Фторированный диен, способ его получения, полимер на его основе, оптическое передающее устройство и оптическое пластмассовое волокно - RU2272020C2
Код документа: RU2272020C2
Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к фторированному диену, содержащему две ненасыщенные связи, к способу его получения и к полимеру на его основе.
УРОВЕНЬ ТЕХНИКИ
В качестве фторированного диена, содержащего две ненасыщенные двойные связи углерод-углерод (здесь и далее в настоящем документе называемые ненасыщенными связями), известен CF2=CF(CF2)kOCF=CF2 (где k представляет собой целое число в диапазоне от 1 до 3) (JP-A-1-131215). В результате циклополимеризации данного соединения можно получить аморфный полимер, и такой полимер отличается высокими модулем упругости, текучестью и удлинением при разрыве, он является жестким и обладает превосходной ударной прочностью. Кроме этого его прозрачность также высока, и его можно использовать в качестве материала для оптики, такого, как материал для оптического волокна или оптического волновода. Однако ему присущ недостаток, который заключается в том, что когда данный материал используют для получения материала для оптики, его температура стеклования (Tg) низка, и если его использовать при высоких температурах в течение длительного периода времени, оптические свойства будут меняться. Соответственно, было бы желательно создать основной материал, отличающийся более высокой Tg.
Цель настоящего изобретения заключается в получении полимера, который бы сохранял механические свойства, которыми обладает вышеупомянутый аморфный полимер, и который бы отличался более высокой температурой стеклования так, чтобы он мог бы быть полимерным материалом для оптики с превосходной теплостойкостью, и в получении фторированного диена, содержащего две ненасыщенные связи, из которого можно получить такой полимер.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фторированному диену, описываемому формулой 1, к способу его получения и к его полимеру.
Фторированный диен, описываемый формулой 1.
Способ получения фторированного диена, описываемого формулой 1, который включает дегалогенирование Z1 и Z2 у фторированного соединения, описываемого формулой 2.
Полимер, содержащий мономерные звенья, полученные в результате полимеризации фторированного диена, описываемого формулой 1.
где R представляет собой атом фтора или трифторметильную группу, каждый из Z1 и Z2, которые не зависят друг от друга, представляет собой атом галогена, отличный от атома фтора, а n представляет собой целое число в диапазоне от 1 до 3.
НАИЛУЧШИЙ ВАРИАНТ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Фторированный, диен, описываемый формулой 1, можно получить в результате дегалогенирования в положениях Z1 и Z2 у фторированного соединения, описываемого формулой 2. Каждый из Z1 и Z2, которые не зависят друг от друга, представляет собой атом галогена, отличный от атома фтора, предпочтительно атом хлора или атом брома, а в особенности предпочтительно, если каждый будет представлять собой атом хлора. В результате дегалогенирования атомов галогена будет образовываться двойная связь, и будет образован фторированный диен, описываемый формулой 1.
Дегалогенирование проводят, используя дегалогенирующий агент, действующий в полярном растворителе. Дегалогенирующий агент представляет собой реакционно-способное соединение, обладающее способностью воздействия на атомы галогена в субстрате, тем самым отрывая атомы галогена. В качестве такого дегалогенирующего агента предпочтительными являются цинк, натрий, магний, олово, медь, железо или другие металлы. С точки зрения условий проведения реакции, где можно было бы использовать относительно низкую температуру реакции, в качестве такого дегалогенирующего агента предпочтителен цинк. В качестве полярного растворителя предпочтительно можно использовать, например, органический полярный растворитель, такой, как диметилформамид, 1, 4-диоксан, диглим или метанол либо вода.
Мольное отношение количества дегалогенирующего агента к количеству фторированного соединения, описываемого формулой 2, предпочтительно заключается в диапазоне от 1 до 10, более предпочтительно от 2 до 3. Температура реакции обычно находится в диапазоне от 40 до 100°С, предпочтительно от 50 до 70°С. Обычно реакцию проводят в результате прибавления по каплям фторированного соединения, описываемого формулой 2, в присутствии дегалогенирующего агента и растворителя, а выделение продукта реакции проводят в результате отвода продукта реакции из реакционной системы при помощи отгонки, проводимой непосредственно сразу же после реакции.
Фторированное соединение, описываемое формулой 2, представляет собой новое соединение, а соединение (формула 2-1), где R представляет собой атом фтора, можно получить, например, из известного фторированного соединения, описываемого формулой 3-1. Кроме этого фторированное соединение (формула 2-2), описываемое формулой 2, где R представляет собой трифторметильную группу, можно получить, например, из известного фторированного соединения, описываемого формулой 4-1.
Прежде всего, будет описан способ получения фторированного соединения, описываемого формулой 2-1. Ненасыщенную группу во фторированном соединении, описываемом формулой 3-1, эпоксидируют с получением эпоксидного соединения (формула 3-2), а данное эпоксидное соединение изомеризуется и превращается во фторированное производное кетона (формула 3-3). К данному фторированному производному кетона добавляют гексафторпропиленоксид до получения фторированного производного простого эфира (формула 3-4), а после этого фторированное производное простого эфира подвергают пиролизу с получением фторированного соединения (формула 2-1), описываемого формулой 2, где R представляет собой атом фтора.
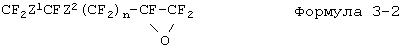
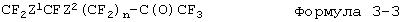

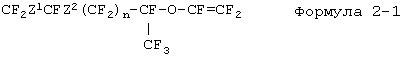
Для получения эпоксидного соединения (формула 3-2) можно воспользоваться способом с применением кислорода, описанным в работе "Chemistry of organic fluorine compound", 1962 edition, pp.168-169, edited by Hudlicky, способом с применением перекиси водорода, описанным в JP-B-44-2963, или же способом с применением водного раствора гипохлорита в присутствии межфазного катализатора.
В случае способа с применением водного раствора гипохлорита температура реакции, по меньшей мере, представляет собой температуру плавления водного раствора гипохлорита, обычно находящуюся в диапазоне от -20 до 60°С, предпочтительно от -20 до 30°С, хотя она может варьироваться в зависимости от использованного межфазного катализатора или от его количества. Количество межфазного катализатора предпочтительно находится в диапазоне от 0,01 до 20% (масс.), в особенности предпочтительно от 0,05 до 10% (масс.) в расчете на количество соединения, описываемого формулой 3-1. В качестве гипохлорита можно упомянуть соль щелочного металла или соль щелочноземельного металла, такие, как NaClO, KClO, Ca(ClO)2 или NaBrO. С промышленной точки зрения, предпочтительно использование NaClO. Эффективная концентрация гипохлорита в водном растворе гипохлорита предпочтительно находится в диапазоне от 1 до 20% (масс.).
В качестве межфазного катализатора могут быть использованы, например, соль четвертичного аммония, соль четвертичного фосфония, соль четвертичного арсония, соль сульфония или краун-эфир, известные в качестве межфазных катализаторов. Среди них предпочтительны соль четвертичного аммония и соль четвертичного фосфония. В качестве органической группы, присоединяемой к атому азота или атому фосфора, могут быть предпочтительны, например, алкильная группа, арильная группа или аралкильная группа, а в качестве аниона предпочтителен ион галогена, такой как ион хлора, или ион минеральной кислоты, такой, как сульфатный ион. В особенности предпочтительным межфазным катализатором является соль тетраалкиламмония.
Эпоксидное соединение (формула 3-2) подвергают реакции изомеризации в газовой фазе или в жидкой фазе с использованием в качестве катализатора соединения металла, такого, как оксид металла, оксигалогенид металла или галогенид металла, в результате чего можно получить фторированное производное кетона (формула 3-3). В качестве входящего в катализатор компонента, являющегося металлом, можно упомянуть, например, Al, Zr, Ti, Fe, Co, Ni, или Cr, а в особенности предпочтителен алюминий. Реакция, в которой фторированный эпоксид изомеризуют в присутствии катализатора, такого, как оксид алюминия или хлорид алюминия, с получением фторированного кетона, известна, и она описывается, например, в патенте США 3391119.
