Полипептиды или их соли и фармацевтическая композиция - RU2083589C1
Код документа: RU2083589C1
Чертежи







Описание
Изобретение относится к полипептидам или их солям, обладающим сильным средством к липополисахаридам, в частности эндотоксинам.
Такой полипептид можно использовать в фармацевтической композиции в качестве антибактериального или противовирусного агента (например, анти-ВИЧ агента).
2. Предпосылки изобретения
Два семейства противомикробных полипептидов были выделены из камчатского краба (см. например,
Shigenaga 1990, j.
Biol.chem т. 265, стр. 21350-21354; Kawara и др. 1990, j.Biol.chem, т. 108, стр. 261-266; Открытая патентная публикация N 167230/1990; Открытая патентная публикация N 152987/1990;
Открытая патентная
публикация N 53799/1990; Опубликованная исследовательская заявка 500194/1990; Miyatfa и др. 1989, j.Biol.chem 106, стр. 663-668; Akaji и др. 1989, chem.Pharm. Bull т. 37, стр.
2661-2664; Taisha
(Метаболизм), т. 26, стр. 301-311 (1989); Shieh и др. 1989, FEBS Lett. т. 252, стр. 121-124; и Nakamura и др. 1988, j.Biol. chem. т. 263, стр. 16709-16713). Одно семейство,
семейство тачиплезина, было
выделено из японского краба Tachypleus. Три тачиплизина I, II и III были идентифицированы, их аминокислотные последовательности приведены на чертеже. Кроме того,
производное пептида тачиплезина с
карбоксилконцевым продолжением гликолализина было обнаружено в Южно-азиатских видах краба, Carcinoscorpius of undicanda (Muta и др. 1990, j. Biochem. т. 108, с.
261-266). Второе семейство, семейство
полуфемузина, было выделено из гемоцитов Американского краба, Limulus polyphemus. Два полифемузина I и II были идентифицированы. Их аминокислотные
последовательности приведены также на чертеже.
Полипептиды обоих семейств состоят из 17 или 18 аминокислотных остатков и имеют четыре сохраняемые общие области и два дисульфидных мостика (см.
чертеж). Было установлено, что как тачиплезины, так и
полифемузины ингибируют как грамотрицательные, так и грамположительные бактерии при низких концентрациях, а также грибы такие, как Candide
albicaus и образуют комплексы с бактериальными
липополисахаридами (Shigenaga и др. 1990, j. Biol.chem, т. 265, с. 21350-21354, и Muta и др. 1990, j. Biochem. т. 108, с. 261-266).
Кроме того, как было установлено, полипептиды в семействе тачиплезина обладают некоторой активностью по ингибированию вируса такого, как вирус гриппа, вирус везикулярного стоматита (marakaui и др. 1991, chemotherapy, т. 37, с. 327-334) или вирус иммунодефицита человека (Morimoto и др. 1991, chemotherapy, т. 37, с. 206-211).
3. Краткое описание изобретения
Изобретение
относится к новым полипептидам, которые получают из
камчатских крабов и которые обладают некоторой гомологичностью с полипептидами высокого сродства относительно эндотоксина, в частности,
тачиплезинами и полифемузинами, но обладают также существенным
отличием. В полипептидах камчатских крабов аминокислота в 6-позиции тачиплезинов или в 7-позиции полифемузинов является валином (Val),
нейтральными алифатическими аминокислотами. В полипептидах,
являющихся предметом изобретения, аминокислотный остаток в 6-позиции является лизином (Lys) или аргинином (Arg); эти основные
аминокислотные остатки существенно отличаются по свойствам от валинового
остатка. Кроме того, аминокислотный остаток в 11-позиции в полипептидах, являющихся предметом изобретения, является тирозином
(Tyr), фенилаланином (Phe) или триптофаном (Trp), ароматическим
аминокислотным остатком, обладающим отличными свойствами от изолейцина (Ile) в 11-позиции тачиплезинов.
