Способ получения пептида - RU2478105C2
Код документа: RU2478105C2
Чертежи















Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения пептида и гликопептида.
Уровень техники
В качестве способа получения пептида полезным является способ лигирования. Среди таких способов лигирования способ природного химического лигирования (способ NCL) является способом, которым можно получать пептид, имеющий природную амидную связь (пептидную связь) в месте лигирования. Такой способ NCL можно применять для лигирования двух незащищенных пептидных цепей. Этот способ является известным в качестве способа, применимого для образования природной амидной связи в месте лигирования (см, например, патентный документ 1). Как показано на фигуре ниже, способ NCL включает в себя химическую селективную реакцию между первым пептидом, имеющим α-карбокситиоэфирную часть у его С-конца, и вторым пептидом, имеющим остаток цистеина у его N-конца. В этой реакции тиоловая группа (группа SH, которую можно также называть сульфгидрильной группой) на боковой цепи цистеина селективно реагирует с атомом углерода карбонила сложной тиоэфирной группы, и в результате реакции обмена тиола образуется связанный сложной тиоэфирной связью начальный промежуточный продукт. Этот промежуточный продукт внутримолекулярно перегруппировывается на произвольной основе с образованием природной амидной связи к месту лигирования. В то же время промежуточный продукт образует тиол на боковой цепи цистеина. С применением данной реакции становится возможным эффективный синтез различных полипептидов.
[Формула 1

Основной недостаток типичного способа NCL состоит в том, что любой из двух пептидных фрагментов, которые лигируют, должен иметь остаток цистеина у его N-конца и что пептид, полученный после лигирования, должен также иметь остаток цистеина у места лигирования по этому способу. Согласно этому этот способ нельзя применять в случае, в котором требуемый пептид, который синтезируют, не содержит остаток цистеина.
Кроме того, в типичном способе NCL два или более пептидных фрагмента, которые лигируют, получают, например, способом твердофазного синтеза. Когда пептид содержит крайне малое количество цистеина (или не содержит цистеин), подобно пептиду, существующему в живом организме, бывает необходимо получить крайне длинный пептидный фрагмент, который подвергают реакции по такому способу NCL. Таким образом, нельзя сказать, что он является эффективным способом.
С другой стороны, известно, что в живом организме присутствуют различные гликопептиды и гликопротеины. Цепи сахаров таких гликопептидов или гликопротеинов в широком варианте классифицируют на два типа; а именно, N-связанные цепи сахаров и O-связанные цепи сахаров. N-связанной цепью сахара обычно является цепь сахара, связывающаяся с атомом азота амида на боковой цепи аспарагина посредством N-гликозидной связи. Обычно такая N-связанная цепь сахара часто связывается с Asn в согласованной последовательности -Asn-X-Ser/Thr- (где X представляет собой аминокислоту, исключая пролин) в природном состоянии. O-связанной цепью сахара является цепь сахара, связывающаяся с гидроксильной группой на боковой цепи серина или треонина посредством O-гликозидной цепи. Примеры таких N-связанных и O-связанных цепей сахаров будут представлены ниже (Gal: галактоза; GlcNAc: N-ацетилглюкозамин; Man: манноза; Fuc: фукоза; GalNAc: N-ацетилгалактозамин). Известно, что природный гликопептид, имеющий такую O-связанную цепь сахара, содержит большое количество пролина, треонина и серина (непатентные документы 1 и 2).
[Формула 2]

Примеры N-связанной цепи сахара и O-связанной цепи сахара
Патентный документ 1: международная публикация WO 96/34878.
Непатентный документ 1: TRENDS in biochemical sciences. Vol.27, No. 3, March 2002.
Непатентный документ 2: Cancer Biology & Therapy 6: 4, 481-486, April 2007.
Раскрытие изобретения
Техническая проблема
Задачей настоящего изобретения является предоставление нового способа получения пептида и гликопептида.
В частности, в общепринятом типичном способе NCL любой из двух пептидных фрагментов, которые лигируют, должен иметь остаток цистеина у его N-конца и, кроме того, пептид, полученный после лигирования, также должен иметь остаток цистеина у места лигирования. Таким образом, способ NCL должен быть предназначен и должен применяться с использованием остатка цистеина требуемого пептида (или гликопептида), чтобы в конце получить его в качестве места лигирования. Поэтому настоящее изобретение относится к новому способу получения пептида и гликопептида, который позволяет разработать способ лигирования, по которому получают требуемый пептид не только с остатком цистеина, но также с частью, соответствующей остатку серина или остатку треонина, который можно применять в качестве места лигирования.
Более определенно, в одном аспекте настоящего изобретения остаток цистеина в пептиде (или гликопептиде) можно превратить в остаток серина. Таким образом, пептид, имеющий остаток цистеина у его N-конца, лигируют с другим пептидом согласно способу NCL и после этого этот остаток цистеина можно превратить в остаток серина. Поэтому согласно настоящему изобретению, даже если остаток цистеина не присутствует в требуемой последовательности, которую получают, если в ней присутствует остаток серина, положение остатка серина может быть определено как место лигирования в способе NCL.
Кроме того, в одном аспекте настоящего изобретения пептид, имеющий у своего N-конца остаток производного треонина, имеющего группу -SH у своего N-конца (или остаток производного треонина, имеющего группу -SH, которая защищена дисульфидной связью или тому подобное), лигируют с другим пептидом согласно способу лигирования и после этого полученный остаток производного треонина можно превратить в остаток треонина. Следовательно, согласно настоящему изобретению, даже если остаток цистеина не присутствует в требуемой последовательности, которую получают, но если в ней присутствует остаток треонина, положение остатка треонина может быть определено как место лигирования в способе лигирования.
Таким образом, настоящее изобретение относится к новому способу получения пептида и гликопептида с применением способа лигирования, в котором серин или треонин, который широко распространен в гликопептидах, можно считать как место лигирования в способе лигирования.
Разрешение проблемы
Для разрешения вышеуказанных проблем настоящее изобретение может иметь следующие характеристики.
Настоящее изобретение может предложить способ получения пептида, отличающийся тем, что он содержит превращение группы -SH пептида, содержащего аминокислотный остаток, имеющий группу -SH, в группу -ОН, где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH в пептиде для реакции с метилирующим агентом;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом и
(c) модификацию условий реакции, чтобы они стали более основными, чем условия стадии (b).
Настоящее изобретение может также предложить способ получения пептида, отличающийся тем, что он содержит превращение группы -SH пептида, содержащего аминокислотный остаток, имеющий группу -SH, в группу -ОН,
где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH в пептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения пептида, отличающийся тем, что он содержит превращение остатка цистеина пептида, содержащего остаток цистеина, в остаток серина, где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH остатка цистеина в пептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток серина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может также предложить способ получения пептида, отличающийся тем, что он содержит превращение остатка производного треонина А, представленного нижеследующей формулой (1) пептида, содержащего остаток производного треонина А в качестве аминокислотного остатка, в остаток треонина.
[Формула 3]

где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH остатка производного треонина А в пептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения пептида, отличающийся тем, что он содержит превращение группы -SMe пептида, содержащего аминокислотный остаток, имеющий группу -SMe, в группу -ОН,
где указанный способ содержит следующие стадии (b) и (с):
(b) предоставление возможности группе -SMe в пептиде для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения пептида, содержащего аминокислотный остаток, имеющий группу -ОН, который содержит следующие стадии:
(о) лигирование первого пептида, содержащего у своего С-конца аминокислотный остаток, в котором карбоксильная группа замещена α-карбокситиоэфирной группой, представленной формулой -C(=O)-SR (где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями), со вторым пептидом, содержащим у своего N-конца аминокислотный остаток, имеющий группу -SH, согласно способу лигирования с получением пептида, содержащего аминокислотный остаток, имеющий группу -SH;
(а) предоставление возможности группе -SH в пептиде, полученном на стадии (о), для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения пептида, содержащего остаток серина, который содержит следующие стадии:
(о) лигирование первого пептида, содержащего у своего С-конца аминокислотный остаток, в котором карбоксильная группа замещена α-карбокситиоэфирной группой, представленной формулой -C(=O)-SR (где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями), со вторым пептидом, содержащим у его N-конца остаток цистеина, согласно способу лигирования с получением пептида, содержащего остаток цистеина;
(a) предоставление возможности группе -SH остатка цистеина в пептиде, полученном на стадии (о), для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток серина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения пептида, содержащего остаток треонина, который содержит следующие стадии:
(о) лигирование первого пептида, содержащего у своего С-конца аминокислотный остаток, в котором карбоксильная группа замещена α-карбокситиоэфирной группой, представленной формулой -C(=O)-SR (где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями), со вторым пептидом, содержащим у своего N-конца остаток производного треонина, согласно способу лигирования с получением пептида, содержащего остаток производного цистеина А, представленного вышеуказанной формулой (1), в качестве аминокислотного остатка;
(a) предоставление возможности группе -SH остатка производного треонина А в пептиде, полученном на стадии (о), для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(с) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может также предложить способ получения гликопептида, отличающийся тем, что он содержит превращение группы -SH гликопептида, содержащего аминокислотный остаток, имеющий группу -SH, в группу -ОН, где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH в гликопептиде для реакции с метилирующим агентом;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом и
(c) модификацию условий реакции, чтобы они стали более основными, чем условия стадии (b).
Настоящее изобретение может также предложить способ получения гликопептида, отличающийся тем, что он содержит превращение группы -SH гликопептида, содержащего аминокислотную последовательность, имеющую группу -SH, в группу -ОН, где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH в гликопептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий аминокислотный остаток, имеющий группу -ОН, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения гликопептида, отличающийся тем, что он содержит превращение остатка цистеина гликопептида, содержащего остаток цистеина, в остаток серина, где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH остатка цистеина в гликопептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток серина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения гликопептида, отличающийся тем, что он содержит превращение остатка производного треонина А, представленного вышеуказанной формулой (1) гликопептида, содержащего остаток производного треонина А в качестве аминокислотного остатка, в остаток треонина, где указанный способ содержит следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH остатка производного треонина А в гликопептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения гликопептида, содержащего аминокислотный остаток, имеющий группу -ОН, который содержит следующие стадии:
(о) лигирование первого пептида или гликопептида, содержащего у своего С-конца аминокислотный остаток, в котором карбоксильная группа замещена а-карбокситиоэфирной группой, представленной формулой -C(=O)-SR (где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями), со вторым пептидом или гликопептидом, содержащим у его N-конца аминокислотный остаток, имеющий группу -SH, согласно способу лигирования, при условии, что по меньшей мере, один из первого пептида или гликопептида и второго пептида или гликопептида является гликопептидом, с получением гликопептида, содержащего аминокислотный остаток, имеющий группу -SH;
(a) предоставление возможности группе -SH в гликопептиде, полученном на стадии (о), для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий аминокислотный остаток, имеющий группу -ОН, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения гликопептида, содержащего остаток серина, который содержит следующие стадии:
(о) лигирование первого гликопептида, С-конец которого представлен следующей формулой
-caxap-Asn-X-C(=O)-SR
где caxap-Asn представляет собой аспарагин, соединенный с цепью сахара,
Х представляет собой часть, другую, чем карбоксильная группа любого данного аминокислотного остатка, другого, чем пролин, и
R выбран из бензильной группы, арильной группы и алкильной группы, которая может быть замещена заместителями, со вторым пептидом, содержащим остаток цистеина у его N-конца, согласно способу лигирования с получением гликопептида, содержащего остаток цистеина;
(a) предоставление возможности группе -SH остатка цистеина в гликопептиде, полученном на стадии (о), для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток серина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение может предложить также способ получения гликопептида, содержащего остаток треонина, который содержит следующие стадии:
(о) лигирование первого гликопептида, С-конец которого представлен следующей формулой
-caxap-Asn-X-C(=O)-SR
где caxap-Asn представляет собой аспарагин, соединенный с цепью сахара,
Х представляет собой часть, другую, чем карбоксильная группа любого данного аминокислотного остатка, другого, чем пролин, и
R выбран из бензильной группы, арильной группы и алкильной группы, которая может быть замещена заместителями, со вторым пептидом, содержащим остаток треонина у его N-конца, согласно способу лигирования с получением гликопептида, содержащего производное треонина А, представленного вышеуказанной формулой (1), в качестве аминокислотного остатка;
(а) предоставление возможности группе -SH остатка производного треонина А в гликопептиде, полученном на стадии (о), для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом с получением промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
Настоящее изобретение предлагает также гликопептид, имеющий структуру, представленную следующей формулой:
-caxap-Asn-X-Y-
где caxap-Asn представляет собой аргинин, связанный с цепью сахара,
Х представляет собой любой данный аминокислотный остаток, другой, чем пролин, и
Y представляет собой остаток производного треонина А, представленный формулой (2):
[Формула 4]
В одном варианте осуществления настоящего изобретения остаток метионина в пептиде или гликопептиде на стадии (а) или стадии (с) может быть предпочтительно остатком защищенного метионина, и способ получения предпочтительно дополнительно содержит следующую стадию (d) после стадии (b) или стадии (с) и особенно после стадии (с), когда требуется:
(d) удаление защитной группы у остатка защищенного метионина.
В одном варианте осуществления настоящего изобретения промежуточный продукт реакции, полученный на стадии (b), может быть предпочтительно в форме сложного эфира.
В одном варианте осуществления настоящего изобретения стадию (b) можно предпочтительно проводить в кислотных условиях и особенно при pH 2-3.
В одном варианте осуществления настоящего изобретения цианирующим агентом, применяемым на стадии (b), предпочтительно является бромид циана.
В одном варианте осуществления настоящего изобретения стадию (с) можно предпочтительно проводить в слабоосновных условиях, например, при pH 7-9, и особенно при pH 7-8. Когда стадию (с) проводят в слабоосновных условиях, в одном варианте осуществления стадию (с) можно предпочтительно проводить в течение приблизительно 10 минут или больше и особенно в течение приблизительно 15 минут или больше (например, в течение приблизительно от 10 минут до 30 часов и особенно в течение приблизительно от 15 минут до 30 часов).
В одном варианте осуществления настоящего изобретения стадию (с) можно предпочтительно проводят в сильноосновных условиях, например, при pH 9-13 и особенно при pH 10-11. Когда стадию (с) проводят в сильноосновных условиях, в одном варианте осуществления стадию (с) можно предпочтительно проводить в течение приблизительно 1 часа или меньше, и особенно в течение приблизительно 10 минут или меньше (например, в течение приблизительно от 5 минут до 1 часа и особенно в течение приблизительно от 5 минут до 10 минут).
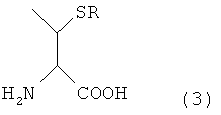
В одном варианте осуществления настоящего изобретения, когда остаток производного треонина содержится у N-конца второго пептида, вышеуказанный остаток производного треонина может быть N-концевым аминокислотным остатком производного треонина, представленным следующей формулой (3)
[Формула 5]

где R представляет собой Н или защитную группу для тиоловой группы, которую легко удаляют в условиях реакции лигирования, и такая группа R предпочтительно представляет собой Н или дисульфидную группу.
В одном варианте осуществления настоящего изобретения любо один, либо особенно оба из первого пептида (или гликопептида) и второго пептида (или гликопептида) предпочтительно могут не содержать цистеин или содержат защищенный цистеин.
В одном варианте осуществления настоящего изобретения либо первый пептид (или гликопептид), либо второй пептид (или гликопептид) может предпочтительно быть пептидом (или гликопептидом), имеющим 80 или меньше, предпочтительно 50 или меньше, и более предпочтительно 30 или меньше аминокислотных остатков.
В одном варианте осуществления настоящего изобретения цепь сахара в гликопептиде может предпочтительно быть N-связаной цепью сахара или O-связанной цепью сахара.
В одном варианте осуществления настоящего изобретения в качестве цепи сахара может быть предпочтительно цепь сахара, представленная следующей формулой (4).

В одном варианте осуществления настоящего изобретения способ получения может предпочтительно дополнительно содержать стадию присоединения цепи сахара после стадии (с) или стадии (d).
В одном варианте осуществления все амидные связи в пептиде или гликопептиде, полученном способом получения настоящего изобретения, могут быть предпочтительно природными амидными связями.
В одном варианте осуществления все конститутивные аминокислоты пептида или гликопептида, полученного способом получения настоящего изобретения, могут быть предпочтительно природными аминокислотами, существующими в качестве конституитивных аминокислот пептида или гликопептида в живом организме.
Благоприятные действия изобретения
Согласно способу получения пептида настоящего изобретения, группу -SH пептида, имеющего такую группу -SH, можно превратить в группу -ОН. Кроме того, группу -SH пептида, содержащего аминокислотный остаток, имеющий такую группу -SH, который получают лигированием первого пептида, имеющего α-карбокситиоэфирную часть, представленную формулой -C(=O)-SR, у его С-конца, со вторым пептидом, содержащим аминокислотный остаток, имеющий группу -SH у его N-конца, согласно способу лигирования можно превратить в группу -ОН. Эти способы можно также применять для гликопептидов.
Так, согласно способу получения пептида настоящего изобретения, остаток цистеина в пептиде можно превратить в остаток серина. В результате этого, если даже остаток цистеина не присутствует в требуемой последовательности, которую получают, но если в ней присутствует остаток серина, можно применять способ NCL.
Кроме того, настоящее изобретение предлагает также способ лигирования, в котором в качестве места лигирования применяют производное треонина, имеющее группу -SH. Поскольку остаток производного треонина, имеющего группу -SH в пептиде, полученном этим способом лигирования, можно превратить в остаток треонина, становится возможным применение способа лигирования с применением остатка треонина в качестве места лигирования для получения пептида, имеющего треонин.
Цистеин, который применяли в качестве места лигирования в обычном способе природного химического лигирования, содержится в малом количестве в пептиде, существующем в живом организме. Согласно способу настоящего изобретения, серин и треонин, которые содержатся в большом количестве в пептиде и особенно в гликопептиде, существующем в живом организме, можно рассматривать в качестве новых мест лигирования в способе лигирования.
Кроме того, вышеуказанный способ применяют для гликопептида и особенно для гликопептида, имеющего O-связанную цепь сахара и содержащего большие количества серина и треонина, или гликопептида, имеющего N-связанную цепь сахара и имеющего в качестве согласованной последовательности последовательность caxap-Asn-X-Ser- или -caxap-Asn-X-Thr (где caxap-Asn представляет собой аспарагин, связанный с цепью сахара, и X представляет собой любой данный аминокислотный остаток, другой, чем пролин), для получения с применением способа лигирования гликопептида, имеющего N-связанную цепь сахара или O-связанную цепь сахара, который имеет такую же структуру, как структура природного гликопептида.
Лучший вариант осуществления изобретения
В первом аспекте настоящее изобретение относится к превращению пептида, содержащего аминокислотный остаток, имеющий группу -SH, по следующим стадиям от (а) до (с) для получения пептида, содержащего аминокислотный остаток, имеющий группу -ОН:
(a) предоставление возможности группе -SH в пептиде для реакции с метилирующим агентом;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом и
(c) модификацию условий реакции, чтобы они стали более основными, чем условия стадии (b).
