Способ очистки азациклогексапептида или его соли - RU2528635C2
Код документа: RU2528635C2
Чертежи







Описание
Область техники
Изобретение относится к области органической химии, в частности к способу очистки азациклогексапептида формулы 1 или его солей.
Уровень техники
Из-за увеличения числа пациентов с иммунным дефицитом, возникающим в результате широкого применения инвазивной терапии и антибиотиков широкого спектра действия, применения химиотерапии у онкологических пациентов и реципиентов трансплантатов органов, а также злокачественных заболеваний крови и СПИДа, в последние десятилетия значительно увеличилось число случаев тяжелых или даже смертельных грибковых инфекций. Применение антимикробных лекарственных средств ограничено из-за их токсичности, лекарственного взаимодействия и устойчивости.
В 1974 было установлено, что соединения эхинокандина обладают повышенной антимикробной активностью. Механизм антимикробной активности представляет собой блокирование синтеза β-(1,3)-0-глюкозана патогенных грибов, таким образом, воздействие на синтез клеточной стенки патогенных грибов, в результате которого достигается противогрибковый эффект. В 2001 каспофунгин был одобрен управлением США по контролю за качеством пищевых продуктов и лекарственных средств, которое задало направление для исследования противогрибковых лекарственных препаратов. Каспофунгин, химическая структура которого представлена формулой 1, изначально был разработан Merck Inc. в качестве противогрибкового/противопневмоцистозного лекарственного препарата с широким спектром действия и представляет собой низкотоксичный агент с уникальным местом воздействия и широким спектром применения.
В 1994 ЕР 0620232 раскрыл способ синтеза и очистки каспофунгина из эхинокандина B0. В дальнейшем US 5552521 раскрыл модифицированный способ синтеза и очистки каспофунгина. В обоих патентах для очистки промежуточных и чистых продуктов применяли препаративную хроматографию на колонках с силикагелем С18 и собранные жидкости лиофилизировали.
Однако применение колонок с силикагелем С18 и лиофильной сушки в случае очистки и высушивания промежуточных и чистых продуктов усложняло воплощение способов самих по себе, задавало высокие требования к потреблению энергии и оборудованию и вызывало серьезное повреждение устройств, таким образом, делая невозможным производство в промышленном масштабе.
Поэтому все еще существует острая необходимость в области техники в предложении нового способа очистки азациклогексапептида формулы 1 или его солей.
Краткое описание изобретения
Цель настоящего изобретения предложить новый способ очистки азациклогексапептида формулы 1 или его солей.
В первом аспекте изобретения предложен способ очистки соединения формулы 1, где указанный способ включает следующие этапы:
(1) загрузку неочищенного соединения формулы 1 в макропористую адсорбционную смолу;
(2) промывание макропористой адсорбционной смолы при помощи водного раствора, органического растворителя или смешанного раствора органического растворителя и воды;
(3) элюирование при помощи водного раствора, органического растворителя или смешанного раствора органического растворителя и воды для получения очищенного соединения формулы 1.

Макропористая адсорбционная смола выбрана из неполярной ароматической адсорбционной смолы, полимеризованной из стирола и дивинилбензола, или умеренно полярной метакриловой смолы со звеньями метакрилата в ее структуре. Предпочтительно макропористая адсорбционная смола выбрана из одной или более следующих смол: XAD-1, XAD-2, XAD-3, XAD-4, XAD-5, XAD-16, XAD-16HP; или из одной или более следующих смол: НР-10, НР-20, HP-20ss, НР-21, НР-30, НР-40, НР-50, SP-825, SP-850, SP-70, SP-700, SP-207; или из одной или более следующих смол: XAD-6, XAD-7, XAD-7HP, XAD-8; или HP-2MG.
Органический растворитель выбран из C1-C4 спирта, C1-C4 кетона, ацетонитрила или тетрагидрофурана. Предпочтительно C1-C4 спирт выбран из одного или более следующих соединений: метанола, этанола, пропанола и бутанола. C1-C4 кетон выбран из одного или более из следующих соединений: ацетон и бутанон.
рН водного раствора, органического растворителя или смешанного раствора органического растворителя и воды составляет ≤7.
