.о. PstI вставка кДHК из плазмиды р69.
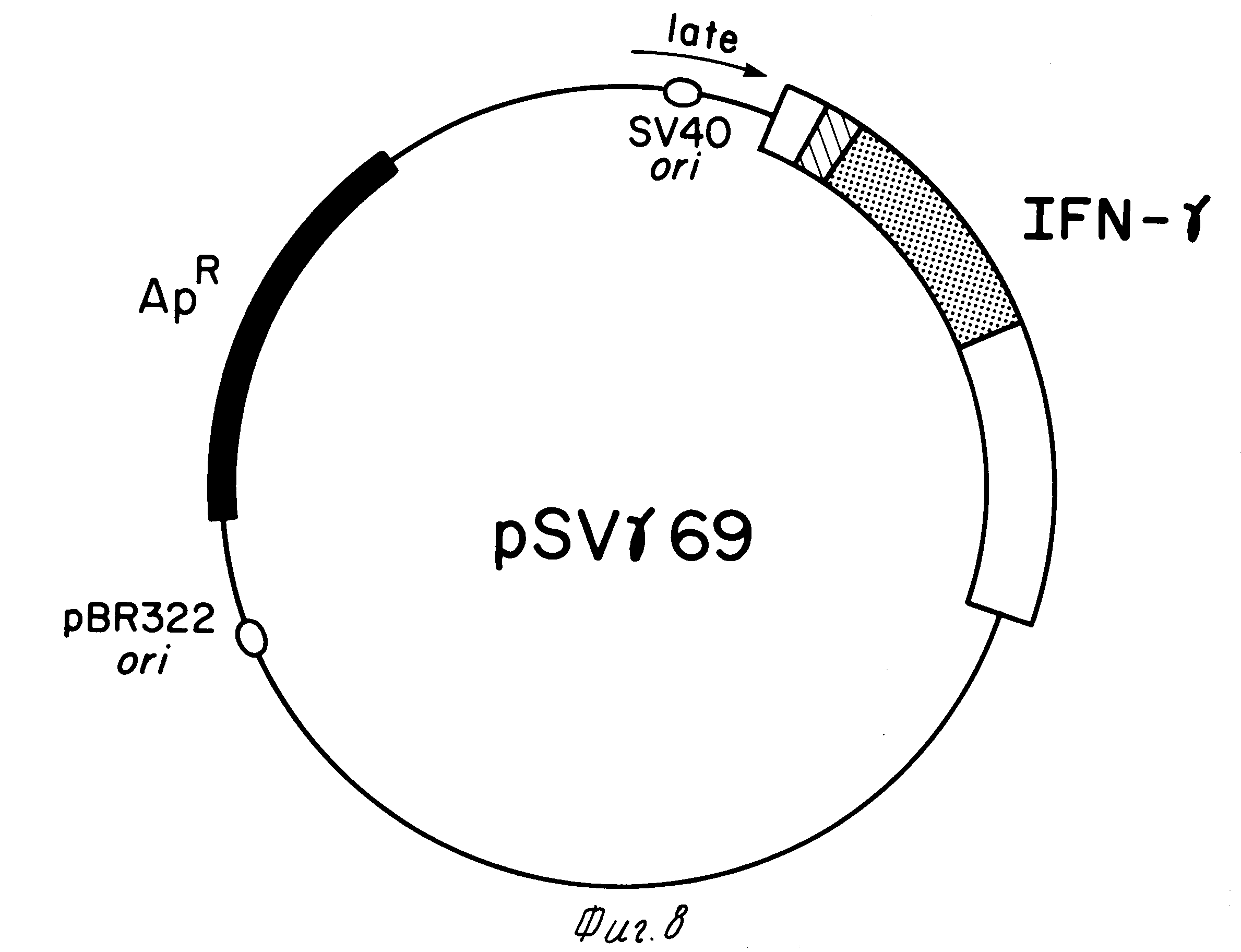
На фиг.8 приведена схема плазмиды, использованной для экспрессии IFN-γ в клетках обезьяны.
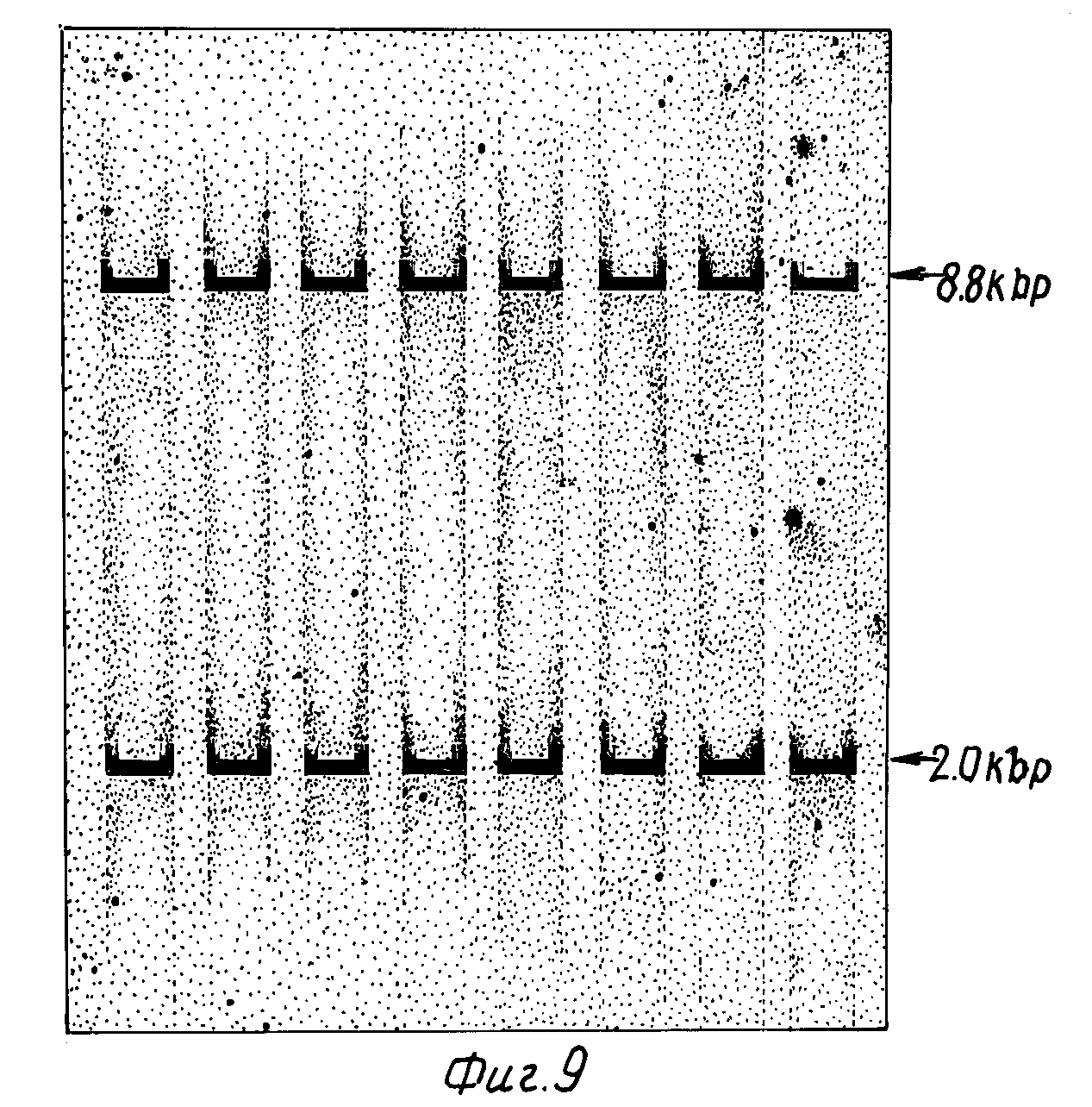
Фиг.9 изображает саузерн-гибридизацию 8 различных EcoRI расщепленных человеческих геномных ДНК, гибридизованных с32р-меченным 60 п.о. DdeI фрагментом из кДНК вставки р69. Два EcoRI фрагмента явно гибридизованы с зондом в каждом образце ДНК.
