Полипептиды, обладающие единственным ковалентно связанным n-концевым водорастворимым полимером - RU2199347C2
Код документа: RU2199347C2
Чертежи












Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к полипептидам, которые связаны на своих N-концах с единственным водорастворимым полимером. Эти полипептиды
обладают свойствами, которые придают им преимущества при использовании в качестве фармацевтических и диагностических агентов. Настоящее изобретение относится также к способам создания этих
полипептидов и связанным с ними фармацевтическим композициям и наборам.
Предпосылки изобретения
В данной заявке приводятся ссылки на различные публикации. Раскрытие этих
публикаций включено, таким образом, в виде ссылок в данную заявку для того, чтобы полнее описать известный уровень техники, в котором применимо данное изобретение.
В последние годы для ковалентной модификации терапевтически и диагностически важных полипептидов стали использовать неантигенные водорастворимые полимеры, такие как полиэтиленгликоль ("PEG"). Сообщали, например, о ковалентном присоединении PEG к терапевтическим полипептидам, таким как интерлейкины (Knauf M.J. с соавт., J. Biol. Chem. 1988, 263, 15,064; Tsutsumi Y. с соавт., J. Controlled Release 1995, 33, 447), интерфероны (Kita Y. с соавт., DrugDes. Delivery 1990, 6,157), каталаза (Abuchowski А. с соавт., J. Biol. Chem. 1977, 252, 3, 582), супероксиддисмутаза (Beauchamp С.О. с соавт. Anal. Biochem. 1983, 131, 25) и аденозиндезаминаза (Chen R. с соавт., Biochim. Biophy. Acta 1981, 660, 293), чтобы увеличить время их полужизни in vivo и/или уменьшить их иммуногенность и антигенность.
Однако такие способы страдают серьезными недостатками. В частности, в большинстве случаев PEG-молекулы присоединяются через аминогруппы на полипептидах с помощью метоксилированных PEG ("mPEG"), обладающих разными реакционноспособными составляющими. Такими полимерами являются mPEG-сукцинимидилсукцинат, mPEG-сукцинимидилкарбонат, mPEG-имидат и mPEG-цианурхлорид. Присоединение с помощью этих полимеров обычно не является специфичным, т.е. оно происходит по разным аминогруппам полипептидов случайным образом, а не исключительно по конкретной аминогруппе. Такое неспецифическое присоединение может модифицировать аминокислотные остатки в активных участках таким образом, что утрачивается биологическая активность этих полипептидов. Полученные в результате конъюгаты могут также содержать гетерогенную смесь модифицированного полипептида, который нежелателен для фармацевтического применения.
Чтобы преодолеть эти трудности, желательно сайт-специфическое присоединение полимера к полипептиду. Данный полипептид делают таким образом, чтобы сохранить биологическую активность, продлить время циркуляции в крови, уменьшить иммуногенность, увеличить растворимость в воде и повысить устойчивость к протеазному расщеплению. Сайт-специфическое присоединение (pegylation) на N-конце, боковой цепи и С-конце мощного аналога фактора, высвобождающего гормон роста, осуществляли путем твердофазного синтеза (Felix А.М. с соавт., Int. J. Peptide Protein Res. 1995, 46, 253). Так как эту специфическую фиксацию осуществляли во время сборки пептида на смоле, данный способ не может быть применен к существующему пептиду.
Используемый дополнительный способ включает в себя сайт-специфическое присоединение пептида к концам присоединенных к поверхности липосомы PEG-цепочек через реакционную альдегидную группу на N-конце, образованном путем натрийперйодатного окисления N-концевого треонина (Zalipsky S. с соавт., Bioconj. Chem. 1995, 6, 705). Однако данный способ ограничен полипептидами с N-концевыми сериновым или треониновым остатками.
Были также описаны способы по специфическому интродуцированию активированных групп на С-конце полипептида с помощью фермента (Schwartz А. с соавт. , Methods Enzymol. 1990, 184, 160; Rose К. с соавт., Bioconjugate Chem. 1991, 2, 154; Gaertner H.F. с соавт., J. Biol. Chem. 1994, 269, 7224). Обычно этими активными группами могут являться гидразид, альдегид и ароматические аминогруппы для последующего присоединения функциональных проб к полипептидам. Однако, поскольку данные способы основаны на специфичности протеаз, они требуют крайней осторожности и объем их применения ограничен.