Предпочтительно на этапе (3) градиентное элюирование проводят при повышении концентрации (об.%) органического растворителя в случае проведения элюирования при помощи водного раствора, органического растворителя или смешанного раствора.
В другом предпочтительном воплощении за этапом (3) следует:
(4) кристаллизация очищенного соединения формулы 1 для получения соединения формулы 1, обладающего чистотой свыше 99%.
Во втором аспекте изобретения предложено соединение формулы 1, обладающее чистотой свыше 99%, где упомянутое соединение получают посредством способа очистки по изобретению, как описано выше.
В третьем аспекте изобретения предложен кристалл соединения формулы 1, обладающего чистотой свыше 99%, как описано выше. Профиль рентгеновской порошковой дифракции (XRPD) кристалла содержит характерные пики при следующих углах дифракции 2θ: 2,940±0,2°, 5,061±0,2°, 5,880±0,2° и 8,960±0,2°. Предпочтительно профиль рентгеновской порошковой дифракции (XRPD) кристалла также содержит характерные пики при следующих углах дифракции 2θ: 6,661±0,2°, 10,299±0,2° и 17,900±0,2°.
Кристалл обладает ИК-спектрограммой, приведенной на Фиг.5. На графике дифференциальной сканирующей калориметрии (DSC) кристалла максимальный эндотермический пик расположен между 140-146°C.
Соответственно, в изобретении предложен новый способ очистки азациклогексапептида формулы 1 или его солей.
Краткое описание графических материалов
На Фиг.1 изображена ВЭЖХ-хроматограмма неочищенного соединения 1, синтезированного посредством пути синтеза согласно US 5552521A.
На Фиг.2 изображена ВЭЖХ-хроматограмма неочищенного соединения 1, синтезированного посредством пути синтеза согласно CN 101648994A.
На Фиг.3 изображена ВЭЖХ-хроматограмма ацетата каспофунгина, полученного по примеру 3 изобретения.
На Фиг.4 изображен профиль XRPD ацетата каспофунгина, полученного согласно примеру 3 изобретения.
На Фиг.5 изображен ИК-спектр ацетата каспофунгина, полученного по примеру 3 изобретения.
На Фиг.6 изображен график DSC ацетата каспофунгина, полученного по примеру 3 изобретения.
На Фиг.7 изображена ВЭЖХ-хроматограмма ацетата каспофунгина, полученного по примеру 4 изобретения.
В вышеупомянутых ВЭЖХ-хроматограммах, "RT" представляет собой время удерживания, "площадь" представляет собой площадь пика, "%площади" представляет собой процент площади пика от общей площади пиков, и "высота" представляет собой высоту пика.
Подробное описание изобретения
Авторы изобретения обнаружили, что соединение 1 может быть полностью изолировано и очищено при помощи макропористой адсорбционной смолы под действием определенных условий.
Как употреблено в данном документе, "соединение формулы 1" и "соединение 1" могут быть применены взаимозаменяемо, оба относятся к соединению, имеющему следующую структуру, или его фармацевтически приемлемым солям:

Как употреблено в данном документе, "фармацевтически приемлемая соль" означает соль, полученную из кислоты, выбранной из соляной кислоты, бромистоводородной кислоты, фосфорной кислоты, серной кислоты, малеиновой кислоты, лимонной кислоты, уксусной кислоты, винной кислоты, янтарной кислоты, щавелевой кислоты, яблочной кислоты, глутаминовой кислоты или других кислот, соответствующих каким-либо фармацевтически приемлемым солям, перечисленным в Journal of Pharmaceutical Science, 66:2 (1977).
Как употреблено в данном документе, "чистота соединения формулы 1", "чистота соединения 1" и "ВЭЖХ-чистота соединения 1" могут быть применены взаимозаменяемо, все относятся к проценту площади пика соединения 1 от общей площади пиков, как определено при условиях детектирования высокоэффективной жидкостной хроматографии (ВЭЖХ) согласно изобретению.
