Производные пептидов, обладающие способностью регулировать пролиферацию клеток, и способы ингибирования клеточной пролиферации человека и животного (варианты) - RU2079509C1
Код документа: RU2079509C1
Чертежи


Описание
Изобретение относится к использованию пептидов, имеющих ингибирующее действие на клеточную пролиферацию, и к новым пептидам, имеющим специфическое и/или общее ингибирующее действие.
Молочная железа содержит клетки, имеющие большое разнообразие структур и функций, и механизмы дифференциации и развития находится в центре внимания многих исследований. Известно, что для систем клеток, имеющих непрерывный обмен, механизм обычно включает резерв полипотентных стволовых клеток, которые делятся и постоянно подают новые клетки в систему. Между тем, первоначально гомогенные стволовые клетки, подаваемые из "резерва", вскоре приобретают ту или иную морфологию и впоследствии развиваются в требуемые функциональные клетки.
Примерами таких систем стволовых клеток являются гемопоетическая (кроветворная) система в костном мозге и эпителиальная, и эпидермальная системы.
Ранее сообщалось, что пептиды, соответствующие ограниченной общей формуле, могут ингибировать гемопоезис (кроветворение) (см. ЕР-А-0112656), в то время как группа димерных пептидов, соответствующих незначительно расширенной общей формуле и связанных дисульфидным мостиком, может стимулировать гемопоезис (см. W 088/03535). В обоих случаях установлено, однако, что никаких эффектов не наблюдают на системах помимо гемопоезиса.
Нами обнаружено, что класс пептидов, включая некоторые пептиды, раскрытые в ЕР-А-01126256, имеет способность ингибировать клеточную пролиферацию и что незначительная модификация в аминокислотной последовательности и/или блокирование существенных групп в боковой цепи могут направлять действие пептидов к специфическим системам, представляющим интерес. Наши данные основаны на наблюдении, что определенные пентапептидные последовательности, раскрытые в наших вышеупомянутых заявках на патент, встречаются в определенных так называемых G-белках, а именно Gαi-белках. G-белки были найдены на внутренней стороне клеточных мембран, и они обеспечивают необходимую связь между трансмембранными рецепторами и эффекторами, расположенными около G-белков внутри клетки. Они участвуют во многих клеточных функциях, в зависимости от эффекторов, с которыми они связаны. Они состоят из 3 связанных субъединиц α,β и γ, причем a субъединица участвует в активации соседнего эффектора. G-белки отличаются по их функции, и субкласс G-белков является теми первоначально найденными белками, которые ингибируют аденилат циклазу. Эти данные приводят к рассмотрению роли пептидов в ингибировании пролиферации эпителиальной и эпидермальной систем и клеток систем вообще.
Таким образом, согласно изобретению мы предлагаем использование соединений формулы (I).
Ra-Rb-Rc-Rd-(Re)n-Rf (I),
где Ra представляет собой

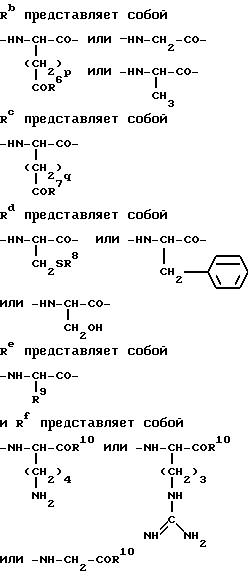
где n представляет собой 0 или 1;
p и q независимо представляют собой 1 или 2;
R1 и R2 являются оба атомами водорода или вместе представляют собой оксогруппу;
R3 и R4 оба атомы водорода;
R5 водород или ацетил, pGlu или Ser;
R6 и R7 независимо представляют гидроксигруппу или аминогруппу;
R8 представляет собой водород;
C7-20 карбоциклическую аралкильную группу или метаболически лабильную S-защищенную группу, выбранную из бензильной, фенильной и пиридильной групп;
R9 представляет водород или метильную группу;
R10 представляет гидрокси или аминогруппу,
при условии, что когда Rd=Ser и n=0, то Rf является иным, чем Gly; за исключением аланина, который может быть в D- или L-форме, и глицерина, все указанные аминокислотные остатки находятся в L-форме при приготовлении лекарственного препарата для ингибирования негемопоетической клеточной пролиферации.
Cпособность пентапептидов ингибировать пролиферацию большого ряда клеток в дополнение к гемопоетической системе или даже исключая гемопоетическую систему является ценной для медицины, когда чрезмерная клеточная пролиферация требует обработки, как при псориазе, либо когда раковая терапия желательна, чтобы повредить отдельную популяцию клеток. Многие типы клеток особенно чувствительны к цитотоксическим лекарственным средствам или облучениям, используемым в противораковой терапии, и одна известная методика состоит в том, чтобы использовать лекарственный препарат, который ингибирует пролиферацию клеток, таких как клетки гемопоетической системы во время противораковой терапии с последующим возобновлением нормальной пролиферации, когда действие ингибирующего лекарственного препарата исчезает. По-видимому, пептиды настоящего изобретения имеют соответствующие короткие биологические полупериоды жизни для такой терапии. Аналогично, пролиферация выбранных популяций клеток, чувствительная к раковой терапии, может быть ингибирована наряду с раковыми клетками сама по себе, и противораковая терапия инициируется только тогда, когда раковые клетки достигают чувствительной фазы пролиферации, в то время как нормальные клетки находятся в менее чувствительной фазе.
