Кольцевая молекула днк, характеризующаяся зависимой от условий точкой начала репликации, способ их получения и их применение в генной терапии - RU2307870C2
Код документа: RU2307870C2
Чертежи














































Описание
Настоящее изобретение относится к новой молекуле ДНК с зависимой от условий репликацией, которая может использоваться в генной терапии или для продукции рекомбинантных белков. Новые молекулы ДНК согласно изобретению здесь и далее обозначены pCOR™.
Генная терапия состоит в коррекции дефицита или аномалии путем введения в пораженный орган или клетку генетической информации. Данная информация может вводиться in vitro в клетку, выделенную из органа и затем снова вводимую в организм, или in vivo непосредственно в ткань-мишень. Как молекула с большой молекулярной массой и отрицательным зарядом, ДНК с трудом преодолевает фосфолипидные клеточные мембраны. Различные векторы, таким образом, используют для обеспечения переноса генов: вирусные векторы, с одной стороны, и природные или синтетические химические и/или биохимические векторы, с другой стороны.
Вирусные векторы (ретровирусы, аденовирусы, аденоассоциированные вирусы и т.д.) очень эффективны, в частности, в пересечении мембран, но связаны с некоторым риском, таким как патогенность, рекомбинация, репликация и иммуногенность.
Химические и/или биохимические векторы позволяют обойти указанный риск (см. обзоры, Behr, 1993, Cotton and Wagner 1993). Они представляют собой, например, катионы (фосфат кальция, DEAE-декстран и т.д.), которые действуют путем образования преципитатов с ДНК, способных "фагоцитироваться" клетками. Они также могут быть липосомами, в которые встроена ДНК и которые сливаются с плазматической мембраной. Синтетические векторы для переноса генов в основном представляют собой липиды или катионные полимеры, которые образуют комплекс с ДНК и формируют вместе с ней частицу, несущую положительные катионные заряды. Для иллюстрации векторов данного типа следует упомянуть, в частности, диоктадециламидоглицилспермин (DOGS, Transfectam™) или N-[1-(2,3-диолеилокси)пропил]-N,N,N-триметиламмоний (DOTMA, Lipofectin™).
Однако применение химических и/или биохимических векторов или голой ДНК предполагает возможность продукции больших количеств ДНК фармакологической чистоты. Причина этого в том, что в способах генной терапии медицинский продукт состоит из ДНК самой по себе, и существенно, чтобы имелась возможность производства в подходящих количествах ДНК, характеризующихся свойствами, которые могут использоваться для лечебных применений у человека.
В случае невирусной векторологии используемые векторы представляют собой плазмиды бактериального происхождения. Плазмиды, в основном используемые в генной терапии, несут (i) точку начала репликации, (ii) маркерный ген, такой как ген устойчивости к антибиотику (канамицину, ампициллину и т.д.), (iii) один или более трансгенов с последовательностями, необходимыми для их экспрессии (энхансер(ы), промотор(ы), последовательности полиаденилирования и пр.). Однако доступная в настоящее время технология не является полностью удовлетворительной.
С другой стороны, остается риск распространения по организму. Так, бактерия, которая присутствует в организме, может с низкой вероятностью получить данную плазмиду. Имеется большая вероятность данного происшествия, если используется лечение путем генной терапии in vivo, при которой ДНК может распространяться по организму пациента и может контактировать с бактериями, которые инфицируют данного пациента, или с бактериями симбиотической флоры. Если бактерия, получающая плазмиду, представляет собой энтеробактерию, такую как E. coli, данная плазмида может реплицироваться. Затем это событие приводит к распространению терапевтического гена. Поскольку терапевтические гены, используемые в лечении путем генной терапии, могут кодировать, например, лимфокин, фактор роста, антионкоген или белок, функция которого дефектна в организме хозяина, и они дают возможность коррекции генного дефекта, распространение некоторых из этих генов может оказывать непредсказуемое и тревожное действие (например, если патогенная бактерия приобрела ген человеческого фактора роста).
С другой стороны, плазмиды, в общем используемые в невирусной генной терапии, также обеспечивают маркер устойчивости к антибиотику (ампициллину, канамицину и т.д.). Бактерия, несущая такую плазмиду, имеет неоспоримое селективное преимущество, поскольку любое лечение антибиотиками, в котором используются антибиотики того же семейства, что и тот, что служит для селекции путем плазмидного гена устойчивости, будет приводить к селекции указанной плазмиды. В данном отношении ампициллин относится к a-лактамам, представляющим семейство антибиотиков, которое наиболее часто применяется в мире. Применение в бактериях маркеров селекции, которые не представляют собой гены устойчивости к антибиотикам, является особенно предпочтительным. Оно позволит избежать селекции бактерий, которые могут получить плазмиду, несущую такой маркер.
Так, особенно важно постараться как можно сильнее ограничить пределы распространения терапевтических генов и генов устойчивости.
Конкретно, предметом настоящего изобретения является представление новых молекул ДНК, которые могут использоваться в генной терапии или для продукции рекомбинантных белков in vitro и которые реплицируются только в клетках, которые могут дополнить некоторые функции данных невирусных векторов.
Изобретение также относится к особенно эффективному способу получения данных молекул ДНК.
Заявленные здесь молекулы ДНК имеют преимущества, позволяющие избежать риск, ассоциированный с распространением плазмиды, такой как (1) репликация и распространение, что может привести к неконтролируемой гиперэкспрессии терапевтического гена, (2) распространение и экспрессия генов устойчивости. Генетическая информация, содержащаяся в молекулах ДНК согласно изобретению, эффективно включает в себя терапевтический(-ие) ген(-ы) и сигналы регуляции его (их) экспрессии, функциональную, зависимую от условий точку начала репликации, которая сильно ограничивает спектр клеток хозяина для плазмиды, маркер селекции сниженного размера, который предпочтительно отличается от гена, который придает устойчивость к антибиотикам, и, где это подходит, фрагмент ДНК, который обеспечивает разрешение мультимеров плазмиды. Вероятность того, что данные молекулы (и, таким образом, генетическая информация, которую они содержат) будут перенесены в микроорганизм и там будут стабильно поддерживаться, очень ограничена.
Наконец, векторы согласно изобретению, также называемые миниплазмидами за счет их кольцевой структуры, сниженного размера и сверхспиральной формы, имеют следующие дополнительные преимущества: за счет их размера, который снижен по сравнению с обычно применяемыми происходящими от ColE1 плазмидами, молекулы ДНК согласно изобретению потенциально характеризуются лучшей биодоступностью in vivo, и данные молекулы ДНК, или pCOR, остаются в стабильной внехромосомной форме прокариотических или эукариотических клетках хозяина, которые не содержат инициирующего белка. В частности, они имеют улучшенные способности проникновения в клетки и распространения в них. Так, допускается, что коэффициент диффузии в тканях обратно пропорционален молекулярной массе (Jain, 1987). Сходным образом, в клетке молекулы с высокой молекулярной массой характеризуются меньшей проницаемостью через плазматическую мембрану. Кроме того, в плане проникновения плазмиды в ядро, что существенно для ее экспрессии, высокая молекулярная масса также является недостатком, причем ядерные поры устанавливают предел размеров для диффузии в ядро (Landford et al, 1986). Снижение размера нетерапевтических частей молекулы ДНК (точки начала репликации и гена селекции, в частности) согласно изобретению также дает возможность для снижения размера молекул ДНК. Часть, которая обеспечивает репликацию и селекцию плазмиды в бактерии (1 т.п.н.), снижена в 3 раза, составляя, например, 3 т.п.н. для части вектора, включающей в себя точку начала репликации и маркерный ген устойчивости. Данное снижение (i) молекулярной массы и (ii) отрицательного заряда обеспечивает улучшенную биодоступность и диффузию молекул согласно изобретению в отношении тканей, клеток и ядер.
Более конкретно, настоящее изобретение относится к кольцевой молекуле ДНК, которая может использоваться в генной терапии, причем данная молекула содержит по меньшей мере одну интересующую последовательность нуклеиновой кислоты и характеризуется тем, что область, которая обеспечивает ее репликацию, содержит точку начала репликации, функциональность которой в клетке хозяина требует наличия по меньшей мере одного конкретного белка, который является чужеродным по отношению к указанной клетке хозяина.
Данная молекула ДНК может быть в одно- или двухцепочечной форме и предпочтительно обладает сверхспиральной формой.
Для целей настоящего изобретения используемые клетки хозяина могут быть различного происхождения. Они могут быть эукариотическими или прокариотическими клетками. Согласно предпочтительному осуществлению настоящего изобретения они являются прокариотическими клетками.
Репликация бактериальных плазмид обычно требует присутствия по меньшей мере одного белка, который кодируется клеткой хозяина, типа РНК-полимеразы, РНКазы, ДНК-полимеразы и т.д. По уже объясненным выше причинам, с данным типом репликации невозможно полностью преодолеть любой вероятный риск распространения в подвергающемся лечению организме. Предпочтительно, функциональность точки начала репликации молекулы ДНК согласно изобретению требует наличия конкретного белка, который является чужеродным для клетки хозяина. Значение данной характеристики состоит в сужении спектра хозяев для заявленной плазмиды до конкретных штаммов, которые экспрессируют инициаторный белок. Молекула ДНК, разработанная в контексте настоящего изобретения, таким образом, предпочтительно обладает так называемой зависимой от условий точкой начала репликации.
Зависимая от условий точка начала репликации, используемая согласно изобретению, может происходить из плазмид или бактериофагов, которые характеризуются следующими характеристиками: они содержат в своей точке репликации последовательности повтора, или итероны, и они кодируют по меньшей мере один инициирующий репликацию белок (Rep), который является специфическим для них. Для примера могут быть указаны зависимые от условий системы репликации следующих плазмид и бактериофагов:
Согласно предпочтительному осуществлению изобретения точка начала репликации, используемая в заявленных молекулах ДНК, происходит из природной плазмиды E. coli, обозначенной как R6K.
Функции репликации R6K сгруппированы во фрагменте ДНК размером 5,5 т.п.н. (фигура 1), содержащем 3 точки начала репликации a, Я и (и a обеспечивают 90% репликации) и оперон, кодирующий инициаторный белок репликации П и белок Bis. Минимальное количество генетической информации, требуемое для поддержания данной плазмиды в числе копий, характерном для нее (15 копий на геном), содержится в двух элементах: 400 п.н. ori и ген pir, продукт которого есть инициаторный белок П.
Ori может разделяться на две функциональные части: коровая часть и активаторный элемент (фигура 1). Коровая часть, существенная для репликации, содержит итероны (7 прямых повторов по 22 п.н.), с которой связывается белок П, представленный в SEQ ID No.1, и фланкирующие сегменты, которые являются мишенями для белков хозяина (IHF, DnaA).
Согласно предпочтительному способу изобретения точка начала репликации заявленного вектора состоит полностью или частично из данной точки начала репликации плазмиды R6K и, более предпочтительно, полностью или частично из SEQ ID No. 1 или одного из ее производных.
Точка начала репликации, описанная выше, которая характеризуется преимуществом очень ограниченного размера, функциональна исключительно в присутствии специфического инициаторного белка, белка Pi, продуцированного геном pir (SEQ ID No. 2). Поскольку данный белок может действовать в транс-положении, возможно физически отделить ori гамма от гена pir, который может вводиться в геном клетки, выбранной в качестве специфического хозяина для данных плазмид. Мутации в П могут изменять его ингибиторные функции (Inuzuka and Wada, 1985) и приводить к снижению числа копий производных R6K, вплоть до количества, более чем в 10 раз превышающего изначальное число копий. Данные замены могут иметь место внутри домена из 40 аминокислот, который поэтому оказывается ответственным за контроль числа копий плазмиды, обеспечиваемый П (фигура 2), или в других областях белка П.
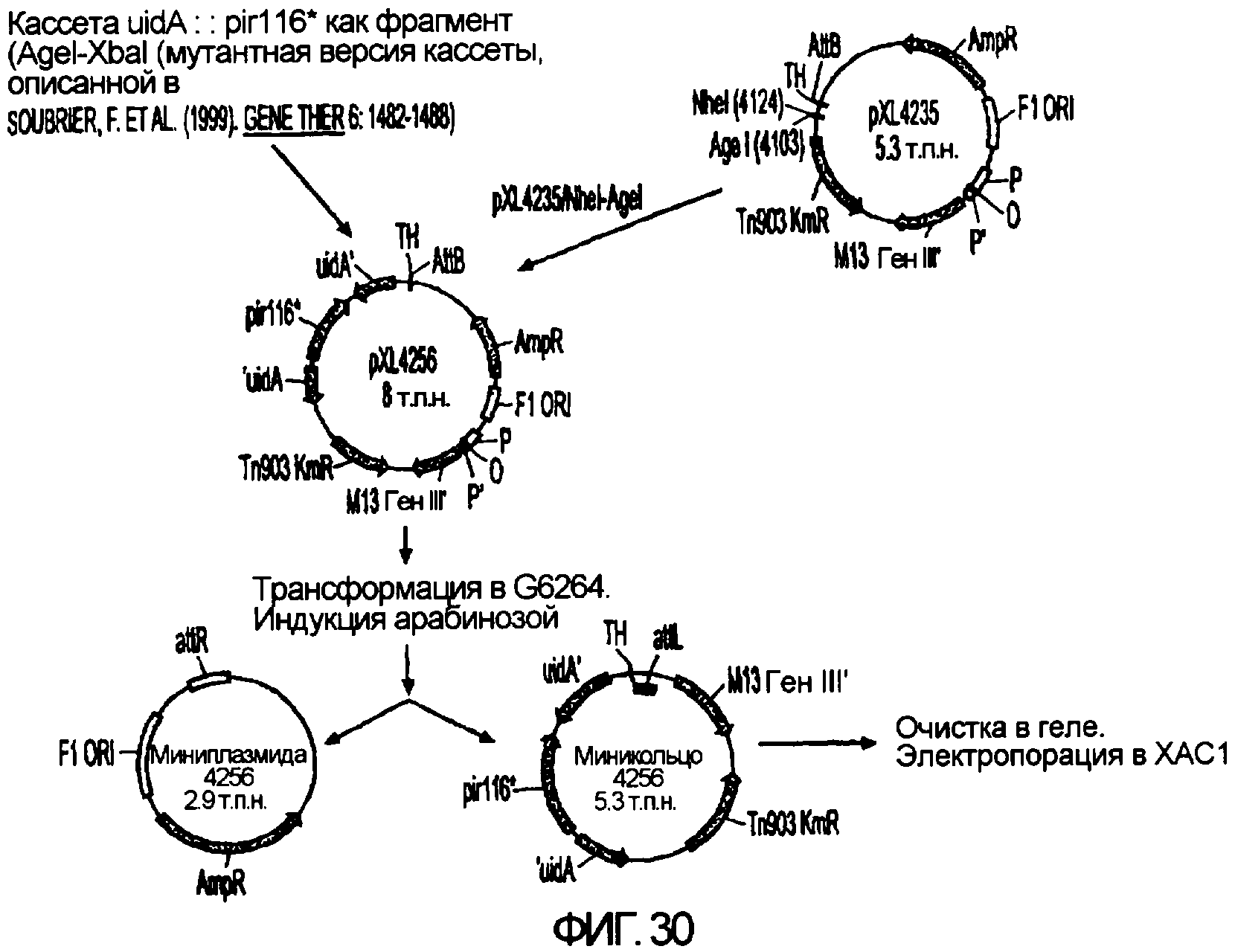
Согласно предпочтительному осуществлению настоящего изобретения белок П, экспрессируемый в клетке хозяина, является результатом экспрессии гена, представленного в SEQ ID No. 2 или одного из его производных, определяемых выше, и, более конкретно, гена pir 116, который содержит мутацию по сравнению с геном pir. Данная мутация соответствует замене пролина на лейцин в положении 106 от стартового кодона. В данном контексте число копий производных R6K составляет примерно 250 на геном.
Для целей настоящего изобретения термин "производное" означает любую последовательность, которая отличается от рассматриваемой последовательности, полученной путем одной или нескольких модификаций генетической и/или химической природы, а также любую последовательность, которая гибридизуется с данными последовательностями или их фрагментами и продукт которых обладает активностью, обозначенной в отношении инициирующего репликацию белка П. Термин "модификаций генетической и/или химической природы" можно понимать как обозначение любой мутации, замены, делеции, добавления и/или модификации одного или нескольких остатков. Термин "производное" также включает в себя последовательности, гомологичные в отношении рассматриваемой последовательности, происходящие из других клеточных источников и, в частности, клеток человеческого происхождения, или из других организмов, и обладающие активностью того же типа. Такие гомологичные последовательности могут быть получены путем экспериментов по гибридизации. Гибридизации могут проводиться, начиная с библиотек нуклеиновых кислот, с использованием природной последовательности или ее фрагмента в качестве зонда, при общепринятых условиях жесткости (Maniatis et al., cf. General techniques of molecular biology), или, предпочтительно, при условиях высокой жесткости.
Кроме зависимой от условий точки начала репликации, определяемой выше, заявленные молекулы ДНК содержат область, включающую в себя один (или несколько) ген (-ов), что дает возможность обеспечения селекции молекулы ДНК в выбранном хозяине.
Это может быть общепринятый маркер типа гена, который придает устойчивость к антибиотику, такому как канамицин, ампициллин, хлорамфеникол, стрептомицин, спектиномицин, ливидомицин или подобные им.
Однако согласно предпочтительному осуществлению изобретения данная область отличается от гена, который придает устойчивость к антибиотику. Это, таким образом, может быть ген, существенный для жизнеспособности рассматриваемого хозяина при определенных условиях культивирования. Это, например, может быть
- ген, кодирующий супрессорную тРНК природного или синтетического происхождения. Это, более предпочтительно, тРНК амбер-кодона (TAG);
- ген, продукт которого необходим для метаболизма клетки в определенных условиях культивирования, а именно, ген, участвующий в биосинтезе метаболита (аминокислоты, витамина и т.д.), или ген катаболизма, который дает возможность для ассимиляции вещества, присутствующего в культуральной среде (конкретного источника азота или углерода) и т.д.
Согласно предпочтительному способу изобретения данная область содержит экспрессирующую кассету гена, кодирующего супрессорную тРНК для конкретных кодонов. Этот последний может быть выбран, в частности, из кодирующих аминокислоты фенилаланин, цистеин, пролин, аланин и гистидин. Более конкретно, это супрессорная тРНК для амбер-кодонов (TAG).
В данном конкретном случае система, используемая для селекции в клетках хозяина молекул ДНК согласно изобретению, включает в себя два элемента: 1) ген на молекуле ДНК, кодирующий супрессорную транспортную ДНК для амбер-кодона (TAG), которая составляет маркер селекции, известный как ген (sup), и 2) конкретный хозяин, один из генов которого, который является существенным при определенных условиях культивирования, содержит амбер-кодон TAG. Данная клетка может расти в условиях культивирования, при которых существенен продукт гена, содержащего кодон TAG, только если плазмида, обеспечивающая экспрессию sup, присутствует в клетке. Условия культивирования, таким образом, обеспечивают пресс селекции молекулы ДНК. Используемые гены sup могут быть природного происхождения (Glass et al., 1982) или могут происходить из синтетической конструкции (Normanly et al., 1986, Kleina et al., 1990).
Такая система предоставляет большую гибкость, поскольку в зависимости от гена, содержащего амбер-мутацию, возможно определить различные селективные среды. В бактерии Lactococcus lactis амбер-кодон, например, расположен в гене биосинтеза пурина. Это обеспечивает селекцию плазмиды, несущей ген, кодирующий супрессорную тРНК, когда бактерии размножаются в молоке. Такой маркер имеет преимущество малого размера и не содержит "чужеродных" последовательностей, происходящих из фагов или транспозонов.
Согласно предпочтительному осуществлению изобретения молекула ДНК также включает в себя фрагмент ДНК, мишень для сайт-специфических рекомбиназ, что обеспечивает разрешение плазмидных мультимеров.
Таким образом, такой фрагмент, введенный в молекулу ДНК, которая является кольцевой и точка начала репликации которой, например, представляет собой, например, ori гамма, обеспечивает разрешение мультимеров такой плазмиды. Такие мультимеры наблюдаются, в частности, когда молекулу ДНК получают в штамме, несущем мутантный аллель pir, такой как pir116, что дает возможность увеличения числа копий производных R6K.
