Гибридные полипептиды с усиленными фармакокинетическими свойствами - RU2279883C2
Код документа: RU2279883C2
Чертежи















Описание
Настоящая заявка является частичным продолжением серийной заявки No. 09/350641, поданной 9 июля 1999 г., которая является частичным продолжением серийной заявки No. 09/315304, поданной 20 мая 1999 г., которая является частичным продолжением серийной заявки No. 09/082279, поданной 20 мая 1998 г., полное содержание которых включено в настоящее описание в качестве ссылки.
1. Введение
Настоящее изобретение относится к энхансерным пептидным последовательностям, первоначально полученным из белковой последовательности оболочки ретровируса (gр41), которые усиливают фармакокинетические свойства любого корового полипептида, с которым они связываются. Частично изобретение основывается на обнаружении того факта, что гибридные полипептиды, включающие энхансерные пептидные последовательности, связанные с коровым полипептидом, обладают усиленными фармакокинетическими свойствами, таким как увеличенный период полужизни. Изобретение также относится к новым антифузогенным и/или противовирусным пептидам, включающим такие пептиды, которые содержат указанные энхансерные пептидные последовательности, и к способам применения таких пептидов. Изобретение также относится к способам усиления фармакокинетических свойств любого корового полипептида посредством связывания энхансерной пептидной последовательности с коровым полипептидом. Используемые в практике осуществления настоящего изобретения коровые полипептиды могут включать любые фармакологически полезные пептиды, которые могут применяться, например, в качестве терапевтического или профилактического средства. В неограничивающем варианте настоящее изобретение иллюстрируется с помощью примера, в котором показано, что гибридный полипептид, включающий, например, коровый полипептид ВИЧ, связанный с энхансерными пептидными последовательностями, представляет мощный, нетоксичный ингибитор ВИЧ-1, ВИЧ-2 и ВИО (SIV) инфекции. Кроме того, энхансерные пептидные последовательности согласно настоящему изобретению связаны с коровым полипептидом респираторно-синцитиального вируса (РСВ) и коровым полипептидом рецептора лютеинизирующего гормона (ЛГ-РГ). В каждом случае показано, что гибридный полипептид обладает усиленными фармакокинетическими свойствами, и гибридный полипептид РСВ проявляет значительную анти-РСВ активность.
2. Предпосылки создания изобретения
Полипептидные продукты имеют широкий диапазон применений в качестве терапевтических и/или профилактических средств для профилактики и лечения заболевания. Многие полипептиды способны регулировать биохимические или физиологические процессы с целью либо профилактики заболевания, либо облегчения связанных с заболеванием симптомов. Например, полипептиды, такие как вирусные или бактериальные полипептиды, успешно применяют в качестве вакцин для профилактики патологических состояний. Кроме того, пептиды успешно применяют в качестве терапевтических средств для лечения симптомов заболеваний. Такие пептиды попадают в различные категории, такие как, например, гормоны, ферменты, иммуномодуляторы, сывороточные белки и цитокины.
Полипептиды, для того чтобы оказывать нужный биологический и терапевтический эффект на целевые участки, должны присутствовать в соответствующих концентрациях в участках применения. Кроме того, должна в основном поддерживаться их структурная целостность. Исходя из этого, показания по применению композиций полипептидов в качестве терапевтических средств определяются химической природой и характеристиками полипептидов, такими как их размер и сложность, конформационные требования, их стабильность, поддерживать которую зачастую бывает сложно, и профили растворимости. Фармакокинетические свойства любого конкретного терапевтического пептида зависят от биологической доступности указанного пептида, характера его распределения в организме и последующего выведения из организма.
Поскольку многие биологически активные вещества, такие как пептиды и белки, быстро разрушаются в организме, очень важно разрабатывать эффективные системы для поддержания стойкой концентрации пептида в кровяном русле, повышать эффективность действия таких пептидов и минимизировать случаи возникновения побочных эффектов и их тяжесть.
3.1. Краткое описание сущности изобретения
Настоящее изобретение относится, во-первых, к энхансерным пептидным последовательностям, первоначально полученным из белковых последовательностей оболочек (gр41) различных ретровирусов, например из ВИЧ-1, ВИЧ-2 и ВИО, которые усиливают фармакокинетические свойства любого корового полипептида, с которым они связываются. В основе изобретения лежит обнаружение того факта, что, когда раскрываемые энхансерные пептидные последовательности связываются с любым коровым полипептидом, получаемый гибридный полипептид обладает усиленными фармакокинетическими свойствами, включающими, например, увеличенный период полужизни и сниженный клиренс, в сравнении с одним только коровым полипептидом. Настоящее изобретение также относится к таким гибридным полипептидам и коровым полипептидам и к новым пептидам, которые проявляют антифузогенную активность, противовирусную активность и/или способность модулировать внутриклеточные процессы, в которых участвуют свернутые спиральные пептидные структуры. В число таких пептидов входят пептиды, которые содержат энхансерные пептидные последовательности.
Коровые полипептиды могут включать любые пептиды, которые могут быть введены в живую систему, например любые пептиды, способные функционировать в качестве терапевтических, профилактических или визуализирующих средств, полезных для лечения или профилактики заболевания или для использования в диагностических или прогностических методах, включая методы визуализации in vivo. Такие пептиды включают, например, факторы роста, гормоны, цитокины, ангиогенные факторы роста, внеклеточные матричные полипептиды, лиганды рецепторов, агонисты, антагонисты или инверсные агонисты, пептидные средства-мишени, такие как визуализирующие средства, или цитотоксичные средства-мишени, или полипептиды, которые проявляют антифузогенную и/или противовирусную активность, и пептиды или полипептиды, которые функционируют в качестве антигенов или иммуногенов, включая, например, вирусные и бактериальные полипептиды.
Изобретение также относится к способам усиления фармакокинетических свойств любого корового полипептида посредством связывания корового полипептида с энхансерными пептидными последовательностями с образованием гибридных полипептидов.
Изобретение далее относится к способам применения раскрываемых в нем пептидов, включая гибридные полипептиды, содержащие энхансерные пептидные последовательности. Например, способы настоящего изобретения включают способы снижения или подавления вирусной инфекции, например инфекции, вызванной ВИЧ-1, ВИЧ-2, РСВ, вирусом кори, гриппа, парагриппа, Эпштейна-Барра и вирусом гепатита, и/или процессов слияния клеток, вызванных вирусом. Энхансерные пептидные последовательности согласно настоящему изобретению могут дополнительно применяться для увеличения in vitro или ex-vivo периода полужизни корового полипептида, к которым присоединяются энхансерные пептидные последовательности, например энхансерные пептидные последовательности могут увеличить период полужизни присоединенных коровых полипептидов в клеточной культуре или образцах клеток или ткани.
Изобретение иллюстрируется примерами, в которых показано, что гибридные полипептиды, содержащие коровый полипептид ВИЧ, связанный с энхансерными пептидными последовательностями, проявляют значительно усиленные фармакокинетические свойства и действуют как мощные нецитотоксичные ингибиторы ВИЧ-1, ВИЧ-2 и ВИО инфекции. Изобретение далее иллюстрируется примерами, в которых показано, что гибридные полипептиды, содержащие коровый полипептид РСВ или полипептид лютеинизирующего гормона, проявляют значительно усиленные фармакокинетические свойства. Кроме того, гибридный полипептид с РСВ обладает существенной активностью против РСВ.
3.2. Определения
В контексте настоящего описания пептиды, полипептиды и белки определяются как органические соединения, включающие две или более аминокислот, ковалентно соединенных, например, с помощью пептидных амидных связей. Пептиды, полипептиды и белки могут также включать неприродные аминокислоты в любой модификации, а также дополнительные аминогруппы и карбоксильные группы, как указано в описании. Термины «пептид», «полипептид» и «белок» взаимозаменяемы при использовании в настоящем описании.
Определенные в настоящем описании пептидные последовательности изображаются с помощью однобуквенных символов, соответствующих конкретным аминокислотным остаткам, как приведено ниже:
А (аланин)
R (аргинин)
N (аспарагин)
D (аспарагиновая кислота)
С (цистеин)
Q (глютамин)
Е (глютаминовая кислота)
G (глицин)
Н (гистидин)
I (изолейцин)
L (лейцин)
К (лизин)
М (метионин)
F (фенилаланин)
Р (пролин)
S (серин)
Т (треонин)
W (триптофан)
Y (тирозин)
V (валин)
Х (любая аминокислота)
Термин "энхансерные пептидные последовательности" в контексте настоящего описания означает пептиды, имеющие следующие консенсусные аминокислотные последовательности:
"WXXWXXXI", "WXXWXXX", "WXXWXX", "WXXWX", "WXXW", "WXXXWXWX", "XXXWXWX", "XXWXWX", "XWXWX", "WXWX", "WXXXWXW", "WXXXWX", "WXXXW", "IXXXWXXW", "XXXWXXW", "XXWXXW", "XWXXW", "XWXWXXXW", "XWXWXXX", "XWXWXX", "XWXWX", "XWXW", "WXWXXXW" или "XWXXXW", где Х может быть любой аминокислотой, W означает триптофан и I означает изолейцин. Как указывается далее, энхансерные пептидные последовательности согласно настоящему изобретению также включают пептидные последовательности, которые в целом такие же, что и консенсусные аминокислотные последовательности, но содержат аминокислотные замещения, вставки или делеции, которые, однако, не устраняют способность пептида усиливать фармакокинетические свойства корового пептида, с которым он связывается, в сравнении с фармакокинетическими свойствами одного корового полипептида.
Термин «коровый полипептид» в контексте настоящего описания относится к любому полипептиду, который может быть введен в живую систему и таким образом представлять собой биологически активную молекулу, например любой полипептид, который может функционировать как фармакологически полезный пептид при лечении или профилактике заболевания.
Термин «гибридный полипептид» в контексте настоящего описания относится к любому полипептиду, включающему амино, карбокси или амино и карбокси концевую энхансерную пептидную последовательность и коровый полипептид. В типичном случае энхансерная пептидная последовательность связана непосредственно с коровым полипептидом. Следует понимать, что энхансерный пептид может также присоединяться к промежуточной аминокислотной последовательности, имеющейся между энхансерной пептидной последовательностью и коровым пептидом.
Термины "антифузогенный" и "препятствующий слиянию мембран" в контексте настоящего описания относятся к способности пептида ингибировать или снижать уровень явлений слияния двух или более структур, например клеточных мембран или вирусных оболочек или жгутиков, в сравнении с уровнем слияния мембран, который имеется между структурами в отсутствие пептида.
Термин "противовирусный" в контексте настоящего описания относится к способности пептида подавлять вирусное инфицирование клеток, протекающее посредством, например, слияния клеток или в виде свободной вирусной инфекции. Такое инфицирование может обуславливаться слиянием мембран, что имеет место в случае вирусов, покрытых оболочкой, или другими процессами слияния, вовлекая вирусную структуру и клеточную структуру, например слияние вирусного жгутика и бактериальной мембраны в ходе бактериальной конъюгации.
4. Краткое описание чертежей
Фиг.1. Гибридные полипептиды. Энхансерные пептидные последовательности, полученные из предположительного N-концевого и С-концевого взаимодействующих участков, изображены связанными с основным коровым полипептидом. Консервативные энхансерные пептидные последовательности затенены. Следует отметить, что указанные энхансерные пептидные последовательности могут использоваться либо как N-концевые, С-концевые или N- и С-концевые добавки. Кроме того, энхансерные пептидные последовательности могут добавляться к коровому полипептиду в прямой или обратной ориентации, индивидуально или в любой из возможных комбинаций, для усиления фармакокинетических свойств пептида.
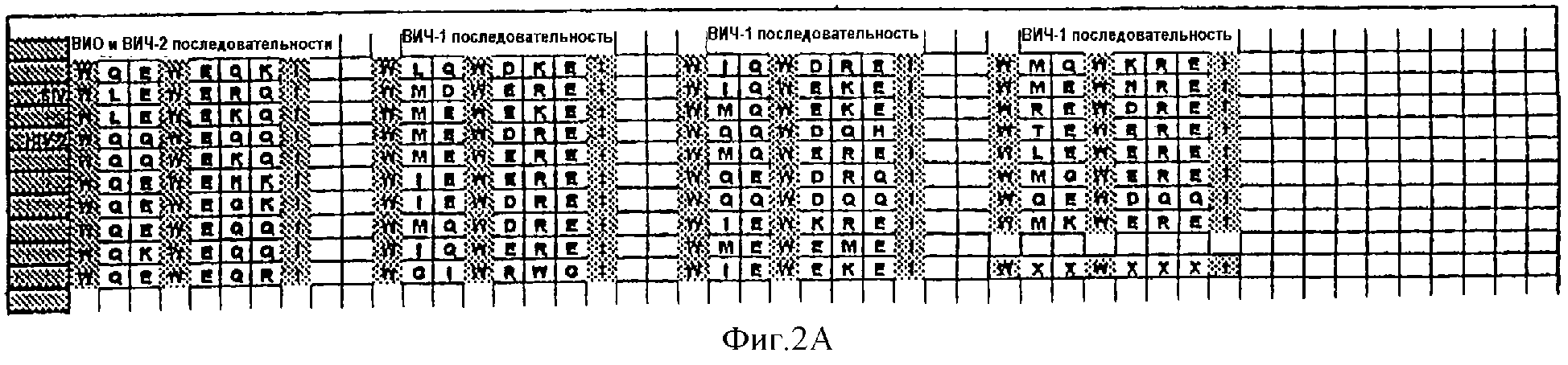
Фиг.2А. Энхансерные пептидные последовательности, полученные из различных белковых последовательностей оболочек (gр41), отражающие N-концевой взаимодействующий участок, наблюдаемый во всех опубликованных к настоящему времени данных о выделенных последовательностях ВИЧ-1, ВИЧ-2 и ВИО. Концевая последовательность "WXXWXXXI" означает консенсусную последовательность.
Фиг.2В. Варианты энхансерной пептидной последовательности, полученные из различных белковых последовательностей оболочек (gр41), отражающие С-концевой взаимодействующий участок, наблюдаемый во всех опубликованных к настоящему времени данных о выделенных последовательностях ВИЧ-1, ВИЧ-2 и ВИО. Концевая последовательность "WXXXWXWX" означает консенсусную последовательность.
Фиг.3. Сравнение титров ВИЧ-1 в тканях мышей SCID-huPBMC, зараженных ВИЧ-1 9320, по данным уровней Р24, полученных в тестах на культуре huPBMC. На фиг.3 показано сравнение уровней ингибирования вирусов Т20 и Т1249 in vivo.
Фиг.4А-4В. Фармакокинетический профиль Т1249 в плазме крови в сравнении с уровнем контроля с коровым вариантом Т1387 у крыс CD после в/в инъекции в течение 2 часов (фиг.4А) и 8 часов (фиг.4В). Полипептид Т1387 представляет собой коровый полипептид и Т1249 представляет собой коровый полипептид, связанный с энхансерными пептидными последовательностями.
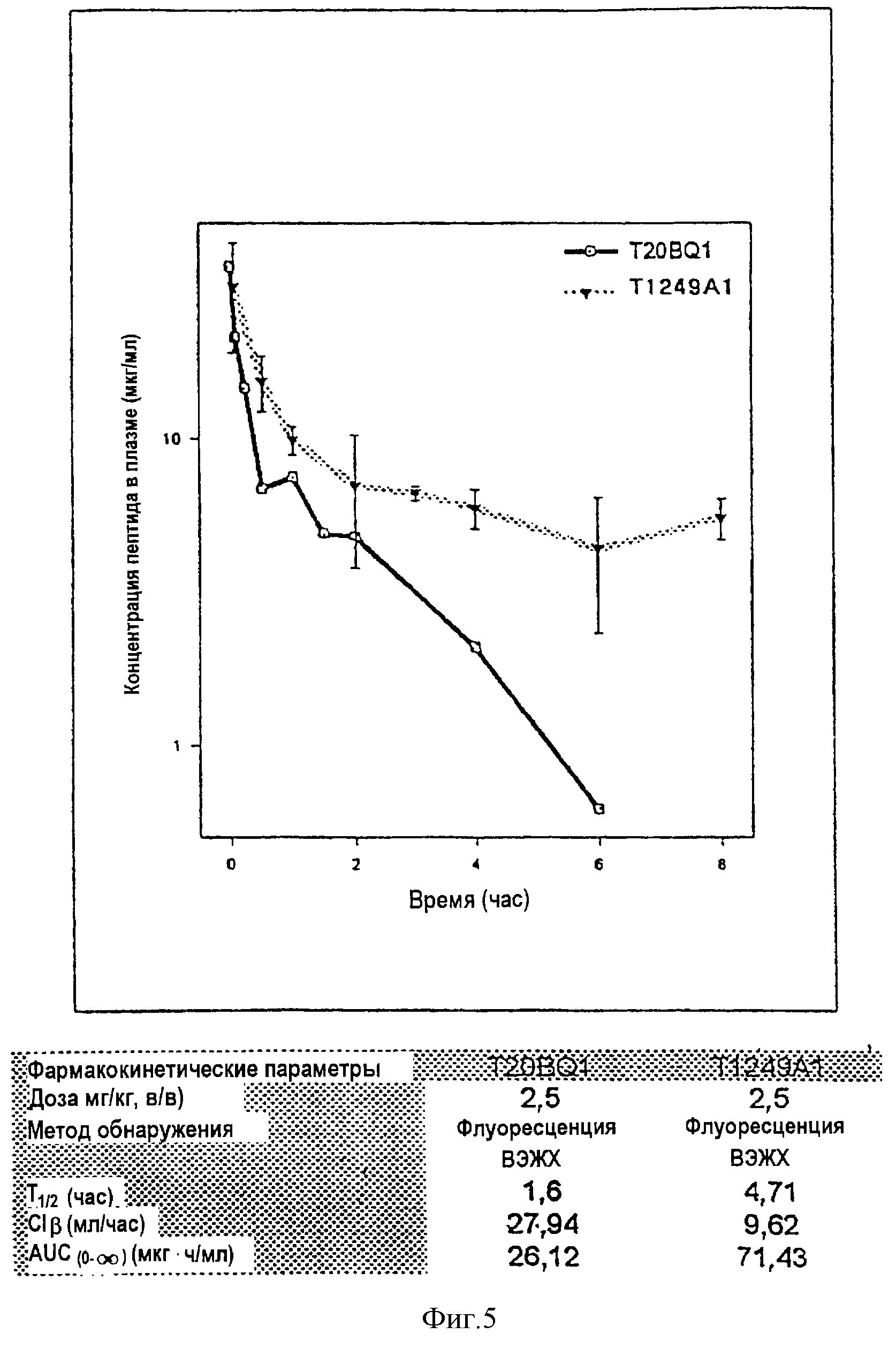
Фиг.5. Фармакокинетический профиль Т1249 в плазме крови в сравнении с контрольным вариантом Т20 у крыс CD после в/в инъекции. Полипептид Т1249 представляет собой гибридный полипептид, образованный из корового полипептида (Т1387), связанного с энхансерными пептидными последовательностями. Т20: n=4; Т1249: n=3.
Фиг.6. Сравнение активности и цитотоксичности Т20/Т1249 против ВИЧ-1/IIIb.
Фиг.7. Непосредственное связывание Т1249 с конструкцией на основе gр41 - М41Δ178.125I-T1249 очищен до максимально достижимой удельной активности методом ВЭЖХ. Показано насыщение при связывании с М41Δ178 (белок слияния из эктодомена gр41, не содержащий аминокислотной последовательности Т20), иммобилизованным на микротитрационном планшете в концентрации 0,5 мг/мл.
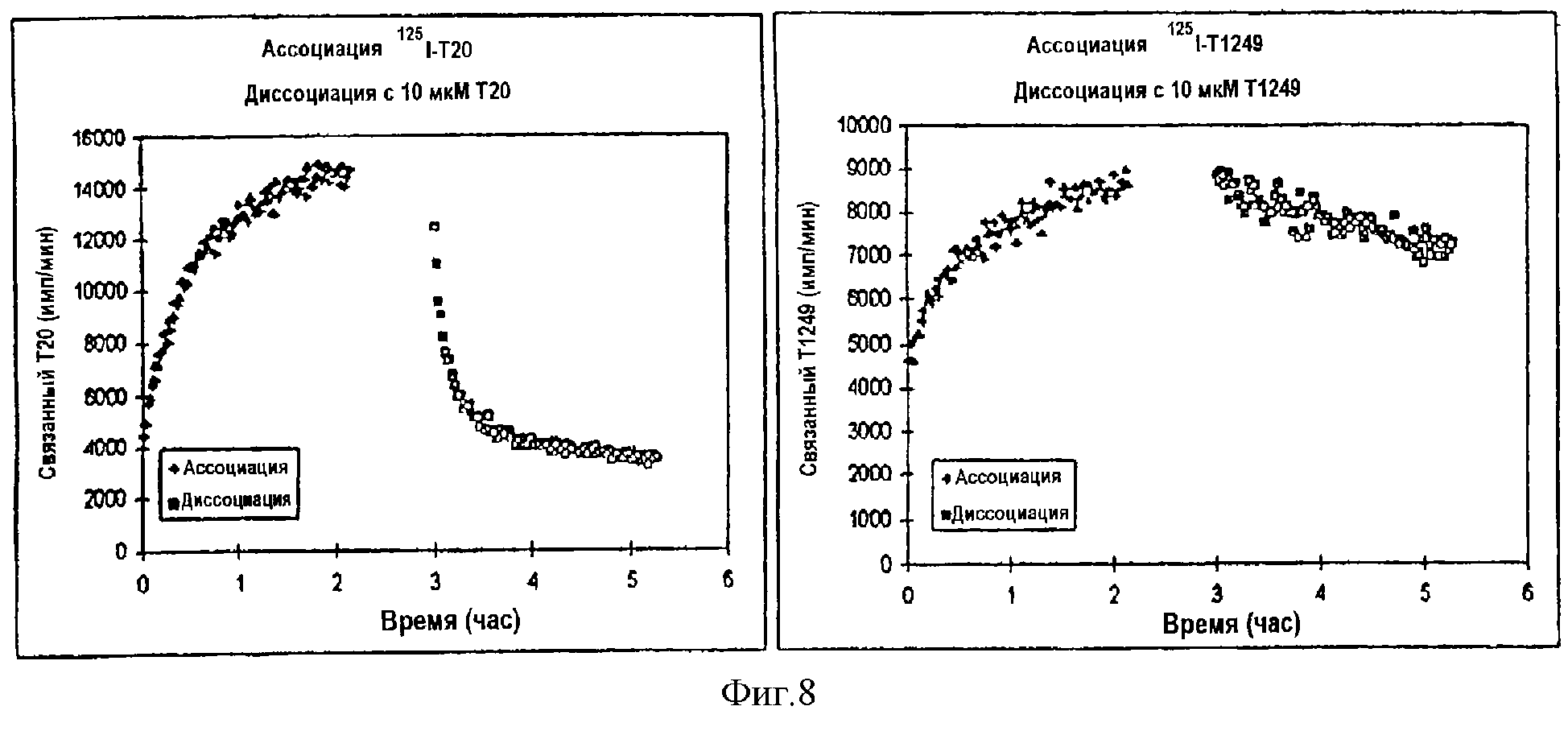
Фиг.8. Процесс ассоциации/диссоциации Т1249 с течением времени. Результаты показывают, что125I-T1249 и125I-T20 обладают сходными значениями аффинности связывания в 1-2 нМ. Начальные скорости ассоциации и диссоциации для125I-T1249 значительно ниже, чем для125I-T20. Диссоциацию связанного лиганда с радиоактивной меткой измеряют при добавлении немеченного пептида до конечной концентрации в 10 мкм в 1/10 общего объема среды для тестирования.
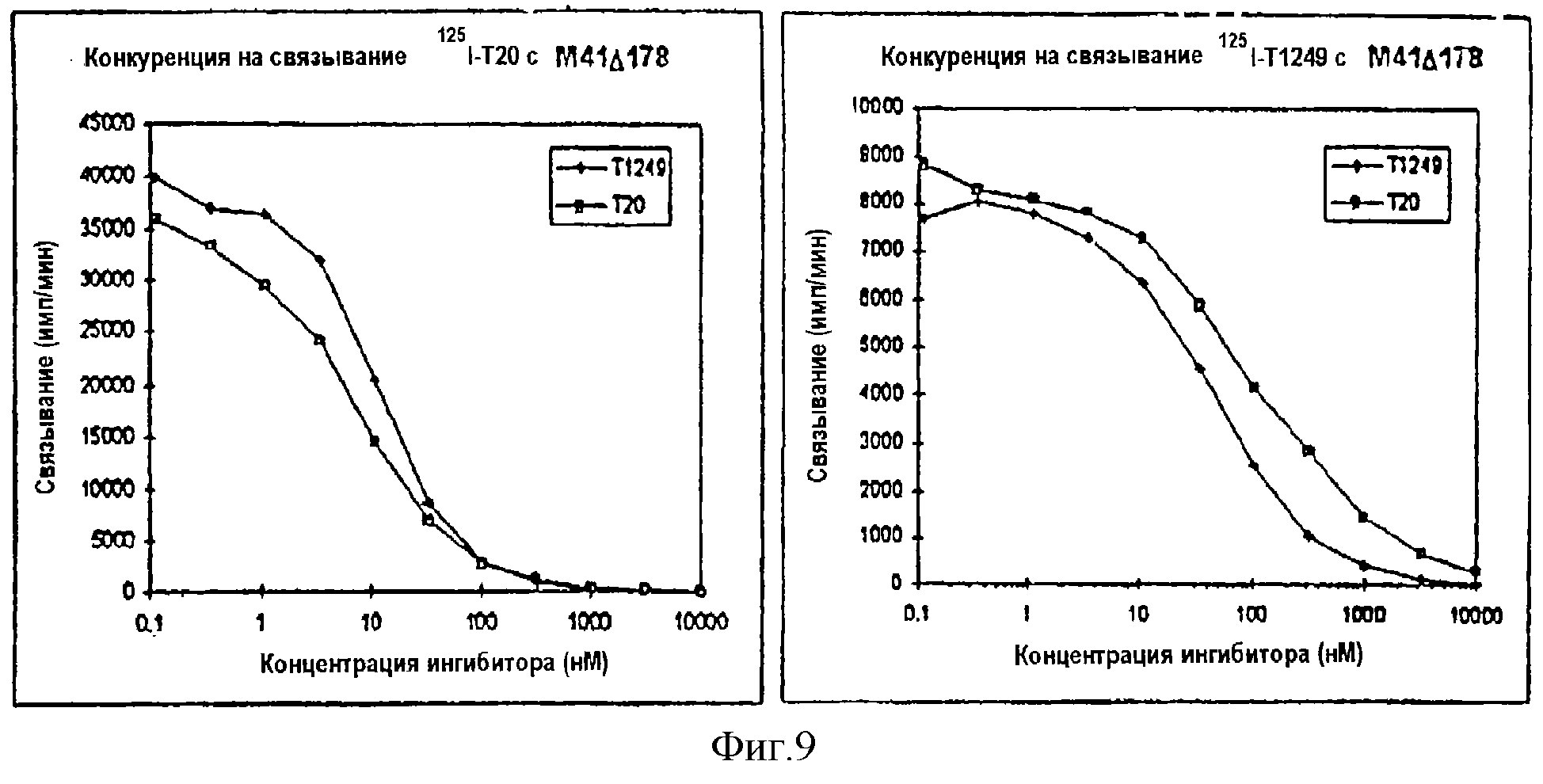
Фиг.9. Конкуренция при связывании Т1249 с М41Δ178. Немеченные Т1249 и Т20 титруют в присутствии одной концентрации125I-T1249 или125I-T20. Лиганд добавляют сразу после начала инкубации немеченного пептида.