Один тип клеточной пролиферации имеет место, когда клетки, такие как клетки костного мозга, фагоциты или гранулоциты, стимулируются CSF лекарственными препаратами во время терапии. Ингибирование роста клеток может восстанавливать такие клетки до нормальных скоростей роста.
При многих аутоиммунных заболеваниях субъект производит лейкоциты, активные против их собственных тканей. Путем ингибирования лейкоцитной функции, по крайней мере на время, такие аутоиммунные реакции могут быть существенно уменьшены.
Путем включения в Gα i -белковый межклеточный сигнальный механизм, другие функции, контролируемые Gαi -белками, могут быть модифицированы с помощью активных пептидов, например подвижность клетки при метаболизме кальция и цитоплазматические клеточные процессы, медиируемые благодаря Gαi-белку.
Таким образом, изобретение также предусматривает новые полипептиды, содержащие полные или частичные последовательности существенно гомологичные с Gαi-белками, обсужденными выше, а именно ---Lys-Ile-Ile-His-Glu-Asp-Glu-Tyr- Ser-Ra′ -Rb-Rc-Rd-Re-Rf-Gln-Tyr--- или части их, или легкие вариации их, такие как полипептиды, описанные в Ann. Rev. Biochem, 56, p. 624-625 (1987) и Prac. Natl-Acad. Sci. USA, 85, p. 3066- 3070 (1988). Такие последовательности предпочтительно содержат по крайней мере один Tyr-остаток, который помогает маркировке полипептида. Как указано ниже, NH2 N-концевой части предпочтительно защищают, например, N-ацилированием, как обсуждено выше. Там, где аминокислотный остаток N-концевой части должен быть Glu в такой гомологической последовательности, он может быть также преимущественно заменен p-Glu.
Соединения по изобретению очень полезны в ингибировании негемопоетической клеточной пролиферации в ходе селективной цитостатической обработки злокачественных гемопоетических клеток, а также в подавлении злокачественных эпидермальных и эпителиальных клеток, которые имеют место, например, в прогрессирующих случаях чешуйчатой карциномы и псориаза.
Наконец, возможно использование пептидов согласно изобретению как "мишени" для других молекул путем связывания молекул с пептидной цепью.
Таким образом, мы предлагаем:
(a) соединения для использования в терапии, диагностике или анализе, включающие радиоизотоп или меченный радиоизотопом лиганд,
ковалентно связанный прямо или непрямо с пептидом согласно
изобретению;
(b) соединения для использования в терапии, включающие цитотоксический лиганд, ковалентно связанный с пептидом
согласно изобретению;
(c) соединения для использования в
диагностике или анализе, включающие флюорохромный лиганд, ковалентно связанный с пептидом согласно изобретению.
В общем, для того, чтобы проявить защитное действие на фоне цитотоксических лекарственных препаратов, пептиды изобретения могут вводиться человеку путем инъекции в области доз 1-10 нг, например 4-5 нг, на вес тела 70 кг в день. При приеме путем инфузии или сходными методиками доза может находиться в диапазоне 30-300 нг на 70 кг веса тела, например около 100 нг, в течение шести дней. В принципе, желательно получить концентрацию пептида около 10-11 М до 10-7 M во внеклеточной жидкости пациента.
В общем, комбинированная терапия с цитотоксическими лекарственными препаратами, такими как цитозин арабинозид, требует тщательного хронометрирования, чтобы гарантировать, что миелопоетическая система защищена до тех пор, пока цитотоксический лекарственный препарат еще присутствует.
Согласно еще одной цели настоящего изобретения обеспечивается разработка фармацевтических композиций, включающих в качестве активного ингредиента, по крайней мере, одно соединение формулы (Ia), (II), (III), (IV), (V) или (VI), как здесь и ранее определено, или физиологически совместимой его соли в сочетании с фармацевтическим носителем или наполнителем. Композиции согласно изобретению могут существовать, например, в форме, подходящей для орального, назального парентерального или ректального введения.
Используемый здесь термин "фармацевтический" включает ветеринарные применения изобретения.