Новые полипептиды, являющиеся предметом изобретения, могут быть использованы в качестве анти-ВИЧ агентов. Как будет подробно описано в разделе 6, см. ниже, полипептиды, являющиеся предметом изобретения, обладают анти-ВИЧ активностью, которая значительно выше по сравнению с известными полипептидами с высоким сродством эндотоксина камчатских крабов.
3.1 Определения
Пептидные
последовательности, определяемые здесь, представляются сокращениями из
трех букв для аминокислотных остатков следующим образом:
Ala (аланин); Arg (аргинин); Cys (цистеин); Gly (гликокол);
Ile (изолейцин); Leu (лейцин); Phe (фенилаланин); Trp (триптофан); Tyr
(титрозин) и Val (валин).
Приводимые термины в настоящем патентном описании имеют следующий смысл:
HIY
(ВИЧ) вирус иммунодефицита человека (все варианты)
MOI
множественность инфекции
S] индекс селективности (отношение CC50 к EC50).
4. Краткое описание чертежей.
На чертеже приведены аминокислотные последовательности Тачиплезина I, Тачиплезина II, Тачиплезина III, Полифемизина и Полифемизина II. Сохраняемые аминокислоты помещены в квадраты. Дисульфидные связи между Cys 3 или -4 и Cys -16 или -17, и Cys -7 или -8 и -12 отмечены жирными линиями.
5. Подробное описание изобретения
Изобретение
относится к новому полипептиду, представляемому следующей
формулой (I):

A1 независимый аминокислотный или пиптидный остаток, выбранный из группы, состоящей из аргинина и аргинилагринина;
A2 независимый аминокислотный остаток, выбранный из группы, состоящей из аргинина или лизина;
A3 независимый аминокислотный остаток, выбранный из группы, состоящей из тирозина и фенилаланина;
A4 =NH2.
В специальном варианте осуществления
изобретения цистеиновые остатки в 3- и 16-позициях и/или цистеиновые остатки в 7- и 12-позициях могут быть связаны через
дисульфидную связь (-S-S-). А также к фармацевтической композиции для
ингибирования ВИЧ-активности у пациента, включающей активный ингредиент и по крайней мере один или несколько фармацевтически
приемлемых носителей, содержащей в качестве активного ингредиента
эффективное количество от 10 до 5000 мг/кг веса человеческого тела в день полипептида формулы I, где
A1=Аргинин
или аргиниларгинин
A2=аргинин или лизин
A3=тирозин или фенилаланин
A4=NH2,
при этом остатки цистеина в положениях 3 и
16 и в положениях 7 и 12 связаны через дисульфидный мостик.
Подобно полипептидам с высоким сродством относительно эндотоксина, выделенным из камчатских крабов, известным в этой области техники, полипептиды, являющиеся предметом изобретения, обладают антипараллельной бета-листовой структурой из-за существования внутримолекулярных водородных связей и четырех цистеиновых остатков в 3-, 7-, 12-и 16-позициях. Поворачивающаяся позиция с возможно структурой бета-поворота расположена в 9- и 10-позициях. Пептидная часть 3-позиции к 8-позиции и пептидная часть 11-позиции к 16-позиции расположены против друг друга.
Однако в контрасте с известными полипептидами, выделенными из камчатских крабов, полипептиды, являющиеся предметом изобретения, являются более основными. В частности, аминокислотный остаток в 6-позиции является аргининовым (Arg) или лизиновым (Lys) остатком. Аминокислотный остаток в 6-позиции в полипептидах, выделенных из камчатских крабов, является валиновым остатком.
Благодаря своей основной природе, полипептиды, являющиеся предметом изобретения, могут образовать соли при помощи присоединения кислоты. Например, полипептид образует соль с неорганической кислотой (хлористоводородной кислотой, бромистоводородной кислотой, фосфорной кислотой, азотной кислотой, серной кислотой и т. п.) или с органической карбоновой кислотой, уксусной кислотой, галоуксусной кислотой такой, как трифторуксусная кислота, пропионовая кислота, малеиновая кислота, янтарная кислота, яблочная кислота, лимонная кислота, винная кислота, салициловая кислота и мочевая кислота такая, как глюкуроновая кислота или хиалуроновая кислота и т.п. (или органической (моно) сульфокислотой (метан/моно/сульфокислотой, пара-тулоол/моно/сульфокислотой и т.п.), включая сложный эфир сахара (моно/сульфокислоты такой, как сульфаты чондроитина).