Вышеописанными стадиями от (а) до (с) более конкретно являются следующие стадии:
(a) предоставление возможности группе -SH в пептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом для получения промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН, в условиях более основных, чем условия стадии (b).
Во втором аспекте настоящее изобретение относится к проведению следующих стадий:
лигирования первого пептида, содержащего у его С-конца аминокислотный остаток, в котором карбоксильная группа замещена α-карбокситиоэфирной группой, представленной формулой -C(=O)-SR (где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями), со вторым пептидом, содержащим у его N-конца аминокислотный остаток, имеющий группу -SH, согласно способу лигирования с получением пептида, содержащего аминокислотный остаток, имеющий группу -SH; и
стадий, включающих в себя вышеуказанные стадии от (а) до (с), с получением пептида, содержащего аминокислотный остаток, имеющий группу -ОН.
В третьем аспекте настоящее изобретение относится к превращению гликопептида, содержащего аминокислотный остаток, имеющий группу -SH, по стадиям, включающим в себя вышеуказанные стадии от (а) до (с), с получением гликопептида, содержащего аминокислотный остаток, имеющий группу -ОН.
В четвертом варианте осуществления настоящее изобретение относится к проведению следующих стадий:
лигирование первого гликопептида, С-конец которого представлен следующей формулой:
-caxap-Asn-X-C(=O)-SR
где caxap-Asn представляет собой аспарагин, присоединенный к цепи сахара,
Х представляет собой часть, другую чем карбоксильная группа любого данного аминокислотного остатка, другого, чем пролин, и R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями со вторым пептидом, содержащим аминокислотный остаток, имеющий группу -SH у его N-конца, согласно способу лигирования с получением гликопептида, содержащего аминокислотный остаток, имеющий группу -SH; и
стадий, включающих в себя вышеуказанные стадии от (а) до (с), с получением гликопептида, содержащего аминокислотный остаток, имеющий группу -ОН.
В настоящем описании термин «пептид» в частности не ограничивается, пока две или более аминокислоты связываются друг с другом посредством их амидной связи. Таким образом, термин «пептид» применяют здесь для включения в него формы известного пептида, нового пептида и модифицированного пептида. В настоящем изобретении соединения, обычно называемые белками, также включены в такой термин пептид. В предпочтительном аспекте в пептиде (или гликопептиде), полученном способом получения настоящего изобретения, две аминокислоты или более аминокислот связаны друг с другом посредством такой же амидной связи (пептидной связи), как и амидная связь природного пептида или гликопептида.
В настоящем описании термин «форма модифицированного пептида» применяют для обозначения соединения, полученного природной или искусственной модификацией пептида. Примеры такой модификации включают в себя алкилирование, ацилирование (например, ацетилирование), амидирование (например, амидирование С-конца пептида), карбоксилирование, этерификацию, образование дисульфидной связи, гликозилирование, липидизацию, фосфорилирование, гидроксилирование и связывание вещества для мечения, причем все такие реакции проводят на одном или многих аминокислотных остатках пептида.
В настоящем описании термин «аминокислота» применяют в самом широком смысле. Поэтому в настоящем описании термин «аминокислота» включает в себя не только природные аминокислоты, такие как серин (Ser), аспарагин (Asn), валин (Val), лейцин (Leu), изолейцин (Ile), аланин (Ala), тирозин (Tyr), глицин (Gly), лизин (Lys), аргинин (Arg), гистидин (His), аспарагиновая кислота (Asp), глутаминовая кислота (Glu), глутамин (Gln), треонин (Thr), цистеин (Cys), метионин (Met), фенилаланин (Phe), триптофан (Thr) и пролин (Pro), но также неприродные аминокислоты, такие мутанты и производные аминокислот. Принимая во внимание такие широкие определения, специалист в данной области должен понимать, что аминокислота в настоящем описании включает в себя, например, L-аминокислоты; D-аминокислоты; химически модифицированные аминокислоты, такие как мутанты и производные аминокислот; аминокислоты, которые не становятся структурными элементами для белков в живом организме, такие как норлейцин, β-аланин и орнитин; и химически синтезированные соединения, имеющие свойства аминокислот, известные специалисту в данной области. Примеры неприродных аминокислот включают в себя α-метиламинокислоты (α-метилаланин и т.д.), D-аминокислоты, подобные гистамину аминокислоты (2-аминогистидин, β-гидроксигистидин, гомогистидин, α-фторметилгистидин, α-метилгистидин и т.д.), аминокислоты, имеющие избыточный метилен на их боковой цепи ("гомо"-аминокислоты), аминокислоты, функциональная группа карбоновой кислоты которых в боковой цепи замещена группой сульфоновой кислоты (цистеиновая кислота и т.д.), а также производное треонина А, описываемое подробно ниже.
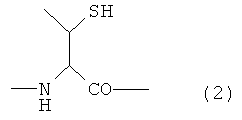
В настоящем описании термин "производное треонина" применяют для обозначения соединения, представленного следующей формулой (3):
[Формула 11]
В формуле (3) R представляет собой Н или защитную группу для тиоловой группы, которая легко удаляется в условиях реакции лигирования, и такая R предпочтительно представляет собой Н или дисульфидную группу. В частности, соединение, представленное нижеследующей формулой (1), где R представляет собой Н в указанной выше формуле (3), называют производным треонина А.
[Формула 12]

Производным треонина формулы (3) является соединение, у которого группой -ОН треонина является группа -SH. Такое производное треонина включает в себя производные, имеющие все типы конфигураций. Считается, что в способе получения настоящего изобретения пространственное обращение имеет место, когда группа -SH аминокислотного остатка в пептиде превращается в группу -ОН. Таким образом, особенно когда нужно получить треонин, существующий в природе, предпочтительно можно применять производное треонина, имеющее группу -SR, конфигурация которой является обращенной по отношению к группе -ОН треонина, существующего в природе.
Вышеуказанное производное треонина можно получить нижеследующим способом при обращении к примерам и примерам синтеза, описываемым, например, ниже.
Сначала получают треонин, содержащий аминогруппу и карбоксильную группу, которые были защищены. Типы таких защитных групп конкретно не ограничиваются, при условии, что представляющий собой интерес пептид можно получить в последующей реакции. Например, можно применять треонин с аминогруппой, защищенной Вос-группой, и карбоксильной группой, защищенной группой TMSE (триметилсилилэтил). После этого гидроксильную группу в β-положении мезилируют известным способом. Затем с применением, например, DBU и тиоуксусной кислоты эту мезильную группу заменяют на тиоацетильную группу (см. D.Crich et al., J. Am. Chem. Soc., 129, 10064 (2007)).
Согласно известному способу эту тиоацетильную группу превращают в тиоловую группу, защищенную защитной группой, известной специалистам в данной области, такой как дисульфидная группа, ацетамидметильная группа, нитробензильная группа или тритильная группа. Например, когда тиоацетильную группу превращают в тиоловую группу, защищенную дисульфидной группой, в качестве ссылки можно применять пример 1 синтеза, описываемый ниже. Дисульфидную группу легко удаляют в условиях реакции для последующего способа лигирования.
В предпочтительном аспекте пептид (или гликопептид), полученный способом получения настоящего изобретения, состоит из аминокислот, которые все присутствуют в качестве конститутивных аминокислот пептида (или гликопептида) в живом организме. Кроме того, в одном аспекте настоящего изобретения пептидом, полученным способом получения настоящего изобретения, предпочтительно является пептид, который не содержит остаток цистеина или содержит малые количества остатков цистеина в конститутивных аминокислотах. Более того, в одном аспекте настоящего изобретения пептид, полученный способом получения настоящего изобретения, имеет 80 или меньше, предпочтительно 50 или меньше и более предпочтительно 30 или меньше аминокислотных остатков между любым данным остатком серина или остатком треонина и следующим остатком серина или остатком треонина или N-концом или С-концом. Например, в одном аспекте настоящего изобретения пептид, полученный способом получения настоящего изобретения, имеет один или несколько остатков серина или остатков треонина в 5-40 аминокислотных остатках и предпочтительно в 20-30 аминокислотных остатках.
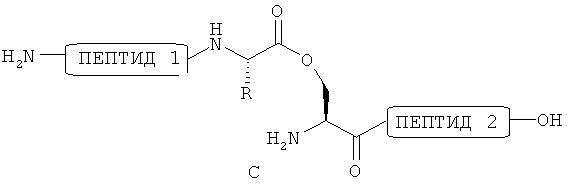
В настоящем описании термин «промежуточный продукт реакции» применяют для обозначения в широком смысле всех соединений, полученных в период между реакцией группы -SMe в пептиде с цианирующим агентом и последующим превращением группы -SMe в группу -ОН. Считается, что схемой реакции настоящего изобретения является нижеследующая схема 1. На схеме 1 форма сложного эфира, представленная С, также является промежуточным продуктом реакции настоящего изобретения. Настоящее описание содержит обозначение "пептид-ОН", например, в следующей схеме 1. Такой "-ОН" означает -ОН С-концевой карбоксильной группы пептида, если не оговорено иначе.
[Формула 13]
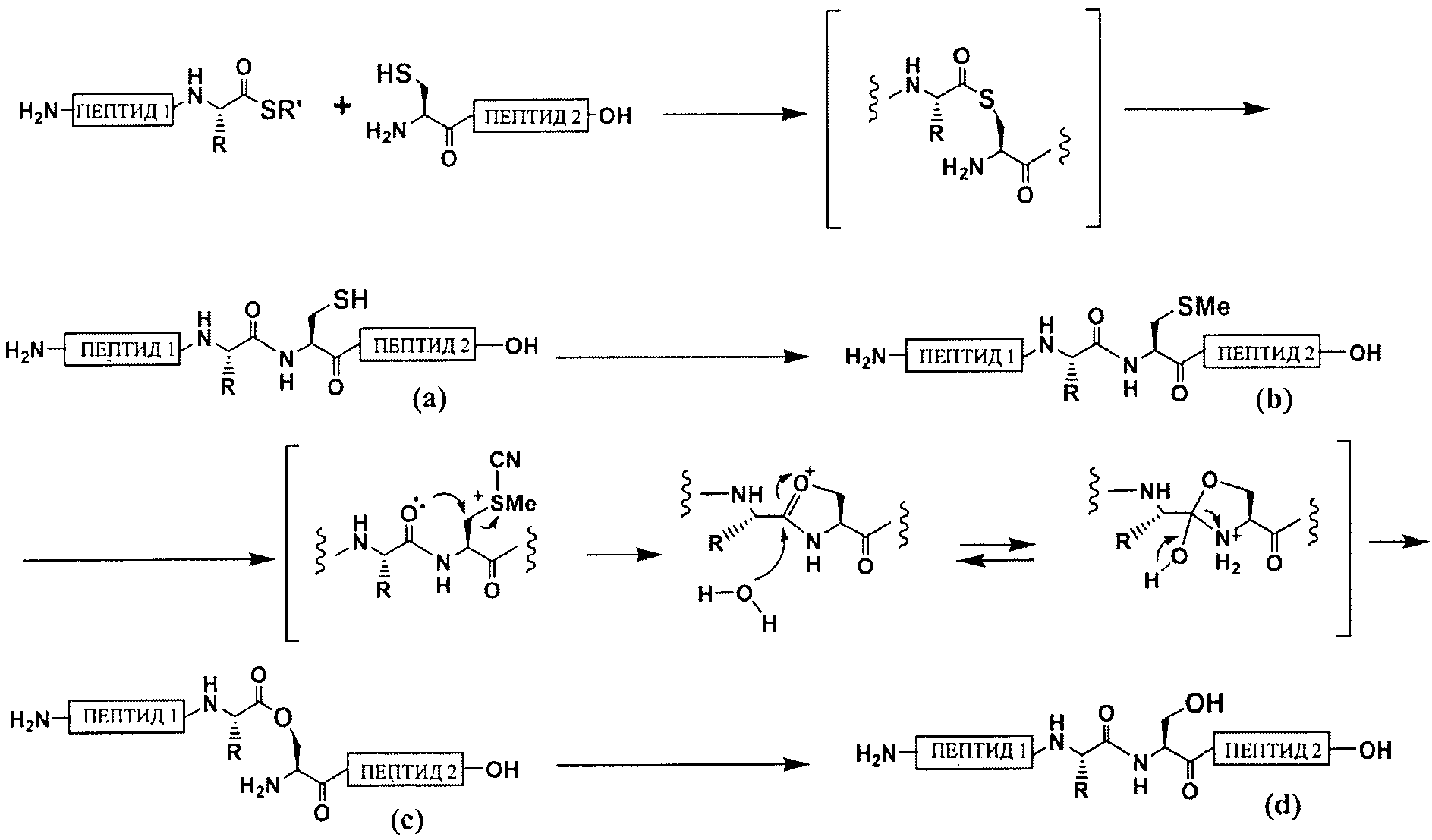
Схема 1
В настоящем описании термин "гликопептид" конкретно не ограничивается, при условии, что он является соединением, образованным присоединением, по меньшей мере, одной цепи сахара к вышеуказанному пептиду. Гликопептид включает в себя известные гликопептиды и новые гликопептиды. Соединения, обычно называемые гликопротеинами, также включены в гликопептиды настоящего изобретения.
В предпочтительном аспекте гликопептид, полученный способом получения настоящего изобретения, является пептидом, имеющим N-связанную цепь сахара или O-связанную цепь сахара. Примеры такого гликопептида включают в себя часть или все из пептидов, такие как эритропоэтин, интерлейкин, интерферон-β, антитело и белок-3 хемотаксического фактора моноцитов (МСР-3).
В одном аспекте настоящего изобретения гликопептид, полученный способом получения настоящего изобретения, имеет 80 или меньше, предпочтительно 50 или меньше и более предпочтительно 30 или меньше аминокислотных остатков между любым данным остатком серина или остатком треонина, к которому не присоединена цепь сахара, и следующим остатком серина или остатком треонина или N-концом или С-концом, к которому не присоединена цепь сахара. Например, в одном аспекте настоящего изобретения гликопептид, полученный способом получения настоящего изобретения, имеет один или несколько остатков серина или остатков треонина в 5-40 аминокислотных остатках и предпочтительно в 20-30 аминокислотных остатках.
В случае такого гликопептида цепь сахара может связываться с аминокислотным остатком в пептиде, непосредственно или через линкер. Место связывания цепи сахара и аминокислоты в частности не ограничивается. Аминокислота предпочтительно связывается с восстанавливающим концом цепи сахара.
Тип аминокислоты, с которым связывается цепь сахара, в частности не ограничивается. Цепь сахара может связываться либо с природной аминокислотой, либо с неприродной аминокислотой. С точки зрения, что настоящий гликопептид должен иметь структуру, идентичную структуре, или сходную со структурой гликопептида (гликопротеина), существующего в живом организме, цепь сахара предпочтительно связывается с Asn, как в случае N-связанной цепи сахара, или с Ser или Thr, как в случае O-связанной цепи сахара. В частности, в случае N-связанной цепи сахара гликопептид, полученный способом получения настоящего изобретения, предпочтительно имеет структуру (-caxap-Asn-X-Thr/Ser-), в которой цепь сахара связывается с Asn, аминокислота (X), другая чем пролин, связывается с С-концевой стороной Asn через амидную связь(пептидную связь) и далее Thr или Ser связывается с С-концевой стороной Х через амидную связь (пептидную связь).
Когда цепь сахара связывается с аминокислотой через линкер, с точки зрения свойств легкости связывания с линкером, предпочтительные аминокислоты, с которыми связывается цепь сахара, включают в себя аминокислоту, имеющую две или более карбоксильных групп в своей молекуле, такую как аспарагиновая кислота или глутаминовая кислота; аминокислоту, имеющую две или более аминогрупп в своей молекуле, такую как лизин, аргинин, гистидин или триптофан; аминокислоту, имеющую гидроксильную группу в своей молекуле, такую как серии, треонин или тирозин; аминокислоту, имеющую тиоловую группу в своей молекуле, такую как цистеин; и аминокислоту, имеющую амидогруппу в свой молекуле, такую как аспарагин или глутамин. В частности, с точки зрения реакционной способности предпочтительными являются аспарагиновая кислота, глутаминовая кислота, лизин, аргинин, серин, треонин, цистеин, аспарагин и глутамин.
В случае, в котором цепь сахара в гликопептиде связывается с аминокислотой через линкер, линкеры, которые широко применяют в настоящей области, можно применять в настоящем изобретении в качестве линкеров. Примеры такого линкера включают в себя
-NH-(CO)-(CH2)a-CH2-
где а равно целому числу, которое конкретно не ограничивается, если только оно не мешает представляющей интерес функции линкера, и а предпочтительно равно целому числу от 0 до 4;
C1-10полиметилен и
-CH2-R3-
где R3 представляет собой группу, образованную удалением одного атома водорода от группы, выбранной из группы, состоящей из алкила, замещенного алкила, алкенила, замещенного алкенила, алкинила, замещенного алкинила, арила, замещенного арила, карбоциклической группы, замещенной карбоциклической группы, гетероциклической группы и замещенной гетероциклической группы.
В настоящем описании термин "цепь сахара" включает в себя не только соединение, образованное соединением двух или более звеньев сахаров (моносахаридов и/или их производных), но также соединение, состоящее из одного звена сахара (моносахарида и/или его производного). Примеры такой цепи сахара включают в себя широкий диапазон цепей сахаров, включающих в себя моносахариды и полисахариды, содержащиеся в живом организме (глюкозу, галактозу, маннозу, фукозу, ксилозу, N-ацетилглюкозамин, N-ацетилгалактозамин, сиаловую кислоту и их комплексы и производные), и цепи сахаров, образованные разложением или индуцированные из комплексных биомолекул, такие как разложенный полисахарид, гликопротеин, протеогликан, гликозаминогликан и гликолипид. Однако примеры не ограничиваются ими. Когда два звена или более звеньев сахаров соединяются друг с другом, звено сахара связывается с другим звеном сахара вследствие конденсации дегидратацией посредством образования гликозидной связи. Цепь сахара может быть либо линейного типа, либо разветвленного типа.
Кроме того, в настоящем описании цепи производных сахаров также включены в термин "цепь сахара". Примеры такой цепи производного сахара включают в себя сахар, составляющий цепь сахара, который имеет карбоксильную группу (например, альдоновую кислоту, положение С-1 которой окислено в карбоновую кислоту (например, D-глюконовую кислоту, получаемую окислением D-глюкозы), и уроновую кислоту, С-атом у конца которой превращен в карбоновую кислоту (D-глюкуроновую кислоту, получаемую окислением D-глюкозы)); сахар, имеющий аминогруппу или ее производное (например, ацетилированную аминогруппу) (например, N-ацетил-D-глюкозамид, N-ацетил-D-галактозамин и т.д.); сахар, имеющий как аминогруппу, так и карбоксильную группу (например, N-ацетилнейраминовую кислоту (сиаловую кислоту), N-ацетилмураминовую кислоту и т.д.); деоксисахар (например, 2-деокси-D-рибозу); сульфатированный сахар, содержащий сульфатную группу; и фосфорилированный сахар, содержащий фосфатную группу. Однако примеры их не ограничиваются указанными.
