Полипептид, способ получения (варианты), днк (варианты), вектор (варианты), клетка, применение полипептида, фармацевтическая композиция (варианты) - RU2177480C2
Код документа: RU2177480C2
Чертежи

























Описание
Изобретение относится к новому белку, названному Компонентом B. Более конкретно, настоящее изобретение относится к новому белку, получаемому из мочи; к получению этого белка из мочи; к его синтезу посредством техники рекомбинантных ДНК с использованием геномной ДНК или кДНК, копирующей указанный новый белок; а также к фармацевтической композиции, содержащей этот белок; и ее использованию в терапевтических целях.
Новый белок имеет относительную низкую молекулярную массу и полипептидную природу, и может быть выделен в процессе экстракции и очистки производных мочи. Для этого мочу человека обрабатывают адсорбирующими материалами, такими как каолин, затем подвергают фильтрации, ионообменной хроматографии и высокоразрешающей хроматографии, предпочтительно в соответствии с нижеописанной техникой; и после лиофилизации получают соединение в виде белого аморфного порошка, который в обращенно-фазовой жидкостной хроматографии высокого давления (ОФ-ЖХВД) перемещается как один пик и который имеет молекулярную массу около 9 кДа, как было показано с помощью электрофореза в полиакриламидном геле с дедецилсульфатом натрия (ДСН-ПААГ), проводимого в восстанавливающих условиях. Этот белок был назван Компонентом B.
Аминокислотная последовательность Компонента B представлена ниже как SEQ ID N 1 (список последовательностей см. в конце описания).
Настоящее изобретение относится к получению нового белка, названного Компонентом B, способом, предусматривающим выделение сырой фракции самого соединения из диализованного концентрата мочи после ее обработки адсорбирующим агентом и очистки с помощью ионообменной хроматографии и высокоразрешающей хроматографии, как описано ниже.
Предпочтительно, если белок настоящего изобретения экстрагируют из мочи человека, поскольку она существует в больших количествах, а поэтому может быть использована в промышленном производстве. Более конкретно, настоящее изобретение относится к полипептиду, имеющему последовательность SEQ ID N 1, к его солям, функциональным производным, предшественникам, активным фракциям, а также к их активным мутантам, т.е. к другим белкам или полипептидам, в которых одна или несколько аминокислот были удалены, замещены другими аминокислотами или добавлены в целях получения полипептидов или белков, имеющих активность, аналогичную активности Компонента B; кроме того, настоящее изобретение также относится к гибридным белкам, т.е. полипептидам, содержащим Компонент B или его мутацию, лигированный с другим белком, и имеющим более продолжительное время полужизни в физиологических жидкостях в организме. Поэтому Компонент В может быть подвергнут слиянию с другим белком, например, таким как иммуноглобулин.
Термин "соли", используемый в настоящем описании, означает соли карбоксильных групп и соли функциональных аминогрупп соединений, синтезируемые известными способами.
Соли карбоксильных групп могут быть неорганическими солями, например, такими как соли натрия, калия и кальция, а также солями, образованными органическими основаниями, такими как амины, например, триэтиламин, аргинин или лизин. Соли аминогрупп могут представлять собой соли, образованные неорганическими кислотами, такими как соляная кислота, и органическими кислотами, такими как уксусная кислота. Термин "функциональные производные", используемый в настоящем описании, относится к производным, которые могут быть получены из Функциональных групп, присутствующих на боковых цепях аминокислотных остатков или на концевых N- или C-группах, в соответствии с известными методами; и которые входят в объем настоящего изобретения в том случае, если они являются фармацевтически приемлемыми, т.е. обладают активностью, аналогичной активности белка, или не наделяют фармацевтические композиции, содержащие указанные производные, токсическими свойствами.
Примерами таких производных являются, например, сложные эфиры или алифатические амиды карбоксильных групп, и N-ацильные производные свободных аминогрупп, O-ацильные производные свободных гидроксильных групп, а также производные, образованные ацильными группами, например алканоильными или ароильными группами.
Термин "предшественники" означает соединения, которые в организме человека или животного превращаются в Компонент B. Термин "активные фракции" белка настоящего изобретения означает любой фрагмент или предшественник полипептидной цепи самого соединения, взятый отдельно или в комбинации со связанными с ним родственными молекулами или остатками, например остатками сахаров или фосфатов, или агрегаты полипептидной молекулы, при условии, что такие фрагменты или предшественники, как лекарственные средства, обладают активностью, аналогичной активности Компонента B.
Настоящее изобретение также относится к смеси полипептидов и производных, определенных выше.
Во втором варианте своего осуществления настоящее изобретение относится к способу получения Компонента B, предусматривающему выделение сырой фракции из диализованного концентрата мочи, после его обработки адсорбирующим агентом и очистки с помощью ионообменной хроматографии и высокоразрешающей хроматографии.
Компонент B предпочтительно получают способом, проиллюстрированным на фиг. 1 и включающим в себя следующие стадии:
a) адсорбция мочи при
кислотном pH на каолине и ее экстракция аммиаком;
b) элюирование фракции (a) на смоле Bio Rex 70 с использованием аммиака;
c) элюирование фракции (b) на DEAE-Сефарозной смоле с
использованием ацетатного буфера;
d) элюирование фракции (c) на CM-Сефарозной смоле с использованием ацетатного буфера;
e) элюирование фракции (d) на C18-смоле (ВЭЖХ) с
использованием смеси ацетатного буфера и ацетонитрила;
f) элюирование фракции (e) на DE-52-смоле с использованием ацетатного буфера;
g) элюирование фракции (f) на D-Zhephyr-смоле с
использованием ацетатного буфера;
h) элюирование фракции (g) на C18-смоле (ВЭЖХ) с использованием смеси водной трифторуксусной кислоты и ацетонитрила;
i) элюирование фракции (h) на
D-Zhephyr-смоле с использованием ацетатного буфера.
Кроме того, настоящее изобретение относится к рекомбинантным ДНК-молекулам, которые содержат нуклеотидную последовательность, кодирующую полипептид настоящего изобретения, его активные мутанты или гибридные белки; а также к содержащим их векторам экспрессии; к клеткам-хозяевам, трансформированным этими векторами; и к способу получения указанных полипептидов, их активных мутантов или гибридных белков путем культивирования указанных трансформированных клеток в соответствующей культуральной среде. Термин "рекомбинантные ДНК-молекулы" относится к геномной ДНК, кДНК, синтетической ДНК и к комбинациям. В частности, настоящее изобретение относится к нуклеотидным последовательностям, проиллюстрированным в SEQ ID N 2 и SEQ ID N 3 соответственно.
SEQ ID N 2 представляет собой геномную ДНК-последовательность, кодирующую Компонент B.
На фиг. 2 показана рестрикционная карта транскрипционной единицы Компонента B.
SEQ ID N 3 представляет собой кДНК-последовательность, кодирующую Компонент B.
На фиг. 8 показана полная нДНК-послецовательность, кодирующая Компонент B, в которой указаны сайты рестрикции. Клонирование Компонента B может быть осуществлено различными способами. В одном из таких способов получают олигонуклеотид или смесь нуклеотидов, последовательность которых, выведенную всходя из последовательности Компонента B или его фрагмента, используют в качестве зонда для клонирования кДНК или геномной ДНК, кодирующей Компонент B.
SEQ ID N 4 представляет собой аминокислотные последовательности, кодируемые геномной ДНК (SEQ ID N 2) и кДНК (SEQ ID N 3).
Настоящее изобретение также относится к рекомбинантным ДНК-молекулам, которые гибридизируются с ДНК-последовательностью, кодирующей Компонент B или его фрагмент.
Нужный ген может содержать или не содержать природные интроны, и может быть получен, например, путем экстракции из соответствующих клеток с последующей очисткой известными методами. Соответствующие препараты ДНК, как человеческая геномная ДНК, разрезают подходящими способами, предпочтительно с использованием рестриктирующих ферментов, и полученные таким образом фрагменты вводят в соответствующие рекомбинантные векторы для создания библиотеки ДНК. Указанные векторы могут быть отобраны с помощью синтетических олигонуклеотидных зондов в целях идентификации последовательности, кодирующей Компонент B настоящего изобретения.
В соответствии с настоящим изобретением, геномную ДНК Компонента B выделяют и клонируют.