Рекомбинация может достигаться посредством различных систем, которые вызывают сайт-специфическую рекомбинацию между последовательностями. Более предпочтительно, сайт-специфическая рекомбинация согласно изобретению достигается средствами специфических последовательностей внутримолекулярной рекомбинации, способных рекомбинировать друг с другом в присутствии специфических белков, в общем называемых рекомбиназами. В данном конкретном случае имеют место рекомбиназы XerC и XerD. По этой причине молекулы ДНК согласно изобретению в общем также включают в себя последовательность, которая обеспечивает данную сайт-специфическую рекомбинацию. Система специфической рекомбинации, присутствующая в генетических конструкциях согласно изобретению (рекомбиназы и участки специфического распознавания), могут быть разного происхождения. В частности, используемые специфические последовательности и рекомбиназы могут принадлежать различным структурным классам, и в частности, к семейству резолвазы транспозона Tn3 или семейству интегразы бактериофага лямбда. Из рекомбиназ, принадлежащих семейству транспозона Tn3, могут быть упомянуты, в частности, резолваза транспозона Tn3 или транспозонов Tn21 и Tn522 (Stark et al., 1992); инвертаза Gin бактериофага мю или, альтернативно, плазмидные резолвазы, такие как из par-фрагмента RP4 (Abert et al., Mol. Microbiol. 12 (1994) 131). Из рекомбиназ, принадлежащих семейству интегразы бактериофага, могут быть упомянуты, в частности, интеграза фагов лямбда (Landy et al., Science 197 (1977) 1147), P22 и Ф80 (Leong et al., J. Biol. Chem. 260 (1985) 4468), HP1 Haemophilus influenzae (Hauser et al., J. Biol. Chem. 267 (1992) 6859), интеграза Cre фага P1, интеграза плазмиды pSAM2 (EP 350 341) или, альтернативно, рекомбиназа FLP плазмиды 2μ и рекомбиназы XerC и XerD из E. coli.
Предпочтительно молекулы ДНК, которые относятся к настоящему изобретению, содержат фрагмент cer из природной плазмиды E. coli ColEl. Используемый фрагмент cer представлен 382 п.н. HpaII из ColEl, который, как было показано, обеспечивает в цис-положении разрешение мультимеров плазмид (Summers et al., 1984; Leung et al., 1985). Также возможно применение фрагмента HpaII-TaqI меньшего размера (280 п.н.) или меньшего фрагмента (примерно 220 п.н.), содержащегося во фрагменте Hpall, причем эти фрагменты обладают одинаковыми свойствами (Summers and Sherratt, 1988). Данное разрешение происходит путем специфической внутримолекулярной рекомбинации, в которой используются четыре белка, кодируемых в геноме E. coli: ArgR, PepA, XerC и XerD (Stirling et al., 1988, 1989; Colloms et al., 1990, Blakely et al., 1993). Было обнаружено, что вставка фрагмента cer из природной плазмиды E. coli ColEl позволяет получить высокую степень разрешения мультимеров плазмид, с получением, таким образом, большой доли мономеров воспроизводимым образом. Это особенно неожиданно, поскольку было показано, что вставка участка cer в мини-кольцо, которое содержит точку начала репликации ColEl из pBluescript SK+, не приводит к эффективному разрешению мультимеров (Kreiss et al., Appl. Microbiol. Biotechnol, 49: 560-567 (1998)), и, таким образом, эффективное разрешение в цис-положении плазмид является непредсказуемым и, как представляется, зависит от конформации плазмиды. В случае плазмиды pCOR эффективное разрешение в цис-положении достигается, когда cer присутствует на pCOR, что приводит к получению непредсказуемо высокого количества мономеров pCOR воспроизводимым образом.
В этой связи особенно предпочтительно использовать весь cer-фрагмент ColEl или его часть, или одно из его производных, как это определено выше.
Согласно некоторому варианту осуществления, молекулы ДНК согласно изобретению могут также содержать последовательность, способную специфически взаимодействовать с лигандом. Предпочтительно, это последовательность, способная образовывать путем гибридизации тройную спираль со специфическими олигонуклеотидами. Данная последовательность, таким образом, дает возможность для очистки молекул согласно изобретению путем селективной гибридизации с комплементарным олигонуклеотидом, иммобилизованным на подложке (см. заявку WO 96/18744 и WO 02/07727). Данная последовательность может присутствовать в природе в точке начала репликации плазмиды, как описано в опубликованной заявке на выдачу патента США 2003/186268, принадлежащей настоящему заявителю, или присутствовать в природе в трансгене, как описано в WO 02/07727, и, альтернативно, может располагаться в любом участке в молекуле ДНК согласно изобретению при обеспечении того, что она не будет влиять на функциональность интересующего гена и точки начала репликации. Образование тройной спирали путем гибридизации, таким образом, происходит между олигонуклеотидом и специфической комплементарной последовательностью, присутствующей в ДНК. В этой связи, для получения лучших выходов и большей избирательности олигонуклеотид и специфичная последовательность, которые являются полностью комплементарными, используют в способе согласно изобретению. Это может быть, в частности, олигонуклеотид поли(CTT) и специфичная последовательность поли(GAA). Например, олигонуклеотиды, содержащие повторяемые мотивы, такие как CTT, способны образовывать тройную спираль с конкретной последовательностью, содержащей комплементарные единицы (GAA). Интересующая последовательность может, в частности, представлять собой область, содержащую 7, 14 или 17 единиц GAA, и в олигонуклеотидах - соответствующие количества повторов CTT. В данном случае олигонуклеотид связывается в антипараллельной ориентации с полипуриновой цепью. Данные тройные спирали стабильны только в присутствии Mg2+ (Vasquez et al., Biochemistry, 34: 7243-7251 (1995); Beal and Dervan, Science, 251: 1360-1363 (1991)).
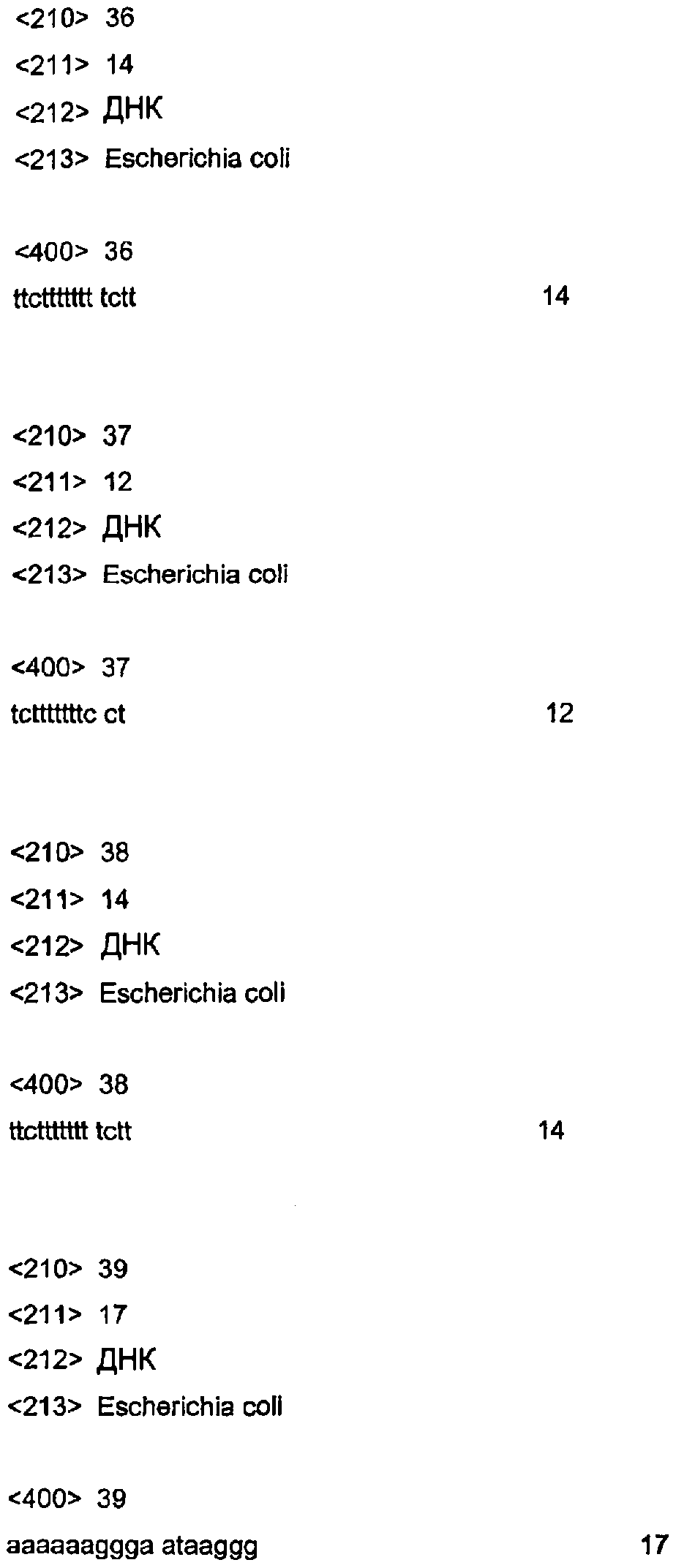
Как указывалось выше, специфичная последовательность может представлять собой последовательность, присутствующую в природе в pCOR, или может представлять собой синтетическую последовательность, искусственно введенную в последнюю. Особенно предпочтительно применение олигонуклеотида, способного образовывать тройную спираль с последовательностью, присутствующей в природе в pCOR, например, в точке начала репликации плазмиды или в маркерном гене. Синтез олигонуклеотидов, способных образовывать тройные спирали с данными природными областями гомопуринов-гомопиримидинов, является особенно предпочтительным, поскольку он может применяться на немодифицированных плазмидах pCOR. Особенно предпочтительные последовательности-мишени, которые могут образовывать тройные структуры с конкретными олигонуклеотидами, идентифицированы в точках начала репликации ColEl и pCOR. Происходящие от ColEl плазмиды содержат 12-членную гомопуриновую последовательность (5'-AGAAAAAAAGGA-3') (SEQ ID NO: 33), картированную выше транскрипта РНК-II, участвующего в репликации плазмиды (Lacatena et al., Nature, 294: 623 (1981)). Данная последовательность образует стабильную тройную структуру с комплементарным 12-членным олигонуклеотидом 5'-TCTTTTTTTCCT-3' (SEQ ID NO: 34). Остов pCOR содержит гомопуриновую полосу из 14 неповторяемых оснований (5'-AAGAAAAAAAAGAA-3') (SEQ ID NO:35), расположенных в A+T-богатом сегменте репликона точки начала репликации pCOR (Levchenko et al., Nucleic Acids Res., 24: 1936 (1996)). Данная последовательность образует стабильную тройную структуру с 14-членным комплементарным олигонуклеотидом 5'-TTCTTTTTTTTCTT-3' (SEQ ID NO: 36). Соответствующие олигонуклеотиды 5'-TCTTTTTTTCCT-3' (SEQ ID NO: 37) и 5'-TTCTTTTTTTTCTT-3' (SEQ ID NO: 38) эффективно и специфически направленно действуют на соответствующие им комплементарные последовательности, расположенные в пределах точки начала репликации ori ColEl или pCOR (ori ). Также применение олигонуклеотида, способного образовывать тройную спираль с последовательностью, присутствующей в точке начала репликации, или маркерного гена является особенно предпочтительным, поскольку это дает возможность с использованием того же олигонуклеотида очищать любую ДНК, содержащую указанную точку начала репликации указанного маркерного гена. Следовательно, не является необходимым модифицировать плазмиду или двухцепочечную ДНК с введением в нее искусственной специфической последовательности.
Хотя предпочтительны полностью комплементарные последовательности, следует понимать, однако, что могут допускаться некоторые несовпадения между последовательностью олигонуклеотида и последовательностью, представленной в ДНК, при обеспечении того, что они не приведут к очень большой потере сродства. Может указываться последовательность 5'-AAAAAAGGGAATAAGGG-3' (SEQ ID NO: 39), присутствующая в гене β-лактамазы E. coli. В данном случае тимин, прерывающий полипуриновую последовательность, может распознаваться гуанином третьей цепи с образованием, таким образом, триплета G*TA, который стабилен при фланкировании двумя триплетами T*AT (Kiessling et al., Biochemistry, 31: 2829-2834 (1992)).
Согласно конкретному осуществлению, используемые олигонуклеотиды могут содержать последовательность (CCT)n, последовательность (CT)n или последовательность (CTT)n, в которых n представляет собой целое число от 1 до 15 включительно. Особенно предпочтительным является применение последовательностей типа (CT)n или (CTT)n. В олигонуклеотидах также могут сочетаться единицы (CCT), (CT) или (CTT).
Используемые олигонуклеотиды могут быть природными (состоящими из немодифицированных природных оснований) или химически модифицированными. В частности, олигонуклеотид может предпочтительно иметь конкретные химические модификации, обеспечивающие повышение его устойчивости или защиты от нуклеаз, или его сродства в отношении специфической последовательности.
В качестве репрезентативной молекулы ДНК согласно изобретению наиболее конкретно может быть заявлена плазмида pXL2774 и ее производные. Для целей изобретения термин "производное", как он понимается, относится к любой конструкции, происходящей от pXL2774 и содержащей один или несколько интересующих генов, отличных от гена люциферазы. Также следует указать на плазмиды pXL3029, pXL3030 и плазмиду pXL3179 или NV1FGF, содержащие экспрессирующие кассеты терапевтического гена. В наиболее предпочтительном осуществлении изобретение относится к pCOR, содержащей ген FGFa или FGF-1, как описано в патенте США 4686113, принадлежащем настоящему заявителю, которая обозначена pXL3179 или NV1FGF.
Настоящее изобретение также относится к разработке способа конструирования специфической клетки хозяина, которая особенно эффективна для продукции данных терапевтических молекул ДНК.
Другой предмет настоящего изобретения относится к способу продукции кольцевой молекулы ДНК, характеризующемуся тем, что культивируется клетка хозяина, содержащая по меньшей мере одну молекулу ДНК, определяемую выше, и белок, который может экспрессироваться in situ или не экспрессируется, причем данный белок обуславливает функциональность точки начала репликации указанной молекулы ДНК, которая является специфической и которая является чужеродной для указанной клетки хозяина, в условиях, которые обеспечивают селекцию клетки хозяина, трансформированного указанными молекулами ДНК.
Более предпочтительно, белок, который обуславливает функциональность точки начала репликации молекулы ДНК, экспрессируется in situ из соответствующего гена. Ген, кодирующий белок инициации репликации, может находиться во вспомогательном репликоне, который совместим с производными используемой общепринятой точки начала репликации или который может вводиться в геном клетки-хозяина путем рекомбинации посредством транспозона, бактериофага или любого другого вектора. В конкретном случае, в котором ген, экспрессирующий белок, помещается во вспомогательный репликон, последний также содержит промоторную область для функциональной транскрипции в клетке, а также область, которая локализована на 3'-конце и которая определяет сигнал терминации транскрипции. В отношении промоторной области это может быть промоторная область, которая в природе ответственна за экспрессию рассматриваемого гена, когда последний способен функционировать в данной клетке. Она также может быть случаем областей отличного происхождения (ответственной за экспрессию других белков) или даже синтетического происхождения. В частности, она может являться случаем промоторных последовательностей прокариотических генов или генов бактериофагов. Например, она может являться случаем промоторных последовательностей, полученных из клеточного генома.
В качестве генов, кодирующих белок инициации репликации, могут применяться гены дикого типа или мутированные аллели, которые дают возможность получения повышенного числа копий плазмид (или производных), специфических для инициаторного белка, которые обуславливают функциональность точки начала репликации, используемой в молекуле ДНК.
Такие мутанты описаны, в частности, для плазмид R6K (Inuzuka and Wada, 1985; Greener et al., (1990), Rtsl (Terawaki and Itoh, 1985, Terawaki et al., 1990; Zeng et al., 1990), F (Seelke et al., 1982; Helsberg et al., 1985; Kawasaki et al., 1991), RK2 (Durland et al., 1990; Haugan et al., 1992, 1995), pSC101 (Xia et al., 1991; Goebel et al., 1991; Fang et al., 1993).
В конкретном случае, в котором используемая молекула ДНК обладает точкой начала репликации, происходящей из плазмиды R6K, инициаторный белок представляет собой производное белка П той же самой плазмиды. Особенно предпочтительной является экспрессия мутантной формы данного белка, которая способна существенно увеличивать число начальных копий. Для того, чтобы сделать это, ген, встроенный в клетку хозяина, предпочтительно, представлен частью или целой последовательностью, указанной в SEQ ID No. 2, или одним из его производных, а более предпочтительно - геном pir116.
Ассоциированная мутация соответствует замене пролина лейцином. Согласно конкретному осуществлению изобретения данный ген pir116 непосредственно встраивается в геном клетки хозяина.
Предпочтительно один из генов специфической клетки хозяина, который является необходимым при выбранных условиях культивирования, содержит специфический кодон, который может распознаваться в молекуле ДНК выбранной супрессорной тРНК. Согласно предпочтительному способу изобретения он представляет собой амбер-кодон TAG. В данном конкретном случае клетка может выращиваться в условиях культивирования, когда необходим продукт гена, содержащего кодон TAG, только если в клетке хозяина присутствует плазмида, обеспечивающая экспрессию sup. Условия культивирования, таким образом, оказывают на молекулу ДНК давление селекции.
Предпочтительно ген, содержащий амбер-кодон, представляет собой ген, участвующий в биосинтезе аминокислоты аргинина. Данный ген argE кодирует N-ацетилорнитиназу (Meinnel et al., 1992) и в данном случае содержит кодон TAG, соответствующий точечной мутации Gln-53 (CAG) TAG; затем давление селекции плазмиды, несущей ген sup, предоставляется в минимальной среде M9 (Maniatis et al., 1989). Однако данный ген также может представлять собой, например, ген биосинтеза витамина или основания нуклеиновой кислоты, или, альтернативно, ген, который обеспечивает использование конкретного источника азота или углерода, или любой другой ген, функциональность которого необходима для жизнеспособности клеток в выбранных условиях культивирования.
Клетка хозяина предпочтительно выбрана из штаммов E. coli, а более предпочтительно, представлена штаммом E.coli XAC-1.
Согласно конкретному осуществлению изобретения клетка хозяина, используемая в заявленном способе, представляет собой клетку E. coli штамма XAC-1, содержащую ген pir116 в ее геноме и трансформированную плазмидой pXL2774 или одной из ее производных.
Согласно предпочтительному варианту изобретения клетка хозяина, используемая в заявленном способе, представляет собой прокариотическую клетку, в которой инактивирован ген endA1 или гомологичный ему ген. Ген endA кодирует эндонуклеазу I E. coli. Данный фермент периплазмы характеризуется неспецифической активностью по расщеплению двухцепочечной ДНК (Lehman, I. R., G. G. Roussos and E. A. Pratt (1962) J. Biol. Chem. 237: 819-828; Wright M. (1971) J. Bacteriol. 107: 87-94). Исследование, проведенное на различных штаммах Escherichia coli (дикого типа или endA), показало, что деградация плазмидной ДНК, инкубируемой в экстрактах данных бактериальных штаммов, имела место в штаммах endA+, но не в мутантах endA.(Wnendt S. (1994) BioTechniques 17: 270-272). Качество плазмидной ДНК, выделенной из штаммов endA+ или из мутантов endA, исследовано компанией Promega с использованием их системы очистки (Shoenfeld, T., J. Mendez, D. Storts, E. Portman, B. Patterson, J.Frederiksen and C. Smith. 1995. Effects of bacterial strains carrying the endA1 genotype on DNA quality isolated with Wizard plasmid purification systems. Promega notes 53). Заключение, полученное исследователями, было следующим: качество ДНК, полученной из мутантов endA, в целом выше, чем качество ДНК, полученной из тестируемых штаммов endA+.
Качество препаратов плазмидной ДНК, таким образом, подвергается воздействию любой контаминации данной эндонуклеазой (относительно долговременная деградация ДНК).
Делеция или мутация гена endA может рассматриваться без проблем, поскольку мутанты, более не имеющие данной эндонуклеазной активности, в целом остаются подобными бактериям дикого типа (Durwald, H. and H. Hoffmann-Berling (1968) J. Mol. Biol. 34: 331-346).
Ген endA1 может инактивироваться мутацией, общей или частичной делецией, разрушением и т.д. Инактивация гена endA штамма E. coli, выбранного для продукции плазмид pCOR, может, более конкретно, достигаться путем переноса посредством бактериофага P1, делеции::TcR, описанной Черепановым и Вакернагелем (Cherepanov, P. P. and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158: 9-14) или путем обмена аллеля дикого типа, представленного в геноме интересующей бактерии, на мутированный или подвергнутый делеции аллель endA путем гомологичной рекомбинации. Применение данного типа штаммов в контексте настоящего изобретения дает возможность для улучшения качества продуцированной ДНК.
Изобретение также относится к любой рекомбинантной клетке, содержащей определенную выше молекулу ДНК. Она может быть клеткой различного происхождения, эукариотического, прокариотического типа и т.д.
Согласно другому осуществлению изобретения клетка хозяина E. coli XAC-1, используемая в заявленном способе, обозначена TEX1 и содержит ген traD или гомологичный ему ген, инактивированный для предотвращения переноса F'. traD представляет собой 5'-конец оперона tra и кодирует мембранный белок массой 81,7 кДа, мембранный белок, который непосредственно участвует в переносе ДНК и метаболизме ДНК (Frost et al., Microbiology Reviews, 1994, 58: 162-210). Мутанты traD не осуществляют перенос ДНК (Panicker et al., J. Bacteriol., 1985, 162:584-590). Эписомный ген traD может инактивироваться мутацией, общей или частичной делецией или разрушением, с использованием способов, хорошо известных специалистам в данной области (см. пример 9). Один из способов инактивации данного гена описан в примере 1, и полученный в результате штамм E. coli XAC-1 pir116 endA- traD-, полученный таким образом, обозначали TEX1 (Soubrier et al., Gene Therapy, 1999, 6: 1482-1488).