Как употреблено в данном документе, "неочищенное соединение формулы 1" и "неочищенное соединение 1" могут быть применены взаимозаменяемо, оба относятся к смеси, содержащей<90% соединения 1, как определено при условиях детектирования высокоэффективной жидкостной хроматографии (ВЭЖХ) согласно изобретению. Неочищенное соединение 1 может быть получено при помощи любого приемлемого способа, известного в области техники, включая, например, но не ограничено, способы, описанные в примере 1 US 5552521A и примерах 1-7 CN 101648994A, где неочищенное соединение 1 получают посредством многоэтапных химических реакций при помощи микробиологически ферментированного продукта, пневмокандина B0, в качестве исходного материала.
Как употреблено в данном документе, "раствор, содержащий неочищенное соединение формулы 1", и "раствор, содержащий неочищенное соединение 1" могут быть употреблены взаимозаменяемо, оба относятся к раствору, содержащему целевое соединение 1 и одно или более нецелевое соединение, и могут быть получены посредством растворения неочищенного соединения 1 в воде или буферном растворе с pH≤7 или посредством смешивания реакционного раствора, содержащего соединение 1 из любого способа, с водой или буферным раствором с pH≤7 для получения смешанного раствора, содержащего органический растворитель. Может быть применен реакционный раствор, содержащий соединение 1 из любого способа, известного в области техники, для приготовления соединения 1, включая, например, но не ограничено, реакционный раствор, полученный посредством многоэтапных химических реакций, при помощи микробиологически ферментированного продукта, пневмокандина B0, в качестве исходного материала. Например, согласно способу синтеза, описанному в US 5552521A, активную амидную группу на пневмокандине B0 восстанавливали при помощи боргидрида для получения амина, и затем активная гидроксильная группа на амине реагировала с соединением, обладающим легко замещаемой группой, такой как тиофенол, для получения фенилсульфида. Затем проводили аммонолиз фенилсульфида этилендиамином в метаноле для получения реакционного раствора соединения 1 в метаноле (см. нижеприведенную схему).

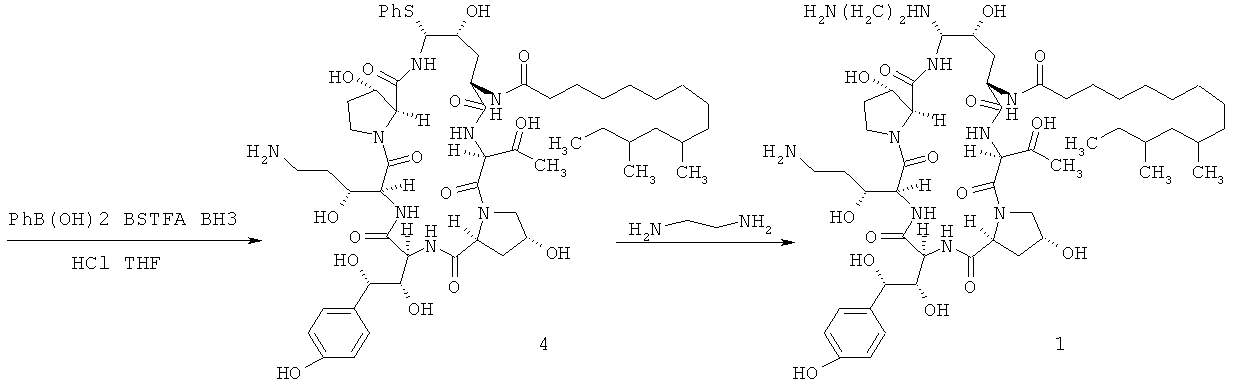
Также может быть применен способ, описанный в патентной заявке КНР CN 101648994A, предусматривающий реакцию активной гидроксильной группы на пневмокандине B0, который представлял собой микробиологически ферментированный продукт и был применен в качестве исходного материала, с соединением, обладающим легко замещаемой группой, такой как тиофенол, для получения фенилсульфида, и аммонолиз фенилсульфида этилендиамином в метаноле для получения амина. Затем амин восстанавливали при помощи боргидрида в тетрагидрофуране для получения реакционного раствора соединения 1 в тетрагидрофуране (см. приведенную ниже схему):


Реакционные растворы соединения 1, упомянутые выше, представляют собой только некоторые примеры, и реакционные растворы соединения 1 по изобретению не должны быть ограничены этими примерами.