С другой стороны, из клеток, экспрессирующих Компонент B, может быть выделена соответствующая мРНК и использована для продуцирования комплентарной ДНК (кДНК) известными методами. Эта кДНК после превращения в двойную спираль может быть введена в соответствующий вектор, который затем используют для трансформации подходящей клетки-хозяина. После этого культуры отбирают с использованием соответствующего зонда в целях получения кДНК, кодирующей целевые последовательности. После выделения нужного клона кДНК подвергают, в основном, таким же манипуляциям, что и геномную ДНК.
кДНК не содержат интроны. Из-за вырожденности генетического кода, могут быть использованы различные кодоны, кодирующие конкретную аминокислоту, так чтобы продуцировался один или несколько олигонуклеотидов, каждый из которых способен кодировать фрагменты Компонента B. Однако лишь один член этого пула имеет нуклеотидную последовательность, идентичную последовательности данного гена. Присутствие этого члена в пуле и его способность гибридизироваться с ДНК также в присутствии других членов пула позволяет использовать группу нефракционированных олигонуклеотидов таким же образом, как один олигонуклеотид, который должен быть использован для клонирования гена, кодирующего целевой пептид. Альтернативно, единственный нуклеотид, содержащий последовательность, относительно которой можно теоретически с наиболее высокой степенью вероятности предположить, что она способна кодировать генные фрагменты Компонента B (согласно описанию, приведенному в "rules for the use of codons" (правила использования кодонов") - Lathe R. et al., Molec. Bid., 183:1-12 (1985)), позволяет идентифицировать комплементарную ДНК, кодирующую Компонент B или в его фрагмент.
Способы гибридизации нуклеиновых кислот известны и описаны в литературе (см. , например, Maniatis T. et al., Molecular Cloning: a Laboratory Manual, Cold Spring Harbor Press, Cold Spring Harbor, NY, 1982); Haymes B. T. et al. , Nucleic Acid Hybridization: A Practical Approach, LRL Press, Oxford, England (1985)). Гибридизация с использованием указанного зонда или группы нуклеотидных зондов позволяет идентифицировать в геномной или кДНК-библиотеке ДНК-последовательности, способные к такой гибридизации, которые после последующего анализа, подтверждали свою способность кодировать полипептид настоящего изобретения (т. е. Компонент B). Олигонуклеотид, содержащий такую комплементарную последовательность, может быть синтезирован и использован в качестве зонда для идентификации и выделения гена полипептида настоящего изобретения т.е. Компонента B (Маниатис и др., см. выше).
После того как соответствующий олигонуклеотид, специфичный для Компонента B, будет отобран в соответствии с вышеописанным методом, он может быть синтезирован и гибридизирован с ДНК или предпочтительно с кДНК, происходящей от клеток, способных экспрессировать нужный ген, предпочтительно после обогащения источника кДНК нужными последовательностями, например путем экстракции РНК из клеток, продуцирующих высокие уровни нужного гена, и последующего превращения этой РНК в соответствующую кДНК с использованием обратной транскриптазы.
Альтернативно, могут быть синтезированы подходящие олигонуклеотиды, специфичные для Компонента B, и затем использованы в качестве праймеров для амплификации кДНК-фрагментов Компонента B с помощью PACE-PCR (M.A. Innit et al. , PCR Protocols A Guide to Methods and Application, Academic Press, 1990).
В частности, в соответствии с настоящим изобретением сначала для идентификации наиболее подходящего источника для мРНК Компонента B осуществляли поиск соответствующих тканей человека и клеточных линий. Для этого скринировали ткани человека, происходящие из головного мозга, почек, печени, легких, сердца, поджелудочной железы, плаценты, селезенки, яичек, тимуса, и матки, а также клеточные линии эпителиоидной карциномы, промиелоцитарного лейкоза, аденокарциномы молочной железы, лимфомы Беркита и миеломы.
Скрининг осуществляли с помощью чувствительного анализа, основанного на полимеразно-цепной реакции с использованием обратной транскриптазы (RT-PCR).
Наилучшим источником мРНК оказались ткани матки человека.
Из этой ткани с использованием метода быстрой амплификации, называемого "быстрой 3'- и 5'-амплификацией кДНК-концов", получали кДНК-лоны Компонента B.
Затем ДНК-молекулы, кодирующие Компонент B и полученные вышеуказанным методом, вводят в экспрессирующие векторы с использованием известной техники (Маниатис и др., см. выше). Двухспиральную кДНК лигируют в плазмидные векторы с помощью, например, метода, в котором используются синтетические ДНК-адапторы, или с помощью метода, в котором проводится "связывание по тупым концам".
Для экспрессии целевого белка экспрессирующий вектор должен также включать в себя специфические нуклеотидные последовательности, содержащие информацию, необходимую для регулирования транскрипции и трансляции ДНК, кодирующей нужный белок, так, чтобы могли осуществляться экспрессия нужного гена и продуцирование белка. Сначала для того, чтобы осуществлялась транскрипция нужного гена, перед этим геном должен быть помещен промотор, который может распознаваться РНК-полимеразой и с которым эта полимераза связывается, способствуя тем самым инициации транскрипции.
Многие из известных промоторов, "работающих" с различной степенью эффективности (сильные промоторы и слабые промоторы), подразделяются на промоторы, используемые в прокариотических клетках, и на промоторы, используемые в эукариотических клетках.
Промоторы, которые используются в настоящем изобретении, могут быть конститутивными (нерегулируемыми) промоторами, например, такими как промотор int лямбда-бактериофага, промотор B1a гена β-лактамазы в pBR322, и промотор CAT гена хлорамфеникол-ацетилтрансферазы pPR325, и т.п., или индуцибельными промоторами, например, такими как промоторы прокариотов, например правый и левый главные промоторы лямбда-бактериофага (Pl и Pr), промоторы trp, rec A, lac Z, lac I, ompF и gal E.coli или гибридный промотор trp - lac, и т.п. (Glic B.R., J. Ind. Microbiol., 1:277-282 (1987)).
Для гарантии эффективной трансляции мРНК необходимо, чтобы вместе с сильными промоторами, способными к продуцированию очень больших количеств мРНК, обеспечивающих высокие уровни экспрессии гена в прокариотических клетках, использовались также сайты связывания с рибосомами. В качестве примера могут служить последовательности Shine-Dalgano (SD), помещенные на соответствующем расстоянии от инициирующего кодона.
Для эукариотических клеток-хозяев могут быть использованы различные последовательности, регулирующие транскрипцию и трансляцию, в зависимости от природы конкретного хозяина.
Эти последовательности могут происходить от вирусных источников, таких как аденовирус, вирус папиломы, обезьяний вирус или т.п., где сигналы регуляции ассоциируются со специфическим геном, имеющим высокий уровень экспрессии. В качестве примеров могут служить промотор TK вируса герпеса, промотор вируса SV 40, промотор гена gal4 дрожжей и т.п. Сигналы инициации транскрипции могут быть выбраны таким образом, чтобы при индуцировании супрессии или активации можно было соответствующим образом модулировать экспрессию генов.
Затем ДНК-молекулу, содержащую нуклеотидную последовательность, кодирующую Компонент B настоящего изобретения, вместе с сигнальными последовательностями, регулирующими транскрипцию и трансляцию, вводят в вектор, который способен интегрировать последовательности целевого гена в хромосоме клетки-хозяина.
Клетки, несущие в своих хромосомах введенную ДНК, могут быть отобраны путем введения одного или нескольких маркеров, позволяющих выбрать клетки, которые содержат экспрессирующий вектор. Такие маркеры могут сообщать клеткам, например, резистентность к антибиотикам или к тяжелым металлам (таким, как медь). Селективный ген может быть непосредственно связан с ДНК-последовательностями, которые должны быть экспрессированы, или он может быть введен в саму клетку путем констрасфекции. Для осуществления более высокой степени экспрессии гена, может оказаться необходимым также присутствие других элементов. Такими элементами могут быть, например, энхансеры транскрипции, сигналы терминации транскрипции и интроны. Экспрессирующие векторы, которые содержат такие элементы описаны Okayama H., Mol. Cell Biol. 3: 280 (1983).
При выборе конкретного плазмидного или вирусного вектора должны учитываться следующие факторы: легкость обнаружения клеток, содержащих данный вектор, т. е. эти клетки должны быть легко отделены от клеток, не содержащих этого вектора; число копий векторов, которые желательно получить в данном конкретном хозяине; и возможность или невозможность перенесения вектора среди различных клеток-хозяев.