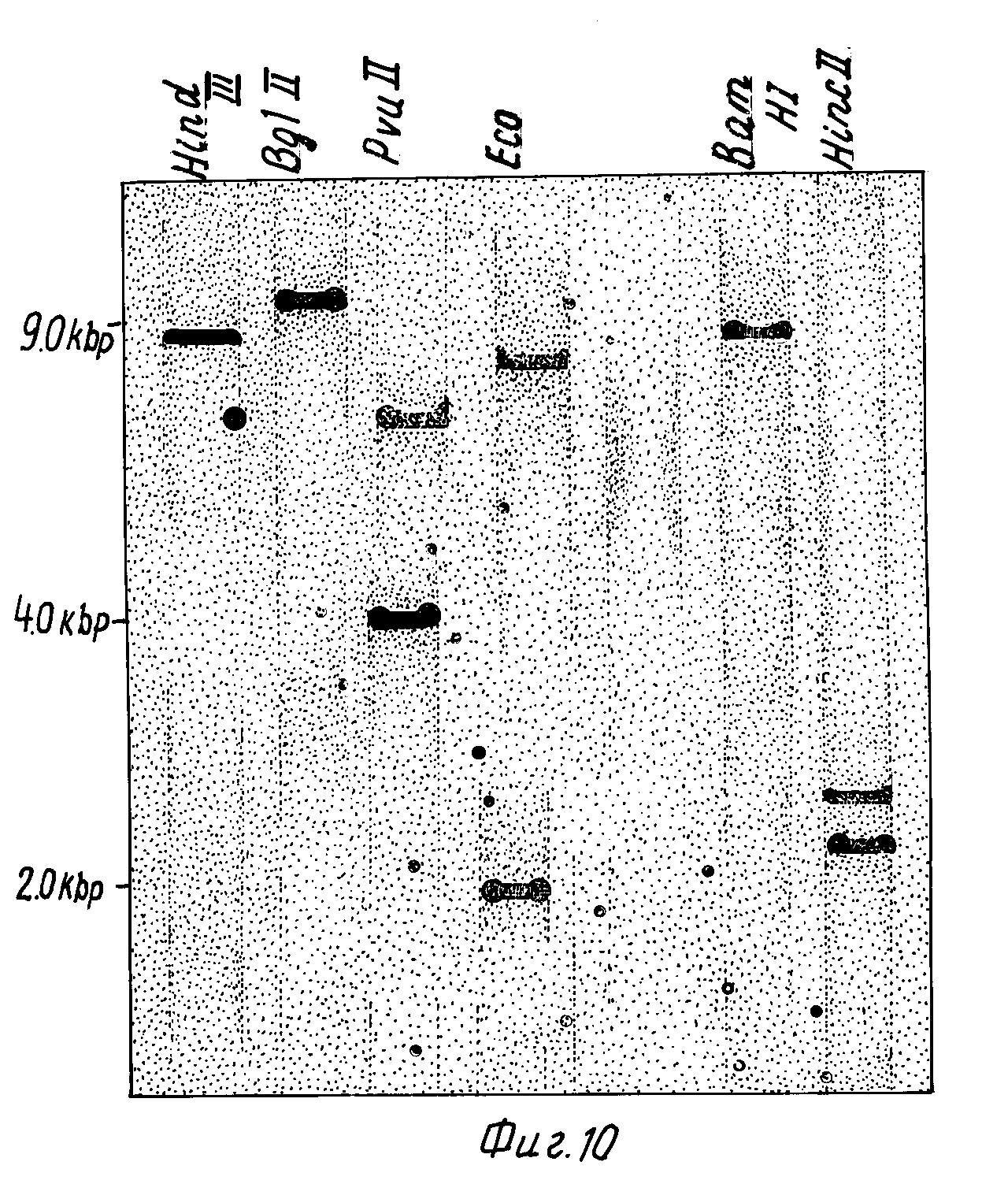
На фиг. 10 представлена саузерн-гибридизация человеческой геномной ДНК, переваренной 6-ю различными рестриктазами, с32р-меченным зондом из р69.
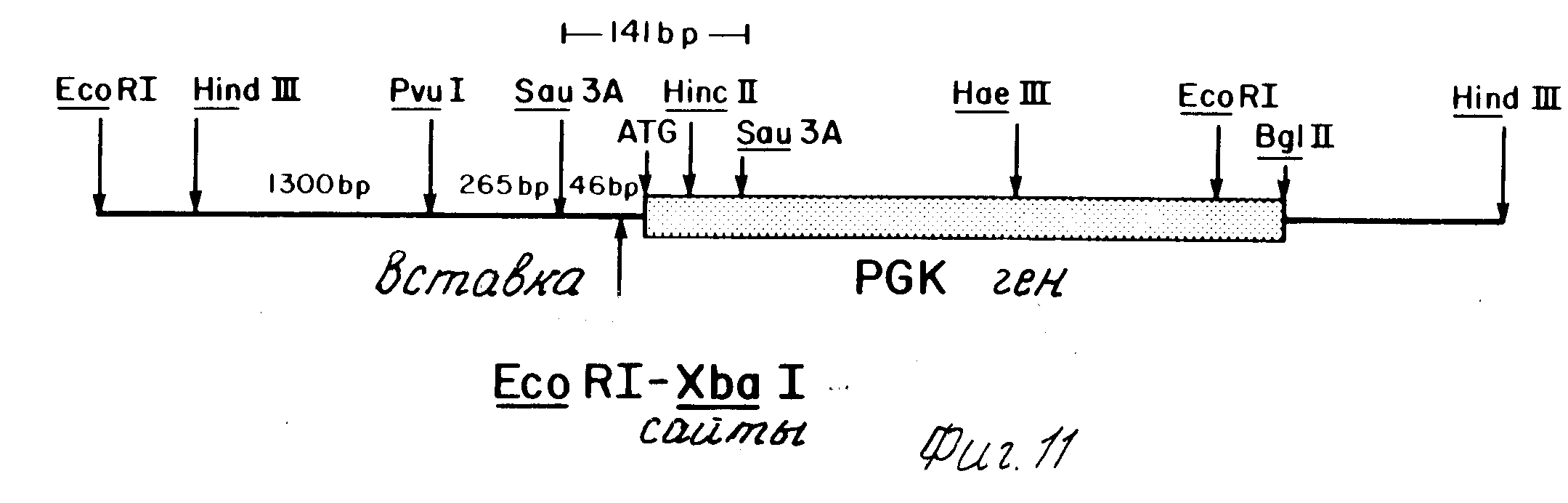
Фиг.11 представляет рестрикционную карту 3,1 ТПН Hind III вставку вектора рB1, из которого выделен PGK промотор. Указаны вставка EcoRI участка и XbaI участка в 5'-фланкирующую ДНК гена PGK.
Фиг. 12 иллюстрирует 5'-фланкирующую последовательность плюс начальную кодирующую последовательность гена PGK перед вставкой участков XbaI и EcoRI.
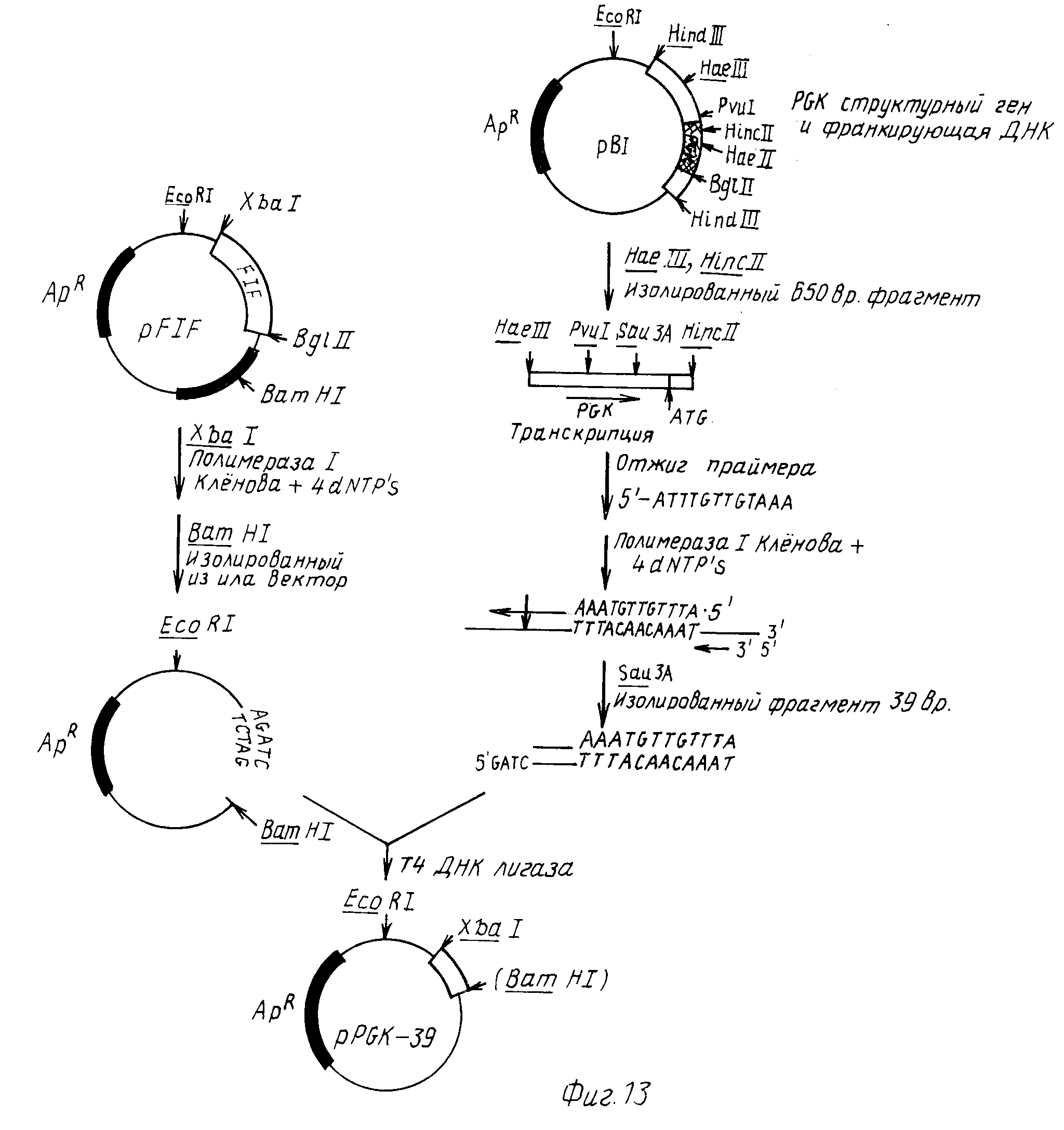
Фиг. 13 изображены методики, использованные для вставок XbaI участка в положение 8 в PGK промотор и для выделения 39 п.о. фрагмента 5'-фланкирующей последовательности PGK, содержащей эти концы XbaI и Sau 3A.