Как употреблено в данном документе, "макроскопическая адсорбционная смола" в основном включает: (а) неполярную ароматическую смолу, полимеризованную из стирола и дивинилбензола, например адсорбционные смолы серии XAD (Rohm & Haas Inc., USA), такие как XAD-1, XAD-2, XAD-3, XAD-4, XAD-5, XAD-16, XAD-16HP или их смеси, и адсорбционные смолы серии Diaion HP (Mitsubishi Inc., Japan), такие как HP-10, НР-20, HP-20ss, НР-21, НР-30, НР-40, НР-50, SP-825, SP-850, SP-70, SP-700, SP-207 или их смеси; и (b) среднеполярную метакриловую адсорбционную смолу с единицами метакрилата в ее структуре, например адсорбционные смолы серии XAD (Rohm & Haas Inc., USA), такие как XAD-6, XAD-7, XAD-7HP, XAD-8 или их смеси, или адсорбционные смолы серии Diaion HP (Mitsubishi Inc., Japan), такие как HP-2MG.
Как употреблено в данном документе, "загрузка" относится к способу приведения раствора, содержащего неочищенное соединение 1, в контакт с макроскопической адсорбционной смолой, так что неочищенное соединение 1 адсорбируется на макроскопическую адсорбционную смолу. "Контактирование" включает помещение макроскопической адсорбционной смолы непосредственно в раствор и затем перемешивание для того, чтобы адсорбция произошла, или размещение макроскопической адсорбционной смолы в хроматографическом устройстве и направление тока раствора через хроматографическую колонку.
"Промывка" макроскопической адсорбционной смолы означает, что приемлемый буферный раствор пропускают через или сквозь макропористую адсорбционную смолу.
Как употреблено в данном документе, "промывочный буферный раствор" относится к буферному раствору, применяемому для промывки макроскопической адсорбционной смолы (в основном для удаления органической фазы) до элюирования целевого соединения 1. Для удобства промывочный буферный раствор и буферный раствор для нанесения образца могут, но необязательно, иметь одинаковый рН.
"Элюирование" молекул из макроскопической адсорбционной смолы означает, что молекулы удаляют из макроскопической адсорбционной смолы посредством изменения полярности буферного раствора вокруг макроскопической адсорбционной смолы. Из-за полярности буферный раствор может конкурировать с молекулами за адсорбционные сайты на макроскопической адсорбционной смоле.
Как употреблено в данном документе, "элюирующий буферный раствор" применяют для элюирования мишенного соединения 1 из стационарной фазы. Мишенное соединение 1 может быть элюировано из макроскопической адсорбционной смолы за счет pH элюирующего буферного раствора.
"Очистка" соединения 1 от композиции, содержащей мишенное соединение 1 и одно или более немишенное соединение, означает, что чистоту соединения 1 в композиции повышают за счет удаления (полного или частичного) по меньшей мере одного немишенного соединения из композиции.
Способ очистки соединения формулы 1 по изобретению предусматривает следующие этапы:
(1) загрузку неочищенного соединения 1 в макропористую адсорбционную смолу;
(2) промывку макропористой адсорбционной смолы при помощи водного раствора, органического растворителя или смешанного раствора органического растворителя и воды;
(3) элюирование при помощи водного раствора, органического растворителя или смешанного раствора органического растворителя и воды для получения очищенного соединения 1 (чистота ≥90%).
За вышеупомянутым этапом (3) способа очистки следует этап кристаллизации для получения соединения 1 с высокой чистотой (чистота ≥99%).
В одном воплощении изобретения способ очистки предусматривает следующие этапы:
во-первых, загрузку неочищенного соединения 1 в макропористую адсорбционную смолу;
во-вторых, промывку макропористой адсорбционной смолы при помощи большого количества водного раствора для удаления органической фазы; и
в-третьих, элюирование при помощи смешанного раствора органического растворителя в градиенте, где органический растворитель применяют в двух или более концентрациях от низкой до высокой в интервале 5%-95% (об.%) и затем собирают и комбинируют соответствующие элюаты (с соединением 1, обладающим чистотой ≥90%) для получения очищенного соединения (чистота ≥90%).