Кроме того, сайт-специфический мутагенез представляет собой подход, который использовали для получения полипептидов для сайт-специфического присоединения полимера. WO 90/12874 описывает сайт-направленное присоединение белков, модифицированных путем вставки цистеиновых остатков или замены других остатков на цистеиновые остатки. Данная публикация описывает также получение mPEG-эритропоэтина ("mPEG-ЕРО") в реакции производного цистеин-специфичного mPEG с рекомбинантно-интродуцированным цистеиновым остатком в ЕРО. Подобным образом интерлейкин-2 присоединяли по его сайту гликозилирования после сайт-направленного мутагенеза (Goodson R.J. с соавт., Вio/Technology 1990, 8, 343).
Гликопротеины снабжены углеводами в качестве дополнительных сайтов-мишеней для модификации. Фермент пероксидаза был модифицирован с помощью PEG-диамина через его углеводную составляющую (Urrotiogoity М. с соавт., Biocata-lysis 1989, 2, 145). WO 94/28024 описывает способы получения mPEG-EPO через перйодатное окисление углевода. Данный химизм подразумевает образование гидразона путем реагирования mPEG-гидразида с альдегидными группами углеводной составляющей ЕРО. Этот вид модификации образует реакционные альдегидные группы через стадию окисления, которые потенциально могут окислять различные виды сахарных остатков в углеводной составляющей и некоторые аминокислотные остатки данного полипептида, такие как метионин. Другой недостаток этого способа происходит от гетерогенности углеводных составляющих ЕРО. ЕРО, экспрессированный в клетках яичника китайского хомячка, обладает четырьмя углеводными цепями, которые включают три N-сцепленные цепочки по аспарагину 24, 38 и 83 и одну 0-сцепленную цепочку по серину 126. В общей сложности было идентифицировано 52 разных N-сцепленных и, как минимум, 6 0-сцепленных олигосахаридных структур (Rush R.S. с соавт., Anal Chem. 1955, 67, 1442; Linsley К. В. с соавт., Anal. Biochem. 1994, 219, 207). Поэтому трудно контролировать число полимерных молекул или присоединение сайтов при модификации ЕРО или других белков через их углеводные цепочки.
Одним словом, способы в данной области по присоединению водорастворимого полимера к полипептиду страдают серьезными недостатками. Эти недостатки включают в себя следующие: (а) отсутствие точности, что касается стехиометрии и позиции присоединения; (b) необходимость осуществлять сложные и трудоемкие методики, такие как сайт-специфический мутагенез; (с) необходимость использовать твердофазный пептидный синтез одновременно с присоединением полимера вместо присоединения полимера к уже существующему полипептиду и (d) жесткое требование тождественности треонина или серина N-концевому аминокислотному остатку.
Некоторое время существовала потребность в общем методе сайт-специфического присоединения водорастворимого полимера к N-концевому аминокислотному остатку полипептида, который бы не страдал от вышеуказанных недостатков. Однако такого метода не существует.
Краткое изложение существа изобретения
В настоящем изобретении разработали две композиции. Первая композиция состоит по существу из полипептида и водорастворимого полимера, ковалентно
связанного с ним по N-концевому α-углеродному атому полипептида через гидразоновую связь или восстановленную гидразоновую связь, при условии, что (а) полимер обладает молекулярным весом
приблизительно от 200 до 200000 дальтон, (b) природная функция полипептида не ограничивается при удалении его N-концевой α-аминогруппы и (с) полипептидный N-концевой аминокислотный остаток не
представляет собой серии или треонин.
Вторая композиция состоит по существу из полипептида и водорастворимого полимера, ковалентно связанного с ним по N-концевому α-углеродному атому полипептида через оксимовую связь или восстановленную оксимовую связь, при условии, что (а) полимер обладает молекулярным весом приблизительно от 200 до 200000 дальтон и (b) природная функция полипептида не ограничивается при удалении его N-концевой α-аминогруппы.