Цепью сахара настоящего изобретения является предпочтительно цепь сахара, которая существует в виде комплексного углевода в живом организме (гликопептид (или гликопротеин), протеогликан, гликолипид и т.д.). Настоящей цепью сахара предпочтительно является N-связанная цепь сахара, O-связанная цепь сахара или тому подобное, которая связывается в виде гликопептида (или гликопротеина) с пептидом (или белком) в живом организме. В связывающем O-связанную цепь сахара гликопептиде N-ацетилгалактозамин (GalNAc), N-ацетилглюкозамин (GlcNAc), ксилоза, фукоза и тому подобное связываются с Ser или Thr пептида посредством O-гликозидной связи, и цепь сахара далее присоединяется к такому связанному соединению. Примеры N-связанной цепи сахара включают в себя тип с высоким содержанием маннозы, комплексный тип и гибридный тип. Из них предпочтительным является комплексный тип.
В настоящем изобретении предпочтительной цепью сахара является цепь сахара, представленная, например, следующей формулой (4):
[Формула 14]
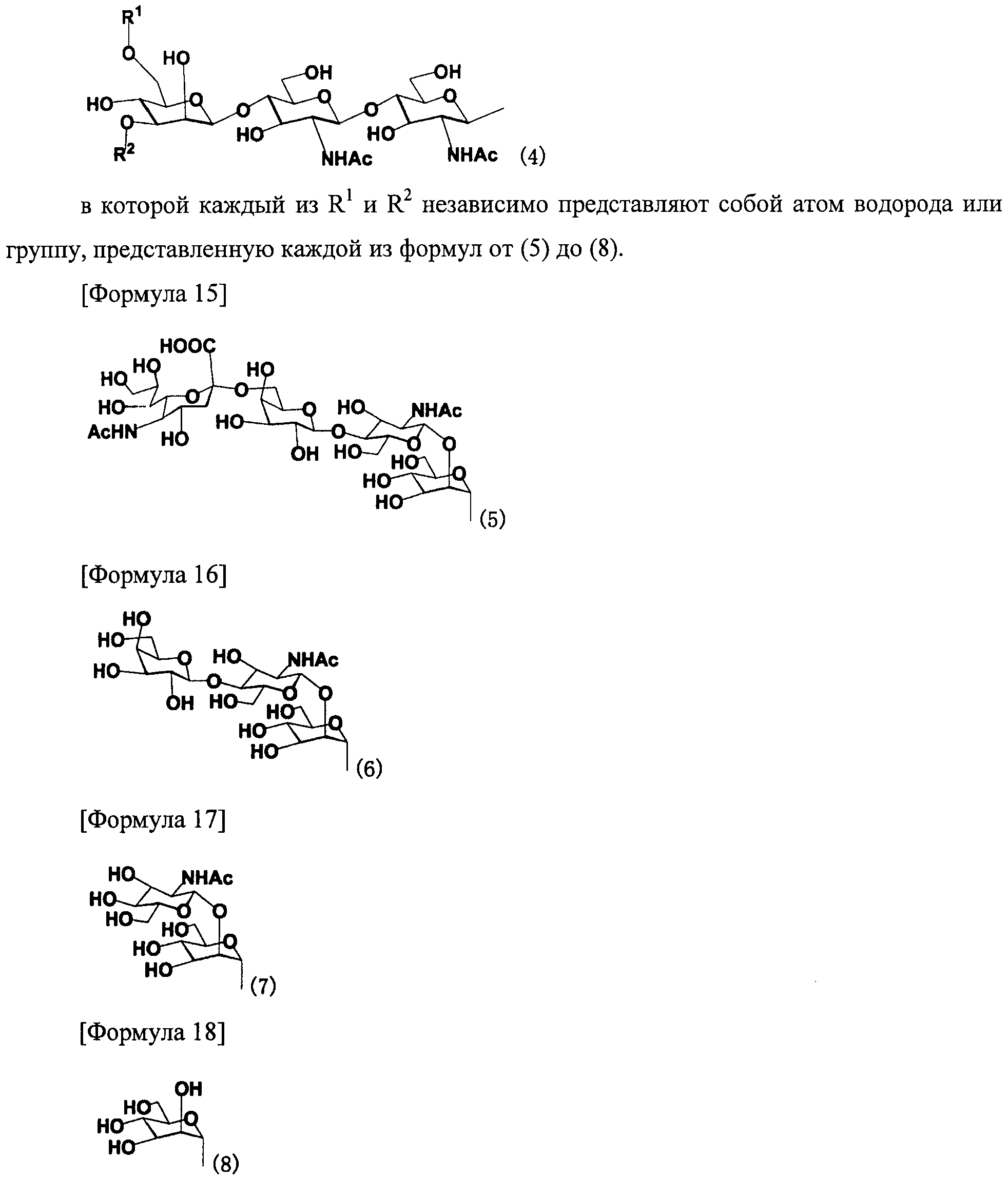
Для предотвращения появления таких проблем, как антигенность, которая может иметь место, когда способ получения гликопептида настоящего изобретения применяют в области получения фармацевтических продуктов и тому подобное, примеры предпочтительной применяемой цепи сахара включают в себя цепь сахара, имеющую структуру, идентичную структуре цепи сахара, существующей в виде гликопротеина, который связывается с белком в организме человека (имеющего такие же типы составляющих сахаров и такие же способы связывания) (например, цепь сахара, описанную в FEBS LETTERS Vol.50, №3, Feb. 1975); и цепь сахара, образованную потерей одного или многих сахаров у невосстанавливающего конца вышеуказанной цепи сахара.
Число цепей сахара, присоединенных к гликопептиду, в частности не ограничивается, при условии, что оно равно 1 или больше 1. С точки зрения получения гликопептида, имеющего структуру, подобную структуре гликопептида, существующего в живом организме, число присоединенных цепей сахара более предпочтительно является почти таким же, как число цепей сахара гликопептида, существующего в организме.
В предпочтительном аспекте настоящего изобретения структуры цепей сахаров в гликопептидах настоящего изобретения являются однородными. В настоящем изобретении такое выражение, как "структуры цепей сахаров в гликопептидах являются однородными" означает, что, когда проводят сравнение между гликопептидами, места присоединения цепей сахаров, типы сахаров, составляющих цепи сахаров, порядок связывания и способ связывания сахаров являются идентичными между ними, и что структуры цепей сахаров имеют процент однородности, по меньшей мере, 90% или больше, предпочтительно 95% или больше и более предпочтительно 99% или больше. Гликопептиды, содержащие однородные цепи сахаров, имеют одинаковое качество (свойства), и такие гликопептиды предпочтительно применяют, в частности в области получения фармацевтических продуктов, анализа и тому подобное.
Пептид, применяемый в качестве исходного вещества в способе получения настоящего изобретения, можно получить с применением способов получения пептидов, известных специалистам в данной области, таких как твердофазный синтез, жидкофазный синтез, синтез с применением клеток и способ разделения и экстракции существующих в природе продуктов. Кроме того, когда в качестве исходного вещества применяют гликопептид, такой гликопептид можно получить включением стадии присоединения цепей сахаров в вышеуказанные известные способы получения пептидов. Что касается способа получения цепей сахаров, применяемых в такой стадии присоединения цепей сахаров, можно сослаться на международные публикации WO 03/008431, WO 2004/058984, WO 2004/058824, WO 2004/070046, WO 2007/011055 и т.д.
В качестве конкретного примера ниже будет описан способ получения пептида или гликопептида, содержащего аминокислотный остаток, имеющий группу -SH, согласно твердофазному способу синтеза. Что касается нижеуказанного способа, можно также сослаться на международную публикацию WO 2004/005330.
Сначала (1) гидроксильную группу смолы, имеющей гидроксильную группу, и карбоксильную группу аминокислоты, аминогруппа которой защищена жирорастворимой защитной группой, подвергают реакции этерификации. В данном случае, поскольку аминогруппа аминокислоты защищена жирорастворимой защитной группой, самоконденсация аминокислот предотвращается, и гидроксильная группа смолы реагирует с карбоксильной группой аминокислоты, так что имеет место этерификация.
Затем (2) жирорастворимую защитную группу получаемого выше эфира отщепляют с образованием свободной аминогруппы,
(3) эту свободную аминогруппу амидируют карбоксильной группой требуемой аминокислоты, аминогруппа которой защищена жирорастворимой защитной группой,
(4) вышеуказанную жирорастворимую защитную группу отщепляют с образованием свободной аминогруппы и
(5) вышеуказанные стадии (3) и (4) повторяют, как необходимо, для получения пептида, в котором лигировано требуемое число требуемых аминокислот и который имеет смолу у одного своего конца и имеет также свободную аминогруппу у другого своего конца.
С применением аминокислоты, имеющей группу -SH (где группа -SH может быть защищена) (например, цистеина или вышеуказанного производного треонина формулы (3)), в вышеуказанных стадиях от (1) до (5) можно получить пептид, содержащий аминокислотный остаток, имеющий группу -SH. Когда группу -SH аминокислоты, имеющей группу -SH, применяют в вышеуказанных стадиях от (1) до (5), группу -SH можно защитить защитной группой, известной специалистам в данной области, такой как дисульфидная группа, ацетамидметильная группа, нитробензильная группа или тритильная группа. После этого, как необходимо, такую защитную группу удаляют. Кроме того, с применением аминокислоты с присоединенной цепью сахара (например, аспарагина с цепью сахара, полученного присоединением цепи сахара к аспарагину, серина с цепью сахара или треонина с цепью сахара, полученного присоединением цепи сахара к серину или треонину, и тому подобное) в качестве аминокислоты в вышеуказанных стадиях от (1) до (5), можно также получить N-связанные и/или O-связанные гликопептиды, имеющие одну или две или больше цепей сахаров в требуемом положении(ях). Как описано выше, такие N-связанные и/или O-связанные гликопептиды могут содержать аминокислотный остаток(и), имеющий группу(ы) -SH в требуемом положении(ях).
(6) После этого сложноэфирную связь между смолой и аминокислотой, образованную в вышеуказанной стадии (1), расщепляют кислотой, получая при этом требуемый пептид (или гликопептид).
Обычно тип твердофазной смолы в частности не ограничивается, пока она является смолой, применяемой в твердофазном синтезе. Можно применять, например, смолу амино-PEGA (изготовлена Merck), смолу Ванга (изготовлена Merck), смолу HMPA-PEGA (изготовлена Merck), смолу Trt-хлорид (изготовлена Merck) и тому подобное.
Кроме того, между такой смолой амино-PEGA и аминокислотой может быть расположен линкер. Примеры такого линкера включают в себя 4-гидроксиметилфеноксиуксусную кислоту (НМРА) и 4-(4-гидроксиметил-3-метоксифенокси)бутилуксусную кислоту (НМРВ).
Примеры жирорастворимой защитной группы включают в себя карбонилсодержащие группы, такие как 9-флуоренилметоксикарбонильная (Fmoc) группа, трет-бутилоксикарбонильная (Boc) группа и аллилоксикарбонильная (Alloc) группа; такие защитные группы, как ацильные группы, такие как ацетильная (Ас) группа, аллильная группа и бензильная группа. Однако примеры их не ограничиваются перечисленными.
При введении жирорастворимой защитной группы в требуемый пептид (или гликопептид), когда вводят, например, группу Fmoc, ее можно ввести добавлением 9-флуоренилметил-N-сукцинимидилкарбоната и бикарбоната натрия к реакционной системе и затем проведением реакции. Реакцию можно проводить при температуре между 0°С и 50°С и предпочтительно при комнатной температуре, в течение приблизительно 1-5 часов.
В качестве аминокислоты, защищенной жирорастворимой защитной группой, можно применять вышеуказанные аминокислоты, защищенные вышеуказанным способом. Кроме того, можно применять также коммерчески доступные продукты. Примеры такой аминокислоты, защищенной защитной группой, включают в себя Fmoc-Ser, Fmoc-Asn, Fmoc-Val, Fmoc-Leu, Fmoc-Ile, Fmoc-Ala, Fmoc-Tyr, Fmoc-Gly, Fmoc-Lys, Fmoc-Arg, Fmoc-His, Fmoc-Asp, Fmoc-Glu, Fmoc-Gln, Fmoc-Thr, Fmoc-Cys, Fmoc-Met, Fmoc-Phe, Fmoc-Trp и Fmoc-Pro.
В качестве катализаторов этерификации можно применять известные дегидратирующие конденсирующие агенты, например, 1-мезитиленсульфонил-3-нитро-1,2,4-триазол (MSNT), дициклогексилкарбодиимид (DCC) и 1,3-диизопропилкарбодиимид (DIPCDI). Что касается применяемого отношения между аминокислотой и дегидратирующим конденсирующим агентом, обычно применяют 1-10 массовых частей и предпочтительно 2-5 массовых частей дегидратирующего конденсирующего агента на 1 массовую часть аминокислоты.
Реакцию этерификации предпочтительно проводят, например, помещением смолы в твердофазную колонку, промыванием смолы растворителем и затем добавлением к ней раствора аминокислоты. Примеры такого промывающего растворителя включают в себя диметилформамид (ДМФА), 2-пропанол и метиленхлорид. Примеры растворителя для растворения аминокислоты включают в себя диметилсульфоксид (ДМСО), ДМФА и метиленхлорид. Такую реакцию этерификации можно проводить при температуре между 0°С и 50°С и предпочтительно при комнатной температуре в течение приблизительно от 10 минут до 30 часов и предпочтительно в течение приблизительно от 15 минут до 24 часов.
Предпочтительно также, чтобы во время реакции непрореагировавшие функциональные группы на твердой фазе были ацетилированы применяемым уксусным ангидридом или тому подобное и блокированы.
Жирорастворимую защитную группу можно отщепить, например, обработкой основанием. Примеры такого основания включают в себя пиперидин и морфолин. Обработку предпочтительно проводят в присутствии растворителя. Примеры растворителя, применяемого в данном изобретении, включают в себя ДМСО, ДМФА и метанол.
Реакцию амидирования между свободной аминогруппой и карбоксильной группой любой данной аминокислоты, атом азота аминогруппы которой защищен жирорастворимой защитной группой, предпочтительно проводят в присутствии активатора и растворителя.
Примеры активатора включают в себя дициклогексилкарбодиимид (DCC), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (WSC/HCl), дифенилфосфорилазид (DPPA), карбонилдиимидазол (CDI), диэтилцианофосфонат (DEPC), 1,3-диизопропилкарбодиимид (DIPCI), гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония (РуВОР), 3-диэтоксифосфорилокси-1,2,3-бензотриазин-4(3Н)-он (DEPBT), 1-гидроксибензотриазол (HOBt), гидроксисукцинимид (HOSu), диметиламинопиридин (DMAP), 1-гидрокси-7-азабензотриазол (HOAt), 3-гидрокси-4-оксо-3,4-дигидро-5-азабензо-1,2,3-триазин (HODhbt), гидроксифталимид (HOPht), пентафторфенол (Pfp-OH), гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU), гексафторфосфонат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU) и тетрафторборат О-бензотриазол-1-ил-1,1,3,3-тетраметилурония (TBTU).
Что касается количества применяемого активатора, применяют 1-20 эквивалентов, предпочтительно 1-10 эквивалентов и более предпочтительно 1-5 эквивалентов активатора относительно любой данной аминокислоты, атом азота аминогруппы которой защищен жирорастворимой защитной группой.
Реакция протекает только с вышеуказанным активатором. Однако в качестве вспомогательного агента предпочтительно добавляют амин. В качестве такого амина можно применять диизопропилэтиламин (DIPEA), N-этилморфолин (NEM), N-метилморфолин (NMM), N-метилимидазол (NMI) и тому подобное. Что касается количества такого применяемого вспомогательного агента, применяют 1-20 эквивалентов, предпочтительно 1-10 эквивалентов и более предпочтительно 1-5 эквивалентов вспомогательного агента относительно любой данной аминокислоты, атом азота аминогруппы которой защищен жирорастворимой защитной группой.
Примеры растворителя включают в себя ДМСО, ДМФА и метиленхлорид. Реакцию проводят при температуре между 0°С и 50°С и предпочтительно при комнатной температуре, в течение приблизительно от 10-30 часов и предпочтительно в течение приблизительно от 15 минут до 24 часов. Предпочтительно также, чтобы во время данной реакции непрореагировавшие аминогруппы на твердой фазе были ацетилированы с применением уксусного ангидрида или тому подобное и блокированы. Жирорастворимую защитную группу можно отщепить таким же способом, как способ, описанный выше.
Для отщепления пептидной цепи от смолы предпочтительной является обработка смолы кислотой. Примеры кислоты, применяемой в данном изобретении, включают в себя трифторуксусную кислоту (TFA) и фторид водорода (HF). Во время обработки из жирорастворимой защитной группы, применяемой для аминокислоты, и линкера на смоле может образовываться очень реакционно-способный тип катионов. Поэтому для захвата катионов такого типа предпочтительно добавляют нуклеофильный реагент. Примеры такого нуклеофильного реагента включают в себя триизопропилсилан (TIS), фенол, тиоанизол и этандитиол (EDT).
Таким образом, можно получить пептид (или гликопептид), содержащий аминокислотный остаток, имеющий группу -SH.
Кроме того, для таким образом полученного пептида или гликопептида, содержащего аминокислотный остаток, имеющий группу -SH, с целью присоединения к нему цепи сахара в качестве типичного примера можно применять способ, использующий обратимую реакцию фермента, включающего в себя трансглутаминазу, чтобы таким образом получить гликопептид, содержащий аминокислоту, имеющую группу -SH.
Кроме того, можно также сочетать реакцию удлинения цепи сахара с применением трансферазы с вышеуказанным способом.
Среди пептидов или гликопептидов, получаемых выше, пептид (или гликопептид), содержащий аминокислотный остаток, имеющий группу -SH у его N-конца, можно лигировать с пептидом (или гликопептидом), имеющим α-карбокситиоэфирную группу у его С-конца, согласно способу лигирования.
В настоящем описании термин "способ лигирования" применяют для включения в него не только способа природного химического лигирования (способа NCL), описанного в патентном документе 1, но также для включения применения такого способа природного химического лигирования для пептидов, содержащих неприродные аминокислоты или производные аминокислот (например, производное треонина А формулы (1), защищенный метионин, аминокислоту, соединенную с цепью сахара, и т.д.), как описано в нижеуказанных примерах. Согласно такому способу лигирования, можно получить пептид, имеющий природную амидную связь (пептидную связь) у места лигирования.
Согласно способу лигирования, лигирование можно проводить во всех случаях между пептидом и пептидом, между пептидом и гликопептидом и между гликопептидом и гликопептидом.