За третьим этапом вышеописанного способа очистки может следовать четвертый этап, где очищенное соединение 1 (чистота ≥90%) смешивают с растворяющим раствором (этанол/вода/уксусная кислота), затем для индуцирования кристаллизации по капле добавляют этилацетат, и после фильтрации получают высокочистое соединение 1 (чистота ≥99%).
На первом этапе «загрузка» означает приведение раствора, содержащего неочищенное соединение 1, в контакт с макропористой адсорбционной смолой, где уровень pH раствора, предпочтительно содержащего соединение 1, составляет ≤7, предпочтительно в диапазоне 4,5-6,0, и более предпочтительно в диапазоне 5,0-5,5. Раствор, содержащий неочищенное соединение 1, может быть получен посредством непосредственно разбавления реакционного раствора, содержащего соединение 1, при помощи воды для получения раствора, содержащего менее 10% органического растворителя, и затем подведения уровня pH до ≤7 при помощи обычной кислоты, такой как уксусная кислота, соляная кислота и т.п.
В другом воплощении изобретения способ очистки предусматривает следующие этапы:
A. приведение раствора, содержащего неочищенное соединение 1, в контакт с макропористой адсорбционной смолой;
B. отделение раствора, содержащего неочищенное соединение 1, от смолы;
C. промывку макропористой адсорбционной смолы после этапа B при помощи промывочного раствора, выбранного из водного раствора, органического растворителя или их смешанного раствора;
D. приведение промытой макропористой адсорбционной смолы, полученной на этапе C, в контакт с элюирующим раствором, выбранным из водного раствора, органического растворителя или их смешанного раствора, и затем сбор элюата, содержащего соединение 1; и
E. концентрирование собранного элюата под действием пониженного давления до сухости, и затем кристаллизация до получения высокочистого соединения 1 (чистота ≥99%).
Упомянутое отделение на этапе B включает фильтрацию.
Требования к способу очистки соединения 1 очень жесткие, поскольку соединение 1 нестабильно. Соединение 1 клинически применяют в виде ацетата, поэтому способ очистки по изобретению проиллюстрирован при помощи его ацетата, где:
во-первых, реакционный раствор, содержащий соединение 1, разбавляют при помощи непосредственно воды для образования раствора, содержащего менее 10% органического растворителя. Уровень pH раствора регулируют при помощи уксусной кислоты до ≤7 и затем адсорбцию проводят при помощи предварительно обработанной макропористой адсорбционной смолы. Макропористую адсорбционную смолу промывают при помощи значительного количества водного раствора уксусной кислоты с pH≤7 для удаления органической фазы. Затем макропористую адсорбционную смолу элюируют в градиенте при помощи водного раствора уксусной кислоты, где pH раствора составляет ≤7 и концентрация органического растворителя составляет 5%-95%. Соответствующие элюаты собирают и объединяют. После концентрации и кристаллизации ацетат соединения 1 с высокой чистотой (чистота ≥99%) получают в качестве кристаллического порошка.
Профиль порошковой рентгеновской дифракции (XRPD) кристалла ацетата для каспофунгина содержит характерные пики при следующих углах дифракции 2θ: 2,940±0,2°, 5,061±0,2°, 5,880±0,2° и 8,960±0,2°. Предпочтительно профиль порошковой рентгеновской дифракции (XRPD) также содержит характерные пики при следующих углах дифракции 2θ: 6,661±0,2°, 10,299±0,2° и 17,900±0,2°.
Во всех способах очистки по изобретению pH водного раствора составляет ≤7, предпочтительно в диапазоне 4,5-6,0 и более предпочтительно в диапазоне 5,0-5,5. Водный раствор включает раствор уксусной кислоты, раствор соляной кислоты и т.п.
Во всех способах очистки по изобретению смешанный раствор органического растворителя и воды содержит 5%-95%, предпочтительно 10%-60% (об.%) органического растворителя от общего объема смешанного раствора.
Во всех способах очистки по изобретению органический растворитель выбран из C1-C4 спирта, C1-C4 кетона, ацетонитрила или тетрагидрофурана. C1-C4 спирт выбран из одного или более следующих соединений: метанол, этанол, пропанол и бутанол. C1-C4 кетон выбран из одного или более следующих соединений: ацетон и бутанон.
