Способ получения акриловой кислоты из гидроксипропионовой кислоты - RU2719485C1
Код документа: RU2719485C1
Чертежи


Описание
ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение по существу относится к способам получения акриловой кислоты, производных акриловой кислоты или их смесей путем приведения в контакт потока поступающих газообразных материалов, содержащих водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с находящимся в реакторе катализатором. Более конкретно, настоящее изобретение относится к применению катализатора, содержащего аморфную и частично дегидратированную фосфатную соль, а реактор представляет собой однослойный реактор, который содержит алюминий, кремний или их смеси и имеет низкую скорость коррозии. В альтернативном варианте осуществления реактор представляет собой двухслойный реактор, который имеет внутренний слой и наружный слой, при этом внутренний слой содержит алюминий и имеет низкую скорость коррозии.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Акриловая кислота, производные акриловой кислоты или их смеси в настоящее время применяют в различных промышленных материалах, таких как адгезивы, связывающие вещества, покрытия, краски, лаки, моющие средства, флокулянты, диспергирующие вещества, тиксотропные вещества, комплексообразователи и суперабсорбирующие полимеры (SAP), которые используют в абсорбирующих изделиях, выполненных с возможностью разового использования, включая подгузники и гигиенические изделия. С точки зрения процесса производства акриловую кислоту в настоящее время, как правило, получают в ходе двухстадийного каталитического окисления пропилена, который, в свою очередь, производят из ископаемых ресурсов, таких как нефть или природный газ. Более подробную информацию об окислении пропилена для получения акриловой кислоты и других способах производства можно найти в публикации Kirk-Othmer Encyclopedia of Chemical Technology, Vol. 1, pgs. 342-369 (5th Ed., John Wiley & Sons, Inc., 2004).
Акриловая кислота ископаемого происхождения способствует выбросам парниковых газов из-за высокого содержания в ней углерода ископаемого происхождения. Кроме того, ископаемые ресурсы не являются возобновляемыми, так как требуется сотня тысяч лет для их естественного формирования и совсем немного времени для их израсходования. С другой стороны, возобновляемые ресурсы относятся к материалам, которые производят с помощью естественного процесса со скоростью, сравнимой со скоростью их потребления (например, в пределах 100-летнего периода времени), и можно восполнять естественным образом или с помощью сельскохозяйственных методов. К примерам возобновляемых ресурсов относятся растения, например, сахарный тростник, сахарная свекла, кукуруза, картофель, цитрусовые, древесные растения, лигноцеллюлоза, углеводы, гемицеллюлоза и отходы целлюлозы, животные, рыбы, бактерии, грибы и продукция лесного хозяйства. Поскольку ископаемые ресурсы становятся все более и более дефицитными, более дорогостоящими и потенциально подлежат регулированию в отношении выбросов CO2, существует растущая потребность в акриловой кислоте не ископаемого происхождения, производных акриловой кислоты не ископаемого происхождения или их смесях, которые могут служить альтернативой акриловой кислоте ископаемого происхождения, производным акриловой кислоты ископаемого происхождения или их смесям.
За последние 80 лет было предпринято множество попыток получения акриловой кислоты не ископаемого происхождения, производных акриловой кислоты или их смесей из возобновляемых источников, таких как молочная кислота (также известная как 2-гидроксипропионовая кислота), производные молочной кислоты (например, алкил-2-ацетокси-пропионат и 2-ацетокси-пропионовая кислота), 3-гидроксипропионовая кислота, глицерин, монооксид углерода и этиленоксид, диоксид углерода и этилен, а также кротоновая кислота. Из этих ресурсов в настоящее время получают только молочную кислоту из сахара с высоким выходом и чистотой (≥90% теоретического выхода или, что эквивалентно, ≥0,9 г молочной кислоты на грамм сахара), при экономических параметрах, которые способны обеспечивать стоимость производства акриловой кислоты, конкурентную по сравнению с акриловой кислотой ископаемого происхождения. Таким образом, молочная кислота или лактат предоставляют реальную возможность использования в качестве сырьевого материала для акриловой кислоты, производных акриловой кислоты на биологической основе или их смесей. Также ожидается, что 3-гидроксипропионовая кислота будет вырабатываться в промышленном масштабе в течение нескольких лет и, соответственно, 3-гидропропионовая кислота будет представлять собой еще одну реальную возможность использования в качестве исходного сырья для акриловой кислоты, производных акриловой кислоты на биологической основе или их смесей. Соли натрия; фосфатные соли; смеси сульфатных и фосфатных солей; основания; цеолиты или модифицированные цеолиты; оксиды металлов или модифицированные оксиды металлов; и сверхкритическая вода являются основными катализаторами, которые с различным успехом применяли в прошлом для дегидратации молочной кислоты или лактата до акриловой кислоты, производных акриловой кислоты или их смесей.
Например, в патенте США №4,786,756 (выданном в 1988 г.) описывают дегидратацию молочной кислоты или лактата аммония в паровой фазе до акриловой кислоты с применением фосфата алюминия (AlPO4), обработанного в реакторе Pyrex водным неорганическим основанием, в качестве катализатора. В патенте ‘756 описывают максимальный выход акриловой кислоты, составляющий 43,3%, когда в реактор подавали молочную кислоту при примерно атмосферном давлении, и соответствующий выход, составляющий 61,1%, когда в реактор подавали лактат аммония. В обоих примерах получали ацетальдегид при значениях выхода, составляющих 34,7% и 11,9% соответственно, и при этом в больших количествах также присутствовали другие побочные продукты, такие как пропионовая кислота, CO и CO2. Отсутствие основной обработки приводило к увеличению количества побочных продуктов. Другой пример представлен Hong et al., Appl. Catal. A: General 396:194-200 (2011), которые разработали и испытали композиционные катализаторы, изготовленные из солей Ca3(PO4)2 и Ca2(P2O7) с применением способа смешивания в суспензии. Катализатор, соответствующий наивысшему выходу акриловой кислоты из метиллактата, составлял 50%-50% (по массе) катализатора. Он обеспечивал выход 68% акриловой кислоты, приблизительно 5% метилакрилата и приблизительно 14% ацетальдегида при 390°C в реакторе Pyrex. При замене сырья с метиллактата на молочную кислоту на том же катализаторе было получено 54% выхода акриловой кислоты, 14% выхода ацетальдегида и 14% выхода пропионовой кислоты. Группа проф. D. Miller в Университете штата Мичиган (MSU) опубликовала множество статей о дегидратации молочной кислоты или сложных эфиров молочной кислоты до акриловой кислоты и 2,3-пентандиона, таких как Gunter et al., J. Catalysis 148:252-260 (1994); и Tam et al., Ind. Eng. Chem. Res. 38:3873-3877 (1999). По сообщениям данной группы максимальные значения выхода акриловой кислоты в ходе дегидратации молочной кислоты в реакторе Pyrex при температуре 350°C над пропитанным NaOH кремнеземом с низкой площадью поверхности и объемом пор составляли приблизительно 33%. В том же эксперименте выход ацетальдегида составлял 14,7%, а выход пропионовой кислоты составлял 4,1%. Примеры других катализаторов, испытываемых группой, представляли собой Na2SO4, NaCl, Na3PO4, NaNO3, Na2SiO3, Na4P2O7, NaH2PO4, Na2HPO4, Na2HAsO4, NaC3H5O3, NaOH, CsCl, Cs2SO4, KOH, CsOH и LiOH. Во всех случаях указанные выше катализаторы тестировали как отдельные компоненты, а не компоненты в составах смесей. В конечном итоге группа пришла к выводу, что выход акриловой кислоты повышен, а выходы побочных продуктов снижены при низкой площади поверхности подложки из кремнезема, высокой температуре реакции, низком давлении реакции и коротком времени пребывания реагирующих веществ в слое катализатора. В конечном итоге, в заявке на патент Китая 200910054519.7 описывают применение молекулярных сит ZSM-5, модифицированных водным раствором щелочи (такой как NH3, NaOH и Na2CO3) или солью фосфорной кислоты (такой как NaH2PO4, Na2HPO4, LiH2PO4, LaPO4 и т.д.). Максимальный выход акриловой кислоты в ходе дегидратации молочной кислоты составил 83,9%, однако данного выхода достигали при очень длительных значениях времени пребывания.
Таким образом, изготовление акриловой кислоты, производных акриловой кислоты или их смесей из молочной кислоты или лактата посредством способов, в частности, описанных в литературе, приведенной выше, продемонстрировало: 1) значения выхода акриловой кислоты, производных акриловой кислоты или их смесей, не превышающие 70% при коротких значениях времени пребывания; 2) низкие показатели избирательности в отношении акриловой кислоты, производных акриловой кислоты или их смесей, то есть существенные количества нежелательных побочных продуктов, таких как ацетальдегид, 2,3-пентандион, пропионовая кислота, CO и CO2; 3) длительные значения времени пребывания в слоях катализатора; 4) деактивацию катализатора за короткое время в потоке (TOS); и 5) проведение реакций в реакторах Pyrex. Побочные продукты могут осаждаться на катализаторе, что приводит к засорению, а также преждевременной и быстрой деактивации катализатора. После осаждения такие побочные продукты могут дополнительно катализировать другие нежелательные реакции, такие как реакции полимеризации. Помимо осаждения на катализаторах такие побочные продукты, даже если они присутствуют лишь в небольших количествах, приводят к дополнительным затратам при обработке акриловой кислоты (когда присутствуют в потоке продуктов реакции), например при получении суперабсорбирующего полимера (SAP). Такие недостатки способов и катализаторов предшествующего уровня техники делают их коммерчески нежизнеспособными.
Соответственно, существует потребность в способах получения акриловой кислоты, производных акриловой кислоты или их смесей из гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей с высоким выходом, селективностью и эффективностью (т. е. коротким временем пребывания); катализаторах с длительным сроком службы; и в промышленных реакторах с низкой скоростью коррозии.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном варианте осуществления настоящего изобретения предложен способ получения акриловой кислоты, производных акриловой кислоты или их смесей. Способ включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, часовой объемной скорости подачи газа (GHSV) и часовой объемной скорости на единицу массы (WHSV) для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей с последующим получением акриловой кислоты, производных акриловой кислоты или их смесей; причем указанный катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой

В другом варианте осуществления настоящего изобретения предложен способ получения акриловой кислоты, производных акриловой кислоты или их смесей. Способ включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей с последующим получением акриловой кислоты, производных акриловой кислоты или их смесей; причем указанный катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой

В еще одном варианте осуществления настоящего изобретения предложен способ получения акриловой кислоты, производных акриловой кислоты или их смесей. Способ включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с катализатором в двухслойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей с последующим получением акриловой кислоты, производных акриловой кислоты или их смесей; причем указанный катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой

КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Хотя описание завершается формулой изобретения, в которой изобретение конкретно определено и четко заявлено, авторы изобретения полагают, что настоящее изобретение будет лучше понято из нижеследующего описания, приведенного в сопровождении описанных ниже чертежей.
ФИГ. 1 представляет собой типичную диаграмму состояния активного катализатора дегидратации (аморфная и частично дегидратированная фосфатная соль) и предшествующего катализатора дегидратации (кристаллическая фосфатная соль, полностью гидратированная либо полностью дегидратированная), на которой представлена зависимость парциального давления водяного пара от температуры. Тройная точка расположена на пересечении трех кривых фазового равновесия. MI - одновалентный катион. Следует отметить, что приведенные значения парциального давления водяного пара и температуры указаны только в качестве иллюстрации.
ФИГ. 2 представляет собой вид сбоку в частичном разрезе трубчатого проточного однослойного реактора с уплотненным слоем, изготовленного в соответствии с одним из вариантов осуществления настоящего изобретения.
ФИГ. 3 представляет собой вид сбоку в частичном разрезе трубчатого проточного двухслойного реактора с уплотненным слоем, изготовленного в соответствии с одним из вариантов осуществления настоящего изобретения и имеющего внутренний слой и наружный слой.
ФИГ. 4 представляет собой схему металлографического метода, используемого компанией Haynes International, inc. (г. Кокомо, штат Индиана, США) для оценки скорости коррозии испытательных образцов, находящихся под воздействием различных сред.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
I. Определения
Применяемый в данном документе термин материал «ископаемого происхождения» относится к материалу, который производят из ресурсов ископаемого происхождения, таких как неочищенная нефть (нефтепродукт), природный газ, уголь, торф и т.д.
Применяемый в данном документе термин «неископаемого происхождения» относится к материалу, который производят из ресурсов неископаемого происхождения. Для ясности и для целей настоящего изобретения термины «возобновляемый» материал, «материал на биологической основе», «ненефтяной» материал и материал «неископаемого происхождения» используют взаимозаменяемо.
Применяемый в данном документе термин «возобновляемый» материал относится к материалу, который производят из возобновляемого ресурса, который представляет собой ресурс, произведенный с помощью естественного процесса со скоростью, сравнимой со скоростью его потребления (например, в пределах 100-летнего временного интервала). Возобновляемый ресурс можно восполнять естественным путем или с помощью сельскохозяйственных методов. К не имеющим ограничительного характера примерам возобновляемых ресурсов относятся растения (такие как сахарный тростник, свекла, кукуруза, картофель, цитрусовые, древесные растения, лигноцеллюлоза, гемицеллюлоза и отходы целлюлозы), животные, рыбы, бактерии, грибы и продукция лесного хозяйства. Такие ресурсы могут представлять собой организмы, встречающиеся в природе, гибриды или организмы, созданные методами генетической инженерии. Формирование ископаемых ресурсов занимает более 100 лет, и, таким образом, их не считают возобновляемыми ресурсами.
Применяемый в данном документе термин «возобновляемое содержание» обозначает количество углерода из возобновляемого ресурса в процентах по весу (по массе) от общего органического углерода в материале согласно определению Методики B стандарта ASTM D6866-10.
Используемый в настоящем документе термин «катализатор» относится либо к активному катализатору, либо к предшествующему катализатору.
Используемый в настоящем документе термин «активный катализатор» относится к катализатору дегидратации in situ, который представляет собой форму катализатора, находящегося в реакторе во время дегидратации и стимулирующего процесс дегидратации. Активный катализатор настоящего изобретения содержит аморфную и частично дегидратированную фосфатную соль, содержащую катион и анион, представленные эмпирической формулой

Используемый в настоящем документе термин «предшествующий катализатор» относится к предварительному катализатору реакции дегидратации, который представляет собой форму катализатора, загруженного в реактор и присутствующего в реакторе до начала реакции дегидратации или перед вхождением потока поступающих газообразных материалов, содержащего водяной пар при парциальном давлении и температуре, превышающих парциальное давление и температуру в тройной точке катализатора, в контакт с предварительным катализатором реакции дегидратации. Предшествующий катализатор превращается в активный катализатор в процессе реакции дегидратации или в условиях технологического процесса, которые могут изменить его физические и химические свойства и превратить его в активный катализатор. Предшествующий катализатор настоящего изобретения содержит кристаллическую фосфатную соль, содержащую катион и анион, представленные эмпирической формулой
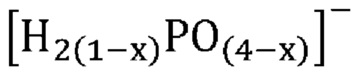

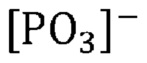
В настоящем документе термин «конденсированный» относится к кристаллическому и полностью дегидратированному материалу.
Используемый в настоящем документе термин «тройная точка» относится к точке на диаграмме состояния катализатора, т. е. графике равновесного парциального давления водяного пара от температуры, которая является точкой пересечения трех линий фазового перехода (также называемых кривыми равновесного состояния) и в которой 3 фазы катализатора сосуществуют в состоянии термодинамического равновесия: 1-я кривая равновесного состояния разделяет кристаллическую и полностью дегидратированную фазу и кристаллическую и полностью гидратированную фазу катализатора; 2-я кривая равновесного состояния разделяет кристаллическую и полностью дегидратированную фазу и аморфную и частично дегидратированную фазу катализатора; и 3-я кривая равновесного состояния разделяет аморфную и частично дегидратированную фазу и кристаллическую и полностью гидратированную фазу катализатора (см. ФИГ. 1).
В настоящем документе термин «парциальное давление водяного пара в тройной точке» относится к парциальному давлению водяного пара в тройной точке катализатора.
В настоящем документе термин «температура в тройной точке» относится к температуре в тройной точке катализатора.
Используемый в настоящем документе термин «фосфатная соль» относится к фосфатной соли, которая является нейтрально заряженной.
Применяемый в настоящем документе термин «монофосфат» или «ортофосфат» относится к любой фосфатной соли, анионная частица [PO4]3- которой образована четырьмя атомами кислорода, расположенными в виде структуры, имеющей форму практически правильного тетраэдра, вокруг центрального атома фосфора.
Применяемый в настоящем документе термин «конденсированный фосфат» относится к любым фосфатным солям, содержащим одну или несколько связей P-O-P, образованных под общим углом тетраэдра PO4.
Применяемый в настоящем документе термин «полифосфат» относится к любому конденсированному фосфату с линейной структурой; т. е. содержащим линейные мостики P-O-P под общим углом тетраэдра PO4, что приводит к образованию конечных цепей.
Применяемый в данном документе термин «циклофосфат» относится к любому конденсированному фосфату с циклической структурой.
Применяемый в настоящем документе термин «гидрат» относится к соли (т. е. соль⋅nH2O), которая содержит некоторое число молекул воды (т. е. nH2O), связанных с ионами в ее кристаллической структуре.
Применяемый в данном документе термин «одновалентный катион» относится к любому катиону с положительным зарядом, составляющим +1.
Применяемый в настоящем документе термин «многовалентный катион» относится к любому катиону с положительным зарядом, большим или равным +2.
Применяемый в настоящем документе термин «гетерополианион» относится к любому аниону с ковалентно связанными полиэдрами XOp и YOr, который, таким образом, содержит связи X-O-Y и, возможно, X-O-X и Y-O-Y; где X и Y представляют собой любые атомы и где p и r представляют собой любые положительные целые числа.
Используемый в настоящем документе термин «гетерополифосфат» относится к гетерополианиону; где X представляет собой фосфор (P), а Y представляет собой любой другой атом.
Применяемый в данном документе термин «фосфатный аддукт» относится к любому соединению с одним или более фосфат-анионами и одним или более анионами, не относящимися к фосфатным, которые не соединены ковалентной связью.
Применяемый в настоящем документе термин «аморфный» относится к состоянию любого материала, при котором отсутствует характеристика дальнего порядка для кристаллического материала. Аморфный материал может представлять собой либо аморфное твердое вещество, либо аморфное жидкое вещество. В контексте настоящего изобретения материалы с содержанием аморфного вещества большим или равным 50 мас. % считаются аморфными материалами. Содержание аморфного вещества в материале может быть определено посредством любого способа, известного специалисту в данной области техники, такого как, например, без ограничений, рентгеноструктурный анализ (XRD), инфракрасная спектроскопия (IR), рамановская спектроскопия, дифференциальная сканирующая калориметрия (DSC) или спектроскопия ядерного магнитного резонанса (NMR) твердого тела. В качестве иллюстрации в способе, основанном на методе XRD, отдельный вклад кристаллической (IC) и аморфной (IA) структур по рентгенограмме рассеяния определяют с применением метода подгонки профиля, т. е. путем деконволюции рентгенограммы рассеяния во вклады отдельных структур с использованием функций Гаусса, Лоренца, Фойхта или связанных функций, известных специалистам в данной области техники. Затем определяют содержание аморфного вещества, XA, в мас. % посредством расчета отношения площади под кривой интенсивности рассеянного излучения для аморфной структуры (IA) к площади под кривой общей интенсивности рассеянного излучения (кристаллической структуры плюс аморфной структуры, IT=IC+IA) для заданного диапазона углов Брэгга (например, 2θ = 5-50°, Cu-излучение, λ = 1,54059 Å в контексте настоящего изобретения), т. е.

Применяемый в настоящем документе термин «кристаллический» относится к состоянию материала, составляющие которого образуют высокоупорядоченную микроскопическую структуру и кристаллическую решетку с дальним порядком. В контексте настоящего изобретения материалы с содержанием аморфного вещества, составляющим менее 50 мас. %, считаются кристаллическими материалами.
Применяемый в данном документе термин «химически инертный» материал относится к материалу, который остается в той же химической форме в условиях равновесия при приведении в контакт с другим материалом или материалами. В контексте настоящего изобретения более приблизительно 90 мас. % материала должно оставаться в той же химической форме для рассмотрения материала в качестве «значительно химически инертного» материала, и более приблизительно 98 мас. % материала должно оставаться в той же химической форме для рассмотрения материала в качестве «по существу химически инертного» материала.
Применяемый в данном документе термин «антиоксидант» относится к молекуле, способной прерывать радикально-цепные процессы либо путем отдачи атома водорода, либо путем обеспечения реакции этиленовой связи с образованием стабилизированного органического радикала и прерыванием, таким образом, радикально-цепных процессов. Не имеющие ограничительного характера примеры антиоксидантов включают в себя тиолы, полифенолы, бутилированный гидрокситолуол (BHT) и бутилированный гидроксианизол (BHA).
Применяемый в данном документе термин «LA» относится к молочной кислоте, термин «AA» относится к акриловой кислоте, термин «AcH» относится к ацетальдегиду, а термин «PA» относится к пропионовой кислоте.
Применяемый в данном документе термин «превращение» в мол. % определяют как [расход на входе гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей (моль/мин) - расход на выходе гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей (моль/мин)]/[расход на входе гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей (моль/мин)] × 100.
Применяемый в данном документе термин «выход» в мол. % определяют как [расход продукта на выходе (моль/мин) / расход на входе гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей (моль/мин)] × 100.
Применяемый в данном документе термин «избирательность» в мол. % определяют как [выход/превращение] × 100.
Применяемый в данном документе термин «общий баланс углерода» определяют как: [((моль монооксида углерода на выходе + моль диоксида углерода на выходе + моль метана на выходе) + (2 × (моль уксусной кислоты на выходе + моль ацетальдегида на выходе + моль этана на выходе + моль этилена на выходе)) + (3 × (моль акриловой кислоты на выходе + моль пропионовой кислоты на выходе + моль гидроксипропионовой кислоты на выходе + моль гидроксиацетона на выходе) + (5 × моль 2,3 пентандиона на выходе) + (6 × моль димера акриловой кислоты на выходе)) / (3 × моль гидроксипропионовой кислоты на входе)] × 100. Если вместо гидроксипропионовой кислоты применяют производное гидроксипропионовой кислоты, вышеуказанную формулу необходимо откорректировать в соответствии с числом атомов углерода в производном гидроксипропионовой кислоты.
Применяемый в настоящем документе термин «часовая объемная скорость подачи газа» или «GHSV» в ч-1 определяют как 60 × [общий расход газа (мл/мин)/объем пустого слоя предшествующего катализатора (мл)]. Общий расход газа рассчитывают в условиях при стандартных температуре и давлении (STP; 0°C и 1 бар).
Применяемый в настоящем документе термин «часовая объемная скорость на единицу массы» или «WHSV» в ч-1 определяют как 60 × [общий расход LA (г/мин)/ масса предшествующего катализатора (г)]. Для целей настоящего описания масса предшествующего катализатора включает в себя только массу кристаллической фосфатной соли и не включает в себя массу какой-либо инертной подложки.
Применяемый в данном документе термин «объемная скорость подачи жидкости» или «LHSV» в ч-1 определяют как 60 × [общий расход жидкости (мл/мин) / объем пустого слоя предшествующего катализатора (мл)]. Для целей настоящего описания масса предшествующего катализатора включает в себя только массу кристаллической фосфатной соли и не включает в себя массу какой-либо инертной подложки.
II. Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных
Неожиданно было обнаружено, что активные катализаторы, содержащие смесь аморфной и частично дегидратированной фосфатной соли, могут стимулировать дегидратацию гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей до акриловой кислоты, производных акриловой кислоты или их смесей, демонстрируя при этом: 1) высокий выход и селективность (т. е. низкое количество и малое число побочных продуктов) и эффективность (т. е. короткое время пребывания); 2) длительный срок службы катализаторов; и 3) низкую скорость коррозии промышленных реакторов. В не имеющем ограничительного характера примере аморфную и частично дегидратированную фосфатную соль можно получать в ходе обратимой реакции при приведении кристаллической фосфатной соли (например, предшествующего катализатора с молярным соотношением фосфора к катионам приблизительно 1) в контакт с водяным паром при парциальном давлении и температуре водяного пара, превышающих тройную точку катализатора. Авторы изобретения также неожиданно обнаружили, что для дегидратации гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей до акриловой кислоты, производных акриловой кислоты или их смесей активный катализатор настоящего изобретения должен находиться в присутствии достаточного количества водяного пара, вопреки распространенному мнению в предшествующем уровне техники, согласно которому реакции дегидратации следует проводить в сухих условиях. Не желая быть связанными какой-либо теорией, заявители предположили, что водяной пар необходим для предотвращения полной дегидратации дигидромонофосфатной соли (которая является полностью гидратированной) до конденсированного фосфата (который является полностью дегидратированным) в данных условиях эксплуатации и сохранения, таким образом, кислотных центров Бренстеда, требуемых либо для избирательной катализируемой кислотой дегидратации, либо для фосфорилирования и дефосфорилирования гидроксипропионовой кислоты и ее производных до акриловой кислоты и ее производных. Способность некоторых фосфатных солей к частичной дегидратации при определенных рабочих условиях показана на ФИГ. 1, где кристаллическая и полностью дегидратированная полифосфатная соль (MIPO3)n в определенном диапазоне парциального давления и температуры водяного пара превращается в аморфную и частично дегидратированную соль MIH2(1-x)PO(4-x), а затем по мере дополнительного возрастания парциального давления или снижения температуры водяного пара снова превращается в кристаллическую структуру в форме дигидромонофосфата MIH2PO4.
В одном варианте осуществления настоящего изобретения активный катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой [H2(1-x)PO(4-x)]¯; где x представляет собой любое действительное число, которое больше 0 и меньше 1. В другом варианте осуществления настоящего изобретения указанная фосфатная соль является аморфной и частично дегидратированной.
В одном варианте осуществления настоящего изобретения активный катализатор содержит аморфную и частично дегидратированную фосфатную соль, содержащую катион и анион, представленные эмпирической формулой [H2(1-x)PO(4-x)]¯; где x представляет собой любое действительное число, которое больше 0 и меньше 1. Не имеющие ограничительного характера примеры катионов в фосфатной соли представляют собой металлические катионы, металлорганические катионы, аммоний, замещенный аммоний, оксикатионы и другие катионы, известные специалистам в данной области техники. Не имеющие ограничительного характера примеры замещенного аммония и других катионов представляют собой изопропиламмоний, этилендиаммоний, саркозиний, L-гистидиний, глициний и 4-аминопиридиний. Не имеющие ограничительного характера примеры оксикатионов представляют собой перванадил- и ванадил-ионы. В другом варианте осуществления настоящего изобретения указанный катион представляет собой одновалентный катион. Не имеющие ограничительного характера примеры одновалентных катионов представляют собой катионы щелочных металлов, металлорганические катионы, аммоний, замещенный аммоний, оксикатионы (например, перванадил) и другие катионы, известные специалистам в данной области техники. В еще одном варианте осуществления настоящего изобретения указанный одновалентный катион выбран из группы, состоящей из Li+, Na+, K+, Rb+, Cs+, Ag+, Tl+ и их смесей. В еще одном варианте осуществления настоящего изобретения указанный одновалентный катион выбран из группы, состоящей из K+, Rb+, Cs+ и их смесей. В одном варианте осуществления настоящего изобретения указанный одновалентный катион выбран из группы, состоящей из K+, Cs+ и их смесей. В другом варианте осуществления настоящего изобретения указанная фосфатная соль является аморфной и частично дегидратированной; причем указанное значение x представляет собой любое действительное число, которое больше 0 и меньше 1; и при этом указанный катион представляет собой одновалентный катион, выбранный из группы, состоящей из K+, Cs+ и их смесей. В еще одном варианте осуществления настоящего изобретения указанная аморфная и частично дегидратированная фосфатная соль выбрана из группы, состоящей из