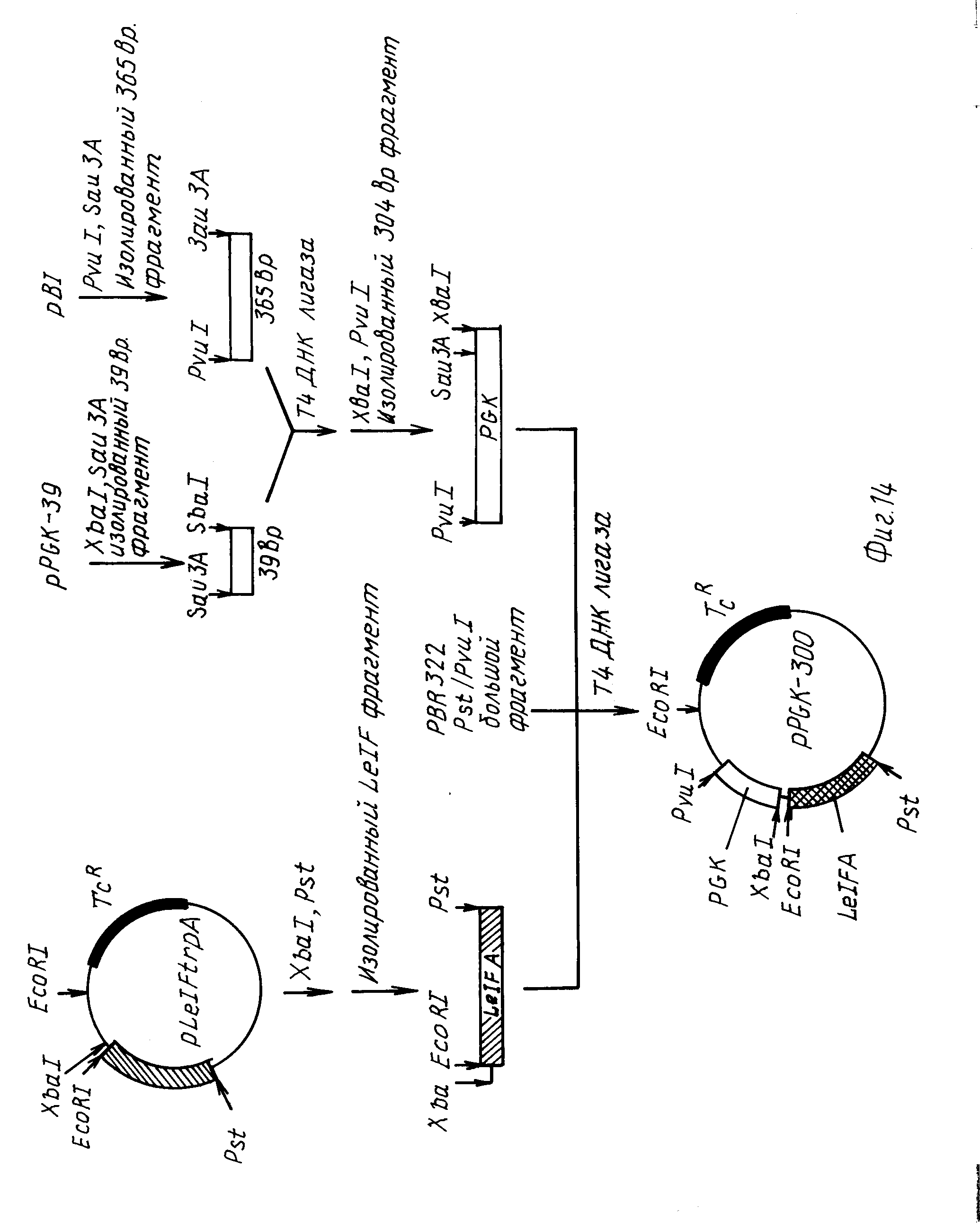
На фиг. 14 изображено конструирование 300 п.о. фрагмента, содержащего указанный фрагмент 39 п.о. дополнительную 5'-фланкирующую последовательность (265 п. о. ) от Pvu I до Sau 3A (см. рис.11), и EcoRI участок, прилежащий к XbaI.
На фиг. 15 изображено конструирование фрагмента 1500 п.о. PGK промотора (Hind III/EcoRI), который содержит, кроме фрагмента, сконструированного, как показано на фиг. 14, 1300 п.о. Hind III до Pvu I фрагмента из PGK 5'-фланкирующей последовательности (см. фиг.11).
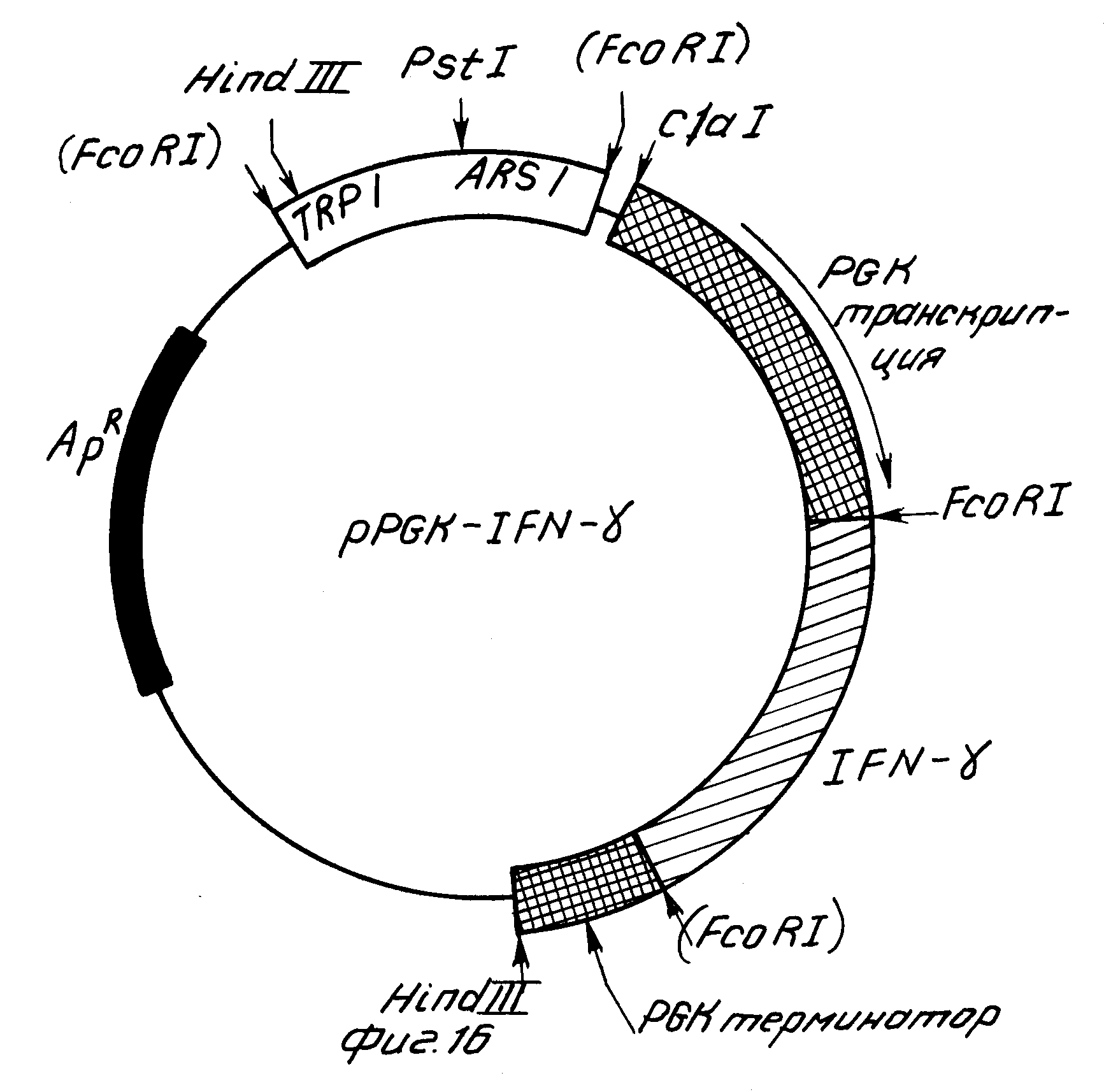
На фиг.16 изображена структура вектора экспрессии иммунного интерферона человека в дрожжах, содержащего модифицированный PGK промотор, IFN-γ кДНК и терминаторный участок дрожжевого PGK гена.
