Способ получения карбопроста и его трометаминовой соли - RU2729626C2
Код документа: RU2729626C2
Чертежи




Описание
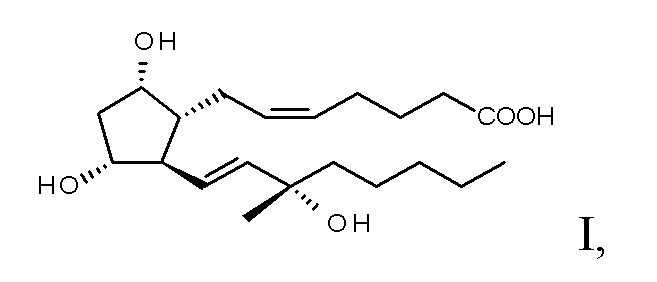
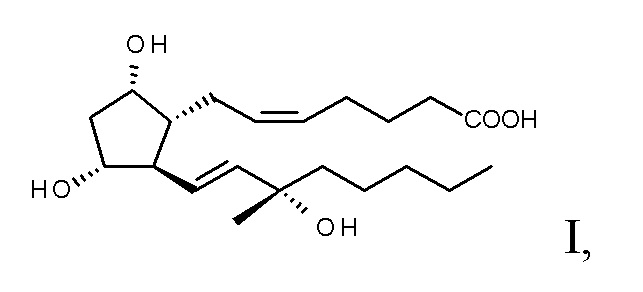
Объектом настоящего изобретения является новый способ получения карбопроста формулы I,

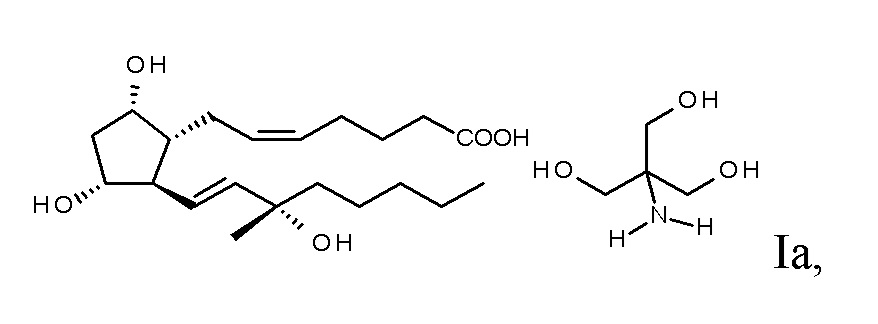
и трометамина карбопроста формулы Ia,

Трометамин карбопроста является оригинальным продуктом Upjohn. Показания к применению: разрешение при беременности и остановка кровотечения после родоразрешения (послеродового кровотечения).
Впервые синтез трометамина карбопроста в экономически эффективном и промышленном масштабах был описан химиками из Upjohn (J. Am. Chem. Soc., 96(18), 5865-5876, 1974).
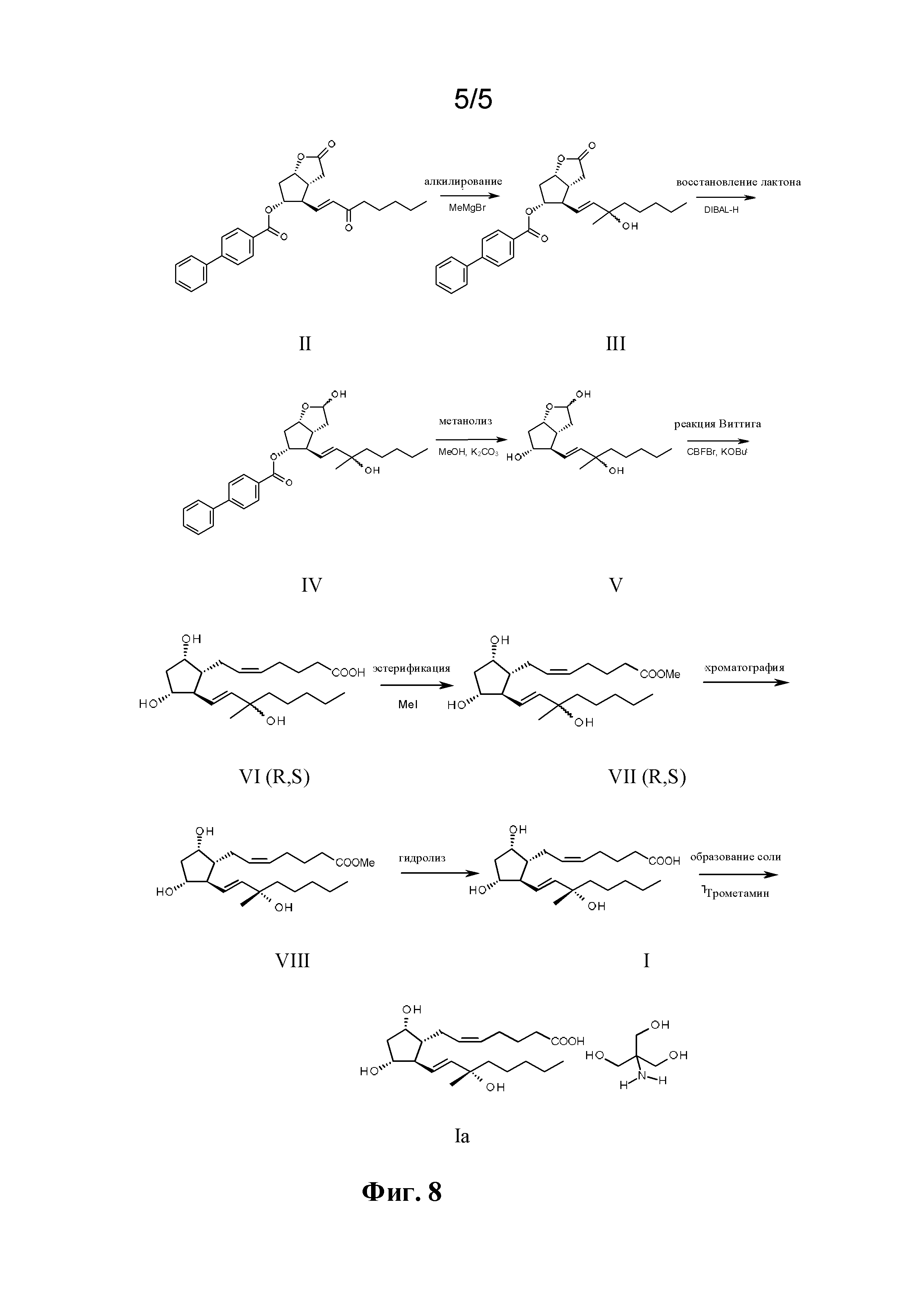
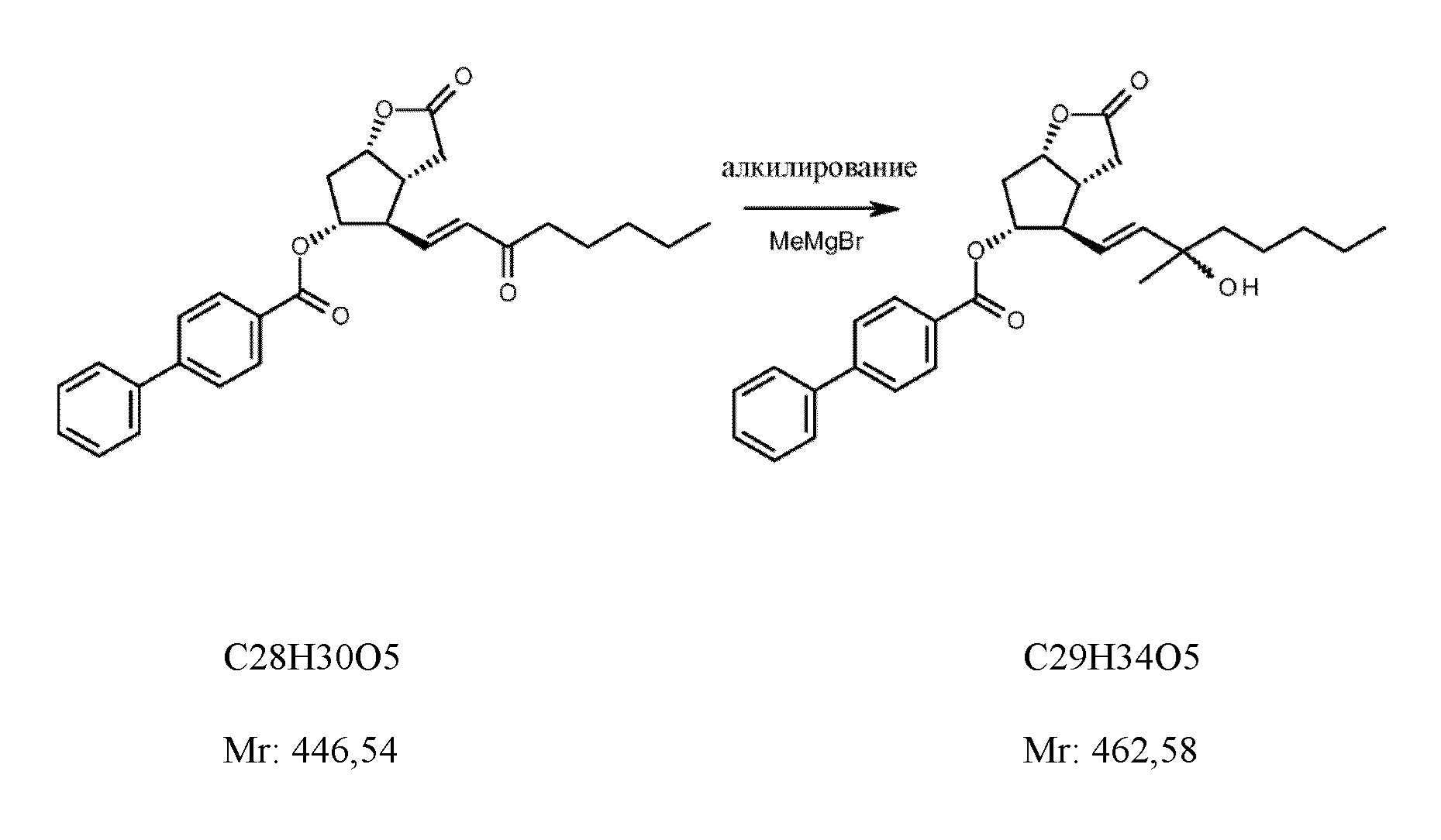
15-метиловый заместитель получали из бензоил-енона с триметилалюминием или с бромметилмагнием (фигура 1).