В настоящем изобретении разработали также четыре способа ковалентного связывания водорастворимого
полимера с N-концевым α-углеродным атомом полипептида. Первый способ, который связывает полимер с углеродным атомом через гидразоновую связь, включает в себя следующие стадии:
(a)
контактирование полипептида с (i) глиоксилатным ионом или его производным в концентрации примерно от 0,1 до 2,0 М, (ii) ионом переходного металла в концентрации примерно от 10 мкМ до 1 М и (iii)
основанием Льюиса в концентрации примерно от 10 мМ до 10 М при рН примерно от 3,0 до 8,0 и температуре примерно от 0 до 100oС так, чтобы образовать трансаминированный полипептид, обладающий
N-концевой α-карбонильной группой;
(b) контактирование полученного трансаминированного полипептида при рН примерно от 1,0 до 7,5 с водорастворимым полимером, обладающим составляющей,
ковалентно связанной с ним, которая реагирует с N-концевой α-карбонильной группой трансаминированного полипептида с образованием гидразоновой связи, ковалентно связывая таким образом полимер с
N-концевым α-углеродным атомом полипептида через гидразоновую связь при условии, что полимер обладает молекулярным весом приблизительно от 200 до 200000 дальтон и природная функция полипептида
не ограничивается при удалении его N-концевой α-аминогруппы.
Второй способ, который связывает полимер с углеродным атомом через оксимовую связь, включает следующие стадии:
(а) контактирование полипептида с (i) глиоксилатным ионом или его производным в концентрации приблизительно от 0,1 до 2,0 М, (ii) ионом переходного металла в концентрации примерно от 10 мкМ до 1
М и (iii) основанием Льюиса в концентрации примерно от 10 мМ до 10 М при рН примерно от 3,0 до 8,0 и температуре примерно от 0 до 100oС так, чтобы образовать трансаминированный полипептид,
обладающий N-концевой α-карбонильной группой;
(b) контактирование полученного трансаминированного полипептида при рН примерно от 1,0 до 7,5 с водорастворимым полимером, обладающим
составляющей, ковалентно связанной с ним, которая реагирует с N-концевой α-карбонильной группой трансаминированного полипептида с образованием оксимовой связи, ковалентно связывая таким образом
полимер с N-концевым α-углеродным атомом полипептида через оксимовую связь при условии, что полимер обладает молекулярным весом примерно от 200 до 200000 дальтон и природная функция полипептида
не ограничивается при удалении его N-концевой α-аминогруппы.
Третий способ включает в себя стадии первого способа, а также дополнительную стадию восстановления гидразоновой связи, образованной на стадии (b). Четвертый способ включает в себя стадии второго способа, а также дополнительную стадию восстановления оксимовой связи, образованной на стадии (b).
В настоящем изобретении создали также фармацевтическую композицию, которая включает в себя эффективное количество первой или второй композиции настоящего изобретения и фармацевтически приемлемый носитель.
Наконец, в настоящем изобретении создали набор, используемый для получения композиций настоящего изобретения. Первый набор для получения первой композиции настоящего
изобретения включает в себя нижеследующее:
(a) глиоксилатный ион или его производное;
(b) ион переходного металла;
(c) основание Льюиса;
(d) водорастворимый полимер,
обладающий молекулярным весом примерно от 200 до 200000 дальтон и обладающий составляющей, ковалентно связанной с ним, которая реагирует с N-концевой α-карбонильной группой трансаминированного
полипептида с образованием гидразоновой связи, ковалентно связывая таким образом полимер с N-концевым α-углеродным атомом полипептида.
Второй набор, используемый для получения
второй композиции настоящего изобретения, включает нижеследующее:
(a) глиоксилатный ион или его производное;
(b) ион переходного металла;
(c) основание Льюиса;
(d)
водрастворимый полимер, обладающий молекулярным весом примерно от 200 до 200000 дальтон и обладающий составляющей, ковалентно связанной с ним, которая реагирует с N-концевой α-карбонильной
группой трансаминированного полипептида с образованием оксимовой связи, связывая таким образом полимер с N-концевым α-углеродным атомом полипептида.
Краткое описание рисунков
На фигуре 1 показана схема получения N-концевого модифицированного ЕРО. Она представляет собой двухстадийную реакцию: трансаминирование и присоединение (pegylation). Использовали четыре
производные mPEG5000 (т.е. mPEG, обладающий м. м. 5000 дальтон) с гидразинкарбоксилатной (HZC), гидразидной (HZ), семикарбазидной (SCZ) и оксиламинной функциональными группами.
