Селективные дипептидные ингибиторы калликреина, фармацевтическая композиция на их основе, их применение и способ лечения - RU2301225C2
Код документа: RU2301225C2
Описание
Настоящее изобретение относится к серии новых соединений, которые являются селективными ингибиторами фермента плазменный калликреин, к фармацевтическим композициям, содержащим эти ингибиторы, и к применению таких композиций в лечении заболеваний человека.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Фермент плазменный калликреин, также известный под классификационным номером ЕС.3.4.21.34, является членом семейства трипсиноподобных сериновых протеаз, которое также включает тканевый калликреин, тромбин, трипсин и плазмин. Он обнаруживается в плазме в виде неактивного зимогена, который активируется фактором XIIa. Фермент обладает широким спектром активности. Плазменный калликреин высвобождает вазоактивный пептид брадикинин из высокомолекулярного кининогена, расщепляя связи Lys-Arg и Arg-Ser. Этот же пептид может быть высвобожден из низкомолекулярного кининогена в присутствии эластазы нейтрофилов. Калликреин также способен к активации проурокиназы и плазминогена, а также, как считают, принимает участие в превращении проренина в ренин. Плазменный калликреин является существенным компонентом внутреннего каскада коагуляции крови, хотя в его роль не входит высвобождение брадикинина или ферментативное расщепление. Для активации в этом каскаде необходим высокомолекулярный кининоген, являющийся предпочтительным субстратом для плазменного калликреина (K.D. Bhoola et al., Pharm. Rev., 1992, 44, 1-80).
Физиологические эффекты плазменного калликреина, вероятно, являются результатом протеолитического расщепления кининогенов, в ходе которого высвобождаются кинины, или других субстратов, например предшественников ростовых факторов. Такие кинины, как брадикинин, являются сильными медиаторами воспаления. Кроме того, они влияют на такие клеточные функции, как кровяное давление, локальный кровоток, транспорт глюкозы и клеточная пролиферация. Это те клеточные процессы, на которые может оказывать влияние высвобождение вторичных мессенджеров, таких как фактор активации тромбоцитов, лейкотриены, простагландины, вещество Р, ацетилхолин и норадреналин.
Несколькими группами были открыты синтетические ингибиторы плазменного калликреина. Они включают кетометиленовые производные аргинина (WO 92/04371 и D.M. Evans et al., Immunopharmacology, 1996, 32, 115-116), производные норагматина и агматина (WO 95/07291, WO 94/29335), производные бензамидина (J. Stürzbecher et al., Brazilian J. Med. Biol. Res. 1994, 27, 1929-1934), производные бороновой кислоты (US 5187157) и производные аминометилциклогексаноила (N. Teno et al., Chem. Pharm. Bull., 1993, 41, 1079-1090). Была показана активность производных аминометилциклогексаноила на моделях коллаген-индуцированного артрита у мышей (Y. Fujimora et al., Agents Actions, 1993, 39, 42-48) и индуцированного эндотоксином диссеминированного внутрисосудистого коагулирования (ДВС-синдром) у крыс (S. Okamoto et al., Agents Actions (Supplement), 1992, 38 (Part 1), 198-205). Производные бороновой кислоты активны на моделях воспалительного заболевания кишечника (А. Stadnicki et al., Digestive Diseases and Sciences, 1996, 41, 912-920 и FASEB, 1998, 12, 325-333).
Избирательность в отношении других членов семейства трипсиноподобных сериновых протеаз является важной проблемой. Об ингибиторах тканевого калликреина, демонстрирующих низкую активность в отношении плазменного калликреина, сообщалось ранее (М. Szelke et al., Brazilian J. Med. Biol. Res. 1994, 27, 1935 и D.M. Evans et al., Immunopharmacology, 1996, 32, 117), но остается потребность в соединениях, которые избирательно ингибируют плазменный, но не тканевой калликреин.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к серии ациламинопиперидин-1-карбоксамидинов, которые являются ингибиторами плазменного калликреина. Эти соединения демонстрируют хорошую избирательность в отношении плазменного калликреина и потенциально полезны в лечении воспалительного заболевания кишечника, артрита, воспаления, септического шока, гипотензии, рака, респираторного дистресс-синдрома у взрослых, диссеминированного внутрисосудистого коагулирования, при операциях в условиях искусственного кровообращения и постоперационных кровотечениях. Изобретение также относится к фармацевтическим композициям ингибиторов, к применению соединений в качестве терапевтических агентов и к способам лечения с использованием композиций.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
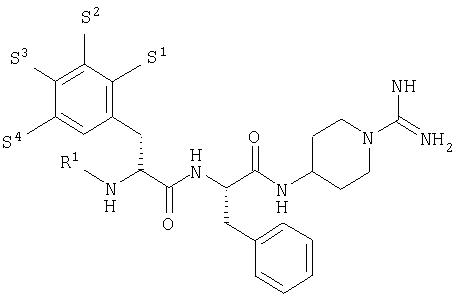
В своем первом аспекте настоящее изобретение включает серию новых 4-(дипептидиламино)-пиперидин-1-карбоксамидинов согласно общей формуле 1