По одному из осуществлений изобретения клетка хозяина, используемая в заявленном способе, представляет собой клетку штамма E. coli XAC-1, содержащую мутацию pir116, в сочетании с мутацией pir42. Мутации pir116 и pir42 влияют на различные домены белка pi. Мутация pir116 влияет на область контроля числа копий, тогда как мутация pir42 влияет на предполагаемый мотив лейциновой застежки, что отражено на фигуре 11. Нуклеотидная и аминокислотная последовательность гена pir, содержащего мутации pir116 и pir42, приведены на фигуре 12 и в SEQ ID NO: 21 и 22, соответственно. Мутация pir42 включает в себя замену C на T в положении 124 от метионинового инициаторного кодона, что, таким образом, приводит к замене пролина в положении 42 на лейцин. Мутация pir42 описана Miron et al. (Proc Natl Acad Sci U S A, 1994.91 (14): p.6438-42; EMBO J, 1992.11 (3): p. 1205-16), и о ней сообщалось, что она увеличивает число копий плазмиды "ori-гамма R6K-KmR-pir42" в 2,5 раза по сравнению с той же плазмидой, несущей ген pir дикого типа. Однако мутация pir42 никогда не была использована или описана с мутацией pir116, и хотя другие увеличивающие копийность мутации, такие как cop21, в гене pir, сочетанные с pir116, не характеризуются увеличением числа копий плазмиды, сочетание мутаций pir116 и pir42 в штамме E. coli XAC-1 endA- traD- неожиданно характеризовалось значительным повышением числа копий плазмиды. Таким образом, настоящими заявителями показаны неожиданные результаты такого сочетания в плане числа копий плазмиды, продуцируемой в штаммах хозяина E. coli, содержащей мутированный ген pir116 и pir42, по сравнению со штаммами, содержащими только pir116, или с клеткой хозяина, содержащей мутацию pir116, сочетанную с другой мутацией гена pir, такой как мутация cop21 (Inuzuka et al., FEBS Lett, 1988. 228(1): p. 7-11). Например, E.coli TEX1 pir42 (= XAC-1 endA- traD- pir116 pir42) характеризовался увеличением числа плазмид в 2-5 раза по сравнению со штаммом pir116, или штаммом, содержащим сочетание мутаций pir116 и cop21 (см. пример 11). В другом осуществлении ген pir содержит по меньшей мере одну мутацию, которая, например, может происходить в области контроля за числом копий, в мотиве, подобном лейциновой застежке, в связывающей ДНК области или в одной или нескольких из данных областей или в другой области белка pi, кодируемого геном pir.
Прокариотическая клетка хозяина согласно изобретению также включает в себя одну или несколько мутаций в одном или разных доменах белка pi, кодируемого копией гена pir, такого как связывающий ДНК домен, и/или область контроля числа копий и/или мотив лейциновой застежки. Прокариотическая рекомбинантная клетка хозяина может включать в себя гетерологичный ген pir в плазмиде или в геноме клетки хозяина.
Такие мутации могут подвергаться скринингу путем использования основанного на флуоресценции метода скрининга согласно одному из аспектов настоящего изобретения, как описано впоследствии. Как показано в примере 13, клетки хозяина, включающие в себя по меньшей мере одну мутацию в гене pir, мутацию pir116 и мутацию в связывающем ДНК домене, подвергали скринингу путем использования метода скрининга согласно изобретению на основе флуоресценции. Клетки хозяина, содержащие мутацию, присутствующую в связывающем ДНК домене в дополнение к pir116, т.е., например, как в конструкции 100B, где тирозин (K) в положении 292 замещен метионином (M), в конструкции 114C, где глутаминовая кислота (E) в положении 130 замещена валином (V), или в конструкции 201C, где аспарагиновая кислота (D) в положении 117 замещена глицином (G) (фиг. 26), тестировали на их способность к продукции большого количества плазмид с использованием способа скрининга, на основе флуоресценции.
Согласно другому осуществлению настоящего изобретения клетка хозяина, используемая в заявленном способе, представляет собой прокариотическую клетку хозяина, в которой ген recA или гомологичный ген инактивирован. Предпочтительно клетка хозяина согласно изобретению представляет собой штамм E. coli XAC-1, включающий в себя мутацию pir116, pir42, endA-, traD-, recA-. Такой штамм обозначен TEX2 pir42. recA может инактивироваться способами, хорошо известными специалистами. recA кодирует главный белок рекомбинации, и мутации в данном гене снижают частоту опосредованного рекомбинацией изменения в плазмидах и внутримолекулярной рекомбинации, которая может приводить к мультимеризации плазмид. Как описано в примере 12, подвергнутый делеции ген recA, содержащий 3 стоп-кодона трансляции (один в каждой рамке) на его 5'-конце, может быть получен путем PCR. Полученный в результате инактивированный ген затем вводят посредством замены гена в геном TEX1 (пример 12.1).
Данные клетки получают любым способом, известным специалистам в данной области, который обеспечивает введение указанной плазмиды в данную клетку. Такой способ может представлять собой, в частности, трансформацию, электропорацию, конъюгацию, слияние протопластов и любой другой способ, известный специалистам в данной области.
Штамм XAC-1 pir116 был помещен на хранение согласно Будапештскому Соглашению в Collection Nationale De Cultures de Micro-organismes (CNCM), Institut Pasteur, 28, rue Dr. Roux, 75724 Paris Cedex 15, Франция, 10 октября 2003 г. под инвентарным № I-3108.
Штамм TEX2 pir42 был помещен на хранение согласно Будапештскому Соглашению в Collection Nationale De Cultures de Micro-organismes (CNCM), Institut Pasteur, 28, rue Dr. Roux, 75724 Paris Cedex 15, Франция, 10 октября 2003 г. под инвентарным № I-3109.
Молекулы ДНК согласно изобретению могут использоваться в любом применении вакцинации или генной и клеточной терапии для переноса гена в данную клетку, ткань или организм или для продукции рекомбинантных белков in vitro.
В частности, они могут использоваться для прямого введения in vivo или для модификации клеток in vitro или ex vivo, для целей имплантации их пациенту.
В данном отношении другой предмет настоящего изобретения относится к любой фармацевтической композиции, включающей в себя по меньшей мере одну молекулу ДНК, как определено выше. Данная молекула может быть ассоциирована с вектором для химической и/или биохимической трансфекции или может не быть связана с вектором. Вектор может, в частности, использовать катионы (фосфат кальция, DEAE-декстран и т.д.) или липосомы. Ассоциированные синтетические векторы могут являться катионными полимерами или липидами. Примерами таких векторов, которые могут быть упомянуты, являются DOGS (Transfectam™) или DOTMA (Lipofectin™).
Фармацевтические композиции согласно изобретению могут быть составлены для целей местного, перорального, парентерального, интраназального, внутривенного, внутримышечного, подкожного, внутриглазного или чрескожного введения. Заявленная плазмида предпочтительно используется в форме для инъекции или в виде компресса. Она может быть смешана с любым носителем, который является фармацевтически приемлемым для инъекционного препарата, в частности, для прямой инъекции в подлежащий лечению участок. Препараты могут включать в себя, в частности, стерильные изотонические растворы или сухие композиции, в частности, высушенные вымораживанием композиции, которые при добавлении, в зависимости от случая, стерилизованной воды или физиологического солевого раствора, обеспечивают получение растворов для инъекций. Они могут, в частности, включать в себя буферы Tris или PBS, разведенные с глюкозой или хлоридом натрия. Прямая инъекция в поврежденную область организма пациента является предпочтительной, поскольку она позволяет терапевтическому действию сосредоточиваться на уровне поврежденных тканей. Используемые дозы могут адаптироваться в качестве функции различных параметров и, в частности, в качестве функции используемого гена, вектора, способа введения, вовлеченной патологии или требуемой длительности лечения.
Молекулы ДНК согласно изобретению могут содержать один или несколько интересующих генов, то есть одну или несколько нуклеиновых кислот (синтетических или полусинтетических ДНК, гДНК, кДНК и т.д.), транскрипция и, возможно, трансляция которых в клетке-мишени приводит к образованию продуктов терапевтической, вакцинной, агрономной или ветеринарной значимости.
Среди генов терапевтической значимости, которые могут упоминаться более конкретно, имеются гены, кодирующие ферменты, производные белков крови, гормоны и лимфокины: интерлейкины, интерфероны, TNF и т.д. (FR 92/03120), факторы роста, нейротрансмиттеры или их предшественники, или синтетические ферменты и трофические факторы (BDNF, CNTF, NGF, IGF, GMF, aFGF, bFGF, NT3, NT5, VEGF-B, VEGF-C и т.д.; аполипопротеины: ApoAI, ApoAIV, ApoE и т.д. (FR 93/05125), дистрофин или аминидистрофин (FR 91/11947), гены подавления опухоли: p53, Rb, RaplA, DCC, k-rev и т.д. (FR 93/04745), гены, кодирующие факторы, участвующие в свертывании: факторы VII, VIII, IX и т.д., суицидные гены: тимидинкиназы, цитозиндезаминазы и т.д.; или, альтернативно, целые природные или искусственные иммуноглобулины или их части (Fab, ScFv и т.д.), лиганд РНК (WO 91/19813) и т.д. Терапевтический ген также может представлять собой антисмысловую последовательность или ген, экспрессия которого в клетке-мишени дает возможность контроля экспрессии генов или транскрипции клеточных мРНК. Такие последовательности могут, например, транскрибироваться в клетке-мишени с образованием РНК, которые комплементарны клеточным мРНК и поэтому блокируют их трансляцию с образованием белка, согласно способу, описанному в патенте EP 140308. Интересующая вставка, которая может переноситься pCOR согласно изобретению, представляет собой PHKi, способную препятствовать трансляции гена-мишени (Wilson et al., Curr Opin Mol Ther. 2003 Aug; 5(4): 389-96) и, таким образом, регулировать экспрессию такого гена.
Интересующий ген может также представлять собой вакцинирующий ген, то есть ген, кодирующий антигенный пептид, способный генерировать иммунный ответ у человека или животных, для цели продукции вакцин. Данные антигенные пептиды могут, в частности, являться конкретными антигенными пептидами вируса Эпштейна-Барр, вируса ВИЧ, вируса гепатита В (EP 185573), или вируса псевдобешенства, или, альтернативно, конкретными антигенными пептидами опухолей (EP 259212).
В общем, в молекулах ДНК согласно изобретению ген терапевтической, вакцинной, агрономной или ветеринарной значимости также содержит промоторную область для функциональной транскрипции в организме или клетке-мишени, а также область, расположенную на 3'-конце, которая определяет сигнал терминации транскрипции и участок полиаденилирования. Что касается промоторной области, то это может быть промоторная область, ответственная в природе за экспрессию рассматриваемого гена, при том, что данная область способна функционировать в указанной клетке или организме. Промоторные области также могут являться промоторными областями отличного происхождения (ответственными за экспрессию других белков) или даже синтетического происхождения. В частности, они могут представлять собой промоторные последовательности из эукариотических или вирусных генов. Например, они могут представлять собой промоторные последовательности, полученные из генома клетки-мишени. Среди эукариотических промоторов, которые могут использоваться, имеются любые промоторы или происходящая от них последовательность, которая стимулирует или подавляет транскрипцию гена специфическим или неспецифическим, индуцируемым или неиндуцируемым, сильным или слабым образом. Эукариотические промоторы могут, в частности, быть всеобщими промоторами (промоторы генов HPRT, PGK, a-актина, тубулина и т.д.), промоторами промежуточных филаментов (промоторы генов GFAP, десмина, виментина, нейрофиламентов, кератин и т.д.), промоторами терапевтических генов (например, промоторы генов MDR, CFTR, фактора VII, ApoAI и т.д.), тканеспецифическими промоторами (промоторы генов пируваткиназы, виллина, кишечного белка, связывающего жирные кислоты, a-актина гладких мышц и т.д.) или, альтернативно, промоторами, которые отвечают на стимул (рецептор стероидных гормонов, рецептор ретиноевой кислоты и т.д.). Сходным образом, они могут представлять собой промоторные последовательности, полученные из генома вируса, такие, например, как промоторы аденовируса E1A и генов MLP, ранний промотор CMV или, альтернативно, LTR-промотор RSV и т.д. Кроме того, данные промоторные области могут модифицироваться путем добавления активирующих или регуляторных последовательностей или последовательностей, которые обеспечивают тканеспецифическую экспрессию, которая является преимущественно тканеспецифической.
Более того, интересующие гены также могут содержать сигнальную последовательность, которая направляет синтезированный продукт в секреторные пути клетки-мишени. Данная сигнальная последовательность может быть природной сигнальной последовательностью синтезированного продукта, но она также может быть любой другой функциональной сигнальной последовательностью или искусственной сигнальной последовательностью. Предпочтительная сигнальная последовательность, используемая согласно изобретению, представляет собой сигнальный пептид секреции человеческого интерферона, описанный Taniguchi et al. (Gene, 1980, 233 (4763): 541-5)
В зависимости от интересующего гена, молекулы ДНК согласно изобретению могут использоваться для лечения или предотвращения некоторых патологий, включая генетические заболевания (дистрофия, кистозный фиброз и т.д.), нейродегенеративные заболевания (болезнь Альцгеймера, болезнь Паркинсона, ALS и т.д.), злокачественные опухоли, патологии, ассоциированные с нарушениями коагуляции или с дислипопротеинемиями, патологии, ассоциированные с вирусными инфекциями (гепатит, СПИД и т.д.), или в областях агрономии и ветеринарии и т.д.
Согласно предпочтительному осуществлению, молекулы ДНК согласно изобретению используют для лечения патологий, связанных с критической ишемией конечностей, таких, например, как как болезнь закупорки периферических артерий и перемежающая хромота.
Более того, настоящее изобретение также относится к применению ДНК молекул зависимой от условий репликации для продукции рекомбинантных белков. Бактерии могут использоваться для продукции белков различного происхождения, эукариотических или прокариотических. Среди бактерий, E. coli представляет собой организм выбора для экспрессии гетерологичных генов за счет простоты манипуляций, большого количества доступных экспрессионных систем и больших количеств белков, которые могут быть получены. Понятно, что система согласно изобретению может использоваться в других организмах, тропизм которых определяется природой точки начала репликации, как указано выше. Для данного применения последовательность нуклеиновой кислоты согласно изобретению включает в себя кодирующую область под контролем сигналов экспрессии, которые подходят для выбранного хозяина, в частности, прокариотического хозяина. Это могут быть, например, промоторы Plac, Ptrp, PT7, Ptrc, Ptac, PL, PBAD или PR, последовательность Шайна-Дальгарно и т.д. (данный набор составляет экспрессирующую кассету). Интересующая нуклеиновая кислота может представлять собой любую последовательность, кодирующую белок, который имеет ценность в областях фармации, сельского хозяйства и пищевой промышленности, химии или агрохимии. Это может быть структурный ген, комплементарная последовательность ДНК, синтетическая или полусинтетическая последовательность и т.д.
Экспрессирующая кассета может вводиться в вектор с зависимой от условий репликацией, который является предметом изобретения, с получением таким образом вектора с зависимой от условий репликацией, который обеспечивает экспрессию интересующего белка в E. coli. Данный вектор имеет несколько преимуществ: отсутствие применения антибиотиков для его селекции в бактерии (сниженная цена, нет необходимости в исследовании, касающемся присутствия антибиотика или потенциально токсичных производных в конечном продукте), фактически отсутствующая вероятность распространения плазмиды в природе (зависимая от условий точка начала репликации), возможность ферментации в полностью определенной среде. Приведенные примеры показывают предпочтительные свойства данных зависимых от условий векторов для продукции рекомбинантных белков.
Как описано выше, молекула ДНК согласно изобретению содержит точку начала репликации ORI, происходящую из R6K, где удален ген pir, и он введен в геном специфической клетки хозяина, который использован для продукции молекул ДНК в большом масштабе. Всегда имеется необходимость продуцировать повышенные количества плазмиды для клинических испытаний и/или для основанной на ДНК генной терапии. Продуцирующие клетки хозяина были сконструированы для переноса гена pir, содержащего по меньшей мере одну мутацию, такую как мутации pir116 и/или pir42. Применение такого мутированного штамма хозяина приводит к увеличению числа копий плазмиды, и, таким образом, значимо повышает выход продукции. Также очень удовлетворительной является конформация плазмид.
В конкретном аспекте предоставлен новый способ скрининга повышения копийности мутанта на основе флуоресценции. Данный способ скрининга на основе флуоресценции заметно превосходит классический способ скрининга, основанный на уровне устойчивости к антибиотикам в бактериях, который не может использоваться, когда начальное число копий плазмиды уже очень высоко, например, как то, которое получается с использованием мутанта pir116, примерно 400 копий плазмиды на клетку. В способе скрининга на основе флуоресценции согласно изобретению предпочтительно используется ген cobA в качестве гена-репортера числа копий на основе красной флуоресценции. Ген cobA, который является геном из Pseudomonas denitrificans (Crouzet et al., J. Bacteriol. 1999, 172:5968-79), кодирует уро-III-метилтрансферазу, фермент пути витамина B12, который добавляет две метильные группы к молекуле урогена III. Wildt et al. (Nature Biotechnology, vol. 17, 1999, pp 1175) описали применение cobA в качестве флуоресцентного гена-репортера транскрипции для E. coli, дрожжей и клеток млекопитающих. Например, такой флуоресцентный ген-репортер использовали для селекции содержащих рекомбинантные плазмиды штаммов E. coli, которые накапливают флуоресцентные порфириноидные соединения вследствие гиперэкспрессии гена cobA, кодирующего уро-III-метилтрансферазу. При освещении УФ-светом клетки флуоресцировали ярко-красным светом (Biotechniques, 1995, vol 19, no. 7, p. 760).
Настоящими заявителями неожиданно обнаружена близкая корреляция между числом копий плазмиды, несущей ген cobA, и уровнем флуоресценции от розового до красного. Способ скрининга копийности мутантов на основе флуоресценции согласно изобретению, таким образом, может использоваться для скрининга различных мутантов, которые могут оцениваться в геноме продуцирующей клетки хозяина, такой как E. coli, или мутантов любых генов, таких как генов, которые встроены в геном продуцирующей клетки хозяина или находятся в плазмиде.
В дополнение к корреляции с числом копий плазмид, способ скрининга на основе флуоресценции согласно изобретению просто и быстро проводится, поскольку он требует только посева на чашки и культивирования трансформированных клеток хозяина в течение ночи и воздействия на них УФ-света для выявления интенсивности продуцируемой флуоресценции, прямо устанавливая при этом число копий плазмид в клетке хозяина.
Таким образом, настоящее изобретение относится к способу детекции мутации, связанной с повышением копийности плазмид, включающему в себя
(a) введение по меньшей мере одной мутации в последовательность-мишень;
(b) трансформацию мутантной последовательности-мишени в клетку хозяина, включающую в себя плазмиду, где плазмида содержит нуклеотидную последовательность, кодирующую уро-III-метилтрансферазу, и число копий плазмиды подвергается воздействию последовательности-мишени;
(c) выращивание клетки-хозяина в условиях, где нуклеотидная последовательность экспрессируется с продукцией культуры клеток хозяина;
(d) воздействие на культуру клеток хозяина УФ-света; и
(e) детекцию флуоресценции, продуцируемой культурой клеток хозяина.
Согласно изобретению способ далее охватывает сравнение флуоресценции, определяемой в (e), с флуоресценцией, продуцируемой культурой клеток хозяина, включающих в себя немутантную последовательность-мишень.
Предпочтительно ген уро-III-метилтрансферазы кодируется геном cobA из Pseudomonas denitrificans.
Мутация может присутствовать в плазмиде, содержащей гетерологичный ген pir, содержащий по меньшей мере одну мутацию. Плазмида может включать в себя по меньшей мере одну мутацию в других областях pir, таких как область контроля числа копий и/или связывающий ДНК домен, и/или мотив лейциновой застежки, и/или другая область гена pir. Также плазмида может включать в себя по меньшей мере одну мутацию в области контроля числа копий и подобном лейциновой застежке мотиве гетерологичного гена. Плазмида может далее включать в себя мутацию в связывающей ДНК области гена pir. Более того, плазмида может включать в себя одну или несколько мутаций в той же или иной области гена pir, кодирующей область контроля числа копий и/или связывающей ДНК домен и/или мотив, подобный лейциновой застежке, или другую область белка П.
Без ограничений, прокариотическая рекомбинантная клетка хозяина согласно изобретению включает в себя мутацию pir116 и вторую мутацию в связывающей ДНК области, такой как pir292, pir130 или pir117 (фиг. 26).