5.1. Получение полипептидов
Новый полипептид
изобретения может быть получен при помощи известных приемов, например,
приемов синтеза в твердой фазе, описанных в книге: "Solid-Plase Peptide Sepethesis" Stew-art u Joung, Pierce chemical Company,
Rochford, Illinois (1984).
Полипептид с линейной цепью,
являющийся предметом изобретения, имеющий приведенную формулу (I), может быть получен при помощи установления связи
карбоксильной группы N-защищенного аргинина в 17-позиции с нерастворимой смолой,
имеющей амино-группы, непосредственно присоединенное или альтернативно присоединенное через "промежуток", имеющую
функциональную группу, способную связываться с карбоксильной группой (например,
группой, способной превратить карбоксильную группу аргинина в паракарбоксиметилбензиловый сложный эфир). Аминовая
группа нерастворимой смолы, содержащей аргининовый (Arg) остаток в 17-позиции, после
деблокирования α -амино (NH2)-защищающей группы способна последовательно связываться при
осуществлении процедуры в твердой фазе через соответствующие защищенные аминокислоты в
16-позиции с 1-позицией аминокислотной последовательности, представленной следующей формулой 1:
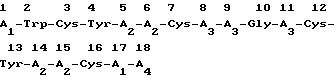
в которой A1, A2, A3, A4, Cys и Giy уже были определены выше), далее осуществляют исключение (удаление) нерастворимой смолы и защищающих групп аминокислот. В этом случае карбоксильные окончания аминокислотного остатка в 17-позиции могут быть либо свободными (A4 соответствует OH), либо превращены в аминокислоты (A4 соответствует - Nh2). Два цистеина в 3- и 16-позициях и 7- и 12-позициях могут образовывать дисульфидную связь (-S-S-) через меркапто группы при окислении воздухом или дисульфидная связь может быть образована по способу Атертона и др. (Atherton, E. и др. J.chem. Soc, Perkin Trans. 1, 1985, стр. 2065), а именно через стадии селективной защиты меркапто групп одной из пар цистеинов в 3- и 16-позициях и 7- и 12-позиций защищающей группой, t-Bus (т-бутилтио), или меркапто групп другой пары цистеинов защищающей группой, Acm (ацетамидометил); затем осуществляют удаление t-Bus, частично окисляя меркапто группы; а затем удаляют Acm-защищающую группу, используя приемы, известные в этой области техники.
Любая нерастворимая смола, содержащая аминогруппу, может быть использована для синтеза нового полипептида, являющегося предметом изобретения, если только она может связываться через свои аминогруппы с карбоксильной группой N-защищенного аргинина или лизина на C-окончании, или, в некоторых случаях, с карбоксильной группой "распорки", соединенной с ней, а затем ее можно удалить (исключить). Примеры таких нерастворимых смол включают (но ими не исчерпывается весь список) амино-метиловые смолы (аминометилированный стирол-дивинилбензол, сополимеры), бензгидриламиновые смолы, метилбензгидриламиновые смолы и аминометилфеноксиметиловые смолы и их производные. Когда используют бензгидриламиновую смолу, метилбензгидриламиновую смолу, диметоксибензгидриламиновую (ДМБГА) смолу или аминометилфеноксиметиловую смолу, амид получают непосредственно при помощи расщепления, но амино-метиловая смола предпочтительна с точки зрения выхода.