Соединения согласно изобретению могут присутствовать в обычных фармакологических формах для приема, таких как таблетки, покрытые таблетки, аэрозоли для носа, растворы, эмульсии, порошки, капсулы или пролонгированные формы. Обычные фармацевтические наполнители, так же как обычные способы получения, могут быть применены для получения этих форм. Таблетки могут быть получены, например, смешиванием активного ингредиента или ингредиентов с известными наполнителями, такими как, например, разбавители, такие как карбонат кальция, фосфат кальция или лактоза, дезинтегрирующие средства, такие как кукурузный крахмал или альгининовая кислота, связующие, такие как крахмал или желатин, смазки, такие как стеарат магния или тальк, и/или агенты для получения пролонгирования, такие как карбоксиполиметилен, карбоксиметил целлюлоза, фталат ацетат целлюлозы, или поливинилацетат.
Таблетки, если требуется, могут состоять из нескольких слоев. Таблетки с покрытием могут быть получены покрытием сердцевин, полученных обычным способом для таблеток, агентами, обычно используемыми для покрытий таблетки, например поливинилпирролидон или шеллак, аравийская камедь, тальк, диоксид титана или сахар. Для того, чтобы получить пролонгирующий эффект или избежать несовместимости, сердцевина может состоять из нескольких слоев. Таблетка с покрытием может состоять из нескольких слоев для того, чтобы получить длительно действующее выделение, и в этом случае могут быть использованы наполнители, упомянутые выше для таблеток.
Растворы для инъекций, например, могут быть получены обычным способом, таким как добавление консервирующих агентов, таких как п-гидроксибензоаты, или стабилизаторов, таких как ЕДТА. Затем растворами заполняют пробирки или ампулы для инъекций. Замедленное освобождение инъекции может быть обеспечено так называемыми мини-насосами.
Аэрозоли для носа могут быть сформированы аналогично в водном растворе и упакованы в контейнеры для распыления либо с аэрозольным распылительным средством или снабжены средствами для ручной компрессии. Капсулы, содержащие один или несколько активных ингредиентов, могут быть получены, например, смешиванием активных ингредиентов с инертными носителями, такими как лактоза или сорбит, и заполнением смеси в желатиновые капсулы.
Подходящие суппозитории могут, например, быть получены смешиванием активного ингредиента или комбинациями активного ингредиента с обычными носителями, предусмотренными для этой цели, такими как натуральные жиры или полиэтиленгликоль, или их производные.
Разовые дозы, содержащие соединения этого изобретения, предпочтительно содержат 1-10 мг, например 4-5 мг, пептида.
Пептиды изобретения могут быть синтезированы любым удобным путем. Обычно, присутствующие химически активные группы боковых цепей (амино, тиол и/или карбоксил) должны быть защищены в ходе взаимодействия на протяжении всего синтеза, но допустимо оставить несколько групп в ответвлениях незащищенными (гидроксигруппы, имидазольные группы, первичные амидные группы, амидные группы в циклических аминокислотах подобно пиро Glu) в течение всей методики синтеза.
Таким образом, конечной стадией должна быть депротекция (снятие защиты) полностью защищенного или частично защищенного производного пептида общей формулы I, и такие способы составляют дальнейшей аспект изобретения.
Наращивание пептидных цепей, в принципе, можно начинать либо с С-концевой части, либо с N-концевой части, хоты обычно используют методику, при которой рост начинается с С-концевой части.
Таким образом, можно начать с С-концевой части с помощью взаимодействия подходящего защищаемого производного, например лизина, с подходящим защищенным производным цистеина. Производное лизина должно иметь свободную α-амино группу, в то время как другой реактант должен иметь либо свободную, либо активированную карбоксильную группу и защищенную аминогруппу. После взаимодействия интермедиат, может быть очищен, например, хроматографически, и затем его селективно N-депротектируют, чтобы допустить присоединение следующего N-защищенного и свободного или активированного аминокислотного остатка. Эта процедура продолжается до тех пор, пока не образуется требуемая аминокислотная последовательность.
Заместители, активирующие карбоновую кислоту, которые, например, могут быть использованы, включают симметричные или смешанные ангидриды, или активированные сложные эфиры, такие как, например, п-нитрофенил сложный эфир, 2,4, 5-трихлорфенил сложный эфир, N-гидроксибензотриазол сложный эфир (OBt), N-гидроксисукцинимидил сложный эфир (OSu) или пентафторфенилсложный эфир (OFFP).
Взаимодействие свободных амино- и карбоксильной групп может, например, осуществляться с использованием дициклогексилкарбодиимида (ДЦК). Другим связывающим агентом, который, например, может быть использован, является N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (ЕЕДО).
Удобно осуществлять реакции взаимодействия при низких температурах, например от -20oC вплоть до температуры окружающей среды, в подходящей системе для растворения, например тетрагидрофуране, диоксане, диметилформамиде, метиленхлориде или смеси этих растворителей.
Может быть более удобным проводить синтез на твердофазной полимерной подложке. Хлорметилированный полистирол (сшитый с 1% дивинилбензола) является одним из пригодных типов подложки, в этом случае синтез будет начинаться с C-концевой части, например, путем связывания N-защищенного лизина с подложкой.