В настоящем изобретении, когда упомянутую выше реакцию изомеризации проводят в газовой фазе, в качестве катализатора можно использовать катализатор на основе оксида металла, такого, как γ-оксид алюминия. Однако более предпочтительным катализатором является оксигалогенид металла. Например, предпочтителен оксигалогенид металла, получаемый в результате активирования упомянутого выше оксида металла или оксида нескольких металлов фторуглеводородом. В качестве фторуглеводорода может быть упомянут, например, хлорфторуглеводород, такой, как трихлортрифторэтан, хлордифторметан, трихлорфторметан или дихлордифторметан.
Реакцию изомеризации по газофазному способу проводят в результате введения газообразного эпоксидного соединения (формула 3-2) в контакт с вышеупомянутым катализатором. Газообразное эпоксидное соединение может быть использовано для реакции разбавленным инертным газом, таким, как газообразный азот. Температура реакции предпочтительно представляет собой, по меньшей мере, температуру, при которой эпоксидное соединение превращается в пар, в особенности температуру, находящуюся в диапазоне от 100 до 300°С.
В настоящем изобретении, когда упомянутую выше реакцию изомеризации проводят в жидкой фазе, в качестве катализатора могут быть использованы вышеупомянутый оксигалогенид металла или вышеупомянутый галогенид металла. Галогенидом металла предпочтительно является галогенид, активированный фторуглеводородом тем же самым способом, что и упомянутый выше. В качестве растворителя - инертный растворитель, такой, как фторированный растворитель, растворитель, относящийся к типу простых эфиров, или апротонный полярный растворитель. Также возможно использование в качестве растворителя применяемого для активирования катализатора жидкого фторуглеводорода как такового. Количество катализатора предпочтительно находится в диапазоне от 0,005 до 20% (моль.), в особенности предпочтительно от 0,1 до 10% (моль.), в расчете на количество эпоксидного соединения (формула 3-2). Температура реакции предпочтительно находится в диапазоне от
-20 до +150°С, в особенности предпочтительно от 20 до 40°С.
Кроме этого, в качестве упомянутого выше растворителя, относящегося к типу простых эфиров, могут быть упомянуты, например, диэтиловый эфир, метил-трет-бутиловый эфир, диметоксиэтан, тетрагидрофуран, диоксан, моноглим, диглим, триглим или тетраглим. В качестве упомянутого выше апротонного полярного растворителя могут быть, например, упомянуты ацетонитрил, бензонитрил, сульфолан, диметилацетамид или диметилсульфоксид. Данные растворители также могут быть использованы в качестве растворителя, относящегося к типу простых эфиров, или апротонного полярного растворителя, которые будут упоминаться в последующем описании.
В растворителе фторид металла воздействует на фторированное производное кетона (формула 3-3), после чего проходит реакция с гексафторпропиленоксидом с получением фторированного производного простого эфира (формула 3-4). Температура реакции предпочтительно, самое большее, равна 50°С, в особенности предпочтительно она находится в диапазоне от 5 до 25°С. В качестве фторида металла могут быть, например, упомянуты фторид калия, фторид цезия или фторид натрия. В качестве растворителя для реакции предпочтительны растворитель, относящийся к типу простых эфиров, или апротонный полярный растворитель. Давление в реакции для гексафторпропиленоксида выгодно выдерживать в диапазоне от 0 до 1 МПа, а предпочтительно использование давления в диапазоне от 0,1 до 0,5 МПа.
Фторированное производное простого эфира (формула 3-4) подвергают пиролизу с получением фторированного соединения (формула 2-1), описываемого формулой 2, где R представляет собой атом фтора. Пиролиз, само собой разумеется, можно проводить путем пиролиза непосредственно фторированного производного простого эфира, или же фторированное производное простого эфира предварительно можно превратить в соль, образующуюся из щелочного металла и соответствующей карбоновой кислоты, а после этого подвергнуть гидролизу. Кроме этого фторированное производное простого эфира (формула 3-4) содержит активную группу (-COF), и после превращения такой активной группы в группу, устойчивую в условиях работы с данным соединением, его можно будет превратить в соль, образующуюся из щелочного металла и карбоновой кислоты. Например, его можно ввести в реакцию с алканолом с образованием алкилового сложного эфира соответствующей карбоновой кислоты, который после этого превращают в соль щелочного металла.
В случае, когда фторированное производное простого эфира подвергают пиролизу непосредственно, предпочитается, чтобы фторированное производное простого эфира находилось бы в газообразном состоянии и в случае необходимости было бы разбавлено инертным газом, таким, как газообразный азот, после чего его вводят в контакт с твердой основной солью или стеклянной дробью при высокой температуре. Температура для пиролиза предпочтительно находится в диапазоне от 200 до 500°С, в особенности предпочтительно от 250 до 350°С. В качестве твердой основной соли могут быть, например, использованы карбонат натрия, карбонат калия или фосфат натрия, а в особенности предпочтителен карбонат натрия.
Фторированное производное простого эфира (формула 3-4) можно ввести в реакцию с гидроксидом щелочного металла с получением соли, образующейся из щелочного металла и соответствующей карбоновой кислоты. Данную соль щелочного металла можно подвергнуть пиролизу при температуре в диапазоне от 100 до 300°С, предпочтительно от 150 до 250°С с получением желательного фторированного соединения. Предпочтительно использовать данный способ пиролиза с участием соли щелочного металла, потому что по сравнению с упомянутым выше газофазным способом пиролиза пиролиз в данном случае можно проводить при низкой температуре, а выход также будет высок. Кроме этого предпочитается, чтобы получение соли щелочного металла проводили бы с использованием воды или спирта в качестве растворителя, а полученную соль щелочного металла подвергали бы пиролизу после высушивания в достаточной степени.
Кроме этого в качестве соли щелочного металла могут быть упомянуты натриевая соль или калиевая соль, но калиевая соль предпочтительна, потому что пиролиз можно будет проводить при более низкой температуре.
Далее будет описан способ получения фторированного соединения (формула 2-2) из фторированного соединения, описываемого формулой 4-1. В положении углеродного атома карбонильной группы фторированного соединения, описываемого формулой 4-1, вводят две трифторметильные группы с получением фторированного спирта (формула 4-2), а к данному фторированному спирту добавляют гексафторпропиленоксид с получением фторированного производного простого эфира (формула 4-3) и после этого данное фторированное производное простого эфира подвергают пиролизу с получением фторированного соединения, описываемого формулой 2-2.



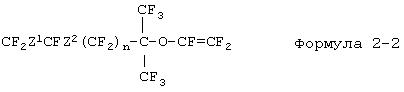
В качестве способа введения двух трифторметильных групп в положении углеродного атома карбонильной группы фторированного соединения, описываемого формулой 4-1, предпочтителен способ проведения реакции трифторметилтриметилсилана с фторированным соединением, описываемым формулой 4-1, в полярном растворителе в присутствии фторида металла или аммониевой соли фтористо-водородной кислоты. В качестве фторида металла предпочтителен фторид щелочного металла, такой, как фторид калия, фторид цезия или фторид натрия. Кроме того, в качестве аммониевой соли фтористо-водородной кислоты предпочтителен фторид тетрабутиламмония. Отношение количества фторида металла или аммониевой соли фтористо-водородной кислоты к количеству фторированного соединения, описываемого формулой 4-1, предпочтительно находится в диапазоне от 2 до 3 в расчете на моли, а отношение количества трифторметилтриметилсилана к количеству фторированного соединения, описываемого формулой 4-1, в расчете на моли предпочтительно находится в диапазоне от 2 до 2,5. Выгодно, чтобы температура для реакции, самое большее, была бы равна 30°С, предпочтительно находилась бы в диапазоне от -78 до +15°С. В качестве полярного растворителя могут быть упомянуты вышеупомянутые растворитель, относящийся к типу простых эфиров, или апротонный растворитель, а в особенности предпочтительны тетрагидрофуран или ацетонитрил.
В результате проведения вышеупомянутой реакции с использованием фторида металла получают алкоксид, образующийся из металла и фторированного спирта (формула 4-2). Данный алкоксид обрабатывают кислотой до получения фторированного спирта. В качестве такой кислоты предпочтительно можно использовать, например, концентрированную серную кислоту, разбавленную серную кислоту, концентрированную хлористо-водородную кислоту или разбавленную хлористо-водородную кислоту. Кроме этого данный алкоксид металла можно подавать на стадию последующей реакции без его превращения во фторированный спирт. А именно, последующую реакцию присоединения гексафторпропиленоксида можно проводить после превращения фторированного спирта в алкоксид металла, и, соответственно, такой алкоксид металла может быть использован как таковой.
