Гены рецептора трансферрина moraxela - RU2235128C2
Код документа: RU2235128C2
Чертежи


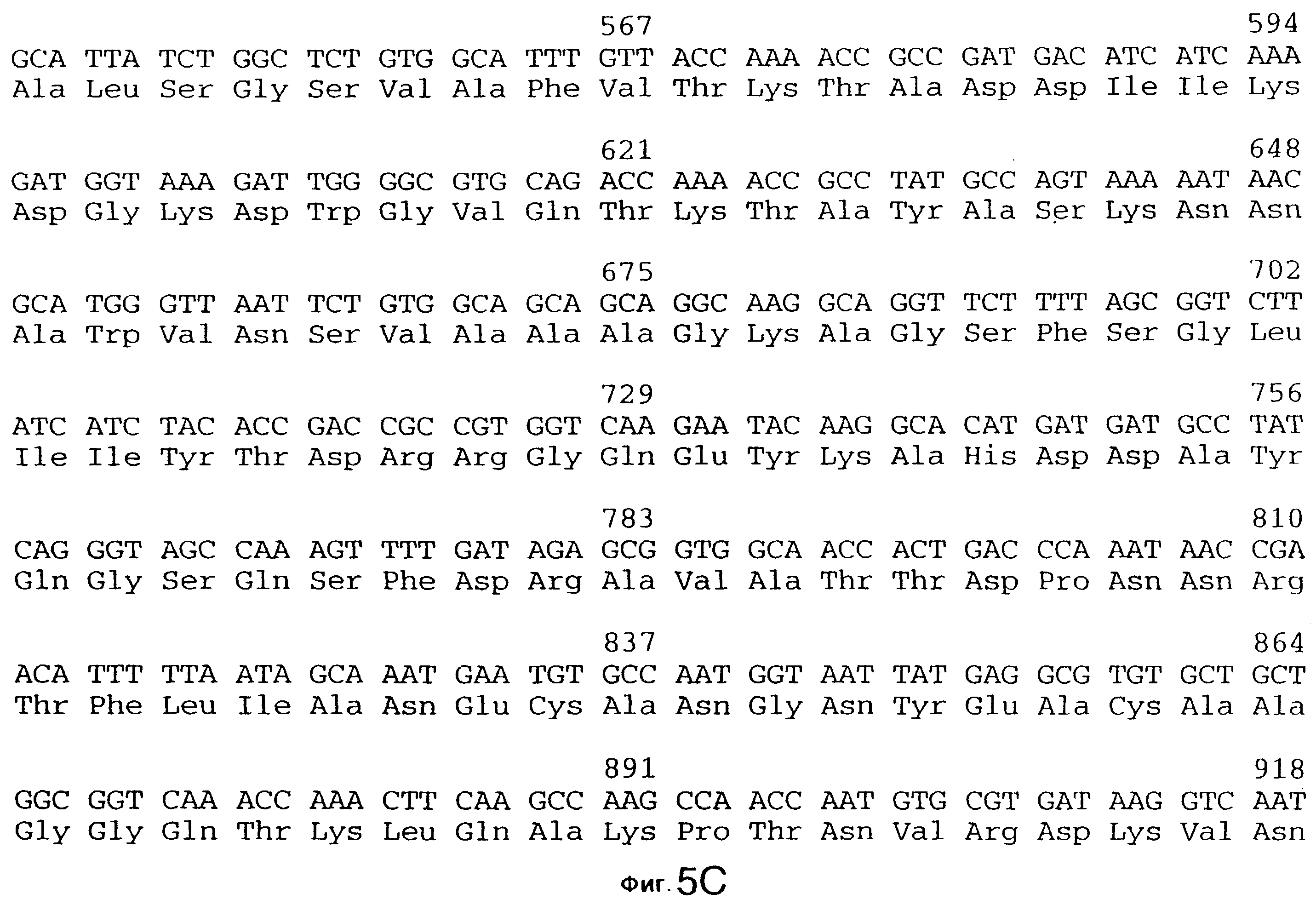
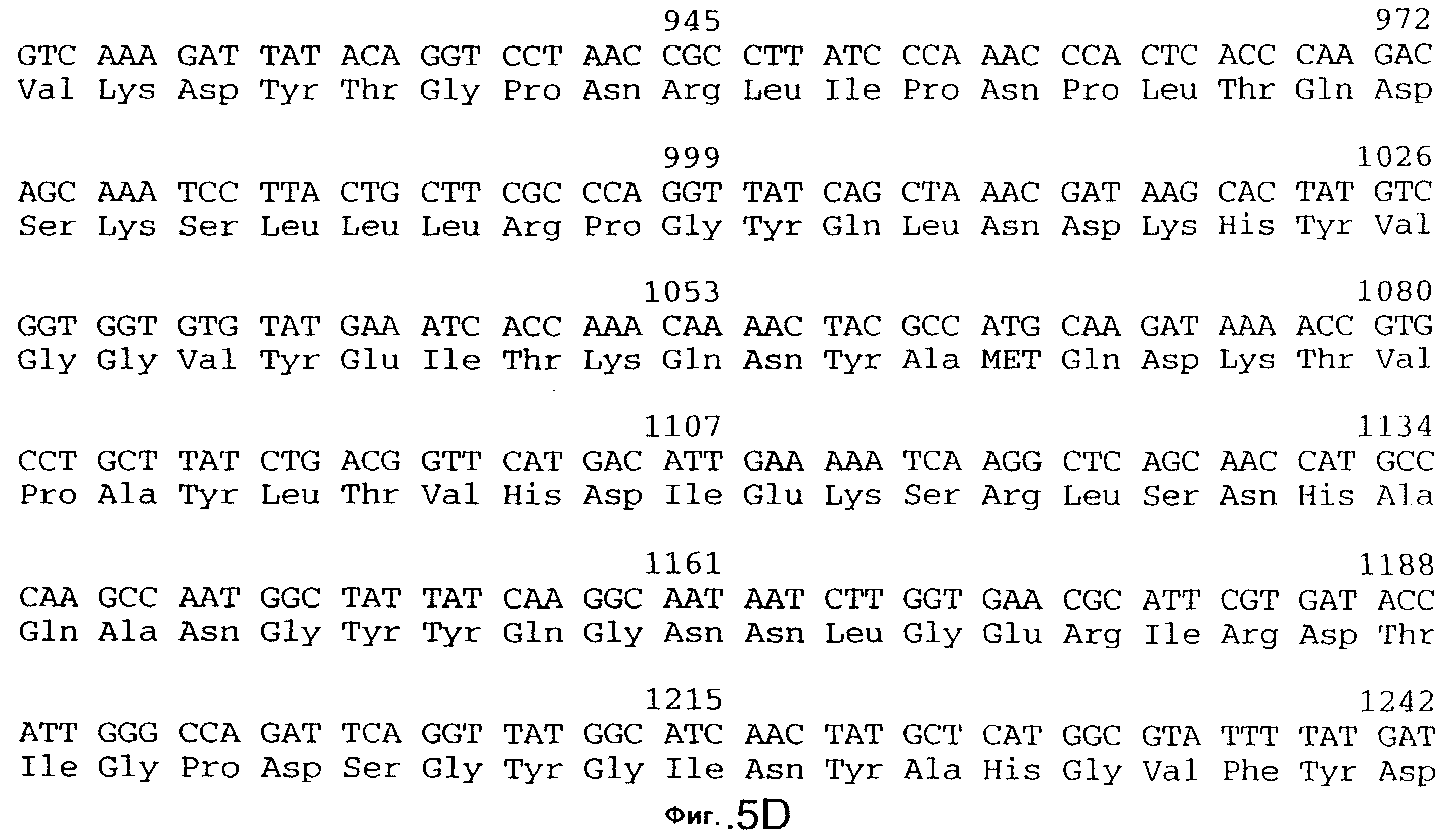

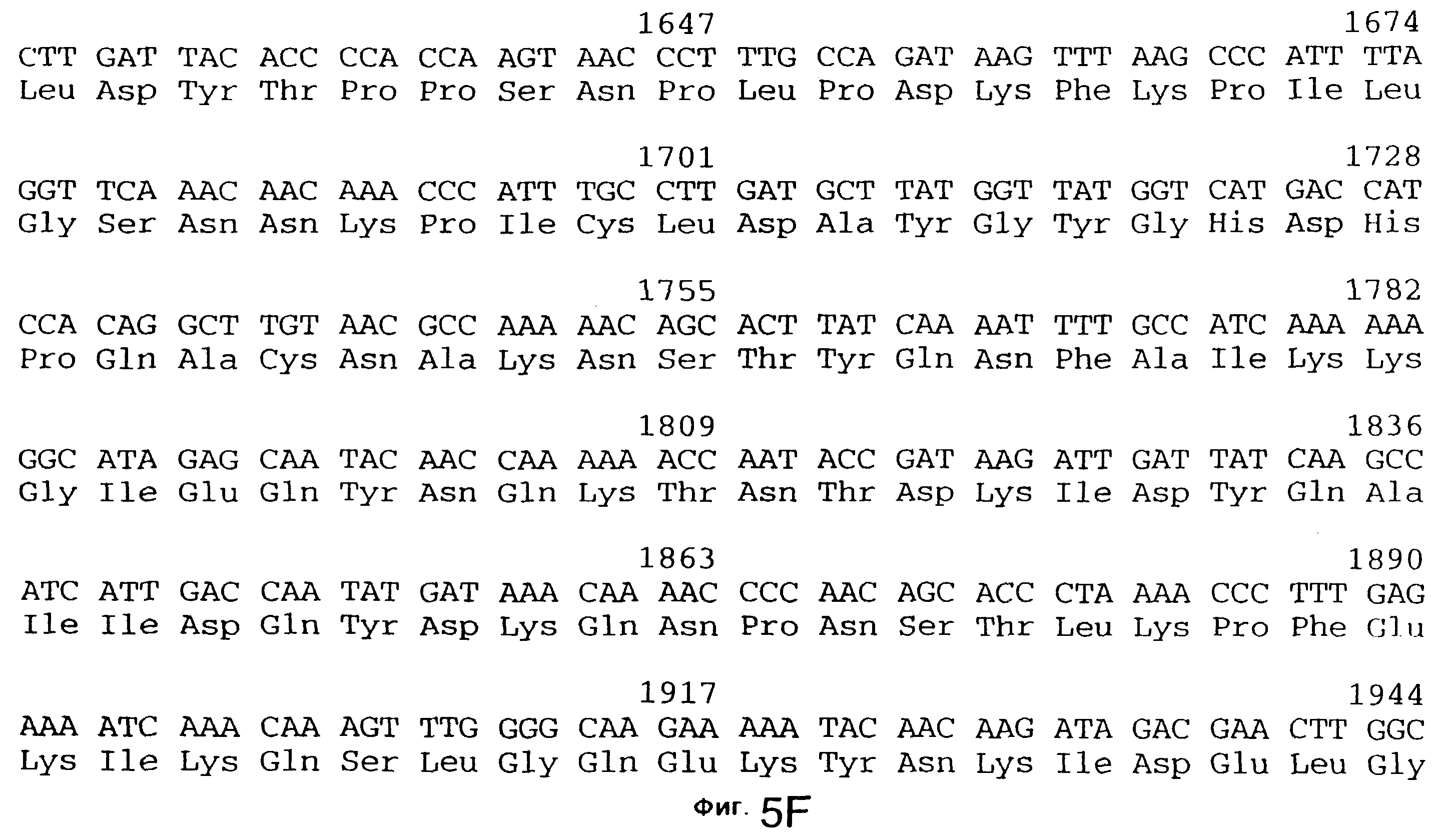
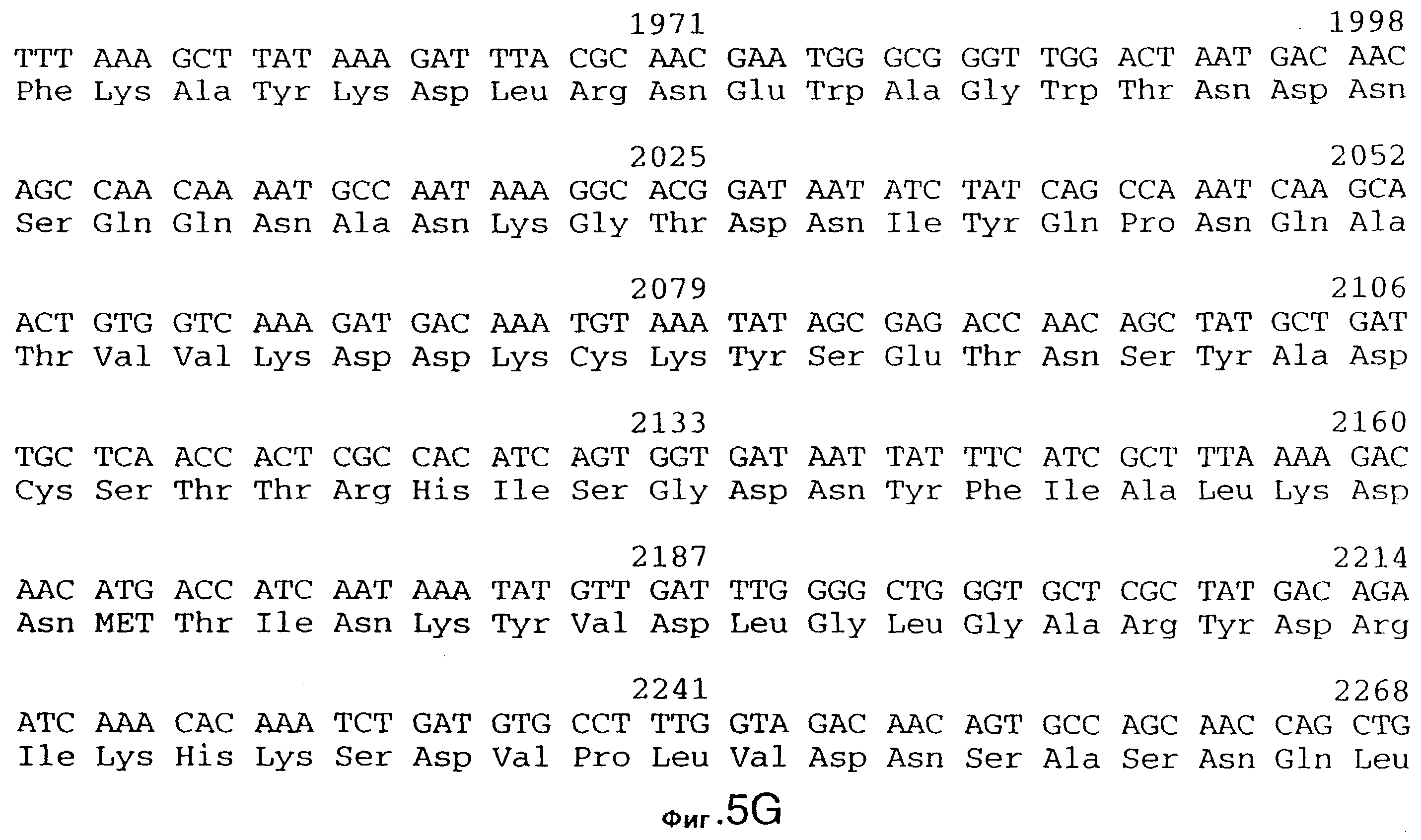
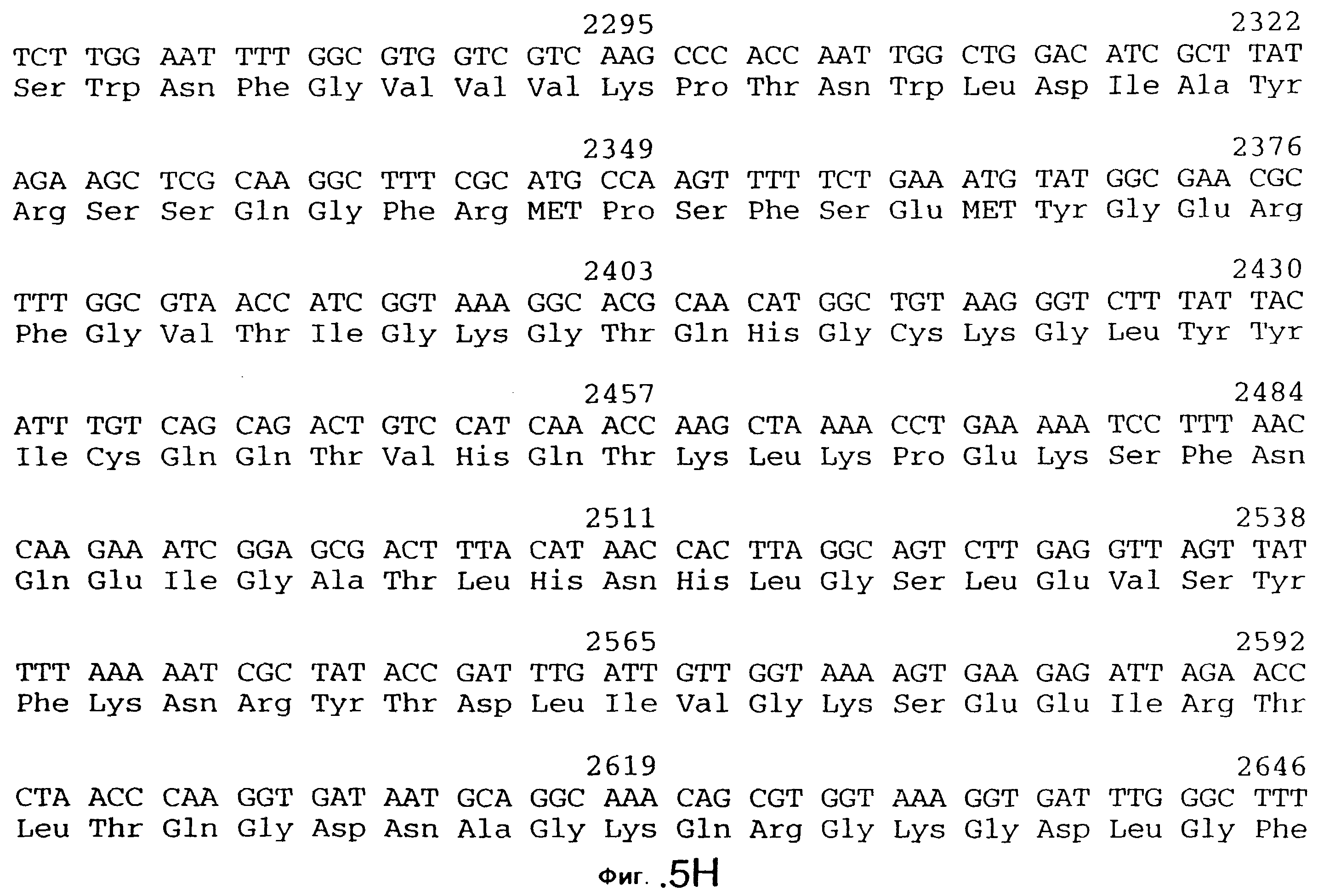








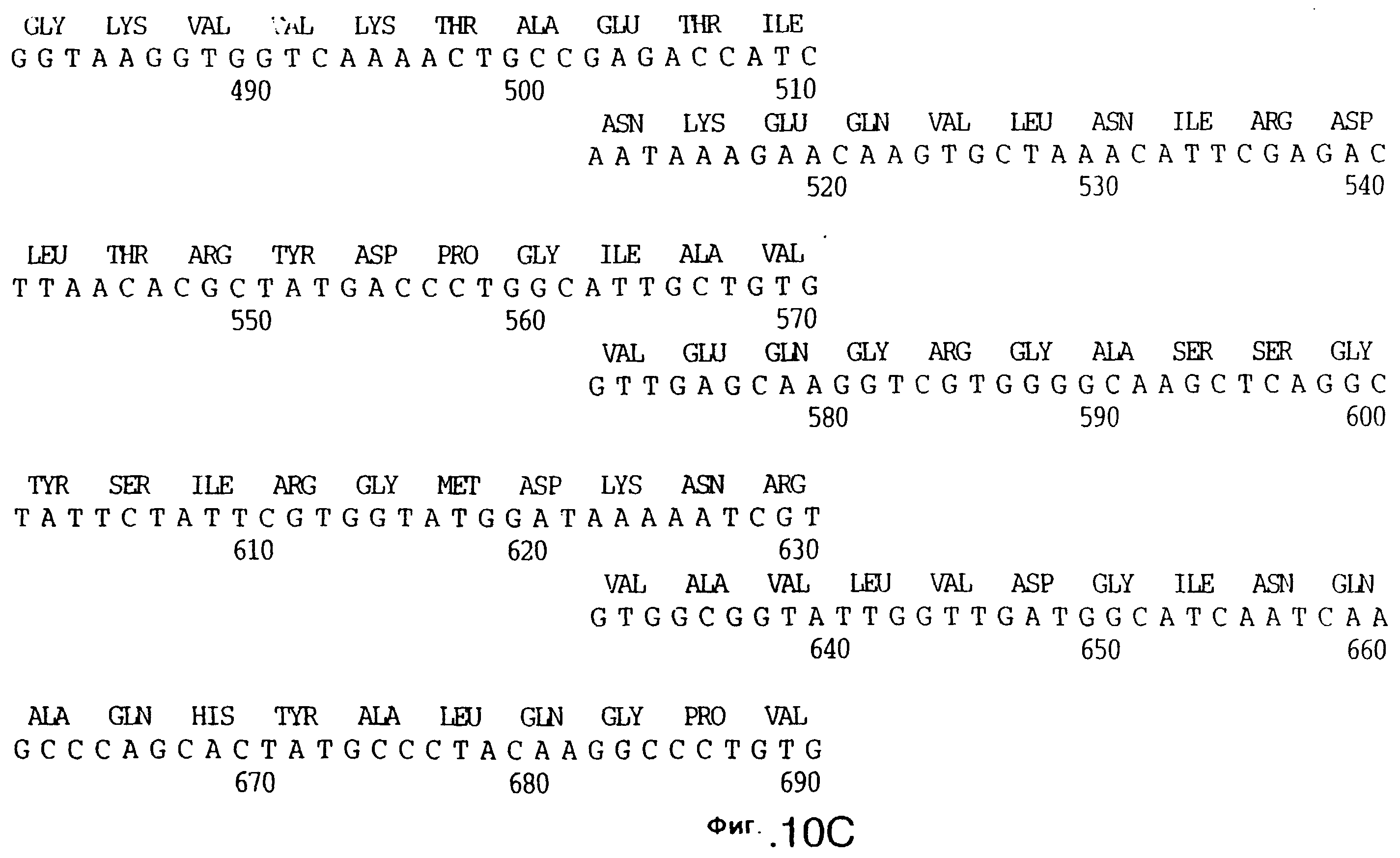




























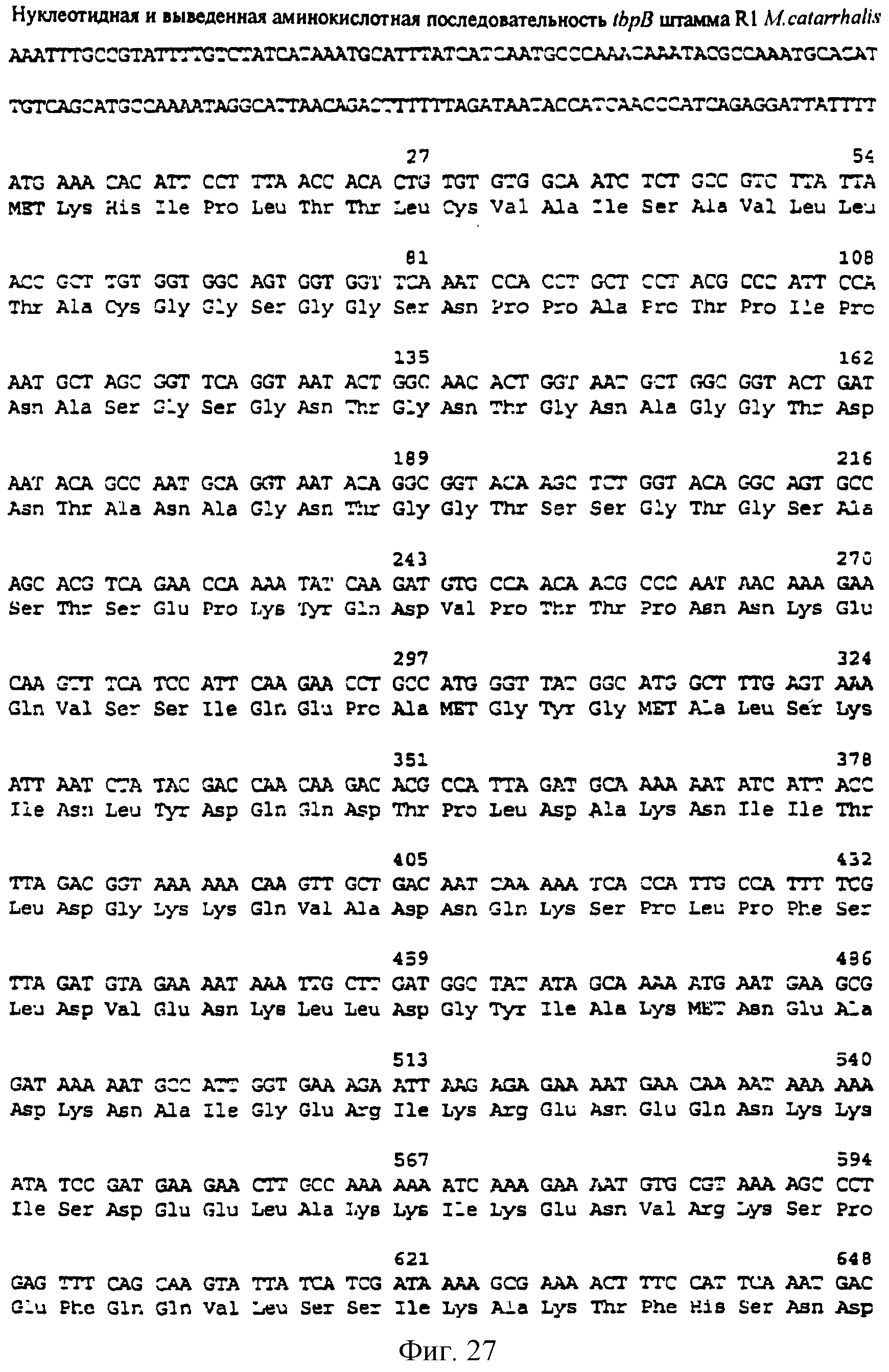
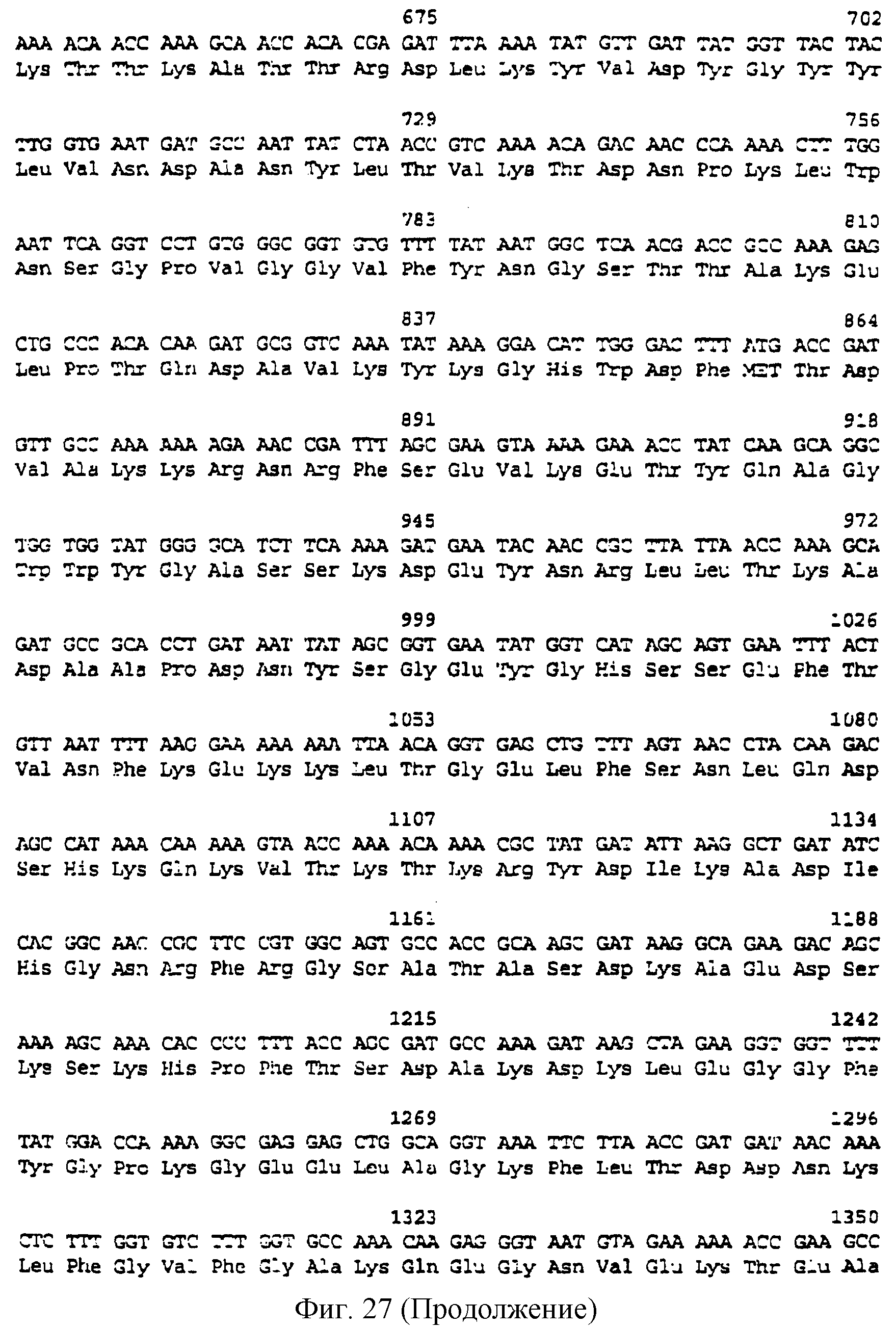



Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к молекулярному клонированию генов, кодирующих белки рецептора трансферрина (TfR), а в частности к клонированию генов рецептора трансферрина Moraxella (Branhamella) catarrhalis.
Эта заявка является частичным продолжением одновременно рассматриваемой заявки на патент США №08/778570, поданной 3 января 1997 г, которая, в свою очередь, является частичным продолжением заявки на патент США №08/613009, поданной 8 марта 1996 г.
Уровень техники
Бактерии Moraxella (Branhamella) catarrhalis являются грамотрицательными диплококковыми патогенами, которые бессимптомно присутствуют в дыхательных путях здорового человека. В последние годы было установлено, что М.catarrhalis является важным патогеном, вызывающим воспаление среднего уха. Кроме того, M.catarrhalis ассоциируется с синуситом, конъюктивитом, и мочеполовыми инфекциями, а также с рядом воспалительных заболеваний нижних дыхательных путей у детей и взрослых, включая пневмонию, хронический бронхит, трахеит, и энфизему (см. 1-8). (В данной заявке для более полного описания состояния той области науки, к которой имеет отношение настоящее изобретение, приводятся различные ссылки на цитируемые работы, которые даны в скобках. Полные библиографические данные для каждой цитируемой работы приводятся в конце описания, непосредственно перед формулой изобретения. Содержание этих работ вводится в настоящее описание посредством ссылки). В некоторых случаях, заражение бактерией М.catarrhalis вызывает септицемию, артрит, эндокардит, и менингит (см. 9-13).
Воспаление среднего уха является одним из наиболее распространенных заболеваний детей в раннем возрасте, и около 80% всех детей в возрасте до трех лет страдает, по крайней мере, одной инфекцией среднего уха (см. 14). Хроническое воспаление среднего уха ассоциируется с нарушением слуха и речи у детей, а в некоторых случаях, с нарушением способности к обучению. Стандартное лечение воспаления среднего уха предусматривает введение антибиотиков и хирургические операции, включая тонзиллэктомию, аденоидэктомию и тимпаноцентез. По оценкам специалистов, стоимость лечения воспаления среднего уха в США составляет от одного до двух миллиардов долларов в год.
В случае воспаления среднего уха, М.catarrhalis обычно выделяют из жидкости среднего уха вместе с бактерией Streptococcus pneumoniae и нетипируемой бактерией Haemophilus influenzae, которые, очевидно, ответственны за 50% и 30% инфекций среднего уха, соответственно. Полагают, что бактерия М.catarrhalis ответственна примерно за 20% инфекций среднего уха (см. 15). Эпидемиологические отчеты сообщают, что число случаев воспаления среднего уха, связанных с М.catarrhalis, возрастает одновременно с увеличением числа резистентных к антибиотикам изолятов М.catarrhalis. Так, например, до 1970 г. не было никаких сообщений о изолятах М.catarrhalis, продуцирующих β -лактамазу, но с середины семидесятых годов было обнаружено возрастающее число изолятов, экспрессирующих β -лактамазу. Последние обзоры дают основание предполагать, что 75% клинических изолятов продуцируют β -лактамазу (см. 16, 26).
Железо является основным питательным элементом, необходимым для роста многих бактерий. Несколько бактериальных видов, включая M.catarrhalis, получают железо от хозяина, используя для захвата трансферрина белки рецепторов трансферрина. Ряд бактерий, включая, Neisseria meningitidis (см. 17), N.gonorrhoeae (см. 18), Haemophilus Influenzae (см. 19), а также M.catarrhalis (см. 20), продуцируют наружные мембранные белки, которые специфически связываются с трансферрином человека. Экспрессия этих белков регулируется количеством железа в окружающей среде.
Два белка рецепторов трансферрина М.catarrhalis, называемых трансферрин-связывающим белком 1 (Tbp1) и трансферрин-связывающим белком 2 (Tbp2), имеют молекулярную массу 115 кДа (Tbp1) и приблизительно 80-90 кДа (Tbp2). В отличие от белков рецепторов трансферрина других бактерий, рецепторы Tbp2 M.catarrhalis обладают предпочтительным сродством к железонасыщенному (то есть, ферри-) трансферрину (см. 21).
Инфекция, вызываемая М.catarrhalis, может приводить к серьезным заболеваниям. Поэтому было бы желательно получить рекомбинантный источник трансферрин-связывающих белков, которые могут быть использованы в иммуногенных препаратах, включая вакцины, в качестве антигенов в качестве носителей для других антигенов, и в качестве иммуногенов, а также для продуцирования диагностических реагентов. Особенно желательны и полезны гены, кодирующие трансферрин-связывающие белки и их фрагменты для специфической идентификации и диагностики инфекций Moraxella, для иммунизации против заболеваний, вызываемых М.catarrhalis, а также для продуцирования диагностических реагентов.
Сущность изобретения
Настоящее изобретение относится к получению очищенных, изолированных молекул нуклеиновой кислоты, кодирующих рецептор трансферрина штамма Moraxella, или фрагмент или аналог белка рецептора трансферрина. Полученные в соответствии с настоящим изобретением молекулы нуклеиновой кислоты могут быть использованы для специфического обнаружения штаммов Moraxella и для диагностики инфекции, вызываемой Moraxella. Очищенные и выделенные молекулы нуклеиновой кислоты настоящего изобретения, такие как ДНК, могут быть также использованы для экспрессии генов Tbp с использованием техники рекомбинантных ДНК в целях продуцирования экономическим способом очищенных и выделенных белков рецептора трансферрина, а также их субъединиц, фрагментов, или аналогов. Рецептор трансферрина, его субъединицы, фрагменты, или аналоги, а также молекулы нуклеиновой кислоты, кодирующие указанный рецептор, его субъединицы, фрагменты или аналоги, и векторы, содержащие такие молекулы нуклеиновой кислоты, могут быть использованы в иммуногенных композициях для вакцинации против заболеваний, вызываемых Moraxella, и для диагностики инфекций Moraxella, a также в качестве инструмента для получения иммунологических реагентов. Моноклональные антитела или моноспецифическая антисыворотка (антитела) против белка рецептора трансферрина, продуцированные в соответствии с аспектами настоящего изобретения, могут быть использованы для диагностики инфекции Moraxella; для специфического обнаружения Moraxella (например, в in vitro и in vivo анализах); и для лечения заболеваний, вызываемых Moraxella.
В соответствии с одним из своих аспектов настоящее изобретение относится к очищенным и выделенным молекулам нуклеиновой кислоты (НК-молекулам), кодирующим белок рецептора трансферрина штамма Moraxella, а более конкретно, штамма M.catarrhalis, в частности, штаммов M.catarrhalis 4223, Q8 или R1, либо фрагмент или аналог белка рецептора трансферрина.