Пептид (или гликопептид), имеющий α-карбокситиоэфирную группу у его С-конца, который применяют в способе лигирования, можно получить способом, известным специалистам в данной области, как описано в патентном документе 1.
Например, как описано в нижеуказанных примерах, способ твердофазного синтеза применяют для получения защищенного пептида (или гликопептида), аминогруппы которого на аминокислотной боковой цепи и у N-конца защищены. Карбоксильную группу на его С-концевой стороне конденсируют с бензилтиолом с применением РуВОР (гексафторфосфат бензотриазол-1-илокситриспирролидинофосфония)/DIPEA в качестве конденсирующего агента в жидкой фазе. После этого цепь аминокислоты освобождают от защитной группы с применением 95% раствора TFA, получая при этом пептид (или гликопептид), имеющий α-карбокситиоэфирную группу у его С-конца.
Способ лигирования можно проводить с применением способа, известного специалистам в данной области, как описано в патентном документе 1, или с применением описаний нижеуказанных примеров. Например, первый пептид, имеющий α-карбокситиоэфирную группу, представленную формулой -C(=O)-SR, у его С-конца, и второй пептид, содержащий аминокислотный остаток, имеющий группу -SH у его N-конца, получают с применением вышеуказанных описаний. В первом пептиде R в частности не ограничивают, при условии, что он не препятствует реакции обмена тиола и он действует в качестве уходящей группы в реакции нуклеофильного замещения на атоме углерода карбонила. Такой R предпочтительно можно выбрать из групп типа бензила, таких как бензилмеркаптан, групп типа арила, таких как тиофенол или 4-(карбоксиметил)тиофенол, групп типа алкила, таких как 2-меркаптоэтансульфонат или амид 3-меркаптопропионовой кислоты, и тому подобное. Группу -SH у N-конца второго пептида можно защитить защитной группой, как описано. Эту защитную группу удаляют в желаемый момент времени перед последующей реакцией лигирования и второй пептид, имеющий группу -SH у его N-конца, реагирует с первым пептидом. Например, если она является защитной группой, которая самопроизвольно удаляется в условиях, в которых имеет место лигирование, второй пептид, защищенный защитной группой, можно непосредственно применять в последующей реакции лигирования.
Два пептида смешивают друг с другом в растворе, таком как 100 мМ фосфатный буфер, когда необходимо, в присутствии каталитического тиола, такого как 4-меркаптофенилуксусная кислота, бензилмеркаптан или тиофенол. Реакцию предпочтительно проводят с применением 0,5-2 эквивалентов второго пептида и приблизительно 5 эквивалентов каталитического тиола относительно 1 эквивалента первого пептида. Реакцию предпочтительно проводят в условиях, состоящих из pH 6,5-7,5 и температуры между 20°С и 40°С в течение приблизительно 1-30 часов. Развитие реакции можно подтвердить известным способом, в котором сочетают ВЭЖХ, МС и тому подобное.
К продукту реакции для подавления побочных реакций добавляют восстанавливающий агент, такой как дитиотреитол (DTT) или гидрохлорид трис-(2-карбоксиэтил)фосфина (ТСЕР), и, если требуется, продукт реакции очищают, так чтобы первый пептид можно было лигировать со вторым пептидом.
В случае, в котором среди пептидов, имеющих карбокситиоэфирную часть (-С=O-SR) у его С-конца, находятся пептиды, имеющие разные группы R, порядок реакций лигирования можно изменить (см. Protein Science (2007), 16: 2056-2064, и т.д.). Поэтому когда лигирование проводят много раз, можно принимать во внимание порядок реакций лигирования. Например, когда в качестве R присутствуют арильная группа, бензильная группа и алкильная группа, реакция лигирования обычно протекает в этом порядке.
В настоящем изобретении будет конкретно описан способ получения пептида (или гликопептида), отличающийся тем, что группа -SH пептида (или гликопептида), содержащего аминокислотный остаток, имеющий группу -SH, превращается в группу -ОН. В качестве исходных веществ применяют пептид или гликопептид, полученный вышеуказанным способом. В одном аспекте настоящего изобретения предпочтительно применяют пептид или гликопептид, полученный способом лигирования. Затем проводят следующие стадии от (а) до (с):
(a) предоставление возможности группе -SH в пептиде для реакции с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) предоставление возможности группе -SMe, образованной на стадии (а), для реакции с цианирующим агентом для получения промежуточного продукта реакции и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН, в условиях более основных, чем условия в стадии (b). Ниже будет приведен в качестве примера случай применения пептида в качестве исходного вещества.
Стадия (а)
Метилирующий агент, применяемый при метилировании стадии (а), в частности не ограничивается, при условии, что он способен превратить группу -SH в пептиде в группу -SMe. Примеры такого метилирующего агента включают в себя иодметан и метил-4-нитробензолсульфонат.
Что касается количества применяемого метилирующего агента, 1-1000 эквивалентов, предпочтительно 10-100 эквивалентов и более предпочтительно 15-30 эквивалентов метилирующего агента можно применять относительно одного остатка группы -SH исходного пептида. Реакцию метилирования желательно проводить при температуре между 0°С и 50°С и предпочтительно между 20°С и 30°С в течение приблизительно от 10 минут до 30 часов и предпочтительно в течение приблизительно от 15 минут до 1 часа.
В качестве растворителя в реакции метилирования предпочтительно применяют буферный раствор. Предпочтительно можно применять буферный раствор, имеющий pH 7-9 и в частности pH 8-9. Например, можно применять 0,25 М буферный раствор трис-HCl (6 М раствор гидрохлорида гуанидина, содержащий 3,3 мМ EDTA, pH 8,6) или тому подобное.
В случае, в котором в качестве аминокислоты в пептиде содержится остаток цистеина и в котором предполагается, что остаток цистеина не превращается в остаток серина и ему дают возможность существовать в виде остатка цистеина в пептиде, полученном способом получения настоящего изобретения, такой цистеин защищают и вводят в форме защищенного цистеина в пептид согласно настоящему способу. Тем самым можно предотвратить метилирование группы -SH цистеина на стадии (а). Принимая во внимание реакцию обмена тиола, обработку кислотой, обработку основанием и т.д. проводимую в каждой стадии способа получения настоящего изобретения, в качестве защитной группы для цистеина можно принимать подходящую защитную группу. Примеры такой защитной группы включают в себя ацетамидметильную (Acm) группу, бензильную группу, ацетамидную группу и тритильную группу. Предпочтительно можно применять группу Acm. После стадий от (а) до (с) остаток защищенного цистеина освобождают от защиты с применением известного способа. Например, когда вводят защищенный цистеин, который был защищен такой защитной группой, как группа Acm, нитробензильная группа или тритильная группа, такой защищенный цистеин превращают в остаток цистеина добавлением стадии применения способа снятия защиты с применением водного раствора ацетата серебра, способа снятия зашиты с применением светового излучения, способа снятия защиты, включающего в себя обработку кислотой, или других способов. Таким образом, остатку цистеина позволяют присутствовать в пептиде, полученном способом получения настоящего изобретения.
В одном аспекте настоящего изобретения можно также получить пептид, имеющий группу -ОН, превращением группы -SMe пептида, имеющего группу SMe, в группу -ОН. В таком случае вышеуказанную стадию (а) можно не включать.
Стадия (b)
С точки зрения безопасности и тому подобное, в качестве цианирующего агента в стадии (b) можно применять, например, бромид циана, фенилцианат или тому подобное. Предпочтительно можно применять бромид циана, который можно легко получить.
Что касается количества применяемого цианирующего агента, можно применять 1-1000 эквивалентов, предпочтительно 10-100 эквивалентов и более предпочтительно 15-30 эквивалентов цианирующего агента относительно одной группы -SMe. Реакцию с цианирующим агентом желательно проводить при температуре между 0°С и 50°С и предпочтительно между 30°С и 40°С в течение приблизительно от 30 минут до 100 часов и предпочтительно в течение приблизительно от 12 часов до 50 часов.
Реакцию с цианирующим агентом проводят в кислотных условиях и особенно предпочтительно ее проводят при pH 2-3. С применением кислотного, водорастворимого вещества и особенно с применением муравьиной кислоты, трифторуксусной кислоты, метансульфоновой кислоты или тому подобное реакцию можно проводить при кислотных условиях. Во время реакции для предотвращения окисления атомов серы особенно предпочтительно, чтобы применяемое кислотное водорастворимое вещество было деаэрировано. Кроме того, с точки зрения стабильности цианирующего агента, реакцию предпочтительно проводят в условиях защиты от света.
В качестве растворителя предпочтительно можно применять вышеуказанный водорастворимый растворитель, имеющий pH 2-3, такой как 80% раствор муравьиной кислоты, 70% раствор муравьиной кислоты или водный раствор, содержащий 2% трифторуксусной кислоты/39% ацетонитрила.
Примером промежуточного продукта реакции, полученного на стадии (b), является форма сложного эфира, имеющего следующую структуру:
[Формула 19]
Когда в качестве аминокислоты в пептиде содержится остаток метионина, предпочтительным является отличие группы -SMe, содержащейся в остатке метионина, от группы -SMe, полученной на стадии (а). В настоящем описании защищенный метионин в частности не ограничивают, при условии, что он является соединением, которое не реагирует с цианирующим агентом на стадии (b). Примером такого защищенного метионина является метионин типа сульфоксида (Met(O): -CH2-CH2-S(=O)-СН3). Как описано в нижеуказанном примере 5, защищенный метионин (например, Met(O)) вводят в пептид с применением известного способа, так чтобы остаток метионина можно было отличить от полученной в стадии (а) группы -SMe, и остаток метионина становился неактивным в реакции с цианирующим агентом на стадии (b).
После этого остаток защищенного метионина превращают в остаток метионина известным подходящим способом, если это необходимо (нижеуказанная стадия (е)). Таким образом, пептид, имеющий остаток метионина, можно также получить способом настоящего изобретения.
Кроме того, окисленный цистеин, который является побочным продуктом, образованным во время реакции с цианирующим агентом в стадии (b), если необходимо, можно удалить. В такой стадии удаления смесь, содержащую промежуточный продукт реакции, полученный на стадии (b), можно подвергнуть реакции при комнатной температуре в течение приблизительно 30 минут в присутствии, например, иодида аммония и диметилсульфида и после этого реакционный раствор можно отделить и промыть. Такую стадию удаления можно проводить в любой временной точке после стадии (b) и ее можно предпочтительно проводить после стадии (с).
Стадия (с)
На стадии (с) в более основных условиях, чем условия на стадии (b), пептид, содержащий аминокислотный остаток, имеющий группу -ОН, получают внутримолекулярным перемещением ацила от О- к N-положению промежуточного продукта реакции, полученного на стадии (b).
Основные условия на стадии (с) могут быть либо кислотными, либо нейтральными, при условии, что они являются более основными условиями, чем условия на стадии (b). Точнее говоря, основные условия на стадии (с) в частности не ограничиваются, при условии, что они являются условиями, при которых группа -NH2 на С-атоме, соседнем со сложноэфирной связью промежуточного продукта реакции, полученного на стадии (b), не протонирована. С точки зрения эффективного превращения промежуточного продукта реакции в пептид, имеющий группу -ОН, можно применять слабоосновные или сильноосновные условия.
Когда основные условия на стадии (с) являются слабоосновными условиями, pH бывает 7-9, и предпочтительно pH 7-8. Слабоосновные условия можно получить, например, добавлением к раствору основного соединения, которое применяют в качестве агента регуляции pH, известного специалистам в данной области, такого как гуанидин, динатрийфосфат, трис или бикарбонат натрия. Что касается количества основного соединения, применяемого в течение этой операции, можно применять 1-1000 эквивалентов, предпочтительно 10-100 эквивалентов и более предпочтительно 15-30 эквивалентов такого основного соединения относительно исходного пептида.
Когда основные условия на стадии (с) являются слабоосновными условиями, реакцию желательно проводить при температуре между 0°С и 50°С и предпочтительно между 20°С и 40°С в течение приблизительно от 10 минут до 30 часов и предпочтительно в течение приблизительно от 15 минут до 30 часов. Реакцию желательно проводить в буферном растворе, имеющем pH 7-9 и предпочтительно pH 7-8. Например, стадию (с) можно проводить в 0,2 М фосфатном буфере (содержащем 6 М раствор гидрохлорида гуанидина, pH 7,2).
Когда основные условия на стадии (с) являются слабоосновными условиями, pH можно дополнительно снизить и стадию (с) можно завершить. В противном случае, эту стадию можно продолжать до стадии очистки с применением ВЭЖХ или тому подобное без изменения pH.
Когда основные условия на стадии (с) являются сильноосновными, pH бывает 9-13 и предпочтительно 10-11. Такими сильноосновными условиями предпочтительно являются условия, при которых соединение, которое избыточно реагировало с гидроксильной группой, можно удалить гидролизом. Сильноосновные условия можно получить добавлением к раствору основного водорастворимого вещества, такого как гидразингидрат, 50 мМ водный раствор гидроксида натрия или тому подобное. Что касается количества применяемого основного водорастворимого вещества, в течение этой операции можно применять 0,5-100 эквивалентов, предпочтительно 0,1-10 эквивалентов и более предпочтительно 0,5-1 эквивалент такого основного водорастворимого вещества относительно исходного пептида. Стадию (с) можно проводить, например, в 5% водном растворе гидразина, имеющем pH 10-11.
Когда основные условия на стадии (с) являются сильноосновными условиями, пептид, содержащий аминокислотный остаток, имеющий группу -ОН, можно получить внутримолекулярной перемещением ацила от О- к N-положению промежуточного продукта реакции, полученного на стадии (b), и в то же время может иметь место децианирование (реакция децианирования для удаления избыточно реагирующих цианирующих агентов гидролизом), деформилирование (реакция деформилирования избыточно реагирующих муравьиных кислот) и тому подобное на избыточно реагирующих группах -ОН на стадиях (а) и (b).
Когда основными условиями на стадии (с) являются сильноосновные условия, стадию (с) желательно проводить при температуре между 0°С и 50°С и предпочтительно между 20°С и 30°С в течение приблизительно от 5 минут до 3 часов, предпочтительно в течение приблизительно от 5 минут до 1 часа и более предпочтительно в течение приблизительно от 5 минут до 10 минут. Когда основные условия на стадии (с) являются сильноосновными условиями, если стадию (с) проводят в течение длительного периода времени, могут иметь место побочные реакции, такие как рацемизация и расщепление пептидной связи.
Когда основные условия на стадии (с) являются сильноосновными условиями, стадию (с) можно завершить уменьшением pH до pH 9 и предпочтительно до pH 5-9, например, около pH 7 или pH 8-9.
Предполагается, что на стадии (с) конфигурация β-положения полученного аминокислотного остатка, имеющего группу -ОН, является обращенной по отношению к конфигурации промежуточного продукта, полученного на стадии (b).
Стадия (d)
Когда пептид, содержащий остаток защищенного метионина, применяют в качестве исходного вещества, после стадии (b) или (с), если требуется, дополнительно проводят следующую стадию (d):
(d) удаление защиты у защищенного метионина.
Удаление защиты можно проводить с применением способа, известного специалистам в данной области, в зависимости от типа применяемого защищенного метионина. Когда в качестве защищенного метионина вводят, например, метионин сульфоксидного типа (Met(O)), такой защищенный метионин превращают в метионин добавлением стадии восстановления с применением смешанного раствора иодида аммония/диметилсульфида/TFA или тому подобное. С точки зрения предотвращения появления побочных реакций, стадию (d) предпочтительно проводят после стадии (с).
Таким образом, в случае получения пептида, имеющего также остаток метионина, можно применять способ получения настоящего изобретения.
Для очистки полученного продукта предпочтительно применяют способ получения очищенного продукта с чистотой 97% или больше в условиях нескольких типов высокоэффективной жидкостной хроматографии. Конкретные примеры такого способа включают в себя кристаллизацию, способ трансакционального распределения, распределительную хроматографию, способ гель-фильтрации, ионообменную хроматографию и высокоэффективную жидкостную хроматографию. Предпочтительно можно применять высокоэффективную жидкостную хроматографию или тому подобное.
Помимо вышеуказанных стадий можно дополнительно проводить стадию присоединения цепи сахара. Такое присоединение цепи сахара можно проводить как на пептиде, так и гликопептиде. Такое присоединение цепи сахара можно проводить в любой временной точке, при условии, что можно получить представляющий интерес гликопептид. Присоединение предпочтительно проводят после стадии (с).
Присоединение цепи сахара можно проводить способом, применяющим обратную реакцию фермента, в том числе трансглутаминазы в качестве типичного примера, или способом, проводимым по описаниям международной публикации WO 2005/010053, как описано ниже.
Сначала получают производное цепи сахара типа комплекса с галогенацетамидом предоставлением ему возможности реагировать с пептидом, получаемым выше (в частности, с пептидом, содержащим аминокислоту, имеющую две или больше карбоксильных групп в его молекуле, такую как аспарагиновая кислота или глутаминовая кислота; аминокислоту, имеющую две или больше аминогрупп в его молекуле, такую как лизин, аргинин, гистидин или триптофан; аминокислоту, имеющую гидроксильную группу в его молекуле, такую как серин, треонин или тирозин; аминокислоту, имеющую тиоловую группу в его молекуле, такую как цистеин; или аминокислоту, имеющую амидогруппу в его молекуле, такую как аспарагин или глутамин, и среди них особенно с пептидом, содержащим аспарагиновую кислоту, глутаминовую кислоту, лизин, аргинин, серин, треонин, цистеин, аспарагин или глутамин). Вышеуказанную реакцию можно проводить при температуре обычно между 0°С и 80°С, предпочтительно между 10°С и 60°С и более предпочтительно между 15°С и 35°С. Время реакции обычно предпочтительно бывает приблизительно от 30 минут до 5 часов. После завершения реакции продукт реакции можно очистить известным способом (например, высокоэффективной жидкостной хроматографией (ВЭЖХ), если это необходимо.
Производное цепи сахара типа комплекса с галогенацетамидом является соединением, образованным замещением гидроксильной группы, связывающейся с атомом углерода в положении 1 комплекса типа цепи сахара, связанной с аспарагином, например, группой -NH-(CO)-(CH2)a-CH2X (где Х представляет собой атом галогена и а равно целому числу, которое в частности не ограничивается, при условии, что оно не мешает представляющей интерес функции линкера, и предпочтительно представляет собой целое число от 0 до 4).
В частности, производному цепи сахара типа комплекса с галогенацетамидом предоставляют возможность реагировать с вышеполученным пептидом в фосфатном буфере при комнатной температуре. После завершения реакции реакционный раствор очищают ВЭЖХ, получая при этом гликопептид с присоединенной цепью сахара.
Можно сочетать реакцию удлинения цепи сахара, в которой применяют трансферазу, с вышеуказанным способом. Полученный таким образом гликопептид также включен в объем настоящего изобретения.
Примеры
Далее настоящее изобретение будет конкретно описано в следующих примерах. Однако предполагается, что эти примеры не ограничивают объем настоящего изобретения.