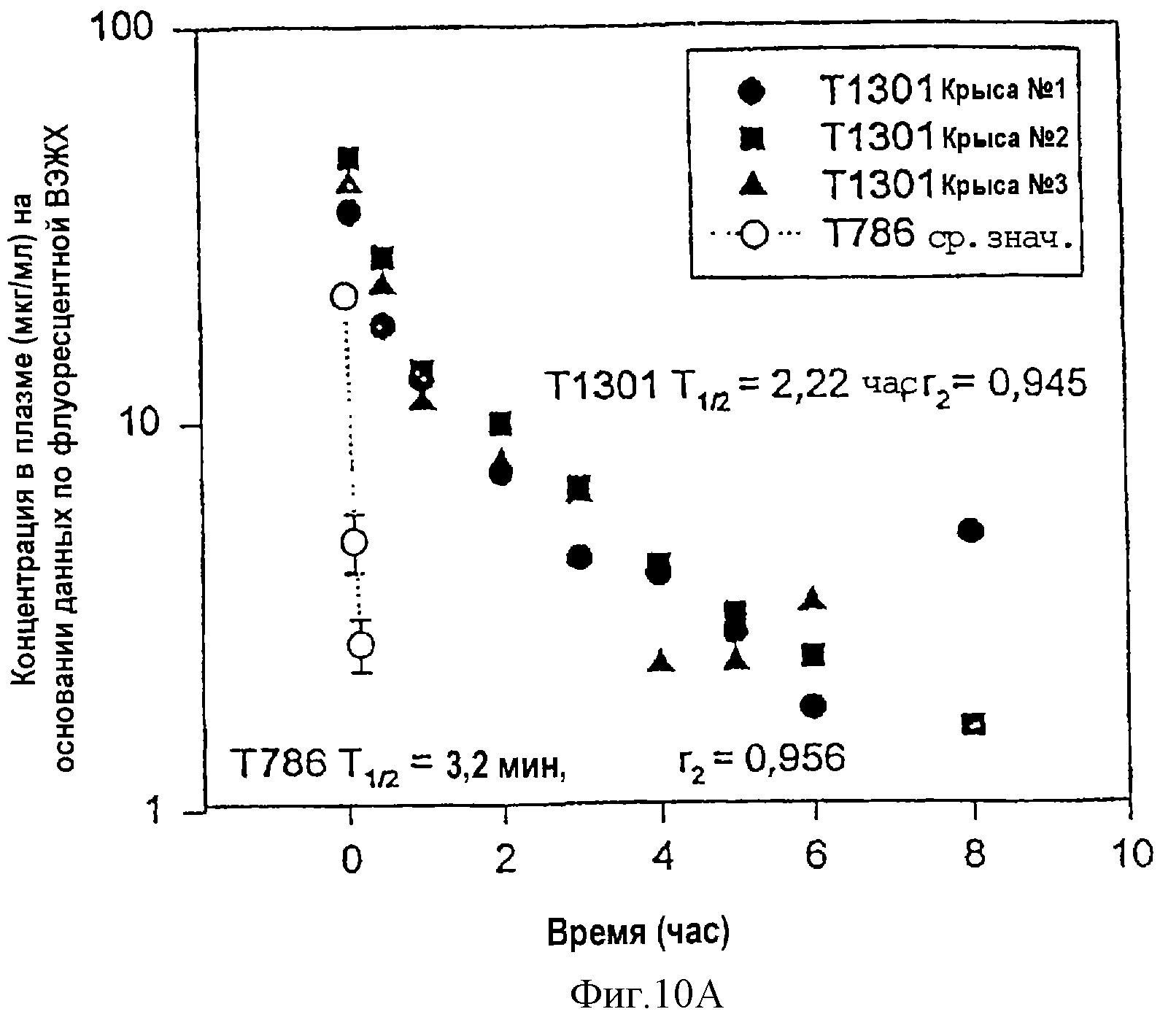
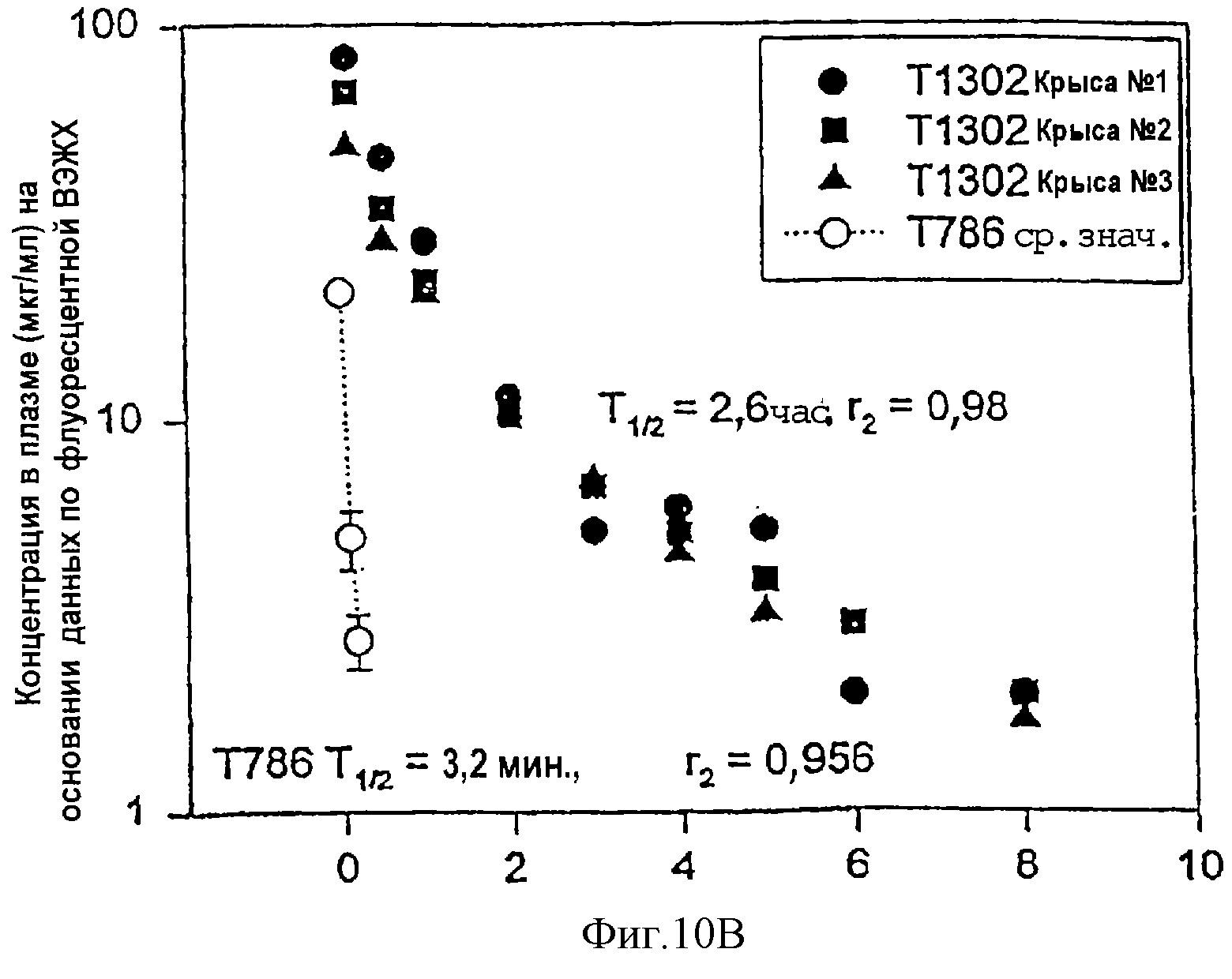
Фиг.10А-10В. Фармакокинетический профиль гибридных полипептидов из РСВ - Т1301 (10А) и Т1302 (10В) в плазме крови крыс CD в сравнении с Т786.
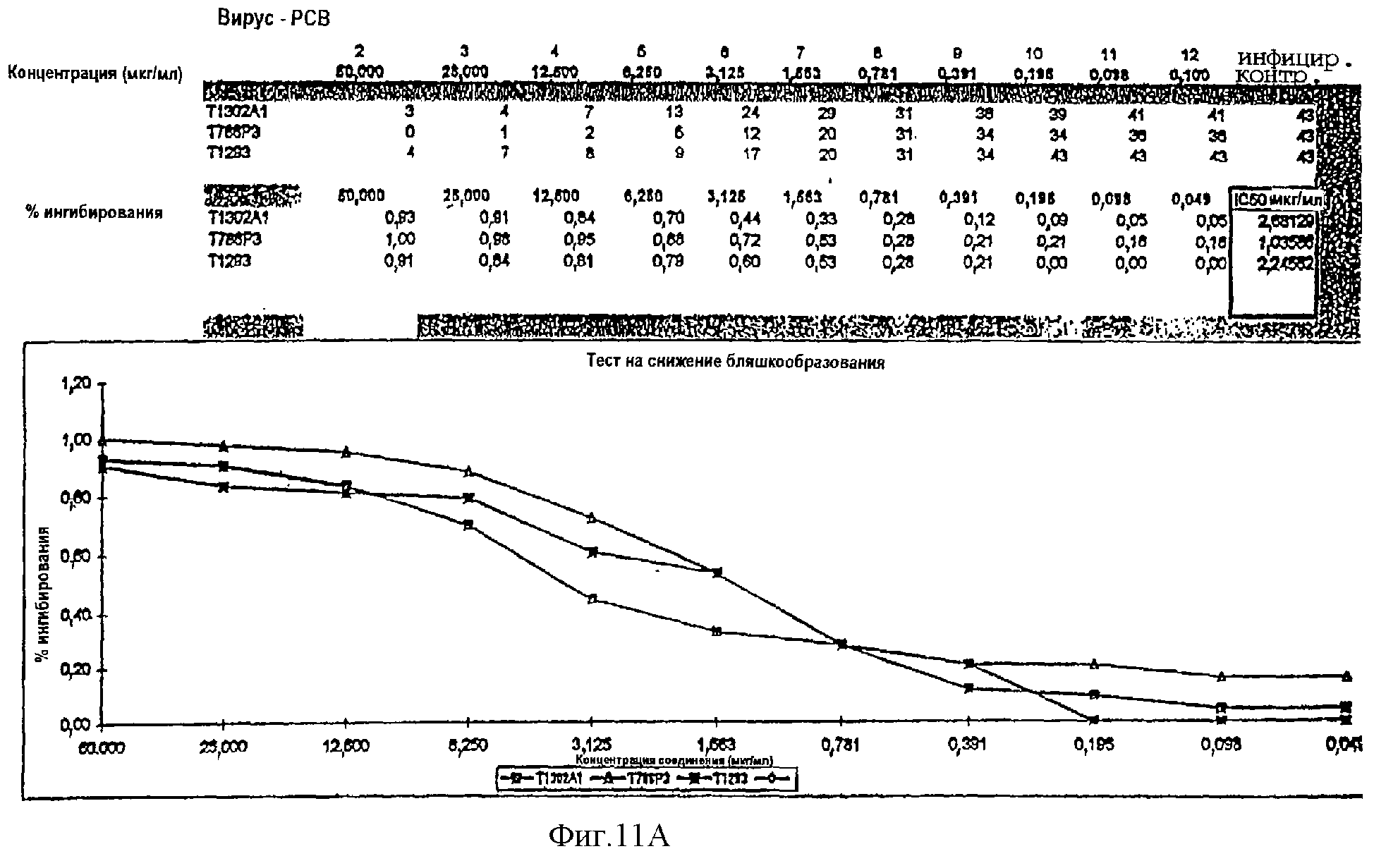
Фиг.11А. Тест на снижение бляшкообразования. Гибридный полипептид Т1293 способен ингибировать инфекцию РСВ со значением ИК50, равным 2,6 мкг/мл.
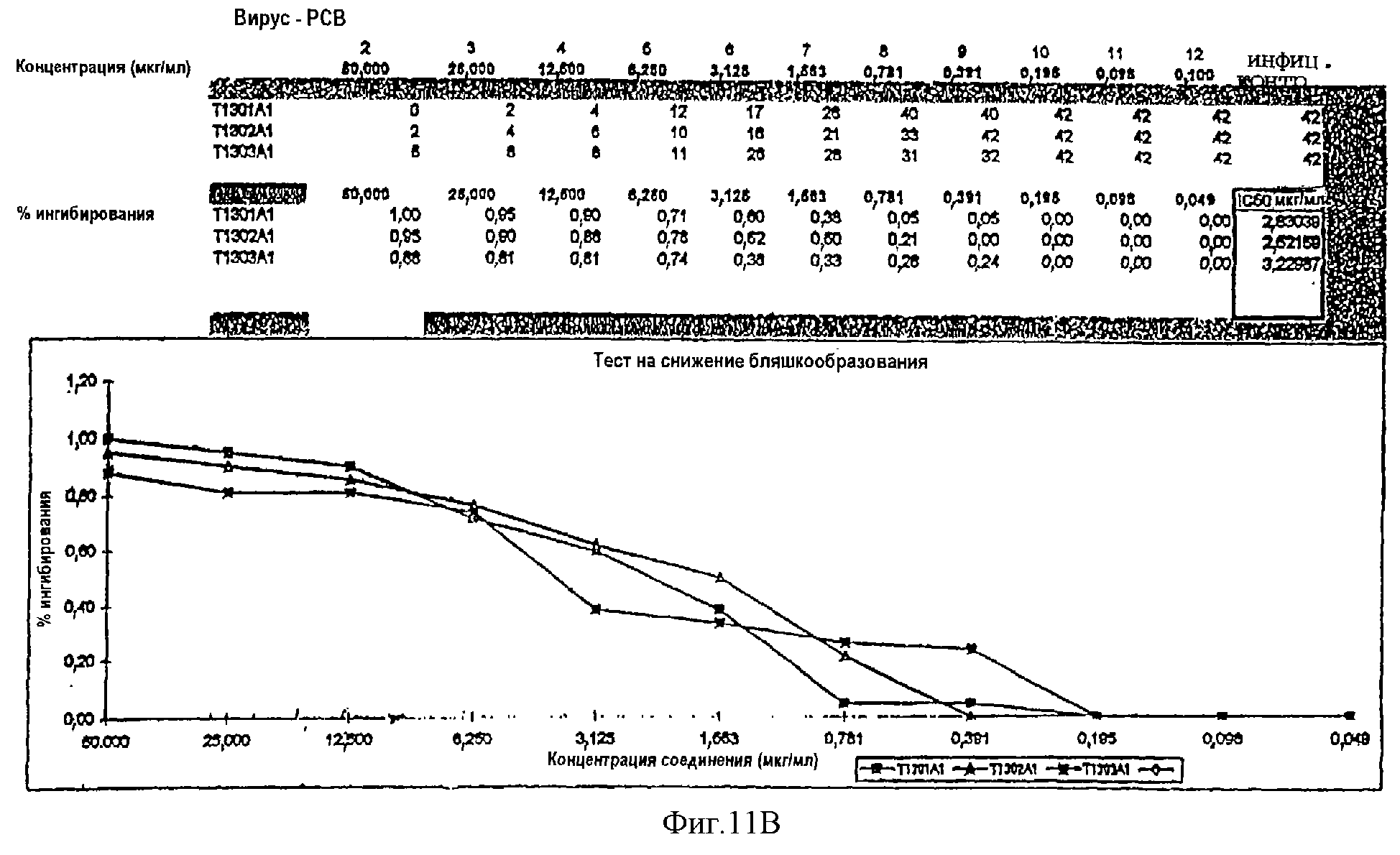
Фиг.11В. Тест на снижение бляшкообразования показывает, что гибридные полипептиды РСВ Т1301, Т1302 и Т1303 ингибируют РСВ инфекцию.
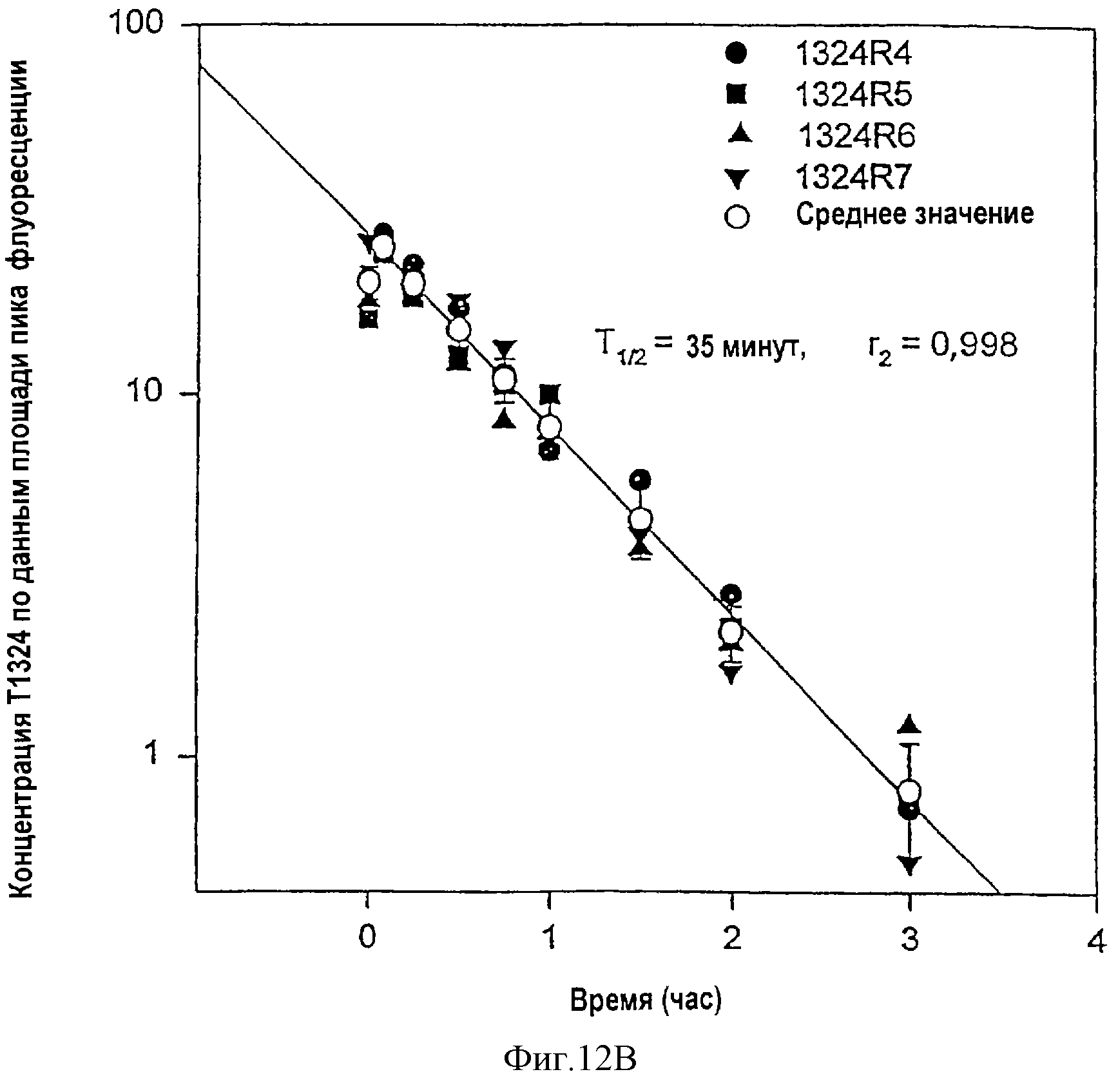
Фиг.12А и 12В. Фармакокинетический профиль гибридного полипептида лютеинизирующего гормона - Т1324 в сравнении с Т1323 в плазме крови самцов крыс CD. Полипептид Т1323 представляет коровый полипептид лютеинизирующего гормона, и полипептид Т1324 представляет гибридный полипептид, включающий коровый полипептид, связанный с энхансерными пептидными последовательностями.
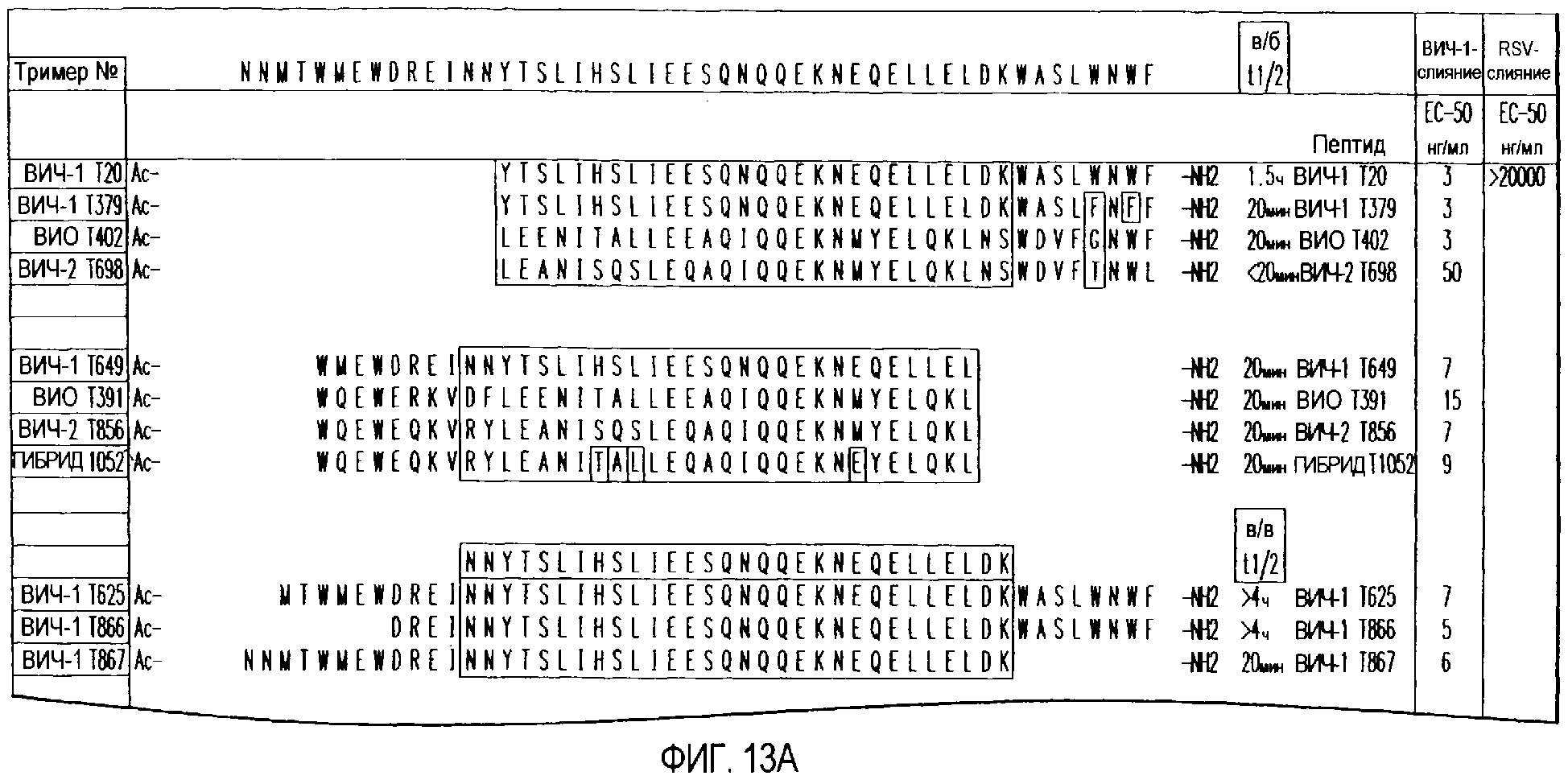
Фиг.13. Гибридные полипептидные последовательности, полученные из различных коровых полипептидов. Коровые полипептидные последовательности показаны в затененном виде. Незатененные последовательности на амино- и карбокси-концах указывают на энхансерные пептидные последовательности.
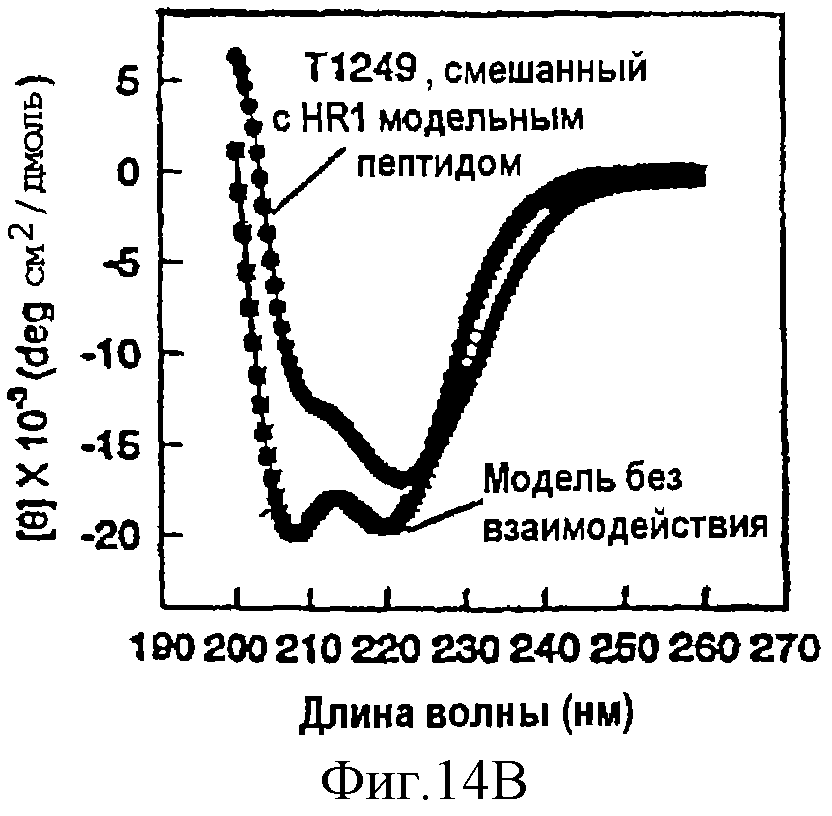
Фиг.14А-В. Спектры кругового дихроизма (КД) для Т1249 в растворе (фосфатно-буферный раствор, рН 7) одного (10 мкМ при 1°С, фиг.14А) и в сочетании с пептидом, включающим 45 остатков, из gр41 HR1 связывающего домена (Т1346); зачерненный квадрат

Фиг.15. Результаты электрофореза в полиакриламидном геле, показывающие защиту со стороны Т1249 конструкции на основе gр41 - М41Δ178 от расщепления протеиназой К; дорожка 1: праймерный маркер; дорожка 2: необработанный М41Δ178; дорожка 3: М41Δ178, инкубированный с протеиназой К; дорожка 4: необработанный Т1249; дорожка 5: Т1249, инкубированный с протеиназой К; дорожка 6: М41Δ178, инкубированный с Т1249; дорожка 7: инкубация Т1249 и М41Δ178 перед добавлением протеиназы К.