В общей формуле 1 R1 представляет собой группу, выбранную из атома водорода (Н), низшей алкильной группы, группы R4-CO, группы R4-O2CCH2, группы R5-OCO и группы R5-SO2. R2 представляет собой группу, выбранную из низшей алкильной группы, циклоалкильной или (С5-С12 )циклоалкилалкильной группы, каждая из которых может быть возможно замещена алкильной или алкоксигруппой, аралкильной группы, которая может быть возможно замещена группами в количестве вплоть до трех, выбранными из F, Cl, Br, I, ОН, низшего алкила, O-(низший алкил), O-бензил, NH2, NO2, NH-ацил, CN и CF3, и аралкилоксиметильной группы, которая может быть возможно замещена группами в количестве вплоть до трех, выбранными из F, Cl, Br, ОН, низшего алкила, O-(низший алкил). Или же R1 и R2 вместе могут составлять орто-ксилиленовую группу (о-С6Н4(СН2)2). Ароматическое кольцо этой ксилиленовой группы может быть возможно замещено группой, выбранной из F, Cl, Br, ОН, низшего алкила и O-(низший алкил).
R3 представляет собой группу, выбранную из Н, ОН и O-(низший алкил).
R4 представляет собой группу, выбранную из Н, низшего алкила и фенила.
R5 представляет собой группу, выбранную из низшего алкила, фенила и бензила.
В контексте настоящего описания изобретения термины "алкильная группа" и "низшая алкильная группа" использованы взаимозаменяемо для обозначения линейных и разветвленных насыщенных углеводородных групп с числом атомов углерода от 1 до 8, таких как метильная, этильная, изопропильная, трет-бутильная, неопентильная и изооктильная группы.
Термин "циклоалкильная группа" использован для обозначения моноциклических или полициклических насыщенных углеводородных групп с числом атомов углерода от 3 до 12, таких как циклопропильная, циклогексильная, бицикло[4.4.0]децильная (то есть декагидронафтильная) и адамантильная группы.
Термин "циклоалкилалкильная группа" использован для обозначения алкильных групп, несущих циклоалкильную группу в качестве заместителя, таких как циклогексилметильная и 1-(циклопентил)этильная группы. Там, где указан интервал значений, как в случае (Са-Сb)циклоалкилалкила, это означает, что циклоалкильная часть группировка от а до b атомов углерода.
Термин "алкоксигруппа" использован для обозначения групп О-(алкил).
Термин "ацильная группа" использован для обозначения групп формил (Н-СО) и алкил-СО.
Термин "аралкильная группа" использован для обозначения алкильных групп, которые в качестве заместителя несут арильную группу, таких как бензильная или 1-нафтилметильная группы. Термин "арильная группа" включает фенильную, нафтильную, фурильную, тиенильную, пирролильную и пиридильную группы.
Термин "аралкилоксиметильная группа" использован для обозначения групп аралкил-ОСН2.
Все соединения по данному изобретению имеют гуанидиновую функциональную группу и поэтому могут образовывать соли присоединения с кислотами. В тех пределах, в которых такие кислоты являются фармацевтически приемлемыми, эти соли включены в объем изобретения. Примерами подходящих кислот являются уксусная кислота, трифторуксусная кислота, фумаровая кислота, яблочная кислота, лимонная кислота, бензойная кислота, бензолсульфоновая кислота, соляная кислота, серная кислота и фосфорная кислота. Некоторые соединения в рамках изобретения имеют кислотную функциональную группу и поэтому могут образовывать соли с щелочными и щелочно-земельными металлами. Как и в первом случае, те из них, которые являются фармацевтически приемлемыми, включены в объем изобретения. Примеры таких солей включают соли натрия, калия и кальция.
Все соединения по данному изобретению имеют по меньшей мере два стереогенных центра (асимметрических углеродных атома) и поэтому могут существовать в форме оптических изомеров, таких как энантиомеры, диастереомеры и эпимеры. Все эти изомеры включены в объем данного изобретения. Смеси таких изомеров, включая рацемические смеси (но не ограничиваясь ими), также включены в объем изобретения.
В своем предпочтительном воплощении настоящее изобретение включает соединения общей формулы 1, в которой R1 выбран из Н, низшего алкила и R4-O2CCH2.
В другом предпочтительном воплощении настоящее изобретение включает соединения общей формулы 1, в которой R2 выбран из (С6-С10)циклоалкилалкила, бензила, возможно замещенного группами в количестве вплоть до трех, выбранными из F, Cl, Br, ОН, низшего алкила и O-(низший алкил), фенетила, возможно замещенного группами в количестве вплоть до трех, выбранными из F, Cl, Br, ОН, низшего алкила, O-(низший алкил), и бензилоксиметила, возможно замещенного группами в количестве вплоть до трех, выбранными из F, Cl, Br, ОН, низшего алкила, O-(низший алкил). Наиболее предпочтительно R2 выбран из циклогексилметила, декагидронафт-2-илметила, бензила, 4-фторбензила, 4-хлорбензила, 4-гидроксибензила, 4-(низший алкил)оксибензила, α-гидроксибензила, α-метоксибензила, фенетила и бензилоксиметила.
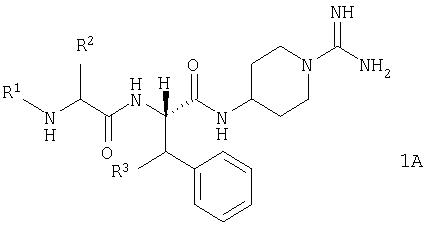
В другом предпочтительном воплощении настоящее изобретение включает соединения общей формулы 1, абсолютная стереохимия которого представлена общей формулой 1А. Более предпочтительно абсолютная стереохимия является такой, как представлено формулой 1В