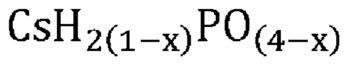
В одном варианте осуществления настоящего изобретения активный катализатор содержит аморфную и частично дегидратированную фосфатную соль, состоящую из одного или более катионов и одного или более фосфат-анионов; причем по меньшей мере один фосфат-анион представлен эмпирической формулой [H2(1-x)PO(4-x)]¯ и при этом x представляет собой любое действительное число, которое больше 0 и меньше 1. Для целей настоящего изобретения термин «один или более катионов» относится к различным типам катионов. В другом варианте осуществления настоящего изобретения указанные катионы представляют собой одновалентные катионы. В еще одном варианте осуществления настоящего изобретения указанные одновалентные катионы выбраны из группы, состоящей из Li+, Na+, K+, Rb+, Cs+, Ag+, Tl+ и их смесей. В еще одном варианте осуществления настоящего изобретения указанные одновалентные катионы выбраны из группы, состоящей из K+, Rb+, Cs+ и их смесей. В одном варианте осуществления настоящего изобретения указанные одновалентные катионы выбраны из группы, состоящей из K+, Cs+ и их смесей.
В одном варианте осуществления настоящего изобретения активный катализатор содержит аморфную и частично дегидратированную фосфатную соль, представленную эмпирической формулой



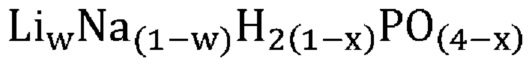





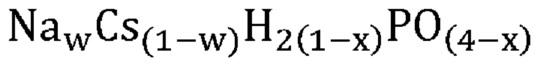
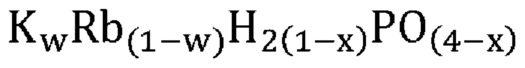


В одном варианте осуществления настоящего изобретения активный катализатор содержит указанную аморфную и частично дегидратированную фосфатную соль, дополнительно содержащую соединение, не относящееся к фосфатам; причем указанное соединение, не относящееся к фосфатам, является по существу химически инертным по отношению к указанной аморфной и частично дегидратированной фосфатной соли. В другом варианте осуществления настоящего изобретения массовое соотношение указанной аморфной и частично дегидратированной фосфатной соли и указанного соединения, не относящегося к фосфатам, составляет от приблизительно 1:10 до приблизительно 4:1.
В одном варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, содержит оксид кремния (также называемый кремнеземом; SiO2). В другом варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, состоит по существу из кремнезема. В еще одном варианте осуществления настоящего изобретения указанный кремнезем выбран из группы, состоящей из аморфного кремнезема (также именуемого в настоящем документе плавленым кремнеземом или плавленым кварцем), кварца, тридимита, кристобалита, моганита, коэсита и их смесей. В еще одном варианте осуществления настоящего изобретения указанный кремнезем представляет собой аморфный кремнезем. В одном варианте осуществления настоящего изобретения указанный кремнезем имеет удельную площадь поверхности, составляющую менее приблизительно 10 м2/г.
В другом варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, содержит катион и анион. Не имеющими ограничительного характера примерами анионов в соединениях не относящихся к фосфатам, являются арсенаты, конденсированные арсенаты, нитраты, сульфаты, конденсированные сульфаты, бораты, карбонаты, хроматы, конденсированные хроматы, ванадаты, ниобаты, танталаты, селенаты, конденсированные силикаты, конденсированные алюминаты, германаты, конденсированные германаты, молибдаты, конденсированные молибдаты, другие мономерные оксианионы, полиоксианионы, гетерополифосфаты, такие как арсенатофосфаты, фосфоалюминаты, фосфобораты, фосфохроматы, фосфомолибдаты, фосфосиликаты, фосфосульфаты, фосфовольфраматы и фосфатные аддукты, такие как фосфатные анионы с теллуровой кислотой, галиды, бораты, карбонаты, нитраты, сульфаты, хроматы, силикаты, оксалаты, их смеси или иные соединения, которые могут быть очевидны специалистам в данной области техники.
В одном варианте осуществления настоящего изобретения указанная аморфная и частично дегидратированная фосфатная соль выбрана из группы, состоящей из





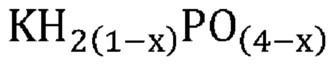

В одном варианте осуществления настоящего изобретения указанная аморфная и частично дегидратированная фосфатная соль выбрана из группы, состоящей из

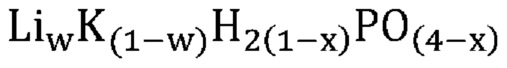


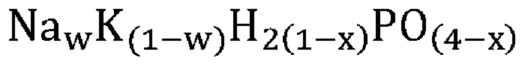



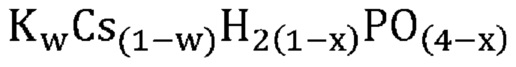

В одном варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, содержит нейтрально заряженную оксисоль, содержащую катион и оксианион, выбранные из группы, представленной молекулярными формулами


В одном варианте осуществления настоящего изобретения указанный катион указанной оксисоли выбран из группы, состоящей из одновалентного катиона, многовалентного катиона и их смесей. Не имеющие ограничительного характера примеры указанного многовалентного катиона указанной оксисоли представляют собой катионы щелочноземельных металлов, переходных металлов, постпереходных или легких металлов и металлоидов; металлоорганические катионы, катионы замещенного аммония, оксикатионы (например, ванадил) и другие катионы, известные специалисту в данной области техники. В другом варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов Be, Mg, Ca, Sr, Ba, Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Re, Al, Ga, In, Tl, Si, Ge, Sn, Pb, Sb, Bi, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu и их смесей. В еще одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов Mg, Ca, Sr, Ba, Y, Mn, Al, Er и их смесей. В еще одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из двухвалентных катионов, трехвалентных катионов, четырехвалентных катионов, пятивалентных катионов и их смесей. В одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из Be2+, Mg2+, Ca2+, Sr2+, Ba2+, Sc3+, Y3+, Ti3+, Ti4+, Zr2+, Zr4+, Hf4+, V3+, V4+, Nb3+, Cr2+, Cr3+, Mo3+, Mo4+, Mn2+, Mn3+, Re4+, Al3+, Ga3+, In3+, Si4+, Ge4+, Sn4+, Pb4+, Sb3+, Sb5+, Bi3+, La3+, Ce3+, Ce4+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+, Ho3+, Er3+, Tm3+, Yb3+, Lu3+ и их смесей. В другом варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из Mg2+, Ca2+, Sr2+, Ba2+, Y3+, Mn2+, Mn3+, Al3+, Er3+ и их смесей. В еще одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли представляет собой Ba2+.
Не имеющие ограничительного характера примеры указанного одновалентного катиона указанной оксисоли представляют собой катионы щелочных металлов. В одном варианте осуществления настоящего изобретения указанный одновалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов Li, Na, K, Rb, Cs, Ag, Tl и их смесей. В другом варианте осуществления настоящего изобретения указанный одновалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов K, Rb, Cs и их смесей.
В одном варианте осуществления настоящего изобретения указанный оксианион указанной оксисоли выбран из группы, представленной молекулярными формулами [SO4]2-, [S2O7]2-, [HSO4]1-, [SO4]2-, [HSO4]-, [Ta2O6]2-, [Ta2O7]4-, [Ta2O9]8-, [Ta2O10]10-, [Ta2O11]12-, [Ta4O11]2-, [Ta4O15]10-, и их смесей. В другом варианте осуществления настоящего изобретения указанный оксианион указанной оксисоли выбран из группы, представленной молекулярными формулами [SO4]2-, [Ta2O6]2-, и их смесей.
Не имеющие ограничительного характера примеры указанной оксисоли представляют собой сульфаты щелочноземельных металлов, танталаты щелочноземельных металлов, сульфаты смесей щелочного и щелочноземельного металлов и танталаты смесей щелочного и щелочноземельного металлов. В одном варианте осуществления настоящего изобретения указанная оксисоль выбрана из группы, состоящей из CaSO4, SrSO4, BaSO4, SrK2(SO4)2, SrRb2(SO4)2, Ca2K2(SO4)3, Ca2Rb2(SO4)3, Ca2Cs2(SO4)3, CaTa4O11, SrTa4O11, BaTa4O11, MgTa2O6, CaTa2O6, SrTa2O6, BaTa2O6, Mg2Ta2O7, Ca2Ta2O7, Sr2Ta2O7, SrK2Ta2O7, Ba2Ta2O7, Ba3Ta2O8, Mg4Ta2O9, Ca4Ta2O9, Sr4Ta2O9, Ba4Ta2O9, Ca5Ta2O10, Ca2KTa3O10, Ca2RbTa3O10, Ca2CsTa3O10, Sr2KTa3O10, Sr2RbTa3O10, Sr2CsTa3O10, Mg5Ta4O15, Sr5Ta4O15, Ba5Ta4O15, Sr2KTa5O15, Ba2KTa5O15, Sr6Ta2O11, Ba6Ta2O11, любых их гидратированных форм и их смесей. В другом варианте осуществления настоящего изобретения указанная оксисоль выбрана из группы, состоящей из CaSO4, CaTa2O6, SrSO4, SrTa2O6, BaSO4, BaTa2O6, любых их гидратированных форм и их смесей. В еще одном варианте осуществления настоящего изобретения указанная оксисоль выбрана из группы, состоящей из BaSO4, BaTa2O6, любых их гидратированных форм и их смесей.
В одном варианте осуществления настоящего изобретения указанная аморфная и частично дегидратированная фосфатная соль выбрана из группы, состоящей из



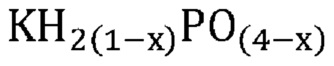

В еще одном варианте осуществления настоящего изобретения указанная аморфная и частично дегидратированная фосфатная соль выбрана из группы, состоящей из

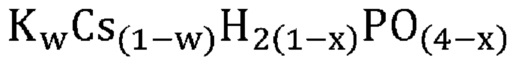

В одном варианте осуществления настоящего изобретения указанное значение x составляет приблизительно 0,8. В другом варианте осуществления настоящего изобретения указанное значение x составляет менее приблизительно 0,8. В еще одном варианте осуществления настоящего изобретения указанное значение x составляет менее приблизительно 0,6. В еще одном варианте осуществления настоящего изобретения указанное значение x составляет менее приблизительно 0,5. В одном варианте осуществления настоящего изобретения указанное значение x составляет от приблизительно 0,1 до приблизительно 0,5. В другом варианте осуществления настоящего изобретения указанное значение x составляет от приблизительно 0,25 до приблизительно 0,45. В еще одном варианте осуществления настоящего изобретения указанное значение x составляет приблизительно 0,4. В еще одном варианте осуществления настоящего изобретения указанное значение x составляет приблизительно 0,4, а указанный одновалентный катион указанной аморфной и частично дегидратированной фосфатной соли представляет собой Cs+.
В одном варианте осуществления настоящего изобретения указанное значение w составляет более приблизительно 0,9. В другом варианте осуществления настоящего изобретения указанное значение w составляет более приблизительно 0,8. В еще одном варианте осуществления настоящего изобретения указанное значение w составляет менее приблизительно 0,2. В еще одном варианте осуществления настоящего изобретения указанное значение w составляет менее приблизительно 0,1.
В одном варианте осуществления настоящего изобретения указанная аморфная и частично дегидратированная фосфатная соль представляет собой гидратированную соль. В другом варианте осуществления настоящего изобретения указанная оксисоль представляет собой гидратированную соль. В еще одном варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, представляет собой гидратированное соединение. Гидратированные соль или соединение содержат конкретное число молекул воды на формульную единицу соли или соединения. Не имеющие ограничительного характера примеры гидратированных солей или соединений представляют собой полугидратированные, моногидратированные, сесквигидратированные, дигидратированные, тригидратированные, тетрагидратированные, пентагидратированные, гексагидратированные, гептагидратированные, октагидратированные, нонагидратированные, нонагидратированные и декагидратированные соли или соединения.
В одном варианте осуществления настоящего изобретения указанный активный катализатор дополнительно содержит инертную подложку. Не имеющие ограничительного характера примеры инертных подложек представляют собой кремнезем, силикат, оксид алюминия, алюминат, алюмосиликат, оксид титана, титанат, оксид циркония, цирконат, уголь (такой как активированный уголь, алмаз, графит или фуллерен), сульфат, фосфат, танталат, оксид церия, другие оксиды металлов и их смеси. В другом варианте осуществления настоящего изобретения указанная инертная подложка состоит по существу из кремнезема. В еще одном варианте осуществления настоящего изобретения указанный кремнезем выбран из группы, состоящей из аморфного кремнезема, кварца, тридимита, кристобалита, моганита, коэсита и их смесей. В еще одном варианте осуществления настоящего изобретения указанный кремнезем представляет собой аморфный кремнезем. В одном варианте осуществления настоящего изобретения указанный кремнезем имеет удельную площадь поверхности, составляющую менее приблизительно 10 м2/г. В другом варианте осуществления настоящего изобретения инертная подложка составляет по количеству от приблизительно 20 мас. % до приблизительно 90 мас. % от общей массы активного катализатора.
В одном варианте осуществления настоящего изобретения масса аморфной и частично дегидратированной фосфатной соли в расчете на общую массу активного катализатора составляет от приблизительно 5 мас. % до приблизительно 90 мас. %. В другом варианте осуществления настоящего изобретения масса аморфной и частично дегидратированной фосфатной соли в расчете на общую массу активного катализатора составляет от приблизительно 8 мас. % до приблизительно 60 мас. %. В еще одном варианте осуществления настоящего изобретения масса аморфной и частично дегидратированной фосфатной соли в расчете на общую массу активного катализатора составляет от приблизительно 12 мас. % до приблизительно 40 мас. %. В еще одном варианте осуществления настоящего изобретения масса аморфной и частично дегидратированной фосфатной соли в расчете на общую массу активного катализатора составляет от приблизительно 12 мас. % до приблизительно 17 мас. %. В одном варианте осуществления настоящего изобретения масса аморфной и частично дегидратированной фосфатной соли в расчете на общую массу активного катализатора составляет от приблизительно 26 мас. % до приблизительно 32 мас. %.
В одном варианте осуществления настоящего изобретения активный катализатор состоит из





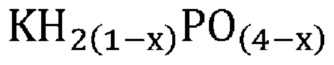



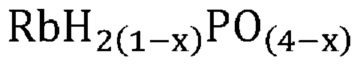
Активный катализатор настоящего изобретения можно использовать для катализа нескольких химических реакций. Не имеющими ограничительного характера примерами реакций являются: дегидратация молочной кислоты, производных молочной кислоты или их смесей до акриловой кислоты; дегидратация 3-гидроксипропионовой кислоты, 3-производных гидроксипропионовой кислоты или их смесей до акриловой кислоты; дегидратация глицерина до акролеина; изомеризация молочной кислоты в 3-гидроксипропионовую кислоту в присутствии воды; восстановление гидроксипропионовой кислоты до пропионовой кислоты или 1-пропанола в присутствии газообразного водорода; дегидратация алифатических спиртов до алкенов или олефинов; дегидрогенизация алифатических спиртов до эфиров; другие реакции дегидрогенизации, гидролиза, алкилирования, деалкилирования, окисления, диспропорционирования, эстерификации, циклизации, изомеризации, конденсации, ароматизации, полимеризации; и другие реакции, которые могут быть очевидны специалисту в данной области техники.
III. Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных
В одном варианте осуществления настоящего изобретения предшествующий катализатор состоит из одного или более катионов; причем отношение общего числа моль всех катионов к общему числу моль фосфора составляет приблизительно 1. В другом варианте осуществления настоящего изобретения указанная фосфатная соль выбрана из группы, состоящей из кристаллической и полностью дегидратированной фосфатной соли, кристаллической и полностью гидратированной фосфатной соли и их смесей.
В одном варианте осуществления настоящего изобретения предшествующий катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой

В одном варианте осуществления настоящего изобретения предшествующий катализатор содержит кристаллическую фосфатную соль, содержащую катион и анион, представленные эмпирической формулой








В одном варианте осуществления настоящего изобретения предшествующий катализатор содержит кристаллическую фосфатную соль, состоящую из одного или более катионов и одного или более фосфат-анионов; при этом по меньшей мере один фосфат-анион представлен эмпирической формулой
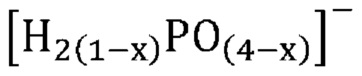
В одном варианте осуществления настоящего изобретения предшествующий катализатор содержит кристаллическую фосфатную соль, представленную эмпирической формулой

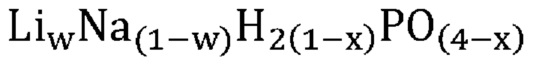


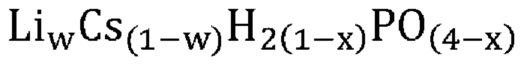






В одном варианте осуществления настоящего изобретения предшествующий катализатор содержит указанную кристаллическую фосфатную соль, дополнительно содержащую соединение, не относящееся к фосфатам; причем указанное соединение, не относящееся к фосфатам, является по существу химически инертным по отношению к указанной кристаллической фосфатной соли. В другом варианте осуществления настоящего изобретения массовое соотношение указанной кристаллической фосфатной соли и указанного соединения, не относящегося к фосфатам, составляет от приблизительно 1 : 10 до приблизительно 4 : 1.
В одном варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, содержит оксид кремния (также называемый кремнеземом; SiO2). В другом варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, состоит по существу из кремнезема. В еще одном варианте осуществления настоящего изобретения указанный кремнезем выбран из группы, состоящей из аморфного кремнезема (также именуемого в настоящем документе плавленым кремнеземом или плавленым кварцем), кварца, тридимита, кристобалита, моганита, коэсита и их смесей. В еще одном варианте осуществления настоящего изобретения указанный кремнезем представляет собой аморфный кремнезем. В одном варианте осуществления настоящего изобретения указанный кремнезем имеет удельную площадь поверхности, составляющую менее приблизительно 10 м2/г.
В другом варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, содержит анион и катион. Не имеющими ограничительного характера примерами анионов в соединениях не относящихся к фосфатам, являются арсенаты, конденсированные арсенаты, нитраты, сульфаты, конденсированные сульфаты, бораты, карбонаты, хроматы, конденсированные хроматы, ванадаты, ниобаты, танталаты, селенаты, конденсированные силикаты, конденсированные алюминаты, германаты, конденсированные германаты, молибдаты, конденсированные молибдаты, другие мономерные оксианионы, полиоксианионы, гетерополифосфаты, такие как арсенатофосфаты, фосфоалюминаты, фосфобораты, фосфохроматы, фосфомолибдаты, фосфосиликаты, фосфосульфаты, фосфовольфраматы и фосфатные аддукты, такие как фосфатные анионы с теллуровой кислотой, галиды, бораты, карбонаты, нитраты, сульфаты, хроматы, силикаты, оксалаты, их смеси или иные соединения, которые могут быть очевидны специалистам в данной области техники.
В одном варианте осуществления настоящего изобретения указанная кристаллическая фосфатная соль выбрана из группы, состоящей из




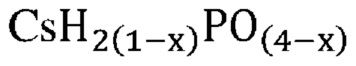


В одном варианте осуществления настоящего изобретения указанная кристаллическая фосфатная соль выбрана из группы, состоящей из
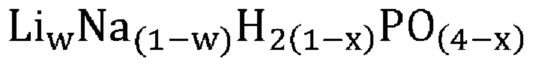
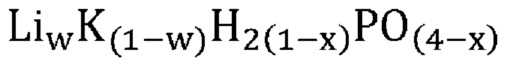






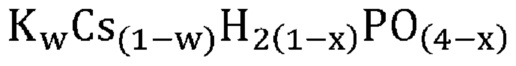

В одном варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, содержит нейтрально заряженную оксисоль, содержащую катион и оксианион, выбранные из группы, представленной молекулярными формулами