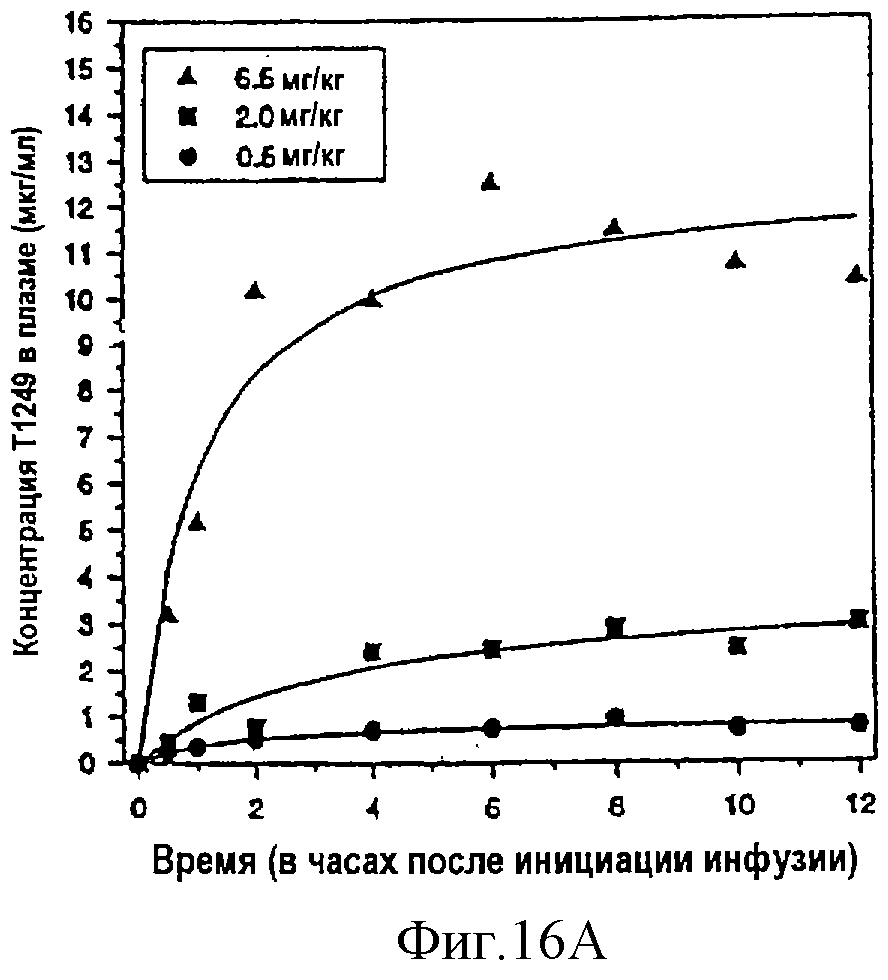
Фиг.16А-С. Фармакокинетика Т1249 у альбиносных крыс линии Спрэг-Доли (Sprague-Dawley); фиг.16А: фармакокинетика Т1249 в случае введения однократной дозы путем непрерывной подкожной инфузии; фиг.16В: фармакокинетика Т1249 в плазме крови при введении его подкожной инъекцией (п/к) или внутривенной инъекцией (в/в); фиг.16С: кинетический анализ Т1249 в лимфе и плазме крови после внутривенной инъекции.
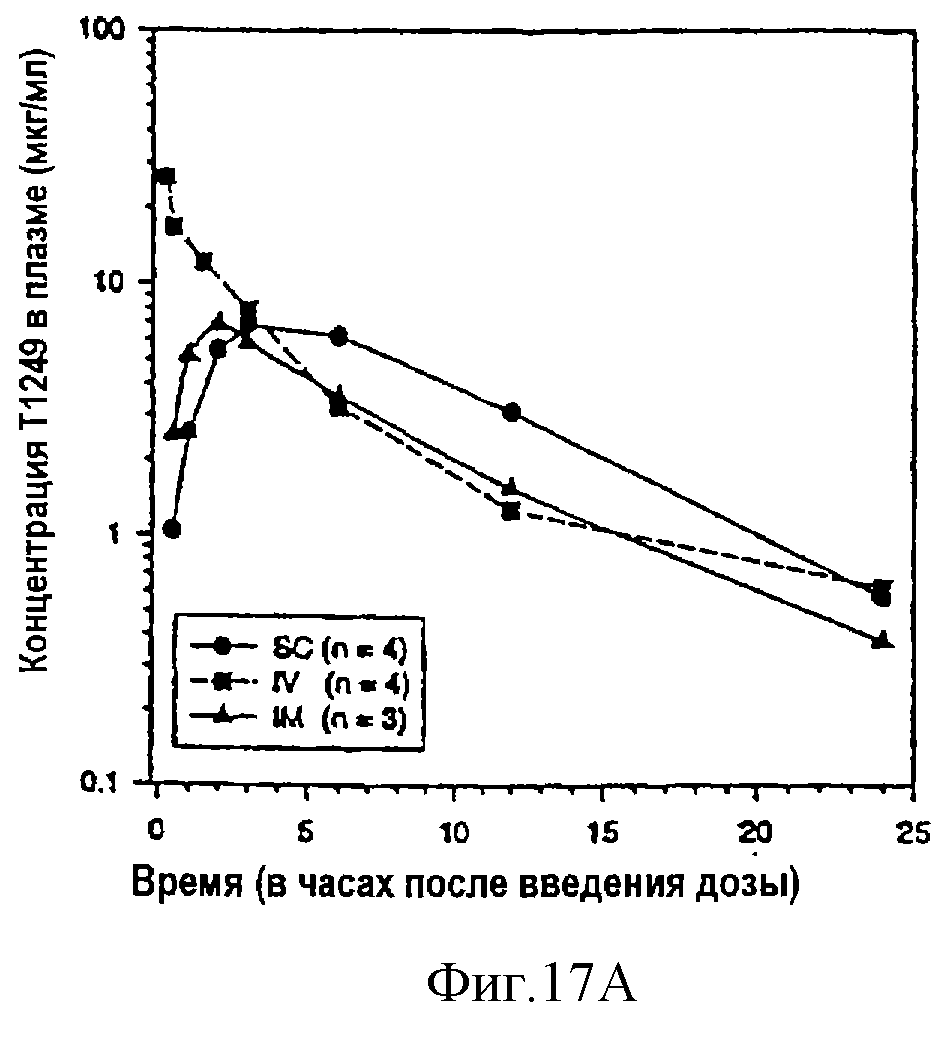
Фиг.17А-В. Фармакокинетика Т1249 у cynomolgus обезьян;
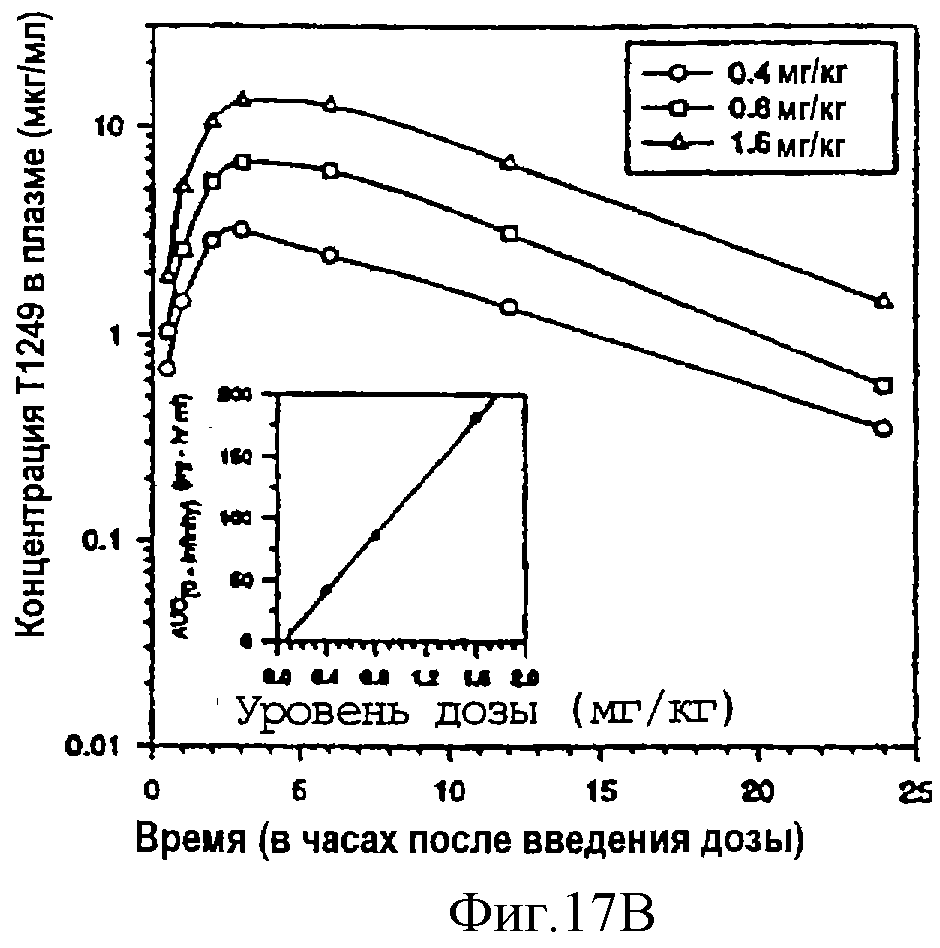
фиг.17А: фармакокинетика в плазме крови однократной дозы Т1249 в 0,8 мг/кг, вводимой подкожной (п/к), внутривенной (в/в) или внутримышечной (в/м) инъекцией; фиг.17В: фармакокинетика в плазме крови Т1249 при подкожном введении трех разных доз (0,4 мг/кг, 0,8 мг/кг и 1,6 мг/кг).
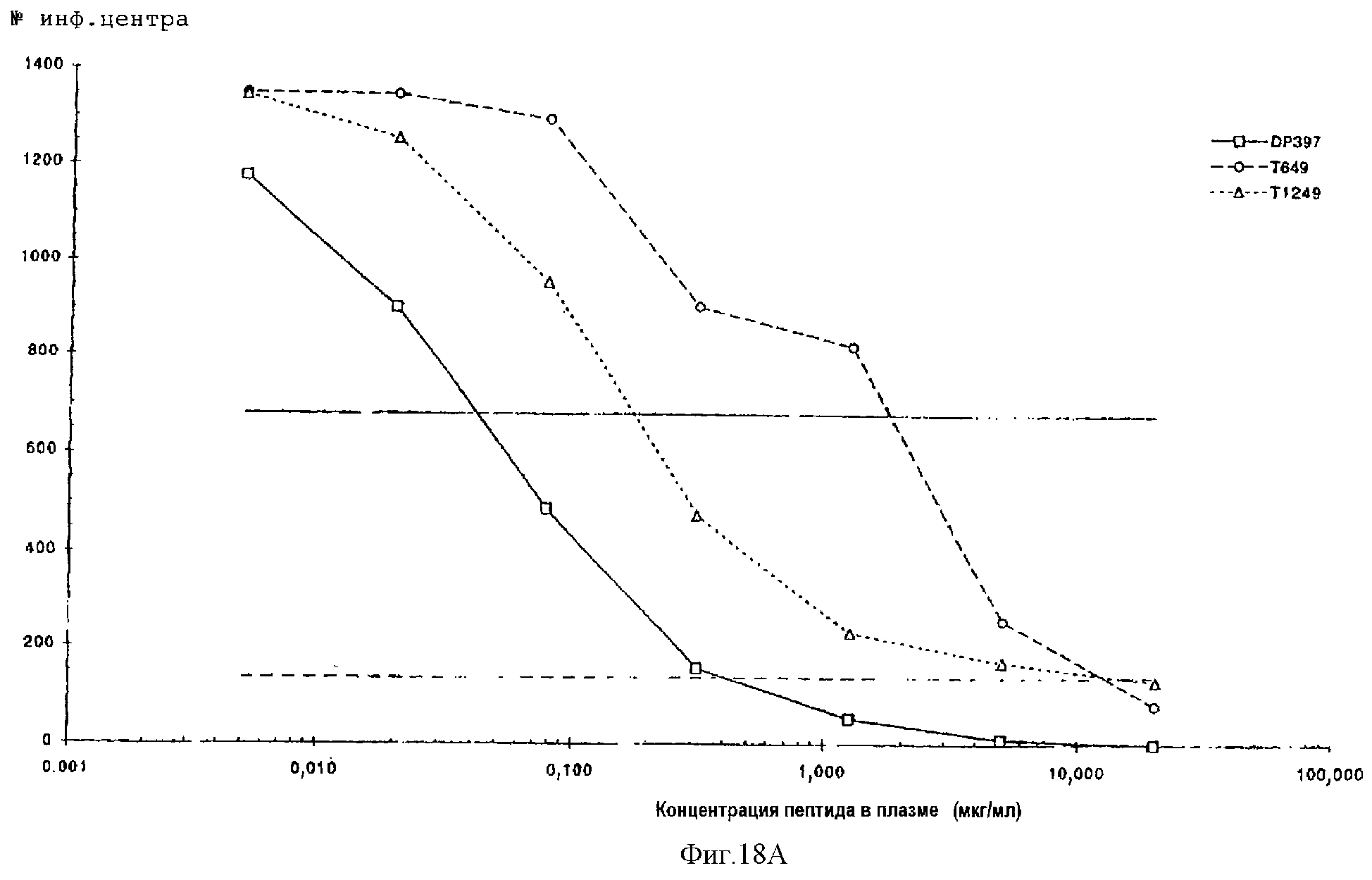
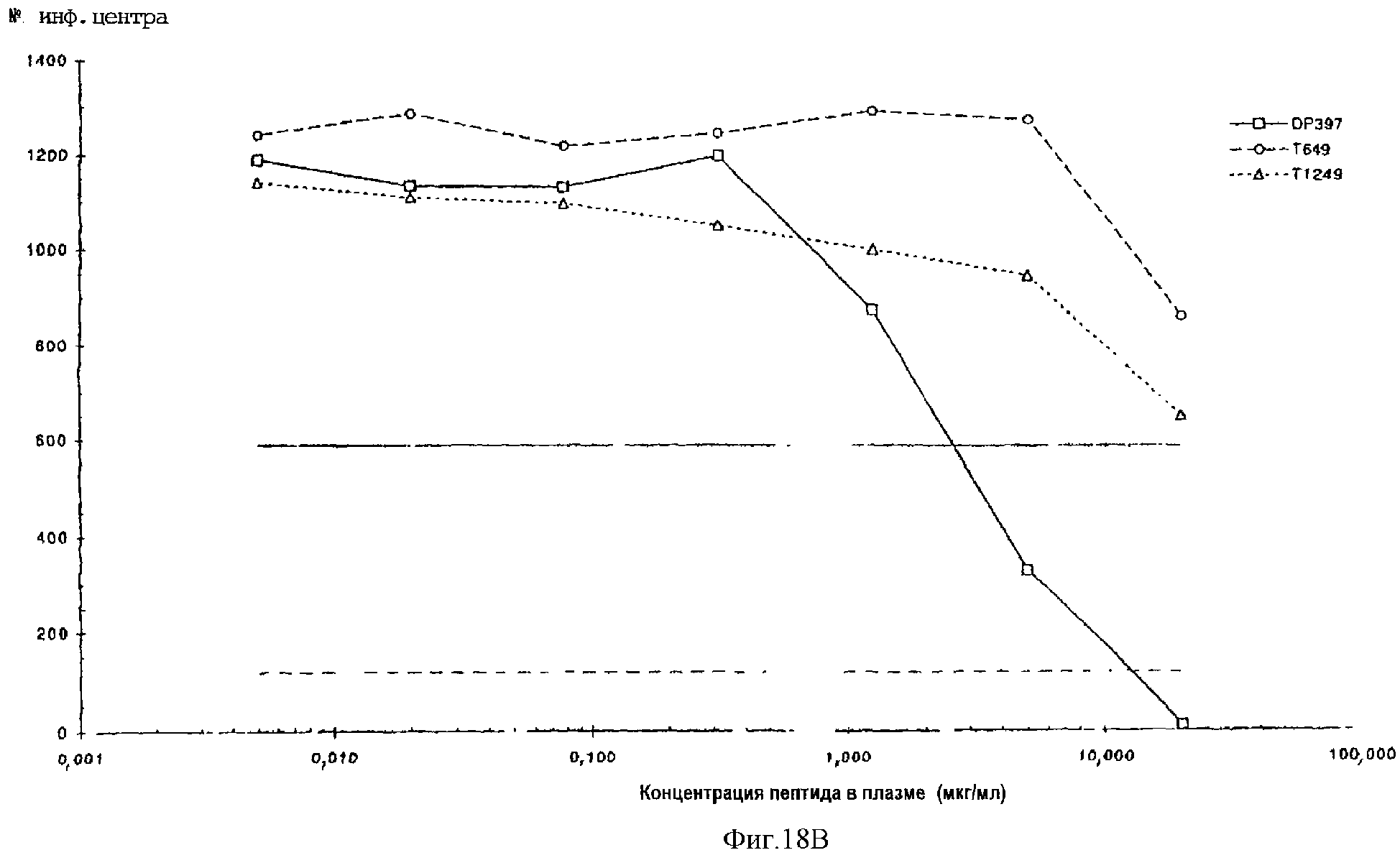
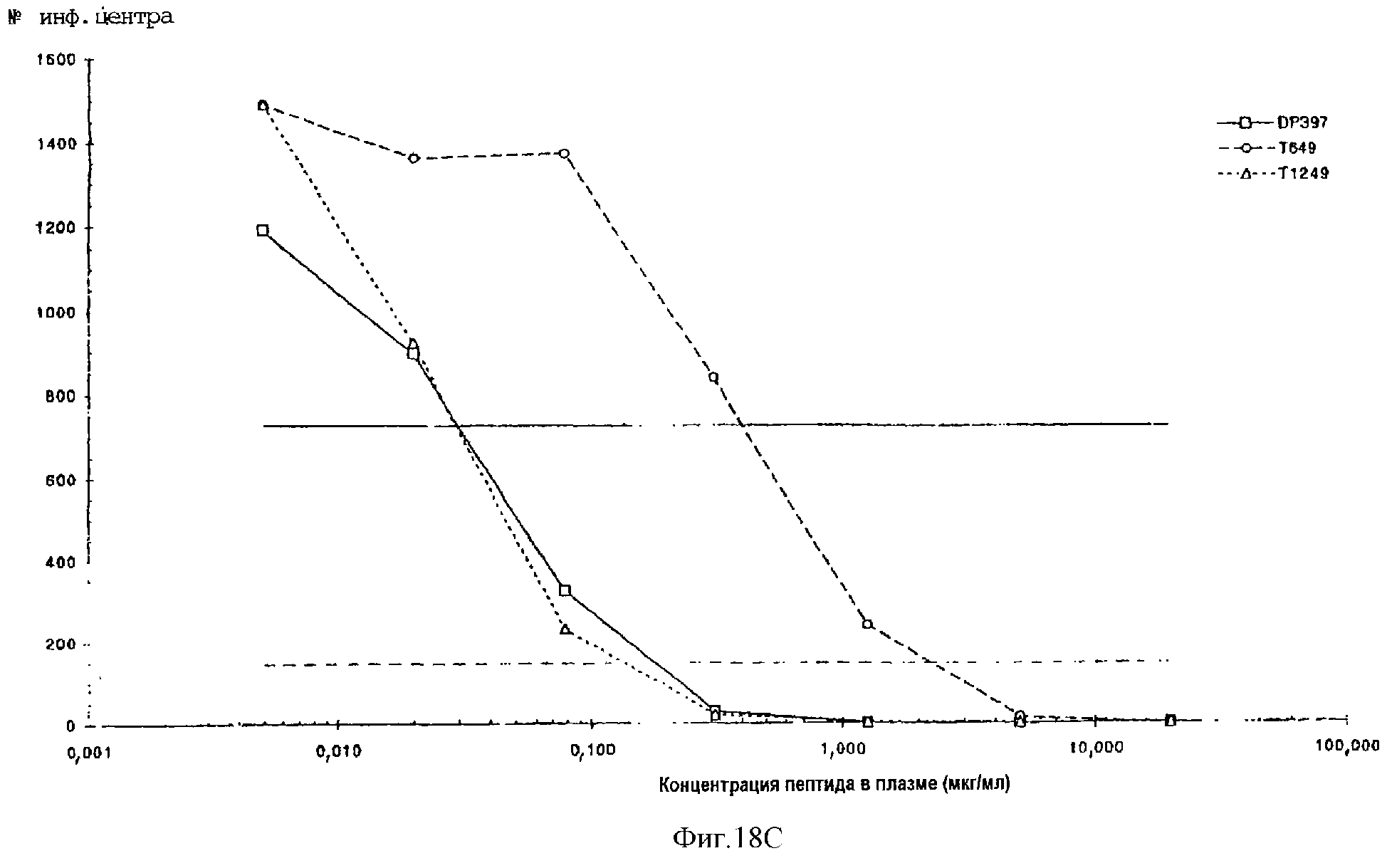
Фиг.18А-18D. Противовирусная активность, проявляемая пептидами DP397

5. Подробное описание изобретения
В настоящем описании раскрыты пептидные последовательности, называемые энхансерными пептидными последовательностями, полученные из различных белковых последовательностей оболочки ретровируса (gр41), которые способны усиливать фармакокинетические свойства коровых полипептидов, к которым они присоединяются. Такие энхансерные пептидные последовательности могут использоваться в методах, направленных на усиление фармакокинетических свойств любого корового полипептида посредством связывания энхансерных пептидных последовательностей с коровым полипептидом с образованием гибридного полипептида с усиленными фармакокинетическими свойствами в сравнении с одним коровым полипептидом. Период полужизни корового полипептида, к которому присоединяются одна или более энхансерных пептидных последовательностей, может быть также увеличен in vitro. Так, например, присоединенные энхансерные пептидные последовательности могут увеличить период полужизни корового полипептида в случае наличия его в клеточной культуре, тканевой культуре или в образце, взятом у пациента, таком как клетка, ткань или другие образцы.
Коровые полипептиды в составе гибридных полипептидов согласно настоящему изобретению могут включать любой пептид, который может быть введен в живую систему, например любой пептид, который может функционировать как терапевтическое или профилактическое средство, полезное для лечения или профилактики заболевания, или как визуализирующий агент, полезный для визуализации структур in vivo.
В описании приведены также пептиды, включая пептиды, которые содержат энхансерные пептидные последовательности, проявляющие антифузогенную и/или противовирусную активность. Кроме того, описаны способы применения таких пептидов, включая способы, применяемые для снижения или подавления вирусной инфекции и/или слияния клеток, вызванного вирусной инфекцией.
5.1. Гибридные полипептиды
Гибридные полипепиды согласно настоящему изобретению включают по меньшей мере одну энхансерную пептидную последовательность и коровый полипептид. Предпочтительно гибридные полипептиды согласно настоящему изобретению включают по меньшей мере две энхансерных пептидных последовательности и коровый полипептид, причем по меньшей мере один энхансерный пептид находится на амино-конце корового полипепида и по меньшей мере одна энхансерная пептидная последовательность, имеющаяся в гибридном полипептиде, находится на карбокси-конце корового полипептида.
Энхансерные пептидные последовательности согласно настоящему изобретению включают пептидные последовательности, первоначально полученные из различных белковых последовательностей оболочки ретровируса (gр41), включая последовательности ВИЧ-1, ВИЧ-2 и ВИО, и внесенные в них специфические вариации или модификации описаны ниже. Коровый полипептид может включать любую пептидную последовательность, предпочтительно любую пептидную последовательность, которая может быть введена в живую систему, включая, например, пептиды, предназначенные для применения с целью лечения, профилактики или визуализации.
В типичном случае длина гибридного полипептида варьирует в диапазоне от примерно 10 до примерно 500 аминокислотных остатков, причем предпочтительной является длина от примерно 10 до примерно 100 аминокислотных остатков, и наиболее предпочтительной является длина от примерно 10 до примерно 40 аминокислотных остатков.
Не ограничивая обсуждение рамками какой-либо конкретной теории, следует тем не менее отметить, что структура белка оболочки такова, что предположительный участок α-спирали, расположенный на С-концевом участке белка, скорее всего, связан с участком «застежки-молнии» лейцина, расположенным на N-концевом участке белка. Сравнительный анализ структуры участков gр41 с N-концевой и С-концевой энхансерными последовательностями, проведенный во всех опубликованных к настоящему времени работах по выделению последовательностей ВИЧ-1, ВИЧ-2 и ВИО, приводит к идентификации консенсусных аминокислотных последовательностей.
В частности, были идентифицированы приведенные ниже консенсусные аминокислотные последовательности, отображающие консенсусные энхансерные пептидные последовательности (консенсусные последовательности перечислены в прямой и обратной ориентациях, поскольку указанные энхансерные пептидные последовательности могут использоваться либо в прямой, либо в обратной ориентации): "WXXWXXXI", "WXXWXXX", "WXXWXX", "WXXWX", "WXXW", "WXXXWXWX", "XXXWXWX", "XXWXWX", "XWXWX", "WXWX", "WXXXWXW", "WXXXWX", "WXXXW", "IXXXWXXW", "XXXWXXW", "XXWXXW", "XWXXW", "XWXWXXXW", "XWXWXXX", "XWXWXX", "XWXWX", "XWXW", "WXWXXXW" или "XWXXXW", где Х может быть любой аминокислотой, W означает триптофан и I означает изолейцин. Прямые ориентации консенсусных аминокислотных последовательностей показаны на фиг.1 и 2.
В типичном случае энхансерная пептидная последовательность составляет примерно 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30 аминокислотных остатков в длину, при этом предпочтительной является длина от примерно 4 до примерно 20 остатков, более предпочтительной является длина от примерно 4 до примерно 10 остатков и наиболее предпочтительной является длина от примерно 6 до примерно 8 остатков.
В предпочтительном варианте осуществления изобретения энхансерные пептидные последовательности, которые могут применяться для усиления фармакокинетических свойств получаемых гибридных полипептидов, включают специфические энхансерные пептидные последовательности, показанные на фиг.2, 13 и в Таблице 1. В число наиболее предпочтительных энхансерных пептидных последовательностей входят такие, которые включают указанные ниже аминокислотные последовательности: "WQEWEQKI" и "WASLWEWF".
С целью иллюстрации, но не с целью ограничения, ниже в Таблице 1 перечислены аминокислотные последовательности, которые отражают предпочтительные варианты энхансерных пептидных последовательностей из числа энхансерных пептидных последовательностей согласно настоящему изобретению. Следует понимать, что хотя показана только прямая ориентация данных последовательностей, обратная ориентация приведенных последовательностей также входит в область настоящего изобретения. Так, например, если ниже показана прямая ориентация энхансерной пептидной последовательности "WMEWDREI", то и ее обратная ориентация, т.е. "IERDWEMW", также включается.
В другом предпочтительном варианте определенные энхансерные пептидные последовательности согласно настоящему изобретению включают энхансерные пептидные последовательности, показанные на фиг.2, 13 и в Таблице 1, несут консервативные замещения аминокислот в одном, двух или трех положениях, причем указанные аминокислотные замещения не устраняют способность энхансерных пептидных последовательностей усиливать фармакокинетические свойства гибридного полипептида в сравнении с соответствующим входящим в его состав коровым полипептидом.
Наиболее предпочтительно такие замещения приводят к получению энхансерных пептидных последовательностей, которые попадают в область консенсусных последовательностей в энхансерной пептидной последовательности. В указанных случаях замещения в основном происходят по аминокислотным остаткам, соответствующим положению «X», обозначенному в указанных выше консенсусных аминокислотных последовательностях и на фиг.1 и 2. Термин «консервативные замещения» относится к таким замещениям, когда указанное замещение осуществляется аминокислотными остатками с близкими значениями заряда, размера и/или гидрофобности/гидрофильности, что и замещаемая аминокислота. Отмеченные характеристики аминокислот хорошо известны специалистам в данной области техники.
