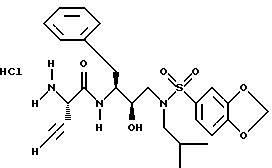
Гидроксиэтиламино сульфонамиды гетероциклокарбонил аминокислоты, ингибирующие ретровирусную протеазу - RU2174519C2
Код документа: RU2174519C2
Чертежи






































Описание
Изобретение относится к ингибиторам ретровирусных протеаз, в частности к новым соединениям, композиции и способу ингибирования ретровирусных протеаз, таких как протеаза вируса иммунодефицита человека (ВИЧ, HIV). Данное изобретение, в частности, относится к гидроксиэтиламин сульфонамидным соединениям гетероциклокарбонил аминокислот, ингибирующим протеазы, композиции и способу ингибирования ретровирусных протеаз, профилактическому предотвращению (профилактике) ретровирусной инфекции или распространения ретровируса и лечению ретровирусной инфекции, например ВИЧ инфекции. Целью изобретения также являются способы получения таких соединений, а также промежуточные соединения, используемые в таких способах.
Во время репликационного цикла ретровирусов продукты транскрипции gag и gag-pol гена транслируются как белки. Эти белки затем процессируются вирусно колированной протеазой (или протеиназой) с получением вирусных ферментов и структурных белков ядра вируса. Чаще всего, белки предшественники gag процессируются в ядро белка, и белки предшественники pol процессируются в вирусные ферменты, например обратную транскриптазу и ретровирусную протеазу. Было показано, что правильное процессирование белков предшественников ретровирусной протеазой необходимо для сборки инфекционных виронов. Например, показано, что мутации сдвига рамки в протеазной области pol гена ВИЧ (HIV) предотвращают процессинг белка предшественника gag. Также было показано, путем сайт-направленного мутагенеза остатка аспарагиновой кислоты в активном сайте ВИЧ протеазы, что процессинг белка предшественника gag предотвращается. Поэтому были сделаны попытки ингибировать вирусную репликацию ингибированием действия ретровирусных протеаз.
Ингибирование ретровирусной протеазы обычно включает переход-состояние миметика, посредством чего ретровирусная протеаза подвергается действию соединения-миметика, которое связывается (обычно обратимым образом) с ферментом, конкурируя с gag и gag-pol белками, тем самым ингибируя специфический процессинг структурных белков и высвобождение ретровирусной протеазы самой по себе. Таким образом, можно эффективно ингибировать репликацию ретровирусных протеаз.
Было предложено несколько классов соединений, в частности, для ингибирования протеаз, как например, для ингибирования ВИЧ протеазы. Такие соединения включают изостеры (isosteres) гидроксиэтиламина и восстановленные изостеры амида. Например, ЕР 0346847; ЕР 0342541; Roberts et al, "Rational Design of Peptide-Based Proteinase Inhibitors", Science, 248, 358 (1990); и Erickson et al, "Design Activity, и 2,8

Известно несколько классов соединений, которые полезны в качестве ингибиторов протеолитического фермента-ренина. Например, US N 4599198; UK 2184730; GB 2209752; EP 0264795; GB 2200115 и US SIR H725. Из них GB 2200115, GB 2209752, EP 0264795, US SIR H725 и US 4599198 раскрывают мочевиносодержащие гидроксиэтиламиновые ингибиторы ренина. В EP 468641 описаны ингибиторы ренина и промежуточные соединения для получения ингибиторов, которые включают сульфонамидсодержащие гидроксиэтиламиновые соединения, такие как 3-(т-бутоксикарбонил) амино-циклогексил-1-(фенилсульфонил)амино-2(5)-бутанол. В Пат. GB 2200115 описаны сульфамоилсодержащие гидроксиэтиламиновые ингибиторы ренина, и в EP 0264795 раскрыты некоторые сульфонамидсодержащие гидроксиэтиламиновые ингибиторы ренина. Однако, известно, что, хотя ренин и ВИЧ протеазы обе классифицируются как аспартил протеазы, соединения, которые эффективны в качестве ингибиторов ренина, обычно нельзя предсказать, будут ли они эффективны для ингибирования ВИЧ протеазы.
Данное изобретение относится к определенным соединениям, являющимся ингибиторами ретровирусных протеаз, их аналогам и их фармацевтически приемлемым солям, сложным эфирам и пролекарствам. Предлагаемые соединения характеризуются как соединения-ингибиторы, представляющие собой гидроксиэтиламин сульфонамиды гетероциклокарбонил аминокислот. Соединения изобретения ингибируют преимущественно ретровирусные протеазы, такие как протеаза вируса иммунодефицита человека (ВИЧ). Поэтому данное изобретение также включает фармацевтические композиции, способы ингибирования ретровирусных протеаз и способы лечения или профилактики ретровирусной инфекции, такой как ВИЧ инфекции. Объектом изобретения также являются способы получения таких соединений, а также промежуточные продукты, используемые в таких способах.
В данном изобретении предложено соединение, ингибирующее ретровирусную протеазу, формулы:

или его фармацевтически приемлемая соль, пролекарство или сложный эфир, где n = 0 или 1;
R1 представляет алкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, цианоалкил, имидазолилметил, -CH2CONH2, -CH2CH2CONH2, -CH2S(O)2NH2, - CH2 SCH2, -CH2S(O)CH3, -CH2S(O)2CH3, -C(CH3)2SCH3, -C(CH3)2S(O)CH3 или -C(CH3)2S(O)2CH3 группы; предпочтительно R1 представляет алкил с 1-5 атомами углерода, алкенил с 2-5 атомами углерода, алкинил с 2-5 атомами углерода, гидроксиалкил с 1-3 атомами углерода, алкоксиалкил, в котором алкил имеет 1-3 атома углерода, и алкокси имеет 1-3 атома углерода, цианоалкил, в котором алкил имеет 1-3 атома углерода, имидазолилметил, -CH2CONH2, -CH2CH2CONH2, -CH2S(O)2NH2, -CH2SCH3, -CH2 S(O)CH3, -CH2S(O)2CH3,
-C(CH3)2SCH3, -C(CH3)2S(O)CH3 или -C(CH3)2S(O)2CH3 группы; более предпочтительно R1 представляет алкил с 1-4 атомами углерода, алкенил с 2-3 атомами углерода, алкинил с 3-4 атомами углерода, цианометил, имидазолилметил, -CH2CONH2, -CH2CH2CONH2, -CH2S(O)2NH2, -CH2SCH3, -CH2 S(O)CH3, -CH2S(O)2CH3, -C(CH3)2SCH3, -C(CH3)2S(O)CH3 или -C(CH3)2 S(O)2CH3 группы; наиболее предпочтительно R1 представляет втор-бутил, трет-бутил, изо-пропил, 3-пропинил или -C(CH3)2S(O)2CH3;
R2 представляет алкил, аралкил, алкилтиоалкил, арилтиоалкил или циклоалкилалкил; предпочтительно R2 представляет алкил с 1-5 атомами углерода, аралкил, в котором алкил имеет 1-3 атома углерода, алкилтиоалкил, в котором алкил имеет 1-3 атома углерода, арилтиоалкил, в котором алкил имеет 1-3 атома углерода, или циклоалкилалкил, в котором алкил имеет 1-3 атома углерода, кольцо имеет 3-6 атома углерода; более предпочтительно R2 представляет алкил с 3-5 атомами углерода, арилметил, алкилтиоалкил, в котором алкил имеет 1-3 атома углерода, арилтиометил или циклоалкилметил, в котором кольцо имеет 5-6 атомов углерода; еще более предпочтительно R2 представляет изобутил, н-бутил, CH3SH2CH2-, бензил, фенилтиометил, (2-нафтилтио)метил, 4-метоксифенилметил, 4-гидроксифенилметил, 4-фторфенилметил или циклогексилметил; еще более предпочтительно R2 представляет бензил, 4-фторфенилметил или циклогексилметил; наиболее предпочтительно R2 представляет бензил; R3 представляет алкил, циклоалкил или циклоалкилалкил; предпочтительно R3 представляет алкил, имеющий 1-5 атомов углерода, циклоалкил с 5-8-членным кольцом или циклоалкилметил, в котором кольцо имеет 3-6 атомов углерода; более предпочтительно R3 представляет пропил, изоамил, изобутил, бутил, циклопентилметил, циклогексилметил, циклогексил или циклогептил; более предпочтительно R3 представляет изобутил или циклопентилметил;
R4 представляет арил, гетероарил или гетероцикл; предпочтительно R4 представляет арил, бензосконденсированный с 5-6 членным гетероарильным кольцом или бензосконденсированный с 5-6 членным гетероциклом; или
R4 представляет группу формулы:

где A и B каждый независимо представляет О, S, SO или SO2; предпочтительно A и B каждый представляет О;
R4 представляет дейтерий, алкил или галоген; предпочтительно
R6 представляет дейтерий, алкил с 1-5 атомами углерода, фтор или хлор; более предпочтительно R6 представляет дейтерий, метил, этил, пропил, изопропил или фтор;
R7 представляет водород, дейтерий, алкил или галоген, предпочтительно R7 представляет водород, дейтерий, алкил с 1-3 атомами углерода, фтор или хлор; более предпочтительно R7 представляет водород, дейтерий, метил или фтор, или R6 или R7 каждый независимо представляет фтор или хлор и предпочтительно R6 или R7 каждый представляет фтор; или
R4 представляет группу формулы:

где Z представляет О, S или NH; и R9 представляет группу формулы:

где Y представляет O, S или NH; X представляет связь, О или NR21;
R20 представляет водород, алкил, алкенил, алкинил, аралкил, гетероаралкил, гетероциклоалкил, аминоалкил, N-монозамещенный или N,N-дизамещенный аминоалкил, где указанные заместители представляют алкил или аралкил, карбоксиалкил, алкоксикарбонилалкил, цианалкил или гидроксиалкил; предпочтительно R20 представляет водород, алкил с 1-5 атомами углерода, алкенил с 2-5 атомами углерода, алкинил с 2-5 атомами углерода, аралкил с 1-5 атомами углерода, гетероаралкил с 5-6 членами в кольце, в котором алкил имеет 1-5 атомов углерода, гетероциклоалкил с 5-6 членами в кольце, в котором алкил имеет 1-5 атомов углерода, аминоалкил с 2- 5 атомами углерода, N-монозамещенный или N,N-дизамещенный аминоалкил, в котором алкил имеет 2-5 атомов углерода, где указанные заместители представляют собой алкил с 1-3 атомами углерода, аралкил, в котором алкил имеет 1-5 атомов углерода, карбоксиалкил с 1-5 атомами углерода, алкоксикарбонилалкил с 1-5 атомами углерода, цианалкил с 1-5 атомами углерода или гидроксиалкил с 2-5 атомами углерода; более предпочтительно R20 представляет водород, алкил с 1-5 атомами углерода, фенилалкил, в котором алкил имеет 1-3 атома углерода, гетероциклоалкил с 5-6 членами в кольце, в котором алкил имеет 1-3 атомов углерода, или N-монозамещенный или N,N-дизамещенный аминоалкил с 2-3 атомами углерода, где указанные заместители представляют алкил с 1-3 атомами углерода; и наиболее предпочтительно R20 представляет водород, метил, этил, пропил, изопропил, изобутил, бензил, 2-(1-пирролидинил) этил, 2-(1-пиперидинил)этил, 2-(1-пиперазинил)этил, 2-(4-метилпиперазин-1-ил)этил, 2-(1-морфолинил)этил, 2-(1-тиаморфолинил)этил или 2-(N,N-диметиламино)этил;
R21 представляет водород или алкил; предпочтительно R21 представляет водород или алкил с 1-3 атомами углерода; более предпочтительно R21 представляет водород или метил; и наиболее предпочтительно R21 представляет водород; или группа формулы -NR20R21 является гетероциклом; предпочтительно группа формулы -NR20R21 является гетероциклом с 5-6 членами в кольце; более предпочтительно группа формулы -NR20R21 является пирролидинилом, пиперидинилом, пиперазинилом, 4-метилпиперазинилом, 4-бензилпиперазинилом, морфолинилом или тиаморфолинилом; и
R22 представляет алкил или R20R21N-алкил; предпочтительно R22 представляет алкил или R20R21N-алкил, где алкил имеет 1-3 атомов углерода; и более предпочтительно R22 представляет алкил с 1-3 атомами углерода;
и предпочтительно R4 представляет фенил, 2-нафтил, 4-метоксифенил, 4-гидроксифенил, 3,4-диметоксифенил, 3-аминофенил, 4-аминофенил, бензотиазол-5-ил, бензотиазол-6-ил, 2-амино-бензотиазол-5-ил, 2-(метоксикарбониламино)бензотиазол-5-ил, 2-аминобензотиазол-6-ил, 2-(метоксикарбониламино)бензотиазол-6-ил, 5-бензоксазолил, 6-бензоксазолил, 6-бензопиранил, 3,4-дигидробензопиран-6-ил, 7-бензопиранил, 3,4-дигидробензопиран-7-ил, 2,3-дигидробензофуран-5-ил, бензофуран-5-ил, 1,3-бензодиоксол-5-ил, 2-метил-1,3-бензодиоксол-5-ил, 2,2-диметил-1,3-бензодиоксол-5-ил, 2,2-дидейтеро-1, 3-бензодиоксол-5-ил, 2,2-дифтор-1,3-бензодиоксол-5-ил, 1,4-бензодиоксан-6-ил, 5-бензимидазолил, 2-(метоксикарбониламино)бензимидазол-5-ил, 6-хинолинил, 7-хинолинил, 6-изохинолинил или 7-изохинолинил; более предпочтительно R4 представляет фенил, 2-нафтил, 4-метоксифенил, 4-гидроксифенил, бензотиазол-5-ил, бензоксазол-6-ил, бензоксазол-5-ил, 2,3-дигидробензофуран-5-ил, бензофуран-5-ил, 1, 3-бензодиоксол-5-ил, 2-метил-1,3-бензодиоксол-5-ил, 2,2-диметил-1,3-бензодиоксол-5-ил, 2,2-дидейтеро-1,3-бензодиоксол-5-ил, 2,2-дифтор-1,3-бензодиоксол-5-ил, 1,4-бензодиоксан-6-ил, 2-(метоксикарбониламино)бензотиазол-5-ил, 2-(метоксикарбониламино) бензотиазол-6-ил или 2-(метоксикарбониламино)бензимидазол-5-ил; и более предпочтительно R4 представляет фенил, 4-метоксифенил, 4-гидроксифенил, бензотиазол-5-ил, бензоксазол-6-ил, 2,3-дигидробензофуран-5-ил, бензофуран-5-ил, 1,3-бензодиоксол-5-ил, 2-метил-1,3-бензодиоксол-5-ил, 2,2-диметил-1, 3-бензодиоксол-5-ил, 2,2-дидейтеро-1, 3-бензодиоксол-5-ил, 2,2-дифтор-1,3-бензодиоксол-5-ил, 1,4-бензодиоксан-6-ил, 2-(метоксикарбониламино)бензотиазол-6-ил или 2-(метоксикарбониламино)бензимидазол-5-ил;
R10 представляет водород, алкил с 1-3 атомами углерода, бензил, фенилметоксикарбонил, трет-бутоксикарбонил или 4-метоксифенилметоксикарбонил; предпочтительно R10 представляет водород, метил или бензил; наиболее предпочтительно R10 представляет водород;
R11 представляет водород, гидроксиалкил или алкоксиалкил, где алкил имеет 1-3 атомов углерода; предпочтительно R11 представляет водород;
R12 и R13 каждый независимо представляет водород, гидрокси, алкокси, 2-гидроксиалкокси, гидроксиалкил или алкоксиалкил, предпочтительно R12 и R13 каждый независимо представляет водород, гидрокси, алкокси, 2-гидроксиэтокси, гидроксиалкил или алкоксиалкил, где алкил имеет 1-3 атомов углерода, более предпочтительно R12 и R13 каждый независимо представляет водород, гидрокси, метокси или этокси; или
R11 и R12 или R12 и R13 вместе с атомами углерода, с которыми они соединены, представляют бензогруппу, которая необязательно замещена по крайней мере одним гидрокси или алкокси с 1-3 атомами углерода; предпочтительно R11 и R12 вместе с атомами углерода, с которыми они соединены, представляют бензогруппу, которая необязательно замещена по крайней мере одним гидрокси или метокси.
Абсолютная стереохимия атома углерода -CH(OH)-группы является предпочтительно (R). Абсолютная стереохимия атома углерода -CH(R1)-группы является предпочтительно (S). Абсолютная стереохимия атома углерода CH(R2)-групп является предпочтительно (S).
Группа
соединений, описанных формулой I, представляющих особый интерес, включает соединения формулы II:

или их фармацевтически приемлемые соли, пролекарства или сложные эфиры, где n, R1, R2, R3, R4 и R10 являются такими, как определено выше.
Группа соединений, описанных формулой II, представляющих особый интерес, включает соединения формулы III:

или их фармацевтически приемлемые соли, пролекарства или сложные эфиры, где n, R1, R2, R3, R4 и R10 являются такими, как определено выше.
Более предпочтительная группа соединений, описанных формулой III, включает соединения или их фармацевтически приемлемые соли, пролекарства или
сложные эфиры, где n равно 0;
R1 представляет втор-бутил, трет-бутил, изо-пропил, 3-пропинил или -C(CH3)2S(O)2CH3;
R2
представляет бензил;
R3 представляет пропил, изоамил, изобутил, бутил, циклогексил, циклогептил, циклопентилметил или циклогексилметил;
R4 является таким, как
определено выше; и
R10 представляет водород, метил или бензил.
Соединения, представляющие интерес, являются следующими: 2S-[[(пирролидин-2-ил)карбонил]
амино]-N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-3,3-диметил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(1,
3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил]-3-метил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(1,
3-бензодиоксол-5-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропил]-3S-метил-пентанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(1,
3-бензодиоксол-5-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропил]-4-пентинамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-[2R-гидрокси-3 [[фенилсульфонил]
(2-метилпропил)амино]-1S-(фенилметил)пропил] -3,3-диметил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-[2R-гидрокси-3- [[фенилсульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил]
-3-метил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-(2R-гидрокси-3- [[фенилсульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] -3S-метил-пентанамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-[2R-гидрокси-3- [[фенилсульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил] -4-пентинамид;
2S-[[(пирролидин-2-ил)карбонил] амино]
-N-[2R-гидрокси- 3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино]-1S- (фенилметил)пропил]-3,3-диметил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил]
амино]-N-[2R-гидрокси-3- [[(4-метоксифенил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил]-3-метил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино]
-N-[2R-гидрокси-3- [[(4-метоксифенил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил] -3S-метил-пентанамид;
2S-[[(пирролидин-2-ил)карбонил] амино]
-N-[2R-гидрокси-3- [[(4-метоксифенил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропил]-4-пентинамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(2,
3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил]-3,3-диметил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(2,
3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил]-3-метил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(2,
3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил]-3S-метил-пентанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(2,
3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил]-4-пентинамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-[2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил]
(2-метилпропил)амино]-1S- (фенилметил)пропил] -3,3-диметил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-[2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил]
(2-метилпропил)амино]-1S- (фенилметил)пропил] -3-метил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-[2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил]
(2-метилпропил)амино]-1S- (фенилметил)пропил] -3S-метил-пентанамид;
2S-[[(пирролидин-2-ил)карбонил]амино]-N-[2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил]
(2-метилпропил)амино]-1S- (фенилметил)пропил] -4-пентинамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(2-нафтил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил) пропил]-3,
3-диметил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(2-нафтил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил) пропил]-3-метил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3- [[(2-нафтил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил) пропил]-3S-метил-пентанамид;
2S-[[(пирролидин-2-ил)карбонил] амино]
-N-[2R-гидрокси-3- [[(2-нафтил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропил]-4-пентинамид;
2S-[[(пирролилин-2-ил)карбонил] амино]-N-[2R-гидрокси-3- [[(1,
4-бензодиоксан-6-ил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил]-3,3-диметил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино]-N-[2R-гидрокси-3- [[(1,
4-бензодиоксан-6-ил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил] -3-метил-бутанамид;
2S-[[(пирролидин-2-ил)карбонил] амино]-N-[2R-гидрокси-3- [[(1,4-бензодиоксан-6-ил)сульфонил]
(2-метилпропил)амино] -1S- (фенилметил)пропил]-3S-метил-пентанамид и
2S-[[(пирролидин-2-ил)карбонил] амино]-N-[2R-гидрокси-3- [[(1,4-бензодиоксан-6-ил)сульфонил] (2-метилпропил)амино]
-1S- (фенилметил)пропил]-4-пентинамид.
Используемый здесь термин "алкил", один или в сочетании, означает прямой или разветвленный алкильный радикал, содержащий предпочтительно от 1 до 8 атомов углерода, более предпочтительно от 1 до 5 атомов углерода, наиболее предпочтительно 1-3 атомов углерода. Примеры таких радикалов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и т.п. Термин "алкенил", один или в сочетании, означает прямой или разветвленный углеводородный радикал, имеющий одну или более двойных связей и содержащий предпочтительно от 2 до 10 атомов углерода, более предпочтительно от 2 до 8 атомов углерода, наиболее предпочтительно от 2 до 5 атомов углерода. Примеры подходящих алкенильных радикалов включают этенил, пропенил, 2-метилпропенил, 1,4-бутадиенил и т.п.
Термин "алкинил", один или в сочетании, означает прямой или разветвленный углеводородный радикал, имеющий одну или более тройных связей и содержащий предпочтительно от 2 до 10 атомов углерода, более предпочтительно от 2 до 5 атомов углерода. Примеры алкинильных радикалов включают этинил, пропинил (пропаргил), бутинил и т.п. Термин "алкокси", один или в сочетании, означает радикал алкилового эфира, где термин алкил имеет такое же значение, как указано выше. Примеры подходящих радикалов алкиловых эфиров включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изо-бутокси, втор-бутокси, трет-бутокси и т.п.
Термин "циклоалкил", один или в сочетании, означает насыщенный или частично насыщенный моноциклический, бициклический или трициклический алкильный радикал, где каждая циклическая часть содержит предпочтительно от 3 до 8 атомов углерода в кольце, более предпочтительно от 3 до 7 атомов углерода в кольце, наиболее предпочтительно от 5 до 6 атомов углерода в кольце, и который может быть необязательно бензосконденсированной системой, которая необязательно замещена, как указано далее в определении "арила". Примеры таких циклоалкильных радикалов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, октагидронафтил, 2,3-дигидро-1Н-инденил, адамантил и т.п. Используемые здесь термины "бициклический" и "трициклический" включают как сконденсированные кольцевые системы, такие как нафтил и β- -карболинил, так и замещенные кольцевые системы, такие как бифенил, фенилпиридил, нафтил и дифенилпиперазинил.
Термин "циклоалкилалкил" означает алкильный радикал, определенный выше, который замещен циклоалкильным радикалом, определенным выше. Примеры таких циклоалкилалкильных радикалов включают циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил, 1-циклопентилэтил, 1-циклогексилэтил, 2-циклопентилэтил, 2-циклогексилэтил, циклобутилпропил, циклопентилпропил, циклогексилбутил и т.п. Термин "бензо", один или в сочетании, означает двухвалентный радикал C6H4=, производный от бензола.
Термин "арил", один или в сочетании, означает фенильный или нафтильный радикал, который необязательно замещен одним или более заместителей, выбранных из алкила, алкокси, галогено, гидрокси, амино, нитро, циано, галоидалкила, карбокси, алкоксикарбонила, циклоалкила, гетероцикло, алканоиламино, амидо, амидино, алкоксикарбониламино, N-алкиламидино, алкиламино, диалкиламино, N-алкиламидо, N, N-диалкиламидо, аралкоксикарбониламино, алкилтио, алкилсульфинил, алкилсульфонил и т.п. Примеры арильных радикалов включают фенил, п-толил, 4-метоксифенил, 4-(трет-бутокси)фенил, 3-метил-4-метоксифенил, 4-фторфенил, 4-хлорфенил, 3-нитрофенил, 3-аминофенил, 3-ацетамидофенил, 4-ацетамидофенил, 2-метил-3-ацетамидофенил, 4-CF3-фенил, 2-метил-3-аминофенил, 3-метил-4-аминофенил, 2-амино-3-метил-фенил, 2,4-диметил-3-аминофенил, 4-гидроксифенил, 3-метил-4-гидроксифенил, 1-нафтил, 2-нафтил, 3-амино-1-нафтил, 2-метил-3-амино-1-нафтил, 6-амино-2-нафтил, 4, 6-диметокси-2-нафтил, пиперазинилфенил и т.п.
Термины "аралкил" и "аралкокси", одни или в сочетании, означают алкил или алкокси радикалы, определенные выше, в которых по крайней мере один атом водорода замещен на арильный радикал, определенный выше, такой как бензил, бензилокси, 2-фенилэтил, дибензилметил, гидроксифенилметил, метилфенилметил, дифенилметил, дифенилметокси, 4-метоксифенилметокси и т.п. Термин "аралкоксикарбонил", один или в сочетании, означает группу формулы аралкил-O-C(О)-, в которой термин "аралкил" имеет значение, данное выше. Примерами аралкоксикарбонильного радикала являются бензилоксикарбонил и 4-метоксифенил-метоксикарбонил. Термин "арилокси" означает группу формулы арил-О-, в которой термин "арил" имеет значение, данное выше. Термин "алканоил", один или в сочетании, означает ацильный радикал, производный от алканкарбоновой кислоты, примеры которого включают ацетил, пропионил, бутурил, валерил, 4-метилвалерил и т.п.
Термин "циклоалкилкарбонил" означает ацильный радикал формулы циклоалкил-C(О)-, в котором термин "циклоалкил" имеет значение, данное выше, такой как циклопропилкарбонил, циклогексилкарбонил, адамантилкарбонил, 1,2,3,4-тетрагидро-2-нафтоил, 2-ацетамидо-1,2,3,4-тетрагидро-2-нафтоил, 1-гидрокси-1,2,3,4-тетрагидро-2-нафтоил и т. п. Термин "аралканоил" означает ацильный радикал, производный арилзамещенной алканкарбоновой кислоты, такой как фенил-ацетил, 3-фенилпропионил (гидроциннамоил), 4-фенилбутурил, (2-нафтил)ацетил, 4-хлорогидроциннамоил, 4-аминогидроциннамоил, 4-метоксигидроциннамоил и т.п.
Термин "ароил" означает ацильный радикал, производный от арилкарбоновой кислоты, причем "арил" имеет значения, указанные выше. Примеры таких ароильных радикалов включают замещенный и незамещенный бензоил или нафтоил, такие как бензоил, 4-хлорбензоил, 4-карбоксибензоил, 4-(бензилоксикарбонил)бензоил, 1-нафтоил, 2-нафтоил, 6-карбокси-2-нафтоил, 6-(бензилоксикарбонил)-2-нафтоил, 3-бензилокси-2-нафтоил, 3-гидрокси-2-нафтоил, 3-(бензилоксиформамидо)-2-нафтоил и т.п.
Термин "гетероцикло", один или в сочетании, означает насыщенный или частично ненасыщенный моноциклический, бициклический или трициклический гетероциклический радикал, содержащий по крайней мере один, предпочтительно 1-4, более предпочтительно 1-2, атомов азота, кислорода или серы в кольце и имеющий предпочтительно 3-8 атомов в каждом кольце, более предпочтительно 3-7 атомов в каждом кольце и наиболее предпочтительно 5-6 атомов в каждом кольце.
"Гетероцикло" группа включает сульфоны, сульфоксиды, N-оксиды третичного азота кольца и карбоциклические сконденсированные и бензосконденсированные кольцевые системы. Такие гетероциклические группы могут быть необязательно замещены по крайней мере по одному, предпочтительно по 1-4, более предпочтительно по 1-2, атомам углерода галогеном, алкилом, алкокси, гидрокси, оксо, арилом, аралкилом, гетероарилом, гетероаралкилом, амидино, N-алкиламидино, алкоксикарбониламино, алкилсульфониламино и т.п., и/или по вторичному атому азота (т. е. -NH-) гидрокси, алкилом, аралкоксикарбонилом, алканоилом, гетероаралкилом, фенилом или фенилалкилом, и/или по третичному атому азота (т.е. =N-) оксидо группой.
"Гетероциклоалкил" означает алкильный радикал, определенный выше, в котором по крайней мере один атом водорода замещен на гетероциклический радикал, определенный выше, такой как пирролидинилметил, тетрагидротиенилметил, пиридилметил и т. п. Термин "гетероарил", один или в сочетании, означает ароматический гетероциклический радикал, определенный выше, который необязательно замещен, как определено выше в отношении определений "арила" и "гетероцикло".
Примерами таких гетероциклических и гетероарильных групп являются пирролидинил, пиперидинил, пиперазинил, морфолинил, тиаморфолинил, пирролил, имидазолил (например, имидазол-4-ил, 1-бензилоксикарбонилимидазол-4-ил и т.д.), пиразолил, пиридил (например, 2-(пиперидинил)пиридил и 2-(4-бензилпиперазин-1-ил-1-пиридинил и т.д.), пиразинил, пиримидинил, фурил, тетрагидрофурил, тиенил, тетрагидротиенил и его сульфоксид- и сульфонпроизводные, триазолил, оксазолил, тиазолил, индолил (например, 2-индолил и т.д.), хинолинил (например, 2-хинолинил, 3-хинолинил, 1-оксидо-2-хинолинил и т.д.), изохинолинил (например, 1-изохинолинил, 3-изохинолинил и т.д.), тетрагидрохинолинил (например, 1,2,3,4-тетрагидро-2-хинолил и т.д.), 1,2,3,4-тетрагидроизохинолинил (например, 1,2,3, 4-тетрагидро-1-оксо-изохинолинил и т.д.), хиноксалинил, β карболинил, 2-бензофуранкарбонил, 1-, 2-, 4- или 5-бензимидазолил, метилендиоксифен-4-ил, метилендиоксифен-5-ил, этилендиоксифенил, бензотиазолил, бензопиранил, бензофурил, 2,3-дигидробензофурил, бензоксазолил, тиофенил и т.д.
Термин "циклоалкилалкоксикарбонил" означает ацильную группу, производную от циклоалкилалкоксикарбоновой кислоты формулы циклоалкилалкил-о-COOH, где циклоалкилалкил имеет значение, данное выше. Термин "арилоксиалканоил" означает ацильный радикал формулы арил-О-алканоил, где арил и алканоил имеют значения, данные выше. Термин "гетероциклоалкоксикарбонил" означает ацильную группу, производную от гетероциклоалкил-О-COOH, где гетероциклоалкил имеет значение, данное выше. Термин "гетероциклоалканоил" означает ацильную группу, производную от гетероциклоалкилкарбоновой кислоты, где "гетероцикло" имеет значение, данное выше.
Термин "гетероциклоалкоксикарбонил" означает ацильный радикал, производный от гетероциклоалкил-О-COOH, где "гетероцикло" имеет значение, данное выше. Термин "гетероарилоксикарбонил" означает ацильный радикал, производный от карбоновой кислоты, представленной формулой гетероарил-О-COOH, где гетероарил имеет значения, данные выше. Термин "аминокарбонил", один или в сочетании, означает аминозамещенную карбонильную (карбамоильную) группу, где аминогруппа может быть первичной, вторичной или третичной аминогруппой, содержащей заместители, выбранные из алкила, арила, аралкила, циклоалкила, циклоалкилалкила и т.п. Термин "аминоалканоил" означает ацильную группу, производную от аминозамещенной алкилкарбоновой кислоты, где аминогруппа может быть первичной, вторичной или третичной аминогруппой, содержащей заместители, выбранные из алкила, арила, аралкила, циклоалкила, циклоалкилалкила и т.п. Термин "галоген" означает фтор, хлор, бром или иод.
Термин "галоалкил" означает алкильный радикал, имеющий данное выше значение, где один или более водородов замещены галогеном. Примеры таких галоалкильных радикалов включают хлорметил, 1-бромэтил, фторметил, дифторметил, трифторметил, 1,1,1-трифторэтил и т.п. Термин "уходящая группа" (L или W) обычно относится к группам, легко замещаемым нуклеофилом, таким как амин, тиол или спирт. Такие уходящие группы хорошо известны в данной области. Примеры таких уходящих групп включают, но ими не ограничиваются, N-гидроксисукцинимид, N-гидроксибензотриазол, галогениды, трифлаты, тозилаты и т.п. Предпочтительные уходящие группы указаны, где это целесообразно.
Способы получения соединений формулы I приведены ниже.
Следует отметить, что общий способ показан на примере получения соединений, имеющих конкретную стереохимию, например, где абсолютная стереохимия относительно гидроксильной группы обозначена как (R). Однако такие способы обычно применимы к соединениям противоположной конфигурации, например, где стереохимия относительно гидроксильной группы обозначена как (S). Кроме того, соединения, имеющие (R) стереохимию, могут быть использованы для получения соединений, имеющих (S) стереохимию. Например, соединение, имеющее (R) стереохимию, может быть превращено в соединение с (S) стереохимией с помощью хорошо известных способов.
Получение
соединений формулы I
Соединения данного изобретения, представленные формулой I, можно получить, используя следующие способы, схематически показанные на Схемах I и II (см. в конце
описания).
N-защищенное производное хлоркетон аминокислоты, имеющее формулу:
где P представляет аминозащитную группу и R2 определен выше, восстанавливают в соответствующий спирт, используя подходящий восстанавливающий агент. Подходящие аминозащитные группы хорошо известны в данной области и к ним относятся карбобензокси, т-бутоксикарбонил и т.п. Предпочтительной аминозащитной группой является карбобензокси. Предпочтительным N-защищенным хлоркетоном является N-бензилоксикарбонил-L-фенилаланин хлор-метил кетон. Предпочтительным восстанавливающим агентом является борогидрид натрия. Реакцию восстановления проводят при температуре от -10oC до около 25oC, предпочтительно при около 0oC, в подходящей системе растворителя, такой как, например, тетрагидрофуран и т.п. N-защищенные хлоркетоны коммерчески доступны, например, от Bachem. Inc., Torrance, California. Альтернативно, хлоркетоны можно получить по методике, представленной S.J. Fittkau, J. Pract. Chem., 315, 1037 (1973), и в дальнейшем применяя методики для N-защиты, которые хорошо известны в данной области.
Галогензамещенный спирт можно использовать непосредственно, как описано ниже, или предпочтительно подвергнуть взаимодействию, предпочтительно
при комнатной температуре, с подходящим основанием в подходящей системе растворителя, с получением N-защищенного аминоэпоксида формулы:
где P и R2 такие, как определено выше. К подходящим системам растворителя для получения аминоэпоксида относятся этанол, метанол, изопропанол, тетрагидрофуран, диоксан и т.п., а также их смеси. К подходящим основаниям для получения эпоксида из восстановленного хлоркетона относятся гидроксид калия, гидроксид натрия, т-бутоксид калия, DBU и т.п. Предпочтительным основанием является гидроксид калия.
Альтернативно, защищенный аминоэпоксид можно получить, как указано в совместно поданной и одновременно
рассматриваемой PCT заявке на патент N PCT/US93/04804 (WO 093/23388) и PCT/US94/12201, и заявке на Пат. США (US Patent Application Attorney Docket) N C-2860, каждая из которых приведена здесь в
качестве ссылки, где раскрыты способы получения хирального эпоксида, хирального цианогидрина, хирального амина и других хиральных промежуточных продуктов, используемых для получения ингибиторов
ретровирусной протеазы, которые используют в качестве исходного реагента DL-, D- или L-аминокислоту, которая реагирует с подходящей аминозащитной группой в подходящем растворителе с получением
сложного эфира аминозащищенной аминокислоты. С целью иллюстрации, для получения ингибиторов данного изобретения можно использовать защищенную L-аминокислоту следующей формулы:
где P3 представляет карбоксилзащищающую группу, например метил, этил, бензил, третичный бутил, 4-метоксифенилметил и т.п.; R2 является таким, как определено выше; и P1 и P2 и/или P3 независимо выбирают из аминозащитных групп, включая, но не ограничиваясь, аралкил, замещенный аралкил, циклоалкенилалкил и замещенный циклоалкенилалкил, аллил, замещенный аллил, ацил, алкоксикарбонил, аралкоксикарбонил и силил. Примеры аралкила включают, но не ограничиваются, бензил, орто-метилбензил, тритил и бензгидрил, которые могут быть необязательно замещены галогеном, C1-C8 алкилом, алкокси, гидрокси, нитро, алкиленом, амино, алкиламино, ациламино и ацилом, или их соли, такие как соли фосфония и аммония. Примеры арильных групп включают фенил, нафталенил, инданил, антраценил, дуренил, 9-(9-фенилфлюоренил) и фенантренил, циклоалкенилалкил или замещенный циклоалкенилалкил, содержащий C6-C10 циклоалкилы. Подходящие ацильные группы включают карбобензокси, т-бутоксикарбонил, изобутоксикарбонил, бензоил, замещенный бензоил, бутирил, ацетил, три-фтороацетил, три-хлороацетил, фталоил и т.п. Предпочтительно P1 и P2 независимо выбирают из аралкила и замещенного аралкила. Более предпочтительно P1 и P2 представляют бензил.
Дополнительно, P1 и/или P2 и/или P3 защитные группы могут образовать гетероциклическое кольцо с азотом, к которому они присоединены, например, 1,2-бис(метилен) бензол, фталимидил, сукцинимидил, малеимидил и т.п., и где эти гетероциклические группы, которые могут дополнительно включать присоединенные арильные и циклоалкильные кольца. Кроме того, гетероциклические группы могут быть моно-, ди- или три- замещенными, например, нитрофталимидил. Термин силил относится к атому кремния, необязательно замещенному одной или более алкильной, арильной или аралкильной группами.
Подходящие силильные защитные группы включают, но не ограничиваются, триметилсилил, триэтилсилил, триизопропилсилил, трет-бутилдиметилсилил, диметилфенилсилил, 1,2-бис(диметилсилил) бензол, 1,2-бис(диметилсилил) этан и дифенилметилсилил. Силилирование аминных функциональных групп с получением моно- или бис-дисилиламина может обеспечить получение производных аминоспирта, аминокислоты, сложных эфиров аминокислот и амидов аминокислот. В случае аминокислот, сложных эфиров аминокислот и амидов аминокислот, восстановление карбонильной функциональной группы дает требуемый моно- или бис-силил аминоспирт. Силилирование аминоспирта может привести к получению N, N,O-три-силил производному. Удаление силильной функциональной группы из силильной функциональной группы простого эфира легко осуществляется обработкой, например, гидроксидом металла или фторидом аммония, либо в отдельной стадии реакции, либо in situ во время получения аминоальдегидного реагента. Подходящими силилирующими агентами являются, например, триметилсилил хлорид, трет-бутил-диметилсилил хлорид, фенилдиметилсилил хлорид, дифенилметилсилил хлорид или продукты их соединения с имидазолом или ДМФ. Способы силилирования аминов и удаления силил защитных групп хорошо известны специалистам в данной области. Способы получения этих аминных производных из соответствующих аминокислот, амидов аминокислот или эфиров аминокислот также хорошо известны специалистам в данной области органической химии, включая химию аминокислот/сложных эфиров аминокислот или аминоспиртов.
Затем эфир аминозащищенную L-аминокислоту восстанавливают в соответствующий спирт.
Например, эфир аминозащищенной L-аминокислоты можно восстановить диизобутилалюминий гидридом при -78oC в подходящем растворителе, таком как толуол. К предпочтительным восстанавливающим
агентам относятся литий алюмогидрид, литий борогидрид, борогидрид натрия, боран, литий три-трет-бутоксиалюмогидрид, комплекс боран/ТГФ. Наиболее предпочтительно восстанавливающий агент представляет
диизобутилалюмогидрид (DiBAL-H) в толуоле. Затем полученный спирт превращают, например, с помощью Swern окисления, в соответствующий альдегид формулы:

где P1, P2 и R2 являются такими, как определено выше. Так, раствор спирта в дихлорметане добавляют к охлажденному (-75 до -68oC) раствору оксалил хлорида в дихлорметане и ДМСО в дихлорметане и перемешивают в течение 35 минут.
К приемлемым окисляющим реагентам относятся, например, комплекс триоксид серы-пиридин и ДМСО, оксалил хлорид и ДМСО, ацетил хлорид или ангидрид и ДМСО, трифторацетил хлорид или ангидрид и ДМСО, метансульфонил хлорид и ДМСО или тетрагидро-тиофен-S-оксид, толуолсульфонил бромид и ДМСО, трифторметан-сульфонил ангидрид (трифлик ангидрид) и ДМСО, пентахлорид фосфора и ДМСО, диметилфосфорил хлорид и ДМСО и изобутилхлороформиат и ДМСО. Условия окисления указаны в Reetz et al [Angew. Chem., 99, p. 1186, (1987)], Angew. Chem. Int. Ed. Engl. , 26, p. 1141, 1987), в котором использовали оксалил хлорид и ДМСО при -78o C.
Предпочтительным способом окисления, описанным в данном изобретении, является способ, использующий комплекс триоксид серы-пиридин, триэтиламин и ДМСО при комнатной температуре. Эта система обеспечивает прекрасные выходы целевого хирального защищенного аминоальдегида, который можно использовать без очистки, а именно, необходимость в очистке килограммов промежуточных соединений с помощью хроматографии отпадает, и операции в большом масштабе становятся менее опасными. Реакция при комнатной температуре также снимает необходимость использования низкотемпературного реактора, что делает способ более приемлемым для коммерческого производства.
Реакцию можно проводить в атмосфере инертного газа, такого как азот или аргон, или в обычном или сухом воздухе, при атмосферном давлении или в герметизированном реакционном сосуде при положительном давлении. Предпочтительной является атмосфера азота. Альтернативные основания аминов включают, например, три-бутил амин, три-изопропил амин, N-метилпиперидин, N-метил морфолин, азабициклононан, диизопропилэтиламин, 2,2,6,6-тетраметилпиперидин, N,N-диметиламинопиридин или смеси этих оснований. Триэтиламин является предпочтительным основанием. Альтернативные чистому ДМСО растворители включают смеси ДМСО с апротонными или галогенированными растворителями, такими как тетрагидрофуран, этилацетат, толуол, ксилол, дихлорметан, этилен дихлорид и т. п. Диполярные апротонные сорастворители включают ацетонитрил, диметилформамид, диметилацетамид, ацетамид, тетраметил мочевину и ее циклический аналог, N-метилпирролидон, сульфолан и т.д. Вместо N,N-дибензил-фенилаланинола в качестве предшественника альдегида, можно использовать производные фенлиаланинола, обсужденные выше, для получения соответствующего N-монозамещенного [либо P1, либо P2 = H] или N,N-дизамещенного альдегида.
Кроме того, для получения альдегидов можно проводить восстановление гидридом амидного или сложно-эфирного производного соответствующего бензил (или другая подходящая защитная группа) азотзащищенного фенилаланина, замещенного фенилаланина или циклоалкильного аналога производного фенилаланина. Гидридный перенос является дополнительным способом синтеза альдегида в условиях, когда избегают конденсаций альдегидов, например, окисление Опенауэра (Oppenauer Oxidation).
Альдегиды данного способа можно также получить восстановлением защищенного фенилаланина и аналогов фенилаланина или их амидных или сложно-эфирных производных, например, обработкой амальгамой натрия с HCl в этаноле или лития, или натрия, или калия, или кальция в аммиаке. Температура реакции может быть от около -20oC до около 45oC и предпочтительно от около 5oC до около 25oC. Два дополнительных способа получения азотзащищенного альдегида включают окисление соответствующего спирта хлорной известью в присутствии каталитического количества 2,2,6,6-тетраметил-1-пиридилокси свободного радикала. Во втором способе окисление спирта в альдегид проводят каталитическим количеством тетрапропиламмоний перрутената в присутствии N-метилморфолин-N-оксида.
Альтернативно, хлорангидридное производное защищенного фенилаланина или производного фенилаланина, раскрытых выше, можно восстановить водородом и катализатором, таким как Pd на карбонате бария или сульфате бария, с или без дополнительного агента, обеспечивающего работу катализатора в нормальных условиях (moderating agent), такого как сера или тиол (Восстановление Розенмунда, Rosenmund Reduction).
Альдегид, полученный при окислении Сверна (Swern), затем подвергают взаимодействию с
галометиллитий-реагентом, который генерируют in situ взаимодействием алкиллитиевого или ариллитиевого соединения с дигалометаном, представленным формулой X1CH2X2, где
X1 и X2 независимо представляют I, Br или Cl. Например, раствор альдегида и хлориодометана в ТГФ охлаждают до -73oC и добавляют раствор н-бутиллития в гексане.
Полученный продукт представляет собой смесь диастереомеров соответствующих аминозащищенных эпоксидов формул:

и

Диастереомеры можно разделить, например, хроматографически, или, альтернативно, сразу, после того как они прореагируют в последующих стадиях. D-аминокислоту можно использовать вместо L-аминокислоты, чтобы получить соединения, имеющие (S) стереохимию на атоме углерода, связанном с R2.
Добавление хлорметиллития или бромметиллития к хиральному аминоальдегиду является высоко диастереоселективным. Предпочтительно, чтобы хлорметиллитий или бромметиллитий генерировался в in-situ реакцией дигалометана и н-бутиллития. К приемлемым галометанам для метиленирования относятся хлориодометан, бромхлорметан, дибромметан, дииодметан, бромфторметан и т.п. Сульфонатный сложный эфир продукта присоединения, например, бромистого водорода к формальдегиду, также является метиленирующим агентом. Тетрагидрофуран является предпочтительным растворителем, однако, альтернативные растворители, такие как толуол, диметоксиэтан, этилен дихлорид, метиленхлорид, могут быть использованы в виде чистых растворителей или в виде их смесей. Диполярные апротонные растворители, такие как ацетонитрил, ДМФ, N-метилпирролидон, используют в качестве растворителей или как часть смеси растворителей. Реакцию можно проводить в атмосфере инертного газа, такого как азот или аргон. н-Бутиллитий можно заменить другими органометаллическими реагентами, таким как метиллитий, трет-бутиллитий, втор-бутиллитий, фениллитий, фенил натрия и т. п. Реакцию можно проводить при температурах от около -80oC до 0oC, но предпочтительно от около -80oC до -20oC. Наиболее предпочтительно температура реакции находится в диапазоне от - 40oC до -15oC. Реагенты можно добавлять в один прием, но многократные добавления в некоторых случаях предпочтительны. Предпочтительным давлением реакции является атмосферное, однако положительное давление при некоторых условиях более преимущественно, например, в случае окружающей среды с высокой влажностью.
Альтернативные способы превращения в эпоксиды данного изобретения включают замещение другого типа заряженного метиленированного предшественника с последующей обработкой его основанием с получением аналогичного аниона. Примеры такого типа соединений включают триметилсульфоксоний тозилат или трифлат, тетраметиламмоний галогенид, метилдифенилсульфоксоний галогенид, в которых галогенидом является хлорид, бромид или иодид.
Превращение альдегидов данного изобретения в их эпоксидное производное можно также проводить многостадийно. Например, добавление аниона тиоанизола, полученного из, например, бутил или ариллитиевого реагента, к защищенному аминоальдегиду, окисление полученного защищенного аминосульфида спирта хорошо известными окисляющими агентами, такими как пероксид водорода, трет-бутилгипохлорит, хлорная известь или периодат натрия, с получением сульфоксида. Алкилирование сульфоксида, например, метил иодидом или бромидом, метил тозилатом, метил мезилатом, метил трифлатом, этил бромидом, изопропил бромидом, бензил хлоридом или т.п., в присутствии органического или неорганического основания. Альтернативно, защищенный аминосульфид спирта может быть алкилирован, например, вышеупомянутыми алкилирующими агентами с получением солей сульфония, которые затем превращают в эпоксиды обработкой трет-амином или минеральными основаниями.
Требуемые эпоксиды образуются, при использовании наиболее предпочтительных условий, диастереоселективно в количественном (весовом) соотношении, по крайней мере около 85:15 (S:R). Продукт может быть очищен с помощью хроматографии с получением диастереомерно и энантиомерно чистого продукта, но чаще всего, для получения ингибиторов ретровирусных протеиназ продукт используют непосредственно без очистки. Вышеприведенный способ применим как для получения смесей оптических изомеров, так и для получения разделенных соединений. Если требуется конкретный оптический изомер, его можно выделить путем подбора исходного вещества, например, L-фенилаланин, D-фенилаланин, L-фенилаланинол, D-фенилаланинол, D-гексагидрофенилаланинол и т.п., или может быть проведено разделение на промежуточной или конечной стадиях. Хиральные вспомогательные вещества, такие как один или два эквивалента камфорсульфоновой кислоты, лимонной кислоты, камфорной кислоты, 2-метоксифенилуксусной кислоты и т. п. , могут быть использованы для получения солей, сложных эфиров или амидов соединений данного изобретения. Эти соединения или производные могут кристаллизоваться или могут быть разделены хроматографически, используя либо хиральную, либо ахиральную колонку, как это хорошо известно специалистам в данной области.
Амино эпоксиды затем подвергают взаимодействию, в подходящей системе растворителя, с
равным количеством или предпочтительно с избытком необходимого амина формулы R3NH2, где R3 является водородом или принимает значения, определенные выше. Реакцию можно
проводить в широком интервале температур, например от около 10oC до около 100oC, и предпочтительно, но необязательно, проводить при температуре, при которой растворитель начинает
кипеть. Подходящие системы растворителя включают протонные, не протонные и диполярные апротонные органические растворители, такие как, например, системы, в которых растворителем является спирт, такой
как метанол, этанол, изо-пропанол и т.п., эфиры, такие как тетрагидрофуран, диоксан и т.п., и толуол, N, N-диметилформамид, диметилсульфоксид и их смеси. Предпочтительным растворителем является
изопропанол. Полученный продукт представляет собой производное 3-(N-защищенный амино)-3-(R2)-1-(NHR3)-пропан-2-ола (здесь и далее обозначаемое как аминоспирт), которое может быть
представлено формулами:
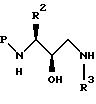

где P, P1, P2, R2 и R2 такие, как указано выше. Альтернативно, вместо аминоэпоксида можно использовать галоспирт.
Определенный выше аминоспирт затем подвергают взаимодействию в подходящем растворителе с сульфонил хлоридом R4SO2Cl, сульфонил бромидом R4SO2Br
или с соответствующим сульфонил ангидридом, предпочтительно в присутствии акцептора кислоты. Подходящие растворители, в которых можно проводить реакцию, включают метиленхлорид, тетрагидрофуран и т.п.
Подходящие акцепторы кислоты включают триэтиламин, пиридин и т.п. Полученное производное сульфонамида может быть представлено, в зависимости от используемого эпоксида, формулами;
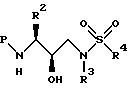

где P, P1, P2, R2, R3 и R4 такие, как указано выше. Эти промежуточные продукты используют для получения соединений-ингибиторов данного изобретения.
Сульфонил галогениды формулы R4SO2X можно получить взаимодействием подходящего арил, гетероарил и бензосконденсированного гетероциклического реагента Гриньяра или литиевого реагента с сульфурилхлоридом или диоксидом серы с последующим окислением галогеном, предпочтительно хлором. Арил, гетероарил и бензосконденсированный гетероциклический реактив Гриньяра или реагент лития можно получить из их соответствующих галогенидных (таких как хлор или бром) соединений, которые коммерчески доступны или их можно легко получить из коммерчески доступных исходных веществ, используя известные в данной области способы. Также, тиолы можно окислить в сульфонил хлориды, используя хлор, в присутствии воды при тщательно контролируемых условиях. Кроме того, сульфоновые кислоты, такие как арилсульфоновые кислоты, можно превратить в сульфонил галогениды, используя реагенты, такие как PCl5, SOCl2, ClC(O)C(O)Cl и т. п., и также в ангидриды, используя подходящие дегидратирующие реагенты. Сульфоновые кислоты, в свою очередь, можно получить, используя хорошо известные в данной области способы. Некоторые сульфоновые кислоты коммерчески доступны. Для получения соединений, в которых -SO2- часть замещена на -SO- или -S- часть соответственно, вместо сульфонил галогенидов, можно использовать сульфинил галогениды (R4 SOX) или сульфенил галогениды (R4SX). Арилсульфоновые кислоты, бензосконденсированные гетероциклические сульфоновые кислоты или гетероарилсульфоновые кислоты можно получить сульфонированием ароматического кольца хорошо известными в данной области способами, такими как реакцией с серной кислотой, SO3, SO3 комплексами, такими как ДМФ (SO3), пиридин (SO3), N,N-диметилацетамид(SO3) и т.п. Предпочтительно арилсульфонил галогениды получают из ароматических соединений взаимодействием с ДМФ (SO3) и SOCl2 или ClC(O)C(O)Cl. Реакции можно проводить постадийно или в одном сосуде.
Арилсульфоновые кислоты, бензосконденсированные гетероцикло сульфоновые кислоты, гетероарил сульфоновые кислоты,
арилмеркаптаны, бензосконденсированные гетероцикло меркаптаны, гетероарилмеркаптаны, арилгалогениды, бензосконденсированные гетероциклогалогениды, гетероарилгалогениды и т.п. коммерчески доступны или
их можно легко получить из коммерчески доступных исходных веществ, используя стандартные, хорошо известные в данной области способы. Например, ряд сульфоновых кислот (R4SO3H),
представленных формулами:

и

где A, B, Z, R6, R7 и R9 такие, как определено выше, можно получить из 1,2-бензолдитиола, 2-меркаптанфенола, 1,2-бензолдиола, 2-аминобензотиазола, бензотиазола, 2-аминобензимидазола, бензимидазола и т.п., которые коммерчески доступны, Carter, Пат. США 4595407; Ehrenfreund et al. Пат. США 4634465; Yoder et al., J. Heterocycl. Chem. 4:166-167 (1967); Cole et al., Aust. J. Chem. 33:675-680 (1980); Cabiddu et al. Synthesis 797-798 (1976); Ncube et al. , Tet. Letters 2345-2348 (1978); Ncube et ai., Tet. Letters 255-256 (1977); Ansink & Cerfontain, Rec. Trav. Chim. Pays-Bas 108:395-403 (1989); и Kajihara & Tsuchiya, EP 638564 Al, которые включены сюда в качестве ссылки. Например, 1, 2-бензолдитиол, 2-меркаптанфенол или 1,2-бензолдиол может взаимодействовать с R6R7C(L')2, где L' такой, как определено ниже, предпочтительно Br или I, в присутствии основания, такого как гидроксид, или с R6R7C= O в присутствии кислоты, такой как толуолсульфоновая кислота, или P2O5, с получением замещенного бензосконденсированного гетероцикла формулы:

который затем может быть сульфирован в вышеупомянутую сульфоновую кислоту. Например, CF2Br2 или CD2Br2 можно подвергнуть взаимодействию с 1,2-бензолдитиолом, 2-меркаптанфенолом или 1,2-бензолдиолом в присутствии основания с получением соединений:

или

соответственно, где A и B являются O или S и D является атомом дейтерия. Кроме того, когда A и/или B представляют S, то серу можно окислить, используя описанные ниже способы, в сульфон или производные сульфоксида.
После получения производного сульфонамида, аминозащитную группу P или P1 и P2 аминозащитные группы удаляют в условиях, которые не будут оказывать действия на остальную часть молекулы. Эти способы хорошо известны в данной области и включают кислотный гидролиз, гидрогенолиз и т.п. Предпочтительный способ включает удаление защитной группы, например, удаление карбобензокси группы, путем гидрогенолиза с использованием палладия на угле в подходящем растворителе, таком как спирт, уксусная кислота и т.п., или в их смеси. Когда защитной группой является т-бутоксикарбонильная группа, то ее можно удалить, используя неорганическую или органическую кислоту, например HCl или трифторуксусную кислоту, в подходящей системе растворителей, например в диоксане или метиленхлориде. Полученный продукт является производным соли амина.
После нейтрализации соли амин
затем связывают с DL-, D-, или L-аминокислотой, соответствующей формуле PNHCH(R1)COOH, где P и R1 такие, как определено выше, с последующим снятием защиты с амина, как описано
выше, и связыванием с циклическими аминокислотами формул:

(когда R10 ≠ H) или

где n, P, R10, R11, R12 и R13 такие, как определено выше, и L представляет уходящую группу, такую как галогенид, ангидрид, активный сложный эфир и т.п. Например, когда R11, R12 и R13 представляют, каждый, водород, то можно использовать N-защищенный или N-замещенный пролин НОВТ активный сложный эфир, N-защищенный или N-замещенный пипеколиновой (pipecolinic) кислоты N-гидроксисукцинамид активный сложный эфир и т.п.
Альтернативно, у промежуточного соединения

можно снять защиту взаимодействием с R10L, где R10 представляет алкил или бензил, или соответствующим альдегидом или кетоном с последующим восстановлением, таким как, например, цианоборогидрид натрия и т.п., с получением

где n, R1, R2, R3, R4, R10, R11, R12 и R13 такие, как определено выше.
Циклические аминокислоты формулы:
являются коммерчески доступными, такие как, например, пролин, 4-гидроксипролин, N-метилпролин, индолин-2-карбоновая кислота и т.п.; или их можно легко получить из коммерчески доступных исходных веществ, используя стандартные, хорошо известные в данной области способы, из таких, как, например, пролин, 4-гидроксипролин, 4-гидроксихинолин-2-карбоновая кислота, 3-гидроксипиколиновая кислота, индолин-2-карбоновая кислота, 5-метоксииндолин-2-карбоновая кислота, каиновая кислота, 4-метокси-2-хинолинкарбоновая кислота и т.п. Альтернативно, циклические аминокислоты можно легко получить циклизацией аминосодержащих альфа-кетон карбоновых кислот или сложных эфиров в циклический имин с последующим восстановлением, например, цианоборогидридом натрия и т.п., в циклический амин, как показано на Схеме III (см. в конце описания), или альтернативно, циклизацией аминокислот, имеющих соответствующую уходящую группу L, такую как хлор, бром, тозилат, мезилат и т.п. , как показано на Схеме IV (см. в конце описания), где n, P3, R10, R11 R12 и R13 такие, как определено выше. Исходные альфа-кетон карбоновая кислота или сложный эфир и аминокислота коммерчески доступны или их можно легко получить из коммерчески доступных веществ, используя хорошо известные способы и методики.
Альтернативно, после нейтрализации соли, амин затем связывают с DL-, D-, или L-аминокислотой, соответствующей формулам:

(когда R10 ≠ H) или

где n, P, R1, R10 R11, R12 и R13 такие, как определено выше, которую можно получить способом, аналогичным способу связывания, описанному выше, из DL-, D- или L-аминокислоты, соответствующей формуле NH2CH(R1)COOP3, где P3 и R1 такие, как определено выше.
DL-, D- или L-аминокислота, соответствующая формуле PNHCH(R1)COOH или NH2CH(R1)COOP3, где Р, P3 и R1 такие, как определено выше, коммерчески доступна (Sigma Chemical Co.) или ее можно легко получить, используя стандартные, известные в данной области способы, из легко доступных исходных веществ. Предпочтительно P представляет бензилоксикарбонил или т-бутоксикарбонил, и P3 представляет бензил или трет-бутил. Стандартные способы связывания можно использовать для связывания аминокислоты и аминов. Группу карбоновой кислоты подвергают взаимодействию с образованием ангидрида, смешанного ангидрида, галоидангидрида, такого как хлор или бромангидрид, или активного сложного эфира, такого как сложные эфиры N-гидроксисукцинимида, НОВТ и т.п., используя хорошо известные способы и условия. Соответствующие системы растворителей включают тетрагидрофуран, этиловый эфир, метил-трет-бутиловый эфир, метиленхлорид, N,N-диметилформамид и т.п., а также их смеси.
Альтернативно, защищенный аминоспирт, полученный раскрытием эпоксида, может быть в дальнейшем защищен по вновь введенной
аминогруппе с помощью защитной группы P', которая не удаляется при удалении аминозащитных групп P или P1 и P2, т.е. P' является селективно удаляемой группой. Специалист в данной
области может подобрать соответствующие комбинации P', P, P1 и P2. Например, подходящими комбинациями являются P=Cbz и P'=Boc; P'=Cbz и P=Boc; P'=Cbz, P2=бензил и
P'=Boc; и P'= P2=бензил и P'=Boc. Полученное соединение, представленное формулой:
или

может быть подвергнуто дополнительным превращениям для получения соединений формул:

или

где n, P, P' R1, R2, R3, R10, R11, R12 и R13 такие, как определено выше. Дополнительные вышеупомянутые превращения могут включать, если необходимо, введение требуемых остатков или групп сразу либо введение в предварительно полученную молекулу более одного остатка или группы в течение одной стадии. Первый вариант представляет последовательный способ синтеза, а последний представляет конвергентный способ синтеза. На этой стадии также возможны синтетические превращения. Защитную группу P' затем селективно удаляют, и полученный амин взаимодействует с сульфонил хлоридом R4SO2Cl, сульфонил бромидом R4SO2Br или с соответствующим сульфонил ангидридом, предпочтительно в присутствии акцептора кислоты, с образованием соединений данного изобретения:

где n, R1, R2, R3, R4, R10, R11, R12 и R13 такие, как определено выше. Селективное снятие защиты и превращение в сульфонамид можно осуществить либо в конце синтеза, либо на любой промежуточной стадии, если требуется.
Описанные выше химические реакции обычно раскрывают, исходя из их наиболее широкого применения для получения соединений данного изобретения. Иногда реакции не могут быть применимы, как описано для каждого соединения, входящего в объем изобретения. Соединения, для которых это имеет место, легко узнаваемы специалистами в данной области. Во всех таких случаях, либо реакции могут быть успешно выполнены с помощью стандартных модификаций, известных специалистам в данной области, например, при помощи соответствующей защиты подверженных влиянию групп, при помощи замены на альтернативные стандартные реагенты, путем обычной модификации условий реакций и т.п., либо могут быть применены другие реакции, раскрытые здесь или нераскрытые стандартные, для получения соединений данного изобретения. Во всех препаративных способах все исходные вещества известны или их можно легко получить из известных исходных веществ.
Очевидно, что специалист в данной области, используя данное описание, может осуществить данное изобретение в наиболее полном объеме. Поэтому представленные ниже предпочтительные конкретные варианты воплощения изобретения следует рассматривать просто как иллюстративные и не ограничивающие каким-либо образом объем настоящего изобретения.
Все реагенты были использованы без очистки. Все протон- и углерод-ЯМР спектры получали либо на Varian VXP-300, либо на VXR-400 ядерном спектрометре магнитного резонанса.
Следующие примеры иллюстрируют получение соединений-ингибиторов данного изобретения и промежуточных соединений, используемых при получении соединений-ингибиторов данного изобретения.
ПРИМЕР 1

Получение 2S-[Бис(фенилметил)амино]бензолпропанола
Способ 1: Получение 2S-[Бис(фенилметил)амино] бензолпропанол Dibal восстановлением фенилметилового эфира N,N-бис(фенилметил)-L-фенилаланина
Стадия 1:
Раствор L-фенилаланина (50, 0 г, 0,302 моль), гидроксида натрия (24,2 г, 0,605 моль) и карбоната калия (83,6 г, 0,605 моль) в воде (500 мл) нагревают до 97oC. Затем медленно добавляют бензил бромид (108,5 мл, 0,605 моль) (время добавления - 25 мин). Смесь перемешивают при 97oC в течение 30 минут в атмосфере азота. Раствор охлаждают до комнатной температуры и экстрагируют толуолом (2 х 250 мл). Объединенные органические слои промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют до масла. Идентификация продукта подтверждается следующим образом. Аналитическая TCX (TLC) (10% этилацетат/гексан, силикагель) показывает основной компонент при Rf значении = 0,32, что отвечает требуемому трибензилированному соединению, фенилметиловому эфиру N, N-(фенилметил)-L-фенилаланина. Это соединение может быть очищено с помощью колоночной хроматографии (силикагель, 15% этилацетат/гексан). Обычно продукт является достаточно чистым для того, чтобы его можно было использовать непосредственно в следующей стадии без дополнительной очистки.1ЯМР спектр соответствует опубликованному в литературе.1ЯМР спектр (CDCl3) δ, 3,00 и 3,14 (ABX-система, 2Н, JAB=14,1 Гц, JAB=7,3 Гц и JBX=5,9 Гц), 3,54 и 3,92 (AB-Система, 4Н, JAB=13,9 Гц), 3,71 (т, 1H, J=7,6 Гц), 5,11 и 5, 23 (AB-Система, 2Н, JAB= 12,3 Гц), и 7,18 (м, 20Н). ЭИМС (EIMS): м/з 434 (М-1).
Стадия 2:
Фенилметиловый эфир бензилированного фенилаланина (0,302 моль), полученный
на предыдущей стадии, растворяют в толуоле (750 мл) и охлаждают до -55oC. 1,5 М раствор DIBAL в толуоле (443,9 мл, 0,666 моль) добавляют с такой скоростью, чтобы поддерживать температуру от
-55 до -50oC (время добавления -1 час). Смесь перемешивают в течение 20 минут в атмосфере азота и затем реакцию прекращают при -55oC медленным добавлением метанола (37 мл). Затем
холодный раствор выливают в холодный (5oC) 1,5N HCl раствор (1,8 л). Выпавшее в осадок твердое вещество (прибл. 138 г) отфильтровывают и промывают толуолом. Твердое вещество суспендируют в
смеси толуола (400 мл) и воды (100 мл). Смесь охлаждают до 5oC и обрабатывают 2,5N NaOH (186 мл) и затем перемешивают при комнатной температуре до тех пор, пока твердое вещество не
растворится. Толуольный слой отделяют от водной фазы и промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют до объема 75 мл (89 г). К остатку добавляют этилацетат (25 мл)
и гексан (25 мл), после чего требуемый спиртовой продукт начинает кристаллизоваться. Через 30 мин дополнительные 50 мл гексана добавляют для того, чтобы ускорить дальнейшую кристаллизацию. Твердое
вещество отфильтровывают и промывают 50 мл гексана, получая 34,9 г первой порции продукта. Вторую порцию продукта выделяют путем повторной фильтрации маточного раствора. Обе порции объединяют и
перекристаллизовывают из этилацетата (20 мл) и гексана (30 мл), получая 40 г β S-2-[Бис(фенилметил)амино] бензолпропанола, 40% выход в расчете на L-фенилаланин. Дополнительные 7 г (7%) продукта
можно получить путем перекристаллизации концентрированного маточного раствора. ТСХ (TLC) продукта Rf=0,23 (10% этилацетат/гексан, силикагель);1ЯМР (CDCl3)δ и 2,44 (м, 1Н),
3,09 (м, 2Н), 3,33 (м, 1Н), 3,48 и 3,92 (AB-Система, 4Н, JAB=13,3 Гц), 3,52 (м, 1Н) и 7,23 (м, 15Н); [α] + 42,4 (с 1,45, CH2Cl2); ДСК (DSC) 77,67oC; Элементный анал. Рассчит. для C23H25ON: C, 83,34; H, 7,60; N, 4,23.
Найдено: C, 83,43; H, 7,59; N, 4,22. ВЭЖХ (HPLC) на хиральной стационарной фазе: Cyclobond I SP колонка (250 х 4,6 мм В.Д. (I.D.)), мобильная фаза: метанол/триэтиламмоний ацетатный буфер pH 4,2 (58:42,
об/об), скорость потока 0,5 мл/мин, обнаружение детектором при 230 нм и температуре 0oC. Время удерживания: 11,25 мин, время удерживания энантиомера требуемого продукта: 12,5 мин.
Способ 2: Получение β S-2-[Бис(фенилметил)амино]бензолпропанола путем N, N-дибензилирования L-фенилаланинола
L-фенилаланинол (176,6 г, 1,168 моль) добавляют к перемешиваемому
раствору карбоната калия (484,6 г, 3,506 моль) в 710 мл воды. Смесь нагревают до 65oC в атмосфере азота. Раствор бензилбромида (400г, 2,339 моль) в 3A этаноле (305 мл) добавляют со
скоростью, которая позволяет поддерживать температуру 60-68oC. Двухфазный раствор перемешивают при 65oC в течение 55 мин и затем дают возможность охладиться до 10oC
при энергичном перемешивании. Масляный продукт отверждается в небольшие гранулы. Продукт разбавляют 2,0 л водопроводной воды и перемешивают в течение 5 минут, чтобы растворить неорганические побочные
продукты. Продукт выделяют фильтрацией при пониженном давлении и промывают водой до тех пор, пока pH не станет равным 7. Полученный неочищенный продукт сушат на воздухе на протяжении ночи, получая
полусухое твердое вещество (407 г), которое перекристаллизовывают из 1,1 л смеси этилацетат/гептан (1:10 по объему). Продукт выделяют фильтрацией (при -8oC), промывают 1,6 л холодной смеси
(-10oC) этилацетат/гептан (1:10 по объему) и сушат на воздухе, получая 339 г β S-2-[Бис(фенилметил)амино]бензолпропанола, Т. пл. = 71,5-73,0oC. При необходимости,
дополнительное количество продукта можно получить из маточного раствора. Другие аналитические характеристики идентичны соединению, полученному как описано в Способе 1.
ПРИМЕР 2
Получение 2S-[Бис(фенилметил)амино]бензолпропанальдегида
Способ 1:
2S-[Бис(фенилметил)амино]бензолпропанол (200 г, 0,604 моль) растворяют в триэтиламине (300 мл, 2,15 моль). Смесь охлаждают до 12oC и добавляют раствор комплекса триоксид серебра/пиридин (380 г, 2,39 моль) в ДМСО (1,6 л) с такой скоростью, чтобы поддерживать температуру между 8-17oC (время добавления - 1,0 ч. ). Раствор перемешивают при комнатной температуре в атмосфере азота в течение 1,5 часов, по истечении которых, по данным анализа ТСХ, реакция завершается (33% этилацетат/гексан, силикагель). Реакционную смесь охлаждают ледяной водой и реакцию прекращают добавлением 1,6 л холодной воды (10-15oC) на протяжении 45 минут. Полученный раствор экстрагируют этилацетатом (2,0 л), промывают 5% лимонной кислотой (2,0 л) и рассолом (2,2 л), сушат над MgSO4 (280 г) и фильтруют. Растворитель удаляют на роторном испарителе при 35-40oC и затем сушат в вакууме, получая 198,8 г 2S-[Бис-(фенилметил)амино] бензолпропанальлегида в виде бледно-желтого масла (99,9%). Полученный неочищенный продукт достаточно чист для того, чтобы непосредственно использоваться в следующей стадии без очистки. Данные анализа соединения находятся в соответствии с опубликованными в литературе. [α] = -92,9o (с 1,87, CH2 Cl2);1H ЯМР (400 МГц, CDCl3) δ, 2,94 и 3,15 (ABX-Система, 2Н, JAX = 13,9 Гц, JAX = 7,3 Гц и JBX = 6,2 Гц), 3,56 (т, 1Н, 7,1 Гц), 3,69 и 3,82 (AB-Система, 4Н, JAB=13,7 Гц), 7,25 (м, 15Н) и 9,72 (с, 1Н); ВЧМС (HRMS) Рассчит. для (М+1) C23H24NO 330,450, найдено: 330,1836. Элем. анал. Рассчит. для C23H23ON: C, 83,86; H, 7,04; N, 4,25. Найдено: C, 83,64; H, 7,42; N, 4,19. ВЭЖХ (HPLC) на хиральной стационарной фазе: (S,S) Pirkle-Whelk-O 1 колонка (250 х 4,6 мм В.Д. (I.D.)), подвижная фаза: гексан/изопропанол (99,5: 0,5, об/об), скорость потока 1,5 мл/мин, обнаружение УФ-детектором при 210 нм. Время удерживания целевого S-изомера: 8,75 мин, время удерживания R-энантиомера 10,62 мин.
Способ 2:
Раствор оксалил хлорида (8,4 мл, 0,096 моль) в дихлорметане (240 мл) охлаждают до -74oC. Раствор ДМСО (DMCO) (12,0 мл, 0,155
моль) в дихлорметане (50 мл) затем медленно добавляют с такой скоростью, чтобы поддерживать температуру -74oC (время добавления ≈ 1,25 час). Смесь перемешивают в течение 5 мин с
последующим добавлением раствора β S-2-[бис(фенилметил)амино]бензолпропанола (0,074 моль) в 100 мл дихлорметана (время добавления 20 мин, темп. от -75oC до -68oC). Раствор
перемешивают при -78oC в течение 35 минут в атмосфере азота. Затем добавляют триэтиламин (41,2 мл, 0,295 моль) в течение 10 мин (темп. от -78oC до -68oC), и в это
время осаждается соль аммония. Холодную смесь перемешивают в течение 30 мин и затем добавляют воду (225 мл). Дихлорметановый слой отделяют от водной фазы и промывают водой, рассолом, сушат над
сульфатом магния, фильтруют и концентрируют. Остаток разбавляют этилацетатом и гексаном и затем фильтруют, чтобы дополнительно удалить соль аммония. Фильтрат концентрируют, получая α
S-[бис(фенилметил)амино]бензолпропанальдегид. Альдегид используют на следующей стадии без очистки.
Способ 3:
К смеси 1,0 г (3,0 ммоль) β
S-2-[бис(фенилметил)амино]бензолпропанола, 0,531 г (4,53 ммоль) N-метил морфолина, 2,27 г молекулярных сит (4A) и 9,1 мл ацетонитрила добавляют 53 мг (0,15 ммоль) тетрапропиламмоний перрутената
(ТРАР). Смесь перемешивают в течение 40 минут при комнатной температуре и концентрируют при пониженном давлении. Остаток суспендируют в 15 мл этилацетата, фильтруют через подложку силикагеля. Фильтрат
концентрируют при пониженном давлении, получая продукт, содержащий приблизительно 50% α S-2-[бис(фенилметил)амино]бензолпропанальдегида в виде бледно-желтого масла.
Способ 4:
К раствору 1,0 г (3,02 ммоль) β S-2-[бис(фенилметил)амино]бензолпропанола в 9,0 мл толуола добавляют 4,69 мг (0,03 ммоль) 2,2,6,6-тетраметил-1-пиперидинилокси в виде свободного радикала
(TEMPO), 0,32 г (3,11 ммоль) бромида натрия, 9,0 мл этилацетата и 1,5 мл воды. Смесь охлаждают до 0oC и водный раствор 2,87 мл 5% хлорной извести для бытовых нужд, содержащей 0,735 г (8,75
ммоль) бикарбоната натрия и 8,53 мл воды, медленно добавляют в течение 25 минут. Смесь перемешивают при 0oC в течение 60 минут. Две дополнительные порции (1,44 мл каждая) смеси добавляют с
последующим перемешиванием в течение 10 минут. Двухфазной смеси дают возможность разделиться. Водный слой экстрагируют дважды 20 мл этилацетата. Объединенный органический слой промывают 4,0 мл
раствора, содержащего 25 мг иодида калия и воды (4,0 мл), 20 мл 10% водного раствора тиосульфата натрия и затем рассолом. Органический раствор сушат над сульфатом магния, фильтруют и концентрируют при
пониженном давлении, получая 1,34 г неочищенного масла, содержащего небольшое количество целевого альдегида, α S-[бис(фенилметил)амино]бензолпропанальдегида.
Способ 5:
Используя методики, описанные в Способе 1 этого Примера, за исключением того, что используют 3,0 эквивалента комплекса триоксид серы-пиридин, выделяют α
S-[бис(фенилметил)амино]бензолпропанальдегид со сравнимыми выходами.
ПРИМЕР 3

