Агонисты и антагонисты уротензина-ii - RU2263679C2
Код документа: RU2263679C2
Описание
Область изобретения
Данное изобретение относится к полипептидным агонистам и антагонистам уротензина-II и способам их использования.
Предпосылки создания изобретения
Уротензин-II (У-II = U-II) является циклическим нейропептидом с сильными сердечно-сосудистыми эффектами. Впервые У-II был выделен из каудальной нейросекреторной системы телеостной рыбы, и его первичная структура была установлена у нескольких видов позвоночных, включая разные виды рыб, лягушек и человека. Анализ последовательности разных пептидов У-II у разных видов выявил, что тогда как N-концевая область высоко вариабельна, С-концевая циклическая область У-II является сильно консервативной. Несомненно, эта циклическая область, которая ответственна за биологическую активность У-II, является полностью консервативной у всех видов, от рыб до человека (Coulouran, et al., Proc. Natl. Acad. Sci. USA (physiology), 95:15803-15808 (1998)). Тот факт, что эволюционное давление действовало так, что полностью сохранялась биологически активная последовательность У-II, наводит на мысль о том, что этот полипептид играет важную роль в физиологии человека.
Циклическая область У-II содержит шесть аминокислотных остатков (-Cys-Phe-Trp-Lys-Tyr-Cys- (SEQ ID NO: 1)) и структурно сходна с биологически важной центральной областью соматостатина-14 (-Phe-Trp-Lys-Thr- (SEQ ID NO: 2)). Однако молекулярное клонирование и анализ последовательности гена препроуротензина II карпа дает возможность предположить, что У-II и соматостатин происходят не от общего источника (Ohsako, S., et al., J. Neurosci., 6:2730-2735 (1986)).
У рыб пептиды У-II, как было показано, проявляют несколько активностей, включая общее действие по сокращению гладкой мускулатуры, хотя реакция отличается у разных видов и зависит от сосудистого ложа (Davenport, A., and Maquire, J., Trends in Pharmacological Sciences, 21: 80-82 (2000); Bern, Y.A., et al., Recent Prog. Horm. Res., 45: 533-552 (1995)). У-II рыб тоже, как было показано, обладает сокращающим действием у млекопитающих, включая главные артерии крыс, но рецептор(ы), опосредующие эти эффекты пептида, полностью не охарактеризованы.
Согласно современным исследованиям, изолированные человеческие соединенные с протеином G рецепторы, гомологичные GРR14 крыс и экспрессируемые преимущественно в сердечно-сосудистой ткани, функционируют как У-II рецепторы (Ames, H., et al., Nature, 401: 282-286 (1999)). У-II рыбы (бычок) и человека, как сообщалось, имеет высокое сродство к рекомбинантным человеческим GРR14, и их связывание функционально сочетается с мобилизацией кальция. Человеческие У-II связываются как с сосудистой тканью, так и с тканью сердца (включая коронарную атерому), и способствует эффективному сужению изолированных артерий приматов, кроме человека (Ames, H., et al., выше). Сила сужения сосудов под действием У-II существенно больше, чем под действием эндотелина-1, что делает человеческий У-II одним из наиболее сильных сосудосуживающих факторов, известных в настоящее время. In vivo человеческий У-II заметно усиливает общее периферическое сопротивление у анестезированных приматов, кроме человека, - реакцию, связанную с сильной дисфункцией сердечного сокращения (Ames, H., et al., выше).
Так как иммунореактивность в отношении человеческого У-II обнаружена в ткани сердца и сосудов (включая коронарную атерому), У-II, как полагают, влияет на сердечно-сосудистые гомеостаз и патологию (например, ишемическую болезнь сердца и застойную сердечную недостаточность). Кроме того, обнаружение иммунореактивности в отношении У-II в спинном мозге и эндокринных тканях наводит на мысль, что У-II может обладать дополнительной активностью, включая модуляцию центральной нервной системы и эндокринную функцию человека (Ames, H., et al., выше). Разумеется, ряд болезней потенциально связывали с избыточной или недостаточной экспрессией активности У-II, включая ишемическую сердечную недостаточность, гипотензию, портальную гипертензию, стенокардию, кровотечение из варикозно расширенных вен, инфаркт миокарда, язвы и некоторые психологические и неврологические расстройства. Таким образом, существует большая потребность в разработке активных соединений, способных к модуляции активности У-II, включая ингибиторы или антагонисты У-II.
Краткое изложение изобретения
Данное изобретение представляет собой новую группу циклических полипептидов, которые обладают активностью антагонистов У-II. Полипептиды согласно изобретению являются октапептидами, имеющими общую формулу: (R1)а-АА1-цикло[АА2-АА3-АА4-АА5-АА6-Cys]-АА7-R2 (формула I), где АА1 является L-изомером ароматической аминокислоты; АА2 является L- или D-изомером Cys; АА3 представляет собой L-изомер ароматической аминокислоты; АА4 является L- или D- изомером Trp; АА5 является L- или D-изомером Lys, N-Ме-Lys, или Orn; АА6 представляет собой L- или D-изомер Val, Thr, Leu, Ile, трет-Leu, Abu, Nle или ароматической аминокислоты; АА7 является L- или D-изомером Val, Thr, Leu, Ile, трет-Leu, Abu, Nle или ароматической аминокислоты; R1 представляет собой Н, низший алкил, низший алканоил или низший ацил; а равно 1 или 2; и R2 представляет собой ОН, OR3, N(R3)2 или NHR3, где R3 представляет собой Н, низший алкил или арилалкил; при условии, что пептид не является Сра-с[D-Cys-Pal-D-Trp-Lys-Val-Cys]-Сра-NH2.