Предпочтительными прокариотическими векторами являются плазмиды, способные реплицироваться в E.coli, например, такие как BR322, CoIEI pSC101, pACYC 184 и т.п. (Maniatis T. и др., см. выше), плазмиды Bacillus, например, такие как pC194, pC221, pT127 и т.п. (Gryczan T.M. The Molecular Biology of the Bacilli, Academic Press, NY, 307-329 (1982)), плазмиды, например, такие как PIY 101 (Kendall K.J. et al., J. Bacteriol. 169:4177-83), плазмиды Peseudomonas (John J.F. et al., Rev. Infect. Dis. 8: 693-704 (1986)) (Izaki K. Jpn. J. Bacteriol. 33: 729-742).
Предпочтительными эукариотическими векторами являются, например, BPV, SV40, бакуловирус и т. п. или их производные. Указанные векторы известны специалистам (Bostein D. et al., Miami Wint Symp. 19: 265-274) (Broach J.R. The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance, Cold Spring Harbor, NY, 455-470 (1981) (Broach J.R., Cell 28: 203-204 (1982)) (Bollon D. P. et al., J. Clin. Hematol. Oncol. 10: 39-48 (1980)) (Maniatis T. Cell Biology: A Comprehensive Treatise vol. 3: Gene Expression, Acad. Press, NY, 563-608 (1980)).
Полученный экспрессирующий вектор вводят в соответствующую хозяйскую клетку стандартными методами, такими как трансформация, трансфекция, липофекция, конъюгация, слияние протопластов, электропорация, осаждение фосфатом кальция, прямое микроинъецирование и т.п. В целях настоящего изобретения могут быть использованы прокариотические или эукариотические клетки-хозяева.
Предпочтительными прокариотами являются бактерии, такие как E.coli, Bacillus, Streptomyces, Pseudomonas, Salmonella, Serratia и т.п.
Особенно предпочтительной является E.coli, например штамм 294 E.coli K12 (ATCC 314446), или E.coli X1776 (ATCC 31537), E.coli W3110 (F, лямбда, ATCC 27325).
Предпочтительными эукариотическими клетками-хозяевами являются клетки млекопитающих, например клетки человека, обезьяны, мыши или хомяка (СНО, клетки яичника китайского хомячка), поскольку эти клетки обеспечивают посттрансляционную модификацию белковых молекул, например правильную укладку и гликозилирование в нужных положениях.
В целях настоящего изобретения могут быть также использованы дрожжевые клетки. Благодаря имеющейся в настоящее время различной технике рекомбинантных ДНК, в которой используются последовательности сильных промоторов и большое число копий плазмид, нужный белок может быть продуцирован и в дрожжах.
После введения вектора в хозяйские клетки эти клетки культивируют в среде, способствующей селективному росту клеток, содержащих указанный вектор.
Экспрессия клонированной ДНК-последовательности обеспечивает продуцирование Компонента B, его мутанта или фрагмента. Продуцированный таким образом белок выделяют или очищают традиционными методами, например путем экстракции, осаждения, хроматографии, электрофореза и т.п., или путем аффинной хроматографии с использованием антител против Компонента B, иммобилизованных на колонке с гелем. Компонент B может быть также продуцирован как белок, секретируемый из молока трансгенных млекопитающих.
В еще одном своем варианте настоящее изобретение относится к использованию Компонента B, его солей, функциональных производных, предшественников или активных фракций в качестве лекарственного средства.
В частности, Компонент B обладает противоспалительными, антиокагулирующими и противоопухолевыми свойствами. Кроме того, Компонент B может быть использован для лечения патологий, связанных с изменением уровней TGF-альфа (фактора роста T-клеток), например поведенческих и гормональных расстройств, антигенеза и т.п.
На практике было показано, что Компонент B ингибирует связывание TGF-альфа с его рецептором с константой аффинности, составляющей Ki = 0,77•10-10 М, и измеренной путем вытеснения I125 - TGF-альфа из его рецептора, полученного из мембраны клеток A 431.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим терапевтически активное количество Компонента B в комбинации с фармацевтически приемлемыми наполнителями или разбавителями. Такие композиции могут быть получены для перорального, ректального, интраназального и предпочтительно для парентерального введения.
Кроме того, в соответствии с настоящим изобретением Компонент B может быть использован для местного применения.
Композиции настоящего изобретения могут быть также изготовлены в виде форм с пролонгированным высвобождением, например в виде подкожных имплантатов, полученных на основе липосом или микрокапсул сополимеров молочной и гликолевой кислот.
Другие варианты осуществления настоящего изобретения будут очевидны из нижеследующего подробного описания изобретения на конкретных примерах.
Пример 1
Способ получения компонента B из мочи
человека
Получение и очистка компонента B из мочи человека полностью
проиллюстрированы на фиг. 1.
a) Стадия 1
К исходному материалу, представляющему собой мочу
человека, добавляли соляную кислоту до тех пор, пока pH не становился равным 3,0.
После декантирования осадка к моче (10 г/1 л исходной мочи) добавляли каолин.
Полученную суспензию оставляли на 16 часов, а затем центрифугировали.
После удаления супернатанта каолин экстрагировали аммиаком 2 М при pH= 11,0.
pH аммиачного элюата доводили до 8,0 и концентрировали с помощью мембранной ультрафильтрации (отрезок 1000 Да). Всю эту процедуру осуществляли при температуре 4oC.
b) Стадия 2
К раствору стадии (a)
добавляли уксусную кислоту до тех пор, пока pH не становился равным 4,0, а затем
добавляли смолу Bio Rex 70, предварительно уравновешенную в ацетатном буфере при pH=4,0.
Полученный раствор оставляли для перемешивания на 4 часа, а затем фильтровали на фильтре-прессе.
Адсорбированный материал элюировали из смолы Bio Rex 70 с использованием аммиака при pH=9,0.
Хроматографический элюат концентрировали с помощью мембранной ультрафильтрации (отрезок 1000 Да).
Всю эту процедуру осуществляли при температуре 4oC.
c) Стадия 3
Материал стадии (b), уравновешенный в
ацетатном буфере, при pH=5,6, адсорбировали на ионообменной смоле, подобной DEAE-Сефарозе, предварительно уравновешенной при pH=5,
6.
После адсорбции, элюирование осуществляли с использованием аммоний-ацетатного буфера 0,5 М при pH=5,6. Затем хроматографический элюат концентрировали с помощью мембранной ультрафильтрации (отрезок 1000 Да). Всю эту процедуру осуществляли при 4oC.
d) Стадия 4
Материал стадии (c) уравновешивали в ацетатном буфере при pH=4,5 и
адсорбировали на ионообменной смоле, подобной CM-Сефарозе и предварительно
уравновешенной при pH=4,5.
После завершения адсорбции элюирование осуществляли аммоний-ацетатным буфером 0, 15 М при pH = 4,5. Хроматографический элюат концентрировали с помощью мембранной ультрафильтрации (отрезок 1000 Да). Всю эту процедуру осуществляли при температуре 4oC.
e)
Стадия 5
Материал стадии (d) очищали при температуре 25oC с помощью обращенно-фазовой жидкостной хроматографии высокого разрешения на смоле C18, уравновешенной в аммоний-ацетатном
буфере 0,05 М при pH = 5,6.
Адсорбированный материал элюировали из смолы с использованием раствора ацетата аммония, содержащего 30% (об./об.) ацетонитрил.
Хроматографический элюат концентрировали путем дистилляции (40oC) в вакууме.
f) Стадия 6
Материал стадии (e) очищали с использованием ионообменной смолы, подобной
DE-52 и уравновешенной в аммоний-ацетатном буфере 0,02 М при pH=5,6.
Элюирование адсорбированного материала осуществляли с использованием 0,25 М буфера. Концентрирование осуществляли с помощью мембранной ультрафильтрации (отрезок 1000 Да). Всю эту процедуру проводили при температуре 4oC.
g) Стадия 7
Материал стадии (f) очищали на колонке,
предварительно упакованной ионообменной смолой, подобной D-zephyr (отвержденной с
помощью Sepracor), и уравновешенной при pH= 6,2 в 20 мМ буферном растворе ацетата натрия (буфер A).
Элюирование абсорбированного материала осуществляли градиентом от 100% буфера A до 100% буферного раствора ацетата натрия, содержащего 1 М NaCl (при pH=6,2).
h) Стадия 8
Материал стадии (g) очищали с помощью обращенно-фазовой жидкостной хроматографии
высокого разрешения на смоле C18 при температуре 25oC.