Соотношение 15-метиловых эпимеров составляло 1:1 в обоих случаях. Эпимеры невозможно было разделить с помощью способа TLC.
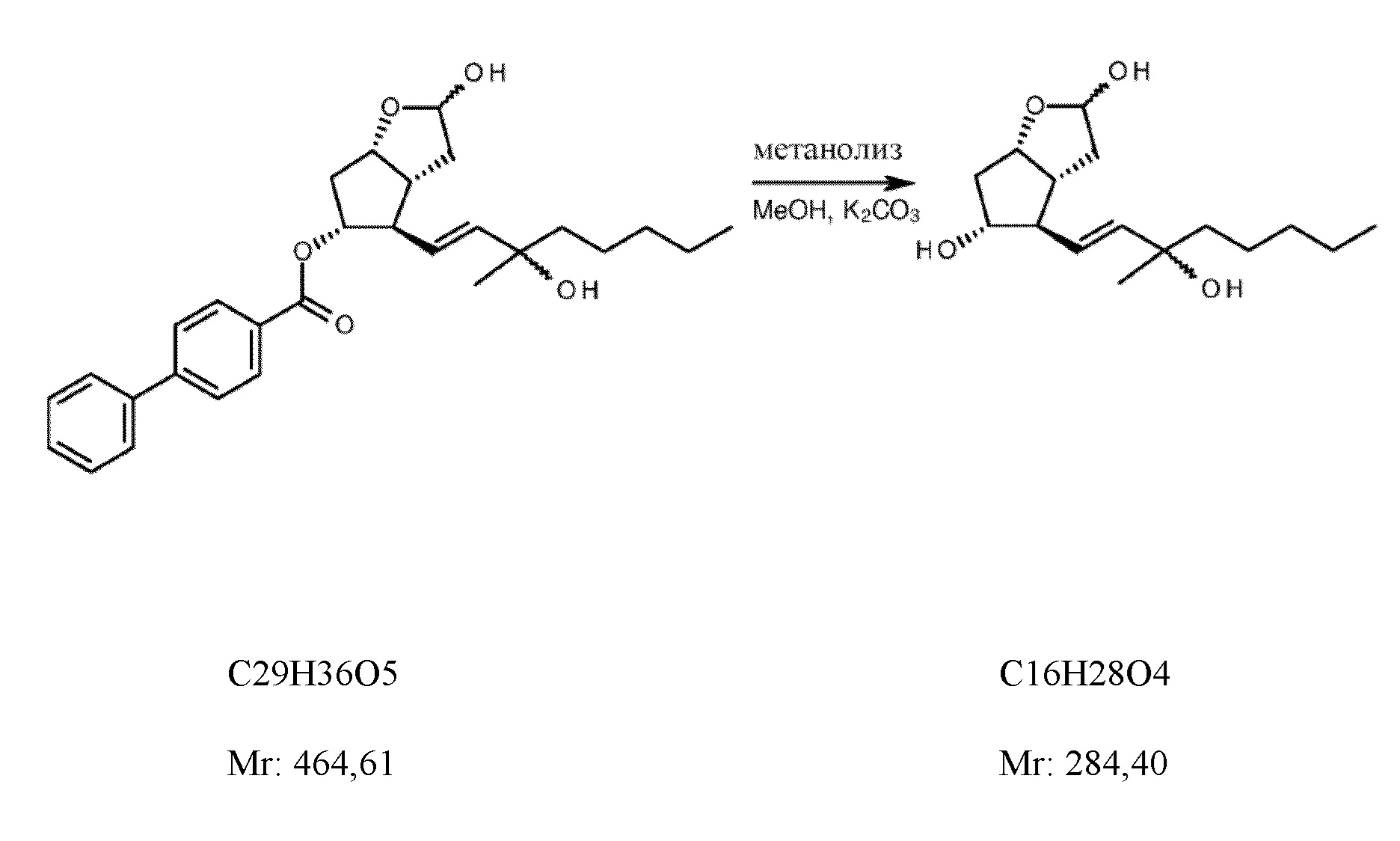
На следующем этапе лактонную группу восстанавливали с помощью гидрида диизобутилалюминия. Восстановление лактона проводили из эпимеров лактона, несущих R = бензоил- или триметилсилил- защитную группу, или R = атом H (фигура 2).
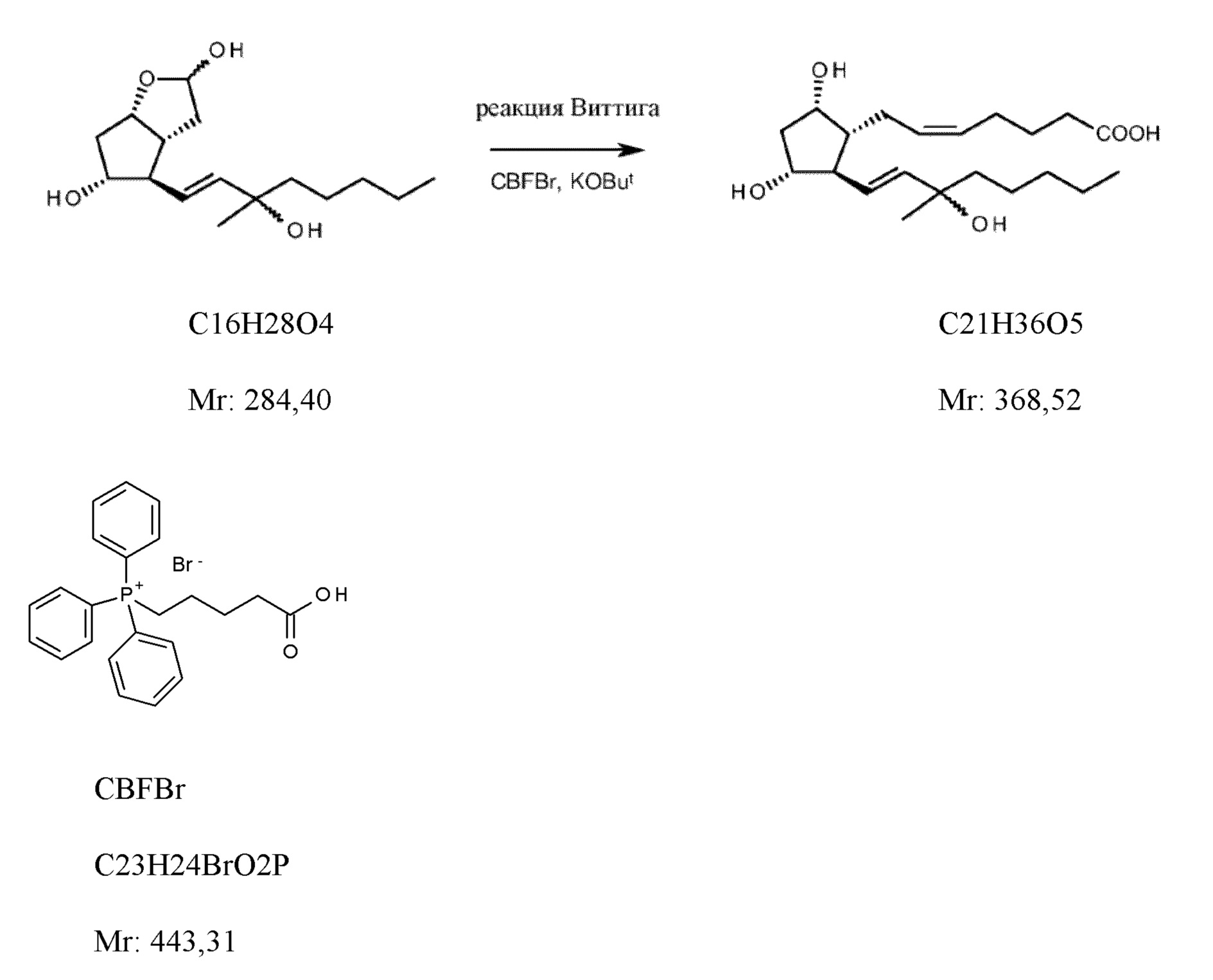
Верхнюю цепь получали с помощью реакции Виттига из всех трех эпимеров лактона (фигура 3). Защитную группу удаляли во время процедуры обработки и в каждом случае получали (R,S) эпимеры карбопроста. Из бромида карбоксибутилфосфония (CBFBr) фосфоран выделяли с помощью реактива NaH/DMSO.
