Усовершенствованные пептидные соединения, высвобождающие гастрин - RU2330859C2
Код документа: RU2330859C2
Чертежи
























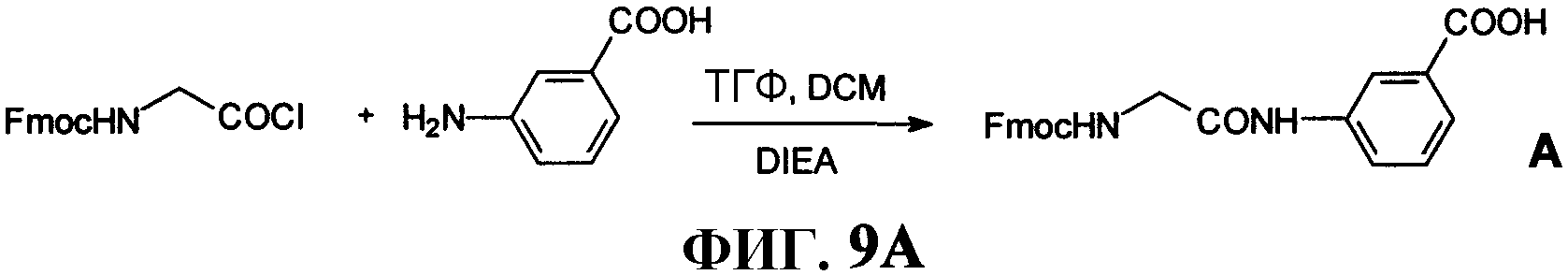






























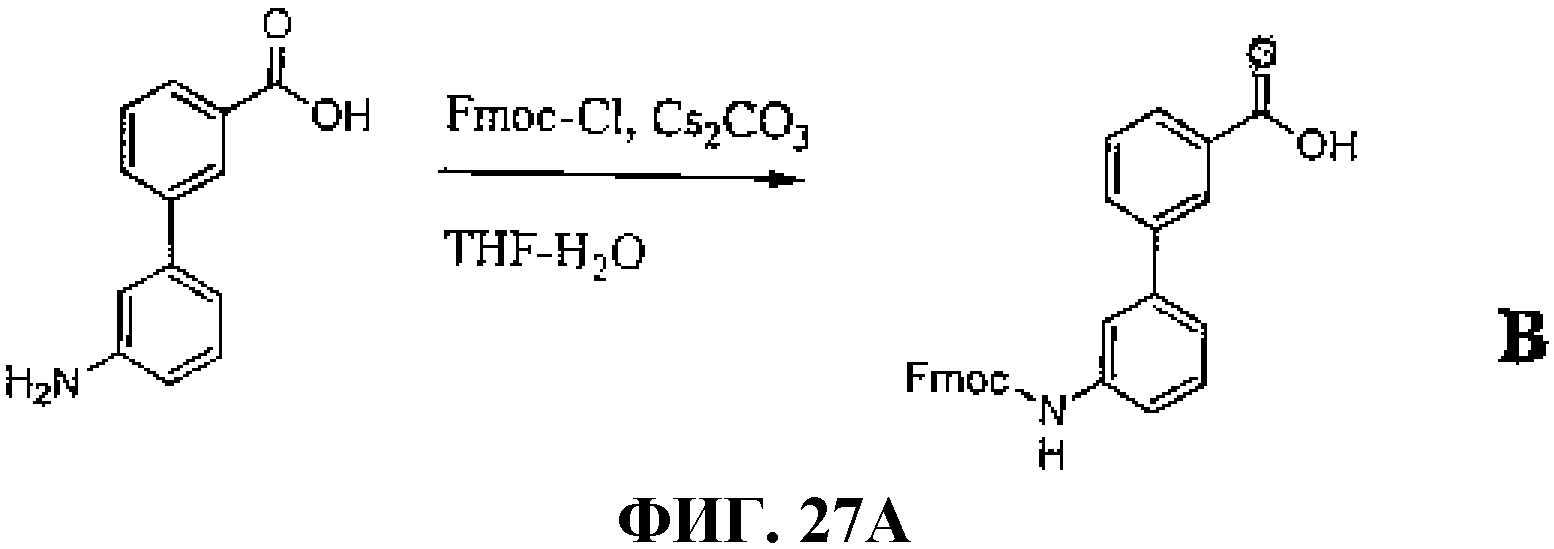























Описание
Перекрестная ссылка на родственные заявки
По настоящей заявке испрашивается приоритет по дате подачи предварительной заявки на патент США № 10/341577 от 13 января 2003 г.
Область изобретения
Настоящее изобретение относится к новым пептидным соединениям, высвобождающим гастрин (GRP), которые можно применять в качестве средств диагностической визуализации или радиотерапевтических средств. Эти соединения GRP метят радионуклидами или метками, выявляемыми световой визуализацией in vivo, и включают применение новых линкеров между меткой и пептидом, обеспечивающим целенаправленную доставку, что обеспечивает улучшенную фармакокинетику.
Предпосылки изобретения
Применение радиофармацевтических препаратов (например, диагностических визуализирующих средств, радиотерапевтических средств) для диагностики и лечения рака хорошо известно. В последние годы открытие направленных на участок поражения радиофармацевтических препаратов для диагностики и/или лечения рака приобрело популярность и продолжает нарастать, по мере того, как специалисты в области медицины лучше понимают специфичность, эффективность и возможность применения таких соединений.
Эти более новые радиофармацевтические средства обычно состоят из агента, обеспечивающего целенаправленную доставку, соединенного с хелатирующим металл агентом, который может образовывать хелатный цикл (например, образовывать комплексы) с диагностическим металлическим радионуклидом, таким как, например, технеций или индий, или терапевтическим металлическим радионуклидом, таким как, например, лютетий, иттрий или рений. Роль агента, хелатирующего металл, состоит в захвате (т.е. хелатировании) металлического радионуклида, по мере того как радиофармацевтическое средство доставляется в желаемый участок. Агент, хелатирующий металл, который непрочно связывается с металлическим радионуклидом, сделал бы радиофармацевтическое средство неэффективным для его желаемого применения, поскольку металлический радионуклид не достиг бы желаемого участка. Таким образом, дальнейшие исследования и разработки привели к открытию агентов, хелатирующих металлы, таких как представлено в патенте США № 5662885, выданном Pollak et al., включенном в настоящее описание в качестве ссылки, которые обладают высоким сродством связывания к металлическим радионуклидам и способностью к конъюгации с агентом, обеспечивающим целенаправленную доставку. В последующем была введена концепция применения "спейсера" для создания физического разделения между агентом, хелатирующим металл, и средством, обеспечивающим целенаправленную доставку, например в патенте США № 5976495, выданном Pollak et al., включенном в настоящее описание в качестве ссылки.
Роль средства, обеспечивающего целенаправленную доставку, ввиду его сродства к определенным участкам связывания является направление диагностического средства, такого как радиофармацевтическое средство, содержащее металлический радионуклид, к желаемому участку для диагностики или лечения. Обычно, средство, обеспечивающее целенаправленную доставку, может включать белок или другую макромолекулу, которая проявляет специфическое сродство к данному рецептору. Другие известные средства, обеспечивающие целенаправленную доставку, включают моноклональные антитела (Mab), фрагменты антител (Fab и (Fab)2) и рецепторно-авидные пептиды Donald J. Buchsbaum, "Cancer Therapy with Radiolabeled Antibodies; Pharmacokinetics of Antibodies and Their Radiolabels; Experimental Radioimmunotherapy and Methods to Increase Therapeutic Efficacy," CRC Press, Boca Raton, Chapter 10, pp. 115-140, (1995); Fischman, et al. "A Ticket to Ride: Peptide Radiopharmaceuticals," The Journal of Nuclear Medicine, vol. 34, No. 12, (Dec. 1993).
В последние годы было установлено, что некоторые раковые клетки содержат рецепторы пептида, высвобождающего гастрин (GRP) (GRP-R), у которых имеется ряд подтипов. В частности, было показано, что несколько типов раковых клеток имеют избыточно экспрессированные или необычно экспрессированные рецепторы GRP. По этой причине было проведено много научных исследований GRP и аналогов GRP, которые связываются с семейством рецепторов GRP. Одним таким аналогом является бомбезин (BBN), пептид из 14 аминокислот (т.е. тетрадекапептид), выделенный из кожи лягушки, который представляет собой аналог человеческого GRP и который связывается с рецепторами GRP с высокой специфичностью и сродством, аналогичным GRP.
Бомбезин и аналоги GRP могут принимать форму агонистов или антагонистов. Связывание агонистов GRP или BBN с рецептором GRP увеличивает скорость клеточного деления этих раковых клеток, и такие агонисты интернализуются клетками, хотя связывание антагонистов GRP или BBN в целом не приводит ни к интернализации клеткой, ни увеличенным скоростям клеточного деления. Такие антагонисты предназначены для полного ингибирования связывания эндогенного GRP с рецепторами GRP и снижения скорости пролиферации раковых клеток (см., например, Hoffken, K.; Peptides in Oncology II, Somatostatin Analogues and Bombesin Antagonists (1993), pp. 87-112). По этой причине проводилась и проводится большая работа, направленная на создание аналогов BBN и GRP, которые являются антагонистами (например, Davis et al., Metabolic Stability and Tumor Inhibition of Bombesin/GRP Receptor Antagonists, Peptides, vol. 13, pp. 401-407, 1992).
При создании эффективного соединения для применения в качестве диагностического или терапевтического средства против рака важно, чтобы препарат имел соответствующие свойства целенаправленной доставки и фармакокинетики in vivo. Например, предпочтительно, чтобы для радиофармацевтического средства меченный радиоактивной меткой пептид имел высокий специфический захват раковыми клетками (например, через рецепторы GRP). Кроме того, также предпочтительно, чтобы после того, как радионуклид локализовался в участке рака, он оставался в нем в течение желаемого количества времени для доставки высоколокализованной дозы облучения к участку.
Более того, создание меченных радиоактивной меткой пептидов, которые эффективно выводятся из нормальных тканей, также является важным фактором для радиофармацевтических средств. Когда биологические молекулы (например, Mab, Fab или пептиды) метятся металлическими радионуклидами (конъюгированным хелатированием) и вводятся животному, такому как человек, то большая процентная доля металлического радионуклида (в любой химической форме) может стать "захваченной" в паренхиме почек или печени (т.е. не выделяется в мочу или желчь). Duncan et al.; Indium-111-Diethylenetriaminepentaacetic Acid-Octreotide Is Delivered in Vivo to Pancreatic, TumorCell, Renal, and Hepatocyte Lysosomes, Cancer Research 57, pp. 659-671, (Feb. 15, 1997). Для малых биологических молекул, меченных радиоактивной меткой (т.е. пептидов или Fab), основным путем выведения активности является выведение через почки, которые также могут удерживать высокие уровни радиоактивного металла (т.е. обычно >10-15% инъецированной дозы). Ясно, что удерживание металлических радионуклидов в почках или печени нежелательно. Напротив, слишком быстрое удаление радиофармацевтического средства из потока крови почками также нежелательно, если требуется более длительная диагностическая визуализация или высокий захват опухолью для лучевой терапии.
В последующей работе, такой как в патенте США № 6200546 и заявке на патент США № 2002/0054855, выданном Hoffman et al., включенном в настоящее описание в качестве ссылки, была предпринята попытка преодолеть эту проблему получением соединения, имеющего формулу X-Y-B, где Х представляет собой группу, способную образовывать комплекс с металлом, Y представляет собой ковалентную связь в спейсерной группе и В представляет собой связывающую часть агониста бомбезина. Сообщалось, что такие соединения имеют высокое сродство связывания с рецепторами GRP, и радиоактивность удерживалась внутри клеток в течение продолжительных периодов времени. Кроме того, исследования in vivo на здоровых мышах показали, что удерживание радиоактивного металла в почках было ниже, чем известно в данной области, причем большая часть радиоактивности выделялась в мочу.
В настоящее время были созданы новые улучшенные радиофармацевтические и другие диагностические соединения с улучшенным выделением почками (т.е. с более низким удерживанием радиоактивного металла в почках) для диагностической визуализации и терапевтических применений. Для диагностической визуализации быстрое выделение почками и низкие удерживаемые уровни радиоактивности имеют решающее значение для улучшенных изображений. Для радиотерапевтического применения решающее значение имеет более медленное выведение из крови для обеспечения возможности более высокого накопления в опухолевой ткани и улучшенной целенаправленной доставки к опухоли при низком удерживании в почках.
Краткое описание изобретения
В одном варианте осуществления настоящего изобретения предоставляются новые улучшенные соединения для применения при диагностической визуализации или лучевой терапии. Соединения включают химическую часть, способную образовывать комплекс с применяемым в медицине ионом металла или радионуклидом (хелатирующим металл), присоединенным к пептиду для целенаправленной доставки к рецептору GPR линкерной или спейсерной группой. В другом варианте осуществления эти соединения включают оптическую метку (например, фотометку или другую метку, выявляемую световой визуализацией, оптико-акустической визуализацией или фотолюминесценцией), присоединенную к пептиду для целенаправленной доставки к рецептору GPR линкерной или спейсерной группой.
В целом, соединения настоящего изобретения могут иметь формулу:
M-N-O-P-G,
где М представляет собой агент, хелатирующий металл (в форме комплекса с радионуклидом или в форме, не образующей комплекс с радионуклидом), или оптическую метку, N-O-P представляет собой линкер, а G представляет собой пептид для целенаправленной доставки к рецептору GPR.
Агент, хелатирующий металл М, может представлять собой любое из агентов, хелатирующих металлы, известных в данной области, для образования комплекса с применяемым в медицине металлом или радионуклидом. Предпочтительные агенты, хелатирующие металлы, включают DTPA, DOTA, DO3A, HP-DO3A, EDTA, TETA, EHPG, HBED, NOTA, DOTMA, TETMA, PDTA, TTHA, LICAM, MECAM или пептиды, образующие хелаты, такие как, например, пептиды, раскрытые в настоящем описании. Агент, хелатирующий металл, может находиться или не находиться в форме комплекса с металлическим радионуклидом и может включать необязательный спейсер, такой как одиночная аминокислота. Предпочтительные металлические радионуклиды для сцинтиграфии или лучевой терапии включают
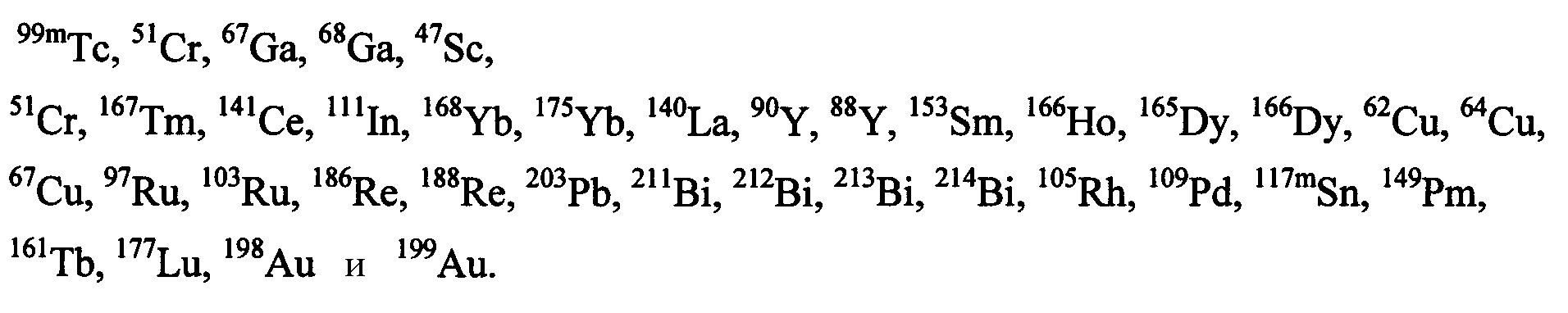
Выбор металла должен определяться на основании желаемого терапевтического или диагностического применения. Например, для диагностических целей предпочтительные радионуклиды включают64Cu,67Ga,68Ga,99mTc и111In, где особенно предпочтительны99mTc и111In. Для терапевтических целей предпочтительные радионуклиды включают

где177Lu и90Y особенно предпочтительны. Наиболее предпочтительным хелатообразующим агентом, используемым в соединениях изобретения, является 1-замещенная 4,7, 10-трикарбоксиметил-1,4,7,10-тетраазациклодекантриуксусная кислота (DO3A).
Оптическая метка М может представлять собой любую из различных оптических меток, известных в данной области. Предпочтительные метки включают без ограничения оптические красители, включая органические хромофоры или флуорофоры, такие как цианиновые красители, поглощающие свет соединения, отражающие и рассеивающие свет соединения и биолюминесцентные молекулы.
В одном варианте осуществления линкер N-O-P содержит, по меньшей мере, одну не альфа-аминокислоту.
В другом варианте осуществления линкер N-O-P содержит, по меньшей мере, одну замещенную желчную кислоту.
В еще одном варианте осуществления линкер N-O-P содержит, по меньшей мере, одну не альфа-аминокислоту с циклической группой.
Пептид для целенаправленной доставки к рецептору GRP может представлять собой GRP, бомбезин или его любые производные или аналоги. В предпочтительном варианте осуществления пептид для целенаправленной доставки к рецептору GRP представляет собой GRP или аналог бомбезина, который действует в качестве агониста. В особенно предпочтительном варианте осуществления пептид для целенаправленной доставки к рецептору GRP представляет собой связывающую часть агониста бомбезина, раскрытую в патенте США № 6200546 и заявке на патент США 2002/0054855, включенных в настоящее описание в качестве ссылки.
Предоставляется также новый способ визуализации с использованием соединений настоящего изобретения.
Набор с одной или несколькими емкостями, который содержит все компоненты, необходимые для получения диагностических или терапевтических средств изобретения, предоставлен в иллюстративном варианте осуществления настоящего изобретения.
Кроме того, предоставляется новый способ получения диагностического визуализирующего средства, включающий стадию добавления к инъецируемой визуализирующей среде вещества, содержащего соединения настоящего изобретения.
Предоставляется также новый способ лучевой терапии с использованием соединений изобретения, как и новый способ получения средства лучевой терапии, включающий стадию добавления к инъецируемой терапевтической среде вещества, включающего соединение изобретения.
Краткое описание чертежей
Фиг.1А представляет собой графическое изображение серии химических реакций для синтеза промежуточного соединения С ((3β,5β )-3-(9Н-флуорен-9-илметокси)аминохолан-24-овой кислоты) из А (метил-(3β,5β)-3-аминохолан-24-ата) и В ((3β,5β)-3-аминохолан-24-овой кислоты), как описано в примере I.
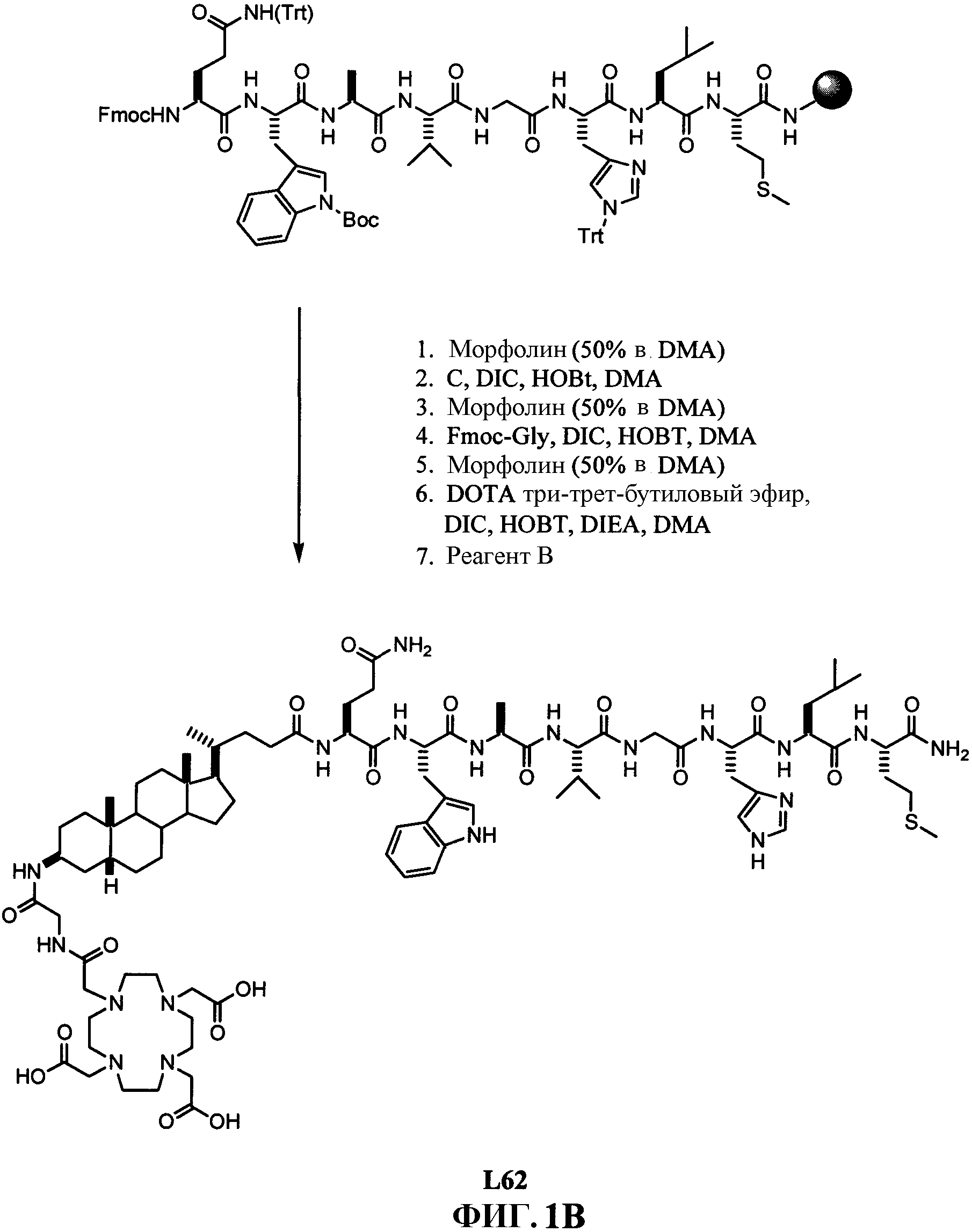
Фиг.1В представляет собой графическое изображение последовательности реакций для синтеза N-[(3β,5β)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]холан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L62), как описано в примере I.
Фиг.2А представляет собой графическое изображение последовательности реакций для синтеза N-[(4-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L70), как описано в примере II.
Фиг.2B представляет собой общее графическое изображение последовательности реакций для синтеза N-[4-[2-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]этокси]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L73), N-[3-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L115) и N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]фенилацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L116), как описано в примере II.
Фиг.2С представляет собой химическую структуру линкера, используемого в реакции, показанной на фиг.2В для синтеза N-[4-[2-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]этокси]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L73), как описано в примере II.
Фиг.2D представляет собой химическую структуру линкера, используемого в реакции, показанной на фиг.2В для синтеза N-[3-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L115), как описано в примере II.
Фиг.2Е представляет собой химическую структуру линкера, используемого в реакции, показанной на фиг.2В для синтеза N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]фенилацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L116), как описано в примере II.
Фиг.2F представляет собой графическое изображение последовательности реакций для синтеза N-[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]глицил-4-пиперидинкарбонил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L74), как описано в примере II.
Фиг.3А представляет собой графическое изображение серии химических реакций для синтеза промежуточного соединения (3β,5β )-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-12-оксохолан-24-овой кислоты (С), как описано в примере III.
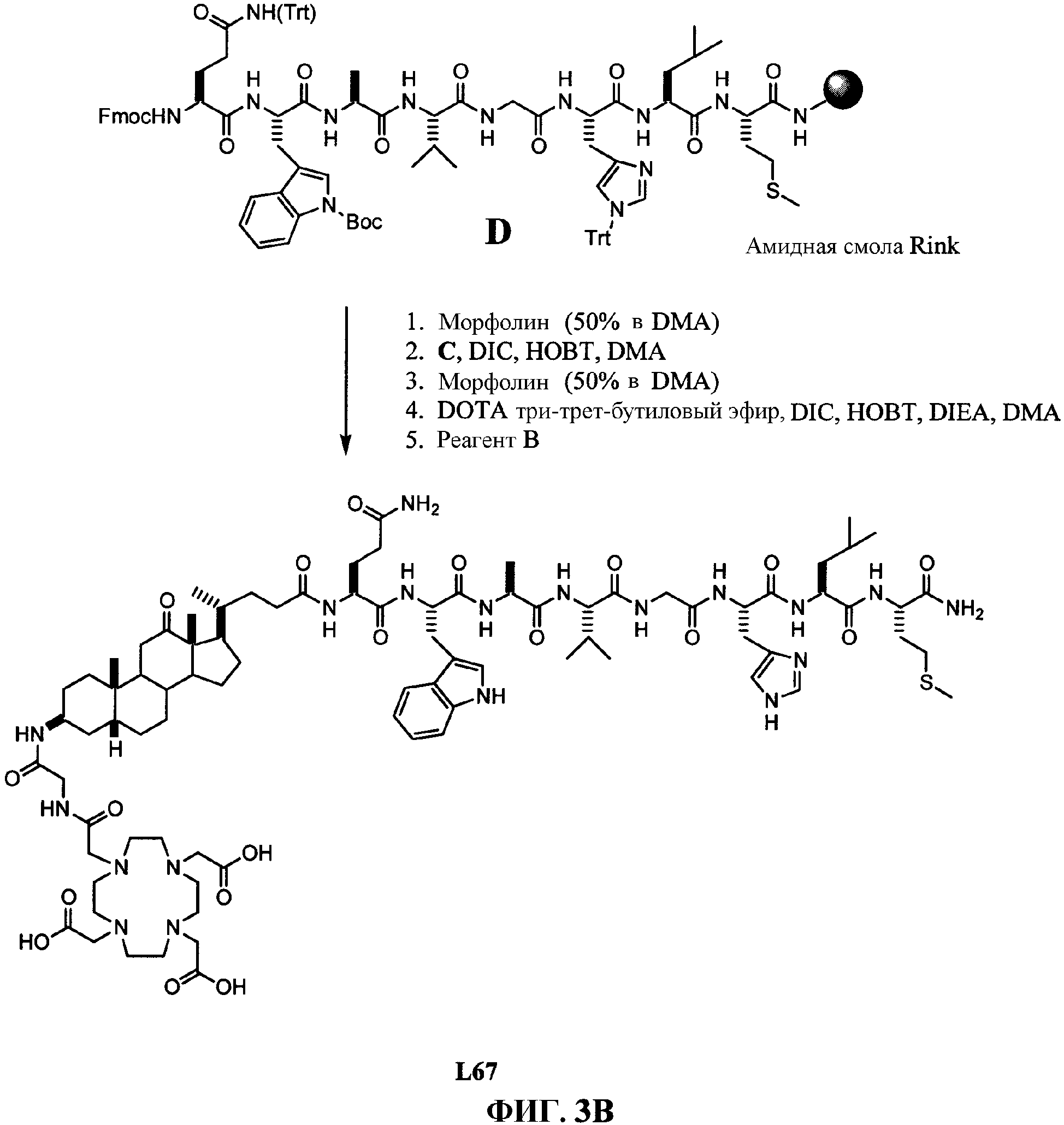
Фиг.3В представляет собой графическое изображение последовательности реакций для синтеза N-[(3β,5β)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-12, 24-диоксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L67), как описано в примере III.
Фиг.3C представляет собой химическую структуру (3β,5β)-3-амино-12-оксохолан-24-овой кислоты (В), как описано в примере III.
Фиг.3D представляет собой химическую структуру (3β, 5β)-3-[[(9H-флуорен-9-илметокси)амино]ацетил]амино-12-оксохолан-24-овой кислоты (С), как описано в примере III.
Фиг.3Е представляет собой химическую структуру N-[(3β,5β)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-12, 24-диоксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L67), как описано в примере III.
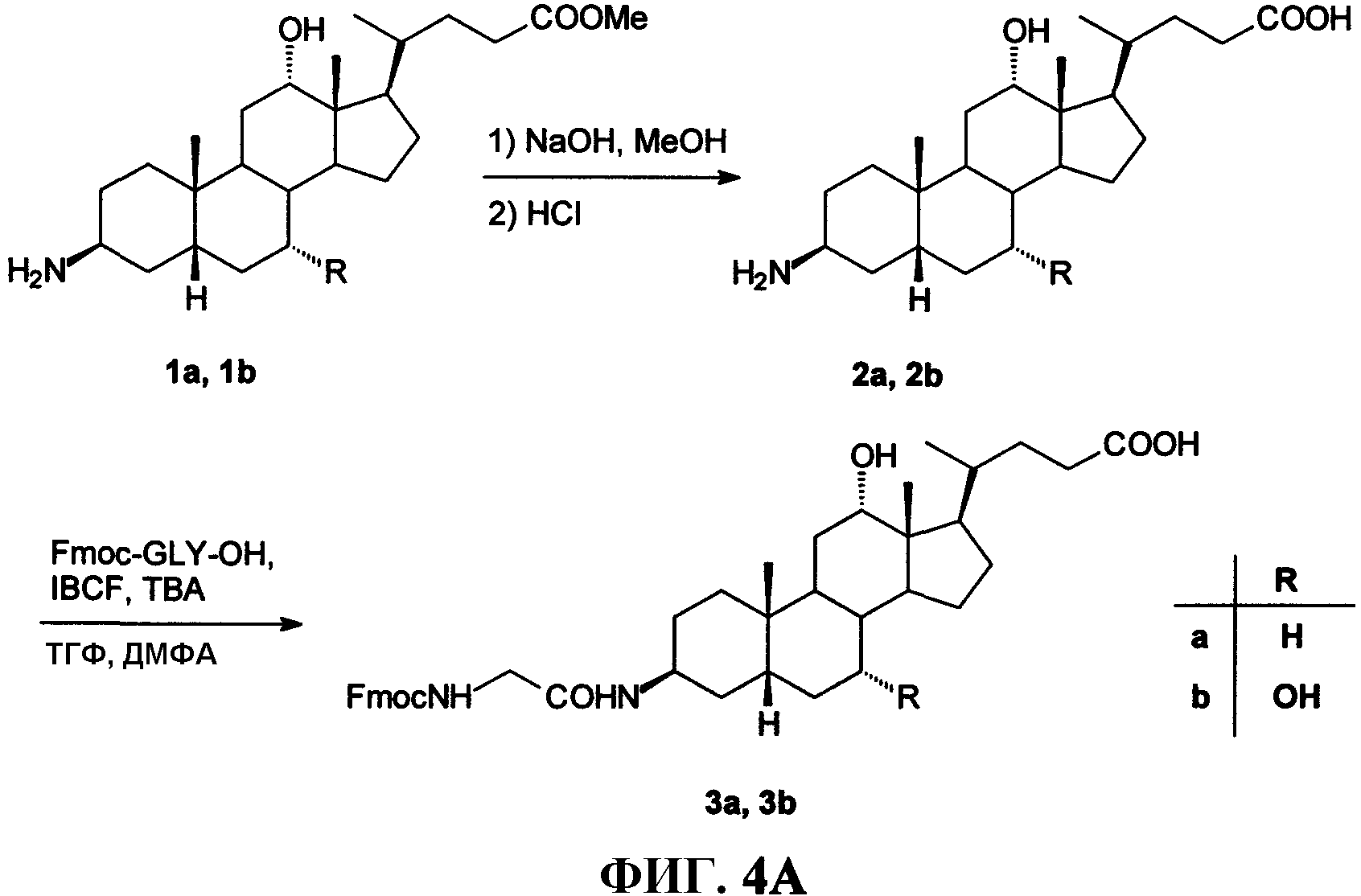
Фиг.4А представляет собой графическое изображение последовательности реакций для получения промежуточных соединений (3β,5β,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-12-гидроксохолан-24-овой кислоты (3а) и (3β,5β,7α,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксохолан-24-овой кислоты (3b), как описано в примере IV.
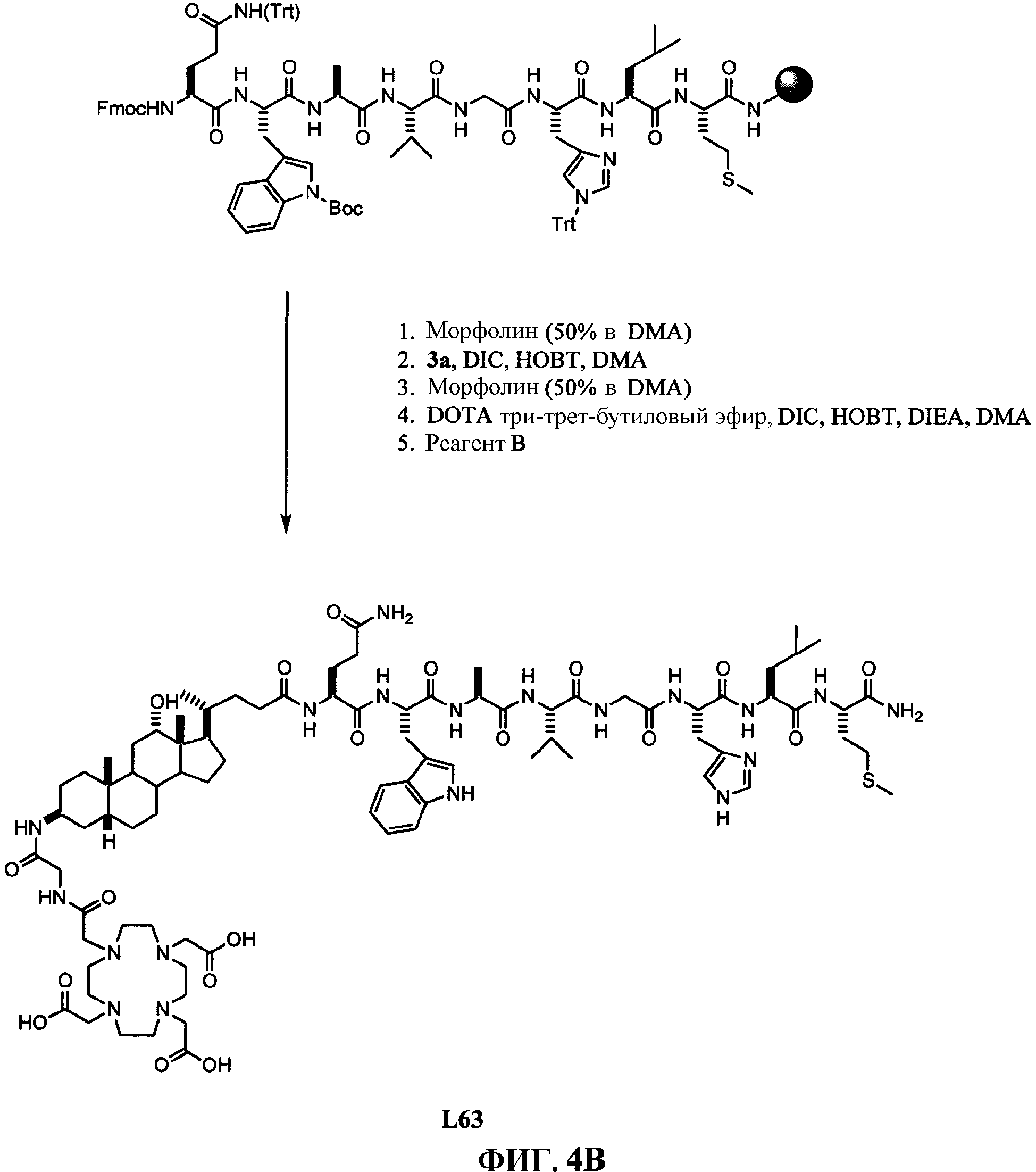
Фиг.4В представляет собой графическое изображение последовательности реакций для синтеза N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-12-гидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L63), как описано в примере IV.
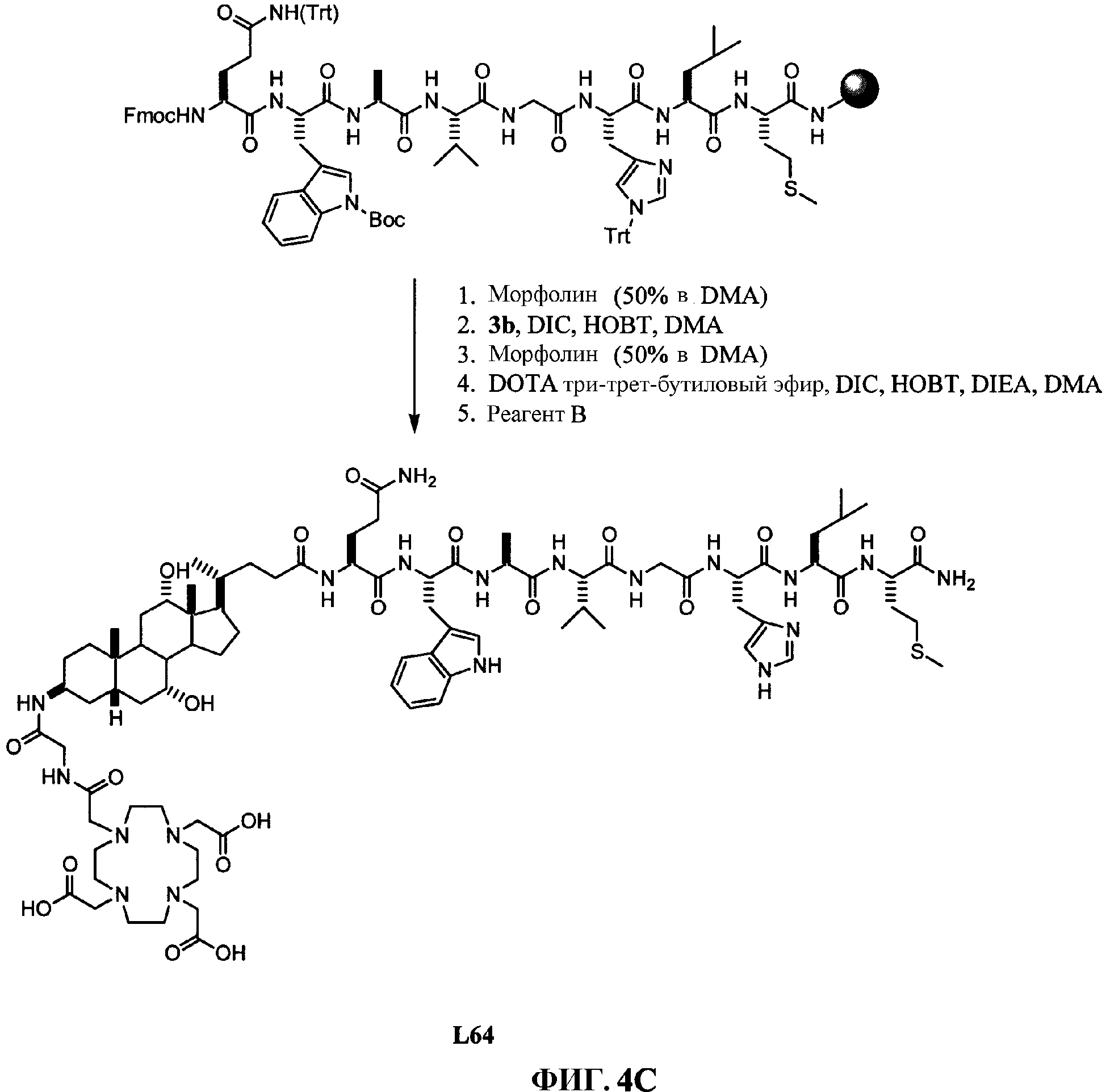
Фиг.4C представляет собой графическое изображение последовательности реакций для синтеза N-[(3β,5β,7α,12α)-3-[[[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-7, 12-дигидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L64), как описано в примере IV.