В одном варианте осуществления настоящего изобретения указанный катион указанной оксисоли выбран из группы, состоящей из одновалентного катиона, многовалентного катиона и их смесей. Не имеющие ограничительного характера примеры указанного многовалентного катиона указанной оксисоли представляют собой катионы щелочноземельных металлов, переходных металлов, постпереходных или легких металлов и металлоидов; металлоорганические катионы, катионы замещенного аммония, оксикатионы (например, ванадил) и другие катионы, известные специалисту в данной области техники. В другом варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов Be, Mg, Ca, Sr, Ba, Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Re, Al, Ga, In, Tl, Si, Ge, Sn, Pb, Sb, Bi, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu и их смесей. В еще одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов Mg, Ca, Sr, Ba, Y, Mn, Al, Er и их смесей. В еще одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из двухвалентных катионов, трехвалентных катионов, четырехвалентных катионов, пятивалентных катионов и их смесей. В одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из Be2+, Mg2+, Ca2+, Sr2+, Ba2+, Sc3+, Y3+, Ti3+, Ti4+, Zr2+, Zr4+, Hf4+, V3+, V4+, Nb3+, Cr2+, Cr3+, Mo3+, Mo4+, Mn2+, Mn3+, Re4+, Al3+, Ga3+, In3+, Si4+, Ge4+, Sn4+, Pb4+, Sb3+, Sb5+, Bi3+, La3+, Ce3+, Ce4+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+, Ho3+, Er3+, Tm3+, Yb3+, Lu3+ и их смесей. В другом варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли выбран из группы, состоящей из Mg2+, Ca2+, Sr2+, Ba2+, Y3+, Mn2+, Mn3+, Al3+, Er3+ и их смесей. В еще одном варианте осуществления настоящего изобретения указанный многовалентный катион указанной оксисоли представляет собой Ba2+.
Не имеющие ограничительного характера примеры указанного одновалентного катиона указанной оксисоли представляют собой катионы щелочных металлов. В одном варианте осуществления настоящего изобретения указанный одновалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов Li, Na, K, Rb, Cs, Ag, Tl и их смесей. В другом варианте осуществления настоящего изобретения указанный одновалентный катион указанной оксисоли выбран из группы, состоящей из катионов металлов K, Rb, Cs и их смесей.
В одном варианте осуществления настоящего изобретения указанный оксианион указанной оксисоли выбран из группы, представленной молекулярными формулами [SO4]2-, [S2O7]2-, [HSO4]1-, [SO4]2-, [HSO4]-, [Ta2O6]2-, [Ta2O7]4-, [Ta2O9]8-, [Ta2O10]10-, [Ta2O11]12-, [Ta4O11]2-, [Ta4O15]10-, и их смесей. В другом варианте осуществления настоящего изобретения указанный оксианион указанной оксисоли выбран из группы, представленной молекулярными формулами [SO4]2-, [Ta2O6]2-, и их смесей.
Не имеющие ограничительного характера примеры указанной оксисоли представляют собой сульфаты щелочноземельных металлов, танталаты щелочноземельных металлов, сульфаты смесей щелочного и щелочноземельного металлов и танталаты смесей щелочного и щелочноземельного металлов. В одном варианте осуществления настоящего изобретения указанная оксисоль выбрана из группы, состоящей из CaSO4, SrSO4, BaSO4, SrK2(SO4)2, SrRb2(SO4)2, Ca2K2(SO4)3, Ca2Rb2(SO4)3, Ca2Cs2(SO4)3, CaTa4O11, SrTa4O11, BaTa4O11, MgTa2O6, CaTa2O6, SrTa2O6, BaTa2O6, Mg2Ta2O7, Ca2Ta2O7, Sr2Ta2O7, SrK2Ta2O7, Ba2Ta2O7, Ba3Ta2O8, Mg4Ta2O9, Ca4Ta2O9, Sr4Ta2O9, Ba4Ta2O9, Ca5Ta2O10, Ca2KTa3O10, Ca2RbTa3O10, Ca2CsTa3O10, Sr2KTa3O10, Sr2RbTa3O10, Sr2CsTa3O10, Mg5Ta4O15, Sr5Ta4O15, Ba5Ta4O15, Sr2KTa5O15, Ba2KTa5O15, Sr6Ta2O11, Ba6Ta2O11, любых их гидратированных форм и их смесей. В другом варианте осуществления настоящего изобретения указанная оксисоль выбрана из группы, состоящей из CaSO4, CaTa2O6, SrSO4, SrTa2O6, BaSO4, BaTa2O6, любых их гидратированных форм и их смесей. В еще одном варианте осуществления настоящего изобретения указанная оксисоль выбрана из группы, состоящей из BaSO4, BaTa2O6, любых их гидратированных форм и их смесей.
В одном варианте осуществления настоящего изобретения указанная кристаллическая фосфатная соль выбрана из группы, состоящей из





В еще одном варианте осуществления настоящего изобретения указанная кристаллическая фосфатная соль выбрана из группы, состоящей из

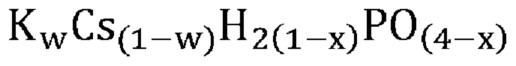

В одном варианте осуществления настоящего изобретения указанное значение w составляет более приблизительно 0,9. В другом варианте осуществления настоящего изобретения указанное значение w составляет более приблизительно 0,8. В еще одном варианте осуществления настоящего изобретения указанное значение w составляет менее приблизительно 0,2. В еще одном варианте осуществления настоящего изобретения указанное значение w составляет менее приблизительно 0,1.
В одном варианте осуществления настоящего изобретения указанная кристаллическая фосфатная соль представляет собой гидратированную соль. В другом варианте осуществления настоящего изобретения указанная оксисоль представляет собой гидратированную соль. В другом варианте осуществления настоящего изобретения указанное соединение, не относящееся к фосфатам, представляет собой гидратированное соединение. Гидратированные соль или соединение содержат конкретное число молекул воды на формульную единицу соли или соединения. Не имеющие ограничительного характера примеры гидратированных солей или соединений представляют собой полугидратированные, моногидратированные, сесквигидратированные, дигидратированные, тригидратированные, тетрагидратированные, пентагидратированные, гексагидратированные, гептагидратированные, октагидратированные, нонагидратированные, нонагидратированные и декагидратированные соли или соединения.
В одном варианте осуществления настоящего изобретения указанный предшествующий катализатор дополнительно содержит инертную подложку. Не имеющие ограничительного характера примеры инертных подложек представляют собой кремнезем, силикат, оксид алюминия, алюминат, алюмосиликат, оксид титана, титанат, оксид циркония, цирконат, уголь (такой как активированный уголь, алмаз, графит или фуллерен), сульфат, фосфат, танталат, оксид церия, другие оксиды металлов и их смеси. В другом варианте осуществления настоящего изобретения указанная инертная подложка состоит по существу из кремнезема. В еще одном варианте осуществления настоящего изобретения указанный кремнезем выбран из группы, состоящей из аморфного кремнезема, кварца, тридимита, кристобалита, моганита, коэсита и их смесей. В еще одном варианте осуществления настоящего изобретения указанный кремнезем представляет собой аморфный кремнезем. В одном варианте осуществления настоящего изобретения указанный кремнезем имеет удельную площадь поверхности, составляющую менее приблизительно 10 м2/г. В другом варианте осуществления настоящего изобретения инертная подложка составляет по количеству от приблизительно 20 мас. % до приблизительно 90 мас. % в расчете на общую массу предшествующего катализатора.
В одном варианте осуществления настоящего изобретения масса кристаллической фосфатной соли составляет от приблизительно 5 мас. % до приблизительно 90 мас. % в расчете на общую массу предшествующего катализатора. В другом варианте осуществления настоящего изобретения масса кристаллической фосфатной соли составляет от приблизительно 8 мас. % до приблизительно 60 мас. % в расчете на общую массу предшествующего катализатора. В еще одном варианте осуществления настоящего изобретения масса кристаллической фосфатной соли составляет от приблизительно 12 мас. % до приблизительно 40 мас. % в расчете на общую массу предшествующего катализатора. В еще одном варианте осуществления настоящего изобретения масса кристаллической фосфатной соли составляет от приблизительно 12 мас. % до приблизительно 17 мас. % в расчете на общую массу предшествующего катализатора. В одном варианте осуществления настоящего изобретения масса кристаллической фосфатной соли составляет от приблизительно 26 мас. % до приблизительно 32 мас. % в расчете на общую массу предшествующего катализатора.
В одном варианте осуществления настоящего изобретения предшествующий катализатор состоит из



В одном варианте осуществления настоящего изобретения предшествующий катализатор состоит из



В одном варианте осуществления настоящего изобретения предшествующий катализатор состоит из двух или более различных фосфатных соединений, выбранных из группы, состоящей из











В другом варианте осуществления настоящего изобретения предшествующий катализатор содержит одну или более фосфатных солей, состоящих по существу из одного или более одновалентных катионов, и одного или более фосфатных анионов, выбранных из группы, представленной молекулярными формулами


Предшествующий катализатор настоящего изобретения можно использовать для катализа нескольких химических реакций. Не имеющими ограничительного характера примерами реакций являются: дегидратация молочной кислоты, производных молочной кислоты или их смесей до акриловой кислоты; дегидратация 3-гидроксипропионовой кислоты, 3-производных гидроксипропионовой кислоты или их смесей до акриловой кислоты; дегидратация глицерина до акролеина; изомеризация молочной кислоты в 3-гидроксипропионовую кислоту в присутствии воды; восстановление гидроксипропионовой кислоты до пропионовой кислоты или 1-пропанола в присутствии газообразного водорода; дегидратация алифатических спиртов до алкенов или олефинов; дегидрогенизация алифатических спиртов до эфиров; другие реакции дегидрогенизации, гидролиза, алкилирования, деалкилирования, окисления, диспропорционирования, эстерификации, циклизации, изомеризации, конденсации, ароматизации, полимеризации; и другие реакции, которые могут быть очевидны специалисту в данной области техники.
IV. Способы получения катализатора
В контексте настоящего изобретения тройная точка фосфатной соли представляет собой точку с такой температурой и парциальным давлением водяного пара, в которой три фазы фосфатной соли (т. е. кристаллическая и полностью гидратированная, кристаллическая и полностью дегидратированная и аморфная и частично дегидратированная) одновременно сосуществуют в термодинамическом равновесии. В качестве примера, без ограничений, тройная точка может быть определена как точка пересечения двух (из трех) линий фазового перехода на диаграмме состояния, на которой представлена зависимость парциального давления водяного пара от температуры (см. ФИГ. 1): кривая A: линия фазового перехода между кристаллической и полностью гидратированной фосфатной солью и кристаллической и полностью дегидратированной фосфатной солью при низкой температуре и низком парциальном давлении водяного пара (например, ниже приблизительно 248°C и 0,85 бар для фосфатных солей калия и ниже приблизительно 267°C и 0,35 бар для фосфатных солей цезия); кривая B: линия фазового перехода между кристаллической и полностью дегидратированной фосфатной солью и аморфной и частично дегидратированной фосфатной солью при высокой температуре и среднем парциальном давлении водяного пара (например, выше приблизительно 248°C и 0,85 бар для фосфатных солей калия и выше приблизительно 267°C и 0,35 бар для фосфатных солей цезия); и кривая C: линия фазового перехода между кристаллической и полностью гидратированной фосфатной солью и аморфной и частично дегидратированной фосфатной солью при высокой температуре и высоком парциальном давлении водяного пара.
Линии фазового перехода могут быть определены любым способом, известным специалистам в данной области техники, таким как, например, без ограничений, рентгеноструктурный анализ (XRD) in-situ, термический анализ (например, термогравиметрический анализ, дифференциальный термический анализ и дифференциальная сканирующая калориметрия), рамановская спектроскопия, инфракрасная спектроскопия (IR), спектроскопия ядерного магнитного резонанса (NMR), или способами, описанными в работе Taninouchi, Y.-k., et al., J. Electrochem. Soc. 156:B572-B579 (2009); или Ikeda, A. and Haile, S. M., Solid State Ionics 2012, 213:63-71 (2012) (все из которых включены в данный документ посредством ссылки). В качестве иллюстрации в способе на основе метода XRD in-situ предшествующий катализатор, содержащий кристаллическую фосфатную соль, приводят в контакт при высокой температуре (например, 450°C) с потоком поступающих газообразных материалов, состоящим из инертного газа (например, азота, гелия или воздуха) и водяного пара, при конкретном парциальном давлении водяного пара до достижения равновесия. Затем температуру постепенно снижают при наблюдении изменений на дифрактограммах, пока не будет наблюдаться фазовый переход. Такую же процедуру повторяют при различных значениях парциального давления воды и фиксируют температуры фазового перехода. Значения парциального давления водяного пара (в логарифмическом масштабе) наносят на график зависимости от температуры фазового перехода (в линейном масштабе) и аппроксимируют по уравнению Аррениуса:

Активные катализаторы настоящего изобретения представляют собой аморфные и частично дегидратированные фосфатные соли, соответствующие области между кривыми B и C (раздвоенная область над тройной точкой) на диаграмме состояния катализатора, т. е. графике равновесного парциального давления водяного пара от температуры (см. Фиг. 1). Эти активные катализаторы могут быть получены путем обработки предшествующих катализаторов, содержащих кристаллическую фосфатную соль, соответствующую области над кривыми A и C либо под кривыми A и B на ФИГ. 1, при достаточной температуре или парциальном давлении водяного пара или комбинации обоих параметров для превращения кристаллической фосфатной соли в аморфную и частично дегидратированную фосфатную соль, соответствующую области между кривыми B и C на диаграмме состояния.
В одном варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего кристаллическую фосфатную соль, в активный катализатор, содержащий аморфную и частично дегидратированную фосфатную соль, включает приведение указанного предшествующего катализатора в контакт с потоком поступающих газообразных материалов, содержащим водяной пар; причем указанное приведение в контакт осуществляют при температуре и парциальном давлении водяного пара достаточных для превращения указанной кристаллической фосфатной соли в указанную аморфную и частично дегидратированную фосфатную соль. В другом варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего кристаллическую фосфатную соль, в активный катализатор, содержащий аморфную и частично дегидратированную фосфатную соль, включает приведение указанного предшествующего катализатора в контакт с потоком поступающих газообразных материалов, содержащим водяной пар, гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси; причем указанное приведение в контакт осуществляют при температуре и парциальном давлении водяного пара достаточных для превращения указанной кристаллической фосфатной соли в указанную аморфную и частично дегидратированную фосфатную соль; и при этом указанное приведение в контакт приводит к получению акриловой кислоты, производных акриловой кислоты или их смесей из указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей.
В одном варианте осуществления настоящего изобретения температура, при которой указанный поток поступающих газообразных материалов находится в контакте с указанным предшествующим катализатором, содержащим кристаллическую фосфатную соль, составляет от приблизительно 120°C до приблизительно 700°C. В другом варианте осуществления настоящего изобретения температура, при которой указанный поток поступающих газообразных материалов находится в контакте с указанным предшествующим катализатором, содержащим кристаллическую фосфатную соль, составляет от приблизительно 150°C до приблизительно 500°C. В еще одном варианте осуществления настоящего изобретения температура, при которой указанный поток поступающих газообразных материалов находится в контакте с указанным предшествующим катализатором, содержащим кристаллическую фосфатную соль, составляет от приблизительно 300°C до приблизительно 450°C. В еще одном варианте осуществления настоящего изобретения температура, при которой указанный поток поступающих газообразных материалов находится в контакте с указанным предшествующим катализатором, содержащим кристаллическую фосфатную соль, составляет от приблизительно 325°C до приблизительно 400°C.
В одном варианте осуществления настоящего изобретения парциальное давление водяного пара в указанном потоке поступающих газообразных материалов превышает или равно приблизительно 0,4 бар. В другом варианте осуществления настоящего изобретения парциальное давление водяного пара в указанном потоке поступающих газообразных материалов превышает или равно приблизительно 0,8 бар. В еще одном варианте осуществления настоящего изобретения парциальное давление водяного пара в указанном потоке поступающих газообразных материалов находится в диапазоне от приблизительно 0,4 бар до приблизительно 20 бар. В еще одном варианте осуществления настоящего изобретения парциальное давление водяного пара в указанном потоке поступающих газообразных материалов находится в диапазоне от приблизительно 0,8 бар до приблизительно 16 бар. В одном варианте осуществления настоящего изобретения парциальное давление водяного пара в указанном потоке поступающих газообразных материалов составляет приблизительно 13 бар.
В одном варианте осуществления настоящего изобретения общее давление указанного потока поступающих газообразных материалов превышает или равно приблизительно 1 бар. В другом варианте осуществления настоящего изобретения общее давление указанного потока поступающих газообразных материалов превышает или равно приблизительно 4 бар. В еще одном варианте осуществления настоящего изобретения общее давление указанного потока поступающих газообразных материалов находится в диапазоне от приблизительно 4 бар до приблизительно 35 бар. В еще одном варианте осуществления настоящего изобретения общее давление указанного потока поступающих газообразных материалов находится в диапазоне от приблизительно 8 бар до приблизительно 30 бар. В одном варианте осуществления настоящего изобретения общее давление указанного потока поступающих газообразных материалов составляет приблизительно 26 бар.
В одном варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего
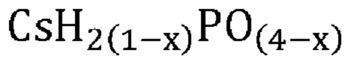


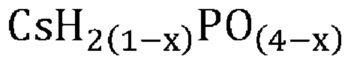
В одном варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего




В одном варианте осуществления настоящего изобретения масса указанной
В одном варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего


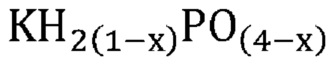


В одном варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего
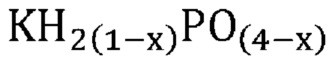

В одном варианте осуществления настоящего изобретения масса указанной
В одном варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего





В одном варианте осуществления настоящего изобретения способ превращения предшествующего катализатора, содержащего