Настоящее изобретение также относится к энхансерным пептидным последовательностям, включающим аминокислотные последовательности, приведенные на фиг.1, 2, 13 и в Таблице 1, которые во всем идентичны им, за тем исключением, что указанные энхансерные пептидные последовательности включают одну или более аминокислотных добавок (в основном не более чем примерно 15 аминокислотных остатков в длину), делеций (например, усечения на аминогруппе или на концевых участках) или неконсервативных замещений, которые тем не менее не устраняют способность полученных энхансерных пептидных последовательностей усиливать фармакокинетические свойства коровых полипептидов, к которым они присоединяются, в сравнении только с коровыми полипептидами без энхансерных пептидных последовательностей.
Добавки в основном не длиннее, чем 15 аминокислотных остатков, и могут включать добавки, включающие 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15 последовательных аминокислотных остатков. Предпочтительно общее число аминокислотных остатков, добавляемых к исходному энхансерному пептиду, не превышает примерно 15 аминокислотных остатков, более предпочтительно составляет не более чем примерно десять аминокислотных остатков и наиболее предпочтительно составляет не более чем примерно пять аминокислотных остатков.
Делеции предпочтительно представляют собой такие делеции, которые включают не более чем примерно 3 аминокислотных остатка (либо последовательных, либо непоследовательных остатка), более предпочтительно делеции, включающие 2 аминокислотных остатка и наиболее предпочтительно делеции из одного аминокислотного остатка. В основном делеции представляют аминокислотные остатки, соответствующие остаткам «X» в консенсусных последовательностях энхансерного пептида.
Энхансерные пептидные последовательности согласно настоящему изобретению также включают определенные энхансерные пептидные последовательности, показанные на фиг.2, 13 и в Таблице 1, содержащие один, два или три неконсервативных аминокислотных замещения, причем предпочтительно наличие двух таких замещений и наиболее предпочтительно наличие одного такого замещения. Термин «неконсервативный» в отношении замещения относится к замещению аминокислотными остатками с несходными показателями заряда, веса и/или гидрофобности/гидрофильности в сравнении с заменяемым аминокислотным остатком. Такие характеристики аминокислот хорошо известны специалистам в данной области техники.
Кроме того, необязательно, чтобы аминокислотные замещения ограничивались генетически кодируемыми аминокислотами, что в равной мере относится и к определенным предпочтительным вариантам осуществления изобретения. Фактически пептиды могут содержать аминокислоты, не кодируемые генетически. Так, в дополнение к природным кодируемым генетически аминокислотам аминокислотные остатки в пептидах могут замещаться природными некодируемыми аминокислотами и синтетическими аминокислотами. Такие замещения могут также иметь место внутри коровых полипептидов в гибридных полипептидах согласно настоящему изобретению, независимо от того, присутствуют ли они в энхансерной последовательности/последовательностях в конкретном гибридном полипептиде.
В число часто встречающихся аминокислот, которые позволяют осуществить полезные замещения, входят, но не ограничиваются приведенным списком, следующие: β-аланин (β-Ala) и другие омега-аминокислоты, такие как 3-аминопропионовая кислота, 2,3-диаминопропионовая кислота (Dpr), 4-аминомасляная кислота и т.д.; α-аминоизомасляная кислота (Aib); ε-аминогексаноевая кислота (Aha); δ-аминовалериановая кислота (Ava); N-метилглицин, или саркозин (MeGly); орнитин (Orn); цитруллин (Cit); т-бутилаланин (t-BuA); т-бутилглицин (t-BuG); N-метилизолейцин (Melle); фенилглицин (Phg); циклогексилаланин (Cha); норлейцин (Nle); нафтилаланин (Nal); 4-хлорфенилаланин (Phe(4-Cl)); 2-фторфенилаланин (Phe(2-F)); 3-фторфенилаланин (Phe(3-F)); 4-фторфенилаланин (Phe(4-F)); пеницилламин (Pen); 1,2,3, 4-тетрагидроизохинолин-3-карбоновая кислота (Tic); β-2-тиенилаланин (Thi); метионинсульфоксид (MSO); гомоаргинин (hArg); N-ацетиллизин (AcLys); 2, 4-диаминомасляная кислота (Dbu); 2, 3-диаминомасляная кислота (Dab); п-аминофенилаланин (Phe(pNH2)); N-метилвалин (MeVal); гомоцистеин (hCys); гомофенилаланин (hPhe) и гомосерин (hSer); гидроксипролин (Hyp); гомопролин (hPro); -метилированные аминокислоты, аналоги циклических аминокислот (используемые, например, для ограничения структуры аминокислотных остатков до определенных конформационных состояний, например, α α'- и ββ'-замещенные циклические аминокислоты, такие как 1-аминоциклопентан-карбоновая кислота (циклолейцин) и ββ-циклопентаметилен-β-меркаптопропионовая кислота (см., например, Hruby et al., 1990, Biochem. J. 268: 249-262) и пептоиды или олигопептоиды (N-замещенные аминокислоты, например N-замещенные глицины; см., например, Simon et al., 1972, Proc. Natl. Acad. Sci. USA 89: 9367-9371).
Несмотря на то, что в большинстве случаев аминокислоты в пептиде замещаются L-энантиомерными аминокислотами, указанные замещения не ограничиваются L-энантиомерными аминокислотами. Так, определение «мутированная» или «измененная» формы в отношении аминокислот охватывает также те ситуации, при которых L-аминокислота замещается идентичной D-аминокислотой (например, L-Arg - D-Arg) или D-аминокислотой из той же категории или подкатегории (например, L-Arg - D-Lys) и наоборот. Такие замещения могут также иметь место на коровых полипептидах в гибридных полипептидах согласно настоящему изобретению, независимо от того, присутствуют ли они в энхансерной последовательности/энхансерных последовательностях конкретного гибридного полипептида.
В дополнение к описанным выше аминокислотным заменам замещения в боковых группах могут быть осуществлены при введении, например, метильной группы или псевдоизостерических групп с разными электронным свойствами (см., например, Hruby et al., 1990, Biochem. J. 268: 249-262). Кроме того, могут быть введены двойные связи между соседними атомами углерода в аминокислотах и циклических пептидах или могут быть образованы аналоги при введении ковалентных связей, таких как в случае образования амидной связи между - и С-концом, между двумя боковыми цепями или между боковой цепью и - или С-концом пептида. Такие замещения могут также иметь место внутри коровых полипептидов в гибридных полипептидах согласно настоящему изобретению, независимо от того, присутствуют они или нет в энхансерной последовательности/энхансерных последовательностях конкретного гибридного полипептида.
Коровый и гибридный полипептиды согласно настоящему изобретению могут быть также конъюгированы с одной или большим числом химических групп. Химические группы, используемые для конъюгирования, предпочтительно не обладают существенной токсичностью или иммуногенностью, т.е. любая токсичность или иммуногенность, наблюдаемые для конъюгата корового или гибридного полипептида, незначительно (т.е меньше чем на 50%) выше, чем любая токсичность или иммуногенность, отмечаемые для немодифицированного корового или гибридного полипептида. Химические модификации корового и/или гибридного полипептидов осуществляются с целью оказания воздействия на фармакокинетические свойства полипептида. Указанные эффекты включают снижение или усиление эффективности лекарственного средства, стабильность, биодоступность, клиренс, иммуногенность и период полужизни in vivo, а также влияние на катаболизм полипептида, его направленную миграцию и локализацию.
В одном варианте осуществления изобретения проводят конъюгацию гибридных полипептидов с одной или большим числом химических групп, осуществляя ее как на коровой, так и на энхансерной частях полипептида. В другом варианте осуществления изобретения такие модификации могут быть осуществлены либо на коровой части полипептида, либо на части энхансерного пептида гибридных полипептидов. В еще одном варианте осуществления изобретения модифицируется только коровая часть полипептида в гибридном полипептиде. В еще одном варианте осуществления изобретения коровый полипептид модифицируют по одной или большему числу химических групп, причем такой коровый полипептид не представляет собой часть гибридного полипептида. Так, например, коровый полипептид, такой как Т1387 (Ac-TALLEQAQIQQEKNEYELQKLDK-NH2) может быть модифицирован с помощью одной или большего числа химических групп.
Примеры химических групп, полезных для осуществления конъюгации, включают полимеры небелковой природы, такие как полиолы. Другие химические группы включают белки, такие как, например, альбумин или иммуноглобулин, а также углеводы, такие, например, как те углеводы, которые естественно встречаются в гликопротеинах. Для модификации белков также используют декстран, DL-аминокислоты и поливинилпирролидон. Обзор полимер-модифицированных пептидов дан в работе Бурнхама (Burnham, Am. J. Hosp. Pharm. 51: 210-18 (1994)), полностью приведенной в настоящем описании в качестве ссылки.
Полиол, например, может быть конъюгирован с коровым или гибридным полипептидом по одному или большему числу аминокислотных остатков, включая, например, остатки лизина, цистеина и гистидина. Используемый полиол может представлять собой любой водорастворимый поли(алкиленоксидный) полимер и может содержать линейную или разветвленную цепь. Подходящие полиолы включают полиолы, замещенные по одному или большему числу гидроксильных положений химической группой, такой как алкильная группа, содержащая от одного до четырех атомов углерода. В типичном случае полиол представляет собой поли(алкиленгликоль), такой как поли(этиленгликоль) (ПЭГ) и в этой связи, для простоты обсуждения, оставшаяся часть описания будет относиться к иллюстративному варианту осуществления изобретения, в котором используемый полиол представляет собой ПЭГ и способ конъюгации полиола с коровым или гибридным полипептидом носит название «пегилирование». Однако специалистам в данной области техники известно, что могут использоваться и другие полиолы, такие как, например, поли(пропиленгликоль) и сополимеры полиэтилен-полипропиленгликоля, с помощью методик конъюгации, указанных в настоящем описании применительно к ПЭГ. Обзор по методам модификации биоактивных молекул дан в работе Инада с соавт. (Inada et al., Trends Biotechnol. 13: 86-91 (1995)), полностью приведенной в настоящем описании в качестве ссылки.
Степень пегилирования корового или гибридного полипептида может быть скорректирована таким образом, чтобы повысить до желаемого уровня период полужизни in vivo в сравнении с соответствующим непегилированным белком. Период полужизни пегилированного корового или гибридного полипептида может увеличиваться постепенно по мере повышения степени пегилирования. Показано, что модификация белков с использованием ПЭГ или производных ПЭГ позволяет увеличить до желаемого уровня эффективность лекарственного средства, его биологическую активность, стабильность (включая устойчивость к нагреванию, действию химических денатурирующих факторов и протеолиза), абсорбцию/поглощение и период полужизни in vivo, а также снижает до требуемого уровня скорость клиренса и иммуногенность (патент США No. 6025325; Westerman et al., Int. J. Cancer 76: 842-50 (1998); Conover et al., Artif. Organs 21: 907-15 (1997); Tsutsumi et al., J. Pharmacol. Exp. Ther. 278: 1006-11 (1996); Kaneda et al., Invasion Metastasis 15: 156-62 (1995); Inada et al., Trends Biotechnol. 13: 86-91 (1995); Paige et al., Pharm. Res. 12:1883-88 (1995); Satake-Ishikawa et al., Cell Struct. Funct. 17: 157-60 (1992); Tanaka et al., Cancer Res. 51: 3710-3714 (1991), включенные полностью в настоящее описание в качестве ссылок).
Среднее значение молекулярной массы ПЭГ может варьировать от примерно 500 до примерно 30000 дальтон (Д), предпочтительно от примерно 1000 до примерно 25000 Д и более предпочтительно от примерно 4000 до примерно 20000 Д. В одном варианте осуществления изобретения пегилирование проводят, используя ПЭГ с молекулярной массой примерно 5000 Д (далее обозначаемый как «ПЭГ(5000)»). В другом варианте осуществления изобретения используют ПЭГ с разветвленной цепью, содержащий две цепи примерно по 10000 Д каждая.
Коммерчески доступные препараты ПЭГ, которые приемлемы для использования в рамках настоящего изобретения, относятся к негомогенным препаратам, продаваемым в соответствии с их молекулярной массой. Так, например, препараты ПЭГ(5000) типично содержат молекулы, варьирующие незначительно по молекулярной массе, обычно в пределах +/-500 Д. Описано множество методов пегилирования белков. См., например, патент США No. 4179337, включенный полностью в настоящее описание в качестве ссылки, который раскрывает конъюгацию большого числа гормонов и ферментов с ПЭГ и полипропиленгликолем с образованием физиологически активных неиммуногенных композиций.
Условия реакции связывания ПЭГ с белком варьируют в зависимости от природы целевого белка, желаемой степени пегилирования, типа используемого ПЭГ или его производного и локализации целевой точки присоединения. Другие факторы включают стабильность, реакционоспособность и антигенность связи с ПЭГ. В основном проводят реакцию ПЭГ, имеющего по меньшей мере одну концевую гидроксильную группу, со связующим веществом с образованием активированного ПЭГ, несущего концевую реакционноспособную группу. Затем проводят реакцию указанной реакционноспособной группы с α- и ε-аминами белков с образованием ковалентной связи. Аминные группы могут конъюгировать с гидроксигруппой ПЭГ с образованием амидной связи. Карбоксильные группы могут конъюгировать с аминными группами корового или гибридного полипептида с образованием амидной связи и с гидроксигруппой ПЭГ с образованием сложного эфира. При этом другой конец молекулы ПЭГ может быть «блокирован» нереакционноспособной химической группой, такой как метоксигруппа, с образованием, например, алкилированных ПЭГ, такой как метокси-ПЭГ (мПЭГ), которая снижает уровень образования перекрестно сшитых комплексов ПЭГ с молекулами белка.
В технике известно несколько типов линкерных групп, применяемых для конъюгации корового, энхансерного или гибридного полипептида с ПЭГ. Примеры линкерных групп описаны, например, в патенте США No. 4609546, в патенте США No. 4847325, в патенте США No. 4902502, в патенте США No. 5034514 и в патенте США No. 5122614. В одном варианте осуществления изобретения в качестве линкера используют энхансерную пептидную последовательность.
Подходящие активированные ПЭГ могут быть получены в результате ряда соответствующих реакций. Так, например, N-гидроксисукцинимидный сложный эфир ПЭГ (M-NHS-ПЭГ) может быть получен из ПЭГ-монометилового эфира (коммерчески доступного от компании Union Carbide) при проведении реакции N,N'-дициклогексилкарбодиимида (ДЦК) с N-гидроксисукцинимидом (NHS) в соответствии с методом Буккманна и Мерра (Buckmann and Merr, Makromol. Chem., 182: 1379-1384 (1981)).
Кроме того, концевая гидроксигруппа ПЭГ может быть превращена в аминогруппу, например, при взаимодействии с тионилбромидом с образованием ПЭГ-Br и при проведении впоследствии аминолиза с избытком аммиака с образованием ПЭГ-NH2. Затем проводят конъюгацию ПЭГ-NH2 с интересующим белком при использовании стандартных связующих реагентов, таких как реагент К Вудварда. Далее концевая группа -СН2ОН в ПЭГ может быть превращена в альдегидную группу, например, при окислении с помощью MnO2. Затем проводят конъюгацию альдегидной группы с белком в ходе реакции восстановительного алкилирования с использованием такого реагента, как цианборгидрид. Аминокислоты могут быть также связаны с ПЭГ по их аминогруппам через соответствующую связку, такую как, например, уретановая группа. Неприродная аминокислота норлейцин с целью связывания с аминогруппами белка может быть активирована по своей карбоксильной группе до сукцинимидильного сложного эфира (Zalipsky et al., Int. J. Peptide Protein Res. 30:740 (1987); Sartore et al., Appl. Biochem. Biotech. 27: 45 (1991)). Общий обзор по пегилированию приведен в работе Zalipsky and Lee in "Poly (Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications", J.M.Harris, Ed., Plenum, NY, Chap 21 (1992), которая включена полностью в настоящее описание в качестве ссылки.
Альтернативно подходящие для применения в рамках настоящего изобретения активированные ПЭГ включают, не ограничиваясь приведенным списком, следующие: акрилат-ПЭГ, альдегид-ПЭГ, аллил-ПЭГ, амино-ПЭГ, аминокислота-ПЭГ, сложные эфиры аминокислоты-ПЭГ, ω-амино α-карбоксил-ПЭГ, бензо-триазолкарбонат, биотин-ПЭГ, т-Бок-ПЭГ, карбонилимидазол-ПЭГ, карбоксиметилированный ПЭГ, эпоксид-ПЭГ, Fmoc-ПЭГ, флуоресцеин-ПЭГ, гидразид-ПЭГ, ω-гидрокси α-амино-ПЭГ, ω-гидрокси α -карбоксил-ПЭГ, изоцианат-ПЭГ, малеимид-ПЭГ, метакрилатный сложный эфир ПЭГ, NHS-малеимид, NHS-винилсульфон, п-нитрофенилкарбонат-ПЭГ, ортопиридилдисульфид, ПЭГ2, фосфолипид-ПЭГ, пропионовая кислота-ПЭГ, силан-ПЭГ, сукцинат-ПЭГ, сукцинимидилбутаноат-ПЭГ (СБА-ПЭГ), сукцини-мидиловый сложный эфир аминокислоты-ПЭГ, сукцинимидиловый сложный эфир карбоксиметилированного ПЭГ, сукцинимидил-пропионат-ПЭГ (СПА-ПЭГ, SPA-PEG), сукцинимидилсукцинат-ПЭГ (СС-ПЭГ, SS-PEG), тиол-ПЭГ, винилсульфон-ПЭГ, которые могут быть приобретены у различных поставщиков. Так, например, Shearwater Polymers, Inc. (Huntsville, Ala.) продает M-NHS-ПЭГ как «SCM-PEG", а также сукцинимидилкарбонат мПЭГ ("SC-PEG") и сукцинимидилпропионат мПЭГ ("SPA-PEG").
Конкретные аминокислоты, включающие гибридный или коровый полипептид, могут быть модифицированы, например, для целей предупреждения и/или облегчения пегилирования определенных аминокислотных остатков. В одном варианте осуществления изобретения потенциальные участки пегилирования могут быть инактивированы за счет модификации тех аминокислотных остатков, которые могут подвергнуться пегилированию. Так, например, все аминокислотные остатки, имеющие свободную аминогруппу в коровом или гибридном полипептиде, могут быть защищены, что предотвратит пегилирование по указанным остаткам. Подходящие защитные группы включают, но не ограничиваются приведенным списком, следующие: трет-бутоксикарбонил (т-Бок) и N-9-флуоренилметоксикарбонил (Fmoc). Может использоваться любая защитная группа для аминогруппы. Несколько таких защитных групп описаны в работах (Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1981, pp.323-334; and Fields and Noble, Int. J. Pept. Protein Res. 35: 161-214 (1990), включенных в настоящее описание полностью в качестве ссылок. При желании, защитная группа может быть впоследствии удалена с использованием рутинных химических процедур, например при обработке пиперидином в диметилформамиде для Fmoc или при обработке трифторуксусной кислотой для т-Бок.
Альтернативно аминокислотные остатки корового или гибридного полипептида, чувствительные к пегилированию, могут быть замещены аминокислотными остатками, устойчивыми к пегилированию, например, посредством направленного мутагенеза. Кроме того, для создания гибридных полипептидов могут использоваться энхансерные пептидные последовательности, которые не содержат аминокислотные остатки, чувствительные к пегилированию.
В другом варианте осуществления изобретения один или большее число аминокислотных остатков корового или гибридного полипептида могут быть модифицированы, с тем чтобы ввести в них дополнительные участки пегилирования. Так, например, один или большее число аминокислотных остатков, чувствительных к пегилированию, могут быть использованы для замещения или добавлены к полипептиду с помощью любого известного в технике метода, такого как стандартная методика синтеза, или в случае применения рекомбинантных методов посредством направленного мутагенеза (Zoller et al., Nucl. Acids Res. 10: 6487 (1987); Carter et al., Nucl. Acids Res. 13: 4331 (1986)).
Пегилирование корового или гибридного полипептида может быть также проведено с тем, чтобы воздействовать на локализацию участка пегилирования или на степень пегилирования. Пегилирование только части возможных участков пегилирования в коровом или гибридном полипептиде может быть проведено с помощью любого известного в технике метода. Так, например, степень пегилирования можно регулировать с помощью условий реакции, таких как изменение коэффициента молярного отношения полипептида к ПЭГ, продолжительность реакции или температура, при которой осуществляется реакция. После очистки пегилированного полипептида, например, с помощью ионообменной хроматографии, степень пегилирования полипептида можно определить, например, при анализе в ДСН-ПААГ. Пегилированный белок может храниться в условиях, в которых используется любая известная в технике среда для хранения, включая хранение при температуре -20°С в ФБР буфере (рН 7,3).
Указанные подходы могут быть объединены для того, чтобы регулировать число и локализацию участков пегилирования вдоль корового или гибридного полипептида. Пегилирование конкретного интересующего аминокислотного остатка может быть осуществлено при объединении методов химической защиты и реакций пегилирования при синтезе корового, энхансерного или гибридного полипептидов. Использование различных защитных групп при защите и депротекции в разных точках в ходе пептидного синтеза позволяет достичь любого(ых) аминокислотного(ых) остатка(ов) для осуществления пегилирования. Таким образом, указанная методика может использоваться для селективной модификации любой химической группой конкретного аминокислотного остатка или любой его части в коровом, энхансерном или гибридном полипептиде. Так, например, можно осуществить химическую защиту частично синтезированного олигопептида с использованием первой защитной группы для предотвращения пегилирования аминокислотных остатков, которые могли бы быть в ином случае чувствительными к пегилированию.
Далее синтез полипептида может быть завершен с использованием второй защитной группы, которая также будет защищать аминокислотные остатки, которые в ином случае были бы чувствительными к пегилированию. Вторая защитная группа должна быть более подвижной, чем первая, так чтобы при завершении синтеза указанная подвижная защитная группа могла бы быть удалена, позволяя осуществлять пегилирование только теперь уже незащищенных интересующих аминокислотных остатков (Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1981).
В одном неограничивающем примере может быть селективно модифицирован любой один аминокислотный остаток из Т1387 (Ас-TALLEQAQIQQEKNEYELQKLDK-NH2), который чувствителен к пегилированию (например, остатки лизина). Так, например, может быть синтезирован KLDK, и ε-NH2 группа может быть подвергнута депротекции и затем пегилирована. Пептидный синтез завершается образованием корового полипептида, который содержит в пегилированном виде только два из трех остатков лизина.
В другом неограничивающем примере большая часть N-концевых остатков лизина в Т1387 может быть модифицирована при осуществлении синтеза NEYELQKLDK в случае защиты ε-NH2 групп лизиновых остатков менее подвижной защитной группой. После завершения синтеза белка, в котором ε-NH2 группа наиболее удаленного к N-концу лизина содержит более подвижную защитную группу, указанный N-концевой лизин подвергают депротекции и пегилированию. Специалистам в данной области понятно, что стратегии, подобные описанной, могут применяться для селективной модификации и достижения аминокислотного(ых) остатка(ов) внутри корового, энхансерного или гибридного полипептида. Информация о таких защитных группах и их подвижности известна из предшествующего уровня техники, в частности в руководстве Грина (Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1981).
Одна или большее число аминокислотных последовательностей также могут быть конъюгированы с коровым или гибридным полипептидами. Используемая аминокислотная последовательность может быть любой аминокислотной последовательностью, которая характеризуется длительным периодом полужизни или может достичь такого в результате слияния белков. В одном варианте осуществления изобретения используемый белок имеет человеческое происхождение. В предпочтительном варианте осуществления изобретения указанный белок представляет собой альбумин человека. Однако специалистам в данной области понятно, что другие белки, такие как, например, иммуноглобулины, также могут использоваться в методе конъюгации, приведенном в описании применительно к альбумину. Так, например, коровый, энхансерный или гибридный полипептид могут быть конъюгированы с представителем семейства иммуноглобулинов или с фрагментами иммуноглобулинов. В одном варианте осуществления изобретения проводят слияние корового, энхансерного или гибридного полипептида с Fc доменом IgG1 человека.
В другом варианте осуществления изобретения полипептид модифицируют путем присоединения модификации непротеиновой природы полиолом, а также модификации аминокислотной последовательности, такой как модификация с помощью ПЭГ и альбумина.
Аминокислотные последовательности могут быть присоединены либо к С-концевому, либо к N-концевому участкам корового, энхансерного или гибридного полипептида, либо в виде боковых цепей полипептида. Аминокислотные последовательности могут быть добавлены к коровому или гибридному полипептиду либо во время синтеза полипептида, либо после его завершения. Альтернативно может быть разработана конструкция, позволяющая получать слитый белок, так чтобы полученную рекомбинантную аминокислотную последовательность присоединить затем к коровому и/или гибридному полипептиду. Так, например, альбумин может быть слит с любым концом корового полипептида. При желании энхансерные пептидные последовательности могут быть затем помещены на свободный конец корового полипептида и/или на конец, содержащий присоединенный рекомбинантный белок. В одном варианте осуществления изобретения аминокислотные последовательности могут служить линкером между коровым и энхансерным полипептидами или могут быть частью его.
Показано, что модификация белков путем слияния с аминокислотными последовательностями, такими как альбумин, повышает до желаемого уровня эффективность лекарственного средства, его стабильность, биологическую активность, а также снижает до требуемого уровня клиренс и токсичность (Kratz et al., Arch. Pharm. (Weinheim) 331: 47-53 (1998); Syed et al., Blood 89: 3243-3252 (1997); Makrides et al., J. Pharmacol. Exp. Ther. 277: 534-42 (1996); Breton et al., Eur. J. Biochem. 231: 563-69 (1995); Paige et al., Pharm. Res.12: 1883-88 (1995)).
Альбумин отличается высокой полиморфностью, так что были идентифицированы многие варианты (Weitkamp et al., Ann. Hum. Genet. 37: 219 (1973)). Последовательности альбумина, например последовательности альбумина человека, хорошо известны специалистам в данной области (см., например, патент США No. 5876969, который полностью включен в настоящее описание в качестве ссылки). Альбуминовые последовательности, которые могут использоваться для целей такой модификации белков, включают, например, пре-про-формы, полноразмерные формы или их фрагменты (см., например, Kratz et al., Arch. Pharm. (Weinheim) 331: 47-53 (1998); Syed et al., Blood 89: 3243-3252 (1997); Makrides et al., J. Pharmacol. Exp. Ther. 277: 534-42 (1996); Breton et al., Eur. J. Biochem. 231: 563-69 (1995); Paige et al., Pharm. Res.12: 1883-88 (1995)). Альбуминовые последовательности могут быть добавлены к полипептидам согласно настоящему изобретению в ходе химического синтеза или с помощью рекомбинантных методов или при использовании их сочетания.
В одном неограничивающем примере полноразмерный альбумин человека может быть слит в рамке считывания с Т1387 (Ас-TALLEQAQIQQEKNEYELQKLDK-NH2) при использовании рекомбинантной технологии.
В другом неограничивающем примере Т1387 (Ас-TALLEQAQIQQEKNEYELQKLDK-NH2), который был пегилирован, может быть конъюгирован с усеченным альбумином человека (доступным от компании Sigma Chemical, St. Louis) с использованием известной в технике методики, такой как методика Пайге с соавт. (Paige et al., Pharm. Res. 12: 1883-88 (1995)).
Модифицированные полипептиды могут быть протестированы на наличие биологической активности, например противовирусной активности, с помощью стандартных методов. Кроме того, с использованием обычных процедур могут быть проанализированы такие особенности модифицированных полипептидов, как фармакокинетические или иммуногенные свойства. Модифицированные полипептиды могут быть также подвергнуты исследованию in vitro и in vivo для выявления наличия изменений в биологической реакции, связанных с модификацией(ями).



