В другом предпочтительном воплощении настоящее изобретение включает соединение, выбранное из:
(2'S,2''R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенил-пропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-карбоксиметиламино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(3''-(4'''-этоксифенил)-2''-метилоксикарбонилметиламино)-пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-амино-3''-циклогексилпропаноиламино)-3'-фенил-пропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-карбоксиметиламино-3''-циклогексилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)-пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-амино-3''-фенилпропаноиламино)-3'-фенилпропаноил-амино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-карбоксиметиламино-3''-фенилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-метилоксикарбонилметиламино)-3''-фенилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-амино-3''-декагидронафт-2'''-илпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(2''-карбоксиметиламино-3''-декагидронафт-2'''-илпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S, 2''R)-4-(2'-(3''-декагидронафт-2'''-ил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S,2''R, 3'R)-4-(2'-(2''-амино-3''-циклогексилпропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S,2''R, 3'R)-4-(2'-(2''-карбоксиметиламино-3''-циклогексилпропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S,2''R, 3'R)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)-пропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S,2''R, 3'R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина;
(2'S,2''R, 3'R)-4-(2'-(2''-карбоксиметиламино-3''-(4'''-этоксифенил)-пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина и
(2'S,2''R, 3'R)-4-(2'-(3''-(4'''-этоксифенил)-2''-(метилоксикарбонилметиламино)-пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина.
Соединения по настоящему изобретению могут быть получены способами, широко известными из уровня техники, и особенно способами, используемыми в области химии пептидов. Пригодным исходным веществом является 4-амино-1-бензилпиперидин (2). В результате защиты первичного амина трет-бутилоксикарбонильной (Boc) группой получают соединение 3, гидрогенолиз которого приводит к производному пиперидина 4. Этот продукт может быть обработан производным изотиомочевины 5 с получением производного карбоксамидинопиперидина 6, в котором аминная и гуанидиновая функциональные группы защищены по-разному.

С производного карбоксамидинопиперидина 6 можно избирательно снять защиту и осуществить его сочетание с N-защищенной аминокислотой 7 с получением промежуточного соединения 8.
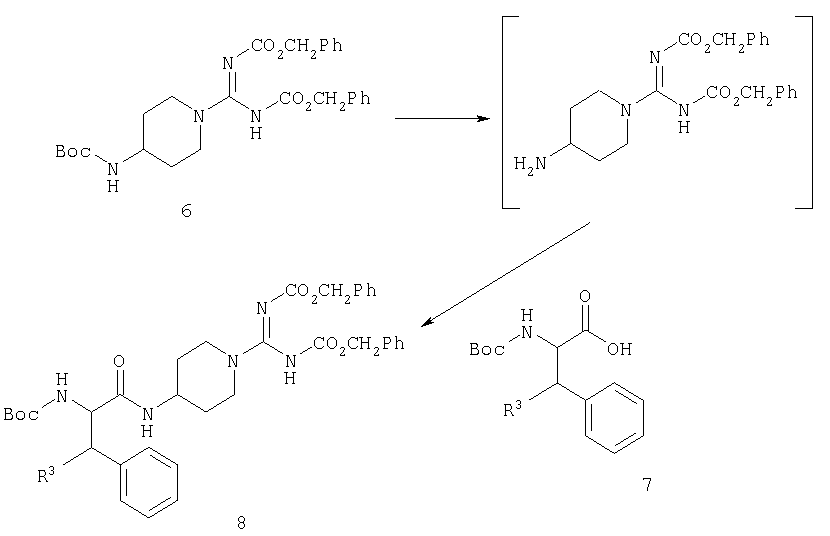
Дальнейшее усложнение промежуточного соединения 8 с получением конечного продукта в определенной степени зависит от природы R1. В случае, когда R1 представляет собой R5-OCO, удобно продолжать синтез через промежуточное соединение 9.
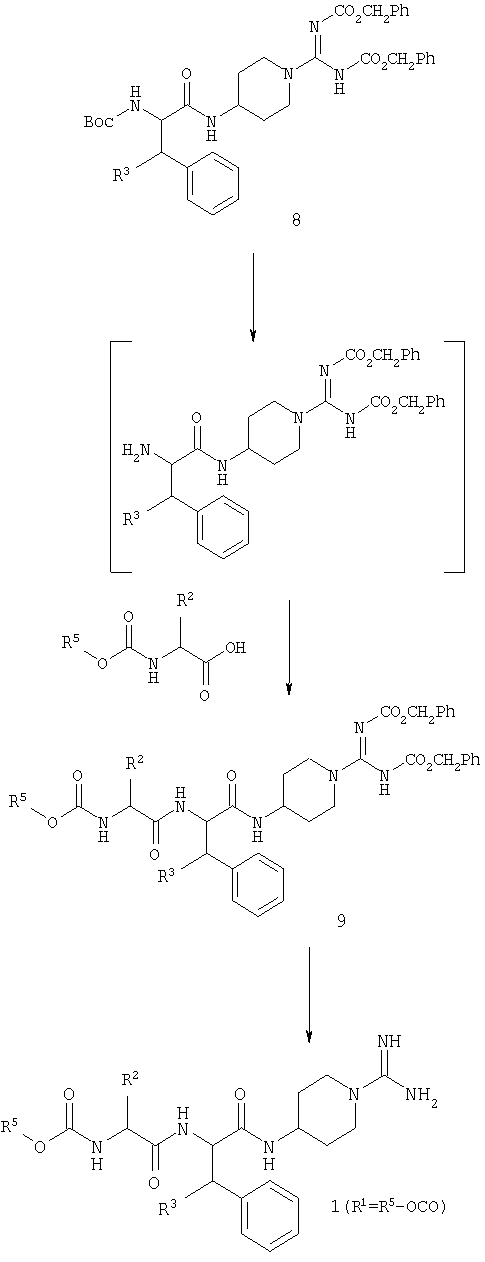
Когда R1 представляет собой H, R4-CO, R4-O2CCH2 или R5-SO2, удобно продолжать синтез через промежуточные соединения 10 и 11.