Такой мутантный продуцирующий штамм хозяина может предпочтительно продуцироваться с использованием универсального плазмидного инструмента, такого как мини-кольцо. Технология мини-кольца описана, среди прочих, в патентах США 6143530 и 6492164 настоящего заявителя или в заявке PCT WO 96/26270.
Мини-кольца являются рекомбинантными молекулами ДНК, которые не содержат какой-либо точки начала репликации, и, таким образом, представляют собой отличный суицидный вектор для переноса гена в геном любых микроорганизмов. В частности, интересующий ген или гены фланкированы двумя последовательностями, обеспечивающими сайт-специфическую рекомбинацию, расположенные в прямой ориентации в мини-кольце. Положение в прямой ориентации указывает на то, что две последовательности следуют в одном и том же направлении 5'-3' в рекомбинантном мини-кольце ДНК. Генетические конструкции мини-кольца в общем представляют собой кольцевые двухцепочечные молекулы ДНК, лишенные точки начала репликации, но также могут находиться в линейной форме и содержат интересующий ген или гены, фланкированные двумя последовательностями, обеспечивающими сайт-специфическую рекомбинацию, расположенные в прямой ориентации. Согласно конкретному осуществлению изобретения, мини-кольцо может использоваться для трансформации любого компетентного микроорганизма с целью перемещения гена в его геном (фигура 31).
Мини-кольцо для перемещения генов получают из родительской плазмиды, по меньшей мере содержащей
a) точку начала репликации и ген-маркер селекции,
b) две последовательности, обеспечивающие сайт-специфическую рекомбинацию, расположенные в прямой ориентации, и
c) один или несколько интересующих генов, помещенные между указанными последовательностями b).
Система специфической рекомбинации, присутствующая в генетических конструкциях, может быть различного происхождения. В частности, используемые специфические последовательности и рекомбиназы могут принадлежать к различным структурным классам, и в частности, семейству интеграз бактериофага или семейству резолваз транспозона Tn3. Из рекомбиназ, принадлежащих семейству интеграз бактериофага, могут быть упомянуты, в частности, интеграза фага лямбда (Landy et al., Science 197: 1147,1977), P22 и Ф80 (Leong et al., J. Biol. Chem. 260: 4468, 1985), HP1 Haemophilus influenza (Hauser et al., J. Biol. Chem. 267 6859, 1992), интеграза Cre фага PI, интеграза плазмиды pSAM2 (EP 350341) или, альтернативно, рекомбиназа FLP плазмиды 2μ. Мини-кольца, таким образом, получают путем рекомбинации посредством сайт-специфической системы семейства интеграз бактериофага, молекулы ДНК согласно изобретению в основном содержат, кроме того, последовательность, являющуюся результатом рекомбинации между двумя последовательностями прикрепления att соответствующего бактериофага или плазмиды.
Из рекомбиназ, принадлежащих семейству транспозона Tn3, могут быть упомянуты, в частности, резолваза транспозона Tn3 или транспозонов Tn21 и Tn522 (Stark et al., Trends Genet, 8, 432-439, 1992); инвертаза Gin бактериофага мю, или, альтернативно, резолваза плазмид, такая как фрагмент par RP4 (Albert et al., Mol. Microbiol. 12: 131, 1994). Когда мини-кольца получают путем рекомбинации посредством сайт-специфической системы семейства транспозона Tn3, они в общем содержат, в дополнение к интересующему гену, который намереваются встраивать в геном микроорганизма, последовательность, которая является результатом рекомбинации между двумя последовательностями распознавания резолвазы указанного транспозона. Последовательности, обеспечивающие сайт-специфическую рекомбинацию, могут также происходить из области loxP фага P1, который составлен по существу из двух последовательностей-повторов, способных распознавать одна другую в присутствии белка, обозначенного Cre (Sternberg et al., J. Mol. Biol. 150: 467, 1971). Плазмида, используемая для продукции мини-кольца, таким образом, содержит (a) бактериальную точку начала репликации и ген-маркер селекции; (b) последовательности-повторы бактериофага P1 (область loxP); и (c) расположенные между указанными последовательностями (b) один или несколько интересующих генов, которые требуется встраивать в геном микроорганизма.
Мини-кольца могут включать в себя последовательности, обеспечивающие сайт-специфическую рекомбинацию, которые происходят от бактериофага, такие как последовательности прикрепления (последовательности attP и attB) бактериофага или последовательности, происходящие от таких последовательностей прикрепления. Данные последовательности способны специфическо рекомбинировать одна с другой в присутствии рекомбиназы, обозначенной как интеграза, с эксцизионазой или без нее. Термин "последовательности, происходящие от таких последовательностей прикрепления" включает в себя последовательности, полученные путем модификации(-й) последовательностей прикрепления бактериофагов, которые сохраняют способность специфически рекомбинировать в присутствии подходящей рекомбиназы. Таким образом, такие последовательности могут представлять собой уменьшенные фрагменты данных последовательностей или, альтернативно, фрагменты, протяженные путем добавления других последовательностей (сайтов рестрикции и тому подобного). Они также могут представлять собой варианты, полученные путем мутации(-й), в частности, путем точечной(-ых) мутации(-й). Термины последовательности attP и attB бактериофага или плазмиды, означают, согласно изобретению, последовательности рекомбинантной системы, специфической для указанного бактериофага или плазмиды, то есть последовательность attP, присутствующую в указанном фаге или плазмиде, и соответствующую хромосомную последовательность attB. Последовательности прикрепления хорошо известны в данной области и включают в себя, среди прочих, последовательности прикрепления фагов, P22, Ф80, P1, и HP1 Haemophilus influenzae, или, альтернативно, плазмиды pSAM2 или плазмиды μ2.
Мини-кольца легко продуцируют из родительской плазмиды, описанной выше. Способ продукции мини-кольца состоит из приведения в контакт культуры клеток, которые трансформируют родительской плазмидой, с интегразой в присутствии эксцизионазы или без нее, так что индуцируется сайт-специфическая рекомбинация. Культура и интеграза с эксцизионазой или без нее приводятся в контакт путем трансфекции или инфекции плазмидой или фагом, содержащим ген указанной интегразы, и, когда это применяется, ген эксцизионазы. Альтернативно, например, индуцируют экспрессию генов, кодирующих указанную интегразу, и, когда это применяется, эксцизионазу, присутствующих в клетке хозяина. Как указано выше, данные гены могут присутствовать в клетке хозяина в интегрированной форме в геноме, в репликативной плазмиде, или, альтернативно, в плазмиде согласно изобретению в не относящейся к терапии части.
Для обеспечения продукции мини-колец согласно изобретению путем сайт-специфической рекомбинации in vivo, интегразу в присутствии или отсутствие эксцизионазы вводят внутрь или индуцируют в клетках или культуральной среде в конкретный момент. Для данной цели могут использоваться разные способы. Согласно первому способу, используют клетку-хозяина, содержащую, например, ген рекомбиназы, т.е. ген интегразы с геном эксцизионазы или без него, в форме, обеспечивающей его регулируемую экспрессию. Ген интегразы с геном эксцизионазы или без него может, например, вводиться под контроль промотора, или системы индуцируемых промоторов, или, альтернативно, в чувствительную к температуре систему.
В частности, ген интегразы может присутствовать в чувствительном к температуре фаге, латентном во время фазы роста и индуцируемом при подходящей температуре (например, лизогенный фаг Xis-cI857).
Альтернативно, ген может находиться под контролем регулируемого промотора, например, промотора placUV5, клетка хозяина обозначается E. coli G6191.
Предпочтительно, интеграза в присутствии или в отсутствие эксцизионазы может находиться под контролем регулируемого промотора, например, промотора PBAD оперона araBAD (арабинозного), который регулируется арабинозой (Guzman et al., J. Bacteriol, 1995, 4121-4130; US 5028530). В частности, применение промотора PBAD обеспечивает достаточную экспрессию эксцизионазы и интегразы в присутствии арабинозы, в качестве индуцирующего средства, и, таким образом, более 90% рекомбинации плазмид, которые присутствуют в бактерии в большом количестве копий, в то время как в отсутствие арабинозы промотор сильно ингибируется. Кассета для экспрессии интегразы в присутствии или отсутствий эксцизиноназы может переноситься в плазмиде, фаге, или даже в плазмиде согласно изобретению в не относящейся к лечению области. Она может интегрироваться в геном клетки хозяина или поддерживаться в репликативной форме. Такие клетки хозяина представляют собой, в частности, E. coli G6264 и E. coli G6289. По другому способу, кассета для экспрессии гена(-ов) переносится плазмидой или фагом, используемыми для трансфекции или инфекции культуры клеток после фазы роста. В данном случае для гена не является необходимым находиться в форме, обеспечивающей его регулируемую экспрессию. В частности, может использоваться любой конститутивный промотор. ДНК также может приводиться в контакт с интегразой и, когда это применяется, с эксцизионазой in vitro, в препарате плазмиды, путем прямой инкубации с белком.
Полученное таким образом мини-кольцо, таким образом, включает в себя экспрессирующую кассету, содержащую один или несколько интересующих генов, подлежащих встраиванию в микроорганизм-мишень, не содержит точку начала репликации и содержит последовательность attR, являющуюся результатом сайт-специфической рекомбинации между последовательностями attB и attP, или последовательность attL, являющуюся результатом сайт-специфической рекомбинации между последовательностями attB и attP. Мини-кольцо, таким образом, может использоваться в качестве универсального суицидного вектора для перемещения генов в любые микроорганизмы. В действительности мини-кольцо, несущее ген для перемещения, фланкированный гомологичными последовательностями и ген устойчивости к антибиотику, легко интегрируется в участок-мишень генома любого микроорганизма путем гомологичной рекомбинации, как представлено на фигуре 31. Второе событие вырезания, которое может быть запущено давлением второго отбора, может затем эффективно отбирать только микроорганизмы, несущие в их геноме новый встроенный ген.
Настоящее изобретение также относится к способу генной инженерии микроорганизма. Данный новый способ может использоваться для инженерии любого микроорганизма независимо от его происхождения. В действительности мини-кольцо не содержит какой либо точки начала репликации, и, таким образом, может использоваться универсально для перемещения генов в любых типах микроорганизмов. Данный способ представляет собой предпочтительную альтернативу применению бактериофага M13 для перемещения генов путем двойной гомологичной рекомбинации в микроорганизме.
Согласно конкретному осуществлению настоящего изобретения мини-кольцо содержит первый подлежащий селекции маркер, такой как ген устойчивости к антибиотику, позволяющий осуществлять отбор первого события рекомбинации. Предпочтительный второй подлежащий селекции маркер представляет собой ген III или ген III' с функциональной делецией. Ген III или его функциональный вариант способен обеспечивать чувствительность к дезоксихолату, как описано в Boecke et al. (Mol. Gen. Genet., 186, 185-92, 1982) и, таким образом, обеспечивает обратную селекцию второго события рекомбинации (фиг.31). Таким образом, способ состоит из введения мини-кольца в микроорганизм любым способом трансформации, хорошо известным в данной области, и, предпочтительно, путем электропорации, отбора события интеграции мини-кольца в культуре, дополненной антибиотиком, или под другим давлением отбора, и отбора второго события вырезания путем обработки дезоксихолатом или другим подходящим давлением отбора.
Настоящее изобретение будет более полно описано с помощью последующих примеров, которые должны рассматриваться в качестве неограничивающих иллюстраций.
Краткое описание чертежей:
Фигура 1. Функциональная организация области R6K, вовлеченной в репликацию.
Фигура 2. Организация функциональных доменов белка П плазмиды R6k.
Фигура 3. Представление протокола введения гена pir в геном E. coli XAC1.
Фигура 4. Схема конструирования векторов pXL2666, 2730 и 2754.
Фигура 5. Конструкция pXL2774.
Фигура 6. Кинетика роста и продукции в ферментере 2L.
Фигура 7. Кинетика роста и продукции в ферментере 800L.
Фигура 8. Конструкция pXL3056.
Фигура 9: Визуализация белка aFGF, продуцируемого E. coli XAC-1 pir116 (pXL3056+PT7pol23) после индукции. Денатурированные общие клеточные экстракты помещали на 12,5%-SDS-полиакриламидный гель. M: маркер молекулярной массы (Biorad, интервал низких масс). Каждую полосу указывали стрелкой и цифрой, которая означает ее массу в кДальтонах. 1: XAC-1 pir116 (pXL3056+pUC4K) не индуцированная; 2: XAC-1 pir116 (pXL3056+pUC4K) индуцированная при 42°C; 3: XAC-1pir116 (pXL3056+PT7pol23) клон 1, не индуцированная; 4: XAC-1 pir116 (pXL3056+PT7pol23) клон 1, индуцированная при 42°C; 5: XAC-1 pir116 (pXL3056+PT7pol23) клон 2, не индуцированная; 6: XAC-1 pir116 (pXL3056+PT7pol23) клон 2, индуцированный при 42°C; t1: 1 мкг очищенного aFGF; t4: 4 мкг очищенного aFGF.
Фигура 10. Схематичное представление векторов pXL3029, pXL3030, и pXL3179 или NV1FGF.
Фигура 11. Схематичное представление функциональных доменов инициаторных белков π R6K.
Фигура 12. Нуклеотидные и аминокислотные последовательности гена pir, содержащего мутации pir116 и pir42.
Фигура 13. Конструкция суицидного вектора pir116 pir42 для гомологичной рекомбинации.
Фигура 14. Схематичное представление PCR-продуктов, полученных при амплификации области uidA:pir116 +/- pir42.
Фигура 15. Электрофорез в агарозном геле, на котором показана топология pCOR-плазмиды pXL3179, продуцированной в TEX1 или TEX1 pir42.
Фигура 16. Схематичное представление суицидной плазмиды pXL3749, несущей ген pir116 cop21.
Фигура 17. Электрофорез в агарозном геле, на котором показано число копий плазмиды pXL2979 при ее продукции в клетке хозяина E. coli TEX1 cop21 (дорожки 1-4), в клетке хозяина E. coli XAC1 pir (дорожки 5-8), в E. coli TEX1 (дорожки 9-12).
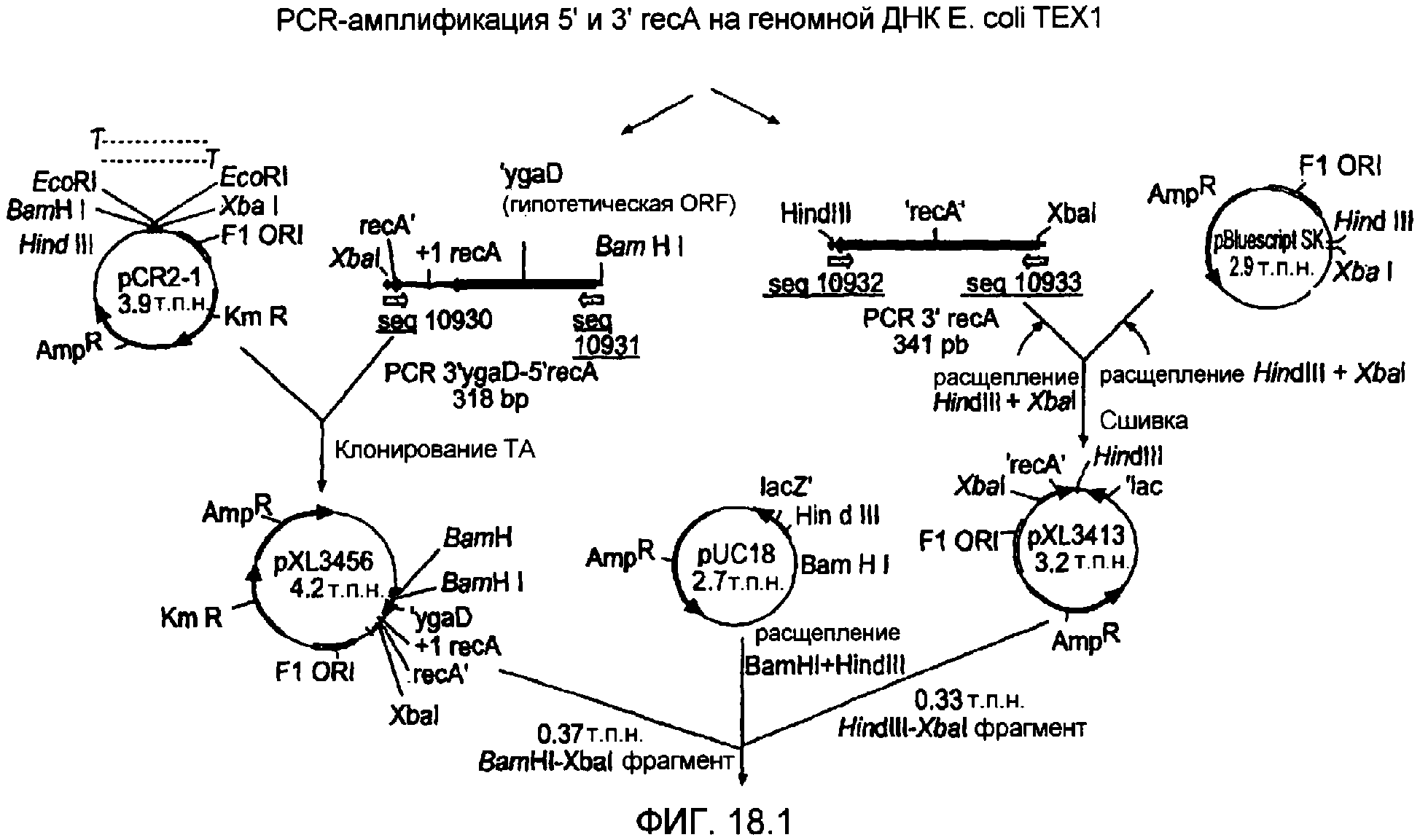
Фигура 18. Представление стратегии клонирования для конструирования суицидного вектора recA-.
Фигура 19. Схематичное представление продуктов PCR, полученных при амплификации областей штамма E. coli TEX2.
Фигура 20. Электрофорез в агарозном геле, на котором показана топология pCOR pXL3179, продуцированной в E. coli TEX2 pir42 (дорожка B), в E. coli TEX1 pir42 (дорожка C), в E. coli TEX1 (дорожка D).
Фигура 21. Анализ плазмиды pXL3179, продуцируемой путем ферментации E. coli TEX2 pir42.
Фигура 22. Основанный на флуоресценции анализ, в котором показано, что при увеличении числа копий плазмиды флуоресценция возрастает.
Фигура 23. Диаграмма плазмид, подвергавшихся скринингу путем анализа на основе флуоресценции.
Фигура 24. Диаграмма плазмиды pXL3830.
Фигура 25. Агаровая чашка, на которой продемонстрирован скрининг на основе флуоресценции мутантов с повышенной копийностью, генерированных путем случайного мутагенеза.
Фигура 26. Оценка мутаций с повышенной копийностью, идентифицированных способом скрининга на основе флуоресценции.
Фигура 27. Диаграмма стратегии оценки мутантов pir116, встроенный в бактериальный геном.
Фигура 28. Оценка числа копий pXL3179 в других штаммах E. coli с мутацией pir116*.
Фигура 29. Конструкция плазмиды, используемой для получения векторов-мини-колец для гомологичной рекомбинации в E. coli.
Фигура 30. Конструкция вектора-мини-кольца, используемого для получения штаммов E. coli с мутацией pir116*.
Фигура 31. Диаграмма перемещения гена путем гомологичной рекомбинации с использованием вектора-мини-кольца.
Фигура 32. Демонстрация клонов-двойных рекомбинантов, растущих на среде, содержащей дезоксихолат натрия.
Фигуры 33A и B. Результаты контрольной PCR на двойных рекомбинантах.
I - МАТЕРИАЛЫ И МЕТОДЫ
A) Материалы
1) Культуральная среда
Использовали полные среды LB, 2XTY и SOC и минимальную среду М9 (Maniatis et al., 1989). Агаровые среды получали путем добавления 15 г агара Difco. Более того, если необходимо, данные среды дополняли антибиотиками (ампициллином или канамицином) в соответствующих концентрациях, равных 100 мг/л и 50 мг/л. Хромогенные субстраты X-Gal и X-Gluc использовали в концентрации 40 мг/л.
2) Штаммы E. coli, плазмиды и бактериофаги
Используемые штаммы E. coli, плазмиды и бактериофаги соответственно указаны в приведенных ниже примерах.
B) Методы
1) Манипуляции с ДНК
Выделение бактериальной ДНК (плазмидной и геномной) и фаговой ДНК (репликативной формы M13), расщепление рестрикционнными эндонуклеазами, лигирование фрагментов ДНК, электрофорез в агарозном геле (в буфере TBE) и другие стандартные способы проводили по рекомендациям производителей для применения ферментов или по процедурам, описанным в "Molecular Cloning: a Laboratory Manual" (Maniatis et al., 1989).
Маркеры размера ДНК, используемые при электрофорезе, являются следующими: лестница 1 т.п.н. (BRL) для линейных фрагментов и сверхспиральный маркер ДНК (Stratagene) для нерасщепленных плазмид.