В случае, когда к фторированному спирту (формула 4-2) для получения фторированного производного простого эфира (формула 4-3) присоединяют гексафторпропиленоксид, фторированный спирт обычно превращают в алкоксид металла, и последний после этого вводят в реакцию с гексафторпропиленоксидом. В качестве входящего в данный алкоксид металла компонента, являющегося металлом, могут быть, например, использованы щелочной металл или серебро. Например, в растворителе для проведения реакции фторированный спирт при комнатной температуре вводят в реакцию с основной солью щелочного металла (такой, как карбонат калия или карбонат натрия) с получением алкоксида металла. После этого полученный алкоксид металла вводят в реакцию с гексафторпропиленоксидом без его выделения из растворителя для проведения реакции или после его выделения и добавления для проведения реакции нового растворителя. В качестве условий для проведения реакции с гексафторпропиленоксидом могут быть использованы те же самые условия проведения реакции, что и в случае, когда к упомянутому выше фторированному производному кетона (формула 3-3) присоединяют гексафторпропиленоксид с получением фторированного производного простого эфира (формула 3-4).
Пиролиз фторированного производного простого эфира (формула 4-3) можно проводить с использованием того же самого способа, в тех же самых условиях проведения реакции, что и пиролиз вышеупомянутого фторированного производного простого эфира (формула 3-4). Например, возможно использование способа проведения пиролиза фторированного производного простого эфира (формула 4-3) в газовой фазе, как упоминалось выше, или способа превращения фторированного производного простого эфира (формула 4-3) в соль карбоновой кислоты, как упоминалось выше, с проведением пиролиза после этого. Также возможно, как упоминалось выше, превращение фторированного производного простого эфира (формула 4-3) в алкиловый сложный эфир соответствующей карбоновой кислоты, который после этого превращают в соль карбоновой кислоты, а данную соль карбоновой кислоты подвергают пиролизу.
Фторированный диен, описываемый формулой 1 настоящего изобретения, может быть заполимеризован, и он пригоден в качестве мономера для получения фторполимера. Такой фторированный диен подвергают циклополимеризации под действием инициатора радикальной полимеризации с получением полимера, содержащего в своей основной цепи мономерные звенья, содержащие фторированные алициклические структуры. Кроме этого его можно заполимеризовать вместе с другими мономерами.
На сополимеризуемые другие мономеры особенных ограничений не накладывается до тех пор, пока они будут представлять собой мономеры, полимеризуемые по радикальному механизму, и можно упомянуть широкий ассортимент фтормономеров, углеводородных мономеров и других мономеров. В особенности предпочтителен олефин, такой, как этилен, или фторолефин, такой, как тетрафторэтилен. Кроме этого также сополимеризуемы могут быть, например, мономер, относящийся к типу фторированных виниловых простых эфиров, такой, как перфтор (алкилвиниловый эфир), сополимеризуемый фторированный диен (отличный от фторированного диена, описываемого формулой 1), такой, как перфтор (бутенилвиниловый эфир) или перфтор (аллилвиниловый эфир), или же мономер, содержащий фторированную алициклическую структуру, такой, как перфтор (2,2-диметил-1,3-диоксол). Такие другие мономеры могут быть сополимеризованы с фторированным диеном индивидуально или в комбинации, состоящей из двух или более их представителей.
Настоящее изобретение также относится к гомополимеру вышеупомянутого фторированного диена настоящего изобретения или сополимеру, образованному из двух или более таких фторированных диенов, и сополимеру вышеупомянутого фторированного диена настоящего изобретения с другими мономерами, сополимеризуемыми с ним. Доля мономерных звеньев, образованных в результате полимеризации фторированного диена настоящего изобретения, в таких полимерах предпочтительно находится в диапазоне от 30 до 100% (моль.), в особенности предпочтительно от 50 до 100% (моль.) в расчете на полное количество мономерных звеньев. Кроме этого, молекулярная масса предпочтительно находится в диапазоне от 500 до 100000, в особенности предпочтительно от 500 до 10000.
В качестве инициатора радикальной полимеризации можно использовать любой инициатор полимеризации, применяемый в обычной радикальной полимеризации, такой, как азосоединение, органический пероксид или неорганический пероксид. В качестве конкретных инициаторов радикальной полимеризации могут быть упомянуты следующие далее соединения. Диизопропилпероксидикарбонат, азосоединение, такое, как 2, 2'-азобис(2-амидинопропан)дигидрохлорид, 4,4'-азобис(4-цианопентановая кислота), 2,2'-азобис(4-метокси-2,4-диметилвалеронитрил) или 1,1'-азобис(1-циклогексанкарбонитрил), органический пероксид, такой, как бензоилпероксид, перфторбензоилпероксид, перфторнонаноилпероксид или пероксид метилэтилкетона, и неорганический пероксид, такой, как K2S2O8 или (NH4 )2S2O8.
На способ полимеризации также особенных ограничений не накладывается, и им могут быть, например, так называемая полимеризация в массе, где фторированный диен подвергают полимеризации непосредственно, растворная полимеризация, которую проводят во фторуглеводороде, хлорированном углеводороде, хлорированном фторуглеводороде, спирте, углеводороде или другом органическом растворителе, который обладает способностью растворять фторированный диен, суспензионная полимеризация, которую проводят в водной среде в присутствии или же в отсутствие подходящего органического растворителя, или эмульсионная полимеризация, которую проводят в водной среде в присутствии эмульгатора. На температуру и давление для проведения полимеризации особенных ограничений не накладывают, но предпочтительно и выгодно их устанавливать, принимая во внимание различные факторы, такие, как температура кипения фторированного диена, требуемый источник тепла и отвод теплоты полимеризации. Например, температуру полимеризации можно установить равной удобной температуре, находящейся в диапазоне от 0 до 200°С, в особенности предпочтительно от 30 до 100°С. Кроме этого в том, что касается давления при проведении полимеризации, то полимеризацию можно проводить при пониженном давлении или при повышенном давлении, а на практике ее с выгодой можно проводить при уровне давления в диапазоне от нормального давления до 10 МПа, более конкретно от нормального давления до 5 МПа.
В качестве характеристик полимера настоящего изобретения можно упомянуть то, что он отличается превосходной прозрачностью, он обладает высокими модулем упругости, текучестью и удлинением при разрыве, он жесток и обладает превосходной ударной прочностью, и он характеризуется высокой температурой стеклования и высокой теплостойкостью. Вследствие наличия таких характеристик полимер настоящего изобретения может быть использован в качестве полимерного материала для оптики, используемого для оптического волокна, оптического волновода или оптического передающего устройства, такого, как линза, которая по своей природе обладает превосходной теплостойкостью. Кроме этого полимер настоящего изобретения также отличается тем, что он оптически прозрачен и характеризуется показателем преломления, меньшим, чем у обычного прозрачного фторполимера. По этой причине его можно смешивать, например, с обычным прозрачным фторполимером, характеризующимся низким показателем преломления, таким, как CYTOP (торговое наименование, изготавливается компанией Asahi Glass Company, Limited) или Teflon AF (торговое наименование, изготавливается компанией Dupont), для получения оптического устройства, такого, как оптическое волокно или оптический волновод с превосходной оптической прозрачностью и высокими эксплуатационными характеристиками.
Оптическое волокно на основе пластика, в котором смесь, содержащую вещество, увеличивающее показатель преломления, примешанное к полимеру настоящего изобретения, используют в качестве сердечника, а полимер настоящего изобретения используют в качестве оболочки, отличается в особенности превосходной теплостойкостью. Такое оптическое волокно на основе пластика может относиться к типу со ступенчатым профилем или к типу с распределением показателя преломления. Полимер настоящего изобретения с выгодой можно использовать для любого типа, но в особенности он пригоден для оптического волокна на основе пластика, которое относится к типу с распределением показателя преломления. В качестве упомянутого выше вещества, увеличивающего показатель преломления, предпочтительно фторированное низкомолекулярное соединение, потому что в результате его использования прозрачность получаемой в результате смеси остается превосходной. В качестве такого фторированного низкомолекулярного соединения могут быть упомянуты, например, перфтор (трифенилтриазин), перфтор(1,3,5-трифенилбензол) или хлортрифторэтиленовый олигомер в качестве предпочтительных примеров. Кроме этого в качестве вещества, увеличивающего показатель преломления, можно использовать смесь двух или более таких соединений.