Фиг.4D представляет собой химическую структуру (3β,5β,7α,12α)-3-амино-7,12-дигидроксихолан-24-овой кислоты (2b), как описано в примере IV.
Фиг.4Е представляет собой химическую структуру (3β,5β,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-12-гидроксохолан-24-овой кислоты (3а), как описано в примере IV.
Фиг.4F представляет собой химическую структуру (3β,5β,7α,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксохолан-24-овой кислоты (3b), как описано в примере IV.
Фиг.4G представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-12-гидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L63), как описано в примере IV.
Фиг.4H представляет собой химическую структуру N-[(3β,5β,7α,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-7,12-дигидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L64), как описано в примере IV.
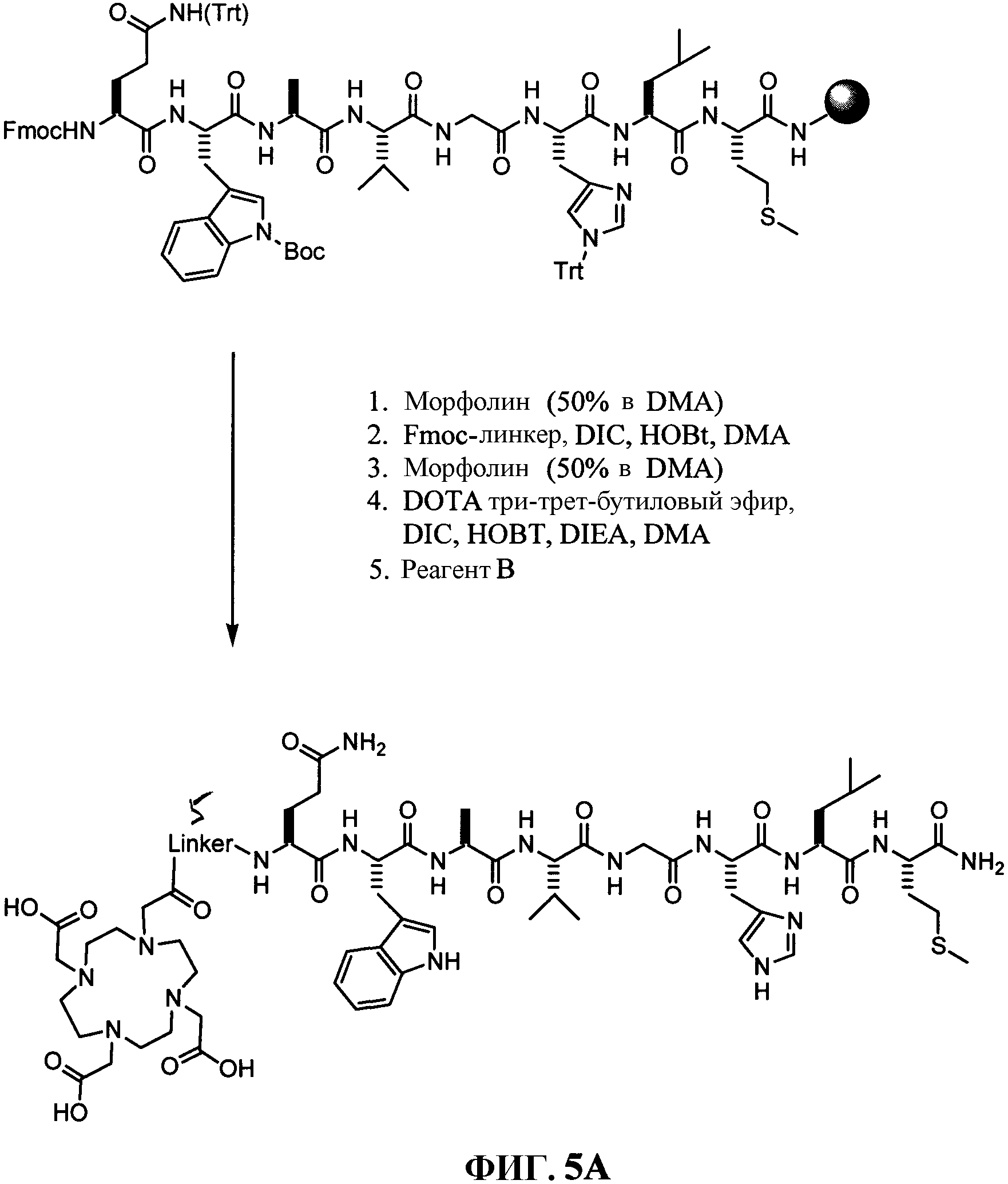
Фиг.5А представляет собой общее графическое изображение последовательности реакций для синтеза 4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]бензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L71) и транс-4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]циклогексилкарбонил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L72), как описано в примере V, где линкер представлен на фиг.5В и 5С, соответственно.
Фиг.5В представляет собой химическую структуру линкера, используемого в соединении L71, как показано на фиг. 5А и как описано в примере V.
Фиг.5С представляет собой химическую структуру линкера, используемого в соединении L72, как показано на фиг.5А и как описано в примере V.
Фиг.5D представляет собой химическую структуру амидной смолы Ринка, функционализированной бомбезином [7-14] (B), как описано в примере V.
Фиг.5Е представляет собой химическую структуру транс-4-[[[(9H-флуорен-9-илметокси)карбонил]амино]метил]циклогексанкарбоновой кислоты (D), как описано в примере V.
Фиг.6А представляет собой графическое изображение последовательности реакций для синтеза промежуточного линкера 2-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]бензойной кислоты (Е), как описано в примере VI.
Фиг.6В представляет собой графическое изображение последовательности реакций для синтеза промежуточного линкера 4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]-3-нитробензойной кислоты (Н), как описано в примере VI.
Фиг.6С представляет собой общее графическое изображение синтеза N-[2-[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]метил]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L75), как описано в примере VI.
Фиг.6D представляет собой общее графическое изображение синтеза N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]-3-нитробензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L76), как описано в примере VI.
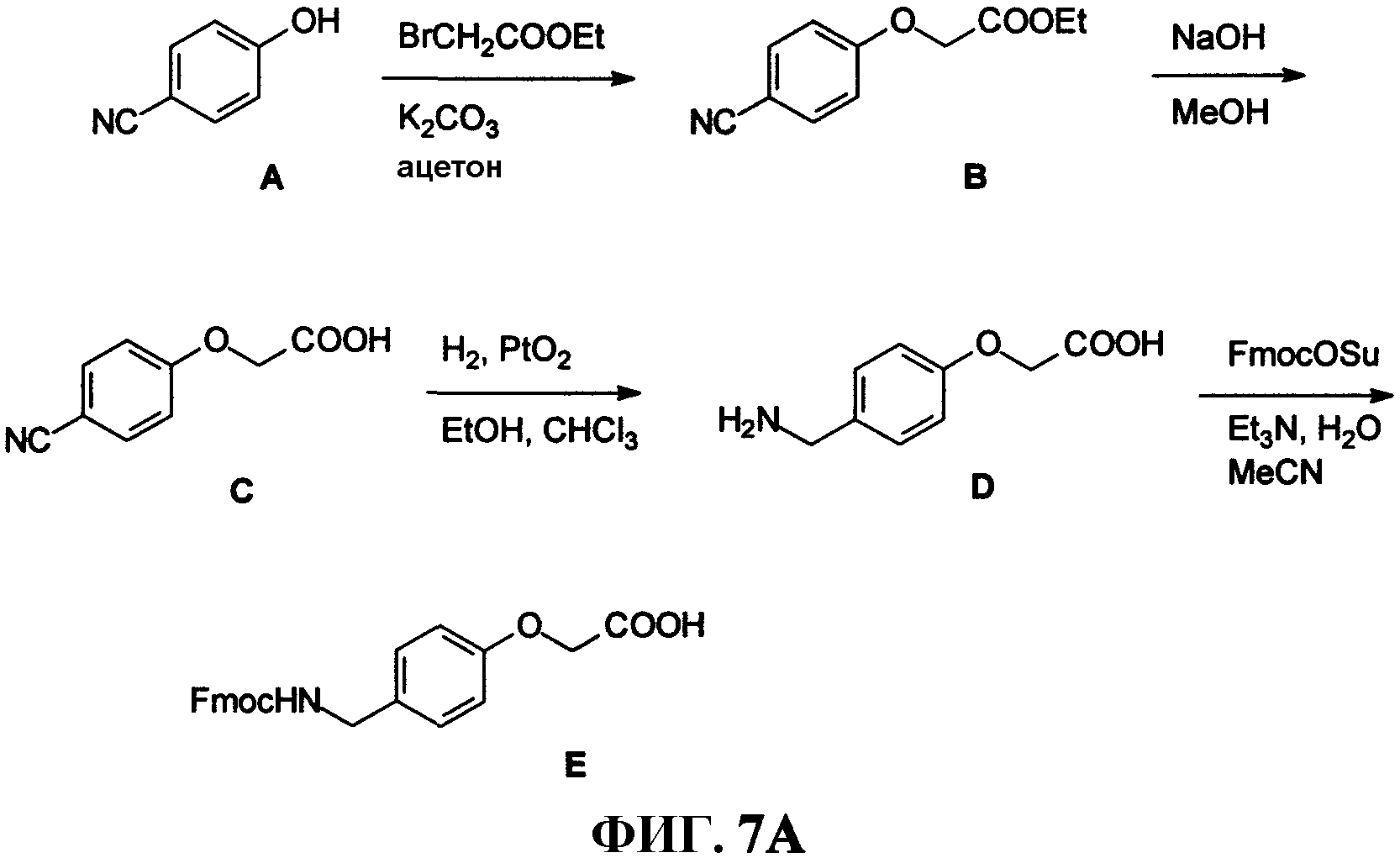
Фиг.7А представляет собой графическое изображение последовательности реакций для синтеза промежуточного линкера [4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]фенокси]уксусной кислоты ( Е), как описано в примере VII.
Фиг.7В представляет собой графическое изображение синтеза N-[[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]фенокси]ацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L124), как описано в примере VII.
Фиг.7C представляет собой химическую структуру N-[[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]фенокси]ацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L124), как описано в примере VII.
Фиг.8А представляет собой графическое изображение последовательности реакций для синтеза промежуточного соединения [4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]-3-метоксибензойной кислоты (Е), как описано в примере VIII.
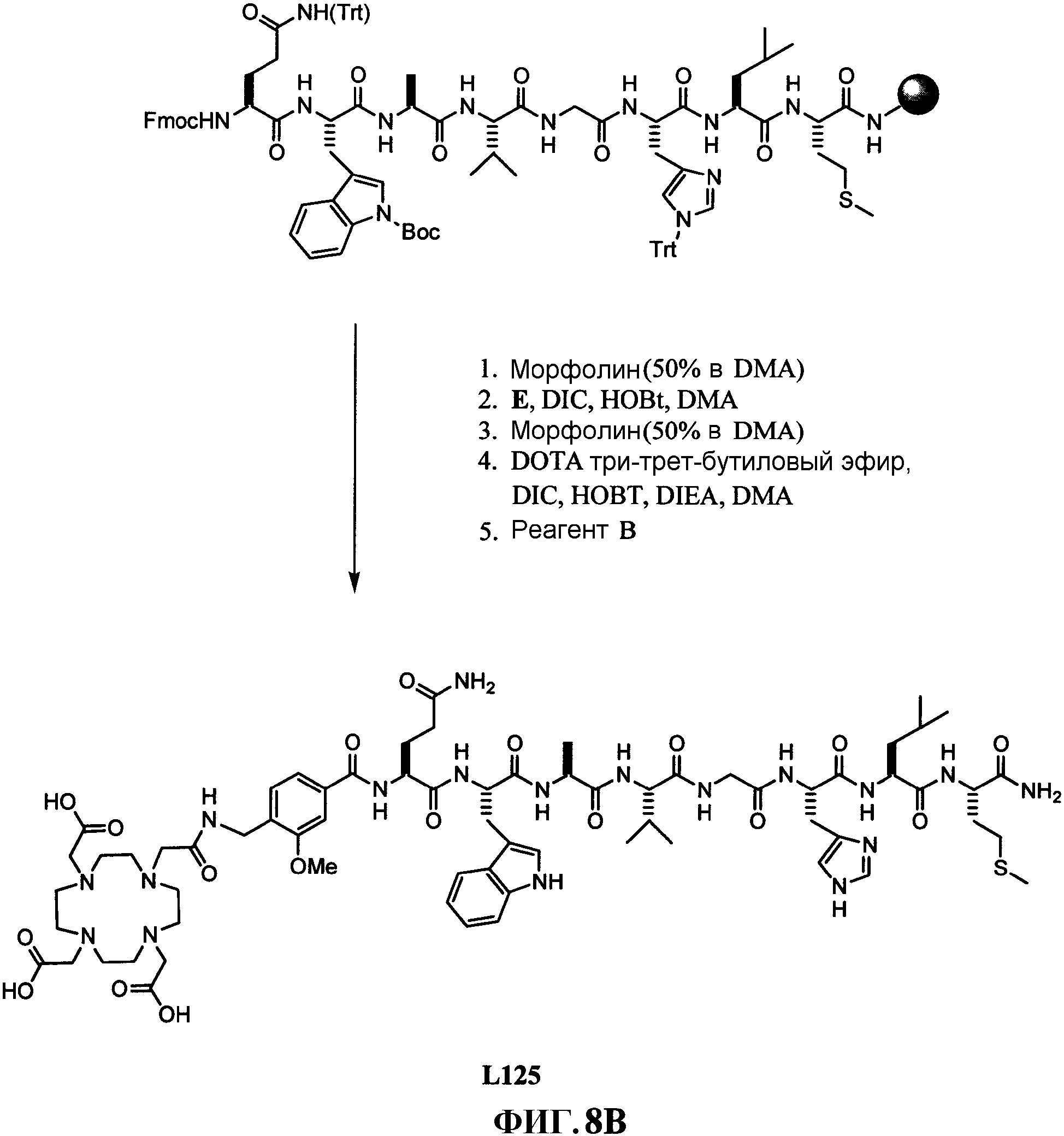
Фиг.8В представляет собой графическое изображение синтеза N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]-3-метоксибензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L125), как описано в примере VIII .
Фиг.8C представляет собой химическую структуру N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]-3-метоксибензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L125), как описано в примере VIII .
Фиг.9А представляет собой графическое изображение реакций для синтеза 3-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]аминобензойной кислоты, (В), как описано в примере IX.
Фиг.9В представляет собой графическое изображение реакций для синтеза 6-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]аминобензойной кислоты, (С), как описано в примере IX.
Фиг.9С представляет собой графическое изображение реакций для синтеза 4-[[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]метиламино]бензойной кислоты, (D), как описано в примере IX.
Фиг.9D представляет собой графическое изображение реакций для синтеза N-[4-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]фенилацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L146); N-[3-[[[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L233); N-[6-[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]нафтоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L234 ) и N-[4-[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]метиламино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L235), как описано в примере IX.
Фиг.10А представляет собой графическое изображение реакций для синтеза 4,10-бис(1,1-диметилэтилового) эфира 7-[[бис(1, 1-диметилэтокси)фосфинил]метил]-1,4,7,10-тетраазациклододекан-1,4,10-триуксусной кислоты Н, как описано в примере X.
Фиг.10В представляет собой графическое изображение реакций для синтеза N-[4-[[[[[4,10-бис(карбоксиметил)-7-(дигидроксифосфинил)метил-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]ацетил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L237), как описано в примере X .
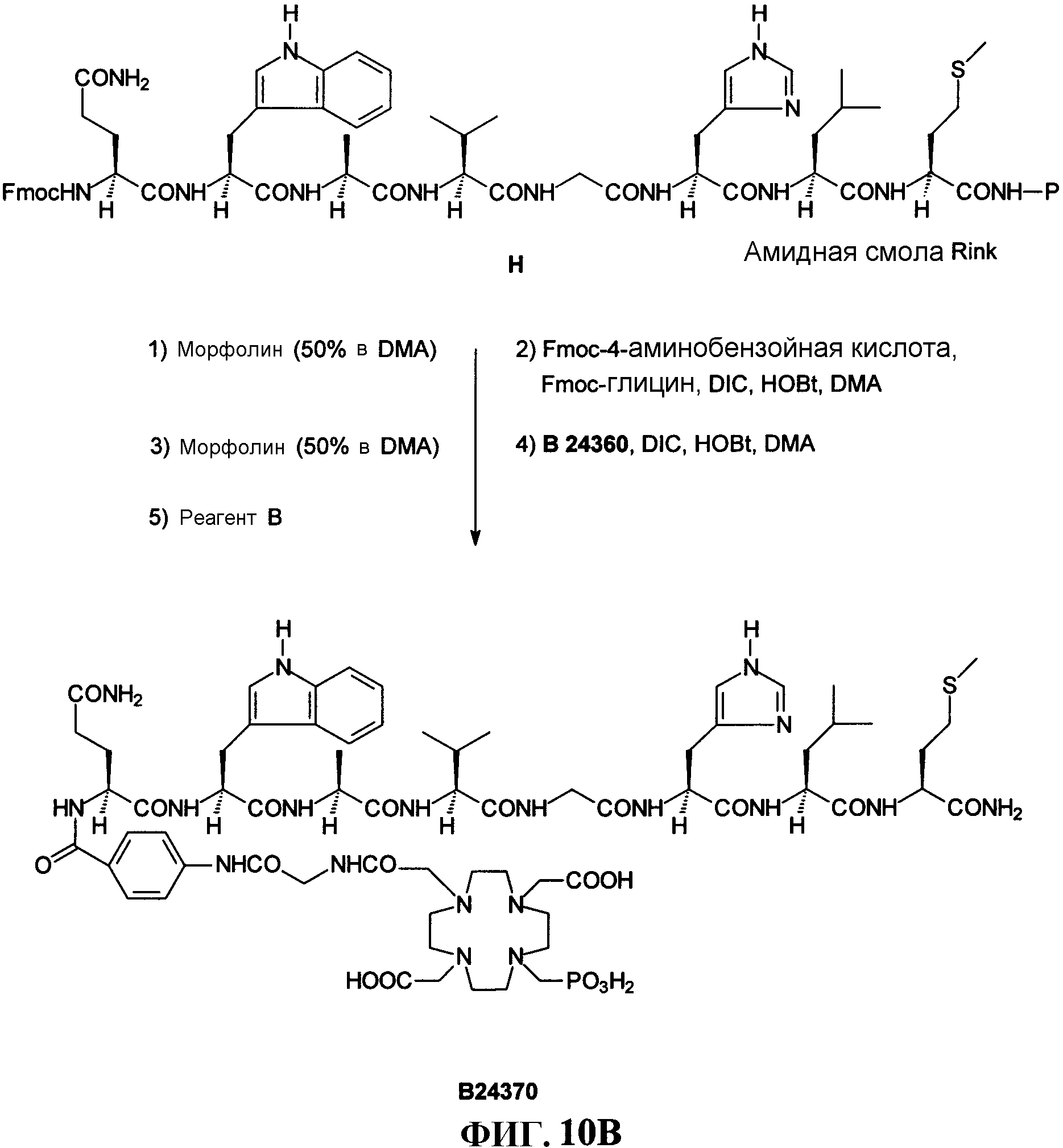
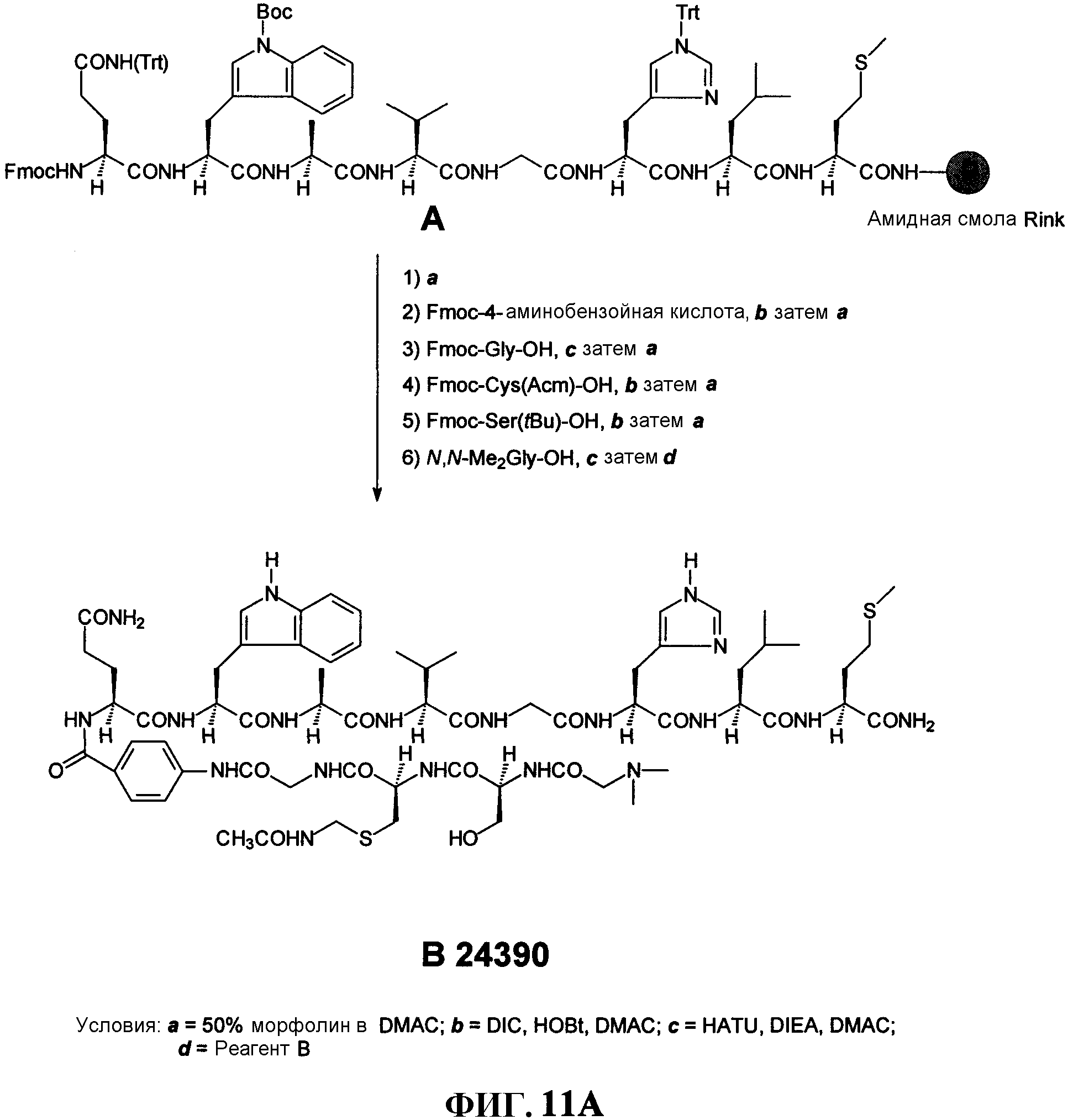
Фиг.11А представляет собой графическое изображение реакций для синтеза N, N-диметилглицил-L-серинил-[S-[(ацетиламино)метил]]-L-цистениилглицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L238), как описано в примере XI.
Фиг.11В представляет собой графическое изображение реакций для синтеза N,N-диметилглицил-L-серинил-[S-[(ацетиламино)метил]]-L-цистениилглицил-(3β,5β, 7α,12α)-3-амино-7,12-дигидрокси-24-оксохолан-24-ил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L239), как описано в примере XI.
Фиг.12А представляет собой графическое изображение реакции для синтеза 4-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-метоксибензойной кислоты (А), как описано в примере XII.
Фиг.12B представляет собой графическое изображение реакции для синтеза 4-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-хлорбензойной кислоты (D), как описано в примере XII.
Фиг.12C представляет собой графическое изображение реакции для синтеза 4-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-метилбензойной кислоты (Е), как описано в примере XII.
Фиг.12D представляет собой химическую структуру N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]глицил]амино]-3-метоксибензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L240), как описано в примере XII .
Фиг.12E представляет собой химическую структуру N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]глицил]амино]-3-хлорбензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L241), как описано в примере XII.
Фиг.12F представляет собой химическую структуру N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]глицил]амино]-3-метилбензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L240), как описано в примере XII.
Фиг.13А представляет собой графическое изображение реакции для синтеза 4-[N,N'-бис[2-[(9H-флуорен-9-илметокси)карбонил]аминоэтил]амино]-4-оксобутановой кислоты (D), как описано в примере XIII.
Фиг.13В представляет собой графическое изображение реакции для синтеза N-[4-[[4-[бис[2-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]этил]амино-1,4-диоксибутил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L244), как описано в примере XIII.
Фиг.13C представляет собой химическую структуру соединения L244, как описано в примере XIII.
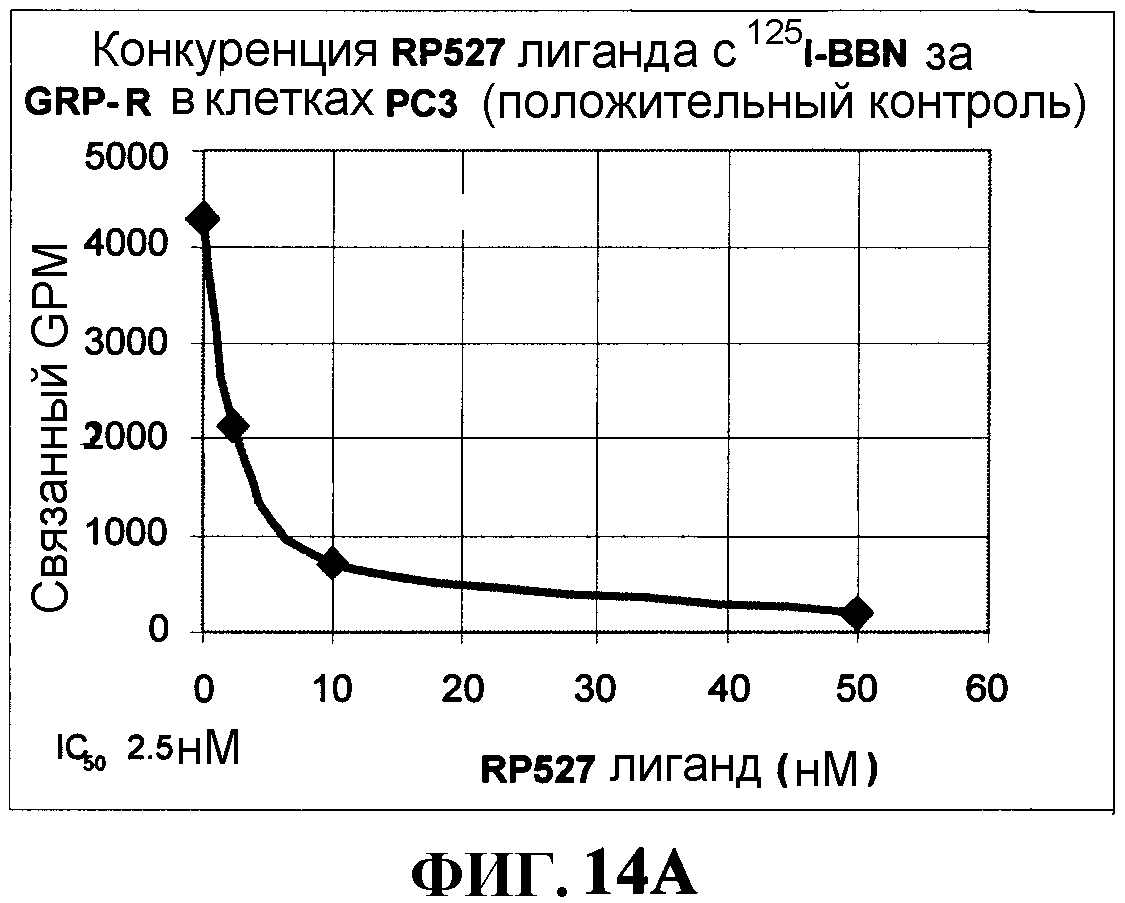
Фиг.14А и Фиг.14В представляют собой графическое изображение кривых связывания и конкуренции, как описано в примере XLIII.
Фиг.15А представляет собой графическое изображение результатов экспериментов лучевой терапии, описанных в примере LV.
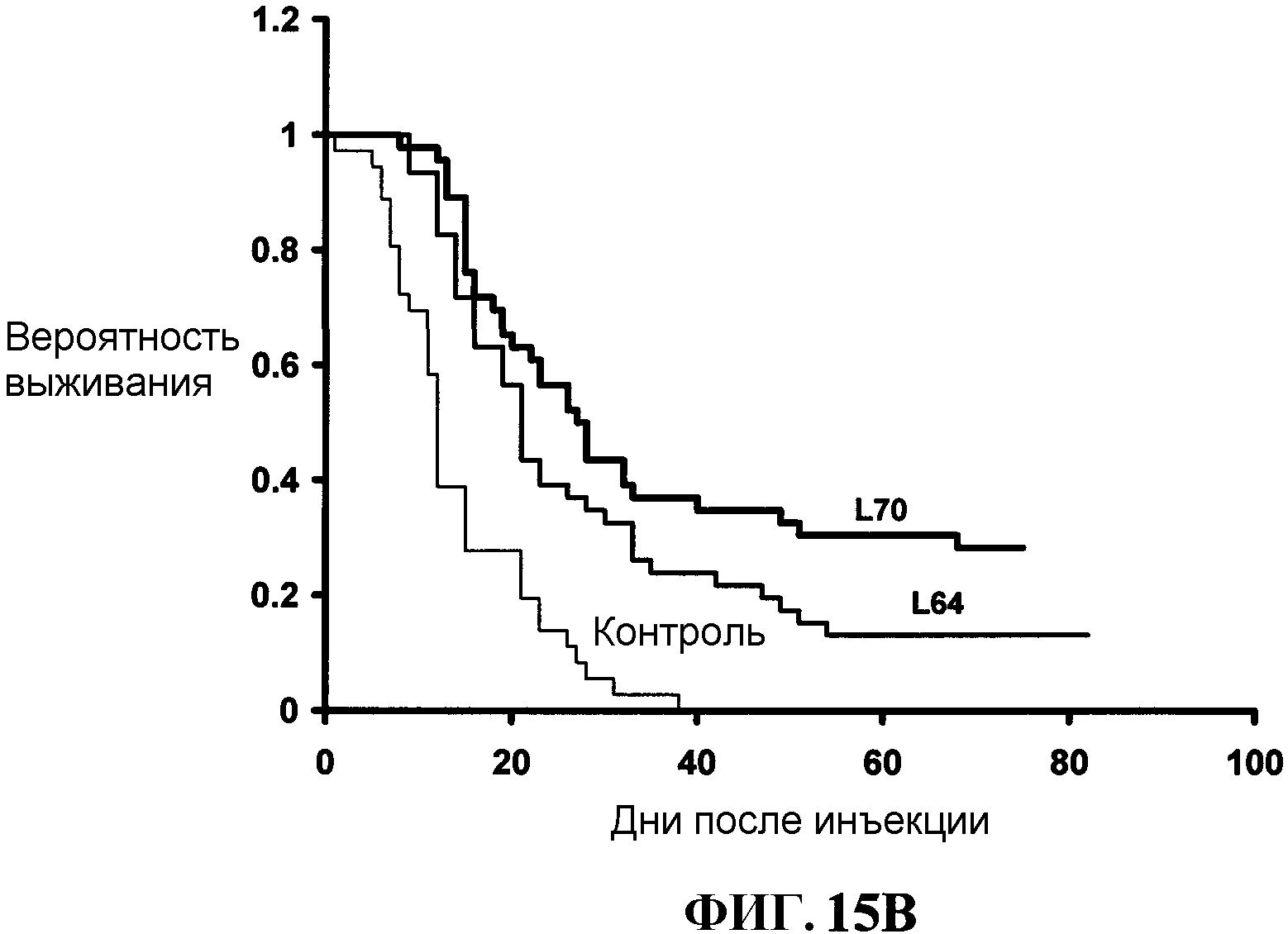
Фиг.15В представляет собой графическое изображение результатов других экспериментов лучевой терапии, описанных в примере LV.
Фиг.16 представляет собой химическую структуру N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]глицил]амино]-L-лизинил-(3,6,9)-триоксаундекан-1,11-дикарбоновая кислота-3, 7-дидеокси-3-аминохолевая кислота)-L-аргинил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L65).
Фиг.17 представляет собой химическую структуру N-[2-S-[[[[[12α-гидрокси-17α-(1-метил-3-карбоксипропил)этиохолан-3β-карбамоилметоксиэтоксиэтоксиацетил]амино-6-[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]гексаноил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L66).
Фиг.18А представляет собой химическую структуру N-[4-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L70).
Фиг.18В представляет собой химическую структуру N-[4-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]-3-карбоксипропионил]амино]ацетил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L114).
Фиг.18С представляет собой химическую структуру N-[4-[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]-2-гидрокси-3-пропокси]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L144).
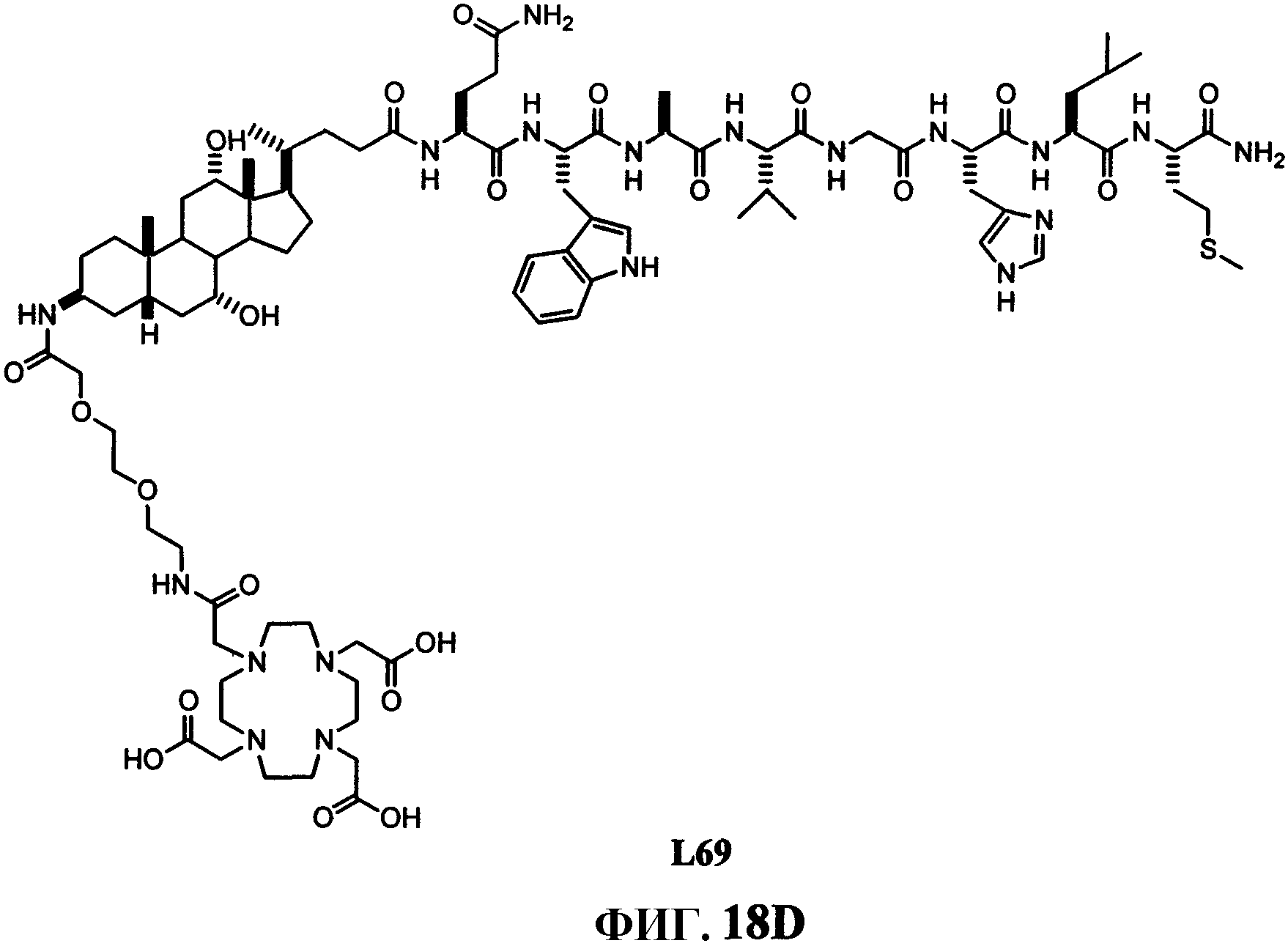
Фиг.18D представляет собой химическую структуру N-[(3β,5β,7α,12α)-3-[[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]этоксиэтокси]ацетил]амино]-7, 12-дигидроксихолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L69).
Фиг.18Е представляет собой химическую структуру N-[4-[[[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]фенилацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L146).
На фиг.19 раскрыты химические структуры промежуточных соединений, которые можно использовать для получения соединений L64 и L70, как описано в примере LVI.
Фиг.20 представляет собой графическое изображение получения L64 с использованием сегментного связывания, как описано в примере LVI.
Фиг.21 представляет собой графическое изображение получения (1R)-1-(бис{2-[бис(карбоксиметил)амино]этил}амино)пропан-3-карбоновая кислота-1-карбоксилглицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L201).
Фиг.22А представляет собой графическое изображение химической структуры промежуточных соединений, используемых для получения L202.
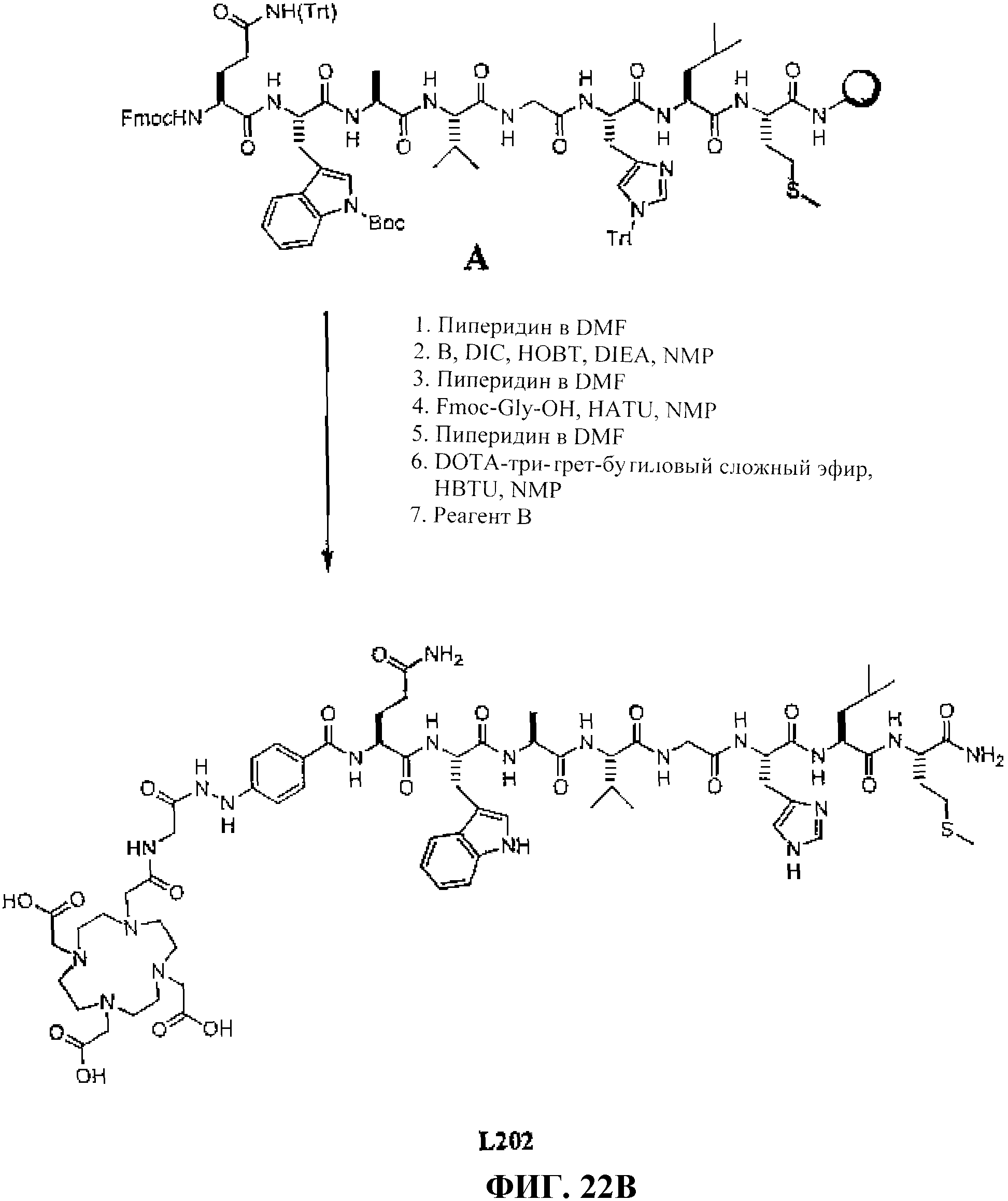
Фиг.22В представляет собой графическое изображение получения N-[(3β,5β, 12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-4-гидразинобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L202).