Секвенирование проводили по способу Сэнгера (Sanger et al., 1977), адаптированному для автоматического метода с использованием флуоресцентных дидезоксинуклеотидов и ДНК-полимеразы Taq (PRISM Ready Reaction DyeDideoxy Terminator Cycle Sequencing Kit, Applied Biosystems).
Использованные олигодезоксинуклеотиды (обозначенные "seq+no.", см. ниже) синтезировали на синтезаторе ДНК/РНК "Applied Biosystems 394 DNA/RNA Synthesizer" фосфорамидитным методом с использованием a-цианоэтильных защитных групп (Sinha et al., 1984). После синтеза защитные группы удаляли обработкой аммиаком. Две преципитации бутанолом обеспечивали очистку и концентрирование олигонуклеотида (Sawadogo et al.,1991).
Последовательности олигонуклеотидов, использованных для PCR-амплификации:

Реакции PCR (Saiki et al., 1985) проводили в следующих условиях, в общем объеме, равных 100 мкл. Реакционная смесь содержала 150 нг геномной ДНК исследуемого штамма, 1 мкг каждого из двух олигонуклеотидных праймеров (24-членные), 10 мкл 10х буфера для PCR, композиция которого была следующей: "500 мМ KCl, 0,1% желатин, 20 мМ MgCl2, 100 мМ Tris-HCl, pH 7,5", а также 2,5 единицы ДНК-полимеразы Taq (Amplitaq Perkin-Elmer). Условия PCR на приборе ДНК-термоциклере Perkin-Elmer Cetus были следующими: 2 мин при 91°С, 30 последовательных циклов денатурации (1 мин при 91°С), гибридизации (2 мин при 42°С) и элонгации (3 мин при 72°С), и, наконец, 5 мин при 72°С. Продукты, полученные таким образом, расщепленные или не расщепленные рестрикционным ферментом, анализировали электрофорезом в агарозном геле.
Анализ различных видов плазмид посредством ДНК-топоизомераз проводили по следующей процедуре: ферменты, очищенные в лаборатории, инкубировали в течение 1 часа при 37°С. Реакционные смеси (общий объем 40 мкл) имеют следующую композицию: 150 нг плазмиды, 300 нг ДНК-топоизомеразы I или 150 нг ДНК-гиразы E. coli, или 160 нг ДНК-топоизомеразы IV S. aureus и 20 мкл буфера, специфического для каждого фермента. Композиция данных буферов указана ниже:
для ДНК-топоизомеразы I:
50 мМ Tris-HCl pH 7,7, 40 мМ KCl, 1 мМ DTT, 100 мкг/мл BSA, 3 мМ MgCl2, 1 мМ EDTA;
для ДНК-топоизомеразы IV:
60 мМ Tris-HCl pH 7,7, 6 мМ MgCl2, 10 мМ DTT, 100 мкг/мл BSA, 1,5 мМ АТФ, 350 мМ глутамата калия;
для ДНК-гиразы: 50 мМ Tris-HCl, pH 7, 7, 5 мМ MgCl2, 1,5 мМ АТФ, 5 мМ DTT, 100 мкг/мл BSA, 20 мМ KCl.
2) Трансформация E. coli
Ее проводили рутинно по способу TSB (буфер для трансформации и хранения), описанным Chung and Miller (1988). Для такого штамма, как TG1 (Gibson et al., 1984), полученная эффективность трансформации составляет около 105-106 трансформантов на мкг pUC4K (Vieira and Messing; 1982). Когда необходима более высокая эффективность трансформации, бактерии трансформировали путем электропорации по процедуре, рекомендованной производителем электропораторов (Biorad). Данный способ дает возможность достижения эффективности от 108 до 1010 трансформантов на мкг pUC4K.
3) Трансфекция клеток, опосредованная катионным липофектантом
Используемые клетки представляют собой мышиные фибробласты NIH 3T3, посеянные за день до эксперимента, в 24-луночные планшеты с плотностью 50 000 клеток на лунку. Используемая культуральная среда представляет собой среду DMEM, содержащую 4,5 г/л глюкозы и дополненную 10% фетальной сыворотки теленка и 1% растворов 200 мМ глутамина, и антибиотиков (5·103 мкг/мл стрептомицина, 5·103 мкг/мл пенициллина) (Gibco). Плазмидную ДНК (1 мкг в 25 мкл 9% NaCl) смешивают на основе объемного соотношения с суспензией липофектанта. Тестируют четыре отношения "заряды липофектанта/заряды ДНК": 0, 3, 6 и 9. Данные отношения рассчитаны в свете того, что 1 мкг плазмидной ДНК несет 3,1 нмоль отрицательных зарядов, и что липофектант содержит 3 положительных заряда на молекулу. По прошествии времени контакта, равного 10 минутам, чтобы обеспечить образование комплекса ДНК/липид, 50 мкл смеси ДНК-липофектант помещали к клеткам в бессывороточной культуральной среде (500 мкл). Клетки предварительно отмывали дважды той же самой средой. Таким образом, предотвращали ингибирование трансфекции сывороткой. После инкубирования (2 часа при 37°C в CO2-инкубаторе), 10% фетальной сыворотки теленка добавляли в среду. Затем клетки повторно инкубировали в течение 24 часов.
4) Измерение люциферазной активности в эукариотических клетках
Его проводили через 24 часа после трансфекции. Люцифераза катализирует окисление люциферина в присутствии АТФ, Mg2+ и О2, сопровождающееся продукцией фотона. Общее количество испускаемого света, измеренное люминометром, пропорционально люциферазной активности образца. Используемые реагенты поставлены Promega (система люциферазного анализа), и их применяли по рекомендованной процедуре. После лизиса клеток нерастворимую фракцию из каждого экстракта элиминировали центрифугированием. Анализ проводили 5 мкл надосадочной жидкости, которую могли разводить или не разводить буфером для лизиса клеток.
5) Измерение концентрации белка в клеточных экстрактах
Его проводили способом BCA (Pierce) с использованием бихинной кислоты (Wiechelman et al., 1988). Стандартный интервал BSA получали в буфере для лизиса (cf. III-B-4). Анализируемые образцы и образцы указанного интервала предварительно обрабатывали на основе объемного отношения 0,1 M иодацетамидом/0,1 M буфером Tris, pH 8,2, в течение 1 часа при 37°C. Эта обработка дает возможность для предотвращения помехи во время анализа вследствие действия восстанавливающего агента (DTT), присутствующего в буфере для лизиса. Результаты анализа считывали при 562 нм.
Пример 1: Конструирование штаммов хозяина XAC-1 pir и pir116 путем гомологичной рекомбинации
Используемый штамм представлял собой штамм E. coli XAC-1 (Normanly et al., 1980). Ген argE данного штамма предпочтительно включал в себя мутацию глутамина-53 (CAG) с образованием амбер-кодона (TAG) (Meinnel et al., 1992). Ген argE относится к дивергентному оперону argECBH и кодирует фермент биосинтеза аргинина N-ацетилорнитиназу. Поэтому XAC-1 не может синтезировать аргинин, и, следовательно, расти в минимальной среде. Данная ауксотрофность может быть деблокирована, если штамм будет нести плазмиду, которая обеспечит экспрессию супрессорной тРНК. Таким образом, возможно за счет культивирования в минимальной среде отбирать бактерии, которые несут такую плазмиду. Для обеспечения репликации в них плазмид, происходящих от R6K, было необходимо вводить путем гомологичной рекомбинации ген pir в геном XAC-1. Ген pir (дикого типа или мутированный) вводят в локус uidA путем обмена между геном uidA дикого типа и копией, прерванной геном pir (или pir116). Ген uidA кодирует

1) Конструирование суицидного вектора, несущего кассету KmR-uidA::pir (или pir 116)
Авторы изобретения применяют стратегию, использующую единственного бактериального хозяина и минимизирующую модификации генома интересующего штамма. Фаг M13mp10 (Messing et Vieira; 1982) использовали в качестве суицидного вектора (Blum et al., 1989). Амбер-мутация в гене II, существенного для репликации, снижает спектр хозяев данного M13 до штаммов, таких как TG1 (supE), которые продуцируют амбер-супрессорную тРНК; поэтому он не может реплицироваться в штаммах E. coli sup+, таких как XAC-1.
Кассеты BamHI размером 3,8 т.п.н., содержащие ген устойчивости к канамицину Tn5 и _uidA::pir или pir116, очищали, соответственно, из M13wm34 и 33 (Metcalf et al., 1994). Их клонировали в M13mp10, линеаризованный посредством BamHI. Рекомбинантные клоны отбирали путем посева на агаровую среду LB, дополненную канамицином, после электропорации смесей лигирования в TG1. Однородность полученных клонов показывали путем анализа профиля рестрикции и путем секвенирования области, соответствующей мутации pir116.
2) Введение генов pir или pir116 в геном E. coli XAC-1 путем гомологичной рекомбинации
Адаптированная стратегия и различные вовлеченные события представлены на фигуре 3.
a) Первое событие рекомбинации
Штамм XAC-1 трансформировали путем электропорации с 10, 100 или 2000 нг каждой RF (mp10-_uidA::pir или pir116). Одну треть каждой экспрессирующей смеси высевали на чашки с LB, содержащей канамицин, и инкубировали в течение ночи при 37°C. Фаги mp10-_uidA::pir или pir116 не могут реплицироваться в штамме XAC-1(sup+). Поэтому маркер устойчивости к канамицину ("KmR") может поддерживаться лишь интеграцией в геном бактерии посредством гомологичной рекомбинации с копией дикого типа гена uidA. Результаты электропорации XAC-1 представлены в таблице 1. Полученная эффективность трансформации составляла 4·109 трансформантов на мкг pUC4K.
При условиях тестирования число интегрантов повышалось в нелинейной манере с повышением количества ДНК. Благодаря эффективности трансформации и размеру RF (11,7 т.п.н.) было возможно примерно предсказать уровень рекомбинации. При рассмотрении точки 100 нг получали частоту рекомбинации, равную 10-6.
b) Второе событие рекомбинации
Затем отбирали второе событие рекомбинации за счет устойчивости штаммов к дезоксихолату ("DocR").
Для выполнения этого пять интегрантов каждой конструкции культивировали в среде 2XTY, дополненной 0,2% дезоксихолатом натрия. Появлялись две различные популяции. Некоторые клоны давали вполне видимое помутнение примерно через 8 часов при 37°C (два клона для конструкции pir и три для конструкции pir116). Другие клоны давали плотную культуру лишь после одной ночи при 37°C. Фактически все были чувствительны к канамицину ("KmS"), как и ожидалось. Для каждого из исследованных штаммов, подвергшихся электропорации, 50 KmS потомков высевали полосами на среду LB, дополненную X-Gluc. Через 48 часов при 37°C клоны UidA+ были бледно-голубыми, тогда как те, которые претерпели перемещение аллелей (случай №1, фигура 3), оставались на этой среде белыми (UidA-). В таблице 2 суммирован фенотипический анализ полученных двойных рекомбинантов. От 18 до 30% двойных рекомбинантов претерпевали перемещение аллелей.
3) Проверка природы признака Pir+ на штаммах, полученных путем рекомбинации
Для гарантии наличия признака Pir+ у штаммов, полученных двойной рекомбинацией, авторы изобретения трансформировали pBW30 три клона каждой конструкции (Metcalf et al., 1994). Тот факт, что трансформанты были получены для всех тестируемых штаммов, дал возможность показать функциональность генов pir и pir116, которые были интегрированы в геном XAC-1. В тех же условиях не было получено трансформантов с родительским штаммом XAC-1. Авторы продолжали исследовать для клона XAC-1 pir (B и C) и два клона XAC-l pir116 (E и D).
4) Проверка путем PCR-амплификации штаммов, полученных путем рекомбинации
Для подтверждения перемещения аллелей авторы изобретения проверяли геномные области на каждой стороне локуса uidA путем PCR-амплификации. Каждая пара олигонуклеотидов состояла из олигонуклеотида, соответствующего внутренней области pir, и второго олигонуклеотида, соответствующего области, близкой к хромосомному uidA, но не находящейся внутри фрагмента, служившего для рекомбинации. Последовательность последнего олигонуклеотида определяли посредством последовательности ECOUIDAA из Genbank (инвентарный номер: M14641). Таким образом, авторы изобретения могли подтвердить точное расположение гена pir в бактериальном геноме. Природа амплифицированных фрагментов, размер которых соответствует тому, который можно ожидать, подтверждали путем расщепления MluI.
Пример 2. Конструирование плазмидных векторов, происходящих из R6K, несущей маркер селекции sup Phe
Конструировали векторы, содержащие ori из R6K и ген устойчивости к канамицину (pXL2666). Наблюдение мультимеров pXL2666 в штамме BW19610 (pir116) 5 (Metcalf et al., 1993) привело авторов изобретения к исследованию влияния на этот феномен наличия cer-фрагмента из ColE1. Затем авторы изобретения вводили экспрессирующую кассету фенилаланиновой супрессорной тРНК (sup Phe) в вектор ori -KmR-cer (pXL2730). Данный вектор pXL2760 служит основой для конструирования векторов, которые могут использоваться в генной терапии.
1) Конструирование и анализ векторов, содержащих ori из R6K и ген устойчивости к канамицину
a) Конструкции
В первой сконструированной плазмиде pXL2666 ген устойчивости к канамицину происходил из pUC4K (Vieira and Messing; 1982), а точка начала репликации, содержащаяся во фрагменте EcoRI-BamHI длиной 417 п.н., происходила из суицидного вектора pUT-T7pol (Herrero et al., 1990) (фигура 4). Перенос pXL2666 в штаммы BW19094 и 19610 (Metcalf et al., 1994) дал возможность показать, что количество плазмиды в штамме pir116 действительно возросло по сравнению с той же плазмидой в штамме pir. Однако электрофоретический анализ нерасщепленных плазмид показал, что данное увеличение идет рука об руку с появлением малого количества мультимерных форм. Данный феномен, возможно, ассоциирован с межмолекулярной рекомбинацией между множественными копиями плазмиды. Таким образом, авторы конструировали pXL2730 путем клонирования фрагмента cer природной плазмиды E. coli, ColE1, который, как было показано, обеспечивает в цис-положении разрешение плазмидных димеров (Summers and Sherrat, 1984) в pXL2666. Использованный фрагмент соответствовал фрагменту HpaII длиной 382 п.н. из ColE1 (Leung et al., 1985). Он содержит специфический участок межмолекулярной рекомбинации; для функционирования он использует только белки хозяина, включая рекомбиназы XerC и XerD и добавочные факторы ArgR и PepA (Stirling et al., 1988,1989 ; Colloms et al., 1990). Для того, чтобы убедиться, что наблюдаемые эффекты действительно связаны с фрагментом cer, авторы изобретения также конструировали контрольную плазмиду pXL2754, в которой фрагмент cer имеет делецию длиной 165 п.н. Данная делеция, как было показано, отменяет действие cer по разрешению мультимеров (Leung et al., 1985). Различные стадии клонирования, которые ведут к конструированию данных плазмид, представлены на фигуре 4.
b) Количественный и качественный анализ плазмидных видов
(i) анализ путем электрофореза в агарозном геле
Электрофоретический анализ различных сконструированных плазмид обеспечивал демонстрацию различных видов плазмид, которые варьировали в зависимости от использованных штаммов. Размер нерасщепленных плазмид оценивали относительно сверхспирального ДНК-маркера. В штамме pir (BW19094) плазмиды pXL2666, 2754 и 2730 были почти полностью в мономерной форме. Полосы выше каждой основной полосы соответствовали различным слегка менее сверхспирализованным топоизомерам, что подтверждалось профилем, наблюдаемым после действия ДНК-гиразы на pXL2730.
В случае штамма pir116 (BW19610) профили были другими: с использованием плазмид pXL2666 и 2754 наблюдали иные виды молекул, варьирующие от мономера до мультимеров (2, 3 или 4 единицы), причем основной формой являлся димер. После расщепления EcoRI обнаруживали только линейную плазмидную ДНК; данные виды плазмид соответствовали только плазмидным мультимерам или различным топоизомерам. Однако поскольку размер определенных с помощью сверхспирального ДНК-маркера форм соответствовал целому продукту мономерной плазмиды, имеется высокая вероятность того, что они являются мультимерами. Образование мультимеров, наиболее вероятно, имеет отношение к мутации pir116, хотя два штамма BW19094 и BW19610 не являются строго изогенными (BW19610 имеет recA). Полученный с использованием pXL2730 профиль отличался: хотя мультимерные формы были еще видны, основной формой являлась мономерная. Фрагмент cer, таким образом, может облегчать разрешение плазмидных мультимеров, которые сконструировали авторы изобретения, независимо от наличия recA в BW19610.
(ii) анализ после обработки ДНК-топоизомеразами
Для опровержения той теории, что формы, наблюдаемые в штаммах, несущих аллель pir116, являются специфическими топоизомерами, каждый плазмидный препарат подвергали действию ДНК-топоизомераз. Активности различных ферментов в экспериментальных условиях были следующими: релаксация ДНК для ДНК-топоизомеразы I E. coli, отрицательное сверхскручивание релаксированной ДНК для ДНК-гиразы E. coli, и распутывание сплетенных ДНК и релаксация сверхспиральной ДНК ДНК-топоизомеразой IV S. aureus. Действие ДНК-топоизомеразы IV дало возможность показать, что формы плазмиды с высокой молекулярной массой не являются результатом запутывания нескольких плазмидных молекул; в данном случае, они были преобразованы в мономерные виды. Функциональность данного фермента, конечно, проверяли на препарате ДНК кинетопласта, состоящего из запутанных молекул ДНК (не показано). Активность по расслаблению также была видна, поскольку получали виды молекул, которые мигрировали слабее, чем необработанный контроль. Действие ДНК-гиразы дало возможность преобразовать слабо релаксированные топоизомеры в более сверхспирализованные виды молекул, выделенные из бактерий (преимущественно, мономер или димер). Более того, была возможность подтвердить, что полученные ДНК преимущественно были в сверхспиральной форме. Обработанные таким образом образцы обеспечивали подтверждение указанных выше результатов в плане основных видов каждой конструкции. ДНК-топоизомераза I действительно релаксировала ДНК, но лишь частично. Это могло быть следствием того факта, что исследуемые плазмиды содержат только несколько однонитевых областей, с которыми предпочтительно связывается данный фермент (Roca, 1995).
2) Введение маркера селекции sup Phe в pXL2730
Авторы изобретения использовали экспрессирующую кассету синтетического гена супрессорной тРНК (Phe) (Kleina et al., 1990). Она вводит фенилаланин в растущую полипептидную цепь в ответ на кодон TAG. Более того, она обеспечивает продукцию в XAC-1 белка ArgE, который достаточно активен, чтобы обеспечить хороший рост в дефицитной по аргинину среде. sup Phe экспрессировалась конститутивно на плазмиде pCT-2-F (Normanly et al., 1986) из синтетического промотора, происходящего из промоторной последовательности Plpp гена E.coli lpp. Ниже данного гена транскрипцию останавливали синтетическим терминатором TrrnC оперона E. coli rrnC (Normanly et al., 1986). Различные стадии клонирования указаны на фигуре 5.
В XAC-1 проводили различные субклонирования. Функциональность экспрессии с экспрессирующей кассеты супрессорной тРНК, таким образом, проверяли посредством a-галактозидазной активности данного штамма, который существует только если имеется супрессия амбер-кодона гена lacZu118am. Начальная стадия состоит из введения экспрессирующей кассеты sup Phe в pXL2730. Результаты, полученные с использованием фрагмента cer (B-1-b), привели к селекции данной плазмиды, а не pXL2666. Авторы изобретения сохраняли ген устойчивости к канамицину для простоты последующего клонирования, в частности, для возможности доступного дополнительного скрининга во время конечного клонирования (потеря KmR).
Пример 3. Подтверждение возможности применений в генной терапии данного плазмидного вектора путем трансфекции фибробластов мыши
1) Конструирование репортерного вектора pXL2774
Для тестирования обоснованности применения в генной терапии системы продукции плазмидной ДНК авторы изобретения вводили репортерный ген, который может использоваться в эукариотических клетках в pXL2760. Авторы изобретения использовали ген luc, который кодирует люциферазу Photinus pyralis, поскольку тест измерения биолюминесценции очень чувствителен и является линейным в большом интервале, и фоновый шум вследствие эндогенной активности эукариотических клеток является очень низким. Ген luc был под контролем промоторных-энхансерных последовательностей раннего гена человеческого цитомегаловируса (промотор CMV), который обеспечивал высокий уровень экспрессии. Имелась нетранслируемая область на 3'-конце luc, происходящая от вируса SV40, которая содержала сигнал полиаденилирования (поли(A)+). После промежуточного клонирования, которое позволяло увеличить число доступных участков рестрикции, кассету "CMV-промотор-luc-поли(A)+" вводили в минимальный вектор ori -cer-sup Phe (pXL2760) вместо маркера KmR. Полученную в результате плазмиду назвали pXL2774. На фигуре 6 показаны различные стадии клонирования. Смеси лигирования трансформировали в XAC-1 pir116 путем электропорации. Проводили инкубацию, позволяющую бактериям экспрессировать маркеры селекции на обогащенной среде (среда SOC); поэтому было необходимо отмывать клетки дважды средой M9 до посева на чашки. Это дало возможность для удаления остаточной среды, которая приводила к фоновому шуму культуры на минимальной среде.