Все характеристики, упомянутые выше или в нижеприведенных примерах изобретения, при желании могут быть скомбинированы. Все характеристики, раскрытые в описании, могут быть применены в любой комбинации. Любая альтернативная характеристика, служащая той же, эквивалентной или похожей цели, может быть заменена каждой характеристикой, раскрытой в описании. Поэтому если не указано иное, раскрытые характеристики являются только общими примерами эквивалентных или сходных характеристик.
Преимущества изобретения главным образом включают:
1. Предложен новый недорогой способ очистки азациклогексапептида, в частности, соединений эхинокандина;
2. Преимущества этапов очистки в способе по изобретению, такие как простота, мягкие условия, высокие выходы, несложные методы обработки и т.п., в значительной степени снижают требования к действиям по способу и оборудованию, а также стоимость;
3. Стабильные мишенные соединения могут быть получены посредством способа по изобретению, таким образом, способствуя контролю качества конечных продуктов и промышленного производства.
В дальнейшем изобретение будет проиллюстрировано при помощи ссылки на следующие конкретные примеры. Следует понимать, что эти примеры предназначены только для иллюстрации изобретения, но не для ограничения его объема. Относительно экспериментальных способов в следующих примерах без конкретных условий, их осуществляют при обычных условиях или как указано производителем. Если не указано иное, все проценты, соотношения, пропорции или части указаны по массе.
Единица массовых/объемных % в изобретении хорошо известна специалистам в области техники, например масса растворенного вещества в 100 мл раствора.
Если не определено иное, все научные и технические термины, применяемые в данном документе, имеют такое же значение, как и обычно понимаемое специалистами в области техники. Кроме того, любой способ или материал сходный с таковым, описанным в данном документе, может быть применен в способе по настоящему изобретению. Предпочтительные воплощения и материалы, описанные в данном документе, приведены только для иллюстрации.
Условия для определения образцов по изобретению (соединение 1) посредством высокоэффективной жидкостной хроматографии:
Хроматограф: водная ВЭЖХ-система
Хроматографическая колонка: Kromasil ODS 250*4,6 мм, 5 мкм
Мобильная фаза A: 0,1% (об.) водный раствор перхлорной кислоты
Мобильная фаза B: ацетонитрил
Процедура:
Вводимый объем: 10 мкл
Температура колонки: 35°C
Длина волны детектирования: 220 нм
Скорость тока: 1,0 мл/мин
Пример 1
Синтез соединения 1 согласно US 5552521A
500 мл реакционного раствора, содержащего соединение 1, получали согласно пути синтеза, раскрытому в US 5552521A из B0 (45,0 г, 42,24 ммоль). Чистота продукта, подлежащего очистке, составляла 78,64%, как установлено посредством ВЭЖХ (Фиг.1).
Пример 2
Синтез соединения 1 согласно CN 101648994A
1,6 л реакционного раствора, содержащего соединение 1, получали согласно пути синтеза, раскрытому в CN 101648994A из B0 (50,0 г, 47 ммоль). Чистота продукта, подлежащего очистке, составила 47,09%, как определено посредством ВЭЖХ (Фиг.2).
Пример 3
Очистка соединения 1
При температуре ниже 20°C 30 мл реакционного раствора, содержащего соединение 1 (полученное в примере 1) разводили в 250 мл очищенной воды. pH подводили до 5,0-5,5 при помощи уксусной кислоты. Адсорбцию проводили при помощи 100 мл предварительно обработанной смолы HP-20ss при скорости тока 1 л/ч. Смолу промывали при помощи 300 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 10%, 20% и 25% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Собирали около четырех объемов колонки элюата и затем концентрировали до сухости.
Полученное соединение 1 с чистотой ≥90% растворяли в 7,3 мл растворяющего раствора этанола/воды/уксусной кислоты=207,8/19,4/1 (об.%) и затем при комнатной температуре индуцировали кристаллизацию посредством добавления по капле этилацетата. После добавления раствор перемешивали в течение 1 часа, пока температура сохранялась постоянной. После фильтрования фильтрационный осадок промывали при помощи раствора воды: этанола:этилацетата=1,0:10,7:17,1 для получения белого кристаллического твердого вещества, т.е. ацетата каспофунгина (0,73 г, чистота=99,85%) (Фиг.3).