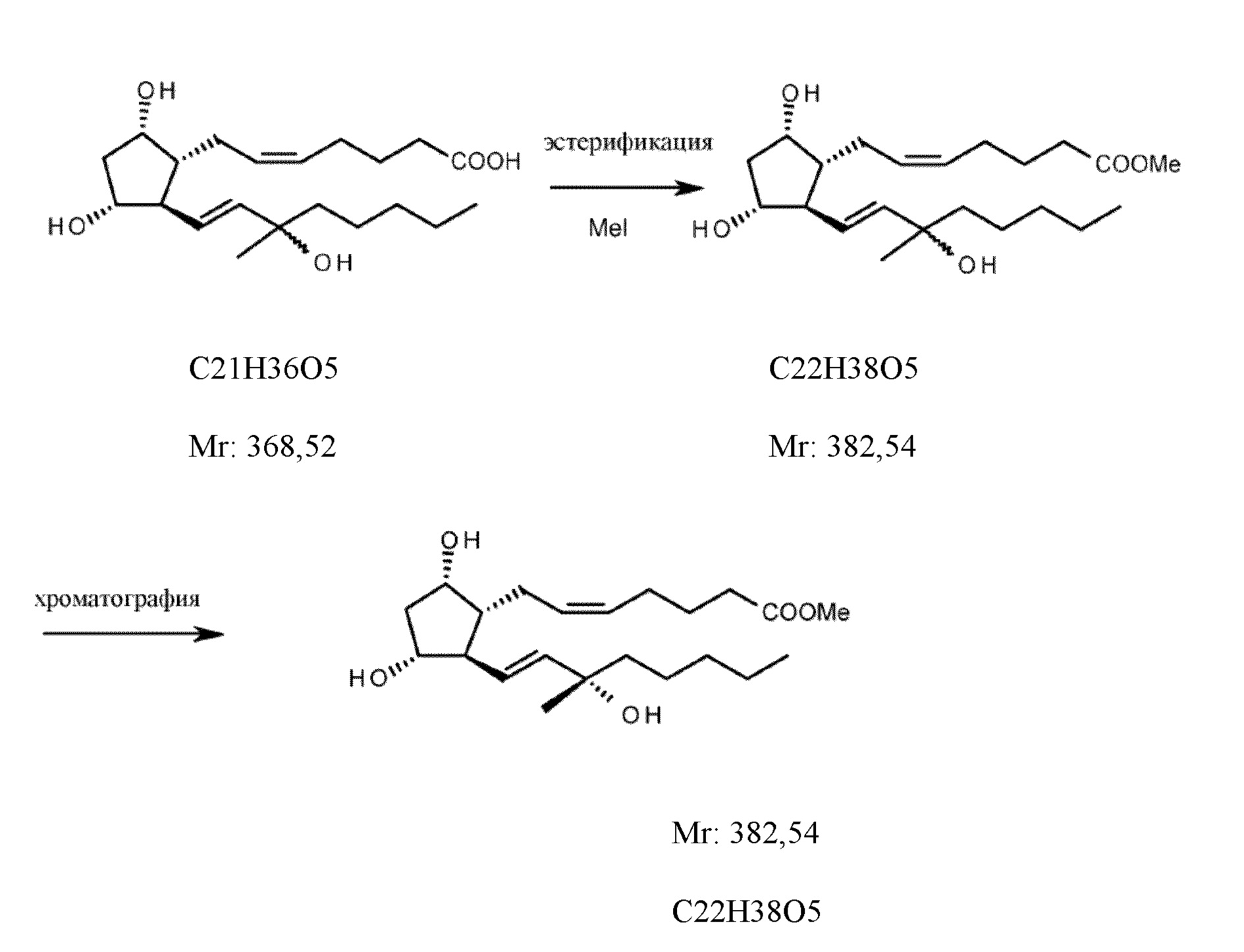
Смесь (R, S) эпимеров карбопроста эстерифицировали с помощью диазометана и (R,S) эпимеры сложного эфира подвергали хроматографии с использованием смеси дихлорметан:ацетон для элюирования с получением сложного метилового эфира карбопроста (фигура 4).
В соответствии с описанием к патенту IN 185790 A1, восстановление оксогруппы PGE-производного с K- или L-селектридом приводит к образованию PGF-производного, которое является значимым промежуточным соединением в синтезе сложного метилового эфира карбопроста.
В описании к патенту IN 185790 A1 описано получение сложного метилового эфира карбопроста, начиная с PGE-производного. В результате селективной каталитической гидрогенизации получают соответствующее PGE2-производное, которое после восстановления оксогруппы приводит в результате к образованию защищенного PGF2a-производного. На последнем этапе силильные защитные группы удаляют с получением сложного метилового эфира карбопроста.
В описании к международной патентной заявке WO 2008/081191 карбопрост получают в соответствии со способом, описанным в J. Am. Chem. Soc., 96(18), 5865-5876, 1974 (фигура 5).
Главные преимущества указанного выше способа получения сложного метилового эфира карбопроста заключаются в следующем.
Реакция Гриньяра
Енон был защищен с помощью триметилсилильной защитной группы,
вместо бромметилмагния использовали более экономически выгодный хлорметилмагний,
количество реактива снижалось с 16 молярных эквивалентов до 5 молярных эквивалентов,
применяемым растворителем был толуол или изомеры ксилена, вместо THF.
В качестве результата этих изменений, соотношение эпимеров лактона возросло от 60:40 до 70:30 в сторону необходимого эпимера.
Восстановление лактона
Количество DIBAL-H было снижено с 4,6-5,4 молярных эквивалентов до 3,5 молярных эквивалентов.
Реакция Виттига
В ходе реакции Виттига растворитель (диметилсульфоксид) не меняли, но для высвобождения фосфорана из бромида карбоксибутилтрифенилфосфония (CBFBr) вместо основания NaH применяли менее воспламеняющийся и более простой в обращении NaNH2.
Температуру в ходе реакции Виттига снижали с 20°C до (-)-25-10°C, что приводило в результате к снижению количества нежелательного транс-эпимера с 6-8% до 3%.
Триэтилсилильные (TES) защитные группы расщеплялись при рабочих условиях реакции Виттига, что являлось дополнительным преимуществом использования TES-защитной группы.
Эстерификация
Вместо менее масштабируемого способа с диазометаном, эпимеры карбопроста эстерифицировали с использованием диметилсульфата или йодистого метила в ацетоне в присутствии карбоната калия.
Вследствие этих изменений выход эпимеров сложного метилового эфира карбопроста, начиная с защищенного енона, увеличивался с 55% до 75%.
Хроматография
Для разделения эпимеров сложного метилового эфира карбопроста применяли способы препаративной HPLC с нормальной и обращенной фазами.
Препаративная HPLC с нормальной фазой: наполнитель: Chiralpak AD;
элюент: смеси гептана или гексана и спиртов. Наилучшую степень разделения достигали с использованием смесей гептан:этанол или гептан:изопропанол.
Препаративная HPLC с обращенной фазой:
наполнитель: Inertsil Prep ODS, элюент: метанол:вода:ацетонитрил, или
наполнитель: YMC C8, элюент: метанол:вода:ацетонитрил.
Было предложено два альтернативных способа синтеза.
В первом способе эпимеры лактона разделяли с помощью способа препаративной HPLC и сложные метиловые эфиры карбопроста получали, начиная с чистых R и S эпимеров. Не наблюдали эпимеризацию в ходе способа, но было отмечено, что препаративное разделение эпимеров является более подходящим на уровне сложного метилового эфира.