В одном из предпочтительных вариантов настоящего изобретения НК-молекула может кодировать только белок Tbp1 штамма Moraxella, или только белок Тbр2 штамма Moraxella. В другом предпочтительном варианте настоящего изобретения НК-молекула может кодировать фрагмент белка рецептора трансферрина штамма Moraxella, имеющий консервативную аминокислотную последовательность.
В другом своем аспекте настоящее изобретение относится к очищенной и выделенной НК-молекуле, имеющей ДНК-последовательность, выбранную из группы, включающей: (а) ДНК-последовательность, представленную на фиг.5, 6, 10, 11 или 27 (SEQ ID Nо: 1, 2, 3, 4, 5, 6, 7, 8, 45 или 46), или комплементарную ей ДНК-последовательность; (b) ДНК-последовательность, кодирующую аминокислотную последовательность, представленную на фиг.5, 6, 10, 11 или 27 (SEQ ID Nо: 9, 10, 11, 12, 13, 14, 15, 16 или 47) или комплементарную ей ДНК-последовательность; и (с) ДНК-последовательность, которая гибридизируется в жестких условиях с любой из ДНК-последовательностей, определенных в пунктах (а) и (b). ДНК-последовательность, определенная в п. (с) имеет предпочтительно последовательность, которая, по крайней мере, примерно на 90% идентична любой одной из ДНК-последовательностей, определенных в пунктах (а) и (b). ДНК-последовательность, определенная в (с), может быть такой, что она будет кодировать эквивалентный белок рецептора трансферрина, происходящий от другого штамма Moraxella.
В другом своем аспекте настоящее изобретение относится к вектору, адаптированному для трансформации хозяина, и содержащему НК-молекулу настоящего изобретения, которая может обладать свойствами нуклеотидной последовательности, содержащейся в векторах LEM3-24, pLEM3, pLEM25, pLEM23, SLRD-А, DS-1698-1-1, DS-1754-1, pSLRD2, pSLRD3, pSLRD4 и pSLRD5.
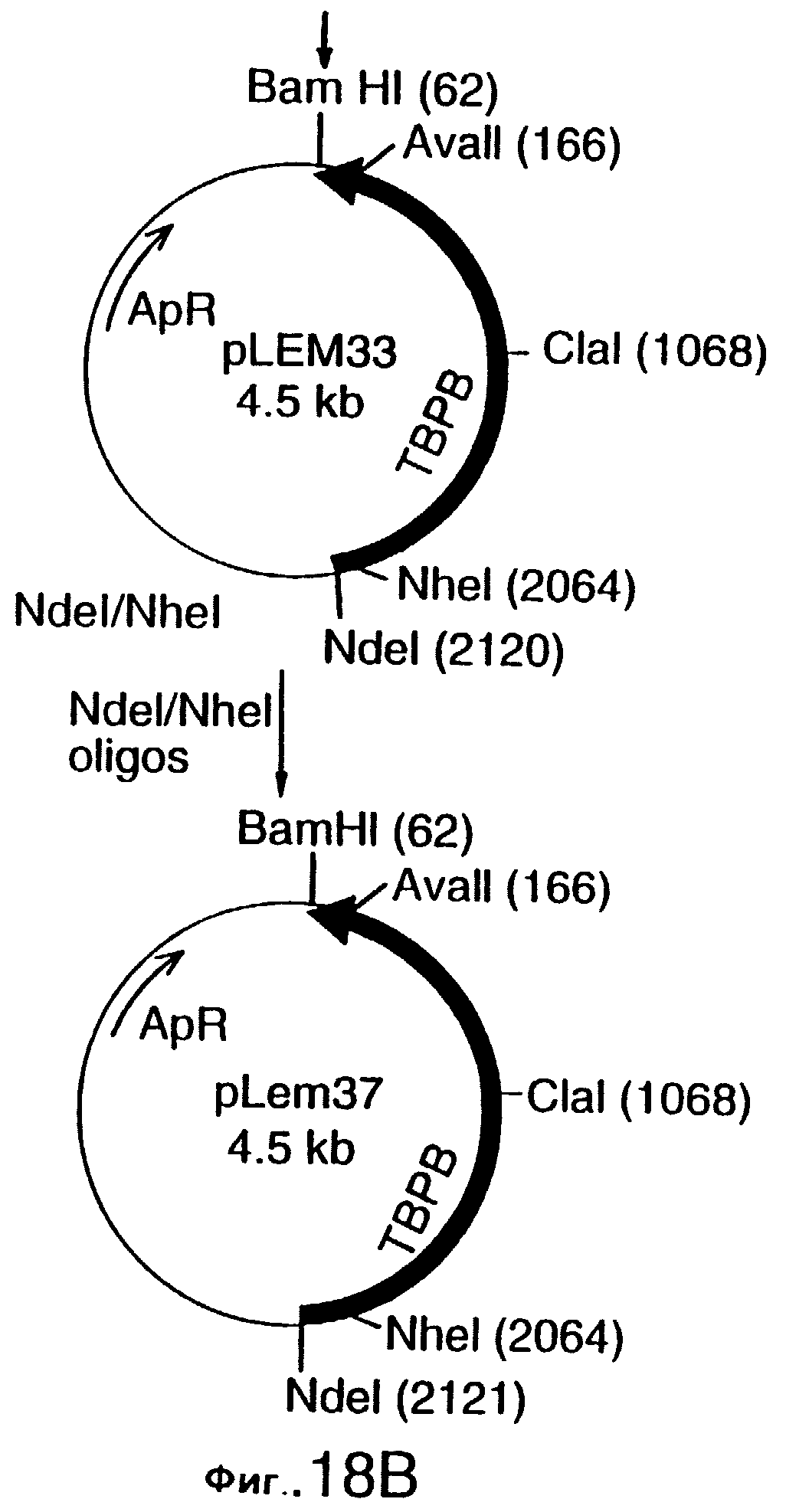
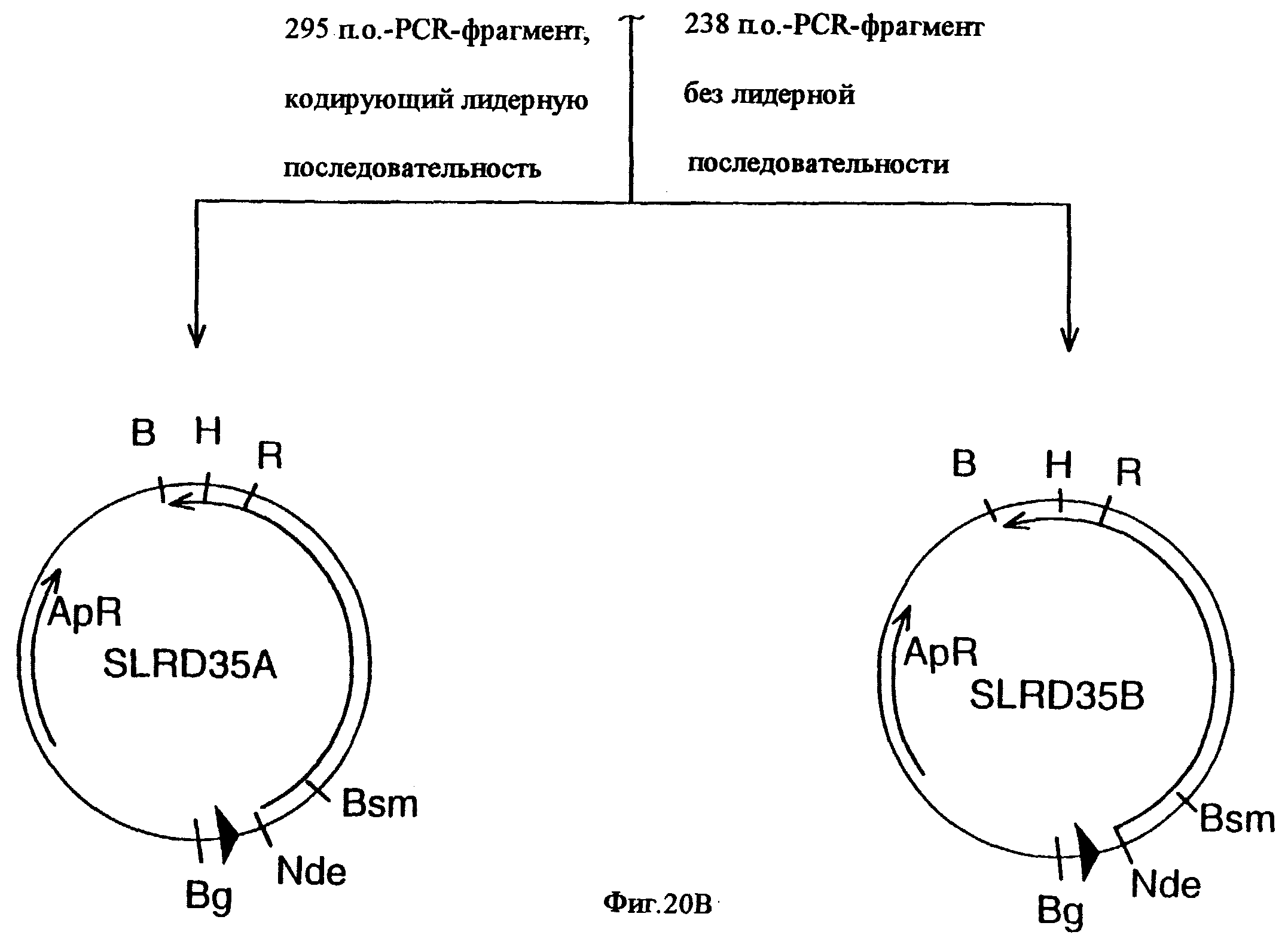
Этот вектор может быть адаптирован для экспрессии кодированного рецептора трансферрина, его фрагментов или аналогов, в гетерологичном или гомологичном хозяине, либо в липидной, либо в нелипидной форме. В соответствии с другим своим аспектом настоящее изобретение относится к экспрессирующему вектору, адаптированному для трансформации хозяина, и содержащему НК-молекулу настоящего изобретения, а также к экспрессионному элементу, оперативно связанному с НК-молекулой, и обеспечивающему экспрессию белка рецептора трансферрина, его фрагмента, или аналога. В конкретном варианте этого аспекта настоящего изобретения НК-молекула может кодировать, в основном, полный белок рецептора трансферрина, или только белок Тbр1, только белок Тbр2 штамма Moraxella, или фрагменты белка Tbp1 или белка Tbp2. Этот экспрессионный элемент может включать промотор и часть нуклеиновой кислоты, кодирующую лидерную последовательность для секреции белка рецептора трансферрина, или его фрагмента, или его аналога из клетки-хозяина. Этот экспрессионный элемент может также включать фрагмент нуклеиновой кислоты, кодирующий сигнал липидизации для экспрессии в клетке-хозяине липидизированной формы белка рецептора трансферрина, или его фрагмента, или его аналога. Хозяин может быть выбран, например, из Escherichia. coli, Bordetella, Bacillus, Haemophilus, Moraxella, а в качестве экспрессирующих систем могут быть использованы грибки, дрожжи или бакуловирус и вирус Semliki Forest (лесов Семлики). В конкретном варианте настоящего изобретения плазмидой, адаптированной для экспрессии Tbp1 является pLEM29, а для экспрессии Tbp2 - рLЕМ33. Другими векторами являются pLEM-37, SLRD35-A и SLRD35-B.
В другом своем аспекте настоящее изобретение относится к трансформированному хозяину, содержащему экспрессирующий вектор настоящего изобретения. Кроме того, настоящее изобретение включает белок рецептора трансферрина, его фрагмент, или аналог штамма Moraxella, продуцируемый трансформированным хозяином.
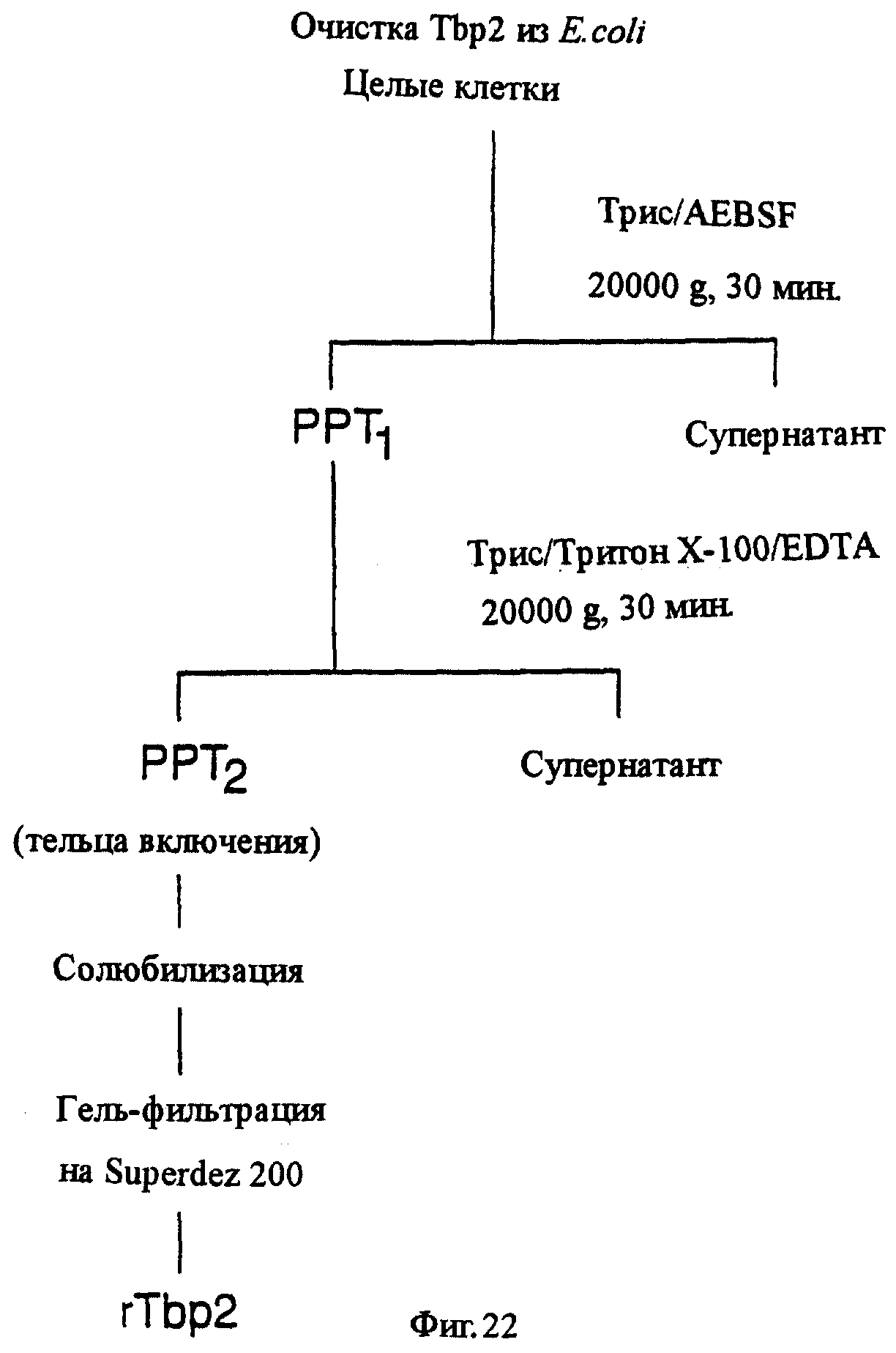
Такой рекомбинантный белок рецептора трансферрина может быть получен, в основном, в чистой форме в соответствии с другим аспектом настоящего изобретения, в котором предлагается способ получения, в основном, чистого рекомбинантного белка рецептора трансферрина, предусматривающий культивирование трансформированного хозяина настоящего изобретения для экспрессии белка рецептора трансферрина в виде телец включения; очистку этих телец включения от клеточного материала и растворимых белков; солюбилизацию белка рецептора трансферрина, полученного из очищенных телец включения; и очистку белка рецептора трансферрина от других солюбилизированных материалов. В основном, чистый рекомбинантный белок рецептора трансферрина может содержать только один Tbp1, только один Тbр2, или их смесь. В основном, чистота этого рекомбинантного белка составляет, по крайней мере, около 70%, а предпочтительно, по крайней мере, около 90%.
Поэтому в других своих аспектах настоящее изобретение относится к рекомбинантно продуцированному белку Tbp1 штамма Moraxella, не содержащему белок Тbр2 штамма Moraxella и любой другой белок штамма Moraxella, и к рекомбинантно продуцированному белку Тbр2 штамма Moraxella, не содержащему белок Tbp1 штамма Moraxella и любой другой белок штамма Moraxella. Штаммом Moraxella может быть штамм 4223 M.catarrhalis, штамм Q8 M.catarrhalis, или штамм R1 M.catarrhalis.
В соответствии с другим своим аспектом настоящее изобретение относится к иммуногенной композиции, которая содержит, по крайней мере, один активный компонент, выбранный, по крайней мере, из одной НК-молекулы настоящего изобретения, и, по крайней мере, одного рекомбинантного белка настоящего изобретения; и его фармацевтически приемлемый носитель или вектор. Этот, по крайней мере, один активный компонент вызывает иммунный ответ при его введении хозяину.
Иммуногенные композиции настоящего изобретения могут быть изготовлены в виде вакцин для in vivo введения хозяину. Для этого, указанные композиции могут быть изготовлены в виде микрочастицы, капсулы, ISCOM, или липосомного препарата. Эта иммуногенная композиция может быть введена в комбинации с молекулой, обеспечивающей доставку этой композиции к специфическим клеткам иммунной системы или к поверхностям слизистых оболочек. Иммуногенные композиции настоящего изобретения (включая вакцины) могут, кроме того, содержать, по крайней мере, один другой иммуногенный или иммуностимулирующий материал, и этот иммуностимулирующий материал может быть, по крайней мере, одним из адъювантов, либо, по крайней мере, одним из цитокинов. Адъювантами, подходящими для использования в настоящем изобретении, являются (но не ограничиваются ими) фосфат алюминия, гидроксид алюминия, QS21, Quil А, их производные и компоненты, матрикс ISCOM, фосфат кальция, гидроксид кальция, гидроксид цинка, гликолипидный аналог, октадециловый сложный эфир аминокислоты, мурамилдипептид, полифосфазен, ISCOPREP, DC-chol, DDBA, и липопротеин. Преимущественные комбинации адъювантов описаны в одновременно рассматриваемых заявках на патент США №08/261194 (поданной 16 июня 1994 г.), и №08/483856 (поданной 7 июня 1995 г.), которые были переуступлены их правопреемнику, и которые вводятся в настоящее описание посредством ссылки (WO 95/34308).
В соответствии со своим другим аспектом настоящее изобретение относится к способу получения иммунного ответа у хозяина, включающему стадию введения восприимчивому хозяину, такому как, человек, эффективного количества иммуногенной композиции настоящего изобретения. Иммунный ответ может быть гуморальным или клеточно-опосредованным иммунным ответом, и может обеспечивать иммунную защиту от заболевания, вызываемого Moraxella. Хозяевами, у которых может быть обеспечена иммунная защита от заболеваний, являются приматы, включая человека.
В другом своем аспекте настоящее изобретение относится к "живому" вектору, обеспечивающему доставку рецептора трансферрина в клетку-хозяина, и содержащему НК-молекулу, описанную выше. Этот вектор может быть выбран из Salmonella, BCG, аденовируса, поксвируса, вируса коровьей оспы и полиовируса.
Молекулы нуклеиновой кислоты настоящего изобретения могут быть использованы в диагностических целях. В соответствии с еще одним своим аспектом настоящее изобретение относится к способу обнаружения присутствия в образце нуклеиновой кислоты, кодирующей белок рецептора трансферрина штамма Moraxella, где указанный способ включает следующие стадии:
(а) контактирование образца с НК-молекулой настоящего изобретения для продуцирования дуплекса, содержащего эту НК-молекулу и любую НК-молекулу, кодирующую белок рецептора трансферрина штамма Moraxella, присутствующего в образце, и специфически гибридизирующуюся с ней; и
(b) определение продуцирования дуплексов.
Кроме того, настоящее изобретение относится к диагностическому набору для обнаружения присутствия в образце нуклеиновой кислоты, кодирующей белок рецептора трансферрина штамма Moraxella, где указанный набор включает:
(a) НК-молекулу настоящего изобретения;
(b) средство для контактирования НК-молекулы с образцом для продуцирования дуплексов, содержащих эту НК-молекулу и любую НК-молекулу, кодирующую белок рецептора трансферрина штамма Moraxella, присутствующего в образце, и специфически гибридизирующуюся с ней; и
(c) средство для определения продуцирования дуплексов.
Кроме того, настоящее изобретение относится к использованию НК-молекул и белков настоящего изобретения в качестве лекарственных средств. Настоящее изобретение также относится к использованию НК-молекул и белков настоящего изобретения в целях изготовления лекарственных препаратов против инфекций, вызываемых штаммами Moraxella.
Преимуществами настоящего изобретения являются:
- выделенная и очищенная молекула нуклеиновой кислоты, кодирующая белок рецептора трансферрина штамма Moraxella, или его фрагмент, или аналог;
- рекомбинантные продуцированные белки рецептора трансферрина, включая Tbp1 и Тbр2, свободные друг от друга и от других белков штамма Moraxella; и
- диагностические наборы и иммунологические реагенты для специфической идентификации Moraxella.
Краткое описание чертежей
Для лучшего понимания настоящего изобретения ниже приводится его подробное описание со ссылками на следующие чертежи, где:
фиг.1 иллюстрирует аминокислотные последовательности (SEQ ID No: 17 и 18) консервативной области белков Tbp1, используемой для синтеза вырожденных праймеров, используемых для PCR-амплификации фрагмента гена tbpA штамма 4223 M.catarrhalis;
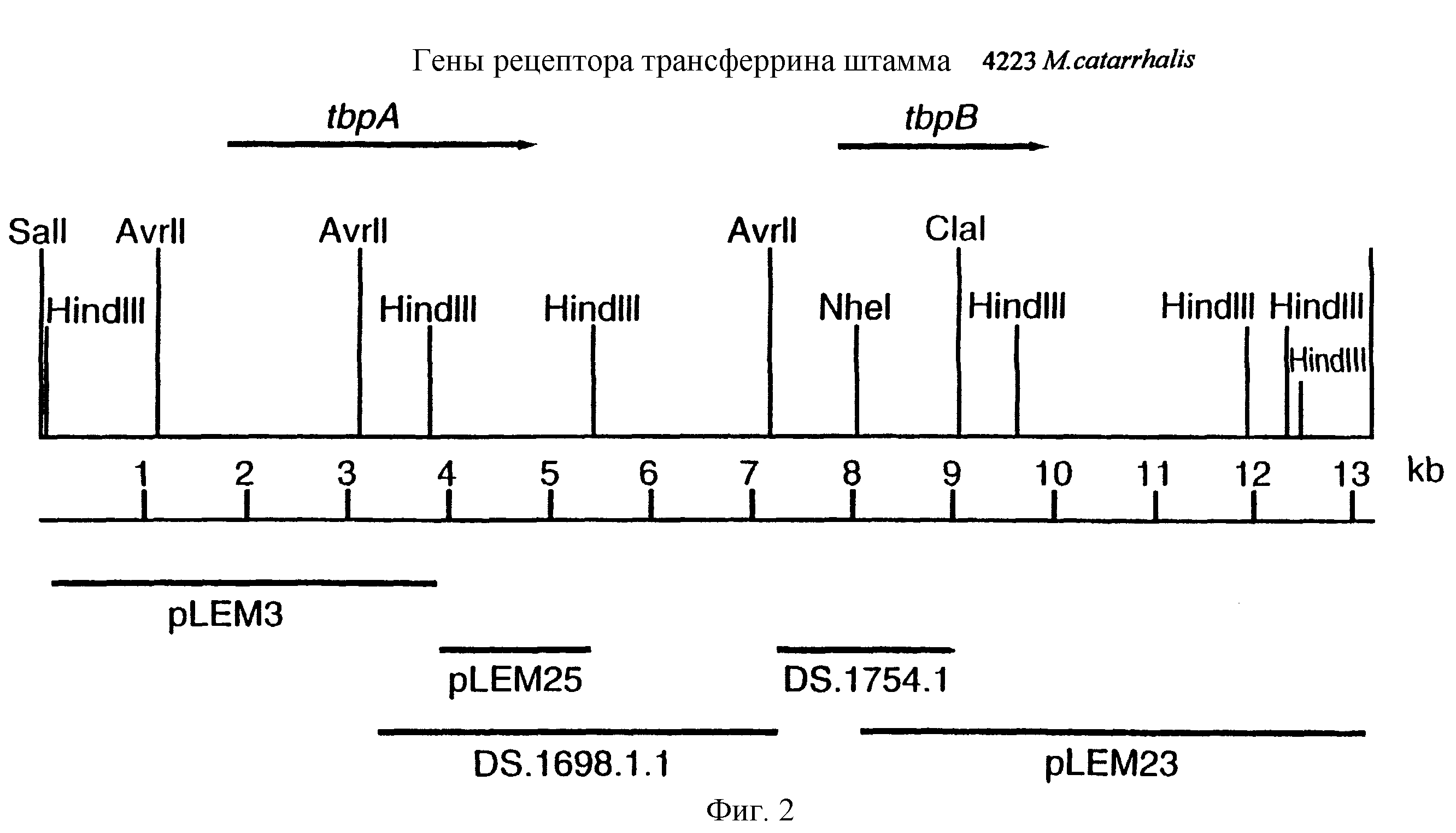
фиг.2 иллюстрирует рестрикционную карту клона LEM3-24, содержащего гены tbpA и tbpB изолята 4223 М.catarrhalis:
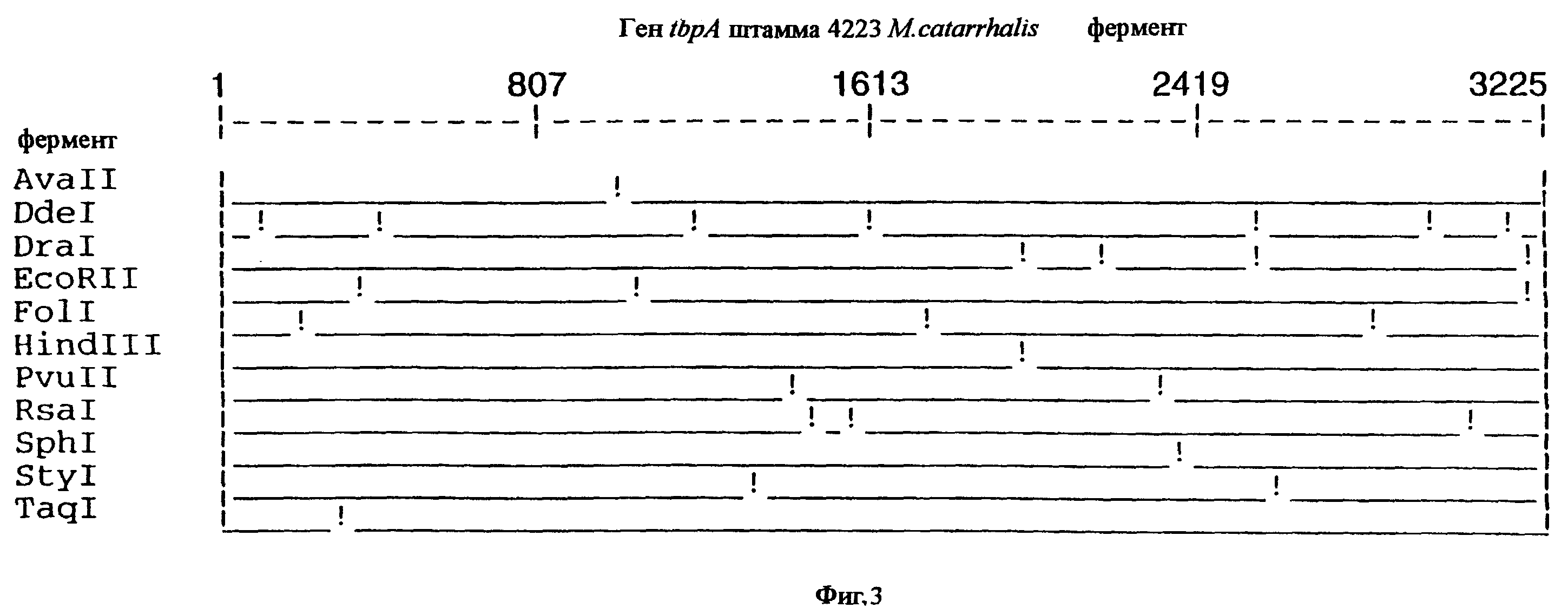
фиг.3 иллюстрирует рестрикционную карту гена tbpA для штамма 4223 M.catarrhalis;
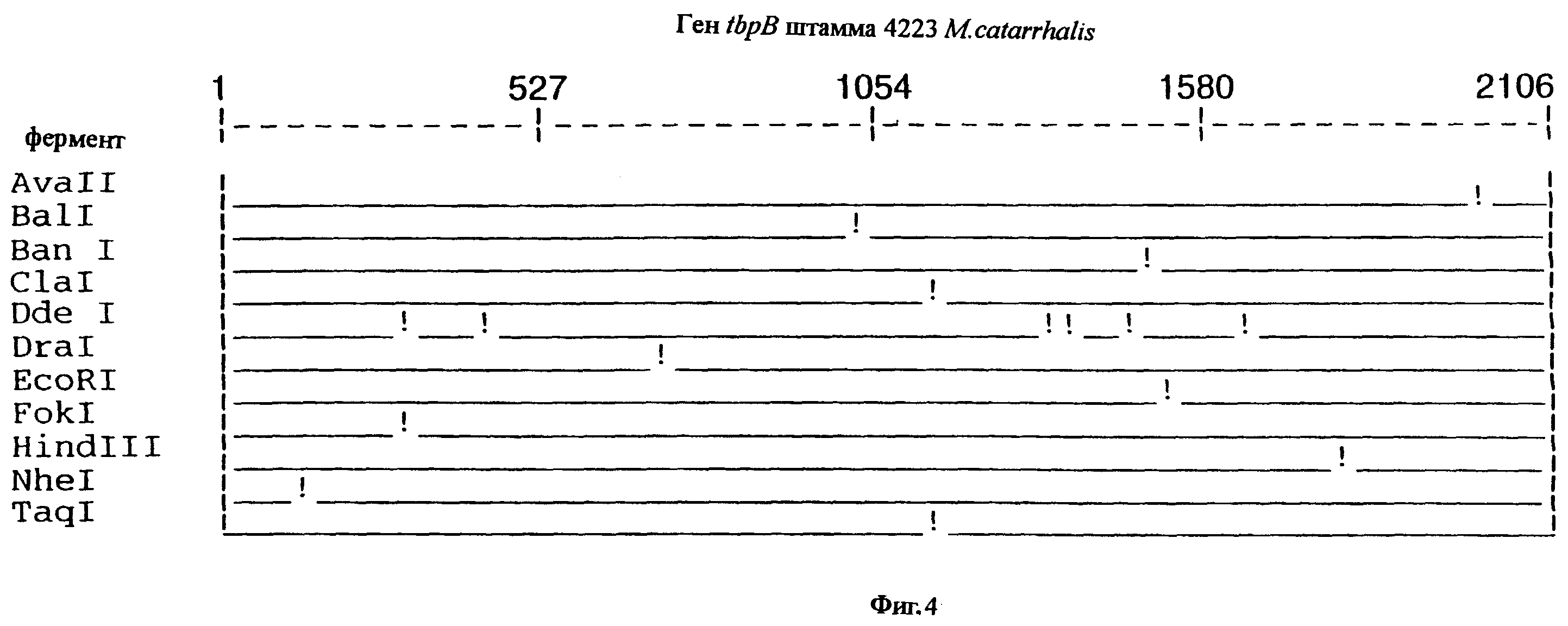
фиг.4 иллюстрирует рестрикционную карту гена tbpB для штамма 4223 M.catarrhalis;
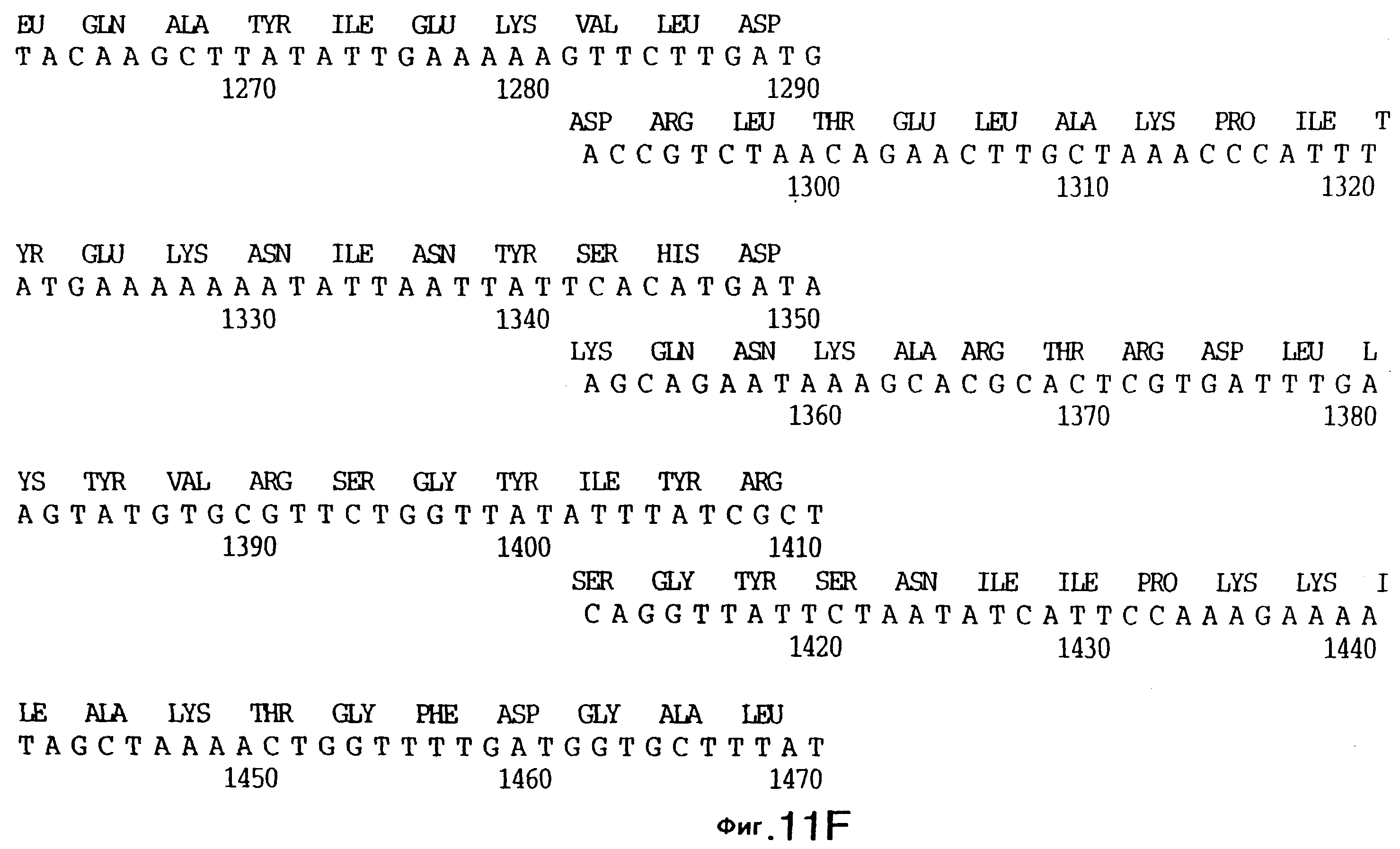
фиг.5 иллюстрирует нуклеотидную последовательность гена tbpA (SEQ ID No: 1 - полная последовательность, и SEQ ID No: 2 - кодирующая последовательность), и выведенную аминокислотную последовательность белка Tbp1 штамма 4223 (SEQ ID No: 9 - полноразмерная последовательность, и SEQ ID No: 10 - последовательность зрелого белка). Лидерная последовательность (SEQ ID No: 19) подчеркнута;
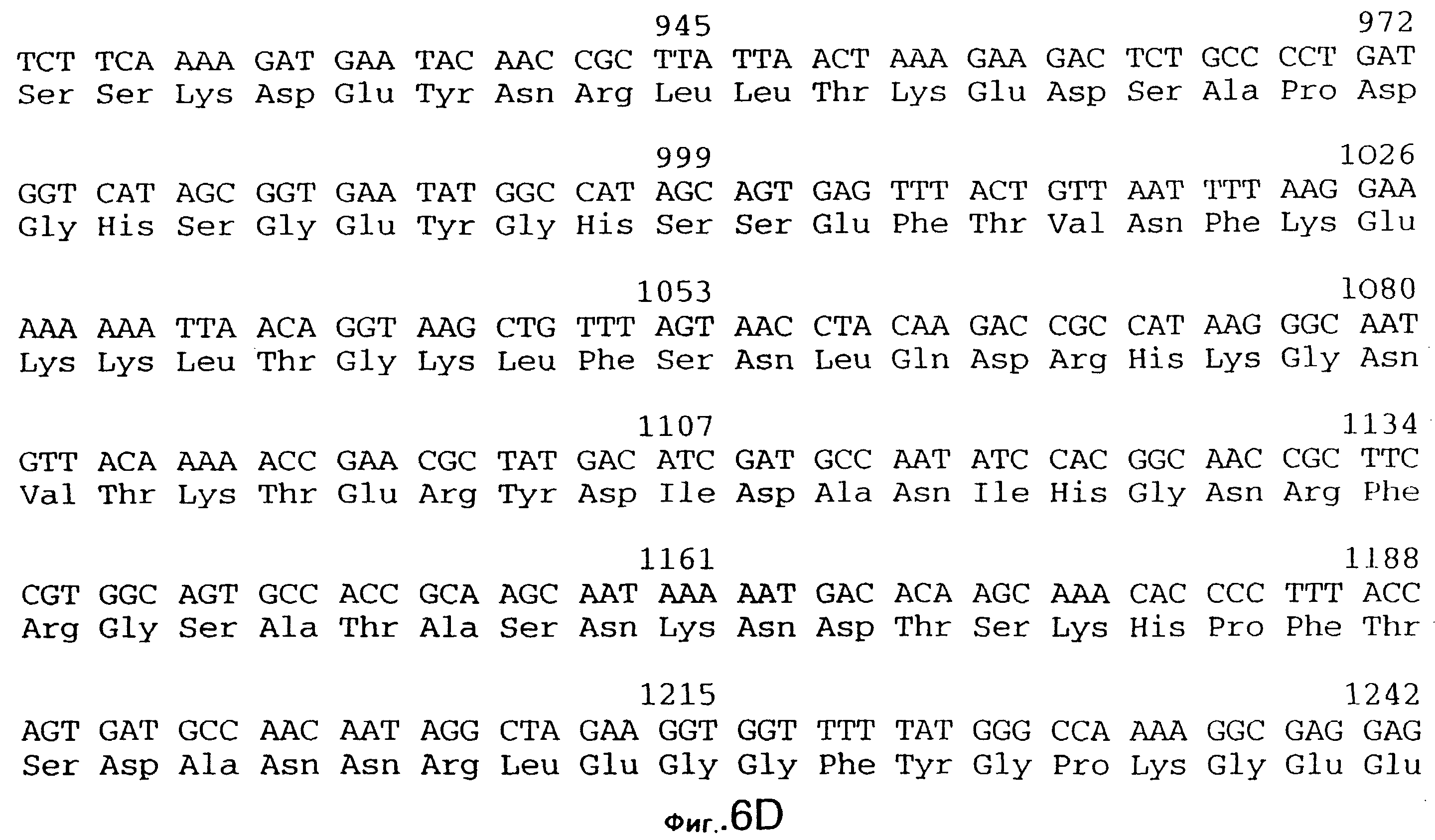
фиг.6 иллюстрирует нуклеотидную последовательность гена tbpB (SEQ ID No: 3 - полная последовательность, и SEQ ID No: 4 - кодирующая последовательность), и выведенную аминокислотную последовательность белка Tbp2 штамма 4223 (SEQ ID No: 11 -полноразмерная последовательность, и SEQ ID No: 12 - последовательность зрелого белка). Лидерная последовательность (SEQ ID No: 20) подчеркнута;
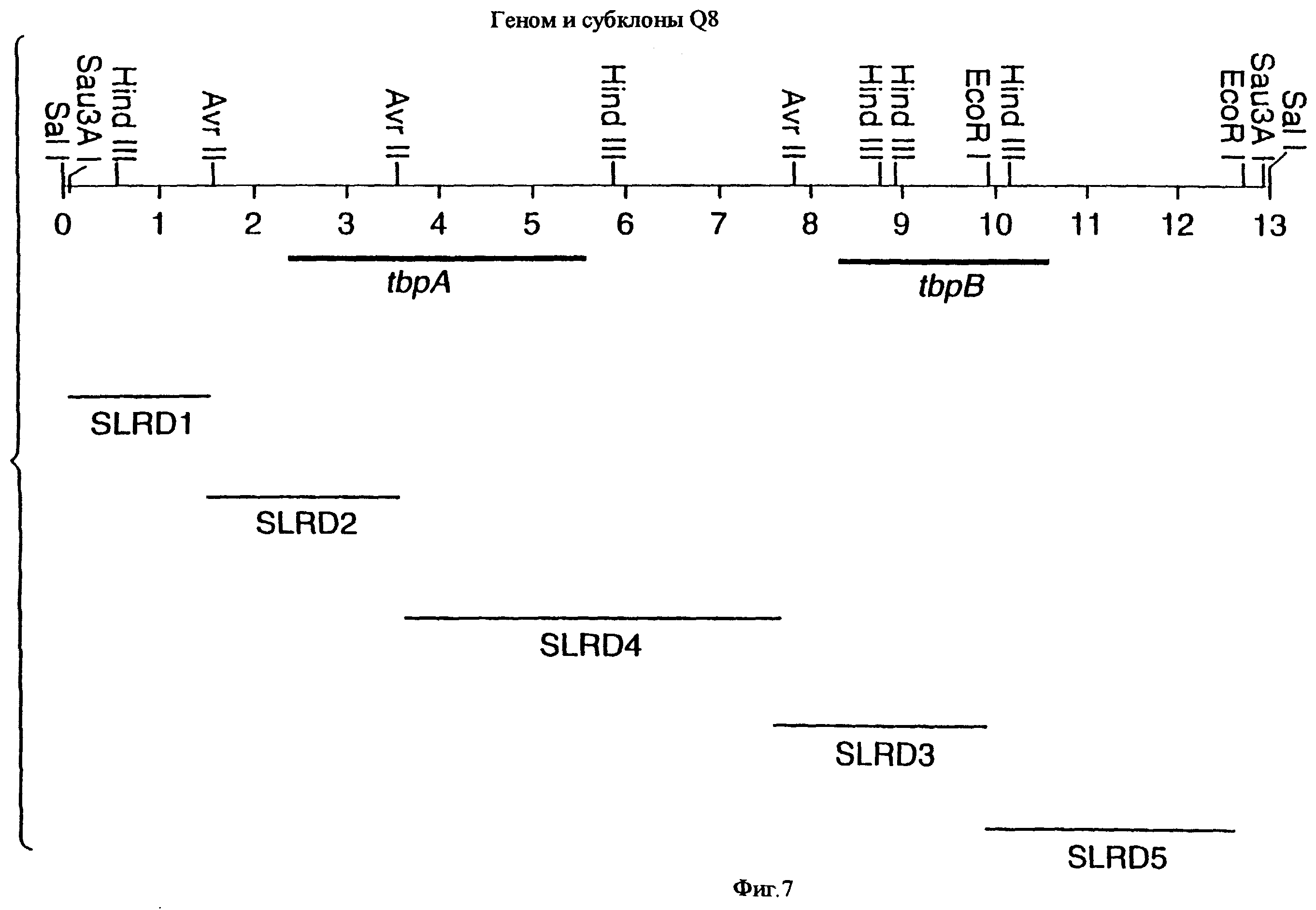
фиг.7 иллюстрирует рестрикционную карту клона SLRD-A, содержащего гены tbpA и tbpB штамма Q8 М.catarrhalis;
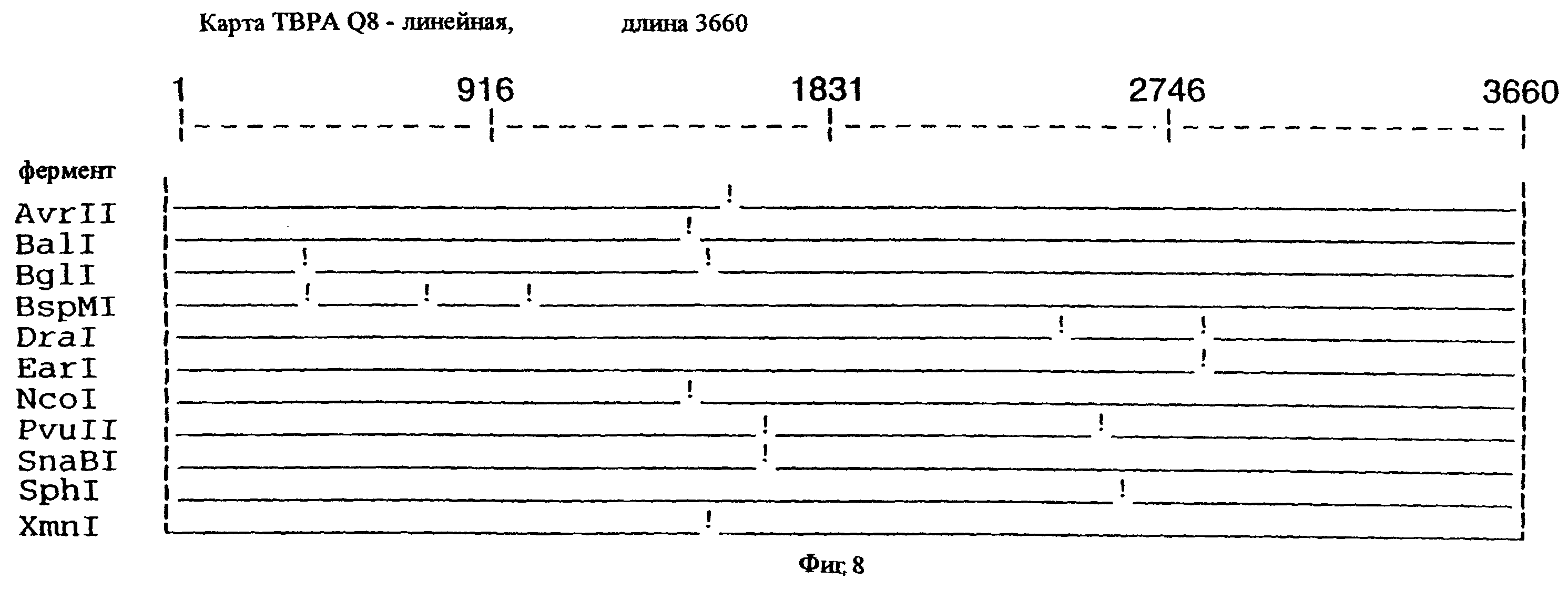
фиг.8 иллюстрирует рестрикционную карту гена tbpA для штамма Q8 М.catarrhalis;
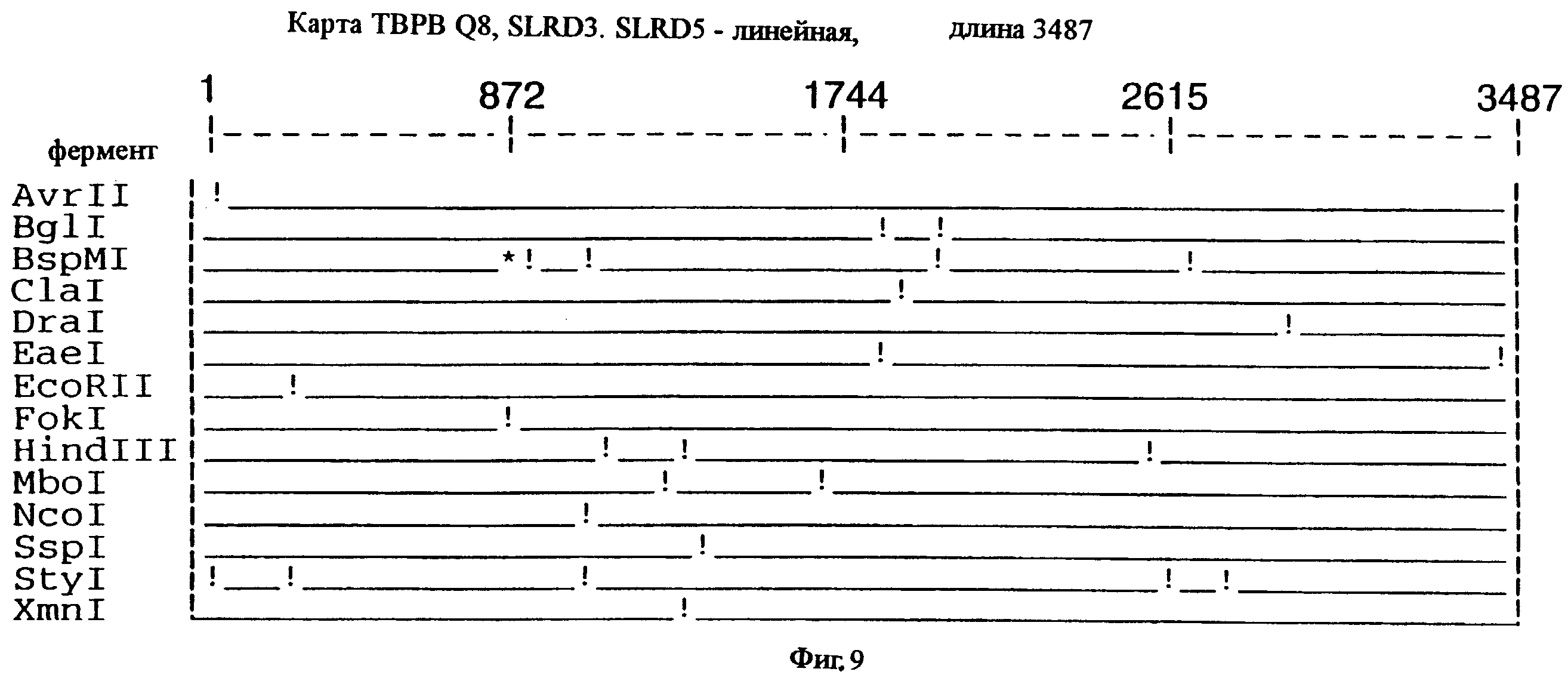
фиг.9 иллюстрирует рестрикционную карту гена tbpB для штамма Q8 М.catarrhalis;
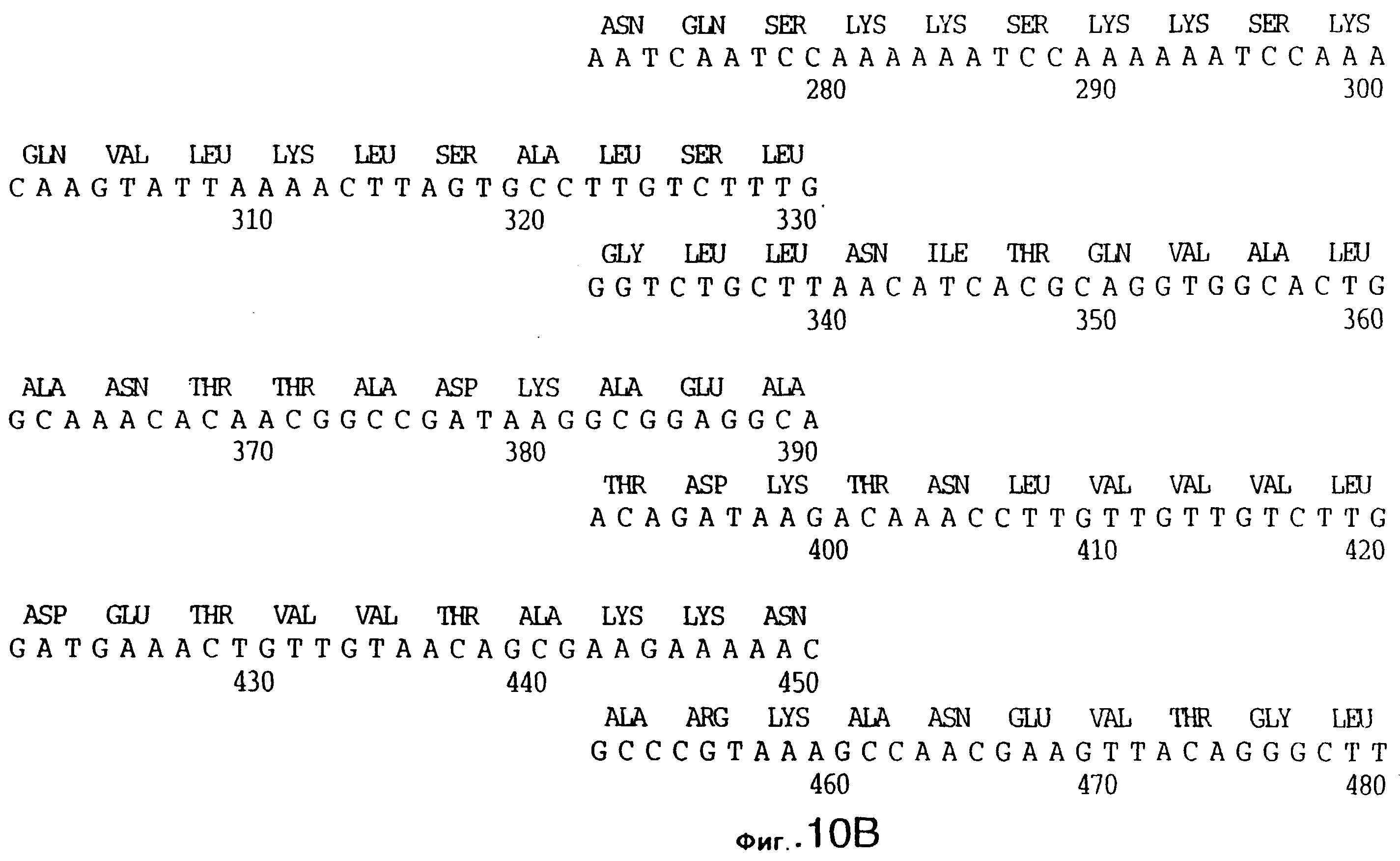
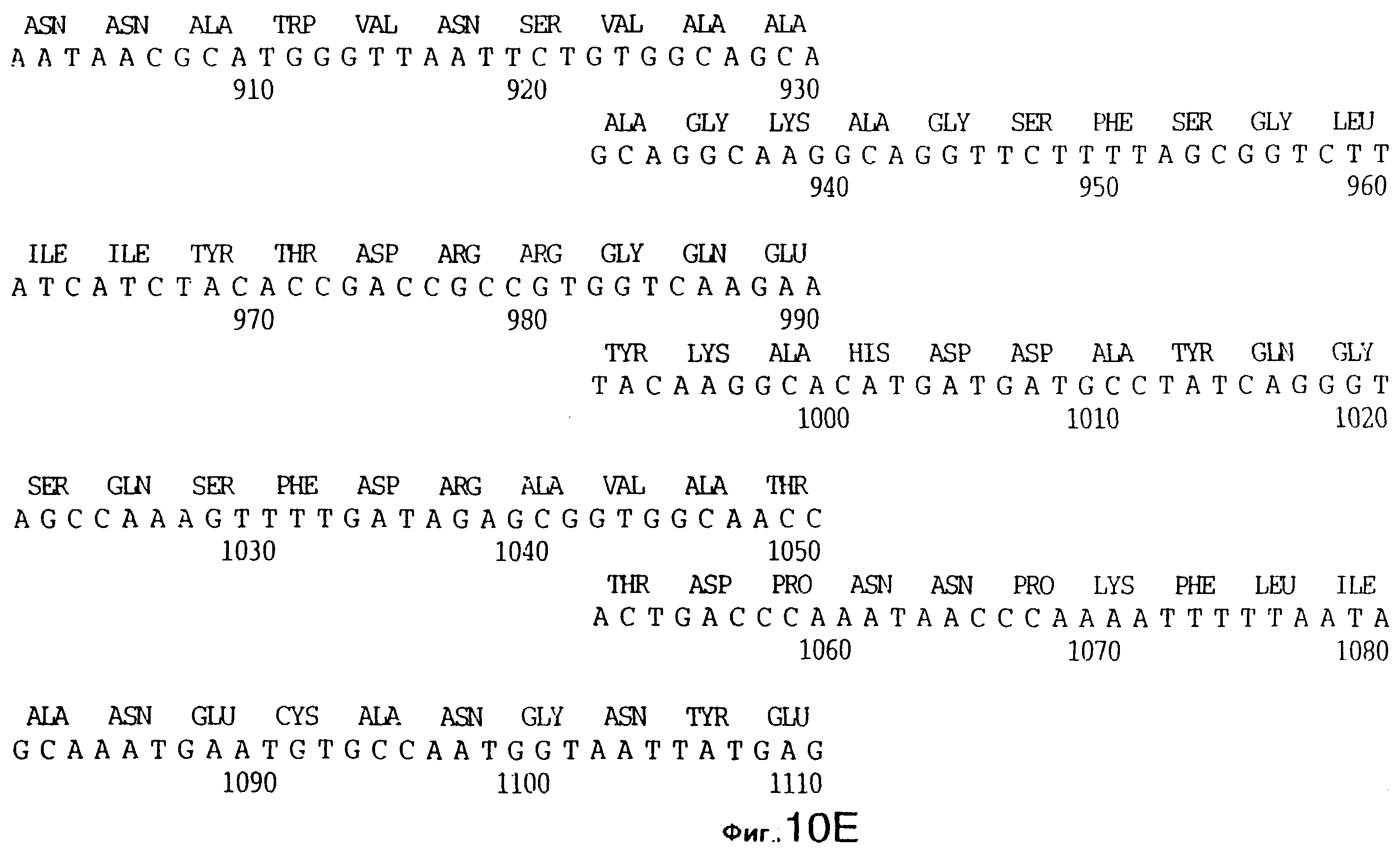
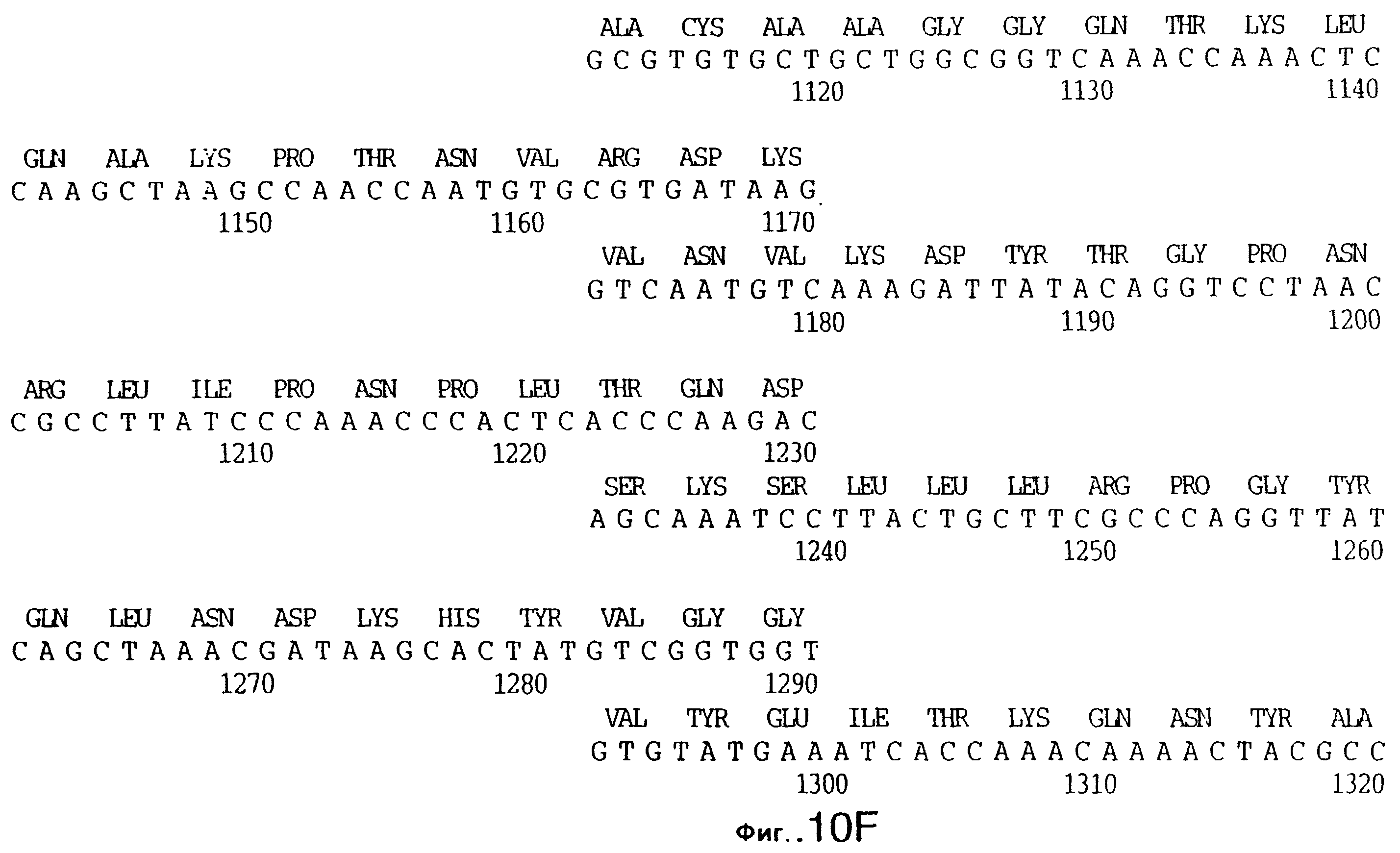
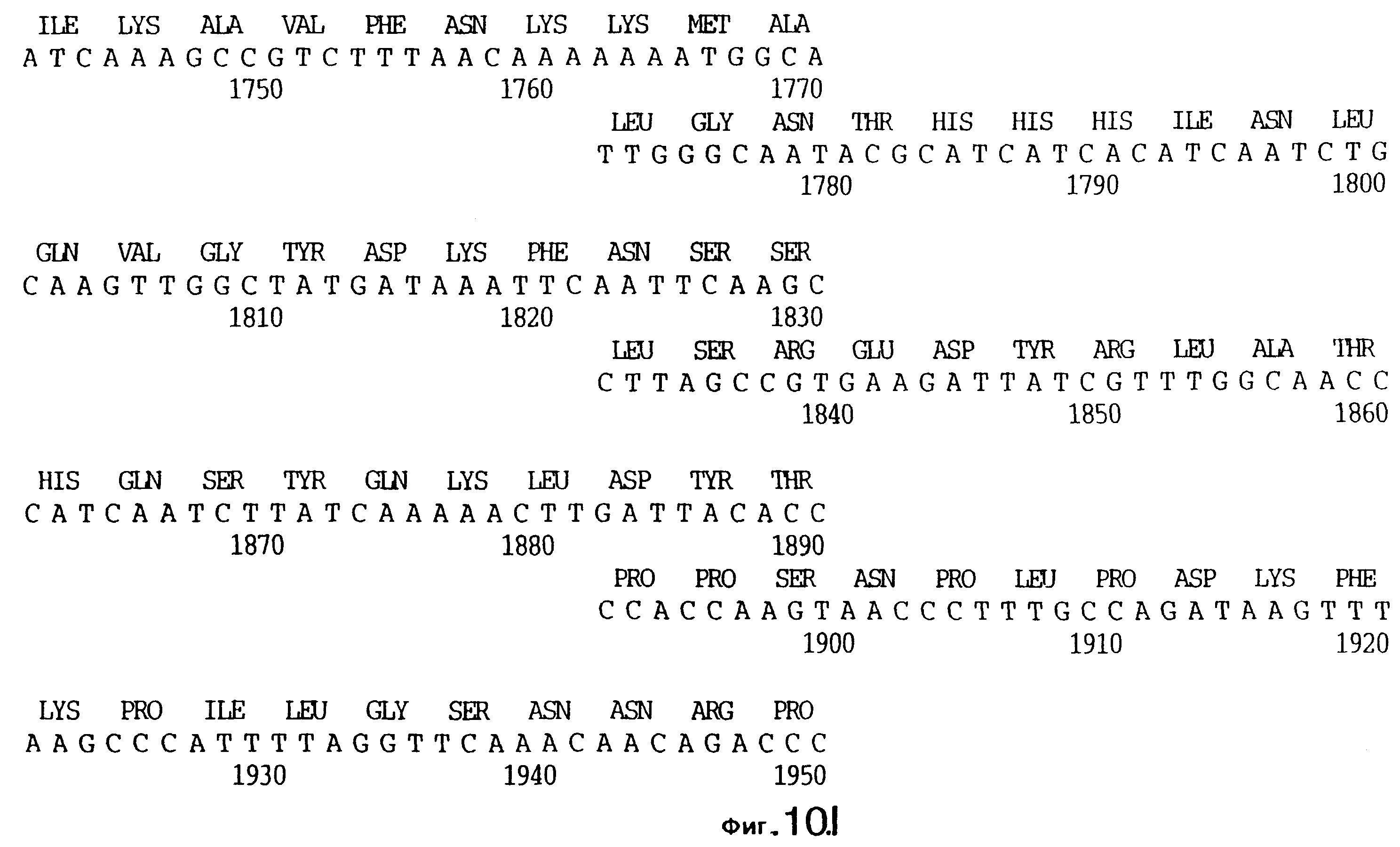
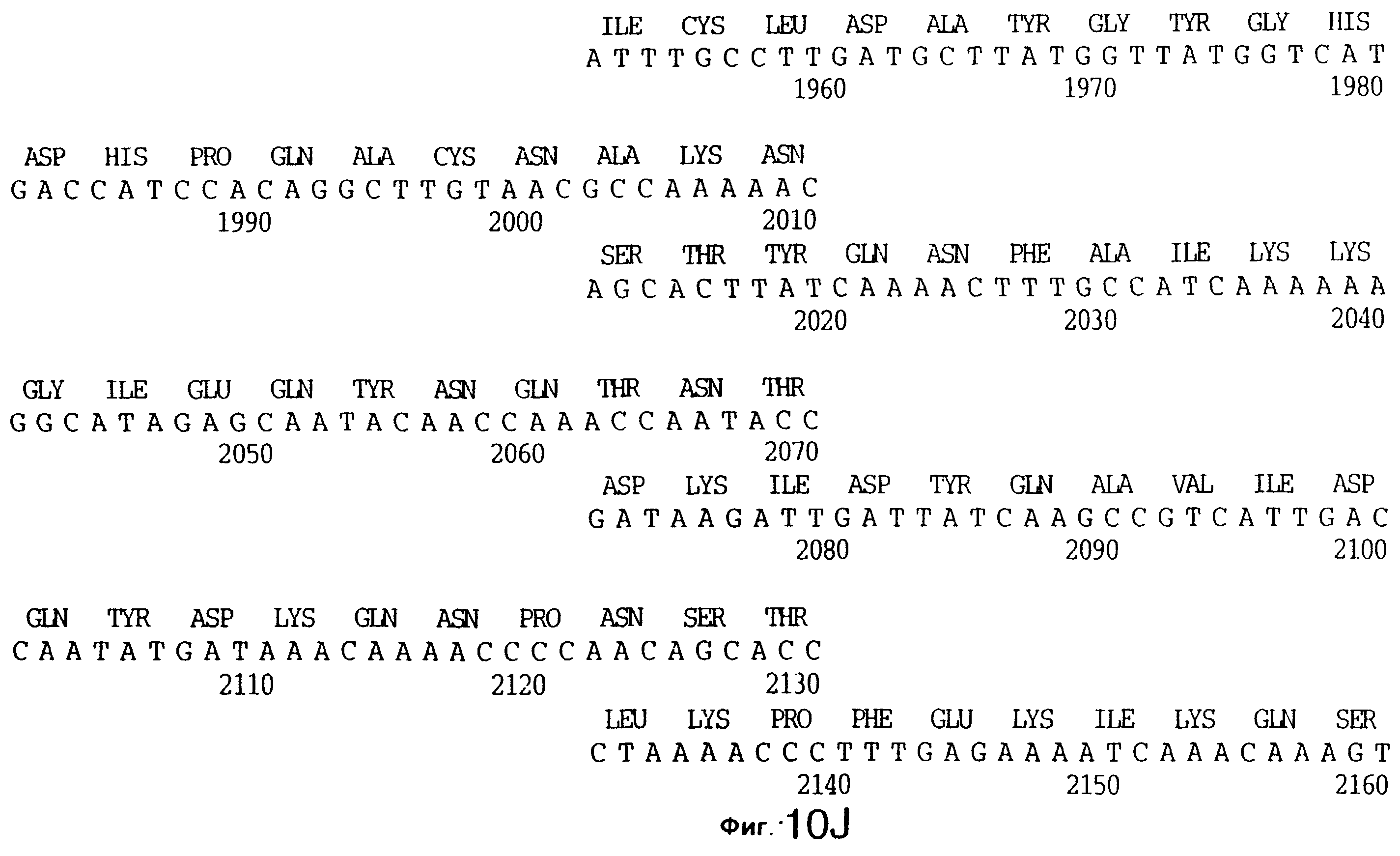
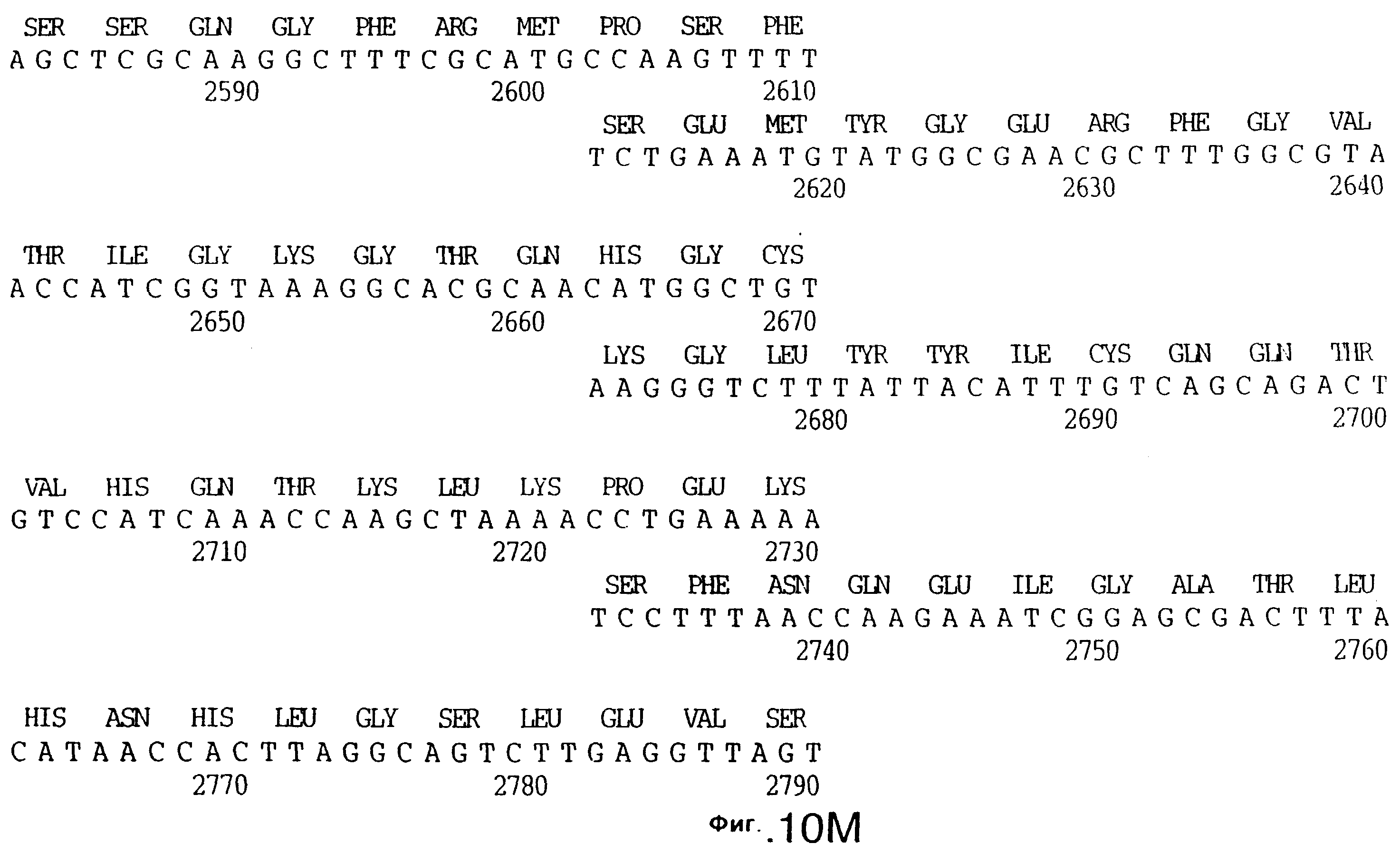
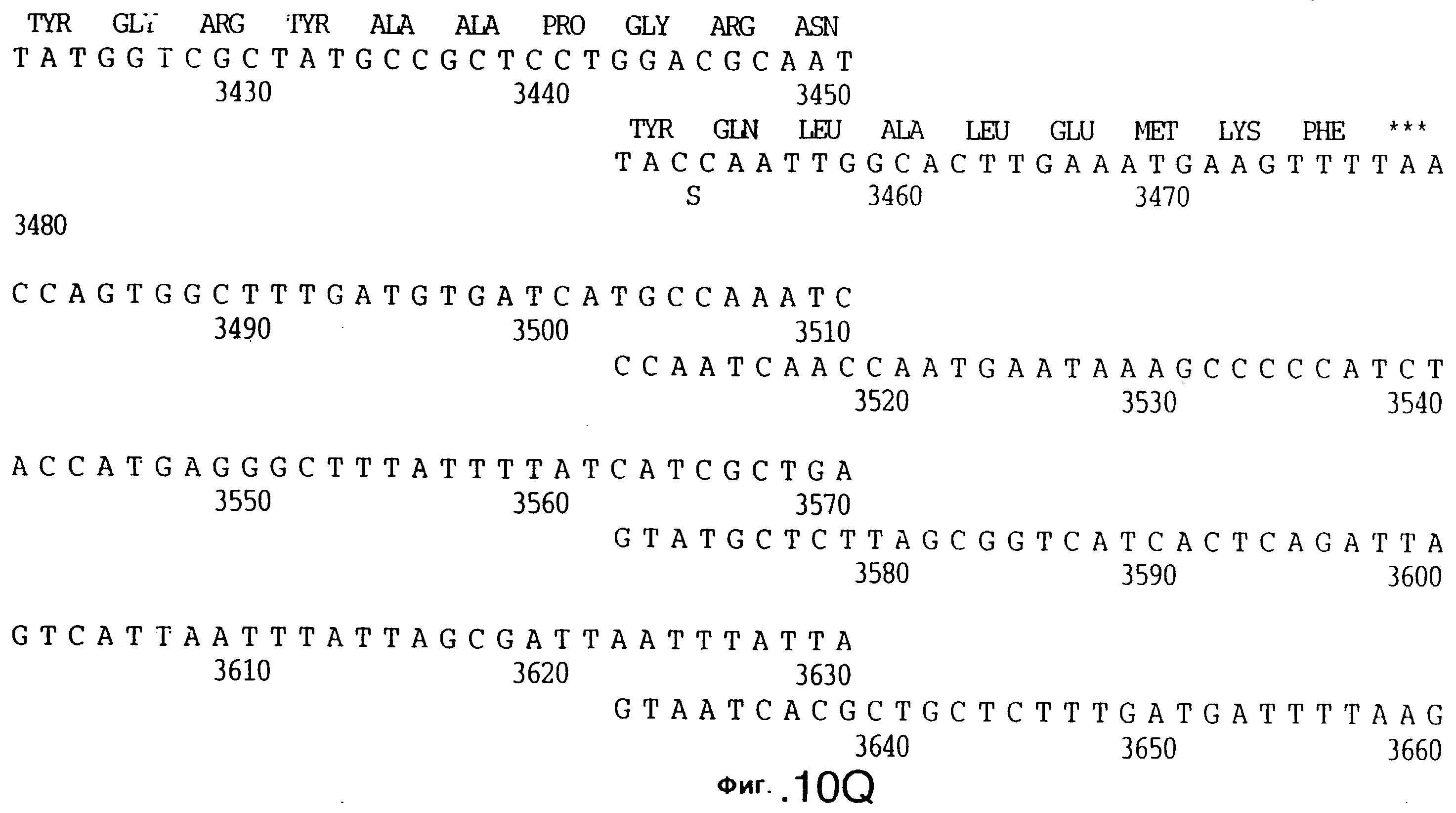
фиг.10 иллюстрирует нуклеотидную последовательность гена tbpA (SEQ ID No: 5 - полная последовательность, и SEQ ID No: 6 - кодирующая последовательность), и выведенную аминокислотную последовательность белка Tbp1 штамма Q8 (SEQ ID No: 13 - полноразмерная последовательность, и SEQ ID No: 14 - последовательность зрелого белка);
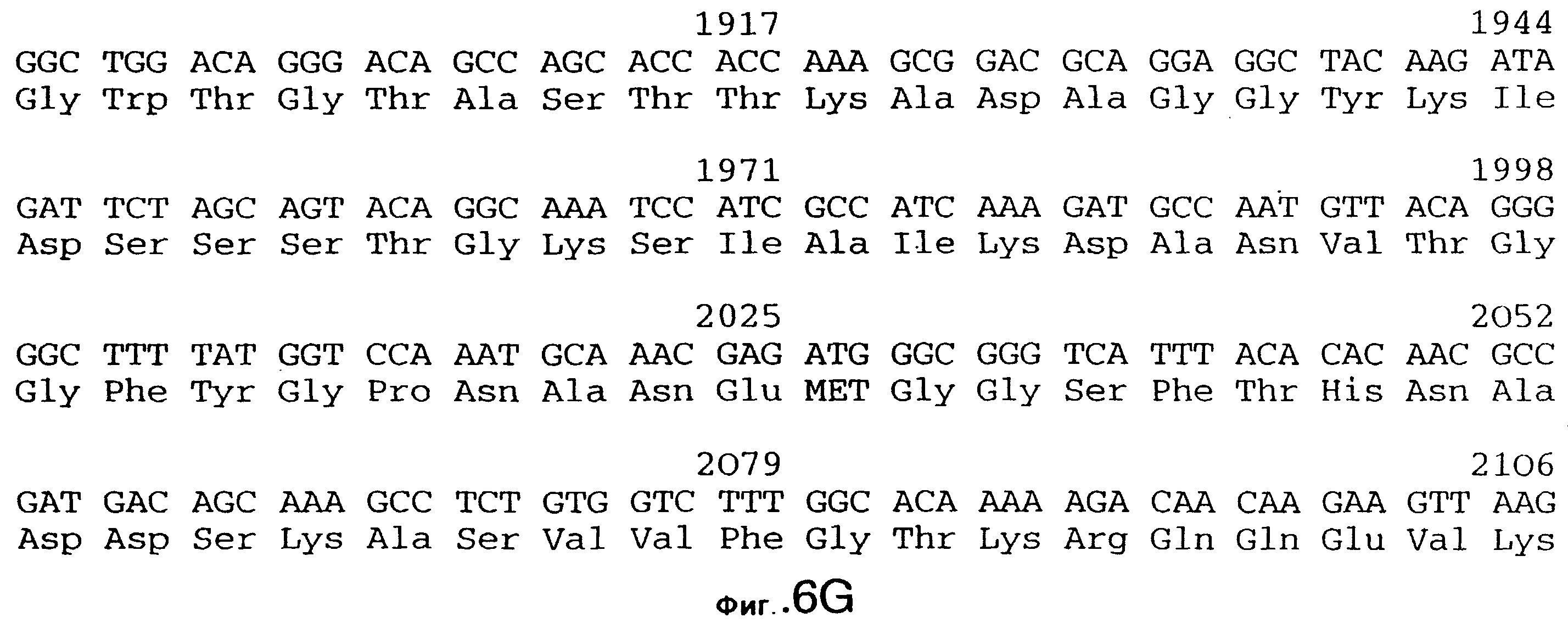
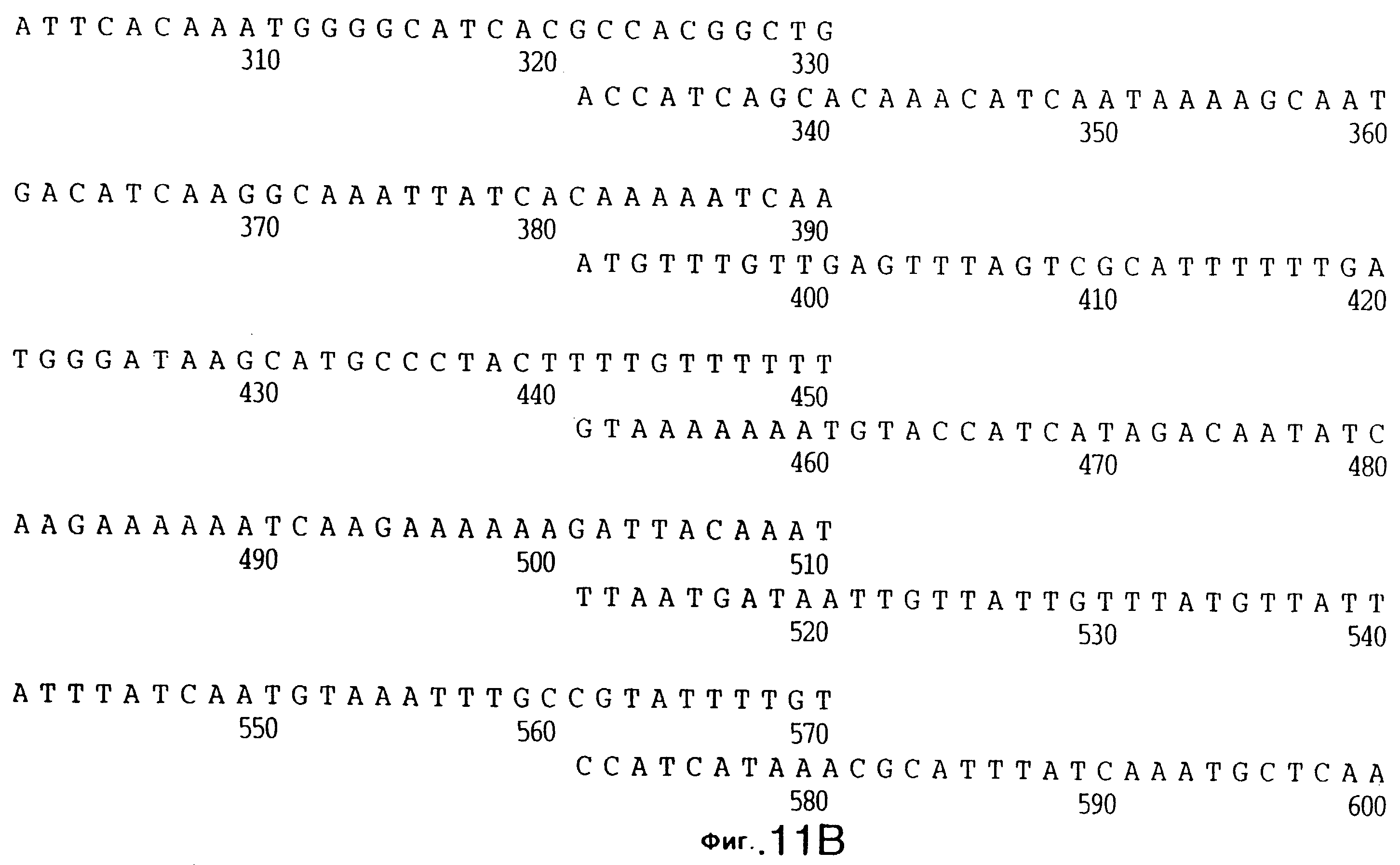
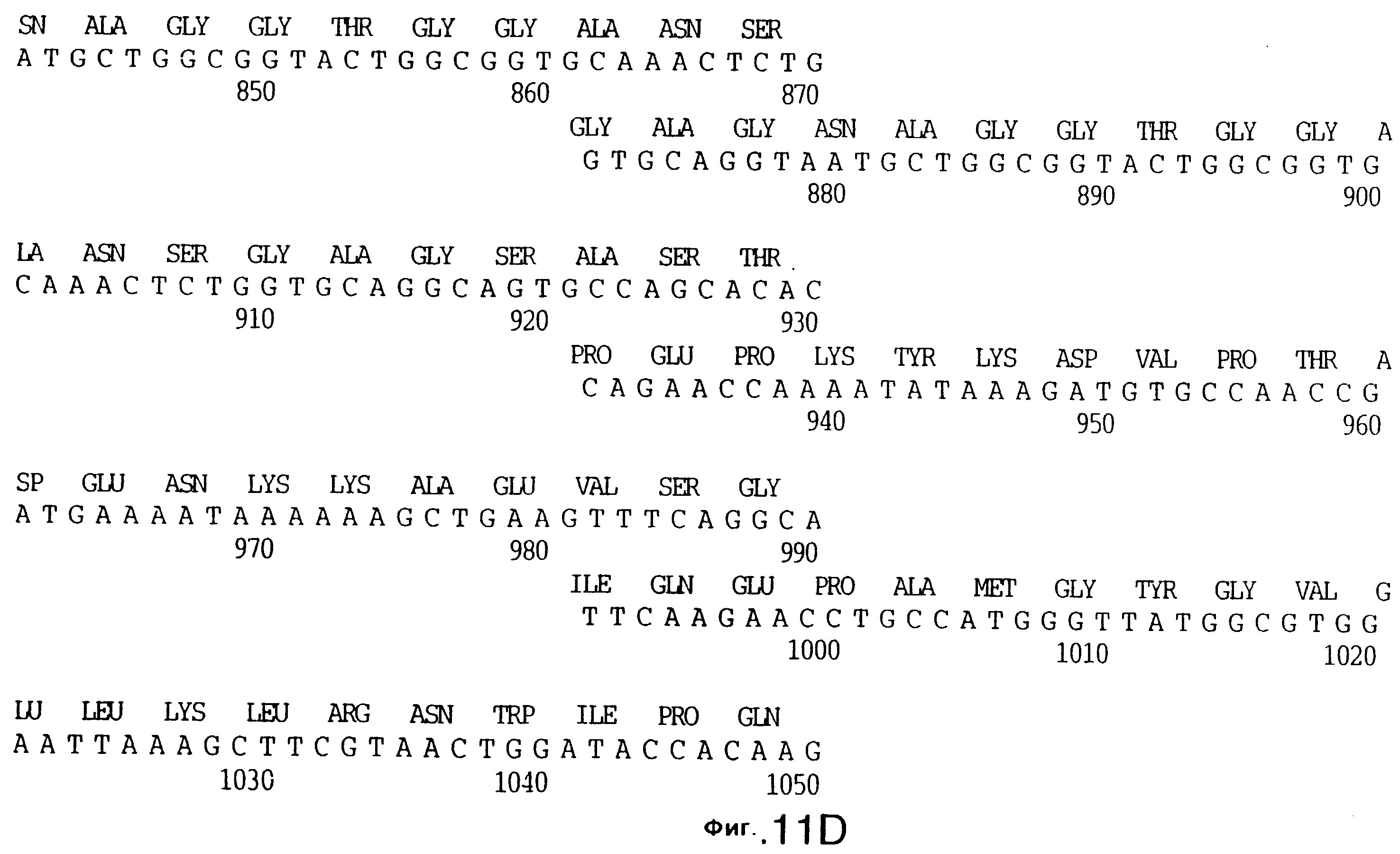
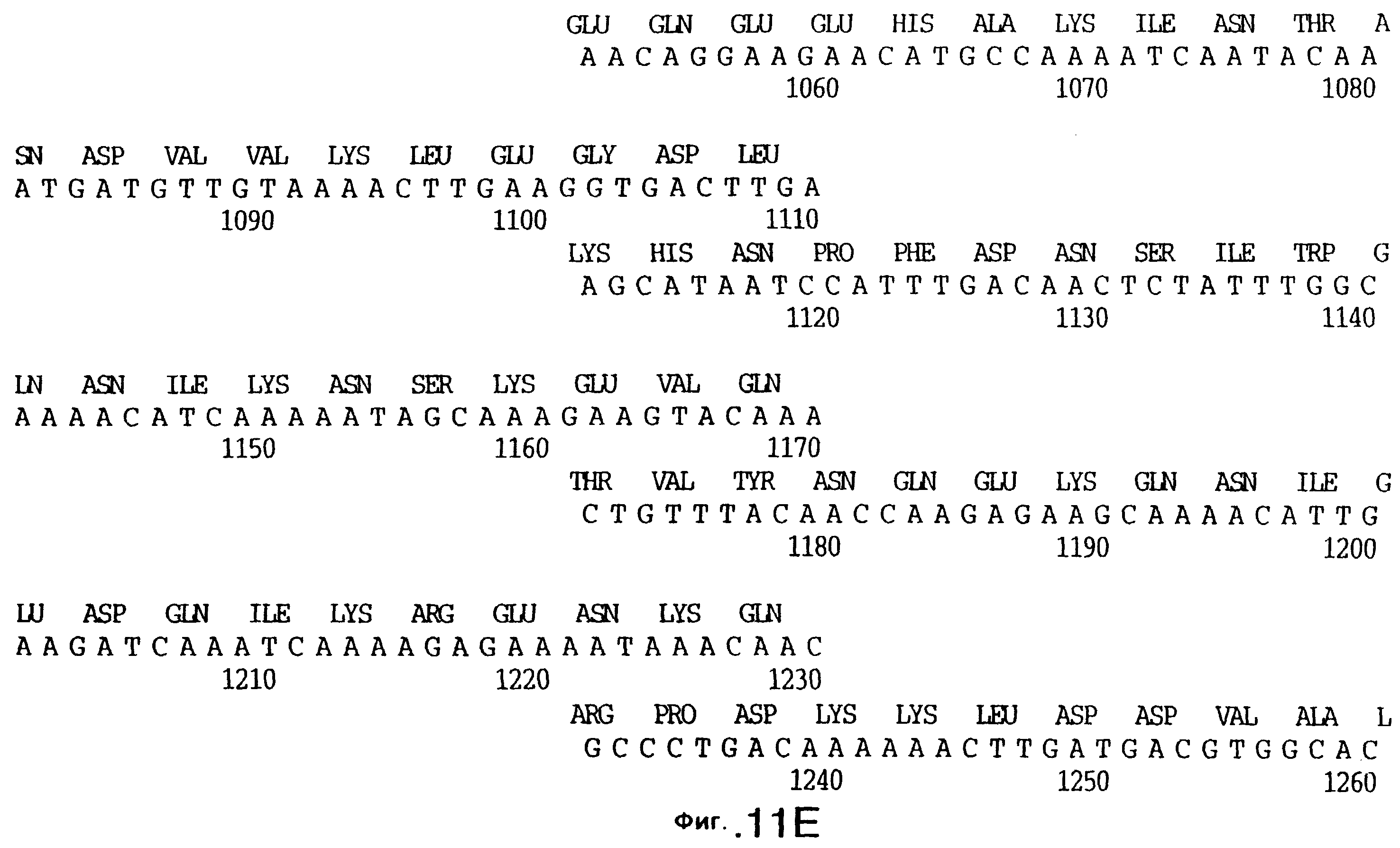
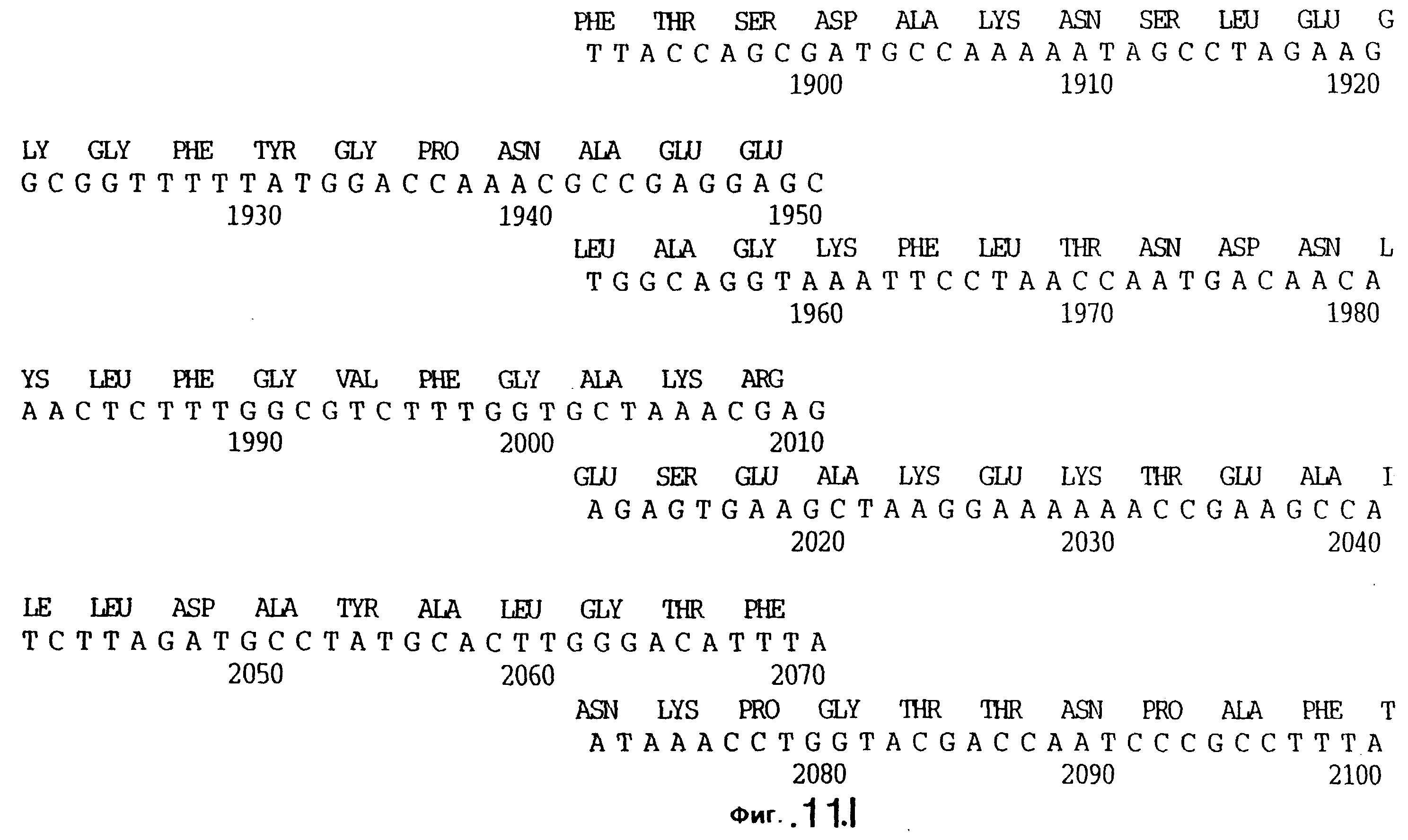
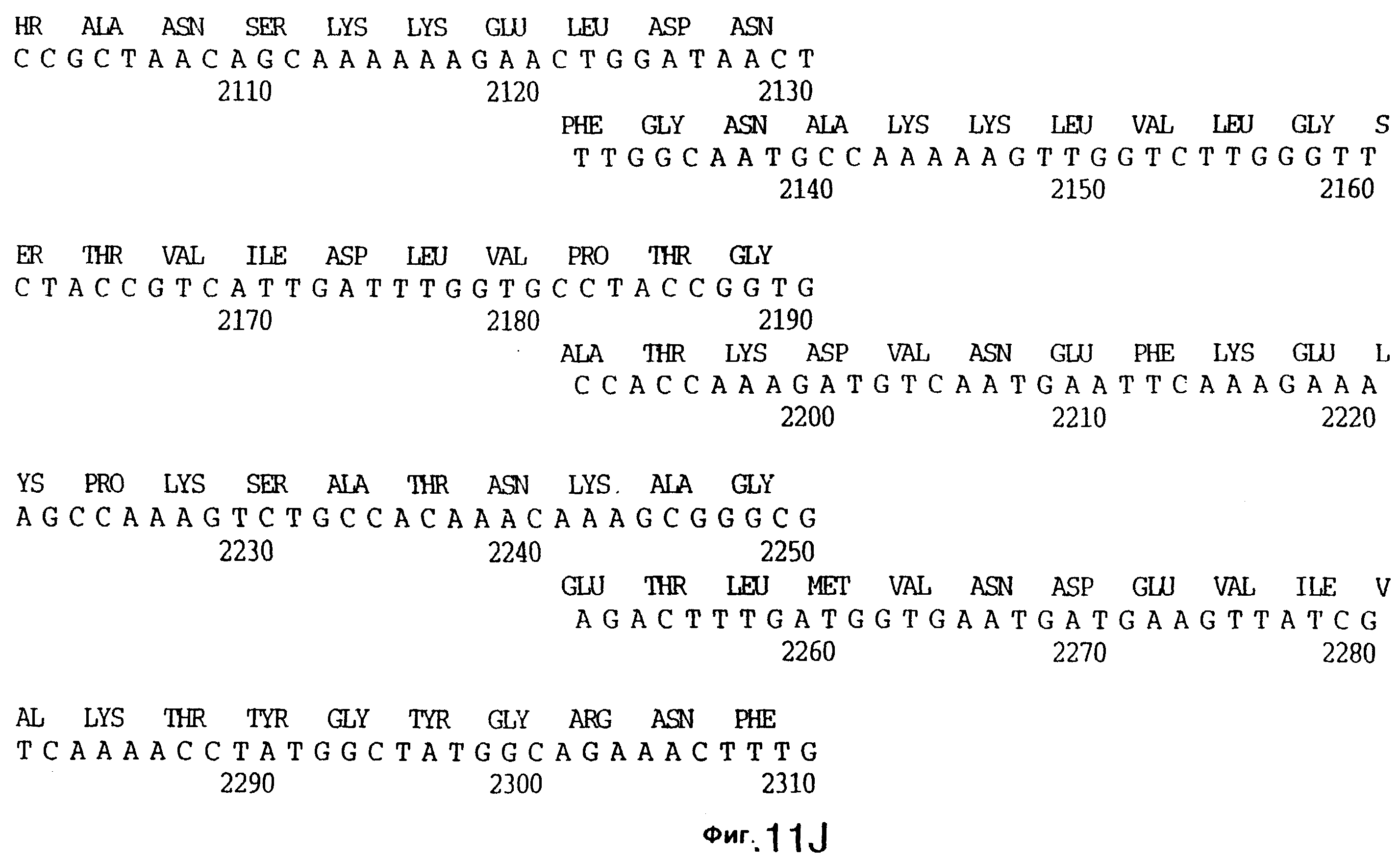
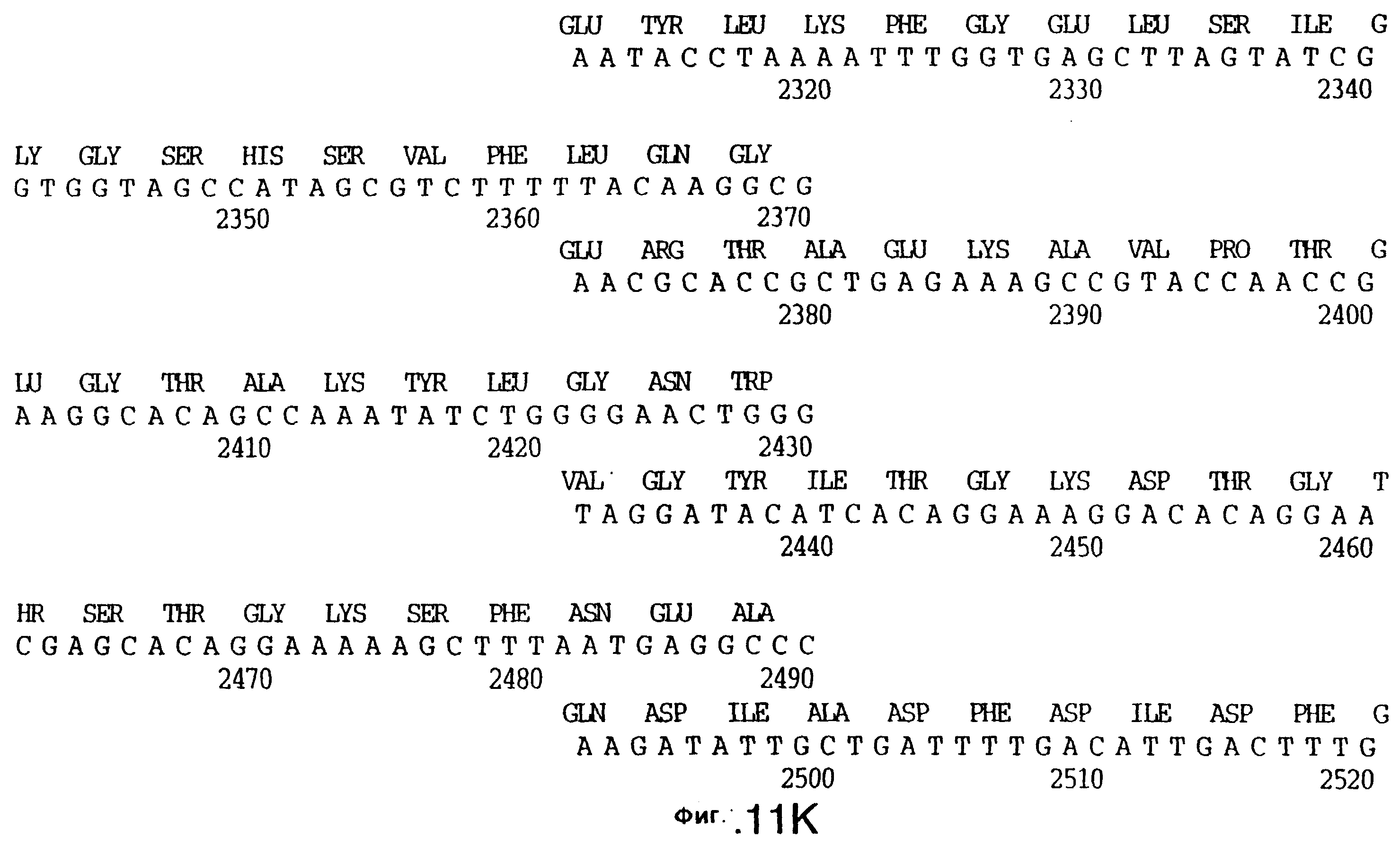
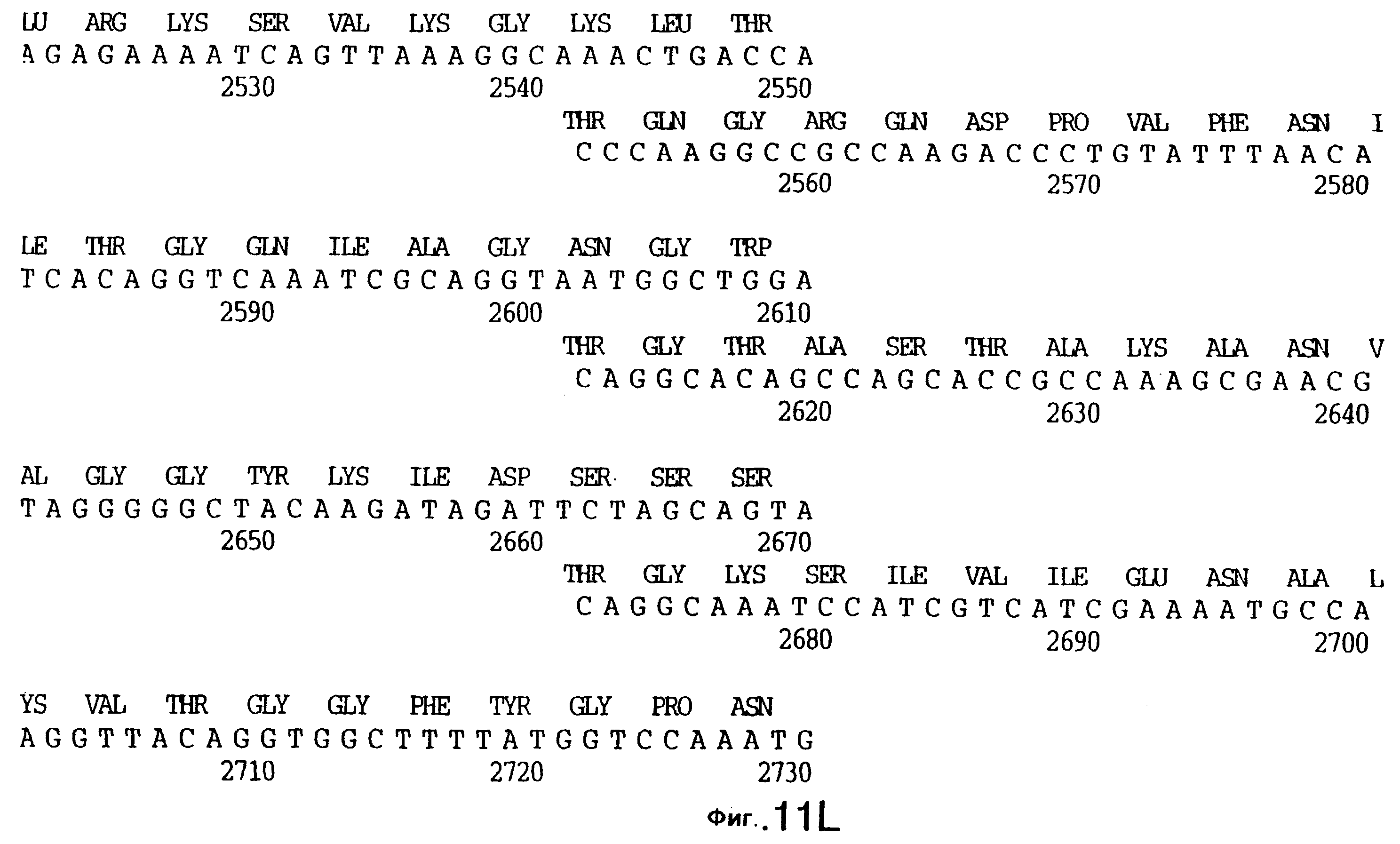
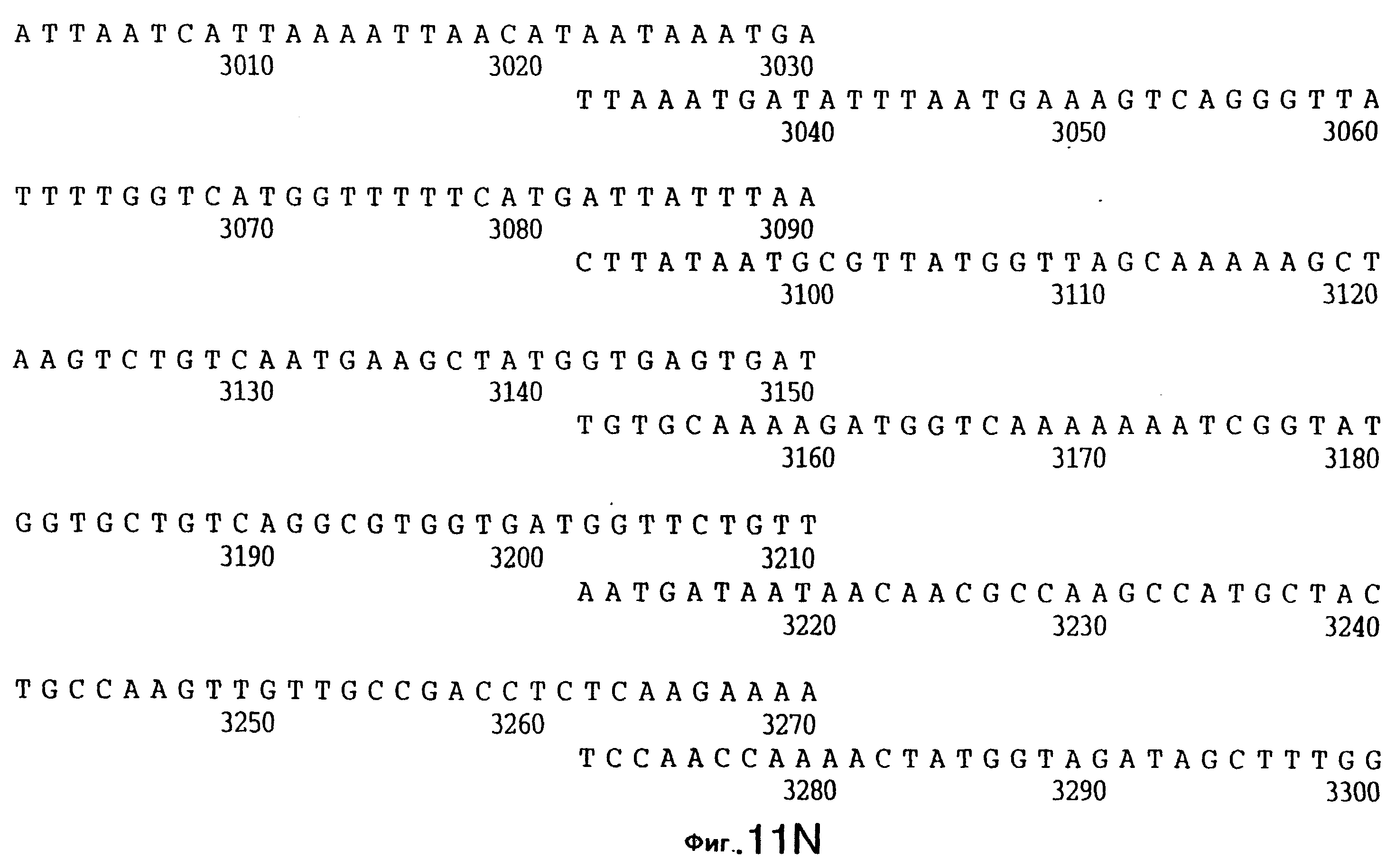
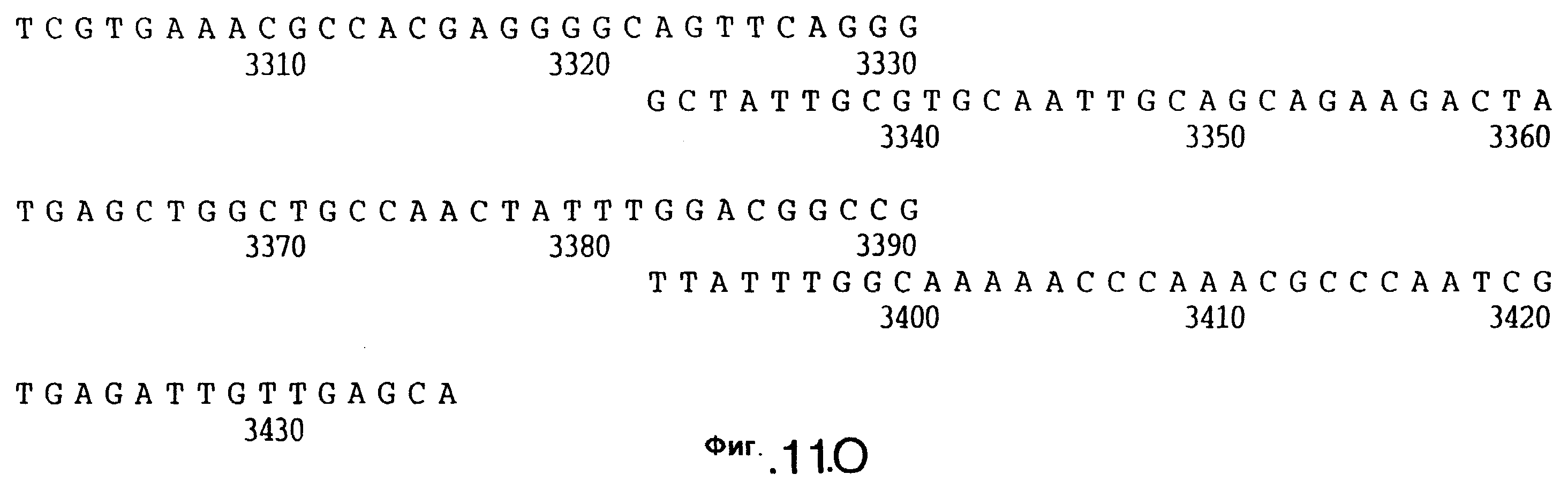
фиг.11 иллюстрирует нуклеотидную последовательность гена tbpB (SEQ ID No: 7 - полная последовательность, и SEQ ID No: 8 - кодирующая последовательность), и выведенную аминокислотную последовательность белка Tbp2 штамма Q8 (SEQ ID No: 15 - полноразмерная последовательность, и SEQ ID No: 16 - последовательность зрелого белка);
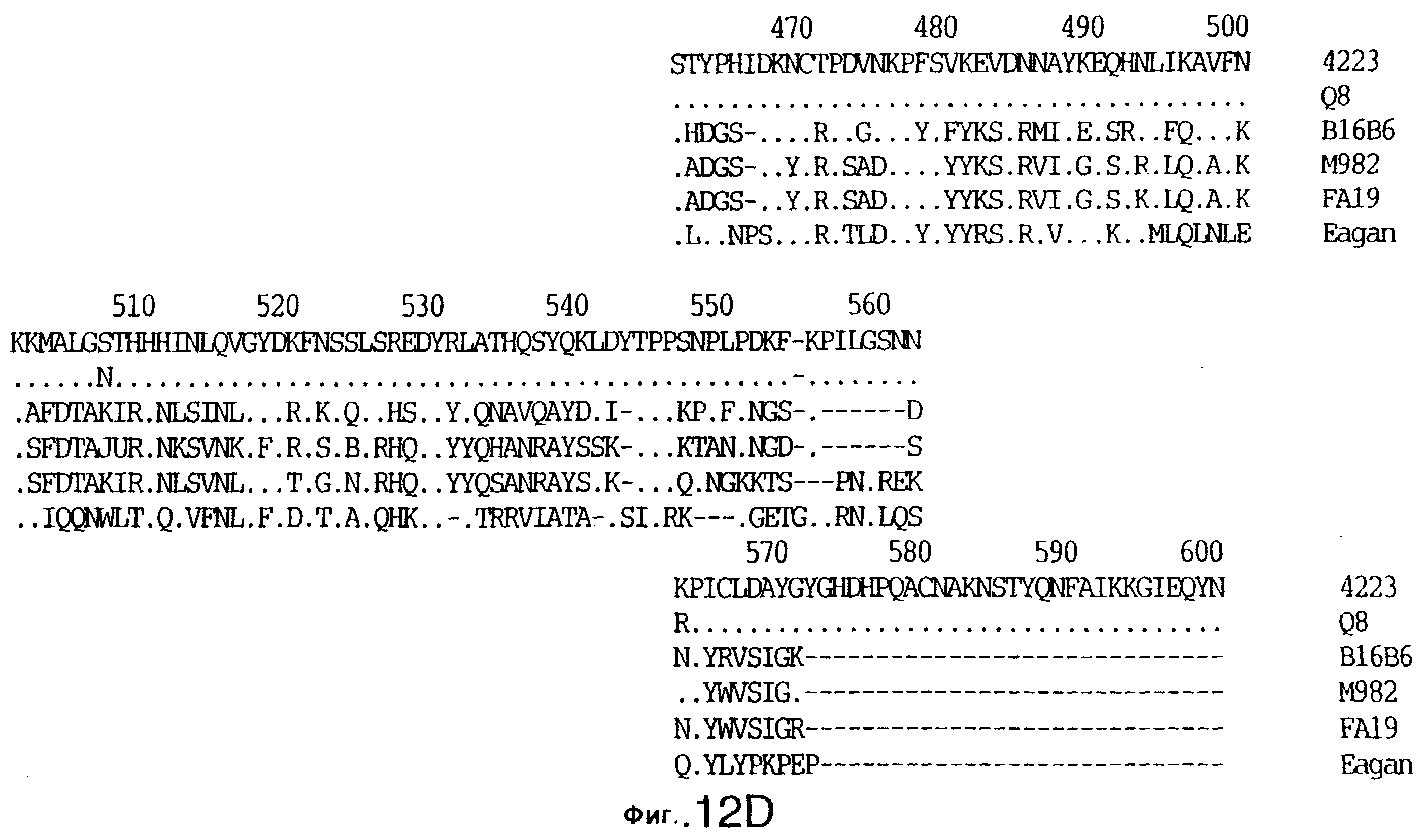
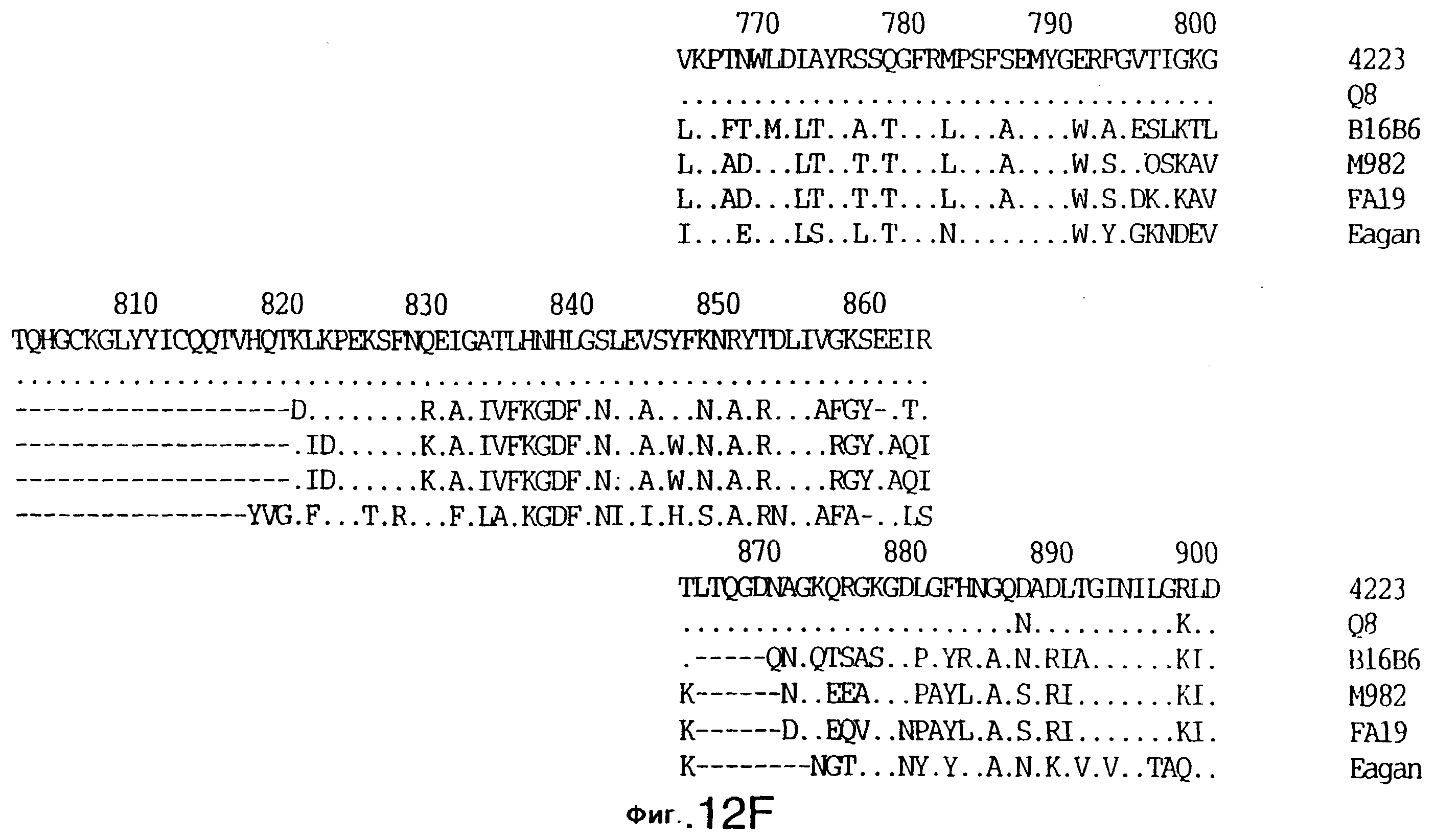
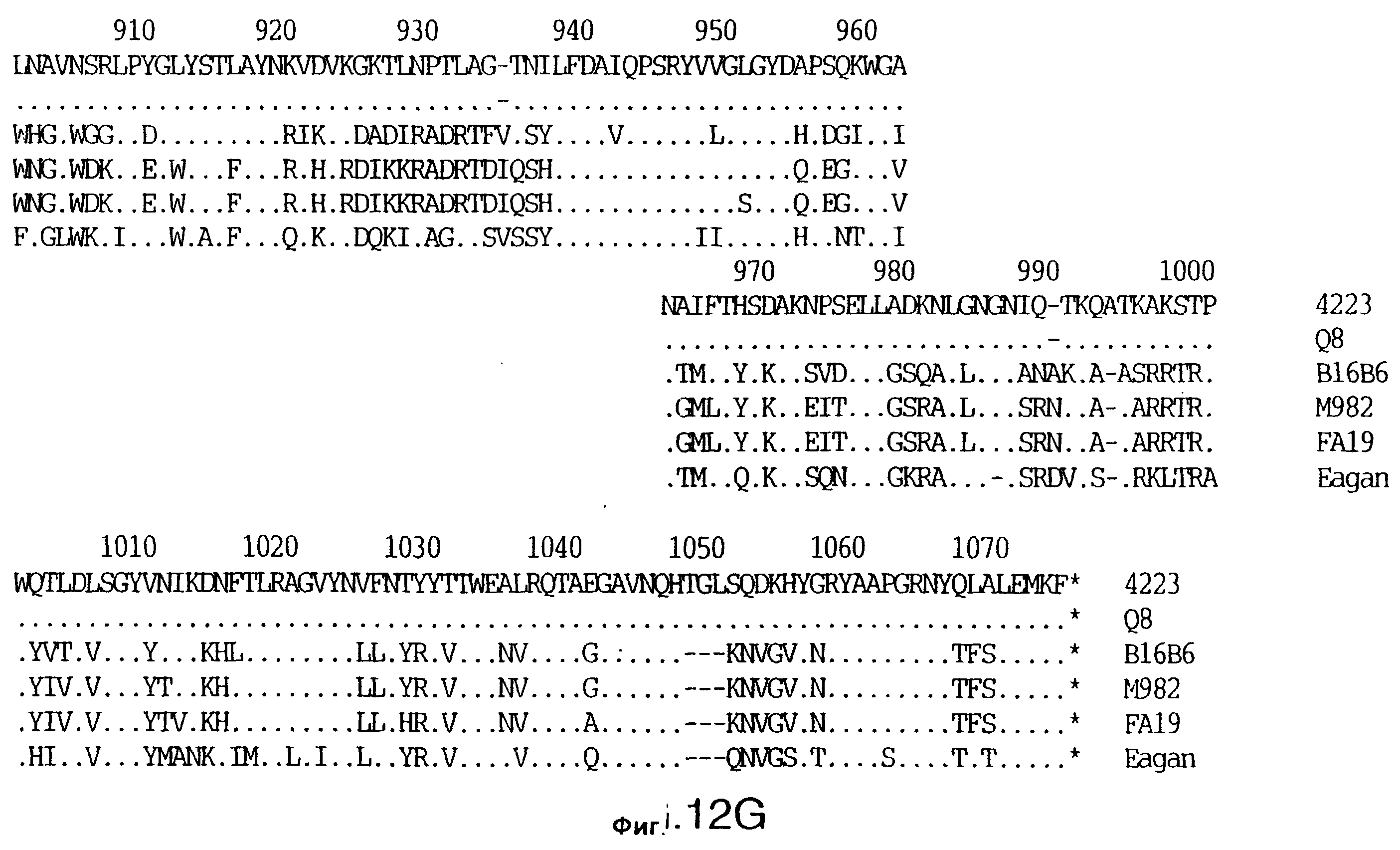
фиг.12 иллюстрирует сравнение аминокислотных последовательностей Tbp1 штамма 4223 М. catarrhalis (SEQ ID No: 9) и Q8 М.catarrhalis (SEQ ID No: 13), штамма Eagan H.influenzae (SEQ ID No: 21), штаммов В16В6 (SEQ ID No: 22) и М982 (SEQ ID No: 23) N.meningitidis, и штамма FA19 N.gonorrhoeae (SEQ ID No: 24). Точки указывают на идентичные остатки, а черточки были введены для максимального соответствия сравниваемых первичных последовательностей;
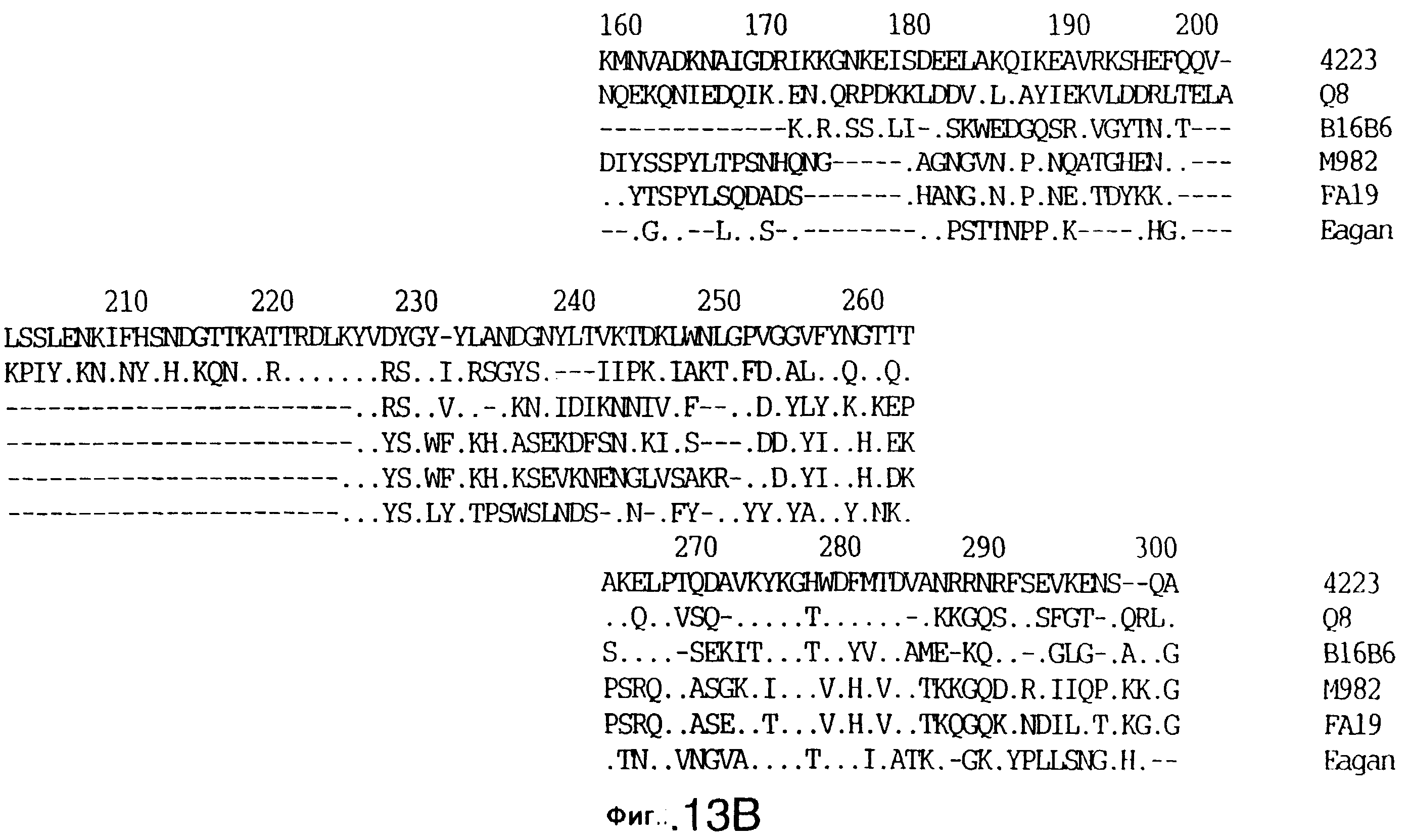
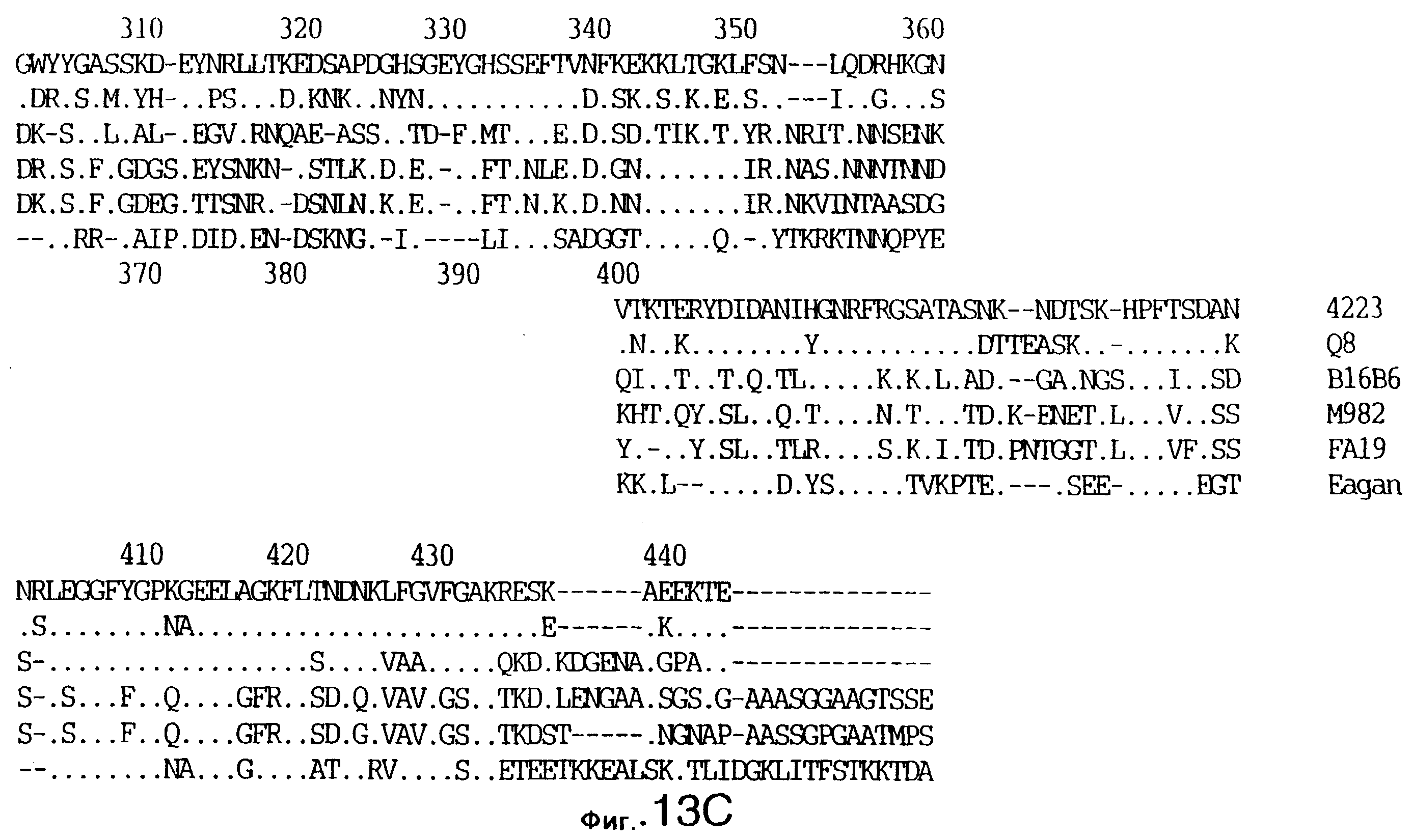
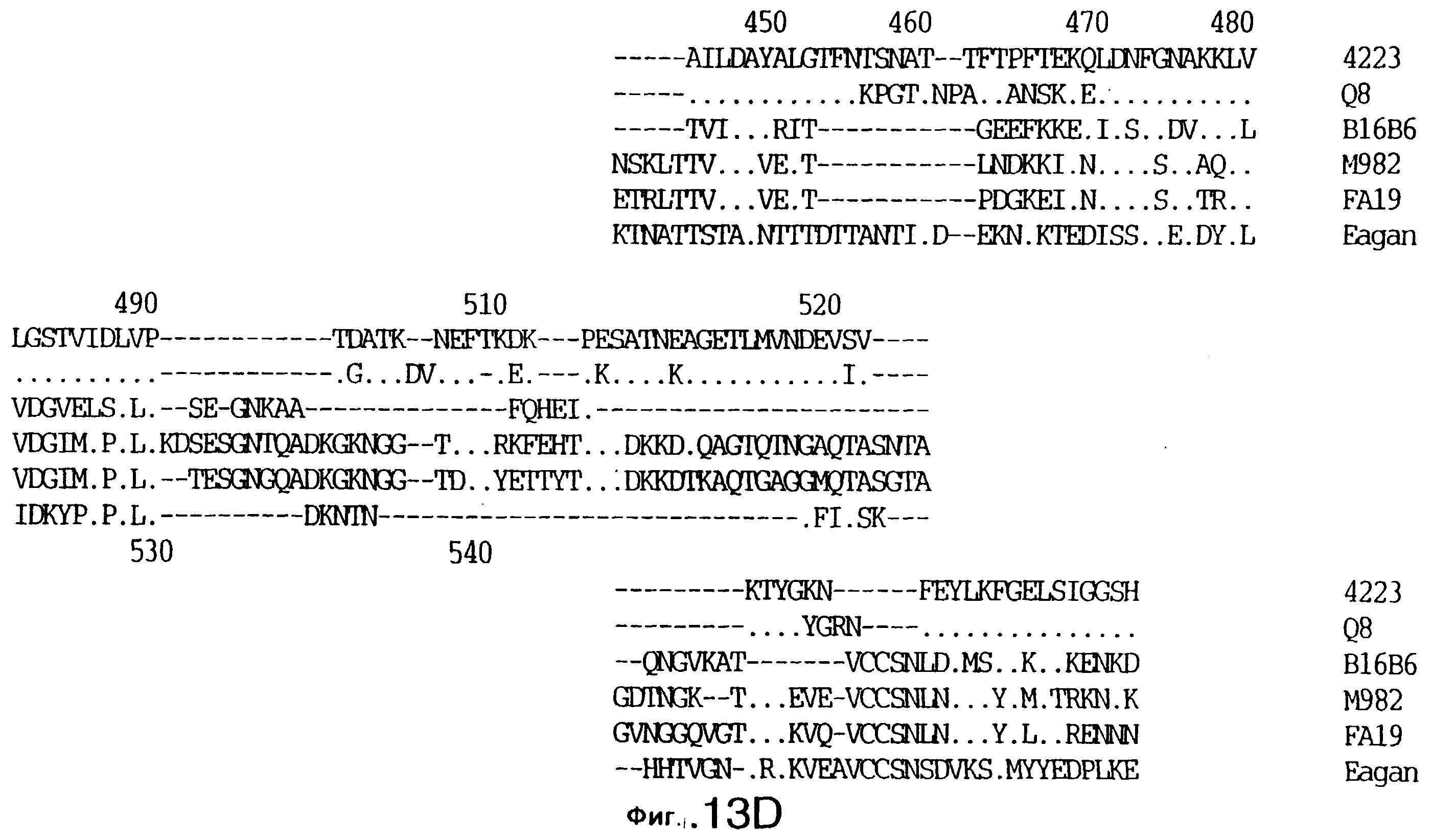
фиг.13 иллюстрирует сравнение аминокислотных последовательностей Тbр2 штамма 4223 М.catarrhalis (SEQ ID No: 11) и Q8 M.catarrhalis (SEQ ID No: 15), штамма Eagan H.influenzae (SEQ ID No: 25), штаммов В16В6 (SEQ ID No: 26) и М982 N.meningitidis (SEQ ID No: 27), и штамма FA19 N.gonorrhoeae (SEQ ID No: 28). Точки указывают на идентичные остатки, а черточки были введены для максимального соответствия сравниваемых первичных последовательностей;
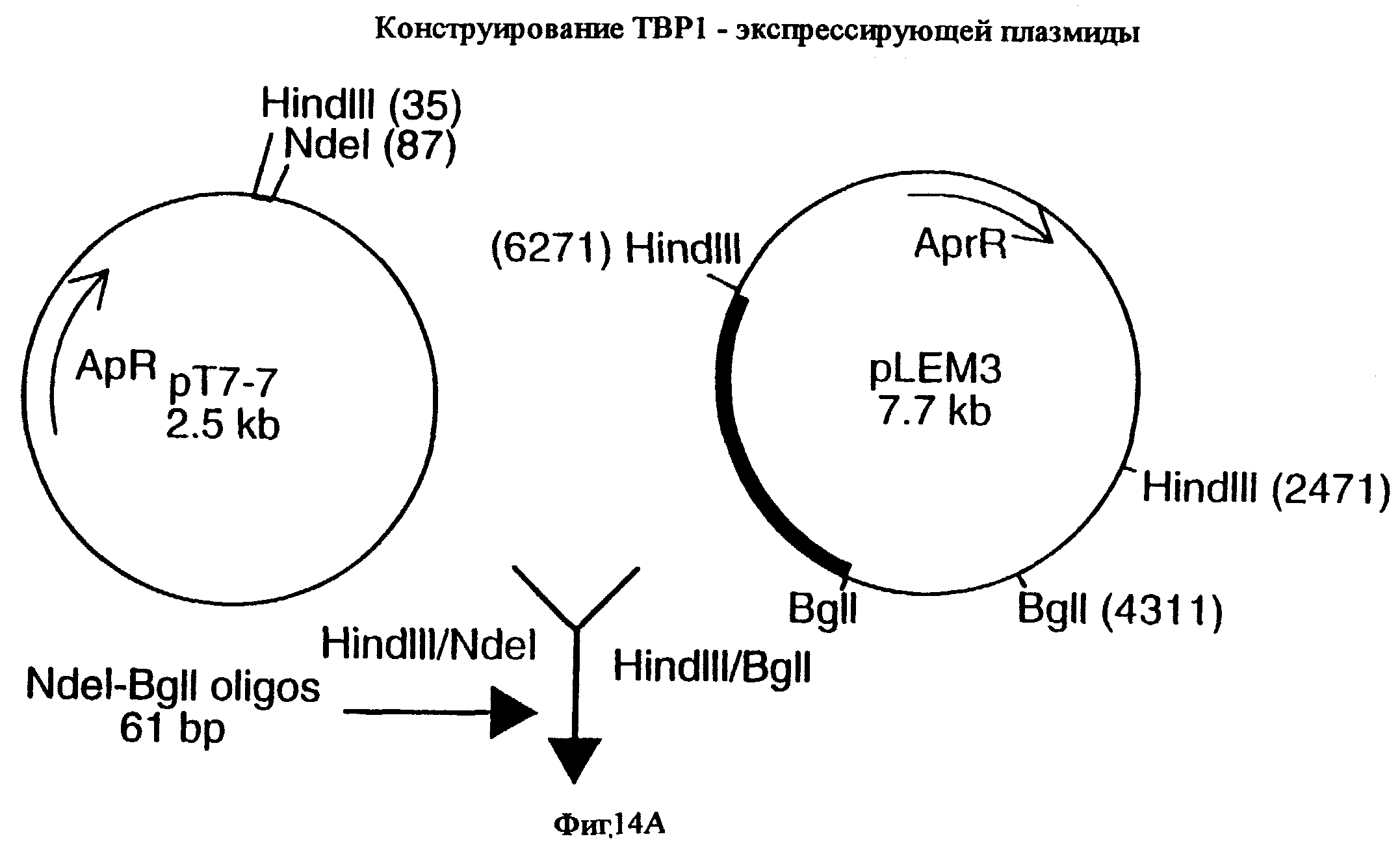
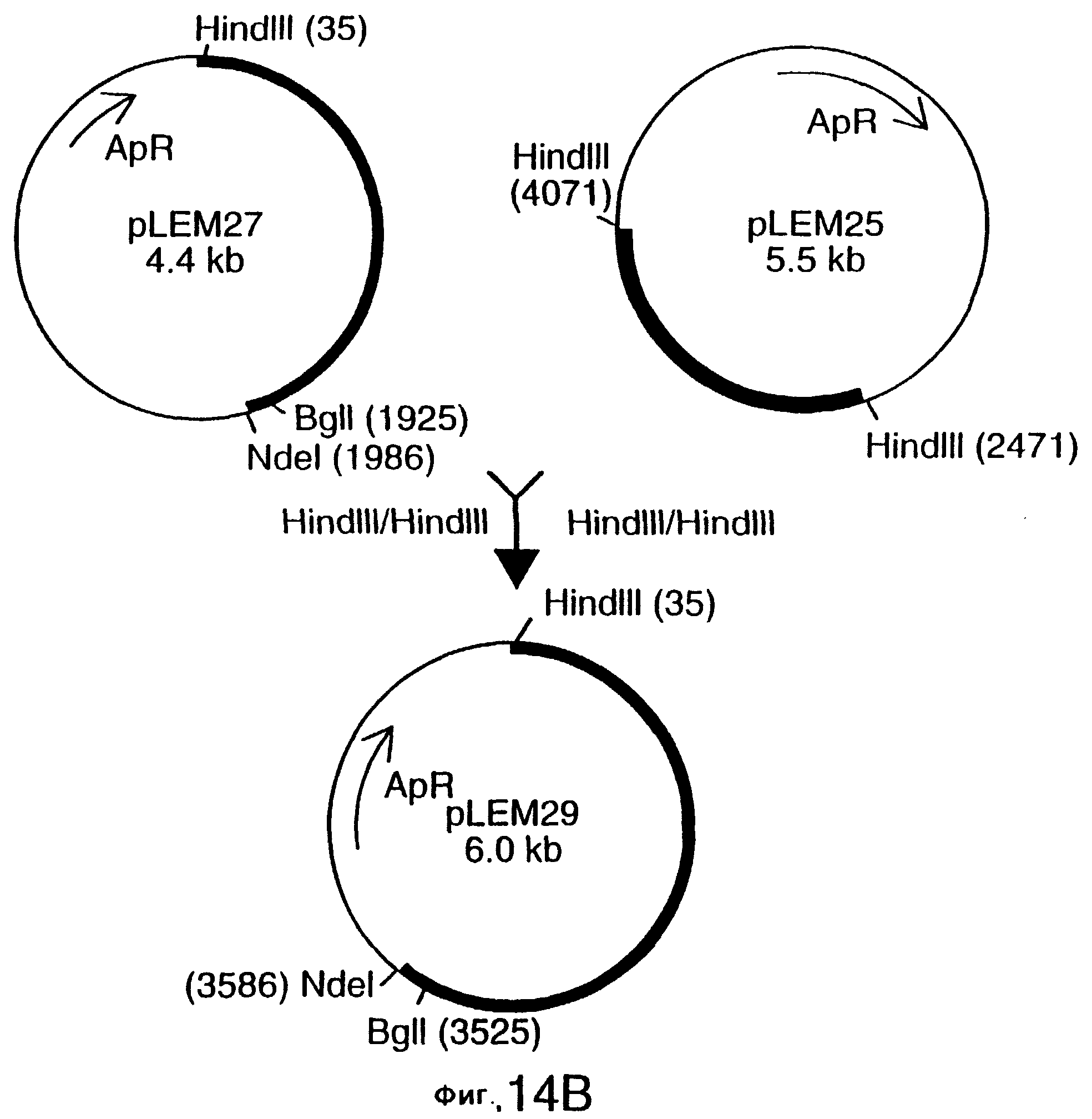
фиг.14 иллюстрирует конструирование плазмиды pLEM29 для экспрессии рекомбинантного белка Tbp1 в E.coli;
фиг.15 иллюстрирует ДСН-ПААГ-анализ экспрессии белка Tbp1 клетками E.coli, трансформированными плазмидой pLEM29;
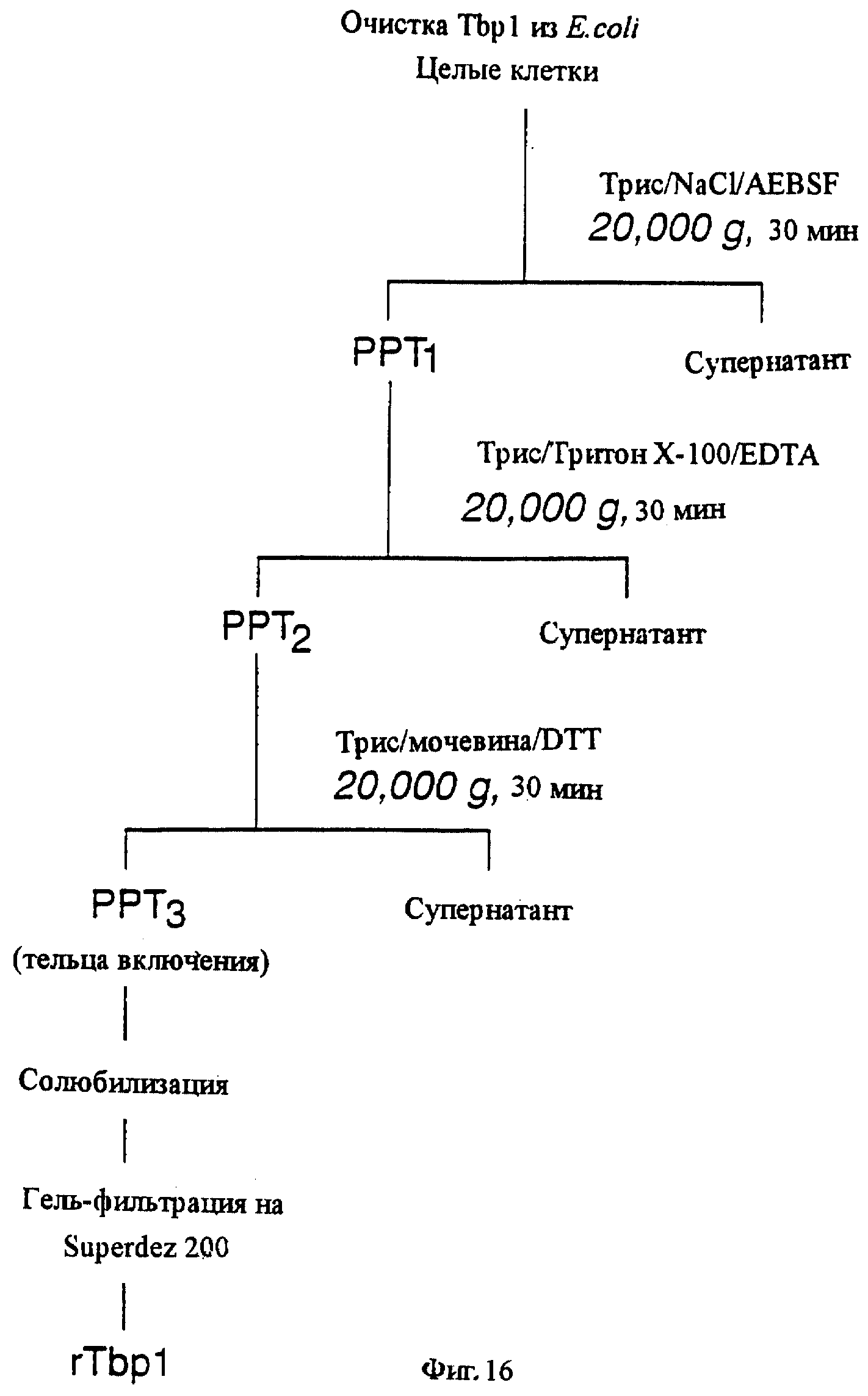
фиг.16 иллюстрирует блок-схему процесса очистки рекомбинантного белка Tbp1;
фиг.17 иллюстриует ДСН-ПААГ-анализ очищенного рекомбинантного белка Tbp1;
фиг.18 иллюстрирует конструирование плазмиды рLЕМ33 и плазмиды pLEM37, которые предназначены для экспрессии гена tbpA штамма 4223 М.catarrhalis в E.coli, и одна из которых не содержит, а другая содержит лидерную последовательность, соответственно;
фиг.19 иллюстрирует ДСН-ПААГ-анализ экспрессии белка rТbр2 клетками E.coli, трансформированными плазмидой pLEM37;
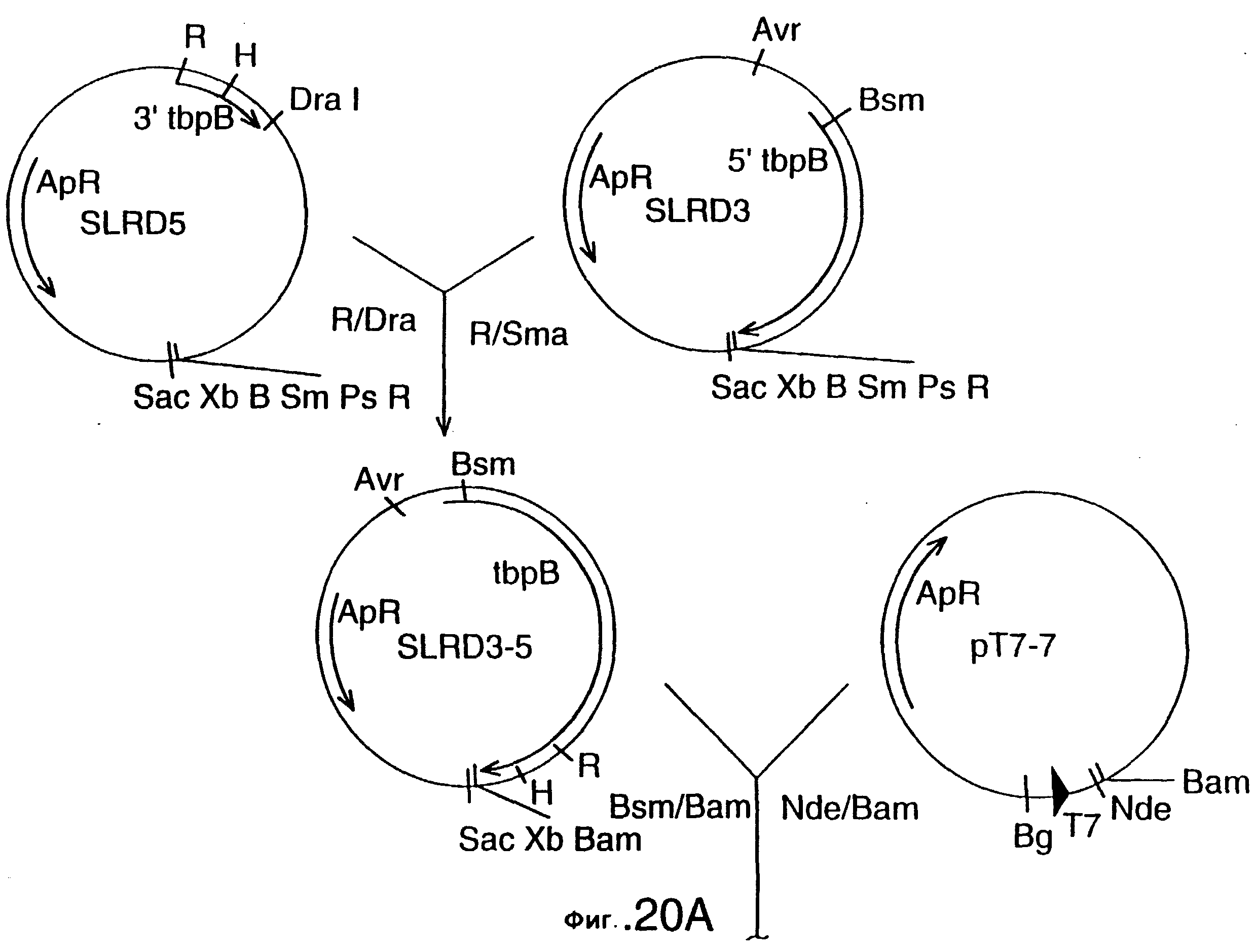
фиг.20 иллюстрирует конструирование плазмиды SLRD35B, предназначенной для экспрессии гена tbpB штамма Q8 M.catarrhalis в E.coli, и не содержащей лидерной последовательности; и конструирование плазмиды SLRD35A, предназначенной для экспрессии гена tbpB штамма Q8 M.catarrhalis в E.coli, и содержащей лидерную последовательность. Рестирикционные сайты: В=BamHI; Вg=BglII; Н=HindIII; R=EcoRI;
фиг.21 иллюстрирует ДСН-ПААГ-анализ экспрессии белка rТbр2 клетками E.coli, трансформированными плазмидами SLRD35A и SLRD35B;
фиг.22 иллюстрирует блок-схему процесса очистки рекомбинантного белка Тbр2 из E.coli;
фиг.23 (панели А и В) иллюстрирует ДСН-ПААГ-анализ очистки рекомбинантного белка Тbр2 штаммов 4223 (панель А) и Q8 (панель В) М.catarrhalis, экспрессированного в E.coli;
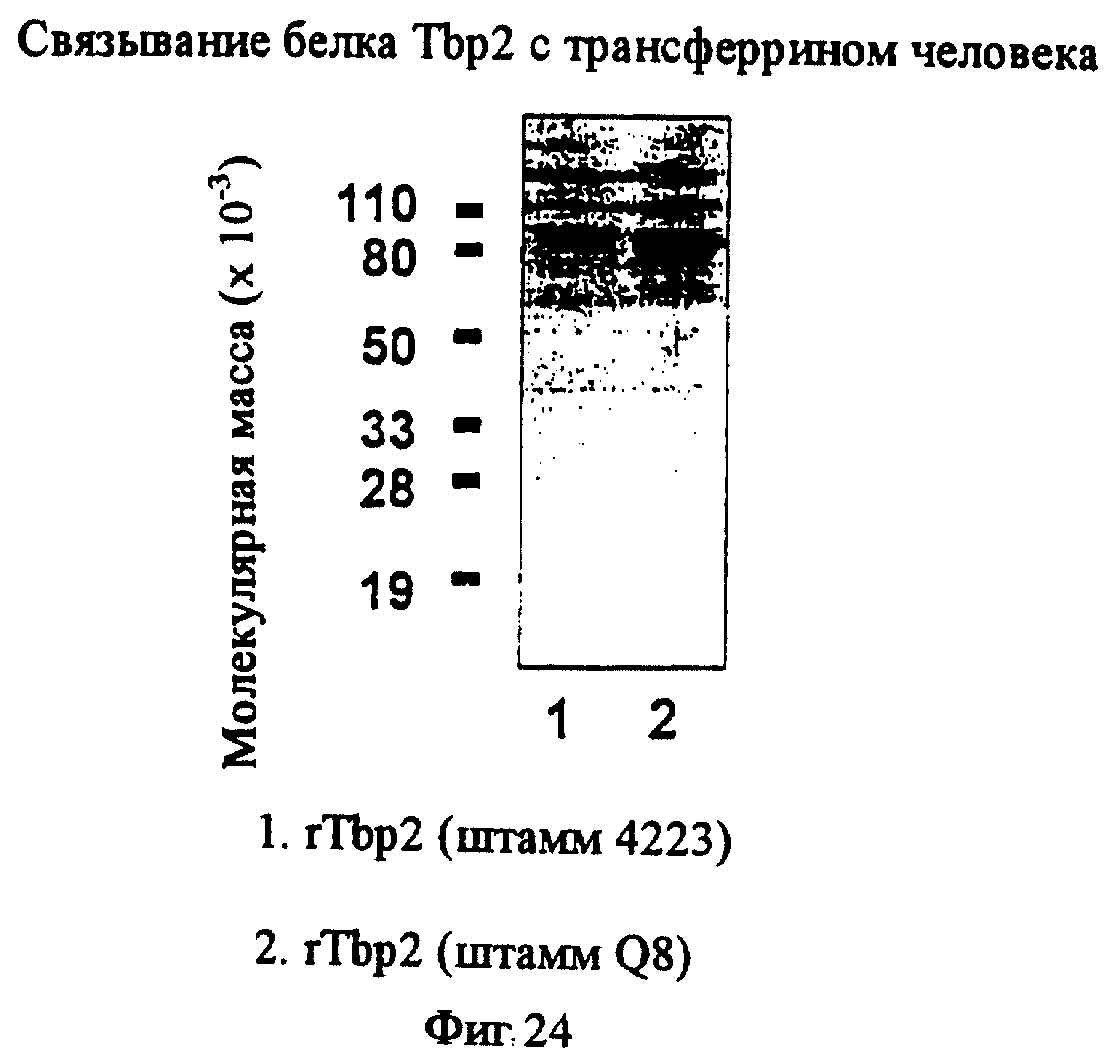
фиг.24 иллюстрирует связывание Тbр2 с трансферрином человека;
фиг.25 (включает панели А, В и С) иллюстрирует антигенную консервативность белка Тbр2 для штаммов М.catarrhalis;
фиг.26 иллюстрирует рестрикционную карту гена tbpB для штамма R1 М.catarrhalis;