Соответствующие аминокислоты, используемые в твердой фазе синтетического метода, могут находиться в L- или D-формах. Защищенной аминокислотой является аминокислота, чьи функциональные группы могут быть защищены защитной группой, используя приемы, известные в этой области техники, или различные защищенные аминокислоты, могут быть получены от фирм-производителей. Любому специалисту в этой области техники известно, что полипептидные синтетические методы требуют использования защищенной группы, чтобы стабилизировать неустойчивую боковую цепь аминокислоты, чтобы предохранить боковую цепь от химического изменения в процессе синтеза. Защищающую группу для α -аминогруппы аминокислоты выбирают из группы, включающей (но ими не исчерпывается полный список) Boc(т-бутилоксикарбонил) или Fmoc(9-флюоренилметилоксикарбонил). Защищающую группу для гуанидино группы аргинина (Arg) выбирают из группы, включающей (но ими не исчерпывается полный список) Tos (толилсульфонил), NO2 (нитро), Mtr (4-метокси-2,3,6-триметилбензол-сульфонил) или Imc (2,3,5,7, 8-пентаметилхроман-5-сульфонил). Защищающая группы для меркапто группы цистеина (Cys) может быть выбрана из группы, включающей (но ими не исчерпывается весь список), Bzl (бензил), MBzl (4-метоксибензил), 4-MeBzl (4-метилбензил), Acm (ацетамидометил), Trt (тритил), Npys (3-нитро-2-пиридинсульфенил), t-Bu (т-бутил) или t-Bus (т-бутилтио) и MBzl, 4-MeBzl, Trt, Acm и Npys используют в предпочтительном варианте. Защищающие группы для гидроксильной группы тирозина (Tyr) выбирают из группы, включающей (но ими не исчерпывается полный список) Bzl, Cl2Bzl (2,6-хлорбензил) или t-Bu. Защищающую группу для e -амино группы лизина (Lys) выбирают из группы, включающей (но ими не исчерпывается полный список (z) бензилоксикарбонил), Clz (2-хлор-бензилоксикарбонил), Boc или Npys.
Сцепление защищенных аминокислот может быть осуществлено в соответствии с различными приемами конденсации, известными в этой области техники, такими, как, например, метод ДЦК (дициклогексилкарбодимида), метод ДИПКДИ (диизопропилкарбодиимида Tartar, А. и др. 1979, J.Org. chem. т. 44, стр. 5000), метод активного сложного эфира, метод смешанных или симметричных ангидридов кислот, метод карбонилдиимидазола, метод ДЦК-НОВ t (1-оксибензотриазола) (konig W. и др. 1970, Chem. Ber т. 103, с. 788, 2024-2034) или метод дифенилфосфорил азида, но в предпочтительном варианте используют ДЦК-метод, ДЦК-НОВ t-метод, ДИПКДИ-НОВ t-метод или метод симметричного ангидрида кислоты. Реакцию конденсации можно осуществить в органическом растворителе таком, как дихлорметан или диметил формамид, или их смеси. Деблокирующий реагент такой, как трифторуксусная кислота (дихлорметан, HCl) диоксан, пиперидин/диметилформамид (ДМФ используют для того, чтобы деблокировать защищающую группу для -аминогруппы. Степень развития реакции конденсации на каждой стадии синтеза определяют при помощи процедуры E.Kaiser и др. [Anal. Biochem. т. 34, с. 595 (1970)] (метод реакции нингидирина).
Когда производное аминометиловой смолы используют в качестве
нерастворимой смолы, защищенный полипептид удаляют из смолы, например, при помощи обработки защищенной
пептидной смолы аммиаком в соответствующем растворителе. Полученный в результате защищенный
пептид затем обрабатывают фтористым водородом, чтобы получить амид полипептида, представленный приведенной
выше формулой, и освобождают от всех защищающих групп. Когда бензгидриламиновую смолу,
метилбензигидриламиновую смолу, аминометилфеноксиметиловую смолу или ДМВНА-смолу (Funa-Koshi. S. и др. J.Chem.
Soc. chem. Commun, 1988, 382) используют в качестве нерастворимой смолы, полипептид
может быть удален из смолы и защищающие группы могут быть удалены одновременно из полипептида при помощи обработки
защищенной пептидной смолы фтористым водородом, МФМСК (трифторметан)
моно/сульфокислотой) (опубликовано издательством Academie Press под редакцией E. Gross Jajima H. и др. "The Peptides", том 5, с. 65
(1983)), ТМСОТФ (триметилсилил трифталатом) (Fuji. N. и др. J.Chem.