Снятие защиты с гуанидиновой функциональной группы дает соединения общей формулы 1, в которой R1 представляет собой Н. Получение производных первичного амина перед снятием защиты с гуанидина дает возможность получения других вариантов R1.

Затем с промежуточных соединений 12, 13 и 14 может быть снята защита с получением соответствующих соединений общей формулы 1.
Когда R1 представляет собой алкил или когда R1 и R2 вместе образуют ксилиленовую группу, синтез удобно продолжать через промежуточное соединение 15.

Два этапа снятия защиты приводят к получению соответствующих соединений общей формулы 1.
Соединения по настоящему изобретению являются сильными и избирательными ингибиторами плазменного калликреина. Поэтому они полезны для лечения болезненных состояний, вызываемых гиперактивностью плазменного калликреина. В общем, для использования в таком лечении соединения готовят в виде препаратов для введения пациенту. Фармацевтический препарат может быть твердым или жидким, таким как таблетка, капсула, раствор или суспензия. Способы получения таких препаратов широко известны из области фармацевтики.
Введение композиций пациентам будет происходить под контролем лечащего врача.
ПРИМЕРЫ
В тексте использованы следующие аббревиатуры:
"Celite" является зарегистрированным товарным знаком корпорации "Celite".
"Vydac" является зарегистрированным товарным знаком W.R. Grace & Со.
ПРИМЕР 1
(2'S,2''R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат

1А. 1-бензил-4-(трет-бутилоксикарбониламино)пиперидин
4-амино-1-бензилпиперидин (3,2 г, 16,8 ммоль) растворяли в CH2Cl2 (100 мл). Добавляли ди-трет-бутил дикарбонат (3,7 г, 17,0 ммоль) и N,N-диизопропилэтиламин (1,9 г, 19 ммоль). Смесь перемешивали в течение 18 часов при комнатной температуре, затем растворитель удаляли в вакууме, а остаток переносили в этилацетат (150 мл). Этот раствор промывали 0,3 М KHSO4 (2×30 мл), насыщ. NaHCO3 (2×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме до получения желтого масла, которое очищали флэш-хроматографией на силикагеле (элюент: 70% хлороформ, 30% циклогексан) с получением желтого твердого вещества, идентифицируемого как 1-бензил-4-(трет-бутилоксикарбониламино)пиперидин (4,9 г, 18,9ммоль, 100%).
1Б. 4-(трет-бутилоксикарбониламино)пиперидин
1-бензил-4-(трет-бутилоксикарбониламино)пиперидин (4,9 г, 18,9 ммоль) растворяли в этаноле (100 мл). Этот раствор гидрогенизировали над 10%-ным палладием на угле, при 60 фунт-сила на кв. дюйм (413,7 кПа). Смесь выдерживали 18 часов при комнатной температуре, после чего фильтровали через Celite и промывали осадок этанолом (100 мл). Объединенные фильтраты выпаривали в вакууме с получением белого твердого вещества, идентифицируемого как 4-(трет-бутилоксикарбониламино)пиперидин (2,3 г, 7,1 ммоль, 51%).
1В. N,N'-ди(бензилоксикарбонил)-4-трет-бутилоксикарбониламино)пиперидин-1-карбоксамидин
4-(трет-бутилоксикарбониламино)пиперидин (1,5 г, 7,5 ммоль) растворяли в этаноле (100 мл). Добавляли N,N'-бис(бензилоксикарбонил)-S-метилизотиомочевину (3,1 г, 8,7 ммоль) и оксид ртути (1,9 г, 8,8 ммоль). Смесь перемешивали при 40°С 4 часа, затем твердую фазу отфильтровывали и промывали этанолом (50 мл). Объединенные фильтраты выпаривали в вакууме с получением бесцветного масла, которое очищали флэш-хроматографией на силикагеле (элюент: 90% петролейный эфир 60-80, 10% этилацетат) до получения бесцветного масла, идентифицируемого как N, N'-ди(бензилоксикарбонил)-4-(трет-бутилоксикарбониламино)пиперидин-1-карбоксамидин (3,3 г, 6,6 ммоль, 87%).
1Г. (2'S)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
N, N'-ди(бензилоксикарбонил)-4-(трет-бутилоксикарбониламино)пиперидин-1-карбоксамидин (3,1 г, 6,1 ммоль) растворяли в смеси 4 М HCl/диоксан (70 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме и растворяли остаток в CH2Cl2 (60 мл). Этот раствор охлаждали до 0°С, добавляли Boc-Phe-ONSu (2,2 г, 6,1 ммоль) и доводили рН до 9 при помощи N-метилморфолина. Смесь перемешивали при комнатной температуре в течение 4 часов, растворитель выпаривали в вакууме и остаток растворяли в этилацетате (200 мл). Этот раствор промывали 0,3 М KHSO4 (2×30 мл), насыщ. NaHCO3 (2×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали флэш-хроматографией на силикагеле (элюент: 70% хлороформ, 30% циклогексан) с получением белого твердого вещества, идентифицируемого как (2'S)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (3,56 г, 5,4 ммоль, 89%).
1Д. (2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-{трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'S)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (2,5 г, 3,85 ммоль) растворяли в 4 М HCl/диоксан (70 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме и остаток растворяли в СН2Cl2/ДМФ (9:1, 50 мл). Этот раствор охлаждали до 0°С, добавляли Boc-DTyr(Et)OH (1,2 г, 3,84 ммоль), затем гидрат 1-гидроксибензотриазола (680 мг, 5,0 ммоль) и водорастворимый карбодиимид (1,0 г, 5,0 ммоль). По прошествии 15 минут рН доводили до 8 при помощи N-метилморфолина. Смесь перемешивали при комнатной температуре в течение 18 часов, после чего растворитель выпаривали в вакууме и растворяли остаток в хлороформе (200 мл). Этот раствор промывали 0,3 М KHSO4 (2×30 мл), насыщ. NaHCO3 (2×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали флэш-хроматографией на силикагеле (элюент: 85% хлороформ, 15% гексан) с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (2,47 г, 3,2 ммоль, 65%).
1Е. (2'S,2''R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
(2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (2,1 г, 2,7 ммоль) растворяли в 4 М HCl/диоксан (50 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме и растворяли остаток в смеси АсОН/вода (95:5, 50 мл). Этот раствор гидрогенизировали над 10%-ным палладием на угле. Смесь выдерживали 2 часа при комнатной температуре, после чего фильтровали через Celite и промывали остаток смесью АсОН/вода (9:1, 30 мл). Объединенные фильтраты выпаривали в вакууме и очищали остаток посредством ЖХСД на VydacC18 (15-25 мкм), используя MeCN/H2О/ТФУ, с получением твердого белого вещества, идентифицируемого как H-DTyr(Et)-Phe-4-амино-1-амидинопиперидина трифторацетат (1,12 г).
[М+Н]+=480,6
ПРИМЕР 2
(2'S, 2''R)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат

2А. (2'S,2''R)-N, N'-ди[бензилоксикарбоксил)-4-(2'-[2''-(трет-бутилоксикарбониламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'S)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (из примера 1Г, 1,9 г, 2,99 ммоль) растворяли в 4 М HCl/диоксан (70 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме и остаток растворяли в СН2Cl2/ДМФ (9:1, 50 мл). Этот раствор охлаждали до 0°С, добавляли Boc-DCha-ОН (900 мг, 3,3 ммоль), затем гидрат 1-гидроксибензотриазола (820 мг, 6,1 ммоль) и водорастворимый карбодиимид (730 мг, 3,6 ммоль). По прошествии 15 минут рН доводили до 8 при помощи N-метилморфолина. Смесь перемешивали при комнатной температуре в течение 18 часов, после чего растворитель выпаривали в вакууме и остаток растворяли в хлороформе (200 мл). Этот раствор промывали 0,3 М KHSO4 (2×30 мл), насыщ. NaHCO3 (2×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали флэш-хроматографией на силикагеле (элюент: 90% хлороформ, 10% гексан) с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(трет-бутилоксикарбониламино)-3''-циклогексилпропаноиламино)-3'-фенилпро-паноиламино)пиперидин-1-карбоксамидин (1,73 г, 2,14 ммоль, 72%).
2Б. (2'S, 2''R)-N,N'-ди(бензилоксикарбонил)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(трет-бутилоксикарбониламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (1,73 г, 2,14 ммоль) растворяли в 4 М HCl/диоксан (50 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме и остаток растворяли в ацетонитриле (100 мл). Добавляли метилбромацетат (400 мг, 2,6 ммоль) и N, N'-диизопропилэтиламин (440 мг, 4,4 ммоль). Реакционную смесь перемешивали при 60°С в течение 5 часов, после чего растворитель выпаривали в вакууме, а осадок растворяли в этилацетате (200 мл). Этот раствор промывали 0,3 М KHSO4 (2×30 мл), насыщ. NaHCO3 (2×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением желтого масла, которое очищали флэш-хроматографией на силикагеле (элюент: 90% хлороформ, 10% гексан) с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-N,N'-ди(бензилоксикарбонил)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (1,65 г, 2,11 ммоль, 98%).
2В. (2'S,2''R)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
(2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноиламино)-пиперидин-1-карбоксамидин (1,62 г, 2,11 ммоль) растворяли в смеси АсОН/вода (95:5, 50 мл). Этот раствор гидрогенизировали над 10%-ным палладием на угле. Смесь выдерживали 2 часа при комнатной температуре, после чего фильтровали через Celite и промывали остаток смесью АсОН/вода (9:1, 30 мл). Объединенные фильтраты выпаривали в вакууме, а остаток очищали при помощи ЖХСД на VydacC18 (15-20 мкм), используя MeCN/H2О/ТФУ, с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат (570 мг).
[М+Н]+=515
ПРИМЕР 3
(2'S,2''R)-4-(2'-(2''-(карбоксиметиламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
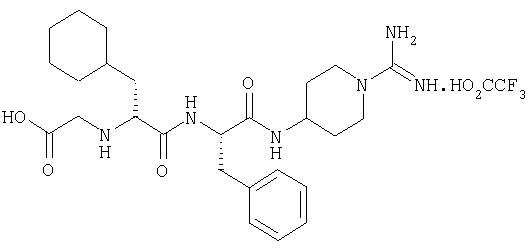
3А. (2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(карбоксиметиламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(3''-циклогексил-2''-(метилоксикарбонилметиламино)пропаноиламино)-3'-фенилпропаноил-амино)пиперидин-1-карбоксамидин (из примера 2Б, 1,6 г, 2,1 ммоль) растворяли в тетрагидрофуране (50 мл). Добавляли 1 М гидроксид лития (3 мл, 3 ммоль). По прошествии 18 часов растворитель выпаривали в вакууме, а остаток растворяли в этилацетате (150 мл). Этот раствор промывали 1 М лимонной кислотой (1×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-N,N'-ди(бензилоксикарбонил)-4-(2''-(карбоксиметиламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноил-амино)пиперидин-1-карбоксамидин (1,62 г; 2,11 ммоль, 100%).
3Б. (2'S,2''R)-4-(2'-(2''-(карбоксиметиламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
(2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(карбоксиметиламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (1,62 г, 2,11 ммоль) растворяли в смеси АсОН/вода (95:5, 50 мл). Этот раствор гидрогенизировали над 10%-ным палладием на угле. Смесь выдерживали 2 часа при комнатной температуре, после чего фильтровали через Celite и промывали остаток смесью АсОН/вода (9:1, 30 мл). Объединенные фильтраты выпаривали в вакууме, а остаток очищали при помощи ЖХСД на VydacC18 (15-25 мкм), используя MeCN/H2О/ТФУ, с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-4-(2'-(2''-карбоксиметиламино)-3''-циклогексилпропаноиламино)-3'-фенилпропаноил-амино)пиперидин-1-карбоксамидина трифторацетат (570 мг).
[М+Н]+=501
ПРИМЕР 4
(2'S,2''R)-4-(2'-(2''-бензоиламино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
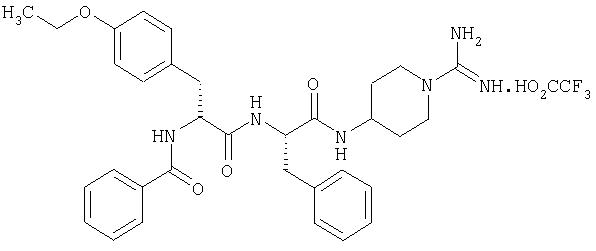
4А. (2'S, 2''R)-4-(2'-(2''-бензоиламино-3''-(4'''-этоксифенип)пропаноиламино)-3'-фенилпропаноиламино)-N,N'-ди(бензилоксикарбонил)пиперидин-1-карбоксамидин