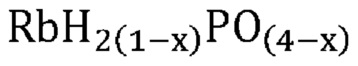

В одном варианте осуществления настоящего изобретения масса указанной
В одном варианте осуществления настоящего изобретения способ получения катализатора дополнительно включает формование частиц указанного предшествующего катализатора до указанного введения в контакт с указанным потоком поступающих газообразных материалов. Не имеющие ограничительного характера примеры операций формования включают в себя гранулирование, агломерирование, уплотнение, пеллетирование и экструдирование. В другом варианте осуществления настоящего изобретения способ получения катализатора дополнительно включает уменьшение размера или измельчение частиц указанного предшествующего катализатора до указанного введения в контакт с указанным потоком поступающих газообразных материалов. В одном варианте осуществления настоящего изобретения способ получения катализатора дополнительно включает просеивание частиц указанного предшествующего катализатора для выбора материала с конкретным гранулометрическим составом до указанного введения в контакт с указанным потоком поступающих газообразных материалов. В другом варианте осуществления настоящего изобретения способ получения катализатора дополнительно включает просеивание частиц указанного предшествующего катализатора для получения медианного размера частиц в диапазоне от приблизительно 50 мкм до приблизительно 500 мкм. В еще одном варианте осуществления настоящего изобретения способ получения катализатора дополнительно включает просеивание частиц указанного предшествующего катализатора для получения медианного размера частиц в диапазоне от приблизительно 100 мкм до приблизительно 200 мкм. В еще одном варианте осуществления настоящего изобретения способ получения катализатора дополнительно включает просеивание частиц указанного предшествующего катализатора для получения размера частиц в диапазоне от приблизительно 106 мкм до приблизительно 212 мкм.
В одном варианте осуществления настоящего изобретения активный катализатор получают посредством: (a) смешивания KH2PO4 и аморфного кремнезема в массовом соотношении от приблизительно 2:1 до приблизительно 1:8 для получения предшествующего катализатора, (b) нагревания указанного предшествующего катализатора при температуре от приблизительно 200°C и до приблизительно 650°C в течение от приблизительно 1 часа до приблизительно 12 часов для получения кальцинированного предшествующего катализатора, (c) необязательно измельчения и просеивания указанного кальцинированного предшествующего катализатора для получения измельченного кальцинированного предшествующего катализатора и (d) приведения в контакт указанного кальцинированного предшествующего катализатора или указанного измельченного кальцинированного предшествующего катализатора с потоком поступающих газообразных материалов, содержащим азот и водяной пар; причем парциальное давление водяного пара в указанном потоке поступающих газообразных материалов составляет от приблизительно 5 бар до приблизительно 15 бар; и при этом указанное приведение в контакт осуществляют при температуре от приблизительно 325°C до приблизительно 425°C для получения указанного активного катализатора.
В другом варианте осуществления настоящего изобретения активный катализатор получают посредством: (a) смешивания KH2PO4 и BaSO4 в массовом соотношении от приблизительно 2:1 до приблизительно 1:8 для получения предшествующего катализатора, (b) нагревания указанного предшествующего катализатора при температуре от приблизительно 200°C и до приблизительно 650°C в течение от приблизительно 1 часа до приблизительно 12 часов для получения кальцинированного предшествующего катализатора, (c) необязательно измельчения и просеивания указанного кальцинированного предшествующего катализатора для получения измельченного кальцинированного предшествующего катализатора и (d) приведения в контакт указанного кальцинированного предшествующего катализатора или указанного измельченного кальцинированного предшествующего катализатора с потоком поступающих газообразных материалов, содержащим азот и водяной пар; причем парциальное давление водяного пара в указанном потоке поступающих газообразных материалов составляет от приблизительно 5 бар до приблизительно 15 бар; и при этом указанное приведение в контакт осуществляют при температуре от приблизительно 325°C до приблизительно 425°C для получения указанного активного катализатора.
В другом варианте осуществления настоящего изобретения активный катализатор получают посредством (a) смешивания K2HPO4, (NH4)2HPO4 и аморфного кремнезема в массовом соотношении от приблизительно 1,3:1,0:16,1 до приблизительно 1,3:1,0:1,2 для получения предшествующего катализатора, (b) нагревания указанного предшествующего катализатора при температуре от приблизительно 200°C и до приблизительно 650°C в течение от приблизительно 1 часа до приблизительно 12 часов для получения кальцинированного предшествующего катализатора, (c) необязательно измельчения и просеивания указанного кальцинированного предшествующего катализатора для получения измельченного предшествующего катализатора и (d) приведения в контакт указанного кальцинированного предшествующего катализатора или указанного измельченного кальцинированного предшествующего катализатора с потоком поступающих газообразных материалов, содержащим азот и водяной пар; причем парциальное давление водяного пара в указанном потоке поступающих газообразных материалов составляет от приблизительно 5 бар до приблизительно 15 бар и при этом указанное приведение в контакт осуществляют при температуре от приблизительно 325°C до приблизительно 425°C для получения указанного активного катализатора.
Способ получения предшествующего катализатора может включать смешивание двух или более различных компонентов. Данную стадию смешивания можно осуществлять посредством любого способа, известного специалисту в данной области техники, такого как, в качестве примера, без ограничений: смешивание в твердой фазе, пропитывание или совместное осаждение. В способе смешивания в твердой фазе различные компоненты физически смешивают вместе с необязательным измельчением посредством любого способа, известного специалисту в данной области техники, такого как, в качестве примера, без ограничений, сдвиговое измельчение, измельчение растяжением, смешивающее измельчение, экструзионное измельчение, шаровое измельчение и другие, а в альтернативном варианте осуществления с последующей любой дополнительной стадией обработки или активации. В способе пропитывания суспензию нерастворимого компонента (например, инертной подложки) обрабатывают раствором растворимых ингредиентов предшествующего катализатора и полученный в результате материал затем обрабатывают или активируют в условиях, при которых происходит переход смеси в более активное или предпочтительное состояние. В способе совместного осаждения гомогенный раствор ингредиентов предшествующего катализатора осаждают посредством добавления дополнительных ингредиентов с последующей необязательной фильтрацией и нагреванием для удаления растворителей и летучих материалов (например, воды, азотной кислоты, диоксида углерода, аммиака или уксусной кислоты).
Смешивание компонентов предшествующего катализатора с поверхностно-активными веществами с последующим нагреванием может приводить к увеличению площади поверхности предшествующего катализатора. В одном варианте осуществления настоящего изобретения способ получения катализатора дополнительно включает смешивание одного или более поверхностно-активных веществ с указанным предшествующим катализатором до указанного введения в контакт с указанным потоком поступающих газообразных материалов. В другом варианте осуществления настоящего изобретения указанные одно или более поверхностно-активных веществ являются катионными или цвиттер-ионными. Не имеющие ограничительного характера примеры поверхностно-активных веществ представляют собой бромид миристилтриметиламмония, бромид гексадецилтриметиламмония, бромид додецилтриметиламмония, бромид децилтриметиламмония и бромид октадецилтриметиламмония.
Нагревание может способствовать протеканию химических реакций, процессов термического разложения, процессов фазового перехода и/или удалению летучих веществ. В одном варианте осуществления настоящего изобретения способ получения активного катализатора дополнительно включает нагревание указанного предшествующего катализатора при температуре, превышающей или равной 180°C, до указанного введения в контакт с указанным потоком поступающих газообразных материалов. В другом варианте осуществления настоящего изобретения способ получения активного катализатора дополнительно включает нагревание указанного предшествующего катализатора при температуре, превышающей или равной 300°C, до указанного введения в контакт с указанным потоком поступающих газообразных материалов. В еще одном варианте осуществления настоящего изобретения способ получения активного катализатора дополнительно включает нагревание указанного предшествующего катализатора при температуре от приблизительно 350°C до приблизительно 650°C до указанного введения в контакт с указанным потоком поступающих газообразных материалов. В еще одном варианте осуществления настоящего изобретения способ получения активного катализатора дополнительно включает нагревание указанного предшествующего катализатора при температуре от приблизительно 400°C до приблизительно 450°C до указанного введения в контакт с указанным потоком поступающих газообразных материалов. Указанное нагревание, как правило, осуществляют с применением любого способа, известного специалистам в данной области техники, такого как, например, без ограничений, конвекционное, контактное, радиационное, микроволновое нагревание и т. п. Нагревание осуществляют с помощью оборудования, такого как, например, без ограничений, печи, распылители или реакторы с различными конструкциями, включающие в себя шахтные печи, вращающиеся печи, подовые печи, реакторы с псевдоожиженным слоем, распылительные сушки. Продолжительность указанного нагревания составляет в одном варианте осуществления настоящего изобретения от приблизительно 1 часа до приблизительно 72 часов. В другом варианте осуществления продолжительность указанного нагревания составляет от приблизительно 2 часов до приблизительно 12 часов. В еще одном варианте осуществления продолжительность указанного нагревания составляет приблизительно 4 часа. В одном варианте осуществления скорость линейного изменения температуры в указанном нагревании составляет от приблизительно 0,5°C/мин до приблизительно 20°C/мин. В другом варианте осуществления скорость линейного изменения температуры в указанном нагревании составляет приблизительно 10°C/мин.
V. Способы получения акриловой кислоты, производных акриловой кислоты или их смесей
Предложен способ дегидратации гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей до акриловой кислоты, производных акриловой кислоты или их смесей. В одном варианте осуществления настоящего изобретения указанная гидроксипропионовая кислота выбрана из группы, состоящей из молочной кислоты (2-гидроксипропионовой кислоты), 3-гидроксипропионовой кислоты и их смесей; а указанные производные гидроксипропионовой кислоты выбраны из группы, состоящей из производных молочной кислоты, производных 3-гидроксипропионовой кислоты и их смесей.
В другом варианте осуществления настоящего изобретения указанная гидроксипропионовая кислота представляет собой молочную кислоту, а указанные производные гидроксипропионовой кислоты представляют собой производные молочной кислоты. Молочная кислота может представлять собой D-молочную кислоту, L-молочную кислоту или их смеси (включая рецемическую смесь). Производные молочной кислоты могут представлять собой соли металлов или аммония с молочной кислотой, алкиловые сложные эфиры молочной кислоты, олигомеры молочной кислоты, циклические сложные диэфиры молочной кислоты, ангидрид молочной кислоты, 2-алкоксипропионовые кислоты или их алкиловые сложные эфиры, 2-арилоксипропионовые кислоты или их алкиловые сложные эфиры, 2-ацилоксипропионовые кислоты или их алкиловые сложные эфиры или их смеси. Не имеющие ограничительного характера примеры металлических солей молочной кислоты представляют собой лактат натрия, лактат калия и лактат кальция. Не имеющие ограничительного характера примеры алкиловых сложных эфиров молочной кислоты представляют собой метиллактат, этиллактат, бутиллактат, 2-этилгексиллактат и их смеси. Не имеющий ограничительного характера пример циклических сложных диэфиров молочной кислоты представляет собой дилактид. Не имеющие ограничительного характера примеры 2-алкоксипропионовых кислот представляют собой 2-метоксипропионовую кислоту и 2-этоксипропионовую кислоту. Не имеющий ограничительного характера пример 2-арилоксипропионовой кислоты представляет собой 2-феноксипропионовую кислоту. Не имеющий ограничительного характера пример 2-ацилоксипропионовой кислоты представляет собой 2-ацетоксипропионовую кислоту. В еще одном варианте осуществления настоящего изобретения производное молочной кислоты представляет собой метиллактат. Метиллактат может быть беспримесным или может находиться в растворе с водой, метанолом или их смесями.
Производные 3-гидроксипропионовой кислоты могут представлять собой соли металла или аммония 3-гидроксипропионовой кислоты, алкиловые сложные эфиры 3-гидроксипропионовой кислоты, олигомеры 3-гидроксипропионовой кислоты, 3-алкоксипропионовые кислоты или их алкиловые сложные эфиры, 3-арилоксипропионовые кислоты или их алкиловые сложные эфиры, 3-ацилоксипропионовые кислоты или их алкиловые сложные эфиры или их смеси. Не имеющие ограничительного характера примеры солей металла 3-гидроксипропионовой кислоты представляют собой 3-гидроксипропионат натрия, 3-гидроксипропионат калия и 3-гидроксипропионат кальция. Не имеющие ограничительного характера примеры алкиловых сложных эфиров гидроксипропионовой кислоты представляют собой метил-3-гидроксипропионат, этил-3-гидроксипропионат, бутил-3-гидроксипропионат, 2-этилгексил-3-гидроксипропионат и их смеси. Не имеющие ограничительного характера примеры 3-алкоксипропионовых кислот представляют собой 3-метоксипропионовую кислоту и 3-этоксипропионовую кислоту. Не имеющий ограничительного характера пример 3-арилоксипропионовой кислоты представляет собой 3-феноксипропионовую кислоту. Не имеющий ограничительного характера пример 3-ацилоксипропионовой кислоты представляет собой 3-ацетоксипропионовую кислоту.
Можно получать гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси путем ферментации сахара или химического превращения сахаров или иных исходных материалов, таких как глицерин. Почти все мировое производство молочной кислоты осуществляют в настоящее время путем ферментации сахара; однако существуют технологии химических превращений, осуществляемые в настоящее время в пробном или демонстрационном масштабе. Кроме того, сахарное сырье может представлять собой сахар поколения 1 (т. е. сахар из кукурузы, сахарного тростника, сахарной свеклы, пшеницы, картофеля, риса и т. п.) или сахар поколения 2 (т. е. сахар, полученный путем гидролиза биомассы или сельскохозяйственных отходов, таких как жмых, кукурузная солома, рисовая шелуха, пшеничная солома, и т. п.).
Производные акриловой кислоты могут представлять собой соли металла или аммония акриловой кислоты, алкиловые сложные эфиры акриловой кислоты, олигомеры акриловой кислоты или их смеси. Не имеющие ограничительного характера примеры солей металла акриловой кислоты представляют собой акрилат натрия, акрилат калия и акрилат кальция. Не имеющие ограничительного характера примеры алкиловых сложных эфиров акриловой кислоты представляют собой метилакрилат, этилакрилат, бутилакрилат, 2-этилгексилакрилат или их смеси.
В одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с любым активным катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), настоящего изобретения в реакторе при определенных значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей, при этом в результате дегидратации в реакторе получают акриловую кислоту, производные акриловой кислоты или их смеси.
В одном варианте осуществления настоящего изобретения указанный поток поступающих газообразных материалов содержит по существу водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси. В другом варианте осуществления настоящего изобретения указанный поток поступающих газообразных материалов дополнительно содержит по существу химически инертный газ (который также именуется в настоящем документе «разбавитель»). В контексте настоящего изобретения по существу химически инертный газ представляет собой любой газ, который является по существу химически инертным по отношению к указанным гидроксипропионовой кислоте, производным гидроксипропионовой кислоты или их смесям, но не обязательно по отношению к указанному катализатору. Не имеющие ограничительного характера примеры по существу химически инертных газов или разбавителей представляют собой азот, гелий, аргон, диоксид углерода, монооксид углерода и их смеси. В еще одном варианте осуществления настоящего изобретения указанный по существу химически инертный газ или разбавитель содержит азот. В еще одном варианте осуществления настоящего изобретения указанный по существу химически инертный газ или разбавитель состоит по существу из азота. В одном варианте осуществления настоящего изобретения указанный поток поступающих газообразных материалов дополнительно содержит газ, выбранный из группы, состоящей из воздуха и кислорода.
В одном варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет от приблизительно 0,5 мол. % до приблизительно 95 мол. % (в стандартных условиях температуры и давления, STP). В другом варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет от приблизительно 1,5 мол. % до приблизительно 20 мол. % (в стандартных условиях STP). В еще одном варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет от приблизительно 2 мол. % до приблизительно 5 мол. % (в стандартных условиях STP). В еще одном варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет приблизительно 2,5 мол. % (в стандартных условиях STP). В одном варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет приблизительно 2,5 мол. % (в условиях STP), концентрация водяного пара в указанном потоке поступающих газообразных материалов составляет приблизительно 50 мол. % (в условиях STP), а концентрация азота в указанном потоке поступающих газообразных материалов составляет приблизительно 47,5 мол. % (в условиях STP). В другом варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет приблизительно 10 мол. % (в условиях STP), а концентрация водяного пара в указанном потоке поступающих газообразных материалов составляет приблизительно 90 мол. % (в условиях STP).
В одном варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет от приблизительно 0,5 мол. % до приблизительно 95 мол. %. В другом варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет от приблизительно 1,5 мол. % до приблизительно 20 мол. %. В еще одном варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет от приблизительно 2 мол. % до приблизительно 5 мол. %. В еще одном варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет приблизительно 25 мол. %. В одном варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет приблизительно 2,5 мол. %, концентрация водяного пара в указанном потоке поступающих газообразных материалов составляет приблизительно 50 мол. %, а концентрация азота в указанном потоке поступающих газообразных материалов составляет приблизительно 47,5 мол. %. В другом варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном потоке поступающих газообразных материалов составляет приблизительно 10 мол. %, а концентрация водяного пара в указанном потоке поступающих газообразных материалов составляет приблизительно 90 мол. %.
Не имеющими ограничительного характера примерами реакторов, подходящих для применения в настоящем изобретении, являются статические реакторы, реакторы с мешалкой, реакторы с рециркуляцией, реакторы с псевдоожиженным слоем катализатора, проточные реакторы с уплотненным слоем катализатора и их комбинации. В одном варианте осуществления настоящего изобретения поток поступающих газообразных материалов движется в реакторе сверху вниз. В другом варианте осуществления настоящего изобретения поток поступающих газообразных материалов движется в реакторе снизу вверх. В еще одном варианте осуществления настоящего изобретения поток поступающих газообразных материалов движется в реакторе горизонтально.
В одном варианте осуществления настоящего изобретения реактор представляет собой проточный реактор с уплотненным слоем катализатора. Как правило, проточный реактор с уплотненным слоем катализатора представляет собой трубчатый реактор. В другом варианте осуществления настоящего изобретения проточный трубчатый реактор с уплотненным слоем катализатора представляет собой однослойный реактор. В еще одном варианте осуществления настоящего изобретения проточный трубчатый реактор с уплотненным слоем катализатора представляет собой двухслойный реактор. На ФИГ. 2 представлен вид в частичном разрезе иллюстративного трубчатого однослойного реактора, а на ФИГ. 3 представлен вид в частичном разрезе иллюстративного трубчатого двухслойного реактора.
Представленный на ФИГ. 2 однослойный реактор 3 состоит из единственного слоя 25 (также именуемого стенкой), который проходит от внутренней поверхности 35 к наружной поверхности 55 и имеет некоторую толщину стенки. Внутренняя поверхность 35 находится в контакте с катализатором 65. В одном варианте осуществления настоящего изобретения однослойный реактор содержит стенку, наружную поверхность и внутреннюю поверхность; причем указанная стенка изготовлена из материала стенки, имеет толщину стенки и проходит от указанной наружной поверхности к указанной внутренней поверхности; и при этом указанная внутренняя поверхность находится в контакте с указанным катализатором. В другом варианте осуществления настоящего изобретения толщина стенки однослойного реактора соответствует размерному ряду номинальных размеров труб (NPS) для бесшовных и сварных стальных труб (ANSI B36.10 от 1979 г.). В еще одном варианте осуществления настоящего изобретения толщина стенки однослойного реактора находится в диапазоне от приблизительно 0,65 дюйма (1,651 мм) до приблизительно 0,5 дюйма (12,7 мм). В еще одном варианте осуществления настоящего изобретения толщина стенки однослойного реактора находится в диапазоне от приблизительно 0,65 дюйма (1,651 мм) до приблизительно 0,432 дюйма (10,973 мм). В одном варианте осуществления настоящего изобретения толщина стенки однослойного реактора находится в диапазоне от приблизительно 0,065 дюйма (1,651 мм) до приблизительно 0,337 дюйма (8,56 мм). В другом варианте осуществления настоящего изобретения толщина стенки однослойного реактора составляет приблизительно 0,133 дюйма (3,378 мм).
Представленный на ФИГ. 3 двухслойный реактор 5 содержит внутреннюю поверхность 30, которая находится в контакте с катализатором 60 и является крайней внутренней поверхностью двухслойного реактора 5. Двухслойный реактор 5 состоит из внутреннего слоя 10, который имеет толщину внутреннего слоя, наружного слоя 20, который имеет толщину наружного слоя, границы 40 раздела между наружным слоем 20 и внутренним слоем 10 и наружной поверхностью 50, которая является крайней наружной поверхностью трубчатого реактора 5. В одном варианте осуществления настоящего изобретения наружный слой 20 двухслойного реактора 5 состоит из двух или более подслоев. В другом варианте осуществления настоящего изобретения двухслойный реактор содержит наружный слой, внутренний слой, наружную поверхность, внутреннюю поверхность и границу между указанным наружным слоем и указанным внутренним слоем; причем указанный наружный слой изготовлен из материала наружного слоя, имеет толщину наружного слоя и отходит от указанной границы к указанной наружной поверхности; при этом указанный внутренний слой изготовлен из материала внутреннего слоя, имеет толщину внутреннего слоя и отходит от указанной внутренней поверхности к указанной границе; и при этом указанная внутренняя поверхность находится в контакте с указанным катализатором. В еще одном варианте осуществления настоящего изобретения указанный наружный слой содержит два или более подслоев.
В одном варианте осуществления настоящего изобретения толщина внутреннего слоя двухслойного реактора составляет от приблизительно 1 мм до приблизительно 20 мм. В другом варианте осуществления настоящего изобретения толщина внутреннего слоя двухслойного реактора составляет от приблизительно 1,5 мм до приблизительно 10 мм. В еще одном варианте осуществления настоящего изобретения толщина внутреннего слоя двухслойного реактора составляет от приблизительно 2 мм до приблизительно 8 мм. В еще одном варианте осуществления настоящего изобретения толщина внутреннего слоя двухслойного реактора составляет от приблизительно 3 мм до приблизительно 6 мм.
В одном варианте осуществления настоящего изобретения толщина наружного слоя двухслойного реактора составляет от приблизительно 1 мм до приблизительно 20 мм. В другом варианте осуществления настоящего изобретения толщина наружного слоя двухслойного реактора составляет от приблизительно 1,6 мм до приблизительно 12,7 мм. В еще одном варианте осуществления настоящего изобретения толщина наружного слоя двухслойного реактора составляет от приблизительно 2 мм до приблизительно 9 мм. В еще одном варианте осуществления настоящего изобретения толщина наружного слоя двухслойного реактора составляет от приблизительно 3 мм до приблизительно 6 мм.
В одном варианте осуществления настоящего изобретения наружный диаметр (НД) однослойного или двухслойного реактора соответствует размерному ряду номинальных размеров труб (NPS) для бесшовных и сварных стальных труб (ANSI B36.10 от 1979 г.). В другом варианте осуществления настоящего изобретения НД однослойного или двухслойного реактора находится в диапазоне от приблизительно 0,84 дюйма (21,34 мм) до приблизительно 9,625 дюйма (244,48 мм). В еще одном варианте осуществления настоящего изобретения НД однослойного или двухслойного реактора находится в диапазоне от приблизительно 1,315 дюйма (33,4 мм) до приблизительно 6,625 дюйма (168,28 мм). В еще одном варианте осуществления настоящего изобретения НД однослойного или двухслойного реактора находится в диапазоне от приблизительно 2,375 дюйма (60,33 мм) до приблизительно 4,5 дюйма (114,3 мм). В одном варианте осуществления настоящего изобретения НД однослойного или двухслойного реактора составляет приблизительно 1,315 дюйма (33,4 мм).
Внутренний слой двухслойного реактора может быть образован различными способами. Не имеющие ограничительного характера примеры способов формирования внутреннего слоя двухслойного реактора включают присоединение внутреннего слоя к наружному слою и нанесение покрытия погружением наружного слоя в ванну, содержащую материал внутреннего слоя. В одном варианте осуществления настоящего изобретения внутренний слой образован посредством способа присоединения внутреннего слоя к наружному слою. В другом варианте осуществления настоящего изобретения способ присоединения выбран из группы, состоящей из наплавки, лазерной наплавки, наплавки взрывом, наплавки электромагнитным плавлением, сварки плавлением, склеивания, впрессовки, прокатывания, соэкструзии, термического напыления, электролитического осаждения и химического осаждения из паровой фазы. Не имеющие ограничительного характера примеры способов присоединения включают наплавку медной трубки (внутренний слой) на внутреннюю поверхность наружного слоя из нержавеющей стали, наплавку взрывом циркониевой трубки (внутренний слой) на внутреннюю поверхность наружного слоя из нержавеющей стали и электролитическое осаждение серебра на наружном слое из нержавеющей стали. В еще одном варианте осуществления настоящего изобретения внутренний слой присоединяют к наружному слою в форме плоских листов, которые затем приваривают внутренним швом к трубе реактора с внутренним слоем и наружным слоем.
В еще одном варианте осуществления настоящего изобретения внутренний слой образован посредством способа нанесения покрытия погружением наружного слоя в ванну, содержащую материал внутреннего слоя. Не имеющие ограничительного характера примеры материалов наружного слоя из нержавеющей стали и материалов внутреннего слоя из алюминия или смеси алюминия и кремния, образованные посредством способа нанесения покрытия погружением, включают в себя следующие имеющиеся в продаже сплавы: Нержавеющая алюминизированная сталь 409 1 типа и нержавеющая алюминизированная сталь 439 1 типа (материал внутреннего слоя представляет собой смесь алюминия и кремния); и алюминизированная сталь типа 2 (материал внутреннего слоя представляет собой только алюминий); все производства компании AK Steel Corp. (г. Вест-Честер, штат Огайо, США). Процесс нанесения покрытия погружением можно реализовать в периодическом, непрерывном или полунепрерывном режимах.
Активные катализаторы настоящего изобретения могут оказывать коррозионное действие на реакторы, поскольку они представляют собой аморфные и частично дегидратированные фосфатные соли. Неожиданно было обнаружено, что некоторые конкретные металлы в материале внутреннего слоя двухслойных реакторов или материале стенок однослойных реакторов могут уменьшать или устранять коррозионные эффекты активных катализаторов, т. е. эти металлы реактора представляют собой металлы, которые обладают устойчивостью к коррозии. Не желая быть связанными какой-либо теорией, заявители предположили, что устойчивые к коррозии металлы реактора: 1) образуют оксиды и тонкий пассивирующий слой, содержащий эти оксиды, на внутренней поверхности реакторов (т. е. на поверхности реактора, которая находится в контакте с катализатором и потоком поступающих газообразных материалов); или 2) представляют собой металлы, которые обладают устойчивостью к коррозии в условиях дегидратации. Пассивирующий поверхность слой на основе оксида может образовываться в результате окисления внутренней поверхности реактора. В одном варианте осуществления настоящего изобретения окисление происходит в процессе дегидратации. В другом варианте осуществления настоящего изобретения окисление происходит перед дегидратацией. В еще одном варианте осуществления настоящего изобретения окисление, которое происходит перед дегидратацией, может быть вызвано стадией предварительной обработки реактора. В любом случае пассивирующий поверхность слой на основе оксида уменьшает или предотвращает окисление других металлов реактора и миграцию образовавшихся ионов в слой катализатора (т. е. коррозию). Миграция ионов металлов в частицы катализатора может оказывать негативное воздействие на выход и селективность полученной акриловой кислоты, производных акриловой кислоты или их смесей, а также на срок службы катализатора, и, кроме того, способствовать снижению прочности самого реактора. Катион металла в оксиде может представлять собой металл, содержащийся в материале внутреннего слоя или материале стенки.
В одном варианте осуществления настоящего изобретения после процесса присоединения происходит окисление внутренней поверхности. В другом варианте осуществления настоящего изобретения после нанесения покрытия погружением происходит окисление внутренней поверхности.
Не имеющим ограничительного характера примером металла, образующего пассивирующий поверхность слой на основе оксида, является алюминий, содержащийся в значительных количествах (например, более приблизительно 1 мас. %) в различных материалах внутреннего слоя или материалах стенки при окислении реактора до или во время протекания реакции дегидратации. В одном варианте осуществления настоящего изобретения внутренняя поверхность подвергнута окислению, и на ней образован пассивирующий поверхность слой на основе оксида, содержащий оксид алюминия. Не имеющими ограничительного характера примерами материалов стенок, имеющих высокое содержание алюминия и образующих оксидный пассивирующий поверхность слой из оксида алюминия на внутренней поверхности реактора, являются следующие имеющиеся в продаже сплавы: KANTHAL APM (сплав ферритного чугуна с хромом и алюминием; сплав FeCrAl) и KANTHAL APMT (сплав ферритного чугуна с хромом, алюминием и молибденом; сплав FeCrAlMo) производства компании Sandvik AB (г. Стокгольм, Швеция); сплавы HAYNES® 214® и HAYNES® HR-224® (оба представляют собой сплавы на основе Ni) производства компании Haynes International, Inc. (г. Кокомо, штат Индиана, США); сплавы INCONEL® alloy 693 и INCONEL® alloy 601 (оба представляют собой сплавы на основе Ni) и сплав INCOLLOY® MA956 (сплав Fe-Cr-Al) производства компании Special Metals Corporation (г. Хантингтон, штат Западная Виргиния, США); и сплавы Fercalloy, Fercalloy 145, Fercalloy 135 и Resistalloy 134 производства компании Resistalloy Trading Limited (г. Шеффилд, Саут-Йоркшир, Великобритания).
Не имеющим ограничительного характера примером металла, образующего пассивирующий поверхность слой на основе оксида, является кремний, содержащийся в значительных количествах (например, более приблизительно 1 мас. %) в различных материалах внутреннего слоя или материалах стенки при окислении реактора до или во время протекания реакции дегидратации. В одном варианте осуществления настоящего изобретения внутренняя поверхность подвергнута окислению, и на ней образован пассивирующий поверхность слой на основе оксида, содержащий оксид кремния. Не имеющими ограничительного характера примерами материалов стенок, имеющих высокое содержание кремния и образующих оксидный пассивирующий поверхность слой из оксида кремния на внутренней поверхности реактора, являются следующие имеющиеся в продаже сплавы: Sandvik SX и Sandvik 253 MA (оба представляют собой сплавы аустенитной стали) производства компании Sandvik AB (г. Стокгольм, Швеция); и HAYNES® HR-160® и Hastelloy® D-205 (оба представляют собой сплавы на основе Ni) производства компании Haynes International, Inc. (г. Кокомо, штат Индиана США). Для целей настоящего изобретения сплавы на основе Ni представляют собой сплавы, которые содержат Ni в количестве более приблизительно 35 мас. %.
Не имеющим ограничительного характера примером смеси металлов, которая представляет собой материал внутреннего слоя двухслойного реактора, является смесь алюминия и кремния, из которой сформирован внутренний слой посредством процесса нанесения покрытия погружением путем погружения наружного слоя из нержавеющей стали в горячую ванну, содержащую алюминий и кремний, и формирования внутреннего слоя из алюминия и кремния. После этого поверхность внутреннего слоя окисляют до или во время протекания реакции дегидратации с образованием на внутренней поверхности реактора пассивирующего поверхность слоя на основе оксида, содержащего оксид алюминия и кремнезем. В одном варианте осуществления настоящего изобретения внутренняя поверхность подвергнута окислению, и на ней образован пассивирующий поверхность слой на основе оксида, содержащий оксид алюминия и кремнезем.
Как правило, толщина пассивирующего поверхность слоя на основе оксида находится в диапазоне от приблизительно 0,1 нм до 1 мкм. В одном варианте осуществления настоящего изобретения толщина пассивирующего поверхность слоя на основе оксида составляет от приблизительно 0,3 нм до приблизительно 100 нм. В другом варианте осуществления настоящего изобретения толщина слоя пассивирующего поверхность слоя на основе оксида составляет от приблизительно 1 нм до приблизительно 50 нм. В еще одном варианте осуществления настоящего изобретения толщина слоя пассивирующего поверхность слоя на основе оксида составляет от приблизительно 5 нм до приблизительно 25 нм.
После формирования внутреннего слоя с помощью процесса присоединения или процесса нанесения покрытия погружением его можно подвергать окислению с образованием на внутренней поверхности пассивирующего поверхность слоя на основе оксида. Процесс окисления может происходить до процесса дегидратации или во время процесса дегидратации, поскольку поток поступающих газообразных материалов содержит водяной пар. Условия процесса окисления зависят от материала внутреннего слоя и, как правило, хорошо известны специалистам в данной области техники. Не имеющие ограничительного характера примеры процесса окисления до дегидратации включают в себя применение атмосферы воздуха или кислорода и: 1) температуры, превышающей приблизительно 500°C, и продолжительности в несколько часов или 2) двухстадийный процесс, в котором на стадии 1 температура составляет приблизительно 600°C и продолжительность составляет приблизительно 1 час, а на стадии 2 температура составляет приблизительно 1000°C и продолжительность составляет приблизительно 1 час.
В одном варианте осуществления настоящего изобретения материал внутреннего слоя выбран из группы, состоящей из алюминия, кремния, меди, серебра, золота, титана, тантала, вольфрама, молибдена, платины, палладия, циркония или их смесей. Из группы перечисленных выше металлов алюминий, кремний, титан, тантал, вольфрам, молибден и цирконий уменьшают или устраняют коррозионные эффекты активных катализаторов за счет образования пассивирующих поверхность слоев оксидов и слоев на основе оксидов; при этом медь, серебро, золото, платина и палладий уменьшают или устраняют коррозионные эффекты активных катализаторов как металлы (не оксиды) благодаря присущей им устойчивости к коррозии. В другом варианте осуществления настоящего изобретения композиция материала реактора содержит металл, выбранный из группы, состоящей из алюминия, кремния и их смесей. В другом варианте осуществления настоящего изобретения материал внутреннего слоя выбран из группы, состоящей из меди, серебра и золота. В еще одном варианте осуществления настоящего изобретения материал внутреннего слоя выбран из группы, состоящей из титана, тантала, вольфрама, молибдена, платины, палладия, циркония и их смесей. В еще одном варианте осуществления настоящего изобретения материал внутреннего слоя выбран из группы, состоящей из алюминия, кремния, меди, серебра и их смесей. В одном варианте осуществления настоящего изобретения материал внутреннего слоя представляет собой медь. В другом варианте осуществления настоящего изобретения материал внутреннего слоя представляет собой серебро. В другом варианте осуществления настоящего изобретения материал внутреннего слоя представляет собой цирконий. В еще одном варианте осуществления настоящего изобретения материал внутреннего слоя представляет собой титан.