Белый порошок ацетата каспофунгина детектировали при помощи рентгеновского дифрактометра RIGAKU D/max 2550VB/PC при скорости сканирования 2°/мин. И применяли медную мишень для облучения. Полученный профиль порошковой рентгеновской дифракции приведен на Фиг.4.
Белый порошок ацетата каспофунгина детектировали при помощи спектрометра РЕ SPEGRUM 1В IR. Полученный ИК спектр приведен на Фиг.5.
Белый порошок ацетата каспофунгина детектировали при помощи дифференциального сканирующего калориметра WATERS Q20. Полученный график DSC приведен на Фиг.6.
Пример 4
Очистка соединения 1
При температуре ниже 20°C 30 мл реакционного раствора соединения 1 (полученного в примере 1) разводили в 250 мл очищенной воды. pH подводили до 5,0-5,5 при помощи уксусной кислоты. Адсорбцию проводили при помощи 100 мл предварительно обработанной смолы HP-20ss при скорости потока 1 л/ч. Смолу промывали при помощи 300 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 25%, 30% и 50% метанола. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Собирали около трех объемов колонки элюата и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (0,41 г, чистота=99,81%), получали при помощи такого же способа кристаллизации, как описанный в примере 3 (Фиг.7).
Пример 5
Очистка соединения 1
При температуре ниже 20°C 30 мл реакционного раствора соединения 1 (полученного в примере 1) разводили в 250 мл очищенной воды. pH подводили до 5,0-5,5 при помощи уксусной кислоты. Адсорбцию проводили при помощи 100 мл предварительно обработанной смолы XAD-1600 при скорости потока 1 л/ч. Смолу промывали при помощи 300 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 20%, 30% и 40% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Около трех объемов колонки элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (0,51 г, чистота=99,46%), получали при помощи того же способа кристаллизации, что и описанный в примере 3.
Пример 6
Очистка соединения 1
При температуре ниже 20°C 30 мл реакционного раствора соединения 1 (полученного в примере 1) разводили в 250 мл очищенной воды. рН подводили до 5,0-5,5 при помощи уксусной кислоты. Адсорбцию проводили при помощи 100 мл предварительно обработанной смолы HZ-803 при скорости потока 1 л/ч. Смолу промывали при помощи 300 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 30%, 40% и 50% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Около трех объемов колонки элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (0,58 г, чистота=99,88%), получали при помощи того же способа кристаллизации, как описанный в примере 3.
Пример 7
Очистка соединения 1
При температуре ниже 20°C 30 мл реакционного раствора соединения 1 (полученного в примере 1) разводили в 250 мл очищенной воды. pH подводили до 5,0-5,5 при помощи уксусной кислоты. Адсорбцию проводили при помощи 100 мл предварительно обработанной смолы LD-605 при скорости тока 1 л/ч. Смолу промывали при помощи 300 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу последовательно элюировали в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 20%, 40% и 50% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Около трех объемов колонки элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (0,65 г, чистота=99,90%), получали при помощи такого же способа кристаллизации как описанный в примере 3.
Пример 8
Очистка соединения 1
При температуре ниже 20°C 160 мл реакционного раствора соединения 1 (полученного в примере 2) разводили в 1440 мл очищенной воды. Адсорбцию проводили при помощи 170 мл предварительно обработанной смолы HP-20ss при скорости потока 1 л/ч. Смолу промывали при помощи 500 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем макропористую адсорбционную смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 10%, 20% и 25% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Около четырех объемов элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (1,76 г, чистота=99,71%), получали при помощи такого же способа кристаллизации, как описанный в примере 3. Пример 9
Очистка соединения 1
При температуре ниже 20°C 160 мл реакционного раствора соединения 1 (полученного в примере 2) разводили в 1440 мл очищенной воды. Адсорбцию проводили при помощи 170 мл предварительно обработанной смолы HP-20ss при скорости потока 1 л/ч. Смолу промывали при помощи 500 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 25%, 30% и 50% метанола. Соответствующие элюаты (чистота>90%) собирали и объединяли. Около трех объемов колонки элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (1,69 г, чистота=99,61%), получали при помощи такого же способа кристаллизации, как описанный в примере 3.