А. Источник IFN-γ иРНК.
Лимфоциты периферической крови (PBL) были получены от доноров (людей) с помощью лейкофореза. Далее PBL были очищены градиентным центрифугированием FicoII-Hypaque, а затем их культивировали при концентрации 5х106 кл/мл в RPMI 1640, 1% L-глутамине, 25 мМ HEPES и 1%-ном растворе пенициллинстрептомицина (Gibco, Grand Island, NY). Эти клетки индуцировали для получения IFN-γ митогенным стафилококковым энтеротоксином В (1 мкг/мл) и культивировали в течение 24-48 ч при 37оС в 5% СО2. К культуре PBL добавили дезацетилтимозин-α-1 (0,1 мкг/мл) для повышения относительного выхода IFN-γ активности.
В. Выделение
информационной РНК
Тотальная РНК из культур PBL экстрагировалась по существу в соответствии с сообщением Berger, S.L. et al. (28). Клетки выделяли центрифугированием, а затем повторно
суспендировали в 10 мМ NaCl, 10 мМ Tris-HCl (рН 7,5), 1,5 мМ MgCl2 и 10 мМ рибонуклеозид-ванадильного комплекса. Клетки лизировали, добавляя NP-40 (конечная концентрация 1%), и нуклеиновые
кислоты выделяли центрифугированием. Надосадочная жидкость содержала тотальную РНК, которую очищали далее многократными экстракциями фенолом и хлороформом. Водную фазу довели до 0,2 м в NaCl, а затем
РНК осадили, добавив два объема этанола. РНК из неиндуцированных культур выделили такими же способами. Для очистки иРНК от остальной РНК использовали Олиго-dT целлюлозную хроматографию (29). Типичные
выходы из 1-2 л культивированных PBL составляли 5-10 мг от общего количества РНК и 50-200 мкг Poly(A)+PHK.
С. Фракционирование иРНК по размерам
Для фракционирования препаратов
иРНК использовали два способа. Эти способы использовали независимо, и каждый из них приводил к значительному обогащению IFN-γ иРНК.
Для фракционирования иРНК использовали центрифугирование в градиенте сахарозы в присутствии денатуранта формамида. Градиенты от 5 до 25% сахарозы в 70% формамиде (27) центрифугировали при 154.000х g в течение 19 ч при 20оС. Последовательные фракции (0,5 мл) выделяли затем с верхней части градиента, осаждали этанолом, а затем аликвотные части инъецировали в Xenopus laevis ооциты для трансляции иРНК (30). Спустя 24 ч при комнатной температуре инкубационную среду исследовали на противовирусную активность в стандартном анализе ингибирования цитопатического эффекта, используя Vesicular Stomatitis Virus вирус (штамм Indiana) или вирус Encephalo myocarditis на клетках WISH (амнион человека) по описанию Стюарта (31), за исключением того, что образцы инкубировали с клетками в течение 24 ч (вместо 4) перед заражением вирусом. Соответственно, наблюдали два пика активности для РНК фракционированных в сахарозном градиенте (фиг.1). Один пик седиментирован с рассчитанным размером 12S и содержит 100-400 ед/мл антивирусной активности (по сравнению со стандартом IFN- α) на мкг введенной РНК. Другой пик активности, седиментированный по размеру 16S, содержит около половины активности более медленно седиментированного пика. Каждый из этих пиков активности, по-видимому, связан с IFN-γ так как для тех же фракций, исследовавшихся на линии бычьих клеток (MD BK), которые не защищены человеческим IFN-γ, не наблюдалось никакой активности. Как активность IFN-α, так и активность IFN-β можно легко определить, исследуя MDBК (31a).
Фракционирование иРНК (200 мкг) проводили также электрофорезом в кислота-мочевина-агарозном геле. Вязкий агарозный гель (37,38) состоял из 1,75% агарозы, 0,025 М цитрата натрия, рН 3,8 и 6М мочевины. Электрофорез проводили в течение 7 ч при 25 мА и 4оС. Затем гель фракционировали лезвием бритвы. Отдельные ломтики расплавили при 70оС, а затем дважды экстрагировали фенолом и один раз хлороформом. Затем фракции осадили этанолом и последовательно анализировали на содержание иРНК IFN-γ введением в Xenopus laevis ооциты, и затем проводили анализ на вирус. Для гель-фракционированных образцов наблюдали только один пик активности (фиг.2). Этот пик соответствует 18S и имеет активность 600 ед/мл на микрограмм введенной РНК. Эта активность также, по-видимому, специфична для IFN-γ, так как не защищает клетки MDBK. Расхождение величин активностей пиков, наблюдавшихся в сахарозных градиентах (12S и 16S) и кислота-мочевинных гелях (18S) можно объяснить тем, что эти независимые методы фракционирования проводились в неодинаковых условиях полного денатурирования.
Д. Получение библиотеки колоний, содержащих последовательности IFN-γ .
3 мкг гельфракционированных иРНК использовали для получения двойных кДНК по стандартным методикам (21,34). кДНК фракционировали по размерам в 6%-ном полиакриламидном геле. Фракции двух размеров электроэлюировали, 800-1500 п. о. (138 нг) и > 1500 п.о (204 нг). Порции 35 нг каждого размера кДНК удлинили дезокси C-остатками, используя терминальную дезоксинуклеотид трансферазу (35), и ренатурировали с 300 нг плазмидой рВР322 (36), которая была аналогично сшита с дезокси G остатками в сайте PstI (35). Каждую ренатурированную смесь трансформировали затем в E.coli K12 штамм 294. Получили приблизительно 8000 трансформантов с 800-1500 п.о. кДНК и 400 трансформантов с кДНК > 1500 п.о.
Е. Анализ библиотеки колоний на индуцированные кДНК.
Полученные колонии отдельно инокулировали в ячейки микротитровальных пластинок, содержащих LB/58+5 мкг/мл тетрациклина и хранившихся при -20оС после добавления DMSO до 7% Две копии библиотеки колоний выращивали на нитроцеллюлозных фильтрах и ДНК каждой колонии фиксировали на фильтре по методу Grunstein-Hogness (37).32р-меченные кДНК зонды приготовили, используя гельфракционированные иРНК размером 18S из индуцированных и неиндуцированных культур PBL. Использовали олиго-dT12-18 в качестве праймера и условия реакции как описано ранее (20б). Фильтры, содержащие 8000 трансформантов с отрезками 600-1500 п.о. кДНК и 400 трансформантов с кДНК более 1500 п.о. гибридизировали с 20х106 срм индуцированных32р-кДНК. Дублицированный набор фильтров гибридизовали с 20х106 кюри/мл неиндуцированных32р-кДНК. Гибридизацию проводили в течение 16 ч, используя условия, описанные Fritsch ed al (28). Фильтры тщательно промыли (28), а затем экспонировали на Кодаковской пленке ХР-5 рентгеновскими лучами с помощью интенсифицирующих экранов в течение 16-48 ч. Картину гибридизации для каждой колонии с двумя зондами сравнивали. Приблизительно 40% колоний явно были гибризидованы с обоими зондами, тогда как приблизительно 50% колоний не подверглись гибридизации ни с одним зондом (фиг. 3). 124 колонии были гибридизованы заметно с индуцированным зондом, но недетектируемо или более слабо с неиндуцированным зондом. Эти колонии индивидуально инокулировали в ячейку микротитровальных пластинок, выращивали, переносили на нитроцеллюлозные фильтры и гибридизовали с теми же двумя зондами, как описано выше. Плазмидная ДНК, выделенная из каждой из этих колоний быстрым способом (39), была также помещена на нитроцеллюлозные фильтры и гибридизована (39) со стимулированными зондами. ДНК из 22 колоний, гибридизованные только с индуцированными зондами, были названы "индуцированными" колониями.
F. Характеристики индуцированных колоний
Плазмидную ДНК получили из 5 индуцированных колоний (41) и использовали для того, чтобы охарактеризовать вставки кДНК. Рестрикционное картирование
пяти индуцированных плазмид (р67, р68, р69, р70 и р71) показало, что четыре из них имеют аналогичные рестрикционные карты. Каждая из этих четырех (р67, р69, р71 и р72) имеют четыре DdeI-сайта, 2
HinfI-сайта и один RsaI-сайт во вставке кДНК. Пятая плазмида (р68) содержит общий DdeI фрагмент и, очевидно, является коротким кДНК клоном, связанным с остальными. Гомологичность, предполагаемая на
основании рестрикционного картрирования, была подтверждена гибридизацией. Приготовили32р-меченный ДНК зонд (42) из 600 п.о. DdeI фрагмента р67 плазмиды и использовали для гибридизации (37)
с остальными индуцированными колониями. Все пять колоний перекрестно гибридизовались с этим зондом, как и с 17 другими колониями из 124 выбранных при отборе индуцированный/неиндуцированный. Длину
вставки кДНК в каждой из этих перекрестногибридизирующихся плазмид определяли после обработки PstI с помощью гельэлектрофореза. Клон с кДНК наибольшей длины, очевидно является клоном 69 с длиной
вставки 1200-1400 п.о. Эту ДНК использовали во всех дальнейших экспериментах, и ее рестрикционная карта приведена на фиг.4.