Пример 1. Получение Ac-Ala-Ser-Gly-Leu
Смолу Ванга (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. Fmoc-Leu (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (2 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 1 часа. Следует отметить, что объем DCM можно также задать как 1,5 мл. После завершения операции перемешивания смолу полностью промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 15 минут. Следует также отметить, что настоящую обработку для удаления защитной группы можно также проводить в течение 10 минут. После промывания ДМФА для последующего удлинения пептидной цепи последовательно конденсировали аминокислоты согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 63,1 мг, 0,50 ммоль) растворяли в ДМФА (1 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. Следует отметить, что объем ДМФА можно также задать как 2 мл. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут для удаления защитной группы Fmoc. Следует отметить, что данную обработку для удаления защиты можно также проводить в течение 10 минут. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, защищенных группами Fmoc, применяли Fmoc-Gly (148,7 мг, 0,50 ммоль), Fmoc-Cys (Trt) (292,9 мг, 0,50 ммоль) и Fmoc-Ala (155,7 мг, 0,50 ммоль) и на твердофазной смоле получали пептид из 4 аминокислотных остатков, имеющий защитную группу, Fmoc-Ala-Cys(Trt)-Gly-Leu (SEQ ID NO: 1). После этого защитную Fmoc-группу удаляли обработкой смолы в течение 20 минут раствором 20% пиперидина/ДМФА (2 мл) и свободные аминогруппы затем защищали ацетилом с применением раствора 20% уксусного ангидрида/ДМФА (2 мл). Следует отметить, что объем раствора 20% уксусного ангидрида/ДМФА можно также задать как 1,7 мл. После промывания ДМФА и DCM предварительно полученный реагент (TFA/вода, фенол/тиоанизол/EDT (1,2-этандитиол)/TIPS=81,5/5/5/5/2,5/1) добавляли до такого количества, чтобы смола была достаточно погружена в него, и смолу затем перемешивали при 25°С в течение 2 часов. Смолу удаляли фильтрованием и реакционный раствор затем концентрировали при пониженном давлении. Полученный остаток очищали ВЭЖХ (колонка Vydac C8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил : вода = 90:10; градиенты А:В=85:15 → 50:50; 60 минут; скорость потока: 3,0 мл/мин), получая при этом пептид из 4 аминокислотных остатков, имеющий защитную группу, Ac-Ala-Cys-Gly-Leu (SEQ ID NO: 2).
30 мг (73 мкмоль) полученного пептида из 4 аминокислотных остатков (SEQ ID NO: 2) помещали в круглодонную колбу и растворяли в 73 мл буфера 0,25 М трис-HCl (pH 8,6, содержит 6 М раствор гидрохлорида гуанидина и 3,3 мМ EDTA) и 24 мл ацетонитрила. После этого к раствору при 25°С добавляли метил-4-нитробензолсульфонат (316 мг). Спустя тридцать минут к нему добавляли 10% раствор TFA (7,3 мл), так чтобы значение pH раствора было установлено до pH 4. После этого к нему добавляли диэтиловый эфир для проведения операции экстракции. После завершения концентрирования полученный остаток помещали в колонку ODS для проведения очистки, тем самым получая 25 мг пептида из 4 аминокислотных остатков, имеющего защитную группу, Ac-Ala-Cys(Me)-Gly-Leu (SEQ ID NO: 3), у которого атом серы остатка цистеина метилирован.
ESI-MC: вычисл. для C17H30N4O6S: [M+1H]1+ 419.2, найдено, 419.1
6,5 мг (15 мкмоль) полученного пептида из 4 аминокислотных остатков (SEQ ID NO: 3), у которого атом серы остатка цистеина метилирован, помещали в эппендорфовскую пробирку и растворяли в 80% растворе муравьиной кислоты (6,5 мл). После этого к раствору при 25°С добавляли 159,0 мг (1,5 ммоль) бромида циана. Реактор защищали от света и реакцию затем проводили при 37°С. Спустя 28,5 часа реакционный раствор замораживали для завершения реакции. Остаток, полученный после лиофилизации реакционного раствора, очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил : вода = 90:10; градиенты А:В=100:0 → 60:40; 30 минут; скорость потока: 1,0 мл/мин), получая при этом 3,8 мг формы сложного эфира в качестве промежуточного продукта реакции.
ESI-MC: вычисл. для C16H28N4O7: [M+1H]1+ 389,4, найдено 389,2.
5 мг полученного промежуточного продукта реакции помещали в эппендорфовскую пробирку таким же способом, как указано выше, и растворяли в 0,6 мл фосфатного буфера (pH 7,2; содержит 6 М гидрохлорид гуанидина) с последующим проведением реакции при 37°С. Спустя один час, после подтверждения ВЭЖХ, что реакция была завершена, реакционный раствор очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил : вода = 90:10; градиенты А:В=100:0 → 60:40; 30 минут; скорость потока: 1,0 мл/мин), получая при этом 3,5 мг представляющего интерес пептида из 4 аминокислотных остатков, имеющего защитную группу, Ac-Ala-Ser-Gly-Leu (SEQ ID NO: 4). Следует указать, что время реакции можно также задать как 30 минут.
ESI-MC: вычисл. для C16H28N4O7: [M+1H]1+ 389.4, найдено, 389.1
Пример 2. Получение Val-Asp-Lys-Ala-Val-Ser-Gly-Leu
Смолу Ванга (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. Fmoc-Leu (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (2 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 1 часа. Следует отметить, что объем DCM можно также задать как 1,5 мл. После завершения операции перемешивания смолу полностью промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 15 минут. Следует отметить, что данную обработку для удаления защитной группы можно также проводить в течение 10 минут. После промывания ДМФА для последующего удлинения пептидной цепи последовательно конденсировали аминокислоты согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 63,1 мг, 0,50 ммоль) растворяли в ДМФА (1 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. Следует отметить, что объем ДМФА можно также задать как 2 мл. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут для удаления защитной группы Fmoc. Следует отметить, что данную обработку для удаления защиты можно также проводить в течение 10 минут. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, защищенных группами Fmoc, применяли Fmoc-Gly (148,7 мг, 0,50 ммоль), Fmoc-Cys (Trt) (292,9 мг, 0,50 ммоль), Fmoc-Val (169,7 мг, 0,50 ммоль), Fmoc-Ala (155,7 мг, 0,50 ммоль), Fmoc-Lys (Воc) (234,3 мг, 0,50 ммоль), Fmoc-Asp (OtBu) (205,8 мг, 0,50 ммоль) и Fmoc-Val (169, 7 мг, 0,50 ммоль) и на твердофазной смоле получали пептид из 8 аминокислотных остатков, имеющий защитную группу, Fmoc-Val-Asp(OtBu)-Lys(Boc)-Ala-Val-Cys(Trt)-Gly-Leu (SEQ ID NO: 5). После этого защитную Fmoc-группу удаляли обработкой смолы в течение 20 минут раствором 20% пиперидина/ДМФА (2 мл) и реакционный раствор затем промывали ДМФА и DCM. После этого предварительно полученный реагент К (ТFА/вода/фенол/тиоанизол/EDT=82,5/5/5/5/2,5) добавляли до такого количества, чтобы смола была достаточно погружена в него, и ее затем перемешивали при 25°С в течение 2 часов. Следует отметить, что вместо реагента К можно было также применять реагент ТFА/вода/фенол/тиоанизол/EDT/TIPS=81,5/5/5/5/2,5/1,0. Смолу удаляли фильтрованием и реакционный раствор затем концентрировали при пониженном давлении. Полученный остаток очищали ВЭЖХ (колонка Vydac C18, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=85:15→50:50; 15 минут; скорость потока: 2,5 мл/мин), получая при этом пептид из 8 аминокислотных остатков, Val-Asp-Lys-Ala-Val-Cys-Gly-Leu (SEQ ID NO: 6).
32 мг (40 мкмоль) полученного пептида из 8 аминокислотных остатков (SEQ ID NO: 6) помещали в круглодонную колбу и растворяли в 40 мл буфера 0,25 М трис-HCl (pH 8,6; содержит 6 М раствор гидрохлорида гуанидина и 3,3 мМ EDTA) и 13 мл ацетонитрила. После этого к раствору при 25°С добавляли метил-4-нитробензолсульфонат (261 мг). Спустя один час к нему добавляли 10% раствор TFA (3,8 мл), так чтобы значение pH раствора было установлено до pH 4. После этого к нему добавляли диэтиловый эфир для проведения операции экстракции. После завершения концентрирования полученный остаток помещали в колонку ODS для проведения очистки, тем самым получая 30 мг пептида из 8 аминокислотных остатков, Val-Asp-Lys-Ala-Val-Cys(Me)-Gly-Leu (SEQ ID NO: 7), у которого атом серы остатка цистеина был метилирован.
ESI-MC: вычисл. для C35H64N9O11S: [M+1H]1+ 819.0, найдено, 818.8.
29 мг (36 мкмоль) полученного пептида из 8 аминокислотных остатков (SEQ ID NO: 7), у которого атом серы остатка цистеина был метилирован, помещали в круглодонную колбу и растворяли в 15 мл 80% раствора муравьиной кислоты. После этого к раствору при 25°С добавляли 381 мг (3,6 ммоль) бромида циана. Реактор защищали от света и реакцию затем проводили при 25°С. Спустя тридцать два часа реакционный раствор замораживали для завершения реакции. Остаток, полученный после лиофилизации реакционного раствора, очищали ВЭЖХ (колонка Vydac C8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил : вода = 90:10; градиенты А:В=90:10→70:30; 60 минут; скорость потока: 4,0 мл/мин), получая при этом 18 мг формы сложного эфира в качестве промежуточного продукта реакции.
ESI-MC: вычисл. для C34H62N9O12: [M+1H]1+ 788,9, найдено 788,5.
5 мг полученного промежуточного продукта реакции помещали в эппендорфовскую пробирку и растворяли в 0,63 мл фосфатного буфера (pH 7,2; содержит 6 М гидрохлорид гуанидина) с последующим проведением реакции при 37°С. Спустя 9,25 часа, после подтверждения, что реакция была завершена, реакционный раствор очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=100:0→60:40; 30 минут; скорость потока: 1,0 мл/мин), получая при этом 4,1 мг представляющего интерес пептида из 8 аминокислотных остатков, Val-Asp-Lys-Ala-Val-Ser-Gly-Leu (SEQ ID NO: 8). Следует указать, что время реакции может быть также задано как 7,25 часа.
ESI-MC: вычисл. для C34H62N9O12: [М+1Н]1+ 788,9, найдено 788,7.
Пример 3. Получение Leu-Phe-Arg-Val-Tyr-Ser-Asn-Phe-Leu-Arg-Gly
Смолу Ванга (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. Fmoc-Gly (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (2 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 1 часа. Следует отметить, что объем DCM можно также задать как 1,5 мл. После завершения операции перемешивания смолу полностью промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 15 минут. Следует также отметить, что данную обработку для удаления защитной группы можно также проводить в течение 10 минут. После промывания ДМФА для последующего удлинения пептидной цепи аминокислоты последовательно конденсировали согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 63,1 мг, 0,50 ммоль) растворяли в ДМФА (1 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут для удаления защитной группы Fmoc. Следует отметить, что данную обработку для удаления защиты можно также проводить в течение 10 минут. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, защищенных группами Fmoc, применяли Fmoc-Arg (Pbf) (324,4 мг, 0,50 ммоль), Fmoc-Leu (176,7 мг, 0,50 ммоль), Fmoc-Phe (193,7 мг, 0,50 ммоль), Fmoc-Asn (177,2 мг, 0,50 ммоль), Fmoc-Cys (Trt) (292,9 мг, 0,50 ммоль), Fmoc-Tyr (tBu) (229,8 мг, 0,50 ммоль), Fmoc-Val (169,7 мг, 0,50 ммоль), Fmoc-Arg (Pbf) (324,4 мг, 0,50 ммоль), Fmoc-Phe (193,7 мг, 0,50 ммоль) и Fmoc-Leu (176, 7 мг, 0,50 ммоль) и на твердофазной смоле получали пептид с 11 аминокислотными остатками, имеющий защитную группу, Fmoc-Leu-Phe-Arg(Pbt)-Val-Tyr(tBu)-Cys(Trt)-Asn-Phe-Leu-Arg(Pbf)-Gly (SEQ ID NO: 9). После этого защитную Fmoc-группу удаляли обработкой смолы в течение 20 минут 2 мл раствора 20% пиперидина/ДМФА. Следует отметить, что данную обработку для удаления защитной группы можно также проводить в течение 10 минут. Реакционный раствор промывали ДМФА и DCM. После этого предварительно полученный реагент К (TFA/вода/фенол/тиоанизол/EDT=82,5/5/5/5/2,5) добавляли до такого количества, чтобы смола была достаточно погружена в него, и затем ее перемешивали при 25°С в течение 2 часов. Следует отметить, что вместо реагента К можно было также применять реагент ТFА/вода/фенол/тиоанизол/EDT/TIPS=81,5/5/5/5/2,5/1,0. Смолу удаляли фильтрованием и реакционный раствор затем концентрировали при пониженном давлении. Полученный остаток очищали ВЭЖХ (колонка Vydac С 18, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=70:30→40:60; 15 минут; скорость потока: 2,0 мл/мин), получая при этом пептид из 11 аминокислотных остатков, Leu-Phe-Arg-Val-Tyr-Cys-Asn-Phe-Leu-Arg-Gly (SEQ ID NO: 10).
21 мг (15 мкмоль) полученного пептида из 11 аминокислотных остатков (SEQ ID NO: 10) помещали в круглодонную колбу и растворяли в 15 мл буфера 0,25 М трис-HCl (pH 8,6, содержит 6 М раствор гидрохлорида гуанидина и 3,3 мМ EDTA) и ацетонитриле (5 мл). После этого к раствору при 25°С добавляли метил-4-нитробензолсульфонат (66 мг). Спустя тридцать минут к нему добавляли 10% раствор TFA (1,5 мл), так чтобы значение pH раствора было регулировано до pH 4. После этого к нему добавляли диэтиловый эфир для проведения операции экстракции. После завершения концентрирования полученный остаток помещали в колонку ODS для проведения очистки, тем самым получая 19 мг пептида из 11 аминокислотных остатков, Leu-Phe-Arg-Val-Tyr-Cys(Me)-Asn-Phe-Leu-Arg-Gly (SEQ ID NO: 11), у которого атом серы остатка цистеина был метилирован.
ESI-MC: вычисл. для C66H100N18O14S: [M+2H]2+ 701,4, найдено 701,5.
18 мг (13 мкмоль) полученного пептида из 11 аминокислотных остатков (SEQ ID NO: 11), у которого атом серы остатка цистеина был метилирован, помещали в круглодонную колбу и растворяли в 5,4 мл 2,4 мМ 80% раствора муравьиной кислоты. После этого к раствору при 25°С добавляли 136 мг (1,3 ммоль) бромида циана. Реактор защищали от света и реакцию затем проводили при 25°С. Спустя приблизительно пятьдесят часов реакционный раствор замораживали для завершения реакции. Остаток, полученный после лиофилизации реакционного раствора, очищали ВЭЖХ (колонка Vydac С8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=85:15→50:50; 60 минут; скорость потока: 3,0 мл/мин), получая при этом 7,5 мг формы сложного эфира в качестве промежуточного продукта реакции.
ESI-MC: вычисл. для C65H100N18O15: [M+2H]2+ 686,4, найдено 686,5.
7 мг полученного промежуточного продукта реакции помещали в эппендорфовскую пробирку и растворяли в 0,50 мл фосфатного буфера (pH 7,2; содержит 6 М гидрохлорид гуанидина) с последующим проведением реакции при 37°С. Спустя 1 час, после подтверждения, что реакция была завершена, реакционный раствор очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=80:20→40:60; 30 минут; скорость потока: 1,0 мл/мин), получая при этом 5,4 мг представляющего интерес пептида из 11 аминокислотных остатков, Leu-Phe-Arg-Val-Tyr-Ser-Asn-Phe-Leu-Arg-Gly (SEQ ID NO: 12).
ESI-MC: вычисл. для C65H100N18O15: [М+2H]2+ 686,4, найдено 686,4.
Пример 4. Получение Ala-Leu-Leu-Val-Asn (цепь олигосахарида)-Ser-Ser-Gln-Pro-Trp-Glu-Pro-Leu-Gln-Leu-His-Val-Asp-Lys-Ala
Смолу амино-PEGA (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. 4-Гидроксиметил-3-метоксифеноксимасляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 моль) и N-этилморфолин (0,25 ммоль) растворяли в ДМФА (2 мл) и полученный раствор затем помещали в колонку с последующим перемешиванием при 25°С в течение 4 часов. После этого смолу полностью промывали ДМФА и DCM, получая при этом HMPB-PEGA-смолу. Полученную HMPB-PEGA-смолу применяли в качестве твердофазного носителя в твердофазном синтезе.
Fmoc-Ser (tBu) (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (2 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим проведением реакции при 25°С в течение 3 часов. Следует отметить, что объем DCM можно также задать как 2,5 мл. После завершения операции перемешивания смолу промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 15 минут. Следует также отметить, что настоящую обработку для удаления защитной группы можно также проводить в течение 10 минут.
После промывания ДМФА смолу, соответствующую 2 мкмоль пептида с 1 аминокислотным остатком переносили в эппендорфовскую пробирку. Связанный с цепью сахара аспарагин (10 мг, 3,6 мкмоль), представленный ниже формулой (9), и DEPBT (2 мг, 6 мкмоль) растворяли в ДМФА (0,12 мл) и полученный раствор затем помещали в эппендорфовскую пробирку. В нее добавляли DIPEA (0,68 мкл, 4 мкмоль) и полученную смесь затем перемешивали при 25°С в течение 18 часов.
[Формула 20]
После завершения операции перемешивания смолу промывали DCM и ДМФА. Для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (1 мл) в течение 15 минут. После промывания ДМФА для последующего удлинения цепи гликопептида аминокислоты последовательно конденсировали согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (1,35 мг, 0,01 ммоль) и DIPCI (1,53 мкл, 1,26 мг, 0,01 ммоль) растворяли в ДМФА (0,02 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. После перемешивания при 25°С в течение 1 часа для удаления защитной группы Fmoc колонку обрабатывали раствором 20% пиперидина/ДМФА (1 мл) в течение 20 минут. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, аминогруппы которых были защищены Fmoc-группами, применяли Fmoc-Val (3,4 мг, 0,01 ммоль), Fmoc-Leu (3,5 мг, 0,01 ммоль), Fmoc-Leu (3,5 мг, 0,01 ммоль) и Fmoc-Ala (3,1 мг, 0,01 ммоль) и на твердофазной смоле получали пептид с 6 аминокислотными остатками с присоединенной цепью сахара, имеющий защитную группу, Fmoc-Ala-Leu-Leu-Val-Asn(цепь олигосахарида)-Ser(tBu) (SEQ ID NO: 13). К этому пептиду с присоединенным сахаром добавляли смесь уксусная кислота : трифторэтанол (1:1) до такого количества, чтобы смола была достаточно погружена в нее. Спустя четыре часа смолу удаляли фильтрованием и фильтратную часть затем добавляли к диэтиловому эфиру, который был приготовлен отдельно, для проведения кристаллизации. После этого часть в виде раствора удаляли с применением мембранного фильтра, тем самым получая остаток, содержащий пептид с 6 аминокислотными остатками с присоединенной цепью сахара, имеющий защитную группу (SEQ ID NO: 13).