В предпочтительном воплощении АА2 и АА4 являются D-Cys и L-Trp соответственно.
В другом предпочтительном полипептиде АА1 является Сра, АА2 представляет собой D-Cys, АА3 является Phe, АА4 является Trp, АА5 является Lys, АА6 является Thr и АА7 является Val.
В особенно предпочтительном воплощении полипептид является октапептидом, имеющим формулу Сра-с[D-Cys-Phe-Trp-Lys-Thr-Cys]-Val-NH2.
Данное изобретение также представляет собой полипептид - агонист уротензина-II- и его варианты, имеющий формулу Asp-с[Cys-Phe-Trp-Lys-Tyr-Cys]-Val-ОН.
Полипептиды согласно изобретению способны к изменению активности У-II и могут воздействовать на связывание У-II с рецептором. Таким образом, эти полипептиды можно вводить субъекту в качестве средств профилактики и лечения медицинских и психологических патологических состояний, характеризующихся избытком или дефицитом или недостаточной экспрессией активности уротензина-II. Такие патологические состояния включают в себя, не ограничиваясь ими, ишемическую болезнь сердца, застойную сердечную недостаточность, портальную гипертензию, кровотечение из варикозных вен, гипертензию, стенокардию, инфаркт миокарда, язвы, беспричинное беспокойство, шизофрению, маниакальную депрессию, делирий, деменцию, задержку умственного развития (олигофрению) и дискинезии.
Данное изобретение также представляет собой фармацевтические композициии, которые содержат терапевтически эффективное количество полипептида формулы I в сочетании с фармацевтически приемлемым носителем. Подходящие носители включают в себя, но не ограничиваясь этим, физиологический раствор, буферный физиологический раствор, декстрозу, воду, глицерин, этанол и их комбинации. Данная композиция может быть адаптирована для способа введения и может быть составлена в виде пилюль, таблеток, капсул, аэрозольного препарата, порошка или жидкости.
Другие характерные черты и преимущества согласно изобретению будут очевидны из следующего его детального описания и из формулы изобретения.
Определения
Под «полипептидом» подразумевается любой пептид (включая циклические пептиды) или белок, состоящий из двух или более аминокислот, соединенных одна с другой пептидными связями или модифицированными пептидными связями. «Полипептидом» называются как короткие цепи, обычно называемые пептидами, олигопептидами или олигомерами, так и более длинные цепи, обычно называемые протеинами (белками). Полипептиды могут содержать другие аминокислоты, кроме 20 кодируемых генами аминокислот. «Полипептиды» включают в себя аминокислотные последовательности, модифицированные или с помощью природных процессов, или с помощью методов химической модификации, которые хорошо известны специалистам. Модификации могут осуществляться где угодно в пептиде, включая основную структуру пептида, боковые цепи аминокислот и амино- или карбоксильные концы.
Система обозначений, использованная здесь для полипептидных аминокислотных остатков, представляет собой те аббревиатуры, которые обычно используются в данной области. Менее обычные аббревиатуры Abu, Сра, Nle, Pal, Tle, Dip, 4-Fpa и Nal означают 2-аминомасляную кислоту, п-хлорфенилаланин, норлейцин, 3-пиридил-2-аланин, трет-лейцин, 2, 2-дифенилаланин, 4-фторфенилаланин и 3-(2-нафтил)-аланин или 3-(1-нафтил)-аланин соответственно.
Под алкилом подразумевается алифатическая углеводородная группа с прямой или разветвленной цепью. Алкил необязательно является замещенным одним или более заместителями, которые могут быть одинаковыми или разными и включают в себя, но не ограничиваясь этим, галоген, циклоалкил, гидроксильную, алкокси, амино, карбамоильную, ациламино, ароиламино, карбоксильную, алкоксикарбонильную, аралкилоксикарбонильную или гетероаралкилоксикарбонильную группы. Типичные примеры алкильных групп включают в себя, не ограничиваясь этим, метил, трифторметил, циклопропилметил, циклопентилметил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-пентил, 3-пентил, метоксиэтил и карбоксиметил. Под «низшим алкилом» подразумевается алкильная группа с прямой или разветвленной цепью, имеющая менее 11 атомов углерода, предпочтительно С1-С8-алкил.
Под «ацилом» подразумевается группа, имеющая структуру

Под «низшим алканоилом» подразумевается ацильная группа, которая описана выше, где R является низшим алкилом.