В качестве способов получения вышеупомянутого оптического волокна на основе пластика, которое относится к типу с распределением показателя преломления, можно упомянуть следующие способы. Способ, в котором получают изготовленный из полимера настоящего изобретения продукт, сформованный в виде цилиндрического столбика, где в области центральной оси имеется предписанная концентрация вещества, увеличивающего показатель преломления, и вещество, увеличивающее показатель преломления, диффундирует под действием термодиффузии из области центральной оси в радиальном направлении с получением распределения показателя преломления, и после этого полученный продукт, сформованный в виде цилиндрического столбика, используют в качестве предварительно сформованной заготовки для формования оптического волокна (JP-A-8-5848). Способ, в котором расплав полимера настоящего изобретения экструдируют и формуют, придавая форму волокна, с получением оптического волокна, при этом высококонцентрированному веществу, увеличивающему показатель преломления, дают возможность находиться в области центральной оси, а оптическое волокно получают в результате прохождения термической диффузии вещества, увеличивающего показатель преломления (JP-A-8-5848). Способ, в котором сформованный в виде цилиндрической трубки продукт получают из полимера настоящего изобретения, предварительно определенное количество вещества, увеличивающего показатель преломления, вводят в центральную область с последующим прохождением термодиффузии с получением заготовки, предварительно сформованной в виде цилиндрической трубки, характеризующейся наличием распределения показателя преломления, из которой формуют оптическое волокно (JP-A-8-334633).
Кроме этого, полимер настоящего изобретения растворим во фторированном растворителе, таком, как перфтор(2-бутилтетрагидрофуран), перфтороктан, перфторгексан, перфтор (трибутиламин), перфтор (трипропиламин), перфторбензол или дихлорпентафторпропан. Раствор, полученный в результате растворения полимера настоящего изобретения в таком растворителе, можно нанести в виде покрытия на субстрат, такой, как стеклянная или кремниевая пластина, используя для нанесения покрытия способы центрифугирования или распыления, а после этого растворитель выпаривают и проводят высушивание до получения тонкой пленки.
Кроме этого у полимера настоящего изобретения можно легко провести замещение концевой группы под действием, например, тепловой обработки или обработки газообразным фтором и, используя определенный способ обработки, можно модифицировать адгезионные свойства по отношению к различным субстратам. Например, полимер настоящего изобретения можно нагревать при температуре, по меньшей мере, равной 200°С, в присутствии воздуха, а после этого подвергать обработке в воде для введения в концевое положение карбоксильной группы. Кроме этого его можно ввести в реакцию с газообразным фтором для удаления концевой реакционно-способной функциональной группы, в результате чего может быть улучшена теплостойкость полимера.
Примеры
Далее будут описаны примеры настоящего изобретения. Настоящее изобретение данными примерами ни в коей мере не ограничивается.
Пример 1
Способ синтеза фторированного эпоксида.
В четырехгорлую стеклянную колбу вместимостью 10 л вводили 6330 г (13,0 моль) 15%-ного водного раствора гипохлорита натрия и 73,8 г хлорида триоктилметиламмония и при тщательном перемешивании проводили охлаждение до тех пор, пока температура внутри колбы не достигала величины, находящейся в диапазоне от 10 до 15°С. После этого сюда по каплям добавляли 1200 г (4,25 моль) CF2ClCFClCF2CF=CF2, синтезированного по способу, известному в литературе, так чтобы температуру внутри колбы выдерживать в диапазоне от 20 до 30°С. После этого, проводя отслеживание хода реакции при помощи газового хроматографа, реакцию проводили до тех пор, пока CF2ClCFClCF2CF=CF2 как исходный материал по существу не был израсходован. В результате разделения двух фаз отбирали и три раза промывали деионизированной водой продукт, образующий нижний слой, для удаления остаточного гипохлорита натрия. Не подвергавшийся переработке продукт дополнительно перегоняли для получения 828,7 г чистого фторированного эпоксида, описываемого следующей формулой (1,2-дихлор-4,5-эпокси-1,1,2,3,3,4,5,5-октафторпентан) (выход: 65%).

Пример 2
Способ синтеза CF2ClCFClCF2 (С=О)CF3.
В четырехгорлую стеклянную колбу вместимостью 2 л вводили 35 г (0,26 моль) хлорида алюминия, а для проведения активирования добавляли 70 г трихлорфторметана. Проводя тщательное перемешивание, по каплям добавляли 1470 г (4,93 моль) фторированного эпоксида, синтезированного в примере 1 так, чтобы температуру внутри колбы выдерживать в диапазоне от 20 до 30°С. После этого, проводя отслеживание хода реакции при помощи газового хроматографа, реакцию проводили при температуре реакции, находящейся в диапазоне от 20 до 40°С, до тех пор, пока исходный материал по существу не был израсходован. После этого не подвергавшийся переработке продукт выделяли при помощи фильтрования и подвергали перегонке для получения 1600 г чистого CF2 ClCFClCF2(С=О)CF3 (4,5-дихлор-1,1,1,3,3,4,5,5-октафтор-2-пентанон) (выход: 91%).
Пример 3
Способ синтеза CF2ClCFClCF2CF(CF3)OCF(CF3)COF.
В автоклав, изготовленный из сплава hastelloy, вместимостью 2 л вводили 18 г (0,31 моль) фторида калия и после этого проводили вакуумирование. После этого вводили 1150 г (3,86 моль) CF2ClCFClCF2(С=О)CF3 и 730 г тетраглима, затем при тщательном перемешивании проводили охлаждение до тех пор, пока температура внутри автоклава не достигала величины, находящейся в диапазоне от 0 до 5°С, и перемешивание продолжали при данной температуре в течение периода времени от 30 минут до 1 часа. После этого присоединяли баллон с гексафторпропиленоксидом и при поддержании внутреннего давления, равного приблизительно 0,2 МПа, и температуры внутри автоклава на уровне величины, самое большее равной 25°С, добавляли 640 г гексафторпропиленоксида. После этого гексафторпропиленоксид выдували и затем перемешивание продолжали при 25°С в течение периода времени от 1 до 2 часов. После этого автоклав открывали и остающуюся твердую фазу удаляли, используя фильтрование, и в результате фазового разделения отбирали не подвергавшийся переработке продукт. Данный не подвергавшийся переработке продукт дополнительно перегоняли с получением 1440 г чистого CF2ClCFClCF2CF(CF3)OCF(CF3)COF (фторангидрид 6,7-дихлор-2,4-бис(трифторметил)-2,4,5,5,6,7, 7-гептафтор-3-оксагептановой кислоты) (выход: 80%).

19F-ЯМР (стандарт CDCl3, CFCl3) δ м.д.: от -126,5 до
-135,5 (Fb, Fd, Fe, 3F), от -109 до -115,5 (Fc, 2F), -81,8 (Fg, 3F), от -77 до 78,5 (Fh, 3F), от -62,0 до -66,0 (Fa, 2F), от 26,9 до 28,4 (Ff, 1F).
Температура кипения: 68°С/5,3 кПа.
Пример 4
Способ синтеза CF2ClCFClCF2CF(CF3)OCF=CF2.