Вставка кДНК в р69, как было показано по продуктам экспрессии в трех независимых системах экспрессии, является к ДНК TFN-γ и приводит к противовирусной активности.
G. Анализ последовательностей вставки кДНК р69
Полная
нуклеотидная последовательность плазмидной р69 кДНК вставки была определена по способу терминации цепи (43) после субклонирования фрагментов в вектор М13 мр7 (44) и химическим методом по Мaxam-Gilbert
(47). Наиболее длинная открытая рамка считывания кодирует белок из 166 аминокислот, представленный на фиг.5. Первый остаток кодируется первым met кодоном на 5' конце кДНК. Первые 20 остатков у
аминоконца вероятно служат сигнальной последовательностью для секреции остальных 146 аминокислот. Эта предполагаемая сигнальная последовательность имеет структуру общую остальным охарактеризованным
сигнальным последовательностям, например, по размерам и гидрофобности. Далее четыре аминокислоты, найденные у предполагаемого сайта расщепления (ser-leu-gly-cys), идентичны четырем остаткам, найденным
в сайте расщепления нескольких лейкоцитарных интерферонов (LeIF B, C, D, F и Н, (20в)). Кодируемая зрелая аминокислотная последовательность из 146 аминокислот имеет молекулярный вес 17140 (именуемая в
дальнейшем рекомбинандным человеческим иммуноинтерфероном). Имеются два потенциальных положения гликозилирования (45) в кодируемой белковой последовательности: у аминокислот от 28 до 30 (asn-gly-thr)
и аминокислот от 100 до 102 (asn-tyr-ser). Существование этих положения согласуется с
наблюдаемым гликозилированием человеческого IFN-γ (1,46). Кроме того, только два цистеиновых остатка
(положения 1 и 3) стерически слишком близки, чтобы образовывать дисульфидный мостик, который связан с наблюдаемой стабильностью IFN-γ в присутствии таких восстанавливающих агентов, как β
-меркаптоэтанол (46). Выведенная зрелая аминокислотная последовательность обычно является полностью основной с 30 лизиновыми, аргининовыми и гистидиновыми остатками и всего 19 остатками аспарагиновой
кислоты и глутаминовой кислоты.
Структура иРНК IFN-γ, выведенная из ДНК последовательности плазмиды р69, заметно отличается от IFN-α (20б, 20в) или IFN-β (31а), иРНК. Как и представлено на фиг.6, кодирующий участок IFN-γ короче, хотя 5' нетранслируемый и 3' нетранслируемый участки гораздо длиннее, чем, соответственно, у IFN-α или IFN-β .
Н. Экспрессия рекомбинантного человеческого иммунного интерферона в E. coli.
В соответствии с фиг.7 50 мкг плазмид р69 были переварены PstI и 1250 п. о. вставка выделена с помощью гельэлектрофореза в 6% полиакриламидном геле. Приблизительно 10 мкг этой вставки электроэлюировали из геля. 5 мкг этого фрагмента PstI частично переварили 3 единицами BstNI в течение 15 мин при 37оС и реакционную смесь очистили в 6%-ном полиакриламидном геле. Приблизительно 0,5 мкг целевого в 1100 п.о. BstNI-PstI фрагмента было выделено. Два указанных дезоксиолигонуклеотида, 5'-dAATTCATGTGTTATTGTC и 5'-dTGACAATAACACATG (фиг.7) были синтезированы фосфотриэфирным методом (48) и фосфорилированы следующим образом. 100 пкмолей каждого дезоксиолигонуклеотида объединили в 30 мкл 60 мМ Tris-HCl (рН 8), 10 мМ MgCl2, 15 мМ β-меркаптоэтанола и 240 мкСi (γ -32p(ATP) Amersham, 5000 Ci (ммоль). 12 единиц Т4 полинуклеотидкиназы добавили, и реакции дали возможность протекать при 37оС в течение 30 мин. Затем добавили 1 Мкл 10 мМ АТР, и реакции дали возможность протекать еще 20 мин. После ОН/CHCl3экстрагирования олигомеры объединили с 0,25 мкг BstNI-PstI 1100 п.о. фрагмента и осадили этанолом. Эти фрагменты лигировали при 20оС в течение 2 ч в 30 мкл 20 мМ Tris-HCl (рН 7,5), 10 мМ MgCl2, 10 мМ дитиотретиола, 0,5 мМ АТР и 10 ед. Т4 ДНК лигазы. Полученную смесь переваривали в течение 1 ч с 30 ед PstI и 30 ед EcoRI (для исключения полимеризации через связывание липких концов), и провели электрофорез в 6%-ном полиакриламидном геле. Электроэлюированием был выделен продукт в 1115 п.о. (110000 кюри/мл).
Плазмида pLeIF Atrp 103 (фиг.7) является производной плазмиды pLeIF А 25 (20б), в которой EcoRI участок, дистальный по отношению LeIF A-гену, был удален (22). 3 мкг pLeIF A trp 103 переваривали 20 ед. EcoRI и 20 ед. PstI в течение 90 мин при 37оС, и провели электрофорез в 6%-ном полиакриламидном геле. С помощью электроэлюирования выделили большой (≈ 3900 п.о.) векторный фрагмент. ДНК фрагмент IFN-γ в 1115, п.о. EcoRI-PstI лигировали с 0,15 мкг этого полученного вектора. Трансформацию E. coli K-12 штамма 294 (АТСС N 31446) дало 120 устойчивых к тетрациклину колоний. Плазмидную ДНК получили из 60 этих трансформантов и переваривали EcoRI и PstI. Три из этих плазмид содержали целевой в 1115 п.о. EcoRI-PstI фрагмент. Анализ последовательности ДНК подтвердил, что эти плазмиды имеют нужную нуклеотидную последовательность в точках связывания между trp промотором, синтетической ДНК и кДНК. Одна из этих плазмид, pIFN-γ trp 48, была выбрана для дополнительного изучения. Эту плазмиду использовали для трансформации E.coli K-12 штамма W 3110 (АТСС N 27325).
1. Структура IFN-γ-кодирующей последовательности.
Структуру гена, кодирующего IFN-γ, анализировали гибридизацией. В этой процедуре (49) 5 мкг микрограмм высокомолекулярной человеческой лимфоцитарной ДНК (полученной по 50) переваривают до завершения различными рестрикционными эндонуклеазами, проводят электрофорез в 1,0%-ном агарозном геле (51) и помещают на нитроцеллюлозный фильтр (49).32р-меченный ДНК-зонд получают (42) из 600 п.о. DdeI фрагмента вставки кДНК р69 и гибридизуют (38) с нитроцеллюлозным пятном ДНК. 107 отсчетов в минуту зонда гибридизовали в течение 16 ч, а затем промыли (38). Восемь образцов геномной ДНК от различных доноров-людей переваривали EcoRI рестриктазой и гибридизовали с32р-меченным зондом р69. Как представлено на фиг.9, наблюдаются два четких сигнала гибридизации с размерами 8,8 т.п.н. и 2,0 т.п.н. что установлено путем сравнения с HindIII обработанной λ ДНК. Это могло быть результатом наличия двух генов IFN-γ или одного гена, расщепленному по EcoRI-сайту. Так как р69 кДНК не содержит EcoRI участков, наличие интрона с внутренним EcoRI-сайтом необходимо в случае одного гена.
Для того чтобы сделать различие между этими двумя возможностями, провели еще одну гибридизацию по Саузерну с тем же зондом, используя пять других переваров одной человеческой ДНК (фиг.10). Два гибридизирующихся фрагмента ДНК наблюдали для двух других переваров, PvuII (6,7 т.п.н. и 4,0 т.п.н.) и HinсII (2,5 т.п.н. и 2,2 т.п.н). Однако три образца переваров дают только один гибридизирующийся ДНK фрагмент: HindIII (9,0 т.п.н.), BglII (11,5 т.п.н.) и BamHI (9,5 т. п.н.). Два IFN-γ гена должны быть связаны на таком необычном коротком расстоянии (менее 9,0 т.п.н.), чтобы они содержались в одном и том же HindIII гибридизирующимся фрагменте. Этот результат предполагает, что только один гомологичный IFN-γ ген (в отличие от многих связанных с IFN-α генов) присутствует в человеческой геномной ДНК, и что этот ген расщепляется на один или более интронов, содержащих EcoRI, PvuII и HincII участки. Это предположение было подтверждено гибридизацией32р-меченного (42) фрагмента, полученного из 3' нетранслируемого участка КДНК из р69 (130 bp DdeI фрагмент из 860 п.о. до 990 п.о. на фиг.5) с EcoRI переварами человеческой геномной ДНК. Только 2,0 т.п.н. EcoRI-фрагмент гибридизуется с этим зондом, предполагая, что этот фрагмент содержит 3' нетранслируемые последовательности, тогда как 8,8 т.п.н. EcoRI-фрагмент содержит 5' последовательности. Структура гена IFN-γ (один ген с по крайней мере одним интроном) существенно отличается от IFN-α (множественные гены (20в) без интронов (51)) или IFN-β (один ген без интронов (52)).