фиг.27 иллюстрирует нуклеотидную последовательность гена tbpB (SEQ ID No: 45 - полная последовательность и SEQ ID No: 46 - кодирующая последовательность) и выведенную аминокислотную последовательность белка Тbр2 штамма R1 М.catarrhalis (SEQ ID No: 47); и
фиг.28 иллюстрирует сравнение аминокислотных последовательностей Tbp2 штамма 4223 М.catarrhalis (SEQ ID No: 21), штамма Q8 M.catarrhalis (SEQ ID No: 15), и штамма R1 (SEQ ID No: 47). Точки указывают на идентичные остатки, а черточки были введены для максимального соответствия сравниваемых первичных последовательностей. Звездочками указаны стоп-кодоны.
Подробное описание изобретения
Для получения очищенной и выделенной нуклеиновой кислоты, которая может иметь форму ДНК-молекул, содержащих, по крайней мере, часть нуклеиновой кислоты, кодирующей рецептор трансферрина, тип которого определен в вариантах настоящего изобретения, может быть использован любой штамм Moraxella. Эти штаммы обычно получают из клинических источников и из коллекций бактериальных культур, таких как Американская коллекция типовых культур.
В настоящей заявке, термины "рецептор трансферрина" (TfR) и "трансферрин-связывающие белки" (Тbр) используются для определения семейства белков Tbp1 и/или Tbp2, которое включает белки, имеющие изменения в своих аминокислотных последовательностях, включая белки различных природных штаммов, например, штаммов Moraxella. Выделенными и очищенными ДНК-молекулами, содержащими, по крайней мере, часть, кодирующую рецептор трансферрина настоящего изобретения, являются также молекулы, кодирующие функциональные аналоги белков рецептора трансферрина Tbp1 и Tbp2 Moraxella. В настоящей заявке первый белок является "функциональным аналогом" второго белка, в том случае, если первый белок является иммунологически родственным второму белку и/или имеет те же самые функции, аналогичные функциям второго белка. Таким функциональным аналогом может быть, например, фрагмент белка, или его мутант, имеющий замену, инсерцию или делецию.
Хромосомную ДНК из штамма 4223 М.catarrhalis гидролизовали ферментом Sаu3А с получением фрагментов размером в пределах от 15-23 т.п.о., и клонировали в BamHI-сайт лямбда-вектора EMBL3. Библиотеку скринировали с использованием антисыворотки морских свинок против Tbp1, и позитивный клон LEM3-24, содержащий вставку размером приблизительно 13,2 т.п.о., отбирали для последующего анализа. Было обнаружено, что лизат от LE392 E.coli, инфицированный LEM3-24, содержит белок размером приблизительно 115 кДа, который реагирует на Вестерн-блотах с антисывороткой против Tbp1. Второй белок размером приблизительно 80 кДа связывается на Вестерн-блотах с антисывороткой морских свинок против Tbp2.
Для того, чтобы определить локализацию гена tbpA на 13,2 т.п.о. - вставке LEM3-24, были использованы вырожденные PCR-праймеры для амплификации небольшой области предполагаемого гена tbpA штамма 4223 М.catarrhalis. Последовательности вырожденных олигонуклеотидных праймеров были выбраны на основе консервативных аминокислотных последовательностей в белках Tbp1 некоторых видов Neisseria и Haemophilus, и показаны на фиг.1 (SEQ ID No: 17 и 18). Был продуцирован амплифицированный 300 п.о. - продукт, и его локализация показана на фиг.5 жирным шрифтом (SEQ ID No: 29). Этот амплифицированный продукт был субклонирован в вектор pCRII, меченный и использованный для зондирования Саузерн-блота, содержащего клон ДНК LEM3-24, гидролизованный рестриктирующей эндонуклеазой. Этот зонд гибридизировали с 3,8 т.п.о -HindIII-HindIII-, 2,0 т.п.о. AvrII-AvrII-, и 4,2 т.п.о. -SаlI-SарhI-фрагментами (фиг.2).
3,8 т.п.о -HindIII-HindIII-фрагмент субклонировали в рАСY-177, и секвенировали. Большую открытую рамку считывания идентифицировали, после чего было обнаружено, что она содержит приблизительно 2 т.п.о - фрагмент предполагаемого гена tbpA. Остальные 1 т.п.о. гена tbpA были получены путем субклонирования находящегося непосредственно ниже смежного HindIII-HindIII-фрагмента в вектор pACY177. Нуклеотидная последовательность гена tbpA штамма 4223 М.catarrhalis (SEQ ID No: 1 и 2), и выведенная аминокислотная последовательность (SEQ ID No: 9 - полноразмерная последовательность; SEQ ID No: 10 - зрелый белок) показаны на фиг.5.
Хромосомную ДНК от штамма Q8 М.catarrhalis гидролизовали ферментом Sau3A I, и 15-23 т.п.о. - фрагменты лигировали с ВamHI-плечами EMBL3. Библиотеку с высоким титром генерировали в клетках LE 392 E.coli и скринировали с использованием олигонуклеотидных зондов, полученных на основе последовательности tbpA штамма 4223. Получали фаговую ДНК, и с помощью рестрикционного анализа было выявлено, что были клонированы вставки размером около 13-15 т.п.о. Для субклонирования фрагментов для анализа последовательностей был использован фаговый клон SLRD-A. Для облегчения клонирования фрагментов был генерирован вектор клонирования (pSKMA), и были получены плазмиды pSLRD1, pSLRD2, pSLRD3, pSLRD4 и pSLRD5, которые содержат все tbpA и большинство tbpB. Нуклеотидная последовательность (SEQ ID No: 5 и 6) и выведенная аминокислотная последовательность (SEQ ID No: 13 - полноразмерная последовательность, SEQ ID No: 14 - последовательность зрелого белка) гена tbpA штамма Q8 показаны на фиг.10.
Было установлено, выведенные аминокислотные последовательности для белка Tbp1, кодированного генами tbpA, имеют некоторую гомологию с аминокислотными последовательностями, кодированными генами ряда видов Neisseria и Haemophtlus (фиг.12; SEQ ID No: 21, 22, 23 и 24)
До разработки настоящего изобретения было установлено, что гены tbpA, идентифицированные в видах Neisseria, Haemophilus и Actinobacillus, предшествуют гену tbpB с несколькими консервативными областями. Эти два гена обычно разделены короткой межгенной последовательностью. Однако ген tbpB не был обнаружен выше гена tbpA в штамме 4223 М.catarrhalis. Для того, чтобы определить локализацию гена tbpB в 13,2 т.п.о. - вставке клона LEM3-24, был синтезирован вырожденный олигонуклеотидный зонд на основе аминокислотной последовательности EGGFYGP (SEQ ID No: 30), которая является консервативной для белков Тbр2 в нескольких видах бактерий. Этот олигонуклеотид был помечен и использован для зондирования Саузерн-блота, содержащего различные эндонуклеазо-рестрикционные фрагменты клона LEM3-24. Этот зонд гибридизировали с 5,5 т.п.о. - NheI-SаlI-фрагментом, который затем субклонировали в pBR328, и секвенировали. Этот фрагмент содержал большую часть предполагаемого гена tbpB за исключением промоторной области. Клон LEM3-24 секвенировали для определения остальной расположенной выше последовательности. Ген tbpB был расположен приблизительно на 3 т. п.о. ниже от конца гена tbpA, в отличие от генетической организации генов tbpA и tbpB в Haemophilus и Neisseria. Нуклеотидная последовательность (SEQ ID No: 3 и 4) гена tbpB от штамма 4223 M.catarrhalis и выведенная аминокислотная последовательность (SEQ ID No: 11, 12) показаны на фиг.6. Был также клонирован и секвенирован ген tbpB штамма Q8 М.catarrhalis. Нуклеотидная последовательность (SEQ ID No: 7 и 8) и выведенная аминокислотная последовательность (SEQ ID No: 15 и 16) показаны на фиг.11. Был также клонирован и секвенирован ген tbpB штамма R1 М.catarrhalis. Нуклеотидная последовательность (SEQ ID No: 45 и 46) и выведенная аминокислотная последовательность (SEQ ID No: 47) показаны на фиг.27. Наблюдались области гомологии между аминокислотными последовательностями Тbр2 штаммов М.catarrhalis, как показано на фиг.28, где иллюстрируется сравнение их первичных последовательностей (SEQ ID No: 11, 15 и 47), и между аминокислотными последовательностями Tbp2 M.catarrhalis и последовательностями Тbр2 ряда видов Neisseria и Haemophilus, как показано на фиг.13, где иллюстрируется сравнение их первичных последовательностей (SEQ ID No: 25, 26, 27, 28).
Клонированные гены tbpA и tbpB были экспрессированы в E.coli для продуцирования рекомбинантных белков Tbp1 и Tbp2, отделенных от других белков Moraxella. Эти рекомбинантные белки очищали и использовали для иммунизации.
Антигенная консервативность белка Tbp2 для других штаммов M.catarrhalis была продемонстрирована путем разделения белков в целых клеточных лизатах штаммов M.catarrhalis или штаммов E.coli, экспрессирующих рекомбинантные белки Tbp2 (rTbp2), с помощью электрофореза на ПААГ с ДСН и иммуноблоттинга с использованием антисыворотки против rТbр2 4223 или антисыворотки против rTbp2 Q8, продуцированной у морских свинок. Таким способом тестировали все штаммы 3, 56, 135, 585, 4223, 5191, 8185 и АТСС 25240 M.catarrhalis, и все они обнаруживали специфическое взаимодействие с антителом против rTbp2 4223 или с антителом против rTbp2 Q8 (фиг.25).