Получение N,N-дибензил-3(S)-амино-1,2-(S)-эпокси-4-фенилбутана
Способ 1:
Раствор α S-[Бис(фенилметил)амино] бензолпропанальдегида (191,7 г, 0,58 моль) и хлориодометана (56,4 мл, 0,77 моль) в тетрагидрофуране (1,8 мл) охлаждают до -30o - -35oC (более низкая температура, такая как -70oC, также приемлема, но более высокие температуры легче достижимы при операциях в промышленном масштабе) в реакторе из нержавеющей стали в атмосфере азота. Раствор н-бутиллития в гексане (1,6 М, 365 мл, 0,58 моль) затем добавляют при скорости, позволяющей поддерживать температуру ниже -25oC. После добавления смесь перемешивают при температуре от -30 до - 35oC в течение 10 минут. Последующее добавление реагентов проводят следующим образом: (1) добавляют дополнительное количество хлориодометана (17 мл), затем н-бутиллитий (110 мл), при < -25oC. После добавления смесь перемешивают при температуре от -30 до - -35oC в течение 10 минут. Операцию повторяют один раз. (2) Добавляют дополнительное количество хлориодометана (8,5 мл, 0,11 моль), затем н-бутиллитий (55 мл, 0,088 моль) при < -25oC. После добавления смесь перемешивают при температуре от -30 до -35oC в течение 10 минут. Операцию повторяют 5 раз. (3) Добавляют дополнительное количество хлориодометана (8, 5 мл, 0,11 моль), затем н-бутиллитий (37 мл, 0,059 моль) при < -25oC. После добавления смесь перемешивают при температуре от -30 до -35oC в течение 10 минут. Операцию повторяют один раз. Прекращают наружное охлаждение, и смесь нагревается до температуры окружающей среды в течение от 4 до 16 часов, когда данные ТСХ (силикагель, 20% этилацетат/гексан) указывают на то, что реакция завершилась. Реакционную смесь охлаждают до 10oC и реакцию останавливают добавлением 1452 г 16% раствора хлорида аммония (полученного растворением 232 г хлорида аммония в 1220 мл воды), поддерживая температуру ниже 23oC. Смесь перемешивают в течение 10 минут и органический и водный слои разделяют. Волную фазу экстрагируют этилацетатом (2 х 500 мл). Этилацетатный слой объединяют с тетрагидрофурановым слоем. Объединенный раствор сушат над сульфатом магния (220 г), фильтруют и концентрируют на роторном испарителе при 65oC. Коричневый масляный остаток сушат при 70oC в вакууме (0,8 х 105 Па, 0,8 бар) в течение 1 часа, получая 222,8 г неочищенного вещества. (Вес неочищенного остатка составлял > 100%. Из-за относительной нестабильности продукта на силикагеле неочищенный продукт обычно непосредственно используют в следующей стадии без очистки). Соотношение диастереоизомеров в неочищенной смеси определяют с помощью протонного ЯМР: (2S)/(2R): 86:14. Минорный (второстепенный) и основной эпоксидные диастереоизомеры в этой смеси идентифицируют с помощью метода ТСХ (силикагель, 10% этилацетат/гексан), Rf=0,29 & 0,32 соответственно. Аналитическую пробу каждого из диастереомеров получают очисткой хроматографией на силикагеле (3% этилацетат/гексан) и характеризуют следующим образом:
N,N, α S-Трис(фенилметил)-2S-оксиранметанамин
1H ЯМР (400 МГц, CDCl3) δ 2,49 и 2,51 (AB-Система, 1H, JAB=2,82), 2,76 и 2,77 (AB-Система, 1H, 2,83 (м, JAB=4,03), 2,83 (м, 2Н), 2,99 & 3,03 (AB-Система, 1H, JAB=10,1 Гц), 3,15 (м, 1H), 3,73 & 3,84 (AB-Система, 4H, JAB= 14,00), 7,21 (м, 15H);13C ЯМР (400 МГц, CDCl3) δ 139,55, 129,45, 128,42, 128,14, 128,09, 126,84, 125,97, 60,32, 54,23, 52,13, 45,99, 33,76; ВЧМС (HRMC) Рассчит. для C24H26NO (M+1) 344,477, Найдено 344,2003.
N,N, α S-Трис(фенилметил)-2R-оксиранметанамин
1H ЯМР (300 МГц, CDCl3) δ 2,20 (м, 1H), 2,59 (м, 1H), 2,75 (м, 2Н), 2,97 (м,
1H), 3,14 (м, 1H), 3,85 (AB-Система, 4Н), 7,25 (м, 15H). ВЭЖХ (HPLC) на хиральной стационарной фазе: Pirkie-Wheik-O 1 колонка (250 х 4,6 мм В.Д.), подвижная фаза: гексан/изопропанол (99,5:0,5, об/об),
скорость потока: 1,5 мл/мин, обнаружение УФ-детектором при 210 нм. Время удерживания (8): 9,38 мин, время удерживания энантиомера (4): 13,75 мин.
Способ 2:
Раствор
неочищенного альдегида 0,074 моля и хлориодометана (7,9 мл, 0,096 моль) в тетрагидрофуране (285 мл) охлаждают до -78oC в атмосфере азота. 1,6 М раствор н-бутиллития в гексане (25 мл, 0,040
моль) затем добавляют с такой скоростью, которая позволяет поддержать температуру -75oC (время добавления - 15 мин). После первого добавления добавляют дополнительное количество
хлориодометана (1,6 мл, 0,022 моль), а затем н-бутиллитий (23 мл, 0,037 моль), поддерживая температуру -75oC. Смесь перемешивают в течение 15 мин. Каждый из реагентов, хлориодометан (0,70
мл, 0,010 моль) и н-бутиллитий (5 мл, 0,008 моль) добавляют еще 4 раза в течение 45 мин при -75oC. Затем охлаждающую баню удаляют и раствор нагревают до 22oC в течение 1,5 часов.
Смесь выливают в 300 мл насыщенного водного раствора хлорида аммония. Тетрагидрофурановый слой отделяют. Водную фазу экстрагируют этилацетатом (1 х 300 мл). Объединенные органические слои промывают
рассолом, сушат над сульфатом магния, фильтруют и концентрируют, получая коричневое масло (27,4 г). Продукт обычно используют в следующей стадии без очистки. Требуемый диастереомер может быть очищен
перекристаллизацией на последующей стадии. Продукт обычно также очищают хроматографией.
Способ 3:
Раствор α S-[Бис(фенилметил)амино]бензолпропанальдегида (178,84 г, 0,
54 моль) и бромхлорметана (46 мл, 0,71 моль) в тетрагидрофуране (1,8 л) охлаждают до -30oC- (-35oC) (более низкая температура, такая как -70oC, также приемлема, но
более высокие температуры легче достижимы при операциях в промышленном масштабе) в реакторе из нержавеющей стали в атмосфере азота. Раствор н-бутиллития в гексане (1,6 М, 340 мл, 0,54 моль) затем
добавляют при скорости, позволяющей поддерживать температуру ниже -25oC. После добавления смесь перемешивают при температуре от -30 до -35oC в течение 10 минут. Последующие
добавления реагентов проводят следующим образом: (1) Добавляют дополнительное количество хлориодометана (14 мл), затем н-бутиллитий (102 мл) при < -25oC. После добавления смесь
перемешивают при температуре от -30 до -35oC в течение 10 минут. Операцию повторяют один раз. (2) Добавляют дополнительное количество хлориодометана (7 мл, 0,11 моль), затем н-бутиллитий
(51 мл, 0,082 моль) при < -25oC. После добавления смесь перемешивают при температуре -30 - (-)35oC в течение 10 минут. Операцию повторяют 5 раз. (3) Добавляют
дополнительное количество бромхлориодометана (7 мл, 0,11 моль), затем н-бутиллитий (51 мл, 0,082 моль) при < -25oC. После добавления смесь перемешивают при температуре от -30 до
-35oC в течение 10 минут. Операцию повторяют один раз. Прекращают наружное охлаждение и смесь нагревается до температуры окружающей среды в течение от 4 до 16 часов, когда данные ТСХ (TLC)
(силикагель, 20% этилацетат/гексан) указывают на то, что реакция завершилась. Реакционную смесь охлаждают до 10oC и реакцию прекращают добавлением 1452 г 16% раствора хлорида аммония
(полученного растворением 232 г хлорида аммония в 1220 мл воды), поддерживая температуру ниже 23oC. Смесь перемешивают в течение 10 минут и органический и водный слои разделяют. Водную фазу
экстрагируют этилацетатом (2 х 500 мл). Этилацетатный слой объединяют с тетрагидрофурановым слоем. Объединенный раствор сушат над сульфатом магния (220 г), фильтруют и концентрируют на роторном
испарителе при 65oC. Коричневый масляный остаток сушат при 70oC в вакууме (0,8 х 105 Па, 0,8 бар) в течение 1 часа, получая 222,8 г неочищенного вещества.
Способ 4:
Используют методики, описанные в Способе 3 этого Примера, за исключением того, что температура реакции составляет -20oC. Полученный N,N, α
S-трис(фенилметил)-2S-оксиранметанамин представляет собой диастереомерную смесь меньшей чистоты, чем смесь по способу 3.
Способ 5:
Используют методики, описанные в Способе 3
этого Примера, за исключением того, что температура реакции составляет от -70 до -78oC. Полученный N,N, α S-трис(фенилметил)-2S-оксиранметанамин представляет собой диастереомерную
смесь, которую непосредственно используют в последующих стадиях без очистки.
Способ 6:
Используют методики, описанные в Способе 3 этого Примера, за исключением того, что
непрерывное добавление бромхлорметана и н-бутиллития проводят при температуре от -30 до -35oC. После реакции и операций обработки, описанных в Способе 3 этого Примера, выделяют N,N, α
S-трис(фенилметил)-2S-оксиранметанамин со сравнимыми выходами и сравнимой чистотой.
Способ 7:
Используют методики, описанные в Способе 2 этого Примера, за исключением того,
что вместо хлориодометана используют дибромметан. После реакции и операций обработки, описанных в Способе 3 этого Примера, получают N,N α S-трис(фенилметил)-2S-оксиранметанамин.
ПРИМЕР 4
Получение N-[3(S)-[N, N-бис(фенилметил)амино]-2(R)- гидрокси-4-фенилбутил]-N-изобутиламина
К раствору неочищенного N,N-дибензил-3(S)-амино-1,2(S)-эпокси-4-фенилбутана (388,5 г, 1,13 моль) в изопропаноле (2,7 л) (или этилацетате) добавляют изобутиламин (1,7 кг, 23,1 моль) в течение 2 мин. Температура повышается от 25oC до 30oC. Раствор нагревают до 82oC и перемешивают при этой температуре в течение 1,5 часов. Теплый раствор концентрируют при пониженном давлении при 65oC. Остаток коричневого масла переносят в 3-литровую колбу и сушат в вакууме (0,8 мм Hg) в течение 16 часов, получая 450 г 3S-[N,N-бис(фенилметил)амино]-4-фенилбутан-2R-ола в виде неочищенного масла.
Аналитическую пробу требуемого основного диастереомерного продукта получают
очисткой небольшой пробы неочищенного продукта хроматографией на силикагеле (40% этилацетат/гексан. ТСХ анализ: силикагель, 40% этилацетат/гексан; Rf=0,28; ВЭЖХ
(HPLC) анализ; колонка с
ультрасферическим ODS, 25% тиениламино-/фосфатный буфер pH 3 - ацетонитрил, скорость потока 1 мл/мин, УФ-детектор, время удерживания 7,49 мин; ВЧМС (HRMC) Рассчит. для C28H27
N2O (М+1)
417,616, найдено 417,2887. Аналитическую пробу диастереомерного продукта, 3S-[N, N-бис(фенилметил)амино] -1-(2-метилпропил) амино-4-фенилбутан-2S-ола, также получают очисткой
небольшой пробы неочищенного продукта хроматографией на силикагеле (40% этилацетат/гексан).
ПРИМЕР 5
Получение соли N-[3(S)-[N,N-бис(фенилметил)амино]-2(R)- гидрокси-4-фенилбутил]-N-изобутиламин щавелевой кислоты
К раствору щавелевой кислоты (8,08 г, 89,72 ммоль) в метаноле (76 мл) добавляют раствор неочищенного 3(S)-[N,N-бис(фенилметил) амино]-1-(2-метилпропил)амино-4-фенилбутан-2(R)-ола (39,68 г), который содержит около 25,44 г (61,06 ммоль) 3(S), 2(R) изомера и около 4,49 г (10,78 ммоль) 3(S), 2(S) изомера, в этилацетате (90 мл) в течение 15 минут. Смесь перемешивают при комнатной температуре в течение приблизительно 2 часов. Твердое вещество отделяют фильтрацией, промывают этилацетатом (2 х 20 мл) и сушат в вакууме в течение приблизительно 1 часа, получая 21,86 г (выделение 70,7% изомера) соли 97% диастереомерной чистоты (исходя из площадей ВЭЖХ пика). ВЭЖХ анализ: Vydec-пептид/белок C18 колонка, УФ детектор 254 нм, скорость потока 2 мл/мин, градиент {А=0,05% трифторуксусная кислота в воде, В=0,05% трифторуксусная кислота в ацетонитриле, 0 мин, 75% A/25% В, 30 мин, 10% A/90% В, 35 мин, 10% A/90% В, 37 мин, 75% A/25% В}; Время удерживания 10,68 мин (3(S), 2(R) изомер) и 9,73 мин (3(S), 2(S) изомер). Т. пл. = 174,99oC; Элем. анализ: Рассчит. C 71,05%, H 7,50%, N 5,53%; Найдено: C 71,71%, H 7,75%, N 5,39%.
Альтернативно, дигидрат щавелевой кислоты (119 г, 0,94 моль) помещают в 5000 мл круглодонную колбу, снабженную механической мешалкой и капельной воронкой. Добавляют метанол (1000 мл) и смесь перемешивают до тех пор, пока растворение не завершится. Раствор неочищенного 3(S)-[N, N-бис(фенил-метил)амино] -1-(-2-метилпропил) амино-4-фенилбутан-2(R)-ола в этилацетате (1800 мл, 0,212 г изомеров аминоспирта/мл, 0,9160 моль) добавляют в течение двадцати минут. Смесь перемешивают в течение 18 часов и твердый продукт выделяют центрифугированием шести порций при 400 об/мин. Каждую порцию промывают 125 мл этилацетата. Затем соль собирают и сушат на протяжении ночи при 1 Торр, получая 336,3 г продукта (71% в расчете на суммарный аминоспирт). ВЭЖХ/МС (электрораспыление) соответствуют требуемому продукту (м/з 417 [М+Н]+).
Альтернативно, неочищенный 3(S)-[N, N-бис(фенилметил)амино] -1-(2-метилпропил)амино-4-фенилбутан-2(R)-ол (5 г) растворяют в метил-трет-бутиловом эфире (MTBE) (10 мл) и добавляют щавелевую кислоту (1 г) в метаноле (4 мл). Смесь перемешивают в течение приблизительно 2 часов. Полученное твердое вещество отфильтровывают, промывают холодным МТВЕ и сушат, получая 2,1 г белого твердого вещества 98,9% диастереомерной чистоты (исходя из площадей ВЭЖХ пика).
ПРИМЕР 6
Получение соли N-[3(S)-[N, N-бис(фенилметил)амино]2(R)- гидрокси-4-фенилбутил]-N-изобутиламин уксусной кислоты
К раствору
неочищенного 3(S)-[N,N-бис(фенилметил)амино]-1-(2- метилпропил)амино-4-фенилбутан-2(R)-ола в метил-трет-бутиловом эфире (МТВЕ) (45 мл, 1,1 г изомеров аминоспирта/мл) добавляют по каплям уксусную
кислоту (6,9 мл). Смесь перемешивают в течение приблизительно 1 часа при комнатной температуре. Растворитель удаляют в вакууме, получая продукт в виде коричневого масла 85% диастереомерной чистоты
(исходя из площадей ВЭЖХ пика). Коричневое масло кристаллизуют следующим образом: 0,2 г масла растворяют в первом растворителе при нагревании, получая прозрачный раствор, второй растворитель добавляют
до тех пор, пока раствор не станет мутным, смесь нагревают снова до прозрачности, вносят затравку приблизительно 99% диастереомерно чистого продукта, охлаждают до комнатной температуры и затем хранят
в холодильнике на протяжении ночи. Кристаллы отфильтровывают, промывают вторым растворителем и сушат. Диастереомерную чистоту рассчитывают, исходя из площадей ВЭЖХ пика. Результаты представлены в
Таблице 1.
Альтернативно, неочищенный 3(S)-[N,N-бис(фенилметил) амино]-1-(2-метилпропил)амино-4-фенилбутан-2(R)-ол { 50,0 г, который содержит около 30,06 г (76,95 ммоль) 3(S), 2(R) изомера и около 5,66 г (13,58 ммоль) 3(S), 2(S) изомера}, растворяют в метил-трет-бутиловом эфире (45,0 мл). К этому раствору добавляют уксусную кислоту (6,90 мл, 120,6 ммоль) в течение приблизительно 10 мин. Смесь перемешивают при комнатной температуре в течение приблизительно 1 часа и концентрируют при пониженном давлении. Масляный остаток очищают перекристаллизацией из метил-трет-бутилового эфира (32 мл) и гептана (320 мл). Твердое вещество выделяют фильтрацией, промывают холодным гептаном и сушат в вакууме в течение приблизительно 1 часа, получая 21,34 г (выделение 58,2% изомера) 96% диастереомерно чистой соли моноуксусной кислоты (исходя из площадей ВЭЖХ пика). Т. пл.= 105-106oC; Элем. анализ: Рассчит. : C 75,53%, H 8,39%, N 5,87%; Найдено: C 75,05%, H 8,75%, N 5, 71%.
ПРИМЕР 7
Получение соли N-[3(S)-[N,N-бис(фенилметил)амино]-2(R)- гидрокси-4-фенилбутил]-N-изобутиламин L-винной кислоты
Неочищенный 3(S)-[N,
N-бис(фенилметил)амино]-1- (2-метилпропил)амино-4-фенилбутан-2(R)-ол (10,48 г, который содержит около 6,72 г (16,13 ммоль) 3(S), 2(R) изомера и около 1,19 г (2,85 ммоль) 3(S), 2(S) изомера) растворяют
в тетрагидрофуране (10,0 мл). К этому раствору добавляют L-винную кислоту (2,85 г, 19 ммоль) в метаноле (5,0 мл) в течение приблизительно 5 мин. Смесь перемешивают при комнатной температуре в течение
приблизительно 10 мин и концентрируют при пониженном давлении. К маслянистому остатку добавляют метил-трет-бутиловый эфир (20,0 мл) и смесь перемешивают при комнатной температуре в течение
приблизительно 1 часа. Твердое вещество отделяют фильтрацией, получая 7,50 г неочищенной соли. Неочищенную соль очищают перекристаллизацией из этилацетата и гептана при комнатной температуре, получая
4,13 г (выделение 45,2% изомера) 95% диастереомерно чистой соли L-винной кислоты (исходя из площадей ВЭЖХ пика). Микроанализ: Рассчит.: C 67,76%, H 7,41%, N 4,94%; Найдено: C 70,06%, H 7,47%, N 5,
07%.
ПРИМЕР 8
Получение соли N-[3(S)-[N,N-бис(фенилметил)амино]-2 (R)-гидрокси-4-фенилбутил]-N-изобутиламин дихлористоводородной кислоты.
Неочищенный 3(S)-[N, N-бис(фенилметил)амино]-1-(2 -метилпропил)амино-4-фенилбутан-2(R)-ол (10,0 г, который содержит около 6,41 г (15,39 ммоль) 3(S), 2(R) изомера и около 1,13 г (2,72 ммоль) 3(S), 2(S) изомера) растворяют в тетрагидрофуране (20,0 мл). К этому раствору добавляют хлористоводородную кислоту (20 мл, 6,0 N) в течение приблизительно 5 мин. Смесь перемешивают при комнатной температуре в течение приблизительно 1 часа и концентрируют при пониженном давлении. Остаток перекристаллизовывают из этанола при 0oC, получая 3,20 г (выделение 42,7% изомера) 98% диастереомерно чистой соли дихлористоводородной кислоты (исходя из площадей ВЭЖХ пика). Элем. анализ: Рассчит.: C 68,64%, H 7,76%, N 5,72%; Найдено: C 68,79%, H 8,07%, N 5,55%.
ПРИМЕР 9
Получение соли
N-[3(S)-[N,N-бис(фенилметил) амино]-2(R)-гидрокси-4-фенилбутил]-N-изобутиламин толуолсульфокислоты
Неочищенный 3(S)-[N, N-бис(фенилметил) амино]-1-(2-метил-пропил)амино-4-фенилбутан-2(R)-ол
(5,0 г, который содержит около 3,18 г (7,63 ммоль) 3(S), 2(R) изомера и около 0,56 г (1,35 ммоль) 3(S), 2(S) изомера) растворяют в метил-трет-бутиловом эфире (10,0 мл). К этому раствору добавляют
раствор толуолсульфокислоты (2,28 г, 12 ммоль) в метил-трет-бутиловом эфире (2,0 мл) и метаноле (2,0 мл) в течение приблизительно 5 мин. Смесь перемешивают при комнатной температуре в течение
приблизительно 2 часов и концентрируют при пониженном давлении. Остаток перекристаллизовывают из метил-трет-бутилового эфира и гептана при 0oC, фильтруют, промывают холодным гептаном и
сушат в вакууме, получая 1,85 г (выделение 40% изомера) 97% диастереомерно чистой соли монотолуолсульфокислоты (исходя из площадей ВЭЖХ пика).
ПРИМЕР 10
Получение соли
N-[3(S)-[N,N- бис(фенилметил)анино]-2(R)-гидрокси-4-фенилбутил]-N-изобутиламин метансульфокислоты
Неочищенный 3(S)-[N, N-бис(фенилметил)амино]-1-(2-метил-пропил)амино-4- фенилбутан-2(R)-ол (1,
68 г, который содержит около 6,85 г (16/44 ммоль) 3(S), 2(R) изомера и около 1,21 г (2,90 ммоль) 3(S), 2(S) изомера) растворяют в тетрагидрофуране (10,0 мл). К этому раствору добавляют
метансульфоновую кислоту (1,25 мл, 19,26 ммоль). Смесь перемешивают при комнатной температуре в течение приблизительно 2 часов и концентрируют при пониженном давлении. Маслянистый остаток
перекристаллизовывают из метанола и воды при 0oC, фильтруют, промывают холодной смесью метанол/вода (1:4) и сушат в вакууме, получая 2,40 г (выделение 28,5% изомера) 98% диастереомерно
чистой соли монометансульфоновой кислоты (исходя из площадей ВЭЖХ пика).
ПРИМЕР 11
Получение N-бензил-L-фенилаланинола
Способ 1:
L-Фенилаланинол (89,51 г, 0,
592 моль) растворяют в 375 мл метанола в атмосфере инертного газа, добавляют 35,52 г (0,592 моль) ледяной уксусной кислоты и 50 мл метанола, а затем раствор 62,83 г (0,592 моль) бензальдегида в 100 мл
метанола. Смесь охлаждают до приблизительно 15oC и добавляют раствор 134,6 г (2,14 моль) цианборогидрида натрия в 700 мл метанола в течение приблизительно 40 минут, поддерживая температуру
от 15oC до 25oC. Смесь перемешивают при комнатной температуре в течение 18 часов. Смесь концентрируют при пониженном давлении и распределяют между 1 л 2 М раствора гидроксида
аммония и 2 л эфира. Эфирный слой промывают 1 л 1 М раствора гидроксида аммония, дважды 500 мл воды, 500 мл рассола и сушат над сульфатом магния в течение 1 часа. Эфирный слой фильтруют, концентрируют
при пониженном давлении и неочищенный твердый продукт перекристаллизовывают из 110 мл этилацетата и 1,3 л гексана, получая 115 г (81% выход) N-бензил-L-фенилаланинола в виде белого твердого
вещества.
Способ 2:
L-Фенилаланинол (5 г, 33 ммоль) и 3,59 г (33,83 ммоль) бензальдегида растворяют в 55 мл 3А этанола в инертной атмосфере в аппарате Парра для встряхивания и
смесь нагревают до 60oC в течение 2,7 часов. Смесь охлаждают до приблизительно 25oC и добавляют 0,99 г 5% платины на угле и смесь гидрируют при давлении водорода 4,219 кг/см2 (60 psi) и 40oC в течение 10 часов. Катализатор отфильтровывают, реакционную смесь концентрируют при пониженном давлении и продукт - неочищенное твердое вещество
перекристаллизовывают из 150 мл гептана, получая 3,83 г (48% выход) N-бензил-L-фенилаланинола в виде белого твердого вещества.
ПРИМЕР 12
Получение
N-(т-Бутоксикарбонил)N-бензил-L- фенилаланинола
N-бензил-L-фенилаланинол (2,9 г, 12 ммоль) растворяют в 3 мл триэтиламина и 27 мл метанола и добавляют 5,25 г (24,1 ммоль) ди-трет-бутил
дикарбоната. Смесь нагревают до 60oC в течение 35 минут и концентрируют при пониженном давлении. Остаток растворяют в 150 мл этилацетата и промывают дважды 10 мл холодной (0-5oC),
разбавленной хлористоводородной кислоты (pH 2,5-3), 15 мл воды, 10 мл рассола, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт в виде масла очищают
хроматографией на силикагеле (этилацетат:гексан, 12:3 в качестве элюирующего растворителя), получая 3,98 г (97% выход) бесцветного масла.
ПРИМЕР 13
Получение
N-(т-Бутоксикарбонил)-N-бензил-L-фенилаланинала
Способ 1:
К раствору 0,32 г (0,94 ммоль) N-(т-бутоксикарбонил)- N-бензил-L-фенилаланинола в 2,8 мл толуола добавляют 2,4 мг (0,015
ммоль) 2,2,6,6-тетраметил-1- пиперидинилокси в виде свободного радикала (TEMPO), 0,1 г (0,97 ммоль) бромида натрия, 2,8 мл этилацетата и 0,34 мл воды. Смесь охлаждают до 0oC и медленно
добавляют водный раствор 4,2 мл 5% хлорной извести для бытовых нужд, содержащей 0,23 г (3,0 мл, 2,738 ммоль) бикарбоната натрия, в течение 30 минут. Смесь перемешивают при 0oC в течение 10
минут. Еще три порции (0,4 мл каждая) хлорной извести добавляют с последующим перемешиванием в течение 10 минут после каждого добавления, чтобы израсходовать все исходное вещество. Двухфазной смеси
дают возможность разделиться. Водный слой экстрагируют дважды 8 мл толуола. Объединенный органический слой промывают 1,25 мл раствора, содержащего 0,075 г иодида калия, бисульфата натрия (0,125 г) и
воды (1,1 мл), 1,25 мл 10% водного раствора тиосульфата натрия, 1,25 мл фосфатного буфера с pH 7 и 1,5 мл рассола. Органический раствор сушат над сульфатом магния, фильтруют и концентрируют при
пониженном давлении, получая 0,32 г (100% выход) N-(т-Бутоксикарбонил)-N-бензил-L-фенилаланинала.
Способ 2:
К раствору 2,38 г (6,98 ммоль)
N-(т-бутоксикарбонил)-N-бензил-L-фенилаланинола в 3,8 мл (27,2 ммоль) триэтиламина при 10oC добавляют раствор 4,33 г (27,2 ммоль) комплекса триоксид серы-пиридин в 17 мл диметилсульфоксида.
Смесь нагревают до комнатной температуры и перемешивают в течение одного часа. Добавляют воду (16 мл) и смесь экстрагируют 20 мл этилацетата. Органический слой промывают 20 мл 5% лимонной кислоты, 20
мл воды, 20 мл рассола, сушат над сульфатом магния и фильтруют. Фильтрат концентрируют при пониженном давлении, получая 2,37 г (100% выход) N-(т-бутоксикарбонил)-N-бензил-L-фенилаланинала.
ПРИМЕР 14
Получение 3(S)-[N-(т-бутоксикарбонил)-N-бензиламино] -1, 2- (S)-эпокси-4-фенилбутaнa
Способ 1:
Раствор 2,5 г (7,37 ммоль) N-(т-бутоксикарбонил)-N-бензил-L-фенилаланинала и 0,72 мл хлориодометана в 35 мл ТГФ охлаждают до -78oC. 4, 64 мл раствора н-бутиллития (1,6 М в гексане, 7,42 ммоль) добавляют медленно, поддерживая температуру ниже -70oC. Смесь перемешивают в течение 10 минут при температуре от -70 до -75oC. Две дополнительные порции 0,22 мл хлориодометана и 1,4 мл н-бутиллития добавляют последовательно и смесь перемешивают в течение 10 минут при поддержании температуры от -70 до -75o C после каждого добавления. Четыре дополнительные порции 0,11 мл хлориодометана и 0,7 мл н-бутиллития добавляют последовательно и смесь перемешивают в течение 10 минут при поддержании температуры от -70 до -75oC после каждого добавления. Смесь нагревают до комнатной температуры в течение 3,5 часов. Реакцию останавливают при температуре ниже 5oC добавлением 24 мл охлажденной льдом воды. Двухфазные слои разделяют и водный слой экстрагируют дважды 30 мл этилацетата. Объединенные органические слои промывают три раза по 10 мл воды, затем 10 мл рассола, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, получая 2,8 г желтого неочищенного масла. Неочищенное масло (> 100% выход) представляет собой смесь диастереомерных эпоксидов N, α S -бис(фенилметил)-N-(т- бутоксикарбонил)-2S-оксиранметанамина и N,α S-бис(фенилметил)-N- (т-бутоксикарбонил)-2R-оксиранметанамина. Неочищенную смесь используют непосредственно в следующей стадии без очистки.
Способ 2:
К суспензии 2,92 г (13,28 ммоль) триметилсульфоксоний иодида в 45 мл ацетонитрила добавляют 1,49 г (13,28 ммоль) т-бутоксида калия.
Добавляют раствор 3,0 г (8,85 ммоль) N-(т-бутоксикарбонил)-N-бензил-L-фенилаланинала в 18 мл ацетонитрила и смесь перемешивают при комнатной температуре в течение одного часа. Смесь разбавляют 150 мл
воды и экстрагируют дважды 200 мл этилацетата. Органические слои объединяют и промывают 100 мл воды, 50 мл рассола, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении,
получая 3,0 г желтого неочищенного масла. Неочищенный продукт очищают хроматографией на силикагеле (этилацетат/гексан: 1:8 в качестве элюирующего растворителя), получая 1,02 г (32,7% выход) смеси двух
диастереомеров N, α S-бис(фенилметил)-N-(т-бутоксикарбонил)-2S- оксиранметанамина и N, α S-бис(фенилметил)-N-(т-бутоксикарбонил) -2R-оксиранметанамина.
Способ 3:
К суспензии 0,90 г (4,42 ммоль) триметилсульфоний иодида в 18 мл ацетонитрила добавляют 0,495 г (4,42 ммоль) т-бутоксида калия. Добавляют раствор 1,0 г (2,95 ммоль)
N-(т-бутоксикарбонил)-N-бензил-L-фенилаланинала в 7 мл ацетонитрила и смесь перемешивают при комнатной температуре в течение одного часа. Смесь разбавляют 80 мл воды и экстрагируют дважды 80 мл
этилацетата. Органические слои объединяют и промывают 100 мл воды, 30 мл рассола, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая 1,04 г желтого неочищенного
масла. Неочищенный продукт представляет собой смесь двух диастереомеров N, α S-бис (фенилметил)-N-(т-бутоксикарбонил)-2S-оксиранметанамина и N, α
S-бис(фенилметил)-N-(т-бутоксикарбонил)-2R-оксиранметанамина.
ПРИМЕР 15
Получение 3(S)-[N-(т-Бутоксикарбонил)-N-(фенилметил) амино] -1-(2-метилпpoпил)аминo-4-фенилбутан-2R-oла
К раствору 500 мг (1,42 ммоль) неочищенного эпоксида (смесь двух стереомеров N, α S-бис(фенилметил)-N-(т-бутоксикарбонил) -2S-оксиранметанамина и N, α S-бис(фенилметил)-N-(т-бутоксикарбонил)-2R-оксиранметанамина) в 98 мл изопропанола добавляют 0,71 мл (7,14 ммоль) изобутиламина. Смесь нагревают с обратным холодильником при 85-90oC в течение 1,5 часов. Смесь концентрируют при пониженном давлении и продукт в виде масла очищают хроматографией на силикагеле (хлороформ: метанол, 100:6 в качестве элюирующих растворителей), получая 330 мг 3S-[N-т-бутоксикарбонил)-N-(фенилметил)амино]-1-(2- метилпропил)амино-4-фенилбутан-2R-ола в виде бесцветного масла (54,5% выход). Так же выделяют 3S-[N-т-бутоксикарбонил)-N- (фенилметил)амино] -1-(2-метилпропил)амино-4-фенилбутан-2S-ол. Когда в качестве исходного вещества используют очищенный N, α S- бис(фенилметил)-N-(т-бутоксикарбонил)-2S-оксиранметанамин, то 3S-[N-т-бутoкcикapбoнил)-N-(фенилметил)амино] -1-(2-метилпропил) амино-4-фенилбутан-2R-ол выделяют после очистки хроматографией с 86% выходом.
ПРИМЕР 16