Ряд подходящих твердофазных методик описан Eric Atherton, Christopher J. London and Robert C. Sheppapd //J. Chem. Soc. Perkin, I538-46 (1981); James P. Tam, Foe S. Tjoeng and R.B. Merrifield J. //Am. Chem. Soc. 102, 6117-27 (1980); James P. Tam, Richard D. Dimarchi and R.B. Merrifield Int. J. //Peptide Protein Res. 16 412-25 (1980); Manfred Mutter and Dieter Bellof //Helvetica Chimica Acta, 67 2009-16 (1984).
Известен широкий выбор защищающих групп для аминокислот, и они иллюстрируются Schroder E. and Lubke K. The Peptides. Vol. 1 and 2. Academic Presms, New York and London, 1965 and 1966; Pettit G.R. Synthetic Peptides. Vol. 1-4. Van Nostrand, Reinhold, New York 1970, 1971, 1975, 1976; Houben-Weyl. Methoden der Organischen Chemic, Synthese von Peptiden. Band 15. Georg Thieme Verlag Stuttgart, NY, 1983; The Peptides, Analysis, Synthesis, Biology 1-7. Ed: Erhard Gross, Johaunes Meienhofer, Academic Press, NY, San Fransisco, London; Solid phase peptide synthesis, 2nd ed. /Hohn M. Stewaet, Janis D. Young, Pierce Chemical Company.
Таким образом, например, группы, защищающие амин, которые могут применяться, включают защищающие группы, такие как карбобензокси (Z-), т-бутоксикарбонил (Бок-), 4-метокси-2,3, 6-триметилбензолсульфонил (Mtr-) и 9-фторенилметоксикарбонил (Fmoc-). Следует принимать во внимание, что когда пептид строят с C-концевой части, группа, защищающая амин, должна присутствовать на a-аминогруппе каждого добавляемого нового остатка и она должна быть селективно удалена до следующей стадии связывания. Одной особенно полезной группой для такой временной защиты амина является Fmoc группа, которая может быть селективно удалена обработкой с пиперидином в органическом растворителе.
Группы, защищающие карбоксил, которые, например, могут применяться, включают легко расщепляемые группы сложного эфира, такие как бензил (-OBZl), п-нитробензил (-ONB) или т-бутил (-tOBu), а также взаимодействие с твердыми подложками, например метильными группами, связанными с полистиролом.
Тиолзащищающие группы включают п-метоксибензил (Mob), тритил (Trt) и ацетамидометил (Acm).
Следует иметь в виду, что существует широкий ряд других таких групп, как, например, описанные в деталях в вышеупомянутых ссылках литературы, и использование всех таких групп в способах, описанных здесь и ранее, находится в пределах объема настоящего изобретения.
Существует ряд методик для удаления амин- и карбоксилзащищенных групп. Они, однако, должны согласовываться с применяемой стратегией синтеза. Защищающие группы в ответвлениях должны быть стабильны в условиях, используемых для удаления временно a-аминозащищающих групп, вплоть до следующей стадии связывания.
Аминзащищающие группы, такие как Boc, и карбоксилзащищающие группы, такие как tOBu, могут удаляться одновременно при обработке кислотой, например трифторуксусной кислотой. Тиолзащищающие группы, такие как Trt, могут быть удалены селективно, с использованием окисляющего агента, такого как йод.
Цистеин, содержащий пептиды, может быть синтезирован способами, описанными в тексте, удалением всех защищающих групп, включая тиолзащищающие группы, в качестве последней стадии синтеза.
Следующие примеры даны только с целью иллюстрации.
Растворители были повторно перегнаны из коммерческого материала и хранились следующим способом: диметилформамид (ДМФ) над молекулярным ситом


TLC-системы были следующими:
S1: Двуокись кремния/CHCl3 MeOH (98:2)
S2: (95:5)
S3: двуокись кремния PP 8/0 1% TFA в 5% EtOH (водн).
Очищенные конечные продукты анализируют с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (HPLC). HPLC-система состоит из HP 1090М хроматографа с встроенным автоматическим пробоотборником и HP 1040 diode array (Hewlett-Packard, Waldbronn, FRG) и колонки supelcosil C-18 (250 x 4,6 мм, 5 мкм частицы). Образцы растворяют в 0,1% (об./об) ТФК (воды) и элюируют с линейным градиентом от 0 до 30% ацетонитрила в 0,1% ТФК (воды). Скорость протока составляла 2 мл/мин. Наблюдение за элюентом проводили при 214 нм при ширине линии 4 нм. Хроматограмма растворителя вычиталась с помощью электроники, и результаты представляли в единицах процента площади.
Аминокислотный анализ
Цистинсодержащие
пептиды окисляют пермуравьиной
кислотой, превращая кислый дабильный цистиновый остаток в кислую стабильную цистеиновую кислоту до кислотного гидролиза в 6M HCl при 110oC в течение 16 часов.
Затем сухие гидролизаты
помещают путем использования фенилизотиоцианата и анализируют, как описано в Heinrikson //Anal. Bioch. 1984, 136, 65-74.