Под «арилом» подразумевается моноциклическая или бициклическая ароматическая группа, содержащая от 6 до 12 атомов углерода в кольцевой части, предпочтительно 6-10 атомов углерода в кольцевой части, такая как фенил, нафтил или тетрагидронафтил. Под «арилалкилом» подразумевается алкильная группа, которая описана здесь, имеющая арильный заместитель, такой как бензил, фенилэтил или 2-нафтилметил.
Под «фармацевтически приемлемой солью» подразумеваются нетоксичные соли присоединения кислоты или комплексы с металлами, которые обычно используются в фармацевтической промышленности. Примеры солей присоединения кислоты включают в себя соли органических кислот, таких как уксусная, молочная, памовая, малеиновая, лимонная, яблочная, аскорбиновая, янтарная, бензойная, пальмитиновая, субериновая, салициловая, винная, метансульфоновая, толуолсульфоновая или трифторуксусная кислоты и тому подобное; полимерные кислоты, такие как дубильная кислота, карбоксиметилцеллюлоза и тому подобное; и неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота и тому подобное. Комплексы металлов включают в себя комплексы цинка, железа и тому подобное.
Под «вариантом» подразумевается полипептид, который отличается от стандартного полипептида, но сохраняет главные свойства. В основном различия ограничены так, что последовательности стандартного полипептида и варианта в целом близко сходны, а во многих областях идентичны. Вариант и стандартный полипептид могут отличаться одной(им) или более из замен, добавлений и/или делеций в аминокислотной последовательности, в любом сочетании. Заменяемый или вставляемый аминокислотный остаток может быть одним из кодируемых или не кодируемых генетическим кодом. Вариант полипептида может быть природно существующим, таким как аллельный вариант, или он может быть вариантом, о существовании которого в природе не известно. Не существующие в природе варианты полипептидов могут быть получены методами мутагенеза или путем прямого синтеза.
В основном вариант отличается от стандартного полипептида заменами консервативных аминокислот, причем остаток заменяется другим со сходными свойствами (например, кислотными, основными, ароматическими и т.д.). Типичными заменами являются замены между Ala, Val, Leu и Ile; между Ser и Thr; между кислыми остатками Asp и Gln; между Asn и Gln и между основными остатками Lys и Arg; или ароматическими остатками Phe и Tyr.
Под «субъектом» подразумевается животное или человек, страдающий связанным с У-II патологическим физиологическим или психологическим состоянием. Субъект может быть млекопитающим, включая, но не ограничиваясь этим, человека и млекопитающих, не являющихся человеком, таких как приматы, собаки, кошки, свиньи, коровы, овцы, козы, лошади, крысы, мыши и тому подобное.