После завершения адсорбции элюирование осуществляли линейным градиентом смеси водного раствора 0,1% трифтороуксусной кислоты и ацетонитрила, подкисленного 0,1% трифтороуксусной кислотой.
Хроматографический элюат концентрировали путем дистилляции (45oC) в вакууме и лиофилизовали.
i) Стадия 9
Повторяли стадию 7. В результате этой процедуры получали конечный продукт,
компонент B, в виде аморфного белого порошка.
Пример 2
Аналитическая
характеризация Компонента B
В целях определения основных физико-химических характеристик
Компонента B полученный очищенный материал из мочи человека подвергали следующей аналитической
обработке.
a) Аминокислотная последовательность
Аминокислотную
последовательность Компонента B определяли в соответствии с методом Эдмана.
Анализ осуществляли с использованием секвенатора Applied Biosystem, модель 477A в соответствии с инструкцией изготовителя.
Указанный анализ дает возможность идентифицировать аминокислотную последовательность Компонента B с 81 аминокислотными остатками, приведенными в SEQ ID N 1.
b) Определение молекулярной массы
Анализ осуществляли с помощью масс-спектроскопии
электронным ударом (МС-ЭД), в результате чего было установлено, что молекулярная масса
составляет 8937,9 Да. Этот анализ выявил пять дисульфидных мостиков и остаток в 80 Да, относящийся к группе
SO4, связанной с Tyr (39).
Пример 3
Выделение геномной
ДНК Компонента B человека
Библиотека геномной ДНК человека в лямбда-шаговом векторе EMBL-3 SP6/T7
поставлялась фирмой Clontech (кат. N HL 1067, J. Lot N 1221). Геномную ДНК экстрагировали из
плаценты человека и частично переваривали ферментом Sau 3A. Перед клонированием в BamHI-сайт вектора EMB-3
Sp6/T7 ДНК-фрагменты выделяли с использованием градиента сахарозы с получением фрагментов
размером от 8 до 22 Kb.
Среда для культивирования
Клетки E. coli K802, поставляемые
фирмой Clontech (кат. N C1004-1) культивировали в LB-среде, дополненной 10 мМ MgSO4 и 0,2% мальтозой (среда для культивирования).
Фаговую библиотеку разводили в 0,1 М NaCl, 8 мМ MgSO4, 50 мМ Трис-Cl (pH 7,5) и 0,01% желатине (SM).
ДНК-библиотеку культивировали на LB-чашках с 1,5% агаром. Верхняя агароза для культивирования библиотеки содержала 0,136 М NaCl, 0,6% агарозу и 1% триптон (Merck кат. N 7213).
Реагенты для гибридизации
20 x SSC - 3 М NaCl, 0,3 М цитрат натрия, pH 7.
Гибридизирующий раствор - 5 x SSC, 0,02% SDS, 0,1% N-лауроилсаркозин, 0,5 блокирующий реагент (Boehringer кат. N 1096176).
Раствор A для промывки (HRP-олигонуклеотиды) - 3 x SSC, 0,1% SDS, мочевина, при различных концентрациях, в зависимости от типа зонда (см. ниже).
Раствор B для промывки (32P-олиго CBEX4L) - 1 x SSC, 0,1% SDS.
Система детекции
Гибриды HRP-олиго/ДНК обнаруживали с помощью ECL-набора и экспонировали с
пленкой (Hyperfilm ECL от фирмы Amersham, кат. N PPM 2106 и 2104 соответственно).
32 P-Олигозондированные фильтры обнаруживали путем экспонироввния с пленкой β-max (Hyperfilm β-max, Amersham, кат. N PRN10).
Олигонуклеотиды
Олигонуклеотиды
синтезировали с помощью автоматического синтезатора ДНК (Applied Biosystem мод. 392).
Олигонуклеотиды очищали с помощью кассет OPC (Applied Biosystem кат. N 400771) или с помощью электрофореза в денатурирующем полиауриламидном геле (ПААГ).
Олигонуклеотиды CB1, CB2 и CBEX2L, используемые в качестве зондов, модифицировали с помощью аминомодификатора MMT-C12 (Clontech кат. N 5206-1) в процедуре последнего цикла синтеза.
Пероксидазу хрена (HRP, Boehringer кат. N 814393) конъюгировали с модифицированным олигонуклеотидом в соответствии с описанием M.S. Urdea (Nuc. Ac. Res. 16, 4937, 1988), используя 1,4-фенилендиизотиоцианат (Aldrich, кат. N 25855-5) в качестве гомобифункционального кросс-линкера.
HRP-Олигонуклеотидные зонды очищали с помощью анионообменной хроматографии высокого разрешения (ВЖХР) на колонке с Nucleopac PA-100 (Dionex кат. N 043010). Затем осуществляли элюирование 20 мМ буферным раствором фосфата натрия при pH = 6,0 и линейным градиентом хлорида натрия (от 0,2 М до 1,0 М) в течение 30 минут.
Очищенные HRP-олигонуклеотиды концентрировали с помощью Centricon 10, промывали фосфатно-буферным раствором (PBS) и хранили в темноте при 4oC.
Концентрацию HRP-олигонулкотида вычисляли при ОП403 (Σ403
= 89,5 см-1•мM-1).
Затем синтезировали олигонуклеотиды, приведенные
в конце описания.
Титрование библиотеки
Геномную ДНК-библиотеку
человека титровали в соответствии со стандартными процедурами (F, Ausubel, Current Protocols in Molecular
Biology) путем инфицирования 0,3 мл культуры клеток E.coli K802, выдержанной в течение ночи,
различными разведениями библиотеки, в пределах от 2 • 10-3 до 2 • 10-7. Смесь клеток библиотеки инкубировали при комнатной температуре в течение 20 минут, а затем в
течение 10 минут смесь инкубировали при 37oC. Инфицированные клетки смешивали с 4
миллилитрами верхней агарозы, предварительно нагретой при температуре 50oC, а затем выливали в
10-сантиметровые планшеты с агаром, предварительно нагретым при 37oC. Эти планшеты
инкубировали в течение ночи при температуре 37oC.
Число бляшек подсчитывали в каждом планшете. Планшеты с дубликатами культуры получали для каждого титра библиотеки. Как это ожидалось, титр геномной ДНК-библиотеки составлял 5 • 109 БОЕ/мл.
Скринирование библиотеки
Клетки E. coli K802 культивировали в течение ночи при
температуре 37oC. 0,6 мл клеточной культуры инфицировали аликвотой библиотеки (6 • 104 БОЕ), суспендированной в СМ. Инфицирование и культивирование осуществляли
вышеописанным способом, за исключением того, что использовали 9 мл верхней агарозы и 15-сантиметровые планшеты.
Полуконфлюэнтные бляшки переносили на найлоновую мембрану Hybond N+ (Amersham) в соответствии с инструкцией руководства фирмы Amersham.
Блотированные ДНК денатурировали путем наложения фильтров колониями кверху на фильтровальную бумагу, пропитанную 1,5 М хлоридом натрия и 0,5 М гидроксидом натрия, на 7 минут.
Затем блотированные ДНК нейтрализовали путем наложения фильтров на фильтровальную бумагу, пропитанную нейтрализующим раствором (1,5 М NaCl, 0,5 М Трис-Cl (pH 7,2), 1 мМ ЭДТА)) (в каждом случае, 2 х 3 мин).
Фильтры промывали в 2 x SSC и осушали воздухом. ДНК закрепляли на мембране путем наложения фильтров на фильтровальную бумагу, погруженную в 0,4 М NaOH, на 20 минут.
После этого фильтры промывали в 5 x SSC в течение одной минуты, а затем хранили в полиэтиленовом мешке при 4oC вплоть до гибридизациии. Человеческую геномную ДНК-библиотеку (1 • 106 клонов) скринировали с высокой плотностью с использованием олигонуклеотидных зондов HRP-CB2. Затем были выбраны 20 положительных клонов.
Шесть положительных клонов повторно скринировали с использованием олигонуклеотидного зонда HRP-CB1 и олигонуклеотидного зонда HRP-CB2. Три клона, обозначенные 4D, 12B и 15, были подтверждены как положительные в отношении гена компонента B.
Гибридизация
Фильтры
предварительно инкубировали в течение 30 минут при 42oC в гибридизирующем растворе, а затем гибридизировали с использованием
соответствующего олигонуклеотидного зонда HRP (5 нг/мл
олигонуклеотидной части в гибридизирующем растворе), при температуре 42oC в течение 45 минут, и наконец, дважды промывали (по 15 минут
каждый) при температуре 42oC в промывающем
растворе, содержащем мочевину в соответствующей концентрации (см. ниже).