Фиг.23А представляет собой графическое изображение химической структуры промежуточных соединений, используемых для получения L203.
Фиг.23В представляет собой графическое изображение получения N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L203).
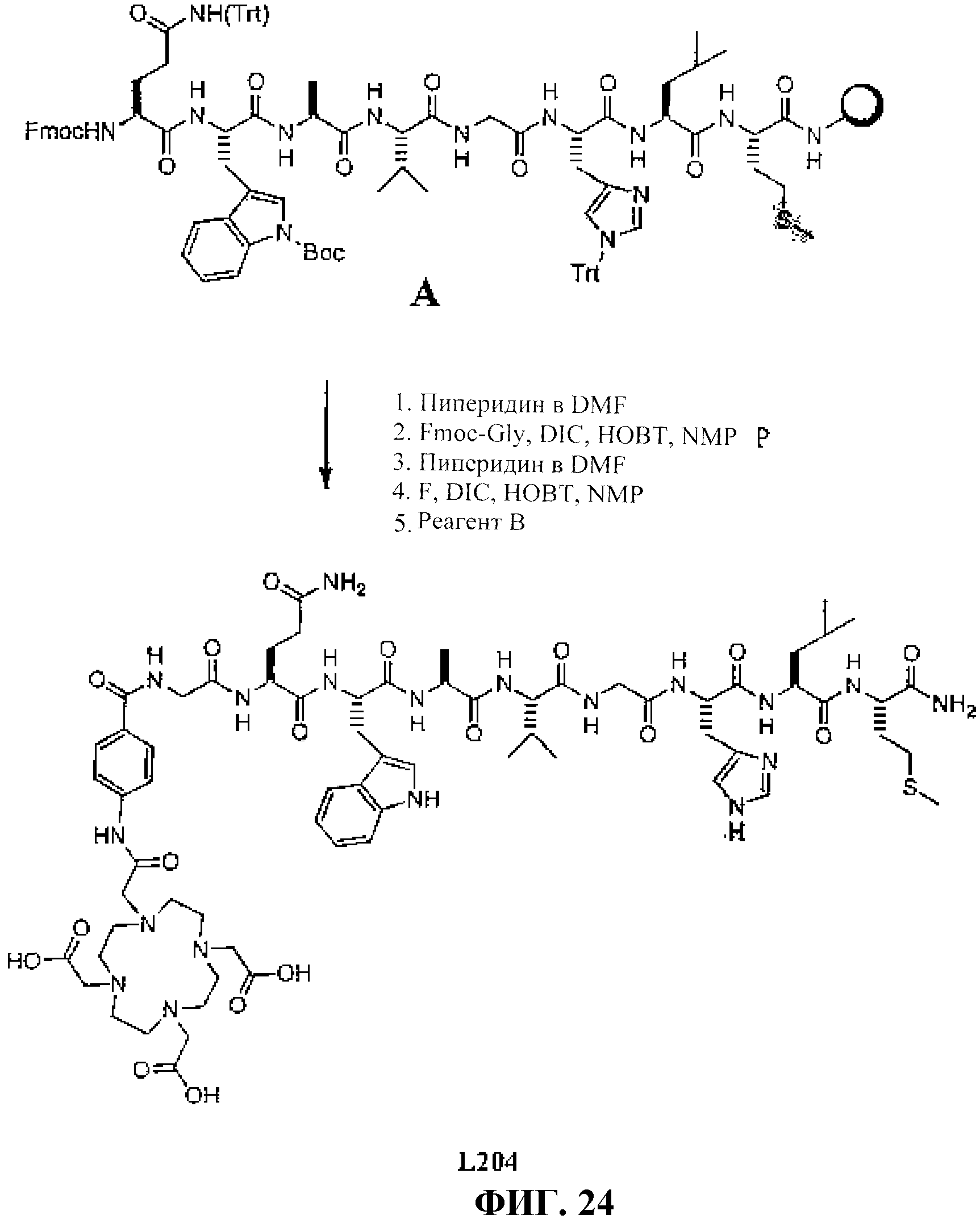
Фиг.24 представляет собой графическое изображение получения N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]-4-аминобензоилглицил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L204).
Фиг.25 представляет собой графическое изображение получения N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]-4-аминобензоилглицил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L205).
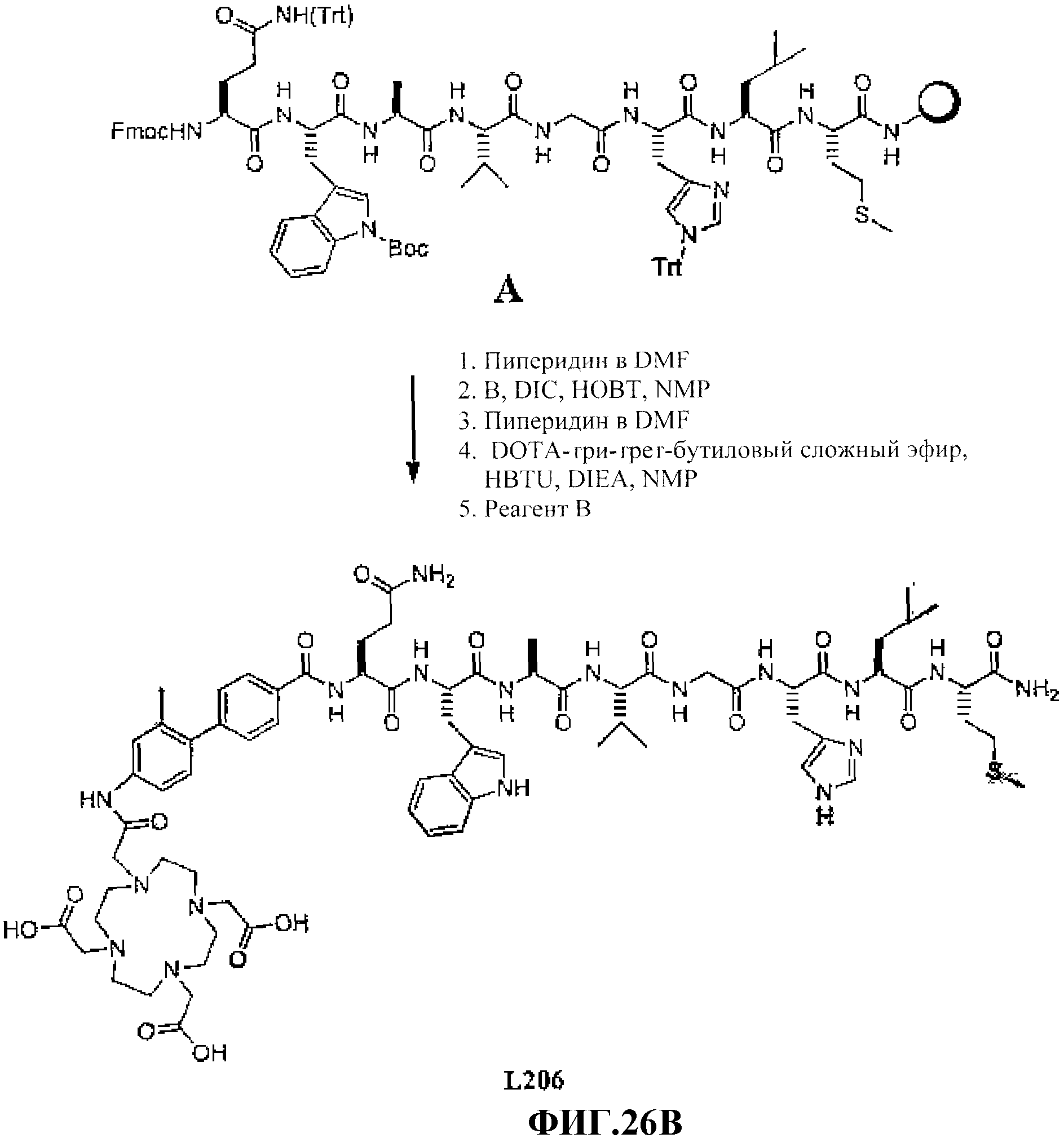
Фиг.26А представляет собой графическое изображение химических структур промежуточных соединений, используемых для получения L206.
Фиг.26В представляет собой графическое изображение получения N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-[4'-амино-2'-метилбифенил-4-карбоксил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L206).
Фиг.27А представляет собой графическое изображение химических структур промежуточных соединений, используемых для получения L207.
Фиг.27В представляет собой графическое изображение получения N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-[3'-аминобифенил-3-карбоксил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L207).
Фиг.27В представляет собой графическое изображение получения N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-[1, 2-диаминоэтилтерефталил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L208).
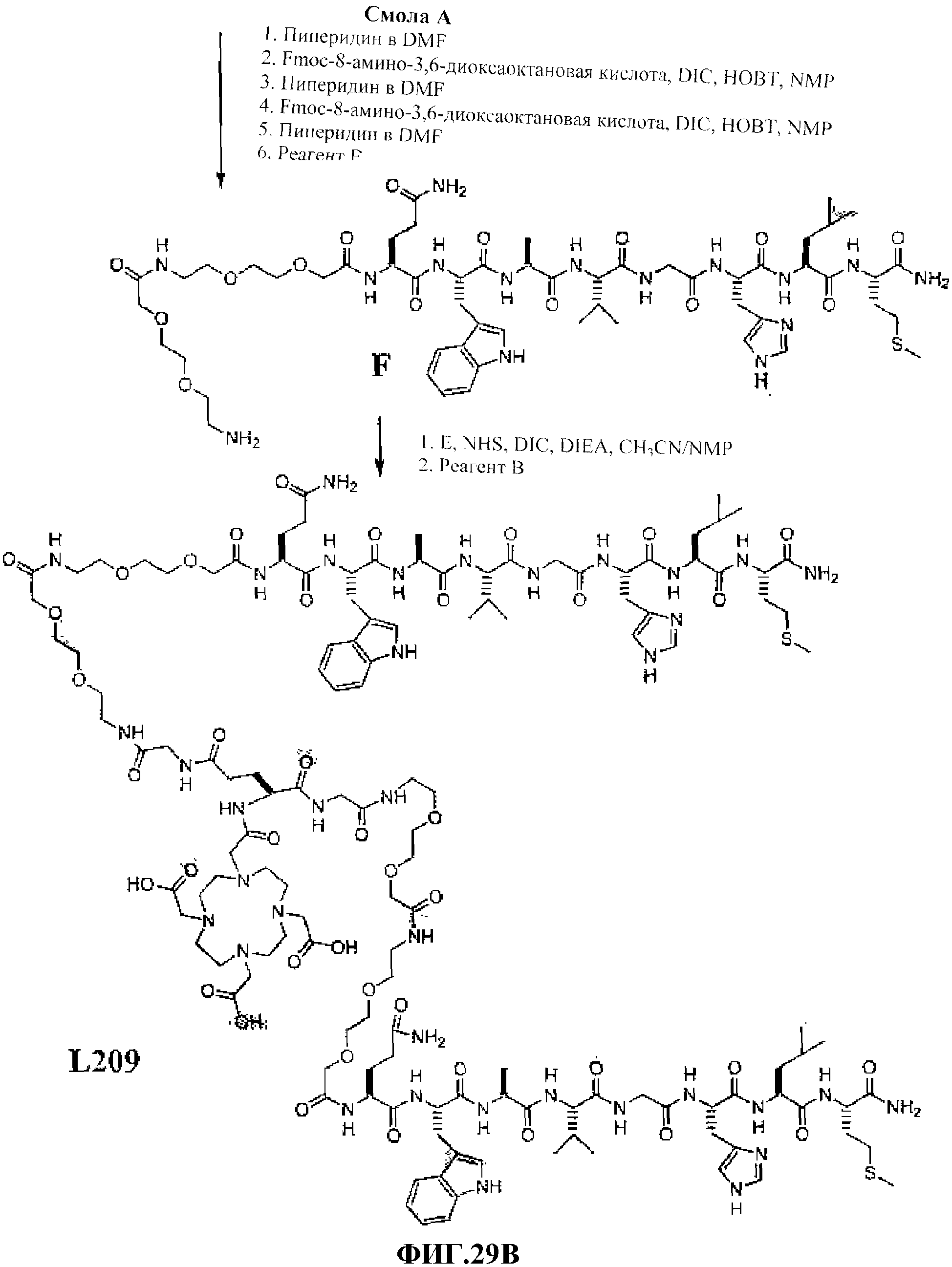
Фиг.29А представляет собой графическое изображение химических структур промежуточных соединений, используемых для получения L209.
Фиг.29В представляет собой графическое изображение получения L209.
Фиг.30А представляет собой графическое изображение химических структур промежуточных соединений, используемых для получения L210.
Фиг.30В представляет собой химическую структуру L210.
Фиг.31 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицилглицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L211).
Фиг.32 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутамил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L212).
Фиг.33 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинкарбоксилата L213.
Фиг.34 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L214.
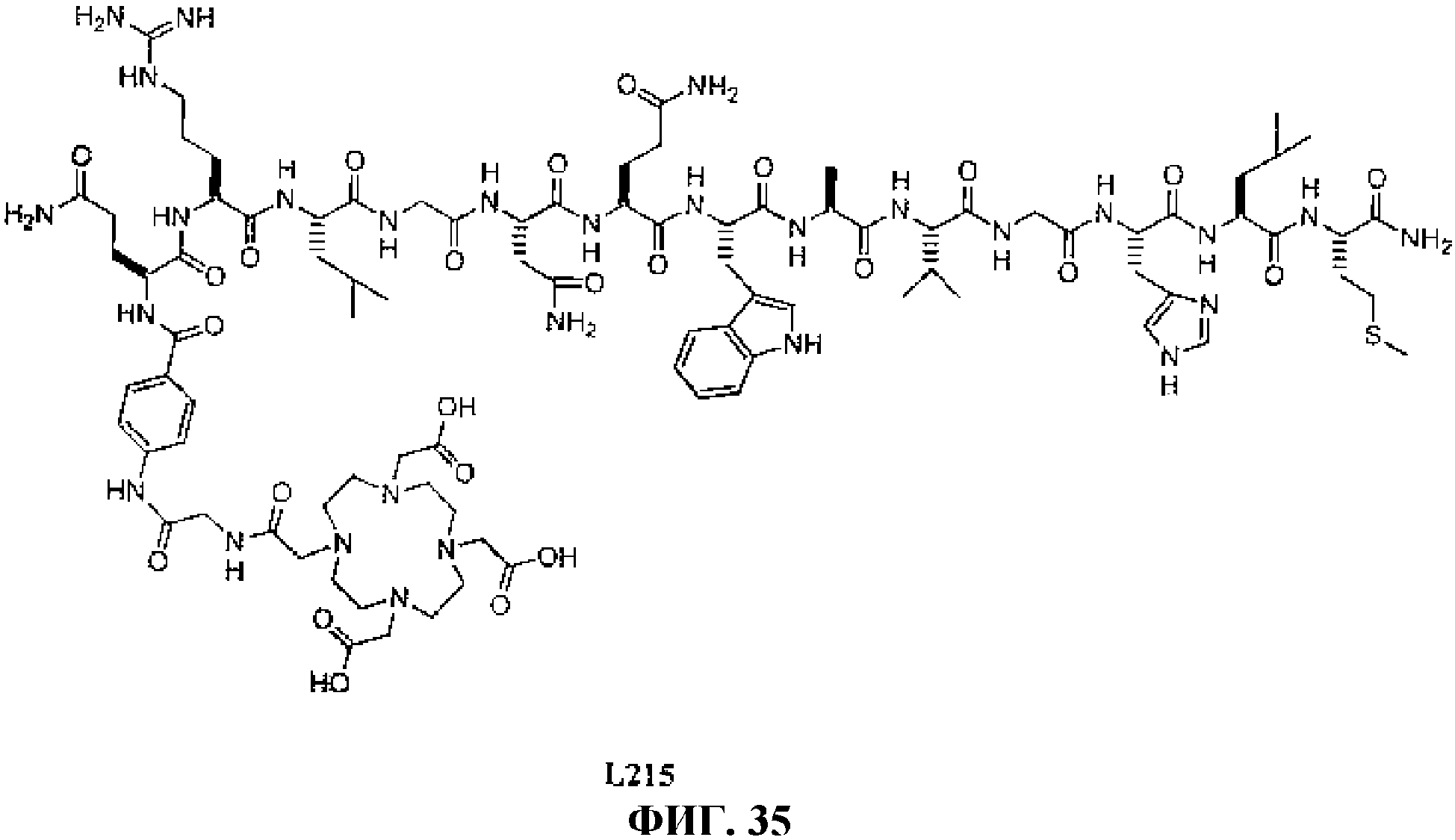
Фиг.35 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-аргинил-L-лейцилглицил-L-аспаргинил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L215.
Фиг.36 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминиларгинил-L-тирозинилглицил-L-аспаргинил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L216.
Фиг.37 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-лизил-L-тирозинилглицил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L217.
Фиг.38 представляет собой химическую структуру L218.
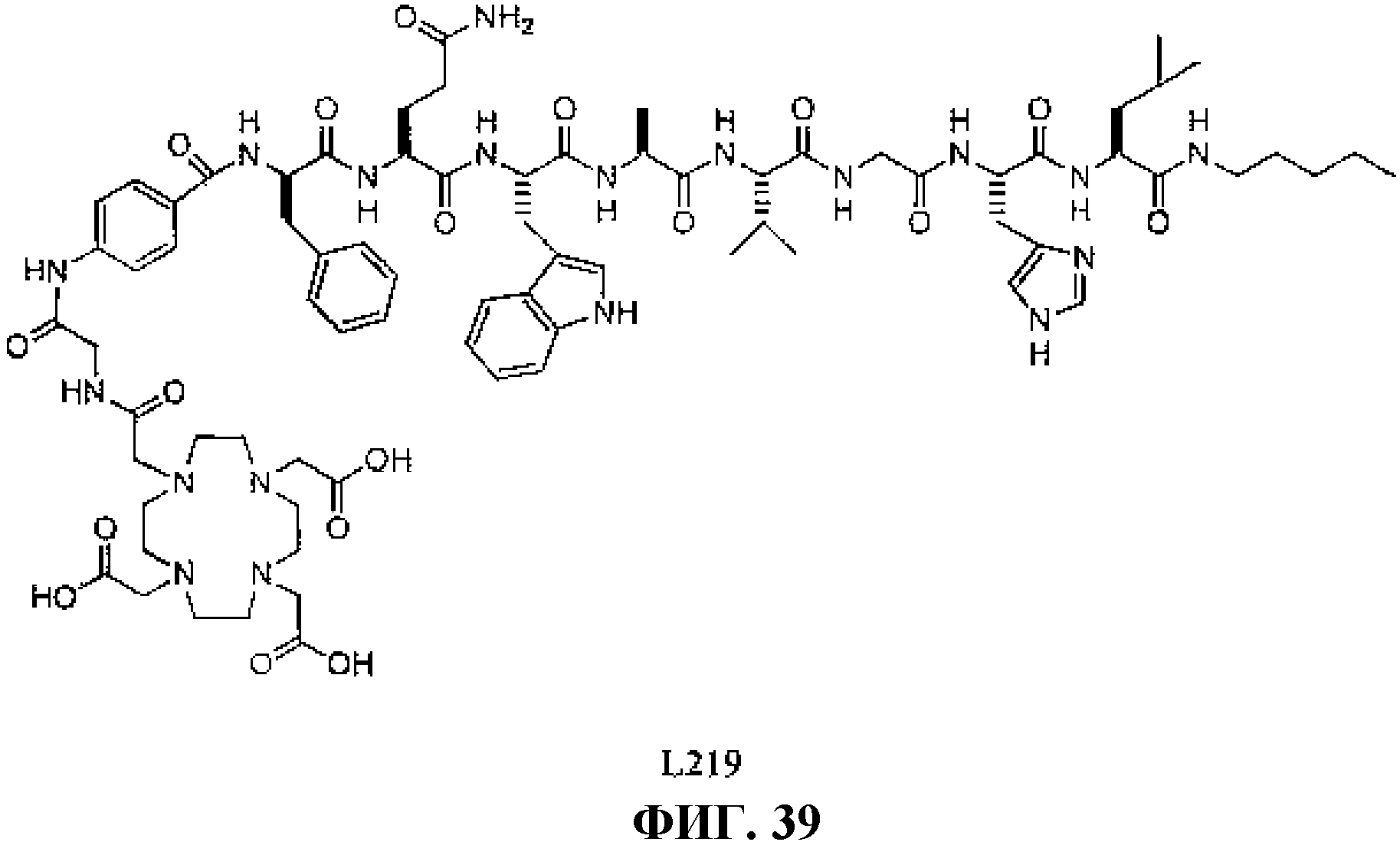
Фиг.39 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейциламинопентинила, L219 .
Фиг.40 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-серенил-L-валил-D-аланил-L-гистидил-L-лейцил-L-метионинамида, L220.
Фиг.41 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-лейцинамида, L221.
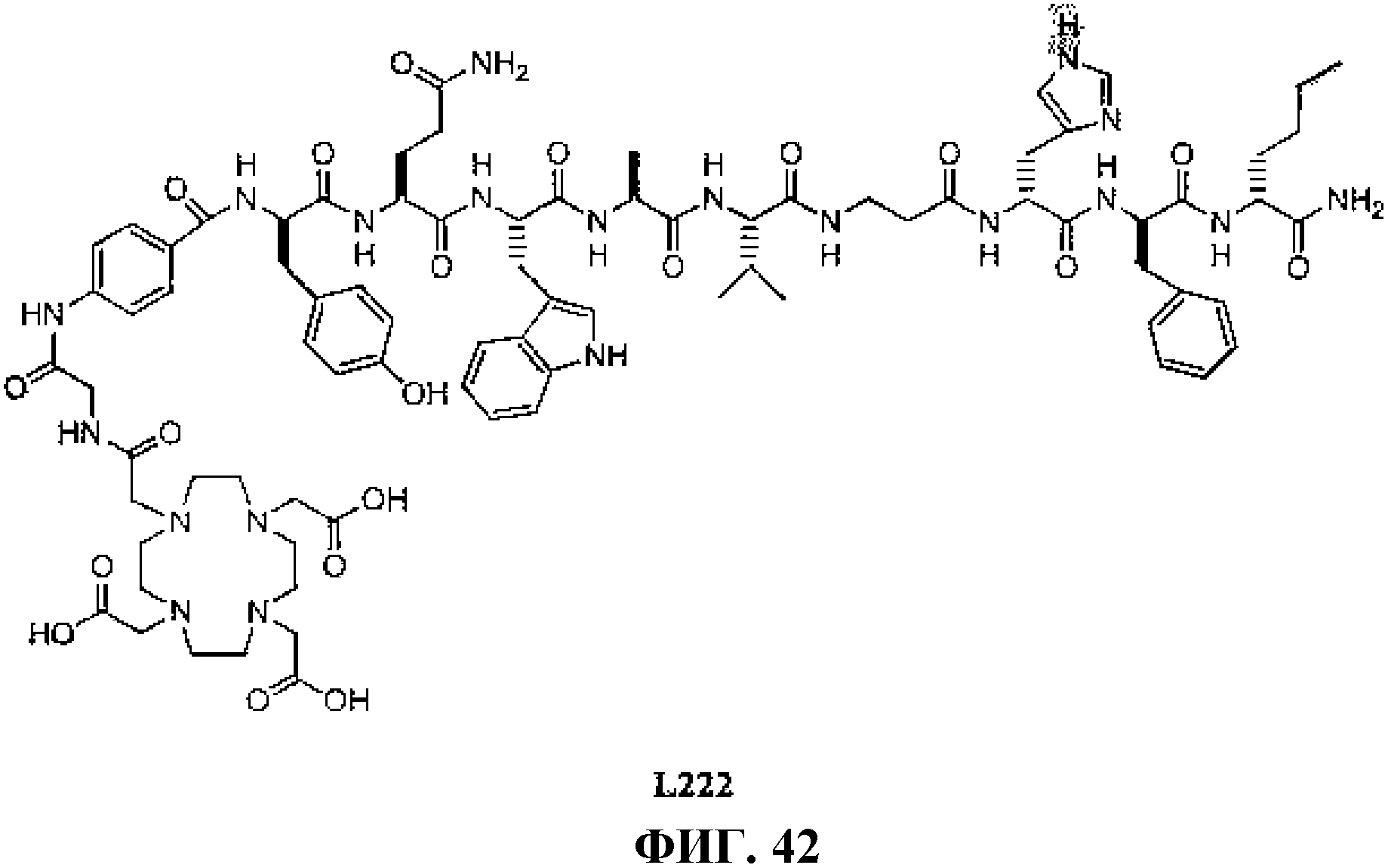
Фиг.42 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-тирозинил-L-глутаминил-L-триптофил-L-аланил-L-валилбетааланил-L-гистидил-L-фенилаланил-L-норлейцинамида, L222.
Фиг.43 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилбетааланил-L-гистидил-L-фенилаланил-L-норлейцинамида, L223.
Фиг.44 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланилглицил-L-гистидил-L-фенилаланил-L-лейцинамида, L224.
Фиг.45 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-лейцил-L-триптофил-L-аланил-L-валинилглицил-L-серинил-L-фенилаланил-L-метионинамида, L225.
Фиг.46 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-гистидил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида, L226.
Фиг.47 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-лейцил-L-триптофил-L-аланил-L-валилглицил-L-серинил-L-фенилаланил-L-метионинамида, L227.
Фиг.48 представляет собой химическую структуру N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-фенилаланил-L-метионинамида, L228.
Фиг.49 представляет собой графическое изображение реакции для синтеза (3β,5β,7α,12α)-3-(9Н-флуорен-9-илметокси)амино-7,12-дигидроксохолан-24-овой кислоты (В), как описано в примере LVII.
Фиг.50 представляет собой графическое изображение реакции для синтеза N-(3β,5β,7α,12α)-3-[[[2-[2-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]этокси]этокси]ацетил]амино]-7,12-дигидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L69), как описано в примере LVII.
Фиг.51 представляет собой графическое изображение реакции для синтеза N-[4-[2-гидрокси-3-[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]пропокси]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L144), как описано в примере LVIII.
Подробное описание изобретения
В следующем описании рассмотрены различные аспекты настоящего изобретения. В целях объяснения представлены определенные конфигурации и детали для обеспечения глубокого понимания настоящего изобретения. Однако специалисту в данной области должно быть очевидно, что настоящее изобретение можно осуществить без определенных деталей. Кроме того, хорошо известные признаки можно опустить или упростить для того, чтобы не запутывать настоящее изобретение.
В одном варианте осуществления настоящего изобретения предоставлены новые улучшенные соединения для применения в диагностической визуализации или лучевой терапии. Соединения включают оптическую метку или химическую часть, способную образовывать комплексы с используемым в медицине ионом металла или радионуклидом (агентом, хелатирующим металл), присоединенным к пептиду для целенаправленной доставки к рецептору GRP линкерной или спейсерной группой.
В целом, соединения настоящего изобретения могут иметь формулу:
M-N-O-P-G,
где М представляет собой агент, хелатирующий металл (в форме комплекса с радионуклидом или в форме, не образующей комплекс с металлическим радионуклидом) или оптическую метку, N-O-P представляет собой линкер, и G представляет собой пептид для целенаправленной доставки к рецептору GPR. Каждый агент, хелатирующий металл, оптическая метка, линкер и пептид для целенаправленной доставки к рецептору GPR, описан в следующем обсуждении.
В другом варианте осуществления настоящего изобретения предоставляется новая улучшенная линкерная или спейсерная группа, которая способна соединять оптическую метку или агент, хелатирующий металл, с пептидом для целенаправленной доставки к рецепторам GPR. В целом, линкеры настоящего изобретения могут иметь формулу:
N-O-P
где каждый из N, O и P определен во всем описании.
Было обнаружено, что соединения, отвечающие определенным в настоящем описании критериям, имеют улучшенные фармакокинетические свойства, по сравнению с другими пептидными конъюгатами для целенаправленной доставки к рецепторам GPR, известными в данной области. Например, соединения, содержащие линкеры настоящего изобретения, дольше удерживались в потоке крови и поэтому имели более длинный период полувыведения, чем ранее известные соединения. Более длинный период полувыведения был более благоприятен в медицинском отношении, потому что он обеспечивал возможность более целенаправленной доставки в опухоль, которую можно применять для диагностической визуализации и, особенно для терапевтических видов применения, при которых раковые клетки и опухоли получают большие количества пептидов, меченных радиоактивной меткой.
1А. Агент, хелатирующий металл
Термин "агент, хелатирующий металл" относится к молекуле, которая образует комплекс с атомом металла, где указанный комплекс стабилен в физиологических условиях. То есть металл остается в виде комплекса с каркасом агента, хелатирующего металл, in vivo. Конкретнее, агент, хелатирующий металл, представляет собой молекулу, которая образует комплексы с радионуклидным металлом, в форме комплекса с металлом, который стабилен в физиологических условиях, и который также имеет, по меньшей мере, одну реакционноспособную функциональную группу для конъюгации с линкером N-O-P. Агент М, хелатирующий металл, может представлять собой любой из агентов, хелатирующих металлы, которые, как известно в данной области, образуют комплексы с используемым в медицине ионом металла или радионуклидом. Агент, хелатирующий металл, может находиться или не находиться в форме комплекса с металлическим радионуклидом. Кроме того, агент, хелатирующий металл, может включать необязательный спейсер, такой, например, как одиночная аминокислота (например, глицин), которая не образует комплекс с металлом, но физически разделяет агент, хелатирующий металл, и линкер.
Агенты, хелатирующие металлы, по изобретению могут включать, например, линейные, макроциклические, терпиридиновые и N3S, N2S2 или N4 хелатирующие соединения (см. также патенты США №№ 5367080, 5364613, 5021556, 5075099, 5886142, описания которых во всей своей полноте включены в данное описание в качестве ссылки), и другие образующие хелаты соединения, известные в данной области, включая, но, не ограничиваясь ими, HYNIC, DPTA, EDTA, DOTA, TETA и бисаминобистиоловые (ВАТ) хелатирующие соединения (см. также патент США № 5720934). Например, хелатирующие N4 соединения описаны в патентах США №№ 6143274, 6093382, 5608110, 5665329, 5656254 и 5688487, описания которых во всей своей полноте включены в данное описание в качестве ссылки. Определенные хелатирующие N2S2 соединения описаны в РСТ/CA94/00395, РСТ/CA94/00479, РСТ/CA95/00249 и в патентах США №№ 5662885, 5976495 и 5780006, описания которых во всей своей полноте включены в данное описание в качестве ссылки. Хелатирующее соединение может также включать производные хелатирующего лиганда меркапто-ацетилглицилглицилглицина (MAG3), который содержит системы N3S и N2S2, такие как МАМА (моноамидмоноаминдитиолы), DADS (N2S диаминдитиолы), CODADS и подобные соединения. Эти системы лигандов и разнообразные другие описаны Liu and Edwards, в Chem Rev. 1999, 99, 2235-2268 и приведенных там ссылках, описания которых во всей своей полноте включены в данное описание в качестве ссылки.
Агент, хелатирующий металл, может также включать комплексы, содержащие атомы лиганда, которые не являются донорными по отношению к металлу в тетрадентатном окружении. Они включают аддукты бороновой кислоты диоксомеров технеция и рения, такие как описано в патентах США №№ 5183653, 5387409 и 5118797, описания которых во всей своей полноте включены в данное описание в качестве ссылки.
Примеры предпочтительных хелатирующих соединений включают, но не ограничиваются ими, диэтилентриаминпентауксусную кислоту (DTPA), 1,4,7,10-тетраазациклотетрадекан-1,4,7,10-тетрауксусную кислоту (DOTA), 1-замещенную 1,4,7-трикарбоксиметил-1,4,7, 10-тетраазациклотетрадекантриуксусную кислоту (DO3A), этилендиаминтетрауксуную кислоту (EDTA), 4-карбонилметил-10-фосфонометил-1,4,7,10-тетраазациклотетрадекан-1,7-диуксусную кислоту (Cm4pm10d2a) и 1, 4,8,11-тетраазациклотетрадекан-1,4,8,11-тетрауксусную кислоту (ТЕТА). Дополнительные хелатирующие лиганды представляют собой этиленбис(2-гидроксифенилглицин) (EHPG) и его производные, включая 5-Cl-EHPG, 5-Br-EHPG, 5-Me-EHPG, 5-трет-Bu-EHPG и 5-втор-Bu-EHPG; бензодиэтилентриаминпентауксусную кислоту (бензо-DTPA) и ее производные, включая дибензо-DTPA, фенил-DTPA, дифенил-DTPA, бензил-DTPA и дибензил-DTPA; бис-2-(гидроксибензил)этилендиаминдиуксусную кислоту (HBED) и ее производные; класс макроциклических соединений, которые содержат, по меньшей мере, 3 атома углерода, предпочтительно, по меньшей мере, 6 и, по меньшей мере, 2 гетероатома (О и/или N), где макроциклические соединения могут состоять из одного кольца, или двух или трех колец, соединенных вместе у гетерокольцевых элементов, например, бензо-DOTA, дибензо-DOTA и бензо-NOTA, где NOTA представляет собой 1,4,7-триазациклононан-N,N',N"-триуксусную кислоту, бензо-ТЕТА, бензо-DOTMA, где DOTMA представляет собой 1,4,7, 10-тетраазациклотетрадекан-1,4,7,10-тетра(метилтетрауксусную кислоту), и бензо-ТЕТМА, где ТЕТМА представляет собой 1,4,8,11-тетраазациклотетрадекан-1,4,8,11-(метилтетрауксусную кислоту); производные 1, 3-пропилендиаминтетрауксусной кислоты (PDTA) и триэтилентетраамингексауксусной кислоты (ТТНА); производные 1,5,10-N,N',N"-трис(2,3-дигидроксибензоил)трикатехолата (LICAM) и 1,3,5-N,N',N"-трис(2, 3-дигидроксибензоил)аминометилбензола (МЕСАМ). Примеры типичных хелатообразующих соединений и хелатообразующих групп, предусмотренных настоящим изобретением, описаны в WO98/18496, WO86/1849606605, WO91/03200, WO95/28179, WO96/23526, WO97/36619, PCT/US98/01473, PCT/US98/20182 и US 4899755, U.S. 5474756, U.S. 5846519 и U.S. 6143274, каждый из которых во всей своей полноте включен в данное описание в качестве ссылки.
В частности, предпочтительные агенты, хелатирующие металлы, включают соединения формулы 1, 2 и 3 (для111In и радиоактивных лантанидов, таких как, например,177Lu,90Y,153Sm и166Ho) и соединения формулы 4, 5 и 6 (для радиоактивного99mTc,186Re и188Re), представленные ниже. Эти и другие группы, хелатирующие металлы, описаны в патентах США №№ 6093382 и 5608110, которые во всей своей полноте включены в данное описание в качестве ссылки. Кроме того, хелатообразующая группа формулы 3 описана, например, в патенте США № 6143274; хелатообразующая группа формулы 5 описана, например, в патентах США №№ 5627286 и 6093382, и хелатообразующая группа формулы 6 описана, например, в патентах США №№ 5662885, 5780006 и 5976495, включенных в данное описание в качестве ссылки. Конкретные хелатирующие металлы соединения формулы 6 включают N, N-диметилGly-Ser-Cys; N,N-диметилGly-Thr-Cys; N,N-диэтилGly-Ser-Cys; N,N-дибензилGly-Ser-Cys и их другие варианты. Например, спейсеры, которые в действительности не образуют комплекс с металлическим радионуклидом, такие как дополнительная одиночная аминокислота Gly, могут быть присоединены к этим хелатирующим металлы агентам (например, N,N-диметилGly-Ser-Cys-Gly; N,N-диметилGly-Thr-Cys-Gly; N, N-диэтилGly-Ser-Cys-Gly; N,N-дибензилGly-Ser-Cys-Gly). Другие полезные хелатирующие металлы агенты, такие как описано в патенте США № 6334996, также включенном в данное описание в качестве ссылки (например, диметилгли-L-трет-бутилгли-L-Cys-Gly; диметилгли-D-трет-бутилгли-L-Cys-Gly; диметилгли-L-трет-бутилгли-L-Cys и т.д.).
Кроме того, защищающие серу группы, такие как Acm (ацетамидометил), тритил или другие известные алкильные, арильные, ацильные, алканоильные, арилоильные, меркаптоацильные и органотиольные группы могут быть присоединены к цистеинаминокислоте этих агентов, хелатирующих металлы.
Кроме того, другие полезные агенты, хелатирующие металлы, включают:


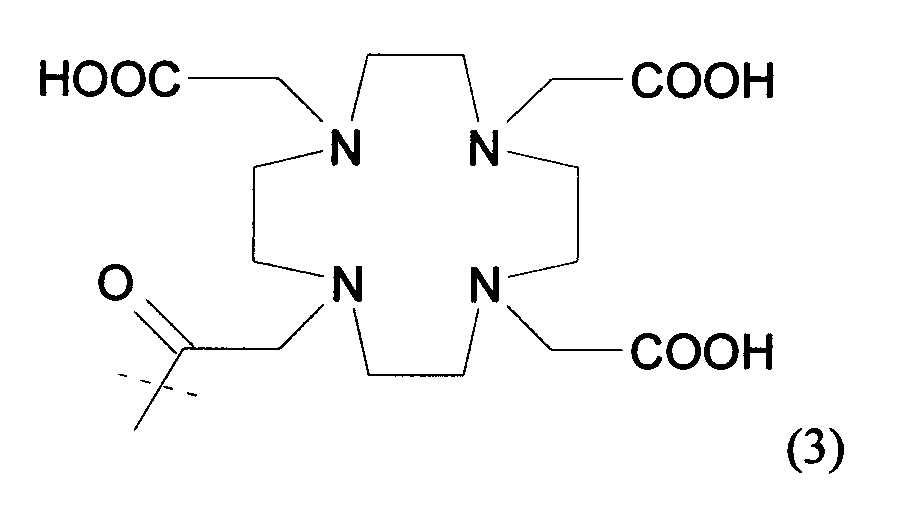




В представленных выше формулах 1 и 2 R представляет собой алкил, предпочтительно метил. В указанных выше формулах 5а и 5b X представляет собой или CH2, или О; Y представляет собой разветвленный или неразветвленный С1-С10алкил; арил, арилокси, ариламино, ариламиноацил; арилакил, где алкильная группа или группы, присоединенные к арильной группе, представляют собой разветвленные или неразветвленные С1-С10алкильные группы, разветвленные или неразветвленные С1-С10гидрокси- или полигидроксиалкильные группы, или полиалкоксиалкильные, или полигидроксиполиалкоксиалкильные группы; J является необязательной, но если она присутствует, то представляет собой C(=O)-, OC(=O)-, SO2-, NC(=O)-, NC(=S)-, N(Y), NC(=NCH3)-, NC(=NH)-, N=N-, гомополиамиды или гетерополиамины, полученные из синтетических или встречающихся в природе аминокислот; где всюду n=1-100. Другие варианты этих структур описаны, например, в патенте США № 6093382. В формуле 6 группа S-NHCOCH3 может быть заменена SH или S-Z, где Z представляет собой любую из известных защищающих серу групп, таких как группы, описанные выше. Формула 7 иллюстрирует один вариант трет-бутильных соединений, которые можно использовать в качестве агентов, хелатирующих металл. Описания каждого из указанных выше патентов, заявок и ссылок во всей своей полноте включены в настоящее описание в качестве ссылки.