На фигуре 2 показана хроматограмма гель-фильтрации mPEG-ЕРО с гидразоновой связью, образованная с использованием гидразинкарбоксилатной (HZC) составляющей, нативного ЕРО и тРЕС5000-гидразинкарбоксилата на колонке TSK G3000SWXL, (7,5 х 30 мм). Подвижная фаза представляет собой 20 мМ цитрат натрия (рН 7,0), содержащий 100 мМ NaCI.
На фигуре 3 показана матрично-ассоциированная лазерная десорбция времяпролетных масс-спектров mPEG5000-гидразинкарбоксилата, нативного ЕРО и mPEG-EPO с гидразоновой связью, образованная с использованием гидразинкарбоксилатной (HZC) составляющей.
Фигура 4 характеризует mPEG-EPO с помощью методов электрофореза: (1) 4-15% SDS-PAGE, окрашивание Кумасси; (2) вестерн-блот; (3) 4-15% SDS-PAGE, окрашивание йодом и (4) изоэлектрическое фокусирование (IFF, рН 3-7). Дорожка 1, маркеры MW или рI; дорожка 2, нативный ЕРО; дорожка 3, трансаминированный ЕРО и дорожки 4 и 5, mPEG-EPO с гидразоновой связью с использованием соответственно гидразинкарбоксилатной (HZC) и гидразидной (HZ) составляющими.
На фигуре 5 графически показаны результаты анализа ELISA для mPEG-EPO с гидразоновыми связями, образованными с помощью гидразидкарбоксилатной (HZC) и гидразидной (HZ) составляющих.
На фигуре 6 графически показаны результаты анализа клеточной пролиферации нативного ЕРО, трансаминированного ЕРО и mPEG-EPO с гидразоновыми связями, образованными с помощью гидразидкарбоксилатной (HZC) и гидразидной (HZ) составляющих.
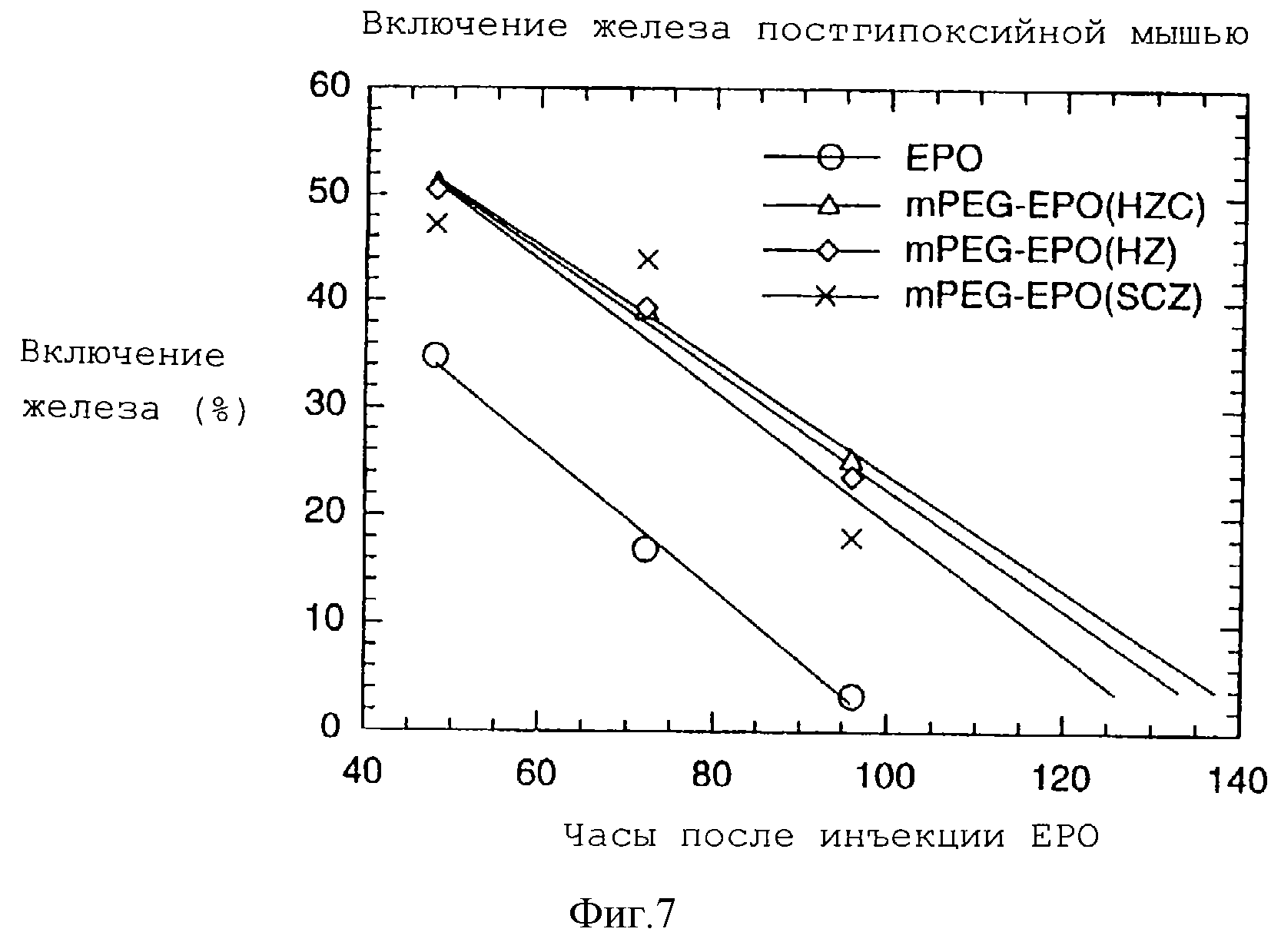
На фигуре 7 графически показаны результаты постгипоксийного биоанализа мыши по mPEG-EPO с гидразоновыми связями, образованными с помощью гидразинкарбоксилатной (HZC) и семикарбазидной (SCZ) составляющих.
Подробное описание изобретения
В настоящем изобретении созданы две композиции. Первая композиция состоит, по существу, из полипептида и водорастворимого полимера, ковалентно связанного с ним
по N-концевому α-углеродному атому данного полипептида через гидразоновую связь или восстановленную гидразоновую связь при условии, что (а) данный полимер обладает молекулярным весом примерно
от 200 до 200000 дальтон, (b) природная функция данного полипептида не утрачивается при удалении его N-концевой α-аминогруппы и (с) N-концевой аминокислотный остаток данного полипептида не