Под «фармацевтически приемлемым носителем» подразумевается носитель, который является физиологически приемлемым для животного, у которого применяется, при сохранении терапевтических свойств соединения, совместно с которым он применяется. Одним из примеров фармацевтически приемлемого носителя является физиологический раствор. Другие физиологически приемлемые носители и соответствующие им композиции известны специалистам и описаны, например, в Remington's Pharmaceutical Sciences, (18th edition), ed. A. Gennaro, 1990, Mack Publishing Company, Easton, PA.
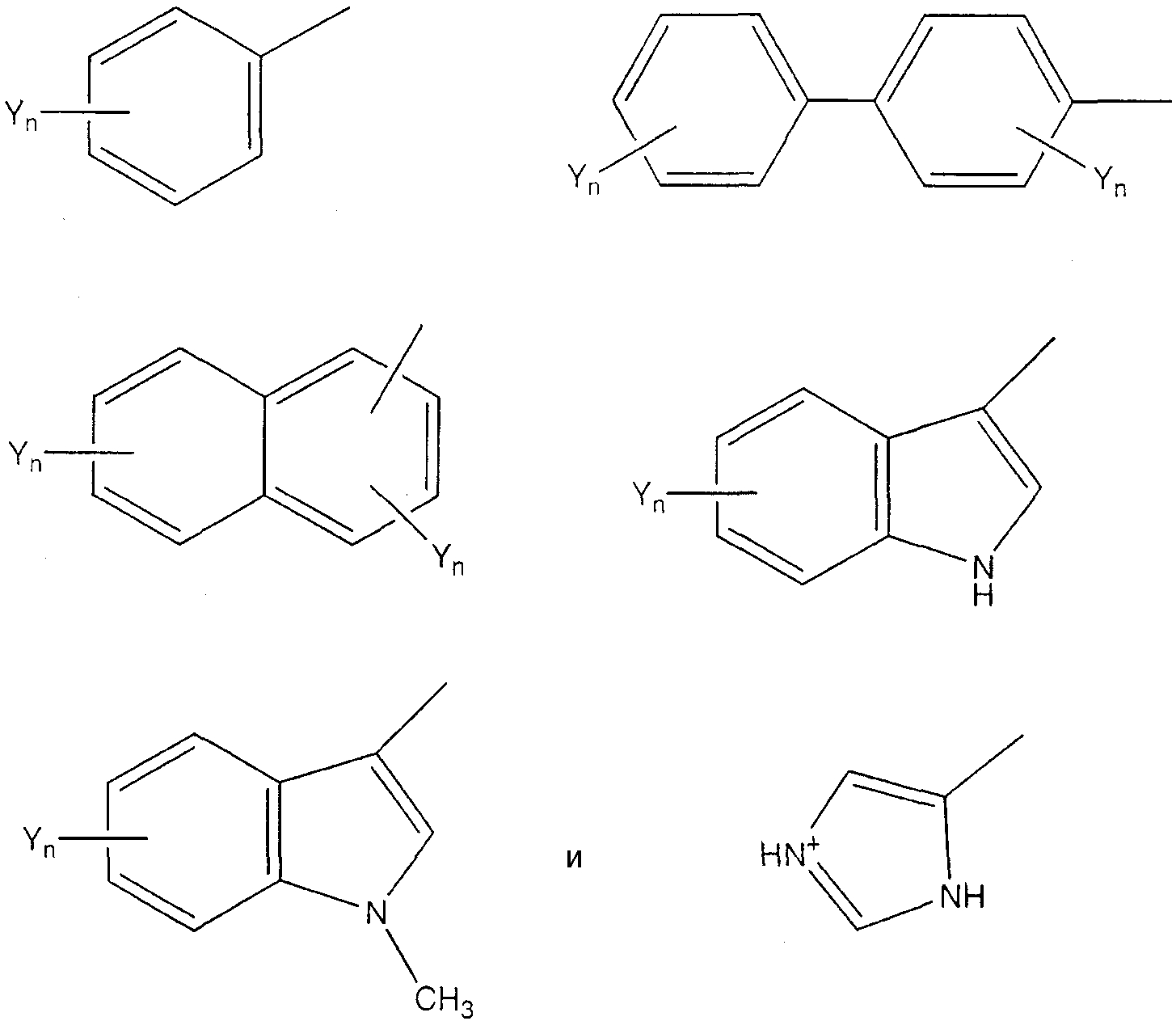
Под «ароматической аминокислотой» подразумевается аминокислота, которая содержит ароматическую группу. При предпочтительных воплощениях ароматическая аминокислота имеет следующую формулу:

(формула II), где Х представляет собой связь или Н, и Ar является частью, содержащей необязательно замещенное ароматическое кольцо. Примеры Ar включают в себя, не ограничиваясь этим, следующие структуры, где Yn представляет собой n необязательных заместителей, а n равно 0, 1, 2 или 3;

При предпочтительных воплощениях каждый заместитель Y независимо представляет собой группы NO2, CN, Cl, Br, I, F, Ме, COR4, COOR4 или OR4, где R4 представляет собой Н или С1-С8 алкил. Примеры ароматических аминокислот включают в себя, не ограничиваясь ими, Phe, Сра, Trp, Pal, His, β-Nal, 3-пиридил-Ala, 4-пиридил-Ala, 2,4-дихлор-Phe, пентафтор-Phe, р-Z-Phe и о-Z-Phe, где Z выбирается из группы, состоящей из Ме, Cl, Br, F, ОН, ОМе и NO2.
Подробное описание
Обнаружено, что минимальная часть У-II- последовательности, которая сохраняет полную биологическую активность, представляет собой октапептид Asp-с[Cys-Phe-Trp-Lys-Tyr-Cys]-Val-ОН (SEQ ID №:3), который соответствует чУII [hUII](4-7). Этот октапептид фактически обладает более высокой активностью, чем полные последовательности У-II человека и рыб, по способности вызывать сужение аорты крыс и связываться с этой тканью.
На основе этой исходной последовательности были синтезированы серии циклических октапептидов, которые обладают активностью антагониста У-II. Эти пептиды, как было открыто, обладают умеренным сродством к рецепторам У-II и способны блокировать вызванные У-II фазовые сокращения кольцевых полосок торакальной аорты крыс. Полипептиды согласно изобретению имеют общую формулу: (R1)а-АА1-цикло[АА2-АА3-АА4-АА5-АА6 -Cys]-АА7-R2 (формула I), где АА1 является L-изомером ароматической аминокислоты; АА2 является L- или D-изомером Cys; АА3 представляет собой L-изомер ароматической аминокислоты; АА4 является L- или D-изомером Trp; АА5 является L- или D-изомером Lys, N-Ме-Lys, или Orn; АА6 представляет собой L- или D-изомер Val, Thr, Leu, Ile, трет-Leu, Abu, Nle или ароматической аминокислоты; АА7 является L- или D-изомером Val, Thr, Leu, Ile, трет-Leu, Abu, Nle или ароматической аминокислоты; R1 представляет собой Н, низший алкил, низший алканоил или низший ацил; а равно 1 или 2; и R2 представляет собой ОН, OR3, N(R3)2 или NHR3, где R3 представляет собой Н, низший алкил или арилалкил.