В предпочтительном варианте осуществления агент, хелатирующий металл, включает циклические или ациклические полиаминокарбоновые кислоты, такие как DOTA (1,4,7,10-тетраазациклотетрадекан-1,4,7, 10-тетрауксусная кислота), DPTA (диэтилентриаминпентауксусная кислота), DPTA-бисметиламид, DTPA-бисморфолинамид, Cm4pm10d2a (4-карбонилметил-10-фосфонометил-1,4,7,10-тетраазациклотетрадекан-1, 7-диуксусная кислота), DO3A N-[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил, HP-DO3A, DO3A-моноамид и его производные.
Предпочтительные металлические радионуклиды для сцинтиграфии или лучевой терапии включают

и их оксиды или нитриды. Выбор металла определяется на основании желаемого терапевтического или диагностического применения. Например, для диагностических целей (например, диагностики и мониторинга результатов лечения при первичных опухолях и метастазах), предпочтительные радионуклиды включают64Cu,67Ga,68Ga,99mTc и111In, где особенно предпочтительны99mTc и111In. Для терапевтических целей (например, для обеспечения лучевой терапии по поводу первичных опухолей и метастазов, связанных с раковыми опухолями предстательной железы, молочной железы, легких и т.д.), предпочтительные радионуклеотиды включают
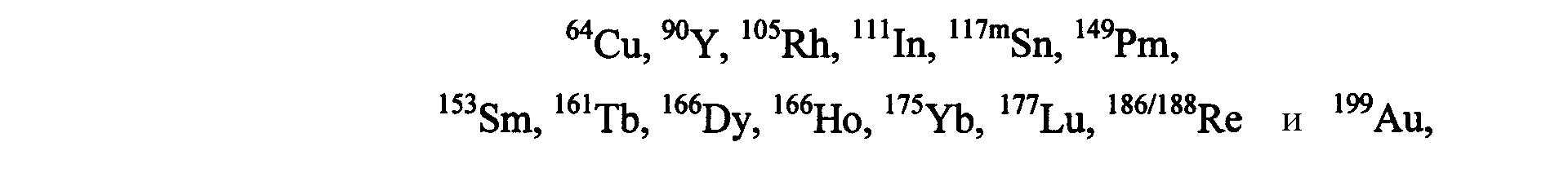
где особенно предпочтительны177Lu и90Y.99mTc особенно полезен и предпочтителен для диагностического радионуклида, благодаря его низкой стоимости, доступности, визуализирующих свойств и высокой удельной активности. Ядерные и радиоактивные свойства99mTc делают этот изотоп идеальным средством сцинтиграфической визуализации. Этот изотоп имеет энергию одиночного фотона 140 кэВ и период радиоактивного полураспада примерно 6 ч и легко доступен из генератора99Mo-99mTc. Например, пептид, меченный99mTc, можно использовать для диагностики и мониторинга хода лечения при первичных опухолях и метастазах. Пептиды, меченные177Lu,90Y или другими терапевтическими радионуклидами, можно использовать для обеспечения лучевой терапии по поводу первичных опухолей и метастазов, связанных с раковыми опухолями предстательной железы, молочных желез, легких и т.д.
В иллюстративном варианте реализации соединения изобретения могут быть конъюгированы с фотометками, такими как оптические красители, включая органические хромофоры или флуорофоры, имеющие обширные, делокализованные кольцевые системы и имеющие максимумы поглощения или эмиссии в диапазоне 400-1500 нм. Соединения изобретения могут быть альтернативно дериватизированы биолюминесцентной молекулой. Предпочтительный диапазон максимумов поглощения для фотометок составляет от 600 до 1000 нм для минимизации помехи сигнала от гемоглобина. Предпочтительно метки поглощения света имеют большие величины молярного коэффициента поглощения, например >105см-1М-1, при этом флюоресцентные оптические красители будут иметь высокие квантовые выходы. Примеры оптических красителей включают, но не ограничиваются ими, красители, описанные в WO/98/18497, WO/98/18496, WO/98/18495, WO/98/18498, WO/98/53857, WO/96/17628, WO/97/18841, WO/96/23524, WO/98/47538 и в приведенных там ссылках. Например, фотометки могут быть ковалентно связаны непосредственно с соединениями изобретения, такими как, например, соединения, состоящие из пептидов, обеспечивающих целенаправленную доставку к рецепторам GRP, и линкеры изобретения. Несколько красителей, которые поглощают и испускают свет в видимой или близкой к инфракрасной области электромагнитного спектра, в настоящее время используются для различных видов биомедицинского применения ввиду их биологической совместимости, высокого молярного коэффициента поглощения и/или высоких квантовых выходов. Высокая чувствительность оптической модальности в сочетании с красителями в качестве контрастирующих средств соответствует требованиям ядерной медицины и обеспечивает возможность визуализации органов и тканей без нежелательного эффекта ионизирующего излучения. Цианиновые красители с интенсивным поглощением и испусканием в близкой к инфракрасной (NIR) области особенно полезны, потому что биологические ткани оптически прозрачны в этой области. Например, индоцианиновый краситель, который поглощает и испускает в области NIR, использовался для мониторинга сердечного выброса, печеночной функции и печеночного кровотока, и его функционализированные производные использовались для конъюгации с биомолекулами для диагностических целей (R.B. Mujumdar, L.A. Ernst, S.R. Mujumdar, et al., Cyanine dye labeling reagents: Sulfoindocyanine succinimidyl esters. Bioconjugate Chemistry, 1993, 4(2), 105-111; Linda G. Lee and Sam L. Woo. "N-Heteroaromatic ion and iminium ion substituted cyanine dyes for use as fluorescent labels", U.S. Pat. No. 5453505; Eric Hohenschuh, et al. "Light imaging contrast agents", WO 98/48846; Jonathan Turner, et al. "Optical diagnostic agents for the diagnosis of neurodegenerative diseases by means of near infra-red radiation", WO 98/22146; Kai Licha, et al. "In-vivo diagnostic process by near infrared radiation", WO 96/17628; Robert A. Snow, et al., Compounds, WO 98/48838). Различные методики и реагенты визуализации описаны в патентах США №№ 6663847, 6656451, 6641798, 6485704, 6423547, 6395257, 6280703, 6277841, 6264920, 6264919, 6228344, 6217848, 6190641, 6183726, 6180087, 6180086, 6180085, 6013243 и опубликованных заявках на патенты США 2003185756,20031656432, 2003158127, 2003152577, 2003143159, 2003105300, 2003105299, 2003072763, 2003036538, 2003031627, 2003017164, 2002169107, 2002164287 и 2002156117.
2А. Линкеры, содержащие, по меньшей мере, одну не альфа-аминокислоту
В одном варианте осуществления изобретения линкер N-O-P содержит, по меньшей мере, одну не альфа-аминокислоту. Таким образом, в этом варианте линкером является N-O-P, где
N представляет собой 0 (где 0 означает отсутствие), альфа- или не альфа-аминокислоту или связывающую группу;
О представляет собой альфа- или не альфа-аминокислоту и
Р представляет собой 0, альфа- или не альфа-аминокислоту или другую связывающую группу,
где, по меньшей мере, один из N, O или Р представляет собой не альфа-аминокислоту.
Так, в одном примере N=Gly, O=не альфа-аминокислота и Р=0.
Альфа-аминокислоты хорошо известны в данной области и включают встречающиеся в природе и синтетические аминокислоты.
Не альфа-аминокислоты хорошо известны в данной области и включают встречающиеся в природе или синтетические не альфа-аминокислоты. Предпочтительные не альфа-аминокислоты включают:
8-амино-3,6-диоксаоктановую кислоту;
N-4-аминоэтил-N-1-уксусную кислоту и
производные полиэтиленгликоля, имеющие формулу NH2-(CH2CH2 O)n-CH2CH2H или NH2-(CH2CH2O)n-CH2CH2CO2H, где n=2-100.
Примеры соединений, имеющих формулу M-N-O-P-G, которые содержат линкеры, по меньшей мере, с одной не альфа-аминокислотой, перечислены в таблице 1. Эти соединения можно получить с использованием способов, описанных в настоящем описании, особенно, в разделе "Примеры", а также аналогичными способами, известными специалисту в данной области.
2В. Линкеры, содержащие, по меньшей мере, одну замещенную желчную кислоту
В другом варианте осуществления настоящего изобретения линкер N-O-P содержит, по меньшей мере, одну замещенную желчную кислоту. Так, в этом варианте осуществления линкера N-O-P
N представляет собой 0 (где 0 означает отсутствие), альфа-аминокислоту, замещенную желчную кислоту или связывающую группу;
О представляет собой альфа-аминокислоту или замещенную желчную кислоту; и
Р представляет собой 0, альфа-аминокислоту, замещенную желчную кислоту или другую связывающую группу,
где, по меньшей мере, один из N, O или Р представляет собой замещенную кислоту.
Желчные кислоты обнаруживаются в желчи (секреции печени) и представляют собой стероиды, имеющие гидроксильную группу и содержащую 5 атомов углерода боковую цепь, заканчивающуюся карбоксильной группой. В замещенных желчных кислотах, по меньшей мере, один атом, такой как атом водорода, желчной кислоты заменен другим атомом, молекулой или химической группой. Например, замещенные желчные кислоты включают кислоты, имеющие 3-амино, 24-карбоксильную функцию, необязательно замещенную в положениях 7 и 12 водородом, гидроксильной или функциональной кетогруппой.
Другие полезные замещенные желчные кислоты в настоящем изобретении включают замещенные холевые кислоты и их производные. Определенные производные замещенной холевой кислоты включают:
(3β,5β)-3-аминохолан-24-овую кислоту;
(3β,5β,12α)-3-амино-12-гидроксихолан-24-овую кислоту;
(3β,5β, 7α,12α)-3-амино-7,12-гидроксихолан-24-овую кислоту;
Lys-(3,6,9)-триоксаундекан-1,11-дикарбонил-3,7-дидеокси-3-аминохолевую кислоту);
(3β,5β,7α)-3-амино-7-гидрокси-12-оксохолан-24-овую кислоту и
(3β,5β,7α)-3-амино-7-гидроксихолан-24-овую кислоту.
Примеры соединений, имеющих формулу M-N-O-P-G, которые содержат линкеры, по меньшей мере, с одной замещенной желчной кислотой, перечислены в таблице 2. Эти соединения можно получить с использованием способов, описанных в настоящем описании, в частности, в разделе "Примеры", а также аналогичными способами, известными специалисту в данной области.
2С. Линкеры, содержащие, по меньшей мере, одну не альфа-аминокислоту с циклической группой
В еще одном варианте осуществления настоящего изобретения линкер N-O-P содержит, по меньшей мере, одну не альфа-аминокислоту с циклической группой. Таким образом, в этом варианте осуществления линкера N-O-P,
N представляет собой 0 (где 0 означает отсутствие), альфа-аминокислоту, не альфа-аминокислоту с циклической группой или другую связывающую группу;
О представляет собой альфа-аминокислоту или не альфа-аминокислоту с циклической группой; и
Р представляет собой 0, не альфа-аминокислоту с циклической группой или другую связывающую группу,
где, по меньшей мере, один из N, O или Р представляет собой не альфа-аминокислоту с циклической группой.
Не альфа-аминокислоты с циклической группой включают замещенный фенил, бифенил, циклогексил или другой амин и карбоксил, содержащий циклические алифатические или гетероциклические части. Примеры таких соединений включают:
4-аминобензойную кислоту,
3-аминобензойную кислоту,
4-аминометилбензойную кислоту,
4-аминооктановую кислоту,
транс-4-аминометилциклогексанкарбоновую кислоту,
4-(2-аминоэтокси)бензойную кислоту,
изонипекотовую кислоту,
2-аминометилбензойную кислоту,
4-амино-3-нитробензойную кислоту,
4-(3-карбоксиметил-2-кето-1-бензимидазоилпеперидин,
6-(пиперазин-1-ил)-4-(3Н)-хиназолинон-3-уксусную кислоту,
(2S,5S)-5-амино-1,2,4,5,6,7-гексагидроазепино[3, 21-hi]индол-4-он-2-карбоновую кислоту,
(4S,7R)-4-амино-6-аза-5-оксо-9-тиабицикло[4.3.0]нонан-7-карбоновую кислоту,
3-карбоксиметил-1-фенил-1,3,8-триазаспиро[4, 5]декан-4-он,
N1-пиперазинуксусную кислоту,
N-4-аминоэтил-N-1-пиперазинуксусную кислоту,
(3S)-3-амино-1-карбоксиметилкапролактам,
(2S,6S, 9)-6-амино-2-карбоксиметил-3,8-диазабицикло[4,3,0]нонан-1,4-дион,
3-амино-3-деоксихолевую кислоту,
4-гидроксибензойную кислоту, 4-аминофенилуксусную кислоту,
3-гидрокси-4-аминобензойную кислоту,
3-метил-4-аминобензойную кислоту,
3-хлор-4-аминобензойную кислоту,
3-метокси-4-аминобензойную кислоту,
6-аминонафтойную кислоту,
N,N'-бис(2-аминоэтил)аминоянтарную кислоту.
Примеры соединений, имеющих формулу M-N-O-P-G, которые содержат линкеры, по меньшей мере, с одной альфа-аминокислотой с циклической группой, перечислены в таблице 3. Эти соединения можно получить с использованием способов, описанных в настоящем описании, в частности, в разделе "Примеры", а также аналогичными способами, известными специалисту в данной области.
Подгруппа соединений, содержащих предпочтительные линкеры, связанные с амино(фенил, бифенил, циклоалкил или гетероциклическими)карбоксилатами и различными пептидами для целенаправленной доставки к рецепторам GRP, представлены в таблице 4. Эти соединения можно получить с использованием способов, раскрытых в настоящем описании, в частности, в разделе "Примеры", а также аналогичными способами, известными специалисту в данной области.
2D. Другие связывающие группы
Другие связывающие группы, которые можно использовать в пределах линкера N-O-P, включают химическую группу, которая служит для сочетания пептида для целенаправленной доставки к рецепторам GRP с хелатирующим металл агентом или с оптической меткой, в то же самое время, не воздействуя на функцию пептида для целенаправленной доставки к рецепторам GRP или функцию комплексообразования с металлами агента, хелатирующего металлы, или возможности детектирования оптической метки. Другие подходящие связывающие группы включают пептиды (т.е. аминокислоты, связанные вместе) как таковые, непептидную группу (например, углеводородную цепь) или комбинацию аминокислотной последовательности и непептидного спейсера.
В одном варианте осуществления другие связывающие группы для использования в пределах линкера N-O-P включают L-глутаминовые или углеводородные цепи или их комбинацию.
В другом варианте осуществления другие связывающие группы для использования в пределах линкера N-O-P включают чисто пептидную связывающую группу, состоящую из ряда аминокислот (например, диглицин, триглицин, gly-gly-glu, gly-ser-gly и т.д.), в котором общее количество атомов между N-концевым остатком пептида для целенаправленной доставки к рецепторам GRP и хелатирующим металл сочетанием или оптической меткой в полимерной цепи составляет ≤12 атомов.
В еще одном варианте осуществления другие связывающие группы для использования в пределах линкера N-O-P могут также включать углеводородную цепь [т.е. R1-(CH2)n-R2], где n=0-10, предпочтительно, n=3-9, R1 представляет собой группу (например, H2N-, HS-, -COOH), которую можно использовать в качестве сайта для ковалентного связывания каркаса лиганда или предварительно полученного агента, хелатирующего металл, или каркаса, образующего комплекс с металлом, или оптической метки; и R2 представляет собой группу, которая используется для ковалентного связывания с N-концом NH2-группы пептида для целенаправленной доставки к рецепторам GRP (например, R2 представляет собой активированную группу СООН). В литературе хорошо описаны несколько химических способов конъюгации лигандов (т.е. хелатообразующих соединений) или предпочтительных хелатов металлов с биомолекулами [Wialbur, 1992; Parker, 1990; Hermanson, 1996; Frizberg et al., 1995]. Один или несколько из этих способов можно использовать для связывания не включенного в комплекс лиганда (хелатообразующего соединения), или хелата радиоактивного металла, или оптической метки с линкером, или для связывания линкера с пептидами для целенаправленной доставки к рецепторам GRP. Эти способы включают образование ангидридов кислот, альдегидов, арилизоцианатов, активированных сложных эфиров или N-гидроксисукцинимидов [Wilbur, 1992; Parker, 1990; Hermanson, 1996; Frizberg et al., 1995].
В предпочтительном варианте осуществления другие связывающие группы для использования в пределах линкера N-O-P могут быть образованы из предшественников линкеров, имеющих электрофилы или нуклеофилы, как представлено ниже:
LP1: предшественник линкера, имеющий, по меньшей мере, в двух участках линкера одинаковый электрофил Е1 или одинаковый нуклеофил Nu1;
LP2: предшественник линкера, имеющий электрофил Е1 и в другом участке линкера другой электрофил Е2;
LP3: предшественник линкера, имеющий нуклеофил Nu1 и в другом участке линкера другой нуклеофил Nu2; или
LP4: предшественник линкера, имеющий один конец, функционализированный электрофилом Е1, и другой - нуклеофилом Nu1.
Предпочтительные нуклеофилы Nu1/Nu2 включают -OH, -NH, -NR, -SH, -HN-NH2, -RN-NH2 и -RN-HNR', где R' и R независимо выбраны из приведенных выше определений для R, но R' не является Н.
Предпочтительные электрофилы E1/E2 включают -COOH, -CH=O (альдегид), -CR=OR' (кетон), -RN-C=S, -RN-C=O, -S-S-2-пиридил, -SO2-Y, -CH2 C(=O)Y и

где Y может быть выбран из следующих групп:


3. Пептид для целенаправленной доставки к рецептору GRP
Пептид для целенаправленной доставки к рецептору GRP (т.е. G в формуле M-N-O-P-G) представляет собой любой пептид, его эквивалент, производное или аналог, который имеет сродство связывания с семейством рецепторов GRP.
Пептид для целенаправленной доставки к рецептору GRP, может принимать форму агониста или антагониста. Известно, что агонист пептида для целенаправленной доставки к рецепторам GRP, "активирует" клетку после связывания с высоким сродством и может быть интернализован клеткой. Напротив, известно, что антагонисты пептида для целенаправленной доставки к рецепторам GRP, связываются только с рецептором GRP на клетке без интернализации клеткой и без "активации" клетки. В предпочтительном варианте осуществления пептид для целенаправленной доставки к рецептору GRP, представляет собой агонист.
В более предпочтительном варианте осуществления настоящего изобретения агонист GRP представляет собой аналог бомбезина (BBN) и/или его производное. Производное BBN или его аналог предпочтительно содержит или одну и ту же первичную структуру области связывания BBN (т.е. BBN(7-14) [SEQ ID NO:1]), или аналогичные первичные структуры с определенными аминокислотными замещениями, которые будут специфически связываться с рецепторами GRP со сродством связывания, лучшим или аналогичным одному BBN (т.е. Kd<25 нМ). Подходящие соединения включают пептиды, пептидомиметики и их аналоги и производные. Присутствие L-метионина (Met) в положении BBN-14 в целом будет придавать агонистические свойства, в то время как отсутствие этого остатка в BBN-14 в целом будет придавать антагонистические свойства [Hoffken, 1994]. Некоторые полезные аналоги бомбезина раскрыты в патентной публикации США 2003/0224998, во всей своей полноте, включенной в данное описание в качестве ссылки.
В данной области хорошо известно, что существует небольшое и селективное количество специфических аминокислотных замещений в области связывания BBN (8-14) (например, D-Ala11 на L-Gly11 или D-Trp8 на L-Trp8), которые можно произвести без уменьшения сродства связывания [Leban et al., 1994; Qin et al., 1994; Jensen et al., 19923]. Кроме того, присоединение некоторых аминокислотных цепей или других групп к N-концу аминогруппы в положении BBN-8 (т.е. остаток Trp8) может резко уменьшить сродство связывания аналогов BBN с рецепторами GRP [Davis et al., 1992; Hoffken, 1994; Moody et al., 1996; Coy, et al., 1988; Cai et al., 1994]. В небольшом числе случаев можно подвесить дополнительные аминокислоты или химические части без уменьшения сродства связывания.
Аналоги пептидов, обеспечивающих целенаправленную доставку к рецепторам BBN, включают молекулы, которые целенаправленны на доставку к рецепторам GRP, со сродством, которое больше или равно BBN, а также мутеины, ретропептиды и ретро-инверсо-пептиды GRP или BBN. Среднему специалисту будет очевидно, что эти аналоги могут также содержать модификации, которые включают замещения и/или удаления и/или добавления одной или нескольких аминокислот, пока эти модификации не вызывают негативного изменения биологической активности описанных в настоящем описании пептидов. Эти замещения можно проводить заменой одной или нескольких аминокислот их аминокислотными синонимами. Аминокислотные синонимы в пределах группы определены как аминокислоты, которые имеют физико-химические свойства, достаточные для обеспечения возможности замещения между членами группы для сохранения биологической функции молекулы.
Удаление или добавление аминокислот можно также произвести в определенных последовательностях при условии, что они не изменяют биологические функции указанных последовательностей. Предпочтительно такие добавления или удаления должны быть ограничены 1, 2, 3, 4 или 5 аминокислотами и не должны удалять или физически нарушать или сменять аминокислоты, которые имеют решающее значение для функциональной конформации. Мутеины пептидов, обеспечивающих целенаправленную доставку к рецепторам GRP, описанные в настоящем описании, могут иметь последовательность, гомологичную последовательности, раскрытой в настоящем описании, в которой аминокислотные замещения, удаления или добавления аминокислот присутствуют в одном или нескольких аминокислотных положениях. Мутеины могут иметь биологическую активность, которая составляет, по меньшей мере, 40%, предпочтительно по меньшей мере, 50%, предпочтительнее 60-70%, наиболее предпочтительно 80-90% описанных в настоящем описании пептидов. Однако они могут также иметь биологическую активность, большую, чем у конкретно проиллюстрированных пептидов, и, таким образом, необязательно должны быть идентичны биологической функции проиллюстрированных пептидов. Аналоги пептидов, обеспечивающих целенаправленную доставку к рецепторам GRP, также включают пептидомиметики или псевдопептиды, включающие изменения амидных связей пептидного каркаса, включая тиоамиды, метиленамины и Е-олефины. В используемый в настоящем описании термин "аналоги" включены также пептиды, основанные на структуре GRP, BBN или их пептидных аналогах с аминокислотами, замененными N-замещенными гидразинокарбонильными соединениями (также известные как азааминокислоты).
Пептид для целенаправленной доставки к рецептору GRP можно получить различными способами, в зависимости от выбранного хелатообразующего соединения. Пептид можно обычно удобно получать общепринятыми и известными в области синтеза пептидов методиками, такими как твердофазный пептидный синтез (SPPS). Твердофазный пептидный синтез (SPPS) включает ступенчатое добавление аминокислотных остатков к растущей пептидной цепи, которая связана с нерастворимой подложкой или матрицей, такой как полистирол. С-концевой остаток пептида сначала прикрепляется к коммерчески доступной подложке его аминогруппой, защищенной N-защитной группой, такой как трет-бутоксикарбонильная группа (Boc) или флуоренилметоксикарбонильная (Fmoc) группа. Аминозащитную группу удаляют подходящими снимающими защиту агентами, такими как TFA, в случае Boc или пиперидин для Fmoc, и добавляют следующий аминокислотный остаток (в N-защищенной форме) с агентом сочетания, таким как N,N'-дициклогексилкарбодиимид (DCC), или N,N'-диизопропилкарбодиимид (DIC), или гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU). После образования пептидной связи реагенты вымывают из подложки. После добавления конечного остатка пептид отщепляют от подложки подходящим реагентом, таким как трифторуксуная кислота (TFA) или фтористый водород (HF).
Затем может быть присоединен линкер для образования конъюгата взаимодействием свободной аминогруппы остатка Trp8 пептида для целенаправленной доставки к рецепторам GRP с соответствующей функциональной группой линкера. Так же весь конструкт хелатообразующего соединения, линкера и нацеливающей части, обсужденный выше, можно также собирать на смоле и затем расщеплять посредством подходящих реагентов, таких как трифторуксусная кислота или HF.
4. Метка и введение радиофармацевтических соединений
Включение металла внутрь радиофармацевтических конъюгатов можно достичь различными способами, общеизвестными в области координационной химии. Когда металл представляет собой99mTc, предпочтительный радионуклид для диагностической визуализации, можно использовать следующую общую методику для образования комплекса технеция. Раствор конъюгата пептида-хелатообразующего соединения образуется первоначальным растворением конъюгата в воде, разбавленной кислоте или водном растворе спирта, такого как этанол. Затем раствор необязательно дегазируют для удаления растворенного кислорода. Когда в пептиде присутствует группа -SH, для защиты тиола от окисления можно необязательно использовать группу, защищающую тиол, такую как Acm (ацетамидометил), тритил или другую группу, защищающую тиол. Группу (группы), защищающие тиол, удаляют подходящим реагентом, например, гироксид натрия, и затем нейтрализуют органической кислотой, такой как уксусная кислота (рН 6,0-6,5). Альтернативно группу, защищающую тиол, можно удалить in situ во время образования хелата технеция. На стадии метки пертехнат натрия, полученный из молибденового генератора, добавляют к раствору конъюгата с достаточным количеством восстанавливающего вещества, такого как хлорид двухвалентного олова, для восстановления технеция и выдерживают при комнатной температуре или нагревают. Меченый конъюгат можно отделить от примесей99mTcO4- и коллоидного99mTcO2 хроматографически, например, на картридже C-18 Sep Pak [Millipore Corporation, Waters Chromatography Division, 34 Maple Street, Milford, Massachusetts 01757] или ВЭЖХ с использованием методов, известных специалистам в данной области.
В альтернативном способе мечение можно осуществить реакцией перехелатирования. При этом способе источник технеция представляет собой раствор технеция, который восстановлен и образовал комплекс с лабильными лигандами перед взаимодействием с выбранным хелатообразующим соединением, способствуя обмену лигандов с выбранным хелатообразующим соединением. Примеры подходящих лигандов для перехелатирования включает тартрат, цитрат, глюконат и гептаглюконат. Следует понимать, что конъюгат можно метить с использованием описанных выше методик или альтернативно можно метить само хелатирующее соединение с последующим сочетанием с пептидом для образования конъюгата; способ именуется способом "предварительно меченого хелата". И Re, и Tc находятся в ряду VIIB периодической таблицы и являются химически однородными. Таким образом, по большей части, химия образования комплексов этих двух металлов с каркасами лигандов, которые проявляют высокую стабильность in vitro и in vivo, является одинаковой [Eckelman, 1995], и для мечения Re можно использовать одинаковые хелатообразующие соединения и методики. Многие комплексы99mTc или186/188Re, которые используются для образования стабильных комплексов радиоактивных металлов с пептидами и белками, образуют хелатный цикл с этими металлами в их состоянии окисления +5 [Lister-James et al., 1997]. Это состояние окисления обеспечивает возможность селективного помещения99mTc или186/188Re в каркасы лигандов, уже конъюгированных с биомолекулой, сконструированной из разнообразных слабых хелатов99m Tc(V) и/или186/188Re(V) (например,99mTc-глюкогептонат, цитрат, глюконат и т.д.) [Eckelman, 1995; Lister-James et al., 1997; Pollak et al., 1996].
5. Диагностические и терапевтические виды применения
При мечении диагностически и/или терапевтически полезными металлами или оптическими метками соединения настоящего изобретения можно применять для лечения и/или диагностики любой патологии, включающей избыточную экспрессию рецепторов GRP (или рецепторов NMB) методами, признанными в области радиологических диагностических средств, радиологических терапевтических средств и оптической визуализации (см., например, Bushbaum, 1995; Fischman et al., 1993; Schubiger et al., 1996; Lowbertz et al., 1994; Krenning et al., 1994; примеры оптических красителей включают, но не ограничиваются ими, красители, описанные в WO 98/18497, WO 98/18496, WO 98/18495, WO 98/18498, WO 98/53857, WO 96/17628, WO 97/18841, WO 96/23524, WO 98/47538 и приведенных там ссылках].
Экспрессия GRP-R характеризуется высокой стимулирующей регуляцией при разнообразных опухолях человека (см., например, WO 99/62563). Таким образом, соединения изобретения можно широко использовать при лечении и диагностике раковых опухолей, включая рак предстательной железы (первичный и метастатический), рак молочной железы (первичный и метастатический), рак толстой кишки, рак желудка, рак поджелудочной железы, немелкоклеточный рак легких, мелкоклеточный рак легких, гастриномы, меланомы, глиобластомы, нейробластомы, лейомиосаркоматозные опухоли матки, внутриэпителиальные неоплазии предстательной железы [PIN] и рак яичников. Кроме того, соединения изобретения можно использовать для дифференцировки состояний, при которых происходит стимулирующая регуляция рецепторов GRP, и состояний, при которых она не происходит (например, хронический панкреатит и карцинома протоков поджелудочной железы соответственно).
Соединения изобретения, которые, как подробнее объясняется в разделе "Примеры", демонстрируют более высокий захват в опухолях in vivo, чем соединения без новых линкеров, раскрытых в настоящем описании, проявляют улучшенную способность к целенаправленной доставке в опухоли, экспрессирующие рецепторы GRP, и, таким образом, визуализировать или доставлять средство лучевой терапии к этим тканям. Действительно, как показано в разделе "Примеры", лучевая терапия более эффективна (и время выживания увеличено) при использовании соединений изобретения.
Диагностическое применение этих соединений может использоваться в качестве первой линии диагностического скрининга для выявления присутствия неопластических клеток с использованием сцинтиграфической, оптической, сонолюминисцентной или фотоакустической визуализации, в качестве средства для целенаправленной доставки в неопластическую ткань с использованием ручных инструментов выявления излучения в области хирургии, управляемой радиоиммунными способами (RIGS), в качестве средства для получения дозиметрических данных перед введением радиотерапевтического соединения из подобранной пары и в качестве средства для оценки популяции рецепторов, как функции лечения с течением времени.
Терапевтическое применение этих соединений можно определить как средство, которое должно использоваться в качестве первой линии терапии при лечении рака, в качестве комбинированного лечения, при котором эти средства можно использовать в сочетании с адъювантной химиотерапией и/или в качестве терапевтического средства из подобранной пары. Концепция подобранной пары относится к одному не металлированному соединению, которое может служить в качестве как диагностического, так и терапевтического средства, в зависимости от радиоактивного металла, который был выбран для связывания с соответствующим хелатом. Если хелатообразующее соединение не может принять требуемые металлы, можно произвести соответствующие замещения для принятия другого металла, в то же самое время сохраняя фармакологию, так что поведение диагностического соединения in vivo можно использовать для прогнозирования поведения радиотерапевтического соединения. При использовании в сочетании с адъювантной химиотерапией можно использовать любое подходящее химиотерапевтическое средство, включая, например, антинеопластические средства, такие как соединения платины (например, спироплатин, цисплатин и карбоплатин), метотрексат, адриамицин, митомицин, ансамитоцин, блеомицин, цитозин, арабинозид, арабинозиладенин, меркаптополилизин, винкристин, бусульфан, хлорамбуцил, мелфалан (например, PAM, a, L-PAM или фенилаланиниприт), меркаптопурин, митотан, гидрохлорид прокарбазина, дактиномицин (актиномицин D), гидрохлорид даунорубицина, гидрохлорид доксорубицина, таксол, митомицин, пликамицин (митрамицин), аминоглутетимид, эстрамустинфосфат натрия, флутамид, ацетат лейпролида, ацетат мегестрола, цитрат тамоксифена, тестолактон, трилостан, амсакрин (m-AMSA), аспарагиназа (L-аспарагиназа) Erwina aparaginase, этопозид-(VP-16), интерферон α-2а, интерферон α-2b, тенипозид (VM-26), сульфат винбластина (VLB) и арабинозил. В определенном варианте осуществления терапевтическое средство может представлять собой моноклональное антитело, такое как моноклональное антитело, способное связываться с антигеном меланомы.
Конъюгат, меченный радионуклидным металлом, таким как99mTc, можно вводить млекопитающему, включая людей-пациентов или индивидуумов, внутривенной, подкожной или внутрибрюшинной инъекцией в фармацевтически приемлемом носителе и/или растворе, таком как солевые растворы, подобные изотоническому солевому раствору. Меченные радиоактивной меткой средства сцинтиграфической визуализации, предоставляемые настоящим изобретением, поставляются имеющими подходящее количество радиоактивности. При образовании радиоактивных комплексов99mTc обычно предпочтительно образовывать радиоактивные комплексы в растворах, содержащих радиоактивность в концентрациях от около 0,01 милликюри (мКи) до 100 мКи на 1 мл. Обычно стандартная доза, которую предстоит ввести, имеет радиоактивность от примерно 0,01 мКи до примерно 100 мКи, предпочтительно от 1 мКи до 30 мКи. Стандартная дозировка раствора, который предстоит инъецировать составляет от около 0,01 мл до около 10 мл. Количество меченого конъюгата, целесообразное для введения, зависит от профиля распределения выбранного конъюгата в том смысле, что быстро выводимый конъюгат может потребовать введения в более высоких дозах, чем тот, который выводится медленнее. Распределение и локализацию in vivo можно проследить стандартными сцинтиграфическими методиками в соответствующее время после введения; обычно от 30 мин до 180 мин, в зависимости от скорости аккумулирования в участке-мишени в отношении скорости выведения в ткани, не являющейся мишенью. Например, после инъекции меченных диагностическими радионуклидами соединений изобретения пациенту для визуализации областей захвата средства и количественной оценки количества радиоактивности, присутствующей в участке, можно использовать гамма-камеру, откалиброванную по энергии гамма-лучей нуклида, включенного в визуализирующее средство. Визуализация участка in vivo может происходить через несколько минут. Однако при желании визуализация может происходить в течение часов или даже дольше, после того как пептид, меченный радиоактивной меткой, инъецирован пациенту. В большинстве случаев достаточное количество введенной дозы будет накапливаться в области, которую предстоит визуализировать в пределах примерно 0,1 ч для обеспечения возможности снять сцинтифотографии.
Соединения настоящего изобретения можно вводить пациенту отдельно или в виде части композиции, которая содержит другие компоненты, такие как эксципиенты, разбавители, нейтрализаторы радикалов, стабилизаторы и носители, хорошо известные в данной области. Соединения можно вводить пациентам или внутривенно, или внутрибрюшинно.
Имеется несколько преимуществ, связанных с настоящим изобретением. Соединения, полученные в соответствии с настоящим изобретением, образуют стабильные, хорошо дифференцированные соединения, меченные99mTc или186/188Re. Аналогичные соединения изобретения можно также получить, используя соответствующие каркасы хелатообразующих соединений для соответствующих радиоактивных металлов для образования стабильных, хорошо дифференцированных продуктов, меченных153Sm,90Y,166Ho,105Rh,199Au,149Pm,177Lu,111In или другим радиоактивным металлом. Меченные радиоактивной меткой пептиды для целенаправленной доставки к рецепторам GRP селективно связываются с неопластическими клетками, экспрессирующими рецепторы GRP, и если используется агонист, становятся интернализованными и удерживаются в опухолевых клетках в течение продолжительного периода времени. Радиоактивный материал, который не достигает (т.е. не связывается) раковых клеток, преимущественно эффективно экскретируется в мочу с минимальным удерживанием радиоактивного метала в почках.
6. Оптическая визуализация, сонолюминесценция, фотоакустическая визуализация и фототерапия
В соответствии с настоящим изобретением для определения локализации мишени со световой визуализацией in vivo после инъекции индивидууму с оптически меченным соединением изобретения можно использовать ряд оптических параметров. Оптические параметры, которые предстоит выявить при получении изображения, могут включать переданное излучение, поглощение, флюоресцентное или фосфоресцирующее испускание, отражение света, изменения амплитуды или максимумов спектра поглощения и упругорассеянное излучение. Например, биологическая ткань относительно прозрачна для света в диапазоне длины волн, близком к инфракрасному спектру (NIR), 650-1000 нм. Излучение NIR может проникать в ткань на глубину до нескольких сантиметров, обеспечивая возможность использования соединений настоящего изобретения для визуализации содержащей мишень ткани in vivo. Использование видимого и близкого к инфракрасному (NIR) света в клинической практике быстро растет. Соединения, поглощающие или испускающие свет в видимой, NIR, или длинноволновой (UV-A, >350 нм) области электромагнитного спектра, можно использовать для оптической томографической визуализации, эндоскопической визуализации и фототерапии.
Большое преимущество биомедицинской оптики заключается в ее терапевтическом потенциале. Было показано, что фототерапия является безопасным и эффективным методом лечения различных поверхностных поражений, как наружных, так и внутренних. Красители важны для усиления выявления сигнала и/или фотосенсибилизации тканей при оптической визуализации и фототерапии. Предыдущие исследования показали, что определенные красители могут локализоваться в опухолях и служить в качестве мощного зонда для диагностики и лечения мелких раковых опухолей (D.A. Bellnier et al., Murine pharmacokinetics and antitumor efficacy of the photodynamic sensitizer 2-[l-hexyloxyethyl]-2-devinyl pyropheophorbide-a, J. Photochem. Photobiol., 1993, 20, pp. 55-61; G.A. Wagnieres et al., In vivo fluorescence spectroscopy and imaging for oncological applications, Photochem. Photobiol., 1998, 68, pp. 603-632; J.S. Reynolds et al., Imaging of spontaneous canine mammary tumors using fluorescent contrast agents, Photochem. Photobiol., 1999, 70, pp. 87-94). Однако эти красители не локализуются предпочтительно в злокачественных тканях.
В иллюстративном варианте осуществления соединения изобретения могут быть конъюгированы с фотометками, такими как оптические красители, включая органические хромофоры или флуорофоры, имеющие значительные по размеру делокализованные кольцевые системы и имеющие максимумы поглощения или эмиссии в диапазоне от 400 до 1500 нм. Соединения изобретения могут альтернативно быть дериватизированы биолюминесцентной молекулой. Предпочтительный диапазон максимумов поглощения для фотометок находится в диапазоне от 600 до 1000 нм для минимизации помех сигнала от гемоглобина. Предпочтительно светопоглощающие метки имеют большие величины молярного коэффициента поглощения, например >105/см/М, в то время как флюоресцентные оптические красители должны иметь высокие квантовые выходы. Примеры оптических красителей включают, но не ограничиваются ими, красители, описанные в WO 98/18497, WO 98/18496, WO 98/18495, WO 98/18498, WO 98/53857, WO 96/17628, WO 97/18841, WO 96/23524, WO 98/47538 и в приведенных там ссылках. Например, фотометки могут быть ковалентно связаны непосредственно с соединениями изобретения, такими как, например, соединения, содержащие пептиды для целенаправленной доставки к рецепторам GRP и линкеры изобретения. Несколько красителей, которые поглощают и испускают свет в видимой или близкой к инфракрасной области электромагнитного спектра, используются в настоящее время для различных биомедицинских видов применения, ввиду их биологической совместимости, высокого молярного коэффициента поглощения и/или высоких квантовых выходов флуоресценции. Высокая чувствительность оптической модальности в сочетании с красителями в качестве контрастных средств соответствует требованиям ядерной медицины и обеспечивает возможность визуализации органов и тканей без нежелательного эффекта ионизирующего излучения. Цианиновые красители с интенсивным поглощением и испусканием в близкой к инфракрасной (NIR) особенно полезны, потому что биологические ткани оптически прозрачны в этой области (B.C. Wilson, Optical properties of tissues. Encyclopedia of Human Biology, 1991, 5, 587-597). Например, индоцианин зеленый, который поглощает и испускает в области NIR, используется для мониторинга сердечного выброса, функций печени и печеночного кровотока (Y-L. He, H. Tanigami, H. Ueyama, T. Mashimo, and I. Yoshiya, Measurement of blood volume using indocyanine green measured with pulse-spectrometry: Its reproducibility and reliability. Critical Care Medicine, 1998, 26(8), 1446-1451; J. Caesar, S. Shaldon, L. Chiandussi, et al., The use of Indocyanine green in the measurement of hepatic blood flow and as a test of hepatic function. Clin. Sci. 1961, 21, 43-57), а его функционализированные производные используются для конъюгации с биомолекулами для диагностических целей (R.B. Mujumdar, L.A. Ernst, S.R. Mujumdar, et al., Cyanine dye labeling reagents: Sulfoindocyanine succinimidyl esters. Bioconjugate Chemistry, 1993, 4(2), 105-111; Linda G. Lee and Sam L. Woo. "N-Heteroaromatic ion and iminium ion substituted cyanine dyes for use as fluorescent labels", U.S. Pat. No. 5,453,505; Eric Hohenschuh, et al. "Light imaging contrast agents", WO 98/48846; Jonathan Turner, et al. "Optical diagnostic agents for the diagnosis of neurodegenerative diseases by means of near infra-red radiation", WO 98/22146; Kai Licha, et al. "In-vivo diagnostic process by near infrared radiation", WO 96/17628; Robert A. Snow, et al., Compounds, WO 98/48838).