Методика твердофазного синтеза
пептидов
Твердофазный
синтез пептидов осуществлялся на приборе ЛКБ Биолинкс 4170. Использованы стандартные методики согласно Инструкции-руководству к прибору ЛКБ Биолинкс 4170 и публикации
ЛКБ "Справочник по синтезу
пептидов на Fmoc-полиамиде", прилагаемым к прибору.
Стадия один. Присоединение полимера к C-концевой аминокислоте
В качестве полимера использован
Ультрасин A, поставляемый
фирмой "Фармация ЛКБ". Он содержит кислотолабильный линкер. Методика присоединения осуществлена в соответствии с инструкциями изготовителя. Полимер подвергают набуханию в
ДМФА (20 мл/г полимера) в
течение одного часа. В дихлорметане 1 моль Fmoc-L-Lys-(N-ε-Boc)-OH/г полимера активируют ДЦК (0,5 ммоль/г полимера). Остаток после испарения растворяют в небольшом
объеме ДМФА и добавляют к
полимеру. Присоединение катализируют действием в течение 60 минут N,N-диметиламинопиридина в ДМФА в количестве 0,1 ммоль/г полимера. Полимер промывают ДМФА, трет-амиловым
спиртом, уксусной кислотой и
трет-амиловым спиртом, диэтиловым эфиром и затем сушат.
Стадия два. Синтез пептида
В колонку Биолинкс загружают Fmoc-L-Lys-(N-e-Boc) полимер,
соответствующий 0,1 моль Lys
(обычно примерно 1 г), и промывают 10 минут ДМФА. Fmoc-защитную группу удаляют обработкой в 10-минутном цикле 20% пиперидина в ДМФА с последующей еще одной промывкой ДМФА.
Полученный PFP-эфир
Fmoc-аминокислоты (0,5 ммоль) активируют обработкой HOBT и присоединяют с рециркулированием в течение 30 минут. После 10-минутного цикла промывают ДМФА. Fmoc-защитную группу
удаляют обработкой в
течение 10 минут 20% пиперидина в ДМФА с последующим 10-минутным промыванием ДМФА. Этапы присоединения повторяют до завершения пептидной последовательности.
Стадия
три. Отщепление и
удаление защитных групп
Пептид отщепляют, и защитные группы удаляют в одну стадию выдерживанием один час с 95%-ной ТФУК. После фильтрования пептид высушивают.
Стадия четыре.
Очистка
Сырой продукт очищают хроматографией на колонке RP-8 на основе двуокиси кремния с применением буфера, содержащего 0,1% ТФУК и 2% или 5% этанола. Отобранные фракции
анализируют с
помощью ВЭЖХ-системы (высокоэффективной жидкостной хроматографии), описанной в описании, и фракции, содержащие наименьшее количество примесей, объединяют и сушат.
Пример
1.
L-пироглутамил-L-глутамил-L-аспартил-L-цистеинил-L-лизин
Соединение (I)
(а) Бензиловый сложный эфир т-Бок-(S-п- метоксибензил)-L-цистеинил-(e-бензилоксикарбонил)-L-лизина (I)
Гидрохлорид бензилового сложного эфира e-бензилоксикарбониллизина растворяют в 3 мл ДМФ, и ТЭА добавляют до трех пор, пока свободный ТЭА будет определяться в паровой фазе влажным куском бумаги
pH
индикатора. К этому раствору добавляют 491 мг сложного эфира т-Бок-(S-п-метоксибензил)-L-цистеин-N-гидроксисукцинимида, растворенного в 3 мл ДМФ. Через соответствующие интервалы времени порции ТЭА
добавляют, чтобы поддержать слабую щелочность раствора. Смесь оставляют на ночь при комнатной температуре, и после проверки на отрицательную нингидриновую реакцию смесь непосредственно подают в 2,5 х
75 см колонку с Sephadex LH-20, уравновешенную ДМФ и калиброванную стандартными реагентами (например, в данном примере - т-Бок-(g-бензил)-L-глутаминовая кислота-п-нитрофениловый сложный эфир и
п-нитрофенол). Поток в колонке поддерживают за счет гравитационного течения, и вытекающий поток анализируют при 280 нм до сбора фракций приблизительно по 10 мл. Продукт может быть идентифицирован
методом тонкослойной хроматографии (т.с.х.) каждой фракции, причем соответствующие фракции собирают и вакуумируют в вакууме, выход: 700 мг (100%) маслянистого продукта, однородного по т.с.х.
(хлороформ/ацетон) (9/1), Rf 0,64.