Согласно второму способу нижнюю цепь получали начиная с кетона с более короткой цепью с использованием бромпентилмагния (фигура 6).
Соотношение эпимеров лактона в данном способе составляло 50:50%, следовательно, данный способ исключали.
В описании к международной патентной заявке WO 2011/008756 A1 описан общий способ синтеза простагландинов путем осуществления метатезиса, сопряженного с замыканием кольца (метатезис с замыканием кольца, реакция RCM).
В случае с карбопростом, начиная с соответствующего промежуточного соединения и используя катализатор Граббса, получали 1-9-лактон, который после удаления защитных групп и открытия лактонного кольца давал сложный метиловый эфир карбопроста (фигура 7).
Преимуществом способа является то, что ключевое промежуточное соединение получают из оптически чистых материалов, следовательно, полученный в результате метиловый сложный эфир карбопроста не содержит R-эпимера.
Недостатки заключаются в том, что в ходе способа проводят трудномасштабируемые реакции, и в них применяют химически чувствительные реактивы.
В описании к патенту US 2013/190404 опубликованы результаты рентгенографии и данные DSC для трометамина карбопроста. В нем описана кристаллизация: карбопрост растворяли в растворителе (ацетонитриле, ацетоне, эфире или C1-4спирте). К раствору по каплям добавляли водный раствор трометамина. Кристаллы собирали. Трометамин карбопроста растворяли в воде и после добавления ацетона кристаллы собирали снова.
В описании к патенту CN 102816099 A раскрыто получение трометамина карбопроста с высокой степенью чистоты.
Очистку неочищенного сложного эфира карбопроста проводили на очень дорогостоящих неподвижных фазах, предпочтительно при размере частиц, составляющем 5-10 мкм, нормальной фазе, цианосвязанном или аминосвязанном или сферическом силикагеле. Применение таких видов силикагеля предусматривало препаративную жидкостную хроматографию высокого давления. Эта технология высокого давления является времязатратной и требующей затрат средств, требует дорогостоящего оборудования, непроницаемого под давлением, элюентов с высокой степенью чистоты и перечисленных дорогостоящих неподвижных фаз.
Чистота согласно HPLC ≥ 99,5%, 15-эпи-эпимер ≤ 0,5%, 5,6-транс-изомер ≤ 0,5%.
Эфир с высокой степенью чистоты гидролизуют и из кислоты получают трометаминовую соль.
Целью авторов настоящего изобретения является разработка способа получения трометаминовой соли карбопроста, где количество трудноудаляемой примеси 15-(R)-изомера (15-эпи-карбопроста, ((R)-III)) не превышает 0,5%.
Соответственно, объектом настоящего изобретения является способ получения карбопроста формулы I,
и
его трометаминовой соли формулы Ia, предусматривающий, что

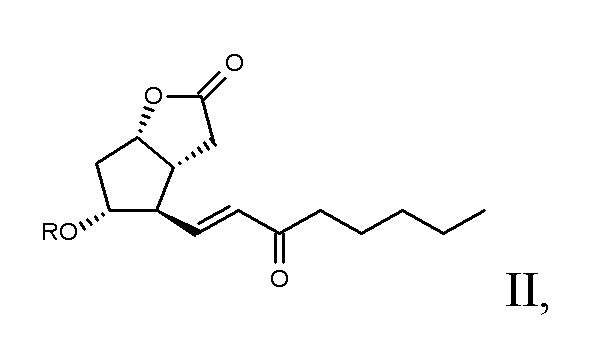
енон общей формулы II,

где R обозначает атом водорода или защитную группу, селективно подвергают алкилированию,
при этом полученный в результате енол общей формулы III,

где R имеет значение, определенное выше, восстанавливают,
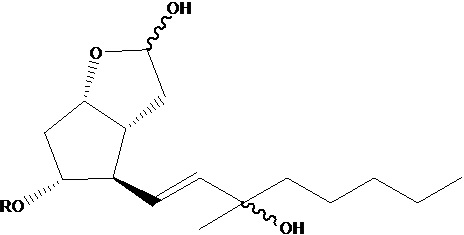
при этом R-защитную группу полученного в результате лактола общей формулы IV,

удаляют
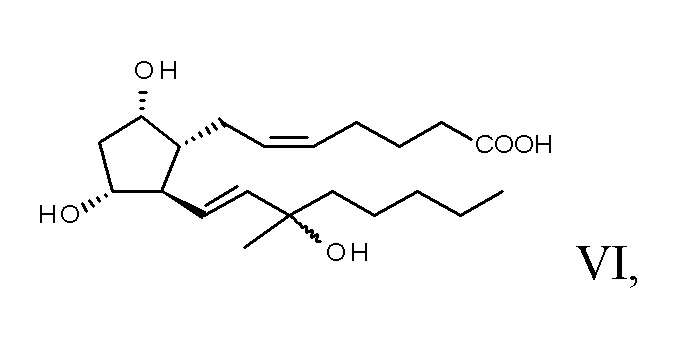
из полученных таким образом эпимеров формулы V лактола,
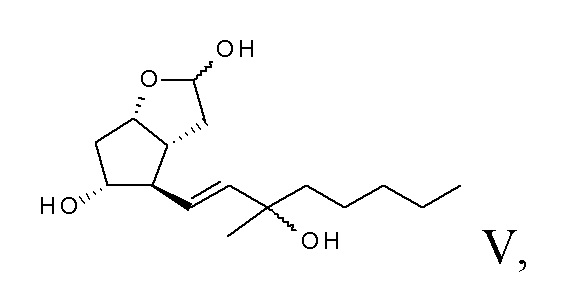
эпимеры формулы VI карбопроста получают с помощью реакции Виттига,

эпимеры карбопроста превращают в сложный метиловый эфир, эпимеры формулы VII сложного метилового эфира разделяют с помощью хроматографии,
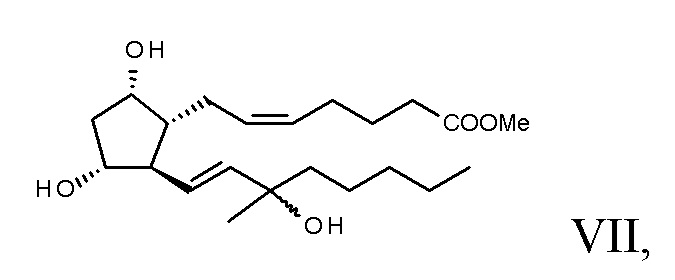
эпимер формулы VIII гидролизуют,

и, если необходимо, превращают в трометаминовую соль,
характеризующийся тем, что
a.) селективное алкилирование осуществляют в присутствии хиральной добавки в апротонном органическом растворителе с реактивом Гриньяра,
b.) хроматографию проводят посредством гравитационной хроматографии на силикагеле,
c.) получение трометаминовой соли осуществляют с помощью твердого трометаминового основания.
В способе согласно настоящему изобретению в качестве реактива Гриньяра применяют хлорметилмагний или бромметилмагний, предпочтительно бромметилмагний, в количестве 3-4 молярных эквивалентов, предпочтительно 3,5 молярного эквивалента.
В качестве образующей комплекс хиральной добавки предпочтительно можно применять (S)-таддол, предпочтительно в количестве 1 молярного эквивалента.
В качестве R-защитной группы можно применять эфирную, силилэфирную, бензильную, замещенную бензильную или ацильную группы, предпочтительно п-фенилбензоиловую группу.
В способе согласно настоящему изобретению, в качестве апротонного органического растворителя применяют эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, диизопропиловый эфир, тетрагидрофуран, метилтетрагидрофуран, диметоксиэтан; ароматические углеводороды, такие как бензол, толуол, ксилол; галогенированные растворители, такие как дихлорметан, или смеси этих растворителей, предпочтительно толуол.
Метилирование осуществляют при температуре от (-)-80 до (-)-20°C, предпочтительно при (-)-50°C.
Согласно настоящему изобретению гравитационную хроматографию на силикагеле можно проводить таким образом, что применяемый элюент содержит основание, или
применяют слабоосновный силикагель /pH=7,5-8,0/.
В качестве основания можно применять органическое основание или аммиак, предпочтительно триэтиламин, где количество основания предпочтительно составляет 0,1%. В качестве элюента можно применять смеси дихлорметан:триэтиламин или дихлорметан:ацетон:триэтиламин.
В качестве слабоосновного силикагеля можно применять, например, сферический силикагель Chromatorex MB с размером частиц, составляющим 40-70 микрометров. В качестве элюента предпочтительно применяют смеси с градиентом ацетон-дихлорметан.
В способе согласно настоящему изобретению получение соли осуществляют в полярном органическом растворителе, не содержащем воду, при этом в качестве полярного органического растворителя можно применять спирты и/или кетоны, предпочтительно изопропиловый спирт и/или ацетон.
Способ согласно настоящему изобретению показан на фигуре 8.