После инъекции оптически меченного соединения производят сканирование тела пациента одним или несколькими источниками света (например, лазером) в диапазоне длин волн, соответствующем фотометке, используемой в средстве. Используемый свет может быть монохроматическим или полихроматическим и постоянным или пульсирующим. Переданный, рассеянный или отраженный свет выявляется фотодетектором, настроенным на одну или несколько длин волн, для определения локализации ткани, содержащей мишень (например, ткани, содержащей GRP) у индивидуума. Изменения оптического показателя можно контролировать в течение времени для выявления накопления оптически меченного реагента в участке-мишени (например, опухоли или другом участке с рецепторами GRP). Стандартные устройства переработки изображения и выявления можно использовать в сочетании с оптическими визуализирующими реагентами настоящего изобретения.
Описанные выше оптические визуализирующие реагенты можно также использовать для акустически-оптической или сонолюминесцентной визуализации, выполняемой оптически меченными визуализирующими средствами (см. патент США № 5171298, WO 98/57666 и приведенных там ссылках). При акустически-оптической визуализации ультразвуковое излучение подается на индивидуума и воздействует на оптические параметры переданного, испускаемого или отраженного света. При сонолюминесцентной визуализации подаваемый ультразвук в действительности генерирует выявляемый свет. Походящие способы визуализации с использованием таких методик описаны в WO 98/57666.
Различные методики и реагенты визуализации описаны в патентах США №№ 6663847, 6656451, 6641798, 6485704, 6423547, 6395257, 6280703, 6277841, 6264920, 6264919, 6228344, 6217848, 6190641, 6183726, 6180087, 6180086, 6180085, 6013243 и опубликованных заявках на патент США 2003185756, 20031656432, 2003158127, 2003152577, 2003143159, 2003105300, 2003105299, 2003072763, 2003036538, 2003031627, 2003017164, 2002169107, 2002164287 и 2002156117.
7. Лучевая терапия
Радиоизотопная терапия включает введение соединения, меченного радиоактивной меткой, в количестве, достаточном для повреждения или разрушения ткани-мишени. После введения соединения (например, внутривенной, подкожной или внутрибрюшинной инъекцией) меченное радиоактивной меткой лекарственное средство локализуется преимущественно в патологическом участке (в данной случае опухолевой ткани или другой ткани, которая экспрессирует рецепторы GRP). После размещения соединение, меченное радиоактивной меткой, затем повреждает или разрушает патологическую ткань энергией, которая высвобождается во время радиоактивного распада введенного изотопа. Как обсуждается в настоящем описании, изобретение также охватывает применение лучевой терапии в комбинации с адъювантной химиотерапией (или в комбинации с любым другим соответствующим терапевтическим средством).
Конструирование успешного радиотерапевтического средства включает несколько решающих факторов:
1) выбор соответствующей группы для целенаправленной доставки радиоактивности к патологическому участку;
2) выбор соответствующего радионуклида, который высвобождает достаточное количество энергии для повреждения этого патологического участка по существу без повреждения прилегающих здоровых тканей; и
3) выбор соответствующей комбинации группы для целенаправленной доставки и радионуклида без неблагоприятного воздействия на способность этого конъюгата локализоваться в патологическом участке. Для радиоактивных металлов это часто включает хелатообразующую группу, которая тесно координирует с радионуклидом, комбинированным с линкером, который присоединяет указанный хелат к группе для целенаправленной доставки и который воздействует на общее биологическое распределение соединения для максимизации захвата в тканях-мишенях и минимизации захвата в здоровых органах, не являющихся мишенью.
Настоящее изобретение предоставляет радиотерапевтические средства, которые соответствуют всем трем указанным выше критериям, посредством соответствующего выбора нацеливающей группы, радионуклида, хелата металла и линкера.
Радиотерапевтические средства могут содержать хелат иона 3+ металла из класса элементов, известных как лантаниды (элементы атомного номера 57-71) и их аналоги (т.е. металлы М3+, такие как иттрий и индий). Типичные радиоактивные металлы в этом классе включают изотопы: иттрий-90, индий-111, прометий-149, самарий-153, диспрозий-166, гольмий-166, иттербий-175 и лютеций-177. Все эти металлы (и другие в серии лантанидов) имеют очень похожие характеристики в отношении того, что они остаются в степени окисления +3 и предпочтительно образуют хелаты с лигандами, которые несут жесткие (кислород/азот) атомы-доноры, как показано на примере производных хорошо известного хелата DTPA (диэтилентриаминпентауксусной кислоты) и макроциклов полиаза-поликарбоксилата, таких как DOTA (1,4,7,10-тетрациклододекан-N,N',N",N'"-тетрауксуная кислота и ее близкие аналоги. Структуры этих хелатообразующих лигандов в их полностью депротонированной форме показаны ниже.

Эти хелатообразующие лиганды инкапсулируют радиоактивный металл связыванием с ним через множественные атомы азота и кислорода, предотвращая, таким образом, высвобождение свободного (несвязанного) радиоактивного металла в организм. Это важно, поскольку диссоциация in vivo радиоактивных металлов +3 из их хелата может привести к захвату радиоактивного металла в печени, костях и селезенке [Brechbiel MW, Gansow OA, "Backbone-substituted DTPA ligands for90Y radioimmunotherapy", Bioconj. Chem. 1991; 2: 187-194; Li, WP, Ma DS, Higginbotham C, Hoffman T, Ketring AR, Cutler CS, Jurisson, SS, "Development of an in vitro model for assessing the in vivo stability of lanthanide chelates." Nucl. Med. Biol. 2001; 28(2): 145-154; Kasokat T, Urich K. Arzneim.-Forsch, "Quantification of dechelation of gadopentetate dimeglumine in rats". 1992; 42(6): 869-76]. До тех пор, пока не осуществляется специальная целенаправленная доставка к этим органам, такой неспецифичный захват крайне нежелателен, поскольку он ведет к неспецифическому облучению тканей, не являющихся мишенями, что может привести к таким проблемам, как подавление гемопоэза ввиду облучения костного мозга.
Для радиотерапевтических видов применения можно применять любое из хелатообразующих соединений для терапевтических радионуклидов, раскрытых в настоящем описании. Однако формы хелата DOTA [Tweedle MF, Gaughan GT, Hagan JT, "1-Substituted-1,4,7-triscarboxymethyl-1,4,7,10-tetraazacyclododecane and analogs." US Patent 4885363, Dec. 5,1989] особенно предпочтительны, поскольку ожидается, что хелат DOTA дехелатируется меньше в организме, чем DTPA или другие линейные хелаты.
Общие способы соединения макроциклов типа DOTA с группами для целенаправленной доставки посредством линкера (например, активацией одного из макроциклов DOTA для образования активного сложного эфира, который затем взаимодействует с аминогруппой на линкере для образования стабильной амидной связи) известны специалистам в данной области (см., например, Tweedle et al. патент США № 4885363). Соединение можно также синтезировать из макроциклов типа DOTA, в которых модифицирован каркас полиазакольца.
Выбор соответствующего нуклида для использования в конкретном радиотерапевтическом применении зависит от многих факторов, включая:
а. Период физического полураспада. Он должен быть достаточно длинным для обеспечения возможности синтеза и очистки радиотерапевтического конструкта из радиоактивного металла и конъюгата и доставки указанного конструкта к участку инъекции без значительного радиоактивного распада перед инъекцией. Предпочтительно радионуклид должен иметь период физического полураспада примерно от 0,5 до 8 д.
b. Энергия испускания из радионуклида. Радионуклиды, которые являются излучателями частиц, такими как альфа-излучатели, бета-излучатели и электронные Оже-излучатели особенно полезны, поскольку они излучают высокоэнергетические частицы, которые осаждают свою энергию на коротких расстояниях, вызывая этим высоколокализованное повреждение. Бета-излучающие радионуклиды особенно предпочтительны, поскольку энергия излучения бета-частиц из этих изотопов расходуется в пределах от 5 до около 150 клеточных диаметров. Радиотерапевтические средства, полученные из этих нуклидов, способны уничтожать патологические клетки, которые относительно близко к участку их локализации, но не могут проходить большие расстояния для повреждения прилежащей нормальной ткани, такой как костный мозг.
С. Удельная активность (т.е. радиоактивность на массу радионуклида). Особенно предпочтительны радионуклиды, которые имеют высокую удельную активность (например, продуцируемые генератором Y-90, In-111, Lu-177). Удельная активность радионуклида определяется способом его получения, определенной мишенью, которая используется для его получения, и свойствами рассматриваемого изотопа.
Многие из лантанидов и лантаноидов включают радиоизотопы, которые имеют ядерные свойства, которые делают их подходящими для использования в качестве радиотерапевтических средств, поскольку они излучают бета-частицы. Некоторые из них перечислены в таблице ниже.
Способы получения радиоактивных металлов, таких как бета-излучающие лантанидные радиоизотопы, известны специалистам в данной области и были описаны в других публикациях [например, Cutler CS, Smith CJ, Ehrhardt GJ.; Tyler TT, Jurisson SS, Deutsch E. "Current and potential therapeutic uses of lanthanide radioisotopes." Cancer Biother. Radiopharm. 2000; 15(6): 531-545]. Многие из этих изотопов можно получать с высоким выходом при относительно низкой стоимости, и многие (например,90-Y,149-Pm,177-Lu) можно получать с величинами удельной активности, близкими к лишенным носителя (т.е. подавляющее большинство атомов являются радиоактивными). Поскольку нерадиоактивные атомы могут конкурировать с их радиоактивными аналогами за связывание с рецепторами на ткани-мишени, использование радиоизотопа с высокой удельной активностью является важным для обеспечения возможности доставки настолько возможно высокой дозы радиоактивности к ткани-мишени.
Радиотерапевтические производные изобретения, содержащие бета-излучающие изотопы рения (186Re и188Re) являются также особенно предпочтительными.
8. Дозировки и добавки
Соответствующие схемы доз для соединений настоящего изобретения известны специалистам в данной области. Соединения можно вводить с использованием многих способов, которые включают, но не ограничиваются ими, однократные или множественные внутривенные или внутрибрюшинные инъекции. Для радиофармацевтических средств вводят количество радиоактивности, которое достаточно для обеспечения возможности визуализации или, в случае лучевой терапии, для того, чтобы вызвать повреждение или уничтожение ткани-мишени, несущей GRP-R, но не так много, чтобы было вызвано существенное повреждение не мишени (здоровой ткани). Количество и доза, требуемые для сцинтиграфической визуализации, обсуждены выше. Количество и доза, требуемые для лучевой терапии, также различны для различных конструктов, в зависимости от энергии и периода полураспада используемого изотопа, степени захвата и выведения средства из тела и массы опухоли. В целом, дозы могут находиться в диапазоне от однократной дозы примерно 30-50 мКи до кумулятивной дозы примерно до 3 кюри.
(Информация о дозировке для оптических соединений)
Композиции изобретения могут включать физиологически приемлемые буферы и могут потребоваться радиационные стабилизаторы для предотвращения радиолитического повреждения соединения перед инъекцией. Радиационные стабилизаторы известны специалистам в данной области и могут включать, например, парааминобензойную кислоту, аскорбиновую кислоту, гентистовую кислоту и им подобные.
Набор, включающий один или несколько емкостей, который содержит все компоненты, необходимые для получения диагностических или терапевтических средств данного изобретения, является неотъемлемой частью изобретения. В случае радиофармацевтических средств такие наборы будут часто включать все необходимые ингредиенты, кроме радионуклида.
Например, набор, включающий одну емкость для приготовления радиофармацевтического средства изобретения, предпочтительно содержит хелатообразующее соединение/линкер/конъюгат пептида для целенаправленной доставки формулы M-N-O-P-G, источник соли двухвалентного олова (если требуется восстановление, например, при использовании технеция) или другое фармацевтически приемлемое восстанавливающее средство, и они соответствующим образом забуферены фармацевтически приемлемой кислотой или основанием для доведения рН до величины от примерно 3 до примерно 9. Количество и тип используемого восстанавливающего средства зависит от природы обменного комплекса, который должен быть образован. Соответствующие условия хорошо известны специалистам в данной области. Предпочтительно, чтобы содержимое набора было в лиофилизированной форме. Такой содержащий одну емкость набор может необязательно содержать лабильные или обменные лиганды, такие как глюкогептонат, глюконат, маннит, малат, лимонная или винная кислота, и может также содержать реактивные модификаторы, такие как диэтилентриаминпентауксусная кислота (DPTA), этилендиаминтетрауксусная кислота (EDTA) или α, β или γ-циклодекстрин, которые служат для повышения радиохимической чистоты и стабильности конечного продукта. Набор может также содержать стабилизаторы, вещества, увеличивающие объем, такие как маннит, которые предназначены для содействия при процессе лиофилизации, и другие добавки, известные специалистам в данной области.
Набор, включающий несколько емкостей, предпочтительно содержит такие же общие компоненты, но использует несколько емкостей при растворении радиофармацевтического средства. Например, одна емкость может содержать все ингредиенты, которые требуются для образования лабильного комплекса Tc(V) после добавления пертехнат (например, источника двухвалентного олова или другого восстанавливающего средства). Пертехнат добавляют в эту емкость, и после ожидания в течение соответствующего периода времени содержимое этой емкости добавляют во вторую емкость, которая содержит хелатообразующее соединение и пептид, обеспечивающий целенаправленную доставку, а также буферы для доведения рН до его оптимальной величины. После времени взаимодействия примерно от 5 до 60 мин образуются комплексы настоящего изобретения. Предпочтительно, чтобы содержимое обеих емкостей этого набора, включающего несколько емкостей, было лиофилизировано. Как указано выше, реактивные модификаторы, обменные лиганды, стабилизаторы, средства, увеличивающие объем и т.д. могут присутствовать в одной из двух или обеих емкостях.
Общее получение соединений
Соединения настоящего изобретения можно получить различными способами в зависимости от выбранного хелатирующего агента. Пептидную часть соединения можно наиболее подходящим способом получить методами, общепринятыми и известными в области синтеза пептидов, такими как твердофазный синтез пептидов (SPPS). Ввиду того что пептидная часть поддается твердофазному синтезу, использование чередующейся защиты FMOC и снятие защиты представляет собой предпочтительный способ получения коротких пептидов. Технология рекомбинантной ДНК является предпочтительной для получения белков и их длинных фрагментов.
Твердофазный синтез пептидов (SPPS) включает поэтапное добавление аминокислотных остатков к растущей пептидной цепи, которая связана с нерастворимой подложкой или матрицей, такой как полистирол. Сначала С-концевой остаток пептида прикрепляется к коммерчески доступной подложке его аминогруппой, защищенной N-защитным агентом, таким как трет-бутилоксикарбонильная группа (Вос) или флуоренилметоксикарбонильная (Fmoc) группа. Аминозащитная группа удаляется подходящими снимающими защиту агентами, такими как TFA в случае Вос или пиридин для Fmoc, а следующий аминокислотный остаток (в N-защищенной форме) добавляется с агентом сочетания, таким как диизопропилкарбодиимид (DIC). После образования пептидной связи реагенты вымывают из подложки. После добавления конечного остатка пептид отщепляется от подложки подходящим реагентом, таким как трифторуксуная кислота (TFA) или гидрофторид (HF).
Альтернативное получение соединений сегментным сочетанием
Соединения изобретения можно также получить способом, известным в данной области как сочетание или конденсация фрагментов (Barlos, K. and Gatos, D.; 2002 "Convergent Peptide Synthesis" in Fmoc Solid Phase Synthesis-A Practical Approach; Eds. Chan, W.C. and White, P.D.; Oxford University Press, New York; Chap. 9, pp 215-228). В этом способе сегменты пептида, обычно в форме защищенной боковой цепи, получают отдельно или синтезом в фазе раствора, или твердофазным синтезом, или комбинацией этих двух способов. Выбор сегментов имеет решающее значение и производится с использованием стратегии деления, которая может обеспечить управляемое количество сегментов, чьи С-концевые остатки и N-концевые остатки выступают для обеспечения самого чистого соединения при синтезе пептидов. С-концевые остатки наилучших сегментов или лишены хиральных альфа-углеродов (глицин или другие части, лишенные хиральных углеродов карбоксильной группы для активации на стадии сочетания) или компрометированы аминокислотами, чья склонность к рацемизации во время активации и сочетания является самой низкой из возможных выборов. Выбор N-концевой кислоты для каждого сегмента основывается на легкости сочетания активированного ацильного промежуточного соединения с аминогруппой. После выбора стратегии деления, выбирается метод сочетания каждого из сегментов на основании доступности синтеза требуемых промежуточных соединений и связанной с ним легкости манипулирования и очистки полученных продуктов (при необходимости). Затем сегменты сочетают друг с другом, оба в растворе или один в твердой фазе, а другой в растворе, для получения конечной структуры в полностью или частично защищенной форме.
Защищенное целевое соединение затем подвергают удалению защитной группы, очищают и выделяют с получением желаемого конечного соединения. Преимущества сегментного сочетания состоят в том, что каждый сегмент можно очистить отдельно, обеспечивая возможность удаления побочных продуктов, таких как последовательности отщепляемых аминокислот в результате неполного сочетания, или последовательности, полученные в результате реакций, таких как дегидрирование амида боковой цепи во время стадий сочетания или внутренней циклизации боковой цепи (такой как циклизации Gln) с альфа-аминогруппой во время снятия защиты групп Fmoc. Все такие побочные продукты присутствовали бы в конечном продукте обычной, основанной на смоле "прямой сквозной" сборки пептидной цепи, в то время как, при необходимости, можно выполнить удаление этих веществ на многих стадия при сегментного сочетания. Другое важное преимущество сегментного сочетания состоит в том, что можно было использовать различные растворители, реагенты и условия для оптимизации синтеза каждого из сегментов до высокой чистоты и выхода, приводя к повышенной чистоте и выходу конечного продукта. Другие осуществленные преимущества представляют собой сниженное потребление реагентов и более низкие затраты.
ПРИМЕРЫ
Следующие примеры предоставлены в качестве примеров различных способов, которые можно использовать для получения различных соединений настоящего изобретения. В пределах каждого примера имеются соединения, идентифицированные одной заглавной буквой, набранной жирным шрифтом (например, А, В, С), которые коррелируют с маркированными таким же образом соответствующими соединениями, обозначенными на фигурах.
Общая экспериментальная методика
А. Определения использованных аббревиатур
Следующие общие аббревиатуры используются по всему данному описанию:
1,1-диметилэтоксикарбонил (Boc или Boc);
9-флуоренилметилоксикарбонил (Fmoc);
1-гидроксибензотриазол (НОВТ);
N,N'-диизопропилкарбодиимид (DIC);
N-метилпирролидон (NMP);
уксусный ангидрид (Ac2O);
(4,4-диметил-2,6-диоксоциклогекс-1-илиден)-3-метилюбутил (iv-Dde);
трифторуксусная кислота (TFA):
реагент В (TFA:H2O:фенол:триизопропилсилан, 88:5:5:2);
диизопропилэтиламин (DIEA);
гексафторфосфат O-(1H-бензотриазол-1-ил)-N,N,N', N'-тетраметилурония (HBTU);
гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU);
N-гидроксисукцинимид (NHS);
твердофазный пептидный синтез (SPPS);
диметилсульфоксид (ДМСО);
дихромметан (DCM);
диметилформамид (ДМФА);
диметилацетамид (DMA):
1,4, 7,10-тетраазациклотетрадекан-1,4,7,10-тетрауксуная кислота (DOTA);
триизопропилсилан (TIPS);
(1R)-1-[1,4,7,10-тетрааза-4,7,10-трис(карбоксиметил)циклододецил]этан-1, 2-дикарбоновая кислота (CMDOTA);
фетальная бычья сыворотка (FBS);
человеческий сывороточный альбумин (HAS);
клеточная линия рака предстательной железы человека (PC3);
изобутилхлорформиат (IBCF);
трибутиламин (TBA);
радиохимическая чистота (RPC) и
высокоэффективная жидкостная хроматография (ВЭЖХ).
В. Материалы
Используемые защищенные Fmoc аминокислоты закупали у Nova-Biochem (San Diego, CA, USA), Advanced Chem Tech (Louisville, KY., USA), Chem-Impex International (Wood Dale 111, USA), and Multiple Peptide Systems (San Diego, CA., USA). Другие химические соединения, реагенты и адсорбенты, требуемые для синтеза, поставлялись Aldrich Chemical Co. (Milwaukee, WI, USA) и VWR Scientific Products (Bridgeport, NJ, USA). Растворители для пептидного синтеза получали от Pharmco Co. (Brookfield CT., USA). Колонки для анализа и очистки ВЭЖХ получали от Waters Co. (Milford, MA, USA). Подробности экспериментов приведены ниже для коммерчески недоступных компонентов.
С. Устройства для пептидного синтеза
Пептиды получали с использованием синтезатора Advanced ChemTech 496 Ω MOS, синтезатора Advanced ChemTech 357 FBS и/или ручным пептидным синтезом. Однако используемые последовательности операций для повторяющегося снятия защиты и удлинения цепи были одинаковы для всех.
D. Автоматизированный синтез с помощью устройства Symphony (произведенное Rainin)
Синтез проводят 3 версией устройства Symphony, поставляемым Protein Technologies Inc. Используют смолу Novagel TGR с замещением 0,25 ммоль/г, и каждая лунка содержит 0,2 г смолы (50 мкмоль). Аминокислоты растворяют в NMP, и концентрация составляет 0,25 М. Готовят 0,25 M раствор HBTU и N-метилморфолина в ДМФА и используют для сочетания. Все сочетания проводят в течение 2,0 ч. Расщепление проводят вне устройства переносом смолы в другой реакционный сосуд и используя реагент В, как при ручном синтезе.
Е. Устройства, используемые для анализа и очистки
Аналитическую ВЭЖХ выполняют с использованием системы градиентного аналитического устройства ЖХ с двойным насосом Shimadzu-LC-10A с использованием версии 4.1 программного обеспечения Shimadzu-Class VP для управления устройством, получения данных и их обработки после рабочего цикла. Масс спектры получают на масс спектрометре Hewlett-Packard Series 1100 MSD, соединенном через интерфейс с градиентным устройством ВЭЖХ с двойным насосом Hewlett-Packard Series 1100, оборудованным автоматическим устройством для отбора проб Agilent Technologies 1100, приспособленным для непосредственного отбора проб в поток или отбора проб на колонку Waters Associates Xterra MS C18 (4,6 мм×50 мм, размер частиц 5 мкм, размер пор 120). Устройство приводится в действие автоматизированной рабочей станцией HP Kayak с использованием программного обеспечения "MSD Anyone" для подачи образца и программного обеспечения HP Chemstation для управления устройством и сбора данных. В большинстве случаев образцы вводят посредством прямой инжекции с использованием пробы 5 мкл раствора образца в концентрации 1 мг/мл и анализируют, используя электрораспыление положительных ионов для получения m/e и m/z (многозарядных) ионов для подтверждения структуры.1Н-ЯМР спектры получают на спектрометре Varian Innova при 500 МГц.13С-ЯМР спектры получают на том же устройстве при 125,73 МГц. Обычно остаточное поглощение1Н или в случае13С-ЯМР поглощение13С применяемого растворителя используют в качестве внутреннего эталона; в других случаях используют тетраметилсилан (μ=0,00 м.д.). Величины резонанса представлены в единицах μ. Данные микроанализа получают по Quantitative Technologies Inc. Whirehouse NJ. Препаративную ВЭЖХ выполняют на градиентном препаративном устройстве ВЭЖХ с двойным насосом Shimadzu-LC-8A с использованием версии 4.3 программного обеспечения Shimadzu-Class VP для управления устройством, сбора данных, сбора фракций и обработки данных после рабочего цикла.
F. Общая методика для пептидного синтеза
В качестве твердой подложки используют смолу Rink Amide-Novagel HL (0,6 ммоль/г).
G. Методика сочетания
В типичном эксперименте первую аминокислоту загружают на 0,1 г смолы (0,06 ммоль). Соответствующую Fmoc-аминокислоту в NMP (0,25 M раствор; 0,960 мл добавляют к смоле с последующим добавлением N-гидроксибензотриазола (0,5 M в NMP; 0,48 мл)) и реакционный блок (в случае автоматизированного пептидного синтеза) или отдельный реакционный сосуд (в случае ручного пептидного синтеза) встряхивают в течение примерно 2 мин. К указанной выше смеси добавляют диизопропилкарбодиимид (0,5 M в NMP; 0,48 мл) и реакционную смесь встряхивают в течение 4 ч при окружающей температуре. Затем реакционный блок или отдельный реакционный сосуда очищают от реагентов подачей положительного давления сухого азота.
Н. Методика промывки
Каждую ячейку реакционного блока заполняют 1,2 мл NMP и блок встряхивают в течение 5 мин. Раствор сливают при положительном давлении азота. Эту процедуру повторяют 3 раза. Такую же методику используют с соответствующим объемом NMP в случае ручного синтеза с использованием отдельных сосудов.
I. Удаление группы Fmoc
Смолу, содержащую аминокислоту, защищенную Fmoc, обрабатывают 1,5 мл 20% пиперидина в ДМФА (об./об.) и реакционный блок или отдельный сосуд ручного синтеза встряхивают в течение 15 мин. Раствор сливают из смолы. Эту процедуру повторяют 1 раз и смолу промывают, используя описанную выше методику промывки.
J. Конечное сочетание лиганда (DOTA и CMDOTA)
N-концевую аминогруппу конструкта связанного со смолой пептидного линкера деблокируют и смолу промывают. Готовят 0,25 M раствор желаемого лиганда и HBTU в NMP и обрабатывают двукратным эквивалентом DIEA. Полученный раствор активированного лиганда добавляют к смоле (1,972 мл; 0,48 ммоль) и реакционную смесь встряхивают при окружающей температуре в течение 24-30 ч. Раствор сливают и смолу промывают. Конечную промывку смолы проводят 1,5 мл дихлорметана (трижды).
К. Снятие защиты и очистка конечного пептида
Раствор реагента В (2 мл; 88:5:5:2-TFA:фенол:вода:TIPS) добавляют к смоле, и реакционный блок или отдельный сосуд встряхивают в течение 4,5 ч при окружающей температуре. Полученный раствор, содержащий пептид со снятой защитой, выливают в емкость. Эту процедуру повторяют еще 2 раза с 1 мл реагента В. Объединенный фильтрат концентрируют при пониженном давлении с использованием центрифужного концентратора Genevac HT-12 серии II. Остаток в каждой емкости затем растирают с 2 мл Et2O, и надосадочную жидкость удаляют. Эту процедуру повторяют дважды с получением пептидов в виде бесцветных твердых веществ. Неочищенные пептиды растворяют в смеси вода/ацетонитрил и очищают, используя колонку препаративной ВЭЖХ Waters Xterra MS C18 (50 мм×19 мм, размер частиц 5 мкм, размер пор 120 ) или колонку Waters-YMC C18 ODS (250 мм×30 мм внутренний диаметр, размер частиц 10 мкм, размер пор 120 ). Фракции с продуктами собирают и анализируют ВЭЖХ. Фракции с чистотой >95% объединяют и пептиды выделяют лиофилизиацией.
Условия препаративной ВЭЖХ (колонка Waters Xterra):
Объемный расход при элюировании: 50 мл/мин.
Детектирование: УФ, λ=220 нм.
Элюент А: 0,1% водн. TFA; Растворитель В: ацетонитрил (0,1% TFA).
Условия анализа ВЭЖХ:
Колонка: Waters Xterra (Waters Co.; 4,6× 50 мм; MS C18; размер частиц 5 мкм, размер пор 120 ).
Объемный расход при элюировании: 3 мл/мин;
Детектирование: УФ, λ=220 нм.
Элюент А: 0,1% водн. TFA; Растворитель В: ацетонитрил (0,1% TFA).
Пример 1 - фиг.1А-В
Синтез L62
Резюме: Как показано на фиг.1А-В, L62 получают с использованием следующих стадий: Гидролиз метилового эфира (3β,5β)-3-аминохолан-24-овой кислоты А NaOH дает соответствующую кислоту В, которая затем реагирует с Fmoc-Cl с получением промежуточного соединения С. Амидная смола Rink, функционализированная октапептидом Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) последовательно реагирует с С, Fmoc-глицином и три-трет-бутиловым эфиром DOTA. После расщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L62. Общий выход: 2,5%. Дополнительные детали представлены ниже:
А. Амидная смола Rink, функционализированная бомбезином [7-14], (A)
В сосуде для твердофазного пептидного синтеза (см. приложение №1) Fmoc-аминокислоту (24 ммоль), N-гидроксибензотриазол (HOBt) (3,67 г; 24 ммоль) и N,N'-диизопропилкарбодиимид (DIC) (3,75 мл; 24 ммоль) последовательно добавляют к суспензии амидной смолы Rink NovaGelTM (10 г; 6,0 ммоль) А в ДМФА (45 мл). Смесь встряхивают в течение 3 ч при комнатной температуре с использованием настольного шейкера, затем раствор сливают и смолу промывают ДМФА (5×45 мл). Смолу встряхивают с 25% пиперидином в ДМФА (45 мл) в течение 4 мин, раствор сливают и добавляют свежий 25% пиперидин в ДМФА (45 мл). Суспензию встряхивают в течение 10 мин, затем раствор сливают и смолу промывают ДМФА (5×45 мл).
Эту методику применяют последовательно для следующих аминокислот: N-α -Fmoc-L-метионин, N-α-Fmoc-L-лейцин, N-α-Fmoc-N-им-тритил-L-гистидин, N-α-Fmoc-глицин, N-α-Fmoc-L-валин, N-α-Fmoc-L-аланин, N-α-Fmoc-N-ин-Boc-L-триптофан.
При последней реакции сочетания N-α-Fmoc-N-γ-тритил-L-глутамин (14,6 г; 24 ммоль), HOBt (3,67 г; 24 ммоль) и DIC (3,75 мл; 24 ммоль) добавляют к смоле в ДМФА (45 мл). Смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают ДМФА (5×45 мл), CH2Cl2 (5×45 мл) и сушат в вакууме.
В. Получение промежуточных соединений В и С (фиг.1А):
1. Синтез (3β,5β)-3-аминохолан-24-овой кислоты (В)
1 M раствор NaOH (16,6 мл; 16,6 ммоль) добавляют по каплям к раствору метилового эфира (3β,5β)-3-аминохолан-24-овой кислоты (5,0 г; 12,8 ммоль) в МеОН (65 мл) при 45°С. После 3 ч перемешивания при 45°С смесь концентрируют до 25 мл и добавляют Н2О (40 мл) и 1 M HCl (22 мл). Осажденное твердое вещество отфильтровывают, промывают Н2О (2×50 мл) и сушат в вакууме с получением В в виде белого твердого вещества (5,0 г; 13,3 ммоль). Выход 80%.
2. Синтез (3β,5β )-3-(9Н-флуорен-9-илметокси)аминохолан-24-овой кислоты (С)
Раствор 9-флуоренилметоксикарбонилхлорида (0,76 г; 2,93 ммоль) в 1,4-диоксане (9 мл) добавляют по каплям к суспензии (3β,5β)-3-аминохолан-24-овой кислоты В (1,0 г; 2,66 ммоль) в 10% водном Na2CO3 (16 мл) и 1,4-диоксане (9 мл), перемешанном при 0°С. После 6 ч перемешивания при комнатной температуре добавляют Н2О (90 мл), водную фазу промывают Et2O (2×90 мл) и затем добавляют 2 M HCl (15 мл) (конечный рН: 1,5). Водную фазу экстрагируют EtOAc (2×100 мл), органическую фазу сушат над Na2SO4 и выпаривают. Неочищенный продукт очищают флэш-хроматографией с получением С в виде белого твердого вещества (1,2 г; 2,0 ммоль). Выход 69%.
С. Синтез L62 (N-[(3β,5β)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-холан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.1В):
Смолу А (0, 5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). (3β,5β)-3-(9Н-флуорен-9-илметокси)аминохолан-24-овую кислоту С (0,72 г; 1,2 ммоль), N-гидроксибензотриазол (HOBT) (0,18 г; 1,2 ммоль), N,N'-диизопропилкарбодиимид (DIC) (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). К смоле добавляют N-α-Fmoc-глицин (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл). Смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл) с последующим добавлением к смоле аддукта трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2, 4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое растирают с Et2O (20 мл) с получением осадка. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл), затем анализируют ВЭЖХ и очищают препаративной ВЭЖХ.
Фракции, содержащие продукт, лиофилизируют с получением L62 (6,6 мг; 3,8×10-3 ммоль) в виде белого твердого вещества. Выход 4,5%.
Пример II - фиг.2А-F
Синтез L70, L73, L74, L115 и L116
Резюме: Продукты получают сочетанием октапептида Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) (с соответствующей защитой боковой цепи) на амидной смоле Rink с различными линкерами с последующей функционализацией три-трет-бутиловым эфиром DOTA. После расщепления и снятия защиты реагентом В конечный продукт очищают препаративной ВЭЖХ. Общий выход 3-9%.
А. Синтез L70 (фиг.2А):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). Fmoc-4-аминобензойную кислоту (0,43 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 моль), DIC (0,19 мл; 1,2 ммоль) и DMA (7мл) добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). Fmoc-глицин (0,36 г; 1,2 ммоль), HATU (0,46 г; 1,2 ммоль) и DIEA (0,40 мл; 2,4 ммоль) перемешивают в течение 15 мин в DMA (7 мл), затем раствор добавляют к смоле, смесь встряхивают в течение 2 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). Аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклодекан-1,4,7, 10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл) добавляют к смоле. Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют и раствор фильтрата выпаривают при пониженном давлении с получением маслянистого неочищенного продукта, который растирают с Et2O (5 мл). Осадок отделяют центрифугированием и промывают Et2O (5×5 мл), затем анализируют ВЭЖХ и очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L70 в виде рыхлого, твердого вещества (6,8 мг; 0,005 ммоль). Выход 3%.
В. Синтез L73, L115 и L116 (фиг.2В-2Е):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). Fmoc-линкер-ОН (1,2 ммоль), НОВТ (0,18 г; 1,2 моль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). Аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл) добавляют к смоле. Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного продукта, который растирают с Et2O (5 мл). Осадок отделяют центрифугированием и промывают Et2O (5×5 мл), затем анализируют ВЭЖХ и очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют.
С. Синтез L74 (фиг.2F)
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). Fmoc-изонипекотовую кислоту (0,42 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 моль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл), и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). Fmoc-глицин (0,36 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл), добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). Аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл) добавляют к смоле. Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл), CH2 Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного продукта, который растирают с Et2O (5 мл). Осадок отделяют центрифугированием и промывают Et2O (5×5 мл), затем анализируют ВЭЖХ и очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L74 в виде белого, рыхлого, твердого вещества (18,0 мг; 0,012 ммоль). Выход 8%.
Пример III - фиг.3А-Е
Синтез L67
Резюме: Гидролиз метилового эфира (3β,5β)-3-амино-12-оксохолан-24-овой кислоты NaOH дает соответствующую кислоту В, которая затем реагирует с Fmoc-глицином с получением промежуточного соединения С. Амидная смола Rink, функционализированная октапептидом Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) последовательно реагирует с С и три-трет-бутиловым эфиром DOTA. После расщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L67. Общий выход: 5,2%.
А. Синтез (3β,5β)-3-аминохолан-12-оксохолан-24-овой кислоты (В) (фиг.3А)
1 M раствор NaOH (6,6 мл; 6,6 ммоль) добавляют по каплям к раствору метилового эфира (3β,5β)-3-амино-12-оксохолан-24-овой кислоты А (2,1 г; 5,1 ммоль) в МеОН (15 мл) при 45°С. После 3 ч перемешивания при 45°С смесь концентрируют до 25 мл и затем добавляют Н2О (25 мл) и 1 M HCl (8 мл). Осажденное твердое вещество отфильтровывают, промывают Н2О (2×30 мл) и сушат в вакууме с получением В в виде белого твердого вещества (1,7 г; 4,4 ммоль). Выход 88%.
В. Синтез (3β,5β )-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-12-оксохолан-24-овой кислоты (С) (фиг.3А)
Трибутиламин (0,7 мл; 3,1 ммоль) добавляют по каплям к раствору N-α -Fmoc-глицина (0,9 г; 3,1 ммоль) в ТГФ (25 мл) и перемешивают при 0°С. Затем добавляют изобутилхлорформиат (0,4 мл; 3,1 ммоль) и через 10 мин добавляют по каплям суспензию трибутиламина (0,6 мл; 2,6 ммоль) и (3β,5β)-3-амино-12-оксохолан-24-овой кислоты В (1,0 г; 2,6 ммоль) в ДМФА (30 мл) в течение 1 ч в охлажденный раствор. Смесь оставляют нагреваться, и через 6 ч раствор концентрируют до 40 мл, затем добавляют Н2О (50 мл) и 1N HCl (10 мл) (конечный рН: 1,5). Осажденное твердое вещество отфильтровывают, промывают Н2О (2×50 мл), сушат в вакууме и очищают флэш-хроматографией с получением С в виде белого твердого вещества (1,1 г; 1,7 ммоль). Выход 66%.
С. Синтез L67 (N-[(3β, 5β)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-12, 24-диоксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.3В и 3Е):
Смолу D (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). (3β,5β)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино]-12-оксохолан-24-овую кислоту С (0,80 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое растирают с Et2O (20 мл).
Пример IV - фиг.4А-Н
Синтез L63 и L64
Резюме: Гидролиз метилового эфира (3β,5β,7α,12α)-3-амино-7,12-дигидроксихолан-24-овой кислоты 1b с NaOH дает промежуточное соединение 2b, которое затем реагирует с Fmoc-глицином с получением 3b. Амидная смола Rink, функционализированная октапептидом Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) реагирует с 3b и три-трет-бутиловым эфиром DOTA. После расщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L64. Такую же методику повторяют, начиная с уже имеющегося промежуточного соединения 2а , с получением L63. Общий выход: соответственно 9 и 4%.
А. Синтез 3β,5β,7α,12α,)-3-амино-7,12-дигидроксихолан-24-овой кислоты 2b (фиг.4А)
1 M раствор NaOH (130 мл; 0,13 ммоль) добавляют по каплям к раствору метилового эфира (3β,5β,7α,12α,)-3-амино-7, 12-дигидроксихолан-24-овой кислоты 1b (42,1 г; 0,10 ммоль) в МеОН (300 мл) при 45°С. После 3 ч перемешивания при 45°С смесь концентрируют до 150 мл и добавляют Н2О (350 мл). После экстракции CH2Cl2(2×100 мл) водный раствор концентрируют до 200 мл и добавляют 1 M HCl (150 мл). Осажденное твердое вещество отфильтровывают, промывают Н2 О (2×100 мл) и сушат в вакууме с получением 2b в виде белого твердого вещества (34,8 г; 0,08 ммоль). Выход 80%.