(b) Бензиловый сложный эфир т-Бок-(b-бензил)-L-аспартил-(S-п-метоксибензил)-L-цистеинил-(e- бензилоксикарбонил)-L-лизина (II)
700
мг блокированного и защищенного дипептида (I) растворяют в 25 мл безводного ДХМ, и 25 мл безводного ТФК добавляют. Через 30 мин кислоту и растворитель удаляют в вакууме. Остаток растворяют в ДХМ
и
снова испаряют. К раствору остатка в ДМФ (3 мл), который слегка подщелачивают ТЭА, добавляют раствор п-нитрофенилового сложного эфира т-Бок-(b-бензил)-L-аспаргиновой кислоты (488 мг) в 3 мл ДМФ.
Щелочность необходимо часто проверять и поддерживать добавлением малых количеств ТЭА. После того, как нингидриновая реакция становится отрицательной (после около 2 часов), реакционную смесь подают в
колонку с Sephadex LH-20 (2,5 x 75 см) и очищают, как описано выше. Выход после вакуумирования в вакууме 900 мг (100%) кристаллического продукта, гомогенного по т.с.х. (хлороформ/ацетон (9/1)), Rf 0,70.
(c) Бензиловый сложный эфир т-Бок-(g-бензил)-L-глутамил-(b-бензил)-L-аспарил-(S-п-метоксибензил)-L-цистеинил-(e- бензилоксикарбонил)-L-лизина (III)
900 мг
блокированного трипептидного производного II деблокируют ТФК, как описано выше, растворяют в 3 мл ДМФ и слабо подщелачивают ТЭА. К этому раствору добавляют 504 мг п-нитроффенилового сложного эфира
т-Бок-(g-бензил)-L-глутаминовой кислоты (в 3 мл ДМФ). Через около 2,5 ч нингидриновая реакция становится отрицательной и смесь подают в колонку с Sephadex LH-20 для очистки, как описано выше.
Разделение компонентов в этой реакционной смеси и ее мониторинг путем т.с.х. могут быть выполнены, как описано выше. Соответствующие фракции (9-15 в этом случае) собирают, выпаривают и сушат. Выход
1140 мг (100%) слабого желтоватого масла, гомогенного по т.с.х. (хлороформ/ацетон (9/1)), Rf 0,53.
(d) Бензиловый сложный эфир
бензилоксикарбонил-L-пироглутамил-(g-бензил)-L-глутамил-(b-бензил)-L-аспарти-(S-п-метоксибензил)-L-цистеинил-(e-бензил-оксикарбонил)-L-лизина (IV)
1140 мг тетрапептидного производного III
деблокируют ТФК в ДХМ, как описано для I, и растворяют в 3 мл ДМФ. Раствор слабо подщелачивают ТЭА, и добавляют 423 мг п-нитрофенилового сложного эфира бензилоксикарбонил-L-пироглутаминовой кислоты в
виде раствора в 3 мл ДМФ. Щелочность реакционной смеси следует неоднократно проверять и, если необходимо, восстанавливать добавлением ТЭА. Через около 3 часов нингидриновый тест становится
отрицательным, и пентапептидное производное IV может быть очищено, как описано выше. Выход 1230 мг (96%), слабо-желтоватое масло, гомогенное по т.с.х. (хлороформ/ацетон (9/1)), Rf 0,44 (с
хвостовой фракцией).
(е) L-пироглутамил-L-глутамил-L-аспартил-L-цистеинил-L-лизин
50 мг защищенного пентапептидного производного IV растворяют в 50 мл жидкого фтористого
водорода при 0oC с добавлением 500 мг метионина в качестве акцептора и оставляют в течение 1 часа. Фтористый водород затем испаряют досуха в вакууме при 0oC, и остаток смешивают
с этилацетатом. Этилацетатную промывку декантируют и отбрасывают. Оставшийся материал растворяют в разбавленной уксусной кислоте и лиофилизируют.
Лиофилизованный материал (21 мг) можно очистить методом HPLC с обращенной фазой, используя C18-колонку 10 мм х 10 см при скорости потока 2,8 мл/мин, используя градиентное элюирование раствором А (0,1%-ный водный раствор трифторуксусной кислоты) и раствором B (0,1%-ный раствор трифторуксусной кислоты в ацетонитриле), причем 0,10%-ный раствор B добавляют в течение 30 минут. Детектирование осуществляют, используя поглощение в ультрафиолете при 214 нм или используя пиридиндисульфидный реагент (для SH-групп).
Ряд следующих пептидов может быть синтезирован по общей методике примера 1. Они идентифицированы и охарактеризованы в табл. 1.
Пептид N 9: пGlu-Glu-Asp(бензил Cys)-Lys
Исходными аминокислотными производными служат: Fmoc-Lys-(N-e-Boc)-OPFP,
Fmoc-Cys-(S-бензил)-OPFP,
Fmoc-Asp-(b-O-трет-Bu)-O-PFP, Fmoc-Glu-(b-O-трет-Bu)-O-PFP, пGlu-O-PCIP. Конечный выход пептида с чистотой 95% составляет примерно 42.