Soc. Chem. Commun. т. 1987, 274) или ТМСБР (триметилсилил бромидом (Fujii. N. и др. Chem. Pharm. Bull. т. 35, с. 3880 (1987) и
т.п.)
В предпочтительном варианте осуществления изобретения
полученный в результате полипептид восстанавливают 2-меркаптоэтанолом, ДТТ (дитиотрейтолом) и т. п. чтобы гарантировать, что
меркапто группы цистеинов находятся в восстановленном виде. Меркапто
группы могут быть затем окислены для того, чтобы получить циклический полипептид. Обработку с целью окисления можно осуществить при
помощи известных приемов. В общем случае, используют такой
окисляющий агент, как воздух или феррицианат (например, феррицианид калия).
В качестве альтернативы полипептиды, являющиеся предметом настоящего изобретения, могут быть получены с использованием техники рекомбинантной ДНК. В соответствии с ней, нуклеотидные кодирующие последовательности для полипептидов могут быть клонированы и экспрессированы с использованием приемов, хорошо известных в этой области техники (см. например, Maniatis и др. Molecular cloniny, a Laboratory manual, Cold spoig Harbor Laboratory, Cold Spoig Harbor, N.y. 1991).
Полипептиды, являющиеся предметом изобретения, могут быть выделены и очищены при помощи средств, хорошо известных в области полипептидов, например, при помощи экстрагирования, рекристаллизации, различных методов хромотографии (гель-проникающей, ионообменной, распределительной, адсорбционной, обращенно-фазовой), электрофореза, распределения при противотоке и т.д. а обращенно-фазовая, высокоэффективная жидкостная хроматография является наиболее эффективной.
5.2. Использование полипептидов
Полипептиды настоящего изобретения,
представленные формулой (1), обладают способностью
связываться с эндотоксинами, антибактериальной активностью и активностью относительно гемолитизирования чувствительных к эндоксину гемоцитов. Кроме
того, полипептиды, являющиеся предметом настоящего
изобретения, обладают противовирусной активностью. В конкретном варианте осуществления настоящего изобретения полипептиды по изобретению обладают
анти-ВИЧ активностью. Как будет подробно описано в
Разделе 6, см. ниже, полипептиды, являющиеся предметом изобретения, проявляют существенно более высокую анти-ВИЧ активность по сравнению с известными
полипептидами с высоким сродством относительно
эндотоксина (например, Тачиплезины I, II или III, или Полифемизины I или II).
Полипептиды, являющиеся предметом изобретения, таким образом, могут быть использованы в фармацевтических композициях, содержащих полипептиды по изобретению или их соли и приемлемый с фармацевтической точки зрения носитель, выбранный в соответствии со способом применения и формой применения фармацевтической композиции. Фармацевтическими носителями могут быть такие физиологически совместимые буферы, как раствор Хенка или Рингера, физиологический соляной раствор, смесь, состоящая из соляного раствора и глюкозы, и обработанный гепарином раствор цитрата натрия-лимонной кислоты-дексторозы. Эту фармацевтическую композицию применяют стоматически или перентерально в соответствии с объектом обработки, и ее можно приготовить в такой форме, как порошок, гранулы, раствор для инъекции или стоматического применения, таблетки, суппозитории, пессарии, мази, кремы или аэрозоль, используя соответствующие носители в соответствии со способом применения.
Когда фармацевтическую композицию применяют непосредственно в форме инъекций к пациенту, полипептид или его соль, являющиеся предметом изобретения, могут быть применены непрерывно или через некоторые промежутки времени в количестве от 10 до 5000 мг на кг массы человеческого тела в день, и при помощи внутривенной капельницы в форме раствора в физиологическом соляном растворе.
6. Примеры.
В примерах, приводимых ниже, описан синтез полипептида (I). Кроме того, описаны результаты анализов на анти-ВИЧ активность полипептидов, являющихся предметом настоящего изобретения, и известных полипептидов с высоким сродством к эндотоксину. Полипептиды по изобретению обладают существенно более высокой анти-ВИЧ активностью по сравнению с известными полипептидами с высоким сродством к эндотоксину.