представляет собой серин или треонин.
Вторая композиция состоит, по существу, из полипептида и водорастворимого полимера, ковалентно связанного с ним по N-концевому α -углеродному атому данного полипептида через оксимовую связь или восстановленную оксимовую связь при условии, что (а) данный полимер обладает молекулярным весом примерно от 200 до 200000 дальтон и (b) природная функция данного полипептида не утрачивается при удалении его N-концевой α-аминогруппы.
В том смысле, как здесь используется, "полипептид" включает и пептиды, и протеины. "Пептид" подразумевает полипептид менее, чем из 10 аминокислотных остатков в длину, а "протеин" подразумевает 10 или более аминокислотных остатков в длину. В настоящем изобретении полипептиды могут быть природными или рекомбинантными (т. е. полученными по технологии рекомбинантной ДНК) и могут содержать мутации (например, точковую, инсерционную или делеционную мутации), а также другие ковалентные модификации (например, гликозилирование и мечение (с помощью биотина, стрептавидина, флуорацина и радиоизотопов, таких как I131). Кроме того, каждая композиция настоящего изобретения может содержать более чем один полипептид, т.е. каждая может быть мономером (один полипептид, связанный с полимером) или мультимер (два или более полипептидов, связанных с полимером или друг с другом).
Для примера, полипептиды включают моноклональные и поликлональные антитела, цитокины, такие как M-CSF и GM-CSF, лимфокины, IL-2, IL-3, факторы роста, такие как PDGF и
EGF, пептидные гормоны, такие как hGH, EPO и его производные, факторы свертывания крови, такие как Фактор VIII, иммуногены, ферменты, ингибиторы ферментов и другие лиганды. В предпочтительном варианте
осуществления композиций настоящего изобретения данный полипептид представляет собой ЕРО или его производное. ЕРО может быть естественно встречаемым или рекомбинантным. Производные ЕРО включают, но не
ограничиваются ими, следующие полипептиды:
GGLYLCRFGPVTWDCGYKGG,
GGTYSCHFGPLTWVCKPQGG,
GGDYHCRMGPLTWVCKPLGG,
VGNYMCHFGPITWVCRPGGG,
GGVYACRMGPITWVCSPLGG,
VGNYMAHMGPITWVCRPGG,
GGTYSCHFGPLTWVCKPQ,
GGLYACHMGPMTWVCQPLRG,
TIAQYICYMGPETWECRPSPKA,
YSCHFGPLTWVCK и
YCHFGPLTWVC,
а также мутанты,
перечисленные ниже в таблице 1.