Среда, выбранная для посева подвергшихся электропорации клеток, представляла собой минимальную среду M9, что дало возможность для отбора бактерий, экспрессирующих супрессорную тРНК и, таким образом, присутствия плазмид авторов изобретения. Добавление X-Gal дало возможность посредством голубого окрашивания визуализировать экспрессию супрессорной тРНК. Чашки анализировали примерно через 20 часов при 37°C. Отсутствие колоний на не содержащем ДНК контроле убедило авторов изобретения, что отбор был корректным, даже при плотном осеменении. Все клоны, оцененные путем рестрикции (8), действительно несли плазмиду, соответствуя ожидаемому профилю. Сконструированную таким образом плазмиду pXL2774 получали из клона, культивируемого в одном литре жидкой среды M9 (примерно 18 часов при 37°C), способом, задействующим, среди прочего, ионообменную стадию (набор Promega, MegaPreps). Количество собранной ДНК составляло 2 мг.
2) Анализ репортерного вектора pXL2774, трансфицированного в клетки млекопитающих
Способность pXL2774 трансфицировать эукариотические клетки и обеспечивать экспрессию люциферазы оценивали трансфекцией в мышиные фибробласты NIH 3T3. Выбранный в качестве вектора сравнения вектор представлял собой плазмиду pXL2622 (это плазмида pGL2 от Promega, промотор SV40 которой заменен на промотор CMV), которая несет ту же люциферазную экспрессирующую кассету, что и pXL2774, только в другом репликоне. Это производное ColE1 длиной 6,2 т.п.н., которое несет ген устойчивости к ампициллину. Данную плазмиду применяли в качестве контроля. Значения люциферазной активности (выраженные в RLU, или относительных единицах люминесценции) приведены в таблице 3.
Наилучшие результаты получены при отношении "заряды липофектанта/заряды ДНК", равном 6; в данных условиях pXL2622 и 2774 оказываются эквивалентными.
Пример 4. Подтверждение природы суицидного вектора плазмид pCOR в E. coli
Нерепликативную природу плазмид типа pCOR, происходящих от R6K, подтверждали экспериментами по электропорации в E. coli JM109 (Yanisch-Perron et al., 1985) плазмид pUC4K (ori ColEI-KmR, (Vieira and Messing, 1982)) и pXL2730 (ori гамма из R6K-KmR, см. пример 2). Использованный электропоратор представлял собой Biorad Gene Pulser, а электрокомпетентные клетки JM109 получали и использовали по процедуре производителя (Инструкция по электротрансформации бактерий и импульсного контроллера, каталожный номер 165-2098).
Электротрансформированные клетки высевали на среду LB, дополненную канамицином (50 мг/л), и инкубировали в течение ночи при 37°C. Полученные результаты представлены ниже.
Результаты
Результаты показывают, что имели место, как минимум, 5 порядков различия между эффективностью трансформации производным ColEI (pUC4K) и производным R6K (pXL2730) в штамме, который не экспрессировал ген pir. В штамме pir+, таком как XAC-1 pir116, эффективность электротрансформации происходящими из R6K плазмидами обычно достигает или превышает 108 транформантов/мкг плазмиды.
Пример 5. Продукция плазмидной ДНК путем культивирования с высокой плотностью штамма E. coli XAC-1 pir116 (pXL2774): процесс ферментации
5.1. Штаммы.
Продукция в E. coli XAC-1 pir116 (пример 1) минимальной плазмиды pXL2774; данная плазмида содержит следующие элементы: ori R6K-cer-тРНК amsupPhe и экспрессирующую кассету гена-репортера luc под контролем промотора CMV (пример 3). Разрабатывали высокопродуктивный процесс получения плазмид данного типа.
5.2. Среды и условия культивирования.
a) Ростовые среды.
Композиция определенной среды, использованной для культур инокуляции (г/л): Na2HPO4 6, KH2PO4 3, NaCl 0,5, NH4Cl 1, NH4H2PO4 3, глюкоза 5, MgSO4·7H2O 0,24, CaCl2·2H2O 0,015, тиамин-HCl 0,010.
Композиция комплексной среды, использованной для подпитываемых культур (г/л): KH2PO4 8, K2HPO4 6,3, Na2HPO4 1,7, (NH4)2 SO4 0,74, NH4Cl 0,12, дрожжевой экстракт 3, глюкоза 2, MgSO4·7H2O 2,4 г/л, CaCl2·2H2O 0,015, тиамин 0,010, раствор солей (Fe, Mn, Co, Zn, Mo, Cu, B, Al).
Композиция среды, определенная для культур в среде для подпитки, идентична комплексной среде, но дрожжевой экстракт заменен 2,5 г/л NH4 Cl.
b) Условия подпитываемой культуры.
Исследования в 2-литровых ферментерах (Setric, Франция), содержащих 1 л среды, проводили для определения оптимальных условий роста и продукции плазмидной ДНК. Ферментер инокулировали 80 мл культуры инокуляции, достигшей начала стационарной фазы роста.
Во время ферментации контролировали pH и доводили ее автоматически до интервала от 6,9 до 7,0 10% (мас./об.) нашатырным спиртом; температуру поддерживали при 37°C; аэрацию устанавливали на 75 л/ч (1,1 vvm) при давлении 0,2 бар, а растворенный кислород доводили до 40% насыщения воздуха путем взаимодействия при скорости перемешивания и, если необходимо, путем насыщения чистым кислородом.
Все параметры (pH, температура, перемешивание, OD, O2 и CO2 в отходящих газах) собирали и рассчитывали на входящей линии через интерфейс HP3852, соединенный с Hewlett-Packard 9000.
Основная композиция среды подачи была следующей: 50% источника углерода, 0,7% сульфата магния, 0,02% тиамина; для комплексной среды добавляли дрожжевой экстракт до концентрации, предпочтительно составляющей от 5 до 10%.
Для адаптации условий культивирования к 800-литровым ферментерам проводили циклы продукции, состоящие из двух последовательных культур инокуляции, в лабораторном масштабе: инокуляция I в перемешиваемой конической колбе и инокуляция II в 2-литровом ферментере (культивирование в замкнутом объеме), с последующим подпитываемым культивированием для продукции в 7-литровом ферментере.
5.3. Результаты
Различные условия культивирования исследовали в комплексной среде, в определенной среде и при различных скоростях роста. Во всех случаях после начального культивирования бактериального штамма в замкнутом объеме и потребления источника углерода в ферментер добавляли среду подачи посредством перистальтического насоса, соединенного с предварительно запрограммированным профилем добавления. Данный профиль формировали на основании предыдущих экспериментов, в которых скорость подачи контролировали по уровню растворенного кислорода или по постоянной скорости роста.
Более того, для экстраполяции без затруднений условий ферментации в 2 литрах на 800-литровый ферментер без избыточной оксигенации среды, максимальную потребность в кислороде в конце культивирования устанавливали на уровне 2,5-3 мМ/мин. Для этого скорость роста микроорганизма снижали, если это необходимо, путем вариации скорости подачи добавочной загрузки.
Как видно из таблицы 4, получали очень хорошие результаты как в комплексной среде, так и в определенной среде, в лабораторном масштабе и в масштабе 800-литрового ферментера; более того, рост плазмидной ДНК и кинетика продукции полностью сравнимы (ср. фигуры 6 и 7).
Х = биомасса (масса сухих клеток)
Из общих результатов вытекает, что:
- изменение масштаба ферментера от 2 литров до 800 литров может проводиться без всяких проблем,
- потребление кислорода строго коррелирует с продукцией биомассы (1,08 г О2/г продуцированной биомассы),
- плазмида является стабильной по меньшей мере для 50 поколений без давления отбора,
- большое количество биомассы, более 40 г сухих клеток/литр, может быть получено в комплексной среде,
- продукция плазмидной ДНК достигает 100 мг сверхспиральной ДНК/л среды,
- имела место хорошая корреляция между продукцией ДНК и количеством биомассы: продукция может оцениваться как 1 мг плазмидной ДНК/единицу OD, или, альтернативно, как 2,7 мг плазмидной ДНК/г биомассы, вне зависимости от длительности ферментации,
- применение определенной среды также дает возможность достичь большого количества биомассы (30 г сухих клеток/л) и высокой продукции плазмидной ДНК (100 мг/л), без какой-либо потери продуктивности.
Пример 6. Перенос pXL2774 в животные клетки in vitro и in vivo
6.1. Перенос pXL2774 в животные клетки in vitro
Способность минимальной плазмиды pXL2774 трансфицировать различные клеточные линии тестировали in vitro на клетках человеческого происхождения и мышиного происхождения. Плазмиду pXL2784 использовали в качестве контроля. Она содержит ту же эукариотическую экспрессирующую кассету (промотор CMV-люцифераза-полиА), что и pXL2774, но это производное ColEI длиной 6,4 т.п.н., которое содержит ген для придания E. coli устойчивости к канамицину.
Тестируемые клетки были следующими:
Условия трансфекции были следующими:
D-1: Инокуляция клеток при плотности 100000 клеток на 2 см2 лунки (24-луночного планшета) в среде DMEM (модифицированная по Дульбекко среда Игла), дополненной 10% фетальной сыворотки теленка (FCS).
D-3: Трансфекция клеток 10 мкл раствора для трансфекции, содержащего: 0,5 мкг ДНК, 150 мМ NaCl, 5% глюкозу и 3 нмоль липофектанта RPR120 535 на мкг ДНК, в 250 мкл культуральной среды, которую дополняли или не дополняли 10% FCS. После инкубации в течение 2 часов среду замещали 500 мкл среды DMEM, дополненной 10% FCS.
D-4: Обновление культуральной среды
D-5: Промывка клеток PBS, с последующим лизисом 100 мкл буфера для лизиса Promega (Promega Cell Lysis Buffer E153 A). Анализ люциферазной активности проводили на люминометре Lumat LB 9501 (Berthold) в 10 мкл лизата, с 10-секундной длительностью интегрирования сигнала. Использованный реагент был от Promega (субстрат для люциферазного анализа Promega). Результаты, объединенные в следующие таблицы 5-8, выражали в RLU (относительных световых единицах) для 10 мкл лизата (среднее от измерения на 4 лунках). Также приведены коэффициенты вариации (CV).
Результаты трансфекции в отсутствие сыворотки представлены ниже.
Результаты трансфекций в присутствии сыворотки (10%) представлены ниже:
Данные результаты выявляют способность pXL2774 эффективно трансфицировать in vitro различные клеточные типы мышиного и человеческого происхождения. Экспрессия гена-репортера luc дала возможность показать, что эффективность трансфекции ею по меньшей мере такая же, как для "стандартной плазмиды", происходящей из ColE1, которая несет ту же экспрессирующую кассету люциферазы.
6.2. Перенос in vivo в животных (мыши) pXL2774
a) Модель 1. Голая ДНК в черепные tibial мышцы
Голую плазмидную ДНК, растворенную в 5% глюкозе, 150 мМ NaCl, инъецировали в черепную tibial мышцу мыши OF1. Мышцы удаляли через 7 суток после инъекции, измельчали, гомогенизировали в 750 мкл буфера для лизиса (буфер для лизиса клеток Promega E153A) и затем центрифугировали при 20000хg в течение 10 минут.
Анализ люциферазной активности проводили на 10 мкл надосадочной жидкости после добавления 50 мкл реагента (субстрат для люциферазного анализа Promega). Считывание проводили на люминометре Lumat LB9501 (Berthold) с 10-секундной длительностью интегрирования сигнала.
Результаты представлены в таблице, приведенной ниже.
Данные результаты показывают, что общепринятая плазмида для репликации, такая как pXL2774, действительно способна трансфицировать мышиные мышечные клетки in vivo со сравнимой или даже превосходящей эффективностью по отношению к "стандартной плазмиде", происходящей из ColE1, которая несет ту же экспрессирующую кассету гена люциферазы.
b) Модель 2. Опухолевая модель 3T3 HER2
Модель является следующей:
- Swiss/голые взрослые самки мышей
- Экспериментальные опухоли, индуцированные после инъекции клеток 107 3T3 HER2 подкожно в бок.
- Смесь трансфекции инъецировали через 7 суток после инъекции клеток.
Инъецированные растворы. ДНК вначале растворяли в буфере. После добавления всех продуктов смесь содержала, кроме ДНК, NaCl (150 мМ) и 5% D-глюкозу в воде или 5 мМ буфера HEPES.
- Через двое суток после инъекции опухолевую ткань удаляли, взвешивали и затем измельчали и гомогенизировали в 750 мкл буфера для лизиса (буфер для лизиса клеток Promega E153 A). После центрифугирования (20000хg в течение 10 минут) 10 мкл надосадочной жидкости удаляли и оценивали люциферазную активность. Данную активность определяли путем измерения общего испускания света, полученного после смешивания с 50 мкл реагента (субстрат для люциферазного анализа Promega) в люминометре Lumat LB 9501 (Berthold) с 10-секундной длительностью интегрирования сигнала.
Полученную в результате активность выражали в RLU (относительных световых единицах), оцененных в надосадочной жидкости лизиса цельной опухоли.
Результаты
Данные результаты показывают, что плазмида с зависимой от условий репликацией, такая как pXL2774, действительно способна трансфицировать мышиные опухолевые клетки in vivo, и эффективность трансфекции ею по меньшей мере сравнима с таковой для "стандартной плазмиды", происходящей из ColE1, которая несет ту же экспрессирующую кассету гена люциферазы.
Данные различные эксперименты демонстрируют, что плазмиды с зависимой от условий репликацией, и, более конкретно, pXL2774, действительно имеют характеристики трансфекции животных клеток, существенные для применения в генной терапии. Более конкретно, показано следующее:
1) способность pXL2774 эффективно транфицировать in vitro различные клеточные типы человеческого или мышиного происхождения;
2) способность pXL2774 трансфицировать in vivo мышиную мышцу;
3) способность pXL2774 трансфицировать in vivo опухолевые клетки, имплантированные мышам.
Эксперименты по электротрансформации, ферментации и трансфекции, таким образом, дали возможность для утверждения плазмид с зависимой от условий репликацией в качестве векторов, которые могут использоваться в генной терапии, причем в данных экспериментах было показано:
i) что данные плазмиды не реплицируются на детектируемом уровне в штамме E. coli, который не экспрессирует ген pir (зависимая от условий точка начала репликации)
ii) что они могут продуцироваться в масштабе, совместимом с промышленной продукцией, в определенной среде, которая не содержит антибиотиков;
iii) что данные плазмиды могут трансфицировать клетки млекопитающих in vitro и особенно in vivo.
Пример 7. Продукция рекомбинантных белков in vitro
7.1. Конструирование экспрессирующих векторов
Для того, чтобы показать осуществимость такого подхода, авторы изобретения сконструировали экспрессирующий вектор по критериям, описанным выше (примеры 2 и 3). Данный вектор pXL3056 содержит:
1) бактериальную часть, которая содержит зависимую от условий точку начала репликации (ori гамма), фрагмент cer ColEI, и ген, который гарантирует отбор в бактериях (sup);
2) экспрессирующую кассету, основанную на системе, описанной Студиером (Studier et al., 1990), включающей в себя промотор гена 10 бактериофага T7, оператор lacO, ген, кодирующий aFGF 154 (кислый фактор роста фибробластов, форма, содержащая 154 аминокислот) (Jaye et al., 1986), и терминатор TF бактериофага T7. Данная экспрессирующая кассета идентична присутствующей в плазмиде pXL2434, которая описана в применении WO 96/08572.
Конструкция pXL3056 представлена на фигуре 8. Фрагмент EcoRI-BglII pXL2434 (1,1 т.п.н.), содержащий кассету, экспрессирующую aFGF, клонировали в вектор pXL2979 с зависимой от условий репликацией (очищенный фрагмент длиной 1,1 т.п.н.) в участки BglII и EcoRI, с образованием pXL3056.
pXL2979 является результатом лигирования 3 фрагментов: i) фрагмент AccI-XbaI pXL2730 (0,8 т.п.н., где предоставлены ori гамма и cer), ii) фрагмент NarI-SalI pXL2755 (0,18 т.п.н., где предоставлен ген sup Phe), iii) фрагмент SalI-SpeI pXL2660 (1,5 т.п.н., где предоставлен ген устойчивости к канамицину).
pXL2660 является результатом клонирования PstI-фрагмента длиной 1,2 т.п.н. из pUC4K (Vieira and Messing, 1982) в pMTL22 (Chambers et al., 1988), линеаризованную PstI.
7.2. Продукция экспрессирующего штамма
Плазмиду pXL3056 вводили путем трансформации в штамм XAC-1 pir116. Полученный в результате штамм затем трансформировали плазмидой PT7 pol23 (Mertens et al., 1995) при 30°C. Для экспрессии интересующего гена под контролем промотора T7 бактерия должна содержать в своем геноме, в плазмиде или в бактериофаге кассету, обеспечивающую экспрессию РНК-полимеразы бактериофага T7. В описанном примере авторы изобретения использовали плазмиду pT7pol23, совместимую с производными R6K, такими как pXL3056, и обеспечивающую индуцируемую температурой экспрессию РНК-полимеразы бактериофага T7. Однако также можно предложить подвергнуть штамм XAC-1 pir116 лизогении лямбдой DE3 (Studier et al., 1990) для сохранения только одной плазмиды и для индукции продукции РНК-полимеразы T7 посредством IPTG, а не температурой.
7.3. Экспрессия aFGF
Штамм XAC-1 pir116 (pXL3056+PT7pol23) культивировали при 30°C в минимальной среде M9, дополненной 0,2% казаминовыми кислотами (DIFCO) и канамицином (25 мкг/мл), до оптической плотности при 600 нм, равной 0,6-1,0. Затем половину культуры помещали на 42°C (индукция РНК-полимеразы T7), тогда как другую половину оставляли при 30°C (отрицательный контроль). Тот же эксперимент проводили на штамме XAC-1 pir116 (pXL3056+pUC4K), который представляет собой контроль для экспрессии aFGF в отсутствие РНК-полимеразы T7.
Полученные результаты представлены на Фиг.9. Они показывают, что продукция aFGF была сравнима или превышала таковую, полученную в системе BL21(DE3)(pXL2434) (WO 96/08572), а это четко указывает на потенциал плазмид с зависимой от условий репликацией в плане экспрессии рекомбинантных белков in vitro, особенно в E. coli.
Пример 8. Конструирование вектора pCOR, которое экспрессирует белок p53 дикого типа или гибридный p53, или человеческий белок FGF1.
В данном примере описано конструирование векторов с зависимой от условий репликацией согласно изобретению, содержащих нуклеиновую кислоту, кодирующую белок p53. Данные векторы могут использоваться для восстановления активности типа p53 в дефицитных клетках (с мутацией, делецией), таких как, например, опухолевые клетки.
Эукариотическая экспрессирующая кассета содержит следующие элементы:
1) "Немедленный ранний" промотор CMV (положения от -522 до +72) с последующей лидерной последовательностью гена тимидинкиназы вируса простого герпеса типа I (положения от -60 до +1 гена со ссылкой на последовательность из статьи McKnight, S.L. (1980) Nucleic Acids Res. 8: 5949-5964);
2a) нуклеиновую кислоту, которая кодирует белок p53 дикого типа или вариант p53, описанный в заявке PCT/FR96/01111 (вариант V325K = V325 с последовательностью Kozak с ATG);
2b) нуклеиновую кислоту, которая кодирует человеческий FGFa или FGF-1, описанные в Jaye M. (Sciences 1986; 233 (4763):451), патенте США № 4686113, и Европейском патенте № 259475;
2c) нуклеиновую кислоту, которая кодирует ген слияния сигнала секреции интерферона человеческих фибробластов (Taniguchi et al.) и встречающейся в природе укороченной формы человеческого FGF-1 от аминокислоты 21 до 154, описанной Jaye et al., и в патенте США 5849538.
3) Последовательность полиаденилирования polyA SV40.
Данные элементы помещали в виде фрагмента AscI-XbaI в вектор pCOR pXL2988 между участками BssHII и SpeI. pXL2988 идентична pXL2979 (пример 7.1.), кроме присутствия добавочного элемента, последовательности, способной образовывать тройную спираль ДНК, составленной из 17 копий тринуклеотида GAA, помещенных около точки начала репликации гамма.
Полученные в результате плазмиды назвали pXL3029, pXL3030, pXL3179 или NV1FGF (фигура 10).
Функциональность данных конструкций подтверждали in vitro на клетках p53-SAOS2 в культуре путем измерения активности активатора транскрипции p53 или p53superWT, или путем измерения секреции FGF1, например, в экспериментах ELISA, которые хорошо известны в данной области.