Пептид N: 15
пGlu-Glu-Asp-(2-пиридилтио-Cys)-Lys
Указанное соединение получено из конечного продукта примера 1 (пGlu-Glu-Asp-Lys-Lys) с помощью реакции в течение 1 часа при pH с пятикратным избытком
дипиридилдисульфида. Полученный продукт очищен хроматографией по вышеприведенной методике. Выход пептида с чистотой 93% составляет 25%
Пептид N 18: пGlu-Glu-Asp-Phe-Lys
Исходными
аминокислотными производными служат: Fmoc-Lys-(N-e-Boc)-O-PFP, Fmoc-Phe-O-PFP, Fmoc-Asp-(b-O-трет-Bu)-O-PFP, Fmoc-Gly-O-PFP, nGly-O-PClP. Конечный выход пептида с чистотой 99% составляет примерно
35%
Пептид N 22: пGlu-Gly-Asp-Cys-Lys
Исходными аминокислотными производными служат: Fmoc-Lys-(N-e-Boc)-O-PFP, Fmoc-Cys-(b-O-трет-Bu)-O-PFP, Fmoc-Asp-(b-O-трет-Bu)-O-PFP,
Fmoc-Gly-O-PFP, пGly-O-PClP. Конечный выход пептида с чистотой 99% составляет примерно 35%
Пептид N 21: пGlu-Glu-Asp-Ser-Lys
Исходными аминокислотными производными служат:
Fmoc-Lys-(N-e-Boc)-O-PFP, Fmoc-Ser-(O-трет-Bu)-O-DBH, Fmoc-Asp-(b-O-трет-Bu)-O-PFP, Fmoc-Glu-(g-O-трет-Bu)-O-PFP, пGlu-O-PClP. Конечный выход пептида с чистотой 92% составляет примерно 44%
Пример 30. pGlu-Glu-Asp-Ser-Lys-OH
Пептид синтезируют на LKB Biolynx 4170 -полностью автоматическом синтезаторе пептидов с мониторингом на УФ 304 нм. Fmoc используют в качестве временной
N-защиты и УФ-индикатора. Аминокислоту C-концевой части связывают с полимером посредством кислого лабильного индикатора anm.
Стандартный протокол:
Связывание с рециркуляцией
30 мин
Промывка ДМФ 10 мин
Удаление защиты 20%-ным пеперидин/ДМФ 10 мин
Промывка ДМФ 10 мин
Аминокислоту C-концевой части активируют как симметричный ангидрид с
ДЦК
и связывают с полимером N,N-диметиламинопиридином в качестве катализатора. После рециркуляции в течение 60 минут используют стандартную методику во время всего синтеза.
Используемые
аминокислотные производные:
Fмос-Lys(ε-N-Boc)-OH
Fmoc-Ser(O-tBu)-ODBH
Fмос-Asp(β-tOBu)-OPfp
Fмос-Glu(γ-tOBu)-OPfp
pGlu-OPClP
После окончательной промывки полностью защищенного
пентапептида полимер промывают диэтиловым эфиром и сушат на воздухе. С пептида полностью снимают защиту и отделяют от полимера
в одной операции
путем обработки с 95% воды. ТФК в течение 1 ч. После фильтрации, промывки ТФК и испарения конечный пептид очищают на RP 8 колонке и элюируют этанолом в 0,1% ТФК.
Выход 44%
Чистота более 92% (HPLC RP18, 214 нм).
Аминокислотный анализ удовлетворительно.
Биологическая активность
Методика испытаний in vivo
Мышей весом 20-24
г инъецировали внутрибрюшинно единичными дозами пептида, растворенного в физиологическом солевом растворе. Зверьков умерщвляли путем сворачивания шеи и в стерильных условиях
извлекали бедренную кость.
Из бедренной кости вымывали костный мозг и с помощью счетчика Coulter подсчитывали общее число клеток. Дифференцированный подсчет типов клеток осуществлен на высушенных на
воздухе мазках костного
мозга, окрашенных красителем Giemsa. Мазки периферийной крови фиксировались в метаноле, окрашивались красителем Giemsa и подсчитывались дифференцированно.
Методика испытаний in
vitro
CFU анализировались с использованием метода с однослойной культурой агара. Клетки костного мозга (1 • 105 клеток) добавляют к 0,3% агара в
ростовой среде и переносят в
чашки для культивирования. Перед нанесением на пластинки клетки костного мозга с пептидом инкубируют 1 час. В качестве фонового стимулятора применяют рекомбинантный
CFU-GM. Чашки инкубируют 7 дней при
37oC в увлажненной атмосфере с 7% CO2 в воздухе. Образовавшиеся колонии подсчитывают с помощью микроскопа.
Влияние (действие) на первичные клеточные культуры оценивалось с помощью измерения активности деления клеток на культурах возрастом 2-3 недель, обрабатывавшихся пептидом в течение последних 48 часов.
Результаты in vitro: CFUc даны в табл. 2.