Пример 10
Очистка соединения 1
При температуре ниже 20°C 160 мл реакционного раствора соединения 1 (полученного в примере 2) разводили в 1440 мл очищенной воды. Адсорбцию проводили при помощи 170 мл предварительно обработанной смолы XAD-1600 при скорости потока 1 л/ч. Смолу промывали при помощи 500 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (о/о) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 20%, 30% и 40% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Около трех объемов колонки элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (1,63 г, чистота=99,01%), получали при помощи такого же способа кристаллизации, как описанный в примере 3.
Пример 11
Очистка соединения 1
При температуре ниже 20°C 160 мл реакционного раствора соединения 1 (полученного в примере 2) разводили в 1440 мл очищенной воды. Адсорбцию проводили при помощи 170 мл предварительно обработанной смолы HZ-803 при скорости потока 1 л/ч. Смолу промывали при помощи 500 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 30%, 40% и 50% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Около трех объемов колонки элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (1,70 г, чистота = 99,71%), получали при помощи такого же способа кристаллизации, как описанный в примере 3.
Пример 12
Очистка соединения 1
При температуре ниже 20°C 160 мл реакционного раствора соединения 1 (полученного в примере 2) разводили в 1440 мл очищенной воды. pH подводили до 5,0-5,5 при помощи уксусной кислоты. Адсорбцию проводили при помощи 170 мл предварительно обработанной смолы LD-605 при скорости потока 1 л/ч. Смолу промывали при помощи 500 мл 0,016% (об.%) раствора уксусной кислоты с pH 5-5,5 для удаления органической фазы. Затем смолу элюировали последовательно в трех градиентах при помощи 0,016% (об.%) водных растворов уксусной кислоты, где pH раствора составлял 5-5,5 и раствор содержал 20%, 40% и 50% ацетона. Соответствующие элюаты (чистота ≥90%) собирали и объединяли. Около трех объемов колонки элюата собирали и затем концентрировали до сухости.
Белое кристаллическое твердое вещество, т.е. ацетат каспофунгина (1,68 г, чистота = 99,88%), получали при помощи такого же способа кристаллизации, как описанный в примере 3.
Вышеупомянутые примеры являются всего лишь предпочтительными примерами для настоящего изобретения, и такие примеры не могут быть применены для ограничения объема изобретения. Техническое содержание настоящего изобретения по существу широко раскрыто в формуле изобретения. И любые объекты или способы, осуществленные иначе, должны рассматриваться в качестве эквивалентов и находиться в объеме изобретения, определенном формулой изобретения, если указанные объекты или способы являются такими же, как определенные формулой изобретения.
Реферат
Предложен способ очистки соединения формулы 1, включающий следующие этапы: (1) загрузку неочищенного соединения 1 в макропористую адсорбционную смолу, (2) промывку макропористой адсорбционной смолы при помощи водного раствора, органического растворителя или смешанного раствора органического растворителя и воды, (3) элюирование при помощи водного раствора, органического растворителя или смешанного раствора органического растворителя и воды. 8 з.п. ф-лы, 7 ил., 12 пр.
Формула
(1) загрузку неочищенного соединения 1 в макропористую адсорбционную смолу;
(2) промывку макропористой адсорбционной смолы водным раствором, органическим растворителем или смешанным раствором органического растворителя и воды;
(3) элюирование водным раствором, органическим растворителем или смешанным раствором органического растворителя и воды с получением очищенного соединения формулы 1

где макропористая адсорбционная смола выбрана из неполярной ароматической адсорбционной смолы, полимеризованной из стирола и дивинилбензола, или умеренно полярной метакриловой адсорбционной смолы, включающей звено метакрилата в своей структуре,
причем органический растворитель выбран из C1-C4 спирта, C1-C4 кетона, ацетонитрила или тетрагидрофурана, и pH водного раствора, органического растворителя или смешанного раствора органического растворителя и воды составляет не более 7.
(4) кристаллизация очищенного соединения формулы 1 с получением кристалла, обладающего чистотой выше 99%.
Комментарии