В четырехгорлую стеклянную колбу вместимостью 2 л вводили 607 г (13,2 моль) этанола и охлаждали до тех пор, пока температура внутри колбы не достигала величины, находящейся в диапазоне от 5 до 10°С. При тщательном перемешивании и поддержании температуры внутри колбы в диапазоне от 5 до 20°С по каплям добавляли 1388 г (2,99 моль) CF2ClCFClCF2CF(CF3)OCF(CF3)COF. После этого в течение некоторого времени продолжали перемешивание при комнатной температуре и добавляли 720 г деионизированной воды с последующим тщательным перемешиванием. В результате разделения двух фаз отбирали продукт, образующий нижний слой. После этого данный продукт переносили в стеклянную делительную склянку вместимостью 5 л и охлаждали до тех пор, пока температура внутри склянки не достигала величины, находящейся в диапазоне от 5 до 10°С. При тщательном перемешивании и поддержании температуры внутри склянки в диапазоне от 5 до 20°С по каплям добавляли 15%-ный раствор гидроксида калия в этаноле. После этого этанол как растворитель отгоняли при пониженном давлении, а полученную твердую соль измельчали в ступке и после этого высушивали при 80°С в течение двух дней в вакуумной сушилке до получения 1480 г (2,96 моль) CF2ClCFClCF2CF(CF3)OCF(CF3)CO2К.
После этого C четырехгорлую стеклянную колбу вместимостью 2 л вводили 970 г (1,94 моль) CF2 ClCFClCF2CF(CF3)OCF(CF3)CO2К и проводили нагревание в вакууме до тех пор, пока температура внутри колбы не достигала величины, находящейся в диапазоне от 190 до 200°С, для проведения пиролитической реакции. Продукт извлекали, используя ловушку с сухим льдом на стороне вакуумного насоса. Не подвергавшийся переработке продукт дополнительно перегоняли для получения 688 г чистого CF2ClCFClCF2CF(CF3)OCF=CF2 (6,7-дСхлор-1,1,2,4,5,5,6,7,7-нонафтор-4-трифторметил-3-окса-1-гептен) (выход: 89%).
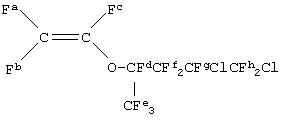
19F-ЯМР (стандарт CDCl3, CFCl3) δ м.д.: от -134,5 до
-130,5 (Fd, Fg, 2F), -134,1 (Fa, 1F, Jac=113 Гц), -121 (Fc, 1F, Jbc=166 Гц), -113,9 (Fb, 1F, Jab=65 Гц), от -111 до 115,5 (Ff, 2F), -78,1 (Fe, 3F), от -62 до -65 (Fh, 2F).
Температура кипения: 63°С/6,7 кПа.
Пример 5
Синтез CF2=CFCF2CF(CF3)OCF=CF2.
В четырехгорлую стеклянную колбу, имеющую вместимость 2 л и оснащенную мешалкой, парциальным конденсатором горячего орошения и капельной воронкой, вводили 207 г (3,17 моль) цинка и в атмосфере инертного газа вводили 975 г диметилформамида. После этого систему вакуумировали до 27 кПа и затем температуру внутри колбы доводили до величины, находящейся в диапазоне от 55 до 60°С, после чего из капельной воронки медленно по каплям добавляли 516 г (1,27 моль) CF2ClCFClCF2CF(CF3)OCF=CF2. В ходе реакции продукт отгоняли и, таким образом, быстро выводили из зоны реакции. После этого не подвергавшийся переработке продукт ректифицировали до получения 348 г чистого CF2=CFCF2CF(CF3)OCF=CF2(1,1,2,4,5,5,6,7,7-нонафтор-4-трифторметил-3-окса-1,6-гептадиен) (выход: 84%).

19F-ЯМР (стандарт CDCl3, CFCl3) δ м.д.: -187,3 (Fg , 1F, Jgh=39 Гц), -140,7 (Fd, 1F), -132,7 (Fc, 1F), -121,1 (Fb, 1F, Jbc=111 Гц), -177,4 (Ff, 2F), -113,5 (Fa, 1F, Jab=83 Гц, Jac=65 Гц), -104,2 (Fi, 1F, Jgi=116 Гц), -87,7 (Fh, 1F, Jhi=50 Гц),
-78,9 (Fe, 3F).
ИК: 1,785 см-1 (CF2=CF-), 1,835 см-1 (CF2=CFO-).
Температура кипения: 54,5°С/33,3 кПа.
Пример 6
Способ синтеза CF2ClCFClCF2С(CF3)2OH.
В четырехгорлую стеклянную колбу, имеющую вместимость 5 л и оснащенную мешалкой, капельной воронкой и трехходовым запорным краном, вводили 134 г (2,3 моль) фторида калия и в атмосфере инертного газа вводили 1500 мл тетрагидрофурана. После этого температуру внутри колбы доводили до -78°С, после чего сюда по каплям прибавляли 200 г (0,8 моль) CF2ClCFClCF2OF, синтезированного по способу, известному из литературы, и 265 г (1,86 моль) трифторметилтриметилсилана. Затем температуру внутри колбы медленно увеличивали до 0°С и при 0°С в течение 12 часов проводили реакцию.
По окончании реакции растворитель и низкокипящие продукты отгоняли при пониженном давлении, а органический компонент в остающейся твердой фазе экстрагировали диэтиловым эфиром. После этого диэтиловый эфир отгоняли при пониженном давлении. Основной компонент остающейся твердой фазы коричневого цвета представлял собой CF2ClCFClCF2С(CF3)2OK, а выход образовавшейся твердой фазы был равен 220 г.
К 220 г полученной твердой фазы при охлаждении на льду медленно по каплям прибавляли 200 мл концентрированной серной кислоты и перемешивание продолжали при комнатной температуре в течение периода времени от 1 до 2 часов. После этого раствор реакционной смеси медленно выливали в 500 мл воды, охлажденной льдом, а затем проводили экстрагирование диэтиловым эфиром. Органический слой отделяли и после этого высушивали над сульфатом магния. Диэтиловый эфир отгоняли при пониженном давлении. Таким образом полученный не подвергавшийся переработке продукт очищали перегонкой до получения 89 г CF2 ClCFClCF2С(CF3)2OH (выход: 30C в расчете на CF2ClCFClF2OF).

19F-ЯМР (стандарт CDCl3, CFCl3) δ м.д.: -131,9 (Fb, 1F),
-107,5 (Fc, 2F), -72, 4 и -71,9 (Fe, каждый 3F), -63,2 (Fa, 2F).
Пример 7
Способ синтеза CF2ClCFClCF2С(CF3)2OCF(CF3 )CO2CHC.
В четырехгорлую стеклянную колбу, имеющую вместимость 200 мл и оснащенную мешалкой, парциальным конденсатором горячего орошения и капельной воронкой, вводили 59 г (181 ммоль) карбоната цезия и 110 мл диметоксиэтана и тщательно перемешивали в атмосфере инертного газа. Выдерживая температуру внутри колбы в диапазоне от 0 до 10°С, по каплям прибавляли 44,5 г (121 ммоль) CF2ClCFClCF2С(CF3)2OH. После завершения прикапывания перемешивание продолжали в течение 4 часов при температуре внутри колбы, равной 25°С. После этого растворитель отгоняли при пониженном давлении до получения твердой соли коричневого цвета CF2ClCFClCF2С(CF3)2OCs.
После этого CF2ClCFClCF2С(CF3)2OCs растворял в 160 мл ацетонитрила, а раствор помещали в автоклав, изготовленный из сплава hastelloy, вместимостью 200 мл, вакуумировали и при тщательном перемешивании охлаждали до тех пор, пока температура внутри автоклава не достигала величины, находящейся в диапазоне от 0 до 5°С. После этого присоединяли баллон с гексафторпропиленоксидом и при поддержании внутреннего давления, равного приблизительно 0,2 МПа, и температуры внутри автоклава на уровне величины, самое большее равной 25°С, добавляли 29,5 г (178 ммоль) гексафторпропиленоксида. После этого перемешивание продолжали в течение одного часа при температуре в диапазоне от 0 до 5°С и в течение периода времени от 1 до 2 часов при 30°С. После этого автоклав открывали и остающуюся твердую фазу удаляли, используя фильтрование, и в результате фазового разделения отбирали не подвергавшийся переработке продукт. Данный продукт представлял собой смесь продуктов, которыми были CF2ClCFClCF2С(CF3)2OCF(CF3)COF и CF2ClCFClCF2С(CF3)2 OCF(CF3)CO2C(CF3)2CF2CFClCF2Cl. Данный продукт обрабатывали метанолом в присутствии фторида калия и экстрагировали при помощи системы растворитель дихлорпентафторпропан (здесь и далее в настоящем документе называемый R225) - вода. Органический слой отделяли и после этого обезвоживали над сульфатом магния с последующей отгонкой растворителя с получением не подвергавшегося переработке продукта. Не подвергавшийся переработке продукт дополнительно перегоняли для удаления низкокипящих примесей до получения 30 г чистого CF2ClCFClCF2С(CF3)2OCF(CF3)CO2CH3 (метил-C,7-дихлор-2,4,4-тристрифторметил-2,5,5,6,7,7-гексафтор-3-оксагептаноат) (выход: 47%).