Получение 3S-(N-т-Бутоксикарбонил)амино-4-фенилбутан-1, 2R-диола
К раствору 1 г (3,39 ммоль) 2S-(N-т-бутоксикарбониламино- 1S-гидрокси-3-фенилбутановой кислоты (коммерчески доступна от Nippon Kayaku, Japan) в 50 мл ТГФ при 0oC добавляют 50 мл комплекса боран-ТГФ (жидкость, 1,0 М в ТГФ), поддерживая температуру ниже 5oC. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 16 часов. Смесь охлаждают до 0oC и медленно добавляют 20 мл воды, чтобы разрушить избыток BH3 и остановить реакцию, поддерживая температуру ниже 12oC. Реакционную смесь перемешивают в течение 20 минут и концентрируют при пониженном давлении. Продукт в виде смеси экстрагируют три раза 60 мл этилацетата. Органические слои объединяют и промывают 20 мл воды, 25 мл насыщенного водного раствора хлорида натрия и концентрируют при пониженном давлении, получая 1,1 г неочищенного масла. Неочищенный продукт очищают хроматографией на силикагеле (хлороформ/метанол, 10: 6 в качестве элюирующих растворителей), получая 900 мг (94,4% выход) 3S-(N-т- бутоксикарбонил)амино-4-фенилбутан-1,2R-диола в виде белого твердого вещества.
ПРИМЕР 17
Получение 3S-(N-т-бутоксикарбонил)амино-2R-гидрокси-4- фенил-бут-1-ил толуолсульфоната
К раствору 744,8 мг (2,65 ммоль) 3S-(N-т-бутоксикарбонил)амино-4-фенилбутан-1,2R-диола в 13 мл пиридина при 0oC добавляют 914 мг толуолсульфонил хлорида одной порцией. Смесь перемешивают при 0oC-5oC в течение 5 часов. Смесь 6,5 мл этилацетата и 15 мл 5% водного раствора бикарбоната натрия добавляют к реакционной смеси и перемешивают в течение 5 минут. Полученную смесь экстрагируют три раза по 50 мл этилацетата. Органические слои объединяют и промывают 15 мл воды, 10 мл насыщенного раствора хлорида натрия и концентрируют при пониженном давлении, получая около 1,1 г желтого твердого вещества в виде комков. Неочищенный продукт очищают хроматографией на силикагеле (этилацетат/гексан 1: 3 в качестве элюирующих растворителей), получая 850 мг (74% выход) 3S-(N-т-бутоксикарбонил)амино-2R- гидрокси-4-фенилбут-1-ил толуолсульфоната в виде белого твердого вещества.
ПРИМЕР 18

Получение 3S-[N-(т-бутоксикарбонил)амино] -1-(2- метилпропил)амино-4-фенилбутан-2R-ола
К раствору 90 мг (0,207 ммоль) 3S-(N-т-бутоксикарбонил)амино-2R-гидрокси-4-фенилбут-1-ил толуолсульфоната в 0,143 мл изопропанола и 0,5 мл толуола добавляют 0,103 мл (1,034 ммоль) изобутиламина. Смесь нагревают до 80-85o C и перемешивают в течение 1,5 часов. Продукт-смесь концентрируют при пониженном давлении при 40-50oC и очищают хроматографией на силикагеле (хлороформ/метанол, 10:1 в качестве элюирующих растворителей), получая 54,9 мг (76,8% выход) 3S-[N-(т- бутоксикарбонил)амино]-1-(2-метилпропил)амино-4-фенилбутан-2R- диола в виде белого твердого вещества.
ПРИМЕР 19
Получение N-[3(S)-бензилоксикарбониламино-2(R)-гидрокси- 4-фенилбутил] -N-изобутиламина
Часть A:
К раствору 75,0 г (0,226 моль) N-бензилоксикарбонил-L-фенилаланин хлорметил кетона в смеси 807 мл метанола и 807 мл тетрагидрофурана при -2oC, добавляют 13,17 г (0,348 моль, 1, 54 экв.) твердого борогидрида натрия в течение 100 минут. Растворители удаляют при пониженном давлении при 40oC и остаток растворяют в этилацетате (прибл. 1 л). Раствор промывают последовательно 1 М раствором кислого сульфата калия, насыщенным раствором бикарбоната натрия и затем насыщенным раствором хлорида натрия. После сушки над безводным сульфатом магния и фильтрования, растворитель удаляют при пониженном давлении. К полученному маслу добавляют гексан (прибл. 1 л) и смесь нагревают до 60oC с вихревым перемешиванием. После охлаждения до комнатной температуры, твердые вещества собирают и промывают 2 л гексана. Полученное твердое вещество перекристаллизовывают из горячего этилацетата и гексана, получая 32,3 г (43% выход) N-бензилоксикарбонил-3(S)-амино-1- хлоро-4-фенил-2(S)-бутанола, т. пл. 150-151oC и M+Li+ = 340.
Часть В:
К раствору 6,52 г (0,116 моль, 1,2 экв.)
гидроксида калия в 968 мл абсолютного этанола при комнатной температуре добавляют 32,3 г (0,097 моль) N-CBZ-3(S)-aмино-1-хлоро-4-фенил- 2(S)-бутанола. После перемешивания в течение пятнадцати минут
растворитель удаляют при пониженном давлении и твердые вещества растворяют в метиленхлориде. После промывания водой, сушки над сульфатом магния, фильтрования и выпаривания, получают 27,9 г белого
твердого вещества. Перекристаллизация из горячего этилацетата и гексана дает 22,3 г (77% выход) N-бензилоксикарбонил-3(S)-амино-1,2(S)-эпокси-4-фенилбутана, т. пл. 102-103oC и МН+
298.
Часть C:
Раствор N-бензилоксикарбонил-3(S)-амино-1,2(S)-эпокси-4-фенилбутана (1,00 г, 3,36 ммоль) и изобутиламина (4,90 г,67,2 ммоль, 20 экв.) в 10 мл изопропилового
спирта нагревают до температуры кипения с обратным холодильником в течение 1,5 часов. Раствор охлаждают до комнатной температуры, концентрируют в вакууме и затем выливают в 100 мл перемешиваемого
гексана, после чего продукт кристаллизуется из раствора. Продукт отделяют фильтрацией и сушат на воздухе, получая 1,18 г, 95% N-[[3(S)-фенилметилкарбамоил) амино-2(R)-гидрокси-4-фенилбутил]
N- [(2-метилпропил)] амин, C23H30N2O3 т. пл. 108,0-109,5oC, MH+ м/з = 371.
ПРИМЕР 20

Получение фенилметил[2R-гидрокси-3-[(3-метилбутил) (фенилсульфонил)амино]-1S-(фенилметил)пропил]карбамата
Реакцией N[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4- фенилбутил] N-изоамиламина (1,47 г, 3,8 ммоль), триэтиламина (528 мкл, 3,8 ммоль) и бензолсульфонилхлорида (483 мкл, 3,8 ммоль) получают фенилметил[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил) амино]-1S-(фенилметил)пропил]карбамат. Колоночная хроматография на силикагеле, при элюировании хлороформом, содержащим 1% этанола, дает чистый продукт. Элем. анал, Рассчит. для C29H36N2O5S: C, 66,39; H, 6,92; N, 5,34. Найдено: C, 66,37; H, 6,93; N, 5,26.
ПРИМЕР 21
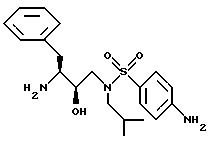
Получение 2R-гидрокси-3-[[(4-аминофенил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламина
Часть A: Получение фенилметилового эфира 2R-гидрокси-3- [[(4-нитрофенил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил) пропилкарбаминовой кислоты.
К раствору 4,0 т (10,8 ммоль) N-[3S- бензилоксикарбониламино-2R-гидрокси-4-фенил] -N-изобутиламина в 50 мл безводного метиленхлорида добавляют 4,5 мл (3,27 г, 32,4 ммоль) триэтиламина. Раствор охлаждают до 0oC и добавляют 2,63 г (11,9 ммоль) 4-нитробензол сульфонилхлорида, перемешивают в течение 30 минут при 0oC, затем в течение 1 часа при комнатной температуре. Добавляют этилацетат, промывают 5% лимонной кислотой, насыщенным бикарбонатом натрия, рассолом, сушат и концентрируют, получая 5,9 г неочищенного вещества. Его перекристаллизовывают из смеси этилацетат/гексан, получая 4,7 г чистого фенилметилового эфира 2R-гидрокси-3-[[(4-нитрофенил)сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропилкарбаминовой кислоты, м/е = 556 (М+Н).
Часть B: Получение
2R-гидрокси-3-[[(4-аминофенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропиламина
Раствор 3,0 г (5,4 ммоль) фенилметилового эфира
2R-гидрокси-3-[[(4-нитрофенил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропилкарбаминовой кислоты в 20 мл этилацетата гидрируют на 1,5 г 10% палладий-на-угле катализаторе при избыточном
давлении водорода 2,461 кг/см2 (35 psig) в течение 3,5 часов. Катализатор удаляют фильтрацией и раствор концентрируют, получая 2,05 г требуемого 2R-гидрокси-3-[[(4-аминофенил)сульфонил]
(2-метилпропил)амино] -1S- (фенилметил)пропиламина, м/е = 392 (М+H).
ПРИМЕР 22
Получение 2R-гидрокси-3[[(3-аминофенил) сульфонил](2-метилпропил) амино-1S-(фенилметил)пропиламина
Часть 1: Получение фенилметилового эфира [2R-гидрокси-3-[(3-нитрофенилсульфонил)(2- метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты
К раствору 1,1 г (3,0 ммоль) N-[3S-бензилоксикарбониламино- 2R-гидрокси-4-фенил] -N-изобутиламина в 15 мл безводного метиленхлорида добавляют 1,3 мл (0,94 г, 9,3 ммоль) триэтиламина. Раствор охлаждают до 0oC и добавляют 0,67 г (3,0 ммоль) 3-нитробензол сульфонилхлорида, перемешивают в течение 30 минут при 0oC, затем в течение 1 часа при комнатной температуре. Добавляют этилацетат, промывают 5% лимонной кислотой, насыщенным бикарбонатом натрия, рассолом, сушат и концентрируют, получая 1,74 г неочищенного вещества. Его перекристаллизовывают из смеси этилацетат/гексан, получая 1,40 г чистого фенилметилового эфира [2R-гидрокси-3-[(3-нитрофенилсульфонил)(2-метилпропил)амино] - 1S-(фенилметил)пропил-карбаминовой кислоты, м/е = 562 (M+Li).
Часть B: Получение
[2R-гидрокси-3-[[(3-аминофенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропиламина
Раствор 1,33 г (2,5 ммоль) фенилметилового эфира
[2R-гидрокси-3- [(3-нитрофенилсульфонил)(2-метилпропил)амино] -1S-(фенилметил) пропилкарбаминовой кислоты в 40 мл 1:1 смеси метанол/тетрагидрофуран гидрируют в присутствии 0,70 г 10% палладия-на-угле
при избыточном давлении водорода 2,812 кг/см2 (40 psig) в течение 1,5 часов. Катализатор удаляют фильтрацией и раствор концентрируют, получая 0,87 г требуемого
[2R-гидрокси-3- [[(3-аминофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина.
ПРИМЕР 23

Получение 2R-гидрокси-3[[(2,3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропиламина
Часть A: Получение 5-(2, 3-дигидробензофуранил)сульфонил хлорида
К раствору 3,35 г безводного N,N-диметилформамида при 0oC в атмосфере азота добавляют 6,18 г сульфурил хлорида, после чего образуется твердое вещество. После перемешивания в течение 15 минут, добавляют 4,69 г 2,3-дигидробензофурана и смесь нагревают при 100oC в течение 2 часов. Реакционную смесь охлаждают, выливают в ледяную воду, экстрагируют метиленхлоридом, сушат над сульфатом магния, фильтруют и концентрируют неочищенное вещество. Его перекристаллизовывают из этилацетата, получая 2,45 г 5-(2, 3-дигидробензофуранил)сульфонил хлорида.
Часть B: Получение фенилметилового эфира 2R-гидрокси-3-[[(2, 3-дигидробензофуран-5-ил)сульфонил](2-метилпропил)амино]- 1S-(фенилметил)пропилкарбаминовой кислоты.
К раствору 1,11 г (3,0 ммоль)N-[3S-бензилоксикарбониламино-2R- гидрокси-4-фенил] -N-изобутиламина в 20 мл безводного метиленхлорида добавляют 1,3 мл (0,94 г, 9,3 ммоль) триэтиламина. Раствор охлаждают до 0oC и добавляют 0,66 г 5-(2,3-дигидробензофуранил)сульфонил хлорида, перемешивают в течение 15 минут при 0oC, затем в течение 2 часов при комнатной температуре. Добавляют этилацетат, промывают 5% лимонной кислотой, насыщенным бикарбонатом натрия, рассолом, сушат и концентрируют, получая 1,62 г неочищенного вещества. Его перекристаллизовывают из диэтилового эфира, получая 1,17 г чистого фенилметилового эфира [2R-гидрокси-3-[[(2, 3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил)пропилкарбаминовой кислоты.
Часть C: Получение [2R-гидрокси-3-[[(2,3-дигидробензофуран-5-ил)
сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина
Раствор 2,86 г фенилметилового эфира [2R-гидрокси-3- [[(2,3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)
пропилкарбаминовой кислоты в 30 мл тетрагидрофурана гидрируют в присутствии 0,99 г 10% палладия-на-угле при избыточном давлении водорода 3,515 кг/см2 (50 psig) в течение 16 часов.
Катализатор удаляют фильтрацией и фильтрат концентрируют, получая 1,99 г требуемого [2R-гидрокси-3-[[(2,3- дигидробензофуран-5-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропиламина.
ПРИМЕР 24
Получение N-[(1,1-диметилэтоксил)карбонил] -N-[2- метилпропил] -3S-(N1-(фенилметоксикарбонил)амино] -2R-гидpoкcи-4-фенилбутиламина
К раствору 7,51 г (20,3 ммоль) N-[3S-[(фенилметоксикарбонил)амино]-2R-гидрокси-4-фенилбутил] -2- метилпропиламина в 67 мл безводного тетрагидрофурана добавляют 2,25 г (22,3 ммоль) триэтиламина. После охлаждения до 0oC, добавляют 4,4 г (20,3 ммоль) ди-трет-бутилдикарбоната и перемешивание продолжают при комнатной температуре в течение 21 часов. Летучие вещества удаляют в вакууме, добавляют этилацетат, затем промывают 5% лимонной кислотой, насыщенным бикарбонатом натрия, рассолом, сушат над сульфатом магния, фильтруют и концентрируют, получая 9,6 г неочищенного продукта. Хроматография на силикагеле с использованием смеси 30% этилацетат/гексан дает 8,2 г чистого N-[[3S-(фенилметилкарбамоил)амино] -2R-гидрокси-4- фенил] -1-[(2-метилпропил)амино-2-(1,1-диметилэтоксил)карбонил] бутана, масс-спектр м/е = 477 (M+Li).
ПРИМЕР 25
Получение N1-[2R-гидрокси-3-[N2-(3-метилбутил) -N2 -(фенилсульфонил)амино]-1S-(фенилметил)пропил]-2S-амино-3,3- диметилбутанамида
Часть A:
К раствору N-CBZ-L-трет-лейцина (450 мг, 1,7 ммоль) и N-гидроксибензотриазола (260 мг, 1,7 ммоль) в ДМФ (10 мл) добавляют EDC (307 мг, 1,6 ммоль). Раствор перемешивают в течение 60 минут при комнатной температуре и затем добавляют 2R-гидрокси-3-[N-(3-метилбутил)-N-(фенилсульфонил)амино] -1S-(фенилметил) пропиламин (585 мг, 1,5 ммоль) в ДМФ (2 мл). Реакционную смесь перемешивают в течение 16 часов при комнатной температуре, затем выливают в 50%-ный насыщенный раствор бикарбоната натрия (200 мл). Водную смесь экстрагируют трижды этилацетатом (50 мл). Объединенные этилацетатные слои промывают водой (50 мл) и насыщенным раствором NaCl (50 мл), затем сушат над сульфатом магния. Фильтрация и концентрирование дает масло, которое хроматографируют на силикагеле (50 гм), элюируя 20% этилацетатом в гексане. Получают фенилметил [1S-[[[2R- гидрокси-3-[(3-метилбутил)фенилсульфонил)амино]-1S-(фенилметил)пропил] амино] карбонил] -2,2-диметилпропил]карбамат в виде твердого вещества. Элем. анал. Рассчит. для C35H47N3O6S: C, 65,91; H, 7,43; N, 6,59. Найдено: C, 65,42; H, 7,24; N, 6,55.
Часть B:
Раствор фенилметил [1S-[[[2R-гидрокси-3-[(3-метилбутил)
(фенилсульфонил)амино] -1S-(фенилметил)пропил] амино] карбонил] -2,2- диметилпропил] карбамата (200 мг, 0,31 ммоль) в метаноле (15 мл) гидрируют в присутствии 10
ПРИМЕР 26
Получение гидрохлоридной соли N1-[2R-гидрокси-3-[N2-(2-метилбутил)-N2 - (фенилсульфонил)амино]-1S-(фенилметил) пропил]-2S-aминo-3S-метилпентамидина
Часть A:
К раствору 2R-гидрокси-3-[N-(3-метилбутил)-N- (фенилсульфонил)амино]-1S-(фенилметил)пропиламина (2,79 г, 7,1 ммоль) в 27 мл диоксана добавляют (2,3 г, 7,1 ммоль) сложный эфир N-трет-бутилкарбонил-L-изолейцин-N-гидроксисукцинамида и реакционную смесь перемешивают в атмосфере азота в течение 16 часов. Реакционную смесь концентрируют в вакууме и остаток растворяют в этилацетате, промывают кислым сульфатом калия (5% водный), насыщенным бикарбонатом натрия и насыщенным хлоридом натрия. Органический слой сушат над сульфатом магния, фильтруют и концентрируют, получая 4,3 г неочищенного вещества, которое хроматографируют, используя 3:1 смесь этилацетат:гексан, получая 3,05 г, 72% выход 2S-[[(1,1-диметилэтокси)карбонил]амино]-N- [2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино] -1S- (фенилметил)пропил] -3-метилпентанамида.
Часть B:
(3,05 г, 5,0 ммоль)
продукта Части A растворяют в 20 мл 4N HCl в диоксане и перемешивают в атмосфере азота в течение 1,5 часов. Содержимое концентрируют в вакууме и промывают диэтиловым эфиром. Неочищенную гидрохлоридную
соль сушат при 1 мм Hg до сухого остатка, получая 2,54 г продукта в виде его гидрохлоридной соли.
ПРИМЕР 27

Получение N1-[2R-гидрокси-3-[N2-(2-метилпропил)-N2 - (4-метоксифенилсульфонил)амино]-1S-(фенилметил)пропил]-2S- амино-3S-метилпентанамида
Часть A:
К раствору 2R-гидрокси-3-[(2-метилпропил)(4-метоксифенилсульфонил) амино] -1S-(фенилметил)пропиламина (1,70 г, 4,18 ммоль) в 40 мл дихлорметана добавляют сложный эфир N-карбобензилокси-N-изолейцин-N- гидроксисукцинамида (1,51 г, 4,18 ммоль) и раствор перемешивают в атмосфере азота в течение 16 часов. Содержимое концентрируют в вакууме и остаток повторно растворяют в этилацетате. Этилацетатный раствор промывают водным раствором 5% KHSO4, насыщенным бикарбонатом натрия, насыщенным хлоридом натрия, сушат над сульфатом магния, фильтруют и концентрируют, получая 2,47 г неочищенного продукта. Продукт очищают хроматографией на силикагеле с использованием элюента - смесь 2:1 гексан:этилацетат, получая 2,3 г (84% выход) 2S-[(карбобензилокси)амино] -N-[2R- гидрокси-3-[(3-метилпропил)(4-метоксифенилсульфонил)амино]-1S- (фенилметил)пропил]-3S-метилпентанамида.
Часть B:
(1,18 г, 1,8 ммоль) продукта Части A растворяют в 50
мл метанола и к нему добавляют 250 мг 10% палладия на угле в потоке азота. Суспензию гидрируют, используя избыточное давление водорода 3,515 кг/см2 (50 psig) в течение 20 часов. Содержимое
продувают азотом и фильтруют через целит и концентрируют в вакууме, получая 935 мг
2S-(амино)-N-[2R-гидрокси-3-[(3- метилпропил)(4-метоксифенилсульфонил)амино]- 1S-(фенилметил)пропил]-3S-метилпентанамида, который используют без дополнительной очистки.
ПРИМЕР 28

Получение фенилметилового эфира 2R-гидрокси-3-[[(2-амино- бензотиазол-6-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты.
Фенилметиловый эфир 2R-гидрокси-3-[[(4-аминофенил)сульфонил]
(2-метилпропил)амино] -1S-(фенилметил)пропилкарбаминовой кислоты 0,30 г (0,571 ммоль) добавляют к хорошо перемешиваемому порошку безводного сульфата меди (1,20 г) и тиоцианата калия (1,50 г), а затем
добавляют сухой метанол (6 мл) и полученную черно-коричневую суспензию нагревают с обратным холодильником в течение 2 часов. Реакционную смесь фильтруют и фильтрат разбавляют водой (5 мл) и нагревают
с обратным холодильником. К реакционной смеси добавляют этанол, охлаждают и фильтруют. После концентрирования фильтрата получают остаток, который хроматографируют (этилацетат:гексан 80:20),
получая 0,26 г (78%) целевого соединения в виде твердого вещества.
ПРИМЕР 29

Получение фенилметилового эфира 2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропилкарбаминовой кислоты
Способ 1:
Фенилметиловый эфир 2R-гидрокси-3-[[(2-аминобензотиазол-6-ил) сульфонил] (2-метилпропил)амино] -1S-(фенилметил) пропилкарбаминовой кислоты (0,25 г, 0,429 ммоль) добавляют к раствору изоамилнитрита (0,116 мл, 0,858 ммоль) в диоксане (5 мл) и смесь нагревают при 85oC. После прекращения выделения азота, реакционную смесь концентрируют и остаток очищают хроматографией (гексан: этилацетат 5: 3), получая 0,130 г (53%) целевого продукта в виде твердого вещества.
Способ 2:
Неочищенный бензотиазол-6-сульфонил хлорид в этилацетате (100 мл) добавляют к
N-[3S-бензилоксикарбониламино-2R- гидрокси-4-фенил]-N-изобутиламину (1,03 г, 2,78 ммоль), а затем N-метилморфолин (4 мл). После перемешивания при комнатной температуре в течение 18 часов, реакционную
смесь разбавляют этилацетатом (100 мл), промывают лимонной кислотой (5%, 100 мл), бикарбонатом натрия (насыщенный, 100 мл) и рассолом (100 мл), сушат (MgSO4) и концентрируют в вакууме.
Остаток хроматографируют (силикагель, этилацетат:гексан 1:1), получая 0,340 г (23%) целевого продукта.
ПРИМЕР 30