В приводимых ниже примерах используют
следующие
аппараты и реагенты:
Аппарат для ВЭЖХ: фирмы Waters Co. США, Модели 600.
Колонна для этого аппарата: типа Asahipar ОДР-90 (фирмы Asahi Chemical Industry Co. Ltd).
Аминокислота Fmoc: производится фирмой Kokusan Kagaku Co.
Амино смола и конденсирующий агент: производятся фирмой Peptide Ken kyusho Co.Ltd
FAB-MS
(FAB-масс-спектрограф): фирмы VC Co, США, модели ZAB-SE.
6.1. Пример 1: Синтез полипептида (I). Синтез полипептида (I), который имеет формулу, приведенную ниже, описан в Разделах 7.1.1-6.1.7, см. ниже. Полипептиды (2-33) (см. табл. 1 выше по поводу структур) синтезировали с использованием аналогичных процедур.

Cys-остатки в 3- и 16-позициях, и в 7- и 12-позициях связаны, соответственно, через дисульфидную связь.
6.1.1. Введение в
аминометиловую смолу соединения Fmoc ДМВНА-CH2CH2COOH [(3-) α-Fmoc-амино-4-метоксибензил)-4-метоксифенил)пропионовая кислота]
270 мг (0,2 ммолей) аминометиловой
смолы (0,74 мэкв/г) и 268,5 мг (0,5 ммолей, 2,5 экв) Fmoc-ДМВНА-CH2CH2COOH (ММ 537) помещали в колонну синтеза в твердой фазе и реакцию конденсации осуществляли в течение 2 ч
при
помощи процедуры ДИПКДИ-НОВ в ДМФ в соответствии с процедурой, предложенной guo L. и др. [Chem. Pharm. Bull. т. 36, с. 4989, 1988).
После завершения реакции связывание осуществляли с целью защиты свободных амино-групп, используя уксусный ангидрид (ДМВНА-смолу).
6.1.2. Введение аргинина в 17-позицию в ДМВНА-смолу.
После удаления Fmoc-групп из ДМВНА-смолы, полученной в разделе 6.1.1. см. выше. 20: пиперидином (ДМФ, добавляли 2,5 эквивалента (экв) Fmoc Arg (Mtr)-OH, в пересчете на ДМВНА-смолу, и реакцию конденсации осуществляли в ДМФ в соответствии с ДИПКДИ-НОВt-процедурой.
Степень развития реакции конденсации контролировали при помощи измерения в соответствии с тестом на нингидрин, предложенным
в Kaiser e и др.
[Anal. Biochem, т. 34, с. 595, (1970)]
6.1.3. Введение цистеина в 16-позицию.
После удаления Fmoc-групп из ДМВНА-смолы при помощи 20% пиперидина (ДМФ, добавляли 2,5 экв Fmoc-Cys (MBzl)-JH, на базе ДМВНА-смолы, и реакцию конденсации осуществляли в ДМФ при помощи ДИПКДИ-НОВt-процедуры. Степень развития реакции конденсации контролировали по аналогии с 6.1.2. см. выше, при помощи измерения в соответствии с испытанием на нингидрин.
6.1.4. введение аминокислот от 15- до 1 позиции.
По аналогии с приведенным описанием, Lys (Boc), Arg (Mtr), Tyr (t-Bu), Cys (MBzl), Tyr [t-Bu] GIy, Lys (Boc), Tyr (t-Bu), Cys (MBzl), Lys (Boc), Arg (Mzl), Tyr (t-Bu), Cys (MBzl), Trp, Arg (Mtr) и Arg (Mtr) последовательно вводили в ДМВНА-смолу, чтобы получить смолу защищающая группа защищенный полипептид (I).
Реакцию конденсации каждой аминокислоты при синтезе в твердой фазе осуществляли в соответствии с
условиями функционирования
из табл. 2
6.1.5. Получение полипептида (I) при помощи удаления защищающих групп, удаления полипептида (I) из смолы и частичная очистка.