Затем фильтры промывали в 2 x SSC при комнатной температуре, а гибридизированные бляшки обнаруживали с помощью ECL-реагентов и экспонировали с пленкой Hyperfilm в течение 60 минут.
Для минимизации неспецифической гибридизации с E.coli и ДНК лямбда-фага были экспериментально определены условия промывки для HRP-олигозондированных фильтров. Серийные разведения в пределах от 500 до 15 аттомолей целевой ДНК были нанесены пятнами на мембрану Hybond N+ в присутствии ДНК лямбда-фага (10 нг). ДНК лямбда-фага и ДНК E.coli (10 нг каждая) были использованы в качестве негативного контроля. После этого получали несколько полосок и использовали их в экспериментах для гибридизации с использованием 5 нг/мл олигонуклеотидного зонда. Промывку осуществляли с использованием промывочного раствора A, содержащего 0%, 9%, 18%, 27% и 36%-ную мочевину.
18% и 27% мочевина оказалась эффективной для CB1 и CB2 соответственно. Фильтры, гибридизированные с использованием CBEX2L, промывки промывочным раствором A, содержащим 18% мочевину.
Гибридизацию с использованием32P-оливонуклеотида CBEX4 осуществляли при 50oC, а фильтры промывали в промывочном растворе B при температуре 45oC.
Субскринирование бляшек
Позитивные колонии собирали в Пастеровскую пипетку и переносили в сосуд, содержащий 1 мл CM + 1 каплю хлороформа. После
2-часовой инкубации при встряхивании при
комнатной температуре, суспензия фага хранилась при 4oC.
Разведение 10-3 суспензии фага наносили на 10-сантиметровые платы и рескринироввли на двух повторяющихся фильтрах с двумя олигозондами, т.е. один используется в первом скрининге, а другой соответствует примыкающему району компонента B.
Независимые клоны, позитивные с обоими зондами, были отобраны и ресуспендированы как указано выше.
Приготовление фаговых стоков
Позитивные клоны были размножены инфекцией
клеток E.coli K802 и выращиванием
на 15-сантиметровых агаровых платах. После инкубации в срезе ON при 37oC сливающийся лизат был собран с агаровых плат с использованием 10 мл SM. Были
добавлены несколько капель хлороформа,
клеточный дебрис удален центрифугированием при 3000 об/мин в течение 5 мин при
4oC и прозрачный супернатант, содержащий фаг, был перенесен в
50% глицерин, разлит на части и
заморожен при -80oC.
Экстракция фаговой ДНК
2 • 109 клеток E.coli K802 были инфицированы отобранным фаговым
клоном (соотношение клетка/фаг
4:1) и выращены в 100 мл жидкой культуральной среды ON при 37oC. В конце инкубации осуществлялся полный лизис клеток добавлением к культуре хлороформа 5
мкл/мл.
Фаговая ДНК экстрагировалась с помощью Quiagen по инструкции изготовителя.
Секвенирование фаговой ДНК
Фаговая ДНК была секвенирована с помощью
циклического секвенирующего набора от
Applied Biosystem (cat. N 401388) с автоматическим секвенатором ДНК (Applied Biosystem мод. 373A). ДНК фага экстрагировали из клонов 4D, 12B и 15, и секвенировали
циклами.
Праймеры для секвенирования подбирали либо исходя из аминокислотной последовательности Компонента B (CBF1, CBF2, CBP1, CBP2), либо исходя из данных секвенирования кДНК или геномной ДНК. Данные секвенирования указывали на три клона, содержащих полную длину гена Компонента B.
Анализ рестрикции фаговой ДНК
Фаговую ДНК подвергали однократному или
многократному перевариванию
рестриктирующим ферментом. ДНК-фрагменты разделяли с помощью электрофореза на 0,6% агарозном геле, а затем блотировали на мембрану Hybond N+. Фильтры повторно
зондировали с использованием
олигонуклеотидов CBEX2L, CB2, и CBEX4L, соответствующих экзонам 1, 2 и 3 соответственно.
Субклонирование гена Компонента B в pBluescript 11SK
Рестрикционный анализ клона 4D
Компонента B, проведенный с использованием ферментов EcoRI, XhoI и SfiI, и Саузернблот-анализ, проведенный с использованием олигонуклеотидных зондов, специфичных для
трех экзонов Компонента B,
обнаруживали наличие полного гена Компонента B, содержащего 5,2 Kb EcoRI-фермент (фиг. 5).
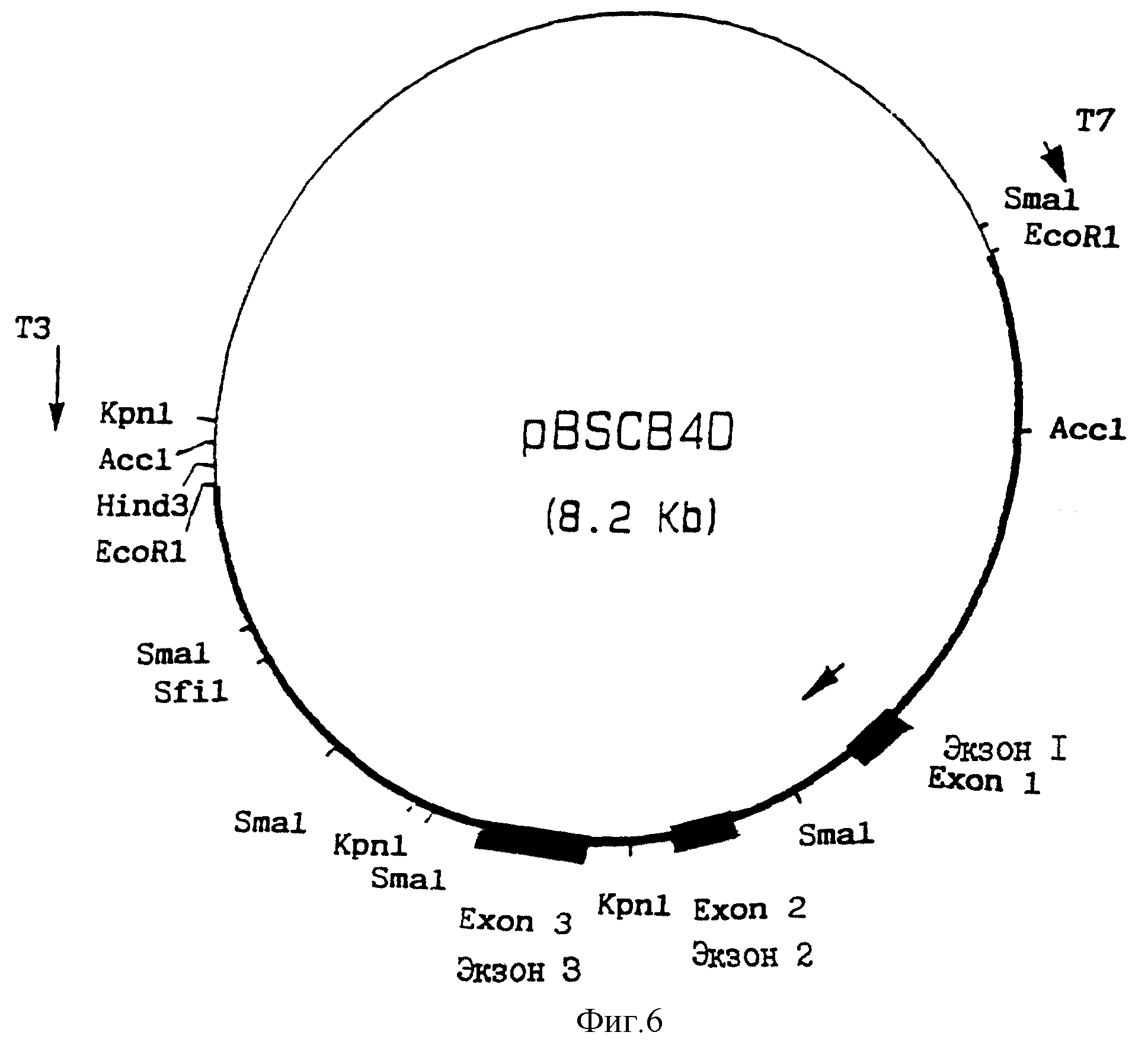
Фаговую ДНК экстрагировали из клона 4D и переваривали ферментом EcoRI. Полученные ДНК-фрагменты разделяли с помощью электрофореза на агарозном геле. 5.2 Kb-фрагмент очищали с помощью Quaex (qiaoen кат. N 20020) и лигировали в EcoRI-линеаризованный pBluescript 11 KC (Stratagene кат. N 212207). Штамм E.coli XLI-Blue (Stratagene кат. N 200268) трансформировали с использованием лигирующей смеси, а тансформированные клетки отбирали на Ap/Tc-чашках. Как показал рестрикционный анализ с использованием фермента EcoRI, был выделен один клон, содержащий нужную плазмиду, который был назван pBS CB4D.