и

Получение фенилметилового эфира 2R-гидрокси-3-[[(2- аминобензотиазол-5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты и фенилметилового эфира 2R-гидрокси-3-[[(2-аминобензотиазол-7-ил)сульфонил](2-метилпропил)амино]- 1S-(фенилметил)пропилкарбаминовой колоты
Фенилметиловый эфир 2R-гидрокси-3[[(3-аминофенилсульфонил](2- метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты 0,36 г (0,685 ммоль) добавляют к хорошо перемешиваемому порошку безводного сульфата меди (1,44 г) и тиоцианата калия (1,80 г) с последующим добавлением сухого метанола (10 мл) и полученную черно-коричневую суспензию нагревают с обратным холодильником в течение 2 часов. Реакционную смесь фильтруют и фильтрат разбавляют водой (5 мл) и нагревают с обратным холодильником. К реакционной смеси добавляют этанол, охлаждают и фильтруют. После концентрирования фильтрата получают остаток, который хроматографируют (этилацетат: гексан 1:1), получая 0,18 г (45%) 7-изомера в виде твердого вещества. Дополнительное элюирование колонки смесью (этилацетат: гексан 3: 2) дает 0,80 г (20%) получаемого 5-изомера в виде твердого вещества.
ПРИМЕР 31

Получение 3S-амино-1-[N-(2-метилпропил)-N-(4-метоксифенил- сульфонил] амино]-4-фенил-2R-бутанола
Часть A: N-бензилоксикарбонил-3(S)-амино-1-хлор-4-фенил-2(S)-бутанол
К раствору N-бензилоксикарбонил-L-фенилаланин хлорметил кетона (75 г" 0,2 моль) в смеси 800 мл метанола и 800 мл тетрагидрофурана добавляют борогидрид натрия (13,17 г, 0,348 моль, 1,54 экв.) в течение 100 мин. Раствор перемешивают при комнатной температуре в течение 2 часов и затем концентрируют в вакууме. Остаток растворяют в 1000 мл этилацетата и проплывают 1N KHSO4, насыщенным водным NaHCO3, насыщенным водным NaCl, сушат над безводным MgSO4, фильтруют и концентрируют в вакууме, получая масло. Неочищенный продукт растворяют в 1000 мл гексана при 60oC и дают возможность охладиться до комнатной температуры, после чего образуются кристаллы, которые отделяют фильтрацией и промывают обильными количествами гексанов. Затем полученное твердое вещество перекристаллизовывают из горячего этилацетата и гексанов, получая 32,3 г 43% N-бензилоксикарбонил-3(S)-амино-1-хлор-4-фенил-2(S)-бутанола, т. пл. 150-151oC, ББА (FAB) MC:MLi+ = 340.
Часть B: 3(S)-[N-(бензилоксикарбонил)амино]-1,2(S)- эпокси-4-фенилбутан
Раствор гидроксида калия (6,52 г, 0,116 моль, 1,2 экв.) в 970 мл абсолютного этанола обрабатывают
N-бензилоксикарбонил-3(S)-амино-1-хлоро-4-фенил-2(S)-бутанолом (32,3 г, 0,097 моль). Раствор перемешивают при комнатной температуре в течение 15 минут и затем концентрируют в вакууме, получая белое
твердое вещество. Твердое вещество растворяют в дихлорметане и промывают водой, сушат над безводным MgSO4 фильтруют и концентрируют в вакууме, получая белое твердое вещество. Твердое
вещество кристаллизуют из гексанов и этилацетата, получая 22,3 г 77% 3(S)-[N-(бензилоксикарбонил)амино]-1,2(S)-эпокси-4- фенилбутана, т.пл. 102-103oC, ББА (FAB) MC:МН+ = 298.
Часть C: N-[3(S)-бензилоксикарбониламино-2(R)-гидрокси- 4-фенил]N-изобутиламин
Раствор N-бензилоксикарбонил-3(S)-амино-1,2-(S) -эпокси-4-фенилбутана (50,0 г, 0,168 моль) и
изобутиламина (246 г, 3,24 моль, 20 эквивалентов) в 650 мл изопропилового спирта нагревают с обратным холодильником в течение 1,25 часов. Раствор охлаждают до комнатной температуры, концентрируют в
вакууме и затем выливают в 1 л перемешиваемого гексана, после чего продукт кристаллизуется из раствора. Продукт отделяют фильтрацией и сушат на воздухе, получая 57,56 г, 92%
N[(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенил] -N-изобутиламина, т. пл. 108,0-109,5oC, МН+ м/з = 371.
Часть D: Фенилметил
[2(R)-гидрокси-3-[N-(2-метилпропил)- N-(4-метоксифенилсульфонил)амино]-1S-(фенилметил)пропил]карбамат
Амин Части C (936,5 мг, 2,43 ммоль) и триэтиламин (288,5 мг, 2,85 ммоль) растворяют в 20
мл дихлорметана и обрабатывают 4-метоксибензолсульфонил хлоридом (461 мг, 2,61 ммоль). Раствор перемешивают при комнатной температуре в течение 16 часов и затем концентрируют в вакууме. Остаток
растворяют в этилацетате и раствор промывают 1N KHSO4, насыщенным водным NaHCO3 рассолом, сушат над безводным MgSO4, фильтруют и концентрируют, получая прозрачное
масло 1,234 г. Масло кристаллизуют из смеси эфира и гексанов, 729,3 мг, 56,5% т. пл. 95-99oC, ББА (FAB) МС:МН+ = 511.
Часть E:
3(S)-амино-1-[N-(2-метилпропил)-N-(4- метоксифенилсульфонил)амино]-4-фенил-2R-бутанол
Раствор фенилметил [2(R)-гидрокси-3-[N-(2-метилпропил)-N-(4- метоксифенилсульфонил)амино]
1-S-(фенилметил)пропил карбамата (671,1 мг, 1,31 ммоль) Части D в 10 мл метанола гидрируют в присутствии 50 мг 10% палладия на угле при избыточном давлении 2,812 кг/см2 (40 psig) при
комнатной температуре в течение 15 часов. Катализатор удаляют фильтрацией через диатомовую землю и фильтрат концентрируют, получая белую пену, 474,5 мг, 96%, ББА (FAB) МС:МН+ = 377.
ПРИМЕР 32

Получение 1,3-бензодиоксол-5-сульфонил хлорида
Способ 1:
К раствору 4,25 г безводного N,N-диметилформамида при 0oC в атмосфере аргона добавляют 7,84 г сульфурил хлорида, после чего образуется твердое вещество. После перемешивания в течение 15 минут, добавляют 6,45 г 1,3-бензодиоксола и смесь нагревают при 100oC в течение 2 часов. Реакционную смесь охлаждают, выливают в ледяную воду, экстрагируют метиленхлоридом, сушат над сульфатом магния, фильтруют и концентрируют, получая 7,32 г неочищенного вещества в виде черного масла. Его хроматографируют на силикагеле, используя 20% метиленхлорид/гексан, получая 1,9 г (1, 3-бензодиоксол-5-ил)сульфонил хлорида.
Способ 2:
В 22-литровую круглодонную колбу, снабженную механической мешалкой, холодильником, нагревающим кожухом и капельной воронкой с
уравновешенным давлением, помещают триоксид серы-ДМФ комплекс (2778 г, 18,1 моль). Затем добавляют дихлорэтан (4 литра) и начинают перемешивание. Затем через капельную воронку в течение пяти минут
добавляют 1,3-бензодиоксол (1905 г, 15,6 моль). Затем температуру повышают до 75oC и поддерживают в течение 22 часов (ЯМР указывает на то, что реакция завершается, спустя 9 часов).
Реакционную смесь охлаждают до 26oC и добавляют оксалил хлорид (2290 г, 18,1 моль) с такой скоростью, чтобы поддерживать температуру ниже 40oC (1,5 часа). Смесь нагревают до
67oC в течение 5 часов, а затем охлаждают до 16oC на бане со льдом. Реакционную смесь гасят водой (5 л) при скорости, которая позволяет поддержать температуру ниже 20o
C. После завершения добавления воды, смесь перемешивают в течение 10 минут. Слои разделяют и органический слой промывают дважды (5 л) H2O. Органический слой сушат сульфатом магния (500 г) и
фильтруют, чтобы удалить осушитель. Растворитель удаляют в вакууме при 50oC. Полученной теплой жидкости дают возможность охладиться, и в это время начинает образовываться твердое вещество.
Спустя один час, твердое вещество промывают гексаном (400 мл), фильтруют и сушат, получая требуемый сульфонил хлорид (2823 г). Промывные гексановые воды концентрируют и полученное твердое вещество
промывают 400 мл гексана, получая дополнительный сульфонил хлорид (464 г). Суммарный выход составляет 3287 г (95,5% в расчете на 1,3-бензодиоксол).
Способ 3:
1,
4-бензодиоксан-6-сульфонил хлорид получают согласно способу, раскрытому в ЕП 583960, который включен сюда в качестве ссылки.
ПРИМЕР 33

Получение 1-[N-[1,3-бензодиоксол-5-ил)сульфонил] -N-(2- метилпропил)амино-3(S)-[бис(фенилметил)амино]-4-фенил-2(R)-бутанола
Способ 1:
В 5000 мл, 3-горлую колбу, снабженную механической мешалкой, помещают соль N-[3(S)-[N,N-бис(фенилметил)амино]-2(R)-гидрокси-4- фенилбутил]-N-изобутиламин щавелевой кислоты (354,7 г, 0,7 моль) и 1,4-диоксан (2000 мл). Затем добавляют раствор карбоната калия (241,9 г, 1,75 моль) в воде (250 мл). Полученную гетерогенную смесь перемешивают в течение 2 часов при комнатной температуре с последующим добавлением 1,3-бензодиоксол-5-сульфонил хлорида (162,2 г, 0,735 моль), растворенного в 1,4-диоксане (250 мл), в течение 15 минут. Реакционную смесь перемешивают при комнатной температуре в течение 18 часов. В реактор загружают этилацетат (1000 мл) и воду (500 мл) и продолжают перемешивание в течение еще 1 часа. Водный слой отделяют и затем экстрагируют этилацетатом (200 мл). Объединенные этилацетатные слои промывают 25% рассолом (500 мл) и сушат над безводным сульфатом магния. После фильтрования и промывки сульфата магния этилацетатом (200 мл), растворитель в фильтрате удаляют при пониженном давлении, получая требуемый сульфонамид в виде вязкого желтого пенистого масла (440,2 г, 105% выход). ВЭЖХ/МС (HPLC/MS) (электрораспыление) (м/з 601 [М+H]+].
ПРИМЕР 34

Получение соли 1-[N-[(1, 3-бензодиоксол-5-ил)сульфонил]-N- (2-метилпропил)амино]-3(S)-амино-4-фенил-2(R)-бутанол метансульфоновой кислоты
Способ 1: Неочищенный 1-[N-[(1, 3-бензодиоксол-5-ил)сульфонил]-N- (2-метилпропил)амино] -3(S)-[бис(фенилметил)амино] -4-фенил-2(R)-бутанол (6,2 г, 0,010 моль) растворяют в метаноле (40 мл). Затем к раствору добавляют метансульфоновую кислоту (0,969 г, 0,010 моль) и воду (5 мл). Смесь помещают в 500 мл реактор Парра для гидрирования, содержащий 20% Pd(OH)2 на угле (255 мг, 50% содержание воды). Реактор помещают в гидрогенизатор и продувают 5 раз азотом и 5 раз водородом. Допускают протекание реакции при 35oC при давлении водорода 4,43 кг/см2 (63 PSI) в течение 18 часов. Добавляют дополнительный катализатор (125 мг) и, после продувания, гидрирование продолжают в течение еще 20 часов. Смесь фильтруют через целит, который промывают метанолом (2 х 10 мл). Приблизительно одну треть метанола удаляют при пониженном давлении. Оставшийся метанол удаляют путем азеотропной дистилляции с толуолом при 80 Торр. Толуол добавляют порциями 15, 10, 10 и 10 мл. Продукт кристаллизуется из смеси и его отфильтровывают и промывают дважды 10 мл порциями толуола. Твердое вещество сушат при комнатной температуре при 1 Торр в течение 6 часов, получая соль амина (4,5 г, 84%). ВЭЖХ/МС (электрораспыление) соответствует требуемому продукту (м/з 421 [М+H]+).
Способ 2:
Часть A: Соль N-[3(S)-[N,
N-бис(фенилметил)амино]- 2(R)-гидрокси-4-фенилбутил] -N-изобутиламин щавелевой кислоты (2800 г, 5,53 моль) и ТГФ (4 л) помещают в 22-литровую круглодонную колбу, снабженную механической мешалкой.
Карбонат калия (1921 г, 13,9 моль) растворяют в воде (2,8 л) и добавляют в ТГФ суспензию. Смесь затем перемешивают в течение одного часа. 1,3-бензодиоксол-5- сульфонил хлорид (1281 г, 5,8 моль)
растворяют в ТГФ (1,4 л) и добавляют в реакционную смесь на протяжении 25 минут. Используют дополнительные 200 мл ТГФ для того, чтобы промыть капельную воронку. Реакционную смесь перемешивают в
течение 14 часов и затем добавляют воду (4 л). Эту смесь перемешивают в течение 30 минут и слоям дают возможность разделиться. Слои удаляют и водный слой промывают дважды ТГФ (500 мл). Объединенные
ТГФ слои сушат сульфатом магния (500 г) в течение одного часа. Раствор затем фильтруют, чтобы удалить осушитель, и его используют в последующих реакциях.
Часть B: К ТГФ раствору неочищенного 1-[N-[(1,3- бензодиоксол-5-ил) сульфонил] -N-(2-метилпропил)амино] -3(S)-[бис (фенилметил) амино]-4-фенил-2(R)-бутанола добавляют воду (500 мл), а затем метансульфоновую кислоту (531 г, 5, 5 моль). Раствор перемешивают, чтобы обеспечить полное смешение, и помещают в 5-галлонный (США, 18,93 л) автоклав. Pearlman катализатор (200 г 20% Pd(ОН)2 на C/50% воды) добавляют в автоклав с помощью ТГФ (500 мл). Реактор продувают четыре раза азотом и четыре раза водородом. Реактор загружают водородом при избыточном давлении 4,219 кг/см2 (60 psig) и начинают перемешивание при 450 об/мин. Спустя 16 часов, ВЭЖХ анализ указывает на то, что небольшое количество монобензилового промежуточного продукта еще присутствует. Добавляют дополнительный катализатор (50 г) и реакцию продолжают в течение ночи. Затем раствор фильтруют через целит (500 г), чтобы удалить катализатор, и концентрируют в вакууме пятью порциями. К каждой порции добавляют толуол (500 мл) и удаляют в вакууме с азеотропно удаляемой остаточной водой. Полученное твердое вещество разделяют на три порции и каждую промывают метил т-бутиловым эфиром (2 л) и фильтруют. Остаточный растворитель удаляют при комнатной температуре в вакуумной печи при менее чем 1 Торр, получая 2714 г целевой соли.
При желании, продукт в дальнейшем может быть очищен следующим способом. Всего 500 мл метанола и 170 г вещества из вышеупомянутого нагревают с обратным холодильником до тех пор, пока оно полностью не растворится. Раствор охлаждают, добавляют 200 мл изопропанола и затем 1000-1300 мл гексана, после чего осаждается белое твердое вещество. После охлаждения до 0oC этот осадок собирают и промывают гексаном, получая 123 г целевого вещества. Благодаря этой процедуре, исходное вещество, которое представляло собой 95:5 смесь спиртовых диастереомеров, представляет собой смесь более чем 99:1 требуемого диастереомера.
ПРИМЕР 35

Получение 2R-гидрокси-3[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропиламина
Часть A: Получение фенилметилового эфира 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропилкарбаминовой кислоты
К раствору 3,19 г (8,6 ммоль) N-[3S-бензилоксикарбонил-амино-2R-гидрокси-4-фенил] -N-изобутиламина в 40 мл безводного метиленхлорида добавляют 0,87 г триэтиламина. Раствор охлаждают до 0oC и добавляют 1,90 т (1, 3-бензодиоксол-5-ил) сульфонил хлорида, перемешивают в течение 15 минут при 0oC, затем в течение 17 часов при комнатной температуре. Добавляют этилацетат, промывают 5%-ной лимонной кислотой, насыщенным бикарбонатом натрия, рассолом, сушат и концентрируют, получая неочищенное вещество. Его перекристаллизовывают из смеси диэтиловый эфир/гексан, получая 4,77 г чистого фенилметилового эфира 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил) сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты.
Часть B: Получение 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)
сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропиламина
Раствор 4,11 г фенилметилового эфира 2R-гидрокси-3-[[(1,3- бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]
-1S- (фенилметил)пропилкарбаминовой кислоты в 45 мл тетрагидрофурана и 25 мл метанола гидрируют в присутствии 1,1 г 10% палладия-на угле при избыточном давлении водорода 3,515 кг/см2 (50
psig) в течение 16 часов. Катализатор удаляют фильтрацией и фильтрат концентрируют, получая 1,82 г требуемого 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)
сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина.
ПРИМЕР 36

Получение Бензотиазол-6-сульфонил хлорида
Часть A: Получение N-(4-сульфонамидофенил)тиомочевины
Смесь сульфаниламида (86 г, 0,5 моль), тиоцианата аммония (76,0 г, 0,5 моль) и разбавленной хлористоводородной кислоты (1,5N, 1 л) механически перемешивают и нагревают с обратным холодильником в течение 2 часов. Около 200 мл воды отгоняют и концентрирование реакционной смеси дает твердое вещество. Твердое вещество отфильтровывают и промывают холодной водой и сушат на воздухе, получая 67,5 г (59%) требуемого продукта в виде белого порошка.
Часть B:
Получение 2-амино-6-сульфонамидобензотиазола
Бром (43,20 г, 0,27 моль) в хлороформе (200 мл) добавляют в течение 1 часа к суспензии N-(4-сульфонамидофенил)тиомочевины (27,72 г, 0,120 моль) в
хлороформе (800 мл). После добавления реакционную смесь нагревают с обратным холодильником в течение 4,5 часов. Хлороформ удаляют в вакууме и остаток повторно дистиллируют с дополнительными
количествами хлороформа. Полученное твердое вещество обрабатывают водой (600 мл), а затем гидроксидом аммония (чтобы сделать его основным), затем нагревают при температуре кипения в течение 1 часа.
Охлажденную реакционную смесь фильтруют, промывают водой и сушат на воздухе, получая 22,0 г (80%) требуемого продукта в виде белого порошка.
Часть C: Получение
Бензотиазол-6-сульфоновой кислоты
Суспензию 2-амино-6-сульфонамидобензотиазола (10,0 г, 43,67 ммоль) в диоксане (300 мл) нагревают с обратным холодильником. Изоамилнитрит (24 мл) добавляют
двумя порциями к реакционной смеси. Наблюдается энергичное выделение газа (реакцию проводят за экраном в целях предосторожности) и спустя 2 часа красный осадок осаждается в реакционном сосуде.
Реакционную смесь фильтруют горячей, твердое вещество промывают диоксаном и сушат. Твердое вещество перекристаллизовывают из смеси метанол-вода. Небольшое количество осадка образуется спустя 2 дня.
Осадок отфильтровывают и маточный раствор концентрируют в вакууме, получая чистый продукт (8,0 г, 85%) в виде бледно-красно-оранжевого твердого вещества.
Часть D: Получение
6-хлорсульфонилбензотиазола
Тионил хлорид (4 мл) добавляют к суспензии бензотиазол-6-сульфоновой кислоты (0,60 г, 2,79 ммоль) в дихлорэтане (15 мл) и реакционную смесь нагревают с обратным
холодильником и к реакционной смеси добавляют диметилформамид (5 мл), получая прозрачный раствор. После 1,5 часов кипячения с обратным холодильником растворитель удаляют в вакууме и избыток HCl и
тионил хлорида удаляют выпариванием с дихлорэтаном.
ПРИМЕР 37

Получение гидрохлоридной соли N-[2R-гидрокси-3-[[(1,3- бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино-1S-(фенилметил) пропил]-2S-[(2S-пирролидинилкарбонил)амино] -3,3- диметилбутанамида
Часть A: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино-1S-(фенилметил)пропил] -2S-[(фенилметоксикарбонил) амино]-3,3-диметилбутанамида

К раствору 118,8 г (0,776 моль) N-гидроксибензотриазола и 137,1 г (0,52 моль) N-карбобензилоксикарбонил-L-третлейцина в 750 мл безводного ДМФ (DMF) при 0oC в атмосфере азота добавляют 109,1 г (0,57 моль) EDC. После перемешивания при 0oC в течение 2 часов добавляют раствор 273 г (0,53 моль) 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил) сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламин метансульфоната, предварительно нейтрализованного 228 мл (210 г, 2,08 моль) 4-метилморфолина, в 250 мл безводного ДМФ. После перемешивания при 0oC в течение 30 минут, смесь перемешивают при комнатной температуре в течение 18 часов. Растворители удаляют при пониженном давлении при 45oC, добавляют 1,5 л этилацетата, промывают 5%-ной лимонной кислотой, насыщенным бикарбонатом натрия, рассолом, сушат над безводным сульфатом магния, фильтруют и концентрируют, получая 400 г неочищенного вещества. Его хроматографируют тремя порциями на приборе Prep 2000 Chromatogram на силикагеле, используя 20-50% этилацетат/гексан в качестве элюента, получая 320 г очищенного вещества, м/е = 674 (M+Li), 98% путем ВЭЖХ.
Часть B: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил) сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]- 2S-амино-3,
3-диметилбутанамида

Раствор 312 г Cbz соединения из предыдущей части в 1 л тетрагидрофурана гидрируют в присутствии катализатора 100 г 4% палладия-на-угле при избыточном давлении водорода 4,219 кг/см2 (60 psig) в течение 6 часов при комнатной температуре. Катализатор удаляют фильтрованием и растворители удаляют при пониженном давлении, получая 240 г требуемого соединения.
Часть C: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5- ил)сульфонил]
(2-метилпропил)амино] -1S-(фенилметил)пропил] - 2S-[[1-фенилметоксикарбонил)пирролидин-2S-илкарбонил]амино]-3,3- диметилбутанамида

250 мл круглодонную колбу снабженную магнитной мешалкой в виде стержня и входом для N2, загружают 1,6 г Cbz-L-Пролина (1,15 экв.) в 40 мл ДМФ. Реакционную смесь охлаждают до 0oC и загружают 0,88 г Hobt (1,5 экв.) и 1,25 г EDC (1,15 экв. ). Реакционную смесь перемешивают 40 минут при комнатной температуре, затем добавляют раствор 3, 0 г 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил амина и 1,85 мл N-метилморфолина (3,0 экв.) в 40 мл ДМФ. Реакционную смесь перемешивают в течение ночи. Реакционную смесь концентрируют в вакууме и остаток распределяют между этилацетатом и насыщенным водным раствором бикарбоната Na. Органические слои промывают 5%-ным водным кислым сульфатом калия и рассолом, сушат над Na2SO4 и концентрируют в вакууме, получая 4,25 г (95%) белой пены; ОФ ВЭЖХ > 97% чистоты.
Часть D: Получение гидрохлоридной соли
N-[2R-гидрокси-3-[[(1,3- бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] - 2S-[(2S-пирролидинилкарбонил)амино]-3,3-диметилбутанамида

300 мл сосуд Фишера-Портера (Fisher-Porter), снабженный магнитной мешалкой в виде стержня, загружают N-[2R-гидрокси-3-[[(1, 3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропил]-2S-[[1-фенилметоксикарбонил)пирролидин- 2S-илкарбонил]амино]-3,3-диметилбутанамидом (1,15 г) и 10% Pd-C (влажный) в 150 мл MeOH. Реакционную смесь гидрируют при 3,515 кг/см2 (50 psi) в течение 6 часов, затем фильтруют через Целит. Фильтрат концентрируют в вакууме до 3,36 г белой пены. Остаток растворяют в 50 мл CH3CN и обрабатывают 0,96 мл (2 экв.) концентрированной HCl. Реакционную смесь концентрируют в вакууме до твердого вещества, растирают с эфиром и фильтруют, получая 3,1 г чистого требуемого продукта.
ПРИМЕР 37A
Получение гидрохлоридной соли N-[2R-гидрокси-3-[[(1,
3-бензодиоксол- 5-ил)сульфонил](2-метилпропил)амино)-1S-(фенилметил)пропил]-2S- [(2S-пирролидинилкарбонил)амино]-3,3-диметилбутанамида

Часть A: Получение N-[2R-гидрокси-3-[(2-метилпропил)амино] 1S-(фенилметил)пропил] -2S-[[1-фенилметоксикарбонил)пирролидин-2S-илкарбонил] амино]-3,3-диметилбутанамида
Бис-защищенное соединение

Примера 43 растворяют в диоксан/HCl и раствор перемешивают в течение приблизительно 2 часов при комнатной температуре. Растворитель удаляют и остаток сушат в вакууме, получая амин
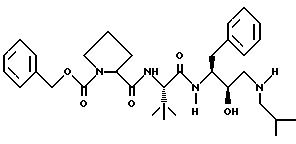
Часть B: Остаток амина Части A перемешивают в этилацетате, добавляют 1,3-бензодиоксол-5-ил сульфонил хлорид Примера 32, а затем триэтиламин. Смесь перемешивают при приблизительно комнатной температуре. Реакционную смесь разбавляют этилацетатом, промывают насыщенным бикарбонатом натрия и рассолом, сушат (MgSO4) и концентрируют, получая целевой продукт. Остаток хроматографируют, если требуется дополнительная очистка

Часть C: 300 мл сосуд Фишера-Портера, снабженный магнитной мешалкой в виде стержня, загружают N-[2R-гидрокси-3-[[(1,3- бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил) пропил] -2S-[[1-(фенилметоксикарбонил)пирролидин-2S-илкарбонил] амино] -3,3-диметилбутанамидом

(1,15 г) и 10% Pd-C (влажный) в 150 мл MeOH. Реакционную смесь гидрируют при 3,515 кг/см2 (50 psi) в течение 6 часов, затем фильтруют через Целит. Фильтрат концентрируют в вакууме до 3,36 г белой пены. Остаток растворяют в 50 мл CH3CN и обрабатывают 0,96 мл (2 экв.) концентрированной HCl. Реакционную смесь концентрируют в вакууме до твердого вещества, растирают с эфиром и фильтруют, получая 3,1 г чистого требуемого продукта