В. Синтез (3β,5β,12α )-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-12-гидроксихолан-24-овой кислоты (3а) (фиг.4А)
Трибутиламин (4,8 мл; 20,2 ммоль) добавляют по каплям к раствору N-α -Fmoc-глицина (6,0 г; 20,2 ммоль) в ТГФ (120 мл) и перемешивают при 0°С. Затем добавляют изобутилхлорформиат (2,6 мл; 20,2 ммоль) и через 10 мин добавляют по каплям суспензию трибутиламина (3,9 мл; 16,8 ммоль) и (3β,5β,12α)-3-амино-12-гидроксихолан-24-овой кислоты 2а (6,6 г; 16,8 ммоль) в ДМФА (120 мл) в течение 1 ч в охлажденный раствор. Смесь оставляют нагреваться и через 6 ч раствор концентрируют до 150 мл, затем добавляют Н2О (250 мл) и 1N HCl (40 мл) (конечный рН: 1,5). Осажденное твердое вещество отфильтровывают, промывают Н2О (2×100 мл), сушат в вакууме и очищают флэш-хроматографией с получением 3а в виде белого твердого вещества (3,5 г; 5,2 ммоль). Выход 31%.
С. Синтез (3β,5β,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксихолан-24-овой кислоты (3b) (фиг.4А)
Трибутиламин (3,2 мл; 13,5 ммоль) добавляют по каплям к раствору N-α-Fmoc-глицина (4,0 г; 13,5 ммоль) в ТГФ (80 мл) и перемешивают при 0°С. Затем добавляют изобутилхлорформиат (1,7 мл; 13,5 ммоль) и через 10 мин добавляют по каплям суспензию трибутиламина (2,6 мл; 11,2 ммоль) и (3β,5β,7α,12α)-3-амино-7,12-дигидроксихолан-24-овой кислоты 3а (4,5 г; 11,2 ммоль) в ДМФА (80 мл) в течение 1 ч в охлажденный раствор. Смесь оставляют нагреваться и через 6 ч раствор концентрируют до 120 мл, затем добавляют Н2О (180 мл) и 1N HCl (30 мл) (конечный рН: 1,5). Осажденное твердое вещество отфильтровывают, промывают Н2О (2×100 мл), сушат в вакууме и очищают флэш-хроматографией с получением 3а в виде белого твердого вещества (1,9 г; 2,8 ммоль). Выход 25%.
В альтернативном способе (3β,5β,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксихолан-24-овую кислоту (3b) можно получить следующим образом:
N-гидроксисукцинамид (1,70 г; 14,77 ммоль) и DIC (1,87 г; 14,77 ммоль) последовательно добавляют к перемешиваемому раствору Fmoc-Gly-OH (4,0 г; 13,45 ммоль) в дихлорметане (15 мл); полученную смесь перемешивают при комнатной температуре в течение 4 ч. Образовавшуюся N,N'-диизопропилмочевину удаляют фильтрованием и твердое вещество промывают эфиром (20 мл). Летучие вещества удаляют и образовавшийся сложный Fmoc-Gly-сукцинимидиловый эфир промывают эфиром (3×20 мл). Затем сложный Fmoc-Gly-сукцинимидиловый эфир повторно растворяют в безводном ДМФА (15 мл) и в прозрачный раствор добавляют 3-аминодеоксихолевую кислоту (5,21 г; 12,78 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 4 ч, добавляют воду (200 мл), и осажденное твердое вещество отфильтровывают, промывают водой, сушат и очищают хроматографией на силикагеле (ТСХ (двуокись кремния): Rf: 0,50, силикагель, CH2Cl2/CH3OH, 9:1) (элюент: CH2Cl2/CH3OH, 9:1)) с получением (3β,5β,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксихолан-24-овой кислоты в виде бесцветного твердого вещества. Выход: 7,46 г (85%).
D. Синтез L63 (N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-12-гидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.4В):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). (3β,5β,12α )-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-12-гидроксихолан-24-овую кислоту 3а (0,82 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (5 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (5×5 мл), затем анализируют и очищают ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L63 в виде белого, рыхлого твердого вещества (12,8 мг; 0,0073 ммоль). Выход 9%.
Е. Синтез L64 (N-[(3β,5β,7α,12α)-3-[[[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-7, 12-дигидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.4С):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение 20 мин, раствор сливают, и смолу промывают DMA (5×7 мл). (3β,5β,7α,12α)-3-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксихолан-24-овую кислоту 3b (0,81 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл), и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1, 2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое растирают с Et2O (5 мл). Осадок отделяют центрифугированием и промывают Et2O (5×5 мл). Затем его растворяют в Н2О (20 мл) и добавляют Na2CO3 (0,10 г; 0,70 ммоль); полученную смесь перемешивают в течение 4 ч при комнатной температуре. Этот раствор очищают ВЭЖХ, фракции, содержащие продукт, лиофилизируют с получением L64 в виде белого, рыхлого твердого вещества (3,6 мг; 0,0021 ммоль). Выход 4%.
Пример V - фиг.5А-Е
Синтез L71 и L72
Резюме: Продукты получают в 2 стадии. Первая стадия представляет собой твердофазный пептидный синтез октапептида Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) (с соответствующими защитными группами боковой цепи) на амидной смоле Rink, обсужденный выше. Вторая стадия представляет собой сочетание с различными линкерами с последующей функционализацией три-трет-бутиловым эфиром DOTA. После отщепления и снятия защиты реагентом В конечный продукт очищают препаративной ВЭЖХ. Общие выходы: 3-9%.
А. Методика функционализации и отщепления бомбезина [7-14] (фиг.5А и 5D)
Смолу В (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). Fmoc-линкер-ОН (1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле. Смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl С (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре. Раствор сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют, и фильтрат выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое растирают с эфиром (5 мл). Осадок отделяют центрифугированием и промывают эфиром (5×5 мл), затем анализируют аналитической ВЭЖХ и очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют.
В. Продукты
1. L71 (4-[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]метил]бензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид)
Продукт получают в виде белого, рыхлого твердого вещества (7,3 мг; 0,005 ммоль). Выход 7,5%.
2. L72 (Транс-4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]циклогексилкарбонил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид)
Продукт получают в виде белого, рыхлого твердого вещества (7,0 мг; 0,005 ммоль). Выход 4,8%.
С. Транс-4-[[[(9H-флуорен-9-илметокси)карбонил]амино]метил]циклогексанкарбоновая кислота (D) (фиг.5Е)
Раствор N-(9-флуоренилметоксикарбонилокси)сукцинимида (4,4 г; 14,0 ммоль) в 1,4-диоксане (40 мл) добавляют по каплям к раствору транс-4-(аминометил)циклогексанкарбоновой кислоты (2,0 г; 12,7 ммоль) в 10% Na2CO3 (30 мл), охлажденному до 0°С. Затем смесь оставляют нагреваться до окружающей температуры и после перемешивания в течение 1 ч при комнатной температуре обрабатывают 1N HCl (32 мл) до тех пор, пока конечный рН не составит 2. Полученный раствор экстрагируют н-BuOH (100 мл); летучие вещества удаляют, и неочищенный остаток очищают флэш-хроматографией с получением D в виде белого твердого вещества (1,6 г; 4,2 ммоль). Выход 33%.
Пример VI - фиг.6А-F
Синтез L75 и L76
Резюме: Указанные 2 продукта получают сочетанием октапептида Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1])(А) на амидной смоле Rink с двумя линкерами Е и Н с последующей функционализацией три-трет-бутиловым эфиром DOTA. После отщепления и снятия защиты реагентом В конечные продукты очищают препаративной ВЭЖХ. Общие выходы: 8, 5% (L75) и 5,6% (L76).
А. 2-[(1,3-дигидро-1,3-диоксо-2Н-изоиндол-2-ил)метил]бензойная кислота, (С) (фиг.6А)
Продукт синтезируют в соответствии с методикой, описанной в литературе (Bornstein, J.; Drummon, P.E.; Bedell, S.F. Org Synth. Coll. Vol.IV 1963, 810-812).
В. 2-(аминометил)бензойная кислота, (D) (фиг.6А)
40% раствор метиламина (6,14 мл; 7,1 ммоль) добавляют к 2-[(1,3-дигидро-1,3-диоксо-2Н-изоиндол-2-ил)метил]бензойной кислоте С (2 г; 7,1 ммоль) и затем добавляют EtOH (30 мл). После 5 мин перемешивания при комнатной температуре реакционную смесь нагревают при 50°С. Через 2,5 ч смесь охлаждают, и растворитель выпаривают при пониженном давлении. Неочищенный продукт суспендируют в 50 мл абсолютного этанола, и суспензию перемешивают при комнатной температуре в течение 1 ч. Твердое вещество отфильтровывают и промывают EtOH с получением 2-(аминометил)бензойной кислоты D (0,87 г; 5,8 ммоль). Выход 81%.
С. 2-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]бензойная кислота, (Е) (фиг.6А)
Продукт синтезируют в соответствии с методикой, описанной в литературе (Sun, J-H.; Deneker, W.F. Synth. Commun. 1998, 28, 4525-4530).
D. 4-(аминометил)-3-нитробензойная кислота, (G) (фиг.6В)
4-(бромметил)-3-нитробензойную кислоту (3,2 г; 12,3 ммоль) растворяют в 8% NH3 в EtOH (300 мл), и полученный раствор перемешивают при комнатной температуре. Через 22 ч раствор выпаривают и остаток суспендируют в Н2О (70 мл). Суспензию перемешивают в течение 15 мин и фильтруют. Собранное твердое вещество суспендируют в Н2О (40 мл) и растворяют добавлением нескольких капель 25% водного NH4OH (рН 12), затем рН раствора доводят до 6 добавлением 6N HCl. Осажденное твердое вещество отфильтровывают и промывают последовательно МеОН (3×5 мл) и Et2O (10 мл) и сушат в вакууме (1,3 кПа; Р2О5) с получением 4-(аминометил)-3-нитробензойной кислоты в виде бледно-коричневого твердого вещества (1,65 г; 8,4 ммоль). Выход 68%.
Е. 4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]-3-нитробензойная кислота, (Н) (фиг.6В)
4-(Аминометил)-3-нитробензойную кислоту G (0,8 г; 4 ммоль) растворяют в 10% водном Na2CO3 (25 мл) и 1,4-диоксане (10 мл), и раствор охлаждают до 0°С. Раствор 9-флуоренилметилхлорформиата (Fmoc-Cl) (1,06 г; 4 ммоль) в 1,4-диоксане (10 мл) добавляют по каплям в течение 20 мин. Через 2 ч при 0-5°С и 1 ч при 10°С реакционную смесь фильтруют и раствор подкисляют до рН 5 добавлением 1N HCl. Осадок фильтруют, промывают Н2О (2×2 мл), сушат в вакууме (1,3 кПа; Р2О5) с получением 4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]-3-нитробензойной кислоты в виде белого твердого вещества (1,6 г; 3, 7 ммоль). Выход 92%.
F. L75 (N-[2-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.6С):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). 2-[[[9Н-флуорен-9-илметокси)карбонил]амино]метил]бензойную кислоту, Е (0,45 г; 1,2 ммоль), N-гидроксибензотриазол (HOBT) (0,18 г; 1,2 ммоль), N,N'-диизопропилкарбодиимид (DIC) (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (три-трет-бутиловый эфир DOTA) (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают, в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и фильтрат выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Полученный осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением L75 (190 мг; 0,13 ммоль) в виде белого твердого вещества. Выход 44%.
G. L76 (N-[4-[[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]метил]-3-нитробензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.6D):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение 20 мин, затем раствор сливают, и смолу промывают DMA (5×7 мл). 4-[[[9Н-флуорен-9-илметокси)карбонил]амино]метил]-3-бензойную кислоту, Н (0, 05 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют сложный три-трет-бутиловый эфир (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое растирают с Et2O (20 мл). Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (141 мг), которое анализируют ВЭЖХ. Порцию 37 мг неочищенного вещества очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L76 (10,8 мг; 7,2×10-3 ммоль) в виде белого твердого вещества. Выход 9%.
Пример VII - фиг.7А-C
Синтез L124
Резюме: 4-цианофенол А вводят в реакцию с бромэтилацетатом и K2CO3 в ацетоне с получением промежуточного соединения В, которое гидролизуют NaOH в соответствующую кислоту С. Последующее гидрирование Н2 и PtO2 при 355 кПа в EtOH/CHCl3 дает соответствующую аминокислоту D, которую защищают FmocOSu с получением Е. Амидную смолу Rink функционализируют октапептидом Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) вводят в реакцию с Е и затем с три-трет-бутиловым эфиром DOTA. После отщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L124. Общий выход: 1,3%.
А. Синтез этилового эфира (4-цианофенокси)уксусной кислоты (В) (фиг.7А)
Продукт синтезируют в соответствии с методикой, описанной в литературе (Archimbault, P.; LeClerc, G.; Strosberg, A.D.; Pietri-Rouxel, F. PCT Int. Appl. WO 980005, 1998).
В. Синтез (4-цианофенокси)уксусной кислоты, (С) (фиг.7А)
1N раствор NaOH (7,6 мл; 7,6 ммоль) добавляют по каплям к раствору этилового эфира (4-цианофенокси)уксусной кислоты В (1,55 г; 7,6 ммоль) в МеОН (15 мл). Через 1 ч раствор подкисляют 1N HCl (7,6 мл; 7,6 ммоль) и выпаривают. Остаток растворяют в воде (20 мл) и экстрагируют CHCl3 (2×30 мл). Органические фазы выпаривают, и неочищенный продукт очищают флэш-хроматографией с получением (4-цианофенокси)уксусной кислоты, С (0,97 г; 5,5 ммоль) в виде белого твердого вещества. Выход 72%.
С. Синтез [4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]фенокси]уксусной кислоты, (Е) (фиг.7А)
PtO2 (150 мг) добавляют к раствору (4-цианофенокси)уксусной кислоты С (1,05 г; 5,9 ммоль) в EtOH (147 мл) и CHCl3 (3 мл). Суспензию перемешивают 30 ч в атмосфере водорода (355 кПа; 20°С). Смесь фильтруют через слой Celite® и раствор выпаривают в вакууме. Остаток очищают флэш-хроматографией с получением D (0,7 г), которую растворяют в Н2О (10 мл), MeCN (2 мл) и Et3N (0,6 мл) при 0°С, затем по каплям добавляют раствор N-(9-флуоренилметоксикарбонилокси)сукцинимида (1,3 г; 3,9 ммоль) в MeCN (22 мл). После перемешивания в течение 16 ч при комнатной температуре реакционную смесь фильтруют и летучие вещества удаляют в вакууме. Остаток обрабатывают 1N HCl (10 мл), и осажденное твердое вещество отфильтровывают и очищают флэш-хроматографией с [4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]фенокси]уксусной кислоты Е (0,56 г; 1,4 ммоль) в виде белого твердого вещества. Общий выход 24%.
D. Синтез L124 (N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]фенокси]ацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.7B):
Смолу А (480 мг; 0,29 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение 20 мин, раствор сливают, и смолу промывают DMA (5×7 мл). [4-[[[9Н-флуорен-9-илметокси)карбонил]амино]метил]фенокси]уксусную кислоту Е (480 мг; 1,19 ммоль), N-гидроксибензотриазол (HOBT) (182 мг; 1,19 ммоль), N,N'-диизопропилкарбодиимид (DIC) (185 мкл; 1,19 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (6 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (6 мл) и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (750 мг; 1,19 ммоль), НОВТ (182 мг; 1,19 ммоль), DIEA (404 мкл; 2,36 ммоль), DIC (185 мкл; 1,19 ммоль), и DMA (6 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор выливают, смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют, и фильтрат выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое растирают с Et2O (5 мл). Осадок отделяют центрифугированием и промывают Et2O (5×5 мл) с получением твердого вещества (148 мг), которое анализируют ВЭЖХ. Порцию 65 мг неочищенного вещества очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L124 (фиг.7С) в виде белого твердого вещества (15 мг; 0,01 ммоль). Выход 7,9%.
Пример VIII - фиг.8А-C
Синтез L125
Резюме: Метиловый эфир 4-(бромметил)-3-метоксибензойной кислоты А подвергают реакции с NaN3 в ДМФА с получением промежуточного азида В, который затем восстанавливают Ph3P и H2O до амина. Гидролиз С NaOH дает кислоту D, которую непосредственно защищают FmocOSu с получением Е. Амидную смолу Rink функционализируют октапептидом Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) (А), подвергают реакции с Е и затем с три-трет-бутиловым эфиром DOTA. После отщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L125. Общий выход: 0,2%.
А. Синтез метилового эфира 4-(азидометил)-3-метоксибензойной кислоты (В) (фиг.8А)
Раствор метилового эфира 4-(бромметил)-3-метоксибензойной кислоты (8 г; 31 ммоль) и NaN3 (2 г; 31 ммоль) в ДМФА (90 мл) перемешивают в течение ночи при комнатной температуре. Летучие вещества удаляют в вакууме, и неочищенный продукт растворяют в EtOAc (50 мл). Раствор промывают водой (2×50 мл) и сушат. Летучие вещества выпаривают с получением метилового эфира 4-(азидометил)-3-метоксибензойной кислоты (6,68 г; 30 ммоль). Выход 97%.
В. Синтез метилового эфира 4-(аминометил)-3-метоксибензойной кислоты (С) (фиг.8А)
Трифенилфосфин (6,06 г; 23 ммоль) добавляют к раствору метилового эфира 4-(азидометил)-3-метоксибензойной кислоты В (5 г; 22 ммоль) в ТГФ (50 мл): наблюдают выделение водорода и образование белого твердого вещества. Смесь перемешивают в атмосфере азота при комнатной температуре. Через 24 ч добавляют еще трифенилфосфин (0,6 г; 2,3 ммоль). Через 24 ч азид расходуется и добавляют Н2О (10 мл). Через 4 ч белое твердое вещество исчезает. Смесь нагревают при 45°С в течение 3 ч и перемешивают в течение ночи при комнатной температуре. Раствор выпаривают досуха и неочищенный продукт очищают флэш-хроматографией с получением метилового эфира 4-(аминометил)-3-метоксибензойной кислоты С (1,2 г; 6,1 ммоль). Выход 28%.
С. Синтез 4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]-3-метоксибензойной кислоты, (Е) (фиг.8А)
1N раствор NaOH (6, 15 мл; 6,14 ммоль) добавляют по каплям к раствору метилового эфира 4-(аминометил)-3-метоксибензойной кислоты С (1,2 г; 6,14 ммоль) в MeOH (25 мл), при нагревании при 40°С. После перемешивания в течение 8 ч при 45°С раствор перемешивают в течение ночи при комнатной температуре. Добавляют 1N раствор NaOH (0,6 мл; 0,6 ммоль), и смесь нагревают при 40°С в течение 4 ч. Раствор концентрируют, подкисляют 1N HCl (8 мл; 8 ммоль), экстрагируют EtOAc (2×10 мл), затем водный слой концентрируют до 15 мл. Раствор (рН 4,5) охлаждают при 0°С и добавляют Et3N (936 мкл; 6,75 ммоль) (рН 11). Раствор N-(9-флуоренилметоксикарбонилокси)сукцинимида (3,04 г; 9 ммоль) в MeCN (30 мл) добавляют по каплям (конечный рН 9), и белое твердое вещество осаждается. После перемешивания в течение 1 ч при комнатной температуре твердое вещество отфильтровывают, суспендируют в 1N HCl (15 мл) и суспензию перемешивают в течение 30 мин. Твердое вещество отфильтровывают с получением 4-[[[9H-флуорен-9-илметокси)карбонил]амино]метил]-3-метоксибензойной кислоты Е в виде белого твердого вещества (275 мг; 0,7 ммоль).
Фильтрат выпаривают в вакууме, полученный белый остаток суспендируют в 1N HCl (20 мл) и перемешивают в течение 30 мин. Твердое вещество отфильтровывают и очищают флэш-хроматографией с получением дополнительного количества кислоты Е (198 мг; 0,5 ммоль). Общий выход 20%.
D. L125 (N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]метил]-3-метоксибензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) (фиг.8B):
Смолу А (410 мг; 0,24 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение 20 мин, раствор сливают, и смолу промывают DMA (5×7 мл). [4-[[[9Н-флуорен-9-илметокси)карбонил]амино]метил]-3-метоксибензойную кислоту Е (398 мг; 0,98 ммоль), HOBT (151 мг; 0,98 ммоль), DIC (154 мкл; 0,98 ммоль) и DMA (6 мл) добавляют к смоле; смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (6 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (6 мл), и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (618 мг; 0,98 ммоль), НОВТ (151 мг; 0,98 ммоль), DIC (154 мкл; 0,98 ммоль), DIEA (333 мкл; 1,96 ммоль) и DMA (6 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое растирают с Et2O (5 мл). Полученный осадок отделяют центрифугированием, промывают Et2O (5×5 мл), анализируют ВЭЖХ и очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L125 (фиг.8С) в виде белого твердого вещества (15,8 мг; 0,011 ммоль). Выход 4,4%.
Пример IX - фиг.9А-9D
Синтез L146, L233, L234 и L235
Резюме: Продукты получают в несколько стадий, начиная с октапептида Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (BBN[7-14] [SEQ ID NO:1]) (А) на амидной смоле Rink. После конечного отщепления и снятия защиты реагентом В неочищенные продукты очищают препаративной ВЭЖХ с получением L146, L233, L234 и L235. Общие выходы: 10%, 11%, 4,5%, 5,7% соответственно.
А. 3 -[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]аминобензойная кислота (D) (фиг.9А)
Раствор 3-аминобезойной кислоты (0,5 г; 3,8 ммоль) и N-этилдиизопропиламина (DIEA) (0, 64 мл; 3,8 ммоль) в ТГФ (20 мл) добавляют по каплям к раствору хлорида Fmoc-глицина (1,2 г; 4,0 ммоль)(3) в ТГФ (10 мл) и 10% CH2Cl2 (10 мл). После 24 ч перемешивания при комнатной температуре добавляют 1 M HCl (50 мл) (конечный рН: 1,5). Осадок отфильтровывают, промывают Н2О (2×100 мл), сушат в вакууме и кристаллизуют из CHCl3/CH3OH (1:1) с получением В в виде белого твердого вещества (0,7 г; 1,6 ммоль). Выход 43%.
В. N-[3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид) L233 (фиг.9D):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл).
Суспензию встряхивают в течение 20 мин, затем раствор сливают, и смолу промывают DMA (5×7 мл). 3-[[[(9Н-флуорен-9-илметокси)карбонил]амино]ацетил]аминобензойную кислоту, В (0, 50 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 6 ч при комнатной температуре, сливают, и смолу промывают DMA (5× 7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт сложного три-трет-бутилового эфира с NaCl (0,79 г; 1,2 ммоль) (5), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (152 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (50 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L233 (17,0 мг; 11,3×10-3 ммоль) в виде белого твердого вещества. Выход 11%.
C. N-[4-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]фенилацетил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид, L146 (фиг.9D):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). Fmoc-4-аминофенилуксусную кислоту (0,45 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 6 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют Fmoc-глицин (0,36 г; 1,2 ммоль), HATU (0,46 г; 1,2 ммоль) и DIEA (0,40 мл; 2,4 ммоль) и перемешивают в течение 15 мин в DMA (7 мл), затем раствор добавляют к смоле, смесь встряхивают в течение 2 ч при комнатной температуре, сливают и смолу промывают DMA (5×7 мл). Смолу затем встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт три-трет-бутилового эфира DOTA с NaCl (0,79 г; 1,2 ммоль) (5), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), DIEA (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (203 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (50 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L146 (11,2 мг; 7,4×10-3 ммоль) в виде белого твердого вещества. Выход 101%.
D. 6-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]аминонафтойная кислота, С (фиг.9В)
Раствор 6-аминонафтойной кислоты (500 мг; 2,41 ммоль) и DIEA (410 мкл; 2,41 ммоль) в ТГФ (20 мл) добавляют по каплям к раствору Fmoc-глицинхлорида (760 мг; 2,41 ммоль) в CH2Cl2/ТГФ 1:1 (10 мл) и перемешивают при комнатной температуре. Через 24 ч растворитель выпаривают в вакууме. Остаток растворяют в 0,5N HCl (50 мл) и перемешивают в течение 1 ч. Белый твердый осадок фильтруют и сушат. Белое твердое вещество суспендируют в метаноле (30 мл) и кипятят в течение 5 мин, затем фильтруют с получением продукта С (690 мг; 1,48 ммоль). Выход 62%.
Е. N-[6-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]нафтоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид) L234
Смолу А (500 мг; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). 6-[[[(9Н-флуорен-9-илметокси)карбонил]амино]ацетил]аминонафтойную кислоту С (560 мг; 1,2 ммоль), HOBT (184 мг; 1,2 ммоль), DIC (187 мкл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 6 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (6 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт три-трет-бутилового эфира DOTA с NaCl (757 мг; 1,2 ммоль), НОВТ (184 мг; 1,2 ммоль), DIC (187 мкл; 1,2 ммоль), DIEA (537 мкл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в колбе, сливают, и смолу промывают DMA (2×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (144 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (54 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L234 (8 мг; 5,1×10-3 ммоль) в виде белого твердого вещества. Выход 4,5%.
F. 4-[[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]метиламино]бензойная кислота, D (фиг.9С)
Раствор 4-N-метиламинонафтойной кислоты (500 мг; 3,3 ммоль) и DIEA (562 мкл; 3,3 ммоль) в ТГФ (20 мл) добавляют к раствору Fmoc-глицинхлорида (1,04 г; 3,3 ммоль) в CH2Cl2/ТГФ 1:1 (10 мл) и перемешивают при комнатной температуре. Через 24 ч растворитель выпаривают в вакууме. Остаток растворяют в 0,5N HCl (30 мл) и перемешивают в течение 3 ч при 0°С. Белый твердый осадок фильтруют и сушат. Белое твердое вещество очищают флэш-хроматографией с получением соединения D (350 мг; 0,81 ммоль). Выход 25%.
G. N-[4-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]метиламино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид) L235 (фиг.9D)
Смолу А (500 мг; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). 4-[[[[(9Н-флуорен-9-илметокси)карбонил]амино]ацетил]-N-метил]аминобензойную кислоту D (510 мг; 1,2 ммоль), HOBT (184 мг; 1,2 ммоль), DIC (187 мкл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 6 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт три-трет-бутилового эфира DOTA с NaCl (757 мг; 1,2 ммоль), НОВТ (184 мг; 1,2 ммоль), DIC (187 мкл; 1,2 ммоль), DIEA (537 мкл; 2, 4 ммоль) и DMA (7 мл). Смесь встряхивают в колбе, сливают, и смолу промывают DMA (2×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок.
Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (126 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (53 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L235 (11 мг; 7,2×10-3 ммоль) в виде белого твердого вещества. Выход 5,7%.
Пример X - фиг.10А-В
Синтез L237
Резюме: 1-формил-1,4,7.10-тетраазациклододекан (А) селективно защищают бензилхлорформиатом при рН 3 с получением В, которое алкилируют трет-бутилбромацетатом и деформилируют гидрохлоридом гидроксиламина с получением D. Взаимодействие с P(OtBu)3 и фармальдегидом дает Е, защиту которого снимают гидрированием, и алкилируют бензилбромацетатом с получением G, которое окончательно гидрируют до Н. Амидную смолу Rink функционализируют октапептидом GlnTrpAlaValGlyHisLeuMetNH2 (BBN[7-14]) (А) последовательно подвергают реакции с Fmoc-4-аминобензойной кислотой, Fmoc-глицином и Н. После отщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L237. Общий выход: 0,21%.
А. Дигидрохлорид фенилметилового эфира 7-формил-1,4,7,10-тетраазациклододекан-1-карбоновой кислоты, В (фиг.10А)
1-формил-1,4,7,10-тетраазациклододекан (А) (14 г; 69,9 ммоль) растворяют в Н2О (100 мл) и добавляют 12N HCl (11 мл) до тех пор, пока рН не будет 3, затем добавляют 1,4-диоксан (220 мл). Раствор бензилхлорформиата (13,8 г; 77 ммоль) в 1,4-диоксане (15 мл) медленно добавляют по каплям в течение 3,5 ч, постоянно поддерживая реакционную смесь при рН 3 непрерывным добавлением 2N NaOH (68,4 мл) аппаратом "pHstat". В конце добавления реакционную смесь перемешивают в течение 1 ч, затем промывают н-гексаном (4×100 мл) и изо-Pr2O (4×100 мл). рН водной фазы доводят до 13 добавлением 10N NaOH (6,1 мл) и экстрагируют CHCl3 (4×100 мл). Органическую фазу промывают раствором соли (100 мл), сушат (Na2SO4), фильтруют и выпаривают. Маслянистый остаток растворяют в ацетоне (200 мл) и добавляют 6N HCl (26 мл). Осажденное твердое вещество отфильтровывают, промывают ацетоном (2×50 мл) и сушат в вакууме с получением соединения В (23,6 г; 58 ммоль) в виде белого кристаллического твердого вещества. Выход 83%.
В. Бис(1,1-диметилэтиловый) эфир 4-(фенилметокси)карбонил-1,4,7,10-тетраазациклододекан-1,7-диуксусной кислоты, D (фиг.10А)
Раствор В (14,4 г; 35,3 ммоль) в Н2О (450 мл) и 1N NaOH (74 мл; 74 ммоль) перемешивают в течение 20 мин, затем экстрагируют CHCl3 (4×200 мл). Органический слой выпаривают с получением маслянистого остатка (12,3 г), который растворяют в CH3CN (180 мл) и N-этилдиизопропиламине (DIEA) (15 мл; 88,25 ммоль). Раствор трет-бутилбромацетата (16,8 г; 86,1 ммоль) в CH3CN (15 мл) добавляют по каплям к предыдущему раствору в течение 2,5 ч. Через 20 ч при комнатной температуре растворитель выпаривают, маслянистый остаток растворяют в CHCl3 (150 мл) и промывают Н2О (5×100 мл). Органический слой сушат (Na2SO4), фильтруют и выпаривают досуха с получением С в виде желтого масла. Неочищенный С (22 г) растворяют в EtOH (250 мл), добавляют NH2OH·HCl (2,93 г; 42,2 ммоль) и раствор кипятят с обратным холодильником. Через 48 ч растворитель выпаривают, остаток растворяют в CH2Cl2 (250 мл), промывают Н2О (3×250 мл), затем раствором соли (3×250 мл). Органический слой сушат (Na2SO4), фильтруют и выпаривают. Маслянистый остаток (18,85 г) очищают хроматографией. Фракции, содержащие продукт, собирают и выпаривают с получением стекловидного белого твердого вещества (17,62 г), которое растворяют в Н2О (600 мл) и 1N NaOH (90 мл; 90 ммоль) и экстрагируют CHCl3 (3×250 мл). Органический слой сушат (Na2SO4) и выпаривают досуха с получением D (16,6 г; 31 ммоль) в виде масла. Выход 88%.
С. Бис(1,1-диметилэтиловый) эфир 4-(фенилметокси)карбонил-10-[[бис(1,1-диметилэтокси)фосфонил]метил]-1, 4,7,10-тетраазациклододекан-1,7-диуксусной кислоты, D (фиг.10А)
Смесь соединения D (13,87 г; 26 ммоль), P(OtBu)3 (7,6 г; 28,6 ммоль) (10) и параформ (0,9 г; 30 ммоль) нагревают при 60°С. Через 16 ч добавляют еще P(OtBu)3 (1 г; 3,76 ммоль) и параформ (0,1 г; 3,33 ммоль). Реакционную смесь нагревают при 60°С в течение еще 20 ч, затем при 80°С в вакууме для устранения летучих примесей. Неочищенное вещество очищают флэш-хроматографией с получением Е (9,33 г; 8 ммоль) в виде масла. Выход 31%.
D. 1-фенилметил-4,10-бис(1,1-диметилэтиловый) эфир 7-[[бис(1,1-диметилэтокси)фосфинил]метил]-1,4,7,10- тетраазациклододекан-1,4,10-триуксусной кислоты, G (фиг.10А)
К раствору Е (6,5 г; 5,53 ммоль) в CH3OH (160 мл) добавляют 5% Pd/C (1 г; 0,52 ммоль) и смесь перемешивают в атмосфере водорода при комнатной температуре. Через 4 ч (израсходованный Н2 165 мл; 6,7 ммоль) смесь фильтруют через фильтр Millipore® (FT 0,45 мкм) и раствор выпаривают при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией с получением F (4,2 г) в виде масла. Бензилбромацетат (1,9 г; 8,3 ммоль), растворенный в CH3CN (8 мл), добавляют по каплям в течение 1 ч к раствору F (4,2 г) в CH3CN (40 мл) и DIEA (1,5 мл; 8,72 ммоль). Через 36 ч при комнатной температуре растворитель выпаривают и остаток (5,76 г) растворяют в CHCl3 (100 мл), промывают Н2О (2×100 мл), затем раствором соли (2×70 мл). Органический слой сушат (Na2SO4), фильтруют и выпаривают. Неочищенное вещество (5,5 г) очищают дважды флэш-хроматографией, фракции собирают и выпаривают досуха с получением G (1,12 г; 1,48 ммоль) в виде масла. Выход 27%.
Е. 4,10-бис(1,1-диметилэтиловый) эфир 7-[[бис(1,1-диметилэтокси)фосфинил]метил]-1,4,7,10-тетраазациклододекан-1,4,10-триуксусной кислоты, Н (фиг.10А)
К раствору G (1,12 г; 1,48 ммоль) в CH3OH (27 мл) добавляют 5% Pd/C (0,2 г; 0,087 ммоль) и смесь перемешивают в атмосфере водорода при комнатной температуре. Через 2 ч (израсходованный Н2 35 мл; 1,43 ммоль) смесь фильтруют через фильтр Millipore® (FT 0,45 мкм) и раствор выпаривают досуха с получением Н (0,94 г; 1,41 ммоль) в виде бледно-желтого масла. Выход 97%.
F. N-[4-[[[[[4, 10-бис(карбоксиметил)-7-(дигидроксифосфинил)метил-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид, L237 (фиг.10B):
Смолу А (330 мг; 0,20 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (5 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (5 мл). Суспензию перемешивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×5 мл). Fmoc-4-аминобензойную кислоту (290 мг; 0, 80 ммоль), HOBT (120 мг; 0,80 ммоль), DIC (130 мкл; 0,80 ммоль) и DMA (5 мл) добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×5 мл). Затем смолу встряхивают с 50% морфолином в DMA (5 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (5 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×5 мл). Fmoc-глицин (240 мг; 0,8 ммоль), HATU (310 мг; 0,8 ммоль) и DIEA (260 мкл; 1,6 ммоль) перемешивают в течение 15 мин в DMA (5 мл), затем раствор добавляют к смоле, смесь встряхивают в течение 2 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×5 мл). Затем смолу встряхивают с 50% морфолином в DMA (5 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (5 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×5 мл). К смоле добавляют Н (532 мг; 0, 80 ммоль), НОВТ (120 мг; 0,80 ммоль), DIC (130 мкл; 0,80 ммоль), DIEA (260 мкл; 1,6 ммоль) и DMA (5 мл). Смесь встряхивают в колбе в течение 40 ч при комнатной температуре, сливают и смолу промывают DMA (5×5мл), CH2Cl2 (5×5 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4 ч. Смолу фильтруют и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (90 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (50 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L237 (6 мг; 3,9×10-3 ммоль) в виде белого твердого вещества. Выход 3,5%.
Пример XI - фиг.11А-В
Синтез L238 и L239
Резюме: Продукты получают в несколько стадий, начиная с октапептида GlnTrpAlaValGlyHisLeuMetNH2 (BBN[7-14]) (А) на амидной смоле Rink. После отщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L238 и L239. Общий выход: 14 и 9% соответственно.
А. N,N-диметилглицил-L-серил-[S-[(ацетил]метил]]-L-цистеинилглицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид, L238 (фиг.11А):
Смолу А (0,5 г; 0,30 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). Fmoc-4-аминобензойную кислоту (0,43 г; 1,2 ммоль), HOBT (0, 18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). Fmoc-глицин (0,36 г; 1,2 ммоль), HATU (0,46 г; 1,2 ммоль) и N-этилдиизопропиламин (0,40 мл; 2,4 ммоль) перемешивают в течение 15 мин в DMA (7 мл), затем раствор добавляют к смоле, смесь встряхивают в течение 2 ч при комнатной температуре, сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют N-α -Fmoc-S-ацетамидометил-L-цистеин (0,50 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл), смесь встряхивают в течение 3 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют N-α-Fmoc-О-трет-бутил-L-серин (0,460 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл), смесь встряхивают в течение 3 ч при комнатной температуре, сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин (7 мл) и смесь встряхивают в течение еще 20 мин.
Раствор сливают, и смолу промывают DMA (5×7 мл). N,N-диметилглицин (0,12 г; 1,2 ммоль), HATU (0,46 г; 1,2 ммоль) и N-этилдиизопропиламин (0,40 мл; 2,4 ммоль) перемешивают в течение 15 мин в DMA (7 мл), затем раствор добавляют к смоле. Смесь встряхивают в течение 2 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (169 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (60 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L238 (22,0 мг; 0,015 ммоль) в виде белого твердого вещества. Выход 14%.
B. N, N-диметилглицил-L-серил-[S-[(ацетиламино)метил]]-L-цистеинилглицил-(3β,5β,7α,12α)-3-амино-7, 12-дигидрокси-24-оксохолан-24-ил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида) L239 (фиг.11В):
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). (3β,5β,7α,12α)-3-[[[9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксихолан-24-овую кислоту В (0,82 г; 1,2 ммоль)(7), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют N-α-Fmoc-S-ацетиламидометил-L-цистеин (0,50 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл), смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). N-α-Fmoc-O-трет-бутил-L-серин (0,46 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, и добавляют свежий 50% морфолин в DMA (7 мл), и смесь встряхивают в течение еще 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). N,N-диметилглицин (0,12 г; 1,2 ммоль), HATU (0,46 г; 1,2 ммоль) и N-этилдиизопропиламин (0,40 мл; 2,4 ммоль) перемешивают в течение 15 мин в DMA (7 мл), затем раствор добавляют к смоле.
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). (3β,5β,7α,12α )-3-[[[9Н-флуорен-9-илметокси)амино]ацетил]амино-7,12-дигидроксихолан-24-овую кислоту В (0,82 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). К смоле добавляют N-α -Fmoc-S-ацетиамидометил-L-цистеин (0,50 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл), смесь встряхивают в течение 3 ч при комнатной температуре, раствор сливают и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). N-α-Fmoc-O-трет-бутил-L-серин (0,46 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 3 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Смолу затем встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл), и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). N,N-диметилглицин (0,12 г; 1,2 ммоль), HATU (0, 46 г; 1,2 ммоль) и N-этилдиизопропиламин (0,40 мл; 2,4 ммоль) перемешивают в течение 15 мин в DMA (7 мл), затем раствор добавляют к смоле.
Пример XII - фиг.12А-F
Синтез L240, L241, L242
Резюме: Продукты получают в несколько стадий, начиная с октапептида GlnTrpAlaValGlyHisLeuMetNH2 (BBN[7-14]) (А) на амидной смоле Rink. После отщепления и снятия защиты реагентом В неочищенные продукты очищают препаративной ВЭЖХ с получением L240, L241 и L242. Общие выходы: 7,4, 3,2, 1,3% соответственно.
А. 4-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-метоксибензойная кислота А (фиг.12А)
Раствор 4-амино-3-метоксибезойной кислоты (1,0 г; 5,9 ммоль) и N-этилдиизопропиламина (1,02 мл; 5,9 ммоль) в ТГФ (20 мл) добавляют по каплям к раствору Fmoc-глицилхлорида (1,88 г; 5,9 ммоль) в CH2Cl2/ТГФ 1:1 (20 мл) и перемешивают при комнатной температуре в атмосфере N2. Через 3 ч растворитель выпаривают в вакууме. Остаток растворяют в 0,5N HCl (50 мл), перемешивают в течение 1 ч при 0°С, затем фильтруют и сушат. Белое твердое вещество суспендируют в MeOH (30 мл) и перемешивают в течение 1 ч, затем фильтруют и суспендируют в растворе CHCl3/гексан 1:4 (75 мл). Суспензию фильтруют с получением соединения А в виде белого твердого вещества (1,02 г; 2,28 ммоль). Выход 39%.