J. Получение экстрактов бактерий
Ночную
культуру E.coli W 3110/pIFN-γ trp 48 в бульоне-Лурия + 5 мкг/мл тетрациклина использовали для инокулирования М9 (53) среды, содержащей 0,2% глюкозы, 0,5% казаминовой кислоты и 5 мкг/л
тетрациклина в разбавлении 1: 100. Индолакриловую кислоту добавили до конечной концентрации до 20 мкг/мл, когда А550 было между 0,1 и 0,2. 10 мл образца собрали центрифугированием при А550 1,0, немедленно повторно суспендировали в 1 мл фосфатного буферного раствора соли, содержащего 1 мг/мл альбумина бычьей сыворотки (pBS-BSA). Клетки разрушали ультразвуком и отделяли от
дебриса центрифугированием. Надосадочную жидкость хранили при 4оС до исследования. Активность интерферона в надосадочной жидкости определили как 250 ед/мл методом сравнения со стандартами
IFN-α с помощью исследования ингибирования цитопатического эффекта (СРЕ).
К. Трансформация штаммов/дрожжей и среды.
Штаммы дрожжей трансформировали, как было описано ранее (54). E.coli штамм JA300 (thr leu B6 thi thy A trp C 1117 hsdm- hsdR- strR) (15) использовали для отбора плазмид, содержащих функциональный TRPI ген. Штамм дрожжей РН218 с генотипом (trp1 gal2 suc2 mal CUPI) (13) использовали в качестве хозяина при трансформации. РН218 был депонирован в Американской коллекции типовых культур АТСС N 44076. Использовали М9 (минимальная среда) с 0,25% казаминовой кислоты (САА) и LB (богатая среда) как описано у Миллера (53), с добавлением 20 мкг/мл ампициллина (сигма) после того, как среду обработали в автоклаве и остудили. Дрожжи выращивали на среде УЕРD, содержащей 1% экстракта дрожжей, 2% пептона и 2% глюкозы ±3% агара Дифко. YNB+CAA содержали 6,7 г дрожжевого азотного основания (без аминокислот) (YNB) (Дифко), 10 мг аденина, 10 мг урацила, 5 г САА, 20 г глюкозы и ±30 г агара на литр.
L. Конструирование дрожжевого вектора экспрессии
1. 10 мкг YRp7 (9, 10, 11) переваривали EcoRI.
Образовавшиеся липкие концы ДНК затупили, используя ДНК полимеразу 1 (фрагмент Кленова) вектор и вставку разогнали на 1% -ном агарозном (SeaKem) геле, срезали с геля, электроэлюировали и
экстрагировали двумя равными объемами хлороформа и фенола перед осаждением этанолом. Полученные тупые концы молекул ДНК лигировали затем вместе в конечном объеме 50 мкл в течение 12 ч при 120оС. Эту лигирующую смесь использовали затем для трансформации E.coli штамма JA300 в устойчивый к ампициллину и прототрофный по триптофану штамм. Плазмиды, содержащие TRPIген в обеих ориентациях,
выделили. pFRWI имел TRPIген в той же ориентации, что и YRp7, тогда как pFRW2 имел TRPIген в противоположной ориентации.
20 мкг pFRW2 сделали линейными с помощью HindIII и провели электрофорез в 1% -ном агарозном геле. Линейные молекулы элюировали из геля и 200 нг лигировали затем с 500 нг 3,1 т.п.н. HindIII вставки плазмиды pBI (8), которая является рестрикционным фрагментом, содержащим дрожжевой ген 3-фосфоглицераткиназы. Лигирующую смесь использовали для трансформации E.coli штамма 294 в устойчивый к ампициллину и чувствительный к тетрациклину. Плазмида, полученная из одного такого рекомбинанта, имела интактный TRPI ген с 3,1 т.п.н. HindIII фрагментом из pBI вставки ДНК в HindIII участок устойчивого к тетрациклину гена. Эта плазмида pFRM31. 5 мкг pFRM31 полностью переварили с помощью EcoRI, дважды экстрагировали фенолом и хлороформом, а затем осадили этанолом. Липкие концы молекулы дополнили, используя ДНК полимеразу 1 в реакции, которая дала 250 мкМ в каждом дезоксинуклеозид фосфате. Реакцию вели в течение 20 мин при 14оС, и за это время ДНК дважды экстрагировали смесью фенолхлороформ, а затем осадили этанолом. Затем повторно суспендированную ДНК полностью переварили ClaI и провели электрофорез в 6%-ном акриламидом геле. Фрагмент вектора элюировали из геля, экстрагировали смесью фенол-хлороформ и осадили этанолом.
6
N-терминальных аминокислот 3-фосфоглицератного фермента, выделенные от человека были следующими:
1 2 3 4 5 6
SER LEU SER HSM LYS LEU
Одна из трансляционных рамок считывания,
полученная из ДНК последовательности 141 п.о. Sau3A до Sau3A рестрикционного фрагмента (содержащего внутренний сайт HincII; см PGK рестрикционную карту фиг.11) продуцирует следующую аминокислотную
последовательность.
1 2 3 4 5 6
MET SER LEU SER SER LYS LEU
После удаления инициатора метионина, видно, что PGK N-терминальная аминокислотная последовательность имеет
гомологию 5-6 аминокислотных остатков с N-терминальной аминокислотной последовательностью человеческого PGK. Это можно объяснить, если предположить, что старт дрожжевого PGK структурного гена
кодируется ДНК 141 и п.о. Sau3A рестрикционного фрагмента рВ1. В предшествующей работе (15) было предположено, что ДНК последовательности, определяющие иРНК PGK, могут находится в этой зоне фрагмента
HindIII. Дальнейшее секвенирование 141 п.о. Sau3A фрагмента обнаруживает ДНК последовательность PGK промотора (фиг.12).
Синтетический олигонуклеотид с последовательностью 5'ATTTGTTGTAAA3' был синтезирован стандартными методами (Crea et al. Nucleic Acids Res. 8, 2331 (1980)). 100 нг этой затравки пометили на 5' конце, используя 10 ед Т4 полинуклеотидкиназы в 20 мкл реакции, также содержащей 200 мк Кюри (Ci) [γ32-p] АТР. Этот меченый затравочный раствор использовали в реакции затравка-пара, предназначенной быть первой стадией многостадийного процесса введения EcoRI рестрикционного участка в PGK 5' фланкирующую ДНК непосредственно перед последовательностью структурного гена PGK.
100 мкг рВ1 (15) полностью переварили HaeIII, а затем разогнали в 6%-ном полиакриламидном геле. Верхнюю полосу на подкрашенном этидумом геле (содержащую область PGK промотора) выделили электроэлюированием. Этот 1200 п. о. HaeIII участок ДНК рестриктировали HincII, затем разогнали в 6%-ном акриламидном геле. Полосу 650 п.о. выделили электроэлюированием. При этом выделили 5 мкг ДНК. Этот 650 п.о. HaeIII-HincII участок ДНК повторно суспендировали в 20 мкл Н2О, затем смешали с 20 мкл фосфорилированного затравочного раствора. Эту смесь экстрагировали их смесью фенол-хлороформ, а затем осаждали этанолом. Высушенную ДНК повторно суспендировали в 50 мкл Н2О а затем нагревали в кипящей водяной бане в течение нескольких минут. Затем этот раствор быстро охладили в бане сухой лед-этанол (10-20 с), а затем перенесли в баню лед-вода. К этому раствору добавили 5Л мкл раствора, содержащего 10 мкл 10Х ДНК полимеразного 1 буфера (Boеhringer Mannheim), 10 мкл раствора, ранее подготовленного 2,5 мМ в каждом деоксинуклеозидтрифосфате (dATP, dTTP, dGTP и dCTР), 25 мкл Н2О и 5 ед ДНК полимеразы 1. Эти 100 мкл реакционной смеси инкуровали при 37оС в течение 4 ч. Затем раствор экстрагировали IX смесью хлороформ-фенол, осадили этанолом, высушили лиофилизированием, а затем полностью рестриктировали 10 ед Sau3A. Этот раствор разогнали затем в 6% -ном акриламидном геле. Полосу, соответствующую размеру 39 п.о. вырезали из геля, а затем выделили электроэлюированием. Эта полоса 39 п.о. имела один тупой конец, и один Sau3A липкий конец. Этот фрагмент клонировали в модифицированный pFIF trp69 вектор (31а). 10 мкг pFIF trp 69 линеализировали XbaI, экстрагировали IX хлороформом, а затем осадили этанолом. XbaI липкий конец заполнили, используя ДНК полимеразу 1 в 50 мкл реакционной среде, содержащей 250 мкМ в каждом нуклеозидном трифосфате. Эту ДНК обработали BamHI, затем разогнали в 6%-ном акриламидном геле. Векторный фрагмент выделили из геля электроэлюированием, а затем суспендировали повторно в 20 мкл Н2О. 20 нг этого вектора легировали с 20 нг 39 п.о. фрагмента, полученного ранее, в течение 4 ч при комнатной температуре. Одну пятую этой лигирующей смеси использовали для трансформации E.coli штамма 294 в устойчивый к ампициллину (на LB + 20 мкг/мл амр. пластинах). Плазмиды из трансформантов исследовали по методике быстрого отбора (39). Одну плазмиду, pHGK-39, выбрали для анализа последовательности. 20 мкг этой плазмиды переварили XbaI, осадили этанолом, а затем обработали 1000 ед бактериальной щелочной фосфазы при 68оС в течение 45 мин. ДНК 3х экстрагировали смесью фенол-хлороформ, затем осадили этанолом. Дефосфорилированные концы пометили затем в 20 мкл реакционной смеси, содержащей 200 мкКюри [γ32-p] АТР и 10 ед Т4полинуклеотидкиназы. Плазмиду обработали SalI и разогнали в 6%-ном акриламидном геле.