ПРИМЕР 38
Получение гидрохлоридной соли N-[2R-гидрокси-3-[[(1,3-бензодиоксол- 5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил) пропил]-2S-амино-3S-метилпенпанамида

Часть A: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5 -ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] - 2S-[[(1, 1-диметилэтокси)карбонил]амино]-3S-метилпентанамида
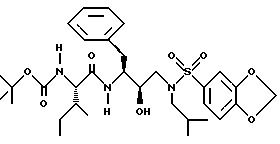
К охлажденному раствору N-т-BOC-L-изолейцина 2,02 г (8,74 ммоль) и 2,00 г (13,11 ммоль) N-гидроксибензотриазола в 17 мл N,N-диметилформамида добавляют 1,84 г (9,61 ммоль) EDC и перемешивают при 0oC в течение одного часа. К реакционной смеси добавляют раствор 3,67 г (8,74 ммоль) 2R-гидрокси-3-[[(1,3-бензодиоксол-5- ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил амина в 6 мл N,N-диметилформамида и раствор перемешивают в течение 16 часов. Растворитель удаляют в вакууме, замещают этилацетатом и промывают насыщенным бикарбонатом натрия, 5% лимонной кислотой и раствором. Органические слои сушат над сульфатом магния, фильтруют и концентрируют, получая 6,1 т неочищенного продукта, который хроматографируют на силикагеле, используя 1: 1 этилацетат:гексан элюент, получая 4,3 г (78% выход) N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] - 1S-(фенилметил)пропил-2S-[[(1,1-диметилэтокси)карбонил] амино] - 3S-метилпентанамида
Часть B: Получение гидрохлоридной соли N-[2R-гидрокси-3- [[(1,3-бензодиоксол-5-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропил]-2S-амино-3S-метилпентанамида

N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2- метилпропил)амино] -1S-(фенилметил)пропил] -2S-[[(1,1-диметилэтокси) карбонил]амино] -3S-метилпентaнaмид (4,29 г, 6,77 ммоль) растворяют в 20 мл 4N HCl в диоксане и перемешивают в течение 20 минут. Высадившийся продукт отпаривают два раза из диэтилового эфира и неочищенную гидрохлоридную соль используют в последующих реакциях.
ПРИМЕР 39
Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]
-1S-(фенилметил)пропил]-2S-[(2S-пирролидинилкарбонил) амино]-3S-метилпентанамида

Часть A: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол- 5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил] -2S-[[1-(фенилметоксикарбонил)пирролидин-2S-илкарбонил]амино]- 3S-метилпентанамида

Раствор 0,5 г (2,2 мМ) M-CEZ-L-пролина в 10 мл безводного ДМФ охлаждают до 0oC и добавляют 0,4 г (2,8 мМ) НОВТ и 0,4 г (2,2 мМ) EDC. Баню со льдом удаляют спустя 20 минут и перемешивание продолжают в течение еще 40 минут. Добавляют раствор 1,0 г (1,9 мМ) гидрохлоридной соли N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5- ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил-2S- амино-3S-метилпентанамида и 0,6 г (5,6 мМ) 4-метилморфолина в 15 мл безводного ДМФ и смесь перемешивают в течение 15 часов. Растворители удаляют в вакууме и остаток распределяют между 150 мл этилацетата и 50 мл 5%-ного раствора кислого сульфата калия. Слои разделяют и органический слой промывают по 50 мл каждого насыщенным раствором бикарбоната натрия, воды и рассола, затем сушат над безводным сульфатом магния, фильтруют и концентрируют в вакууме, получая 1,5 г неочищенного вещества. Очистку проводят при помощи флэш хроматографии на силикагеле, используя 70-80% этилацетат/гексан, получая 1,3 г (90%) требуемого продукта в виде белого твердого вещества, м/е = 771 (M+Li).
Часть B: Получение N-[2R-гидрокси-3-[[(1,3-бензодиоксол- 5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил]
-2S-[(2S-пирролидинилкарбонил)амино]-3S-метилпентанамида
В колбу Фишера-Портера, снабженную магнитной мешалкой в виде стержня, загружают 1,2 г (1,5 мМ) продукта Части A и 25 мл ТГФ. Раствор
гидрируют в присутствии 1 г 10% катализатора палладия-на-угле (50% воды по весу) при избыточном давлении водорода 3,515 кг/см2 (50 psig) в течение 16 часов при комнатной температуре.
Катализатор удаляют фильтрацией и растворитель удаляют при пониженном давлении, получая 0,9 г неочищенного вещества. Очистку проводят, используя флэш хроматографию на силикагеле, используя 1-4%
метанол/метиленхлорид, и получают 0,8 г (80%) требуемого продукта, м/е = 637 (М+Н).
ПРИМЕР 40
Получение N-[2R-гидрокси-3-[(1,3-бензодиоксол-5-ил)сульфонил]
(2-метилпропил)амино]-1S-(фенилметил)пропил]-2S-амино-3-метилбутанамида

Часть A: Получение N-[2R-гидрокси-3-[(1,3-бензодиоксол-5-ил) сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-2S- [(фенилметоксикарбонил)амино]-3-метилбутанамида

В 250-мл круглодонную колбу, снабженную магнитной мешалкой в виде стержня, загружают N-Cbz-L-Валин (4,22 г, 16,8 ммоль) в 20 мл ДМФ. Раствор охлаждают до 0oC и добавляют HoBt (2,96, 21,9 ммоль) и EDC (3,22 г, 16,8 ммоль) и перемешивают 1 час. В реакционную смесь затем добавляют N-метилморфолин (1,7 г, 16,8 ммоль), 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил)пропиламин (7,55 г, 14,6 ммоль) в 30 мл ДМФ. Реакционную смесь перемешивают на протяжении ночи при комнатной температуре, затем концентрируют в вакууме и распределяют между этилацетатом и 5% лимонной кислотой. Объединенные органические слои промывают насыщенным бикарбонатом натрия и рассолом и сушат над сульфатом натрия. Концентрирование в вакууме дает 10 г неочищенного продукта. Очистка Преп (Prep) ВЭЖХ (20-40% этилацетат/гексан) дает 5,8 г (61%) требуемого соединения.
Часть B:
Получение N-[[2R-гидрокси-3-[(1,3-бензодиоксол-5- ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]- 2S-амино-3-метилбутанамида
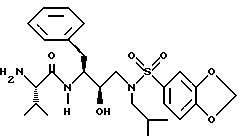
В 300-мл круглодонный сосуд, снабженный магнитной мешалкой в виде стержня, загружают N-[[2R-гидрокси-3-[(1,3-бензодиоксол-5- ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]- 2S-[(фенилметоксикарбонил)амино] -3-метилбутанамид (5,8 г), 2,3 г 10% Pd-C в 75 мл тетрагидрофурана. В реакционную смесь пропускают 3,515 кг/см2 (50 psi) H2 и гидрируют на протяжении ночи. Реакционную смесь фильтруют через Целит и концентрируют в вакууме, получая 4,4 г белой пены, которую используют в последующих реакциях без дополнительной очистки.
ПРИМЕР 41
Получение гидрохлоридной соли N-[[2R-гидрокси- 3-[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил]
-2S-амино-3-(метилсульфонил)пропанамида

Часть A: Получение N-[[2R-гидрокси-3-[(1, 3-бензодиоксол- 5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S-[[(1,1-диметилэтокси)карбонил]амино-3-(метилтио)пропанамида

N-т-Вос-S-метил-(L)-цистеин (2,80 г, 11,9 ммоль), 1-гидроксибензотриазол гидрат (1,92 г, 12,5 ммоль), и 1-(3-диметиламинопропил)-3-3-этилкарбодиимид гидрохлорид (2,27 г, 11,9 ммоль) смешивают в N,N-диметилформамиде (30,0 мл) при 0oC в течение 10 мин. Добавляют N-метилморфолин (3,03 г, 33,0 ммоль) и раствор перемешивают дополнительные 10 мин при 0oC. Добавляют 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил](2- метилпропил)амино]-1S-(фенилметил)пропиламин (5,00 г, 11,9 ммоль) и раствор нагревают до комнатной температуры и перемешивают в течение 2 часов. Реакционную смесь выливают в этилацетат (500 мл) и промывают 10%-ной водной хлористоводородной кислотой (3 х 100 мл), насыщенным водным бикарбонатом натрия (3 х 100 мл) и рассолом (2 х 100 мл). Органический слой сушат над сульфатом натрия и фильтруют через слой силикагеля (50 г). Получают требуемый продукт (7,13 г, 11,9 ммоль, 93% выход) в виде белого твердого вещества удалением растворителя при пониженном давлении; м/е рассчит. 637; найдено (M+Li) 644.
Часть B: Получение N-[[2R-гидрокси-3-[(1,
3-бензодиоксол- 5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S-[[(1,1-диметилэтокси)карбонил]амино-3-(метилсульфонил) пропанамида

N-[[2R-гидрокси-3-[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S- [[(1,1-диметилэтокси)карбонил]амино] -3-(метилтио)пропанамид (7,10 г, 11,1 ммоль) растворяют в метаноле (150 мл). Раствор оксона® (20,8 г, 33,9 ммоль) в воде (150 мл) добавляют по каплям к раствору при комнатной температуре на протяжении 1,5 часов. Раствор становится мутным, и во время добавления образуется осадок. Реакционную смесь перемешивают в течение дополнительного часа и добавляют тетрагидрофуран (200 мл). После перемешивания в течение дополнительного часа, раствор выливают в этилацетат (1000 мл) и промывают водой (3 х 200 мл), затем рассолом (2 х 300 мл). Органический слой сушат над безводным сульфатом натрия и растворитель удаляют при пониженном давлении. Получают требуемый продукт (5,75 г, 8,86 ммоль, 79% выход) в виде беловатого твердого вещества; м/е рассчит. 669; найдено (М+Н) 670.
Часть C: Получение гидрохлоридной соли N-[[2R-гидрокси-3- [(1,3-бензодиоксол-5-ил) сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропил]-2S-амино-3-(метилсульфонил)пропанамида

N-[[2R-гидрокси-3-[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S-[[(1,1- диметилэтокси)карбонил]амино] -3-(метилсульфонил)пропанамид (5,5 г, 8,20 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре. Безводную хлористоводородную кислоту барботируют через раствор в течение 15 мин. Раствор перемешивают при комнатной температуре в течение 2 часов и растворитель удаляют при пониженном давлении. Получают требуемый продукт (4,91 г, 8,10 ммоль, 99% выход) в виде белого твердого вещества; м/е рассчит. 569; найдено (M+Li) 576.
ПРИМЕР 42
Получение гидрохлоридной соли N-[[2R-гидрокси-3- [(1,
3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропил]-2S-амино-3-метил-3-(метилсульфонил) пропанамида

Часть A: Получение N-[[2R-гидрокси-3-[(1,3-бензодиоксол-5 ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] - 2S-[[(1, 1-диметилэтокси)карбонил]амино]-3-(метилтио)бутанамида

Соль N-т-boc-3-метил-L-пеницилламин дициклогексиламина (4,00 г, 9,00 ммоль), 1-гидроксибензотриазол гидрат (1,69 г, 11,00 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (1,71 г, 9,00 ммоль) смешивают в диметилформамиде (60,0 мл) при комнатной температуре. Гетерогенную смесь перемешивают в течение 1 часа и добавляют 2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил) сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламин (3, 78 г, 9,00 ммоль) и гетерогенную смесь перемешивают в течение 16 часов. Раствор выливают в этилацетат (600 мл) и промывают 10%-ной водной лимонной кислотой (2 х 300 мл), насыщенным водным раствором бикарбоната натрия (2 х 300 мл) и рассолом (300 мл). Раствор сушат над сульфатом натрия и растворитель удаляют в вакууме. Требуемый продукт чистят флэш хроматографией (0-80% этилацетат-гексаны на силикагеле). Получают продукт (5,21 г, 7,83 ммоль, 87% выход) в виде белой пены; м/е рассчит. 665; найдено (M+Li) 672.
Часть B: Получение N-[[2R-гидрокси-3-[(1,
3-бензодиоксол- 5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S-[[(1,1-диметилэтокси)карбонил]амино]-3-метил-3- (метилсульфонил)бутанамида

N-[[2R-гидрокси-3-[(1,3-бензодиоксол-5-ил)сульфонил] (2- метилпропил)амино] -1S-(фенилметил)пропил] -2S-[[(1, 1- диметилэтокси)карбонил]амино] -3-метил-3-(метилтио)бутанамид (5,01 г, 7,53 ммоль) растворяют в тетрагидрофуране (250 мл). Раствор оксона® (13,8 г, 22,6 ммоль) в воде (250 мл) добавляют по каплям к раствору при комнатной температуре в течение 2 часов. Раствор становится мутным, и во время добавления образуется осадок. Раствор выливают в этилацетат (500 мл) и промывают водой (3 х 200 мл), затем рассолом (2 х 300 мл). Органический слой сушат над безводным сульфатом натрия и растворитель удаляют в вакууме. Получают продукт (4,72 г, 6,77 ммоль, 89% выход) в виде белой пены; м/е рассчит. 697; найдено (M+Li) 704.
Часть C: Получение гидрохлоридной соли N-[[2R-гидрокси-3- [(1,
3-бензодиоксол-5-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропил]-2S-амино-3-метил-3-(метилсульфонил)бутанамида
N-([2R-гидрокси-3-[(1,3-бензодиоксол-5-ил)сульфонил] (2- метилпропил)амино] -1S-(фенилметил)пропил] -2S-[[ (1,1-диметилэтокси)карбонил]амино] -3-метил-3-(метилсульфонил) бутанамид (4,51 г, 6,46 ммоль) растворяют в дихлорметане (200 мл) при комнатной температуре. Безводную хлористоводородную кислоту барботируют через раствор в течение 30 мин. Раствор перемешивают при комнатной температуре в течение 1 часа и растворитель удаляют в вакууме. Получают продукт (4,02 г, 6,35 ммоль, 99% выход) в виде белого твердого вещества; м/е рассчит. 697; найдено (M+Li) 704.
ПРИМЕР 43
Получение N-[2R-гидрокси-3-[[(1,1-диметилэтокси) карбонил](2-метилпропил)амино] -1S-(фенилметил)пропил]
-2S- [[1-(фенилметоксикарбонил)пирролидин-2S-илкарбонил]амино]-3,3- диметилбутанамида

Часть A: Получение N-[(1,1-диметилэтоксил)карбонил]-N- [2-метилпропил] -3S-[N1-(фенилметоксикарбонил)амино]-2R- гидрокси-4-фенилбутиламина

Раствор N-[3S-[N1-(бензилоксикарбонил)амино]-2R- гидрокси-4-фенилбутил] -N-(2-метилпропил)амина (18,5 г, 50 ммоль), BOC-ON (12,35 г, 50 ммоль) и триэтиламина (7 мл) в тетрагидрофуране (400 мл) перемешивают при комнатной температуре в течение 18 часов и затем концентрируют в вакууме. Остаток растворяют в дихлорметане (1 л) и промывают гидроксидом натрия (5%, 2 х 200 мл) и рассолом, сушат (MgSO4) и затем концентрируют в вакууме, получая 23,5 г (количественный выход) чистого требуемого продукта.

Часть B: Получение N-[2R-гидрокси-3-[[(1,1- диметилэтокси)карбонил](2-метилпропил)амино] -1S-(фенилметил) пропил]-2S-[(фенилметоксикарбонил)амино] -3,3-диметилбутанамида

N-[(1, 1-диметилэтоксил)карбонил]-N-[2-метилпропил]-3S- [N1-(фенилметоксикарбонил)амино] -2R-гидрокси-4-фенилбутиламин в этаноле гидрируют при избыточном давлении водорода 3,164 кг/см2 (45 psig) в присутствии 5% Pd(C) катализатора, получая N-[(1,1-диметилэтоксил)карбонил]-N-[2-метилпропил] -3S-[N1- (фенилметоксикарбонил)амино]-2R-гидрокси-4-фенилбутиламин. После стандартной обработки, неочищенный амин (12,24 г, 36, 42 ммоль) добавляют к смеси N-карбобензилоксикарбонил-L-трет-лейцина (9,67 г, 36,42 ммоль), НОВТ (4,92 г, 36,42 ммоль) и EDC (6,98 г, 36,42 ммоль) в ДМФ (300 мл), после того как смесь перемешивают при комнатной температуре в течение 1 часа. Смесь перемешивают в течение дополнительных 18 часов. ДМФ удаляют в вакууме, остаток растворяют в дихлорметане (500 мл), промывают гидроксидом натрия (5%, 2 х 200 мл) и рассолом (200 мл), сушат и концентрируют, получая 21 г (количественный) требуемого продукта.
Часть C: Получение
N-[2N-гидрокси-3-[[(1,1- диметилэтокси)карбонил][2-метилпропил]амино]-1S-(фенилметил) пропил]-2S-амино-3,3-диметилбутанамида

N-[2R-гидрокси-3-[[(1,1-диметилэтокси)карбонил] [2- метилпропил]амино]-1S-(фенилметил)пропил] -2S- [(фенилметоксикарбонил)амино] -3,3-диметилбутанамид (20 г, 34, 29 ммоль) в метаноле (250 мл) гидрируют при комнатной температуре в присутствии Pd/C (10%, 5 г). Катализатор отфильтровывают и фильтрат концентрируют, получая 13,8 г (90%) чистого требуемого продукта.
Часть D: Получение N-[2R-гидрокси-3-[[(1,1-диметилэтокси)
карбонил][2-метилпропил]амино]-1S-(фенилметил)пропил]-2S-[[1- (фенилметоксикарбонил)пирролидин-2S-илкарбонил]амино]-3,3- диметилбутанамида
Cbz-L-пролин растворяют в ДМФ в атмосфере азота при
перемешивании при приблизительно 0oC. Добавляют гидроксибензотриазол (ГБТ, НВТ), а затем добавляют EDC. Реакционную смесь перемешивают при комнатной температуре, добавляют амин Части C и
добавляют N-метил-морфолин. Реакционную смесь перемешивают в течение приблизительно одного дня. Реакционную смесь концентрируют в вакууме и остаток распределяют между этилацетатом и насыщенным водным
бикарбонатом натрия. Органический слой промывают 5%-ным водным кислым сульфатом калия и рассолом, сушат над Na2SO4 и концентрируют в вакууме, получая

ПРИМЕР 44
Получение фенилметилового эфира 2R-гидрокси-3- [[(1,4-бензодиоксан-6-ил)сульфонил] (2-метилпропил)амино] -1S- (фенилметил)пропилкарбаминовой кислоты

К раствору N-[3S-[(фенилметоксикарбонил)амино]-2R-гидрокси- 4-фенилбутил]-N-(2-метилпропил)амина (0,5 г, 1,35 ммоль) в CH2Cl2 (5,0 мл), содержащему Et3N (0,35 мл, 2,5 ммоль), добавляют 1,4-бензодиоксан-6-сульфонил хлорид (0,34 г 1,45 ммоль) и перемешивают при 0oC в течение 30 мин. После перемешивания при комнатной температуре в течение 1 часа, реакционную смесь разбавляют CH2Cl2 (20 мл), промывают холодной 1N HCl (3 х 20 мл), водой (2 х 20 мл), насыщенным NaHCO3 (2 х 20 мл) и водой (3 х 20 мл), сушат (Na2SO4) и концентрируют при пониженном давлении. Полученный остаток очищают флэш хроматографией, используя 35% EtOAc в гексане, получая требуемый продукт в виде белого аморфного твердого вещества, который кристаллизуется из MeOH в виде белого порошка (0,65 г, 84% выход); т. пл. 82-84oC, ВРМС-ББА (HRMC-FAB): рассчит. для C30H37N2O7S 569,2321 (МН+), найдено 569,2323.
ПРИМЕР 45
Получение [2R-гидpoкcи-3-[(бензoтиaзoл-6- cульфoнил)(2-метил-пропил)амино]-1S-(фенилметил)пропиламин гидрохлорида

Часть A: Получение т-бутилового эфира [2R-гидрокси-3-[(4- аминофенилсульфонил)(2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты

Смесь [2R-гидрокси-3-[(4-аминофенилсульфонил)(2-метилпропил) амино] -1S-(фенилметил)пропиламина 3,7 г (9,45 ммоль) и BOC-ON (2,33 г, 9,45 ммоль) и триэтиламина (0,954 г, 9,45 ммоль) в тетрагидрофуране (60 мл) перемешивают в течение 16 часов и концентрируют в вакууме. Остаток растворяют в дихлорметане (200 мл), промывают гидроксидом натрия (1N, 100 мл) и лимонной кислотой (5%, 100 мл), сушат (MgSO4) и концентрируют, получая 1,18 г (94%) требуемого продукта в виде белого твердого вещества.
Часть B: Получение т-бутилового эфира
[2R-гидрокси-3-[[(2- аминобензотиазол-6-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропилкарбаминовой кислоты

т-Бутиловый эфир [2R-гидрокси-3-[(4-аминофенилсульфонил) (2-метилпропил)амино] -1S-(фенилметил)пропилкарбаминовой кислоты (1,12 т, 2,279 ммоль) добавляют к хорошо перемешиваемому порошку безводного сульфата меди (4,48 г) и тиоцианата калия (5,60 г) с последующим добавлением сухого метанола (35 мл) и полученную черно-коричневую суспензию нагревают с обратным холодильником в течение 2 часов. Реакционная смесь становится серой. Реакционную смесь фильтруют и фильтрат разбавляют водой (50 мл) и нагревают с обратным холодильником. К реакционной смеси добавляют этанол, охлаждают и фильтруют. Фильтрат после концентрирования дает остаток, который хроматографируют (этилацетат/метанол 90:10), получая 0,80 г (78%) незащищенного (после удаления защиты) соединения в виде твердого вещества. Соединение повторно прямо защищают следующим способом: (2,25 г, 5,005 ммоль) BOC-ON (1,24 г) и триэтиламин (0,505 г, 5,005 ммоль) в тетрагидрофуране (20 мл) перемешивают при комнатной температуре в течение 18 часов. Реакционную смесь концентрируют и остаток растворяют в дихлорметане (200 мл) и промывают гидроксидом натрия (1N, 100 мл) и лимонной кислотой (5%, 100 мл), сушат (MgSO4) и концентрируют, получая остаток, который хроматографируют (этилацетат:гексан 3:1), получая 1,8 г (65%) требуемого продукта в виде твердого вещества.
Часть C: Получение т-бутилового эфира [2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропилкарбаминовой кислоты

т-Бутиловый эфир [2R-гидрокси-3-[[(2-аминобензотиазол-6-ил) сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропилкарбаминовой кислоты (1,80 г, 3,2755 ммоль) добавляют к раствору изоамилнитрита (0,88 мл) в диоксане (20 мл) и смесь нагревают при 85oC. После прекращения выделения азота реакционную смесь концентрируют и остаток очищают с помощью хроматографии (гексан: этилацетат 1: 1), получая 1,25 г (78%) требуемого продукта в виде твердого вещества.
Часть D: Получение гидрохлоридной соли
[2R-гидрокси-3 [[(бензотиазол-6-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропиламина
т-Бутиловый эфир [2R-гидрокси-3-[[(бензотиазол-6-ил) сульфонил](2-метилпропил)амино] -1S-(фенилметил)пропилкарбаминовой кислоты (1,25 г, 2,3385 ммоль) добавляют к смеси диоксан/HCl (4N, 10 мл) и перемешивают при комнатной температуре в течение 2 часов и концентрируют. Избыток HCl вытесняют толуолом, получая 1,0 г (количественный выход) требуемого продукта.
ПРИМЕР 46

Получение 2S-Амино-N-[2R-гидрокси-3-[[{1,3-бензодиоксол-5- ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропилпент-4-инамида
Часть A: Получение 2S-[[(1,1-диметилэтокси)карбонил]амино] -N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил) амино] -1S-(фенилметил)пропил]пент-4-инамида

К охлажденному раствору N-т-Boc-L-пропаргил глицина (5,0 г 23,4 ммоль) и 4,7 г (1,5 экв.) N-гидроксибензотриазола в 40 мл N,N-диметилформамида добавляют 4,6 г (23,4 ммоль) EDC и перемешивают при 0oC в течение одного часа. К реакционной смеси добавляют раствор 12,10 г (23,4 ммоль) 2R-гидрокси-3-[[(1,3- бензодиоксол-5-ил)сульфонил](2-метилпропил)амино]-1S- (фенилметил)пропиламина в 6 мл N,N-диметилформамида и раствор перемешивают в течение 16 часов. Растворитель удаляют на роторном испарителе, добавляют этилацетат, промывают насыщенным бикарбонатом натрия, 5%-ной лимонной кислотой и рассолом. Органический слой сушат над сульфатом магния, фильтруют и концентрируют, получая 13,3 г неочищенного продукта, который кристаллизуют из смеси диэтиловый эфир: этилацетат, получая 6,9 г 2S-[[(1,1-диметилэтокси)карбонил] амино] -N-[2R- гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]пент-4-инамида.
Часть B: Получение 2S-амино-N-[2R-гидрокси-3-[[(1,
3- бензодиоксол-5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропил]пент-4-инамида

5, 0 г (8,12 ммоль) продукта Части A растворяют в 20 мл 4 N HCl в диоксане и перемешивают в течение 30 минут. Высадившийся продукт два раза отпаривают диэтиловым эфиром и эту неочищенную гидрохлоридную соль используют в последующих реакциях.
ПРИМЕР 47
Получение дигидробромидной соли N-[2R-гидрокси- 3-[[(бензотиазол-6-ил)сульфонил] (2-меатилпропил)амино]
-1S- (фенилметил)пропил] -2S-(амино) - 3,3-диметилбутанамида

Часть A: Получение N-[2R-гидрокси-3-[[(бензотиазол-6-ил) сульфонил](2-метилпропил)амино] -1S-(фенилметил)пропил] -2S- [[(N-бензилокси)карбонил] амино]-3,3-диметилбутанамида