(2'S,2''R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)-пиперидин-1-карбоксамидин (из примера 1Е, 100 мг, 0,12 ммоль) растворяли в 4 М HCl/диоксан (20 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме, а остаток растворяли в СН2Cl2 (20 мл). Этот раствор охлаждали до 0°С и добавляли бензоилхлорид (19,7 мг, 0,141 ммоль) и триэтиламин (36 мг, 0, 36 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, после чего растворитель выпаривали в вакууме, а остаток растворяли в этилацетате (70 мл). Этот раствор промывали 0,3 М KHSO4 (2×20 мл), насыщ. NaHCO3 (2×20 мл), водой (2×20 мл) и рассолом (1×20 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали флэш-хроматографией на силикагеле (элюент: 85% хлороформ, 15% циклогексан) с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-4-(2'-(2''-бензоиламино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)-N, N'-ди(бензилоксикарбонил)пиперидин-1-карбоксамидин (65 мг, 0,072 ммоль, 60%).
4Б. (2'S, 2''R)-4-(2'-(2''-бензоиламино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)-пиперидин-1-карбоксамидина трифторацетат
(2'S, 2''R)-4-(2'-(2''-бензоиламино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)-N,N'-ди(бензилоксикарбонил)пиперидин-1-карбоксамидин (65 мг, 0,072 ммоль) растворяли в смеси АсОН/вода (95:5, 25 мл). Этот раствор гидрогенизировали над 10%-ным палладием на угле. Смесь выдерживали 1 час при комнатной температуре, после чего фильтровали через Celite, а остаток промывали смесью АсОН/вода (9:1, 20 мл). Объединенные фильтраты выпаривали в вакууме, а остаток очищали при помощи ЖХСД на VydacC18 (15-25 мкм), используя MeCN/Н2O/ТФУ, с получением белого твердого вещества, идентифицируемого как (2'S,2''R)-4-(2'-(2''-бензоиламино-3''-(4'''-этоксифенил)пропаноиламино)-3'-фенилпропаноиламино)-пиперидин-1-карбоксамидина трифторацетат (44 мг).
[М+Н]+=585
ПРИМЕР 5
(2'S,2''R,3'R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
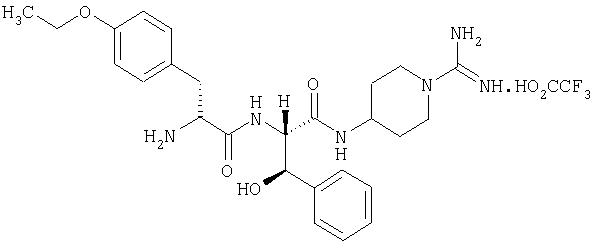
5А. (2S, 3R)-2-(трет-бутилоксикарбониламино)-3-гидрокси-3-фенилпропионовая кислота
H-thPse-OH (J. Biol. Chem., 1953, 204, 323) (1,4 г, 7,73 ммоль) растворяли в диоксане (75 мл). Добавляли гидроксид натрия (820 мг, 20,5 ммоль) в воде (75 мл), затем ди-трет-бутил дикарбонат (2,1 г, 9,6 ммоль). Смесь перемешивали в течение 18 часов при комнатной температуре, затем диоксан удаляли в вакууме, а остаток промывали диэтиловым эфиром (1×100 мл), подкисляли до рН 4 при помощи 1 М HCl и экстрагировали при помощи CHCl3 (3×100 мл). Объединенные экстракты промывали водой (1×50 мл) и рассолом (1×50 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, идентифицируемого как (2S, 3R)-2-(трет-бутилоксикарбониламино)-3-гидрокси-3-фенилпропионовая кислота (1,6 г, 5,7 ммоль, 74%).
5Б. (2'S,3'R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
N, N'-ди(бензилоксикарбонил)-4-(трет-бутилоксикарбониламино)пиперидин-1-карбоксамидин (из примера 1В, 2,3 г, 4,5 ммоль) растворяли в 4 М HCl/диоксан (70 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме, а остаток растворяли в смеси СН2С12/ДМФ (9:1, 50 мл). Этот раствор охлаждали до 0°С и добавляли (2S, 3R)-2-(трет-бутилоксикарбониламино)-3-гидрокси-3-фенилпропионовую кислоту (1,6 г, 5,6 ммоль), затем гидрат 1-гидроксибензотриазола (1,1 г, 8,1 ммоль) и водорастворимый карбодиимид (1,4 г, 75,0 ммоль). По прошествии 15 минут рН доводили до 8 при помощи N-метилморфолина. Смесь перемешивали при комнатной температуре в течение 18 часов, после чего растворитель выпаривали в вакууме, а остаток растворяли в этилацетате (200 мл). Этот раствор промывали 0,3 М KHSO4 (2×30 мл), насыщ. NaHCO3 (2×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали флэш-хроматографией на силикагеле (элюент: 50% этилацетат, 50% петролейный эфир) с получением белого твердого вещества, идентифицируемого как (2'S,3'R)-N,N'-ди(бензилоксикарбонил)-4-(2'-трет-бутилоксикарбониламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (1,1 г, 1,6 ммоль, 36%).
5В. (2'S,2''R,3'R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'S,3'R)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (1,1 г, 1,6 ммоль) растворяли в 4 М HCl/диоксан (70 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме, а остаток растворяли в смеси СН2Cl2/ДМФ (9:1, 50 мл). Этот раствор охлаждали до 0°С и добавляли Boc-DTyr(Et)-OH (620 мг, 2,0 ммоль), затем гидрат 1-гидроксибензотриазола (270 мг, 2,0 ммоль) и водорастворимый карбодиимид (420 мг, 2,1 ммоль). По прошествии 15 минут рН доводили до 8 при помощи N-метилморфолина. Смесь перемешивали при комнатной температуре в течение 18 часов, после чего растворитель выпаривали в вакууме, а остаток растворяли в этилацетате (200 мл). Этот раствор промывали 0,3 М KHSO4 (2×30 мл), насыщ. NaHCO3 (2×30 мл), водой (2×30 мл) и рассолом (1×30 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали флэш-хроматографией на силикагеле (элюент: 60% этилацетат, 40% петролейный эфир), с получением белого твердого вещества, идентифицируемого как (2'S, 2''R,3'R)-N,N'-ди(бензилоксикарбонил)-4-(2'-(2''-трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (1,2 г, 1,3 ммоль, 80%).