Затем осуществляли рестрикционный анализ с использованием SmaI, KpnI, HindIII, SfiI, AccI, NotI, SalI, XhoI, EcoRI, ClaI, HincII, HindII, ScaI, BglII, Aat2, NcoI, NheI, HpaI и MluI. Кроме того, осуществляли Саузерн-блот-анализ с использованием олигонуклеотидных зондов, специфичных для Компонента B и плазмиды pBS CB4D после однократного и двойного переваривания.
На фиг. 6 показана рестрикционная карта плазмиды pBSCB4D.
На фиг. 4 показана рестрикционная карта гена Компонента B. Ген Компонента B содержит 3 экзона, разделенные друг от друга двумя интронами. Эти экзоны фланкированы соответствующими сайтами.
Экзон 1, имеющий длину 84 п.о., содержит 26 нуклеотидов нетранслированной мРНК и последовательность, кодирующую 19 аминокислот предполагаемого сигнального пептида. Этот экзон отделен от экзона 2 интроном, длина которого составляет 410 п.о.
Экзон 2, имеющий длину 120 п.о., кодирует 3 аминокислоты предполагаемого сигнального пептида и 37 аминокислот зрелого белка. Этот экзон отделен от экзона 3 интроном, длина которого составляет около 550 п.о.
Экзон 3, имеющий длину 326 п.о., кодирует C-концевые 44 аминокислоты Компонента B и содержит 192 нуклеотида нетранслируемой мРНК, которая имеет сигнал полиаденилирования (TATAAA), расположенный на расстоянии 14 п.о. ("вверх по течению") от сайта 3'-процессинга, к концу которого добавляется polyA-хвост.
В частности, было установлено, что в трех геномных клонах последовательность, кодирующая сигнальный пептид, содержит кодон Leu в положении II предполагаемого сигнального пептида.
Было установлено, что аминокислотная последовательность Компонента B, продуцированная геномным геном, идентичная последовательности, экспериментально определенной методом расщепления по Эдману.
Анализ последовательности, расположенной перед экзоном 1, выявил промоторную область (фиг. 3), содержащую TATA-блок (в - 28), и различные, расположенные "вверх по течению" элементы промотора и энхансеры, включая GC-богатый участок в -58, AP-1-сайт в -83, AP-2-сайт в -360, и несколько E-блоков.
TATA-блок является предпочтительным сайтом связывания для инициации транскрипции фактора TFIII. GC-богатый участок представляет собой сайт связывания для sp-1, главного фактора транскрипции, участвующего в транскрипции широкого ряда генов (Transcription and Splicing B.D.Hames & D.M. Clover Eds. , IRL Press, 1988).
AP-1-сайт является сайтом связывания для AP-1, комплекса факторов транскрипции, образованного c-fos и c-jun. AP-1-сайт присутствует в нескольких генах, ответственных за рост и дифференцировку клеток. AP-1 является одним из нескольких cis-элементов, опосредующих ответную реакцию по отношению к активаторам протеинкиназы C (The hormonal control of gene transcription P. Cohen & J.G.Foulkes End. Elsevier, 1991).
AP-2-сайт является мишенью для AP-2, фактора транскрипции, активированного PMA и cAMP (ibidem).
E-блоки являются обобщающими последовательностями, обнаруженными в нескольких энхансерных областях, и играют важную роль для определения тканеспецифической экспрессии генов. E-блок содержит последовательность CANNTG, где два основания, расположенные в середине, могут меняться для каждого конкретного E-блока. (R. E. Kingston Current Opinion Cell Biol. 1989; 1, 1081-1087).
Промотор Компонента B содержит потенциальный восприимчивый элемент для рецептора фактора глюкокортикоидной реакции (GPE) что указывает на то, что ген Компонента B может быть индуцирован глюкокортикоидами.
Субклонирование гена Компонента B в вектор для экспрессии в клетках млекопитающих
Известно,
что продуцирование
rec-белков в клетках млекопитающих может быть увеличено благодаря присутствию интронов. Геномная ДНК Компонента B может быть экспрессирована в клетках млекопитающих.
Для этого 1364 п. о. - фрагмент, простирающийся от +50 до +1413 гена Компонента B, вырезали из плазмиды pBSCB4D путем переваривания ферментами PvuII и NarI. На фиг. 2 показана рестрикционная карта транскриптона Компонента B, где PvuII- и NarI-сайты расположены по концам. Полный ген Компонента B восстанавливали путем лигирования этого фрагмента с синтетическим олигонуклеотидом, воспроизводящим 5'-конец гена, фланкированного соответствующим рестрикционным сайтом для последующего клонирования гена в эукариотической экспрессирующей плазмиде.
Пример 4
Выделение кДНК-клонов Компонента
B
Для получения частичных кДНК-клонов, соответствующих 5'- и 3'-концам мРНК Компонента B, использовали способ быстрой амплификации
кДНК-концов, описанной Frohman et al. (1988) Proc. Natl.
Acad. Sci., USA 85; 8998. Частичные клоны содержат перекрывающуюся ДНК-последовательность, а поэтому они могут быть использованы для
конструирования полной кДВК-последовательности Компонента B. Схема,
иллюстрирующая общую стратегию для PAGE-клонированзя, показана на фиг. 7.
В 3'-PAGE ДНК-последовательность второго экзона гена Компонента B использовали для конструирования генного специфического праймера CKCB1 (5'-TCAACTCCTACACCTCCAACCAC-3') (SEQ ID N 21). Синтез кДНК инициировали от poly A-конца poly A+ РНК матки человека с использованием в качестве праймера олигонуклеотид 5-CGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT-3' (SEQ ID N 24), называемый адапторным праймером AP. кДНК использовали в качестве матрицы для полимеразно-цепной реакции (PCR), где в качестве праймеров использовали CKCB1 и AP, которые продуцировали фрагмент, имеющий приблизительно 450 п.о. и соответствующий 3'-концу кДНК Компонента B.
В 5'-PAGE, из ДНК-последовательности 450 п.о. 3'-PAGE-фрагмента конструировали праймер CKCB7 (5'-CGTCAGAGAGGAGGTC-3) (SEQ ID N 22), который использовали в качестве праймера для синтеза кДНК из poly A+ РНК матки человека. После очистки в целях удаления мРНК и праймера CKCB7 конец олигодезоксицитидана добавляли к 3'-концу кДНК. Эту кДНК с "хвостом" использовали в качестве матрицы в PCR, где в качестве праймеров служили "гнездовой" праймер CKCB2 (5'-ACCGTCACCAGCGTGGTC-3') (SEQ ID N 23) и "якорный" праймер (ACP, 5'-CTACTACTACTAGGCCACGCGTCGACTAGTACGGGIIGGGIIGGGIIG-3') (SEQ ID N 25), или смесь ACP и универсального праймера амплификации (UAP, 5'-CTACTACTACTAGGCCACGCGTCGACTAGTAC-3') (SEQ ID N 26). CKCB2 гибридизировали с 3'-концов последовательности второго экзона, а ACP гибридизировали с олигодезоксицитидиновым "хвостом". В результате получали фрагмент приблизительно в 230 п.о., который содержал ДНК-последовательность, соответствующую 5'-концу мРНК Компонента B.
Используемые общие схемы эксперимента (например, электрофорез в полиакриламидном геле, осаждение этанолом, лигирование и переваривание рестриктирующими эндонуклеазами), бактериальные культуральные среды (например, LB) и химические растворы (например, фенол) подробно описаны Sambrook et al. (1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press, New York, если это не указано особо.