2 мг (0,55 мкмоль) полученного пептида с 6 аминокислотными остатками с присоединенной цепью сахара, имеющий защитную группу (SEQ ID NO: 13), молекулярное сито (MS) 4 А (10 мг) и бензилмеркаптан (2 мкл, 16,4 мкмоль) перемешивали в растворителе ДМФА (85 мкл) в атмосфере аргона при -20°С в течение 1 часа. После этого к реакционному раствору добавляли РуВОР (1,4 мг, 2,7 мкмоль) и DIPEA (0,46 мкл, 2,7 мкмоль) и полученную смесь затем перемешивали в течение 2,5 часа. После этого в реакционному раствору добавляли диэтиловый эфир (5 мл) для осаждения соединения. Осажденное соединение отделяли фильтрованием и осадок затем выделяли с.применением 50% водного раствора ацетонитрила. Выделенный продукт лиофилизировали и к полученному лиофилизированному продукту добавляли 95% водный раствор TFA. Полученную смесь перемешивали при 25°С в течение 2 часов. Смолу удаляли фильтрованием. Реакционный раствор концентрировали и затем растворяли в 50% водном растворе ацетонитрила с последующей лиофилизацией. Лиофилизированнный продукт очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=80:20→40:60; 60 минут; скорость потока: 1,0 мл/мин), получая при этом пептид с 6 аминокислотными остатками с присоединенной цепью сахара, имеющий бензилтиоэфирную группу у С-конца, Ala-Leu-Leu-Val-Asn(цепь олигосахарида)-Ser-SBn (SEQ ID NO: 15).
Нижеследующий способ также применяли в качестве вышеуказанной стадии получения пептида с 6 аминокислотными остатками с присоединенной цепью сахара, показанного в SEQ ID NO: 15, реакцией с аспарагином, связанным с цепью сахара, в эппендорфовской пробирке.
После промывания ДМФА смолу, соответствующую 4,3 мкмоль пептида с 1 аминокислотным остатком, переносили в эппендорфовскую пробирку. Аспарагин, связанный с цепью сахара (17 мг, 8,6 мкмоль), представленный формулой (9), и DEPBT (6 мг, 8,6 мкмоль) растворяли в смеси ДМФА:ДМСО=4:1 (0,29 мл) и полученный раствор затем помещали в эппендорфовскую пробирку. В нее добавляли DIPEA (1,5 мкл, 8,6 мкмоль) и полученную смесь затем перемешивали при 25°С в течение 18 часов.
После завершения операции перемешивания смолу промывали DCM и ДМФА. Для удаления защитной Fmoc-группы смолу обрабатывали раствором 20% пиперидин/ДМФА (1 мл) в течение 10 минут. После промывания ДМФА для последующего удлинения гликопептидной цепи аминокислоты последовательно конденсировали согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (2,9 мг, 0,022 ммоль) и DIPCI (3,3 мкл, 0,022 ммоль) растворяли в ДМФА (0,54 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (1 мл) в течение 20 минут для удаления защитной группы Fmoc. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, аминогруппы которых были защищены группами Fmoc, применяли Fmoc-Val (7,3 мг, 0,022 ммоль), Fmoc-Leu (7,6 мг, 0,022 ммоль), Fmoc-Leu (7,6 мг, 0,022 ммоль) и Boc-Ala (4, 0 мг, 0,022 ммоль) и на твердофазной смоле получали пептид с 6 аминокислотными остатками, соединенный с сахаром и имеющий защитную группу, Boc-Ala-Leu-Leu-Val-Asn(цепь олигосахарида)-Ser(tBu) (SEQ ID NO: 14). К этому соединенному с сахаром пептиду добавляли смесь уксусная кислота:трифторэтанол (1:1) до такого количества, чтобы смола была достаточно погружена в нее. Спустя двадцать четыре часа смолу удаляли фильтрованием и фильтратную часть затем добавляли к диэтиловому эфиру, который приготовляли отдельно, для проведения кристаллизации. После этого часть в виде раствора удаляли с применением мембранного фильтра, тем самым получая остаток, содержащий пептид из 6 аминокислотных остатков, соединенный с цепью сахара и имеющий защитную группу (SEQ ID NO: 14).
18 мг (7,5 мкмоль) полученного пептида с 6 аминокислотными остатками, соединенного с цепью сахара и имеющего защитную группу (SEQ ID NO: 14), молекулярное сито (MS) 4 А (190 мг) и бензилмеркаптан (26 мкл, 37,5 мкмоль) перемешивали в растворителе ДМФА (1.9 мл) в атмосфере аргона при -20°С в течение 1 часа. После этого к реакционному раствору добавляли РуВОР (20 мг, 37,5 мкмоль) и DIPEA (6,7 мкл, 37,5 мкмоль) и полученную смесь затем перемешивали в течение 2 часов. После этого к реакционному раствору добавляли диэтиловый эфир (10 мл) для осаждения соединения. Осажденное соединение отделяли фильтрованием и осадок затем растворяли в ДМФА. Полученный раствор концентрировали при пониженном давлении и к полученному остатку добавляли 95% водный раствор TFA. Полученную смесь перемешивали при 25°С в течение 2 часов. Реакционный раствор концентрировали и затем очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=100:0→40:60; 60 минут; скорость потока: 1,0 мл/мин), получая при этом пептид из 6 аминокислотных остатков, соединенный с цепью сахара пептида и имеющий бензилтиоэфирную группу у его С-конца, Ala-Leu-Leu-Val-Asn(цепь олигосахарида)-Ser-SBn (SEQ ID NO: 15).
По другому варианту, смолу Ванга (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. Fmoc-Ala (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (2 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 1 часа. Следует отметить, что объем DCM можно также задать как 1,5 мл. После завершения операции перемешивания смолу полностью промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 15 минут. Следует также отметить, что настоящую обработку для удаления защитной группы можно также проводить в течение 10 минут. После промывания ДМФА для последующего удлинения пептидной цепи последовательно конденсировали аминокислоты согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 63,1 мг, 0,50 ммоль) растворяли в ДМФА (1 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. Следует отметить, что объем ДМФА можно также задать как 2 мл. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут для удаления защитной группы Fmoc. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, защищенных группами Fmoc, применяли Fmoc-Lys (Воc) (234,3 мг, 0,50 ммоль), Fmoc-Asp (OtBu) (205,8 мг, 0,50 ммоль), Fmoc-Val (169,7 мг, 0,50 ммоль), Fmoc-His (Trt) (309,9 мг, 0,50 ммоль), Fmoc-Leu (176,7 мг, 0,50 ммоль), Fmoc-Gln (184,2 мг, 0,50 ммоль), Fmoc-Leu (176,7 мг, 0,50 ммоль), Fmoc-Pro (168,7 мг, 0,50 ммоль), Fmoc-Glu (OtBu) (212,8 мг, 0,50 ммоль), Fmoc-Trp (Boc) (263,3 мг, 0,50 ммоль), Fmoc-Pro (168,7 мг, 0,50 ммоль), Fmoc-Gln (184,2 мг, 0,50 ммоль) и Fmoc-Cys (Trt) (292,9 мг, 0,50 ммоль) и на твердофазной смоле получали пептид с 14 аминокислотными остатками, имеющий защитную группу, Fmoc-Cys(Trt)-Gln-Pro-Trp(Boc)-Glu(OtBu)-Pro-Leu-Gln-Leu-His(Trt)-Val-Asp(OtBu)-Lys(Boc)-Ala (SEQ ID NO: 16). После этого защитную Fmoc-группу удаляли обработкой смолы в течение 20 минут раствором 20% пиперидина/ДМФА (2 мл) и полученный продукт затем промывали ДМФА и DCM. После этого предварительно полученный реагент К (ТFА/вода/фенол/тиоанизол/EDT=82,5/5/5/5/2,5) добавляли до такого количества, чтобы смола была достаточно погружена в него, и смесь затем перемешивали при 25°С в течение 2 часов. Следует отметить, что вместо реагента К можно было также применять реагент ТFА/вода/фенол/тиоанизол/EDT/TIPS=81,5/5/5/5/2,5/1,0. Смолу удаляли фильтрованием и реакционный раствор затем концентрировали при пониженном давлении. Полученный остаток очищали ВЭЖХ (колонка Vydac C8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода=90:10; градиенты А:В=80:20→40:60; 30 минут; скорость потока: 4 мл/мин), получая при этом пептид из 14 аминокислотных остатков, Cys-Gln-Pro-Trp-Glu-Pro-Leu-Gln-Leu-His-Val-Asp-Lys-Ala (SEQ ID NO: 17).
1,1 мг полученного таким образом пептида из 14 аминокислотных остатков (SEQ ID NO: 17) и 1,3 мг предварительно синтезированного пептида из 6 аминокислотных остатков, соединенного с цепью сахара и имеющего бензилтиоэфирную группу у его С-конца (SEQ ID NO: 15), помещали в одну эппендорфовскую пробирку и растворяли в 275 мкл фосфатного буфера (pH 7,2; содержит 6 М гидрохлорид гуанидина). После этого к раствору при 25°С добавляли тиофенол (1 мкл) и бензилмеркаптан (1 мкл) и реакцию затем проводили при 37°С. Спустя двадцать шесть часов, после подтверждения ВЭЖХ, что реакция завершилась, реакционный раствор очищали ВЭЖХ (колонка Vydac C18, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=80:20→50:50; 60 минут; скорость потока: 1,0 мл/мин), получая при этом 1,5 мг пептида из 20 аминокислотных остатков, соединенного с цепью сахара, Ala-Leu-Leu-Val-Asn(цепь олигосахарида)-Ser-Cys-Gln-Pro-Trp-Glu-Pro-Leu-Gln-Leu-His-Val-Asp-Lys-Ala (SEQ ID NO: 18).
1,5 мг (0,39 мкмоль) полученного пептида из 20 аминокислотных остатков, соединенного с цепью сахара (SEQ ID NO: 18) помещали в круглодонную колбу и растворяли в 39 мл буфера 0,25 М трис-HCl (pH 8,6, содержит 6 М раствор гидрохлорида гуанидина и 3,3 мМ EDTA) и 0,13 мл ацетонитрила. После этого к раствору при 25°С добавляли метил-4-нитробензолсульфонат (1,7 мг). Спустя сорок минут к нему добавляли 10% раствор TFA (1,5 мл), так чтобы значение pH раствора было установлено до pH 4. После этого к нему добавляли диэтиловый эфир для проведения операции экстракции. После концентрирования водного слоя полученный остаток очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=80:20→55:45; 60 минут; скорость потока: 1,0 мл/мин) (где при очистке можно применять также колонку C18 вместо вышеуказанной колонки C4), получая при этом 1,6 мг пептида из 20 аминокислотных остатков, соединенного с цепью сахара, Ala-Leu-Leu-Val-Asn(цепь олигосахарида)-Ser-Cys(Me)-Gln-Pro-Trp-Glu-Pro-Leu-Gln-Leu-His-Val-Asp-Lys-Ala (SEQ ID NO: 19), у которого атом серы остатка цистеина метилирован.
ESI-MC: вычисл. для C165H265N31O74S: [М+2Н]2+ 1300.7, найдено, 1300.4.
1,6 мг (0,4 мкмоль) полученного пептида из 20 аминокислотных остатков, соединенного с цепью сахара (SEQ ID NO: 19), у которого атом серы остатка цистеина метилирован, помещали в эппендорфовскую пробирку и растворяли в 0,4 мл 80% раствора муравьиной кислоты. После этого к раствору при 25°С добавляли 4,3 мг (0,04 ммоль) бромида циана. Реактор защищали от света и реакцию затем проводили при 37°С. Спустя тридцать пять часов реакционный раствор замораживали для завершения реакции. К остатку, полученному после лиофилизации реакционного раствора, добавляли 5% гидразингидрат (200 мкл) и pH внутри реакционной системы устанавливали на уровне до pH 10-11. Спустя пять минут к реакционной системе добавляли уксусную кислоту (5 мкл), так чтобы pH внутри ее установить на уровне 8-9. Образованный продукт очищали ВЭЖХ (колонка Vydac C4, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=80:20→50:50; 30 минут; скорость потока: 1,0 мл/мин), получая при этом 0,7 мг пептида из 20 аминокислотных остатков, соединенного с цепью сахара, Ala-Leu-Leu-Val-Asn(цепь олигосахарида)-Ser-Ser-Gln-Pro-Trp-Glu-Pro-Leu-Gln-Leu-His-Val-Asp-Lys-Ala (SEQ ID NO: 20), который был представляющим интерес гликопептидом.
Спустя тридцать четыре часа после реакции с бромидом циана реакционный раствор концентрировали при пониженном давлении и к полученному остатку добавляли 5% гидразингидрат (200 мкл) с последующим перемешиванием смеси в течение 10 минут. После этого к реакционному раствору добавляли 5 мкл уксусной кислоты и полученную смесь затем очищали ВЭЖХ. В этом случае можно было также получить тот же самый пептид из 20 аминокислотных остатков, соединенный с цепью сахара (SEQ ID NO: 20).
ESI-MC: вычисл. для C164H263N31O75: [М+2H]2+ 1290.6, найдено, 1290.7
Пример 5. Получение Ac-Gly-Ser-Gly-Met-Ala
Смолу Ванга (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. Fmoc-Ala (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (1,5 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 2 часов. После завершения операции перемешивания смолу полностью промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 15 минут. После промывания ДМФА для последующего удлинения пептидной цепи аминокислоты последовательно конденсировали согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 63,1 мг, 0,50 ммоль) растворяли в ДМФА (2 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 10 минут для удаления защитной группы Fmoc. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, защищенных группами Fmoc, применяли Fmoc-Met(O) (193,8 мг, 0,50 ммоль), Fmoc-Gly (148,9 мг, 0,50 ммоль), Fmoc-Cys (Trt) (292,9 мг, 0,50 ммоль) и Fmoc-Gly (148,9 мг, 0,50 ммоль), и на твердофазной смоле получали пептид из 5 аминокислотных остатков, имеющий защитную группу, Fmoc-Gly-Cys(Trt)-Gly-Met(O)-Ala (SEQ ID NO: 21). После этого защитную Fmoc-группу удаляли обработкой смолы в течение 10 минут раствором 20% пиперидина/ДМФА (2 мл) и свободные аминогруппы затем защищали ацетилом с применением раствора 20% уксусного ангидрида/ДМФА (1,25 мл). После промывания смолы ДМФА и DCM предварительно полученный реагент (ТFА/вода/TIPS=95/2,5/2,5) добавляли до такого количества, чтобы смола была достаточно погружена в него, и ее затем перемешивали при 25°С в течение 2 часов. Смолу удаляли фильтрованием и к фильтраты для осаждения затем добавляли 20 мл диэтилового эфира. Осадок отделяли фильтрованием и фильтрат затем растворяли в 0,1% водном растворе TFA. Полученный раствор концентрировали при пониженном давлении и образовавшийся продукт затем лиофилизировали, получая при этом смесь, содержащую пептид из 5 аминокислотных остатков, Ac-Gly-Cys-Gly-Met(O)-Ala (SEQ ID NO: 22), у которого атом серы метионина в положении 4 окислен.
5 мг полученной смеси пептида из 5 аминокислотных остатков (SEQ ID NO: 22), у которого атом серы метионина в положении 4 был окислен, помещали в круглодонную колбу и растворяли в 10 мл буфера 0,25 М трис-HCl (pH 8,6; содержит 6 М раствор гидрохлорида гуанидина и 3,3 мМ EDTA). К полученному раствору добавляли 2-меркаптоэтанол (7 мкл) и полученную смесь затем перемешивали в течение 10 минут. После этого к реакционному раствору добавляли 3,3 мл раствора ацетонитрила, содержащего метил-4-нитробензолсульфонат (66 мг). Спустя двадцать пять минут к нему добавляли 10% раствор TFA (1,0 мл) для нейтрализации. После этого к нему добавляли диэтиловый эфир для проведения операции экстракции 3 раза. После концентрирования водного слоя при пониженном давлении полученный остаток очищали ВЭЖХ (колонка Vydac C18, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=100:0→100:0→60:40; 0 минут → 5 минут → 35 минут; скорость потока: 4,0 мл/мин), получая при этом 4 мг пептида из 5 аминокислотных остатков, Ac-Gly-Cys(Me)-Gly-Met(O)-Ala (SEQ ID NO: 23), у которого атом серы остатка цистеина в положении 2 метилирован и остаток метионина в положении 4 окислен.
3,8 мг полученного пептида из 5 аминокислотных остатков (SEQ ID NO: 23), у которого атом серы остатка цистеина в положении 2 метилирован и остаток метионина в положении 4 окислен, помещали в эппендорфовскую пробирку и растворяли в 3,1 мл 80% раствора муравьиной кислоты. После этого к раствору добавляли 79,0 мг бромида циана и реактор затем защищали от света. Реакционный раствор перемешивали в условиях атмосферы аргона при 37°С в течение 39 часов. После завершения реакции образовавшийся продукт подвергали вакуумному концентрированною, получая при этом остаток, содержащий форму сложного эфира в качестве промежуточного продукта реакции.
Полученный остаток помещали в эппендорфовскую пробирку и растворяли в 1,25 мл буфера фосфата натрия (содержащего 6 М гидрохлорид гуанидина; pH 7,2), с последующим перемешиванием в течение 45 минут. После этого к реакционному раствору добавляли 3,6 мл трифторуксусной кислоты, 22 мг иодида аммония и 11 мкл диметилсульфида и полученную смесь затем перемешивали при комнатной температуре. Спустя тридцать минут к реакционному раствору добавляли 10 мл воды и полученный раствор затем отделяли и промывали тетрахлоридом углерода. Водный слой концентрировали при пониженном давлении и полученный остаток затем очищали ВЭЖХ (колонка Vydac С 18, 250×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=100:0→100:0→60:40; 0 минут → 5 минут → 65 минут; скорость потока: 1,0 мл/мин), получая при этом 2,6 мг представляющего интерес пептида из 5 аминокислотных остатков, имеющего защитную группу, Ac-Gly-Ser-Gly-Met-Ala (SEQ ID NO: 24).
ESI-MC: вычисл. для C16H28N4O7: [M+1H]1+ 464.2, найдено, 464.5.