Защищенную полипептидную (I) смолу подвергали обработке 20% пиперидином (ДМФ, чтобы удалить группу Fmoc, а затем подвергали реакции при температуре 25oC в течение 2 ч в системе 1 М ТМСОТФ-тиоанизол (ТФК) трифторуксусная кислота) (10 мл трифторуксусной кислоты в присутствии м-крезола (100 экв) и) тандитиола (300 экв) на 100 мг смолы. Смолу отделяли фильтрацией из реакционной смеси и дважды промывали 1 мл трифторуксусной кислоты. В смесь фильтрата и промывочной жидкости добавляли последовательно 100 мл охлажденного льдом сухого простого эфира. Образовавшийся осадок центрифугировали и остаток отделяли от верхнего слоя декантированием. Полученный в результате остаток промывали холодным простым эфиром, растворяли в 10 мл 4н. раствора AcOH, добавляли 830 мг, 80 экв дитиотрейтола и смесь перемешивали при комнатной температуре в течение ночи.
Реакционный раствор центрифугировали, верхний слой обрабатывали Сефадексом Г-10 (sephadex G-10, 3,7х5 см), подвергали гель-проникающей фильтрации с 4н. раствором уксусной кислоты (AcOH), а вытекающий поток собирали в виде основной части элюата и подвергали лиофилизации, чтобы получить порошок частично очищенного нециклизованного полипептида (I).
6.1.6. Получение полипептида (I) окислением воздухом.
У половины вытекшей фракции pH обеспечивали на уровне 7,5 при помощи концентрированного водного раствора аммиака, и ее подвергали окислению воздухом при помощи аэрации, чтобы осуществить реакцию циклизации. После завершения окисления воздухом циклизованный полипептид (I) адсорбировали на 10 г смолы HP-20, а затем элюировали 60% CH3CN (в 1н. растворе AcOH). Элюат концентрировали при комнатной температуре при пониженном давлении, чтобы удалить CH3CN, а затем подвергали лиофилизации, чтобы получить порошок. Порошок растворяли в небольшом количестве воды и раствор сливали на Asahipak и подвергали очистке при помощи высокоэффективной жидкостной хроматографии (ВЭЖХ Модель 600, производимая фирмой Waters Co), используя градиентное элюирование при помощи CH3CN, чтобы получить полипептид (I) с единственным пиком с выходом 27% (величина, рассчитанная на основе смолы защищающая группа защищенный полипептид (I).
6.1.7. Анализ полипептида.
Ценность аминокислотной композиции определяли при помощи переваривания леукции аминопептидазой полипептида, очищенного, как это делали в Разделе 6.1.6, см. выше, при этом получали, что оно согласуется с рассчитанным значением для композиции, основанной для аминокислотной последовательности формулы (1).
Специфическое вращение [α] полученного полипептида составляло +8,4o (C=0,1,1н. раствор уксусной кислоты).
6.2. Пример 2: Противовирусная активность против вируса иммунодефицита человека (ВИЧ).
Противовирусная активность относительно ВИЧ полипептида (I), синтезированного в примере 1, а также полипептидов (2), (3), (4), (7), (12), (13), (14), (20), (21) и (26) испытывали и оценивали в соответствии со следующей процедурой.
ВИЧ-инфицированные клетки МТ-4 (2,5•104 клеток) углубление, множественность инфекции (MOI):0,001) сразу же после инфицирования добавляли вместе с испытываемым материалом с различными изменениями концентрации в микротитрованный планшет с 96 углублениями. После инкубирования при температуре 37oC в течение 5 дней в CO2-инкубаторе измеряли число живых клеток при помощи МТТ-метода (Pouwels и др. J. Virol. methods т. 20, с. 309-321 (1988). Противовирусная активность выражается в виде концентрации, при которой гибель клеток, вызванная ВИЧ-инфекцией, ингибирована на 50% (EC50:50% эффективная концентрация). С другой стороны, для того, чтобы определить цитотоксичность испытываемого материала на МТ-4-клетках, инкубировали не инфицированные вирусом клетки так же, как это описано выше, вместе с испытываемым соединением при различных изменениях концентрации. Цитотоксичность выражается в виде 50% цитотоксичной концентрации (TC50), свойственной испытуемому материалу. Кроме того, грубое отношение CC50 к EC50 /CC50/EC50 выражается как эффективное отношение (SI=показатель селективности).