Полосу меченой вставки выделили из геля и определили последовательность методом химической деградации (47). Последовательность ДНК у 3'-конца этого промоторного отрезка соответствовала ожидаемой.
2. Конструирование 312 bp
PvuI-EcoRI PGK промоторного фрагмента
25 мкг pPGK-39 (фиг. 13) одновременно переваривали SalI и XbaI (5 ед каждого), затем разогнали электрофоретически в 6%-ном геле. Полосу 390 п.о.
содержащую участок 39 п.о. промотора, выделили электроэлюированием. Повторно суспендированную ДНК рестриктировали Sau3A, затем разогнали электрофоретически в 8%-ном акриламидном геле. Полосу 39 п.о.
PGK промотора выделили электроэлюированием. ДНК содержала 39 п.о. 5' конец PGK промотора на фрагменте Sau3A-XbaI.
25 мкг pB1 рестриктировали PvuI и KphI, затем разогнали электрофоретически в 6%-ном акриламидном геле. 0,8 т.п.н. полосу ДНК выделили электроэлюированием, затем рестриктировали Sau3A и разогнали электрофоретически в 6%-ном акриламидном геле. Полосу 265 п.о. из PGK промотора (фиг.11) выделили электроэлюированием.
Затем ДНК сшили с 39 п.о. промоторным фрагментом, описанным выше, в течение двух часов при комнатной температуре. Лигирующую смесь рестриктировали XbaI и PvuI, затем разогнали электрофоретически в 6%-ном акриламидном геле. 312 п. о. Xba-PvuI рестрикционный фрагмент выделили электроэлюированием, затем добавили к лигирующей смеси, содержащей 200 нг pBR322 (36) (выделенный ранее с опущенным 162 п.о. PvuI-PstI рестрикционным фрагментом) и 200 нг XbaI-PstI LeIFA кДНК гена, ранее выделенного из 20 мкг pLeIF trp A25. Эту трехфакторную лигирующую смесь использовали для трансформации E.coli штамма 294 в устойчивый к тетрациклину. Колонии трансформантов минискринировали (39) и одну из колоний, pPGK-300, выделили, она имела 304 п.о. PGK 5' фланкирующую ДНК, слитую с LeIF A геном в составе вектора на основе pBR322. 5' конец LeIF A гена имел следующую последовательность: 5'-CTAGAATTC-3'. Таким образом, слияние XbaI участка из PGK промоторного фрагмента в эту последовательность позволяет добавление к участку XbaI участка EcoRI. Таким образом, pPGK-300 содержит часть PGK промотора, выделенного в PvuI-EcoRI фрагмент.
3. Конструирование 1500 п.о. EcoRI-EcoRI PGK промоторного фрагмента
10 мкг pBI переваривали PvuI и EcoRI, разогнали в 6%-ном акриламидном геле. 1,3 т.п.н. PvuI-EcoRI
ДНК полосу из PGK 5'-фланкирующей ДНК выделили электроэлюированием. 10 мкг pPGK-300 переварили EcoRI и PvuI и 312 п.о. промоторный фрагмент выделили электроэлюированием после электрофореза
переваренной смеси в 6%-ном акриламидном геле. 5 мкг pFRL4 обработали EcoRI, осадили этанолом, а затем обработали бактериальной щелочной фосфатазой при 68оС в течение 45 мин. После трех
экстракций ДНК смесью фенол/хлороформ, провели осаждение этанолом, и повторное суспендирование в 20 мл Н2О; 200 нг вектора сшили со 100 нг 312 п.о. EcoRI-PvuI ДНК из pPGK-300 и 100 нг
EcoRI-PvuI ДНК из рВ1. Лигирующую смесь использовали для трансформации E.coli штамма 294 в устойчивый к ампициллину. Одним из полученных трансформатов была pPGK-1500. Эта плазмида содержит 1500 п.о.
PGK промоторный фрагмент как EcoRI-EcoRI или HindIII-EcoRI фрагмент ДНК.
10 мкг pPGK-1500 полностью переварили ClaI и EcoRI, затем переваренную смесь разогнали электрофоретически в 6%-ном акриламидном геле. Фрагмент 900 п. о. содержащий PGK промотор, выделили электроэлюированием. 10 мкг pIFN-γ trp 48 было полностью переварено EcoRI и HincII и разогнано электрофоретически в 6%-ном акриламидном геле. Полосу 938 п.о. содержащую прямоэкспрессируемую IFN-γ кДНК, выделили из геля электроэлюированием.
Вектор экспрессии дрожжей сконструировали в трех факторной редакции, сшивая вместе фрагмент PGK промотора (на ClaI-EcoRI участке), делетированный pFRM-31 и выделенной ранее IFN-γ кДНК. Лигирующую смесь инкубировали при 14оС в течение 12 ч. Лигирующую смесь использовали далее для трансформации E.coli штамма 294 в устойчивый к ампициллину. Трансформанты анализировали на присутствие соответствующим образом сконструированной экспрессионной плазмиды, pPGK-IFN-γ (фиг. 16). Плазмиды, содержащие экспрессионную систему, использовали для трансформации сферопластов дрожжевого штамма RH218 в триптофан прототропный на агаре, пропускающем триптофан. Затем эти рекомбинантные дрожжи анализировали на предмет присутствия рекомбинантного иммунного интерферона человека.
Дрожжевые экстракты получили следующим образом: 10 мл культур выращивали в YNB+CAA до достижения А660 ≃ 1-2, собрали центрифугированием, затем повторно суспендировали в 500 мкл PBS буфера (20 мМ NaH2PO4, рН 7,4 150 мМ NaCl). Добавили равный объем стеклянных шариков (0,45-0,5 мм), и полученную смесь вращали в течение 2 мин. Эти экстракты вращали затем 30 с при 14000 об/мин и надосадочную жидкость удалили. Активность интерферона в надосадочной жидкости определили как 16000 ед/мл путем сравнения с IFN-α стандартом, используя СРЕ ингибирующий анализ.
М. Конструирование вектора клеточной культуры pSVγ 69.
Фрагмент 342 п.о. HindIII-PvuII, включающий ori SV40, превратили во фрагмент, связывающий EcoRI рестрикционный участок. HindIII участок конвертировали добавлением синтетического олигомера 95'dAGCTGAATTC) и PvuII сайт конвертировали лигированием по тупому концу в EcoRI сайт с использованием полимеразы 1. Полученный EcoRI фрагмент вставки в EcoRI участок pML-1 (23). Плазмиду с SV40 поздним промотором, ориентированным в сторону от амрR гена, далее модифицировали, удалив EcoRI участок, ближайший к амрR гену pML-1 (22).
Фрагмент 1023 п.о. Hpal-BglII, клонированной HBV ДНК (55), был выделен и Hpal сайт вируса гепатита. В (HBV) превратили в EcoRI участок синтетическим олигомером (5'dGCGAATTCGC). Этот EcoRI-BglII связанный фрагмент непосредственно клонировали в EcoRI-BamHI участок плазмиды, описанной ранее, несущей ori SV40.
В оставшийся EcoRI участок вставили IFN-γ ген на фрагмент р69 в 1250 п. о. PstI после конверсии PstI концов в EcoRI концы. Выделили те клоны, в которых SV40 поздний промотор предшествовал структурному гену IFN-γ. Полученные плазмиды были затем введены в клетки культур тканей (24), используя ДЕАЕ-декстран методику (56), модифицированную таким образом, что трансфекцию в присутствии ДЕАЕ-декстрана проводили в течение 8 ч. Клеточную среду меняли каждые 2-3 дня. Ежедневно отбирали 200 мкл на интерфероновый биоанализ. Типичные выходы составили 50-100 ед/мл в образцах, проанализированных три или четыре дня спустя трансфекции.