Значительным преимуществом способа согласно настоящему изобретению является то, что в результате его осуществления получают трометаминовую соль карбопроста, где количество трудноудаляемой примеси 15-(R)-изомера (примеси 15-эпи-карбопроста, образующейся из эпимерной примеси (R)-III) составляет не более 0,5%.
Примесь 15-(R) образуется на первом этапе синтеза во время алкилирования при реакции Гриньяра.
Исходный материал способа по настоящему изобретению представляет собой енон формулы II, содержащий п-фенилбензоиловую защитную группу.
Ключевым этапом способа является алкилирование енона формулы II. Более высокая степень стереоселективности может быть достигнута в ходе реакции алкилирования, при этом образуется меньшее количество нежелательного эпимера ((R)-III), и очистка необходимого эпимера ((S)-III) и дополнительных промежуточных соединений, полученных из него, также будет проще и более экономически выгодной.
В способе по настоящему изобретению авторы стремились сделать алкилирование (метилирование) как можно более селективным и удалить нежелательный эпимер как можно более экономически выгодным способом.
Поскольку исходное соединение, защищенный PG-енон, является само по себе хиральным соединением, теоретически возможно, чтобы алкилирование протекало с хорошей селективностью без добавления хиральной добавки, даже если центр реакции находится относительно далеко от центров асимметрии. Хороший пример этого приведен в описании к международной патентной заявке WO 2008/081191 A1, где триэтилсилил-защищенный PG-енон вступает в реакцию в ксилоле, толуоле или в смеси этих растворителей при -78°C с 5 эквивалентами хлорметилмагния. При алкилировании достигали весьма выгодного соотношения эпимеров (S: R) = 70:30.
Селективность реакции предположительно зависит от условий реакции (растворителя, температуры реакции, реактива, последовательности добавления) и от структуры исходного материала.
Промежуточное соединение PG-енон, полученное в ходе производства простагландина CHINOIN, содержит п-фенилбензоильную защитную группу (II). Однако, реакция енона с бромметилмагнием приводит в результате к соотношению эпимеров (S)-III: (R)-III, составляющему 55:45.
При осуществлении алкилирования с помощью бромметилмагния в толуоле, в присутствии триэтиламина, селективность в степени, являющейся низкой (55:45), не изменилась, и, что удивительно, в присутствии хирального S-диметил-1-фенилэтиламинового основания селективность исчезла.
Путем присоединения к PG-енону реактива, полученного из триметилалюминия и 2,6-ди-трет-бутил-4-метилфенона, селективность возросла до 63:37.
Для повышения селективности алкилирование проводили в присутствии различных хиральных добавок.
Возможные хиральные добавки (таблица 1)



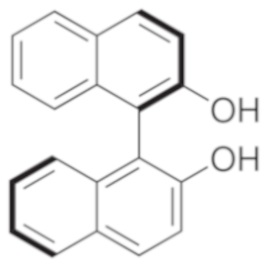
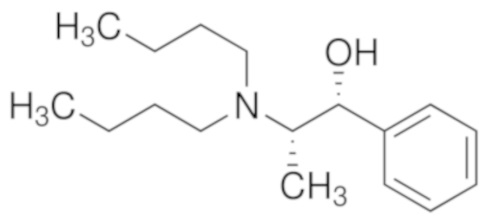
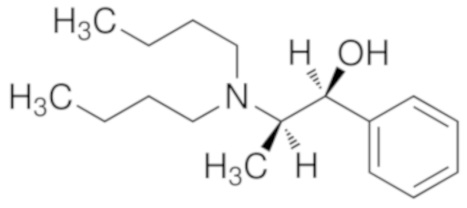
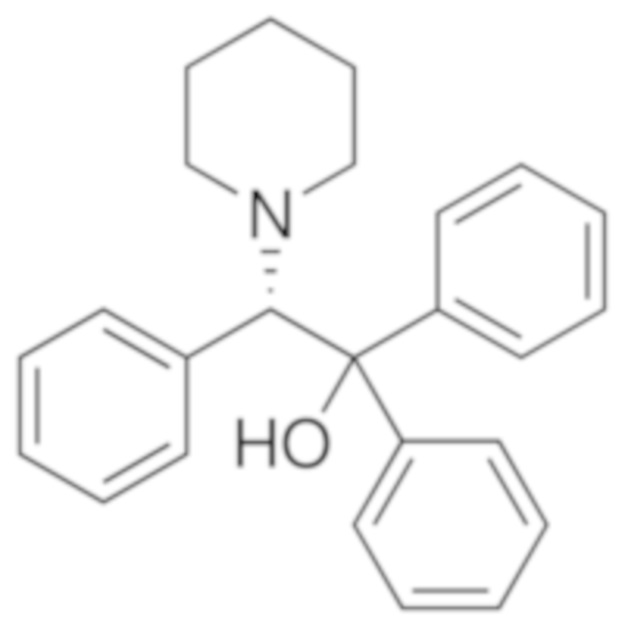



Таблица 1.
В случае алкилирующего средства бромметилмагния (S)-таддол оказывается наиболее эффективной добавкой, с помощью которой в результате получают соотношение (S)-III : (R)-III = 70:30, по этой причине авторы настоящего изобретения подробно изучили эту реакцию. Алкилирование осуществляли в растворителях типа эфира, и в галогенированных растворителях, и в толуоле.
Реакции осуществляли при -50°C, поскольку при более низких температурах, при (-)-70 и при (-)-80°C, реакция замедлялась, в то время как соотношение эпимеров не улучшалось. При более высокой температуре, при (-)-25°C, образовывалось много побочных продуктов.
Влияние количества (S)-таддола на соотношение эпимеров (S)-III : (R)-III (таблица 2)
(Растворитель: толуол, температура реакции: (-)-50°C)
Таблица 2
Из приведенных выше результатов видно, что оптимальное количество хирального вспомогательного средства (S)-таддола составляет 1 молярный эквивалент. При использовании меньшего количества соотношение эпимеров является менее подходящим, в то время как более высокий избыток не имеет никакого дополнительного влияния.
Влияние растворителей на соотношение эпимеров (S)-III : (R)-III (таблица 3)
Количество ((S)-таддола 1 молярный эквивалент, количество MeMgBr, составляющее 3,5 молярного эквивалента,
температура реакции: (-)-50°C)
Таблица 3
Неожиданно, наилучшее соотношение эпимеров с очень хорошим выходом было достигнуто не в растворителе типа эфира, характерного для реакций Гриньяра, а в толуоле (S)-III : (R)-III = 70:30.
Особенно интересно то, что в хлороформе селективность S/R преобразовалась в обратную.
Для осуществления реакций был выбран избыток в количестве 3,5 молярного эквивалента реактива Гриньяра, бромметилмагния. Таким образом, в способе по настоящему изобретению применяли меньшее количество реактива Гриньяра, чем в способе, приведенном в описании к международной патентной заявке WO 2008/081191, где реактив Гриньяра (MeMgCl) применяли в избытке, составляющем 5 молярных эквивалентов.
Дополнительным преимуществом является то, что реакцию проводят при более высокой температуре, при (-)-50°C, вместо (-)-78°C, применяемой в указанном описании к патентной заявке.
Авторы настоящего изобретения также изучили влияние концентрации. Однако, в изучаемой области (5, 8 и 10-кратный избыток растворителя) концентрация не влияла на соотношение эпимеров. Наиболее подходящим избытком растворителя был 8-кратный избыток. Более концентрированные реакционные смеси было сложно перемешивать, в то время как в более разбавленных растворах реакция замедлялась.
Авторы настоящего изобретения неожиданно обнаружили, что в присутствии ахирального триметиламина с помощью реакции Гриньяра, проходящей в толуоле, получали в результате соотношение эпимеров (S)-III : (R)-III = 55:45, вместо ожидаемого соотношения 50:50%. Однако, комбинированный эффект триэтиламина и (S)-таддола дополнительно не улучшал селективность 70:30, достигнутую без применения основания.
В ходе реакции Гриньяра наилучшее соотношение эпимеров (70:30) достигали в толуоле при (-)-50°C с применением 3,5 молярного эквивалента реактива бромметилмагния и 1 молярного эквивалента (S)-таддола, материала на основе хирального вспомогательного средства.
Разделение эпимеров (S)-III и (R)-III, в соответствии с имеющимся знанием, возможно только с помощью очень дорогостоящей препаративной HPLC, следовательно на данном промежуточном уровне для авторов настоящего изобретения разделение не было целью. В конце реакции реакционную смесь расслаивали с помощью разбавленной кислоты. После обработки значительное количество (S)-таддола кристаллизовалось из смеси. Остальную часть (S)-таддола удаляли посредством фильтрования на колонке с силикагелем путем промывания с помощью элюентов гексан-этилацетат и этилацетат.
Восстановленный (S)-таддол может быть повторно использован в стереоселективной реакции Гриньяра.