Пример 9. Конструирование TEX 1 (XAC1 pir116, endA-, traD-)
E. coli XAC-1 pir116 содержит F'-эписому, кольцевую молекулу ДНК длиной примерно 100 т.п.н., которая несет proB+ lacI373 lacZu118am. Многие мужские лабораторные штаммы E. coli несут мутацию traD36 на своей эпиосоме, но не было описано мутаций, воздействующих на перенос F' в XAC-1. Ген traD находится на 5'-конце оперонов tra (переноса) и кодирует мембранный белок, непосредственно участвующий в переносе ДНК и метаболизме ДНК (Frost et al., BBRC, 1994, 58: 162-210). Центральный фрагмент ДНК длиной 2 т.п.н. из traD, содержащий 92% гена, замещали омега-элементом длиной 2 т.п.н. (инвентарный номер Genbank M60473) из pHP45(Prentki and Krisch, 1984, Gene, 29: 303-313) путем гомологичной рекомбинации в XAC-1 pir116 endA-. Омега-элемент содержит ген устойчивости к антибиотикам aadA, фланкированный короткими инвертированными повторами. Ген aadA кодирует аминогликозид-3-аденилтрансферазу и придает устойчивость к стрептомицину и спектиномицину ("SpR"). Омега-фрагмент использовали, поскольку он приводит к преждевременному завершению РНК и синтеза белка, ведя к инактивации целого оперона traD. Данный новый штамм pCOR XAC-1 pir116 endA- traD-::SpR обозначали TEX1. Перенос любых постоянных плазмид, pCOR или pUC, не выявлялся в случае, когда донор представлял собой TEX1.
Новый штамм хозяина для pCOR TEX1 оценивали в экспериментах по ферментации. Комплексные среды, содержащие дрожжевой экстракт, использовали для подпитываемой ферментации с XAC-1 pir116. Стабильность pCOR (более 50 поколений) дала возможность использования неселективных сред. В данных условиях XAC-1 pir116 продуцировал более 40 г/л массы сухих клеток и 100 мг/л pCOR pXL2774 было получено из 2-литровых ферментерах. Число копий pCOR оценивали как 400-500 копий на клетку, и скорость синтеза плазмидной ДНК была постоянной во время ферментации. Данные результаты экстраполировали на применение 800-литрового ферментера, подходящего для производства. Ферментацию также проводили в отсутствие дрожжевого экстракта или любого сырого материала животного происхождения. Сходные результаты (30 г/л сухой массы клеток и 100 мг/л плазмидной ДНК) получали с использованием определенной среды в 2-литровых культурах без потери продуктивности.
Пример 10. Конструирование штаммов хозяина XAC-1 pir116 pir42 путем гомологичной рекомбинации
1) Конструирование суицидного вектора, несущего кассету "KmR-uidA::pir116:pir42"
Кассету KmR-uidA::pir116 из M13wm33, описанную в примере 1 (Metcalf W. et al. Gene, 1994, 138(1-2): p. 1-7), модифицировали путем сайт-специфического мутагенеза с использованием PCR (набор для сайт-специфического мутагенеза QuickChange, Stratagene, La Jolla, Калифорния) для введения мутации pir42 в ген pir116. Различные стадии клонирования/мутагенеза описаны на фигуре 13.
Используемые для мутагенеза олигонуклеотиды содержат мутацию pir42 вместе с молчащей мутацией, которая создает участок ClaI, чтобы легко обозначить процессинг pir42 путем анализа рестрикции, если это требуется.
Используемые смысловые и антисмысловые олигонуклеотиды являются следующими:
смысловой олигонуклеотид номер 11076 (SEQ ID NO: 7)

антисмысловой олигонуклеотид номер 11077 (SEQ ID NO: 8)

Способ, используемый для замены pir116 на pir116 pir42 в геноме хозяев E. coli pCOR TEX1, был основан на способе Blum et al. (J. Bacteriol. 1989, 171, pp 538-46). Рекомбинантный бактериофаг pXL3723, показанный на фигуре 13, представляет собой суицидный вектор во всех несупрессорных штаммах E. coli, поскольку он имеет несмысловую мутацию в гене II, кодирующем никазу M13, которая предотвращает репликацию вирусного генома.
Двойную рекомбинацию проводили, как описано для конструкции XAC-1 pir116 (пример 1, пункт 2). Клоны, претерпевшие события двойной гомологичной рекомбинации, подвергали скринингу путем PCR для тестирования присутствия мутации pir42 в геноме TEX1. Геномную ДНК, выделенную из кандидатов на двойную рекомбинацию, использовали в качестве матрицы для PCR. Во-вторых, проводили секвенирование каждого уникального амплифицированного фрагмента, причем все они были ожидаемого размера. PCR-фрагменты показаны на фигуре 14.
Праймеры PCR были следующими:
праймер 11088 (SEQ ID NO: 9): 5'-GAGATCGCTGATGGTATCGG-3'
праймер 11089 (SEQ ID NO: 10): 5'-TCTACACCACGCCGAACACC-3'
Данный анализ показал, что один из шести двойных рекомбинантов претерпел аллельный обмен. Данный новый штамм, называемый TEX1 pir42, далее оценивали на его способность реплицировать плазмиды pCOR по сравнению с родительским штаммом TEX1.
2) Оценка TEX1 pir42
Плазмиды pCOR трансформировали параллельно TEX1 и TEX1 pir42 и выращивали в течение ночи в 2 мл селективной среды M9. Затем плазмидную ДНК экстрагировали наборами для получения минипрепаратов Wizard SV plus (Promega) для оценки относительного числа копий плазмиды и топологии плазмид pCOR в обоих штаммах.
2-кратное увеличение числа копий воспроизводимо получали в TEX1 pir42, трансформированных плазмидой pCOR pXL3516 (2,56 т.п.н.). Для дальнейшей характеристики TEX1 pir42 число копий и топология плазмид pCOR, таких как pXL3179 и pXL2774, оценивали путем анализа электрофореза в агарозном геле после очистки плазмидной ДНК в малом масштабе (от 4 до 6 клонов/штамм). Число копий оценивали на плазмидах, линеаризованных ферментом рестрикции EcoRI. Тест топологии проводили на нерасщепленных плазмидах, в отсутствие этидий-бромида. Полученный в результате агарозный гель показан на фигуре 15, и на нем ясно видно более высокое число плазмид при продукции pXL3179 в TEX1 pir42 по сравнению с продукцией штамма TEX1. На фигуре 15 также показана топология плазмиды pXL3179, и там показано, что повышение числа копий плазмиды, которая, по существу, была в виде мономеров, с новыми плазмидами в виде мультимеров. Результаты, полученные с данными плазмидами pCOR, также суммированы в таблице 5. Относительное число копий рассчитывали по сравнению с той же плазмидой, что находилась в TEX1. 2-3-кратное повышение в числе копий плазмиды наблюдали с плазмидами pXL3179 и 2774, продуцированными в TEX1 pir42.
Пример 11. Сравнительные эксперименты: конструирование TEX1 cop21 (XAC-1 endA- traD pir116 cop21)
1) Конструирование TEX1 cop21
Штамм TEX1 cop21 конструировали так же, как описано в примере 10 для TEX1 pir42. Олигонуклеотиды, использованные для введения cop21 в ген pir116 путем сайт-специфического мутагенеза, были следующими:
смысловые олигонуклеотиды 11153 (SEQ ID NO: 11)

антисмысловой олигонуклеотид 11154 (SEQ ID NO: 12)

Мутацию cop21 вводили в виде серинового кодона TCC вместо серинового кодона TCA для удаления участка рестрикции HindIII вблизи мутации.
Используемой для направленного мутагенеза матрицей была pXL3395 (см. фигуру 13). Полученную в результате плазмиду, названную pXL3432, использовали для конструирования суицидного вектора M13 так же, как это показано для pir42 на фигуре 13. Суицидный вектор pXL3749 показан на фигуре 16.
Клоны E. coli, полученные после гомологичной рекомбинации с pXL3749, подвергали скринингу посредством PCR и последующей рестрикцией посредством HindIII, а также секвенированию для мониторинга мутаций cop21 и pir116. Один клон из шести тестированных двойных рекомбинантов претерпел перемещение генов. Полученный в результате штамм назвали TEX1 cop21.
2) Оценка TEX1 cop21
TEX1 cop21 трансформировали различными плазмидами pCOR, включающими pXL2979, pCOR-вектор KmR длиной 2,5 т.п.н. (см. пример 7.1) и анализировали на предмет повышения числа копий путем гель-электрофореза. Такой эксперимент с pXL2979 показан на фигуре 17. Плазмидную ДНК из четырех независимых клонов для каждого штамма, полученную с использованием набора для минипрепаратов Promega линеаризовали с использованием EcoRI, подвергали электрофорезу на агарозном геле и затем окрашивали с использованием этидий-бромида. Каждый образец представлял собой сходное количество бактерий, что измерено за счет оптической плотности при 600 нм. Электрофорез в агарозном геле, полученный для плазмиды pCOR pXL2979, продуцированной в TEX1 cop21, XAC1 pir и TEX1 E.coli, показан на фигуре 17. Из нее ясно видно, что в числе копий плазмиды не имеется повышения, когда плазмиды продуцируются в штамме TEX1 cop21 по сравнению с TEX1.
Пример 12. Конструирование TEX2 pir42 (XAC-1 pir116 pir42 recA- )
Вначале конструировали recA- производное TEX1. Затем мутацию pir42 вводили в полученный в результате штамм, обозначенный TEX2, с образованием TEX2 pir42.
1) Конструирование E. coli TEX2, recA- производного TEX1
Ген recA с делецией, содержащий 3 стоп-кодона трансляции (по одному в каждой рамке) на его 5'-конце, получали путем PCR. Данный ген recA с делецией вводили путем замены гена (Blum et al. J. Bacteriol., 1989, 171, pp. 538-46) в геном TEX1. Конструкция суицидного вектора для гомологичной рекомбинации показана на фигуре 18. Праймеры PCR, использованные для амплификации фрагментов recA, показаны в следующей таблице 6.


Участки рестрикции, добавленные к последовательности recA, подчеркнуты.
Для поддержания фенотипа RecA+, необходимого для того, чтобы произошла гомологичная рекомбинация, функция recA была обеспечена для E. coli TEX1 посредством плазмиды, содержащей гетерологичный ген recA, который может дополнять мутанты recA E. coli, такие как, например, ген recA бактерии Agrobacterium radiobacter, и ген устойчивости к антибиотикам, такой как ген устойчивости к ампициллину. После замены гена плазмиду элиминировали из рекомбинантного штамма путем разведения культуры в неселективной среде (LB). Отсутствие плазмиды подвергали скринингу на предмет потери устойчивости к антибиотику.
Полученный в результате штамм назвали TEX2. За заменой гена следили путем PCR, как показано на фигуре 19. Праймеры PCR описаны в следующей таблице 7.
Первый праймер основывался на последовательности гена recA. Второй основывался на последовательности, находящейся близко, но вне участка гомологии, присутствующего в суицидном векторе pXL3457 (непосредственно 5'- или 3'-конец recA), для того, чтобы амплификация могла произойти только на геномном фрагменте. Последовательность обоих олигонуклеотидов выбирали по последовательности E. coli, которая включает в себя локус recA (Genbank ECAE000354).
PCR-фрагменты, полученные на штамме с делецией recA, были короче по сравнению с полученными на штаммах дикого типа, как представлено на следующей таблице 8.
Таблица 8
Праймеры PCR для амплификации recA

Полученный профиль PCR был ожидаемым и демонстрировал присутствие укороченного гена recA в геноме TEX2. Проверяли recA- фенотип (чувствительность к УФ-свету), а также фенотипические характеристики TEX2. Фенотипические характеристики TEX2 были теми же, что и для штамма TEX1, т.е. ara-, RifR, NalR, SpR, UidA-, Arg-, KmSAmpS, как и ожидалось.
B) Конструирование E. coli TEX2 pir42
Штамм TEX2 pir42 конструировали путем двойной гомологичной рекомбинации по стратегии, описанной в примере 10, за исключением того, что рекомбинацию в TEX2 проводили в присутствии плазмиды, несущей гетерологичный ген recA, способный дополнять мутантов recA E.coli, для поддержания recA+ фенотипа, требуемого для гомологичной рекомбинации.
За заменой гена следили путем рестрикционного анализа PCR-продукта, расщепленного ClaI (см. фигуру 14). Замена гена происходила в двух из четырех исследованных клонов с двойной рекомбинацией.
C) Оценка E. coli TEX2 pir42
1) Оценка продукции плазмиды в лабораторном масштабе
TEX2 pir42 трансформировали pCOR-плазмидой pXL3179 (2,4 т.п.н.). Продукцию pXL3179 в TEX2 pir42 интенсивно исследовали в лабораторном масштабе, в плане воспроизводимости увеличения числа копий плазмиды, условий культивирования, а также стабильности (число поколений). Все исследования, соответственно, показали 2-5-кратное увеличение числа копий плазмиды по сравнению с продукцией pXL3179 в TEX1 при тех же условиях. Число копий плазмиды оценивали далее в плане продукции pXL3179 в TEX2 pir42, TEX1 pir42 и TEX1, в сравнительных экспериментах. В данном эксперименте плазмиды выделяли из идентичного количества бактериальной биомассы, основываясь на OD при 600 нм, и анализировали путем электрофореза в агарозном геле. Гель окрашивали этидий-бромидом после электрофореза. Электрофорез в агарозном геле, который показан на фигуре 20, ясно демонстрирует, что плазмиды продуцированы в TEX2 pir42 с высокой копийностью, и он показывает, что, как преимущество, число плазмидных мультимеров снижено, когда продукция происходит в TEX2 pir42 вместо TEX1 pir42.
2) Оценка в ферментерах
Данные результаты подтверждали в большем масштабе в 7-итровых ферментерах, как описано ниже.
a) Композиция среды ферментации
Композиция среды, использованной для культур инокуляции, представляла собой: Na2HPO4 6 г/л, KH2PO4 3 г/л, NaCl 0,5 г/л, NH4Cl 1 г/л, NH4H2PO4 3 г/л, глюкоза 5 г/л, MgSO4·7H2О 0,24 г/л, CaCl2·2H2O 0,015 г/л, тиамин-HCl 0,010 г/л.
Композиция среды, использованной для подпитываемой культуры, была следующей: KH2PO4 8 г/л, K2HPO4 6,3 г/л, Na2HPO4 1,7 г/л, NH4Cl 2,5 г/л, глюкоза 10 г/л, MgSO4·7H2О 2,6 г/л, тиамин 0,011 г/л, противопенное средство Biospumex36 antifoam 0,1 мл/л, солевая смесь (см. таблицу 9) 2,5 мл/л.
Композиция среды подачи была следующей: 50% глюкозы, 0,7% магния, 0,02% тиамина-HCl, 1% противопенного средства Biospumex36.
b) Параметры ферментации
7-литровый ферментер, содержащий 3 литра среды подпитки, инокулировали 1,2% культуры инокуляции. Культуру для инокуляции получали следующим образом: 250 мл среды инокуляции в 2-литровой колбе инокулировали 0,25 мл замороженной суспензии клеток штамма E. coli TEX2 pir42 (pXL3179).
Флаконы инкубировали в течение 24 часов при 37°C и 220 об./мин. Через 24 часа измеряли различные параметры: остаточная глюкоза составляла 0 г/л, OD600нм составляла 2,7, а pH был равен 6,24. Во время ферментации контролировали pH, и доводили его автоматически от 6,9 и 7 NH3. Температуру поддерживали при 37°C, и растворенный кислород доводили до 45% pO2 путем противодействия скорости перемешивания.
После начального культивирования бактериального штамма в замкнутом объеме примерно в течение 17 часов и затраты источника углерода (глюкозы) добавляли среду подачи. Глюкозу и кислоты, такие как лактат и ацетат, поддерживали в концентрации, близкой к 0.
c) Результаты
Конечные результаты представлены в таблице 10, по сравнению с продукцией в 100-литровом ферментере E. coli TEX1 (pXL3179) в оптимизированных условиях.
Как и для XAC-1 pirll6, не имелось отличий между 7-литровым и 800-литровым ферментерами в плане числа копий плазмиды pXL3179, продуцированной в TEX1.
Плазмиды pXL3179, продуцированные в 7-литровом ферментере с использованием E. coli TEX2 pir42 сравнивали с продукцией pXL3179 в 100-литровом ферментере с E. coli TEX1 в оптимизированных условиях. Было продемонстрировано, что, как и для XAC-1 pir116 (см. пример 5.3), имела место стабильная скорость продукции плазмиды в 7-литровом, 100-литровом, или 800-литровом ферментере в TEX1.
По сравнению с TEX1 имелось в 3 раза больше копий плазмиды pXL3179 на бактерию в TEX2 pir42.
Плазмиды, соответствующие различным временным точкам ферментации, экстрагировали из идентичного количества бактериальной биомассы, основываясь на OD при 600 нм, и анализировали путем электрофореза в агарозном геле. На фигуре 21 ясно показано увеличение числа копий плазмиды в течение ферментации. На фигуре 21 также показана топология плазмиды pXL3179, продуцируемой в Op1328S5 TEX2pir42, которая находится почти исключительно в мономерной форме.
Таким образом, штамм хозяина E. coli TEX2 pir42 согласно изобретению предоставлял неожиданно большое увеличение числа копий плазмид pCOR, таких как pXL3179, от 2 до 5-кратного в TEX2 pir42 по сравнению с TEX1, в лабораторном масштабе и в ферментерах. Более того, при том, что число копий плазмид сильно увеличилось, продуцированные таким образом плазмиды характеризовались мономерной топологией не только в лабораторном масштабе, но также и в большем масштабе (7-литровый ферментер), совместимом с промышленной продукцией.
Пример 13. Новый мутант с повышенной копийностью pir116, идентифицированный новым способом скрининга, основанном на флуоресценции
Для увеличения числа копий плазмиды pCOR в бактериальных клетках хозяина авторы изобретения подвергали мутагенезу ген pir116, который кодирует версию гена pir с повышенной копийностью. К настоящему моменту все мутации, повышающие число копий, происходящих от R6K плазмид, таких как осуществления pCOR, обнаружены в гене pir.
После случайного мутагенеза путем PCR мутантные гены pir116 вводили в вектор pCOR, содержащий ген-репортер cobA, который описан ниже. После основанного на флуоресценции скрининга оценивали число копий и топологию выбранной мутантной плазмиды. Авторы получали три различных мутанта гена pir116, которые повышают число копий плазмиды. Данные новые мутации не были описаны ранее.
Классический способ скрининга мутаций повышения копийности основан на устойчивости к антибиотикам. В данном способе уровень устойчивости бактерии хозяина к антибиотику является функцией числа копий гена устойчивости к антибиотику, расположенному на плазмиде внутри клетки. Как только увеличивается число копий плазмиды и, следовательно, гена устойчивости к антибиотику, также возрастает уровень устойчивости к антибиотику. Однако данный способ не применим для плазмид, происходящих из R6K, в клетках хозяина, содержащих мутацию pir116, вследствие слишком высокой фоновой копийности плазмиды (примерно 400 копий/клетку) в данных клетках. В соответствии с этим разрабатывали новый способ скрининга, основанный на флуоресценции, для идентификации мутаций гена pir116 по повышению копийности.
Для данного нового способа ген cobA вводили в вектор pCOR для обеспечения простого средства мониторинга увеличения копийности плазмиды. Ген cobA получали из Pseudomonas denitrificans (Crouzet et al., J. Bacteriol, 172: 5968-79 (1990)). Он кодирует уро-III-метилтрансферазу, фермент пути витамина B12, который добавляет две метильные группы к молекуле урогена III. При гиперэкспрессии в E. coli, cobA приводит к накоплению красных продуктов, которые флуоресцируют при освещении, близком к УФ. При воздействии УФ бактериальные колонии, гиперэкспрессирующие данный ген, становятся розовыми до красных. Авторы изобретения тестировали данный ген на предмет определения того, может ли он служить геном-репортером для копийности плазмиды в системе pCOR.
Для оценки отношений между числом копий плазмиды и уровнем флуоресценции трансформированных бактерий, на которых воздействует УФ-свет, конструировали контрольную плазмиду (pXL3767), содержащую cobA с делецией его собственного промотора (фиг. 22). Данную плазмиду трансформировали в три различных штамма хозяина (XAC1 pir, XAC1 pir116 и TEX1 pir42). Данные штаммы отбирали на основе предыдущих экспериментов, где показано, что среднее число копий плазмиды pCOR в XAC1 pir составляет 1, это число примерно в пять-десять раз выше в TEX1, и в 15-30-раз выше в TEX1 pir42.
Рекомбинантные колонии высевали полосами на минимальную среду M9 и подвергали воздействию УФ-света на трансиллюминаторе, как показано на фигуре 22. Авторы изобретения наблюдали, что интенсивность флуоресценции колоний положительно коррелировала с числом копий плазмиды при том, что XAC1 pir116 характеризуется более интенсивной флуоресценцией, чем XAC pir, и TEX1 pir42 характеризуется более интенсивной флуоресценцией, чем XAC1 pir116.