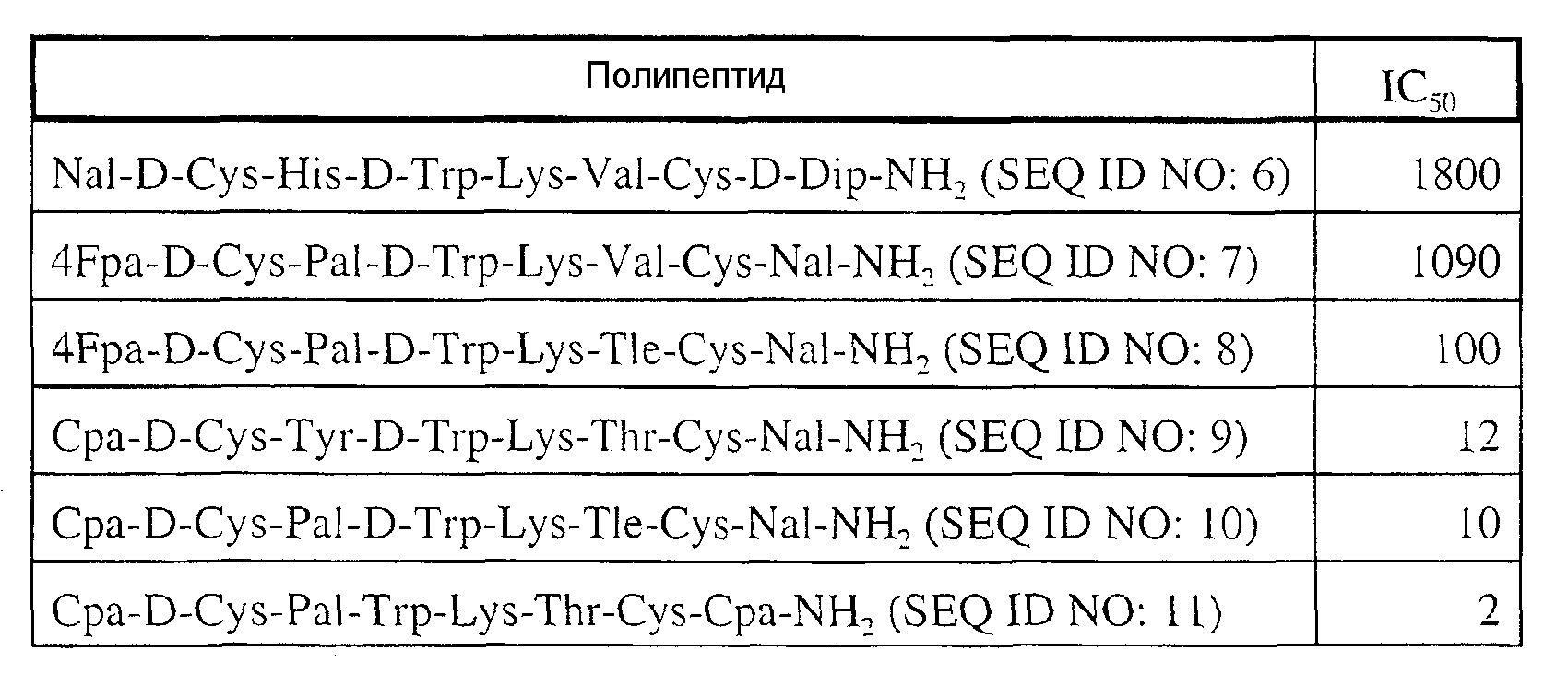
Одним из наиболее сильных испытанных ингибиторов У-II был антагонист SRIF Сра-с[D-Cys-Pal-D-Trp-Lys-Val-Cys]-Сра-амид (SEQ ID NO: 4), который имел IC50, равную примерно 100 нМ, и Kd, равный 240. Другим сильным антагонистом У-II был Сра-с[D-Cys-Phe-Trp-Lys-Thr-Cys]-Val-NH2 (SEQ ID NO: 5), который имел IC50, равную примерно 2 нМ. Другие антагонисты SRIF, которые были испытаны, обобщены в примере 2 ниже (см. таблицу 1).
Полипептиды согласно изобретению способны к модуляции активности У-II и поэтому пригодны для лечения физиологических и психологических патологических состояний, связанных или с избытком, или с недостатком экспрессии активности У-II у субъекта. Такие патологические состояния включают в себя, например, острую сердечную недостаточность, гипотензию, гипертензию, стенокардию, кровотечение из варикозных вен, инфаркт миокарда, язвы и некоторые психологические и неврологические расстройства, включая беспричинное беспокойство, шизофрению, маниакальную депрессию, делирий, деменцию, олигофрению и дискинезии.
Если патологическое состояние происходит от избытка активности У-II, одним из подходов к лечению является введение субъекту, нуждающемуся в этом, ингибиторного соединения (антагониста), необязательно в сочетании с фармацевтически приемлемым носителем, в количестве, эффективном для подавления функции У-II. Альтернативно, для лечения патологических состояний, связанных с недостаточной экспрессией активности У-II, применяется соединение, которое активирует У-II (агонист).