Поскольку разделение эпимеров III возможно только с помощью дорогостоящего способа препаративной HPLC, авторы настоящего изобретения искали другие способы разделения эпимеров.
Эпоксидирование по Шарплессу в общем можно применять для кинетического оптического расщепления аллиловых спиртов. (Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity?, V. S. Martin, S. S. Woodard, T. Katsuki, Y. Yamada, M. Ikeda, K. B. Sharpless, JACS, 103, 6237-6240 (1981).)
Основой разделения является то, что в присутствии материала на основе хирального вспомогательного средства эпоксидирование двух эпимерных аллиловых спиртов может быть осуществлено таким образом, что только нежелательный эпимер образует эпоксид, в то время как желательный эпимер остается непрореагировавшим. Начиная с эпимерной смеси 1:1 и используя 0,5 молярного эквивалента реактива для эпоксидирования, в идеальном случае необходимый эпимер может быть получен с 50% выходом, с чистотой, составляющей 100%.
В ходе эпоксидирования по Шарплессу обычное средство для окисления представляет собой трет-бутилгидроксипероксид (TBHP), материал на основе хирального вспомогательного средства представляет собой сложный диэтиловый (DET) или диизопроипловый эфир (DIPT) D- или L-винной кислоты, при этом катализатор представляет собой тетраизопропилат титана.
В данном случае, в присутствии материала на основе хирального вспомогательного средства, представляющего собой сложный диизопропиловый эфир (D)-винной кислоты ((D)-DIPT), эпоксидирование нежелательного эпимера (R)-III) происходило на более высоком уровне. Исходя из смеси 50:50% эпимеров енола, эпимерная чистота полученного соединения формулы III составляла 70%.
Хотя эпимерный состав енола формулы III составлял 70:30, реакция эпоксидирования не приводила к изменению этого соотношения. Следовательно, данный способ разделения эпимеров был исключен.
Восстановление лактона
Лактонную группу со смесью эпимеров (S) 70: (R) 30 енола III, полученную в присутствии материала на основе хирального вспомогательного средства (S)-таддола, восстанавливали в условиях, часто используемых в химическом производстве простагландинов, с помощью реактива DIBAL-H в тетрагидрофуране при (-)-75°C.
Разделение эпимеров IV PPB-лактол (смеси из 4 изомеров), полученных после обработки, не было успешным посредством кристаллизации в ацетоне, этилацетате, метил-трет-бутиловом эфире, смеси толуола-гексана или в смесях из вышеуказанных растворителей.
Метанолиз
п-Фенилбензоиловую (PPB) защитную группу эпимеров формулы IV лактола удаляли в растворе метанола в присутствии основания (карбоната калия).
Разделение эпимеров формулы V лактола (смеси из 4 изомеров), полученных после обработки, не было успешным посредством кристаллизации в ацетоне, этилацетате, метил-трет-бутиловом эфире, смеси толуола-гексана или в смесях из вышеуказанных растворителей.
Реакция Виттига
Реакцию Виттига осуществляли в тетрагидрофуране. Для образования верхней цепи осуществляли реакцию эпимеров формулы V лактола с фосфораном, высвобожденным из бромида (карбоксибутил)трифенилфосфония (CBFBr) с трет-бутилатом калия в растворителе, представляющем собой тетрагидрофуран. Карбопрост (R,S) формулы I, полученный после обработки, подавали на следующий этап эстерификации, без выделения эпимеров.
Эстерификация
Разделение 15-эпимеров казалось наиболее перспективным на уровне сложного эфира карбопроста, поэтому авторы настоящего изобретения подробно исследовали возможность разделения эпимеров.
Целью для авторов настоящего изобретения являлось осуществление разделения с помощью экономически выгодной гравитационной хроматографии, вместо очень дорогостоящей методики препаративной HPLC.
На основании исследований с применением TLC и HPLC полученных эфиров, сложный метиловый эфир был более подходящим для разделения эпимеров.
Хроматографическую очистку проводили с использованием растворителей типа сложного эфира, эфира и кетона и галогенированных растворителей.
Дополнительно к способу, описанному в J. Am. Chem. Soc., 96(18), 5865-5876, 1974, в котором использовали смесь дихлорметан:ацетон = 2:1 для элюирования, также достигали хорошего разделения с помощью смесей этилацетат:метил-этил-кетон, изопропилацетат:метил-этил-кетон и метил-трет-бутиловый эфир:ацетон.
Для разделения эпимеров была выбрана смесь дихлорметан:ацетон = 2:1, но на основании наблюдения распадания сложного метилового эфира карбопроста на слабокислой поверхности силикагеля, были предложены изложенные ниже инновационные изменения.
В элюенты для хроматографической очистки добавляли 0,1% триэтиламина, чтобы предотвратить распадание чувствительного к кислоте третичного спирта на колонке с силикагелем.
Технологические примеси неочищенного сложного метилового эфира карбопроста удаляли с помощью хроматографии на колонке с силикагелем с использованием смесей метил-трет-бутиловый эфир:0,1% триэтиламин и метил-трет-бутиловый эфир:ацетон:0,1% триэтиламин для элюирования.
Разделение эпимеров сложного метилового эфира карбопроста осуществляли с использованием смесей дихлорметан:0,1% триэтиламин и дихлорметан:ацетон:триэтиламин = 2:1:0,1% посредством повторной хроматографии на такой же хроматографической колонке.
При повторной хроматографии количество нежелательного эпимера может быть снижено до определенного предела (≤0,5%).
Выпаренную основную фракцию, которая содержала нежелательный эпимер на уровне, соответствующем указанному пределу, перемещали на следующий этап.
Между двумя хроматографиями колонку с силикагелем регенерировали путем промывания с помощью элюентов 0,1% триэтиламин:ацетон и 0,1% триэтиламин:дихлорметан.
Можно также успешно выполнять хроматографию, если применяют слабоосновный силикагель. Наилучшего разделения достигали на сферическом силикагеле Chromatorex MB с размером частиц, составляющим 40-70 (значение pH: 7,5-8,0), с применением смесей с градиентом ацетон:дихлорметан в качестве элюентов. В этом случае добавление 0,1% триэтиламина к смеси для элюирования не является необходимым.
Хотя стоимость сферического силикагеля Chromatorex MB с размером частиц, составляющим 40-70, на один порядок выше, чем силикагеля Geduran Si 60 неправильной формы с размером частиц, составляющим 0,063-0,200 мм, при применении более дорогого силикагеля (R, S) эпимеры могут быть разделены за один прогон хроматографии с выходом, составляющим 57%, и полученный в результате сложный метиловый эфир карбопроста содержит примесь 15-эпи-изомера в количестве не более 0,5%.
Гидролиз
Сложный метиловый эфир карбопроста гидролизовали до карбопроста в растворе метанола посредством обработки с помощью раствора гидроксида натрия. Повышение кислотности для получения карбопроста необходимо осуществлять быстро для предотвращения эпимеризации в кислой среде (Eur. J. Pharm. Sci., 3, 27-38 (1995).
Получение соли
Для получения трометаминовой соли карбопрост растворяли в изопропаноле, к раствору добавляли твердое трометаминовое основание и смесь перемешивали. Когда завершалось образование соли, реакционную смесь фильтровали. Трометаминовую соль карбопроста кристаллизовали посредством добавления ацетона, этилацетата и гексана.
Трометаминовую соль можно перекристаллизовывать с хорошим выходом.
Преимуществом способа по настоящему изобретению в сравнении со способом, раскрытым в описании к патенту US 2013/0190404 A, является то, что авторы настоящего изобретения не используют воду, следовательно, не существует необходимости в применении больших количеств растворителей для осаждения карбопроста из воды.
В способе по настоящему изобретению в присутствии хирального вспомогательного средства (S)-таддола достигают соотношения эпимеров, составляющего 70:30. Селективность является такой же, как показано в описании к международной патентной заявке WO 2008/081191.
Все выходы двух способов, считая от защищенных PG-енонов (TES-PG-енон и PPB-PG-енон, соответственно) до эпимерных (R,S) смесей сложных эфиров, составляли 75 и 86% соответственно, что означает, что выход, достигнутый с помощью способа по настоящему изобретению, является на 10% выше.
Предварительную очистку неочищенной эпимерной смеси сложных эфиров осуществляют в обоих способах посредством гравитационной колоночной хроматографии, однако в описании к международной патентной заявке WO 2008/081191 не раскрыты условия хроматографирования.
Разделение изомеров в способе согласно описанию к международной патентной заявке WO 2008/081191 проводят посредством дорогостоящей препаративной хроматографии высокого давления, в то время как в способе согласно настоящему изобретению изомеры разделяют посредством гравитационной хроматографии, которая является времясберегающей, и экономичной в отношении затрат, и масштабируемой.