5Г. (2'S,2''R,3'R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
(2'S,2''R, 3'R)-N,N'-ди(бензилоксикарбонил)-4-(2'-(2''-трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-гидрокси-3'-фенил-пропаноиламино)пиперидин-1-карбоксамидин (1,2 г, 1,3 ммоль) растворяли в 4 М HCl/диоксан (50 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме, а остаток растворяли в смеси АсОН/вода (95:5, 50 мл). Этот раствор гидрогенизировали над 10%-ным палладием на угле. Смесь выдерживали 2 часа при комнатной температуре, после чего фильтровали через Celite и промывали остаток смесью АсОН/вода (9:1, 30 мл). Объединенные фильтраты выпаривали в вакууме, а остаток очищали при помощи ЖХСД на VydacC18 (15-25 мкм), используя MeCN/Н2О/ТФУ, с получением белого твердого вещества, идентифицируемого как (2'S,2''R,3'R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат (540 мг).
[М+Н]+=497,0
ПРИМЕР 6
(2'S,2''R,3'R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
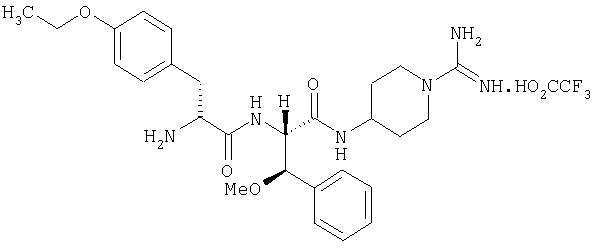
6А. (2'SR,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'SR,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-трет-бутилоксикарбониламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин получали способом, описанным в Примере 5, но начиная с рацемического Н-thPse-OH.
6Б. (2'SR,3'RS)-N,N'-ди(бензилоксикарбонил)-4-[2'-[трет-бутилоксикарбониламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'SR,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-гидрокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (180 мг, 0,27 ммоль) растворяли в СН2Cl2 (30 мл). Затем добавляли иодометан (190 мг, 1,3 ммоль) и оксид серебра (132 мг, 0,8 ммоль). Смесь выдерживали 18 часов при 60°С, фильтровали и фильтрат выпаривали в вакууме с получением коричневого масла, которое очищали флэш-хроматографией на силикагеле (элюент: 50% этилацетат, 50% петролейный эфир) с получением белого твердого вещества, идентифицируемого как (2'SR,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (112 мг, 0,16 ммоль, 61%).
6В. (2'SR,2''R,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин
(2'SR,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-(трет-бутилоксикарбониламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (112 мг, 0,16 ммоль) растворяли в 4 М HCl/диоксан (20 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме, а остаток растворяли в CH2Cl2 (30 мл). Этот раствор охлаждали до 0°С и добавляли Boc-DTyr(Et)-OH (50 мг, 0,16 ммоль), затем PyBrop (76 мг, 0,16 ммоль). рН доводили до 9 при помощи N,N-диизопропилэтиламина. Смесь перемешивали при комнатной температуре в течение 18 часов, после чего растворитель выпаривали в вакууме, а остаток растворяли в этилацетате (70 мл). Этот раствор промывали 0,3 М KHSO4 (1×20 мл), насыщ. NaHCO3 (1×20 мл), водой (1×20 мл) и рассолом (1×20 мл), высушивали (Na2SO4) и выпаривали в вакууме с получением белого твердого вещества, которое очищали флэш-хроматографией на силикагеле (элюент: 65% хлороформ, 15% гексан) с получением белого твердого вещества, идентифицируемого как (2'SR,2''R,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (108 мг, 0,12 ммоль, 75%).
6Г. (2'S,2''R,3'R)-4-(2'-(2''-амино-3''-(4'''-этоксифенил)пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат
(2'SR,2''R,3'RS)-N, N'-ди(бензилоксикарбонил)-4-(2'-(2''-(трет-бутилоксикарбониламино)-3''-(4'''-этоксифенил)пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидин (108 мг, 0,12 ммоль) растворяли в 4 М HCl/диоксан (20 мл). Выдерживали 30 минут при комнатной температуре, после чего растворитель выпаривали в вакууме, а остаток растворяли в смеси АсОН/вода (95:5, 20 мл). Этот раствор гидрогенизировали над 10%-ным палладием на угле. Смесь выдерживали 2 часа при комнатной температуре, после чего фильтровали через Celite и промывали остаток смесью АсОН/вода (9:1, 30 мл). Объединенные фильтраты выпаривали в вакууме, а остаток очищали при помощи ЖХСД на VydacC18 (15-25 мкм), используя MeCN/Н2О/ТФУ, с получением белого твердого вещества, идентифицируемого как (2'S,2''R,3'R)-4-(2'-(2''-амино-3''-(4''-этоксифенил)пропаноиламино)-3'-метокси-3'-фенилпропаноиламино)пиперидин-1-карбоксамидина трифторацетат (18 мг).
[М+Н]+=511,3
Следующие соединения были получены с использованием аналогичных способов.
Примеры 7-17