В одном варианте осуществления настоящего изобретения после процесса присоединения происходит окисление внутренней поверхности, когда материал внутреннего слоя выбран из группы, состоящей из алюминия, кремния, титана, тантала, вольфрама, молибдена, циркония и их смесей. В другом варианте осуществления настоящего изобретения после нанесения покрытия погружением происходит окисление внутренней поверхности, когда материал внутреннего слоя выбран из группы, состоящей из алюминия, кремния, титана, тантала, вольфрама, молибдена, циркония и их смесей.
В одном варианте осуществления настоящего изобретения материал наружного слоя выбран из группы, состоящей из углеродистой стали, нержавеющей стали, титана, сплавов на основе Ni и их смесей. В другом варианте осуществления настоящего изобретения материал наружного слоя представляет собой сплав на основе Ni. В еще одном варианте осуществления настоящего изобретения материал наружного слоя выбран из группы, состоящей из нержавеющей стали и углеродистой стали. В еще одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой углеродистую сталь. В одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь.
В одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, а материал внутреннего слоя двухслойного реактора выбран из группы, состоящей из алюминия, кремния и их смесей. В другом варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, а материал внутреннего слоя двухслойного реактора представляет собой алюминий. В еще одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой углеродистую сталь, а материал внутреннего слоя двухслойного реактора выбран из группы, состоящей из алюминия, кремния и их смесей. В еще одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой углеродистую сталь, а материал внутреннего слоя двухслойного реактора представляет собой алюминий.
Не имеющие ограничительного характера примеры нержавеющих сталей включают в себя ферритные нержавеющие стали, мартенситные нержавеющие стали, аустенитные нержавеющие стали и двухфазные нержавеющие стали. Все нержавеющие стали содержат по меньшей мере 10,5 мас. % хрома. Ферритные нержавеющие стали, относящиеся к серии 400, содержат очень мало никеля, и наиболее распространенными среди них являются марки 409, 439, 18Cr-2Mo, 26Cr-1Mo, 29Cr-4Mo и 29Cr-4Mo-2Ni. Мартенситные нержавеющие стали, относящиеся к серии 400, имеют более высокие содержания углерода, чем ферритные стали, и наиболее распространенными среди них являются марки 410 и 420. Марки аустенитных нержавеющих сталей относятся к сериям 200 и 300, составляют более 70% выпускаемой нержавеющей стали, содержат минимум 16 мас. % хрома и от 2 мас. % до 20 мас. % никеля, и наиболее распространенными среди них являются марки 201, 301, 304, 316 и 316L. Наконец, двухфазные нержавеющие стали имеют смешанную микроструктуру аустенитных и ферритных нержавеющих сталей и содержат от 19 мас. % до 32 мас. % хрома, до 5 мас. % молибдена, а содержание никеля в них меньше, чем в аустенитных нержавеющих сталях. Основными производителями марок нержавеющей стали являются компании Sandvik AB (г. Стокгольм, Швеция), Outokumpu Group (г. Эспо, Финляндия), ThyssenKrupp AG (г. Эссен, Германия), Acerinox S.A. (г. Мадрид, Испания) и AK Steel Corp. (г. Вест-Честер, штат Огайо, США).
В одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, а материал внутреннего слоя двухслойного реактора представляет собой медь. В другом варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, а материал внутреннего слоя двухслойного реактора представляет собой серебро. В еще одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, а материал внутреннего слоя двухслойного реактора представляет собой титан. В еще одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, а материал внутреннего слоя двухслойного реактора представляет собой цирконий.
В одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой углеродистую сталь, а материал внутреннего слоя двухслойного реактора представляет собой медь. Углеродистая сталь содержит углерод в диапазоне от 0,12 мас. % до 2 мас. %, медь в диапазоне от 0,4 мас. % до 0,6 мас. %, марганец в количестве не более 1,65 мас. % и кремний в количестве не более 0,6 мас. %. В другом варианте осуществления настоящего изобретения материал наружного слоя представляет собой углеродистую сталь, а материал внутреннего слоя двухслойного реактора представляет собой серебро. В еще одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой углеродистую сталь, а материал внутреннего слоя двухслойного реактора представляет собой титан. В еще одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой углеродистую сталь, а материал внутреннего слоя двухслойного реактора представляет собой цирконий.
В одном варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, материал внутреннего слоя сформирован посредством процесса погружения материала наружного слоя в ванну, содержащую алюминий и кремний, а внутренняя поверхность двухслойного реактора подвергнута окислению и на ней образован пассивирующий поверхность слой на основе оксида, содержащий оксид алюминия и кремнезем. В другом варианте осуществления настоящего изобретения материал наружного слоя представляет собой нержавеющую сталь, материал внутреннего слоя сформирован посредством процесса погружения материала наружного слоя двухслойного реактора в ванну, содержащую алюминий, а внутренняя поверхность двухслойного реактора подвергнута окислению и на ней образован пассивирующий поверхность слой на основе оксида, содержащий кремнезем.
В одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора выбран из группы, состоящей из алюминия, кремния, меди, серебра, золота, титана, тантала, вольфрама, молибдена, платины, палладия, циркония и их смесей. В другом варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора выбран из группы, состоящей из алюминия, кремния, титана, тантала, вольфрама, молибдена, циркония и их смесей. В еще одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора выбран из группы, состоящей из меди, серебра, золота, платины, палладия и их смесей. В еще одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора выбран из группы, состоящей из алюминия, кремния и их смесей. В одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора представляет собой алюминий. В другом варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора представляет собой кремний. В еще одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора выбран из группы, состоящей из алюминия, кремния и их смесей. В еще одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора представляет собой медь. В одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора представляет собой серебро.
В одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора присутствует в материале реактора в количестве более приблизительно 1 мас. %. В другом варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора присутствует в материале реактора в количестве более приблизительно 2 мас. %. В еще одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора присутствует в материале реактора в количестве более приблизительно 3 мас. %. В еще одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора присутствует в материале реактора в количестве более приблизительно 4 мас. %. В одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора присутствует в материале реактора в количестве от приблизительно 1 мас. % до приблизительно 6 мас. %. В другом варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора присутствует в материале реактора в количестве от приблизительно 3 мас. % до приблизительно 5 мас. %.
В одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве более приблизительно 1 мас. %. В другом варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве более приблизительно 2 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве более приблизительно 3 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве более приблизительно 4 мас. %. В одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве от приблизительно 1 мас. % до приблизительно 50 мас. %. В другом варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве от приблизительно 1 мас. % до приблизительно 6 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве от приблизительно 1 мас. % до приблизительно 4 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве от приблизительно 3 мас. % до приблизительно 5 мас. %.
В одном варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве более приблизительно 1 мас. %. В другом варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве более приблизительно 2 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве более приблизительно 3 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве более приблизительно 4 мас. %. В одном варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве от приблизительно 1 мас. % до приблизительно 50 мас. %. В другом варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве от приблизительно 1 мас. % до приблизительно 6 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве от приблизительно 3 мас. % до приблизительно 5 мас. %.
В одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит никель в количестве от приблизительно 50 мас. % до приблизительно 75 мас. %. В одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит хром в количестве от приблизительно 15 мас. % до приблизительно 20 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит железо в количестве от приблизительно 3 мас. % до приблизительно 30 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит никель в количестве от приблизительно 50 мас. % до приблизительно 75 мас. %, хром в количестве от приблизительно 15 мас. % до приблизительно 20 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 30 мас. %. В одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве более приблизительно 1 мас. %, никель в количестве от приблизительно 50 мас. % до приблизительно 75 мас. %, хром в количестве от приблизительно 15 мас. % до приблизительно 20 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 30 мас. %. Не имеющие ограничительного характера примеры имеющихся в продаже материалов стенок с композициями, соответствующими приведенному выше описанию, представляют собой сплавы HAYNES® 214® и HAYNES® HR-224® производства компании Haynes International, Inc. (г. Кокомо, штат Индиана, США).
В одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит никель в количестве приблизительно 60 мас. %. В другом варианте осуществления настоящего изобретения материал стенки дополнительно содержит хром в количестве от приблизительно 20 мас. % до приблизительно 30 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит железо в количестве от приблизительно 3 мас. % до приблизительно 20 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит никель в количестве приблизительно 60 мас. %, хром в количестве от приблизительно 20 мас. % до приблизительно 30 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 20 мас. %. В одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве более приблизительно 1 мас. %, никель в количестве приблизительно 60 мас. %, хром в количестве от приблизительно 20 мас. % до приблизительно 30 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 20 мас. %. Не имеющие ограничительного характера примеры имеющихся в продаже материалов стенок с композициями, соответствующими приведенному выше описанию, представляют собой сплав INCONEL® alloy 693 и сплав INCONEL® alloy 601 производства компании Special Metals Corporation (г. Хантингтон, штат Западная Виргиния, США).
В одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит железо в количестве приблизительно 70 мас. %. В другом варианте осуществления настоящего изобретения материал стенки дополнительно содержит хром в количестве приблизительно 22 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит железо в количестве приблизительно 70 мас. % и хром в количестве приблизительно 22 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит алюминий в количестве более приблизительно 1 мас. %, железо в количестве приблизительно 70 мас. % и хром в количестве приблизительно 22 мас. %. Не имеющие ограничительного характера примеры имеющихся в продаже материалов стенок с композициями, соответствующими приведенному выше описанию, представляют собой сплавы KANTHAL APM и KANTHAL APMT производства компании Sandvik AB (г. Стокгольм, Швеция); и сплав INCOLLOY® MA956 (сплав Fe-Cr-Al) производства компании Special Metals Corporation (г. Хантингтон, штат Западная Виргиния, США).
В одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит никель в количестве от приблизительно 10 мас. % до приблизительно 65 мас. %. В другом варианте осуществления настоящего изобретения материал стенки дополнительно содержит хром в количестве от приблизительно 15 мас. % до приблизительно 30 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки дополнительно содержит железо в количестве от приблизительно 2 мас. % до приблизительно 65 мас. %. В еще одном варианте осуществления настоящего изобретения материал стенки содержит кремний в количестве более приблизительно 1 мас. %, никель в количестве от приблизительно 10 мас. % до приблизительно 65 мас. %, хром в количестве от приблизительно 15 мас. % до приблизительно 30 мас. % и железо в количестве от приблизительно 2 мас. % до приблизительно 65 мас. %. Не имеющие ограничительного характера примеры имеющихся в продаже материалов стенок с композициями, соответствующими приведенному выше описанию, представляют собой сплавы Sandvik SX и Sandvik 253 MA производства компании Sandvik AB (г. Стокгольм, Швеция) и сплавы HAYNES® HR-160® и Hastelloy® D-205 производства компании Haynes International, Inc. (г. Кокомо, штат Индиана, США).
В одном варианте осуществления настоящего изобретения однослойный реактор имеет скорость коррозии менее приблизительно 1,3 мм/год. В другом варианте осуществления настоящего изобретения двухслойный реактор имеет скорость коррозии менее приблизительно 1,3 мм/год. Для целей настоящего изобретения скорость коррозии измеряют с использованием металлографического метода, как показано на ФИГ. 4 (скопировано из брошюры компании Haynes International, Inc.). Более конкретно, в этом методе (известном специалистам в данной области техники) в слой катализатора внедряют металлический испытательный образец и проводят реакцию дегидратации в течение периода TOS (в часах). После завершения реакции дегидратации металлический испытательный образец извлекают из слоя катализатора и подвергают обычному металлографическому анализу, который включает в себя фиксацию образца в термоотверждающейся смоле, отверждение смолы, обтачивание образца для отображения его поверхности, а также анализ образца на сканирующем электронном микроскопе (SEM) для вычисления потери металла (A - B) / 2 в мм, как показано на ФИГ. 4. Затем рассчитывают скорость коррозии (CR) в мм в год (мм/год) по формуле CR = {[(A - B) / 2] * 24 * 365} / TOS, где 24 обозначает число часов в сутках, а 365 обозначает число дней в году. Следует отметить, что эта формула предполагает, что коррозия, наблюдаемая в течение периода TOS (как правило, от нескольких часов до приблизительно 72 часов), линейно экстраполируется на год, что необязательно является точным.
В одном варианте осуществления настоящего изобретения указанная скорость коррозии составляет менее приблизительно 1 мм/год. В другом варианте осуществления настоящего изобретения указанная скорость коррозии составляет менее приблизительно 0,5 мм/год. В еще одном варианте осуществления настоящего изобретения указанная скорость коррозии составляет менее приблизительно 0,13 мм/год. В еще одном варианте осуществления настоящего изобретения указанная скорость коррозии составляет менее приблизительно 0,05 мм/год.
В одном варианте осуществления настоящего изобретения указанный устойчивый к коррозии металл реактора имеет стандартную свободную энергию образования оксидов более приблизительно 700 кДж/моль O2 при температуре приблизительно 400°C. Стандартную свободную энергию образования оксидов можно найти с помощью диаграммы Эллингема, поскольку она хорошо известна специалистам в данной области техники. В другом варианте осуществления настоящего изобретения указанный устойчивый к коррозии металл реактора имеет стандартную свободную энергию образования оксидов более приблизительно 950 кДж/моль O2 при температуре приблизительно 400°C. В еще одном варианте осуществления настоящего изобретения указанный устойчивый к коррозии металл реактора имеет стандартную свободную энергию образования оксидов более приблизительно 1050 кДж/моль O2 при температуре приблизительно 400°C. В еще одном варианте осуществления настоящего изобретения устойчивый к коррозии металл реактора выбран из группы, состоящей из Ca, Mg и их смесей.
В одном варианте осуществления настоящего изобретения указанный устойчивый к коррозии металл реактора имеет электродный потенциал относительно стандартного водородного электрода (SHE) менее приблизительно 0,2 В при температуре приблизительно 25°C, pH менее приблизительно 7 и концентрации окисленных частиц в воде приблизительно 10-6 моль/кг. Электродный потенциал металла реактора можно найти на диаграмме Пурбе для конкретного металла или его можно оценить с помощью уравнения Нернста, поскольку он хорошо известен специалистам в данной области техники. В другом варианте осуществления настоящего изобретения указанный устойчивый к коррозии металл реактора имеет электродный потенциал относительно стандартного водородного электрода (SHE) менее приблизительно 0 В при температуре приблизительно 25°C, pH менее приблизительно 7 и концентрации окисленных частиц в воде приблизительно 10-6 моль/кг. В еще одном варианте осуществления настоящего изобретения указанный устойчивый к коррозии металл реактора имеет электродный потенциал относительно стандартного водородного электрода (SHE) от приблизительно 0,2 В до приблизительно 0,3 В при температуре приблизительно 25°C, pH менее приблизительно 7 и концентрации окисленных частиц в воде приблизительно 10-6 моль/кг.
В одном варианте осуществления настоящего изобретения температура в ходе указанной дегидратации составляет более приблизительно 100°C. В другом варианте осуществления настоящего изобретения температура в ходе указанной дегидратации составляет от приблизительно 120°C до приблизительно 700°C. В еще одном варианте осуществления настоящего изобретения температура в ходе указанной дегидратации составляет от приблизительно 150°C до приблизительно 500°C. В еще одном варианте осуществления настоящего изобретения температура в ходе указанной дегидратации составляет от приблизительно 300°C до приблизительно 450°C. В одном варианте осуществления настоящего изобретения температура в ходе указанной дегидратации составляет от приблизительно 325°C до приблизительно 400°C. В другом варианте осуществления настоящего изобретения температура в ходе указанной дегидратации составляет приблизительно 350°C. В еще одном варианте осуществления настоящего изобретения температура в ходе указанной дегидратации составляет приблизительно 375°C. В еще одном варианте осуществления настоящего изобретения температура во время указанной дегидратации больше температуры тройной точки указанного катализатора или равна ей. В одном варианте осуществления настоящего изобретения температура во время указанной дегидратации по меньшей мере на 10°C выше температуры тройной точки указанного катализатора. В другом варианте осуществления настоящего изобретения температура во время указанной дегидратации по меньшей мере на 50°C выше температуры тройной точки указанного катализатора. В еще одном варианте осуществления настоящего изобретения температура во время указанной дегидратации по меньшей мере на 100°C выше температуры тройной точки указанного катализатора.
В одном варианте осуществления настоящего изобретения указанное парциальное давление водяного пара во время указанной дегидратации превышает или равно приблизительно 0,4 бар. В другом варианте осуществления настоящего изобретения указанное парциальное давление водяного пара во время указанной дегидратации превышает или равно приблизительно 0,8 бар. В еще одном варианте осуществления настоящего изобретения указанное парциальное давление водяного пара во время указанной дегидратации превышает или равно приблизительно 4 бар. В еще одном варианте осуществления настоящего изобретения указанное парциальное давление водяного пара во время указанной дегидратации составляет от приблизительно 5 бар до приблизительно 35 бар. В одном варианте осуществления настоящего изобретения указанное парциальное давление водяного пара во время указанной дегидратации составляет приблизительно 13 бар. В одном варианте осуществления настоящего изобретения парциальное давление водяного пара во время указанной дегидратации больше парциального давления водяного пара в тройной точке указанного катализатора или равно ему. В другом варианте осуществления настоящего изобретения парциальное давление водяного пара во время указанной дегидратации по меньшей мере на 1 бар больше парциального давления водяного пара в тройной точке указанного катализатора. В еще одном варианте осуществления настоящего изобретения парциальное давление водяного пара во время указанной дегидратации по меньшей мере на 2 бар больше парциального давления водяного пара в тройной точке указанного катализатора. В еще одном варианте осуществления настоящего изобретения парциальное давление водяного пара во время указанной дегидратации по меньшей мере на 5 бар больше парциального давления водяного пара в тройной точке указанного катализатора.
Процесс дегидратации можно осуществлять под вакуумом, при атмосферном давлении или при давлении, более высоком по сравнению с атмосферным давлением. В одном варианте осуществления настоящего изобретения процесс дегидратации осуществляют при общем давлении, составляющем по меньшей мере приблизительно 1 бар. В другом варианте осуществления настоящего изобретения процесс дегидратации осуществляют при общем давлении от приблизительно 2 бар до приблизительно 100 бар. В еще одном варианте осуществления настоящего изобретения процесс дегидратации осуществляют при общем давлении от приблизительно 5 бар до приблизительно 40 бар. В еще одном варианте осуществления настоящего изобретения процесс дегидратации осуществляют при общем давлении от приблизительно 10 бар до приблизительно 35 бар. В одном варианте осуществления настоящего изобретения процесс дегидратации осуществляют при общем давлении, составляющем приблизительно 1,6 бар. В другом варианте осуществления настоящего изобретения процесс дегидратации осуществляют при общем давлении, составляющем приблизительно 8 бар. В еще одном варианте осуществления настоящего изобретения процесс дегидратации осуществляют при общем давлении, составляющем приблизительно 26 бар.
В одном варианте осуществления настоящего изобретения указанная GHSV в процессе дегидратации составляет от приблизительно 720 ч-1 до приблизительно 36 000 ч-1. В другом варианте осуществления настоящего изобретения указанная GHSV составляет от приблизительно 1440 ч-1 до приблизительно 18 000 ч-1. В еще одном варианте осуществления настоящего изобретения указанная GHSV составляет от приблизительно 2300 ч-1 до приблизительно 6000 ч-1. В еще одном варианте осуществления настоящего изобретения указанная GHSV составляет от приблизительно 2300 ч-1 до приблизительно 3600 ч-1. В одном варианте осуществления настоящего изобретения указанная GHSV составляет приблизительно 2300 ч-1. В другом варианте осуществления настоящего изобретения указанная GHSV составляет приблизительно 3600 ч-1.
В одном варианте осуществления настоящего изобретения указанная WHSV составляет от приблизительно 0,02 ч-1 до приблизительно 10 ч-1. В другом варианте осуществления настоящего изобретения указанная WHSV составляет от приблизительно 0,2 ч-1 до приблизительно 2 ч-1. В еще одном варианте осуществления настоящего изобретения указанная WHSV составляет от приблизительно 0,3 ч-1 до приблизительно 1,4 ч-1. В еще одном варианте осуществления настоящего изобретения указанная WHSV составляет от приблизительно 0,3 ч-1 до приблизительно 0,4 ч-1. В одном варианте осуществления настоящего изобретения указанная WHSV составляет приблизительно 0,4 ч-1.
В контексте настоящего изобретения термин «приведение в контакт» относится к действию, приводящему указанный поток поступающих газообразных материалов, содержащий водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, в непосредственную близость к поверхности указанного катализатора. Гидроксипропионовая кислота, производные гидроксипропионовой кислоты или их смеси должны находиться в контакте с поверхностью катализатора на скорости, достаточно низкой для завершения реакции дегидратации, но достаточно высокой для предотвращения разложения гидроксипропионовой кислоты, акриловой кислоты или их производных до нежелательных продуктов при температуре указанной стадии приведения в контакт. Для описания скорости указанной стадии приведения в контакт можно применять несколько параметров, такие как, например, без ограничений, WHSV, GHSV, LHSV и массовый расход на единицу доступной площади поверхности катализатора (WVSA), которую можно рассчитывать как отношение WHSV к удельной площади поверхности катализатора (SA), (WVSA = WHSV/SA); причем в единицах г/м2⋅ч.; где г относится к г гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей. Можно применять ряд способов, основанных на поглощении инертного газа, для определения доступной площади поверхности, включая, без ограничений, статические способы объемного и гравиметрического определения и динамический способ, которые хорошо известны специалистам в данной области техники.
В одном варианте осуществления настоящего изобретения гидроксипропионовая кислота, производные гидроксипропионовой кислоты или их смеси находятся в контакте с катализатором при WVSA, составляющей от приблизительно 10-4 г∙м-2⋅ч-1 до приблизительно 104 г⋅м-2⋅ч-1. В другом варианте осуществления настоящего изобретения гидроксипропионовая кислота, производные гидроксипропионовой кислоты или их смеси находятся в контакте с катализатором при WVSA, составляющей от приблизительно 10-2 г⋅м-2⋅ч-1 до приблизительно 102 г⋅м-2⋅ч-1. В еще одном варианте осуществления настоящего изобретения гидроксипропионовая кислота, производные гидроксипропионовой кислоты или их смеси находятся в контакте с катализатором при WVSA, составляющей от приблизительно 0,1 г⋅м-2⋅ч-1 до приблизительно 10 г⋅м-2⋅ч-1.
В одном варианте осуществления настоящего изобретения указанная гидроксипропионовая кислота представляет собой молочную кислоту; причем указанный поток поступающих газообразных материалов содержит разбавитель; при этом указанный разбавитель состоит по существу из азота; причем указанная температура составляет от приблизительно 300°C до приблизительно 450°C; при этом указанное парциальное давление водяного пара превышает или равно 0,8 бар; и причем указанная GHSV находится в диапазоне от приблизительно 2300 ч-1 до приблизительно 3600 ч-1, а указанная WHSV находится в диапазоне от приблизительно 0,2 ч-1 до приблизительно 2 ч-1.
В одном варианте осуществления настоящего изобретения TOS составляет более приблизительно 2 ч. В другом варианте осуществления настоящего изобретения TOS составляет от приблизительно 2 ч до приблизительно 24 ч. В еще одном варианте осуществления настоящего изобретения TOS составляет от приблизительно 24 ч до приблизительно 48 ч. В еще одном варианте осуществления настоящего изобретения TOS составляет от приблизительно 24 ч до приблизительно 72 ч. В одном варианте осуществления настоящего изобретения TOS составляет приблизительно 72 ч. В другом варианте осуществления настоящего изобретения TOS составляет более приблизительно 1000 ч. В еще одном варианте осуществления настоящего изобретения TOS составляет приблизительно 1 год.
В одном варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты, или их смеси производят с выходом по меньшей мере 50 мол. %. В другом варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 70%. В еще одном варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80 мол. %.
В одном варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с избирательностью по меньшей мере приблизительно 50 мол. %. В другом варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с избирательностью по меньшей мере приблизительно 70 мол. %. В еще одном варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с избирательностью по меньшей мере приблизительно 80 мол. %.
В одном варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 70 мол. %, и с избирательностью по меньшей мере приблизительно 70 мол. %. В другом варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80 мол. %, и с избирательностью по меньшей мере приблизительно 80 мол. %.
В одном варианте осуществления настоящего изобретения пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 5 мол. %. В другом варианте осуществления настоящего изобретения пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 1 мол. %.
В одном варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 70 мол. % и с избирательностью по меньшей мере приблизительно 70 мол. % в течение TOS приблизительно 72 ч; причем пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 5 мол. % в течение указанного TOS приблизительно 72 ч. В другом варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80 мол. % и с избирательностью по меньшей мере приблизительно 80 мол. % в течение TOS приблизительно 72 ч; причем пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 1 мол. % в течение указанного TOS приблизительно 72 ч.
В одном варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с превращением гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей, составляющим более приблизительно 50 мол. %. В другом варианте осуществления настоящего изобретения указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с превращением гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей, составляющим более приблизительно 80 мол. %.
В одном варианте осуществления настоящего изобретения уксусную кислоту, пировиноградную кислоту, 1,2-пропандиол, гидроксиацетон, димер акриловой кислоты и 2,3-пентандион производят вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями с выходом менее приблизительно 2 мол. % каждый. В другом варианте осуществления настоящего изобретения уксусную кислоту, пировиноградную кислоту, 1,2-пропандиол, гидроксиацетон, димер акриловой кислоты и 2,3-пентандион производят вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями с выходом менее приблизительно 0,5 мол. % каждый. В еще одном варианте осуществления настоящего изобретения ацетальдегид производят вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями с выходом менее приблизительно 8 мол. %. В еще одном варианте осуществления настоящего изобретения ацетальдегид производят вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями с выходом менее приблизительно 4 мол. %. В одном варианте осуществления настоящего изобретения ацетальдегид производят вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями с выходом менее приблизительно 3 мол. %.
В одном варианте осуществления настоящего изобретения указанная фосфатная соль является кристаллической; причем указанное значение x составляет 1; при этом указанный катион представляет собой K+; причем указанный материал стенки дополнительно содержит никель в количестве от приблизительно 50 мас. % до приблизительно 75 мас. %, хром в количестве от приблизительно 15 мас. % до приблизительно 20 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 30 мас. %; при этом указанное количество указанного алюминия составляет от приблизительно 3 мас. % до приблизительно 5 мас. %; причем указанная гидроксипропионовая кислота представляет собой молочную кислоту; при этом указанная температура составляет приблизительно 375°C, а указанное парциальное давление водяного пара составляет приблизительно 13 бар; причем указанная GHSV составляет приблизительно 2300 ч-1, а указанная WHSV составляет от приблизительно 0,3 ч-1 до приблизительно 0,4 ч-1; при этом указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80% и с избирательностью по меньшей мере приблизительно 80% в течение TOS приблизительно 72 ч; причем пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 1 мол. % в течение указанного TOS приблизительно 72 ч.
В другом варианте осуществления настоящего изобретения указанная фосфатная соль является кристаллической; причем указанное значение x составляет 1; при этом указанный катион представляет собой Cs+; причем указанный материал стенки дополнительно содержит никель в количестве от приблизительно 50 мас. % до приблизительно 75 мас. %, хром в количестве от приблизительно 15 мас. % до приблизительно 20 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 30 мас. %; при этом указанное количество указанного алюминия составляет от приблизительно 3 мас. % до приблизительно 5 мас. %; причем указанная гидроксипропионовая кислота представляет собой молочную кислоту; при этом указанная температура составляет приблизительно 375°C, а указанное парциальное давление водяного пара составляет приблизительно 13 бар; причем указанная GHSV составляет приблизительно 2300 ч-1, а указанная WHSV составляет от приблизительно 0,3 ч-1 до приблизительно 0,4 ч-1; при этом указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80% и с избирательностью по меньшей мере приблизительно 80% в течение TOS приблизительно 72 ч; причем пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 1 мол. % в течение указанного TOS приблизительно 72 ч.
В еще одном варианте осуществления настоящего изобретения: указанная фосфатная соль является кристаллической; причем указанное значение x составляет 1; при этом указанный катион представляет собой K+; причем указанный материал стенки дополнительно содержит никель в количестве приблизительно 60 мас. %, хром в количестве от приблизительно 20 мас. % до приблизительно 30 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 20 мас. %; при этом указанное количество указанного алюминия составляет от приблизительно 1 мас. % до приблизительно 4 мас. %; причем указанная гидроксипропионовая кислота представляет собой молочную кислоту; при этом указанная температура составляет приблизительно 375°C, а указанное парциальное давление водяного пара составляет приблизительно 13 бар; причем указанная GHSV составляет приблизительно 2300 ч-1, а указанная WHSV составляет от приблизительно 0,3 ч-1 до приблизительно 0,4 ч-1; при этом указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80% и с избирательностью по меньшей мере приблизительно 80% в течение TOS приблизительно 72 ч; при этом указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80 мол. % и с избирательностью по меньшей мере приблизительно 80 мол. % в течение TOS приблизительно 72 ч; причем пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 1 мол. % в течение указанного TOS приблизительно 72 ч.
В еще одном варианте осуществления настоящего изобретения: указанная фосфатная соль является кристаллической; причем указанное значение x составляет 1; при этом указанный катион представляет собой Cs+; причем указанный материал стенки дополнительно содержит никель в количестве приблизительно 60 мас. %, хром в количестве от приблизительно 20 мас. % до приблизительно 30 мас. % и железо в количестве от приблизительно 3 мас. % до приблизительно 20 мас. %; при этом указанное количество указанного алюминия составляет от приблизительно 1 мас. % до приблизительно 4 мас. %; причем указанная гидроксипропионовая кислота представляет собой молочную кислоту; при этом указанная температура составляет приблизительно 375°C, а указанное парциальное давление водяного пара составляет приблизительно 13 бар; причем указанная GHSV составляет приблизительно 2300 ч-1, а указанная WHSV составляет от приблизительно 0,3 ч-1 до приблизительно 0,4 ч-1; при этом указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80% и с избирательностью по меньшей мере приблизительно 80% в течение TOS приблизительно 72 ч; при этом указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80 мол. % и с избирательностью по меньшей мере приблизительно 80 мол. % в течение TOS приблизительно 72 ч; причем пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 1 мол. % в течение указанного TOS приблизительно 72 ч.
В одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; причем указанный катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой [H2(1-x)PO(4-x)]¯; где x представляет собой любое действительное число, которое больше или равно 0 и меньше или равно 1; причем указанное парциальное давление водяного пара превышает или равно 0,4 бар; при этом указанный однослойный реактор содержит стенку, наружную поверхность и внутреннюю поверхность; причем указанная стенка изготовлена из материала стенки, имеет толщину стенки и проходит от указанной наружной поверхности к указанной внутренней поверхности; при этом указанный материал стенки содержит алюминий в количестве более приблизительно 1 мас. %; причем указанная внутренняя поверхность находится в контакте с указанным катализатором; при этом скорость коррозии указанного однослойного реактора во время указанной дегидратации составляет менее приблизительно 1,3 мм/год; и причем в результате указанной дегидратации в указанном однослойном реакторе получают акриловую кислоту, производные акриловой кислоты или их смеси.