Очистка посредством хроматографии оказывается более сложной из-за того факта, что сложный метиловый эфир карбопроста распадается на силикагеле, который имеет кислотную природу. Успешное разделение может быть достигнуто только в том случае, если скорость элюирования выше, чем скорость распадания. Этот критерий удовлетворяют путем применения дорогостоящей препаративной хроматографии, в которой относительно небольшое количество вводимого материала проходит через колонку с силикагелем с высокой скоростью.
Применимость гравитационной хроматографии в способе по настоящему изобретению является возможной благодаря инновационной реализации, предусматривающей, что в элюент, используемый для хроматографии, добавляют 0,1% основной добавки, предпочтительно органического основания с низкой температурой кипения, триэтиламина, который путем связывания с кислыми участками силикагеля препятствует распаданию материала, подлежащего очистке, на хроматографической колонке. Хроматография также может быть выполнена с хорошей эффективностью, в случае применения силикагеля слабоосновной (рН =7,5-8,0) природы. В данном случае нейтральный элюент также является пригодным для разделения эпимеров сложного метилового эфира карбопроста.
Дополнительные преимущества способа по настоящему изобретению.
Хиральный катализатор является дорогостоящим, но он может быть регенерирован и повторно использован в 90-95%, в то время как использование триэтилхлорсилана, которое также является дорогостоящим, значительно увеличивает издержки производства.
Реакцию Гриньяра проводят при более высокой температуре (при -50°C, вместо -78°C), при этом количество реактива Гриньяра является меньшим (3,5 экв., вместо 5 экв.).
При применении способа хроматографии высокого давления с обращенной фазой, приведенного в описании к международной патентной заявке WO 2008/081191, как наиболее благоприятного способа очистки, очищенный продукт находится в водной фазе, и для получения продукта необходимы дополнительные этапы экстракции, увеличивающие время производства В способе по настоящему изобретению гравитационной колоночной хроматографии с нормальной фазой фракции, удовлетворяющие требования по качеству, объединяют и выпаривают с получением очищенного продукта.
Дополнительные подробности относительно настоящего изобретения представлены примерами без ограничения настоящего изобретения данными примерами.
Применение по меньшей мере 0,01% основной добавки является эффективным, и применение более 1% основной добавки не является целесообразным.
Примеры
1.a Сложный (3aR,4R,5R,6aS)-гексагидро-2-оксо-4-[(1E)-3-гидрокси-3-метил-1-октен-1-ил]-2H-циклопента[b]фуран-5-иловый эфир [1,1'-бисфенил]-4-карбоновой кислоты
4,66 кг (S)-таддола добавляли к 25,4 л дистиллированного толуола в атмосфере азота. Практически гомогенный раствор охлаждали и добавляли 25 л раствора 1,4 M бромметилмагния при (-)-50°C. После перемешивания в течение 30 минут добавляли 4,46 кг сложного (3aR,4R,5R,6aS)-гексагидро-2-оксо-4-[(1E)-3-оксо-1-октен-1-ил]-2H-циклопента[b]фуран-5-илового эфира [1,1'-бисфенил]-4-карбоновой кислоты в дистиллированном толуоле при (-)-50°C. После завершения реакции смесь выливали в смесь 1 M хлористоводородной кислоты и толуола и тщательно перемешивали. Фазы разделяли, водную фазу экстрагировали с помощью толуола. Органическую фазу промывали с помощью 1 M раствора гидрокарбоната натрия и с помощью насыщенного солевого раствора. Органическую фазу выпаривали.
Сухой остаток растворяли в метаноле при 50°C и затем охлаждали до 0°C. Осажденный (S)-таддол удаляли с помощью фильтрации.
Фильтрат выпаривали, остаток растворяли в толуоле, а оставшуюся часть (S)-таддола удаляли с помощью хроматографии на колонке с силикагелем с фильтрованием, используя элюенты гексан:этилацетат и этилацетат.
Основную фракцию, содержащую продукт, полученную с помощью хроматографии, выпаривали.
Выход: 4,40 кг (95%).
Восстановление (S)-таддола
4,6 кг (S)-таддола, повторно полученного посредством кристаллизации и посредством хроматографического разделения, растворяли в ацетоне при 50°C и кристаллизовали при 0°C после добавления гексана. Кристаллы собирали с помощью фильтрации, промывали и высушивали.
Выход: 4,3 кг (93,5%), чистота согласно HPLC: 99,96%.
1.b Сложный (3aR,4R,5R,6aS)-гексагидро-2-гидрокси-4-[(1E)-3-гидрокси-3-метил-1-октен-1-ил]-2H-циклопента[b]фуран-5-иловый эфир [1,1'-бисфенил]-4-карбоновой кислоты

9,7 кг сложного (3aR,4R,5R,6aS)-гексагидро-2-оксо-4-[(1E)-3-гидрокси-3-метил-1-октен-1-ил]-2H-циклопента[b]фуран-5-илового эфира [1,1'-бисфенил]-4-карбоновой кислоты растворяли в 62 кг не содержащего воду тетрагидрофурана в атмосфере азота. При (-)-75°C добавляли раствор 9,8 кг гидрида диизобутилалюминия в толуоле. В конце восстановления реакционную смесь переносили посредством отсасывания в 2 M раствор бисульфата натрия, фазы тщательно перемешивали и отделяли после седиментации. Водную фазу экстрагировали с помощью толуола, объединенную органическую фазу промывали с помощью 1 M раствора гидрокарбоната натрия и с помощью насыщенного солевого раствора. Органическую фазу выпаривали.
Выход: 9,74 кг (99,96%).
1c. 2H-Циклопента[b]фуран-2,5-диол, гексагидро-4-(3-гидрокси-3-метил-1-октен-1-ил)
10,3 кг сложного (3aR,4R,5R,6aS)-гексагидро-2-гидрокси-4-[(1E)-3-гидрокси-3-метил-1-октен-1-ил]-2H-циклопента[b]фуран-5-илового эфира [1,1'-бисфенил]-4-карбоновой кислоты растворяли в 46 л метанола, добавляли 1,5 кг карбоната калия и осуществляли реакцию при 40°C. После завершения реакции смесь охлаждали до 0°C и нейтрализовали с помощью разбавленной фосфорной кислоты. Осажденные кристаллы отфильтровывали, промывали смесью метанол:вода и фильтрат концентрировали. К концентрированному раствору добавляли воду и хлорид натрия. Продукт экстрагировали с помощью этилацетата, объединенную органическую фазу обесцвечивали с помощью активированного угля, уголь отфильтровывали и фильтрат выпаривали.
Выход: 6,1 кг (97%).
1d. Неочищенный карбопрост
(5Z,9α,11α,13E)-15-метил-9,11,15-тригидрокси-проста-5,13-диен-1-карбоновая кислота, неочищенная (R,S)
20 кг бромида карбоксибутилтрифенилфосфония (CBFBr) добавляли к 133 л не содержащего воду тетрагидрофурана в инертной атмосфере, охлаждали до 0°C и к смеси добавляли несколько порций 17 кг трет-бутилата калия. Суспензию, окрашенную в оранжевый цвет, охлаждали до (-)-5-(-)-10°C и добавляли раствор 5,9 кг 2H-циклопента[b]фуран-2,5-диола, гексагидро-4-(3-гидрокси-3-метил-1-октен-1-ил) в не содержащем воду терагидрофуране. После завершения реакции добавляли воду к реакционной смеси и с помощью 2 M раствора бисульфата натрия pH доводили до 10-11. Реакционную смесь концентрировали и охлаждали до 20°C. Осажденные кристаллы отфильтровывали, промывали с помощью 1 M раствора гидрокарбоната натрия и с помощью воды. Фильтрат экстрагировали с помощью дихлорметана. pH водной фазы доводили до нейтрального с помощью 2 M раствора бисульфата натрия, затем после добавления этилацетата кислотность повышали до pH=2. Осажденные кристаллы отфильтровывали, промывали с помощью этилацетата. Фазы фильтрата разделяли. Водную фазу экстрагировали этилацетатом. Объединенную органическую фазу промывали с помощью насыщенного солевого раствора, высушивали над сульфатом натрия и концентрировали. Концентрат охлаждали до 20°C и кристаллизовали путем добавления диизопропилового эфира. Кристаллы отфильтровывали и промывали с помощью смеси диизопроипловый эфир:ацетон. Фильтрат выпаривали.
Выход: 7,1 кг, (93%).
1e1. Сложный метиловый эфир карбопроста
Сложный метиловый эфир (5Z,9α,11α,13E,15S)-15-метил-9,11,15-тригидрокси-проста-5,13-диен-1-карбоновой кислоты
7,7 кг неочищенного карбопроста (R,S) растворяли в 28 л дистиллированного ацетона, к раствору добавляли 9 кг карбоната калия и 9,1 кг метилйодида и реакционную смесь перемешивали при 50°C. В конце реакции смесь переносили посредством отсасывания в смесь метил-трет-бутилового эфира и 1 M раствора бисульфата натрия. После перемешивания и осаждения фазы разделяли и водную фазу экстрагировали метил-трет-бутиловым эфиром. Объединенную органическую фазу промывали 1 М раствором гидрокарбоната натрия и насыщенным солевым раствором, высушивали над сульфатом натрия и выпаривали. Неочищенный продукт: 8 кг (100%).