Пример 6. Получение Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly-Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly
Смолу амино-PEGA (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем ее полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в ДМФА смолу. 4-Гидроксиметил-3-метоксифеноксимасляную кислоту (НМРВ) (0,25 ммоль), TBTU (0,25 ммоль) и N-этилморфолин (0,25 ммоль) растворяли в ДМФА (2 мл) и полученный раствор затем помещали в колонку с последующим перемешиванием при 25°С в течение 4 часов. После этого смолу полностью промывали ДМФА и DCM и получали HMPB-PEGA-смолу. Полученную HMPB-PEGA-смолу применяли в качестве твердофазного носителя в твердофазном синтезе.
Fmoc-Gly (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (4,5 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 3 часов. После завершения операции перемешивания смолу промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 10 минут. После промывания смолы ДМФА для последующего удлинения пептидной цепи аминокислоты последовательно конденсировали согласно нижеописанному способу.
Аминокислоту, аминогруппа которой была защищена группой Fmoc, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 63,1 мг, 0,50 ммоль) растворяли в ДМФА (4 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 10 минут для удаления защитной группы Fmoc. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве аминокислот, защищенных группами Fmoc, применяли Fmoc-Pro, Fmoc-Ala, Fmoc-Pro, Fmoc-Arg (Pbf), Fmoc-Thr (tBu), Fmoc-Asp (OtBu), Fmoc-Pro, Fmoc-Ala, Fmoc-Ser (tBu), Fmoc-Thr (tBu), Fmoc-Val, Fmoc-Gly, Fmoc-His (Trt), Fmoc-Ala, Fmoc-Pro, Fmoc-Pro и Fmoc-Ala. На твердофазной смоле получали пептид из 18 аминокислотных остатков, имеющий защитную группу, Fmoc-Ala-Pro-Pro-Ala-His(Trt)-Gly-Val-Thr(tBu)-Ser(tBu)-Ala-Pro-Asp(OtBu)-Thr(tBu)-Arg(Pbf)-Pro-Ala-Pro-Gly (SEQ ID NO: 25).
Этот пептид из 18 аминокислотных остатков, имеющий защитную группу (SEQ ID NO: 25), обрабатывали раствором 20% пиперидина/ДМФА (2 мл) на смоле в течение 10 минут для удаления защитной Fmoc-группы. После того как образованную смолу промывали ДМФА и DCM, Fmoc-Thr (GalNAc) (0,20 ммоль), HOBt (0,50 ммоль) и DIPCI (0,50 ммоль), которые были растворены в ДМФА (3,6 мл) и активированы в течение 15 минут, добавляли к полученной смоле. После проведения реакции в течение 5 часов смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут для удаления защитной Fmoc-группы, получая при этом на твердофазной смоле пептид из 19 аминокислотных остатков, связанный с сахаром и имеющий защитную группу, Thr(GalNAc)-Ala-Pro-Pro-Ala-His(Trt)-Gly-Val-Thr(tBu)-Ser(tBu)-Ala-Pro-Asp(OtBu)-Thr(tBu)-Arg(Pbf)-Pro-Ala-Pro-Gly (SEQ ID NO: 26).
0,02 ммоль полученного на твердофазной смоле пептида из 19 аминокислотных остатков, связанного с сахаром и имеющего защитную группу (SEQ ID NO: 26), переносили в другую колонку твердофазного синтеза. Boc-Ser(tBu) (0,1 ммоль), DIPCI (0,1 ммоль) и HOBt (0,1 ммоль), которые были растворены в 0,5 мл ДМФА и затем активированы в течение 15 минут, добавляли в колонку и реакцию затем проводили в течение 1 часа. После этого реакционный раствор фильтровали и добавляли смесь уксусная кислота:трифторэтанол = 1:1 до такого количества, чтобы смола была достаточно погружена в нее. Спустя четырнадцать часов смолу удаляли фильтрованием и фильтрат затем концентрировали при пониженном давлении, получая при этом остаток, содержащий пептид из 20 аминокислотных остатков, связанный с сахаром и имеющий защитную группу, Boc-Ser(tBu)-Thr(GalNAc)-Ala-Pro-Pro-Ala- His(Trt)-Gly-Val-Thr(tBu)-Ser(tBu)-Ala-Pro-Asp(OtBu)-Thr(tBu)-Arg(Pbf)-Pro-Ala-Pro-Gly (SEQ ID NO: 27).
75 мг (35 мкмоль) полученного пептида из 20 аминокислотных остатков, связанного с сахаром и имеющего защигную группу (SEQ ID NO: 27), и 123 мкл (105 мкмоль) бензилмеркаптана добавляли к 3,5 мл ДМФА и полученную смесь затем перемешивали в атмосфере аргона при -20°С в течение 1 часа. После этого РуВОР (91 мг, 175 мкмоль) и DIPEA (30 мкл, 175 мкмоль) добавляли к реакционному раствору и полученную смесь затем перемешивали в течение 2,5 часа. После этого к реакционному раствору добавляли диэтиловый эфир для осаждения кристаллов с последующим отделением их фильтрованием. К полученному остатку добавляли смесь ТРА/вода/TIPS=95/2,5/2,5 до такого количества, чтобы смола была достаточна погружена в нее, и полученную смесь затем перемешивали при 25°С в течение 2 часов. Смолу удаляли фильтрованием и фильтрат затем концентрировали, так чтобы выделить полученный остаток. Полученный остаток очищали ВЭЖХ (колонка Vydac C8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=90:10→60:40; 30 минут; скорость потока: 4,0 мл/мин), получая при этом пептид из 20 аминокислотных остатков, связанный с сахаром и имеющий бензилтиоэфирную группу у С-конца, Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly-SBn (SEQ ID NO: 28).
По другому варианту 0,02 ммоль ранее полученного пептида из 19 аминокислотных остатков, связанного с сахаром и имеющего защитную группу (SEQ ID NO: 26), на твердофазной смоле переносили в другую колонку твердофазного синтеза. Вос-Thia (0,04 ммоль), DIPCI (0,1 ммоль) и HOBt (0,1 ммоль), которые были растворены в 1,5 мл ДМФА и активированы в течение 15 минут, добавляли в колонку и реакцию затем проводили в течение 20 минут. После этого реакционный раствор фильтровали и к нему добавляли реагент (ТFА/вода/TIPS=95/2,5/2,5) до такого количества, чтобы смола была достаточно погружена в него. Полученную смесь перемешивали при 25°С в течение 2 часов. Смолу удаляли фильтрованием и фильтрат затем концентрировали. Полученный остаток растворяли в 2,1 мл 0,2 М метоксиамина и 0,1 М буфере фосфата натрия (pH 4; содержит 6 М гидрохлорид гуанидина), так чтобы тиазолин у N-конца подвергался размыканию цикла. После этого реакционный раствор очищали ВЭЖХ (колонка Vydac C8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,09% TFA; ацетонитрил:вода = 90:10; градиенты А:В=90:10→60:40; 30 минут; скорость потока: 4,0 мл/мин), получая при этом пептид из 20 аминокислотных остатков, связанный с сахаром и имеющий бензилтиоэфирную группу у С-конца, Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala- His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly-SBn (SEQ ID NO: 28).
По другому варианту 0,02 ммоль ранее полученного пептида из 19 аминокислотных остатков, связанный с сахаром и имеющий защитную группу (SEQ ID NO: 26), на твердофазной смоле переносили в другую колонку твердофазного синтеза. Вос-Thia (0,04 ммоль), DIPCI (0,1 ммоль) и HOBt (0,1 ммоль), которые были растворены в 1,5 мл ДМФА и активированы в течение 15 минут, добавляли в колонку и реакцию затем проводили в течение 20 минут. После этого реакционный раствор фильтровали и к нему добавляли реагент (ТFА/вода/TIPS=95/2,5/2,5) до такого количества, чтобы смола была достаточно погружена в него. Полученную смесь перемешивали при 25°С в течение 2 часов. Смолу удаляли фильтрованием и фильтрат затем концентрировали. Полученный остаток растворяли в 2,1 мл 0,2 М метоксиамина и 0,1 М буфере фосфата натрия (pH 4; содержит 6 М гидрохлорид гуанидина), так чтобы тиазолин у N-конца подвергался размыканию цикла. После этого реакционный раствор очищали ВЭЖХ (колонка Vydac С8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=90:10→60:40; 30 минут; скорость потока: 4,0 мл/мин), получая при этом пептид из 20 аминокислотных остатков, связанный с сахаром, Cys-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly (SEQ ID NO: 29).
13 мг полученного таким образом пептида из 20 аминокислотных остатков, связанного с сахаром (SEQ ID NO: 29), и 12 мг ранее полученного пептида из 20 аминокислотных остатков, имеющего бензилтиоэфирную группу у его С-конца (SEQ ID NO: 28), растворяли в 2,9 мл фосфатного буфера (pH 7,2; содержит 6 М гидрохлорид гуанидина). После этого к вышеуказанному раствору добавляли 30 мг 4-меркаптофенилуксусной кислоты и 17 мг трис-(2-карбоксиэтил)фосфина и реакцию затем проводили в течение 3 часов. Реакционный раствор очищали ВЭЖХ (колонка Vydac С 8, 250×10 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,09% TFA; ацетонитрил:вода = 90:10; градиенты А:В=90:10→60:40; 30 минут; скорость потока: 4,0 мл/мин), получая при этом 18 мг пептида из 40 аминокислотных остатков, связанного с сахаром, Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly-Cys-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly (SEQ ID NO: 30).
16 мг полученного пептида из 40 аминокислотных остатков, связанного с сахаром (SEQ ID NO: 30), помещали в круглодонную колбу и растворяли в 3,8 мл буфера 0,25 М трис-HCl (pH 8,6; содержит 6 М гидрохлорид гуанидина и 3,3 мМ EDTA). После этого к полученному раствору добавляли 2-меркаптоэтанол (3 мкл) и полученную смесь затем перемешивали в течение 10 минут. Затем к реакционному раствору добавляли 1,27 мл ацетонитрила, содержащего 25 мг метил-4-нитробензолсульфоната. Спустя двадцать пять минут 0,45 мл 10% раствора TFA добавляли к реакционному раствору для нейтрализации и затем в нему добавляли 2 мл диэтилового эфира с последующим разделением и промыванием 3 раза. После концентрирования водного слоя при пониженном давлении полученный остаток очищали ВЭЖХ (колонка Vydac C-4, 250×4,5 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,08% TFA; ацетонитрил:вода = 90:10; градиенты А:В=100:0→100:0→90:10→60:40; 0 минут → 10 минут → 40 минут; скорость потока: 1,2 мл/мин), получая при этом 13 мг смеси, содержащей пептид из 40 аминокислотных остатков, связанный с сахаром, Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly-Cys(Me)-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly (SEQ ID NO: 31), у которого атом серы остатка цистеина в положении 21 метилирован.
12 мг полученной смеси, содержащей пептид из 40 аминокислотных остатков, связанный с сахаром (SEQ ID NO: 31), у которого атом серы остатка цистеина в положении 21 метилирован, помещали в круглодонную колбу и растворяли в 2,9 мл 80% водного раствора муравьиной кислоты. После этого к раствору при 25°С добавляли 152 мг бромида циана. Реактор защищали от света и реакцию затем проводили при 37°С. Спустя тридцать шесть часов реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в 2,9 мл трифторуксусной кислоты и после этого к раствору добавляли 2,4 мг иодида аммония и 24 мкл диметилсульфида с последующей реакцией смеси при комнатной температуре в течение 30 минут. Спустя тридцать минут к реакционной системе добавляли 6 мл воды и разделение и промывание затем проводили с применением тетрахлорида углерода. Водный слой концентрировали при пониженном давлении и остаток затем растворяли в 1,4 мл 5% водного раствора гидразина с последующей реакцией смеси при комнатной температуре в течение 10 минут. Спустя десять минут к реакционному раствору добавляли 0,14 мл уксусной кислоты и полученную смесь затем очищали ВЭЖХ (колонка Vydac C-4, 250×4,6 мм; растворители для проявления А: 50 мМ водный раствор AcONH4 и В: ацетонитрил; градиенты А:В=90:10 → 70:30; 60 минут; скорость потока: 1,0 мл/мин), получая при этом 2,3 мг представляющего интерес пептида из 40 аминокислотных остатков, связанного с сахаром, Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly-Ser-Thr(GalNAc)-Ala-Pro-Pro-Ala-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly (SEQ ID NO: 32).
ESI-MC: вычисл.: [М+3Н]3+ 1388.5, найдено, 1388.6
Пример 7. Получение Glu-Ser-Asn-Gly-Thr-Leu-Thr-Leu
Trt-хлорид-смолу (изготовлена Merck) (100 мкмоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. После фильтрования Fmoc-Gly (0,20 ммоль) и DIPEA (0,4 ммоль) растворяли в DCM (0,66 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 2 часов. После завершения операции перемешивания смолу достаточно промывали смесью DCM:MeOH:DIPEA=17:2:1, DCM и ДМФА и для удаления защитной Fmoc-группы смолу обрабатывали раствором 20% пиперидина/ДМФА (1 мл) в течение 20 минут. После промывания смолы ДМФА для дальнейшего удлинения пептидной цепи аминокислоты последовательно конденсировали согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 63,1 мг, 0,50 ммоль) растворяли в ДМФА (2 мл) и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут для удаления защитной группы Fmoc. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве защищенных аминокислот применяли Fmoc-Asn (177,2 мг, 0,50 ммоль), Fmoc-Ser (tBu) (191,6 мг, 0,50 ммоль) и Boc-Glu (OtBu) (151,6 мг, 0,50 ммоль). На твердофазной смоле получали пептид из 4 аминокислотных остатков, имеющий защитную группу, Boc-Glu(OtBu)-Ser(tBu)-Asn-Gly (SEQ ID NO: 33).
После промывания полученной смолы ДМФА и DCM к ней добавляли 2 мл смеси AcOH:MeOH:DCM=5:4:1 с последующей реакцией при комнатной температуре в течение 2 часов. Спустя два часа реакционный раствор выделяли фильтрованием и затем концентрировали при пониженном давлении с получением остатка. К этому остатку добавляли 1 мл бензола, затем с раствором проводили 2 раза операцию азеотропной перегонки и полученный продукт затем растворяли в 0,81 мл ДМФА. К полученному раствору в атмосфере аргона при 0°С добавляли 42,1 мг РуВОР, 14 мкл DIPEA и 57 мкл бензилмеркаптана и смесь затем перемешивали в течение 30 минут. После завершения реакции реакционный раствор нейтрализовали насыщенным водным раствором хлорида аммония и затем отделяли и промывали водой и насыщенным раствором соли. Органический слой сушили над сульфатом магния и затем фильтровали. После этого фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (проявляющие растворители: этилацетат:МеОН:вода = 20:2:1), собирая при этом фракции, содержащие представляющие интерес пептиды, и собранные фракции затем концентрировали при пониженном давлении. Полученный остаток растворяли в 95% водном растворе TFA и реакцию затем проводили в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении, получая при этом пептид из 4 аминокислотных остатков, имеющий бензилтиоэфирную группу у его С-конца, Glu-Ser-Asn-Gly-SBn (SEQ ID NO: 34).
По другому варианту, смолу Ванга (изготовлена Merck) (0,1 ммоль) помещали в колонку твердофазного синтеза и затем полностью промывали метиленхлоридом (DCM) и ДМФА. После этого получающуюся в результате смолу превращали в полностью набухшую в DCM смолу. Fmoc-Leu (0,50 ммоль), MSNT (0,50 ммоль) и N-метилимидазол (0,375 ммоль) растворяли в DCM (2 мл) и полученный раствор затем помещали в колонку твердофазного синтеза с последующим перемешиванием при 25°С в течение 2 часов. Спустя два часа смолу полностью промывали DCM и ДМФА и для удаления защитной группы Fmoc смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут. После промывания ДМФА и DCM для последующего удлинения пептидной цепи аминокислоты последовательно конденсировали согласно нижеуказанному способу.
Аминокислоту, аминогруппа которой была защищена, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 0,50 ммоль) растворяли в 2 мл ДМФА и полученный раствор затем активировали в течение 15 минут. После этого раствор помещали в колонку твердофазного синтеза. После перемешивания при 25°С в течение 1 часа смолу обрабатывали раствором 20% пиперидина/ДМФА (2 мл) в течение 20 минут для удаления защитной группы Fmoc. Эти операции повторяли, так чтобы аминокислоты были последовательно конденсированы. В качестве защищенных аминокислот применяли Fmoc-Thr (tBu) (198,8 мг, 0,50 ммоль) и Fmoc-Leu (176,8 мг, 0,50 ммоль) и на твердофазной смоле получали пептид из 3 аминокислотных остатков, имеющий защитную группу, Fmoc-Leu-Thr(tBu)-Leu. После этого защитную Fmoc-группу удаляли обработкой смолы в течение 20 минут раствором (2 мл) 20% пиперидина/ДМФА. Затем производное треонина, имеющее защитную группу, Boc-Thr(S-S-втор-Bu), синтезированное отдельно в примере синтеза 1, HOBt (67,6 мг, 0,50 ммоль) и DIPCI (77,0 мкл, 0,50 ммоль), которые растворяли в 2 мл ДМФА и затем активировали в течение 15 минут, добавляли к раствору. Полученную смесь затем подвергали реакции в течение 3 часов, получая при этом на твердофазной фазе пептид из 4 аминокислотных остатков, имеющий защитную группу, Boc-Thr(S-S-втор-Bu)-Leu-Thr(tBu)-Leu (SEQ ID NO: 35). К вышеуказанному пептиду из 4 аминокислотных остатков добавляли 95% водный раствор TFA и реакцию затем проводили при комнатной температуре в течение 2 часов. После этого смолу удаляли фильтрованием и фильтрат затем концентрировали при пониженном давлении, получая при этом пептид из 4 аминокислотных остатков, имеющий защитную группу, Thr(S-S-втор-Bu)-Leu-Thr-Leu (SEQ ID NO: 36).