Терапевтически эффективное количество полипептида формулы I или варианта, или их фармацевтически приемлемых солей можно вводить перорально, парентерально (например, путем внутримышечной, внутрибрюшинной, внутривенной или подкожной инъекции, или посредством имплантата), назально, вагинально, ректально, под язык или местно в смеси с фармацевтически приемлемым носителем, адаптированным для пути введения.
Способы изготовления лекарственных форм, хорошо известные в данной области, можно найти, например, в Remington's Pharmaceutical Sciences (18th edition), ed. A. Gennaro, 1990, Mack Publishing Company, Easton, PA. Композиции, предназначенные для перорального применения, можно изготовить в твердой или жидкой формах в соответствии с любым способом, известным в области производства фармацевтических композиций. Данные композиции могут необязательно содержать подсластители, вкусовые вещества, красители, отдушки и/или консерванты для получения более приятных препаратов. Твердые дозированные формы для перорального введения включают в себя капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых формах активное соединение смешано по меньшей мере с одним инертным фармацевтически приемлемым носителем или наполнителем. Они могут включать, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактозу, сахарозу, крахмал, фосфат кальция, фосфат натрия или каолин. Могут также использоваться связывающие вещества, буферные вещества и/или улучшающие скольжение вещества (например, стеарат магния). Таблетки и пилюли могут быть дополнительно изготовлены с энтеросолюбильными покрытиями.
Жидкие дозированные формы для перорального применения включают в себя фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и мягкие желатиновые капсулы. Эти формы содержат инертные растворители, обычно используемые в данной области, такие как вода или масляная среда. Помимо таких инертных растворителей, композиции могут также включать в себя вспомогательные вещества, такие как увлажняющие вещества, эмульгирующие вещества и суспендирующие вещества.
Лекарственные формы для парентерального введения включают в себя стерильные водные или неводные растворы, суспензии или эмульсии. Примеры подходящих носителей включают в себя пропиленгликоль, полиэтиленгликоль, растительные масла, желатин, гидрированные нафталины и инъекционные органические сложные эфиры, такие как этилолеат. Такие препараты могут также содержать вспомогательные вещества, такие как консерванты, увлажнители, эмульгаторы и диспергирующие вещества. Биосовместимые, биоразрушаемые лактидный полимер, лактид/гликолидный сополимер или полиоксиэтилен-полиоксипропиленовый сополимеры могут использоваться для регуляции высвобождения соединений. Другие потенциально пригодные системы парентеральной доставки полипептидов согласно изобретению включают в себя частицы этилен-винилацетатного сополимера, осмотические насосы, имплантируемые инфузионные системы и липосомы.
Жидкие лекарственные формы могут быть стерилизованы, например, путем фильтрования через задерживающий бактерии фильтр, включением стерилизующих веществ в композиции или облучением или нагреванием композиций. Альтернативно, они могут быть также изготовлены в виде стерильных твердых композиций, которые могут быть растворены в стерильной воде или какой-то другой стерильной инъекционной среде непосредственно перед использованием.
Композиции для ректального или вагинального применения предпочтительно являются суппозиториями, которые могут содержать, кроме активных веществ, наполнители, такие как масло какао или основу для суппозитория. Композиции для назального или сублингвального применения также изготавливают со стандартными наполнителями, известными специалистам. Препараты для ингаляции могут содержать наполнители, например лактозу, или могут быть водными растворами, содержащими, например, полиоксиэтилен-9-лауриловый эфир, гликохолат и дезоксихолат, или могут быть масляными растворами для введения в виде капель в нос или назального аэрозоля, или в виде геля.
Количество активного ингредиента в композициях согласно изобретению может меняться. Специалист поймет, что точные индивидуальные дозы могут быть до некоторой степени скорректированы в зависимости от ряда факторов, включая полипептид, который нужно вводить, время введения, путь введения, природу лекарственной формы, путь выведения, природу патологических состояний у субъекта, а также возраст, вес, состояние здоровья и пол пациента. Кроме того, тяжесть патологического состояния, связанного с У-II, которое нужно лечить, также будет оказывать влияние на уровень дозирования. В основном применяются уровни дозировки между 0,1 мкг/кг и 100 мг/кг веса тела в сутки в виде однократной дозы или многократных доз. Предпочтительно, основной интервал дозировки заключен между 250 мкг/кг и 5,0 мг/кг веса тела в сутки. Широкие вариации требуемой дозировки связаны с различной эффективностью при разных путях введения. Например, при пероральном введении обычно нужно ожидать, что потребуются более высокие уровни дозировки, чем при введении путем внутривенной инъекции. Изменения этих уровней дозировки могут быть уточнены с помощью стандартных рутинных эмпирических способов оптимизации, которые хорошо известны специалистам. В основном точная терапевтически эффективная дозировка будет определяться лечащим врачом с учетом указанных выше факторов.
Полипептиды согласно изобретению можно вводить в составе композиции с длительным непрерывным высвобождением, такой как описанные, например, в патентах США №№5672659 и 5595760. Использование композиций с немедленным или длительным непрерывным высвобождением зависит от типа патологического состояния, которое нужно лечить. Если патологическое состояние представляет собой острое или сверхострое заболевание, лечение лекарственной формой с немедленным высвобождением будет предпочтительнее, чем композицией с продолжительным высвобождением. Альтернативно, для профилактического или долговременного лечения в основном будет предпочтительнее композиция с длительным непрерывным высвобождением.