Кроме того, способность антител против rTbp2 от одного штамма распознавать нативный или рекомбинантный белок от гомологичного или гетерологичного штамма в ELISA-анализе, продемонстрирована в таблице 1.
Было проведено аминокислотное секвенирование N-конца и бромциановых фрагментов рецепторов трансферрина от штамма 4223 M.catarrhalis. Оба N-конца Tbp1 и Tbp2 были блокированы. Предполагаемые сигнальные последовательности Tbp1 и Tbp2 подчеркнуты на фиг.5 и 6 (SEQ ID Nos: 19 и 20), соответственно. Выведенные аминокислотные последовательности для N-концевой области Tbp2 дают основание предположить, что они имеют липопротеиновую структуру.
Результаты, представленные в таблице 1 и 2, иллюстрируют способность антисывороток против rTbp1 и против rTbp2, продуцированных у морских свинок путем их иммунизации белками Tbp1 или Tbp2, лизировать M.catarrhalis. Эти результаты показывают, что антисыворотки, продуцированные путем иммунизации белками Tbp1 и Тbр2, выделенными из М.catarrhalis, обладают бактерицидным действием против гомологичного неагглютинирующего штамма RH408 М.catarrhalis [штамм, который, согласно Будапештскому договору, был депонирован 13 декабря 1994 г. под per. №55637 Американской коллекцией типовых культур (находящейся в США по адресу 1301 Parklawn Drive, Rockville, Maryland 20852, USA) в связи с заявкой на патент США №08/328589, переуступленной ее правопреемнику (WO 96/12733)], происходящего от изолята 4223. Кроме того, антисыворотка, продуцированная путем иммунизации белком Tbp1, выделенным из штамма 4223 M.catarrhalis, обладала бактерицидной активностью против неагглютинирующего штамма Q8 (любезно предоставленного Dr. M.Bergeron, Centre Hospitalier de I'Universite Laval, St. Foy, Quebec). Кроме того, антисыворотка, продуцированная против рекомбинантного белка Тbр2 (rТbр2), обладала бактерицидным действием против гомологичного штамма M.catarrhalis.
Способность выделенных и очищенных трансферрин-связывающих белков генерировать бактерицидные антитела in vivo является свидетельством того, что эти белки могут быть использованы в качестве вакцин для вырабатывания иммунитета против заболевания, вызываемого Moraxella.
Таким образом, в соответствии с другим своим аспектом, настоящее изобретение относится к вакцине против инфекции, вызываемой штаммами Moraxella, где указанная вакцина содержит иммуногенно эффективное количество трансферрин-связывающего белка, происходящего от штамма Moraxella, и его физиологически приемлемый носитель. Вакцинные препараты могут содержать трансферрин-связывающие белки, которые отличаются по своей антигенности или по своим последовательностям.
Трансферрин-связывающий белок настоящего изобретения может быть использован в качестве диагностического реагента, в качестве антигена для генерирования антител против белков, связывающихся с трансферрином, в качестве антигена для вакцинации против заболевания, вызываемого видами Moraxella, и для обнаружения инфекции, вызываемой Moraxella и другими такими бактериями.
Трансферрин-связывающий белок настоящего изобретения может быть также использован в качестве белка-носителя для гаптенов, полисахаридов или пептидов при получении вакцины конъюгата против антигенных детерминант, не родственных трансферрин-связывающим белкам. Поэтому в других вариантах осуществления настоящего изобретения, указанный трансферрин-связывающий белок может быть использован в качестве молекулы-носителя для получения химерных молекул и вакцин конъюгатов (включая гликоконъюгаты) против патогенных бактерий, включая инкапсулированные бактерии. Так, например, гликоконъюгаты настоящего изобретения могут быть использованы для вырабатывания иммунитета против заболеваний и инфекций, вызываемых любыми бактериями, имеющими полисахаридные антигены, включая липоолигосахариды (ЛОС) и PRP. Такими бактериальными патогенами могут быть, например, Haemophilus influenzae, Streptococcus pneumoniae, Escherichia coli, Neisseria meningitidis, Salmonella typhi, Streptococcus mutans, Cryptococcus neoformans, Klebsiella, Staphytococcus aureus и Pseudomonas aeruginosa. Конкретные антигены, которые могут быть конъюгированы с трансферрин-связывающим белком, и методы осуществления такого конъюгирования описаны в заявке на патент США №08/433522 (поданной 23 ноября 1993 г (WO 94/12641), и переуступленной ее правопреемнику), которая вводится в настоящее описание посредством ссылки.
В другом варианте настоящего изобретения функция трансферрин-связывающего белка как носителя может быть использована для индуцирования иммунного ответа против аномальных полисахаридов опухолевых клеток, или для продуцирования противоопухолевых антител, которые могут быть конъюгированы с химиотерапевтическими или биологически активными агентами.
Настоящее изобретение относится к трансферрин-связывающим белкам от Moraxella catarrhalis, которые могут быть использованы в качестве активного ингредиента в вакцине против заболеваний, вызываемых инфекцией Moraxella. Настоящее изобретение также относится к фармацевтической вакцинной композиции, содержащей трансферрин-связывающие белки Moraxella catarrhalis, и необязательно, фармацевтически приемлемый носитель и/или разбавитель.
В другом своем аспекте настоящее изобретение относится к использованию трансферрин-связывающих белков для получения фармацевтической вакцинной композиции, предназначенной для иммунизации против заболевания, вызываемого инфекцией Moraxella.
Для каждого специалиста очевидно, что различные варианты настоящего изобретения могут иметь множество применений в области вакцинации, диагностики, лечения, например, инфекций Moraxella, и получения иммунологических или других диагностических реагентов. Ниже приводится обсуждение неограничивающее таких применений.
1. Получение и использование вакцины
Иммуногенные композиции, подходящие для использования в качестве вакцин, могут быть получены из иммуногенных белков рецепторов трансферрина, их аналогов и фрагментов, кодированных молекулами нуклеиновой кислоты, а также молекулами нуклеиновых кислот, описанными в настоящей заявке. Эта вакцина вырабатывает иммунный ответ, который продуцирует антитела, включая антитела против рецептора трансферрина, и антитела, которые являются опсонизирующими или бактерицидными. Если вакцинированный индивидуум будет заражен Moraxella, то антитела будут связываться с рецептором, и, тем самым, преграждать доступ бактерий к источнику железа, который необходим для их жизнеобеспечения. Кроме того, опсонизирующие или бактерицидные антитела против рецептора трансферрина могут также обеспечивать защиту посредством альтернативных механизмов.
Иммуногенные композиции, включая вакцины, могут быть получены в виде инъецируемых жидких растворов или эмульсий. Трансферрин-связывающие белки, их аналоги и фрагменты, и кодирующие молекулы нуклеиновых кислот могут быть смешаны с фармацевтически приемлемыми носителями, которые являются совместимыми с трансферрин-связывающими белками, их фрагментами и аналогами, или молекулами нуклеиновых кислот. Такими носителями могут быть вода, физиологический раствор, декстроза, глицерин, этанол, и их комбинации. Для повышения эффективности вакцин, эти иммуногенные композиции и вакцины могут, кроме того, содержать вспомогательные ингредиенты, такие как смачивающие или эмульгирующие агенты, рН-забуферивающие агенты, или адъюванты. Иммуногенные композиции и вакцины могут быть введены парентерально путем подкожной, внутрикожной или внутримышечной инъекции. Альтернативно, иммуногенные композиции настоящего изобретения могут быть изготовлены и введены так, чтобы в результате такого введения продуцировался иммунный ответ на поверхности слизистой оболочки. Таким образом, иммуногенная композиция может быть нанесена на поверхность слизистой оболочки, например, путем интраназального или перорального (через желудок) введения. Иммуногенная композиция может быть введена в комбинации с молекулой, обеспечивающей ее направленную доставку к специфическим клеткам иммунной системы или к поверхности слизистой оболочки. Некоторыми из таких обеспечивающих доставку молекул являются витамин В12 и фрагменты бактериальных токсинов, описанных в WO 92/17167 (Biotech Australia Pty. Ltd.), а также моноклональные антитела, описанные в патенте США №5194254 (Barber et al.). Альтернативно, может оказаться предпочтительным другой способ введения, включая суппозитории или пероральные препараты. Для получения суппозиториев, могут быть использованы связующие вещества и носители, например, полиалкаленгликоли или триглицериды. Пероральные составы могут включать обычно используемые наполнители, такие как, например, фармацевтически чистые сахарин, целлюлоза и карбонат магния. Эти композиции могут быть также изготовлены в виде растворов, суспензий, таблеток, пилюль, капсул, препаратов пролонгированного действия, или порошков, которые содержат от около 1 до 95% белков рецепторов трансферрина, их фрагментов, аналогов, и/или НК-молекул.