Цитированные ссылки
1) Akai, К., Yamaguchi, К. and Ueda, М., Modified forms of human erythropoietin and DNA sequences encoding genes which can
express them, EP 0427 189 Al.
2) Bittorf, Т., Jaster, R. and Brock, J. (1993) FEBS Letts. 336: 133-136.
3) Results of Bunn, H.F. et al.
4) Byrne, Т.Е. and Elliott, S.G., Erythropoietin isoforms, EP 0 668 351 Al.
5) Chern, Y., Chung, T. and Sytkowski, A.J. (1991) Eur. J. Biochem. 202: 225-229.
6) Funakoshi, A. , Muta, H., Baba, Т. and Shimizu, S. (1993) Biochem. Biophys. Res. Cononun. 195: 717-722.
7) Okasinski, G. , Devries, P. J. , Mellovitz, B.S., Meuth, J.L. and Schaefer, V.G., Erythropoietin analog compositions and methods, WO 94/25055.
8) Grodberg, J., Davis, K.L. and Sytkowski, A.J. (1993) Eur. J. Biochem. 218: 597-601.
9) Results of Pulito, V. et al.
10) Shoemaker, С.В., Erythropoietin composition, U.S. 4,835,260.
В том смысле, как здесь используется, "природная функция" полипептида подразумевает его функцию до ковалентной модификации его N-концевой α-аминогруппы. Природные функции включают, например, ферментативную активность, связывание рецептора (например, антител), связывание лиганда и иммуногенность.
Способы настоящего изобретения, описанные более полно ниже, вызывают потерю N-концевой α-аминогруппы ковалентно модифицированного полипептида. В соответствии с этим данный полипептид должен обладать первичной структурой, такой что его природная функция после ковалентной модификации сохраняется и не может быть утрачена. Природная функция полипептида утрачивается при удалении его N-концевой α-аминогруппы, если такое удаление уменьшает более чем на 99% способность полипептида осуществлять свою природную функцию. В одном варианте осуществления настоящего изобретения указанное удаление не уменьшает способность данного полипептида осуществлять свою природную функцию более чем на 90%. В предпочтительном варианте осуществления настоящего изобретения указанное удаление не уменьшает способность данного полипептида осуществлять свою природную функцию более чем на 50%.
В том смысле, как здесь используется, "гидразоновая связь" представляет собой связь, включающую ковалентную структуру NH-N=C, "оксимовая связь" представляет собой связь, включающую ковалентную структуру О-N=C, "восстановленная гидразоновая связь" представляет собой связь, включающую ковалентную структуру NH-NH-C, а "восстановленная оксимовая связь" представляет собой связь, включающую ковалентную структуру О-NH-C. Здесь созданы соединения, содержащие восстановленную гидразоновую и оксимовую связи, поскольку эти связи обладают повышенной химической стабильностью.
Как рассмотрено выше, в данной области известны способы для связывания водорастворимых полимеров с N-концевым α-углеродным атомом полипептида через гидразоновую связь при условии, что N-концевой аминокислотный остаток представляет собой серин или треонин. Эти известные способы не пригодны в отношении полипептидов, обладающих каким-либо иным N-концевым остатком. Хотя эти известные способы коренным образом отличаются от способов настоящего изобретения, они результативны для N-концевого серина и треонина полипептидов, обладающих полимерной связью по N-концевому α-углеродному атому через гидразоновую связь. По этой причине первая композиция настоящего изобретения не включает полипептид, связанный с полимером через гидразоновую связь, где N-концевой аминокислотный остаток полипептида представляет собой серин или треонин.
Водорастворимые полимеры, используемые в настоящем изобретении, включают, не ограничиваясь ими, (а) декстран и производные декстрана, включающие декстрансульфат, поперечно сшитый декстрин и карбоксиметилдекстрин; (b) целлюлозу и производные целлюлозы, включающие метилцеллюлозу и карбоксиметилцеллюлозу; (с) крахмал и декстрины и его производные; (d) полиалкиленгликоль и его производные, включая PEG, mPEG, гомополимеры PEG, гомополимеры полипропиленгликоля, сополимеры этиленгликоля с пропиленгликолем, где указанные гомополимеры и сополимеры представляют собой незамещенные или замещенные по одному концу алкильной группой; (е) гепарин и фрагменты гепарина; (f) поливиниловый спирт и поливинилэтилэфиры; (д) поливинилпирролидон; (h) а, b-поли[(2-гидроксиэтил)-DL-аспартамид и (i) полиоксиэтилированные полиолы. Эти полимеры могут быть линейными, разветвленными или звездообразными с широким диапазоном молекулярных весов. В предпочтительном варианте осуществления настоящего изобретения полимер представляет собой mPEG.