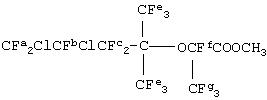
19F-ЯМР (стандарт CDCl3, CFCl3) δ м.д.: от -133 до -129,5 (Fb и Ff, 2F), от -110 до -97 (Fc, 2F), -82,1 (Fg, 3F), -66,9 и -65,9 (Fe, каждый 3F), от -65,0 до -62,5 (Fa, 2F).
1H-ЯМР (стандарт CDCl3, Si(CH3)4 δ м.д.: 3,95 (3H).
Пример 8
Способ синтеза CF2 ClCFClCF2С(CF3)2OCF=CF2.
В стеклянную делительную склянку, имеющую вместимость 200 мл и оснащенную мешалкой, парциальным конденсатором горячего орошения и капельной воронкой вводили 28 г (53 ммоль) CF2ClCFClCF2С(CF3)2OCF(CF3)CO2CH3 и проводили охлаждение до тех пор, пока температура внутри склянки не достигала величины, находящейся в диапазоне от 5 до 10°С, и при тщательном перемешивании и поддержании температуры внутри склянки в диапазоне от 5 до 20°С по каплям прибавляли 20 г 15%-ного раствора гидроксида калия в этаноле. После этого этанол как растворитель отгоняли при пониженном давлении, а полученную твердую соль измельчали в ступке и высушивали в течение двух дней при 80°С в вакуумной сушилке до получения 27,5 г (50 ммоль) CF2ClCFClCF2С(CF3)2OCF(CF3)CO2 К.
После этого в трехгорлую стеклянную колбу вместимостью 100 мл вводили 15 г (27 ммоль) CF2ClCFClCF2С(CF3)2OCF(CF3)CO2 К и проводили нагревание в вакууме до тех пор, пока температура внутри колбы не достигала величины, находящейся в диапазоне от 150 до 170°С, для проведения пиролитической реакции. Продукт извлекали, используя ловушку с сухим льдом на стороне вакуумного насоса. Не подвергавшийся переработке продукт перегоняли дополнительно до получения 3,7 г чистого CF2ClCFClCF2 С(CF3)2OCF=CF2 (6,7-дихлор-1,1,2,5,5,6,7,7-октафтор-4,4'-бис(трифторметил)-3-окса-1-гептен) (выход: 30%).
После этого считается возможным синтезировать CF2=CFCF2С(CF3)2OCF=CF2 в результате дехлорирования продукта тем же самым способом, что и в примере 3.
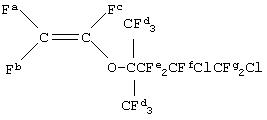
19F-ЯМР (стандарт CDCl3, CFCl3) δ м.д.: -131,7 (Fa, 1F, Jac=83 Гц), -131,5 (Ff, 1F), -119,6 (Fc, 1F, Jbc= 111 Гц),
-114,8 (Fb, 1F, Jac= 65 Гц), от -110 до -100 (Fe, 2F), -68,0 и -67,6 (Fd, каждый 3F), -65,0 и -62,0 (Fg, 2F).
Температура кипения: 63°С/2,7 кПа.
Пример 9
Полимеризация CF2=CFCF2CF(CF3)OCF=CF2.
2 г CF2=CFCF2CF(CF3)OCF=CF2 и 6,2 мг диизопропилпероксидикарбоната помещали в стеклянную ампулу, замораживали в жидком азоте, деаэрировали в вакууме и после этого герметично запаивали. После нагревания при 40°С в течение 20 часов в печи отвержденное содержимое вынимали и высушивали при 200°С в течение одного часа. Выход полученного полимера (здесь и далее в настоящем документе называемого полимером А1) составлял 99%. Часть полимера А1 растворяли в перфтор (2-бутилтетрагидрофуране) (здесь и далее в настоящем документе называемом PBTHF) и проводили измерение характеристической вязкости, которая оказалась равна 0,44 дл/г. Молекулярная масса полимера была равна 131500 в случае среднечисленной молекулярной массы (Mn) и 263000 в случае среднемассовой молекулярной массы (Mw).
Пленка полимера А1, полученная в результате прессования, характеризовалась показателем преломления, равным 1,327 при измерении на рефрактометре Аббе, и температурой стеклования, равной 124°С при измерении по методу динамического термомеханического анализа (ДМА). Измеряли механические свойства полимера А1 при растяжении, при этом модуль упругости при растяжении был равен 1430 МПа, предел текучести был равен 36 МПа, а относительное удлинение при разрыве было равно 4,2%. Кроме этого при помощи аппарата для измерения вязкоупругих свойств у вращающегося расплава при 230°С измеряли вязкость при нулевом сдвиге, которая была получена равной 89000 Па*сек. Температура стеклования полимера, полученного в результате полимеризации мономера CF2=CFCF2CF2OCF=CF2 (перфторбутенилвиниловый эфир, «здесь и далее в настоящем документе называемый PBVE») при тех же самых условиях, была равна 108°С при измерении по методу динамического термомеханического анализа (ДМА), таким образом, для случая настоящего полимера А1 в сопоставлении с обычно используемым полимером было подтверждено улучшение температуры стеклования.
Кроме этого измеряли спектр инфракрасного поглощения полимера, при этом было обнаружено, что поглощения в области 1785 см-1, приписываемое CF2=CF-, и в области 1835 см-1, приписываемое CF2=CFO-, которые наблюдались у мономера, исчезали. Данный полимер А1 не содержал двойных связей в боковых группах и не подвергался реакции сшивания, и было обнаружено, что он представляет собой полимер, полученный в результате циклизации, потому что его можно было полностью растворить в R225 (дихлорпентафторпропан) даже при высокой степени превращения. Кроме этого согласно данным анализа по методу19 F-ЯМР было установлено, что он представляет собой полимер, содержащий повторяющиеся звенья, описываемые следующей структурой:

Пример 10
2 г CF2=CFCF2CF(CF3)OCF=CF2 и 5,0 мг перфторбензоилпероксида помещали в стеклянную ампулу, замораживали в жидком азоте, деаэрировали в вакууме и после этого герметично запаивали. После нагревания при 70°С в течение 20 часов в печи отвержденное содержимое вынимали и высушивали в вакууме при 100°С в течение 10 часов. Выход полученного полимера (здесь и далее в настоящем документе называемого полимером А2) составлял 96%. Часть полимера А2 растворяли в PBTHF и проводили измерение характеристической вязкости, которая оказалась равна 0,77 дл/г. Пленка полимера А2, полученная в результате прессования, характеризовалась показателем преломления, равным 1,329 при измерении на рефрактометре Аббе, и температурой стеклования, равной 124°С при измерении по методу динамического термомеханического анализа (ДМА). Измеряли механические свойства полимера А2 при растяжении, при этом модуль упругости при растяжении был равен 1370 МПа, предел текучести был равен 38 МПа, а относительное удлинение при разрыве было равно 7,0%. Кроме этого полимер CF2=CFCF2CF(CF3)OCF=CF2 обладал превосходной прозрачностью, и он оказался пригодным в качестве полимерного материала для оптики, например для оптического волокна или оптического волновода.
Пример 11
В автоклав из нержавеющей стали вместимостью 200 мл вводили 80 г воды, 15 г (45,7 ммоль) CF2=CFCF2CF(CF3 )OCF=CF2, 38 мг перфторбензоилпероксида и 2,4 г метанола. Автоклав продували азотом и после этого нагревали до тех пор, пока температура внутри автоклава не становилась равной 70°С, после чего полимеризацию проводили в течение 20 часов. Полученный полимер промывали деионизированной водой и метанолом и после этого высушивали при 200°С в течение одного часа. Выход полученного полимера (здесь и далее называемого полимером А3) был равен 83%.