В другом варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и молочную кислоту, производные молочной кислоты или их смеси, с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации молочной кислоты, производных молочной кислоты или их смесей; причем указанный катализатор содержит фосфатную соль, содержащую одновалентный катион и анион, представленные эмпирической формулой [H2(1-x)PO(4-x)]¯; где x представляет собой любое действительное число, которое больше или равно 0 и меньше или равно 1; при этом указанный катион выбран из группы, состоящей из K+, Cs+ и их смесей; причем указанная температура составляет от приблизительно 300°C до приблизительно 450°C; причем указанное парциальное давление водяного пара превышает или равно 0,4 бар; при этом указанная GHSV составляет приблизительно 2300 ч-1; причем указанная WHSV составляет от приблизительно 0,3 ч-1 до приблизительно 0,4 ч-1; при этом указанный однослойный реактор содержит стенку, наружную поверхность и внутреннюю поверхность; причем указанная стенка изготовлена из материала стенки, имеет толщину стенки и проходит от указанной наружной поверхности к указанной внутренней поверхности; при этом указанный материал стенки содержит алюминий в количестве более приблизительно 1 мас. %; причем указанная внутренняя поверхность находится в контакте с указанным катализатором; при этом скорость коррозии указанного однослойного реактора во время указанной дегидратации составляет менее приблизительно 1,3 мм/год; при этом указанную акриловую кислоту, производные акриловой кислоты или их смеси производят с выходом по меньшей мере приблизительно 80 мол. % и с избирательностью по меньшей мере приблизительно 80 мол. % в течение TOS приблизительно 72 ч; причем пропионовую кислоту получают в виде примеси вместе с указанной акриловой кислотой, производными акриловой кислоты или их смесями; и при этом избирательность для указанной пропионовой кислоты составляет менее приблизительно 1 мол. % в течение указанного TOS приблизительно 72 ч.
В еще одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; причем указанный катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой [H2(1-x)PO(4-x)]¯; где x представляет собой любое действительное число, которое больше или равно 0 и меньше или равно 1; причем указанное парциальное давление водяного пара превышает или равно 0,4 бар; при этом указанный однослойный реактор содержит стенку, наружную поверхность и внутреннюю поверхность; причем указанная стенка изготовлена из материала стенки, имеет толщину стенки и проходит от указанной наружной поверхности к указанной внутренней поверхности; при этом указанный материал стенки содержит кремний в количестве более приблизительно 1 мас. %; причем указанная внутренняя поверхность находится в контакте с указанным катализатором; при этом скорость коррозии указанного однослойного реактора во время указанной дегидратации составляет менее приблизительно 1,3 мм/год; и причем в результате указанной дегидратации в указанном однослойном реакторе получают акриловую кислоту, производные акриловой кислоты или их смеси.
В еще одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает приведение в контакт потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, с катализатором в двухслойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; причем указанный катализатор содержит фосфатную соль, содержащую катион и анион, представленные эмпирической формулой [H2(1-x)PO(4-x)]¯; где x представляет собой любое действительное число, которое больше или равно 0 и меньше или равно 1; причем указанное парциальное давление водяного пара превышает или равно 0,4 бар; при этом указанный двухслойный реактор содержит наружный слой, внутренний слой, наружную поверхность, внутреннюю поверхность и границу раздела между указанным наружным слоем и указанным внутренним слоем; причем указанный наружный слой изготовлен из материала наружного слоя, имеет толщину наружного слоя и отходит от указанной границы к указанной наружной поверхности; при этом указанный внутренний слой изготовлен из материала внутреннего слоя, имеет толщину внутреннего слоя и отходит от указанной внутренней поверхности к указанной границе; причем указанный материал внутреннего слоя выбран из группы, состоящей из алюминия, кремния, меди, серебра, золота, титана, тантала, вольфрама, молибдена, платины, палладия, циркония и их смесей; причем указанная внутренняя поверхность находится в контакте с указанным катализатором; при этом скорость коррозии указанного двухслойного реактора во время указанной дегидратации составляет менее приблизительно 1,3 мм/год; и при этом в результате указанной дегидратации в указанном двухслойном реакторе получают акриловую кислоту, производные акриловой кислоты или их смеси.
В одном варианте осуществления настоящего изобретения указанный поток поступающих газообразных материалов, содержащий водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, получают посредством приведения потока поступающих жидких материалов, содержащего гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, в контакт с водяным паром. В другом варианте осуществления настоящего изобретения указанный поток поступающих газообразных материалов, содержащий водяной пар и молочную кислоту, производные молочной кислоты или их смеси, получают посредством приведения потока поступающих жидких материалов, содержащего молочную кислоту, производные молочной кислоты или их смеси, в контакт с водяным паром. В еще одном варианте осуществления настоящего изобретения указанный поток поступающих жидких материалов, содержащий гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, может дополнительно содержать одну или более по существу химически инертных жидкостей. Не имеющие ограничительного характера примеры по существу химически инертных жидкостей представляют собой воду, углеводороды, хлорированные углеводороды, фторированные углеводороды, бромированные углеводороды, сложные эфиры, простые эфиры, кетоны, альдегиды, кислоты, спирты или их смеси. Не имеющие ограничительного характера примеры углеводородов представляют собой линейные и разветвленные C5-C8 алканы. Не имеющий ограничительного характера пример сложных эфиров представляет собой этилацетат. Не имеющий ограничительного характера пример эфиров представляет собой дифениловый эфир. Не имеющий ограничительного характера пример кетонов представляет собой ацетон. Не имеющие ограничительного характера примеры спиртов представляют собой метанол, этанол и линейные и разветвленные C3-C8 спирты. В одном варианте осуществления настоящего изобретения указанные одна или более по существу химически инертных жидкостей содержат воду. В одном варианте осуществления настоящего изобретения указанные одна или более по существу химически инертных жидкостей состоят по существу из воды.
В одном варианте осуществления настоящего изобретения поток поступающих жидких материалов, содержащий воду и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, подают в испаритель, расположенный перед каталитическим реактором, для по меньшей мере частичного превращения потока поступающих жидких материалов в поток поступающих газообразных материалов, содержащий водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, перед приведением в контакт с указанным катализатором. В другом варианте осуществления настоящего изобретения поток поступающих жидких материалов, содержащий гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, подают напрямую в каталитический реактор и приводят в контакт с указанным катализатором. В еще одном варианте осуществления настоящего изобретения по существу химически инертный газ или по существу химически инертную жидкость подают в испаритель или в каталитический реактор. Поток поступающих жидких материалов, содержащий гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси и по существу химически инертный газ или по существу химически инертную жидкость, можно подавать в указанный испаритель или указанный каталитический реактор вместе или по отдельности. Не имеющие ограничительного характера примеры по существу химически инертных газов представляют собой азот, гелий, воздух, аргон, диоксид углерода, монооксид углерода, водяные пары и их смеси. Не имеющие ограничительного характера примеры по существу химически инертных жидкостей представляют собой воду, углеводороды, хлорированные углеводороды, фторированные углеводороды, бромированные углеводороды, сложные эфиры, простые эфиры, кетоны, альдегиды, кислоты, спирты или их смеси.
В одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает: a) обеспечение потока поступающих жидких материалов, содержащего гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси; b) необязательно объединение указанного потока поступающих жидких материалов с по существу химически инертным газом для образования смеси жидкость/газ; и c) приведение указанного потока поступающих жидких материалов или указанной смеси жидкость/газ в контакт с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения при парциальном давлении водяного пара, составляющем приблизительно 0,4 бар или более, для получения указанных акриловой кислоты, производных акриловой кислоты или их смесей.
В другом варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает: a) обеспечение потока поступающих жидких материалов, содержащего водный раствор гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; b) необязательно объединение указанного потока поступающих жидких материалов с по существу химически инертным газом для образования смеси жидкость/газ; и c) приведение указанного потока поступающих жидких материалов или указанной смеси жидкость/газ в контакт с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения при парциальном давлении водяного пара, составляющем приблизительно 0,4 бар или более, для получения указанных акриловой кислоты, производных акриловой кислоты или их смесей.
В еще одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает: a) получение потока поступающих жидких материалов, содержащего водный раствор гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; b) необязательно объединение указанного потока поступающих жидких материалов с по существу химически инертным газом для образования смеси жидкость/газ; c) испарение указанного потока поступающих жидких материалов или указанной смеси жидкость/газ для получения потока поступающих газообразных материалов; и d) приведение указанного потока поступающих газообразных материалов с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения при парциальном давлении водяного пара, составляющем приблизительно 0,4 бар или более, для получения указанных акриловой кислоты, производных акриловой кислоты или их смесей.
В еще одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает: a) обеспечение потока поступающих жидких материалов, содержащего водный раствор гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; причем гидроксипропионовая кислота находится в водном растворе по существу в мономерной форме; b) необязательно объединение указанного потока поступающих жидких материалов с по существу химически инертным газом для образования смеси жидкость/газ; c) испарение указанного потока поступающих жидких материалов или указанной смеси жидкость/газ для получения потока поступающих газообразных материалов; и d) приведение указанного потока поступающих газообразных материалов с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения при парциальном давлении водяного пара, составляющем приблизительно 0,4 бар или более, для получения указанных акриловой кислоты, производных акриловой кислоты или их смесей.
В одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает: a) обеспечение потока поступающих жидких материалов, содержащего водный раствор гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; причем гидроксипропионовая кислота находится в водном растворе по существу в мономерной форме и при этом гидроксипропионовая кислота, производные гидроксипропионовой кислоты или их смеси составляют от приблизительно 10 мас. % до приблизительно 25 мас. % водного раствора; b) необязательно объединение указанного потока поступающих жидких материалов с по существу химически инертным газом для образования смеси жидкость/газ; c) испарение указанного потока поступающих жидких материалов или указанной смеси жидкость/газ для получения потока поступающих газообразных материалов; и d) приведение указанного потока поступающих газообразных материалов с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения при парциальном давлении водяного пара, составляющем приблизительно 0,4 бар или более, для получения указанных акриловой кислоты, производных акриловой кислоты или их смесей.
В другом варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает: a) обеспечение потока поступающих жидких материалов, содержащего водный раствор гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; причем в водном растворе гидроксипропионовой кислоты содержатся ее олигомеры; b) нагревание указанного потока поступающих жидких материалов при температуре от приблизительно 50°C до приблизительно 100°C для обеспечения гидролиза олигомеров гидроксипропионовой кислоты и получения потока поступающих жидких материалов, содержащего мономерную гидроксипропионовую кислоту; c) необязательно объединение указанного потока поступающих жидких материалов, содержащего мономерную гидроксипропионовую кислоту, с по существу химически инертным газом для образования смеси жидкость/газ; d) испарение указанного потока поступающих жидких материалов, содержащего мономерную гидроксипропионовую кислоту, или указанной смеси жидкость/газ для получения потока поступающих газообразных материалов; и e) приведение указанного потока поступающих газообразных материалов с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения при парциальном давлении водяного пара, составляющем приблизительно 0,4 бар или более, для получения указанных акриловой кислоты, производных акриловой кислоты или их смесей.
В еще одном варианте осуществления настоящего изобретения способ получения акриловой кислоты, производных акриловой кислоты или их смесей включает: a) обеспечение потока поступающих жидких материалов, содержащего водный раствор гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей; b) необязательно объединение потока поступающих жидких материалов с по существу химически инертным газом для образования смеси жидкость/газ; c) испарение указанного потока поступающих жидких материалов или указанной смеси жидкость/газ для получения потока поступающих газообразных материалов; d) приведение указанного потока поступающих газообразных материалов с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных»), или любым предшествующим катализатором, описанным в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения при парциальном давлении водяного пара, составляющем приблизительно 0,4 бар или более, для получения потока акриловой кислоты; и e) охлаждение указанного потока акриловой кислоты для получения потока жидкой акриловой кислоты, содержащего указанную акриловую кислоту, производные акриловой кислоты или их смеси.
В одном варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих жидких материалов составляет от приблизительно 2 мас. % до приблизительно 95 мас. %. В другом варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих жидких материалов составляет от приблизительно 5 мас. % до приблизительно 60 мас. %. В еще одном варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих жидких материалов составляет от приблизительно 10 мас. % до приблизительно 40 мас. %. В еще одном варианте осуществления настоящего изобретения концентрация гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей в указанном потоке поступающих жидких материалов составляет приблизительно 20% мас.
В одном варианте осуществления настоящего изобретения поток поступающих жидких материалов содержит водный раствор гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей. В другом варианте осуществления настоящего изобретения поток поступающих жидких материалов содержит водный раствор молочной кислоты, производных молочной кислоты или их смесей. В еще одном варианте осуществления настоящего изобретения указанные производные молочной кислоты в указанном водном растворе выбраны из группы, состоящей из солей металлов или аммония молочной кислоты, алкиловых сложных эфиров молочной кислоты, олигомеров молочной кислоты, циклических сложных диэфиров молочной кислоты, ангидрида молочной кислоты, 2-алкоксипропионовых кислот или их алкиловых сложных эфиров, 2-арилоксипропионовых кислот или их алкиловых сложных эфиров, 2-ацилоксипропионовых кислот или их алкиловых сложных эфиров или их смесей.
В одном варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном водном растворе составляет от приблизительно 2 мас. % до приблизительно 95 мас. %. В другом варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном водном растворе составляет от приблизительно 5 мас. % до приблизительно 60 мас. %. В еще одном варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном водном растворе составляет от приблизительно 10 мас. % до приблизительно 40 мас. %. В еще одном варианте осуществления настоящего изобретения концентрация молочной кислоты, производных молочной кислоты или их смесей в указанном водном растворе составляет приблизительно 20 мас. %. В одном варианте осуществления настоящего изобретения поток поступающих жидких материалов содержит водный раствор молочной кислоты и производных молочной кислоты. В другом варианте осуществления настоящего изобретения поток поступающих жидких материалов содержит менее приблизительно 30 мас. % производных молочной кислоты в расчете на общую массу потока поступающих жидких материалов. В еще одном варианте осуществления настоящего изобретения поток поступающих жидких материалов содержит менее приблизительно 10 мас. % производных молочной кислоты в расчете на общую массу потока поступающих жидких материалов. В еще одном варианте осуществления настоящего изобретения поток поступающих жидких материалов содержит менее приблизительно 5 мас. % производных молочной кислоты в расчете на общую массу потока поступающих жидких материалов.
Молочная кислота может быть представлена в мономерной форме или в виде олигомеров в указанном водном растворе молочной кислоты, производных молочной кислоты или их смесей. В одном варианте осуществления настоящего изобретения олигомеры молочной кислоты в указанном водном растворе молочной кислоты, производных молочной кислоты или их смесей составляют менее приблизительно 30 мас. % в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей. В другом варианте осуществления настоящего изобретения олигомеры молочной кислоты в указанном водном растворе молочной кислоты, производных молочной кислоты или их смесей составляют менее приблизительно 10 мас. % в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей. В еще одном варианте осуществления настоящего изобретения олигомеры молочной кислоты в указанном водном растворе молочной кислоты, производных молочной кислоты или их смесей составляют менее приблизительно 5 мас. % в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей. В еще одном варианте осуществления настоящего изобретения в указанном водном растворе молочной кислоты, производных молочной кислоты или их смесей молочная кислота представлена по существу в мономерной форме.
Способ для удаления олигомеров из водного раствора молочной кислоты, производных молочной кислоты и их смесей может включать стадию очистки или гидролиз, вызванный стадией нагревания. В одном варианте осуществления настоящего изобретения стадия нагревания может включать нагревание водного раствора молочной кислоты, производных молочной кислоты или их смесей при температуре от приблизительно 50°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты. В другом варианте осуществления настоящего изобретения стадия нагревания может включать нагревание водного раствора молочной кислоты, производных молочной кислоты или их смесей при температуре от приблизительно 95°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты. В еще одном варианте осуществления настоящего изобретения стадия нагревания может включать нагревание водного раствора молочной кислоты, производных молочной кислоты или их смесей при температуре от приблизительно 50°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты и для получения водного раствора на основе мономерной молочной кислоты, содержащего по меньшей мере 80 мас. % молочной кислоты в мономерной форме в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей. В еще одном варианте осуществления настоящего изобретения стадия нагревания может включать нагревание водного раствора молочной кислоты, производных молочной кислоты или их смесей при температуре от приблизительно 50°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты и для получения водного раствора на основе мономерной молочной кислоты, содержащего по меньшей мере 95 мас. % молочной кислоты в мономерной форме в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей. В одном варианте осуществления настоящего изобретения приблизительно 88 мас. % водного раствора молочной кислоты, производных молочной кислоты или их смесей разбавляют водой и гидролизируют олигомеры для произведения водного раствора на основе приблизительно 20 мас. % молочной кислоты. Возможно снижение избирательности в отношении акриловой кислоты из-за олигомеров молочной кислоты, что связано с их высокой температурой кипения. По мере снижения содержания воды в водном растворе потеря материалов, поступающих на каталитическую реакцию, увеличивается из-за потерь на стадии выпаривания. Кроме того, из-за олигомеров молочной кислоты может происходить спекание, деактивация катализатора и засорение реактора.
В другом варианте осуществления настоящего изобретения поток поступающих жидких материалов, содержащий гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, может дополнительно содержать один или более антиоксидантов. В другом варианте осуществления настоящего изобретения поток поступающих жидких материалов, содержащий гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, дополнительно содержит бутилированный гидрокситолуол (BHT), бутилированный гидроксианизол (BHA) или их смеси. В еще одном варианте осуществления настоящего изобретения поток поступающих жидких материалов, содержащий гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, дополнительно содержит этиленгликоль, этандитиол, метанол, метантиол или их смеси.
Поток поступающих жидких материалов можно вводить в испаритель или в каталитический реактор с помощью простой трубы или через распылительные форсунки. Не имеющие ограничительного характера примеры распылительных форсунок включают веерные форсунки, работающие под давлением вихревые форсунки, форсунки с аэродинамическим распылом, пневмораспылители, ротационные форсунки и форсунки с диоксидом углерода в сверхкритическом состоянии. В одном варианте осуществления настоящего изобретения диаметр капель водного раствора составляет менее приблизительно 500 мкм. В другом варианте осуществления настоящего изобретения диаметр капель водного раствора составляет менее приблизительно 200 мкм. В еще одном варианте осуществления настоящего изобретения диаметр капель водного раствора составляет менее приблизительно 100 мкм.
На стадии выпаривания указанный поток поступающих жидких материалов или указанную смесь жидкость/газ нагревают для получения потока газообразных поступающих материалов. В одном варианте осуществления настоящего изобретения температура во время стадии выпаривания составляет от приблизительно 165°C до приблизительно 450°C. В другом варианте осуществления настоящего изобретения температура во время стадии выпаривания составляет от приблизительно 200°C до приблизительно 400°C. В еще одном варианте осуществления настоящего изобретения температура во время стадии выпаривания составляет от приблизительно 250°C до приблизительно 375°C. В еще одном варианте осуществления настоящего изобретения температура во время стадии выпаривания составляет от приблизительно 300°C до приблизительно 375°C. В одном варианте осуществления настоящего изобретения температура во время стадии выпаривания составляет приблизительно 250°C. В другом варианте осуществления настоящего изобретения температура во время стадии выпаривания составляет приблизительно 375°C.
В одном варианте осуществления настоящего изобретения время пребывания в испарителе во время указанной стадии выпаривания составляет от приблизительно 0,2 с до приблизительно 10 с. В другом варианте осуществления настоящего изобретения время пребывания в испарителе во время указанной стадии выпаривания составляет от приблизительно 0,5 с до приблизительно 5 с. В еще одном варианте осуществления настоящего изобретения время пребывания в испарителе во время указанной стадии выпаривания составляет от приблизительно 1 с до приблизительно 3 с. В еще одном варианте осуществления настоящего изобретения время пребывания в испарителе во время указанной стадии выпаривания составляет от приблизительно 0,5 с до приблизительно 0,6 с.
Стадию выпаривания можно осуществлять под вакуумом, при атмосферном давлении или при давлении, более высоком по сравнению с атмосферным давлением. В одном варианте осуществления настоящего изобретения стадию выпаривания осуществляют при общем давлении, составляющем по меньшей мере приблизительно 1 бар. В другом варианте осуществления настоящего изобретения стадию выпаривания осуществляют при общем давлении, составляющем от приблизительно 2 бар до приблизительно 100 бар. В еще одном варианте осуществления настоящего изобретения стадию выпаривания осуществляют при давлении, составляющем от приблизительно 5 бар до приблизительно 40 бар. В еще одном варианте осуществления настоящего изобретения стадию выпаривания осуществляют при общем давлении, составляющем от приблизительно 10 бар до приблизительно 35 бар. В одном варианте осуществления настоящего изобретения стадию выпаривания осуществляют при общем давлении, составляющем приблизительно 1,6 бар. В одном другом варианте осуществления настоящего изобретения стадию выпаривания осуществляют при общем давлении, составляющем приблизительно 8 бар. В еще одном варианте осуществления настоящего изобретения стадию выпаривания осуществляют при общем давлении, составляющем приблизительно 26 бар.
Стадию выпаривания можно осуществлять в испарителях различных типов, таких как, без ограничений, распылитель, пластинчатый теплообменник, пустой проточный реактор и проточный реактор с неподвижным слоем катализатора. Стадию выпаривания можно осуществлять в испарителе, в котором поток поступающих жидких материалов протекает с направлением сверху вниз, снизу вверх или горизонтально. В одном варианте осуществления настоящего изобретения стадию выпаривания осуществляют в испарителе, в котором поток поступающих жидких материалов протекает с направлением сверху вниз. Также стадию выпаривания можно выполнять в периодическом режиме.
В одном варианте осуществления настоящего изобретения материал внутренней поверхности испарителя выбран из группы, состоящей из аморфного кремнезема, кварца, других оксидов кремния, боросиликатного стекла, кремния и их смесей. В еще одном варианте осуществления настоящего изобретения материал внутренней поверхности испарителя представляет собой аморфный кремнезем или боросиликатное стекло.
В одном варианте осуществления настоящего изобретения стадии выпаривания и дегидратации объединяют в одну стадию. В другом варианте осуществления настоящего изобретения стадии выпаривания и дегидратации осуществляют последовательно в одном реакторе. В еще одном варианте осуществления настоящего изобретения стадии выпаривания и дегидратации осуществляют последовательно в тандемном реакторе.
Поток акриловой кислоты, полученной при указанной дегидратации, охлаждают для получения потока жидкой акриловой кислоты в качестве потока продукта. Следует регулировать время, необходимое для охлаждения потока акриловой кислоты, для устранения полимеризации акриловой кислоты или ее разложения до этилена. В одном варианте осуществления настоящего изобретения время пребывания потока акриловой кислоты на стадии охлаждения составляет менее приблизительно 30 с. В другом варианте осуществления настоящего изобретения время пребывания потока продукта на стадии охлаждения составляет от приблизительно 0,1 с до приблизительно 10 с.
Поток жидкой акриловой кислоты, содержащий акриловую кислоту, производные акриловой кислоты или их смеси, полученный в соответствии с настоящим изобретением, можно очищать с применением некоторых или всех способов экстракции, высушивания, дистилляции, охлаждения, частичного плавления и декантирования, описанных в документе US 20130274518 A1 или US 20150329462 A1 (включенном в данный документ посредством ссылки), для получения сырой и ледяной акриловой кислоты. После очистки можно полимеризовать сырую и ледяную акриловую кислоту для получения суперабсорбирующего полимера с применением способов, которые аналогичны таковым, описанным в документе US 20130274697 A1 или US 20130273384 A1 (включенном в данный документ посредством ссылки).
В одном варианте осуществления настоящего изобретения указанную сырую акриловую кислоту этерифицируют спиртом для получения акрилатного мономера. Не имеющие ограничительного характера примеры спиртов представляют собой метанол, этанол, бутанол (н-бутиловый спирт), 2-этилгексанол, изобутанол, трет-бутиловый спирт, гексиловый спирт, октиловый спирт, изооктиловый спирт, лауриловый спирт, пропиловый спирт, изопропиловый спирт, гидроксиэтиловый спирт, гидроксипропиловый спирт и полиолы, такие как гидроксиалкил и алкилалканоламин. В другом варианте осуществления настоящего изобретения указанную сырую акриловую кислоту этерифицируют метанолом, этанолом, н-бутиловым спиртом или 2-этилгексанолом для получения метилакрилатного мономера, этилакрилатного мономера, н-бутилакрилатного мономера или 2-этилгексилакрилатного мономера соответственно. В еще одном варианте осуществления настоящего изобретения указанные метилакрилатный мономер, этилакрилатный мономер, н-бутилакрилатный мономер или 2-этилгексилакрилатный мономер полимеризуют для получения метилакрилатного полимера, этилакрилатного полимера, н-бутилакрилатного полимера или 2-этилгексилакрилатного полимера соответственно. В еще одном варианте осуществления настоящего изобретения указанный метилакрилатный мономер, этилакрилатный мономер, н-бутилакрилатный мономер или 2-этилгексилакрилатный мономер сополимеризуют с другим мономером для получения метилакрилатного сополимера, этилакрилатного сополимера, н-бутилакрилатного сополимера или 2-этилгексилакрилатного сополимера соответственно. Не имеющие ограничительного характера примеры других мономеров представляют собой винилацетат и этилен. В одном варианте осуществления настоящего изобретения указанный метилакрилатный полимер, этилакрилатный полимер, н-бутилакрилатный полимер или 2-этилгексилакрилатный полимер смешивают с метилметакрилатом (MMA) для получения смесей MMA и метилакрилатного полимера, смесей MMA и этилакрилатного полимера, смесей MMA и н-бутилакрилатного полимера или смесей MMA и 2-этилгексилакрилатного полимера соответственно. Не имеющие ограничительного характера области применения полимеров, сополимеров или смесей относятся к покрытиям для защиты поверхностей, краскам, смолам, адгезивам, пластмассе и дисперсиям. В другом варианте осуществления настоящего изобретения указанный спирт представляет собой спирт на биологической основе. В еще одном варианте осуществления настоящего изобретения указанный другой мономер представляет собой мономер на биологической основе. В еще одном варианте осуществления настоящего изобретения указанный MMA представляет собой MMA на биологической основе.
В одном варианте осуществления настоящего изобретения способ получения акриловой кислоты включает:
a) разбавление водой приблизительно 88 мас. % водного раствора молочной кислоты для получения приблизительно 20 мас. % водного раствора молочной кислоты;
b) нагревание приблизительно 20 мас. % водного раствора молочной кислоты при температуре от приблизительно 95°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты с получением раствора мономерной молочной кислоты, содержащего по меньшей мере приблизительно 95 мас. % молочной кислоты в мономерной форме в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей;
c) объединение раствора мономерной молочной кислоты с азотом для получения смеси жидкость/газ;
d) выпаривание смеси жидкость/газ в испарителе при времени пребывания, составляющем от приблизительно 0,5 с до приблизительно 0,6 с, при температуре от приблизительно 300°C до приблизительно 375°C для получения потока поступающих газообразных материалов, содержащего приблизительно 2,5 мол. % молочной кислоты и приблизительно 50 мол. % водяного пара;
e) приведение указанного потока поступающих газообразных материалов с любым катализатором, описанным в разделе II («Активные катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») или в разделе III («Предшествующие катализаторы для дегидратации гидроксипропионовой кислоты или ее производных до акриловой кислоты или ее производных») настоящего изобретения в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной молочной кислоты и получения потока акриловой кислоты; причем указанное парциальное давление водяного пара превышает или равно 0,4 бар; при этом указанный однослойный реактор содержит стенку, наружную поверхность и внутреннюю поверхность; причем указанная стенка изготовлена из материала стенки, имеет толщину стенки и проходит от указанной наружной поверхности к указанной внутренней поверхности; при этом указанный материал стенки содержит алюминий в количестве более приблизительно 1 мас. %; причем указанная внутренняя поверхность находится в контакте с указанным катализатором; при этом скорость коррозии указанного однослойного реактора во время указанной дегидратации составляет менее приблизительно 1,3 мм/год; и
f) охлаждение указанного потока акриловой кислоты со временем пребывания от приблизительно 0,1 с до приблизительно 10 с для получения указанного потока жидкой акриловой кислоты, содержащего указанную акриловую кислоту.
В другом варианте осуществления настоящего изобретения способ получения акриловой кислоты включает:
a) разбавление водой приблизительно 88 мас. % водного раствора молочной кислоты для получения приблизительно 20 мас. % водного раствора молочной кислоты;
b) нагревание приблизительно 20 мас. % водного раствора молочной кислоты при температуре от приблизительно 95°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты с получением раствора мономерной молочной кислоты, содержащего по меньшей мере приблизительно 95 мас. % молочной кислоты в мономерной форме в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей;
c) объединение раствора мономерной молочной кислоты с азотом для получения смеси жидкость/газ;
d) выпаривание смеси жидкость/газ в испарителе при времени пребывания, составляющем от приблизительно 0,5 с до приблизительно 0,6 с, при температуре от приблизительно 300°C до приблизительно 375°C для получения потока поступающих газообразных материалов, содержащего приблизительно 2,5 мол. % молочной кислоты и приблизительно 50 мол. % водяного пара;
e) приведение указанного потока поступающих газообразных материалов в контакт с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной молочной кислоты и получения потока акриловой кислоты; причем указанный катализатор содержит фосфатную соль
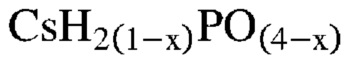
f) охлаждение указанного потока акриловой кислоты со временем пребывания от приблизительно 0,1 с до приблизительно 10 с для получения потока жидкой акриловой кислоты, содержащего указанную акриловую кислоту.
В еще одном варианте осуществления настоящего изобретения способ получения акриловой кислоты включает:
a) разбавление водой приблизительно 88 мас. % водного раствора молочной кислоты для получения приблизительно 20 мас. % водного раствора молочной кислоты;
b) нагревание приблизительно 20 мас. % водного раствора молочной кислоты при температуре от приблизительно 95°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты с получением раствора мономерной молочной кислоты, содержащего по меньшей мере приблизительно 95 мас. % молочной кислоты в мономерной форме в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей;
c) объединение раствора мономерной молочной кислоты с азотом для получения смеси жидкость/газ;
d) выпаривание смеси жидкость/газ в испарителе при времени пребывания, составляющем от приблизительно 0,5 с до приблизительно 0,6 с, при температуре от приблизительно 300°C до приблизительно 375°C для получения потока поступающих газообразных материалов, содержащего приблизительно 2,5 мол. % молочной кислоты и приблизительно 50 мол. % водяного пара;
e) приведение указанного потока поступающих газообразных материалов в контакт с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной молочной кислоты и получения потока акриловой кислоты; причем указанный катализатор содержит фосфатную соль
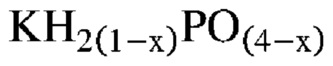
f) охлаждение указанного потока акриловой кислоты со временем пребывания от приблизительно 0,1 с до приблизительно 10 с для получения потока жидкой акриловой кислоты, содержащего указанную акриловую кислоту.
В еще одном варианте осуществления настоящего изобретения способ получения акриловой кислоты включает:
a) разбавление водой приблизительно 88 мас. % водного раствора молочной кислоты для получения приблизительно 20 мас. % водного раствора молочной кислоты;
b) нагревание приблизительно 20 мас. % водного раствора молочной кислоты при температуре от приблизительно 95°C до приблизительно 100°C для обеспечения гидролиза олигомеров молочной кислоты с получением раствора мономерной молочной кислоты, содержащего по меньшей мере приблизительно 95 мас. % молочной кислоты в мономерной форме в расчете на общее количество молочной кислоты, производных молочной кислоты или их смесей;
c) объединение раствора мономерной молочной кислоты с азотом для получения смеси жидкость/газ;
d) выпаривание смеси жидкость/газ в испарителе при времени пребывания, составляющем от приблизительно 0,5 с до приблизительно 0,6 с, при температуре от приблизительно 300°C до приблизительно 375°C для получения потока поступающих газообразных материалов, содержащего приблизительно 2,5 мол. % молочной кислоты и приблизительно 50 мол. % водяного пара;
e) приведение указанного потока поступающих газообразных материалов в контакт с катализатором в двухслойном реакторе при некоторых значениях температуры, парциального давления водяного пара, GHSV и WHSV для дегидратации указанной молочной кислоты и получения потока акриловой кислоты; причем указанный катализатор содержит фосфатную соль