Анализ показывает, что продукт экспресси не имеет CYS-TYS-CYS N-концевой части рекомбинантного человеческого иммуноинтерферона (см. фиг.5), обеспечивающей расщепление сигнального пептида по связи CYS-GLN (аминокислоты 3 и 4 на фиг. 5), чтобы зрелый полипептид фактически состоял бы из 143 аминокислот.
N. Частичная очистка
иммунного рекомбинантного интерферона des-CYS-TYR-CYS, полученного из клеток обезьяны
Для получения больших количеств человеческого интерферона IFN-γ в клетках обезьяны свежие монослои
COS-7 клеток в десяти 10 см пластинах были трансфицированы 30 мкг pDLIF3 в 110 мл ДЕАЕ-декстран (200 мкг/мл ДЕАЕ декстран 500000 MW, 0,05 М трис рН 7,5 в ДМЕМ). Спустя 16 ч при 37оС
пластины промыли дважды ДМЕМ. 15 мл свежего ДМЕМ с 10% f.b.s. 2 мМ глутамина, 50 мк/мл пенициллина G и 50 мг/мл стрептомицина добавили затем на каждую пластину. Среду заменили на следующий день
освобожденной от сыворотки ДМЕМ. Свежую свободную от сыворотки среду добавили затем ежедневно. Собранную среду хранили при 4оС до тех пор, пока не использовали для анализа или связывали с
CPG. Было обнаружено, что фракции, собранные спустя 3 и 4 дня после трансфекции, сохраняли практически всю активность.
0,5 г CPG (контролируемое пористое стекло, Electronucleonics, CPG 350, с размером пор 120/200) добавили к 100 мл клеточной надосадочной жидкости и полученную смесь перемешивали в течение 3 ч при 4оС. После недолгого центрифугирования осевшие шарики набили в колонку и тщательно промывали 20 мМ NaPO4, 1 M NaCl 0,1% β-меркаптоэтанолом, рН 7,2. Затем активности элюировали тем же самым буфером, содержащим 30% этиленгликоля с последующим элюированием указанным буфером, содержащим 50% этиленгликоля. Практически вся активность связана с СРG. 75% элюированной активности нашли во фракциях, элюированных 30% этиленгликолем. Эти фракции собрали и разбавили 20 мМ NaPO4, 1 M NaCl, рН 7,2, до конечной концентрации 10% этиленгликоля и непосредственно вводили в 10 мл Con A Sepharose (Pharmacia) колонку. После тщательной промывки 20 мМ NaPO4, 1M NaCl, рН 7,2, активности элюировали 20 мМ NaPO4, 1M NaCl, 0,2 М α-метил-Д-маннозидом. Существенное количество активности (55%) не связано с этим лецитином. 45% активности элюировано α-метил-Д-маннозидом.
Данные биоисследований
А. Характеристика противовирусной активности
Для нейтрализации антител
образцы разбавляли, в случае необходимости, до концентрации 500-1000 ед/мл PBS-BSA. Равные объемы образца инкубировали в течение 2-12 ч при 4оС с рядом разбавлений кроличьими
антисыворотками человеческого лейкоцитарного, фибробластного или иммунного интерферона. Анти-IFN-α и β получили из Национального института аллергических и инфекционных заболеваний.
Анти-IFN γ-α были получены с использованием аутентичного IFN-γ (5-20% чистоты), очищенного из стимулированных лимфоцитов периферической крови. Образцы центрифугировали в течение 3
мин при 12000х g в течение 3 мин перед исследованием. Для проверки стабильности при рН 2 образцы доводили до рН 2, добавляя 1н. HCl, инкубировали в течение 2-12 ч при 4оС и нейтрализовали
добавлением 1н. NaCl перед исследованием. Для тестирования чувствительности к натрию додецилсульфату (SDS) образцы инкубировали в равном объеме 0,2% SDS в течение 2-12 ч при 4оС перед
анализом.
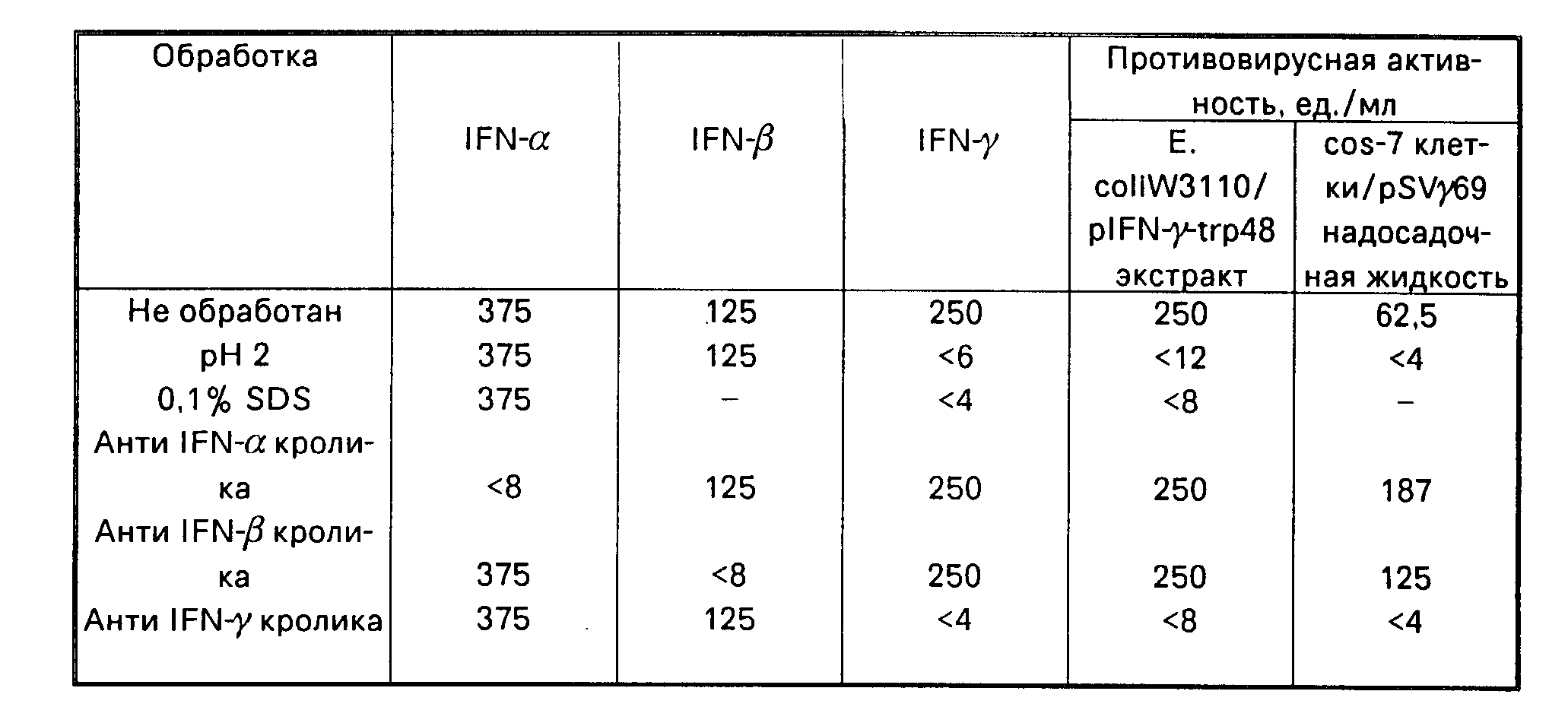
В. Характеристики IFN- γ, полученного с помощью E.coli и COS-7 клеток приведены в таблице
Таблица характеризует IFN-α, β,γ после
различных обработок. Активность интерферона, полученного в E. coli W3110/pIFN γ trp 48 и COS-7/pSV γ 69, кислотно-чувствительная, SDS-чувствительная, нейтрализуется антисывороткой
иммунного интерферона и не нейтрализуется антителами к IFN-α или β. Эти данные подтверждают, что интерферон, полученный в этих системах, является IFN-γ и что кДНК вставка плазмиды
р69 кодирует IFN-γ.
Иммунный интерферон, полученный согласно изобретению, был определен с помощью секвенирования гена и выведения из нуклеотидной последовательности аминокислотной последовательности (фиг.5).
Реферат
Использование: изобретение относится к генетической инженерии, в частности к способу получения рекомбинантной плазмидной ДНК, способной экспрессировать иммунный интерферон человека. Сущность изобретения: способ получения плазмидной ДНК заключается в том, что в вектор pBR 332 или его производное встраивают к ДНК, кодирующую иммунный интерферон человека, имеющий аминокислотную последовательность, начинающуюся от аминокислот GLN- ASp - pRO в положениях 4,5 и 6 или CYS - TYR - CYS в положениях 1,2 и 3 до конца последовательности. 1 табл, 16 ил.
Комментарии