2,2 мг полученного таким образом пептида из 4 аминокислотных остатков, имеющего защитную группу (SEQ ID NO: 36), и 2,2 мг ранее полученного пептида из 4 аминокислотных остатков, имеющего тиоэфирную группу (SEQ ID NO: 34), растворяли в хх мл буфера 0,2 М фосфата натрия (pH 7,3; содержит 6 М гидрохлорид гуанидина и 20 мг 4-меркаптофенилуксусной кислоты) с последующей реакцией смеси в течение 3,5 часа. После завершения реакции к реакционному раствору добавляли дитиотреитол и полученную смесь затем перемешивали в течение 10 минут. После этого реакционный раствор очищали ВЭЖХ (колонка Cadenza С 18, 75×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,1% TFA; ацетонитрил:вода = 90:10; градиенты А:В=90:10→35:65; 15 минут; скорость потока: 1,0 мл/мин), получая при этом пептид из 8 аминокислотных остатков, Glu-Ser-Asn-Gly-Thr(SH)-Leu-Thr-Leu (SEQ ID NO: 37), у которого гидроксильная группа боковой цепи остатка треонина в положении 5 замещена тиоловой группой. Полагают, что остаток производного треонина, содержащийся в полученном пептиде, имеет конфигурацию, в основном представленную следующей группой:
[Формула 21]

3,5 мг полученного пептида из 8 аминокислотных остатков (SEQ ID NO: 37), у которого гидроксильная группа боковой цепи остатка треонина в положении 5 была замещена тиоловой группой, помещали в круглодонную колбу и растворяли в 4,1 мл буфера 0,25 М трис-HCl (pH 8,6; содержит 6 М раствор гидрохлорида гуанидина и 3,3 мМ ЭДТА). После этого к полученному раствору добавляли 2-меркаптоэтанол (2,9 мкл) и полученную смесь затем перемешивали в течение 15 минут. Затем к реакционному раствору добавляли 1,37 мл ацетонитрила, в котором был растворен метил-4-нитробензолсульфонат (26,2 мг), и реакцию затем проводили в течение 50 минут. После завершения реакции к реакционному раствору добавляли 10% раствор TFA (1,0 мл) для нейтрализации и реакционный раствор затем очищали ВЭЖХ (колонка Cadenza С 18, 75×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,1% TFA; ацетонитрил:вода = 90:10; градиенты А:В=90:10→35:65; 15 минут; скорость потока: 1,0 мл/мин), получая при этом пептид из 8 аминокислотных остатков, Glu-Ser-Asn-Gly-Thr(SMe)-Leu-Thr-Leu (SEQ ID NO: 38), у которого атом серы, замещенной тиоловой группы остатка треонина в положении 5, был метилирован.
4 мг полученного пептида из 8 аминокислотных остатков (SEQ ID NO: 38), у которого атом серы, замещенной тиоловой группы остатка треонина в положении 5, был метилирован, помещали в круглодонную колбу и растворяли в 4,63 мл 70% водного раствора муравьиной кислоты. После этого к раствору добавляли 49,0 мг бромида циана и полученную смесь затем перемешивали в условиях защита от света в атмосфере аргона при 37°С. Спустя сорок восемь часов реакционный раствор концентрировали при пониженном давлении и к остатку добавляли 0,93 мл 5% водного раствора гидразина с последующим проведением реакции в течение 15 минут. К реакционному раствору добавляли 60 мкл уксусной кислоты и реакционный раствор затем очищали ВЭЖХ (колонка Cadenza С 18, 75×4,6 мм; растворители для проявления А: 0,1% водный раствор TFA и В: 0,1% TFA; ацетонитрил:вода = 90:10; градиенты А:В=85:15→45:55; 15 минут; скорость потока: 1,0 мл/мин), получая при этом представляющий интерес пептид из 8 аминокислотных остатков, Glu-Ser-Asn-Gly-Thr-Leu-Thr-Leu (SEQ ID NO: 39). На основании механизма реакции предполагалось, что конфигурация остатков треонина, содержащихся в полученном пептиде, может быть такой же, как конфигурация природного остатка треонина.
ESI-MC: вычисл. для C34H59N9O15: [М+1Н]1+ 834.41, найдено, 835.1.
Пример 1 синтеза: Синтез Boc-Thr(S-S-втор Bu)
Boc-Thr (300 мг, 1,37 ммоль) растворяли в перегнанном ДМФА (6,85 мл, 200 мМ) и затем к раствору добавляли гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (786 мг, 4,1 ммоль) и HOBt (370 мг, 2,74 ммоль). Полученную смесь перемешивали при 0°С в течение 15 минут. После этого к реакционному раствору добавляли TMSEtOH (1,95 мл, 13,7 ммоль) и полученную смесь затем перемешивали при 0°С. Спустя двадцать часов, после подтверждения ТСХ окончания реакции, реакционный раствор разбавляли этилацетатом и затем нейтрализовали насыщенным раствором бикарбоната натрия. Раствор затем отделяли и промывали дважды водой и насыщенным раствором соли один раз. Органический слой сушили над сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (проявляющий растворитель:этилацетат:гексан = 1:3), получая при этом триметилсилиловый (TMS) эфир Boc-Thr (выход: 342 мг, выход: 78%).
Полученный TMS-эфир Boc-Thr подвергали азеотропной перегонке с бензолом 2 раза и остаток затем растворяли в 20,8 мл DCM. К раствору добавляли 322 мл (4,16 ммоль) метансульфонилхлорида и 1,16 мл (8,32 ммоль) триэтиламина и полученную смесь затем перемешивали при 0°С. Спустя десять минут, после подтверждения ТСХ окончания реакции, реакционный раствор разбавляли DCM и затем нейтрализовали насыщенным водным раствором хлорида аммония. После этого раствор отделяли и промывали дважды водой и один раз насыщенным раствором соли. Органический слой сушили над сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (проявляющий растворитель:этилацетаттексан = 1:4), получая при этом TMS-эфир Boc-Thr(OMs) (выход по массе: 724,6 мг, выход в процентах: 88%).
1,06 г полученного TMS-эфир Boc-Thr(OMs) подвергали азеотропной перегонке с бензолом 2 раза и затем сушили в эксикаторе на протяжении ночи. Полученный продукт растворяли в 8,9 мл ДМФА. После этого раствор, полученный растворением 0,57 мл (7,95 ммоль) тиоуксусной кислоты и 0,79 мл (5,3 ммоль) DBU в 4,4 мл ДМФА и последующей реакцией в течение 20 минут, добавляли к вышеуказанному раствору. Полученную смесь перемешивали при 45°С. Спустя восемнадцать часов, после подтверждения ТСХ окончания реакции, реакционный раствор нейтрализовали насыщенным водным раствором хлорида аммония и затем отделяли и промывали дважды водой и один раз насыщенным раствором соли. Органический слой сушили над сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (проявляющий растворитель:этилацетаттексан = 1:20), получая при этом TMS-эфир Boc-Thr(SAc) (выход по массе: 536,8 мг, выход в процентах: 53,7%).
1,1 г полученного TMS-эфира Boc-Thr(SAc) растворяли в 100 мл метанола и после этого к этому раствору добавляли раствор, полученный растворением дипиридилдисульфида (3,21 г, 14,6 ммоль) и 1,75 мл (16,1 ммоль) втор-BuSH в 5,5 мл метанола. После этого к раствору добавляли 11,7 мл метанольный раствор гидроксида натрия (500 мМ). Спустя восемнадцать часов, после подтверждения ТСХ окончания реакции, реакционный раствор нейтрализовали 1% водным раствором уксусной кислоты, затем концентрировали при пониженном давлении и затем разбавляли этилацетатом. Образовавшийся продукт отделяли и промывали дважды водой и затем один раз насыщенным раствором соли. Органический слой сушили над сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (проявляющий растворитель:этилацетаттексан = 1:40). Полученную смесь подвергали азеотропной перегонке с бензолом 2 раза и затем ее растворяли в 29 мл (100 мМ) ДМФА. К этому раствору добавляли 2,28 мг (7,3 ммоль) тригидрата фторида тетрабутиламмония и полученную смесь перемешивали при 0°С. После перемешивания в течение 15 минут, после подтверждения ТСХ окончания реакции, реакционный раствор нейтрализовали насыщенным водным раствором хлорида аммония. Этот реакционный раствор промывали дважды водой и затем один раз насыщенным раствором соли. Органический слой сушили над сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (проявляющий растворитель:этилацетаттексан = 1:4 → этилацетат:метанол:вода = 20:2:1), получая при этом представляющий интерес Boc-Thr(S-S-втор-Bu) (выход по массе: 650 мг, выход в процентах: 69%).
Свободный текст перечня последовательностей
SEQ ID NO: 1 является аминокислотной последовательностью, имеющей защитную группу примера 1.
SEQ ID NO: 2 является ацетилированной аминокислотной последовательностью примера 1.
SEQ ID NO: 3 является аминокислотной последовательностью, имеющей метилированный цистеин примера 1.
SEQ ID NO: 4 является ацетилированной аминокислотной последовательностью примера 1.
SEQ ID NO: 5 является аминокислотной последовательностью, имеющей защитную группу, примера 2.
SEQ ID NO: 6 является аминокислотной последовательностью примера 2.
SEQ ID NO: 7 является аминокислотной последовательностью, имеющей метилированный цистеин, примера 2.
SEQ ID NO: 8 является аминокислотной последовательностью примера 2.
SEQ ID NO: 9 является аминокислотной последовательностью, имеющей защитную группу, примера 3.
SEQ ID NO: 10 является аминокислотной последовательностью примера 3.
SEQ ID NO: 11 является аминокислотной последовательностью, имеющей метилированный цистеин, примера 3.
SEQ ID NO: 12 является аминокислотной последовательностью примера 3.
SEQ ID NO: 13 является связанной с цепью сахара аминокислотной последовательностью, имеющей защитную группу, примера 4.
SEQ ID NO: 14 является связанной с цепью сахара аминокислотной последовательностью, имеющей защитную группу, примера 4.
SEQ ID NO: 15 является связанной с цепью сахара аминокислотной последовательностью, имеющей бензилтиоэфирную группу, примера 4.
SEQ ID NO: 16 является аминокислотной последовательностью, имеющей защитную группу, примера 4.
SEQ ID NO: 17 является аминокислотной последовательностью примера 4.
SEQ ID NO: 18 является связанной с цепью сахара аминокислотной последовательностью примера 4.
SEQ ID NO: 19 является связанной с цепью сахара аминокислотной последовательностью, имеющей метилированный цистеин, примера 4.
SEQ ID NO: 20 является связанной с цепью сахара аминокислотной последовательностью примера 4.
SEQ ID NO: 21 является аминокислотной последовательностью, имеющей защитную группу и сульфоксид метионина, примера 5.
SEQ ID NO: 22 является ацетилированной аминокислотной последовательностью, имеющей сульфоксид метионина, примера 5.
SEQ ID NO: 23 является ацетилированной аминокислотной последовательностью, имеющей метилированный цистеин и сульфоксид метионина, примера 5.
SEQ ID NO: 24 является ацетилированной аминокислотной последовательностью примера 5.
SEQ ID NO: 25 является аминокислотной последовательностью, имеющей защитную группу, примера 6.
SEQ ID NO: 26 является связанной с цепью сахара аминокислотной последовательностью, имеющей защитную группу, примера 6.
SEQ ID NO: 27 является связанной с цепью сахара аминокислотной последовательностью, имеющей защитную группу, примера 6.
SEQ ID NO: 28 является связанной с цепью сахара аминокислотной последовательностью, имеющей бензилтиоэфирную группу, примера 6.
SEQ ID NO: 29 является связанной с цепью сахара аминокислотной последовательностью примера 6.
SEQ ID NO: 30 является связанной с цепью сахара аминокислотной последовательностью примера 6.
SEQ ID NO: 31 является связанной с цепью сахара аминокислотной последовательностью, имеющей метилированный цистеин, примера 6.
SEQ ID NO: 32 является связанной с цепью сахара аминокислотной последовательностью примера 6.
SEQ ID NO: 33 является аминокислотной последовательностью, имеющей защитную группу, примера 7.
SEQ ID NO: 34 является аминокислотной последовательностью, имеющей бензилтиоэфирную группу, примера 7.
SEQ ID NO: 35 является аминокислотной последовательностью, имеющей защитную группу и производное треонина, примера 7.
SEQ ID NO: 36 является аминокислотной последовательностью, имеющей производное треонина, примера 7.
SEQ ID NO: 37 является аминокислотной последовательностью, имеющей производное треонина, примера 7.
SEQ ID NO: 38 является аминокислотной последовательностью, имеющей производное треонина, примера 7.
SEQ ID NO: 39 является аминокислотной последовательностью примера 7.
Промышленная применимость
Настоящее изобретение относится к способу получения пептида и гликопептида. Согласно настоящему изобретению можно применять способ дотирования даже в случае, в котором нужно получить пептид, который не содержит цистеин. Кроме того, настоящее изобретение является также применимым для получения N-связанного гликопептида и O-связанного гликопептида.
Реферат
Настоящее изобретение относится к способу получения пептида, отличающемуся тем, что он содержит превращение группы -SH пептида, содержащего аминокислотный остаток, имеющий группу -SH, в группу -ОН, где указанный способ содержит следующие стадии от (а) до (с): (а) реакция группы -SH в пептиде с метилирующим агентом для превращения группы -SH в группу -SMe; (b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира; (с) превращение промежуточного продукта реакции полученного на стадии (b) в пептид, содержащий аминокислотный остаток, имеющий группу -ОН в более основных условиях, чем условия стадии (b). 7 н. и 13 з.п. ф-лы, 7 пр.
Формула
-ОН группу, отличающийся тем, что он включает превращение группы -SH пептида, содержащего аминокислотный остаток, имеющий группу -SH, в группу -ОН, где указанный способ включает следующие стадии от (а) до (с):
(a) реакция группы -SH в пептиде с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН в более основных условиях, чем условия стадии (b).
(а) реакция группы -SH остатка цистеина в пептиде с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток серина, в более основных условиях, чем условия на стадии
(b).
[Формула 1]

где указанный способ включает следующие стадии от (а) до (с):
(a) реакция группы -SH остатка производного треонина А в пептиде с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
(d) удаление защитной группы с защищенного остатка метионина.
(b) реакция группы -SMe в пептиде с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира; и
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН, в более основных условиях, чем условия на стадии (b).
(d) удаление защитной группы с защищенного остатка метионина.
(о) лигирование первого пептида, содержащего у своего С-конца аминокислотный остаток, в котором карбоксильная группа замещена α-карбокситиоэфирной группой, представленной формулой -C(=O)-SR (где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями), со вторым пептидом, содержащим у своего N-конца аминокислотный остаток, имеющий группу -SH, согласно способу лигирования с получением пептида, содержащего аминокислотный остаток, имеющий группу -SH;
(a) реакция группы -SH в пептиде, полученном на стадии (о), с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
(с) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -ОН, в более основных условиях, чем условия на стадии (b).
в котором стадия (о) представляет собой стадию лигирования первого пептида, содержащего у своего С-конца аминокислотный остаток, в котором карбоксильная группа замещена α-карбокситиоэфирной группой, представленной формулой -C(=O)-SR, где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями, со вторым пептидом, содержащим у его N-конца остаток цистеина, согласно способу лигирования с получением пептида, содержащего остаток цистеина;
в котором стадия (а) представляет собой стадию реакции группы -SH остатка цистеина в пептиде, полученном на стадии (о), с метилирующим агентом для превращения группы -SH в группу -SMe;
в котором стадия (b) представляет собой стадию реакции группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира; и в котором стадия (с) представляет собой стадию превращения промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток серина, в более основных условиях, чем условия на стадии (b).
в котором стадия (о) представляет собой стадию лигирования первого пептида, содержащего у своего С-конца аминокислотный остаток, в котором карбоксильная группа замещена α-карбокситиоэфирной группой, представленной формулой -C(=O)-SR, где R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями, со вторым пептидом, содержащим у своего N-конца остаток производного треонина, согласно способу лигирования с получением пептида, содержащего остаток производного треонина А, представленного следующей формулой (1):
[Формула 2]
в качестве аминокислотного остатка;
в котором стадия (а) представляет собой стадию реакции группы -SH остатка производного треонина А в пептиде, полученном на стадии (о), с метилирующим агентом для превращения группы -SH в группу -SMe;
в котором стадия (b) представляет собой стадию реакции группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
в котором стадия (с) представляет собой стадию превращения промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
(d) удаление защитной группы с защищенного остатка метионина.
(d) удаление защитной группы с защищенного остатка метионина, где указанный первый пептид является пептидом, не содержащим остатка цистеина, или пептидом, содержащим защищенный остаток цистеина, и указанный второй пептид является пептидом, не содержащим остатка цистеина в каких-либо участках, иных, чем его N-конец, или пептидом, чьи остатки цистеина в каких- либо участках, иных, чем его N-конец, все представляют собой защищенные остатки цистеина.
(a) реакция группы -SH в гликопептиде с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в пептид, содержащий аминокислотный остаток, имеющий группу -SH в более основных условиях, чем условия стадии (b).
в котором аминокислотный остаток, имеющий группу -SH, является остатком цистеина, и остаток цистеина гликопептида превращают в остаток серина,
в котором стадия (а) представляет собой стадию реакции группы -SH остатка цистеина в гликопептиде с метилирующим агентом для превращения группы -SH в группу -SMe;
в котором стадия (b) представляет собой стадию реакции группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции;
в котором стадия (с) представляет собой стадию превращения промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток серина, в более основных условиях, чем условия на стадии (b).
в котором аминокислотный остаток, имеющий группу -SH, является остатком производного треонина А, представленного нижеследующей формулой (1), и остаток производного треонина А гликопептида превращают в остаток треонина
[Формула 3]

в котором стадия (а) представляет собой стадию реакции группы -SH остатка производного треонина А в гликопептиде с метилирующим агентом для превращения группы -SH в группу -SMe;
в котором стадия (b) представляет собой стадию реакции группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
в котором стадия (с) представляет собой стадию превращения промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
(о) лигирование первого гликопептида, С-конец которого представлен следующей формулой:
-caxap-Asn-X-C(=O)-SR,
где caxap-Asn представляет собой аспарагин, соединенный с цепью сахара,
Х представляет собой часть, другую чем карбоксильная группа любого данного аминокислотного остатка, другого, чем пролин, и
R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями, со вторым пептидом, содержащим остаток цистеина у его N-конца, согласно способу лигирования с получением гликопептида, содержащего остаток цистеина;
(a) реакция группы -SH остатка цистеина в гликопептиде, полученном на стадии (о), с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции в форме сложного эфира;
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток серина, в более основных условиях, чем условия на стадии (b).
(о) лигирование первого гликопептида, С-конец которого представлен следующей формулой
-caxap-Asn-X-C(=O)-SR,
где сахар-Asn представляет собой аспарагин, соединенный с цепью сахара,
Х представляет собой часть, другую чем карбоксильная группа любого данного аминокислотного остатка, другого, чем пролин;
R выбран из бензильной группы, арильной группы и алкильной группы, которые могут быть замещены заместителями, со вторым пептидом, содержащим остаток производного треонина у его N-конца, согласно способу лигирования с получением гликопептида, содержащего остаток производного треонина А, представленного следующей формулой (1):
[Формула 4]

в качестве аминокислотного остатка;
(a) реакция группы -SH остатка производного треонина А в гликопептиде, полученном на стадии (о), с метилирующим агентом для превращения группы -SH в группу -SMe;
(b) реакция группы -SMe, образованной на стадии (а), с цианирующим агентом с получением промежуточного продукта реакции;
(c) превращение промежуточного продукта реакции, полученного на стадии (b), в гликопептид, содержащий остаток треонина, в более основных условиях, чем условия на стадии (b).
-caxap-Asn-X-Y-,
где caxap-Asn представляет собой аспарагин, связанный с цепью сахара, Х представляет собой любой данный аминокислотный остаток, другой, чем пролин, и
Y представляет собой остаток производного треонина А, представленный формулой (2):
[Формула 5]

Комментарии