Часть полимера А3 растворяли в PBTHF и проводили измерение характеристической вязкости, которая оказалась равна 0,31 дл/г. Пленка полимера А3, полученная в результате прессования, характеризовалась показателем преломления, равным 1,328 при измерении на рефрактометре Аббе, и температурой стеклования, равной 124°С при измерении по методу дифференциальной сканирующей калориметрии (ДСК). Измеряли механические свойства полимера А3 при растяжении, при этом модуль упругости при растяжении был равен 1280 МПа, предел текучести был равен 38 МПа, а относительное удлинение при разрыве было равно 5,1%. Кроме этого при помощи аппарата для измерения вязкоупругих свойств у вращающегося расплава при 230°С измеряли вязкость при нулевом сдвиге, которая была получена равной 5200 Па*сек.
Пример 12
Сополимеризация CF2=CFCF2CF(CF3 )OCF=CF2 с тетрфторэтиленом.
В автоклав из нержавеющей стали вместимостью 200 мл вводили 80 мл R225, 5,6 г (17 ммоль) CF2=CFCF2CF(CF3 )OCF=CF2 и 0,02C г пероксида перфторбензойной кислоты. Автоклав вакуумировали вакуумным насосом при охлаждении жидким азотом, затем вакуумный насос отсоединяли для того, чтобы дать температуре возможность вернуться к значению комнатной температуры, а после этого автоклав снова вакуумировали вакуумным насосом при охлаждении жидким азотом. Данную операцию повторяли три раза. После этого температуру внутри автоклава возвращали к значению комнатной температуры и затем вводили 32 г (320 ммоль) тетрафторэтилена. А автоклав нагревали до тех пор, пока температура внутри автоклава не становилась равной 70°С, и проводили полимеризацию в течение 3 часов. После этого остаточный тетрафторэтилен выдували, а остаточный мономер отгоняли при пониженном давлении до получения 30 г белого полимера (здесь и далее в настоящем документе называемого полимером В1). Анализировали структуру полученного полимера В1, при этом было установлено, что структура, полученная из CF2 =CFCF2CF(CF3)OCF=CF2, была введена в количестве 2% (моль.) в часть, образуемую политетрафторэтиленом.
Пример 13
Сополимер CF2 =CFCF2CF(CF3)OCF=CF2 и PBVE.
В автоклав из нержавеющей стали вместимостью 200 мл вводили 80 г воды, 15 г CF2=CFCF2CF(CF3)OCF=CF2, 15 г CBVE, 75 мг перфторбензоилпероксида и 2,4 г метанола. Автоклав продували азотом и после этого нагревали до тех пор, пока температура внутри автоклава не становилась равной 70°С, после чего полимеризацию проводили в течение 20 часов. Полученный полимер (здесь и далее в настоящем документе называемый полимером В2) промывали деионизированной водой и метанолом и после этого высушивали при 200°С в течение одного часа. Выход полученного полимера В2 был равен 85%.
Часть полимера В2 растворяли в PBTHF и проводили измерение характеристической вязкости, которая оказалась равна 0,35 дл/г. Пленка полимера В2, полученная в результате прессования, характеризовалась показателем преломления, равным 1,336 при измерении на рефрактометре Аббе, и температурой стеклования, равной 116°С при измерении по методу динамического термомеханического анализа (ДМА).
Пример 14
Сополимеризация CF2=CFCF2CF(CF3)OCF=CF2 с перфтор (2,2-диметил-1,3-диоксолом).
В автоклав из нержавеющей стали вместимостью 200 мл вводили 80 г воды, 21 г CF2=CFCF2CF(CF3)OCF=CF2, 9 г перфтор (2,2-диметил-1,3-диоксола) (здесь и далее в настоящем документе называемого "PDD"), 75 мг диизопропилпероксидикарбоната и 2,4 г метанола. Автоклав продували азотом и после этого нагревали до тех пор, пока температура внутри автоклава не становилась равной 40°С, после чего полимеризацию проводили в течение 20 часов. Полученный полимер (здесь и далее в настоящем документе называемый полимером В3) промывали деионизированной водой и метанолом и после этого высушивали при 200°С в течение одного часа. Выход полученного полимера В3 был равен 90%.
Часть полимера В3 растворяли в PBTHF и проводили измерение характеристической вязкости, которая оказалась равна 0,40 дл/г. Пленка полимера В3, полученная в результате прессования, характеризовалась показателем преломления, равным 1,315 при измерении на рефрактометре Аббе, и температурой стеклования, равной 167°С при измерении по методу динамического термомеханического анализа (ДМА).
Пример 15
93 части полимера А1, полученного в примере 9, и 7 частей перфтор(трифенилтриазина) помещали в стеклянную ампулу, герметично запаивали и после этого смешивали в расплаве при 240°С до получения однородной полимерной смеси (здесь и далее в настоящем документе называемой смесью С1). Пленка смеси С1, полученная в результате прессования, характеризовалась показателем преломления, равным 1,349 при измерении на рефрактометре Аббе, и температурой стеклования, равной 102°С при измерении по методу динамического термомеханического анализа (ДМА).
После этого в соответствии со способом, описанным в JP-A-8-5848, изготавливали оптическое волокно, используя смесь С1 и полимер А1. А именно, сначала в герметично запаянной стеклянной трубке расплавляли смесь С1 и получали продукт С1а, сформованный в виде цилиндрического столбика. После этого из расплава исключительно полимера А1 формовали цилиндрическую трубку и в полую область данной цилиндрической трубки вставляли сформованный продукт С1а, а для получения единой предварительно сформованной заготовки проводили нагревание при 220°С. Расплав данной предварительно сформованной заготовки при 240°С формовали с получением оптического волокна, в котором показатель преломления постепенно уменьшался от центральной области в направлении области периферии.
По способу сокращения измеряли коэффициент ослабления для полученного оптического волокна, который оказался равным 192 дБ/км при 650 нм, 109 дБ/км при 850 нм и 81 дБ/км при 1300 нм, тем самым было подтверждено, что данное оптическое волокно способно превосходно передавать свет в диапазоне от видимого света до света из ближней инфракрасной области.
Данное оптическое волокно нагревали и хранили в печи при 70°С в течение 10000 часов, а затем вынимали, после чего измеряли распределение показателя преломления при помощи интерференционного микроскопа interfaco и сравнивали его с распределением показателя преломления до тепловой обработки, и при этом никаких изменений не наблюдалось. Кроме этого импульсным методом измеряли ширину полосы пропускания для оценки характеристик пропускания. Ширину полосы пропускания измеряли после нагревания и хранения оптического волокна при 70°С в течение 10000 часов, при этом она соответствовала 360 МГц*км как до, так и после хранения при нагревании, и никакого уменьшения ширины полосы пропускания не происходило, таким образом, было подтверждено, что теплостойкость превосходна.
Пример 16
При помощи экструдера формовали оптическое волокно, относящееся к типу сердечник-оболочка, проводя концентрично двухтоновое экструдирование так, чтобы полимер PBVE (характеристическая вязкость: 0,27 дл/г, показатель преломления: 1,342) располагался бы в серединной части, а полимер А3 располагался бы в области периферии. Наружный диаметр полученного оптического волокна составлял 510 мкм, а диаметр сердечника был равен 490 мкм. Кроме этого по способу сокращения измеряли коэффициент ослабления, который оказался равным 146 дБ/км при 650 нм, 85 дБ/км при 850 нм и 71 дБ/км при 1300 нм, тем самым было подтверждено, что данное оптическое волокно способно превосходно передавать свет в диапазоне от видимого света до света из ближней инфракрасной области.