Эти вакцины вводят способом, совместимым с данной формой лекарственного препарата, и в таком количестве, которое обеспечивает эффективность, протективное действие и иммуногенность вакцины. Количество вводимого препарата зависит от индивидуума, подвергаемого лечению, включая, например, способность иммунной системы этого индивидуума синтезировать антитела, и, если необходимо, продуцировать клеточно-опосредованный иммунный ответ. Точное количество активного ингредиента, необходимое для введения, может быть установлено лечащим врачом. Однако интервал подходящих доз может быть легко установлен любым специалистом, и составляет порядка нескольких микрограммов белков рецептора трансферрина, его аналогов, фрагментов, и/или НК-молекул. Подходящие схемы для введения первичных и бустер-доз также варьируются, и могут включать первоначальное введение с последующим введением бустер-доз. Доза вакцины может также зависеть от способа введения, и будет варьироваться в зависимости от массы тела хозяина.
Молекулы нуклеиновой кислоты, кодирующие рецептор трансферрина Moraxella, могут быть использованы непосредственно для иммунизации путем прямого введения ДНК, например, посредством инъекции для генетической иммунизации, или путем конструирования "живого вектора", такого, как Salmonella, BCG, аденовирус, поксвирус, вирус коровьей оспы или полиовирус, содержащего НК-молекулы. Обсуждение некоторых "живых" векторов, которые могут быть использованы для представления гетерологичных антигенов иммунной системы, приводится, например, в работе O'Наgаn (см. 22). Способы прямой инъекции ДНК исследуемым индивидуумам в целях их генетической иммунизации описаны, например, Ulmer et al. (см. 23).
Иммуногенность может быть значительно повышена путем совместного введения антигенов с адъювантами, обычно используемыми в виде 0,05-1,0%-ного раствора в забуференном фосфатом физиологическом растворе. Адъюванты усиливают иммуногенность антигена, но сами по себе, они необязательно должны быть иммуногенными. Адъюванты могут действовать путем удерживания антигена вблизи участка введения для продуцирования депо-эффекта, облегчающего медленное пролонгированное высвобождение антигена по направлению к клеткам иммунной системы. Адъюванты могут также привлекать клетки иммунной системы к "антигенному депо" и стимулировать такие клетки на продуцирование иммунного ответа.
Иммуностимулирующие агенты или адъюванты использовались в течение многих лет для усиления иммунных ответов хозяина, например, в ответ на введение вакцины. Внутренние адъюванты, такие как липосахариды, обычно являются компонентами инактивированных или аттенюированных бактерий, используемых в качестве вакцин. Внешние адъюванты являются иммуномодуляторами, которые обычно нековалентно связываются с антигенами, и которые используют в препаратах для усиления иммунного ответа. Так, например, были идентифицированы адъюванты, которые усиливают иммунный ответ на антиген, вводимый парентерально. Однако некоторые из этих адъювантов являются токсичными, и могут оказывать нежелательное побочное действие, что делает их непригодными для введения человеку и многим животным. Действительно, обычно, в качестве адъювантов для введения человеку и животным, в вакцинах используется только гидроксид алюминия и фосфат алюминия (имеющий распространенное название "квасцы"). Эффективность квасцов в повышении гуморального ответа против дифтерийного и столбнячного токсоидов хорошо известна, а поэтому в HBsAg-вакцине в качестве адъюванта использовали квасцы. Хотя эффективность квасцов в некоторых случаях является хорошо установленным фактом, однако, их применение ограничено. Так, например, квасцы являются неэффективными при вакцинации против гриппа и продуцируют неустойчивый клеточно-опосредованный иммунный ответ. Антитела, которые продуцируются у мыши антигенами, стимулированными квасцами, имеют, в основном, изотип IgG1, и не могут обеспечивать оптимальной защиты при использовании некоторых вакцинных агентов.
Внешние адъюванты широкого ряда могут стимулировать сильный иммунный ответ против антигенов. Такими адъювантами являются сапонины, образующие комплексы с антигенами мембранных белков (иммуностимулирующие комплексы), блок-сополимеры полиоксиэтилена и полиоксипропилена с минеральным маслом, инактивированные микобактерии с минеральным маслом, полный адъювант Фрейнда, бактериальные продукты, такие как мурамилдипептид (МДП) и липополисахарид (ЛПС), а также липид А, и липосомы.
Для эффективного индуцирования гуморального иммунного ответа (HIR) и клеточно-опосредованного иммунитета (CMI), иммуногены часто эмульгируют в адъювантах. Многие адъюванты являются токсичными, и индуцируют грануломы, острые и хронические воспалительные реакции (полный адъювант Фрейнда, FCA), цитолиз (сапонины и блок-сополимеры полиоксиэтилена и полиоксипропилена), повышение температуры тела, артрит, и передний увеит (ЛПС и МДП). Хотя FCA является прекрасным адъювантом и широко используется в исследованиях, однако, он не лицензирован для введения человеку или для использования в ветеринарных вакцинах из-за его токсичности.
Желательными свойствами идеальных адъювантов являются:
(1) отсутствие токсичности;
(2) способность стимулировать продолжительный иммунный ответ;
(3) простота изготовления и стабильность при длительном хранении;
(4) способность продуцировать клеточно-опосредованный (CMI) и гуморальный (HIR) ответы против антигена, вводимого различными способами, если это необходимо;
(5) синергическое действие в сочетании с другими адъювантами;
(6) способность к селективному взаимодействию с популяцией антиген-представляющих клеток (АРС);
(7) способность к специфическому продуцированию соответствующего Тн1- или Тн2-клеточно-специфического иммунного ответа; и
(8) способность к селективному увеличению уровней соответствующего изотипа антитела (например, IgA), продуцируемого против антигенов.
В патенте США 4855283, выданном Lockhoff et al. 8 августа 1989 г. и вводимом в настоящее описание посредством ссылки, описаны гликолипидные аналоги, включая N-гликозиламиды, N-гликозилмочевины и N-гликозилкарбаматы, каждый из которых замещен в сахарном остатке аминокислотой, и которые используются в качестве иммуномодуляторов или адъювантов. Так, например, Lockhoff и др., 1991 (см. 24) сообщают, что н-гликолипидные аналоги, имеющие структурное сходство с природными гликолипидами, такими как гликофосфолипиды и гликоглицеролипиды, способны продуцировать сильный иммунный ответ при введении вакцины, полученной на основе вируса простого герпеса, и вакцины, полученной на основе псевдорабивируса. Некоторые гликолипиды были синтезированы из длинноцепочечных алкиламинов и жирных кислот, которые непосредственно связаны с сахарами посредством аномерного атома углерода в целях имитации функций природных липидных остатков.
В патенте США 4258029, выданном Moloney et al., и переуступленном его правопреемнику, который вводится в настоящее описание посредством ссылки, указывается, что гидрохлорид октадецилтирозина (ОТН), при образовании его комплекса со столбнячным токсоидом и вакциной, полученной на основе инактивированного формалином вируса полимиелита типа I, II и III, функционирует как адъювант. Кроме того, Nixon-George et al., 1990, (см. 25) указывают, что октадециловые сложные эфиры ароматических аминокислот, образующие комплексы с рекомбинантным поверхностным антигеном вируса гепатита В, усиливают иммунный ответ хозяина против вируса гепатита В.