В табл. 3 приведены значения EC50, CC50 и SI для полипептидов (1), (2), (3), (4), (7), (12), (13), (14), (20), (21) и (26) и известных полипептидов, имеющих высокое сродство с эндотоксинами, в частности, Тачиплезинов I и II и Полифемузинов I и II, и известного Анти-ВИЧ-агента A T.
В табл. 4 приведены физические свойства полипептидов (1), (3), (13), (20) и (21).
Примеры составления фармацевтических композиций.
Пример 1. Капсула для орального применения
полипептид (гидрохлорид примера 1, табл. 3) данного изобретения (активный ингредиент) 100 мг
Зерновой крахмал (разбавители и
связующие) 200 мг
Стеарат магния (смазочные
материалы) 20 мг
Макрокристаллическая целлюлоза (растворители) 680 мг
1000 мг
Перечисленные компоненты смешивают до
однородной массы и заполняют твердые желатиновые капсулы,
200 мг смеси в каждой капсуле.
Пример 2. Композиция для инъекции.
Полипептид (13, табл. 3) данного
изобретения 10 мг
Сульфат хондроитин натрия 10 мг
Указанные компоненты растворяют в 2 мл 5%-ного водного раствора маннитола (для внутривенного вливания) и этот получившийся раствор
стерильно фильтруют.
Затем фильтрат растворяют в 200 мл 5%-ного водного раствора маннитола (для внутривенного вливания) и получившийся раствор используют для внутривенного капельного вливания.
Результаты, приведенные в табл. 3, указывают на то, что полипептиды, синтезированные в примере 1, обладают существенно более высокой анти-ВИЧ активностью, которую определяли эффективной концентрацией. Соединения (1), (3), (12), (13), (14) и (26), в частности, были особенно селективными при подавлении (уничтожении) ВИЧ-инфицированных клеток. Показатель селективности соединений (1), (3), (12), (13), (14) и (26) был по крайней мере в 7 раз выше по сравнению с любым известным соединением с высоким сродством к эндотоксину.

Реферат
Назначение: в медицине в качестве антибактериального и противовирусного агента (например, анти-ВИЧ агент). Сущность изобретения: полипептиды формулы 1: A1 -Tгр-Cys-Tyr-A2-Cys-A3-A2-Gly- A3-Cys-Tyr-A2-A2-Cys-A1-NH2 или их соли, где A1=Аргинин, аргиниларгинин; A2=аргинин, лизин; A3=тирозин, фенилаланин, причем остатки цистеина в положениях 3 и 16 и в положениях 4 и 12 могут быть связаны через дисульфидный мостик; фармацевтическая композиция, содержащая в качестве активного ингредиента эффективного количества от 10 до 5000 мг/кг веса человеческого тела в день полипептида 1, в котором остатки цистеина в положениях 3 и 16, и 7 и 12 связаны через дисульфидный мостик. 3 с.п. ф-лы, 4 табл., 1 ил.

Формула
где A1 независимая аминокислота или пептидный остаток, выбранные из группы, состоящей из аргинина и аргиниларгинина;
A2 независимый аминокислотный отстаток, выбранный из группы, состоящей из аргинина и лизина;
A3 независимый аминокислотный остаток, выбранный из группы, состоящей из тирозина и фенилаланина;
A4 NH2,
или их соли.
где A1 независимый аминокислотный или пептидный остаток, выбранный из группы, состоящей из аргинина и аргиниларгинина;
A2 независимый аминокислотный отстаток, выбранный из группы, состоящей из аргинина и лизина;
A3 аминокислотный остаток, выбранный из группы, состоящей из тирозина и фенилаланина;
A4 NH2,
при этом остатки цистеина в положениях 3 и 16 и в положениях 7 и 12 связаны через дисульфидный мостик,
или их соли.
Комментарии