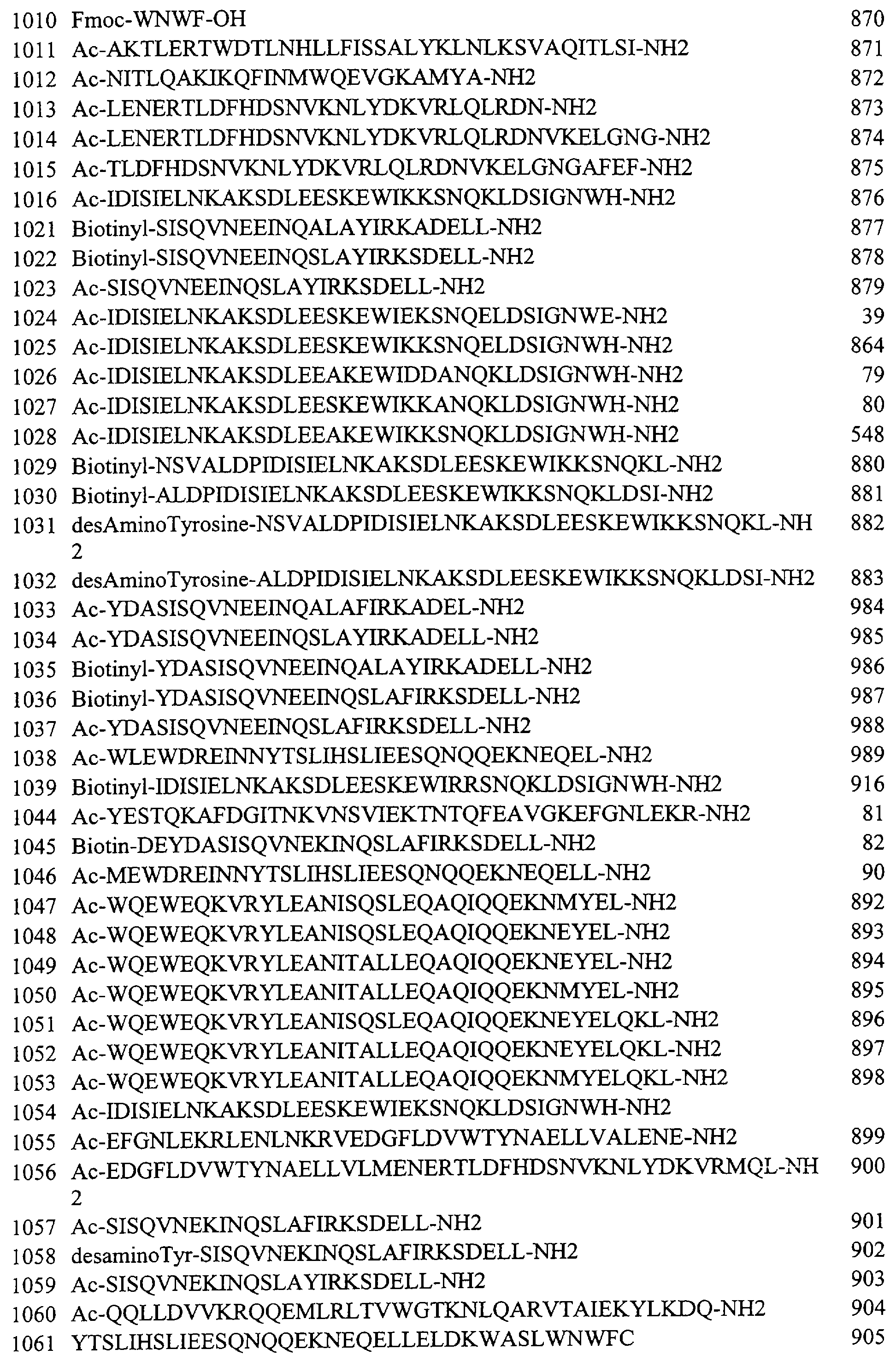



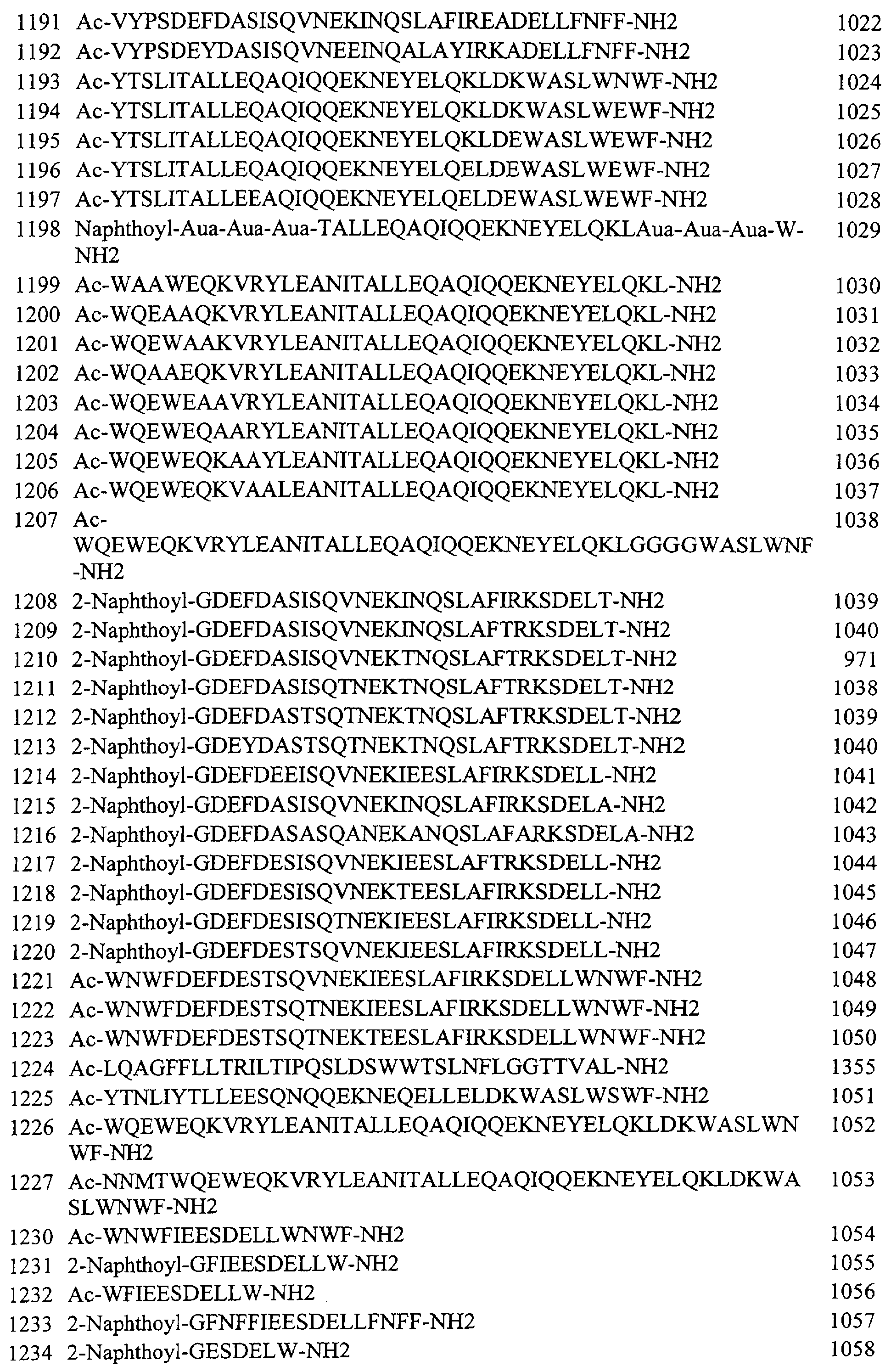










Следует понимать, что пептиды, перечисленные в Таблице 2 и в примере, приведенном ниже в разделе 11, также охватываются рамками настоящего изобретения. Как отмечалось ранее, те из пептидов, которые перечислены в Таблице 2 и в примере, приведенном ниже в разделе 11, и которые еще не содержат энхансерные пептидные последовательности (то есть которые не являются гибридными полипептидами), могут применяться в сочетании с энхансерными пептидными последовательностями и в настоящем описании раскрываются способы получения гибридных полипептидов. Кроме того, коровые полипептиды и коровый полипептид в составе гибридных полипептидов, показанные в Таблице 2, фиг.13 и в примере, представленном ниже в разделе 11, могут применяться в сочетании с любыми указанными в описании энхансерными пептидными последовательностями с целью получения с помощью рутинных процедур дополнительных гибридных полипептидов, которые также входят в область настоящего изобретения.
Так, например, пептид DP397, приведенный в примере раздела 11, обозначает коровый полипептид и также входит в область настоящего изобретения. Кроме того, гибридные полипептиды, включающие коровый полипептид DP397 в сочетании с одним или большим числом энхансерных полипептидных последовательностей, приведенных в настоящем описании, также входят в область настоящего изобретения.
Следует отметить, что несмотря на то, что в Таблице 2 и на фиг.13 приведено множество полипептидов с модифицированными аминокислотами, например, с блокированными амино- и/или карбокси-концами или с включением d-изомеров аминокислот (обозначаемых в виде остатков, заключенных в скобки), тем не менее любой полипептид, включающий первичную аминокислотную последовательность, такую как показано в Таблице 2 и на фиг.13, также входит в качестве составной части в область настоящего изобретения.
Коровые полипептидные последовательности, в частности показанные в Таблице 2, на фиг.13 и в приведенном ниже в разделе 11 примере, а также гибридные полипептиды, включающие такие коровые полипептиды, могут проявлять противовирусную и/или антифузогенную активность и/или могут обладать способностью модулировать внутриклеточные процессы, в которых участвуют свернутые спиральные пептидные структуры. Кроме того, такие пептиды могут также применяться отчасти в методах скрининга соединений, включая пептиды, для обнаружения такой активности. В число коровых полипептидных последовательностей входят, например, такие, которые получены из белковых последовательностей индивидуальных вирусов. В число коровых полипептидных последовательностей также входят такие, например, аминокислотные последовательности, которые получают из белковой последовательности более чем одного вируса (например, коровый полипептид, полученный из ВИЧ-1, ВИЧ-2 и ВИО).
Кроме того, указанные коровые полипептиды могут представлять указанные выше аминокислотные замещения, делеции и/или вставки для энхансерных полипептидных последовательностей. В тех случаях, когда коровый полипептид проявляет противовирусную и/или антифузогенную активность, такие модификации предпочтительно не устраняют (либо самого по себе, либо в составе гибридного полипептида) указанную активность.
В отношении аминокислотных делеций предпочтительно, чтобы получаемый коровый полипептид содержал по меньшей мере 4-6 аминокислотных остатков в длину. В отношении аминокислотных вставок предпочтительные вставки должны быть не больше, чем примерно 50 аминокислотных остатков, и более предпочтительно, не более чем примерно 15 аминокислотных остатков. Предпочтительно также, чтобы вставки корового полипептида представляли собой амино- и/или карбокси-концевые вставки.
В число аминокислотных замещений, делеций и/или вставок корового или гибридного полипептидов согласно настоящему изобретению входят такие, которые соответствуют тем замещениям, делециям и/или вставкам, которые обнаружены в мутантных формах, например в природных мутантных формах эндогенной белковой последовательности, из которой конкретный коровый полипептид был получен.
Так, например, если коровый полипептид получен из вирусного белка и указанный коровый полипептид (либо сам по себе, либо в составе гибридного полипептида) проявляет противовирусную активность в отношении того или другого вируса, возможно, что могут существовать или могут появиться в конечном итоге такие варианты вируса (например, варианты штамма), которые обладают некоторым уровнем устойчивости к пептиду в сравнении с уровнем противовирусного эффекта указанного пептида в отношении того вирусного штамма, из которого была получена исходная эндогенная коровая полипептидная последовательность.
Для получения коровых полипептидов, которые проявляют противовирусную активность в отношении таких устойчивых вирусных штаммов, могут быть введены модификации в исходный коровый полипептид. В частности, любым специалистом со средним уровнем знаний в данной области с помощью стандартных методов могут быть легко выделены изоляты устойчивого вируса. При этом может быть легко осуществлено определение последовательности внутри устойчивого вируса, соответствующего исходному коровому полипептиду, с последующим сравнением с исходным коровым полипептидом.
В том случае, когда соответствующая последовательность получена из мутанта, устойчивый штамм имеет отличия от последовательности корового полипептида, и модификации в коровый полипептид могут быть введены так, чтобы полученный модифицированный коровый полипептид имел ту же самую последовательность, что и соответствующий участок в устойчивом вирусе.
Полученный модифицированный коровый полипептид либо сам по себе, либо в составе гибридного полипептида будет проявлять противовирусные свойства в отношении того вирусного штамма, который был устойчивым к исходному коровому полипептиду. В этой связи указанные способы могут использоваться для идентификации коровых полипептидов, проявляющих противовирусную активность в отношении вирусных штаммов, которые обладают устойчивостью или приобрели указанную устойчивость к противовирусной активности других коровых полипептидов.
Ниже в примере раздела 11 описан один конкретный, неограничивающий вариант применения такого способа для получения модифицированного корового полипептида, который проявляет противовирусную активность в отношении вирусного штамма, устойчивого к «родительскому» коровому полипептиду.
В одном варианте осуществления изобретения указанные модифицированные коровые полипептиды, которые проявляют противовирусную активность в отношении штаммов, устойчивых к «родительскому» коровому полипептиду, включают такие, в которые введены аминокислотные замещения, делеции и/или вставки, модифицирующие «родительский» коровый полипептид таким образом, что в получаемом модифицированном коровом полипептиде удаляется консенсусная последовательность для N-гликозилирования и O-гликозилирования, присутствовавшая в «родительском» коровом полипептиде.
Так, например, консенсусная последовательность для сайта N-гликозилирования представляет собой -N-X-S/T, где S/T обозначает либо серин, либо треонин, и Х обозначает любую аминокислоту, за исключением пролина или аспарагиновой кислоты. Таким образом, в одном варианте осуществления изобретения родительский коровый полипептид, содержащий такую консенсусную последовательность, может быть модифицирован с помощью вставки, замещения и/или делеции аминокислот, так что в модифицированном коровом полипептиде указанная консенсусная последовательность удаляется.
В число указанных амино- и карбокси-концевых вставок входят такие, которые включают аминокислотные последовательности, расположенные на амино- и/или карбокси-концах относительно эндогенной белковой последовательности, из которой был получен коровый полипептид. Так, например, если коровый полипептид получен из белка gр41, такая вставка должна включать амино- и/или карбокси-концевую вставку, содержащую аминокислотную последовательность gр41, соседнюю с коровой полипептидной последовательностью gр41. Размер таких амино- и/или карбокси-концевых вставок варьирует и в типичном случае составляет примерно 1, 5, 10, 15, 20, 25, 30, 35, 40, 45 или 50 аминокислотных остатков в направлении к амино- и/или карбокси-концу исходного корового полипептида.
Гибридные полипепидные последовательности согласно настоящему изобретению могут также включать дополнительные модификации, которые позволяют без труда обнаруживать полипептид. Так, например, гибридные полипептиды могут быть помечены либо непосредственно, либо опосредованно. Методы мечения пептидов хорошо известны специалистам в данной области и включают, не ограничиваясь приведенным списком, радиоактивный, флуоресцентный и колориметрический методы. Методы опосредованного мечения также хорошо известны и включают, не ограничиваясь приведенным списком, мечение биотином/стрептавидином и опосредованное мечение с помощью антител.
Изобретение также относится к сочетанию энхансерных полипептидных последовательностей с типами молекул, отличных от пептидов. Так, например, энхансерные пептидные последовательности могут быть соединены с молекулами нуклеиновых кислот (например, с ДНК или РНК) или с любым типом малых органических молекул с целью усиления фармакокинетических свойств указанных молекул.
5.2. Синтез пептидов
Энхансерные, коровые и гибридные полипептиды согласно настоящему изобретению могут быть синтезированы или получены с помощью процедур, известных в данной области. См., например, руководство Крейтона (Creighton, 1983, Proteins: Structures and Molecular Principles, W.H.Freeman and Co., NY), которое полностью включено в настоящее описание в качестве ссылки. Гибридные полипептиды могут быть получены с использованием стандартного ступенчатого синтеза в растворе или в твердой фазе, а также фрагментной конденсации или методов Fmoc или Бок химии (см., например, Chemical Approaches to the Synthesis of Peptides and Proteins, Williams et al., Eds., 1997, CRC Press, Boca Raton Florida и содержащиеся в работе ссылки; а также Solid Phase Peptide Synthesis: A Practical Approach, Atherton & Sheppard, Eds., 1989, IRL Press, Oxford, England и содержащиеся в работе ссылки). Аналогично может быть осуществлен синтез с модификациями по амино- и/или карбокси-концам.
В целом, указанные методы могут включать последовательное добавление одной или более аминокислот или соответствующим образом защищенных аминокислот к растущей пептидной цепи. В норме либо амино, либо карбоксильную группу первой аминокислоты защищают с помощью подходящей защитной группы. Защищенная или дериватизированная аминокислота может быть затем либо присоединена к инертной твердой подложке, либо может быть использована в виде раствора при добавлении следующей аминокислоты в последовательность, содержащую комплементарную (амино или карбоксильную) группу, защищенную соответствующим образом, в условиях, способствующих образованию амидной связи. Затем защитную группу удаляют из указанного, вновь добавленного аминокислотного остатка, после чего добавляют следующую аминокислоту (соответствующим образом защищенную) и так далее. После того как все требуемые аминокислоты будут связаны в соответствующей последовательности, удаляют оставшиеся защитные группы и любую твердую подложку либо в последовательном режиме, либо одновременно с процессом создания желаемого конечного полипептида. При введении несложной модификации в указанную общую процедуру она может быть применена для добавления более чем одной аминокислоты, в ходе наращивания цепи, например, при связывании (в условиях, не способствующих рацемизации хиральных центров) защищенного трипептида с защищенным соответствующим образом дипептидом с образованием, после удаления защитных групп, пентапептида.
Типичные защитные группы включают Т-бутилоксикарбонил (Бок), 9-флуоренилметоксикарбонил (Fmoc), бензилоксикарбонил (Cbz), п-толуолсульфонил (Тос), 2,4-динитрофенил, бензил (Bzl), бифенилизопропилоксикарбоксикарбонил, циклогексил, изопропил, ацетил, о-нитрофенилсульфонил и др. Из приведенных групп предпочтительными являются Бос и Fmoc.
Типичные твердые носители относятся в основном к перекрестно сшитым полимерным материалам. Они, в свою очередь, включают, не ограничиваясь приведенным списком, перекрестно сшитые дивинилбензоловые полимеры на основе стирола, например сополимеры дивинилбензола-гидроксиметилстирола, сополимеры дивинилбензола-хлорметилстирола и сополимеры дивинилбензола-бензгидриламинополистирола. Такие сополимеры имеют преимущество, связанное с возможностью непосредственного введения концевой амидной функциональной группы в пептидную цепь, так что функция указанной группы сохраняется в цепи и после того, как цепь отделяют от основы.
Указанным способом могут быть синтезированы полипептиды, содержащие либо L-, либо D-аминокислоты.
Состав полипептида может быть подтвержден количественным аминокислотным анализом и наличие конкретной последовательности в каждом пептиде может быть определено с помощью методов, применяемых для анализа последовательности.
Энхансерный, коровый и гибридный полипептиды согласно настоящему изобретению могут быть очищены с помощью любых известных в технике методов, таких как высокоэффективная жидкостная хроматография в нормальной фазе или с обращением фаз, ионообменная хроматография, гель-электрофорез, аффинная хроматография, гранулометрический метод, осаждение и др. Фактические условия, используемые для очистки конкретного полипептида, зависят, в частности, от стратегии синтеза и от таких факторов, как заряд, гидрофобность, гидрофильность, растворимость, стабильность и др. параметров, очевидных для специалистов в данной области.
Гибридный, энхансерный и коровый полипептиды могут быть также получены с помощью рекомбинантных ДНК методов. В настоящем изобретении нуклеотидные последовательности, кодирующие полипептиды согласно настоящему изобретению, могут быть синтезированы и/или клонированы и далее экспрессированы в соответствии с известными специалистам в данной области процедурами. См., например, руководство Самбрука (Sambrook et al., 1989, Molecular Cloning, A Laboratory Manual, Vols. 1-3, Cold Spring Harbor Press, NY).
Фрагмент ДНК, кодирующий интересующий полипептид, можно получить с помощью множества методов молекулярной биологии, известных специалистам в данной области. Так, например, для образования фрагмента ДНК, кодирующего интересующий белок, может применяться полимеразная цепная реакция (ПЦР).
Альтернативно фрагмент ДНК может быть получен из коммерческого источника.
ДНК, кодирующая интересующие полипептиды, может быть введена с использованием рекомбинантной технологии в различные хозяйские векторные системы, которые позволяют осуществлять крупномасштабную репликацию ДНК. Могут быть разработаны такие векторы, которые содержат необходимые элементы для осуществления направленной транскрипции и/или трансляции последовательности ДНК, кодирующей гибридный полипептид.
Векторы, которые могут при этом использоваться, включают, не ограничиваясь приведенным списком, векторы, получаемые из рекомбинантной ДНК бактериофага, плазмидной ДНК или космидной ДНК. Так, например, могут использоваться плазмидные векторы, такие как векторы серии pcDNA3, pBR322, pUC 19/18, pUC 118, 119 и М13 mp. Векторы бактериофагов могут включать векторы бактериофагов серии λgt10, λgt11, λgt18-23, λZAP/R и EMBL. Космидные векторы, которые могут использоваться, включают, не ограничиваясь приведенным списком, pJB8, pCV 103, pCV 107, pCV 108, pTM, pMCS, pNNL, pHSG274, COS202, COS203, pWE15, pWE16 и хароид 9 серии векторов.
Альтернативно, генноинженерными методами могут быть созданы рекомбинантные векторы вирусов, которые включают, не ограничиваясь приведенным списком, векторы, полученные из вирусов, таких как вирус герпеса, ретровирусы, вирусы коровьей оспы, аденовирусы, адено-ассоциированные вирусы или вирусы папилломы быка и вирусы растений, такие как вирус табачной мозаики и бакуловирус.
Для целей экспрессии биологически активного полипептида нуклеотидная последовательность, кодирующая белок, может быть вставлена в соответствующий вектор экспрессии, например в вектор, который содержит необходимые элементы для транскрипции и трансляции вставленных кодирующих последовательностей. При этом могут применяться известные в технике методы для конструирования векторов экспрессии, содержащих последовательность, кодирующую гибридный полипептид, которая функционально связана с соответствующими сигналами регуляции транскрипции/трансляции. Указанные методы включают технологию использования in vitro рекомбинантной ДНК и методы синтеза. См., например, работы Sambrook et al., 1992, Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory, N.Y. and Ausubel et al., 1989, Current Protocols in Molecular Biology, Greene Publishing Associates & Wiley Interscience, N.Y., каждая из которых полностью включена в настоящее описание в качестве ссылки.
Молекула нуклеиновой кислоты, кодирующая интересующие гибридный, энхансерный и коровый полипептиды, может быть функционально связана с множеством различных промоторных/энхансерных элементов. Указанные промоторные/энхансерные элементы могут быть выбраны для целей оптимизации экспрессии терапевтических количеств белка. Элементы экспрессии указанных векторов могут варьировать по активности и специфичности. В зависимости от природы используемой хозяйской/векторной системы, может применяться один любой элемент из множества подходящих элементов транскрипции и трансляции. Промотор может быть представлен таким промотором, который в естественном состоянии находится в связи с интересующим геном. Альтернативно, ДНК может находиться под контролем рекомбинантного или гетерологичного промотора, т.е. промотора, который в норме не связан с указанным геном. Так, например, тканеспецифичные промоторные/энхансерные элементы могут использоваться для регуляции экспрессии ДНК, перенесенной в клетки специфического типа.
Примеры участков регуляции транскрипции, характеризующихся тканевой специфичностью, которые описаны в литературе и могут использоваться, включают, не ограничиваясь приведенным списком, участок регуляции гена эластазы I, активный в гроздевидных клетках поджелудочной железы (Swift et al., 1984, Cell 38: 639-646; Ornitz et al., 1986, Cold Spring Harbor Symp. Quant. Biol. 50: 399-409; MacDonald, 1987, Hepatology 7: 42S-51S); участок регуляции гена инсулина, активный в бета-клетках поджелудочной железы (Hanahan, 1985, Nature 315: 115-122); участок регуляции гена иммуноглобулина, активный в лимфоидных клетках (Grosschedl et al., 1984, Cell 38: 647-658; Adams et al., 1985, Nature 318: 533-538; Alexander et al., 1987, Mol. Cell Biol. 7: 1436-1444); участок регуляции гена альбумина, активный в печени (Pinkert et al., 1987, Genes and Devel. 1: 268-276); участок регуляции гена альфа-фетопротеина, активный в печени (Krumlauf et al., 1985, Moll. Cell Biol. 5: 1639-1648; Hammer et al., 1987, Science 235: 53-58); участок регуляции гена альфа-1-антитрипсина, активный в печени (Kelsey et al., 1987, Genes and Devel. 1: 161-171); участок регуляции гена бета-глобина, активный в миелоидных клетках (Magram et al., 1985, Nature 315: 338-340; Kollias et al., 1986, Cell 46: 89-94); участок регуляции гена миелинового основного белка, активный в олигодендроцитных клетках мозга (Readhead et al., 1987, Cell 48: 703-712); участок регуляции гена легкой цепи 2 миозина, активный в скелетной мышце (Shani, 1985, Nature 314: 283-286) и участок регуляции гена гонадотропного рилизинг-гормона, активный в гипоталамусе (Mason et al., 1986, Science 234: 1372-1378). Могут использоваться промоторы, выделенные из генома вирусов, растущих в клетках млекопитающих (например, промоторы вируса коровьей оспы 7.5К, SV40, HSV, аденовирусов MLP, MMTV, LTR и CMV), а также промоторы, получаемые с помощью рекомбинантной ДНК и методов синтеза.
В некоторых случаях промоторные элементы могут представлять собой конститутивные или индуцибельные промоторы и могут использоваться в соответствующих условиях для достижения экспрессии на высоком уровне или для осуществления регулируемой экспрессии интересующей нуклеотидной последовательности. Экспрессия генов, находящихся под контролем конститутивных промоторов, не требует наличия специфического субстрата для индукции генной экспрессии и происходит при всех условиях роста клеток. И, наоборот, экспрессия генов, контролируемых индуцибельными промоторами, отзывается на наличие или отсутствие индуктора.
Для осуществления на достаточном уровне трансляции вставленных последовательностей, кодирующих белок, также требуются специфические инициирующие сигналы. Указанные сигналы включают инициирующий кодон ATG и примыкающие последовательности. В тех случаях, когда в соответствующие векторы экспрессии вводят полную кодирующую последовательность, включая инициирующий кодон и примыкающие последовательности, могут не понадобиться дополнительные сигналы для регуляции трансляции. Однако в тех случаях, когда вводят лишь часть кодирующей последовательности, должно быть обеспечено наличие экзогенных сигналов трансляции, включая инициирующий кодон ATG. Кроме того, инициирующий кодон должен находиться в одной фазе с рамкой считывания последовательностей, кодирующих белок, для обеспечения трансляции всей вставки. Указанные экзогенные сигналы регуляции трансляции и инициирующие кодоны могут иметь различное происхождение, как естественное, так и синтетическое. Эффективность экспрессии может усиливаться при включении последовательностей, ослабляющих транскрипцию, энхансерных элементов и др.