Пример 17
У предварительно сформованной заготовки, полученной в примере 15, дополнительно создавали покрытие, используя полую трубку, изготовленную из полимера В3, с последующим формованием из расплава при 240°С с получением оптического волокна, в котором показатель преломления постепенно уменьшается от центральной области в направлении области периферии. По способу сокращения измеряли коэффициент ослабления для полученного оптического волокна, который оказался равным 142 дБ/км при 650 нм, 59 дБ/км при 850 нм и 31 дБ/км при 1300 нм, тем самым было подтверждено, что данное оптическое волокно способно превосходно передавать свет в диапазоне от видимого света до света из ближней инфракрасной области. Кроме этого при 850 нм для данного оптического волокна при радиусе изгиба 10 мм измеряли увеличение коэффициента ослабления, которое оказалось равным 0,13 дБ, тем самым было установлено, что для оптического волокна имеют место небольшие потери на изгибах.
Оптическое волокна нагревали и хранили в печи при 70°С в течение 10000 часов, а затем измеряли коэффициент ослабления, и при этом никаких изменений не наблюдалось. Кроме этого импульсным методом измеряли ширину полосы пропускания для оценки характеристик пропускания. Ширину полосы пропускания измеряли после нагревания и хранения оптического волокна при 70°С в течение 10000 часов, при этом она соответствовала 280 МГц*км как до, так и после хранения при нагревании, и никакого уменьшения ширины полосы пропускания не происходило, таким образом, было подтверждено, что теплостойкость хорошая.
Пример 18
PDD и тетрафторэтилен подвергали радикальной полимеризации при массовом соотношении 80:20 с использованием в качестве растворителя PBTHF и получали полимер, у которого Tg была равна 160°С, а среднечисленная молекулярная масса составляла приблизительно 1,7х105. Данный полимер подвергали тепловой обработке при 250°С в течение 5 часов в атмосфере газовой смеси фтор/азот (концентрация газообразного фтора: 20% (об.)) с получением полимера (здесь и далее в настоящем документе называемого полимером D1) с хорошими коэффициентом пропускания света и теплостойкостью. Полимер D1 бесцветен и прозрачен, а его показатель преломления равен 1,305.
Пример 19
При помощи экструдера формовали оптическое волокно, относящееся к типу сердечник-оболочка, проводя концентрично двухтоновое экструдирование так, чтобы полимер А1 располагался бы в серединной части, а полимер D1 располагался бы в области периферии. Наружный диаметр полученного оптического волокна составлял 980 мкм, а диаметр сердечника был равен 900 мкм. Кроме этого по способу сокращения измеряли коэффициент ослабления, который оказался равным 186 дБ/км при 650 нм, 95 дБ/км при 850 нм и 71 дБ/км при 1300 нм, тем самым было подтверждено, что данное оптическое волокно способно превосходно передавать свет в диапазоне от видимого света до света из ближней инфракрасной области.
Пример 20
92,5 части полимера А1 и 7,5 части перфтор(1,3,5-трифенилбензола) помещали в стеклянную ампулу, герметично запаивали и после этого смешивали в расплаве при 250°С до получения однородной полимерной смеси (здесь и далее в настоящем документе называемой смесью С2). Пленка смеси С2, полученная в результате прессования, характеризовалась показателем преломления, равным 1,358 при измерении на рефрактометре Аббе, и температурой стеклования, равной 98°С при измерении по методу динамического термомеханического анализа (ДМА).
После этого изготавливали оптическое волокно, используя смесь С2 и полимер А1. А именно, прежде всего в герметично запаянной стеклянной трубке расплавляли смесь С2 и получали продукт С2а, сформованный в виде цилиндрического столбика. Затем из расплава исключительно полимера А1 формовали цилиндрическую трубку и в полую область данной цилиндрической трубки вставляли сформованный продукт С2а и для получения единой предварительно сформованной заготовки проводили нагревание при 220°С. Расплав данной предварительно сформованной заготовки при 240°С формовали с получением оптического волокна, в котором показатель преломления постепенно уменьшался от центральной области в направлении области периферии.
По способу сокращения измеряли коэффициент ослабления для полученного волокна, который оказался равным 180 дБ/км при 650 нм, 92 дБ/км при 850 нм и 80 дБ/км при 1300 нм, тем самым было подтверждено, что данное оптическое волокно способно превосходно передавать свет в диапазоне от видимого света до света из ближней инфракрасной области.
Данное оптическое волокно нагревали и хранили в печи при 70°С в течение 2000 часов, а затем вынимали, после чего измеряли распределение показателя преломления при помощи интерференционного микроскопа Interfaco и сравнивали его с распределением показателя преломления до хранения при нагревании, и при этом никаких изменений не наблюдалось. Кроме этого импульсным методом измеряли ширину полосы пропускания для оценки характеристик пропускания. Ширину полосы пропускания измеряли после нагревания и хранения оптического волокна при 70°С в течение 2000 часов, при этом она соответствовала 320 МГц*км как до, так и после хранения при нагревании, и никакого уменьшения ширины полосы пропускания не происходило, таким образом, было подтверждено, что теплостойкость хорошая.
Пример 21
85 частей полимера А1 и 15 частей хлортрифторэтиленового олигомера помещали в стеклянную ампулу, герметично запаивали и после этого смешивали в расплаве при 250°С до получения однородной полимерной смеси (здесь и далее в настоящем документе называемой смесью С3). Пленка смеси С3, полученная в результате прессования, характеризовалась показателем преломления, равным 1,356 при измерении на рефрактометре Аббе, и температурой стеклования, равной 90°С при измерении по методу динамического термомеханического анализа (ДМА).
После этого изготавливали оптическое волокно, используя смесь С3 и полимер А1. А именно, прежде всего в герметично запаянной стеклянной трубке расплавляли смесь С3 и получали продукт С3а, сформованный в виде цилиндрического столбика. Затем из расплава исключительно полимера А1 формовали цилиндрическую трубку и в полую область данной цилиндрической трубки вставляли сформованный продукт С3а и для получения единой предварительно сформованной заготовки проводили нагревание при 220°С. Расплав данной предварительно сформованной заготовки при 240°С формовали с получением оптического волокна, в котором показатель преломления постепенно уменьшался от центральной области в направлении области периферии.
По способу сокращения измеряли коэффициент ослабления для полученного волокна, который оказался равным 120 дБ/км при 650 нм, 68 дБ/км при 850 нм и 50 дБ/км при 1300 нм, тем самым было подтверждено, что данное оптическое волокно способно превосходно передавать свет в диапазоне от видимого света до света из ближней инфракрасной области.
Данное оптическое волокно нагревали и хранили в печи при 70°С в течение 1000 часов, а затем вынимали, после чего измеряли распределение показателя преломления при помощи интерференционного микроскопа interfaco и сравнивали его с распределением показателя преломления до хранения при нагревании, и при этом никаких изменений не наблюдалось. Кроме этого, импульсным методом измеряли ширину полосы пропускания для оценки характеристик пропускания. Ширину полосы пропускания измеряли после нагревания и хранения оптического волокна при 70°С в течение 1000 часов, при этом она соответствовала 330 МГц*км как до, так и после хранения при нагревании, и никакого уменьшения ширины полосы пропускания не происходило, таким образом, было подтверждено, что теплостойкость хорошая.
ВОЗМОЖНОСТЬ ПРИМЕНЕНИЯ В ПРОМЫШЛЕННОСТИ
По сравнению с обычно используемым полимером фторированного диена, не содержащим боковой цепи, полимер настоящего изобретения отличается высокой температурой стеклования и такими же или более высокими оптическими свойствами, такими, как прозрачность. Соответственно этому, полимер настоящего изобретения пригоден в качестве полимерного материала для оптики с превосходной теплостойкостью, и он представляет собой полимерный материал с превосходными свойствами в особенности в том, что касается материала для оптического волокна на основе пластика.
Реферат
Использование: получение оптических материалов и полимерных соединений. Сущность: описан новый фторированный диен CF2=CF(CF2)nС(CF3)ROCF=CF2(где R представляет собой атом фтора или трифторметильную группу, а n представляет собой целое число в диапазоне от 1 до 3) и способ его получения дегалогенированием различных соединений. Также описан полимер, мономером которого является вышеописанный фторированный диен. Далее заявленно оптическое предающее устройство, содержащее вышеописанный полимер и оптическое пластмассовое волокно, содержащее вышеописанный полимер. Технический результат: получение полимера, пригодного в качестве полимерного материала для оптики, обладающего высокой температурой стеклования. 5 н. и 5 з.п. ф-лы.
Комментарии