Полипептиды согласно изобретению могут быть получены любым подходящим способом. Полипептиды могут быть выделены из существующих в природе источников, получены путем рекомбинации или получены синтетически, или произведены с помощью комбинации этих методов. Синтез коротких пептидов хорошо известен специалистам. См., например, Stewart et al., Solid Phase Peptide Synthesis (Pierce Chemical Co., 2d ed., 1984). Пептиды согласно изобретению могут быть синтезированы стандартными способами синтеза пептидов, известными специалистам, и примеры таких способов приведены в примере 1, ниже.
Данное изобретение иллюстрируется следующими примерами, которые никоим образом не предназначены для ограничения настоящего изобретения.
Пример 1: Получение Сра-с[D-Cys-Pal-D-Trp-Lys-Val-Cys]-Сра-амида
Стадия 1: Получение Вос-4-хлорфенилаланин-S-метилбензил-D-цистеин-3-пиридил-2-аланин-D-триптофан-N6 -бензилоксикарбонил-лизин-валин-S-метилбензил-цистеин-4-хлорфенилаланин-бензгидриламиновой смолы.
Бензгидриламинполистирольную смолу (Advanced ChemTech, Inc., Louisville, KY) (1, 2 г, 0, 5 ммоль) в хлоридной ионной форме помещали в реакционный сосуд пептидного синтезатора Advanced ChemTech (модель 200), запрограммированного на выполнение следующего реакционного цикла: (а) метиленхлорид; (b) 33% трифторуксусная кислота в метиленхлориде (2 раза в течение 1 мин и 25 мин каждый); (с) метиленхлорид; (d) этанол; (е) метиленхлорид; (f) 10% триэтиламин в хлороформе.
Нейтрализованную смолу перемешивают с Вос-4-хлорфенилаланином и диизопропилкарбодиимидом (1,5 ммоль, каждого) в метиленхлориде в течение 1 часа, и полученную аминокислотную смолу в нем затем проводят через цикл стадий с (а) по (f) в вышеприведенной программе промывания. Следующие аминокислоты (1,5 ммоль) затем последовательно соединяют с помощью той же самой процедуры: Вос-S-метилбензил-Cys, Вос-Val, Вос-N6-бензилоксикарбонил-лизин, Вос-D-Trp, Вос-Pal и Вос-S-метилбензил-D-Cys и Вос-4-хлорфенилаланин. После промывания и сушки завершенная смола весила примерно 2,0 г.
Стадия 2: Удаление защиты и отщепление от смолы.
Смолу, описанную на стадии 1 (1,0 г, 0,25 ммоль), смешивали с анизолом (5 мл), дитиотрейолом (100 мг) и безводным фтористым водородом (35 мл) примерно при 0°С и перемешивали в течение 45 мин. Избыток фтористого водорода быстро выпаривали в потоке сухого азота, после чего свободный пептид осаждался, и его промывали эфиром. Неочищенный пептид затем растворяли в 500 мл 90% уксусной кислоты. Затем добавляли концентрированный раствор I2/МеОН до тех пор, пока не станет наблюдаться постоянное коричневое окрашивание. Избыток I2 удаляли добавлением аскорбиновой кислоты, и раствор выпаривали до небольшого объема, который наносили на колонку (2,5 х 90 см) октадецилсилан-двуокиси кремния (10-15 мкм) VYDAC™. Эту колонку элюировали линейным градиентом ацетонитрила в 0,1% растворе трифторуксусной кислоты в воде. Фракции исследовали методом тонкослойной хроматографии и аналитической высокоэффективной жидкостной хроматографии и объединяли с получением максимальной чистоты. Повторная лиофилизация раствора из воды давала 125 мг желаемого продукта в виде белого пушистого порошка. Продукт был гомогенным, как обнаружено, по данным ВЭЖХ и ТСХ. Аминокислотный анализ кислотного гидролизата и матричная лазерная десорбционная масс-спектрометрия (MALDI) подтвердили композицию октапептида. Другой пептид согласно изобретению может быть получен с применением аналогичной методики с подходящими реактивами.
Пример 2: Использование кольцевой полоски аорты крысы для испытания антагонистов У-II
Самцов крыс Sprague-Dawley (250-350 г), которые прошли карантин в течение 5-7 дней перед экспериментами, забивали путем декапитации (эксперименты были санкционированы Advisory Committee for Animal Resources, Tulane University School of Medicine). Торакальную аорту иссекали, освобождали от соединительной ткани и разрезали на кольца шириной примерно 1,5 мм. Кольца суспендировали в 15-мл бане для органов, содержащей раствор Кребса с высоким содержанием калия (9,15 г/л хлорида калия, 2,1 г/л бикарбоната натрия, 1,0 г/л глюкозы, 0,16 г/л одноосновного фосфата калия, 0,14 г/л сульфата магния (безводного) и 0,22 г/л хлорида кальция (безводного)).