2. Иммуноанализы
Белки рецептора трансферрина настоящего изобретения, их аналоги и/или фрагменты могут быть использованы в качестве иммуногенов и в качестве антигенов в иммуноанализах, включая твердофазный иммуноферментный анализ (ELISA), радиоиммуноанализ (РИА), и другие не-иммуноферментные анализы, проводимые с использованием связывающих антител, или процедуры, известные специалистам, и применяемые для детекции антител против белка рецептора трансферрина Moraxella. В анализах ELISA белок рецептора трансферрина, его аналоги или фрагменты, соответствующие фрагментам белка TfR, иммобилизируют на выбранной поверхности, например, на поверхности, способной связываться с белком или пептидами, такой как стенки полистиролового планшета для микротитрования. С выбранной поверхностью после ее промывки для удаления не полностью адсорбированного рецептора трансферрина, его аналогов или фрагментов может быть связан неспецифический белок, такой как, раствор альбумина бычьей сыворотки (BSA) или казеина, который, как известно, является антигенно нейтральным по отношению к испытуемому образцу. Это позволяет блокировать сайты неспецифической адсорбции на иммобилизирующей поверхности, и тем самым снизить "фон", продуцируемый неспецифическим связыванием антисыворотки с поверхностью.
Затем иммобилизирующую поверхность подвергают контакту с образцом, таким как клинические или биологические материалы, тестируемые на способность образовывать иммунный комплекс (антиген/антитело). Эта процедура может включать разведение образца разбавителями, такими как, BSA, бычий гамма-глобулин (BGG) и/или забуференный фосфатом физиологический раствор (РВ8)/Твин. Затем образец инкубируют примерно в течение 2-4 часов при температуре порядка от около 25° С до 37° С. После инкубирования контактирующую с образцом поверхность промывают для удаления материала, не образующего иммунный комплекс. Процедуру промывки можно осуществлять с использованием раствора, такого как, РВS/Твин или боратный буфер.
После образования специфических иммунокомплексов между испытуемым образцом и связанным белком рецептора трансферрина, его аналогами и/или фрагментами, и после их промывки, частота, и даже уровень образования иммунокомплексов могут быть определены способом, в котором иммунокомплекс подвергают взаимодействию со "вторым" антителом, обладающим специфичностью к "первому" антителу. В случае, если тестируемый образец происходит от человека, то "вторым" антителом является антитело, обладающее специфичностью к иммуноглобулинам человека, а в частности, IgG. Для осуществления такого обнаружения, "второе" антитело может иметь ассоциированную активность, такую как, ферментативная активность, которая способствует генерированию, например, развитию окраски после инкубирования с соответствующим хромогенным субстратом. Количественная оценка может быть затем проведена путем измерения степени развития окраски с использованием, например, спектрофотометра.
3. Использование последовательностей в качестве гибридизационных зондов
Нуклеотидные последовательности настоящего изобретения, содержащие последовательность гена рецептора трансферрина, могут быть использованы для идентификации и клонирования генов рецептора трансферрина для любых видов Moraxella.
Нуклеотидные последовательности, содержащие последовательность генов рецептора трансферрина настоящего изобретения, могут быть использованы для анализа на их способность селективно образовывать дуплексные молекулы с комплементарными фрагментами других генов TfR. В зависимости от целей применения могут быть использованы различные условия гибридизации для достижения различных степеней селективности зондов по отношению к другим генам TfR. Для достижения высокой степени селективности используются относительно жесткие условия для образования дуплексов, такие как условия с низким содержанием соли и/или с высокой температурой, например, 0,02 М-0,15 М NaCl при температурах от около 50° С до 70° С. В некоторых случаях требуются менее жесткие условия гибридизации, такие как, присутствие 0,15 М-0,9 М соли при температурах в пределах от около 20° С до 50° С. Условия гибридизации могут быть также сделаны более жесткими путем добавления возрастающих количеств формамида для дестабилизации гибридного дуплекса. Таким образом, конкретные условия гибридизации могут быть легко достигнуты путем определенных манипуляций, и их выбор, в основном, зависит от желаемых результатов. Обычно, подходящие температуры гибридизации в присутствии 50% формамида составляют: 42° С для зонда, который является на 95-100% гомологичным фрагменту-мишени: 37° С для зонда, гомологичного на 90-95%, и 32° С для зонда, гомологичного на 85-90%.
В клиническом диагностическом варианте настоящего изобретения для оценки гибридизации, НК-последовательности генов TfR настоящего изобретения могут быть использованы в комбинации с соответствующими компонентами, такими как, метка. Специалистам известен широкий ряд соответствующих индикаторов для мечения, включая радиоактивные, ферментные или другие лиганды, такие как, авидин/биотин и дигоксигенин, которые способны продуцировать детектируемый сигнал. В некоторых диагностических вариантах вместо радиоактивной метки может быть использована ферментная метка, такая как, щелочная фосфотаза или пероксидаза. В случае применения ферментных меток для идентификации специфической гибридизации с образцами, содержащими последовательности гена TfR, могут быть использованы колометрические индикаторные субстраты, которые как известно, обеспечивают продуцирование сигнала, фиксируемого человеческим глазом или с помощью спектрофотометрического прибора.
НК-последовательности генов TfR настоящего изобретения могут быть использованы в качестве гибридизационных зондов при гибридизации растворов и в случае применения твердофазных методов. В вариантах осуществления изобретения, предусматривающих использование твердофазных методов, тестируемые ДНК (или РНК) от таких образцов, как клинические образцы, включая экссудаты, физиологические жидкости (например, сыворотку, амниотическую жидкость, выделения из среднего уха, мокрота, бронхоалвеолярный лаваж), или даже ткани, адсорбируют или каким-либо другим способом присоединяют к выбранной матрице или поверхности. Затем фиксированную одно-цепочечную нуклеиновую кислоту подвергают специфической гибридизации с выбранными зондами, содержащими нуклеиново-кислотные последовательности генов TfR настоящего изобретения или их фрагментов в нужных условиях гибридизации. Выбор условий гибридизации зависит от конкретных факторов, и может быть осуществлен на основе конкретно требуемых критериев в зависимости, например, от содержания G+C, типа нуклеиновой кислоты-мишени, источника нуклеиновой кислоты, размера гибридизационного зонда и т.п. После промывания поверхности для гибридизации в целях удаления молекул неспецифически связанных зондов, специфическую гибридизацию или даже ее количественный уровень оценивают с помощью метки. При этом предпочтительно выбирать те части НК-последовательностей, которые являются консервативными для различных видов Moraxella. Выбранный зонд может иметь размер, по крайней мере, 18 п.о., и этот размер может варьироваться в пределах от около 30 до 90 п.о.
4. Экспрессия генов рецептора трансферрина
Для экспрессии генов рецептора трансферрина в экспрессирующих системах могут быть использованы плазмидные векторы, содержащие репликон и регуляторные последовательности, которые происходят от видов, совместимых с клеткой-хозяином. Обычно вектор несет сайт репликации, а также последовательности-метки, которые способны обеспечивать фенотипическую селекцию в трансформированных клетках. Так, например, E.coli может быть трансформирована с использованием плазмиды pBR322, которая содержит гены устойчивости к ампициллину и тетрациклину, и тем самым обеспечивает легкую идентификацию трансформированных клеток. Плазмида pBR322 или другие микробные плазмиды или фаги также должны содержать (либо они должны быть модифицированы так, чтобы они содержали) промоторы, которые могут быть использованы клеткой-хозяином для экспрессии их собственных белков.
Кроме того, фаговые векторы, содержащие репликон и регуляторные последовательности, которые являются совместимыми с хозяином, могут быть использованы в качестве трансформирующего вектора для этих хозяев. Так, например, фаг в лямбда-GEM™ -11 может быть использован для конструирования рекомбинантных фаговых векторов, которые могут быть использованы для трансформации клеток, таких как, LE392 E.coli.
Промоторами, обычно используемыми в рекомбинантных ДНК-конструкциях, являются промоторные системы β -лактамазы (пенициллиназы) и лактозы, а также другие бактериальные промоторы, такие как, промоторная система Т7, описанная в патенте США 4952496. Нуклеотидные последовательности промоторов детально известны специалистам, что позволяет им осуществлять функциональное лигирование этих промоторов с генами. Выбор конкретного промотора обычно зависит от желаемых результатов. В качестве хозяев, подходящих для экспрессии генов рецептора трансферрина, их фрагментов, аналогов, или вариантов, могут служить экспрессирующие системы E.coli, виды Bacillus, Наеmоphilus, грибки, дрожжи, Moraxella, Bordetella, или бакуловирусы.
В соответствии с настоящим изобретением особенно предпочтительными способами получения белка рецептора трансферрина, его фрагмента или аналога являются рекомбинантные методы, поскольку природный белок TfR, выделенный из культуры видов Moraxella, может включать следовые количества токсических материалов или других примесей. Эту проблему можно решить путем использования рекомбинантно продуцированного белка TfR в гетерологичных системах, которые могут быть выделены из хозяина так, чтобы присутствие примесей в очищенном материале было минимальным. В связи с этим особенно предпочтительными хозяевами для экспрессии являются грам-положительные бактерии, которые не имеют ЛПС, а поэтому не содержат эндотоксинов. Такими хозяевами являются виды Bacillus, и они с успехом могут быть использованы для продуцирования апирогенного рецептора трансферрина, его фрагментов или аналогов. Кроме того, рекомбинантные методы продуцирования позволяют получать Tbp1 или Тbр2 или их соответствующие аналоги или фрагменты, которые отделены друг от друга, что отличает их от нормальных комбинированных белков, присутствующих в Moraxella.
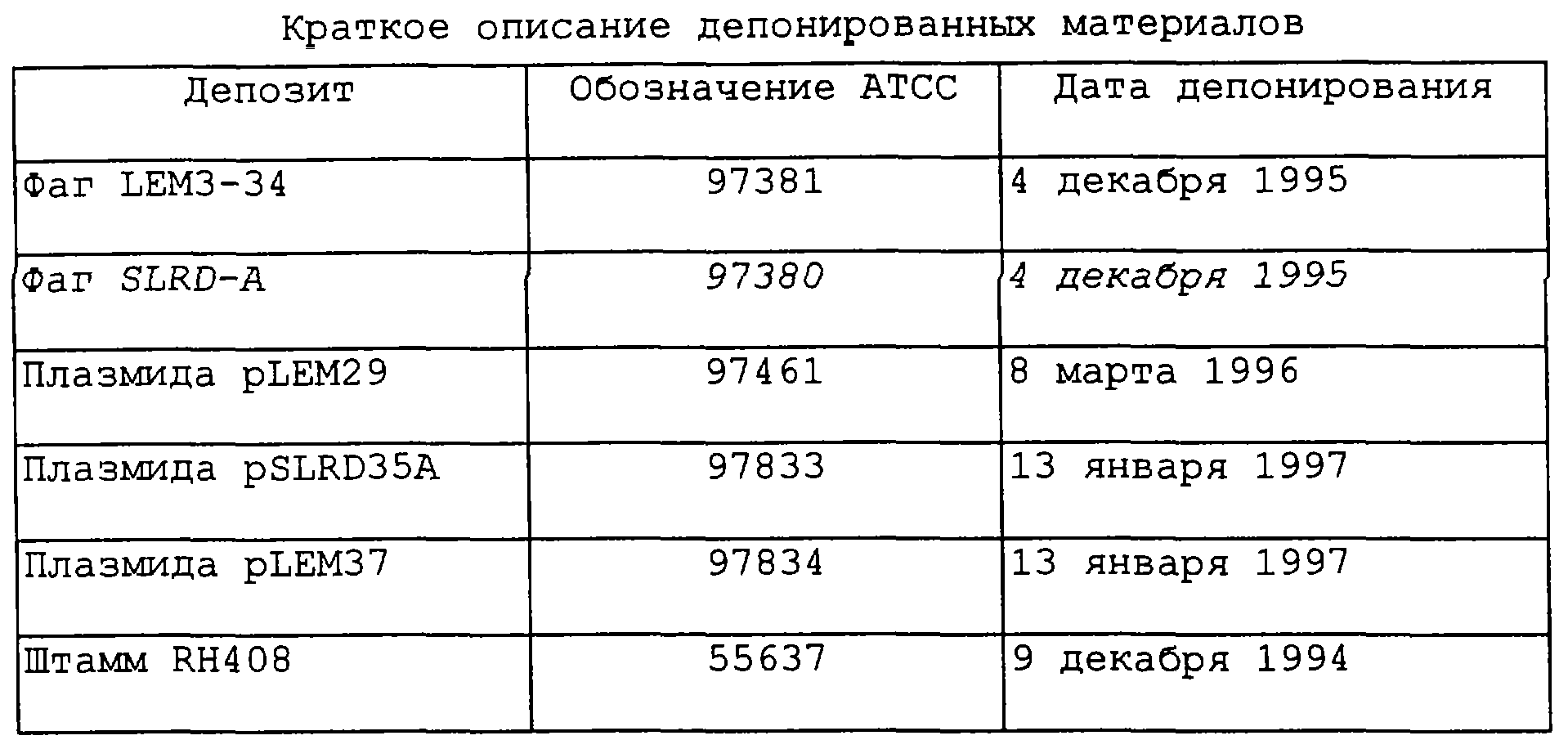
Биологические депозиты
Некоторые векторы, которые содержат, по крайней мере, фрагмент, кодирующий белок рецептора трансферрина, происходящий от штаммов 4223 и Q8 Moraxella catarrhalis и штамма RH408 М.catarrhalis, и которые описаны и обозначены в настоящей заявке, были перед подачей настоящей заявки депонированы, согласно Будапештскому договору, в Американской коллекции типовых культур (АТСС), находящейся в США по адресу 1301 Parklawn Drive, Rockville, Maryland 20852, USA. Образцы депонированных векторов и бактериальных штаммов будут доступны любому специалисту, и ограничения, наложенные на доступ к ним, будут сняты после выдачи патента США на эту заявку. Кроме того, этот депозит будет заменен в том случае, если хранилище не сможет выдать жизнеспособные образцы. Объем настоящего изобретения, заявленного и описанного в настоящей заявке, не ограничивается депонированными биологическими материалами, поскольку эти варианты депонированного материала представлены лишь для иллюстрации настоящего изобретения. Любые эквивалентные или аналогичные векторы или штаммы, кодирующие аналогичные или эквивалентные антигены, описанные в настоящей заявке, входят в объем настоящего изобретения.
Краткое описание депонированных материалов
Примеры
Выше представлено, в основном, общее описание настоящего изобретения. Более полное представление о настоящем изобретении может быть получено при ознакомлении с ниже приведенными конкретными примерами. Эти примеры описаны лишь в целях иллюстрации и не должны рассматриваться, как некое ограничение объема изобретения. Предусматривается и может считаться целесообразным изменение формы и замена эквивалентов. Хотя в настоящем описании используются специфические термины, однако, эти термины носят лишь описательный характер, и не имеют своей целью ограничение настоящего изобретения.
Описание методов молекулярной генетики, биохимии белков, и иммунологии, используемых, но не описанных подробно в выше приведенном описании и нижеследующих примерах, можно найти в научной литературе, и эти методы могут быть осуществлены любым специалистом.
Пример 1
Этот пример иллюстрирует получение и иммунизацию морских свинок белками Tbp1 и Тbр2, происходящими от М.catarrhalis.
Белки Tbp1 и Тbр2 получали следующим образом.
Неочищенные цельные мембранные препараты, не содержащие железа, разводили до концентрации 4 мг белка/мл в 50 мМ Трис-НСl-1М NaCl, pH 8, в полном объеме 384 мл. Мембраны солюбилизировали путем добавления 8 мл 0,5 М EDTA и 8 мл 30%-ного саркозила, а затем образцы инкубировали в течение 2 часов при комнатной температуре и при легком помешивании. Солюбилизированные мембраны центрифугировали в течение 20 минут при 10 тыс. об/мин. К супернатанту добавляли 15 мл апо-hTf-Сефарозы 4В, а затем инкубировали в течение 2 часов при комнатной температуре и при мягком встряхивании. После этого смесь вводили в колонку. Колонку промывали 50 мл 50 мМ Трис-НСl-1М NaCl - 250 мМ гидрохлорида гуанидина для удаления примесных белков. Тbр2 элюировали из колонки путем добавления 100 мл 1,5 М гидрохлорида гуанидина. Tbpl элюировали путем добавления 100 мл 3 М гидрохлорида гуанидина. Первые 20 мл фракции диализовали против 3 замен 50 мМ Трис-HCl, рН 8,0. Образцы хранили при -20° С, либо диализовали против бикарбоната аммония, и лиофилизовали.
Морских свинок (Charles River) иммунизировали на день +1 путем внутримышечного введения 10 мкг дозы белка Tbpl или белка Тbр2, эмульгированного в полном адъюванте Фрейнда. Животных подвергали бустер-иммунизации на +14-й день и +29-й день путем введения аналогичной дозы белка, эмульгированного в неполном адъюванте Фрейнда. Пробы крови брали на +42 день, и сыворотки использовали для анализа на бактерицидную активность антитела. Кроме того, все антисыворотки оценивали путем иммуноблот-анализа на реактивность с белками штамма 4223 М.catarrhalis.
Бактерицидную активность антисывороток морских свинок против Tbp1- и Тbр2 4223 М.catarrhalis определяли следующим образом. Неагглютинирующий штамм RH408 М.catarrhalis, происходящий от изолята 4223, инокулировали в 20 мл бульона BHI, и культивировали на шейкере при 170 об/мин в течение 18 часов при 37° С. Один миллилитр этой культуры использовали для инокуляции 20 мл бульона BHI, в который была добавлена 25 мМ этилендиамин-ди-гидроксифенилуксусная кислота (EDDA; Sigma). Эту культуру выращивали до оптической плотности OП578=0,5. Клетки разводили в отношении 1:200000 в буфере, содержащем 140 мМ NaCl, 93 мМ МаНСО3, 2 мМ барбитурат натрия, 4 мМ барбитуровую кислоту, 0,5 мМ MgCl2×6Н2O, 0,4 мМ CaCl2×2H2O, pH 7,6 (вероналовом буфере), и включающем 0,1% альбумина бычьей сыворотки (VBS), и помещали на лед. Антисыворотки морских свинок против Tbp1 и Тbр2 4223 М.catarrhalis наряду с предварительно взятыми контрольными антисыворотками нагревали до 56° С в течение 30 минут для инактивации эндогенного комплемента. В каждую лунку 96-луночного планшета для микротитрования Nunclon (Nunc, Roskilde, Denmark) добавляли 2-кратные серийные разведения каждой антисыворотки в VBS. Разведения начинали при 1:8 и доводили до конечного объема 25 мкл в каждой лунке. Затем в каждую лунку добавляли 25 мкл разведенных бактериальных клеток. Комплемент морской свинки (Biowhittaker, Walkers-ville, MD) разводили 1:10 в VBS, и 25 мкл аликвоты добавляли в каждую лунку. Планшеты инкубировали на роторной платформе при легком встряхивании при 70 об/мин в течение 60 минут при 37° С. 50 мкл каждой реакционной смеси засевали на планшеты с агаром Mueller Hinton (Becton-Dickinson, Cockeysville, MD). Планшеты инкубировали при 37° С в течение 72 часов, а затем подсчитывали число колоний на планшет. Бактерицидные титры оценивали как обратную величину наивысшего разведения антисыворотки, способной к уничтожению более, чем 50% бактерий по сравнению с контролем, содержащим неиммунные сыворотки. Результаты, показанные в таблице 1, иллюстрируют способность антисывороток морских свинок против Tbp1 и Тbр2 осуществлять лизис M.catarrhalis.
Пример 2
Этот пример иллюстрирует получение хромосомной ДНК из штаммов 4223 и Q8 M.catarrhalis
Изолят 4223 M.catarrhalis инокулировали в 100 мл бульона BHI, а затем инкубировали на шейкере в течение 18 часов при 37° С. Клетки собирали путем центрифугирования при 10000× g в течение 20 минут. Осадок после центрифугирования использовали для экстракции хромосомной ДНК из штамма 4223 M.catarrhalis.
Клеточный осадок ресуспендировали в 20 мл 10 мМ Трис-НСl (рН 7,5) - 1,0 мМ EDTA (ТЕ). Проназу и ДСН добавляли до конечных концентраций 500 мкг/мл и 1,0%, соответственно, после чего, суспензию инкубировали в течение 2 часов при 37° С. После нескольких последовательных экстракций фенолом, фенолом:хлороформом (1:1), и смесью хлороформа:изоамилового спирта (24:1), водный экстракт диализовали при 4° С против 1,0 M NaCl в течение 4 часов, и против ТЕ (рН 7,5) в течение еще 48 часов с тремя заменами буфера. К диализату добавляли 2 объема этанола, и ДНК наматывали на стеклянную палочку. Затем ДНК сушили воздухом и растворяли в 3,0 мл воды. Концентрация, определенная с помощью УФ-спектрофотометрии, составляла около 290 мкг/мл.
Штамм Q8 М.catarrhalis культивировали в бульоне BHI, как описано в примере 1. Клетки осаждали из 50 мл культуры путем центрифугирования при 5000 об/мин в течение 20 минут при 4° С. Клеточный осадок ресуспендировали в 10 мл ТЕ (10 мМ Трис-HCl, 1 мМ EDTA, рН 7,5), а затем добавляли протеиназу К и ДСН до конечных концентраций 500 мкг/мл и 1%, соответственно. Образец инкубировали при 37° С в течение 4 часов до тех пор, пока не был получен прозрачный лизат. Этот лизат дважды экстрагировали насыщенной Трис смесью фенола и хлороформа (1:1) и дважды хлороформом. Конечную водную фазу диализовали в течение 24 часов против 2× 1000 мл 1 М NaCl при 4° С с одной заменой буфера, и в течение 24 часов против 2× 1000 мл ТЕ при 4° С с одной заменой буфера. Конечный продукт диализа осаждали двумя объемами 100%-ного этанола. ДНК наматывали на стеклянную палочку, сушили и ресуспендировали в 5-10 мл ТЕ-буфера.
Пример 3
Этот пример иллюстрирует конструирование хромосомных библиотек М.catarrhalis в EMBL3.
Серию Sau 3А-гидролиза хромосомной ДНК с получением гидролизатов в конечных объемах 10 мкл (каждого) проводили для оптимизации условий, необходимых для генерирования максимальных количеств рестрикционных фрагментов размерами в пределах от 15 до 23 т.п.о. С использованием оптимизированных условий гидролиза крупномасштабный гидролиз проводили в объеме 100 мкл, включающем: 50 мкл хромосомной ДНК (290 мкг/мл), 33 мкл воды, 10 мкл 10× Sаu3А-буфера (New England Biolabs), 1,0 мкл BSA (10 мг/мл, New England Biolabs), и 6,3 мкл Sаu3А (0,04 ед./мкл). После инкубирования в течение 15 минут при 37° С реакцию гидролиза прекращали путем добавления 10 мкл смеси 100 мМ Трис-НСl (рН 8,0) - 10 мМ EDTA - 0,1% бромфенолового синего - 50% глицерина (буфера для загрузки). Гидролизованную ДНК подвергали электрофорезу на 0,5% агарозном геле в 40 мМ Трис-ацетате - 2 мМ Na2EDTA-2H2O (рН 8,5) (ТАЕ-буфере) при 50 В в течение 6 часов. Область, содержащую рестрикционные фрагменты с размером молекулы в пределах от 15 до 23 т.п.о., вырезали из геля, и помещали в трубку для диализа, содержащую 3,0 мл ТАЕ-буфера. ДНК подвергали электроэлюции из гелевого фрагмента путем подачи напряженности поля 1,0 В/см в течение 18 часов. Электроэлюированную ДНК экстрагировали один раз фенолом и один раз фенолом-хлороформом (1:1), а затем осаждали этанолом осушенную ДНК растворяли в 5,0 мкл воды.
Фракционированную по размеру хромосомную ДНК лигировали с BamHI-гидролизованными плечами EMBL3 (Promega) с использованием ДНК-лигазы Т4 в конечном объеме 9 мкл. Цельную лигированную смесь упаковывали в лямбда-фаг с использованием коммерчески доступного набора для упаковки (Amersham) в соответствии с инструкциями производителей.
"Упакованную" библиотеку ДНК амплифицировали на твердой среде. 0,1 мл аликвоты штамма NM539 Escherlchia coli в 10 мМ MgSO4 (ОП260=0,5) инкубировали при 37°С в течение 15 минут вместе с 15-25 мкл "упакованной" библиотеки ДНК. Образцы смешивали с 3 мл 0,6% агарозы, содержащей 1,0% пептон триптиказы BBL - 0,5% NaCl (верхняя агароза BBL), после чего смеси засевали на чашки с 1,5% агаром, содержащие 1,0% пептон триптиказы BBL - 0,5% NaCl, и инкубировали в течение 18 часов при 37° С. В каждую чашку добавляли 3 мл количество 50 мМ Трис-НСl (рН 7,5) - 8 мМ гептагидрата сульфата магния-100 мМ NaCl - 0,01% (мас./об.) желатина (буфер SM), и эти чашки оставляли на 7 часов при 4° С. Буфер SM, содержащий фаг, собирали с чашек, объединяли, и хранили в пробирке с завинчивающейся крышкой при 4° С в присутствии хлороформа.
Хромосомную ДНК из штамма Q8 М.catarrhalis гидролизовали ферментом Sаu3А 1 (0,1 ед./30 мкг ДНК) при 37° С в течение 30 минут, а затем фракционировали по размеру на 0, 6% агарозном геле с низкой температурой плавления. ДНК-фрагменты размером 15-23 т.п.о. вырезали, а ДНК подвергали электроэлюированию в диализной трубке, содержащей ТАЕ (40 мМ Трис-ацетат, рН 8,5, 2 мМ EDTA), при 150 В в течение 25 минут. ДНК один раз экстрагировали фенолом/хлороформом (1:1), осаждали, и ресуспендировали в воде. Затем ДНК лигировали в течение ночи с BamHI-плечами EMBL3 (Promega), и эту лигированную смесь упаковывали с использованием in vitro набора для упаковки в лямбда-фаге (Stratagene), а затем засевали на клетки LE392 E.coli. Библиотеку титровали и хранили при 4° С в присутствии 0,3% хлороформа.
Пример 4
Этот пример иллюстрирует скрининг библиотек М.catarrhalis.
10-Микролитровые аликвоты фагового исходного материала от образца EMBL3/4223, полученного, как описано выше в примере 3, объединяли (каждый) со 100 мкл штамма LE392 E.coli в 10 мМ МgSO4 (ОП260=0,5) (засеянные клетки), и инкубировали при 37° С в течение 15 минут. Образцы смешивали с 3 мл (каждой) верхней агарозы BBL, и смеси выливали на планшеты с 1,5% агарозой, содержащие 1% бактотриптон - 0,5% бактодрожжевой экстракт - 0, 05% NaCl (агароза LB; Difco), и содержащие в качестве добавки 200 мкМ EDDA. Планшеты инкубировали в течение 18 часов при 37° С. Бляшки переносили на нитроцеллюлозные фильтры (Аmеrscham Hybond-C Extra) стандартным методом, и эти фильтры погружали в 5% раствор альбумина бычьей сыворотки (BSA; Boehringer) в 20 мМ Трис-HCl (рН 7,5) - 150 мМ NaCl (TBS) на 30 минут при комнатной температуре или на ночь при температуре 4° С. Фильтры инкубировали, по крайней мере, в течение 1 часа при комнатной температуре, или в течение 18 часов при 4° С, в TBS, содержащем 1:1000 разведение антисыворотки морской свинки против Tbp1 4223 M.catarrhalis. После проведения четырех последовательных 10 минутных промывок в растворе ТВS-0,05% Твин-20 (TBS-Твин), фильтры инкубировали в течение 30 минут при комнатной температуре в растворе TBS-Твин, содержащем разведение 1/4000 рекомбинантного белка G, меченного пероксидазой хрена (rProtein G-HRP; Zymed). Фильтры промывали, как описано выше, и погружали в субстратный раствор CN/DAB (Pierce). Проявление окраски прекращали путем погружения фильтров в воду. Позитивные бляшки удаляли из планшетов, и каждую помещали в 0,5 мл буфера SM, содержащего несколько капель хлороформа. Процедуру скрининга повторяли еще 2 раза до тех пор, пока 100% перенесенных бляшек не становились позитивными, с использованием антисыворотки морской свинки против Tbp1 4223 М.catarrhalis.
Библиотеку EMBL3/Q8 засевали на клетки LE392 в планшеты с YT с использованием 0,7% верхнего агара в YT в качестве покрывающего слоя. Бляшки переносили на нитроцеллюлозные фильтры, и эти фильтры зондировали с помощью олигонуклеотидных зондов, меченных32Pα -dCTP (набор для рандомизированного мечения праймированной ДНК, Random Primed DNA labeling kit, Boehringer Mannheim). Предварительную гибридизацию осуществляли в хлоридно-натриевом/цитратно-натриевом (SSC) буфере (см. 27) при 37° С в течение 1 часа, а гибридизацию осуществляли в течение ночи при 42° С. Зонды получали на основе внутренней последовательности tbpA 4223:
IRDLTRYDPG
(SEQ ID No: 31)
4236-RD 5'- ATTCGAGACTTAACACGCTATGACCCTGGC -3'
(SEQ ID No: 32)
4237-RD 5'- ATTCGTGATTTAACTCGCTATGACCCTGGT -3'
(SEQ ID No: 33).
Предполагаемые бляшки повторно засевали и подготавливали для второго и третьего циклов скрининга с использованием аналогичных процедур. Фаговый клон SLRD-A использовали в целях субклонирования генов tfr для анализа последовательности.
Пример 5
Этот пример иллюстрирует иммуноблот-анализ фаговых лизатов с использованием антисыворотки против Tbp1 и Тbр2 4223 М-саtarrhalis.
Белки, экспрессированные фаговыми элюатами, отобранными, как описано выше в примере 4, осаждали следующим образом. 60 мкл каждого фагового элюата объединяли с 200 мкл засеянных клеток LE392 E.coli, и инкубировали в течение 15 минут при 37° С. Смесь инокулировали в 10 мл бульона, который содержит 1,0% NZ-амин А - 0,5% NaCl - 0,1% казамино-кислоты - 0,5% дрожжевой экстракт - 0,2% гептагидрат сульфата магния (бульон NZCYM), и в который была добавлена 200 мМ EDDA, и культивировали при 37° С в течение 18 часов при встряхивании. К 1,0 мл культуры добавляли ДНКазу до конечной концентрации 50 мкг/мл, и образец инкубировали при температуре 37° С в течение 30 минут. После этого добавляли трихлоруксусную кислоту до конечной концентрации 12,5%, и смесь оставляли на льду на 15 минут. Белки осаждали путем центрифугирования при 13000× g в течение 10 минут, и осадок после центрифугирования промывали 1,0 мл ацетона. Осадок сушили воздухом и ресуспендировали в 50 мкл 4% ДСН - 20 мМ Трис-НСl (рН 8,0) - 0,2 мМ EDTA (буфер для лизиса).
После электрофореза на 11,5% полиакриламидном геле с ДСН белки переносили на фильтры Immobilon-P (Millipore) в 25 мМ Трис-HCl - 220 мМ глицине - 20% метаноле (буфер для переноса) на 18 часов при постоянном напряжении 20 В. Мембраны блокировали в 5% BSA в TBS в течение 30 минут при комнатной температуре. Блоты экспонировали либо антисывороткой морских свинок против Tbp1 4223 М.catarrhalis, либо антисывороткой морских свинок против Тbр2 4223 M.catarrhalis, разведенной 1:500 в TBS-Твин, в течение 2 часов при комнатной температуре. После проведения трех последовательных 10 минутных промывок в TBS-Твин, мембраны инкубировали в ТВS-Твин, содержащем разведение 1:4000 рекомбинантного белка G, меченного перокси-дазой хрена (rProtein G-HRP), в течение 30 минут при комнатной температуре. Мембраны промывали как описано выше, и погружали в субстратный раствор CN/DAB. Проявление окраски прекращали путем погружения блотов в воду.
Три фаговых клона EMBL3 экспрессировали 115 кДа-белок, который реагировал с антисывороткой против Tbp1, и 80 кДа-белок, который реагировал с антисывороткой против Тbр2 на Вестерн-блотах, что подтверждало наличие в этих клонах генов, кодирующих белки рецептора трансферрина Moraxella catarhallis.
Пример 6
Этот пример иллюстрирует субклонирование гена tbpA белка Tbp1 4223 M.catarrhalis.
Засеянные лизаты культур рекомбинантного фага, описанного в примере 5, получали путем объединения фагового элюата и засеянных клеток LE392 E.coli в целях продуцирования конфлюэнтного лизиса на чашках с LB-агаром. Фаговую ДНК экстрагировали из засеянных лизатов с использованием системы очистки ДНК Wizard Lambda Preps DNA Purification System (Promega) в соответствии с инструкциями производителей.
Было обнаружено, что клон LM3-24 EMBL3 содержит вставку размером 13,2 т.п.о., фланкированную двумя SalI-сайтами. Был получен зонд для гена tbpA, который состоял из амплифицированного 300 п.о. - продукта, генерированного посредством PCR с использованием двух вырожденных олигонуклеотидных праймеров, соответствующих аминокислотной последовательности части белка Tbp1 (фиг.1). Последовательности праймеров были получены на основе аминокислотных последовательностей NEVTGLG (SEQ ID No: 17) и GAINEIE (SEQ ID No: 18), которые, как было обнаружено, являлись консервативными для аминокислотных последовательностей, выведенных исходя из последовательностей генов tbpA от нескольких различных штаммов N.meningitidis и Наеmоphilus influenzae. Амплифицированный продукт клонировали в pCRII (Invitrogen, San Diego, CA) и секвенировали. Выведенная аминокислотная последовательность являлась гомологичной другим предполагаемым аминокислотным последовательностям, происходящим от генов tbpA N.meningitidis и Н.influenzae (фиг.12). Субклон линеаризовали ферментом Notl (New England Biolabs) и метили с использованием набора для рандомизированного мечения дигоксигенином (Boehringer Mannheim) в соответствии с инструкциями производителей. Концентрация зонда, как было оценено, составляла 2 нг/мкл.
ДНК от фагового клона гидролизовали ферментами HindIII, AvrII, SalI/SphI, или SalI/AvrII, и подвергали электрофорезу на 0,8% агарозном геле ДНК переносили на найлоновую мембрану (Genescreen Plus, Dupont) с использованием устройства для вакуумного переноса LKB VacuGene XL (Pharmacia). После переноса блот сушили воздухом, и предварительно гибридизировали в растворе, содержащем 5Х SSC - 0,1% N-лауроилсаркозин - 0,02%, додецилсульфат натрия - 0,1% блокирующий реагент (Boehringer Mannheim) в 10 мМ малеиновой кислоте - 15 мМ NaCl (pH 7,5) (раствор для предварительной гибридизации). К раствору для предварительной гибридизации добавляли меченный зонд до конечной концентрации 6 нг/мл, и блот инкубировали в растворе зонда в течение 18 часов при 42° С. Блот дважды промывали 2Х SSC - 0,1% ДСН в течение 5 минут при комнатной температуре, а затем дважды промывали в 0,1X SSC - 0,1% ДСН в течение 15 минут при температуре 60° С. После промывок мембрану уравновешивали в 100 мМ малеиновой кислоте - 150 мМ NaCl (pH 7,5) (буфер 1) в течение 1 минуты, а затем оставляли в буфере 1 (буфере 2), содержащем 1,0% блокирующий реагент (Boehringer Mannheim) в течение 60 минут при комнатной температуре. Блот экспонировали конъюгатом антитела против DIG со щелочной фосфатазой (Boehringer Mannheim), разведенным 1/5000 в буфере 2, в течение 30 минут при комнатной температуре. После проведения двух 15-минутных промывок в буфере 1, блот уравновешивали в 100 мМ Трис-НСl (pH 9,5), 100 мМ NaCl, 50 мМ MgCl2 (буфер 3) в течение 2 минут. Блот смачивали субстратом PPD Lumigen (Boehringer Mannheim), разведенным 1/100 в буфере 3, а затем заворачивали в оберточную бумагу Saran, и экспонировали с рентгеновской пленкой в течение 30 минут. Зонд гибридизировали с 3,8 т.п.о. HindIII-HindIII-, 2,0 т.п.о. AvrII-AvrII-, и 4,2 т.п.о. SalI-SphI-фрагментом.