5.3. Использование энхансерных пептидных последовательностей, коровых полипептидов и гибридных полипептидов согласно настоящему изобретению
Как указывалось выше, энхансерные пептидные последовательности согласно настоящему изобретению могут использоваться для усиления фармакокинетических свойств любого корового полипептида посредством связывания корового полипептида с энхансерными пептидными последовательностями с образованием гибридных полипептидов. Наблюдаемое усиление фармакокинетических свойств оценивается в отношении фармакокинетических свойств одного корового полипептида. Стандартный набор характерных фармакокинетических параметров и методы определения и оценки фармакокинетических свойств агента, такого как полипептид, известны специалистам в данной области техники. Неограничивающие иллюстративные варианты таких методов приведены ниже, в разделе «Примеры».
Энхансерные пептидные последовательности согласно настоящему изобретению могут, кроме того, применяться для усиления in vitro или ex vivo периода полужизни корового полипептида, к которому присоединяют энхансерные пептидные последовательности. Так, например, энхансерные пептидные последовательности могут увеличивать период полужизни присоединяемых коровых полипептидов в случае наличия получаемых гибридных, полипептидов в клеточной культуре, культуре ткани или в образцах, взятых у пациента (например, в образцах клеток, биопсийных образцах ткани или других образцах, содержащих жидкости тела).
Коровые полипептиды и гибридные полипептиды согласно настоящему изобретению могут также применяться в составе способов, направленных на модулирование (например, на снижение, ингибирование, разрушение, стабилизацию или усиление) явлений слияния. Предпочтительно такие пептиды проявляют антифузогенную или противовирусную активность. Пептиды согласно настоящему изобретению могут также обладать способностью модулировать внутриклеточные процессы, в которые вовлечены взаимодействия свернутых спиральных пептидов.
В определенных вариантах осуществления изобретения гибридные полипептиды и коровые полипептиды согласно настоящему изобретению, которые проявляют противовирусную активность, могут использоваться в составе способов, направленных на снижение вирусной инфекции. Такие способы противовирусного действия могут применяться, например, против ретровирусов человека, в частности ВИЧ (вируса иммунодефицита человека), например ВИЧ-1 и ВИЧ-2, и Т-лимфоцитных вирусов человека (HTLV-I и HTLV-II) и ретровирусов других организмов, отличных от человека, таких как бычий вирус лейкоза, вирусы саркомы и лейкоза кошек, вирусы иммунодефицита обезьяны (ВИО), вирусы саркомы и лейкоза, вирусы прогрессирующей пневмонии овец.
Способы противовирусного действия согласно настоящему изобретению могут также применяться против иных вирусов, отличных от ретровирусов, включая, но не ограничиваясь приведенными примерами, респираторно-синцитиальный вирус (РСВ), вирус чумы собак, вирус псевдочумы птиц, вирус парагриппа человека, вирусы гриппа, вирусы кори, вирусы Энштейна-Барра, вирусы гепатита В и вирусы Мэйсон-Пфайзера.
Указанные выше вирусы относятся к вирусам, содержащим оболочку. Противовирусные способы согласно настоящему изобретению могут также применяться против вирусов, не содержащих оболочку, включая, но не ограничиваясь приведенным списком, пикорнавирусы, такие как вирусы полиомиелита, вирус гепатита А, энтеровирус, эховирусы и вирусы коксаки, папо-вавирусы, такие как вирус папилломы, парвовирусы, аденовирусы и реовирусы.
Другие виды антифузогенной активности, которые могут модулироваться с помощью способов, использующих пептиды согласно настоящему изобретению, включают, не ограничиваясь, модулирование обмена нейротрансмиттеров посредством клеточного слияния и слияния по типу сперматозоид-яйцеклетка. К числу внутриклеточных нарушений, включающих взаимодействия свернутых спиральных структур, которые могут ослабляться с помощью способов, использующих пептиды согласно настоящему изобретению, относятся, например, заболевания, связанные с действием бактериальных токсинов.
Антифузогенная или противовирусная активность данного корового полипептида или гибридного полипептида может быть без труда установлена стандартными методами in vitro, ex vivo и с использованием модели животных, причем в том, что касается определения противовирусной активности, они могут быть специфичными или частично специфичными в отношении интересующего вируса, и все они хорошо известны специалистам в данной области.
Приведенное выше описание относится прежде всего к противовирусной активности и к активности, связанной с антифузогенным действием корового и гибридного полипептидов согласно настоящему изобретению. Гибридные полипептиды согласно настоящему изобретению могут также применяться в составе любого способа, в рамках которого может рассматриваться введение или иное использование одного корового полипептида. Использование гибридных полипептидов в качестве составной части указанных способов особенно предпочтительно в тех случаях, когда желательно добиться усиления фармакокинетических свойств корового полипептида. Так, например, инсулин используют как составной элемент лечения при некоторых типах диабета. И в этой связи как составной элемент способов, направленных на ослабление симптомов тех форм диабета, при которых используют и/или предполагается использовать инсулин, может использоваться гибридный полипептид, включающий инсулин или фрагмент инсулина в качестве корового полипептида.
В дополнение к указанным выше способам пептиды согласно настоящему изобретению могут также применяться в качестве составного элемента прогностических методов, направленных на профилактику заболеваний, включая, но не ограничиваясь, заболевания, связанные с процессом слияния, внутриклеточные процессы, в которых участвуют свернутые спиральные пептиды, и вирусную инфекцию, в которой имеет место процесс слияния по типу клетка-клетка и/или вирус-клетка. Так, например, коровый и гибридный полипептиды согласно настоящему изобретению могут использоваться в составе профилактических методов, используемых для предотвращения вирусной инфекции.
Гибридные полипептиды согласно настоящему изобретению могут также применяться в составе диагностических методов. Такие методы могут относиться либо к методам in vitro, либо к методам in vivo. Любой диагностический метод, в рамках которого может использоваться конкретный коровый полипептид, может применяться также с использованием гибридного полипептида, включающего коровый полипептид и модификацию первичной аминокислотной последовательности, позволяющую обнаруживать гибридный полипептид. Такие методики отражают усовершенствование диагностических методов, определяемое тем, что увеличение периода полужизни гибридного полипептида в сравнении с коровым полипептидом может приводить к повышению чувствительности той диагностической процедуры, в составе которой он используется. Указанные диагностические процедуры включают, не ограничиваясь, методы визуализации, например, методы визуализации in vivo. В неограничивающем примере методов визуализации структура, которая связывает коровый полипептид гибридного полипептида может быть обнаружена посредством связывания с гибридным полипептидом и визуализацией (либо непосредственной, либо опосредованной) связанного гибридного полипептида.
5.4. Фармацевтические композиции, дозировки и способы введения
Пептиды согласно настоящему изобретению могут вводиться с помощью известных в технике методов. Предпочтительно рассматриваемые средства включают в состав композиции и вводят системно. Процедуры, используемые для приготовления композиций и их введения, описаны, в частности, в руководстве Ремингтона ("Remington's Pharmaceutical Sciences", последнее издание, Mack Publishing Co., Easton, PA). Подходящие пути введения включают пероральное, ректальное, внутривагинальное, внутрилегочное (например, путем ингаляции), чрезкожное, чрезслизистое или внутрикишечное введение, парентеральные способы доставки, включая внутримышечную, подкожную, интрамедуллярную инъекции, а также внутриоболочечную, непосредственно внутрижелудочковую, внутривенную, внутрибрюшинную, интраназальную или внутриглазную инъекции, если называть лишь некоторые варианты. В случае внутривенной инъекции средства согласно настоящему изобретению могут изготавливаться в виде водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Хенкса, раствор Рингера или физиологический солевой раствор, если называть лишь некоторые варианты. Кроме того, для доставки пептидов согласно настоящему изобретению могут применяться инфузионные насосы. Для чрезслизистого введения при изготовлении композиции используют усилители проницаемости, подходящие для того барьера, через который следует пройти. Такие усилители проницаемости в целом известны в технике.
В тех случаях, когда предпочтительно внутриклеточное введение пептидов согласно настоящему изобретению или других ингибиторов, могут применяться известные в технике методы. Так, например, указанные средства могут быть инкапсулированы в липосомы или микросферы и затем введены с помощью указанных выше способов. Липосомы представляют собой сферические липидные бислойные частицы с внутренней водной средой. Все молекулы, присутствующие в водном растворе к моменту образования липосом, включаются во внутреннюю водную среду. Содержимое липосом защищено от внешней микросреды, и, в связи с тем, что липосомы слиты с клеточными мембранами, способны эффективно достигать клеточной цитоплазмы. Кроме того, благодаря гидрофобности липосом в случае использования малых молекул можно добиться непосредственного внутриклеточного введения.
Нуклеотидные последовательности, кодирующие пептиды согласно настоящему изобретению при их внутриклеточной доставке, могут быть экспрессированы в интересующих клетках с помощью известных в технике методов. Так, например, векторы экспрессии, полученные из вирусов, таких как ретровирусы, вирусы коровьей оспы, аденоассоциированные вирусы, вирусы герпеса или вирусы папилломы быка, могут применяться для доставки и экспрессии таких нуклеотидных последовательностей в популяции целевых клеток. Методы конструирования таких векторов и экспрессионных конструкций также известны. См., например, Sambrook, et al., 1989, Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Press, Cold Spring Harbor NY and Ausubel et al., 1989, Current Protocols in Molecular Biology, Greene Publishing Associates & Wiley Interscience, N.Y.
Эффективные дозы пептидов согласно настоящему изобретению, которые следует вводить, могут быть определены с помощью известных в технике процедур, учитывающих такие параметры, как биологический период полужизни, биодоступность и токсичность. В особенно предпочтительных вариантах осуществления изобретения диапазон эффективных дозировок гибридного полипептида может быть определен любым специалистом в данной области с использованием рутинных исследований in vitro и in vivo, известных в данной области техники. Так, например, оценка противовирусной активности в клеточной культуре in vitro с использованием методов, примеры которых приведены ниже в разделе 7 для Т1249, предоставляет данные, на основе которых любой специалист в данной области может легко определить среднюю ингибирующую концентрацию (ИК) пептида согласно настоящему изобретению, необходимую для блокирования определенного уровня вирусной инфекционности (например, на 50% - ИК50 или на 90% - ИК90). И затем соответствующие дозы могут быть выбраны любым специалистом в данной области на основании фармакокинетических данных, полученных с использованием одной или большего числа обычных моделей животных, таких как фармакокинетические данные, описанные ниже в примерах раздела 10 для Т1249, с получением значения минимальной концентрации в плазме (Смин) пептида, которое равно или превосходит определенное значение ИК.
Примерные значения доз полипептида могут включать низкие дозы, такие как 0,1 мкг/кг веса тела, и высокие дозы, такие как 10 мг/кг веса тела. Более предпочтительно диапазон эффективных дозировок варьирует от 0,1 до 100 мкг/кг веса тела. Другие иллюстративные примеры дозировок пептидов согласно настоящему изобретению включают 1-5 мг, 1-10 мг, 1-30 мг, 1-50 мг, 1-75 мг, 1-100 мг, 1-125 мг, 1-150 мг, 1-200 мг или 1-250 мг пептида. Терапевтически эффективная доза означает такое количество соединения, которого достаточно для ослабления симптомов или для увеличения выживания пациента. Токсичность и терапевтическая эффективность таких соединений может быть определена с использованием стандартных фармацевтических процедур в культуре клеток экспериментальных животных, например, при определении ЛД50 (летальная доза для 50% популяции) и ЭД50 (доза, терапевтически эффективная для 50% популяции). Соотношение доз, вызывающих токсический и терапевтический эффекты, представляет терапевтический индекс, который может быть выражен в виде коэффициента ЛД50/ЭД50. Предпочтительны соединения с высоким терапевтическим индексом. Данные, получаемые с помощью таких методов анализа клеточных культур и исследования животных, могут быть затем использованы при установления дозировок при использовании на людях. Дозировка таких соединений находится предпочтительно в диапазоне циркулирующих в крови концентраций, который включает ЭД50 с небольшой токсичностью или вовсе без токсичности. Доза может варьировать в пределах указанного диапазона в зависимости от формы используемой дозировки и от пути ее введения. Терапевтически эффективную дозу любого соединения, применяемого в рамках способа согласно настоящему изобретению, оценивают вначале по результатам анализа клеточной культуры. Далее на моделях животных может быть определена доза, необходимая для достижения диапазона циркулирующих в плазме концентраций, который включает ИК50 (например, концентрацию исследуемого соединения, которая позволяет достичь половины от максимального уровня ингибирования фузогенного эффекта, такого как половина от максимального уровня ингибирования вирусной инфекции в сравнении с уровнем указанного явления в отсутствие исследуемого соединения), определенную в клеточной культуре. Такая информация может применяться с целью более точного определения доз для использования на людях. Уровни концентрации в плазме можно определить, например, с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) или любого биологического или иммунологического метода, позволяющего измерять уровни пептидов.
Гибридные полипептиды согласно настоящему изобретению могут вводиться в виде однократного приема, дробного, периодического или непрерывного приема. Так, например, полипептиды согласно настоящему изобретению могут вводиться в виде однократной дозы, такой как однократная подкожная, однократная внутривенная инфузия или однократное проглатывание. Полипептиды согласно настоящему изобретению могут вводиться с использованием множества форм дробного введения, включая периодическое введение. Так, например, в некоторых вариантах полипептиды согласно настоящему изобретению могут вводиться один раз в неделю, один раз в день, два раза в день (т.е. каждые 12 часов), каждые шесть часов, каждые четыре часа, каждые два часа или каждый час. Полипептиды согласно настоящему изобретению могут быть назначены для постоянного введения, такого как постоянная подкожная или внутривенная инфузия с помощью насоса или с использованием подкожного или другого имплантата, который позволяет осуществлять постоянное всасывание полипептидов в организм пациента.
Гибридные полипептиды согласно настоящему изобретению могут также вводиться в сочетании по меньшей мере еще с одним терапевтическим средством. Хотя этот вариант не является предпочтительным при лечении ВИЧ, в случае терапии других заболеваний (например, при лечении рака) введение может осуществляться в сопутствующем или последовательном режиме, включая циклический режим терапии (что включает введение первого соединения в течение определенного периода времени, затем введение второго противовирусного соединения в течение определенного периода времени и повтор последовательного введения в указанном порядке для снижения развития толерантности, возникающей к одному виду лечения).
В случае вирусных, например, ретровирусных, инфекций может быть введено эффективное количество гибридного полипептида или его фармацевтически приемлемого производного в сочетании по меньшей мере с одним, предпочтительно по меньшей мере с двумя, другими противовирусными средствами.
Если рассматривать в качестве примера ВИЧ инфекцию, то указанные противовирусные средства могут включать, не ограничиваясь приведенными примерами, DP-107 (T21), DP-178 (Т20), любой другой коровый полипептид, показанный в Таблице 2, который был получен из ВИЧ-1 или ВИЧ-2, любой другой гибридный полипептид, коровый полипептид которого, по меньшей мере частично, получен из ВИЧ-1 или ВИЧ-2, цитокины, например, rIFN α, rIFN β, rIFN λ, ингибиторы обратной транскриптазы, включая нуклеозидные и ненуклеозидные ингибиторы, например AZT, ЗТС, D4T, ddI, адефовир, абакавир и другие дидезоксинуклеозиды или дидезоксифторнуклеозиды или мезилат делавиридина, невирапин, эфавиренц, ингибиторы кэппинга вирусной мРНК, такие как рибавирин, ингибиторы ВИЧ протеазы, такие как ритонавир, нелфинавира мезилат, ампренавир, саквинавир, саквинавира мезилат, индинавир или АВТ378, АВТ538 или МК639, амфотерицин В как связывающаяся с липидами молекула с анти-ВИЧ активностью, и кастаноспермин как ингибитор процессинга гликопротеина.
Гибридные и/или коровые полипептиды согласно настоящему изобретению могут также применяться с профилактической целью для предупреждения развития заболевания. Гибридные и/или коровые полипептиды могут предупреждать возникновение заболевания за счет их непосредственного действия или альтернативно могут применяться в виде вакцины, когда в организме хозяина повышается выработка антител против гибридных полипептидов согласно настоящему изобретению, которые затем служат для нейтрализации патогенных организмов, включая, например, ингибирование вирусной, бактериальной и паразитарной инфекции.
Для всех таких указанных выше видов лечения конкретный вид композиции, путь ее введения и доза выбираются лечащим врачом на основании состояния пациента (см., например, Fingl et al., 1975, in "The Pharmacological Basis of Therapeutics", Ch.1, p.1).
Следует отметить, что лечащий врач должен знать, как и когда нужно закончить, приостановить или откорректировать введение, в случае развития токсичности или дисфункции органов. И, наоборот, лечащий врач должен также знать о необходимости откорректировать лечение за счет увеличения вводимой дозы, если имеет место неадекватный клинический эффект (не вызывая токсичности). Величина вводимой дозы при лечении онкогенного заболевания варьирует в зависимости от тяжести состояния, которое предстоит лечить, и от пути введения. Доза и, возможно, частота введения дозы также зависят от возраста, веса тела и индивидуальной реакции пациента. Программа, сравнимая с обсуждавшейся выше, может также применяться в ветеринарии.
Использование фармацевтически приемлемых носителей для создания композиций на основе соединений, раскрываемых в настоящем описании, в виде определенных дозированных форм, подходящих для системного введения, также входит в область настоящего изобретения. При выборе подходящего носителя и с использованием соответствующей технологии производства композиции согласно настоящему изобретению, в частности, те из них, которые изготавливаются в виде растворов, могут вводиться парентерально, с помощью таких способов, как подкожная инъекция, внутривенная инъекция, подкожная инфузия или внутривенная инфузия, например, с использованием инфузионного насоса. С использованием фармацевтически приемлемых известных носителей соединения могут быть легко приготовлены в виде композиции, в дозировке, приемлемой для перорального введения. Указанные носители позволяют изготавливать на основе соединений согласно настоящему изобретению такие формы, как таблетки, пилюли, капсулы, жидкие формы, гели, сиропы, формы в виде кашицы, суспензии и др., для перорального приема пациентом, проходящим лечение.
Фармацевтические композиции, подходящие для применения в рамках настоящего изобретения, включают композиции, в которых активные ингредиенты содержатся в эффективном количестве, достаточном для достижения поставленной цели. Определение эффективных количеств в состоянии провести любой специалист в данной области, особенно в связи с наличием детального описания настоящего изобретения.
Кроме активных ингредиентов, указанные фармацевтические композиции могут содержать подходящие фармацевтически приемлемые носители, включающие наполнители и добавки, облегчающие процесс изготовления на основе активных соединений препаратов, которые могут использоваться с фармацевтической целью. Препараты, приготовленные для перорального введения, могут иметь вид таблеток, драже, капсул или растворов. Для перорального введения пептидов может использоваться процедура, такая как, например, практикуется в Emisphere Technologies и которая хорошо известна специалистам в данной области.
Фармацевтические композиции согласно настоящему изобретению могут изготавливаться с помощью методов, которые сами по себе хорошо известны, например, при осуществлении традиционных процессов смешивания, растворения, гранулирования, получения драже, растирания в порошок, распылительной сушки, эмульгирования, инкапсулирования, улавливания или лиофилизации.
Фармацевтические композиции для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Кроме того, эмульсии и суспензии активных соединений могут быть приготовлены в виде соответствующих масляных смесей для инъекции. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло или синтетические эфиры жирных кислот, такие как этилолеат, или триглицериды, липосомы, или другие известные в технике вещества, применяемые для получения липидных или липофильных эмульсий. Водные суспензии для инъекции могут содержать вещества, повышающие вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Суспензия может также необязательно содержать подходящие стабилизаторы или средства, которые повышают растворимость соединений, позволяя получать высококонцентрированные растворы.
Фармацевтические композиции для перорального введения могут быть получены при объединении активных соединений с твердым наполнителем, необязательном измельчении полученной смеси и обработке смеси гранул, после добавления при желании соответствующих добавок, с получением таблеток или ядер драже. Приемлемые носители представляют, например, наполнители, такие как сахара, включая лактозу, сахарозу, трегалозу, маннит или сорбит, целлюлозные препараты, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропил-метилцеллюлоза, натрий-карбоксиметилцеллюлоза и/или поливинил-пирролидон (ПВП). При желании могут быть добавлены средства, способствующие разложению, такие как перекрестно сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
На ядра драже наносят подходящие покрытия. Для этой цели могут использоваться концентрированные растворы сахара, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, гель карбопол, полиэтиленгликоль и/или диоксид титана, лакирующие растворы и подходящие органические растворители или их смеси. К таблеткам или покрывающим оболочкам драже могут быть добавлены красители или пигменты для идентификации или для указания различных сочетаний доз активного ингредиента.
Фармацевтические препараты, которые могут применяться перорально, включают капсулы с насечкой, изготовленные из желатина, а также мягкие запечатанные капсулы, выполненные из желатина и пластификатора, такого как глицерин или сорбит. Капсулы с насечкой могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими веществами, такими как крахмалы, и необязательно со стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Дополнительно могут вноситься стабилизаторы.
В тех случаях, когда желательно добиться усиления иммунной реакции хозяина, гибридные полипептиды могут приготавливаться в виде композиции в сочетании с подходящим адъювантом для целей указанного усиления иммунной реакции. Такие адъюванты могут включать, не ограничиваясь приведенным списком, минеральные гели, такие как гидроксид алюминия; поверхностно-активные вещества, такие как лизолецитин, плуроновые полиоли, полианионы; другие пептиды; масляные эмульсии и потенциально полезные адъюванты, такие как БЦЖ и Corynebacterium parvum. Для введения композиций вакцин используется множество методов. Указанные методы включают, не ограничиваясь, пероральный, внутрикожный, внутримышечный, внутрибрюшинный, внутривенный, подкожный и интраназальный пути введения.
6. Пример: Идентификация консенсусных аминокислотных последовательностей, которые включают энхансерные пептидные последовательности
Белок ретровируса gр41 содержит структурные домены, относящиеся к участку α-спирали, расположенному в С-концевом регионе белка, и к участку «застежки-молнии», расположенному в N-концевом регионе белка. Сравнение участков энхансерной пептидной последовательности, содержащихся внутри gр41 (фиг.2А и 2В), в gр41 из всех выделенных последовательностей ВИЧ-1, ВИЧ-2 и ВИО, публикация о которых имеется к настоящему времени, позволило идентифицировать консенсусные аминокислотные последовательности, показанные на фиг.1.
Как будет описано более подробно в приведенных ниже примерах, такие последовательности представляют энхансерные пептидные последовательности в указанном связывании данных пептидных последовательностей с множеством различных коровых полипептидов, что усиливает фармакокинетические свойства получаемых гибридных полипептидов.
7. Пример: Гибридные полипептиды, которые функционируют как мощные ингибиторы ВИЧ-1 инфекции
Как показано на фиг.13, Т1249 представляет собой гибридный полипептид, включающий энхансерные пептидные последовательности, связанные с коровым полипептидом ВИЧ. Как будет показано ниже, гибридный полипептид Т1249 проявляет усиленные фармакокинетические свойства и мощную активность in vitro в отношении изолятов ВИЧ-1, ВИЧ-2 и ВИО, причем отмечается повышенная активность в отношении клинических изолятов ВИЧ-1 в тестах на инфекционность в отношении huPBMC in vitro, а также для huPBMC при SCID (тяжелом комбинированном иммунодефиците) на мышиной модели ВИЧ-1 инфекции in vivo. В описанных ниже биологических тестах активность Т1249 сравнивают с мощным противовирусным полипептидом Т20. Полипептид Т20, также известный как DP-178, получен из последовательности белка gр41 ВИЧ-1, который был раскрыт и заявлен в патенте США No. 5464933.
7.1. Материалы и методы
7.1.1. Синтез и очистка пептидов
Пептиды синтезируют с использованием методов Fast Мос химии. В основном, если особо не оговорено иное, пептиды содержат амидированные карбоксильные концы и ацетилированные амино концы. Очистку проводят методом ВЭЖХ с обращением фаз.
Т1249 (Ac-WQEWEQKITALLEQAQIQQEKNEYELQKLDKWASLWEWF-NH2) представляет собой пептид из 39 аминокислот (М.в.=5036,7), состоящий полностью из натуральных аминокислот и блокированный по амино концу ацетильной группой, а по карбоксильному концу амидо группой с целью повышения стабильности. Т1387 является пептидом, состоящим из 23 аминокислот и не содержащим энхансерных пептидных последовательностей (Ас-TALLEQAQIQQEKNEYELQKLDK-NH2). Таким образом, Т1387 представляет коровый полипептид гибридного полипептида Т1249. Т1387 блокирован по своим амино- и карбокси-концам таким же образом, как и Т1249.
В частности, Т1249 синтезируют с использованием стандартных методов синтеза в твердой фазе. Идентичность основного пика в профиле ВЭЖХ Т1249 подтверждают данные масс-спектроскопии.
Т1249 без труда очищают хроматографией с обращением фаз на 6-дюймовой колонке, заполненной С18, 10 микрон, с подложкой 120А.
7.1.2. Вирус
ВИЧ вирус HIV-1LAI (Popovic, M. et al., 1984, Science 224: 497-508) размножают в клетках СЕМ, культивируемых в среде RPMI 1640, содержащей 10% фетальной сыворотки теленка. Супернатант от инфицированных клеток СЕМ пропускают через фильтр 0,2 мкм и оценивают титр инфекционности в микроинфекционном тесте с использованием клеточной линии АА5 для поддержки репликации вируса. Для указанной цели добавляют 20 мкл серийно разведенного вируса к 20 мкл клеток СЕМ в концентрации 6×105/мл на 96-луночном микротитрационном планшете. Каждое разведение вируса исследуют в трех повторах. Клетки культивируют в течение семи дней при добавлении через день свежей среды. На 7 день после инфицирования образцы супернатанта тестируют на наличие репликации вируса на основании определения активности обратной транскриптазы, высвобождаемой из супернатанта. Величину TCID50 вычисляют по формуле Рида и Мюнха (Reed and Muench; Reed, L.J. et al., 1938, Am. J. Hyg. 27: 493-497).
7.1.3 Тест на слияние клеток
Примерно 7×104 клеток Molt-4 инкубируют с 1×104клетками СЕМ при хроническом инфицировании ВИЧ вирусом HIV-1LAI на 96-луночном планшете для культуры тканей в культуральной среде в общем объеме 100 мкл (RPMI 1640, содержащей 10% инактивированный нагреванием ФБР с добавками 1% L-глютамина и 1% пенициллина-стрептомицина), как описано ранее (Matthews, T.J. et al., 1987, Proc. Natl. Acad. Sci. USA 84: 5424-5428). Добавляют ингибиторы пептидов в объеме 10 мкл и клеточные смеси инкубируют в течение 24 часов при 37°С в атмосфере 5% CO2. По истечении указанного времени подсчитывают количество многокоровых гигантских клеток (синцитий, по ширине соответствующий пяти клеткам или более) при микроскопировании с увеличением 10× и 40×, которые позволяют осматривать в поле зрения полностью лунку. Обработанные клетки сравнивают с инфицированными необработанными контролями и полученные результаты выражают в виде процента ингибирования инфицированного контроля.