Результаты, показанные на фиг.22, демонстрируют, что данный основанный на флуоресценции способ анализа легко различает число копий тестируемых плазмид, особенно копийность плазмид, обнаруженную в штаммах TEX1 и TEX1 pir42. То есть, интенсивность красной флуоресценции, наблюдаемая в данном анализе, возрастает с копийностью плазмиды pCOR-cobA.
Продемонстрировав позитивную корреляцию между флуоресценцией и числом копий cobA, авторы изобретения конструировали плазмиду, в которую были введены мутантные гены pir116 для скрининга. Четыре плазмиды с различными комбинациями конститутивных модулей, как показано на фиг. 23, конструировали и тестировали. Одна из данных плазмид характеризовалась значительно отличающимся уровнем флуоресценции при трансформации изогенных штаммов pir116 и pirl116 pir42. Имеющая отношение к делу часть данной плазмиды pXL3830 показана на фиг.24.
Во время экспериментов по скринингу и оценке использовали контрольные плазмиды. Вначале использовали контрольную плазмиду фонового уровня pXL3830, содержащую pir116 "дикого типа", для установки фонового уровня флуоресценции. Во-вторых, в качестве положительного контроля использовали pXL3795, содержащую двойную мутацию pirll6- pir42, которая повышает число копий плазмиды в 4-6 раз, по сравнению с pXL3830.
Случайный мутагенез проводили на гене pir116 с использованием набора для случайного мутагенеза Diversify PCR (BD Biosciences Clontech, Palo Alto, Калифорния, США). Использовали условие 1, при котором в среднем вводятся 2 мутации на 1000 оснований. Предварительный запуск эксперимента с использованием "условия 1" показал путем секвенирования 12 мутантов, что скорость мутации действительно составляла примерно 2 мутации в гене pir116. Ген pir116 амплифицировали как фрагмент EcoRI-SstI с олигонуклеотидами C8832 (5-CTTAACGGCTGACATGGGAATTC-3') (SEQ ID NO: 23) и C8833 (5'- CGATGGGCGAGCTCCACCG-3') (SEQ ID NO: 24). После расщепления EcoRI и SstI мутированный фрагмент, содержащий pirl16, клонировали в pXL3830 вместо гена pir116 "дикого типа".
Плазмидами, несущими мутантный pir116 ("pirll6*"), трансформировали штамм E. coli XAC-1, родительский для хозяина pCOR XAC1 pir116. Трансформантов подвергали скринингу на предмет увеличенной флуоресценции при УФ-свете и сравнивали с контролем XAC1 (pXL3830) и XAC1 (pXL3795). Чашку-дубликат не подвергали УФ для минимизации вторичных мутаций. Чашка репрезентативного скрининга при УФ-освещении показана на фиг. 25.
Следующая блок-схема суммирует результаты экспериментов по скринингу.
Случайный мутагенез pir (белок pi) на плазмиде pCOR, содержащей ген-репортер cobA
Тестировали 2200 клонов
24 кандидата
16 подтверждали и оценивали на предмет числа копий плазмиды/топологии/последовательности pir* (гель)
Три лучших выбирали для дальнейшего исследования.
Оценка трех выбранных мутантов суммирована на фиг. 26. Каждый мутант характеризовался повышением числа копий по сравнению с плазмидой pir116. В случае мутантов 114C и 100B плазмида была, по существу, в мономерной форме. Это могло быть преимуществом по сравнению с плазмидой pir116 pir42, которая характеризовалась повышенным числом копий и высоким содержанием мономера, как и мутант 201C.
Ген pirll6* каждого мутанта секвенировали. Каждый клон содержит единственную не изогенную мутацию в ORF pir116. Все три мутации влияют на C-конец белка pi, который вовлечен в связывание ДНК. Ни одна из данных мутаций не была описана до этого.
После выявления путем скрининга плазмидной системы данные мутации оценивали в системе продукции, то есть там, где ген pir116* был введен в геном штамма E.coli - хозяина pCOR. Стратегия этой оценки суммирована на фиг.27.
Для данной оценки плазмидой pXL3179 трансформировали каждый из трех штаммов E. coli, несущих мутации, идентифицированные на фиг.26, и оценивали на предмет копийности и топологии плазмиды. Результаты данных экспериментов представлены на фиг.28. Имело место наблюдение, что число копий плазмиды было значительно снижено по отношению к XAC1 pir116 только для мутанта 201C.
Пример 14. Мини-кольцо с геном III M13 как инструмент для интеграции в E. coli путем гомологичной рекомбинации
1. Суицидные векторы
Замена генов путем двойной гомологичной рекомбинации в E. coli требует применения суицидного вектора. Данные векторы конструируют и продуцируют в хозяине, способном реплицировать их, и впоследствии используют для рекомбинации в хромосому хозяина, не способного их реплицировать.
Бактериофаг M13 представляет собой очень полезный генетический инструмент, который может применяться в мутантах rep (Metcalf, W., W. Jiang, et al., Gene 138: 1-7 (1994)) или в несупрессорных штаммах E. coli, когда используются M13 mp8-11 (Blum, P. et al., J. Bacteriol. 171: 538-46 (1989)). Часто встречаются некоторые ограничения, касающиеся конструкции, размера вставки и нестабильности. Плазмиды, несущие точку начала репликации ДНК гамма R6K, являются хорошо известными суицидными векторами (Miller, V. and J. Mekalanos, J Bacteriol., 170: 2575-83 (1988)), но они не могут использоваться для модификации штаммов E. coli, которые экспрессируют белок pi, обеспечивающий репликацию таких плазмид.
Конструировали универсальный суицидный вектор с новым маркером обратной селекции и использовали его для конструирования штаммов E. coli, в бактериальный геном которых встроены мутанты гена pir116 (pir116*) путем гомологичной рекомбинации. Представленная здесь стратегия продемонстрирована для мутанта 114C, но также она использовалась для продукции штаммов, несущих другие мутанты pirll6*.
2. Маркер обратной селекции
Другие маркеры могут использоваться для выбора бактерий, претерпевающих второе событие рекомбинации. Данное событие приводит к потере данного маркера и, в некоторых случаях, к замене гена после рекомбинации между хромосомой и суицидным вектором. Например, ген SacB из Bacillus летален при высеве бактерий, экспрессирующих данный ген, на среду, содержащую сахарозу (Ried, J. L. and A. Collmer, Gene 57: 239-46 (1987)). В другом примере, ген устойчивости к тетрациклину придает чувствительность к фузариновой кислоте (Bochner, B. R., et al., J Bacteriol. 143 (2): 926-3 (1980)). Инфекция бактериофагом M13 придает чувствительности к детергенту дезоксихолату (Blum, P., et al., J Bacteriol. 171: 538-46 (1989)).
Вследствие дефицита эффективности в некоторых штаммах E. coli разработали способ положительной селекции двойных рекомбинантов. Ген III из бактериофага M13 оценивали в качестве маркера обратной селекции. Данный ген кодирует малый компонент вириона, ответственный за инфекционность частиц. При гиперэкспрессии мультикопийной плазмиды pBR322 ген III придает чувствительности к дезоксихолату клеткам вследствие вставки белка гена III в мембрану бактерий (Boeke, J. D. et al., Mol Gen Genet 186: 185-92 (1982)). Ни в одном из отчетов не указано, может ли данный ген использоваться в качестве эффективного маркера обратной селекции, когда он присутствует в одной копии в геноме E. coli. Поэтому авторы тестировали данную гипотезу с мини-кольцевым суицидным вектором.
3. Амплификация путем PCR версии гена III из M13 с делецией
Для снижения размера мини-кольцевого вектора, подлежащего конструкции, выбрали версию гена III с делецией (ген III'), которая, тем не менее, способна придавать чувствительности к дезоксихолату (Boeke, J. D., P. Model, et al., Mol Gen Genet 186: 185-92 (1982)). Данный ген амплифицировали путем PCR вместе с его собственным промотором из M13 mp18 (Yanisch-Perron, C., J. Vieira, et al., Gene 33: 103-19 (1985)) в виде фрагмента BglII-XhoI fragment (см. фигуру 29).
Олигонуклеотиды были следующими:

Амплифицированный фрагмент клонировали путем клонирования T-A в pGEMT-easy (Promega Corporation, Madison, Висконсин, США) с образованием pXL4230 (фиг.29). Нуклеотидная последовательность вставки, как было обнаружено, согласуется с той, что описана в GenBank под инвентарным номером VB0018. pXL4230 обеспечивает чувствительность к дезоксихолату при трансформации штамма E. coli DH10B (Invitrogen), подтверждая свою ожидаемую функцию.
4. Суицидный вектор на основе мини-кольца
Поскольку плазмида-мини-кольцо не содержала никакой точки начала репликации, она может использоваться в качестве универсального суицидного вектора. С данной целью для отбора первого события рекомбинации должен быть добавлен маркер селекции, такой как ген устойчивости к канамицину Для обратной селекции бактерий, которые не претерпевают второго события рекомбинации, к мини-кольцевому вектору добавляют ген III'. Конструкция плазмиды, использованной при продукции мини-кольца для рекомбинации, показана на фиг.29.
Мини-кольцо образовано из плазмиды, такой как pXL4235, после индукции интегразы бактериофага лямбда, которая рекомбинирует между attP и attB на плазмиде (Darquet, A. Met al., Gene Ther 4 (12): 1341-9 (1997)). Данная рекомбиназа экспрессируется под контролем PBAD не зависимым от арабинозы способом в штамме E. coli G6264, который описан в заявке на выдачу патента США № 09/981803. Полученное в результате мини-кольцо содержит attL, образующую TH (тройную спираль) последовательность для очистки, маркер для селекции ген-устойчивости к канамицину Tn903, маркер обратной селекции маркерный ген III' и интересующий фрагмент для гомологичной рекомбинации, клонированный в участок множественного клонирования pXL4235 (фиг.29).
В качестве примера конструкции, использованные для получения штаммов E. coli, экспрессирующие повышающих копийность мутантов pir116, описаны на фиг.30. Данные штаммы могут использоваться для продукции плазмид pCOR (Soubrier, F. et al., Gene Ther 6: 1482-1488 (1999)). Поскольку не имеется гомологии между pir и бактериальным геномом последовательности pir116* встраивали в хромосомный ген uidA E. coli, который кодирует
Протокол для очистки мини-кольца и рекомбинации осуществлялся следующим образом. Плазмидой pXL4256 (фиг.30) трасформировали штамм E. coli G6264 для получения G6656. Пятьдесят мл среды LB, дополненной ампициллином (100 мг/л), инокулировали 0,5 мл инкубированной в течение ночи культуры G6656 и инкубировали при 37°C, с перемешиванием при 200 об./мин до достижения оптической плотности при 600 нм значения 0,7. Продукцию мини-кольца индуцировали добавлением к среде 250 мкл стерильного раствора 10% арабинозы. Через 30 минту при 37°C, 200 об./мин, общую плазмидную ДНК выделяли с использованием системы для очистки мидипрепаратов ДНК Wizard Plus Midipreps DNA Purification (Promega Corporation, Madison, Висконсин, США).
Шесть мкг препарата плазмидной ДНК загружали на 0,8% агарозный препаративный гель. "Лестницу" сверхспиральной ДНК (Promega Corporation, Madison, Висконсин, США) использовали в качестве стандарта молекулярной массы. После электрофореза в течение ночи при 50 В сверхспиральную конструкцию мини-кольца (5,1 т.п.н.) экстрагировали и очищали из геля с использованием набора для очистки из геля SV (Promega Corporation, Madison, Висконсин, США).
5. Двойная гомологичная рекомбинация мини-кольцом 4256 (мини-кольцевого суицидного вектора uidA::pir116*)
Стадии рекомбинации для конструирования штаммов и соотвествующие фенотипы показаны на фиг.31.
Для первого события рекомбинации (интеграция) 0,2, 1 и 5 мкл очищенным мини-кольцом 4256 вводили электропорацией в штамм E. coli XAC1 (Normanly, J et al., Proc Natl Acad Sci USA 83: 6548-52 (1986)), которая представляет собой родительский штамм для хозяев pCOR. Колонии, устойчивые к канамицину, получали на агаре LB, дополненной канамицином (50 мг/л), после инкубации в течение ночи при 37°C.
Для оценки числа колоний, потенциально кодирующих контаминантные колонии не подвергшейся рекомбинации pXL4256, 50 колоний KmR в параллели высевали полосами на агар LB, дополненный канамицином или ампициллином. Лишь 4 колонии из 50 были устойчивы к канамицину и ампициллину и, как было показано анализом рестрикции плазмиды, содержали pXL4256, не подвергшуюся рекомбинации. Это указывало на то, что 46 колоний из 50, полученным путем электропорации, действительно содержат интегранты с мини-кольцом 4256.
Для второго события рекомбинации (вырезания) все 46 интегрантов KmR выделяли на свежеприготовленных чашках с агаром LB, содержащих 1,5% дезоксихолата натрия ("Doc"; Sigma) и инкубировали при 37°С в течение ночи. Лишь немного устойчивых к дезоксихолату (DocR) колоний (от 1 до 15) получали для каждого интегранта, как показано фиг.32. Данный результат согласовывался с отбором относительно редкого события, такого как второго события рекомбинации. 100 колоний DocR, полученных из 15 интегрантов, подвергали реплике на агар LB с 1, 5% Doc и на агар LB плюс канамицин для скрининга двойных рекомбинантов DocR и KmS. 86% подвергнутых скринингу колоний были KmS, и это указывало на потерю ими суицидного вектора.
Для скрининга замены аллеля хромосомный локус uidA амплифицировали путем PCR. Если замена аллеля произошла, ожидаемый размер PCR-фрагмента составляет 1,3 т.п.н. Размер фрагмента, соответствующего локусу uidA дикого типа, то есть без интегрированной мутации pirll6*, составляет 0,85 т.п.н. Результаты, представленные на фиг.33, панель A, показывают, что замена аллеля произошла в 30% двойных рекомбинантов. Это подтверждали фенотипическим анализом, поскольку данные клоны также являлись UidA- (бета-глюкуронидаза -) и давали белые колонии на агаре LB, дополненном Xgluc.
Целостность бактериального генома в области, близкой к участку гомологичной рекомбинации, проверяли путем PCR на двух независимых рекомбинантах. Первый праймер (seq6113 или seq6115) основывался на последовательности гена pir и второй (seq6112 или seq6116) характеризовался последовательностью, основанной на последовательности, расположенной близко к области гомологии, но вне ее (непосредственно в 5'- или 3'-направлении от uidA). ДНК XAC1 использовали в качестве отрицательного контроля, в то время как XAC1 pir116 или TEX1 (Soubrier, F. et al., Gene Ther 6: 1482-1488 (1999)) использовали в качестве положительных контролей.
Олигонуклеотиды, использованные в качестве праймеров PCR, были следующими:


Ожидаемый размер продукта PCR составляет 0,83 т.п.н. при использовании праймеров seq6112 и seq6113, и 0,88 т.п.н. при использовании праймеров seq6114 и seq6115. Результаты представлены на фиг.33, панель B. Два двойных рекомбинанта, полученных с мини-кольцевым суицидным вектором, характеризовались ожидаемым профилем PCR. Это демонстрирует, что двойная гомологичная рекомбинация может легко достигаться в E. coli с применением мини-кольцевых плазмид в качестве суицидного вектора и гена III' M13 в качестве маркера обратной селекции. Данный способ замены генов может неспосредственно и универсально осуществляться на генетическом фоне любых микроорганизмов.
Библиография
Alting-Mees, M.A., J.A. Sorge, and J.M. Short. 1992. Methods Enzymol. 216:483-495.
Blum, P., D. Holzschu, H.S. Kwan, D. Riggs, and S. Artz. 1989. J. Bacteriol. 171:538-546.
Brosius, J. 1989. Metliods Enzymol. 216:469-483.
Chambers, S.P., S.E. Prior, D.A. Barstow, and N.P. Minton. 1988. Gene 68:139-149.
Chung, C.T., and R.H. Miller. 1988. Nucleic Acids Res. 16:3580.
Colloms, S.D., P. Sykora, G. Szatmari, and D.J. Sherrat. 1990 J. Bacteriol. 172:6973-6980.
Datta, N., and P. Kontomichalou. 1965. Nature 208:239-241.
Dickely, F., D. Nilsson, E.B. Hansen, and E. Johansen. 1995. Mol. Microbiol. 15:839-847.
Filutowicz, M., S. Dellis, I. Levchenko, M. Urh, F. Wu, and D. York. 1994. Prog, in Nucleic Acid Res. and Mol. Biol. 48:239-273.
Gibson, Т J. 1984. Ph.D Thesis. University of Cambridge.
Greener, A., M. Filutowicz, M. McEachern, and D. Helsinki. 1990. Mol. Gen. Genet. 224:24-32.
Herrero, M., V. de Lorenzo, and K.N. Timmis. 1990. J. Bacteriol. 172:6557-6567.
Hodgson, C.P. 1995. Bio/Technology 13:222-225.
Inuzuka, M., and Y. Wada. 1985. EMBO J. 4:2301-2307.
Jaye, M. et al., (1986) Science 233:541-5.
Kleina, L.G., J.M. Masson, J. Normanly, J. Abelson, and J.H. Miller. 1990. J. Mol. Biol. 213:705-717.
Kowalczykowski, S.C., and A.K. Eggleston. 1994. Annu. Rev. Biochem. 63:9991-10043.
Leung, D.W., E. Chen, G. Cachianes, andD.V. Goeddel. 1985. DNA 4:351-355.
Maniatis, Т., E.F. Fritsch, and J. Sambrook. 1989. Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y.
Meinnel, Т., E. Schmitt, Y. Mechulam, and S. Blanquet. 1992. J. Bacteriol. 174:2323-2331.
Mertens, N., E. Remant and W. Fiers. (1995) Bio/Technology 13:175-179.
Messing, J., and J. Vieira. 1982. Gene 19: 269-276.
Metcalf, W.W., W. Jiang, and B.L. Wanner. 1994. Gene 138:1-7.
Miller, V.L., and JJ. Mekalanos. 1988. J. Bacteriol. 170:2575-2583.
Normanly, J., J.M. Masson, L.G. Kleina, J. Abelson, and J.H. Miller. 1986. Proc. Natl. Acad. Sci. USA 83:6548-6552.
Normanly, I, L.G. Kleina, J.M. Masson, J. Abelson, and J.H. Miller. 1990. J. Mol. Biol. 213:719-726.
Roca, J. 1995. TIBS 20:156-160.
Saiki, R.K., S. Scharf, F. Faloona, K.B. Mullis, G.T. Horn, H.A. Erlich, and N. Arheim. 1985. Science 230:1350-1354.
Sanger, F., S. Nicklen, and A.R. Coulson. 1977. Proc. Natl. Acad. Sci. USA 74:5463-5467.
Sawadogo, M., and M.W. Van Dyke. 1991. Nucleic Acids Res. 19:674.
Scott, J.R. 1984. Microbiol. Rev. 48:1-23.
Simoes, D.A., M. Dal Jensen, E. Dreveton, M.O. Loret, S. Blanchin-Roland, J.L. Uribelarrea, and J.M. Masson. 1991. Ann. N.Y. Acad. Sci. 646:254-258.
Simon, R., U. Priefer, and A. Punier. 1983. Bio/Technology 1:784-791.
Sinha, N.D., J. Biernat, J. McManus, and H. Koster. 1984. Nucleic Acids Res. 12:4539-4557.
Stirling, C.J. G. Stewart, and D.J. Sherrat 1988. Mol. Gen. Genet. 214:80-84.
Stirling, C.J., S.D. Colloms, J.F. Collins, G. Szatmari, and D.J. Sherrat. 1989. EMBO J. 8:1623-1627.
Studier, F.W., A.H. Rosenberg., JJ. Dunn and J.W. Dubendorff (1990). Methods Enzymol 185:60-89.
Summers, D.K., and D.J. Sherrat. 1984. Cell 36:1097-1103.
Takahashi, K., Y. Sawasaki, J. Hata, K. Mukai and T. Goto. (1990) In Vitro Cell Dev. Biol. 26:265-74.
Vieira, J., and J. Messing. 1982. Gene 19:259-268.
Wiechelman, K., R. Braun, and J. Fitzpatrick. 1988. Anal. Biochem. 175:231-237.
Yanisch-Perron, C. Vieira and J. Messing (1985) Gene 33:103-119 13.
Реферат
Изобретение относится к биотехнологии и представляет собой прокариотическую рекомбинантную клетку хозяина, содержащую гетерологичный белок инициации репликации, который активирует зависимую от условий точку начала репликации, и внехромосомную молекулу ДНК, содержащую гетерологичный терапевтический ген и зависимую от условий точку начала репликации. Также изобретение относится к способу продуцирования плазмиды, содержащей гетерологичный терапевтический ген и зависимую от условий точку начала репликации. А также к самой плазмиде, представляющей собой суицидный вектор для переноса генов, содержащей гетерологичный терапевтический ген и зависимую от условий точку начала репликации. Данное изобретение позволяет избежать неконтролируемой гиперэкспрессии терапевтического гена и генов устойчивости. 3 н. и 41 з.п. ф-лы, 33 ил., 10 табл.

Комментарии