Для субклонирования 3,8 т.п.о. HindIII-HindIII-фрагмента в pACYC177, фаговую ДНК из клона EMBL3, и плазмидную ДНК из вектора pACYC177 (New England Biolabs) гидролизовали ферментом HindIII, и фракционировали путем электрофореза на 0, 8%-ном агарозном геле 3,8 т.п.о -HindIII-HindIII-ДНК-фрагмент фага и 3,9 т.п.о. -HindIII-HindIII фрагмент pACYC177 вырезали из геля и очищали с использованием набора Geneclean kit (Bio 101 Inc., La-Jolla, CA) в соответствии с инструкциями производителей. Очищенную вставку и вектор лигировали вместе с использованием ДНК-лигазы Т4 (New England Biolabs), и трансформировали в НВ101 E.coli (Gibco BRL). Для экстракции и высококачественной с точки зрения секвенирования очистки ДНК от одного из устойчивых к ампициллину/восприимчивых к канамицину трансформантов, который, как было обнаружено, несет 3,8 т.п.о. HindIII-HindIII-вставку, был использован набор Qiagen Plasmid Midi-kit (Qiagen). Этот субклон был обозначен рLЕМ3. Как описано ниже в примере 7, последующее секвенирование выявило, что рLЕМ3 содержит первые приблизительно 2,0 т.п.о последовательности tbpA (фиг.2 и 5).
Для субклонирования оставшихся 1 т.п.о. гена tbpA, 1,6 т.п.о -HindIII-HindIII-фрагмент субклонировали в pACYC177, как описано выше, и трансформировали путем электропорации в НВ101 E.coli (Gibco BRL). Для экстракции плазмидной ДНК из предполагаемого трансформанта, восприимчивого к канамицину и несущего плазмиду с 1,6 т.п.о. -HindIII-HindIII-вставкой, использовали набор, содержащий плазмидную ДНК, Midi-kit (Qiagen). Этот субклон был обозначен pLEM25. Как описано ниже в примере 7, секвенирование выявило, что pLEM25 содержит оставшиеся 1 т.п.о. гена tbpA (фиг.2 и 5).
Пример 7
Этот пример иллюстрирует субклонирование гена tbpB 4223 M.catarrhalis.
Как описано выше, во всех видах Neisseriae и Haemophilus, исследованных до настоящего изобретения, было обнаружено, что гены tbpB находятся непосредственно выше генов tbpA, которые имеют гомологию с геном tbpA 4223 M.catarrhalis. Однако последовательность, расположенная выше 4223 M.catarrhalis, не соответствует другим последовательностям, кодирующим tbpB.
Для определения локализации гена tbpB в фаговом клоне EMBL3 проводили Саузерн-блот-анализ с использованием вырожденного зонда из высоко консервативной области аминокислотной последовательности белка Tbp2. Был сконструирован вырожденный олигонуклеотидный зонд, соответствующий последовательности, кодирующей EGGFYGP (SEQ ID No: 30), которая является консервативной в белке Tbp2 в различных видах Neisseriae и Наеmоphilus. Зонд метили дигоксигенином с использованием набора для присоединения олигонуклеотидов (Boehringer Mannheim) в соответствии с инструкциями производителей. Клон ДНК EMBL3, гидролизованный ферментом HindIII, фракционировали на 0,8% агарозном геле, и переносили на найлоновую мембрану Geneclean Plus, как описано в примере 6. После проведения гибридизации, как описано выше, мембрану дважды промывали 2Х SSC - 0,1% ДСН (каждый раз) в течение 5 минут при комнатной температуре, а затем дважды промывали в 0,1% SSC - 0,1% ДСН (каждый раз) в течение 15 минут при температуре 50°С. Детекцию меченного зонда осуществляли, как описано выше. Зонд гибридизировали с 5,5 т.п.о. -NheI-SalI-фрагментом.
5,5 т.п.о. -NheI-SalI-фрагмент субклонировали в pBR328 следующим образом. ДНК LEM3-24 и ДНК pBR328 гидролизовали ферментом NheI-SalI, и подвергали электрофорезу на 0,8%-ной агарозе. 5,5 т.п.о. -NheI-SalI-фрагмент и 4,9 т.п.о. -NheI-SalI-фрагменты pBR328 вырезали из геля и очищали с использованием набора Geneclean kit, как описано в примере 6. Фрагменты лигировали вместе с использованием ДНК-лигазы Т4, и трансформировали в DH5 E.coli. Для экстракции ДНК из резистентного к ампициллину/чувствительного к тетрациклину клона, содержащего 5,5 т.п.о. -NheI-SalI-вставку, использовали набор Midi-Plasmid, DNA kit (Quagen). Этот субклон обозначали pLEM23. В результате секвенирования было выявлено, что pLEM23 содержит 2 т.п.о. гена tbpB от 4223 М.catarrhalis (фиг.2).
Пример 8
Этот пример иллюстрирует субклонирование генов tfr Q8 М.catarrhalis.
Гены tfr Q8 М.catarrhalis субклонировали следующим образом. Фаговую ДНК получали из планшетов. Для этого верхний слой агарозы из трех планшетов с конфлюэнтной культурой соскребали в 9 мл буфера SM (0,1 М NaCl, 0,2% MgSO4, 50 мМ Трис-HCl, рН 7,6, 0,01% желатина) и добавляли 100 мкл хлороформа. Полученную смесь подвергали вихревому перемешиванию в течение 10 секунд, а затем инкубировали при комнатной температуре в течение 2 часов. Клеточный дебрис удаляли путем центрифугирования при 8000 об/мин в течение 15 минут при 4° С на роторе SS34 (модель Sorvall RC5C). Затем фаг осаждали путем центрифугирования при 35000 об/мин на роторе 70.1 Ti при 10° С в течение 2 часов (модель Beckman L8-80), и ресуспендировали в 500 мкл буфера SM. Образец инкубировали в течение ночи при 4° С, а затем добавляли РНКазу и ДНКазу до конечных концентраций 40 мкг/мл и 10 мкг/мл, соответственно, и эту смесь инкубировали в течение одного часа при 37° С. К смеси добавляли 10 мкл 0,5 М EDTA и 5 мкл 10% ДСН, после чего, образец инкубировали в течение 15 минут при температуре 6° С. Полученную смесь дважды экстрагировали фенолом/хлороформом (1:1) и дважды хлороформом, а затем ДНК осаждали путем добавления 2,5 объема абсолютного этанола.
Путем частичного гидролиза получали рестрикционную карту, и фрагменты субклонировали с использованием внешних SalI-сайтов от EMBL3 и внутренних AvrII- или EcoRI-сайтов, как указано на фиг.4. В целях облегчения субклонирования плазмидную pSKMA конструировали путем введения нового сайта множественного клонирования в pBluescript SK (Stratagene). Для введения рестрикционных Mst II-, Sfi I- и AvrII-сайтов между SalI- и HindIII-сайтами pBluescript SK были использованы олигонуклеотиды:

Плазмида pSLRDI содержала ~1,5 т.п.о. -SalI-AvrII-фрагмент, клонированный в рSКМА; плазмиды p3LRD2 и pSLRD4 содержали ~2 т.п.о. - и 4 т.п.о. -АvrII-AvrII-фрагменты, клонированные в рSКМА, соответственно, и содержали полный ген tbpA. Плазмида pSLRDS содержала ~2,3 т.п.о. -AvrII-EcoRV1-фрагмент, клонированный в рSКМА, а плазмида SLRD5 представляла собой 22,7 т.п.о. -EcoRI-ЕсоRI-фрагмент, клонированный в рSКМА. Эти два клона содержали полный ген tbpB (фиг.7).
Пример 9
Этот пример иллюстрирует секвенирование генов tbp М.catarrhalis.
Обе цепи генов tbp, субклонированных как описано в примерах 6-8, секвенировали с использованием ДНК-секвенатора Applied Biosystems. Последовательности генов tbpA Q8 и 4223 М.catarrhalis показаны на фиг.5 и 10, соответственно. Полученную аминокислотную последовательность сравнивали с другими аминокислотными последовательностями Tbp1, включая последовательности Tbp1 Neisseriae meningitidis, Neisseriae gonorrhoeae, и Haemophilus influenzae (фиг.12). Последовательности генов tbpB Q8 и 4223 М.catarrhalis показаны на фиг.6 и 11, соответственно. Для получения предполагаемого начала последовательности гена tbpB 4223 M. catarrhalis, данные для этой последовательности были получены непосредственно из ДНК-клона LEM3-24. Эту последовательность подтверждали путем секвенирования клона DS-1754-1. Последовательность транслированных генов tbpB от 4223 и Q8 М.catarrhalis была гомологична выведенным аминокислотным последовательностям Тbр2 от Neisseria meningitidis, Neisseria gonorrhoeae, и Haemophilus influenzae (фиг.13).
Пример 10
Этот пример иллюстрирует генерирование экспрессионного вектора для продуцирования рекомбинантного белка Tbp1. Схема конструирования показана на фиг.14.
Плазмидную ДНК из субклона рLЕМ3, полученную как описано в примере 6, гидролизовали ферментами HindIII и BglI для генерирования 1,84 т.п.о. -ВglI-HindIII-фрагмента, содержащего приблизительно две трети гена tbpA. Во избежание ко-имиграции 1,89 т.п.о. -ВglI-HindIII-фрагмента вектора, к гидролизату добавляли BamHI. Кроме того, плазмидную ДНК из вектора рТ7-7 гидролизовали ферментами NdeI и HindIII. Для конструирования начала гена tbpA синтезировали олигонуклеотид на основе первых 61 оснований гена tbpA до BglI-сайта; Ndel-сайт включали в 5'-конец. Очищенную вставку, вектор и олигонуклеотид лигировали вместе с использованием лигазы Т4 (New England Biolabs), и трансформировали в DH5α E.coli. ДНК очищали из одного из 4,4 т.п.о. - трансформантов, резистентных к ампициллину, и содержащих нужные рестрикционные сайты (pLEM27).
Очищенную ДНК pLEM27 гидролизовали ферментом HindIII, лигировали в 1,6 т.п.о. - HindIII-HindIII-фрагмент-вставку pLEM25, полученную как описано в примере 6, и трансформировали в DH5α E.coli. ДНК очищали из резистентного к ампициллину трансформанта, содержащего нужные рестрикционные сайты (pLEM-29), и трансформировали путем электропорации в BL21 (DE3) (Novagen; Madison, WI) с продуцированием pLEM29B-1 E.coli.
Выделенную трансформированную одиночную колонию использовали для инокуляции 100 мл бульона YT, содержащего 100 мкг/мл ампициллина, и культуру выращивали на шейкере при 200 об/мин в течение ночи при 37° С. 200 мкл ночной культуры инокулировали в 10 мл бульона YT, содержащего 100 мкг/мл ампициллина, и культуру выращивали при температуре 37° С до достижения ОП578=0,35. Культуру индуцировали путем добавления 30 мкл 100 мМ IPTG, а затем культивировали при 37° С в течение еще 3 часов. Один миллилитр культуры удаляли ко времени индуцирования (0 часов), и через 1 час (t=1) и 3 часа (t=3) после индуцирования. Один миллилитр образцов осаждали путем центрифугирования, и ресуспендировали в 4% ДСН 20 мМ Трис-HCl, рН 8, 200 мкМ EDTA (буфере для лизиса). Образцы фракционировали путем электрофореза на 11,5% ПААГ с ДСН, и переносили на фильтры Immunobilon (Amersham). Блоты проявляли с использованием антисыворотки против Tbp1 (4223 М.catarrhalis) в качестве "первого" антитела при разведении 1:1000, и рекомбинантного белка G, конъюгированного с пероксидазой хрена (Zymed), в качестве "второго" антитела. Для детекции был использован хемилюминесцентный субстрат (Lumiglo; Kirkegaard and Perry Laboratories, Gaithersburg, MD) Индуцированные рекомбинантные белки были видимыми на геле, окрашенном Кумасси (фиг.15). Антисыворотка против Tbp1 (4223) распознавала рекомбинантные белки на Вестерн-блотах.
Пример 11
Этот пример иллюстрирует экстракцию и очистку рекомбинантного белка Tbp1 от штамма 4223 М.catarrhalis.
Рекомбинантный белок Tbp1, который содержался в тельцах включения, очищали из клеток E.coli, экспрессирующих ген tbpA (пример 10), способом, показанным на фиг.16. Клетки E.coli из 500 мл культуры, полученной как описано в примере 10, ресуспендировали в 50 мл 50 мМ Трис-HCl, рН 8,0, содержащего 0,1 М NaCl и 5 мМ AEBSF (ингибитора протеазы), и разрушали путем обработки ультразвуком (3× 10 мин, рабочий цикл 70%). Экстракт центрифугировали при 20000× g в течение 30 минут, и полученный супернатант, который содержал >85% растворимых белков от E.coli, отбрасывали.
После этого оставшийся осадок (фиг.16, PPT1) экстрагировали в 50 мл 50 мМ Триса, рН 8,0, содержащего 0,5% Тритона X-100 и 10 мМ EDTA. После центрифугирования при 20000× g в течение 30 минут, супернатант, содержащий остаточные растворимые белки и большинство мембранных белков, отбрасывали.
Затем оставшийся осадок (фиг.16, PPT2) экстрагировали в 50 мл 50 мМ Трис (рН 8,0), содержащего 2 М мочевину и 5 мМ дитиотреитол (DTT). После центрифугирования при 20000× g в течение 30 минут, осадок (фиг.16, РРТ3), полученный после вышеуказанной экстракции, содержал очищенные тельца включения.
Белок Tbp1 солюбилизировали из РРТ3 в 50 мМ Трис, рН 8,0, содержащем 6 М гидрохлорид гуанидина и 5 мМ DTT. После центрифугирования полученный супернатант, кроме того, очищали путем гель-фильтрации на колонке Superdex 200, уравновешенной в 50 мМ Трис (рН 8,0), содержащем 2 М гидрохлорид гуанидина и 5 мМ DTT. Фракции анализировали путем электрофореза на ПААГ с ДСН, и те фракции, которые содержали очищенный белок Tbp1, объединяли. К объединенной фракции белка Tbp1 добавляли Тритон Х-100 до конечной концентрации 0,1%. Затем фракции диализовали в течение ночи при 4° С против 50 мМ Трис, рН 8,0, а затем центрифугировали при 20000× g в течение 30 минут. Белок, оставшийся растворимым в этих условиях, и очищенный белок Tbp1, хранили при -20° С. В результате очистки, показанной на фиг.16, был продуцирован белок Tbp1, который имел, по крайней мере, 70%-ную чистоту, как было определено путем электрофореза на ПААГ с ДСН (фиг.17).
Пример 12
Этот пример иллюстрирует конструирование экспрессионной плазмиды для rTbp2 от 4223 М.catarrhalis без лидерной последовательности.
Схема конструирования плазмиды, экспрессирующей rTbp2, показана на фиг.18. Для конструирования первых приблизительно 58 п.о. гена tbpB 4223 М.catarrhalis, кодирующих зрелый белок, были использованы олигонуклеотиды. В 5'-конец этих олигонуклеотидов был включен NdeI-сайт:
5'TATGTGTGGTGGCAGTGGTGGTTCAAATCCACCTGCTCCTACGCCCATTCCAAATG (SEQ IO NO: 36) 3'
3'ACACACCACCGTCACCACCAAGTTTAGGTGGACGAGGATGCGGGTAAGGTTTACGATC (SEQ ID NO: 37) 5'
NheI-ClaI-фрагмент, содержащий приблизительно 1 т.п.о. гена tbpB от pLEM23, полученного как описано в примере 7, лигировали с вышеуказанными олигонуклеотидами, и встраивали в рТ7-7, разрезанную ферментом NdeI-ClaI, что приводило к образованию плазмиды pLEM31, которая содержала 5'-половину tbpB. Олигонуклеотиды также были использованы для конструирования последних приблизительно 104 п.о. гена tbpB от AvaII-сайта до конца гена. В 3'-конец олигонуклеотидов был включен BamHI-сайт:
5'GTCCAAATGCAAACGAGATGGGCGGGTCATTTACACACAACGCCGATG ACAGCAAAGCCTCTGTGGTCTTTGGCACAAAAAGACAACAAGAAGTTAAGTAGTAG (SEQ ID NO: 38) 3'
3'GTTTACGTTTGCTCTACCCGCCCAGTAAATGTGTGTTGCGGCTACTGTC GTTTCGGAGACACCAGAAACCGTGTTTTTCTGTTGTTCTTCAATTCATCATCCTAG (SEQ ID NO: 39) 5'
ClaI-AvaII-фрагмент из pLEM23, содержащий приблизительно 0,9 т.п.о. 3'-конца гена tbpB, лигировали с AvaII-BamHI-олигонуклеотидами, и встраивали в плазмиду рТ7-7, разрезанную ферментом ClaI-BamHI, что приводило к образованию pLEM32. После этого 1,0 т.п.о. -NdeI-ClaI-вставку из pLEM31 и 1,0 т.п.о. -СlaI-ВamHI-вставку из pLEM32, встраивали в рТ7-7, разрезанную ферментами NdeI-BamHI, и получали pLEM33, которая имела полноразмерный ген tbpB под контролем промотора Т7.
ДНК очищали из pLEM33 и трансформировали путем электропорации в электрокомпетентные клетки BL21 (DЕ3) (Novagen; Madison, WI) с последующим генерированием штамма pLEM33B-1. Штамм pLEM33B-1 культивировали и индуцировали с использованием IPTG, как описано выше в примере 10. Экспрессированные белки разделяли путем электрофореза в ПААГ с ДСН, и переносили в мембраны, подходящие для иммуноблоттинга. Блоты проявляли с использованием антисыворотки против Тbр2 4223, разведенной 1:4000, в качестве "первого" антитела, и рекомбинантного белка G, конъюгированного с пероксидазой хрена (Zymed), в качестве "второго" антитела. Для детекции был использован хемилюминесцентный субстрат (Lumiglo; Kirkegaard and Perry Laboratories, Gaithersburg, MD). Индуцированные рекомбинантные белки были видимыми на гелях, окрашенных Кумасси синим (фиг.19). Антисыворотка против Тbр2 4223 распознавала рекомбинантные белки на Вестерн-блотах.
Пример 13
Этот пример иллюстрирует конструирование экспрессионной плазмиды для rТbр2 от 4223 М.catarrhalis, имеющей лидерную последовательность.
Схема конструирования показана на фиг.18. Для конструирования первых приблизительно 115 п.о. гена tbpB до NheI-сайта были использованы олигонуклеотиды, содержащие природную лидерную последовательность гена tbpB 4223 M.catarrhalis. В 5'-конец этих олигонуклеотидов включали NdeI-сайт:
5'TATGAAACACATTCCTTTAACCACACTGTGTGTGGCAATCTCTGCCGTC TTATTAACCGCTTGTGGTGGCAGTGGTGGTTCAAATCCACCTGCTCCTACGCCCATTCCAAATG (SEQ ID NO: 40) 3'
3' ACTTTGTGTAAGGAAATTGGTGTGACACACACCGTTAGAGACGGCAGAA TAATTGGCGAACACCACCGTCACCACCAAGTTTAGGTGGACGAGGATGCGGGTAAGGTTTACGATC (SEQ ID NO: 41) 5'
NdeI-NheI-олигонуклеотиды лигировали с pLEM33, разрезанной ферментами NdeI-NheI, что приводило к генерированию pLEM-37, которая содержала полноразмерный ген tbpB 4223, кодирующий белок Тbр2 с его лидерной последовательностью, под контролем промотора Т7.
ДНК из pLEM37 очищали и трансформировали путем электропорации в электрокомпетентные клетки BL21 (DE3) (Novagen; Madison, WI) с последующим генерированием штамма pLEM37-B-2. pLEM37B-2 культивировали и индуцировали с использованием IPTG, как описано выше в примере 10. Экспрессированные белки разделяли путем электрофореза в ПААГ с ДСН, и переносили на мембраны, подходящие для иммуноблоттинга. Блоты проявляли с использованием антисыворотки против Тbр2 4223, разведенной 1:4000, в качестве "первого" антитела; и рекомбинантного белка G, конъюгированного с пероксидазой хрена (Zymed), в качестве "второго" антитела. Для детекции использовали хемилюминесцентный субстрат (Lumiglo; Kirkegaard and Perry Laboratories, Gaithersburg, MD). Индуцированные рекомбинантные белки были видимы на гелях, окрашенных Кумасси синим (фиг.21).
Антисыворотка против Tbp2 4223 распознавала рекомбинантные белки на Вестерн-блотах.
Пример 14
Этот пример иллюстрирует конструирование экспрессионной плазмиды для rТbр2 от Q8 М.catarrhalis без лидерной последовательности.
Схема конструирования для гТbр2 показана на фиг.20. 5'-конец гена tbpB Q8 М.catarrhalis был PCR-амплифицирован от кодона Сys1 зрелого белка до рестрикционного Bsm 1-сайта. В 5'-конец для последующего клонирования в рТ7-7, вводили рестрикционный Ndel-сайт, и этот конечный PCR-фрагмент имел длину 238 п.о. PCR-праймеры указаны ниже:

Ген tbpB Q8 субклонировали в два фрагмента, находящиеся на плазмидах SLRD3 и SLRD5, полученных как описано в примере 8. Плазмиду SLRD3-5 конструировали так, чтобы она содержала полноразмерный ген tbpB, путем гидролиза плазмиды SLRD5 ферментами EcoRI и Dra I, что приводило к высвобождению 3'-конца tbpB, и встраиванию этого ~619 п.о. - фрагмента в плазмиду SLRD3, которая была гидролизована ферментами EcoRI и Smal. 1,85 т.п.о -BsmI-BamHI-фрагмент из SLRD3-5 лигировали с 238 п.o.- PCR-фрагментом, и встраивали в рТ7-7, гидролизованную ферментами NdeI и BamHI, в результате чего была образована плазмида SLRD35B. Эта плазмида содержала полноразмерный ген tbpB, без его лидерной последовательности, под контролем промотора Т7. ДНК из SLRD35B очищали и трансформировали путем электропорации в электрокомпетентные клетки BL21 (DE3) для генерирования штамма SLRD35BD, который был культивирован и индуцирован с использованием IPTG, как описано выше в примере 10. Экспрессированные белки разделяли путем электрофореза на ПААГ с ДСН, и индуцированный белок Тbр2 был хорошо заметен после окрашивания Кумасси синим (фиг.19).
Пример 15
Этот пример иллюстрирует конструирование экспрессионной плазмиды для rТbр2 от Q8 М.catarrhalis вместе с его лидерной последовательностью.
Схема конструирования для гТbр2 показана на фиг.20. 5'-конец гена tbpB Q8 был PCR-амплифицирован от старт-кодона ATG до рестрикционного BsmI-сайта. Для облегчения клонирования в экспрессионный вектор рТ7-7 у 5'-конца встраивали NdeI-сайт, после чего, конечный PCR-фрагмент имел длину 295 п.о. PCR-праймеры указаны ниже:

SLRD3-5 (пример 14) гидролизовали ферментами BsmI и BamHI с генерированием 1,85 т.п.о. - фрагмента, который лигировали с 295 п.о. - PCR-фрагментом, а затем лигировали в вектор рТ7-7, гидролизованный ферментами NdeI и BamHI. Полученная плазмида SLRD35A содержала полноразмерный ген tbpB Q8 вместе с его эндогенной лидерной последовательностью под контролем промотора Т7. ДНК из SLRD35A очищали и трансформировали путем электропорации в электрокомпетентные клетки BL21 (DE3), в результате чего был создан штамм SLRD35AD, который затем культивировали и индуцировали с использованием IPTG, как описано выше в примере 10. Экспрессированные белки разделяли путем электрофореза на ПААГ с ДСН, и индуцированный белок Тbр2 был хорошо заметен визуально после окрашивания Кумасси синим (фиг.19).
Пример 16
Этот пример иллюстрирует экстракцию и очистку rТbр2 4223 и Q8 М.catarrhalis из E.coli.
Трансформанты pLEM37B (4223) и SLRD35AD (Q8) культивировали для продуцирования Тbр2 в тельцах включения, а затем Тbр2 очищали в соответствии со схемой, показанной на фиг.22. Клетки E.coli из 500 мл культуры ресуспендировали в 50 мл 50 мМ раствора Трис-HCl, рН 8,0, содержащего 5 мМ AEBSF (ингибитор протеазы), и разрушали путем обработки ультразвуком (3× 10 мин, рабочий цикл 70%). Этот экстракт центрифугировали при 20000× g в течение 30 минут, и полученный супернатант, который содержал >95% растворимых белков от E.coli, отбрасывали.
Затем оставшийся осадок (PPT1) экстрагировали в 50 мл 50 мМ Трис, рН 8,0, содержащего 0, 5% Тритон Х-100 и 10 мМ EDTA. Смесь перемешивали при 4° С в течение, по крайней мере, 2 часов, а затем центрифугировали при 20000× g в течение 30 минут, и супернатант, содержащий остаточные растворимые белки и большинство мембранных белков, отбрасывали.
Образовавшийся осадок (РРТ2), полученный в результате вышеописанной экстракции, содержал тельца включения. Белок Тbр2 солюбилизировали в 50 мМ Трис, рН 8,0, содержащем 6 М гуанидин и 5 мМ DTT. После центрифугирования полученный супернатант очищали с помощью гель-фильтрации на колонке с Superdex 200, уравновешенной в 50 мМ Трис, рН 8,0, содержащем 2 М гуанидин и 5 мМ DTT. Фракции анализировали путем электрофореза на ПААГ с ДСН, и те фракции, которые содержали очищенный белок Тbр2, объединяли. К этой объединенной фракции Тbр2 добавляли Тритон Х-100 до конечной концентрации 0,1%. После этого фракцию диализовали против PBS в течение ночи при температуре 4° С и центрифугировали при 20000× g в течение 30 минут. Белок, сохранивший растворимость в этих условиях, и очищенный белок Тbр2, хранили при -20° С. На фиг.22 показан ДСН-ПААГ-анализ фракций после процедуры очистки rТbр2 из штамма 4223 (Панель А) и штамма Q8 (Панель В). rТbр2 имел, по крайней мере, 70% чистоту.
Группам из пяти мышей BALB/c три раза подкожно (s.с.) инъецировали на 1-, 29- и 43-й день очищенный гТbр2 (0,3 мг-10 мг) из штаммов 4223 и Q8 М. catarrhalis в присутствии или отсутствии АlРO4 (1/5 мг на дозу). Пробы крови брали на день 14, 28, 42 и 56 для анализа на титры антитела против rТbр2 посредством ИФА.
Группы из двух кроликов и двух морских свинок (Charles River, Quebec) иммунизировали внутримышечно (i.m.) на 1-й день путем введения 5 мг дозы очищенного белка rТbр2, эмульгированного в полном адъюванте Фрейнда (CFA). Животных подвергали бустер-иммунизации на 14-й и 29-й день путем введения аналогичной дозы белка, эмульгированного в неполном адъюванте Фрейнда (IFA). Пробы крови брали на 42-й день для анализа титров антитела против rTbp2 и бактерицидной активности. В таблице 2 показана бактерицидная активность антител, продуцированных против трансферрин-связывающих рекомбинантных белков rTbp1 (4223), rTbp2 (4223) и rTbp2 (Q8), полученных, как описано в этих примерах, против штаммов 4223 и Q8 М.catarrhalis.
Пример 17
Этот пример иллюстрирует связывание Тbр2 с трансферрином человека in vitro.
Трансферрин-связывающую активность Тbр2 оценивали способами, описанными Schryvers and Lee (см. 28), в которые были внесены некоторые модификации. Для этого очищенный rTbp2 подвергали электрофорезу в прерывистом двухслойном 12,5%-ном ПААГ-ДСН. Белки электрофоретически переносили на мембрану PVDF и инкубировали с трансферрином человека, конъюгированным с пероксидазой хрена (HRP-трансферрин человека, разведение 1:50) (Jackson Immuno Research Labs Inc., Mississauga, Ontario), в течение ночи при 4° С. Для хемилюминесцентной детекции HRP-активности использовали субстрат LumiGLO (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD) в соответствии с инструкциями производителей. В этих условиях как rTbp2 4223, так и rTbp2 Q8 связывались с трансферрином человека, как показано на фиг.24
Пример 18
Этот пример иллюстрирует антигенную консервативность белка Тbр2 для штаммов М.catarrhalis.
Цельные клеточные лизаты штаммов М.catarrhalis и штаммов E.coli, экспрессирующих рекомбинантные белки Тbр2, выделяли путем электрофореза на ПААГ с ДСН, и электрофоретически переносили на мембрану PVDF. Для детекции Тbр2 в качестве "первого" антитела была использована антисыворотка морских свинок против rTbp2 4223 или против rTbp2 Q8, а в качестве "второго" антитела было использовано конъюгированное с щелочной фосфатазой козье антитело против иммуноглобулинов морских свинок. Были протестированы штаммы 3, 56, 135, 585, 4223, 5191, 8185 и АТСС 25240 M.catarrhalis, и все они обнаруживали способность к специфическому связыванию с антителом против rTbp2 4223 или против rTbp2 Q8 (фиг.25).
В таблице 3 проиллюстрирована способность антител против rTbp2 от одного штамма M.catarrhalis распознавать нативный или рекомбинантный белок, происходящий от гомологичного или гетерологичного штамма M.catarrhalis.
Пример 19
Этот пример иллюстрирует PCR-амплификацию гена tbpB от штамма R1 M.catarrhalis и характеризацию амплифицированного гена tbpB R1.
Хромосомную ДНК из штамма R1 M.catarrhalis получали стандартными методами. Олигонуклеотидный смысловой праймер конструировали исходя из области, составляющей приблизительно 274 оснований, расположенных выше гена tbpB 4223 M.catarrhalis, а антисмысловой праймер конструировали исходя из области, составляющей приблизительно 11 оснований, расположенных ниже конца гена tbpB 4223. Были использованы следующие праймеры:
Смысловой праймер (4940): 5'-GATATAAGCACGCCCTACTT-3' (SEQ ID No: 48)
Антисмысловой праймер (4967): 5'-CCCATCAGCCAAACAAACATTGTGT-3' (SEQ ID No: 49)
Каждая реакционная пробирка содержала 10 мМ Трис-HCl (рН 8,85), 25 мМ КСl, 5 мМ (NH4)2SO4, 2 мМ MgSO4, 800 мМ dNTP, 1,0 мг каждого из праймеров 4940 и 4967, 10 нг ДНК R1, и 2,5 единиц ДНК-полимеразы Pwo (Boehringer Mannheim) в полном объеме 100 мкл. Устанавливали следующий температурный цикл: первый цикл - 5 минут при 95° С, последующие 25 циклов: 30 секунд при 95° С, 45 секунд при 50° С, и 2 минуты при 72° С, и дополнительный цикл - 10 минут при 72°С. Амплифицированный продукт очищали с использованием набора Geneclean (BIO 101) в соответствии с инструкциями производителей, а затем секвенировали.
Рестрикционная карта tbpB штамма R1 M.catarrhalis, полученная путем частичного гидролиза, как описано выше, показана на фиг.26. Нуклеотидные и выведенные аминокислотные последовательности PCR-амплифицированного гена tbpB R1 показаны на фиг.27. Ген tbpB R1 кодирует белок с 714 аминокислотами, имеющий молекулярную массу 76,8 кДа. Лидерная последовательность белка Tbp2 R1 идентична лидерной последовательности белков Тbр2 штаммов 4223 и Q8. При сравнении выведенной первичной последовательности Tbp2 R1 с первичной последовательностью Tbp2 4223 было установлено, что они имеют идентичность 83% и гомологию 88% (фиг.28). Как было обнаружено, в белках Tbp2 от других штаммов M.catarrhalis, а также в белках Tbp2 от H.influenzae и N.meningitidis присутствует консервативный эпитоп LEGGFYG (SEQ ID No: 50).
Краткое изложение сущности изобретения
По своему существу, настоящее изобретение относится к получению очищенных и выделенных ДНК-молекул, содержащих гены белка рецептора трансферрина Moraxella catarrhalis; к последовательностям этих генов, кодирующих белок рецептора трансферрина; и к их выведенным аминокислотным последовательностям. Эти гены и ДНК-последовательности могут быть использованы для диагностики, иммунизации, и получения диагностических и иммунологических реагентов. Для предупреждения заболеваний, вызываемых Moraxella могут быть получены иммуногенные композиции, включая вакцины, продуцированные на основе экспрессированных рекомбинантных белков Tbp1 и Tbp2, их фрагментов, или аналогов. Возможны модификации, не выходящие за рамки объема настоящего изобретения.


Титры антител выражали как log2 разведения антисыворотки, способной к уничтожению 50% клеток. NT = не тестировали.
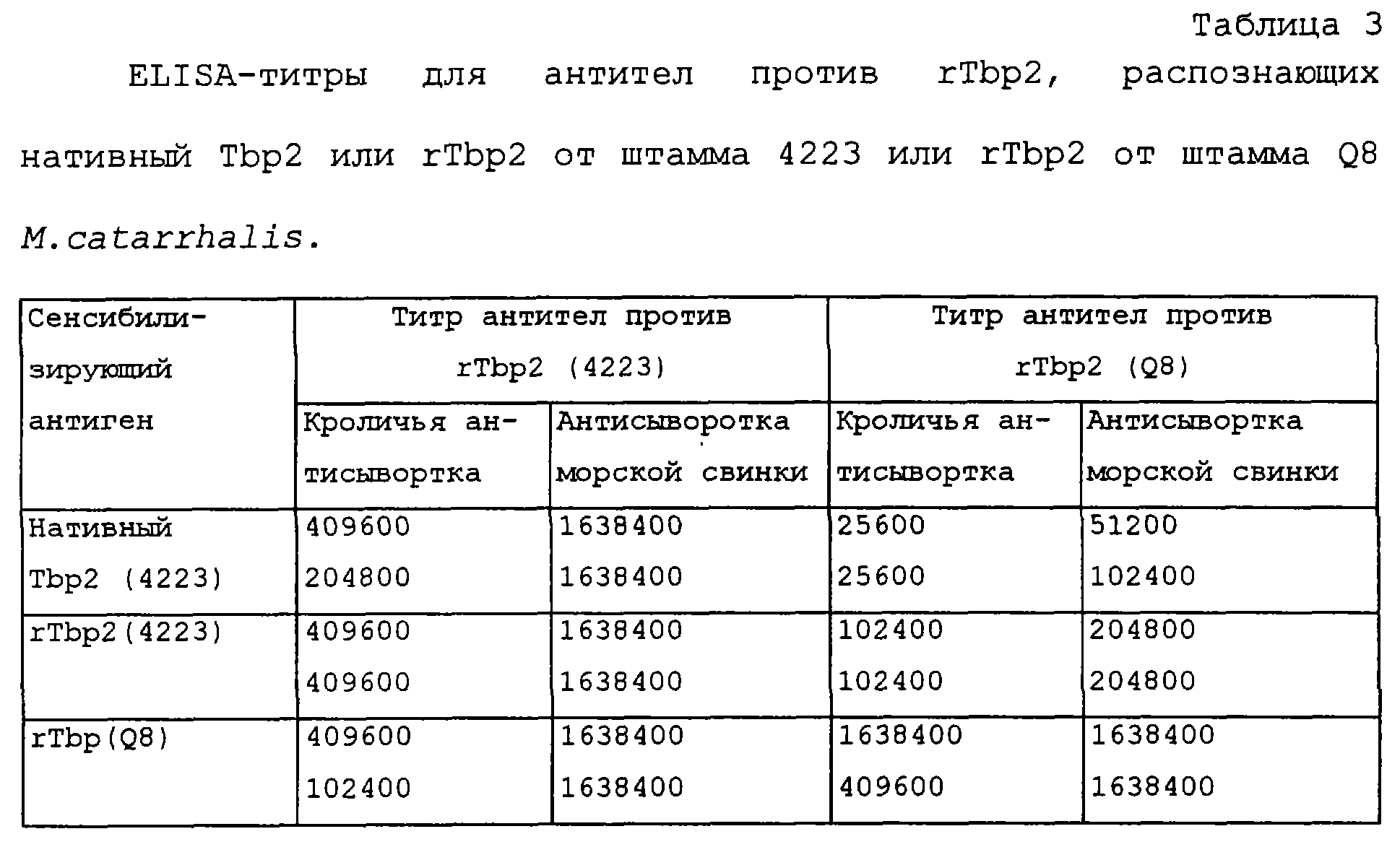


Реферат
Изобретение относится к биотехнологии и представляет собой очищенную и выделенную молекулу нуклеиновой кислоты, которая кодирует белок бактерии Moraxella catarrhalis или иммуногенный фрагмент рецептора трансферрина, а также способ получения рекомбинантного белка рецептора трансферрина. Нуклеиновая кислота может быть использована в составе иммуногенной композиции, применяемой при вакцинации против заболеваний, вызываемых бактерией Moraxella catarrhalis, для диагностики инфекций Moraxella, а также в качестве инструмента для получения иммунологических реагентов. 8 н. и 6 з.п. ф-лы, 90 ил., 3 табл.

Комментарии