f) охлаждение указанного потока акриловой кислоты со временем пребывания от приблизительно 0,1 с до приблизительно 10 с для получения потока жидкой акриловой кислоты, содержащего указанную акриловую кислоту.
VI. Примеры
Следующие примеры приведены для иллюстрации настоящего изобретения, но не предназначены для ограничения его объема.
ПРИМЕР 1. Предшествующий катализатор, содержащий 13 мас. % KPO3, 37 мас. % Ba2P2O7 и 50% плавленого кремнезема
Нитрат бария (Ba(NO3)2 (150,00 г, 550,5 ммоль; Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 202754), фосфат калия K2HPO4 (31,96 г, 183,5 ммоль; Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 60347) и двухосновный фосфат аммония (NH4)2HPO4 (99,21 г, 734,0 ммоль; Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 379980) объединяли и совместно измельчали с помощью планетарной шаровой мельницы (Retsch GmbH, г. Хан, Германия; модель PM 100; № по каталогу 20.540.0003) для получения мелкодисперсного порошка. Затем материал переносили в стеклянный лабораторный стакан объемом 1 л и кальцинировали с использованием лабораторной печи с циркуляцией воздуха (Nabertherm GmbH, г. Лилиенталь, Германия; № по каталогу N30/85 HA; в условиях: 450°C, 12 ч, скорость линейного изменения температуры 2°C/мин и открытый выпускной клапан). Кальцинированное твердое вещество измельчали и просеивали для получения твердого вещества с размером частиц от 106 мкм до 212 мкм. Затем 4,18 г этого твердого вещества и 4,18 г плавленого кремнезема (Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 342831; измельченного и просеянного до размера частиц 106-212 мкм) добавляли в стеклянный сцинтилляционный флакон и перемешивали в вихревом смесителе до полного смешивания. Полученный предшествующий катализатор содержал 13 мас. % KPO3, 37 мас. % Ba2P2O7 и 50% плавленого кремнезема.
ПРИМЕР 2. 20 мас. % водный раствор молочной кислоты
455 г 88 мас. % раствора L-молочной кислоты (Corbion Purac Co., г. Ленекса, штат Канзас, США) разводили с применением 1,508 г воды. Разбавленный раствор нагревали до 95°C и выдерживали при этой температуре при перемешивании в течение приблизительно 12 часов. Затем раствор охлаждали до комнатной температуры и измеряли концентрацию в нем мономеров молочной кислоты с помощью ВЭЖХ-системы Agilent 1100 (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенной детектором на диодной матрице (DAD) и колонкой Atlantis T3 (Waters Corp., г. Милфорд, штат Массачусетс, США); № по каталогу 186003748), с использованием методов, по существу известных специалистам в данной области техники, для получения 20 мас. % водного раствора L-молочной кислоты, по существу не содержащего олигомеров.
ПРИМЕР 3. Испытание предшествующего катализатора
Футерованный стеклом трубчатый реактор из нержавеющей стали (SGE Analytical Science Pty Ltd., г. Рингвуд, Австралия; номер продукта: 08277028) с НД 12,7 мм (½ дюйма), ВД 9,5 мм и длиной 60 см заполняли в 3 зонах следующим образом: 1) нижняя зона: 0,4 г кварцевой ваты, 24,1 г плавленого кремнезема с размером частиц 4-20 меш, (Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 342831) и далее набивали 0,2 г кварцевой ваты для получения длины нижней зоны, составляющей 30,5 см (12 дюймов); 2) средняя зона / зона дегидратации: набивали 8,81 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора длиной 10,2 см (4 дюйма; объем слоя катализатора составил 7,2 мл); и 3) верхняя зона / зона испарителя: 0,1 г кварцевой ваты помещали в верхнюю часть зоны дегидратации, а поверх нее помещали 3,6 г плавленого кремнезема (4-20 меш) для получения зоны испарителя длиной 5,1 см (2 дюйма).
Сначала реактор помещали в алюминиевый блок, а затем помещали в двустворчатую печь серии 3210 (Applied Test Systems, г. Батлер, штат Пенсильвания, США) таким образом, чтобы верхняя часть зоны испарителя была расположена на одной линии с верхней частью алюминиевого блока. Реактор устанавливали по нисходящей и оснащали питающим насосом Knauer Smartline 100 (Knauer GmbH, г. Берлин, Германия), регулятором расхода газа Brooks 0254 (Brooks Instrument LLC, г. Хатфилд, штат Пенсильвания, США), регулятором обратного давления Brooks и покрытой тефлоном ловушкой. Верхнюю часть реактора оборудовали 3,2 мм (1/8 дюйма) трубопроводом из нержавеющей стали, который использовали в качестве трубопровода для подачи азота, и 1,6 мм (1/16 дюйма) трубой из полиэфирэфиркетона (PEEK™) (Supelco Inc., г. Беллефонт, штат Пенсильвания, США), которую использовали в качестве трубопровода для подачи поступающих жидких материалов, соединенного с питающим насосом. Нижнюю часть реактора соединяли с ловушкой с применением 3,2 мм (1/8 дюйма) футерованной плавленым кремнеземом трубки из нержавеющей стали и фитингов Swagelok™. Двустворчатую печь нагревали таким образом, чтобы поддерживать постоянной температуру стенки реактора при приблизительно 375°C во время протекания реакции.
В реактор подавали отдельные потоки поступающей жидкости и газа, которые смешивали вместе перед достижением слоя катализатора. Инертный газ представлял собой азот под давлением 24,8 бар изб. (360 фунтов на кв. дюйм изб.), который подавали в реактор со скоростью 130 мл/мин (в условиях STP). Поступающий поток жидкости представлял собой водный раствор молочной кислоты (20 мас. % L-молочной кислоты из примера 2), который подавали в реактор со скоростью 0,13 мл/мин. После зоны испарителя полученный в результате поток поступающих газообразных материалов имел следующий состав: 49,6 мол. % воды, 47,9 мол. % азота и 2,5 мол. % молочной кислоты. В зоне дегидратации GHSV составляла приблизительно 2262 ч-1, WHSV составляла приблизительно 0,38 ч-1, а парциальное давление водяного пара составляло приблизительно 13 бар (186 фунтов на кв. дюйм).
Газообразный поток продукта охлаждали и анализировали на линии посредством Agilent 7890A GC (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Varian CP-PoraBond Q (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7351). Жидкий поток продукта собирали в ловушку и анализировали автономно (с применением способов, по существу известных специалисту в данной области техники) с применением прибора для ВЭЖХ Agilent 1100 (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного детектором на диодной матрице (DAD) и колонкой Atlantis T3 (Waters Corp., г. Милфорд, штат Массачусетс, США; № по каталогу 186003748), и прибора для GC Hewlett Packard HP6890 series (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Agilent CP-Wax 58 FFAP CB (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7717).
Поток жидких продуктов охлаждали и собирали за период приблизительно 72 ч. Общий выход акриловой кислоты составлял 84,4 мол. %, избирательность по акриловой кислоте составляла 87,2 мол. %, превращение молочной кислоты составляло 96,7 мол. %, а избирательность по пропионовой кислоте составляла 0,74 мол. %.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 4. Испытательный образец из нержавеющей стали 316L (316L SS)
Испытательный образец 316L SS шлифовали наждачной тканью (сначала с зернистостью 320 грит, затем с зернистостью 500 грит) и очищали с помощью растворителя (ацетон, гексаны, а затем хлороформ). Размеры испытательного образца, измеренные с использованием микрометра, составили 30,03 мм × 8,01 мм × 1,5 мм, а масса испытательного образца, измеренная перед помещением в реактор, составила 2819,80 мг. Номинальный химический состав этого сплава включает в себя приблизительно 65 мас. % железа, приблизительно 17 мас. % хрома, приблизительно 12 мас. % никеля и 2-3 мас. % молибдена.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 5. Коррозионные испытания испытательного образца 316L SS
Футерованный стеклом трубчатый реактор из нержавеющей стали (SGE Analytical Science Pty Ltd., г. Рингвуд, Австралия; номер продукта: 08277028) с НД 12,7 мм (½ дюйма), ВД 9,5 мм и длиной 60 см заполняли в 3 зонах следующим образом: 1) нижняя зона: 0,4 г кварцевой ваты, 25,15 г плавленого кремнезема с размером частиц 4-20 меш, (Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 342831) и далее набивали 0,1 г кварцевой ваты для получения длины нижней зоны, составляющей 29,5 см (11,6 дюймов); 2) средняя зона / зона дегидратации: набивали 1,44 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с длиной 2,54 см (1 дюйм) в нижней части этой зоны, затем поверх этого предшествующего катализатора помещали испытательный образец 316L SS, изготовленный в сравнительном примере 4, и затем вокруг и поверх этого испытательного образца помещали дополнительные 7,31 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с общей длиной 10,2 см (4 дюйма) (общая масса предшествующего катализатора составила 8,75 г, а объем слоя катализатора составил 7,2 мл); и 3) верхняя зона / зона испарителя: 0,1 г кварцевой ваты помещали в верхнюю часть зоны дегидратации, а поверх нее помещали 3,8 г плавленого кремнезема (4-20 меш) для получения зоны испарителя длиной 5,1 см (2 дюйма).
Сначала реактор помещали в алюминиевый блок, а затем помещали в двустворчатую печь серии 3210 (Applied Test Systems, г. Батлер, штат Пенсильвания, США) таким образом, чтобы верхняя часть зоны испарителя была расположена на одной линии с верхней частью алюминиевого блока. Реактор устанавливали по нисходящей и оснащали питающим насосом Knauer Smartline 100 (Knauer GmbH, г. Берлин, Германия), регулятором расхода газа Brooks 0254 (Brooks Instrument LLC, г. Хатфилд, штат Пенсильвания, США), регулятором обратного давления Brooks и покрытой тефлоном ловушкой. Верхнюю часть реактора оборудовали 3,2 мм (1/8 дюйма) трубопроводом из нержавеющей стали, который использовали в качестве трубопровода для подачи азота, и 1,6 мм (1/16 дюйма) трубой из полиэфирэфиркетона (PEEK™) (Supelco Inc., г. Беллефонт, штат Пенсильвания, США), которую использовали в качестве трубопровода для подачи поступающих жидких материалов, соединенного с питающим насосом. Нижнюю часть реактора соединяли с ловушкой с применением 3,2 мм (1/8 дюйма) футерованной плавленым кремнеземом трубки из нержавеющей стали и фитингов Swagelok™. Двустворчатую печь нагревали таким образом, чтобы поддерживать постоянной температуру стенки реактора при приблизительно 375°C во время протекания реакции.
В реактор подавали отдельные потоки поступающей жидкости и газа, которые смешивали вместе перед достижением слоя катализатора. Инертный газ представлял собой азот под давлением 24,8 бар изб. (360 фунтов на кв. дюйм изб.), который подавали в реактор со скоростью 130 мл/мин (в условиях STP). Поступающий поток жидкости представлял собой водный раствор молочной кислоты (20 мас. % L-молочной кислоты из примера 2), который подавали в реактор со скоростью 0,13 мл/мин. После зоны испарителя полученный в результате поток поступающих газообразных материалов имел следующий состав: 49,6 мол. % воды, 47,9 мол. % азота и 2,5 мол. % молочной кислоты. В зоне дегидратации GHSV составляла приблизительно 2262 ч-1, WHSV составляла приблизительно 0,38 ч-1, а парциальное давление водяного пара составляло приблизительно 13 бар (186 фунтов на кв. дюйм).
Газообразный поток продукта охлаждали и анализировали на линии посредством Agilent 7890A GC (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Varian CP-PoraBond Q (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7351). Жидкий поток продукта собирали в ловушку и анализировали автономно (с применением способов, по существу известных специалисту в данной области техники) с применением прибора для ВЭЖХ Agilent 1100 (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного детектором на диодной матрице (DAD) и колонкой Atlantis T3 (Waters Corp., г. Милфорд, штат Массачусетс, США; № по каталогу 186003748), и прибора для GC Hewlett Packard HP6890 series (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Agilent CP-Wax 58 FFAP CB (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7717).
Поток жидких продуктов охлаждали и собирали за период, составляющий приблизительно 72 ч. Общий (т. е. за период 72 ч) выход акриловой кислоты составлял 81,7 мол. %, избирательность по акриловой кислоте составляла 83,8 мол. %, превращение молочной кислоты составляло 97,5 мол. %, а избирательность по пропионовой кислоте составляла 2,02 мол. %. В конце эксперимента испытательный образец извлекали из зоны дегидратации, и рассчитывали скорость коррозии, которая составила 3,04 мм/год.
ПРИМЕР 6. Термообработанный испытательный образец из сплава HAYNES® 214®
Испытательный образец из сплава HAYNES® 214® (Haynes International Inc., г. Кокомо, штат Иллинойс, США) шлифовали наждачной тканью (сначала с зернистостью 320 грит, затем с зернистостью 500 грит), очищали с помощью растворителя (ацетон, гексаны, а затем хлороформ) и подвергали термообработке (т. е. окисляли) в муфельной печи (1 ч при 600°C, а затем 1 ч при 1000°C; Wilt Industries Inc., г. Лейк Плезант, штат Нью-Йорк, США) перед использованием. Размеры испытательного образца, измеренные с использованием микрометра, составили 30,38 мм × 7,97 мм × 1,48 мм, а масса испытательного образца, измеренная перед помещением в реактор, составила 2768,44 мг. Номинальный химический состав этого сплава включает в себя приблизительно 75 мас. % никеля, приблизительно 16 мас. % хрома, приблизительно 3 мас. % железа и приблизительно 4,5 мас. % алюминия. Термическая обработка испытательного образца (т. е. окисление, осуществляемое перед дегидратацией) проводилось с целью окисления алюминия, включенного в сплав, и формирования пассивирующего поверхность слоя на основе оксида, содержащего кремнезем.
ПРИМЕР 7. Коррозионные испытания термообработанного испытательного образца из сплава HAYNES® 214®
Футерованный стеклом трубчатый реактор из нержавеющей стали (SGE Analytical Science Pty Ltd., г. Рингвуд, Австралия; номер продукта: 08277028) с НД 12,7 мм (½ дюйма), ВД 9,5 мм и длиной 60 см заполняли в 3 зонах следующим образом: 1) нижняя зона: 0,4 г кварцевой ваты, 25,19 г плавленого кремнезема с размером частиц 4-20 меш, (Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 342831) и далее набивали 0,1 г кварцевой ваты для получения длины нижней зоны, составляющей 29,5 см (11,6 дюймов); 2) средняя зона / зона дегидратации: набивали 2,18 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с длиной 2,54 см (1 дюйм) в нижней части этой зоны, затем поверх этого предшествующего катализатора помещали термообработанный испытательный образец из сплава HAYNES® 214®, изготовленный в примере 6, и затем вокруг и поверх этого испытательного образца помещали дополнительные 6,18 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с общей длиной 10,2 см (4 дюйма) (общая масса предшествующего катализатора составила 8,36 г, а объем слоя катализатора составил 7,2 мл); и 3) верхняя зона / зона испарителя: 0,1 г кварцевой ваты помещали в верхнюю часть зоны дегидратации, а поверх нее помещали 4,32 г плавленого кремнезема (4-20 меш) для получения зоны испарителя длиной 5,1 см (2 дюйма).
Сначала реактор помещали в алюминиевый блок, а затем помещали в двустворчатую печь серии 3210 (Applied Test Systems, г. Батлер, штат Пенсильвания, США) таким образом, чтобы верхняя часть зоны испарителя была расположена на одной линии с верхней частью алюминиевого блока. Реактор устанавливали по нисходящей и оснащали питающим насосом Knauer Smartline 100 (Knauer GmbH, г. Берлин, Германия), регулятором расхода газа Brooks 0254 (Brooks Instrument LLC, г. Хатфилд, штат Пенсильвания, США), регулятором обратного давления Brooks и покрытой тефлоном ловушкой. Верхнюю часть реактора оборудовали 3,2 мм (1/8 дюйма) трубопроводом из нержавеющей стали, который использовали в качестве трубопровода для подачи азота, и 1,6 мм (1/16 дюйма) трубой из полиэфирэфиркетона (PEEK™) (Supelco Inc., г. Беллефонт, штат Пенсильвания, США), которую использовали в качестве трубопровода для подачи поступающих жидких материалов, соединенного с питающим насосом. Нижнюю часть реактора соединяли с ловушкой с применением 3,2 мм (1/8 дюйма) футерованной плавленым кремнеземом трубки из нержавеющей стали и фитингов Swagelok™. Двустворчатую печь нагревали таким образом, чтобы поддерживать постоянной температуру стенки реактора при приблизительно 375°C во время протекания реакции.
В реактор подавали отдельные потоки поступающей жидкости и газа, которые смешивали вместе перед достижением слоя катализатора. Инертный газ представлял собой азот под давлением 24,8 бар изб. (360 фунтов на кв. дюйм изб.), который подавали в реактор со скоростью 130 мл/мин (в условиях STP). Поступающий поток жидкости представлял собой водный раствор молочной кислоты (20 мас. % L-молочной кислоты из примера 2), который подавали в реактор со скоростью 0,13 мл/мин. После зоны испарителя полученный в результате поток поступающих газообразных материалов имел следующий состав: 49,6 мол. % воды, 47,9 мол. % азота и 2,5 мол. % молочной кислоты. В зоне дегидратации GHSV составляла приблизительно 2262 ч-1, WHSV составляла приблизительно 0,38 ч-1, а парциальное давление водяного пара составляло приблизительно 13 бар (186 фунтов на кв. дюйм).
Газообразный поток продукта охлаждали и анализировали на линии посредством Agilent 7890A GC (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Varian CP-PoraBond Q (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7351). Жидкий поток продукта собирали в ловушку и анализировали автономно (с применением способов, по существу известных специалисту в данной области техники) с применением прибора для ВЭЖХ Agilent 1100 (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного детектором на диодной матрице (DAD) и колонкой Atlantis T3 (Waters Corp., г. Милфорд, штат Массачусетс, США; № по каталогу 186003748), и прибора для GC Hewlett Packard HP6890 series (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Agilent CP-Wax 58 FFAP CB (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7717).
Поток жидких продуктов охлаждали и собирали за период, составляющий приблизительно 72 ч. Общий (т. е. за период 72 ч) выход акриловой кислоты составлял 86,2 мол. %, избирательность по акриловой кислоте составляла 88,5 мол. %, превращение молочной кислоты составляло 97,3 мол. %, а избирательность по пропионовой кислоте составляла 0,58 мол. %. В конце эксперимента испытательный образец извлекали из зоны дегидратации, и рассчитывали скорость коррозии, которая составила 0,79 мм/год.
ПРИМЕР 8. Испытательный образец из сплава HAYNES® HR-160®
Испытательный образец из сплава HAYNES® HR-160® (Haynes International Inc., г. Кокомо, штат Иллинойс, США) шлифовали наждачной тканью (сначала с зернистостью 320 грит, затем с зернистостью 500 грит) и очищали с помощью растворителя (ацетон, гексаны, а затем хлороформ) перед использованием. Размеры испытательного образца, измеренные с использованием микрометра, составили 29,13 мм × 8,20 мм × 1,51 мм, а масса испытательного образца, измеренная перед помещением в реактор, составила 2710,33 мг. Номинальный химический состав этого сплава включает в себя приблизительно 37 мас. % никеля, приблизительно 29 мас. % кобальта, приблизительно 28 мас. % хрома, приблизительно 2 мас. % железа и приблизительно 2,75 мас. % кремния. Следует отметить, что перед использованием испытательный образец купон не подвергали термообработке (т. е. не подвергали окислению перед дегидратацией), и поэтому считается, что перед дегидратацией на нем не был сформирован пассивирующий поверхность слой на основе оксида, содержащий кремнезем.
ПРИМЕР 9. Коррозионные испытания испытательного образца из сплава HAYNES® HR-160®
Футерованный стеклом трубчатый реактор из нержавеющей стали (SGE Analytical Science Pty Ltd., г. Рингвуд, Австралия; номер продукта: 08277028) с НД 12,7 мм (½ дюйма), ВД 9,5 мм и длиной 60 см заполняли в 3 зонах следующим образом: 1) нижняя зона: 0,4 г кварцевой ваты, 24,64 г плавленого кремнезема с размером частиц 4-20 меш, (Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 342831) и далее набивали 0,1 г кварцевой ваты для получения длины нижней зоны, составляющей 29,5 см (11,6 дюймов); 2) средняя зона / зона дегидратации: набивали 2,41 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с длиной 2,54 см (1 дюйм) в нижней части этой зоны, затем поверх этого предшествующего катализатора помещали испытательный образец из сплава HAYNES® HR-160®, изготовленный в примере 8, и затем вокруг и поверх этого испытательного образца помещали дополнительные 6,63 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с общей длиной 10,2 см (4 дюйма) (общая масса предшествующего катализатора составила 9,04 г, а объем слоя катализатора составил 7,2 мл); и 3) верхняя зона / зона испарителя: 0,1 г кварцевой ваты помещали в верхнюю часть зоны дегидратации, а поверх нее помещали 4,38 г плавленого кремнезема (4-20 меш) для получения зоны испарителя длиной 5,1 см (2 дюйма).
Сначала реактор помещали в алюминиевый блок, а затем помещали в двустворчатую печь серии 3210 (Applied Test Systems, г. Батлер, штат Пенсильвания, США) таким образом, чтобы верхняя часть зоны испарителя была расположена на одной линии с верхней частью алюминиевого блока. Реактор устанавливали по нисходящей и оснащали питающим насосом Knauer Smartline 100 (Knauer GmbH, г. Берлин, Германия), регулятором расхода газа Brooks 0254 (Brooks Instrument LLC, г. Хатфилд, штат Пенсильвания, США), регулятором обратного давления Brooks и покрытой тефлоном ловушкой. Верхнюю часть реактора оборудовали 3,2 мм (1/8 дюйма) трубопроводом из нержавеющей стали, который использовали в качестве трубопровода для подачи азота, и 1,6 мм (1/16 дюйма) трубой из полиэфирэфиркетона (PEEK™) (Supelco Inc., г. Беллефонт, штат Пенсильвания, США), которую использовали в качестве трубопровода для подачи поступающих жидких материалов, соединенного с питающим насосом. Нижнюю часть реактора соединяли с ловушкой с применением 3,2 мм (1/8 дюйма) футерованной плавленым кремнеземом трубки из нержавеющей стали и фитингов Swagelok™. Двустворчатую печь нагревали таким образом, чтобы поддерживать постоянной температуру стенки реактора при приблизительно 375°C во время протекания реакции.
В реактор подавали отдельные потоки поступающей жидкости и газа, которые смешивали вместе перед достижением слоя катализатора. Инертный газ представлял собой азот под давлением 24,8 бар изб. (360 фунтов на кв. дюйм изб.), который подавали в реактор со скоростью 130 мл/мин (в условиях STP). Поступающий поток жидкости представлял собой водный раствор молочной кислоты (20 мас. % L-молочной кислоты из примера 2), который подавали в реактор со скоростью 0,13 мл/мин. После зоны испарителя полученный в результате поток поступающих газообразных материалов имел следующий состав: 49,6 мол. % воды, 47,9 мол. % азота и 2,5 мол. % молочной кислоты. В зоне дегидратации GHSV составляла приблизительно 2262 ч-1, WHSV составляла приблизительно 0,38 ч-1, а парциальное давление водяного пара составляло приблизительно 13 бар (186 фунтов на кв. дюйм).
Газообразный поток продукта охлаждали и анализировали на линии посредством Agilent 7890A GC (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Varian CP-PoraBond Q (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7351). Жидкий поток продукта собирали в ловушку и анализировали автономно (с применением способов, по существу известных специалисту в данной области техники) с применением прибора для ВЭЖХ Agilent 1100 (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного детектором на диодной матрице (DAD) и колонкой Atlantis T3 (Waters Corp., г. Милфорд, штат Массачусетс, США; № по каталогу 186003748), и прибора для GC Hewlett Packard HP6890 series (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Agilent CP-Wax 58 FFAP CB (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7717).
Поток жидких продуктов охлаждали и собирали за период, составляющий приблизительно 72 ч. Общий (т. е. за период 72 ч) выход акриловой кислоты составлял 88 мол. %, избирательность по акриловой кислоте составляла 89,4 мол. %, превращение молочной кислоты составляло 98,4 мол. %, а избирательность по пропионовой кислоте составляла 0,42 мол. %. В конце эксперимента испытательный образец извлекали из зоны дегидратации, и рассчитывали скорость коррозии, которая составила 1,22 мм/год.
ПРИМЕР 10. Медный испытательный образец
Медный испытательный образец шлифовали наждачной тканью (сначала с зернистостью 320 грит, затем с зернистостью 500 грит). Размеры испытательного образца, измеренные с использованием микрометра, составили 29,46 мм × 7,66 мм × 1,18 мм, а масса испытательного образца, измеренная перед помещением в реактор, составила 2400,59 мг.
ПРИМЕР 11. Коррозионные испытания медного испытательного образца
Футерованный стеклом трубчатый реактор из нержавеющей стали (SGE Analytical Science Pty Ltd., г. Рингвуд, Австралия; номер продукта: 08277028) с НД 12,7 мм (½ дюйма), ВД 9,5 мм и длиной 60 см заполняли в 3 зонах следующим образом: 1) нижняя зона: 0,4 г кварцевой ваты, 22,33 г плавленого кремнезема с размером частиц 4-20 меш, (Sigma-Aldrich Co., г. Сент-Луис, штат Миссури, США; № по каталогу 342831) и далее набивали 0,1 г кварцевой ваты для получения длины нижней зоны, составляющей 29,5 см (11,6 дюймов); 2) средняя зона / зона дегидратации: набивали 2,05 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с длиной 2,54 см (1 дюйм) в нижней части этой зоны, затем поверх этого предшествующего катализатора помещали медный испытательный образец, изготовленный в примере 10, и затем вокруг и поверх этого испытательного образца помещали дополнительные 5,63 г предшествующего катализатора, полученного в примере 1, для получения слоя катализатора с общей длиной 10,2 см (4 дюйма) (общая масса предшествующего катализатора составила 7,68 г, а объем слоя катализатора составил 7,2 мл); и 3) верхняя зона / зона испарителя: 0,1 г кварцевой ваты помещали в верхнюю часть зоны дегидратации, а поверх нее помещали 3,93 г плавленого кремнезема (4-20 меш) для получения зоны испарителя длиной 5,1 см (2 дюйма).
Сначала реактор помещали в алюминиевый блок, а затем помещали в двустворчатую печь серии 3210 (Applied Test Systems, г. Батлер, штат Пенсильвания, США) таким образом, чтобы верхняя часть зоны испарителя была расположена на одной линии с верхней частью алюминиевого блока. Реактор устанавливали по нисходящей и оснащали питающим насосом Knauer Smartline 100 (Knauer GmbH, г. Берлин, Германия), регулятором расхода газа Brooks 0254 (Brooks Instrument LLC, г. Хатфилд, штат Пенсильвания, США), регулятором обратного давления Brooks и покрытой тефлоном ловушкой. Верхнюю часть реактора оборудовали 3,2 мм (1/8 дюйма) трубопроводом из нержавеющей стали, который использовали в качестве трубопровода для подачи азота, и 1,6 мм (1/16 дюйма) трубой из полиэфирэфиркетона (PEEK™) (Supelco Inc., г. Беллефонт, штат Пенсильвания, США), которую использовали в качестве трубопровода для подачи поступающих жидких материалов, соединенного с питающим насосом. Нижнюю часть реактора соединяли с ловушкой с применением 3,2 мм (1/8 дюйма) футерованной плавленым кремнеземом трубки из нержавеющей стали и фитингов Swagelok™. Двустворчатую печь нагревали таким образом, чтобы поддерживать постоянной температуру стенки реактора при приблизительно 375°C во время протекания реакции.
В реактор подавали отдельные потоки поступающей жидкости и газа, которые смешивали вместе перед достижением слоя катализатора. Инертный газ представлял собой азот под давлением 24,8 бар изб. (360 фунтов на кв. дюйм изб.), который подавали в реактор со скоростью 130 мл/мин (в условиях STP). Поступающий поток жидкости представлял собой водный раствор молочной кислоты (20 мас. % L-молочной кислоты из примера 2), который подавали в реактор со скоростью 0,13 мл/мин. После зоны испарителя полученный в результате поток поступающих газообразных материалов имел следующий состав: 49,6 мол. % воды, 47,9 мол. % азота и 2,5 мол. % молочной кислоты. В зоне дегидратации GHSV составляла приблизительно 2262 ч-1, WHSV составляла приблизительно 0,38 ч-1, а парциальное давление водяного пара составляло приблизительно 13 бар (186 фунтов на кв. дюйм).
Газообразный поток продукта охлаждали и анализировали на линии посредством Agilent 7890A GC (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Varian CP-PoraBond Q (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7351). Жидкий поток продукта собирали в ловушку и анализировали автономно (с применением способов, по существу известных специалисту в данной области техники) с применением прибора для ВЭЖХ Agilent 1100 (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного детектором на диодной матрице (DAD) и колонкой Atlantis T3 (Waters Corp., г. Милфорд, штат Массачусетс, США; № по каталогу 186003748), и прибора для GC Hewlett Packard HP6890 series (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США), оснащенного ионизационным детектором и колонкой Agilent CP-Wax 58 FFAP CB (Agilent Technologies, Inc., г. Санта-Клара, штат Калифорния, США; № по каталогу CP7717).
Поток жидких продуктов охлаждали и собирали за период, составляющий приблизительно 72 ч. Общий (т. е. за период 72 ч) выход акриловой кислоты составлял 86 мол. %, избирательность по акриловой кислоте составляла 88,1 мол. %, превращение молочной кислоты составляло 97,6 мол. %, а избирательность по пропионовой кислоте составляла 0,53 мол. %. В конце эксперимента испытательный образец извлекали из зоны дегидратации и не наблюдали на нем признаков коррозии, т. е. скорость коррозии составила 0 мм/год.
Представленные в виде таблицы результаты из примеров можно видеть в таблице 1 ниже.