In vitro: митоз эпидермальных клеток Число митозов
Контроль 143±15
пGlu-Gly-Asp-Cys-Lys 159±12
Пептид 21 (пGlu-Glu-Asp-Ser-Lys
99±15
Замена серина в положении 4 приводит к изменению специфичности, проявляющемся в том, что пептид N 21 не оказывает воздействия на клетки
костного мозга или параметры периферийной
крови, но оказывает значительное ингибирующее действие на митозы эпидермальных клеток.
Замена бензилцистеина или фенилаланина не оказывает неблагоприятного воздействия на ингибирующую активность пептида.
Действие in vivo
Все результаты представлены в виде процента от контрольных испытаний (табл.3).
Дериватизация тиольной группы цистеина метаболически подвижной пиридильной группы приводит к замедлению снижения GFUc со значительным эффектом спустя 3 дня. Кроме того, пептид N 15 сохраняет краткосрочное действие на периферийные лимфоциты.
Замена глутаминовой кислоты в положении 2 глицином приводит к довольно быстрому ингибирующему действию на CFU в костном мозге. Действие на периферийную кровь чрезвычайно быстро и кратковременно.
Эффективная доза ингибирования пролиферации клеток
Мономер (pEEDCK*) преимущественно
ингибирует пролиферацию миеломных клеток.
Ингибирование может быть определено in vitro с помощью анализа колонии CFU-GM на агаре, и эффективные дозы в этих опытах составляют величины в интервале от
10-13 до 10-6 M (см.
ОД Laerum etal. International Journal of Cell Cloning 8:431-444 (1990)).
Ингибирование in vivo пролиферации CFU-GM и уменьшение числа CFU-GM и числа гранулоцитов в периферийной крови было обнаружено после инъекции (впрыскивания) или непрерывного вливания (собственные наблюдения и работы ОД Laerum and W. Paukovits in "The Inhibitors of Hematopoiesis" Ed. A. Najman, M. Guigonetal. INSERM/ John Libley Eurotext Ltd. 1987, том 162, с. 21-30). Эффективные дозы составляют величины в интервале 0,01 нг/кг 30 мкг/кг. Поскольку данные in vitro показывают аналогичную чувствительность клеток и мышиного и человеческого происхождения, данный факт дает основания предполагать сходную чувствительность к in vivo. Из указанных данных следует, что человека весом 70 кг следует получать от 1 нг до 2 мг за инъекцию.
Предклинические модели.
Соответствующие клинические данные были получены с использованием подавления миеломы на мышах, после применения арабинофуранозилцитозина или п-Musiard, и после ионизирующего облучения. В этих исследованиях было продемонстрировано значительное подавление миеломы после инъекции доз pEEDCK 30 мкг/кг (Собственные наблюдения и работы Paukovits WR et Al. Blood, т. 77, 1991, с. 1313-1319).
Реферат
Использование: в медицине, как соединения, обладающие способностью регулировать пролиферацию клеток. Сущность изобретения: производные пептидов формулы I A-B-C-D-E при определенных их значениях. Способ ингибирования клеточной пролиферации у человека и животных путем введения эффективного количества соединения I. 5 с. и 6 з.п. ф-лы, 3 табл.
Формула
Ra Rb Rc Rd (Re)n - Rf
где Ra -

Rb -

Rc -

Rd -

Re -

R1 -
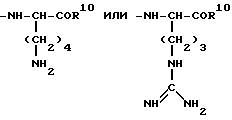
или -NH-CH2-COR10
где n 0 или 1;
р и q независимо 1 или 2;
R1 и R2 оба водород или вместе оксогруппа;
R3 и R4 H;
R5 водород, ацетил, p Glu или Ser;
R6 и R7 независимо гидроксигруппа или аминогруппа;
R8 водород;
С7 С20-кабоциклическая аралкильная группа или метаболически лабильная S-защищающая группа, выбранная из бензильной, фенильной и пиридильной групп
R9 водород или метильная группа;
R10 гидрокси-или аминогруппа, причем если Rd Ser, n 0, то Rf иной, чем Gly, и, за исключением аланина, который может быть в D- или L-форме, и глицина, все указанные аминокислотные остатки находятся в L-форме, кроме соединений:
pGIu Asp Asp Cys Lys; pGIu GIu Asp Cys Lys; pGIu GIn - Asp Cys Lys; pGIu GIu Asp Cys GIy Lys; pGIu GIu Asp Cys - AIa Lys; pGIu GLu Asp Cys Lys LysNH2
обладающие способностью регулировать пролиферацию клеток.

где R8 С7-20 -карбоциклическая аралкильная группа,
и все аминокислоты имеют хиральную форму, определенную в п.1.

и все аминокислоты имеют хиральную форму, определенную в п.1.

где R12 бензильная, фенильная или пиридильная группа, и все аминокислоты имеют хиральную форму, определенную в п.1.
и все аминокислоты имеют харальную форму, определенную в п.1.
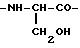
и все аминокислоты имеют хиральную форму, определенную в п.1.
Комментарии