7.1.4. Тесты на инфекционность Magi-CCR-5
Примерно 1×104 клеток Magi-CCR-5 (полученных от NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID; Chackerian, B. et al., 1997, J. Virol. 71: 3932-3939) высевают на 48-луночный планшет для культуры тканей (примерно 2×104 клеток/лунку в селективной ростовой среде в объеме 300 мкл/лунку, состоящей из DMEM с добавлением 10% инактивирован-ного нагреванием ФБР, 1% L-глютамина, 1% пенициллина-стрептомицина, гигромицина В, генетицина и пуромицина) и оставляют для контактирования в течение ночи при 37°С в атмосфере 5% CO2. Слияние клеток достигало примерно 30% к следующему дню. Посевную среду удаляют и добавляют разбавленный пептидный ингибитор в количестве 50 мкл/лунку (для сред, вносимых только в необработанные контроли), затем 100 мкл/лунку с разбавленным вирусом (желательный титр вводимого вируса составляет 100-200 КОЕ/лунку). И, наконец, к каждой ячейке добавляют 250 мкл селективной ростовой среды и планшеты инкубируют в течение 2 дней при 37°С в атмосфере 5% CO2. Фиксирование и окрашивание проводят в соответствии с протоколом Национального института аллергии и инфекционных заболеваний (США), разработанным для клеток MAGI-CCR5. Вкратце, процедура состоит в том, что из планшета удаляют среду и к каждой лунке добавляют 500 мкл фиксатора. Планшеты оставляют на 5 минут при комнатной температуре для фиксации. Фиксатор удаляют, каждую лунку промывают дважды DPBS и к каждой лунке добавляют 200 мкл раствора красителя. Далее планшеты инкубируют в течение 50 минут при 37°С в атмосфере 5% CO2, раствор красителя удаляют и каждую лунку промывают два раза DPBS. Планшеты оставляют высыхать на воздухе, после чего под микроскопом, осматривая всю лунку, подсчитывают количество окрашенных синих клеток. Данные, полученные для обработанных клеток, сравнивают с таковыми для инфицированных, необработанных контролей и результаты выражают в виде процента ингибирования инфицированных контролей.
7.1.5. Тест с обратной транскриптазой
Для определения обратной транскриптазы (ОТ) в условиях микротеста адаптируют процедуру Гоффа с соавт. (Goff, et al., 1981, J.Virol. 38: 239-248) и Вилли с соавт. (Willey, R. et al., 1988, J.Virol. 62: 139-147). В супернатантах из вирусной/клеточной культур доводят содержание тритона Х-100 до 1%. К 50 мкл ОТ коктейля (75 мМ KCl, 2 мМ реагента Кливеланда, 5 мМ MgCl2, 5 мкг/мл поли А, 0,25 единиц/мл олиго дТ, 0,05% NP40, 50 мМ Трис-HCl, рН 7,8, 0,5 мкМ нерадиоактивного дТТФ и 10 cCi/мл32P-дТТФ) на 96-луночном микротитрационном планшете с U-образным дном добавляют 10 мкл каждого образца супернатанта/тритона Х-100 и инкубируют при 37°С в течение 90 минут. После инкубации 40 мкл реакционной смеси из каждой лунки переносят в аппарат Шляйхера и Шуэлла (Schleicher and Schuell, S+S) для проведения дот-блот-анализа в условиях частичного вакуума, содержащий сетчатый 96-гнездный сепаратор (Wallac, каталожный номер 1450-423) и фильтр-подложку, насыщенную 2×SSC буфером (0,3 М NaCl и 0,003 М цитрат натрия). Каждую лунку промывают 4 раза по меньшей мере 200 мкл буфера 2xSSC в условиях полного вакуума. Устройство для микротестирования разбирают, удаляют сетчатую фильтровальную бумагу и промывают 3 раза буфером 2× SSC. В конце процедуры пропитывают фильтровальной мембраной абсорбирующую бумагу, позволяют ей высохнуть на воздухе и запечатывают в пакеты, запечатываемые при повышенной температуре. Образцы помещают в люминесцирующую кассету, вносят в нее счищенный люминесцентный экран по крайней мере на 8 мин и закрывают. Экспозиция длится в течение 16 часов. Величину пиксел-индекса (PIV), получаемую в указанном формате по результатам оценки фосфоро-люминесцирующих блотов (Molecular Dynamics Phosphorimager), используют далее для определения задействованной или ингибированной фракции (Fa) для всех доз ингибитора(ов) при сравнении с необработанными, инфицированными контролями (анализ проводят по программе ImageQuant при вычете фонового значения).
7.1.6. Тест на инфекционность/нейтрализацию по РВМС
В прототипе указанного теста используют клеточные линии, в которых используются РВМС, полученные из Межнационального банка крови (Interstate Blood Bank), активированные в течение 2-3 дней с помощью сочетания ОКТЗ (0,5 мкг/мл) и CD28 антител (0,1 мкг/мл). Целевые клетки объединяют со средой для выделения лимфоцитов (LSM), промывают и замораживают. При возникновении потребности клетки оттаивают и активируют, как указано выше, в течение минимум 2-3 дней до тестирования. В указанном варианте определения клетки берут в концентрации 2×106/мл в 5% IL-2 среде и в конечном объеме 100 мкл. Готовят стандартные пептидные растворы в DPBS (1 мг/мл). Разведения пептидов делают в полной среде, содержащей 20% ФБР RPMI 1640/5% IL-2.
7.1.7. Модель инфицирования in vivo ВИЧ-1 с использованием huPBMC и мышей линии SCID
Самкам мышей линии SCID (в возрасте 5-7 недель) инъецируют внутрибрюшинно 5-10×107 РВМС взрослого человека. Через две недели после восстановления мышей инфицируют внутрибрюшинно на 0 день 103 TCID50 ВИЧ-1 9320 (AZT-чувствительный изолят А018). Лечение пептидами проводят внутрибрюшинной инъекцией 2 раза в день, начиная с - 1 дня и продолжают до 6 дня. Распространение инфекции в клетках крови, спленоцитах, лимфатических узлах и перитонеальных клетках оценивают при совместном культивировании с бластными монокоровыми клетками периферической крови человека еженедельно в течение трех последовательных недель с последующими обескровливанием животных и отбором тканей (день 7, примерно через 12-18 часов после введения последней инъекции). Супернатанты от культуры при совместном культивировании оценивают с точки зрения образования р24 антигена ВИЧ-1 в качестве показателя развития вирусной инфекции (наборы Immunotek Coulter и соответствующий протокол).
7.1.8. Фармакокинетические исследования на крысах
Используют самцов крыс линии CD весом 250-300 г с двойным катетером в яремной вене, полученных из Чарльз Ривер Лаборатории (Charles River Laboratories). Пептиды инъецируют в виде пептидного раствора в один яремный катетер в количестве 200 мкл (примерно 3,75 мг/мл), дозу раствора соответствующей концентрации определяют по методу Эдельхока (Edelhoch, 1967, Biochemistry 6: 1948-1954) и корректируют его в соответствии с весом животных, так чтобы каждое животное получало дозу в 2,5 мг/кг. Отбирают приблизительно 250-300 мкл крови в заданные периоды времени (0, 15, 30 мин и 1, 2, 4, 6 и 8 часов) и добавляют к пробиркам с ЭДТА. Плазму отбирают от осевших при центрифугировании клеток и либо замораживают, либо сразу начинают подготавливать для проведения флуоресцентного ВЭЖХ анализа.
7.1.9. Флуоресцентный ВЭЖХ анализ образцов плазмы
100 мкл образца плазмы добавляют к 900 мкл осаждающего буфера (ацетонитрил, 1,0% ТФУ, детергент) с получением осадка, в котором находится большая часть белков плазмы. После центрифугирования при 10000 об/мин в течение 10 минут отбирают 400 мкл супернатанта и добавляют к 600 мкл воды для ВЭЖХ. Основываясь на концентрации пептида, имеющегося в каждом образце, делают серийные разведения разбавляющим буфером, включающим 40% осаждающего буфера и 60% воды для ВЭЖХ. Кроме разведений образца, делают серийные разведения раствора дозы в буфере и в плазме и используют их для построения стандартной кривой, связывающей значения площади пика с известной концентрацией пептида. Указанную кривую затем используют для вычисления концентрации пептида в плазме с учетом всех сделанных разведений и количества, введенного в колонку.
7.1.10. ХТТ протокол
Для измерения цитотоксического/цитостатического эффектов пептидов проводят ХТТ тест (Weislow O.S. et al.,1989, J. Natl. Cancer Inst. 81: 577-586) в присутствии варьирующих концентраций пептида для целей эффективного определения индекса селективности (ИС). TK50 определяют в указанном методе при инкубировании клеток в присутствии и в отсутствие серийных разведений пептида с последующим добавлением ХТТ. В выживших/метаболизирующих клетках ХТТ восстанавливается до растворимого коричневого красителя - ХТТ-формазана. Определяют величину поглощения и сравнивают значения, полученные в присутствии и в отсутствие пептида для определения величины TK50 по методу Карбера (см., например, Lennette E.H. et al., Eds., 1969, "Diagnostic Procedures for Viral and Rickettsial Infections", American Public Health Association, Inc., fourth ed., pp.47-52). Клетки Molt 4, СЕМ (80000 клеток/лунку) и сочетания двух типов клеток (70000 и 10000 соответственно) вносят на микротитрационный планшет и инкубируют с серийными разведениями пептида в течение 24 часов в общем объеме смеси 100 мкл. После инкубирования к каждой лунке добавляют 25 мкл рабочего стандартного раствора (1 мг/мл ХТТ, 250 мкМ ФМС в полной среде, содержащей 5% ДМСО) и планшеты инкубируют при 37°С. Измеряют развившееся окрашивание и результаты используют для выражения значений, полученных в лунках, содержащих пептиды, в виде процента от значений, полученных для лунок с необработанными контролями.
7.2. Результаты
7.2.1. Противовирусная активность - тесты на слияние
Проводят прямое сравнение Т1249 с Т20 в тесте на слияние по типу клетка-клетка, опосредованное вирусом, с использованием хронически инфицируемых клеток СЕМ, смешанных с неинфицированными клетками Molt-4, как показано ниже в Таблице 3. Сравнение уровня ингибирования слияния Т1249 в присутствии лабораторных изолятов, таких как IIIb, MN и RF, с аналогичными результатами для Т20 указывает на примерно 2,5-5-кратное превосходство первого относительно Т20. Т1249 был также более активным (превышение в 3-28 раз), чем Т20, в отношении нескольких клинических изолятов, индуцирующих синцитии, включая AZT-устойчивый изолят (G691-2), изолят, полученный до обработки AZT (G762-3) и 9320 (изолят, использованный в исследованиях huPBMC-SCID). Наиболее примечательно то, что Т1249 был в 800 раз активнее, чем Т20, против ВИЧ-2 NIHZ.
7.2.2. Противовирусная активность - тесты на инфекционность для Magi-CCR-5
Тесты на инфекционность для Magi-CCR-5 позволяют проводить прямое сравнение вирусных изолятов, индуцирующих и неиндуцирующих синцитии, а также сравнение лабораторных и клинических изолятов. Указанный тест представляет также непосредственный показатель вирусного инфицирования (экспрессия ТАТ после инфицирования, трансактивация образования бета-галактозидазы под действием повторяющихся последовательностей LTR), в отличие от обычно применяемых опосредованных измерений инфекционности, таких как продукция антигена р24 или обратной транскриптазы. Тесты на инфекционность с использованием Magi-CCR-5 (см. Таблицу 4, ниже) показал, что Т1249 постоянно более эффективен, чем Т20, в отношении всех испытанных изолятов, как с точки зрения значения ЭК50, так и ингибирования величины Vn/Vo=0,1. Т1249 демонстрирует значительное повышение активности против клинического изолята ВИЧ-1 301714 (>25 раз), который является одним из наиболее чувствительных изолятов по отношению к Т20. Т1249 по меньшей мере в 100 раз более активен, чем Т20, против изолята ВИО В670. Указанные результаты вместе с данными по слиянию клеток позволяют предположить, что Т1249 является мощным пептидным ингибитором ВИЧ-1, ВИЧ-2 и ВИО.

7.2.3. Противовирусная активность - тесты на инфекционность для huPBMC
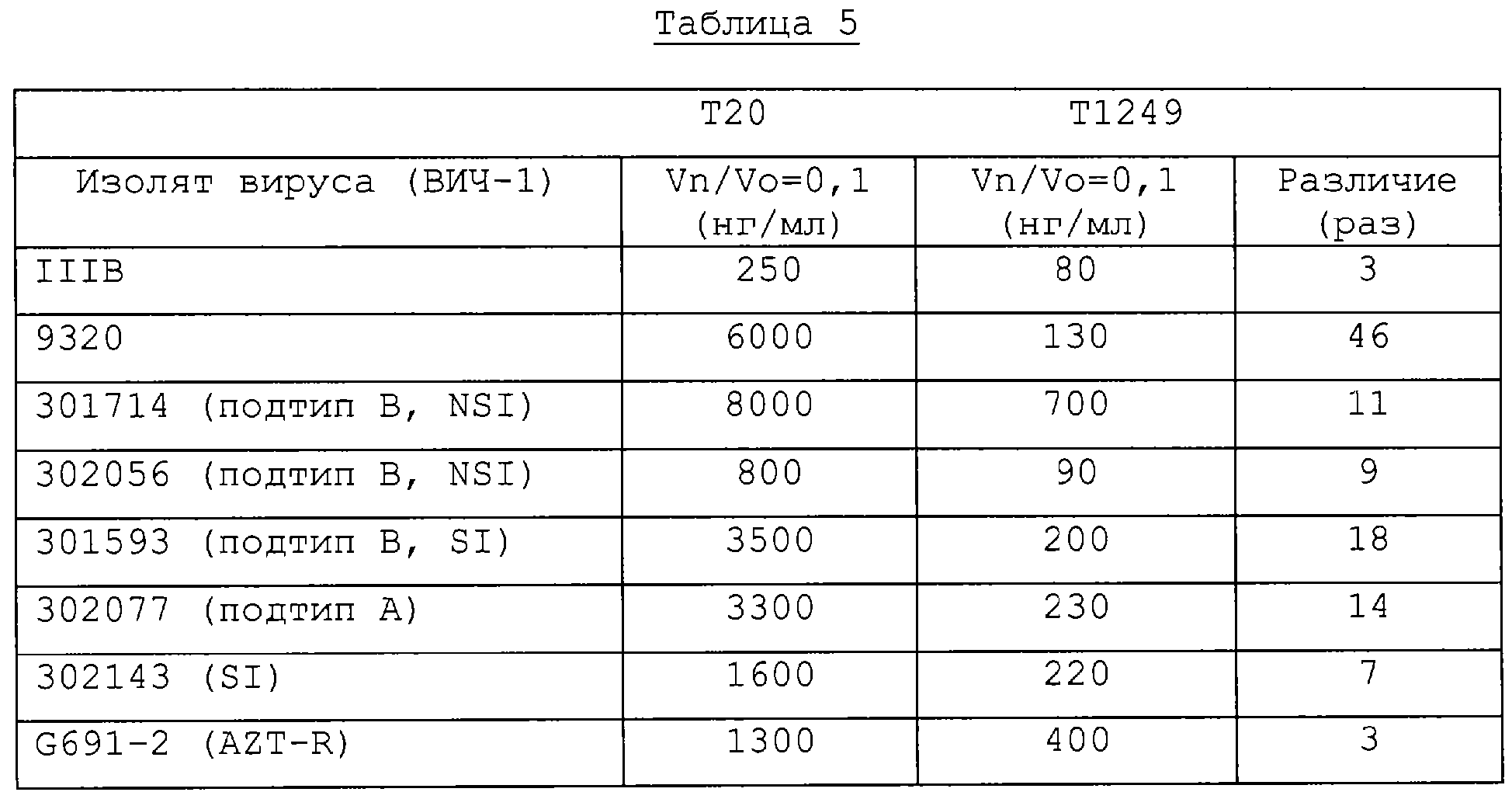
7.2.4. Противовирусная активность - лабораторные изоляты, резистентные к Т20
Проводят непосредственное сравнение Т1249 с Т20 в тестах на слияние по типу клетка-клетка, опосредованное вирусом, с использованием хронически инфицируемых клеток СЕМ, смешанных с неинфицированными клетками Molt-4 (Таблица 6, ниже). Т1249 был почти в 200 раз более активен, чем Т20, в отношении Т20-устойчивого изолята.
В тестах с клетками Magi-CCR-5 (см. Таблицу 7, ниже) Т1249 был в 50000 раз более активным, чем Т20, в отношении Т20-резистентных изолятов, таких как pNL4-3 SM и pNL4-3 STM (Rimsky L. and Matthews Т., 1998, J. Virol. 72: 986-993).
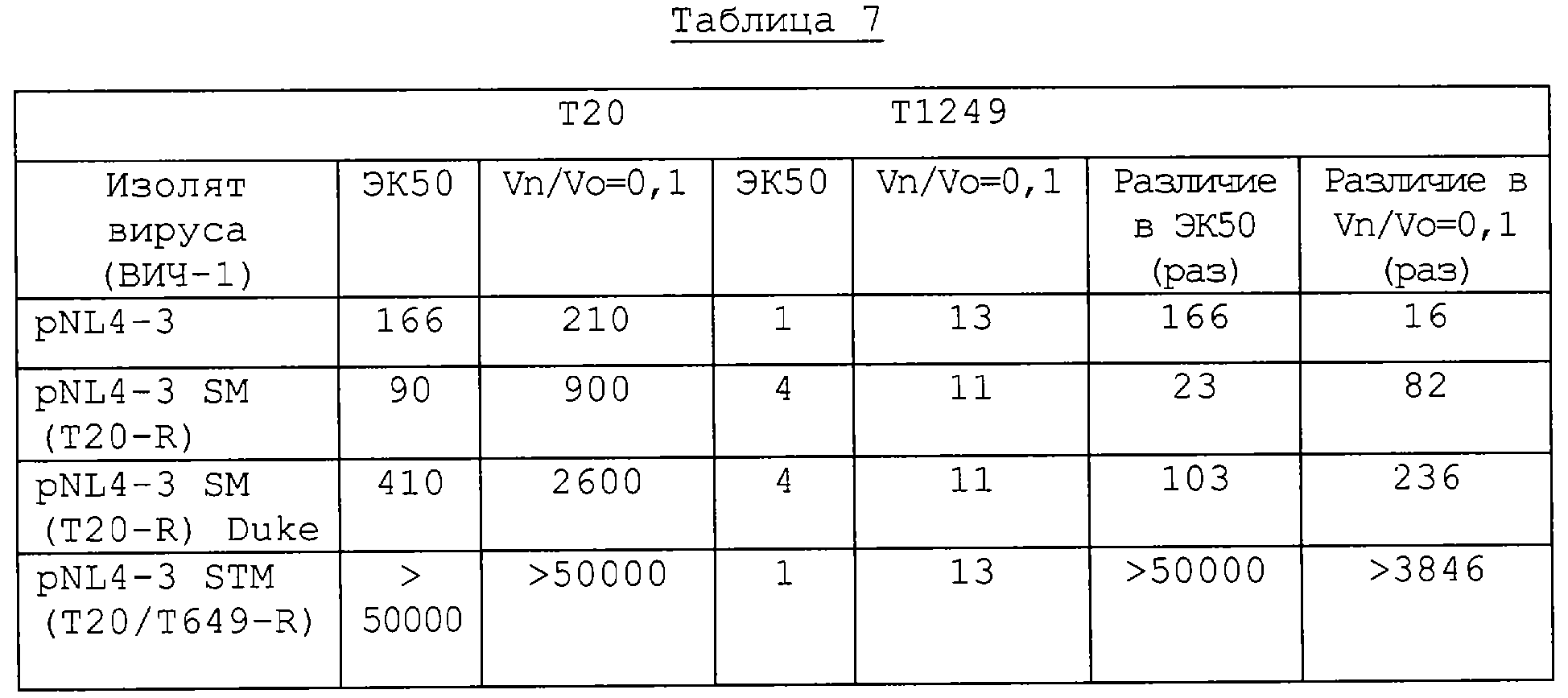
Проводят непосредственное сравнение Т1249 с Т20 в тестах на инфицирование huPBMC (см. Таблицу 8, ниже) и оценивают различия в активности против резистентных изолятов. Т1249 более чем в 250 раз, превосходит Т20 по активности против резистентного изолята pNL4-3 SM.

7.2.5. Противовирусная активность - модель in vivo SCID-huPBMC
Противовирусную активность Т1249 in vivo сравнивают непосредственно с активностью Т20 в модели инфицирования ВИЧ-1 9320 с использованием мышей с huPBMC-SCID (фиг.3). Через две недели после восстановления huPBMC мышей инфицируют 103 TCID50ВИЧ-1 9320 после пассажей в РВМС (AZT-чувствительный изолят А018). Лечение пептидами проводят внутрибрюшинной инъекцией два раза в день ежедневными общими дозами 67 мг/кг (Т20), 20 мг/кг (Т1249), 6,7 мг/кг (Т1249), 2,0 мг/кг (Т1249) и 0,67 мг/кг (Т1249) в течение 8 дней, начиная с -1 дня. Распространение инфекции в клетках крови, спленоцитах, лимфатических узлах и перитонеальных клетках оценивают при совместном культивировании с бластными монокоровыми клетками периферической крови человека еженедельно в течение трех последовательных недель с последующими обескровливанием животных и отбором тканей (день 7, примерно через 12-18 часов после последней инъекции лекарства). Супернатанты от культуры при совместном культивировании оценивают с точки зрения продукции р24 антигена ВИЧ-1 в качестве показателя развития вирусной инфекции. Инфекционный вирус не обнаруживался в крови или лимфатических тканях у животных, которым вводили Т20, хотя вирус обнаруживался в перитонеальных смывах и препарате селезенки. Все сегменты были негативными на наличие инфекционного вируса при введении дозы 6,7 мг/кг Т1249, указывая по меньшей мере на 10-кратное усиление активности в сравнении с результатами лечения Т20. При введении дозы 2,0 мг/кг Т1249 и лимфа, и селезенка были полностью свободны от обнаруживаемого инфекционного вируса с показателем снижения титра вируса в перитонеальном смыве 2 log10 и показателем снижения титра вируса в крови 1 log10 в сравнении с инфицированными контролями. При наименьшей дозе Т1249 - 0,67 мг/кг результаты анализа перитонеальных смывов и крови были эквивалентны таковым инфицированного контроля, однако по меньшей мере 1 log10 снижение титра инфекционного вируса наблюдается в лимфе и в тканях селезенки. В целом, результаты показывают, что Т1249 от 30 до 100 раз более активен в отношении ВИЧ-1 9320 in vivo при указанных условиях.
7.2.6. Фармакокинетические исследования на крысах
Для дальнейшего изучения фармакокинетического профиля Т1249 используют крыс с канюлями. Самцам крыс CD весом 250-300 г вводят Т1249 и Т20 внутривенно через катетер в яремной вене (фиг.4А-5). Полученные образцы плазмы оценивают с помощью флуоресцентной ВЭЖХ для определения количеств пептида в экстрагированной плазме. Бета-фаза периода полужизни и общее значение AUC у Т1249 были почти в три раза выше, чем у Т20 (фиг.5).
7.2.7. Цитотоксичность
Как видно из фиг.6, не обнаружено явных признаков цитотоксичности Т1249 in vitro.
Кроме того, Т1249 не имеет острой токсичности (смертность в течение 24 часов) в дозе 167 мг/кг (наивысшая испытанная доза) при внутривенном введении через канюлю в яремной вене (0,3 мл в течение 2-3 минут).
7.2.8. Непосредственное связывание с конструкцией М41Δ178 из gр41
Т1249 подвергают радиоактивному мечению с помощью125I и далее проводят очистку методом ВЭЖХ до достижения максимальной удельной активности. Таким же способом иодируют Т20. Проводят насыщающее связывание М41Δ178 (усеченный слитый белок слияния из эктодомена gp41 без аминокислотной последовательности Т20), иммобилизованного на микротитрационных планшетах в количестве 0,5 мг/мкл, как показано на фиг.7. Неспецифическое связывание определяют как связывание радиолиганда в присутствии 1 мкМ немеченного пептида. Специфическое связывание представляет разницу между общим и неспецифическим связыванием. Полученные результаты показывают, что125I-T1249 и125I-T20 обладают сходными аффинностями по связыванию при 1-2 нМ. Анализ обратно-пропорциональной функции по графику Скатчарда (Scatchard plots) позволяет полагать, что каждый лиганд связывается с гомогенным классом сайтов.
Кинетику связывания125I-Т1249 и125I-T20 определяют на сцинтилляционных планшетах для микротитрации, покрытых 0,5 мкг/мл М41D178. Период, в течение которого осуществляются ассоциация и диссоциация, показан на фиг.8. Диссоциацию связанного радиолиганда определяют после добавления немеченного пептида в конечной концентрации 10 мкМ в одну десятую часть общего объема, используемого в тесте. Исходные и достигаемые по прошествии времени скорости для125I-T1249 значительно ниже, чем для125I-T20. Параметры диссоциации обоих лигандов не меняются, если диссоциацию инициируют другим немеченным пептидом (т.е.125I-T1249 с Т20).
Для дальнейшей демонстрации того факта, что оба лиганда конкурируют за один и тот же целевой сайт, титруют немеченные Т1249 и Т20 в присутствии единичной концентрации125I-T1249 или125I-T20. Лиганд добавляют сразу же после начала инкубации немеченного пептида. Кривые конкурентного ингбирования, приведенные на фиг.9, дают основания полагать, что хотя оба лиганда имеют сходные аффинности, требуется более высокая концентрация немеченного Т1249 или Т20 для полного замещения125I-T1249 конкурентном связывании.
7.2.9. Непосредственное связывание с HR1 участком gр41
Используют метод спектроскопии кругового дихроизма (КД) для определения вторичной структуры одного Т1249 в растворе (фосфатно-буферный раствор, рН 7) и в сочетании с пептидом, содержащим 45 остатков (Т1346), из HR1 (гептадный повтор 1) связывающего участка gр41. На фиг.14А показан спектр КД для одного Т1249 в растворе (10 мкМ, 1°С). Спектр типичен для пептидов, имеющих альфа-спиральную структуру. В частности, развертка указанного спектра с помощью одного показателя разложения, проанализированного для набора спектров 33 белков, указывает на предположительное содержание спирали в структуре Т1249 (одного в растворе) до 50%. На фиг.14В показан спектр Т1249 в смеси с Т1346. Зачерненные квадраты (■) означают теоретический вариант спектра, предсказанный для случая «модели невзаимодействия», в рамках которой допускается, что пептиды в растворе не взаимодействуют. Реальный спектр, получаемый в эксперименте (•), заметно отличается от указанного теоретического варианта спектра в «модели невзаимодействия», указывая на то, что два пептида действительно взаимодействуют, в результате чего возникают выявляемые при измерении структурные изменения, которые наблюдаются на спектре КД.
7.2.10. Защита от протеазы связывающего участка Т1249 в gр41
Чувствительность химерного белка М41Δ178, описанного ранее в разделе 7.2.8, к расщеплению протеиназой К, определяют и анализируют с помощью электрофореза в полиакриламидном геле. Результаты представлены на фиг.15.
В случае отдельного инкубирования либо М41Δ178 (необработанный; фиг.15, дорожка 2), либо Т1249 (необработанный; фиг.15, дорожка 4) с протеиназой К (фиг.15, дорожки 3 и 5 соответственно) оба расщепляются. Однако в том случае, когда Т1249 инкубируют с М41Δ178 перед добавлением протеиназы К (фиг.15, дорожка 7) выявляется защищенный HR-1 фрагмент размером примерно 6500 дальтон. Результаты секвенирования защищенного фрагмента показывают, что он соответствует участку первичной последовательности, расположенному внутри эктодомена gр41. Защищенный фрагмент заключает в себе растворимый HR1 пептид (Т1346), использованный выше в разделе 7.2.9 при изучении спектра КД, а также содержит семь аминокислотных остатков, расположенных на амино конце. Указанная защита может быть определена связыванием Т1249 со специфической последовательностью gр41, которая содержится в конструкции М41Δ178.