Вышеприведенное описание представлено только для ясности понимания, и под ним не следует подразумевать какие-либо ненужные ограничения, поскольку модификации в пределах объема настоящего изобретения могут быть очевидны специалисту в данной области техники.
Размеры и величины, описанные в настоящем документе, не следует понимать как строго ограниченные перечисленными точными числовыми значениями. Напротив, если не указано иное, каждый такой размер должен обозначать как указанное значение, так и функционально эквивалентный диапазон, в который входит это значение. Например, размер, описанный как «40 мм», подразумевает «приблизительно 40 мм».
Каждый документ, указанный в настоящем документе, включая перекрестные ссылки или родственный патент или заявку, настоящим включен в полном объеме в настоящий документ путем ссылки, если только не исключен в явной форме или не ограничен иным образом. Упоминание любого документа не является признанием, что он представляет собой предшествующий уровень техники в отношении любого изобретения, описанного или заявленного в настоящем документе, или что в нем самом или в любой комбинации с любой другой ссылкой или ссылками представлено, предложено или описано любое такое изобретение. Кроме того, если какое-либо значение или определение термина в этом документе противоречит какому-либо значению или определению этого же термина в документе, включенном в настоящий документ путем ссылки, преимущество имеет значение или определение, закрепленное за этим термином в настоящем документе.
Хотя в настоящем документе показаны и описаны конкретные варианты осуществления настоящего изобретения, специалистам в данной области будет понятно, что допустимы и другие различные изменения и модификации без отступления от сущности и объема изобретения. Таким образом, подразумевается, что прилагаемая формула изобретения охватывает все такие изменения и модификации в пределах объема настоящего изобретения.
Реферат
Изобретение относится к способу получения акриловой кислоты, включающему приведение потока поступающих газообразных материалов, содержащего водяной пар и гидроксипропионовую кислоту, производные гидроксипропионовой кислоты или их смеси, в контакт с катализатором в однослойном реакторе при некоторых значениях температуры, парциального давления водяного пара, часовой объемной скорости подачи газа (GHSV) и часовой объемной скорости на единицу массы (WHSV) для дегидратации указанной гидроксипропионовой кислоты, производных гидроксипропионовой кислоты или их смесей с последующим получением акриловой кислоты; причем указанный катализатор содержит фосфатную соль, содержащую катион и анион, представленный эмпирической формулой: [НPO], где х представляет собой любое действительное число, которое больше или равно 0 и меньше или равно 1; причем указанное парциальное давление водяного пара превышает или равно 13 бар; при этом указанный однослойный реактор содержит стенку, наружную поверхность и внутреннюю поверхность; причем указанная стенка изготовлена из материала стенки, имеет толщину стенки и проходит от указанной наружной поверхности к указанной внутренней поверхности; при этом указанный материал стенки содержит алюминий в количестве от примерно 1 мас. % до примерно 50 мас. %; причем указанная внутренняя поверхность находится в контакте с указанным катализатором; и при этом скорость коррозии указанного однослойного реактора во время указанной дегидратации составляет менее приблизительно 1,3 миллиметра в год (мм/год). 14 з.п. ф-лы, 4 ил., 1 табл., 11 пр.
Формула
Документы, цитированные в отчёте о поиске
Каталитическая конверсия гидроксипропионовой кислоты или ее производных в акриловую кислоту и ее производные
Комментарии