К тканям применялась оптимальная нагрузка (0,2 г), и среда в бане поддерживалась при 37°С и продувалась смесью 95% О2/5% СО2. Перед помещением в баню для органов выбранные препараты протирали увлажненным ватным тампоном, чтобы удалить эндотелиальный клеточный слой, и эффект этой процедуры проверяли, применяя тест ацетилхолиновой релаксации (Gibson, А., Br. J. Pharmacol. 91:205 (1987)). Кольца аорты уравновешивались в течение 90 мин при оптимальном напряжении. Во время периода уравновешивания раствор в бане заменяли каждые 15 мин. Реакции сокращения колец аорты в ответ на различные концентрации пептидов выражали в вольтах. Изменения в напряжении гладких мышц артерии регистрировали изометрически, используя датчик изменения силы (Radnoti) и AcqKnowledge ACK100 Version 3.2 (BIOPAC Systems, Inc., Santa Barbara, CA).
В силиконизированных стеклянных пробирках пептиды растворяли в деионизированной воде при концентрации 1 мкг/мкл (стандартизованный раствор) и затем разбавляли 1:10 стерильным БСА-физиологическим раствором (0,1% БСА, фракция V, Sigma, St. Louis в 0,9% NaCl). Все пептидные растворы готовили свежими непосредственно перед экспериментами. Пептиды в интервалах концентраций от 10-6 до 10-12 М/л в конечном объеме 16-80 мкл вводили непосредственно в баню с испытуемым органом, содержащую буфер Кребса и непрерывно насыщаемую 95% О2 и 5% СО2, и на кольца аорты при оптимальном напряжении покоя (1 - 0,2 г). Вызванные пептидом изменения в напряжении колец аорты регистрировали с помощью датчиков изменения напряжения и обрабатывали с помощью компьютерной системы BIOPAC Inc., как описано выше. Каждое кольцо подвергали действию пептида только в одной концентрации.
При использовании методик исследования, известных специалистам, было обнаружено, что минимально полностью активная последовательность У-II представляет собой октапептид Asp-с[Cys-Phe-Trp-Lys-Tyr-Cys]-Val-ОН (SEQ ID NO: 3), который фактически был более активным, чем полные последовательности человека и рыб по стимуляции сужения аорты крыс. Было открыто, что различные антагонисты соматостатина (SRIF) обладают способностью блокировать вызванные У-II фазовые сужения у кольцевых полосок торакальной аорты крыс. Одним из наиболее сильных ингибиторов был антагонист SRIF Сра-с[D-Cys-Pal-D-Trp-Lys-Val-Cys]-Сра-амид (SEQ ID NO: 4), который имел IC50, равную 100 нМ, и Kd, равный 240 нМ. Полипептид Сра-с[D-Cys-Phe-Trp-Lys-Thr-Cys]-Val-NH2 также был сильным антагонистом У-II с IC50 примерно 2 нМ. Другие соединения, которые были испытаны, обобщены в таблице 1 ниже.
Таблица 1. IC50 антагонистов SRIF (нМ) в отношении стимуляции фазовых сужений аорты крыс
Эквиваленты
Хотя данное изобретение было описано со ссылками на предпочтительные воплощения, квалифицированный специалист может легко установить его существенные признаки и, не выходя за рамки сущности и объема, может произвести различные изменения и модификации согласно изобретению для адаптации его к разному применению и разным условиям. Опытные специалисты могут себе представить или могут реализовать без избыточного экспериментирования многие эквиваленты конкретных воплощений настоящего изобретения, описанные здесь. Такие эквиваленты, как подразумевается, входят в объем согласно изобретению.
Все публикации и патенты, упомянутые в данном описании, включены сюда в виде ссылки.
Реферат
Изобретение относится к новым антагонистам уротензина-II. Представлена группа циклических полипептидов общей формулы: (R1)а-АА1-цикло[АА2-АА3-АА4-АА5 -АА6-Cys]-АА7-R2, где АА1 означает L-изомер ароматической аминокислоты; АА2 означает L- или D-изомер Cys; АА3 означает L-изомер ароматической аминокислоты; АА4 означает L- или D-изомер Trp; АА5 означает L- или D-изомер Lys, N-Ме-Lys, или Orn; АА6 означает L- или D-изомер Val, Thr, Leu, Ile, Tle, Abu, Nle или ароматической аминокислоты; АА7 означает L- или D-изомер Val, Thr, Leu, Ile, Tle, Abu, Nle или ароматической аминокислоты; R1 означает Н, низший алкил, низший алканоил или низший ацил; а равно 1 или 2; и R2 означает ОН, OR3, N(R3)2 или NHR3, где R3 означает Н, низший алкил или арилалкил, в качестве антагониста уротензина-II. 4 н. и 20 з.п.ф-лы, 1 табл.
Формула

Документы, цитированные в отчёте о поиске
Циклопептиды, способ их получения, содержащая их фармацевтическая композиция, способ ее получения
Комментарии