Примеры 18-52

Примеры 53-54

Примеры 55-58

Пример 59
Определение константы ингибирования Кi для плазменного калликреина
Ингибирование активности плазменного калликреина in vitro оценивали с помощью стандартных опубликованных способов (см., например, Johansen et al., Int. J. Tiss. Reac. 1986, 8, 185; Shori et al., Biochem. Pharmacol., 1992, 43, 1209; Stürzebecher et al., Biol. Chem. Hoppe-Seyler, 1992, 373, 1025). Плазменный калликреин человека (Callbiochem) инкубировали при 37°С с тремя разными концентрациями хромогенного субстрата S-2302 (Chromogenix AB) и различными концентрациями тестируемого соединения. Остаточную ферментативную активность (начальную скорость реакции) определяли измеряя изменение оптического поглощения при 405 нм, а константу ингибирования Ki для тестируемого соединения - как определено графиком Диксона (Dixon, Biochem. J., 1953, 55, 170). Типичные результаты представлены в Таблице 1.
Пример 60
Определение избирательности фермента
Был проведен дополнительный скрининг отобранных соединений на ингибиторную активность в отношении других трипсиноподобных протеаз, следуя методике из примера 59 и с использованием соответствующего фермента и хромогенного субстрата (Cromogenix AB). Показательные результаты представлены в Таблице 2. Избирательность определяется следующим образом:
Избирательность = (Кi для тестируемого фермента) / (Кi для плазменного калликреина)

Таблетка для перорального применения
Таблетки, содержащие 100 мг соединения Примера 1 в качестве активного агента, получали из следующих ингредиентов:
Вещества смешивали и затем прессовали с получением 2000 таблеток массой 250 мг, причем каждая содержала 100 мг соединения Примера 1.
Реферат
Изобретение относится к новым 4-(дипептидиламино)-пиперидин-1-карбоксамидинов общей формулы (1) или к их оптическим изомерам или фармацевтически приемлемым солям

где R1 выбран из Н, низшего алкила, R4-CO, R4 -О2CCH2, R5-OCO и R5-SO2; R2 выбран из низшего алкила, циклоалкила, (С5-С12)циклоалкилалкила, фенилалкила и др.; R3 выбран из Н, ОН и группы O-низший алкил; R4 выбран из Н, низшего алкила и фенила; R5 выбран из низшего алкила, фенила и бензила. Указанные соединения являются ингибиторами калликреина и могут быть использованы для лечения воспалительного заболевания кишечника, артрита, септического шока, гипотензии или рака. 4 н. и 8 з.п. ф-лы, 2 табл.
Формула


Комментарии