Смесь N-бензилоксикарбонил-т-бутилглицина (2,0 г, 7,538 ммоль), НОВТ (1,02 г, 7,55 ммоль) и EDC (1,45 г, 7,55 ммоль) в ДМФ (20 мл) перемешивают при комнатной температуре в течение 1 часа. Затем добавляют 2R-гидрокси-3-[[(бензотиазол-6-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропиламин гидрохлорид (3,825 г, 7,54 ммоль) и N-метилморфолин (3,80 г) и продолжают перемешивание в течение 18 часов. ДМФ удаляют в вакууме, остаток растворяют в дихлорметане (500 мл) и промывают лимонной кислотой (1N, 100 мл), бикарбонатом натрия (100 мл), рассолом (200 мл), сушат, фильтруют и концентрируют, получая 4,69 г (91%) чистого N-[2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил] (2-метилпропил)амино]-1S- (фенилметил)пропил] -2S-[N-(фенилметоксикарбонил)амино]-3,3- диметилбутанамида.
Часть B: Получение дигидробромидной соли N-[2R-гидрокси-3- [[(бензотиазол-6-ил)сульфонил]
(2-метилпропил)амино]-1S- (фенилметил)пропил] -2S-(амино)-3,3-диметилбутанамида
Раствор N-[2R-гидрокси-3-[[(бензотиазол-6-ил)сульфонил] (2-метилпропил)амино]
-1S-(фенилметил)пропил)-2S-[N-(фенил-метоксикарбонил) амино] -3,3-диметилбутанамида (4,69 г, 6,89 ммоль) в дихлорэтане (200 мл) обрабатывают HBr (48% в уксусной кислоте, 7,1 мл) и реакционную смесь
перемешивают в течение 2 часов при комнатной температуре. Реакционную смесь концентрируют и остаток промывают несколько раз диэтиловым эфиром, получая 4,88 г требуемого дигидробромидного продукта в
виде порошка: ББА-МС высокого разрешения: рассчит. для C27H38N4O4S2; 547,2413, найдено: 547,2429 (М+Н).
ПРИМЕР 48
Получение 5-хлоросульфонил-2-карбометоксиамино-бензимидазола

Раствор 2-карбометоксиамино-бензимидазола (5,0 г, 0,026 ммоль) в хлорсульфоновой кислоте (35,00 мл) перемешивают при 0oC в течение 30 минут и при комнатной температуре в течение 3 часов. Полученную темноокрашенную реакционную смесь выливают в смесь лед-вода (200 мл) и перемешивают при комнатной температуре в течение 30 минут. Полученный осадок фильтруют и промывают холодной водой (500 мл). Твердое вещество сушат на протяжении ночи в высоком вакууме в эксикаторе над гранулами NaOH, получая 5-хлоросульфонил-2-карбометоксиамино-бензимидазол (5,9 г, 78%) в виде серого порошка.1Н ЯМР (ДМСО-d6) δ : 3,89 (с, 3Н), 7,55 (д, J=8,4 Гц,1Н), 7,65 (д, J=8,4 Гц, 1Н), 7,88 (с, 1H). (German Patent DE 3826036).
ПРИМЕР 49
Получение фенилметилового
эфира N-[2R-гидрокси-3-[N1-[(2-карбометоксиамино-бензииидазол-5-ил)сульфонил] -N1-(2-метилпропил)амино] -1S-(фенилметил)пропил]карбаминовой кислоты
К холодному раствору N-[3S-[(фенилметоксикарбонил)амино]- 2R-гидрокси-4-фенилбутил] -N-(2-метилпропил)амина (5,0 г, 13,5 ммоль) в дихлорметане (70 мл) добавляют триэтиламин (5,95 т, 54,0 ммоль) с последующим добавлением 5-хлоросульфонил-2- карбометоксиаминобензимидазола (4,29 г, 14, 85 ммоль) небольшими порциями в виде твердого вещества. Реакционную смесь перемешивают при 0oC в течение 30 минут и при комнатной температуре в течение 2,5 часов после того, как реакция аминоспирта завершится. Смесь охлаждают и фильтруют и фильтрат концентрируют. Полученный остаток растворяют в EtOAc (200 мл), промывают последовательно холодной 5%-ной лимонной кислотой (3 х 50 мл), насыщенным водным бикарбонатом натрия (3 х 50 мл) и водой (3 х 100 мл), затем сушат (Na2SO4), концентрируют и сушат в вакууме. Остаток растирают с метанолом, охлаждают, фильтруют, промывают MeOH-EtOAc (1:1, об/об) и сушат в эксикаторе, получая чистый фенилметиловый эфир N-[2R-гидрокси-3- [[(2-карбометоксиамино-бензимидазол-5-ил)сульфонил] (2- метилпропил)амино] -1S-(фенилметил)пропил)карбаминовой кислоты (6,02 г, 72%) в виде светло-коричневого порошка: ББАМС (FABMC): м/з = 630 (M+Li); ВРМС (HRMS): рассчит. для C31H38N5O5S (М+Н) 624,2492, найдено 624,2488.
ПРИМЕР 50
Получение 2R-гидрокси-3-[[(2-амино-бензимидазол-5-ил) сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина
Раствор фенилметилового эфира N-[2R-гидрокси-3-[[(2- карбометоксиамино-бензимидазол-5-ил)сульфонил](2-метилпропил) амино]-1S-(фенилметил)пропил)карбаминовой кислоты (0,36 г, 0,58 ммоль) в 2,5N растворе КОН в метаноле (2,00 мл) нагревают при 70oC в атмосфере азота в течение 3 часов. Реакционную смесь разбавляют водой (10 мл) и экстрагируют EtOAc (3 х 15 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют. Полученный остаток очищают ВЭЖХ с обращенной фазой, используя 10-90% CH

ПРИМЕР 51
Получение фенилметилового эфира
N-[2R-гидрокси- 3-[[(2-амино-бензимидазол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил]карбаминовой кислоты
К раствору 2R-гидрокси-3-[[(2-амино-бензимидазол-5-ил) сульфонил](2-метилпропил)амино] -1S-(фенилметил)пропиламина (0,22 г, 0,33 ммоль) в ТГФ (3,00 мл) добавляют триэтиламин (0,11 г, 1,1 ммоль) и бензилоксикарбонил сукцинимид (0,09 г, 0,36 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Раствор концентрируют и остаток распределяют между EtOAc (15 мл) и насыщенным водным раствором бикарбоната натрия. Органическую фазу промывают рассолом, сушат (Na2SO4) и концентрируют. Полученный остаток очищают ВЭЖХ с обращенной фазой, используя 10-90% CH3CN/H2O градиент (30 мин) при скорости потока 70 мл/мин. Соответствующие фракции объединяют и сушат вымораживанием, получая чистый фенилметиловый эфир N-[2R-гидрокси-3-[[(2-амино-бензимидазол-5-ил)сульфонил](2-метилпропил) амино] -1S- (фенилметил)пропил)карбаминовой кислоты (0,12 г, 61%) в виде белого порошка: ББА-МС м/з = 566 (М+Н); ВРМС (HMRS): рассчит. для C29H36N5O5S 3566, 2437 (М+Н), найдено 566, 2434.
ПРИМЕР 52
Получение
2R-гидрокси-3-[[(2-кapбoметoкcиaминoбензимидaзoл 5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина
Раствор фенилметилового эфира N-[2R-гидрокси-3-[[(2- карбометоксиаминобензимидазол-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил)пропил] карбаминовой кислоты (2,5 г, 0,4 ммоль) в MeOH (10 мл) и ТГФ (50 мл) гидрируют в присутствии 10% Pd/C (1,2 г) при комнатной температуре при 4,219 кг/см2 (60 psi) в течение 16 часов. Катализатор удаляют фильтрованием и фильтрат концентрируют при пониженном давлении. Затем полученное твердое вещество промывают эфиром и сушат в вакууме, получая 2R-гидрокси-3-[[(2-карбометоксиаминобензимидазол-5-ил)сульфонил] (2- метилпропил)амино]-1S-фенилметил)пропиламин (1,5 г, 77%) в виде беловатого порошка: Rt = 12,8 мин; ББА-МС м/з = 490 (М+Н); ВРМС (HMRS): рассчит. для C23H32N5O5S 490,2124 (М+Н), найдено 490,2142.
ПРИМЕР 53
Получение
N-[2R-гидрокси-3-[[(2- карбометоксиамино-бензимидазол-5-ил)сульфонил] (2-метилпропил)
амино]-1S-(фенилметил)пропил]-2S-амино-3,3-диметилбутанамида
Часть A: Получение N-[2R-гидрокси-3-[N1-[(2- кapбoметокcиaмино-бензимидазол-5-ил) сульфонил]-N1 -(2- метилпропил)амино]-1S-(фенилметил)пропил]-2S- [(фенилметоксикарбонил)амино]-3,3-диметилбутанамида

К раствору N-карбобензилоксикарбонил-L-трет-лейцина (0,65 г, 2,45 ммоль) в ДМФ (10 мл) добавляют HOBt (0,5 г, 3,22 ммоль) и EDC (0,49 г, 2,55 ммоль) и полученную смесь перемешивают при 0oC в течение 2 часов. Затем добавляют раствор 2R-гидрокси-3{ [(2- карбометоксиамино-бензимидазол-5-ил) сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропиламина (1,2 г, 2,45 ммоль) в ДМФ (4 мл) и N-метилморфолин (0,74 г, 7,3 ммоль) и смесь перемешивают при комнатной температуре в течение 16 часов. Затем ДМФ отгоняют в вакууме и оставшийся остаток распределяют между холодной 1N водной HCl (100 мл) и EtOAc (200 мл). Органическую фазу промывают последовательно холодной 1N HCl (2 х 50 мл), рассолом (2 х 50 мл), 0,25N NaOH (3 х 50 мл), рассолом, сушат (Na2SO4) и концентрируют в вакууме. Полученный остаток очищают флэш хроматографией на колонке с силикагелем, используя EtOAc в ка естве элюента, получая 1,5 г (83%) чистого N-[2R-гидрокси-3- [[(2-карбометоксиамино-бензимидазол-5-ил) сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S- [(фенилметоксикарбонил)амино] -3,3-диметилбутанамида: Rt = 21,2 мин; ББА-МС м/з = 737 (М+Н); ВРМС (HMRS): рассчит. для C37H49N6O8S 737,3333 (М+Н), найдено 737,3334.
Часть B: Получение
N-[2R-гидрокси-3-[[(2-карбометоксиамино-бензимидазол-5-ил)сульфонил] (2-метилпропил)амино] -1S-(фенилметил)пропил] -2S-амино-3,3-диметилбутанамида
Раствор
N-[2R-гидрокси-3-[[(2-карбометоксиамино-бензимидазол-5-ил) сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил]-2S- [(фенилметоксикарбонил)амино]-3,3-диметилбутанамида (4,0 г, 5,4 ммоль) в MeOH (15
мл) и ТГФ (65 мл) гидрируют в присутствии 10% Pd/C (2,0 г) при комнатной температуре при 3,515 кг/см2 (50 psi) в течение 16 часов. Катализатор удаляют фильтрацией и фильтрат концентрируют
при пониженном давлении. Полученный остаток растирают с эфиром и фильтруют. Твердый остаток промывают эфиром и сушат в вакууме, получая
N-[2R-гидрокси-3-[[(2-карбометоксиамино-бензимидазол-5-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-2S-амино-3,3-диметилбутанамид (2,9 г, 88%) в виде бледно-желтого порошка. Порцию
продукта очищают ВЭЖХ (HPLC) с обращенной фазой, используя 10-90% CH3N/H2O градиент (30 мин) при скорости потока 70 мл/мин. Соответствующие фракции объединяют и сушат
вымораживанием с получением чистого N-[2R-гидрокси-3-[[(2-карбометоксиамино-бензимидазол-5-ил) сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил-2S- амино-3,3-диметилбутанамида в виде белого
порошка: Rt = 13,9 мин; ББА-МС м/з = 609 (M+Li); 603 (М+Н); ВРМС (HMRS): рассчит. для C29H43N6O6S 603,2965 (М+Н); найдено 603,2972.
ПРИМЕР 54
По методикам предыдущих Примеров можно получить соединения, представленные в Таблицах 2-9 (см. в конце описания).
ПРИМЕР 55
Соединения данного
изобретения являются эффективными ингибиторами ВИЧ протеазы. Согласно данным ферментативного анализа, описанного ниже, соединения, представленные в описанных здесь примерах, ингибируют ВИЧ фермент.
Предпочтительные соединения данного изобретения и рассчитанные для них IC50 значения (50%-ная ингибирующая концентрация, т.е. концентрация, при которой соединение-ингибитор уменьшает
активность фермента на 50%) представлены в Таблице 10. Способ ферментативного анализа описан ниже. Субстратом является 2-Ile-Nle-Phe(n-NO2)-Gln-ArgNH2. Положительным контролем
является MVT-101 (Miller, М. et al. Science, 246, 1149 (1989)]. Условия анализа следующие:
Буфер для анализа: 20 мМ фосфат натрия, pH 6,4; 20% глицерин; 1 мМ ЭДТК; 1 мМ ДДТ; 0,1% CHAPS.
Вышеуказанный субстрат растворяют в ДМСО, затем разбавляют в 10 раз буфером для анализа. Конечная концентрация субстрата при анализе равна 80 мкМ. ВИЧ протеазу разбавляют буфером для анализа до конечной 12,3 наномолярной концентрации фермента, в расчете на молекулярную массу 10780.
Конечная концентрация ДМСО составляет 14%, и конечная концентрация глицерина составляет 18%. Испытуемое соединение растворяют в ДМСО и разбавляют ДМСО в 10х относительно концентрации теста; добавляют 10 мкл препарата фермента, вещества смешивают и затем смесь инкубируют при температуре окружающей среды в течение 15 минут. Ферментативную реакцию инициируют добавлением 40 мкл субстрата. Увеличение флуоресценции фиксируют в 4 точках по времени (0, 8, 16 и 24 минуты) при температуре окружающей среды. Каждый анализ проводят в двух лунках.
Предшествующие примеры могут быть воспроизведены с аналогичным успехом путем замены обычных и конкретно описанных реагентов и/или условий способа данного изобретения на реагенты и/или условия обработки, используемые в предшествующих примерах.
ПРИМЕР 56
Эффективность различных соединений
определялась с помощью вышеописанного ферментативного анализа и СЕМ клеточного анализа.
Способ анализа ВИЧ ингибирования остро инфицированных клеток представляет собой по существу автоматический колориметрический анализ с использованием соединения тетразолиния, который описан Pauwles et al, J. Virol. Methods, 20, 309-321 (1988). Анализы проводят в 96-луночных планшетах для тканевой культуры. СЕМ клетки, CD4+ клеточную линию, выращивают в RPMI-1640 среде (Gibco), дополненной 10%-ной фетальной телячьей сывороткой, и затем обрабатывают полибреном (2 мкг/мл). 80-мкл объем среды, содержащей 1 • 104 клеток, диспергируют в каждой лунке планшета для тканевой культуры. В каждую лунку добавляют 100 мкл испытываемого соединения, растворенного в среде тканевой культуры (или среду без испытываемого соединения, в качестве контроля), до достижения требуемой конечной концентрации, и клетки инкубируют при 37oC в течение 1 часа. Замороженную культуру ВИЧ-1 разбавляют в культуральной среде до концентрации 5 х 104 TCID50 на мл (TCID50 = доза вируса, которая инфицирует 50% клеток в тканевой культуре), и 20 мкл пробы вируса (содержащей 1000 TCID50 вируса) добавляют в лунки, содержащие испытываемое соединение, и в лунки, содержащие только среду (инфицированные контрольные клетки). Несколько лунок содержат культуральную среду без вируса (неинфицированные контрольные клетки). Аналогично, токсичность испытываемого соединения определяют добавлением среды без вируса в несколько лунок, содержащих испытываемое соединение. В общем, планшеты с тканевой культурой содержали следующие эксперименты. (см. табл. A).
В экспериментах 2 и 4 конечные концентрации испытываемых соединений составляли 1, 10, 100 и 500 мкл/мл. В качестве положительного лекарственного контроля включают либо азидотимидин (AZT), либо дидезоксиинозин (ddI). Испытываемые соединения растворяют в ДМСО и разбавляют тканевой культуральной средой так, чтобы в любом случае конечная концентрация ДМСО не превышала 1,5%. ДМСО добавляют во все контрольные лунки в соответствующей концентрации.
После введения вируса клетки инкубируют при 37oC в увлажненной 5% CO2 атмосфере в течение 7 дней. Испытываемые соединения обычно добавляют на день 0, 2 и 5, при необходимости. На 7-й день после инфицирования, клетки в каждой лунке повторно суспендируют и отбирают 100 мкл пробы каждой клеточной суспензии на анализ. К каждой 100 мкл клеточной суспензии добавляют 20 мкл 5 мг/мл раствора 3-(4,5-диметилтиазол-2-ил)-2,5- дифенилтетразолий бромида (ММТ) и клетки инкубируют в течение 4 часов при 27oC в окружающей среде, содержащей 5% CO2. Во время инкубации МТТ метаболитически восстанавливается живыми клетками, приводя к продуцированию в клетке окрашенного продукта формазана. К каждой пробе добавляют 100 мкл 10% додецилсульфата Na в 0,01N HCl для лизирования клеток и пробы инкубируют в течение ночи. Для каждой пробы определяют поглощение при 590 нм, используя Molecular Devices микропланшет-ридер. Значения поглощения для каждого ряда лунок сравнивают для того, чтобы оценить вирусную контрольную инфекцию, ответ неинфицированных контрольных клеток, а также испытываемое соединение на цитотоксичность и антивирусную эффективность.
Соединения данного изобретения являются эффективными противовирусными соединениями и, в частности, эффективными ретровирусными ингибиторами, как показано выше. Так, заявляемые соединения являются эффективными ингибиторами ВИЧ протеазы. Предполагают, что заявляемые соединения могут также ингибировать другие ретровирусы, такие как другие лентивирусы, в частности другие штаммы ВИЧ, например, В14Ч-2, вирус Т-клеток лейкемии человека, респираторный синцитиальный вирус, вирус иммунодефицита обезьяны, кошачий вирус иммунодефицита, цитомегаловирус и пикорнавирус. Таким образом, заявляемые со единения эффективны для лечения, профилактики ретровирусных инфекций и/или предотвращения распространения ретровирусных инфекций.
Заявляемые соединения являются также эффективными для предотвращения роста ретровирусов в растворе. Как клеточные культуры человека, так и клеточные культуры животного, такие как Т-лимфоцитные культуры, используют для целого ряда хорошо известных целей, таких как методы исследования и диагностики, включая калибровку и контроль. До и во время роста и хранения клеточной культуры, заявляемые соединения можно добавить в среду клеточной культуры при концентрации, эффективной для предотвращения неожиданной или нежелательной репликации ретровируса, который может случайно, непреднамеренно или преднамеренно присутствовать в клеточной культуре. Вирус может присутствовать изначально в клеточной культуре, например, известно, что ВИЧ присутствует в Т-лимфоцитах человека задолго до его обнаружения в крови, или на протяжении экспозиции с вирусом. Такое использование заявляемых соединений предотвращает неизвестную или случайную экспозицию потенциально летального ретровируса исследователю или практикующему врачу.
Соединения данного изобретения могут содержать один или более асимметрических атомов углерода и поэтому могут существовать в форме оптических изомеров, а также в форме рацемических или нерацемических их смесей. Оптические изомеры можно получить разделением рацемических смесей стандартными способами, например, образованием диастереоизомерных солей обработкой оптически активной кислотой или основанием и затем разделением смеси диастереоизомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуоилвинная и камфорсульфоновая кислота. Другой способ разделения оптических изомеров включает использование колонки для хиральной хроматографии, оптимально подобранной, чтобы максимально увеличить разделение энантиомеров. Следующий приемлемый способ включает синтез ковалентных диастереоизомерных молекул взаимодействием соединений формулы I с оптически чистой кислотой в активированной форме или оптически чистым изоцианатом. Синтезированные диастереоизомеры можно разделить обычным способом, таким как хроматография, дистилляция, кристаллизация или сублимация, и затем гидролизовать, чтобы высвободить энантиомерно чистое соединение. Оптически активные соединения формулы I можно также получить, используя оптически активные исходные вещества. Эти изомеры могут быть в форме свободной кислоты, свободного основания, сложного эфира или соли.
Соединения данного изобретения могут быть использованы в форме солей, полученных из неорганических и органических кислот. Такие соли включают, но ими не ограничиваются, следующие: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидрокси-этансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат, мезилат и ундеканоат. Кроме того, основные азотсодержащие группы могут быть кватернизованы такими агентами, как низшие алкил галогениды, такие как метил, этил, пропил и бутил хлориды, бромиды и иодиды; диалкил сульфаты, такие как диметил, диэтил, дибутил и диамил сульфаты, галогениды с длинной цепью, такие как децил, лаурил, миристил и стеарил хлориды, бромиды и иодиды, аралкил галогениды, такие как бензил и фенэтил бромиды, и другие. При этом получают водо- или маслорастворимые, или диспергируемые продукты.
Примеры кислот, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения кислот, включают такие неорганические кислоты, как хлористоводородная кислота, серная кислота и фосфорная кислота, и такие органические кислоты, как щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота. Другие примеры включают соли со щелочными металлами или щелочноземельными металлами, такими как натрий, калий, кальций или магний, или с органическими основаниями.
Суммарная суточная доза, вводимая пациенту в разовой или разделенной общей дозе (повторяемой через определенные интервалы времени) может составлять количества, например, от 0,001 до 10 мг/кг веса тела ежедневно, и чаще всего от 0,01 до 1 мг. Композиции стандартной дозы могут содержать такие количества суб-многократных доз, которые составляют суточную дозу.
Количество активного ингредиента, которое можно сочетать с веществами-носителями для того, чтобы получить разовую дозированную форму, обычно варьируется в зависимости от пациента, подлежащего лечению, и конкретного способа введения.
Схему приема лекарственного средства для лечения болезненного состояния соединениями и/или композициями данного изобретения выбирают в соответствии с рядом факторов, включающих тип, возраст, вес, пол, диету и медицинское состояние пациента, тяжесть заболевания, способ введения, фармакологические характеристики, такие как активность, эффективность, фармакокинетические и токсикологические профили конкретного используемого соединения, используют ли систему доставки лекарственного средства или соединение вводят как часть комбинации лекарственных средств. Таким образом, используемая в действительности схема приема лекарственного средства может широко варьироваться и поэтому может отклоняться от предпочтительной схемы приема, представленной выше.
Соединения данного изобретения можно вводить перорально, парентерально, путем ингаляционного распыления, ректально или местно, в препаративных формах в виде единичных доз, содержащих стандартные нетоксичные фармацевтически приемлемые носители, адъюванты и разбавители, при необходимости. Местное введение может также включать использование трансдермального введения, такого как трансдермальные пластыри или устройства для лекарственного электрофореза. Используемый здесь термин парентеральный включает подкожные инъекции, внутривенную, внутримышечную, внутристернальную (intrasternal) инъекцию или методики вливания.
Препараты для инъекций, например стерильные водные или маслянистые суспензии для инъекций, можно приготовить согласно известным в данной области способам, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты. Стерильный препарат для инъекций может также представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например в 1,3-бутандиоле. К приемлемым носителям или растворителям, которые можно использовать, относятся вода, раствор Рингера (Ringer) и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные, нелетучие масла. Для этой цели может быть использовано любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, находят применение при приготовлении препаратов для инъекций.
Суппозитории для ректального введения лекарственного средства можно получить смешиванием лекарственного средства с подходящим, не вызывающим раздражение наполнителем, таким как масло какао и полиэтиленгликоли, которые являются твердыми при обычных температурах, но жидкими при ректальной температуре и поэтому могут расплавляться в прямой кишке и высвобождать лекарственное средство.
Твердые лекарственные формы для перорального введения могут включать капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение может быть смешано с по крайней мере одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также включать, при обычной практике, дополнительные вещества, помимо инертных разбавителей, например смазки, такие как стеарат магния. В случае капсул, таблеток и пилюль, дозированные формы могут также включать забуферивающие агенты. Таблетки и пилюли можно, кроме того, изготовлять с энтеросолюбильным покрытием.
Жидкие лекарственные формы для перорального введения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно используемые в данной области, такие как вода. Такие композиции могут также включать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, и подслащивающие агенты, средства, придающие вкус и аромат.
Хотя соединения данного изобретения можно вводить в качестве единственного активного фармацевтического агента, их можно также использовать в комбинации с одним или более иммуномодуляторов, противовирусных агентов или других противоинфекционных средств. Например, соединения данного изобретения можно вводить в сочетании с AZT, DDI, DDC или с ингибиторами глюкозидазы, такими как N-бутил-1-деоксиноджиримицин, или в виде их пролекарств, для профилактики и/или лечения СПИД. При применении в виде комбинации терапевтические средства могут быть сформулированы в виде раздельных композиций, которые дают в одно и то же время или в различные времена, или терапевтические средства можно вводить в виде единой композиции.
Вышеперечисленное является только иллюстрацией данного изобретения и подразумевается, что данное изобретение не ограничивается раскрытыми соединениями. Подразумевается, что вариации и изменения, которые очевидны специалистам в данной области, не выходят за объем и сущность данного изобретения, которые определены в нижеследующей формуле изобретения.
Из вышеприведенного описания специалистам в данной области можно легко установить существенные характеристики данного изобретения и, не выходя за рамки существа и объема изобретения, можно сделать различные изменения и модификации данного изобретения, чтобы адаптировать его к различным использованиям и условиям.
Реферат
Описываются новые гидроксиэтиламино сульфонамиды гетероциклокарбонил аминокислот, представленные общей формулой (I), или их фармацевтически приемлемая соль, где n = 0; R1 - алкил с 1 - 5 атомами углерода; R2 - аралкил, в котором алкил имеет 1 - 3 атома углерода; R3 - алкил с 1 - 5 атомами углерода; R4 - группа формулы (II), где А и В каждый независимо - кислород; R6 - водород; R7 - водород. Новые соединения эффективны в качестве ингибиторов ретровирусных протеаз и, в частности, в качестве ингибитора ВИЧ-протеазы. 7 с. и 5 з.п.ф-лы, 40 табл.


Формула
или их фармацевтически приемлемая соль,
где n = 0;
R1 - алкил с 1-5 атомами углерода;
R2 - аралкил, в котором алкил имеет 1-3 атома углерода;
R3 - алкил с 1-5 атомами углерода;
R4 - группа формулы

где А и В каждый независимо - кислород;
R6 - водород или дейтерий;
R7 - водород или дейтерий.
2S-[[пирролидин-2-ил)карбонил] амино] -N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил)амино]-1S(фенилметил)пропил]-3S-метил-пентанамид.
07.06.1995 по пп.1,2,5,6 и 8-12;
10.03.1995 по пп.3,4 и 7.
Комментарии