В. N-[4-[[[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]глицил]амино]-3-метоксибензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид L240
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). 4-[[[(9Н-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-метоксибензойную кислоту, А (0,50 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 5 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), N-этилдиизопропиламин (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (5×20 мл) с получением твердого вещества (152 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (52 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L240 (12,0 мг; 7,8×10-3 ммоль) в виде белого твердого вещества. Выход 7,4%.
С. 4-амино-3-хлорбензойная кислота С (фиг.12В)
1N NaOH (11 мл; 11 ммоль) добавляют к раствору 4-амино-3-хлорбензоата метила (2 г; 10,8 ммоль) в MeOH (20 мл) при 45°С. Реакционную смесь перемешивают в течение 5 ч при 45°С и в течение ночи при комнатной температуре. Добавляют еще 1N NaOH (5 мл; 5 ммоль), и реакционную смесь перемешивают при 45°С в течение 2 ч. После отгонки растворителя добавляют 1N HCl (16 мл). Твердый осадок фильтруют и сушат с получением 4-амино-3-хлорбензойной кислоты, С в виде белого твердого вещества (1,75 г; 10,2 ммоль). Выход 94,6%.
D. 4-[[[(9Н-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-хлорбензойная кислота, D (фиг.12В)
Раствор 4-амино-3-хлорбензойной кислоты (1,5 г; 8,75 ммоль) и N-этилдиизопропиламина (1,46 мл; 8,75 ммоль) в ТГФ (50 мл) добавляют по каплям к раствору Fmoc-глицилхлорида (2,76 г; 8,75 ммоль) в CH2Cl2/ТГФ 1:1 (30 мл) и перемешивают при комнатной температуре в атмосфере N2. Через 3 ч растворитель выпаривают в вакууме. Остаток растворяют в 0,5N HCl (50 мл), фильтруют и сушат.
Белое твердое вещество суспендируют в MeOH (30 мл) и перемешивают в течение 1 ч, затем фильтруют и сушат с получением 4-[[[(9Н-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-хлорбензойной кислоты (2,95 г; 6,5 ммоль). Выход 75%.
Е. N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]глицил]амино]-3-хлорбензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид, L241 (фиг.12Е):
Смолу А (0, 5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). 4-[[[(9Н-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-хлорбензойную кислоту (0,54 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 5 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл).
Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,79 г; 1,2 ммоль), HOBT (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), N-этилдиизопропиламин (0,40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 40 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (5×20 мл) с получением твердого вещества (151 мг), которое анализируют ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L241 (5,6 мг; 3,6×10-3 ммоль) в виде белого твердого вещества. Выход 3,2%.
F. 4-[[[(9H-флуорен-9-илметокси)карбонил]амино]ацетил]амино-3-метилбензойная кислота, Е (фиг.12С)
Раствор 4-амино-3-метилбензойной кислоты (0,81 г; 5,35 ммоль) и N-этилдиизопропиламин (0,9 мл; 5,35 ммоль) в ТГФ (30 мл) добавляют по каплям к раствору Fmoc-глицилхлорида (1,69г; 5,35 ммоль) в CH2 Cl2/ТГФ 1:1 (20 мл) и перемешивают при комнатной температуре в атмосфере N2. Через 3 ч растворитель выпаривают в вакууме. Остаток растворяют в 0,5N HCl (50 мл) и перемешивают в течение 3 ч при 0°С, затем фильтруют и сушат. Белое твердое вещество суспендируют в МеОН (50 мл) и перемешивают в течение 1 ч, затем фильтруют и сушат с получением соединения Е (1,69 г; 3,9 ммоль). Выход 73%.
G. N-[4-[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]глицил]амино]-3-метилбензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид) L242
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). 4-[[(9Н-флуорен-9-илметокси)амино]ацетил]амино-3-метилбензойную кислоту, Е (0,52 г; 1,2 ммоль), HOBT (184 мг; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 5 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (6 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают и смолу промывают DMA (5×7 мл). К смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl (0,76 г; 1,2 ммоль), НОВТ (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), N-этилдиизопропиламин (0,40 мл; 2,4 ммоль) и DMA (7 мл).
Смесь встряхивают в течение 40 ч при комнатной температуре, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (5×20 мл) с получением твердого вещества (134 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (103 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L242 (4,5 мг; 2,9×10-3 ммоль) в виде белого твердого вещества. Выход 1,3%.
Пример XIII - фиг.13А-C
Синтез L244
Резюме: Продукт получают в несколько стадий, начиная с октапептида GlnTrpAlaValGlyHisLeuMetNH2 (BBN[7-14]) на амидной смоле Rink (А). Конечную стадию сочетания с три-трет-бутиловым эфиром DOTA проводят в фазе раствора после отщепления и снятия защиты реагентом В линкера-бомбезина [7-14]. Неочищенный продукт очищают препаративной ВЭЖХ с получением L244. Общий выход: 0,4%.
А. N,N'-(иминоди-2,1-этандиил)бис[2,2,2-трифторацетамид], A (фиг.13А)
Этиловый эфир трифторуксусной кислоты (50 г; 0,35 ммоль) по каплям выливают в раствор диэтилентриамина (18 г; 0,175 ммоль) в ТГФ (180 мл) при 0°С в течение 1 ч. Через 20 ч при комнатной температуре смесь выпаривают до маслянистого остатка (54 г). Масло кристаллизуют из Et2O (50 мл), фильтруют, промывают охлажденным Et2O (2×30 мл) и сушат с получением А в виде белого твердого вещества (46 г; 0,156 ммоль). Выход: 89%.
В. N,N'-бис[2-(трифторацетил)аминоэтил]амино]-4-оксобутановая кислота, В (фиг.13А)
Янтарный ангидрид (0,34 г; 3,4 ммоль) добавляют в раствор А (1 г; 3,4 ммоль) в ТГФ (5 мл) при комнатной температуре. Через 28 ч неочищенный продукт концентрируют до получения остатка (1,59 г), промывают EtOAc (2×10 мл) и 1N HCl (2×15 мл). Органический слой сушат над Na2SO4, фильтруют и выпаривают с получением маслянистого остатка (1,3 г), который очищают флэш-хроматографией (5) с получением В в виде масла (0,85 г; 2,15 ммоль). Выход 63%.
С. 4-[N,N'-бис[2-[(9H-флуорен-9-илметокси)карбонил)аминоэтил]амино]-4-оксобутановая кислота, D (фиг.13А)
Янтарный ангидрид (2 г; 20 ммоль) добавляют в раствор А (5 г; 16,94 ммоль) в ТГФ (25 мл) при комнатной температуре. Через 28 ч неочищенный продукт концентрируют до получения остатка (7 г), промывают в этилацетате (100 мл) и 1N HCl (2×50 мл). Органический слой сушат над Na2SO4, фильтруют и выпаривают с получением В в виде маслянистого остатка (6,53 г). 2N NaOH (25 мл) добавляют к суспензии неочищенного В (5 г) в EtOH (35 мл), получая полный раствор через 1 ч при комнатной температуре. Через 20 ч растворитель выпаривают с получением С в виде масла (8,48 г). Раствор 9-флуоренилметилхлорформиата (6,54 г; 25,3 ммоль) в 1,4-диоксане (30 мл) по каплям добавляют в раствор С в 10% водном Na2CO3 (30 мл) через 1 ч при 0°С. Через 20 ч при комнатной температуре получают желеобразную суспензию и фильтруют с получением белого твердого вещества (3,5 г) и желтого раствора. Раствор выпаривают, остающийся водный раствор разбавляют Н2О (150 мл) и экстрагируют EtOAc (70 мл). Свежий EtOAc (200 мл) добавляют к водной фазе, получая суспензию, которую охлаждают до 0°С и подкисляют до рН 2 концентрированной HCl. Органический слой промывают Н2О (5×200 мл) до достижения нейтрального рН, затем сушат с получением стекловидного твердого вещества (6,16 г). Соединение суспендируют в кипящем н-гексане (60 мл) в течение 1 ч, фильтруют с получением D в виде белого твердого вещества (5,53 г, 8,54 ммоль). Общий выход 50%.
D. N-[4-[[4-[бис-[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]этил]амино-1, 4-диоксибутил]амино]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид, L244 (фиг.13В)
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию встряхивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). 4-[N,N'-бис[2-[9Н-флуорен-9-илметокси)карбонил]аминоэтил]амино]-4-оксобутановую кислоту (777,3 мг; 1,2 ммоль), HOBT (184 мг; 1,2 ммоль), DIC (187 мкл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 40 ч при комнатной температуре, раствор сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение 20 мин. Раствор сливают, и смолу промывают DMA (2×7 мл) и CH2Cl2 (5×7 мл), затем встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (5×20 мл) с получением F в виде белого твердого вещества (140 мг). Три-трет-бутиловый эфир DOTA (112 мг; 0,178 ммоль), HATU (70 мг; 0,178 ммоль) и DIEA (60 мкл; 0,356 ммоль) добавляют к раствору F (50 мг; 0,0455 ммоль) в DMA (3 мл) и CH2Cl2 (2 мл) и перемешивают в течение 24 ч при комнатной температуре. Неочищенный продукт выпаривают до уменьшенного объема (1 мл) и встряхивают с реагентом В (25 мл) в течение 4,5 ч. После выпаривания растворителя остаток обрабатывают Et2O (20 мл) с получением осадка. Осадок отделяют центрифугированием и промывают Et2O (5×20 мл) с получением бежевого твердого вещества (132 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного вещества (100 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L244 (фиг.13С) (3,5 мг; 1,84×10-3 ммоль) в виде белого твердого вещества. Выход 0,8%.
Общие экспериментальные методики для примеров XIV-XLII
L201-L228
Неавтоматизированное сочетание
6,0 эквивалентов соответствующим образом защищенной аминокислоты обрабатывают 6,0 эквивалентами каждого из соединений HOBT и DIC и активируют вне реакционного сосуда. Эту активированную карбоновую кислоту в NMP затем переносят в смолу, содержащую амин, и реакцию проводят в течение 4-6 ч, затем смолу подвергают дренажу и промывают.
Специальное сочетание Fmoc-Gly-OH с 4-аминобензойной кислотой и амидами аминобифенилкарбоновой кислоты:
Fmoc-Gly-OH (10,0 эквивалентов) обрабатывают HATU (10,0 эквивалентов) и DIEA (20,0 эквивалентов) в NMP (10 мл NMP используют на 1 г аминокислоты по массе), и раствор перемешивают в течение 10-15 мин при комнатной температуре перед переносом в сосуд, содержащий смолу, нагруженную амином. Объем раствора доводят до 15,0 мл для каждого грамма смолы. Сочетание продолжают в течение 20 ч при комнатной температуре и из смолы удаляют все реагенты. Эту процедуру повторяют еще 1 раз и затем промывают NMP перед переходом к следующей стадии.
Получение моноамида DO3A:
8,0 эквивалентов монокислоты DOTA растворяют в NMP и обрабатывают 8,0 эквивалентами HBTU и 16,0 эквивалентами DIEA. Раствор перемешивают в течение 15 мин при комнатной температуре, затем переносят в амин на смоле и сочетание продолжают в течение 24 ч при комнатной температуре. Затем смолу подвергают дренажу, промывают, и затем пептид отщепляют и очищают.
Отщепление неочищенного пептида от смолы и очистка:
Смолу суспендируют в реагенте В (15,0 мл/г) и встряхивают в течение 4 ч при комнатной температуре. Затем смолу подвергают дренажу и снова промывают 2×5 мл реагента В и объединяют с предыдущим фильтратом. Фильтрат затем концентрируют при пониженном давлении до состояния пасты/жидкости при комнатной температуре и растирают с 25,0 мл безводного эфира (на каждый грамм используемой смолы). Затем суспензию центрифугируют и слой простого эфира удаляют. Эту процедуру повторяют еще 2 раза и бесцветный осадок после промывки эфиром очищают препаративной ВЭЖХ.
Пример XIV - фиг.21
Синтез L201
Используют 0,5 г Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолы (0,4 ммоль/г, 0,5 г, 0,2 ммоль) (смола А). Добавляют остальные аминокислотные звенья, как описано в общей методике, с получением (1R)-1-(бис{2-[бис(карбоксиметил)амино]этил}амино)пропан-3-карбоновая кислота-1-карбоксилглицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L201). Выход 17,0 мг (5,4%).
Пример XV - фиг.22А и 22В
Синтез L202
4-Fmoc-гидразинобензойная кислота (фиг.22А):
Суспензию 4-гидразинобензойной кислоты (5,0 г, 32,9 ммоль) в воде (100 мл) обрабатывают карбонатом цезия (21,5 г, 66,0 ммоль). К указанному выше раствору по каплям добавляют Fmoc-Cl (9,1 г, 35,0 ммоль) в ТГФ (25 мл) при перемешивании в течение периода 1 ч. Раствор перемешивают еще в течение 4 ч после добавления, реакционную смесь концентрируют примерно до 75 мл и экстрагируют эфиром (2×100 мл). Эфирный слой удаляют, и водный слой подкисляют 2N HCl. Выделенное твердое вещество отфильтровывают, промывают водой (5×100 мл) и затем перекристаллизовывают из ацетонитрила с получением продукта (соединение В) в виде бесцветного твердого вещества. Выход: 11,0 г (89%).
1H ЯМР (ДМСО-d6): δ 4,5 (м, 1H, Ar-CH2-CH), 4,45 (м, 2H, Ar-CH2), 6,6 (уш.с, 1H, Ar-H), 7,4-7,9 (м, 9, Ar-H и Ar-CH2), 8,3 (с, 2H, Ar-H), 9,6 (с, 2H, Ar-H). МС: m/z 373,2 [M-H].
Используют 0,5 г Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолы (0,4 ммоль/г, 0,5 г, 0,2 ммоль) (смола А). Аминокислотные звенья добавляют, как описано в общей методике, включая соединение В, с получением N-[(3β,5β,12α)-3-[[[[[4, 7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-4-гидразинобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L202 ) (фиг.22В), Выход: 25,0 мг (8,3%).
Пример XVI - фиг.23А и фиг.23В
Синтез L203
Получение 4-Fmoc-аминомензилбензоата, соединения В, фиг.23А:
Суспензию 4-boc-аминобензойной кислоты (0,95 г, 4,0 ммоль) в безводном ацетонитриле (10,0 мл) обрабатывают порошкообразным карбонатом цезия (1,3 г, 4,0 ммоль) и энергично перемешивают в атмосфере азота. Добавляют бензилбромида (0,75 г, 4,4 ммоль), и реакционную смесь кипятят с обратным холодильником в течение 20 ч в атмосфере азота. Затем реакционную смесь выливают в ледяную воду (200 мл), выделенное твердое вещество отфильтровывают и промывают водой (5×50 мл). Неочищенное вещество затем перекристаллизовывают из водного метанола с получением продукта в виде бесцветного твердого вещества (соединение В). Выход: 0,8 г (61%).
1H ЯМР (CDCl3): δ 1,5 (с, 9H, третичные метилы), 5,4 (с, 2H, Ar-CH2), 7,4 (м, 7H, Ar-H) и 8,0 (м, 2H, Ar-H). МС: m/z 326,1 [M+H].
4-Аминобензилбензоат - Соединение С, фиг.23А:
4-Boc-аминобензилбензоат (0,8 г; 2,5 ммоль) растворяют в DCM (20 мл), содержащем TFA (25% по объему) и перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь выливают в 100,0 г колотого льда и нейтрализуют насыщенным раствором бикарбоната натрия до тех пор, пока рН не достигнет примерно 8,5. Органический слой отделяют, и водный слой экстрагируют DCM (3×20 мл), и все органические слои объединяют. Затем слой DCM промывают 1×50 мл насыщенного бикарбоната натрия, водой (2×50 мл) и сушат (сульфат натрия). Удаление растворителя дает бесцветное твердое вещество (соединение С), которое направляют на следующую стадию без дальнейшей очистки. Выход: 0,51 г (91%).
1H ЯМР (CDCl3): δ 5,3 (с, 2H, Ar-CH2), 6,6 (д, 2H, Ar-H, J=1,0 Гц), 7,4 (м, 5H, Ar-H, J=1,0 Гц) и 7,9 (д, 2H, Ar-H, J=1,0 Гц).
4-(2-Хлорацетил)аминобензилбензоат, соединение D (фиг.23А):
Амин (0,51 г; 2,2 ммоль) растворяют в безводном диметилацетамиде (5,0 мл) и охлаждают на льду. По каплям через шприц добавляют хлорацетилхлорида (0,28 г, 2,5 ммоль), раствор оставляют нагреваться до комнатной температуры и перемешивают в течение 2 ч. Добавляют дополнительные 2,5 ммоль хлорацетилхлорида, и перемешивание продолжают еще 2 ч. Затем реакционную смесь выливают в ледяную воду (100 мл). Осажденное твердое вещество отфильтровывают, промывают водой и затем перекристаллизовывают из смеси гексан/эфир с получением бесцветного твердого вещества (соединение D). Выход: 0,38 г (56%).
1H ЯМР (CDCl3): δ 4,25 (с, 2H, CH2-Cl), 5,4 (с, 2H, Ar-H), 7,4 (м, 5H, Ar-H), 7,6 (д, 2H, Ar-H), 8,2 (д, 2H, Ar-H) и 8,4 (с, 1H, -CONH).
трет-бутил 2-{1,4,7,10-тетрааза-7, 10-бис{[(трет-бутил)оксикарбонил]метил}-4-[(N-{4-[бензилоксикарбонил]фенил}карбамоил]циклододецил}ацетат, соединение Е, фиг.23А:
Три-трет-бутиловый эфир DO3A.HCl (5,24 г, 9, 5 ммоль) суспендируют в 30,0 мл безводного ацетонитрила, добавляют безводный карбонат калия (2,76 г, 20 ммоль) и перемешивают в течение 30 мин. Затем к указанной выше смеси в течение 10 мин по каплям добавляют хлорацетамид D (2,8 г, 9,2 ммоль) в безводном ацетонитриле (20,0 мл). Затем реакционную смесь перемешивают в течение ночи. Раствор фильтруют, и затем концентрируют при пониженном давлении до состояния пасты. Пасту растворяют примерно в 200 мл воды и экстрагируют 5×50 мл этилацетата. Объединенный органический слой промывают водой (2×100 мл) и сушат (сульфат натрия). Раствор фильтруют, выпаривают при пониженном давлении до состояния пасты, и пасту подвергают флэш-хроматографии на силикагеле (600,0 г). При элюировании 5% метанолом в DCM элюируется продукт. Все фракции, которые однородны по данным ТСХ, объединяют и выпаривают с получением бесцветной смолы. Смолу перекристаллизовывают из изопропилового эфира и DCM с получением соединения Е. Выход: 4,1 г (55%).
1H ЯМР (CDCl3): δ 1,5 (с, 27H, метилы), 2,0-3,75 (м, 24H, группы NCH2), 5,25 (д, 2H, Ar-CH2), 7,3 (м, 5H, Ar-H), 7,8 (д, 2H, Ar-H) и 7,95(д, 2H, Ar-H). МС: m/z 804,3 [M+H].
Восстановление указанной выше кислоты Е с получением соединения F, фиг.23А:
Сложный бензиловый эфир Е из указанной выше стадии (1,0 г, 1,24 ммоль) растворяют в смеси метанол-вода (10,0 мл, 95:5) и добавляют палладий на угле (10%, 0,2 г). Раствор затем гидрируют, используя аппарат Парра, при 50 фунт/кв.дюйм в течение 8 ч. Раствор отфильтровывают от катализатора и затем концентрируют при пониженном давлении с получением бесцветного рыхлого твердого вещества F. Его далее не очищают и сразу направляют на следующую стадию. МС: m/z 714,3 [M+Na].
Получение L203 (фиг.23В)
Указанную выше кислоту F сочетают с амином на смоле А [H-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смола] с использованием описанных выше стандартных методик сочетания. 0,5 г (0,2 ммоль) смолы дает 31,5 мг конечного очищенного пептида (10,9%), N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L203) (фиг.23В).
Пример XVII - фиг.24
Синтез L204
Используют Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,5 г, 0,2 ммоль) (смола А). Сначала загружают Fmoc-Gly-OH и затем F по указанной выше методике (фиг.23А), используя стандартные условия сочетания. Выход: 24,5 мг (8,16%) N-[(3β,5β,12α)-3-[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]-4-аминобензоилглицил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L204) (фиг.24).
Пример XVIII - фиг.25
Синтез L205
Fmoc-6-аминоникотиновую кислоту1 получают, как описано в литературе ("Synthesis of diacylhydrazine compounds for therapeuic use". Hoelzemann, G.; Goodman, S. ( Merk Patent G.m.b.H. Germany). Ger.Offen. 200, 16 pp. CODEN: GWXXBX DE 19831710 A1 20000120) и сочетают с предварительно загруженной Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолой (0,5 г, 0,2 ммоль) (смола А) с последующей другой аминогруппой, как указано выше, с получением N-[(3β, 5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]-4-аминобензоилглицил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L205). Выход: 1,28 мг (0,4%).
Пример XIX - фиг.26A и 26В
Синтез L206
4'-Fmoc-амино-3'-метилбифенил-4-карбоновая кислота В:
Аминокислоту (0,41 г, 1,8 ммоль) растворяют в растворе карбоната цезия (0,98 г, 3,0 ммоль) в 10,0 мл воды. См. "Rational Desing of Diflunisal Analoge with Reduced Affinity for Human Serum Albumin" Mao, H. et al., J. Am. Chem. Soc., 2001, 123(43), 10429-10435. Раствор охлаждают на ледяной бане и по каплям добавляют раствор Fmoc-Cl (0,52 г, 2,0 ммоль) в ТГФ (10,0 мл) при энергичном перемешивании. После добавления реакционную смесь перемешивают при комнатной температуре в течение 20 ч. Раствор затем подкисляют 2N HCl. Осажденное твердое вещество отфильтровывают, промывают водой (3×20 мл) и сушат на воздухе. Затем неочищенное твердое вещество перекристаллизовывают из ацетонитрила с получением бесцветного рыхлого твердого вещества В (фиг.26А). Выход: 0,66 г (75%).
1H ЯМР (ДМСО-d6): δ 2,2 (с, Ar-Me), 4,25 (т, 1H, Ar-CH, J=5 Гц), 4,5 (д, 2H, O-CH2, J=5,0 Гц), 7,1 (уш.с, 1H, CONH), 7,4-8,0 (м, 8H, Ar-H) и 9,75 (уш.с, 1H, -COOH). МС: m/z 472,0 [M-H].
Кислоту В из указанной выше стадии сочетают с Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолой (0,2 г, 0,08 ммоль) (смола А ) в стандартных условиях сочетания. Дополнительные группы добавляют, как указано выше, с получением N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-[4'-амино-2'-метилбифенил-4-карбоксил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида (L206). Выход 30, 5 мг (24%).
Пример XX - фиг.27A-В
Синтез L207
3'-Fmoc-аминобифенил-3-карбоновую кислоту получают из соответствующего амина с использованием описанной выше методики (см. "Synthesis of 3'-methyl-4'-nitrobiphenylcarboxylic acid by reaction of 3-methyl-4-nitrobenzenediazonium acetate with methyl benzoate", Bayland, E. and Gorrod, J., J. Chem. Abstracts (1962), 2209-2211). 0,7 г амина дает 0,81 г FMoc-производного (58%) (Соединение В, фиг.27А).
1H ЯМР (ДМСО-d6): δ 4,3 (т, 1H, Ar-CH), 4,5 (д, 2H, O-CH2), 7,25-8,25 (м, 16H, Ar-H) и 9,9 (с, 1H, -COOH). МС: m/z 434 [M-H].
Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,2 г, 0,08 ммоль) (смола А) сочетают с указанной выше кислотой В и дополнительными группами, как указано выше (фиг.27В). Получают 29,0 мг N-[(3β,5β,12α)-3-[[[[[4,7, 10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-[3'-аминобифенил-3-карбоксил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида ( L207) (23%).
Пример XXI - фиг.28
Синтез L208
Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,2 г, 0,08 ммоль) (смола А) деблокируют и сочетают с терефталевой кислотой, используя HATU в качестве агента сочетания. Полученную кислоту на смоле активируют DIC и NHS и затем сочетают с этилендиамином. И наконец, монокислоту DOCA сочетают с амином на смоле. Получают N-[(3β,5β,12α)-3-[[[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]ацетил]амино]-[1, 2-диаминоэтилтерефталил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид (L208) с выходом 17,5 мг (14%).
Пример XXII - фиг.29A-B
Синтез L209
Boc-Glu(G-OBn)-G-OBn:
Boc-глутаминовую кислоту (5,0 г, 20,2 моль) растворяют в ТГФ (50,0 мл) и охлаждают до 0°С на ледяной бане. Добавляют HATU (15,61 г, 41,0 ммоль) с последующим добавлением DIEA (6,5 г, 50,0 ммоль). Реакционную смесь перемешивают при 0°С в течение 30 мин. Добавляют сложный бензиловый эфир глицина [8,45 г, 50 ммоль, полученный в результате нейтрализации гидрохлорида бензилглицина карбонатом натрия, экстракцией DCM и удалением растворителя] в ТГФ (25,0 мл). Реакционную смесь оставляют охлаждаться до комнатной температуры и перемешивают в течение 20 ч при комнатной температуре. Все летучие вещества удаляют при пониженном давлении. Остаток обрабатывают насыщенным раствором карбоната натрия (100 мл) и экстрагируют этилацетатом (3×100 мл). Органические слои объединяют и промывают 1N HCl (2×100 мл) и водой (2×100 мл) и сушат (сульфат натрия). Раствор фильтруют, и растворитель удаляют при пониженном давлении с получением пасты, которую подвергают флэш-хроматографии на силикагеле (500,0 г). Элюирование 2% метанолом в DCM дает продукт в виде бесцветной пасты (соединение В, фиг.29А). Выход: 8,5 г (74,5%).
1H ЯМР (CDCl3): δ 1,4 (с, 9H, -CH3S), 2,0-2,5 (м, 4H,-CH-CH2 и CO-CH), 4,2 (м, 5H, N-CH2-CO), 5,15 (с, 4H, Ar-СН2), 5,45 (уш.с, 1H, Boc-NH), 7,3 (м, 10H, Ar-H) и 7,6 (2уш.с, 2H, CONH). МС: m/z 564,1 [M+H].
Время удерживания аналитической ВЭЖХ: 8,29 мин (чистота >97%, 20-65% В за 15 мин).
H-Glu(G-OBn)-G-OBn:
Полностью защищенное производное глутаминовой кислоты (1,7 г, 3,2 ммоль) В из указанной выше стадии растворяют в DCM/TFA (4:1, 20 мл) и перемешивают до тех пор, пока по данным ТСХ не исчезнет исходное вещество (2 ч). Реакционную смесь выливают в ледяной насыщенный раствор бикарбоната натрия (200 мл), органический слой отделяют, водный слой экстрагируют 2×50 мл DCM и объединяют с органическим слоем. Слой DCM промывают насыщенным бикарбонатом натрия (2×100 мл), водой (2×100 мл) и сушат (сульфат натрия). Раствор фильтруют и выпаривают при пониженном давлении, и остаток сушат в вакууме с получением стекловидного вещества (соединение С, фиг.29А), которое направляют на следующую стадию без дальнейшей очистки. Выход: 0,72 г (95%). МС: m/z 442,2 [M+H].
(DOTA-три-трет-бутил)-Glu-(G-OBn)-G-OBn:
Амин C из указанной выше стадии (1,33 г, 3 ммоль) в безводном DCM (10,0 мл) добавляют к активированному раствору три-трет-бутилового эфира DOTA [2,27 г, 3,6 ммоль обрабатывают HBTU, 1,36 г, 3,6 ммоль и DIEA (1,04 г, 8 ммоль) и перемешивают в течение 30 мин при комнатной температуре в 25 мл безводного DCM] и перемешивают при комнатной температуре в течение 20 ч. Реакционную смесь разбавляют 200 мл DCM, промывают насыщенным карбонатом натрия (2×150 мл) и сушат (сульфат натрия). Раствор фильтруют, и растворитель удаляют при пониженном давлении с получением коричневой пасты. Неочищенный продукт подвергают флэш-хроматографии на силикагеле (500,0 г). Элюирование 2% метанолом в DCM дает продукт в виде бесцветной смолы (соединение D, фиг.29А). Выход: 1,7 г (56,8%).
1H ЯМР (CDCl3): δ 1,3 и 1,4 (2с, 9H, три метила, каждый из свободного основания и натриевого аддукта DOTA), 2,0-3,5 (м, 20H, группы N-CH2 и -CH-CH2 и CO-CH2), 3,75-4,5 (м, 13H, N-CH2-CO), 5,2 (м, 4H, Ar-CH2) и 7,25 (м, 10H, Ar-H). МС m/z 1018,3 [M+Na] и 996,5 [M+H] и 546,3 [M+Na+H]/2.
Время удерживания ВЭЖХ:11,24 мин (>90%, 20-80% D за 30 мин).
(DOTA-три-трет-бутил)-Glu-(G-OH)-G-OH:
Сложный бис-бензиловый эфир (0,2 г, 0.2 ммоль) с указанной выше стадии растворяют в смеси метанол-вода (20 мл, 9:1) и гидрируют при 50 фунт/кв.дюйм в присутствии катализатора 10% Pd/C (0,4 г, 50% масс. воды). После исчезновения исходного вещества по данным ВЭЖХ и ТСХ (4 ч) раствор отфильтровывают от катализатора, растворитель удаляют при пониженном давлении и остаток сушат в высоком вакууме в течение примерно 20 ч (<0,1 мм) с получением продукта в виде бесцветной пены (соединение Е, фиг.29А). Выход: 0,12 г, (73, 5%).
1H ЯМР (ДМСО-d6): δ 1,3 и 1,4 (2с, 9H соответствующие метилам свободного основания и натриевого аддукта DOTA), 1,8-4,7 (м, 33H, группы NCH2, COCH2 и CH-CH2 и NH-CH-CO), 8,1, 8,2 и 8,4 (3уш.с, NHCO). МС: m/z 816,3 [M+H] и 838,3 [M+Na].
Время удерживания ВЭЖХ: 3,52 мин (20-80% В за 30 мин, чистота >95%).
Н-8-амино-3,6-диоксиоктаноил-8-амино-3,6-диоксаоктаноил-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2:
Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,5 г, 0,2 ммоль) А деблокируют и сочетают дважды последовательно с 8-амино-3,6-диоксаоктановой кислотой с получением указанного выше лишенного защиты пептида (соединение F, фиг.29В) после очистки препаративной ВЭЖХ. Выход: 91,0 мг (37%).
Время удерживания ВЭЖХ: 8,98 мин (чистота >95%, 10-40% В более чем за 10 мин). МС: m/z 1230,6 [M+H], 615,9 [М+2H]/2.
Сочетание в фазе раствора бис-кислоты Е и указанного выше амина F (фиг.29В)
Бис-кислоту (13,5 мг, 0,0166 ммоль) Е растворяют в 100 мкл безводного ацетонитрила и обрабатывают NHS (4,0 мг, 0,035 ммоль) и DIC (5,05 мг, 0,04 ммоль) и перемешивают в течение 24 ч при комнатной температуре. К указанной выше активированной кислоте добавляют свободный амин F (51,0 мг, 0,41 ммоль) [полученный из соли TFA обработкой насыщенным бикарбонатом натрия и лиофилизацией раствора с получением амина в виде рыхлого твердого вещества] с последующим добавлением 100 мкл NMP, и перемешивание продолжают в течение 40 ч при комнатной температуре. Раствор разбавляют безводным эфиром (10 мл), и осадок отделяют центрифугированием и снова промывают 2×10 мл безводного простого эфира. Затем неочищенное твердое вещество очищают препаративной ВЭЖХ с получением продукта в виде бесцветного рыхлого твердого вещества L209, как показано на фиг.29В с выходом 7,5 мг (14,7%).
Пример XXIII - фиг.30A-B
Синтез L210
Н-8-аминооктаноил-8-аминооктаноил-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2:
Получают точно таким же образом, как в случае соединения F (фиг.29В), но с использованием 1-аминооктановой кислоты, и амин (соединение В, фиг.30А) очищают препаративной ВЭЖХ. Выход: 95,0 мг (38,9%). Время удерживания ВЭЖХ: 7,49 мин (чистота >95%; 10-40%В за 10,0 мин). МС: m/z 1222,7 [M+H], 611,8 [М+2H]/2.
(DOTA-три-трет-бутил)-Glu-(G-OH0-G-OH (0,0163 г, 0,02 ммоль) превращают в его сложный бис-NHS эфир, как в случае L209, в 100 мкл ацетонитрила и обрабатывают свободным основанием, соединением В (60,0 мг, 0,05 ммоль) в 100 мкл NMP и реакцию продолжают в течение 40 ч, затем обрабатывают и очищают с получением L210 (фиг.30В) с выходом 11,0 мг (18%).
Пример XXIV - фиг.31
Синтез L211
Получают на 0,2 г Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смоле (0,08 ммоль) с использованием стандартных методик. N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицилглицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид L211 получают с выходом 4,7 мг (3,7%) (фиг.31).
Пример XXV - фиг.32
Синтез L212
Получают на смоле Rink Amide Novagel (0,47 ммоль/г, 0,2 г, 0,094 ммоль) построением последовательности стандартных методик. N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутамил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид L212 получают с выходом 25,0 мг (17,7%) (фиг.32).
Пример XXVI - фиг.33
Синтез L213
Получают на Fmoc-Met-2-хлортритилхлоридной смоле (NovaBioChem, 0,78 ммоль/г, 0,26 г, 0,2 ммоль) и остальную часть последовательности строят с использованием стандартной методики. N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионин L213 получают с выходом 49,05 мг (16,4%) (фиг.33).
Пример XXVII - фиг.34
Синтез L214
Используют Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,2 г, 0,08 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L214, используя стандартные условия. Получают 8,5 мг продукта (6,4%) (фиг.34).
Пример XXVIII - фиг.35
Синтез L215
Используют Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,2 г, 0,08 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-аргинил-L-лейцилглицил-L-аспаргинил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L215. Получают 9,2 мг (5,5%) (фиг.35).
Пример XXIX - фиг.36
Синтез L216
Используют Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,2 г, 0,08 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминиларгинил-L-тирозинилглицил-L-аспаргинил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L216. Получают 25,0 мг (14,7%) (фиг.36).
Пример XXX - фиг.37
Синтез L217
Используют Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу (0,2 г, 0,08 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-лизил-L-тирозинилглицил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида L217. Получают 58,0 мг (34,7%) (фиг.37).
Пример XXXI - фиг.38
Синтез L218
Используют Fmoc-Q(Trt)-W(Boc)-A-V-G-H(Trt)-L-M-смолу А (0,2 г, 0,08 ммоль). Для введения лизина используют Fmoc-Lys(ivDde). После завершения линейной последовательности защитную группу лизина удаляют с использованием 10% гидразина в ДМФА (2×10 мл; 10 мин каждый и затем промывают). Остальную часть аминокислот затем вводят с использованием методик, описанных в "общем" разделе для завершения требуемой последовательности пептида. L218, показанный на фиг.38, получен с выходом 40,0 мг (23,2%).
Пример XXXII - фиг.39
Синтез L219
Используют смолу 4-сульфамилбутирил АМ Novagel (1,1 ммоль/г; 0,5 г; 0,55 ммоль). Первую аминокислоту загружают на смолу при -20°С в течение 20 ч. Остальную часть последовательности завершают с использованием обычных методик сочетания. После промывки смолу алкилируют 20,0 эквивалентами йодацетонитрила и 10,0 эквивалентами DIEA в течение 20 ч. Затем смолу освобождают от жидкости и промывают, затем расщепляют 2,0 эквивалентами пентиламина в 5,0 мл ТГФ в течение 20 ч. Затем смолу промывают 2×50 мл ТГФ и все фильтраты объединяют. Затем ТГФ выпаривают при пониженном давлении, остаток затем деблокируют 10,0 мл реагента В и очищают пептид, N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейциламинофенил, L219, как описано выше. Выход составил 28,0 мг (2,8%) (фиг.39).
Пример XXXIII - фиг.40
Синтез L220
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-серенил-L-валил-D-аланил-L-гистидил-L-лейцил-L-метионинамида, L220. Выход составил 31,5 мг (41,4%) (фиг.40).
Пример XXXIV - фиг.41
Синтез L221
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-лейцинамида, L221. Выход составил 28,0 мг (34, 4%) (фиг.41).
Пример XXXV - фиг.42
Синтез L222
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-D-тирозинил-L-глутаминил-L-триптофил-L-аланил-L-валилбетааланил-L-гистидил-L-фенилаланил-L-норлейцинамида, L222. Выход составил 34, 0 мг (40,0%) (фиг.42).
Пример XXXVI - фиг.43
Синтез L223
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-фенилаланил-L-глутаминил-L-триптофил-L-аланил-L-валилбетааланил-L-гистидил-L-фенилаланил-L-норлейцинамида, L223. Выход составил 31,2 мг (37,1%) (фиг.43).
Пример XXXVII - фиг.44
Синтез L224
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0, 05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланилглицил-L-гистидил-L-фенилаланил-L-лейцинамида, L224. Выход составил 30,0 мг (42,2%) (фиг.44).
Пример XXXVIII - фиг.45
Синтез L225
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-лейцил-L-триптофил-L-аланил-L-валинилглицил-L-серинил-L-фенилаланил-L-метионинамида, L225. Выход составил 15,0 мг (20,4%) (фиг.45).
Пример XXXIX - фиг.46
Синтез L226
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-гистидил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамида, L226. Выход составил 40,0 мг (52,9%) (фиг.46).
Пример XL - фиг.47
Синтез L227
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-лейцил-L-триптофил-L-аланил-L-валилглицил-L-серинил-L-фенилаланил-L-метионинамида, L227. Выход составил 28,0 мг (36,7%) (фиг.47).
Пример XLI - фиг.48
Синтез L228
Используют смолу NovaSyn TGR (0,25 ммоль/г; 0,15 г, 0,05 ммоль) А с получением N-[(3β,5β,12α)-3-[[[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]ацетил]амино]глицил-4-аминобензоил-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-фенилаланил-L-метионинамида, L228. Выход составил 26,0 мг (33,8%) (фиг.48).
Пример XLII - Синтез дополнительных соединений GRP
A. Общая методика получения 4,4'-аминометилбифенилкарбоновой кислоты (В2) и 3,3'-аминометилбифенилкарбоновой кислоты (В3):
1. Метилгидроксиметилбифенилкарбоксилаты:
Коммерчески доступную (Aldrich Chemical Co) 4-гидроксиметилфенилборную кислоту или 3-гидроксиметилфенилборную кислоту (1,0 г, 6,58 ммоль) перемешивают с изопропанолом (10 мл) и 2 М карбонатом натрия (16 мл) до тех пор, пока раствор не станет гомогенным. Раствор дегазируют пропусканием азота через раствор и затем обрабатывают твердым метил-3-бромбензоатом или метил-4-бромбензоатом (1,35 г, 6,3 ммоль) с последующим катализом Pd (0) {[(C6H5)3P]4Pd; 0,023 г, 0,003 ммоль}. Реакционную смесь кипятят с обратным холодильником, пока не будет израсходован исходный бромбензоат по данным ТСХ (2-3 ч). Затем реакционную смесь разбавляют 250 мл воды и экстрагируют этилацетатом (3×50 мл). Органические слои объединяют и промывают насыщенным раствором бикарбоната натрия (2×50 мл) и сушат (Na2SO4). Растворитель удаляют при пониженном давлении и остаток подвергают флэш-хроматографии на силикагеле (100 г). Элюирование 40% этилацетатом дает продукт в виде твердого вещества или масла.