Процедуры 3' RACE-клонирования
3' RACE-система для
быстрой амплификации кДНК-концов была закуплена у Life Technologies, Inc., Grand Island, NY, Poly A+-РНК
матки человека закупали у Clontech Laboratories, Inc., Palo Alto, CA. Синтез первой
нити кДНК осуществляли с использованием протокола и реагентов, поставляемых вместе с 3'-RACE-системой. Для этого 1
мкл (1 мкг) poly A+-РНК матки объединяли с 1 икл 10 мкМ раствора AP и 12
мкл воды, обработанной диэтилпирокарбонатом (DEPC), и полученную смесь нагревали 10 минут до 65oC. После
охлаждения смеси на льду реакционные компоненты добавляли так, чтобы конечная
композиция имела приблизительно следующий состав: 20 мМ Трис-HCl (pH 8,4, 50 мМ KCl, 2,5 мМ KCl, 2,5 мМ MgCl2,
100 мкг-мл альбумина бычьей сыворотки, 500 нМ AP, 500 мкМ каждого из dATP,
dCTP, dGTP и dTTP, и 50 нг/мкл РНК, в объеме 19 мкл). Реакционную смесь нагревали до 42oC, а затем добавляли 1 мкл
(20 ед.) обратной транскриптазы superScript. После 30-минутного
инкубирования при 42oC реакционную смесь охлаждали на льду, и добавляли 1 мкл (2 ед.) РНКазы Н. Затем проводили РНКаза
Н-переваривание в течение 10 минут при 42oC. Полученную
реакционную смесь хранили при -20oC вплоть до осуществления PCR.
Для осуществления PCR были получены четыре идентичные смеси (40 мкл), каждая из которых имела следующий состав: 1 мкл poly A+- к ДНК матки в 40 мМ KCl, 70 мМ Трис-HCl (pH 8,8) 0,1% Тритон X-100, 1 мМ MgCl2, 0,25 мкМ CKCB1, и 0,5 мкМ AP. Реагенты из 3'-RACE-системы не использовались для PCR. CKCB1- и AP-праймеры синтезировали на олигонуклеотидном синтезаторе модели 392 (Applied Biosystems, Inc.). После разблокирования лиофилизации и ресуспендирования в DEPC-обработанной воде измеряли оптическую плотность каждого раствора при длине волны 260 нм. Исходя из измерений оптической плотности, в DEPC-обработанной воде приготавливали 10 мкМ раствор каждого из олигонуклеотидов. Неочищенные олигонуклеотидные растворы использовали для PCR-реакции без дополнительной очистки. Концентрацию неочищенного AP, который давал результаты, идентичные результатам, полученным с использованием 3' RACE-системы, определяли экспериментально: 0,4 мкМ неочищенного AP эквивалентны 0,2 мкМ AP, закупленного у Life Technologies для PCR-реакции.
40 мкл PCR-реакционной смеси нагревали в термоячейке до температуры 94oC, а затем к каждой смеси добавляли 10 мкл смеси, содержащей следующие компоненты: 1,25 ед. ДНК-полимеразы AmpliTag (Perkin Elmer Cetus, Norwalk, CT), 40 мМ KCl, 70 мМ Трис-HCl (pH 8,8), 0,1% Тритон-X-100, 1 мМ MgCl2 и 1 мМ каждого из dATP, dTTP, dGTP и dCTP. Конечная концентрация каждого реагента в PCP составляла приблизительно 1 мкл кДНК матки на 50 мкл, 1,25 ед. ДНК-полимеразы AmpliTag на 50 мкл; 40 мМ KCl, 70 мМ Трис-HCl (pH 8,8), 0, 1% Тритона X-100, 1 мМ MgCl2, 0,2 мкм CKCB1, 0,4 мкМ AP и 0,2 каждого из dATP, dTTP, dGTP и dCTP. После завершения 5-минутного инкубирования при 94oC, осуществляли программу термоциклической PCR "Touchdown" в соответствии с описанием Don R.H., Cox P. T., Wainwright B.J., Baker K., & Mattick J.S. (1991) Nucl. Acids Res. 19, 4008, варьируя температуру отжига от 73oC до 63oC.
После PCR-ампилификации четыре реакционных смеси объединяли, и ДНК-продукты фракционировали по размерам с помощью электрофореза на 5% полиакриламидном геле. ДНК-продукт, имеющий длину приблизительно 450 п.о., вырезали из геля, и очищали путем электроэлюирования в диализной трубке. Элюат экстрагировали смесью фенола и хлороформа (50:50, об./об.), осаждали этанолом, осушали и ресуппендировали в 10 мкл стерильной воды.
Вследствие независимой от матрицы концевой трансферазной активности ДНК-полимеразы Tag, и ее в высокой степени избирательности по отношению dATP (Clark J.M. (1988) Nucl. Acids Res. 16, 9677, и Mole S.E., Iggo, R.D., & Lae D. P. (1989) Nucl. Acids Res. 17, 3319), очищенный PCR-фрагмент в 450 п.о., имел, как и ожидалось, один дезоксиаденозиновый остаток у каждого 3'-конца. Для субклонирования и характеризации PCR-фрагмента, получали "T-вектор pBluescriptsk +" (Stratagene, La jolla, CA) в соответствии с описанием Marchuk и др. (1991) Nucl. Acids Res. 19, 1154. Плазмиду pBluescript (20 мкг) переваривали рестриктирующей эндонуклеазой EcoRV, а затем очищали путем экстракции смесью (50: 50, об./об.) фенола и хлороформа. После осаждения этанолом, ДНК обрабатывали 9 единицами Tag-ДНК-полимеразы в течение 2 часов при 70oC в 50 мкл реакции, содержащей 50 мМ KCl, 10 мМ Трис-HCl (pH 9,0), 0,1% Тритон-X100, 1,5 мМ MgCl2, и 2 мМ dTTP. После этого вектор снова очищали путем экстракции фенолом и хлороформом (50: 50, об./об.) и осаждали этанолом. Результатом этой процедуры было добавление одиночного дезокситимидинового остатка к каждому 3'-концу, что позволяло использовать этот вектор для инсерции ДНК-фрагментов, синтезированных посредством Tag-ДНК-полимеразы.
Нефосфорилированный PCR-фрагмент в 450 п.о. вставляли в T-вектор посредством реакции с ДНК-дигазой T4 (New England Biolabs, Beverly, MA), используя условия, рекомендуемые изготовителем. Лигирующую реакционную смесь инкубировали приблизительно 72 часа при 16oC, а затем использовали для трансформации компетентных клеток XLI-Blue E.coli (Stratagene, La jolla, CA). Трансформированные клетки высеивали на LB-агар, содержащий 50 мкг/мл ампицилина. Перед посевом клеток на поверхность агара каждой чашки наносили путем распыления 100 мкл 2% X-gal (Life Technologies, Inc., Grand Island, NY) и 40 мкл 100 мМ IPTG (Life Technologies, Inc., Grand Island, NY) и оставляли для осушки. После инкубирования в течение ночи при 37oC были видны колонии, окрашенные голубым пигментом, и неокрашенные колонии (белые). Из культур с 12 белыми колониями выделяли плазмидную ДНК. Все 12 изолятов содержали вставку в 450 п.о. Для последующего анализа выбирали 5 клонов: 3CB4, 3CB6, 3CB7, 3CB8 и 3CB9. Анализ ДНК-последовательности проводили с использованием секвеназного набора "Sequenase version 2,0" (United States Biochemical, Cleveland, Ohio).
Процедура
5'-RACE-клонирования
5'-RACE-систему для быстрой амплификации кДНК-концов закупали у Life Technologies, Inc. , Grand Island, NY. Poly A+ РНК матки
человека закупали у Clontech
Laboratories, Palo alto, CA. Эксперименты по 5'-RACE-клонированию осуществляли с использованием протокола и реагентов, поставляемых вместе с 5'-RACE-системой, за
исключением того, что: (a) CKCB7-,
ACP- и UAP-праймеры синтезировали на олигонуклеотидном синтезаторе модели 392, Applied Biosystem. Inc. , и использовали как описано для 3'-RACE-клонирования; и (b)
термоциклическую программу PCR
"Touchdown", описанную для 3'-RACE-клонирования использовали для кДНК-амплификации. Первую нить кДНК синтезировали следующим образом: 1 мкл (1 мкг) poly A+
РНК матки объединяли с 0,5 мкл
10 мМ раствора CKCB7 и 13,5 мкл DEPC-обработанной воды, и полученную смесь нагревали при 70oC в течение 10 минут. После охлаждения смеси на льду добавляли
реакционные компоненты так, чтобы
конечный состав приблизительно содержал: 20 нМ Трис-HCl (pH 8,4) 50 мМ KCl, 3 мМ MgCl2, 10 мМ DTT, 200 мМ CKCB7, 400 мкМ каждого из dATP, dCTP, dGTP и dTTP,
и 40 нг/мкл РНК, в объеме 24
мкл. Реакционную смесь нагревали до 42oC, и добавляли 1 мкл (220 ед.) обратной транскриптазы II. После 30-минутного инкубирования при 42oC и
15-минутного инкубирования при
70oC реакционную смесь помещали в условия 55oC и добавляли 1 мкл (2 ед.) РНказы Н. Переваривание РНКазой Н осуществляли в течение 10 минут при
температуре 55oC.