На основании указанного выше, выход неочищенного сложного метилового эфира карбопроста, рассчитанного для PG-енона, составляет 86%.
В начале выпаривания в раствор добавляли 70 мл триэтиламина. Концентрат очищали посредством хроматографии на колонке с силикагелем с использованием метил-трет-бутилового эфира:триэтиламина (0,1%), затем смесей метил-трет бутиловый эфир:ацетон:триэтиламин = 20:1:0,1% для элюирования. Основную фракцию, содержащую эпимеры (R,S) сложного метилового эфира карбопроста выпаривали. Эпимеры разделяли на колонке с силикагелем с использованием смесей дихлорметан:триэтиламин (0,1%) и дихлорметан:ацетон:триэтиламин = 2:1:0,1% для элюирования посредством повторной хроматографии. Между двумя циклами хроматографии колонку с силикагелем регенерировали смесью ацетон:триэтиламин (0,1%) для элюирования, а затем смесью дихлорметан:триэтиламин (0,1%) для элюирования.
Основную фракцию выпаривали.
Выход: для сложного метилового эфира карбопроста (VII) 2,35 кг (42%) (с учетом того, что неочищенный сложный метиловый эфир карбопроста содержит эпимеры в соотношении 70:30).
1e2. Альтернативный способ очистки
Неочищенный сложный метиловый эфир карбопроста растворяли в дихлорметане и очищали посредством хроматографии на колонке с силикагелем Chromatorex MB70-40/75 с использованием смесей дихлорметан-ацетон с градиентами = 4:1, 2:1 и затем элюентов на основе ацетона. Фракции, содержащие сложный метиловый эфир карбопроста, исследовали с помощью HPLC, фракции надлежащего качества выпаривали.
Выход: 2,97 кг (57,5%).
1f. Карбопрост
(5Z,9α,11α,13E)-15-метил-9,11,15-тригидрокси-проста-5,13-диен-1-карбоновая кислота)

550 г сложного метилового эфира карбопроста растворяли в 5 л дистиллированного метанола и добавляли 5 л 2 н. раствора гидроксида натрия. После завершения гидролиза добавляли воду к реакционной смеси и раствор концентрировали. К концентрированной реакционной смеси добавляли воду и метил-трет-бутиловый эфир, тщательно перемешивали, затем фазы разделяли. К водной фазе добавляли хлорид натрия и метил-трет-бутиловый эфир и pH доводили до 4 с помощью 2 M раствора бисульфата натрия. Фазы разделяли, водную фазу экстрагировали с помощью метил-трет-бутилового эфира, органическую фазу промывали с помощью насыщенного солевого раствора, высушивали над сульфатом натрия и выпаривали.
Выход: 519 г, (98%).
1g. Трометамин карбопроста
Соль (5Z,9α,11α,13E)-15-метил-9,11,15-тригидрокси-проста-5,13-диен-1-карбоновой кислоты, полученной с помощью 2-амино-2-(гидроксиметил)-1,3-пропандиола
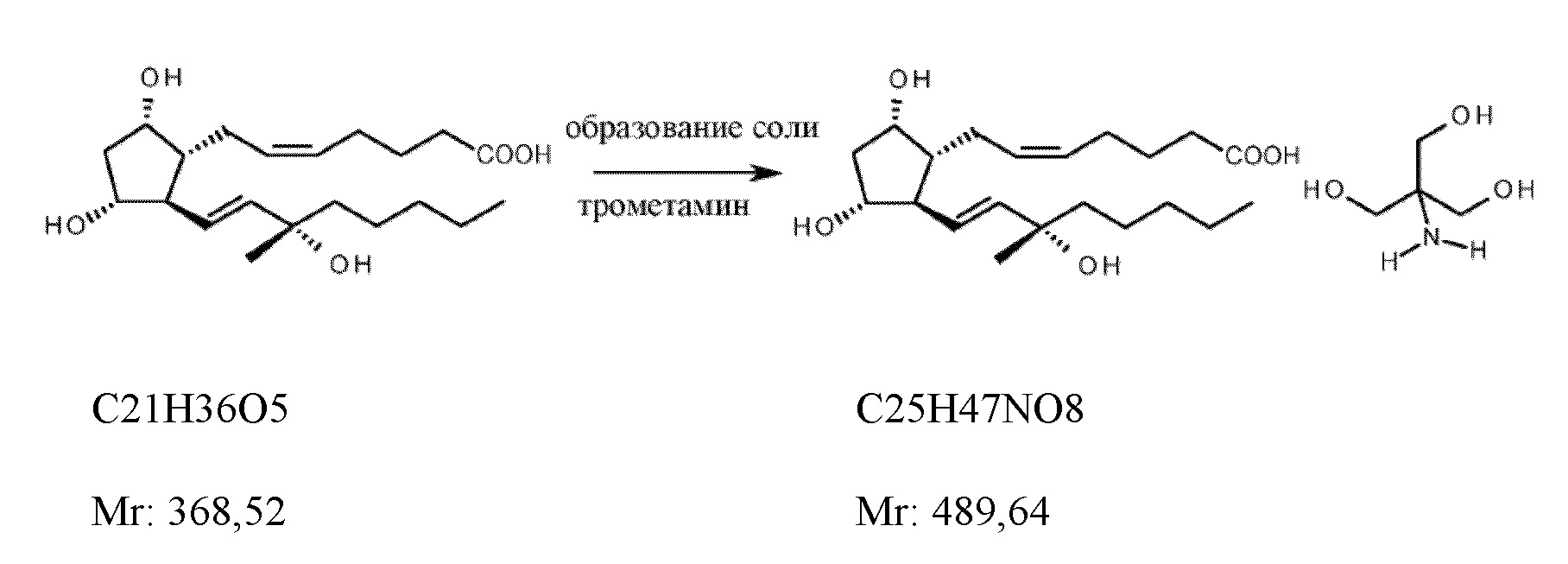
509 г карбопроста растворяли в 2,7 л фильтрованного, дистиллированного изопропанола, затем добавляли 170,8 г трометамина и реакционную смесь перемешивали при комнатной температуре в течение прим. 1 часа. Раствор фильтровали и концентрировали. Добавляли к концентрату изопропанол (фильтрованный, дистиллированный) и ацетон (фильтрованный, дистиллированный). Реакционную смесь перемешивали при 20°C при осаждении кристаллов. К суспензии кристаллов добавляли фильтрованный, дистиллированный этилацетат, затем фильтрованный, дистиллированный гексан и перемешивание продолжали в течение дополнительного часа. Кристаллы отфильтровывали, промывали с помощью смеси гексан:ацетон:этилацетат и высушивали.
Выход: 593 г, (86%).
1h. Перекристаллизация трометамина карбопроста
500 г трометаминовой соли карбопроста растворяли в фильтрованном, дистиллированном изопропаноле. К полученному раствору добавляли по каплям фильтрованный, дистиллированный ацетон при 20°C. После осаждения большинства кристаллов, добавляли этилацетат (фильтрованный, дистиллированный), затем гексан (фильтрованный, дистиллированный) и суспензию кристаллов дополнительно перемешивали. После прим. 1 часа перемешивания кристаллы отфильтровывали, промывали с помощью смеси гексан:ацетон:этил ацетат и высушивали.
Выход: 480 г, 96%.
Реферат
Изобретение относится к способу получения карбопроста формулы I и его трометаминовой соли формулы Ia посредством селективного алкилирования енона общей формулы II, где R обозначает защитную группу, посредством восстановления полученного в результате енола общей формулы III, где значение R определено выше, посредством удаления R-защитной группы полученного в результате лактола общей формулы IV, посредством осуществления реакции эпимеров формулы V лактола в ходе реакции Виттига, с получением эпимеров формулы VI карбопроста, посредством преобразования эпимеров карбопроста в их сложный метиловый эфир, посредством хроматографического разделения эпимеров формулы VII сложного метилового эфира, посредством гидролиза эпимера формулы VIII, и, если необходимо, посредством получения трометаминовой соли карбопроста, предусматривающий, что селективное алкилирование осуществляют в присутствии хирального вспомогательного средства в апротонном органическом растворителе с реактивом Гриньяра, хроматографию осуществляют посредством гравитационной хроматографии на силикагеле, трометаминовую соль получают путем применения твердого трометаминового основания, элюент, применяемый для гравитационной хроматографии на силикагеле, содержит основание или в ходе гравитационной хроматографии на силикагеле применяют слабоосновный силикагель. 16 з.п. ф-лы, 8 ил., 8 пр.
Формула




Комментарии