8. Пример: гибридные полипептиды респираторно-синцитиального вируса
В указанном ниже примере описываются гибридные полипептиды респираторно-синцитиального вируса (РСВ) с усиленными фармакокинетическими свойствами. Кроме того, представлены данные, показывающие, что гибридные полипептиды РСВ являются мощными ингибиторами РСВ инфекции.
8.1. Материалы и методы
8.1.1. Синтез и очистка пептидов
РСВ пептиды синтезируют с использованием стандартных методов Fast Мос химии. В основном, если особо не оговорено иное, рассматриваемые пептиды содержат амидированные карбоксильные концы и ацетилированные амино концы. Очистку проводят с использованием ВЭЖХ с обращенной фазой.
8.1.2. Тест на респираторно-синцитиальный вирус по снижению бляшкообразования
Все необходимые разведения пептидов делают на чистом стерильном 96-луночном ТС планшете. Готовят в общей сложности одиннадцать разведений для каждого пептида и оставляют одну контрольную лунку без пептида. Диапазон конечных концентраций пептида начинается с 50 мкг/мл или со 100 мкг/мл, всего одиннадцать разведений в двух повторах. РСВ получают в концентрации 100 КОЕ/лунку в 100 мл 3% буфера ЕМЕМ, которая была определена на основе известного титра РСВ. Затем указанный вирус добавляют ко всем лункам.
Среды отбирают из лунок 96-луночного планшета с клетками Нер2 накануне стадии слияния. Материал из планшета с разведениями переносят на планшеты для клеточных культур, начиная с ряда 1 и затем переносят в ряд 12, 11 и т.д. до тех пор, пока не будет сделан перенос во все ряды. Планшеты снова возвращают в инкубатор на 48 часов.
Клетки проверяют для подтверждения наличия синцитий в контрольных ячейках. Отбирают среды и в каждую лунку добавляют примерно 50 мкл 0,25% кристаллического фиолетового красителя в метаноле. Лунки сразу же промывают водой для удаления избытка красителя и оставляют высыхать. С помощью препаровальной лупы подсчитывают число синцитий в каждой лунке.
8.2. Результаты
Фармакокинетические исследования, как видно из фиг.10А-10В, с гибридными пептидами из РСВ - Т1301 (Ас-WQEWDEYDASISQVNEKINQALAYIREADELWA WF-NH2) и Т1302 (Ас-WQAWDEYDASISQVNEKINQALAYIREADELW AWF-NH2), содержащими энхансерные пептидные последовательности, продемонстрировали в указанных случаях значительно увеличенный период полужизни в сравнении с коровым пептидом Т786 (Ас-VYPSDEYDASISQVNEEINQALAYIRKADELLENV-NH2). Для гибридных полипептидов Т1301, Т1302 и Т1303 (Ас-WQAWDEYDASISDVNEKINQALAYIREADELWEWF-NH2) также показан существенно увеличенный период полужизни в сравнении с коровым полипептидом Т1476 (Ac-DEYDASISQVNEKINQALAYIREADEL-NH2).
Гибридные полипептиды Т1301, Т1302 и Т1303, а также полипептиды Т786 и Т1293 анализируют на их способность ингибировать бляшкообразование в Нер2 клетках под действием РСВ. Как показано на фиг.11А и 11В, оба исследованных гибридных полипептида РСВ, а также коровый полипептид Т786 способны ингибировать инфицирования РСВ. Неожиданно было обнаружено, что гибридный полипептид Т1293 также является мощным анти-РСВ соединением (фиг.13).
9. Пример: гибридные полипептиды лютеинизирующего гормона
В приведенном ниже примере описываются гибридные белки из лютеинизирующего гормона (ЛГ) с усиленными фармако-кинетическими свойствами. Указанные гибридные пептиды ЛГ были синтезированы и очищены с помощью описанных выше методов: коровый пептид Т1323 (Ac-QHWSYGLRPG-NH2) и гибридный полипептид Т1324 (Ac-WQEWEQKIQHWSYGLRPGWASLWEWF-NH2), который включает аминокислотную последовательность корового полипептида Т1323, объединяют с энхансерными полипептидами по амино- и карбокси-концам. Как показано на фиг.12А и 12В, гибридный пептид Т1324 имеет значительно увеличенный период полужизни в сравнении с коровым пептидом Т1323, который не содержит энхансерных пептидных последовательностей.
10. Пример: фармакология гибридного полипептида Т1249
Т1249, изображенный на фиг.13, представляет собой гибридный полипептид, включающий энхансерные пептидные последовательности, соединенные с коровым полипептидом, полученным из смеси вирусных последовательностей. Как было показано выше, в примере раздела 7, гибридный полипептид Т1249 проявляет усиленные фармакокинетические свойства и активность in vitro и in vivo в отношении ВИЧ-1. В приведенном ниже примере описаны фармакологические свойства Т1249 на моделях как грызунов, так и приматов.
10.1. Материалы и методы
10.1.1. Введение однократной дозы грызунам
Т1249 вводят альбиносным крысам Спрэг-Доли (Sprague-Dawley) в виде однократной дозы путем постоянной подкожной инфузии (ПКИ), подкожной инъекции (п/к) или внутривенной инъекции (в/в). Каждая экспериментальная группа включает по девять крыс каждого пола на группу. Группам животных вводят стерильные препараты лекарственного вещества в массе Т1249 в дозе 0,5, 2,0 или 6,5 мг/кг путем ПКИ. Одна группа, служащая в качестве контроля, получает 50 мМ карбонат-бикарбоната, рН 8,5. Пептиды вводят в течение 12 часов через поливинил-хлоридный/полиэтиленовый катетер, имплантированный подкожно хирургическим путем в задней части шеи. Две группы получают в виде подкожной инъекции в интракапсулярный участок однократную дозу Т1249, равную 1,2 или 1,5 мг/кг. Две другие группы получают в виде внутривенной инъекции однократную дозу Т1249, равную 1,5 или 5 мг/кг. Фактическое количество Т1249 в миллиграммах вычисляют на основе содержания пептида, определяемого для каждой вводимой партии препарата.
Параметры, существенные для анализа, включают наблюдение животных в клетке (два раза в день учет случаев смертности и заболеваний), клинический осмотр, проведение лабораторных анализов, определение веса тела и проведение некропсии. Образцы крови получают с помощью метода случайного отбора образцов в течение 12-часового периода от трех крыс каждого пола и из каждой группы в каждой из следующих временных точек: 0,5, 1, 2, 4, 6, 8, 10 и 12 часов после введения дозы. Проводят анализ образцов с использованием PcAb ECLIA теста (Blackburn, G. et al., 1991, Clin. Chem. 37: 1534-1539; Deaver, D., 1995, Nature 377: 758).
Для проведения фармакокинетического анализа Т1249 в крови и лимфатической системы крыс Т1249 готовят в виде стерильного раствора в бикарбонатном буфере и вводят его в виде однократной дозы внутривенной инъекцией болюсом в латеральную хвостовую вену в дозе 20 мг/кг. Кровь отбирают с помощью постоянного катетера, помещенного в яремную вену животного. Образцы собирают сразу же после введения дозы и через 5, 15 и 30 минут, а также через 1, 2, 4 и 6 часов после введения лекарства. Для анализа лимфатических жидкостей образцы отбирают непосредственно перед введением дозы и каждые 20 минут в течение шести часов после введения дозы. Лимфатическую жидкость собирают с помощью катетера, помещенного непосредственно в грудной лимфатический проток, как описано ранее (Kirkpatrick and Silver, 1970, The Journal of Surgical Research 10: 147-158). Концентрации Т1249 в плазме и лимфатической жидкости определяют с помощью стандартного Т1249 по конкурентному ИФТФА методу (Hamilton, G. 1991, р.139 in "Immunochemistry of Solid-Phase Immunoassay", Butler, J., ed., CRC Press, Boston).
10.1.2. Введение однократной дозы приматам
Стерильные препараты нерасфасованного лекарственного вещества Т1249 вводят обезьянам cynomolgus в виде однократных доз путем подкожной (п/к), внутримышечной (в/м) или внутривенной (в/в) инъекции. В последовательном перекрестном опыте одной группе животных, включающей по два животных каждого пола, вводят однократную дозу Т1249 в виде болюса в/в (0,8 мг/кг), в/м (0,8 мг/кг) или п/к (0,4, 0,8 и 1,6 мг/кг) инъекцией. Между дозами делают перерыв для очищения длительностью по меньшей мере три дня. Лиофилизованный Т1249 восстанавливают в стерильном фосфатно-буферном растворе при рН 7,4 непосредственно перед введением дозы. Фактическое количество исследуемого продукта в миллиграммах вычисляют на основании содержания пептида, известного для вводимой партии.
Параметры, существенные для анализа, включают наблюдение животных в клетке, физический осмотр и учет веса тела. В ходе в/в фазы исследования проводят отбор образцов крови в пробирки с гепарином в следующие временные точки: сразу после введения дозы и через 0,25, 0,5, 1,5, 3, 6, 12 и 24 часа после введения дозы. В ходе в/м и п/к фаз исследования проводят отбор образцов крови от каждого животного в пробирки с гепарином в следующие временные точки: через 0,5, 1, 2, 3, 6, 12 и 24 часа после введения дозы. Образцы плазмы готовят в течение одного часа в процессе отбора и быстро замораживают в жидком азоте. Анализ образцов проводят с использованием PcAb ECLIA теста (Blackburn G. et al., 1991, Clin. Chem. 37: 1534-1539; Deaver, D., 1995, Nature 377: 758).
10.1.3. Объединяющие фармакокинетические исследования
Шесть самцов обезьян cynomolgus распределяют случайным образом по трем группам, включающим по два животных на группу. Все дозы Т1249 вводят в виде подкожной инъекции болюсом. Исследование разбивают на два сеанса. В сеансе 1 животным в группах 1, 2 и 3 вводят стерильный препарат нерасфасованного лекарственного вещества Т1249 (например, нерасфасованный Т1249, растворенный в карбонат-бикарбонатном буфере, рН 8,5) два раза в день в течение четырех дней подряд (дни исследования 1-4) в дозах 0,2, 0,6 и 2,0 мг/кг/дозу соответственно. Далее следует десятидневный период очищения организма, разделяющий сеанс 1 и сеанс 2. В ходе сеанса 2 животным в группах 1, 2 и 3 вводят стерильный препарат лекарственного продукта Т1249 (т.е. водный раствор рН 6,5 плюс маннит) два раза в день в течение четырех дней подряд (дни исследования 15-18) в дозах 0,2, 0,6 и 2,0 мг/кг/дозу соответственно.
Образцы крови для фармакокинетического анализа отбирают в 1 и 15 дни исследования для оценки фармакокинетических параметров эффекта однократной дозы и в 4 и 18 дни исследования для оценки фармакокинетических параметров плазмы крови в стационарном состоянии. Образцы отбирают в следующие временные точки: сразу после введения дозы и через 0,5, 1,5, 3, 6, 8 и 12 часов после введения дозы. За животными наблюдают в ходе 1 и 2 сеансов с целью выявления клинически значимых признаков и изменений в весе тела.
10.2. Результаты
10.2.1. Фармакокинетика Т1249 при введении крысам
Для проведения первичной оценки фармакокинетических параметров плазмы крови и характера распределения в организме Т1249 используют модель крыс. Во всех группах дозирования при введении Т1249 не отмечается изменений в весе тела животных, отклонений, выявляемых при физическом осмотре, в гематологических показателях, в результатах клинического химического анализа и при макроскопическом обследовании.
Крысы, получавшие Т1249 при ПКИ, достигают стационарного состояния по концентрациям пептида в плазме примерно через четыре часа после введения. И концентрация в плазме, характерная для стационарного состояния (Кпсс), и вычисленное значение площади пика на графике зависимости концентрации в плазме от времени (AUC) прямо пропорциональны введенной дозе, указывая на то, что Т1249 подчиняется линейной фармакокинетике в исследуемом диапазоне доз от 0,5 до 6,5 мг/кг. Кривые зависимости вычисленных фармакокинетических параметров и значений концентраций в плазме от времени при введении способом ПКИ представлены в Таблице 9 и на фиг.16А соответственно.
В случае введения Т1249 в виде болюса в/в инъекцией имеет место линейный характер зависимости фармакокинетики от дозы в диапазоне исследованных доз. В отличие от него при введении Т1249 п/к инъекцией не наблюдается зависимости от дозы в изученном диапазоне доз. Кривые зависимости вычисленных фармакокинетических параметров и значений концентраций в плазме от времени при п/к и в/в введении представлены в Таблице 10 и на фиг.16В соответственно.
Биодоступность Т1249, вводимого крысам подкожно, определяют в сравнении с ее величиной при в/в введении. Результаты приведены ниже, в Таблице 11. В низкой дозе (1,2 мг/кг) Т1249 демонстрирует при подкожном введении относительную биодоступность (FR), равную 73%. Относительная биодоступность составляет 30% в том случае, когда при высокой дозе (15 мг/кг) введения Т1249 достигается концентрация, большая той концентрации, которая ингибирует на 90% (ИК90) инфекционность ВИЧ-1 в течение всех 12 часов испытания всех изученных доз.
Кинетические данные для концентраций Т1249 в плазме крови и в лимфе приведены на фиг.16С и далее отражены в Таблице 12. Т1249 быстро проникает в лимфатическую систему и уравновешивается с плазменным уровнем лекарственного вещества в течение примерно одного часа после введения. После достижения равновесия между двумя системами уровни лекарственного вещества в плазме и в лимфе становятся сравнимыми за период до трех часов после введения дозы у четырех из пяти животных. Одно животное имеет постоянно более низкие концентрации Т1249 в лимфе, чем другие животные, однако профиль выведения из лимфы у указанного животного не отличается от данного показателя у других животных в той же группе. Сравнение полужизни фазы выведения (t1/2) из плазмы и лимфы позволяет предположить, что перемещение Т1249 между указанными двумя системами управляется процессом диффузии. Через три часа развивается вторая, более быстрая, фаза выведения из лимфатической системы. Указанное различие может быть определено механизмом действия (например, в связи с перераспределением или ускоренного разрушения пептида в лимфе) или другими факторами. Концентрация Т1249 в лимфатической жидкости через шесть часов после инъекции выше, чем значение ИК90 для поддержания вирусной инфекционности обычных лабораторных штаммов и первичных клинических изолятов ВИЧ-1.
Оценивается также уровень проникновения Т1249 в спинномозговую жидкость (СМЖ). Концентрации Т1249 были ниже предела обнаружения (LOD: 2,0 мкг Т1249/мл СМЖ) во всех измеряемых временных точках, указывая на то, что Т1249 не проникает в центральную нервную систему после введения однократной дозы.
10.2.2. Фармакокинетика Т1249 при введении приматам
Модели приматов используют для оценки отношений уровня дозы и различных фармакокинетических параметров при парентеральном введении Т1249. Концентрации Т1249 в плазме, более высокие, чем 6,0 мкг/мл, достигаются при всех путях введения, и количественно определяемые уровни (т.е. уровни, большие 0,5 мкг/мл) обнаруживаются к 24 часам после п/к и в/в введения. Значения полужизни t1/2 выведения сравнимы при всех путях введения (5,4 часа, 4,8 часа и 5,6 часов - для в/в, п/к и в/м введения соответственно). Во всех временных точках в течение 24-часового периода отбора образцов наблюдаются плазменные концентрации Т1249, превышающие значения ИК90 для лабораторных штаммов и клинических изолятов ВИЧ-1.
Сравнение данных, полученных в случае парентерального введения 0,8 мг/кг Т1249 с использованием всех путей введения (п/к, в/в и в/м) представлено на фиг.17А. На фиг.17В показано сравнение данных, полученных при п/к инъекции трех различных доз Т1249 (0,4 мг/кг, 0,8 мг/кг и 1,6 мг/кг). На вставке, приведенной на фиг.17В, показан график зависимости вычисленной концентрации AUC от введенной дозы.
Т1249 демонстрирует линейную фармакокинетику в организме обезьян cynomolgus при п/к введении в диапазоне исследованных доз, что свидетельствует об отсутствии в указанном диапазоне насыщения механизма или механизмов клиренса. Краткое обобщение фармакокинетических данных при парентеральном введении Т1249 обезьянам cynomolgus дано ниже в Таблице 13. Сравнение плазменных значений AUC показывает, что, относительно внутривенного режима введения, биодоступность Т1249 составляет примерно 64% при введении внутримышечной инъекцией и 92% при введении подкожной инъекцией.
10.2.3. Объединяющие фармакокинетические исследования
Объединяющие фармакокинетические исследования проводят с целью сравнения фармакокинетических профилей в плазме нерасфасованных лекарственных веществ Т1249, используемых в описанных выше неклинических испытаниях, с лекарственным продуктом Т1249, который вводится определенному субъекту или пациенту, например, для лечения ВИЧ инфекции. Исследование проводят в параллельных группах, в виде однонаправленного перекрестного сравнения трех значений нерасфасованных доз лекарственного вещества Т1249 и трех значений доз приготовленного лекарственного продукта. Фармакокинетику в плазме оценивают после введения однократной дозы и после достижения стационарного состояния.
После введения Т1249 подкожной инъекцией во всех группах дозирования отмечается наличие выявляемых при измерении уровней пептида. Кривые зависимости концентрации от времени в плазме были в общем параллельны внутри всех групп дозирования после приема исходной дозы (дни 1 и 15) и в стационарном состоянии (дни 4 и 18) как для нерасфасованного лекарственного вещества Т1249, так и для лекарственного продукта Т1249. Кроме того, значения AUC(0-12 час) менялись прямо пропорционально в зависимости от уровня дозы каждой формы лекарственного средства. Вычисленные значения AUC(0-12 час) для лекарственного продукта варьируют от 43% до 80% от значения AUC(0-12 час), вычисленной для лекарственного вещества после введения однократной дозы, и от 36% до 71% в стационарном состоянии.
Т1249 как в виде нерасфасованного лекарственного вещества, так и в виде лекарственного продукта демонстрирует сходные фармакокинетические профили в обезьянах cynomolgus после подкожного введения болюсом в дозе исследованного уровня и объема. Результаты непосредственного сравнения формы кривых зависимости концентрации от времени в плазме в настоящем исследовании и форм кривых в предыдущем исследовании на обезьянах cynomolgus позволяют предположить наличие депо-эффекта в случае введения Т1249 подкожной инъекцией. Данное предположение вытекает из факта повышения времени, в ходе которого достигается максимальная концентрация в плазме (tmax) и значения t1/2.
Полученные результаты указывают на то, что форма нерасфасованного лекарственного вещества, используемая в фармакологической программе, дает сравнимые величины AUC и других фармакокинетических параметров с теми, что наблюдаются после введения готового лекарственного продукта. Приведенные наблюдения показывают, что введение Т1249 по клиническим показаниям приводит к достижению всеобъемлющего воздействия Т1249 на весь организм пациента.
11. Пример: выделение новых коровых полипептидов с противовирусной активностью в отношении изолята ВИЧ-1, резистентного к Т649
В одном конкретном, но не ограничивающем примере настоящего описания рассматривается получение модифицированного корового полипептида, который проявляет противовирусную активность в отношении ВИЧ штаммов, резистентных к немодифицированному, «родительскому» коровому пептиду.
Пептид Т649, указанный в Таблице 2, представляет собой пептид, полученный из участка gр41 белка ВИЧ-1, известного как HR2. В исследованиях вариантов ВИЧ-1, резистентных к Т649, при выделении и секвенировании нуклеиновой кислоты, кодирующей HR2 участок резистентных вариантов gр41 белка, выявлена мутация, которая приводит к появлению единичной мутации внутри указанного участка: замене остатка аспарагина (N) на лизин (К).
На основании указанного результата был синтезирован новый полипептид, фигурирующий в настоящем описании как DP397, который содержит аминокислотную последовательность Т649, в которую введена указанная выше мутация N на К. Пептиды Т649 и DP397 показаны ниже, где видно, что между ними имеется различие по единственной аминокислоте, выделенной жирным шрифтом:
Т649: WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL
DP397: WMEWDREINKYTSLIHSLIEESQNQQEKNEQELL
Следует отметить, что различие между Т649 и DP397 попадает в потенциальный сайт N-гликозилирования (подчеркнут). Таким образом, мутация в gр41 у Т649-резистентных штаммов устраняет указанный потенциальный сайт N-гликозилирования.
Коровый полипептид DP397 проявляет противовирусную активность в отношении вариантов ВИЧ-1, которые резистентны к пептиду Т649. В частности, пептид DP397 проявляет заметно повышенную противовирусную активность в отношении четырех вариантов ВИЧ-1, по данным теста на инфекционность для Magi-CCR-5, описанного ранее в разделе 7.1.7. Кроме того, было также показано в ряде экспериментов, что пептид DP397 проявляет повышенную противовирусную активность в отношении указанных штаммов в сравнении с пептидом Т1249.
На фиг.18А-D показано количество инфицированных клеток при воздействии на них Т649-резистентных вариантов как функции концентрации пептида для Т649, DP397 и Т1249. Конкретно, на фиг.18А-В приведены данные экспериментов с использованием Т649-резистентных штаммов ВИЧ-1, названных RF-649 и DH012-649 соответственно. Указанные штаммы получают из изолятов ВИЧ-1RF и ВИЧ-1DHO12 соответственно, которые были пассированы на клеточных культурах в присутствии Т649 с целью продукции Т649-резистентных вариантов.
На фиг.18C-D приведены данные экспериментов с использованием полученных генно-инженерными методами Т649-резистентных штаммов ВИЧ-1, называемых 3'ETVQQQ и SIM-649 соответственно. Штамм 3'ETVQQQ получен из клона ВИЧ-1LAI, подвергнутого молекулярной мутации, в результате которой он содержит аминокислотную последовательность ETVQQQ вместо GIVQQQ в HR1 домене gр41 белка. HR1 представляет собой участок gр41 белка ВИЧ-1, с которым связываются HR2 домен и Т649 пептид. Штамм SIM-649 был получен из клона ВИЧ-1LAI, подвергнутого молекулярной мутации, в результате которой он содержит аминокислотную последовательность SIM вместо GIV в HR1 домене gр41 белка, и впоследствии пассированного на клеточных культурах в присутствии Т649 с целью продукции Т649-резистентных вариантов.
Пептид DP397 демонстрирует заметно усиленное ингибирование ВИЧ-1 инфекции в сравнении с Т649 для всех четырех исследованных штаммов. Кроме того, пептид DP397 демонстрирует усиленное ингибирование ВИЧ-1 инфекции в сравнении с Т1249 для RF-649 штамма (фиг.18А) и, при более высоких концентрациях, для DH012-649 штамма (фиг.18В).
Настоящее изобретение не ограничивается приведенными в описании конкретными вариантами его осуществления, которые даны лишь для иллюстрации отдельных аспектов изобретения, и функционально эквивалентные методы и компоненты входят в область настоящего изобретения. В действительности, для специалистов в данной области очевидны различные модификации, которые могут быть введены в изобретение на основании приведенного описания и сопровождающих его рисунков. Такие модификации также попадают в область изобретения и прилагаемой формулы изобретения.
Реферат
Изобретение относится к области медицины и касается гибридных полипептидов с усиленными фармакокинетическими свойствами. Сущность изобретения заключается в том, что гибридные полипептиды, включающие энхансерные пептидные последовательности, связанные с коровым полипептидом, обладают усиленными фармакокинетическими свойствами, такими как увеличенный период полужизни. Изобретение также относится к способам усиления фармакокинетических свойств любого корового полипептида посредством связывания энхансерных пептидных последовательностей с коровым полипептидом. Применяемые при осуществлении настоящего изобретения коровые полипептиды могут включать любой фармакологически полезный пептид, который может использоваться, например терапевтическое или профилактическое средство. Преимущество изобретения заключается в усилении фармакокинетических свойств. 6 н. и 46 з.п. ф-лы, 18 ил., 13 табл.
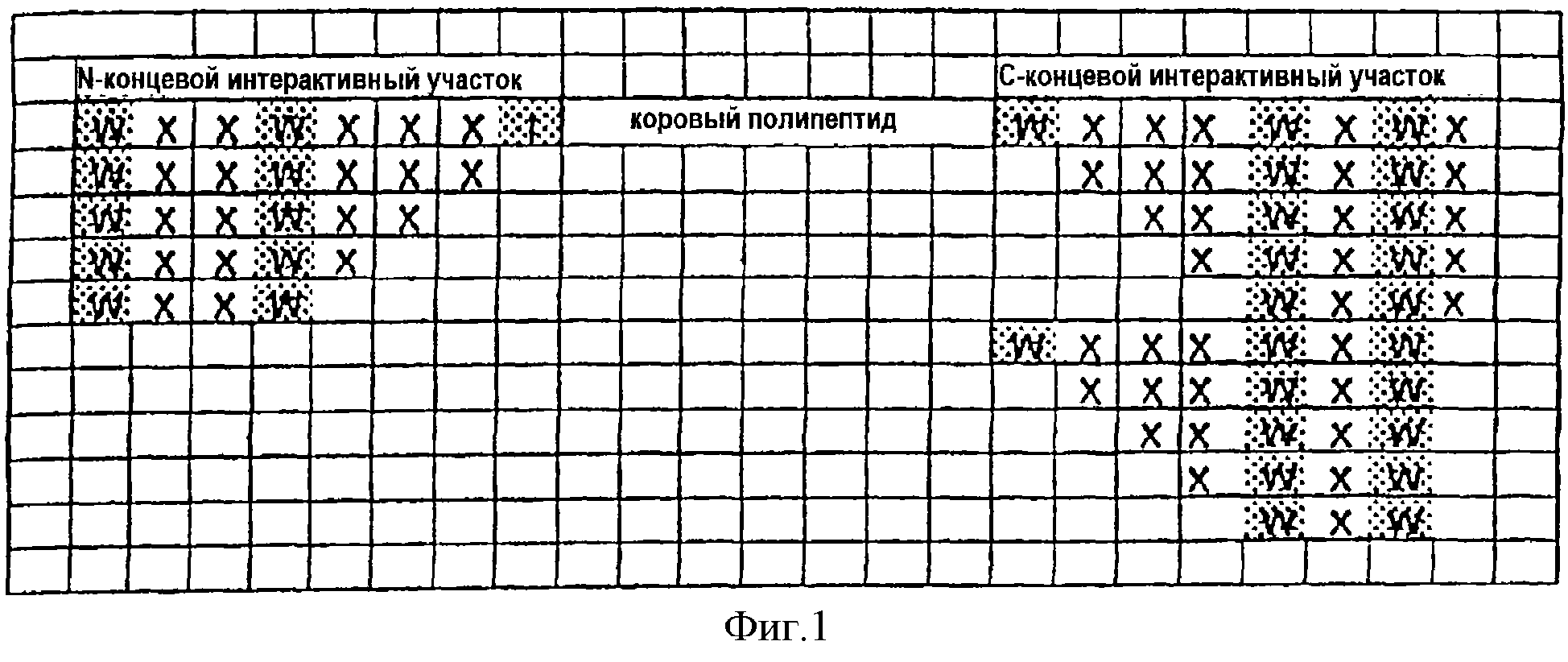
Комментарии