кДНК отделяли от невключенных dNTPS, CKCB7 и белков с использованием Glassmax DNA Isolation Spin Cartridge (входящего в 5'-RACE-систему). В частности, 120 мкл раствора для связывания (6 М NaI) добавляли к реакционной смеси первой нити, и кДНК/NaI-раствор переносили в центрифужную пробирку GIASS MAX. После центрифугирования при 13000 • g в течение 20 с добавляли 0,4 мл холодного (4oC) промывочного буфера. Затем центрифуговали еще 20 с при 13000 • g. Эту стадию повторяли еще два раза. После двухкратной промывки 400 мкл холодного (4o C) 70% этанола кДНК элюировали путем добавления в центрифужную пробирку 50 мкл стерилизованной дистиллированной воды, после чего центрифугировали еще 20 секунд при 13000 • g. Гомополимерный "хвост" добавляли к 3'-концу кДНК с использованием терминальной дезоксинуклеотидилтрансферазы (TdT) и dCTP. Эту реакцию осуществляли в PCR-совместимом буфере. 10 мкл очищенной кДНК объединяли с 7,5 мкл DEPC-обработанной воды, 2,5 мкл 10 x реакционного буфера, 1,5 мкл 25 мМ раствора MgCl2 и 2,5 мкл 2 мМ раствора dCTP. Реакционную смесь инкубировали в течение 2 - 3 минут при 94oC. После охлаждения в течение 1 минуты на льду добавляли 1 мкл TdT (10 ед. /мкл). Конечная смесь имела следующий состав: 10 мкл кДНК в 20 мМ Трис-HCl (pH 8,4), 50 мМ KCl, 1,5 мМ MgCl2, 200 мкМ dCTP, 0,4 ед./мкл TdT. Реакционную смесь инкубировали в течение 10 минут при 37oC, а затем в течение 10 минут при 70oC для инактивации TdT.
Для осуществления PCR были приготовлены четыре различные реакционные смеси, имеющие следующие конечные концентрации праймера (на 50 мкл):
1. 400 нМ ACP
2. 800
нМ ACP
3. 360 нМ UAP и 40 нМ ACP (UAP:ACP = 9:1)
4. 720 нМ UAP и 80 нМ ACP (UAP:ACP = 9:1)
Конечные концентрации (на 50 мкл) остальных компонентов во всех четырех реакциях
были
идентичными: 5 мкл poly A+ oC-концевой кДНК в 20 мМ Трис-HCl (pH 8,4); 50 мМ KCl: 1,5 мМ MoCl2: 400 нМ CKCB2, и 200 мкМ каждого из dATP, dCTP, dGTP и dTTP. Компоненты,
включая
ACP- и UAP-праймеры, смешивали в первоначальном объеме 45 мкл и нагревали до 94oC в термоячейке. Затем к каждой реакции добавляли 5 мкл смеси 1,25 ед. ДНК-полимеразы AmpliTag
(Perkin Elmer
Cetus, Norwalk, CT) в 20 мМ Трис-HCl (pH 8,4) с доведением конечного объема до 50 мкл. Для амплификации 5'-кДНК-фрагмента использовали программу термоциклической PCR "Touchdown".
После PCR-амплификации все четыре реакционные смеси объединяли и ДНК-продукты фракционировали по размерам с помощью электрофореза на 8% полиакриламидном геле. ДНК-продукт, составляющий примерно 230 п.о., вырезали из геля, очищали и субклонировали в "T-вектор", как было описано выше для 3'-RACE-фрагмента в 450 п.о. Для дальнейшего анализа выделяли 5 клонов: 5CB2, 5CB3, 5CB5, 5CB6, 5CB11. Анализ ДНК-последовательности осуществляли с использованием секвеназного набора "Sequenase version 2.0" (United States Biochemical, Cleveland, Ohio).
На фиг. 8 показана полная кДНК-последовательность Компонента B, собранная из RACE-клонов 5CB3 и 3CB7, в которой указываются рестрикционные сайты.
Анализ первичных структур кДНК-последовательности клонов 5CB3, 5CB6, 5CB11 и 3CB4, 3CB7, 3CB9 показал полное соответствие этих последовательностей экзону Компонента B (геномный клон 4D Примера 3).
Хотя настоящее изобретение описано выше на конкретных примерах его осуществления, однако специалистам в данной области очевидно, что в него могут быть внесены различные изменения и модификации, не выходящие за рамки существа и объема нижеследующей формулы изобретения.
Надписи к чертежам
Фиг. 1. Рабочая схема для очистки Компонента B из мочи.
Фиг. 2. Рестрикционная карта геномного транскриптона Компонента B (геномная ДНК указанного компонента B показана в SEQ ID N 2). Стрелками показаны сайты сплайсинга.
Фиг. 3. Последовательность области промотора Компонента B (область промотора указанного Компонента B показана в SEQ ID N 2), сайты связывания для факторов транскрипции AP-1, AP-2, Sp-1 и E-блоков указываются. Показаны также TATA-блок и GPE.
Фиг. 4. Рестрикционная карта гена Компонента B. Выведенная мРНК показана строкой ниже геномного гена: области, обведенные в рамку, обозначают последовательность, кодирующую белок.
Фиг. 5. Рестрикционная карта вставки клона 4D.
Фиг. 6. Рестрикционная карта плазмиды pBSCB4D.
Фиг. 7. Общая схема, используемая для RACE-клонирования ДНК-последовательности Компонента B.
Фиг. 8. Полная кДНК-последовательность Компонента B, в которой указаны сайты рестрикции (кДНК указанного Компонента B представлена в SEQ ID N 3).
Реферат
Изобретение относится к генной инженерии и медицине, а именно к новому полипептиду. Полипептид ингибирует связывание TGF-альфа (фактор роста Т-клеток) с его рецепторами. Аминокислотная последовательность приведена в описании. Адсорбируют мочу при кислотном рН на каолине. Экстрагируют ее аммиаком. Элюируют полученные фракции на смоле Bio Rex 70 с использованием аммиака. Полученные после элюирования фракции эллюируют на DEAE-Сефарозной смоле с использованием ацетатного буфера, затем на СМ-Сефарозной смоле с использованием ацетатного буфера, затем на смоле HPLC С 18 (ВЭЖХ) c использованием смеси ацетатного буфера и ацетонитрила, затем на смоле D-Zephyr с использованием ацетатного буфера, затем на смоле HPLC С 18 (ВЭЖХ) с использованием смеси водной трихлоруксусной кислоты и ацетонитрила. Затем на смоле D-Zephyr с использованием ацетатного буфера. Получают полипептид или содержащий его в основном чистый белок. Полипептид кодируется ДНК с нуклеотидной последовательностью, приведенной в материалах заявки. Экспрессирующий вектор включает фрагмент ДНК, кодирующий полипептид. Штамм E.coli трансформирован экспрессирующим вектором и продуцирует полипептид с аминокислотной последовательностью, приведенной в описании. Полипептид также получают при культивировании прокариотической или эукариотической клетки, трансформированной экспрессирующим вектором и выделением его из культуральной среды. Фармкомпозиция, обладающая свойством ингибировать связывание TGF-альфа, содержит терапевтически приемлемое количество полипептида или в основном чистого белка в комбинации с одним или несколькими терапевтически приемлемыми накопителями или разбавителями. Лекарственное средство, обладающее противовоспалительной, и/или антикоагулирующей, и/или противоопухолевой активностью содержит в своем составе полипептид с аминокислотной последовательностью, приведенной в материалах заявки. Изобретение позволяет получить новый белок с относительно низкой молекулярной массой, который может быть использован при получении противовоспалительных, антикоагулирующих средств лечения, а также средств, обладающих противоопухолевой активностью. 19 с. и 1 з.п. ф-лы, 8 ил.

Комментарии