Выход:
В2 - 0,45 г (31%); т.пл. 170-171°С.
В3 - 0,69 г (62%); масло.
B2 -1H ЯМР (CDCl3) δ 3,94 (s, 3H, -COOCH3), 4,73 (с, 2H, -CH2-Ph), 7,475 (д, 2H, J=5 Гц), 7,6 (д, 2H, J=10 Гц), 7,65 (д, 2H, J=5 Гц) и 8,09 (д, 2H, J=10 Гц).
МС: m/e 243,0 [M+H].
B3 -1H ЯМР (CDCl3) δ 3,94 (с, 3H, -COOCH3), 4,76 (с, 2H, -CH2-Ph), 7,50 (м, 4H), 7,62 (с, 1H), 7,77 (с, 1H), 8,00 (с, 1H) и 8,27 (с, 1H).
МС: m/e 243,2 [M+H].
2. Азидометилфенилкарбоксилаты
Указанные выше бифениловые спирты (2,0 ммоль) в безводном дихлорметане (10 мл) охлаждают на льду, обрабатывают дифенилфосфорилазидом (2,2 моль) и DBU (2,0 ммоль) и перемешивают в атмосфере азота в течение 24 ч. Реакционную смесь разбавляют водой и экстрагируют этилацетатом (2×25 мл). Органические слои объединяют и последовательно промывают 0,5 M раствором лимонной кислоты (2×25 мл), водой (2×25 мл) и сушат (Na2SO4). Раствор фильтруют и выпаривают при пониженном давлении с получением неочищенного продукта. 4,4'-изомер кристаллизуют из смеси гексан/эфир, а 3,3'-изомер растирают с изопропиловым эфиром для удаления всех примесей; продукт гомогенен по данным ТСХ, и дальнейшая очистка не требуется.
Выход:
Метил-4-азидометил-4-бифенилкарбоксилат - 0,245 г (46%); т.пл. 106-108°С.
Метил-3-азидометил-3-бифенилкарбоксилат - 0,36 г (59%, масло).
4,4'-изомер -1H ЯМР (CDCl3) δ 3,95 (с, 3H, -COOCH3), 4,41 (с, 2H, -CH2N3), 7,42 (д, 2H, J=5 Гц), 7,66 (м, 4H) и 8,11 (д, 2H, J=5 Гц),
3, 3'-изомер -1H ЯМР (CDCl3) δ 3,94 (с, 3H, -COOCH3), 4,41 (с, 2H, -CH2N3), 7,26-7,6 (м, 5H), 7,76 (д, 1H, J=10 Гц), 8,02 (д, 1H, J=5 Гц) и 8,27 (с, 1H).
3. Гидролиз метилбифенилкарбоксилатов:
Примерно 4 ммоль сложных метиловых эфиров обрабатывают 20 мл 2 М раствора гидроксида лития и перемешивают до тех пор, пока раствор не становится гомогенным (20-24 ч). Водный слой экстрагируют 2×50 мл эфира, и органический слой отбрасывают. Водный слой затем подкисляют 0,5 M лимонной кислотой, и осажденное твердое вещество отфильтровывают и сушат. Другая очистка не требуется, и кислоты направляют на следующую стадию.
Выход:
сложный метиловый эфир 4, 4'-изомера - 0,87 г дает 0,754 г кислоты (86,6%); т.пл. 205-210°С.
сложный метиловый эфир 3,3'-изомера - 0,48 г дает 0,34 г кислоты (63,3%); т.пл. 102-105°С.
4,4'-изомер -1H ЯМР (ДМСО-d6) δ: 4,52 (с, 2H, -CH2N3), 7,50 (д, 2H, J=5 Гц), 7,9 (м, 4H) и 8,03 (д, 2H, J=10 Гц),
3, 3'-изомер -1H ЯМР (ДМСО-d6) δ: 4,54 (с, 2H, -CH2N3), 7,4 (д, 1H, J=10 Гц), 7,5-7,7 (м, 4H), 7,92 (ABq, 2H) и 8,19 (с, 1H).
4. Восстановление азидов в амин:
Его проводят в твердой фазе и амин никогда не выделяют. Смолу нагружают азидокарбоновой кислотой с использованием стандартных методов сочетания пептидов. После промывки смолу, содержащую азид, встряхивают с 20 эквивалентами трифенилфосфина в смеси ТГФ/вода (95:5) в течение 24 ч. Раствор сливают при положительном давлении азота, затем промывают стандартной методикой промывки. Полученный амин используют в следующем сочетании.
5. (3β,5β,7α,12α )-3-[{(9Н-флуорен-9-илметокси)амино]ацетил}амино-7,12-диоксихолан-24-овая кислота:
Трибутиламин (3,2 мл; 13,5 ммоль) добавляют по каплям в раствор Fmoc-глицина (4,0 г, 13,5 ммоль) в ТГФ (80 мл) и перемешивают при 0°С. Затем добавляют изобутилхлорформиат (1,7 мл; 13,5 ммоль) и через 10 мин по каплям добавляют суспензию трибутиламина (2,6 мл; 11,2 ммоль) и (3β, 5β,7α,12α)-3-амино-7,12-диоксихолан-24-овой кислоты (4,5 г; 11,2 ммоль) в ДМФА (80 мл) в течение 1 ч в охлажденный раствор. Смесь оставляют нагреваться до окружающей температуры, и через 6 ч раствор концентрируют до 120 мл, затем добавляют воду (180 мл) и 1N HCl (30 мл) (конечный рН 1,5). Осажденное твердое вещество отфильтровывают, промывают водой (2×100 мл), сушат в вакууме и очищают флэш-хроматографией. Элюирование смесью хлороформ/метанол (8:2) дает продукт в виде бесцветного твердого вещества.
Выход: 1,9 г (25%). ТСХ: Rf 0,30 (CHCl3/MeOH/NH4OH-6:3:1).
ТЕСТИРОВАНИЕ СОЕДИНЕНИЙ IN VITRO И IN VIVO
Пример XLIII: Анализ связывания in vitro с рецепторами GRP в линиях клеток РС3 - фиг.14А-В
Для идентификации потенциальных соединений-лидеров используют анализ in vitro, который идентифицирует соединения с высоким сродством к GRP-R. Поскольку известно, что линия клеток РС3, полученная из рака предстательной железы человека, проявляет высокую экспрессию GRP-R на клеточной поверхности, был разработан и обоснован формат анализа связывания радиоактивного лиганда в 96-луночном планшете для измерения связывания125I-BBN с GRP-R положительными клетками РС3 и способности соединений изобретения ингибировать это связывание. Этот анализ используют для измерения IC50 для лиганда RP527, лиганда L134 (контроли) и соединений изобретения, которые ингибируют связывание125I-BBN с GRP-R. (RP527 является N,N'-диметилглицин-Ser-Cys(Acm)-Gly-5-аминопентановой кислотой-BBN (7-14) [SEQ. ID. NO:1], которая имеет МС: 1442,6 и IC50: 0,84). Van de Wiele C, Dumont F et al., Technetium-99m RP527, a GRP analogue for visualization of GRP receptor-expressing malignancies: a feasibility study. Eur.J.Nucl.Med., (2000) 27; 1694-1699. L134 является DO3A-моноамид-8-аминооктановой кислотой-BBN (7-14) [SEQ. ID. NO:1], которая имеет МС: 1467,0. L134 представляет собой DO3A моноамид-аминооктанил-BBN [7-14].
Анализ связывания радиоактивного лиганда на планшете был обоснован для BBN и аналогов BBN (включая коммерчески доступные BBN и L1), а также с использованием99mTc RP527 в качестве радиоактивного лиганда.
А. Материалы и методы:
1. Клеточная культура:
РС3 (линию клеток рака предстательной железы человека) получали из Американской коллекции типовых культур и культивировали во сосудах для культуры ткани RPMI 1640 (Corning). В эту ростовую среду добавляют 10% инактивированную нагреванием FBS (Hylicone, SH30070.03), 10 мМ HEPES (GibcoBRL, 15630-080) и антибиотик/антимикотическое средство (GibcoBRL, 15240-062) до конечной концентрации пенициллина-стрептомицина (100 единиц/мл) и фунгизона (0,25 мкг/мл). Все культуры поддерживали в увлажненной атмосфере, содержащей 5% СО2/95% воздух при 37°С и обычным образом пересевают с использованием 0,05% трипсина/EDTA (GibcoBRL, 25300-054). Клетки для экспериментов высевают в концентрации 2,0×104/лунка в 96-луночные титровальные микропланшеты с белым/прозрачным дном (Falcon Optilux-I) или в 96-луночные клеточные планшеты черные/прозрачные с коллагеном I (Beckton Dickinson Biocat). Планшеты используют для исследований связывания на 1-й или 2-ой день после посева.
2. Буфер связывания :
RPMI 1640 с добавками 20 мМ HEPES, 0,1% BSA (масс./об.), 0,5 мМ PMSF (AEBSF), бацитрацина (50 мг/50 мл), рН 7,4.
125I-BBN (без носителя, 2200 Ки/ммоль) получают от Perkin-Elmer.
В. Анализ конкуренции с 125I-BBN за GRP-R в клетках РС3:
Анализ в 96-луночном планшете используют для определения IC50 различных соединений изобретения ингибирования связывания125I-BBN с GRP-R человека. Выполняют следующую общую методику:
Все испытуемые соединения растворяют в буфере связывания и соответствующие разведения также производят в буфере связывания. Клетки РС3 (линия клеток рака предстательной железы человека) для анализа высевают в концентрации 2,0×104/лунка в 96-луночные титровочные микропланшеты с белым/прозрачным дном (Falcon Optilux-I) или в 96-луночные клеточные планшеты черные/прозрачные из коллагена I (Beckton Dickinson Biocat). Планшеты используют для исследований связывания на 1-й или 2-й день после высевания. Планшеты проверяют на слияние (>90% слившихся) перед анализом. Для анализа RP527 или лиганд L134 (контроли) и соединения изобретения в концентрации в диапазоне от 1,25×10-9M до 5×10-9M совместно инкубируют с125I-BBN (25000 имп.мин/лунка). Эти исследования проводят с аналитическим объемом 75 мкл на лунку. По 3 лунки используют для каждой точки данных. После добавления соответствующих растворов планшеты инкубируют в течение 1 ч при 4°С для предотвращения интернализации комплекса лиганд-рецептор. Инкубацию заканчивают добавлением 200 мкл ледяного инкубационного буфера. Планшеты промывают 5 раз и подвергают блоттингу в сухом виде. Радиоактивность выявляют с использованием счетчика LKB CompuGamma или сцинитилляционного счетчика микропланшет.
Кривые конкурентного связывания с RP527 (контроль) и L70, соединения изобретения, можно найти на фиг.14А-В. Эти данные показывают, что IC50 контроля RP527 составляет 2,5 нМ и IC50 соединения изобретения L70 составляет 5 нМ. IC50 контроля L134 составляет 5 нМ. Величины IC50 для испытуемых соединений изобретения можно найти в таблице 1-3 выше, и они показывают, что они сравнимы с величинами контролей и, следовательно, можно ожидать, что они имеют достаточное сродство к рецептору для того, чтобы обеспечить возможность захвата несущими рецептор клетками in vivo.
С. Анализ интернализации и выходящего тока
Эти исследования проводят в 96-луночном планшете. После промывки для удаления сывороточных белков клетки РС3 инкубируют вместе с125I-BBN,177 Lu-134 или меченными радиоактивной меткой соединениями данного изобретения в течение 40 мин при 37°С. Инкубации прекращают добавлением 200 мкл ледяного буфера связывания. Планшеты промывают дважды буфером связывания. Для удаления связанного с поверхностью радиоактивного лиганда клетки инкубируют с 0,2 M уксусной кислотой (в солевом растворе), рН 2,8 в течение 2 мин. Планшеты центрифугируют и кислую промывную среду собирают для определения количества радиоактивности, которая не интернализовалась. Клетки собирают для определения количества интернализованного125 I-BBN и все образцы анализируют в гамма-счетчике. Данные анализа интернализации нормализуют сравнением количества импульсов, полученного в различные точки времени, с количествами импульсов, полученными в конечную точку времени (Т 40 мин).
Для исследований выходящего тока после загрузки клеток125I-BBN или меченными радиоактивной меткой соединениями данного изобретения в течение 40 мин при 37°С несвязанный материал вымывают и % интернализации определяют, как указано выше. Клетки затем ресуспендируют в буфере связывания при 37°С до 3 ч. Через 0,5, 1, 2 или 3 ч количество остающегося интернализированного материала относительно исходного уровня загрузки определяют, как описано выше, и используют для расчета процента выходящего тока, представленного в таблице 5.
Эти данные показывают, что соединения настоящего изобретения интернализуются и удерживаются клетками РС3 в степени, аналогичной контролям.
Пример XLIV - Получение меченных Tc соединений GRP
Растворы пептидов соединений изобретения, представленных в таблице 6, получают в концентрации 1 мг/мл в 0,1% водной TFA. Раствор хлорида двухвалентного олова получают растворением SnCl2·2H2O (20 мг/мл) в 1N HCl. Растворы глюконата двухвалентного олова, содержащие 20 мкг SnCl2/100 мкл, получают добавлением аликвоты раствора SnCl2 (10 мкл) к раствору глюконата натрия, полученного растворением 13 мг глюконата натрия в воде. Раствор гидроксипропил-гамма-циклодекстрина [HP-γ-CD] получают растворением 50 мг HP-γ-CD в 1 мл воды.
Идентифицированные ниже соединения, меченные99mTc, получают смешиванием 20 мкл раствора немеченых соединений (20 мкг), 50 мкл раствора HP-γ-CD, 100 мкл раствора Sn-глюконата и 20-50 мкл99mTc пертехната (5-8 мКи, Syncor). Конечный объем составляет примерно 200 мкл и конечный рН составляет 4,5-5. Реакционную смесь нагревают при 100°С в течение 15-20 мин и затем анализируют ВЭЖХ с обращенной фазой для определения радиохимической чистоты (RCP). Пики желаемых продуктов выделяют ВЭЖХ, собирают в стабилизирующий буфер, содержащий 5 мг/мл аскорбиновой кислоты, 16 мг/мл HP-γ-CD и 50 мМ фосфатного буфера, рН 4, 5 и концентрируют с использованием быстро создаваемого вакуума для удаления ацетонитрила. Система ВЭЖХ, используемая для анализа и очистки, представляет собой следующую: колонка C18 Vydac, 4,6× 250 мм, водная фаза: 0,1% TFA в воде, органическая фаза: 0,085% TFA в ацетонитриле. Объемный расход: 1 мл/мин. Используют изократическое элюирование смесью 20%-25% ацетонитрил/0,085% TFA, в зависимости от природы отдельного пептида.
Результаты мечения сведены в таблице 6.
Пример XLV - Получение 177Lu-L64 для исследований связывания с клетками и биораспределения
Это соединение синтезируют инкубацией 10 мкг лиганда L64 (10 мкл раствора 1 мг/мл в воде), 100 мкл буфера ацетата аммония (0,2 М, рН 5,2) и ˜1-2 мКи177LuCl3 в 0,05N HCl (MURR) при 90°С в течение 15 мин. Свободный177 Lu нейтрализуют добавлением 20 мкл 1% раствора Na2EDTA·2H2O (Aldrich) в воде. Полученная радиохимическая чистота (RCP) составляет ˜95%. Меченный радиоактивной меткой продукт отделяют от немеченого лиганда и других примесей ВЭЖХ с использованием колонки YMC Basic C8 [4,6×150 мм], при температуре колонки 30°С и объемном расходе 1 мл/мин при градиенте от 68%А/32%B до 66%А/34%B за 30 мин, где А представляет собой цитратный буфер (0,02 М, рН 3,0), и В представляет собой 80% CH3CN/20% CH3OH. Выделенное соединение имеет RCP ˜100% и время удерживания ВЭЖХ 23,4 мин.
Образцы для исследований биологического распределения и клеточного связывания получают сбором желаемого пика ВЭЖХ в 1000 мкл цитратного буфера (0,05 М, рН 5,3, содержащего 1% аскорбиновую кислоту и 0,1% HSA). Органический элюент в собранном элюате удаляют центрифужным концентрированием в течение 30 мин. Для исследований клеточного связывания очищенный образец разбавляют связывающей клетки средой до концентрации 1,5 мКи/мл в пределах 30 мин исследования in vitro. Для исследований биологического распределения образец разбавляют цитратным буфером (0,05 М, рН 5,3, содержащим 1% аскорбинат натрия и 0,1% HSA) до конечной концентрации 50 мКи/мл в пределах 30 мин исследования in vivo.
Пример XLVI - Получение 177Lu-L64 для исследований лучевой терапии
Это соединение синтезируют инкубацией 70 мкг лиганда L64 (70 мкл раствора 1 мг/мл в воде), 200 мкл буфера ацетата аммония (0,2 М, рН 5,2) и ˜30-40 мКи177LuCl3 в 0,05N HCl (MURR) при 85°С в течение 10 мин. После охлаждения при комнатной температуре свободный177Lu нейтрализуют добавлением 20 мкл 2% раствора Na2EDTA·2H2O (Aldrich) в воде. Полученная радиохимическая чистота (RCP) составляет ˜95%. Меченный радиоактивной меткой продукт отделяют от немеченого лиганда и других примесей ВЭЖХ с использованием колонки 300VHP Anion Exchange (7,5×50 мм), которую последовательно элюируют при объемном расходе 1 мл/мин водой, смесью 50% ацетонитрил/вода и затем 1 г/л водного раствора ацетата аммония. Желаемое соединение элюируют из колонки 50% CH3CN и смешивают ˜1 мл цитратного буфера (0,05 М, рН 5,3), содержащего 5% аскорбиновую кислоту, 0,2% HSA и 0,9% (об./об.) бензилового спирта. Органическую часть выделенной фракции удаляют в вакууме водоструйного насоса в течение 40 мин, и концентрированный раствор (˜20-25 мКи) доводят в течение 30 мин исследования in vivo до концентрации 7,5 мКи/мл с использованием цитратного буфера (0,05 М, рН 5,3), содержащего 5% аскорбиновую кислоту, 0,2% HSA и 0,9% (об./об.) бензилового спирта. Полученное соединение имеет RCP >95%.
Пример XLVII - Получение 111In-L64
Это соединение синтезируют инкубацией 10 мкг лиганда L64 (5 мкл раствора 2 мг/мл в 0,01N HCl), 60 мкл этанола 1,12 мКи111InCl3 в 0,05N HCl (80 мкл) и 155 мкл буфера ацетата натрия (0,5 М, рН 4,5) при 85°С в течение 30 мин. Свободный111In нейтрализуют добавлением 20 мкл 1% раствора Na2EDTA·2H2O (Aldrich) в воде. Полученная радиохимическая чистота (RCP) составляет 87%. Меченный радиоактивной меткой продукт отделяют от немеченого лиганда и других примесей ВЭЖХ с использованием колонки Vydac C18 [4,6×250 мм], при температуре колонки 50°С и объемном расходе 1 мл/мин, при градиенте от 75%А/25%B до 65%А/35%B за 20 мин, где А представляет собой 0,1% TFA в воде, а В представляет собой 0,085% TFA в ацетонитриле. При этой системе время удерживания для111In-L64 составляет 15,7 мин. Полученное соединение имеет RCP 96,7%.
Пример XLVIII - Получение 177 Lu-L134 (контроль)
Маточный раствор пептида получают растворением лиганда L134 (полученного, как описано в заявке на патент США № 2002/0054855 и WO 02/87637, обе включенные в данное описание в качестве ссылки) в 0,01N HCl до концентрации 1 мг/мл.177Lu-L134 получают смешиванием следующих реагентов в показанном порядке.
Реакционную смесь инкубируют при 85°С в течение 10 мин. После охлаждения до комнатной температуры в водяной бане добавляют 20 мкл 1% раствора EDTA и 20 мкл EtOH. Соединение анализируют ВЭЖХ с использованием колонки С18 (VYDAC Cat # 218TP54), которую элюируют при объемном расходе 1 мл/мин при градиенте от 21 А до 25% В за 20 мин, где А представляет собой 0,1% TFA/H2O, а В представляет собой 0,1% TFA/CH3CN.177Lu-L134 образуется с выходом 97,1% (RCP) и имеет время удерживания ˜16,1 мин в данной системе.
Пример XLIX - Получение 177Lu-L63
Это соединение получают, как описано для177Lu-L134. Соединение анализируют ВЭЖХ с использованием колонки С18 (VYDAC Cat # 218TP54), которую элюируют при объемном расходе 1 мл/мин при градиенте от 30 до 34% В за 20 мин (где растворитель А представляет собой 0,1% TFA/H2O, а В представляет собой 0,1% TFA/CH3CN).177Lu-L63, который образуется, имеет RCP 97,8% и имеет время удерживания ˜14,2 мин в этой системе.
Пример L - Получение 177 Lu-L70 для исследований клеточного связывания и биологического распределения
Это соединение получают, следуя описанным выше методикам, но заменяя L70 (лиганд примера II). Очистку выполняют с использованием колонки YMC Basic C8 (4,6×150 мм), при температуре колонки 30°С и объемном расходе 1 мл/мин при градиенте от 80%А/20%B до 75%А/25%B за 40 мин, где А представляет собой цитратный буфер (0,02 М, рН 4,5), а В представляет собой 80% CH3CN/20% CH3OH. Выделенное соединение имеет RCP ˜100% и время удерживания ВЭЖХ 25,4 мин.
Пример LI - Получение 177Lu-L70 для исследований лучевой терапии
Это соединение получают, как описано выше для L64.
Пример LII - Получение 111In-L70 для исследований клеточного связывания и биологического распределения
Это соединение синтезируют инкубацией 10 мкг лиганда L70 (10 мкл раствора 1 мг/мл в 0,01N HCl), 180 мкл буфера ацетата аммония (0,2 М, рН 5,3), 1,1 мКи111InCl3 в 0,05N HCl (61 мкл, Mallinckrodt) и 50 мкл солевого раствора при 85°С в течение 30 мин. Свободный111In нейтрализуют добавлением 20 мкл 1% раствора Na2EDTA·2H2O (Aldrich) в воде. Полученная радиохимическая чистота (RCP) составляет 86%. Меченный радиоактивной меткой продукт отделяют от немеченого лиганда и других примесей ВЭЖХ с использованием картриджа Raters XTerra C18, соединенного с колонкой сильного анионного обмена Vydac [7,5×250 мм], при температуре колонки 30°С и объемном расходе 1 мл/мин, при градиенте, приведенном в представленной ниже таблице, где А представляет собой 0,1 мМ NaOH в воде, рН 10,0, В представляет собой раствор ацетата аммония в воде с концентрацией 1 г/л, рН 6,7, и С представляет собой ацетонитрил. При этой системе время удерживания для111In-L70 составляет 15 мин, тогда как время удерживания для лиганда L70 составляет от 27 до 28 мин. Выделенное соединение имеет RCP 96%.
Образцы для исследований биологического распределения и клеточного связывания получают сбором желаемого пика ВЭЖХ в 500 мкл цитратного буфера (0,05 М, рН 5,3, содержащего 5% аскорбиновую кислоту, 1 мг/мл L-метионина и 0,2% HSA). Органическую часть сбора удаляют спиновым вакуумом в течение 30 мин. Для исследований клеточного связывания очищенный, концентрированный образец используют в пределах 30 мин исследования in vitro. Для исследований биологического распределения образец разводят цитратным буфером (0,05 М, рН 5,3, содержащим 5% аскорбинат натрия и 0,2% HSA) до конечной концентрации 10 мКи/мл в пределах 30 мин исследования in vivo.
Пример LIII - Фармакокинетические исследования in vivo
А Биологическое распределение следовых доз
Фармакокинетические исследования низких доз (например, исследования биологического распределения) выполняют с использованием идентифицированных ниже
соединений изобретения у получивших ксенотрансплантат, несущих опухоль РС3 "голых" мышей ([Ncr]-Foxn1
В то время как распределение радиоактивности в крови, печени и почках после инъекции L64 и L70 аналогично контрольному соединению L134, захват в опухоли гораздо выше через 1 и 24 ч как для L64, так и для L70. L63 также проявляет высокий захват опухолью, хотя и при увеличенных величинах в крови и печени в ранние периоды времени. Захват поджелудочной железой мышей, который, как известно, в норме имеет рецепторы GRP, гораздо выше для L64, L70 и L63, чем для L134.
Пример LIV - Специфичность подтипов рецепторов
В настоящее время известны 4 члена семейства рецепторов GRP млекопитающих: рецептор, предпочитающий GRP (GRP-R), рецептор, предпочитающий нейромедин-В (NMB-R), подтип 3 рецептора бомбезина (BB3-R) и подтип рецептора ВВ4. Исследовали специфичность177Lu-L245 в отношении подтипов рецепторов. Результаты показывают, что177Lu-L245 специфически связывается с GRP-R и NMB-R и имеет небольшое сродство к BB3-R.
Специфичность подтипа комплекса лютетия L245 [177Lu-L24] (полученного, как описано выше) определяют авторадиографией рецепторов in vitro с использованием методики, описанной Reubi et al., "Bombesin Receptor Subtypes in Human Cancers: Detection with the Universal Radioligand (125)I-[D-TYR(6), beta-ALA, PHE(13), NLE(14)] Bombesin (6-14)", Clin. Cancer Res. 8:1139-1146 (2002), и образцы ткани, которые, как было обнаружено ранее, экспрессируют только один подтип рецептора GRP. Карциноидную ткань подвздошной кишки человека используют в качестве источника для подтипа рецепторов NMB-R, карцинома предстательной железы человека - для GRP-R и бронхиальный карциноид человека - для BB3-R. Соединение, известное как универсальный лиганд,125I-[Dtyr6, β Ala11, Phe13, Nle14]-BBN(6-14), которое связывается со всеми тремя подгруппами GRP-R, используют в качестве положительного контроля. Наблюдали следующие результаты.
Эти результаты указывают на то, что, как ожидается, производные L245 хорошо связываются с карциномой предстательной железы человека, которая в первую очередь экспрессирует GRP-R. Они также указывают на то, что не ожидается, что производные L245 хорошо связываются со здоровой предстательной железой человека (которая в первую очередь экспрессирует рецептор BB3-R) или с раковыми опухолями, которые в первую очередь экспрессируют подтип рецепторов BB3-R.
Пример LV - Исследования лучевой терапии
А. Исследования кратковременной эффективности:
Исследования лучевой терапии выполняли, используя модель "голых" мышей, несущих опухоль РС3. Соединения изобретения L64, L70, L63, меченные Lu-177, и соединение контроля лечения L134 сравнивали с не леченой контрольной группой (n=12 для каждой группы лечения и n=36 для не леченой контрольной группы). Для первого исследования мышам вводили 100 мкл соединения изобретения, меченного177Lu, при 30 мКи/кг внутривенно или подкожно в стерильных условиях. Исследуемых мышей выдерживали в огражденной среде до 30 дней. Массу тела и размер опухоли (измерением циркулем) регистрировали у каждой мыши 3 раза в неделю в течение исследования. Критерии раннего прекращения включают: смерть; потерю общей массы тела (TBW), равную или большую чем 20%; размер опухоли, равный или больший, чем 2 см3. Результаты представлены на фиг.15А. Эти результаты показывают, что у животных, леченных L70, L64 или L63, было увеличенная выживаемость, по сравнению с контрольными животными, не получавшими лечение, и в сравнении с теми животными, которые получали такую же дозу L134.
Повторное исследование выполняли с L64 или L70 с использованием той же дозы, как и ранее, но используя большее количество животных на соединение (n=46) и при более длительном наблюдении. Результаты повторного исследования представлены на фиг.15В. Относительно тех же контролей, что и ранее (n=36), лечение как L64, так и L70 дает значительно увеличенную выживаемость (p<0,0001), причем L70 лучше, чем L64 (p<0,079).
Пример LVI
Альтернативное получение L64 и L70 с использованием сегментного сочетания
Соединения L64 и L70 можно получить, используя библиотеку промежуточных соединений, в целом представленных A-D (фиг.19), которые получают стандартными способами, известными в области синтеза пептидов в твердой фазе и фазе раствора (Synthetic Peptides - A User's Guide 1992, Grant, G., Ed. WH. Freeman Co., NY, Chap 3 and Chap 4 pp 77-258; Chan, W.C. and White, P.D. Basic Procedures in Fmoc Solid Phase Peptide Synthesis - A Practical Approach 2002, Chan, W.C. and White, P.D. Eds Oxford University Press, New York, Chap. 3 pp 41-76; Barlos, K and Gatos, G. Convergent Peptide Synthesis in Fmoc Solid Phase Peptide Synthesis - A Practical Approach 2002, Chan, W.C. and White, P.D. Eds Oxford University Press, New York, Chap. 9 pp 216-228), которые включены в настоящее описание в качестве ссылки.
Эти способы включают методы пептидного синтеза, основанные на Aloc, Boc, Fmoc или бензилоксикрбонила, или разумно выбранных комбинациях этих методов в твердой фазе или в фазе раствора. Промежуточные соединения, которые предстоит использовать для данной стадии, выбирают на основании выбора соответствующих защитных групп для каждого положения в молекуле, которые можно выбрать из перечня групп, показанного на фиг.1. Среднему специалисту в данной области будет очевидно, что можно также использовать промежуточные соединения, совместимые с методом пептидного синтеза, состоящие из альтернативных защитных групп, и что перечисленные варианты показанных выше защитных групп служат в качестве иллюстративных, а не эксклюзивных, и что такие альтернативы хорошо известны в данной области.
Это достаточно проиллюстрировано на фиг.20, которая схематически представляет данный подход. Замена промежуточного соединения С1 соединением С2, показанная при синтезе L64, обеспечивает получение L70, когда используют такие же методы синтеза.
Пример LVII - фиг.49 и 50
Синтез L69
Резюме: Взаимодействие (3β,5β, 7α,12α)-3-амино-7,12-дигидроксихолан-24-овой кислоты А с Fmoc-Cl дает промежуточное соединение B. Амидная смола Rink, функционализированная октапептидом GlnTrpAlaValGlyHisLeuMetNH2 (BBN[7-14]) (А) последовательно реагирует с В, F-moc-8-амино-3,6-диоксаоктановой кислотой и три-трет-бутиловым эфиром DOTA. После отщепления и снятия защиты реагентом В неочищенный продукт очищают препаративной ВЭЖХ с получением L230 В 24280. Общий выход: 4,2%.
А. (3β,5β,7α, 12α)-3-(9Н-флуорен-9-илметокси)амино-7,12-дигидроксихолан-24-овая кислота, В (фиг.49)
Раствор 9-флуоренилметоксикарбонилхлорида (1,4 г; 5,4 ммоль) в 1,4-диоксане (18 мл) добавляют по каплям к суспензии (3β,5β,7α,12α)-3-амино-7,12-дигидроксихолан-24-овой кислоты А (2,0 г; 4,9 ммоль) (3) в 10% водный раствор Na2CO3 (30 мл) и 1,4-диоксан (18 мл), при перемешивании при 0°С. После 6 ч перемешивания при комнатной температуре добавляют Н2О (100 мл), водную фазу промывают Et2O (2× 90 мл) и затем добавляют 2 M HCl (15 мл) (конечный рН: 1,5). Осажденное твердое вещество отфильтровывают, промывают Н2О (3×100 мл), сушат в вакууме а затем очищают флэш-хроматографией с получением В в виде белого твердого вещества (2,2 г; 3,5 ммоль). Выход 71%.
B. N-[3β,5β,7α,12α )-3-[[[2-[2-[[[4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклододец-1-ил]ацетил]амино]этокси]ацетил]амино]-7, 12-дигидрокси-24-оксохолан-24-ил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид, L69 (фиг.50)
Смолу А (0,5 г; 0,3 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение еще 20 мин, затем раствор сливают и смолу промывают DMA (5×7 мл). (3β,5β,7α,12α)-3-(9Н-флуорен-9-илметокси)амино-7,12-дигидроксихолан-24-овую кислоту В (0,75 г; 1,2 ммоль), N-гидроксибензотриазол (HOBT) (0,18 г; 1,2 ммоль), N,N'-диизиопропилкарбодиимид (DIC) (0,19 мл; 1,2 ммоль) и DMA (7 мл) добавляют к смоле, смесь встряхивают в течение 24 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают, добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл). К смоле добавляют Fmoc-8-амино-3,6-диоксаоктановую кислоту (0,79 г; 1,2 ммоль), HOBt (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль) и DMA (7 мл). Смесь встряхивают в течение 3 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл). Затем смолу встряхивают с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл) и смесь встряхивают в течение еще 20 мин. Раствор сливают, и смолу промывают DMA (5×7 мл), к смоле добавляют аддукт трис(1,1-диметилэтилового) эфира 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты с NaCl С (0,79 г; 1,2 ммоль), HOBt (0,18 г; 1,2 ммоль), DIC (0,19 мл; 1,2 ммоль), N-этилдиизопропиламин (0, 40 мл; 2,4 ммоль) и DMA (7 мл). Смесь встряхивают в течение 24 ч при комнатной температуре, сливают и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (248 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного продукта (50 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L69 (6,5 мг; 3,5×10-3 ммоль) (фиг.50) в виде белого твердого вещества. Выход 5,8%.
Пример LVIII - фиг.51
Синтез L144
Резюме: Амидную смолу Rink, функционализированную октапептидом GlnTrpAlaValGlyHisLeuMetNH2 (BBN[7-14]) (А) вводят в реакцию с 4-[2-гидрокси-3-[4,7,10-трис[2-(1,1-диметилэтокси)-2-оксоэтил]-1,4,7, 10-тетраазациклододец-1-ил]пропокси]бензойной кислотой. После отщепления и снятия защиты реагентом В (2) неочищенный продукт очищают препаративной ВЭЖХ с получением L144. Общий выход: 12%.
N-[4-[2-гидрокси-3-[4,7,10-трис(карбоксиметил)-1,4,7, 10-тетраазациклододец-1-ил]пропокси]бензоил]-L-глутаминил-L-триптофил-L-аланил-L-валилглицил-L-гистидил-L-лейцил-L-метионинамид, L144 (фиг.51):
Смолу А (0,4 г; 0,24 ммоль) встряхивают в сосуде для твердофазного пептидного синтеза с 50% морфолином в DMA (7 мл) в течение 10 мин, раствор сливают и добавляют свежий 50% морфолин в DMA (7 мл). Суспензию перемешивают в течение еще 20 мин, затем раствор сливают, и смолу промывают DMA (5×7 мл). 4-[2-гидрокси-3-[4,7,10-трис(2-(1,1-диметилэтокси)-2-оксоэтил]-1,4,7,10-тетраазациклододец-1-ил]пропокси]бензойную кислоту В (0,5 г; 0,7 ммоль), HOBT (0,11 г; 0,7 ммоль), DIC (0,11 мл; 0,7 ммоль) N-этилдиизопропиламин (0,24 мл; 1,4 ммоль) и DMA (7 мл) добавляют к смоле. Смесь встряхивают в течение 24 ч при комнатной температуре, сливают, и смолу промывают DMA (5×7 мл), CH2Cl2 (5×7 мл) и сушат в вакууме. Смолу встряхивают в колбе с реагентом В (25 мл) (2) в течение 4,5 ч. Смолу фильтруют, и раствор выпаривают при пониженном давлении с получением маслянистого неочищенного вещества, которое после обработки Et2O (20 мл) дает осадок. Осадок отделяют центрифугированием и промывают Et2O (3×20 мл) с получением твердого вещества (240 мг), которое анализируют ВЭЖХ. Некоторое количество неочищенного продукта (60 мг) очищают препаративной ВЭЖХ. Фракции, содержащие продукт, лиофилизируют с получением L144 (10,5 мг; 7,2×10-3 ммоль) в виде белого твердого вещества. Выход 12%.

Реферат
Предложены новые усовершенствованные соединения для применения при диагностической визуализации или терапии, имеющие формулу M-N-O-P-G, где М представляет собой агент, хелатирующий металл (в форме комплекса с металлическим радионуклидом или нет), N-O-P представляет собой линкер, и G представляет собой пептид для целенаправленной доставки к рецептору GRP. Также представлены способы визуализации у пациента с использованием соединений изобретения, способы получения диагностического визуализирующего средства и радиотерапевтического средства. 33 н. и 16 з.п. ф-лы, 51 ил., 8 табл.
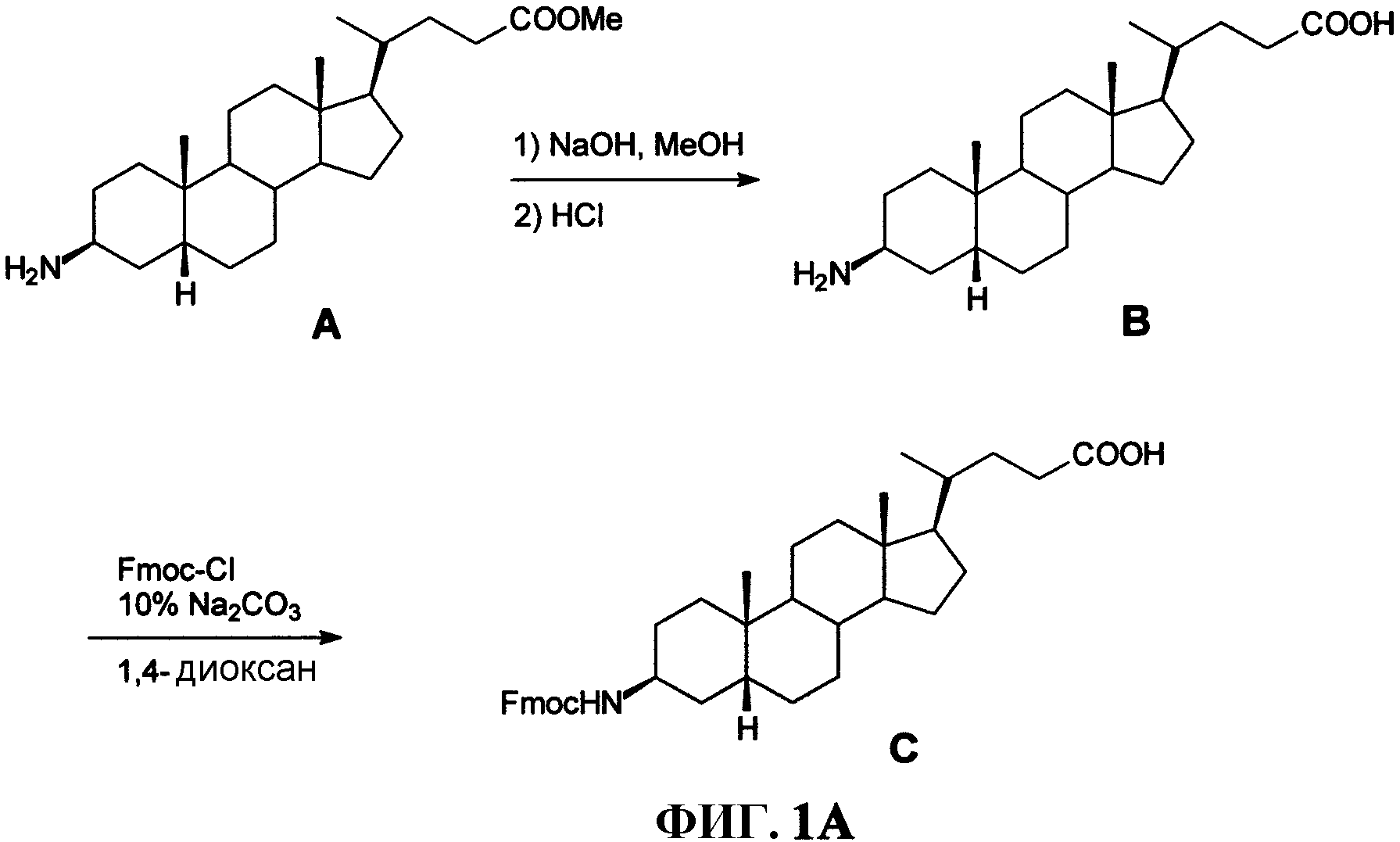
Комментарии