При использовании композиций настоящего изобретения в качестве фармацевтических препаратов полимер не является токсичным. Более того, если говорят, что указанный полимер обладает данным молекулярным весом, этот молекулярный вес является лишь приблизительным, отражающим средний молекулярный вес популяции полимерных молекул, отличающихся друг от друга по числу субъединиц, представленных в каждой молекуле.
В одном варианте осуществления настоящего изобретения PEG или его производные обладают молекулярным весом примерно от 700 до 20000 дальтон. В предпочтительном варианте осуществления настоящего изобретения PEG или его производные обладают молекулярным весом около 5000 дальтон. Также в предпочтительном варианте осуществления композиций настоящего изобретения полипептид представляет собой ЕРО, а полимер представляет собой mPEG, обладающий молекулярным весом около 5000 дальтон.
В настоящем изобретении создали также четыре способа ковалентного связывания водорастворимого полимера с N-концевым α-углеродным атомом
полипептида. Первый способ, который связывает полимер с указанным углеродным атомом через гидразоновую связь, включает следующие стадии:
(a) контактирование полипептида с (i) глиоксилатным
ионом или его производным в концентрации примерно от 0,1 до 2,0 М, (ii) ионом переходного металла в концентрации примерно от 10 мкМ до 1 М и (iii) основанием Льюиса в концентрации примерно от 10 мМ до
10 М при рН примерно от 3,0 до 8,0 и температуре примерно от 0 до 100oС так, чтобы образовать трансаминированный полипептид, обладающий N-концевой α-карбонильной группой;
(b) контактирование полученного трансаминированного полипептида при рН примерно от 1,0 до 7,5 с водорастворимым полимером, обладающим составляющей, ковалентно связанной с ним, которая реагирует с
N-концевой α-карбонильной группой трансаминированного полипептида с образованием гидразоновой связи, ковалентно связывая таким образом полимер с N-концевым α-углеродным атомом
полипептида через гидразоновую связь при условии, что полимер обладает молекулярным весом примерно от 200 до 200000 дальтон и природная функция полипептида не утрачивается при удалении его N-концевой
аминогруппы.
Второй способ, который связывает полимер с углеродным атомом через оксимовую связь, включает следующие стадии:
(a) контактирование полипептида с (i) глиоксилатным
ионом или его производным в концентрации примерно от 0,1 до 2,0 М, (ii) ионом переходного металла в концентрации примерно от 10 мкМ до 1 М и (iii) основанием Льюиса в концентрации примерно от 10 мМ до
10 М при рН примерно от 3,0 до 8,0 и температуре примерно от 0 до 100oС так, чтобы образовать трансаминированный полипептид, обладающий N-концевой α-карбонильной группой;
(b) контактирование трансаминированного полипептида при рН примерно от 1,0 до 7,5 с водорастворимым полимером, обладающим составляющей, ковалентно связанной с ним, которая реагирует с N-концевой
α-карбонильной группой трансаминированного полипептида с образованием оксимовой связи, ковалентно связывая таким образом, полимер с N-концевым α-углеродным атомом полипептида через
оксимовую связь при условии, что полимер обладает молекулярным весом примерно от 200 до 200000 дальтон и природная функция полипептида не утрачивается при удалении его N-концевой α
-аминогруппы.
Третий способ включает в себя стадии указанного первого способа, а также дополнительную стадию восстановления гидразоновой связи, образованной на стадии (b). Четвертый дополнительный способ включает в себя стадии указанного второго способа, а также дополнительную стадию восстановления оксимовой связи, образованной на стадии (b). Восстановительную стадию осуществляли путем использования, например, натрийборогидрида (NaBH4) и натрийцианоборогидрида (МаВН3СN) в соответствии с известными способами.
Производные глиоксилатного иона включают, не ограничиваясь ими, глиоксиламидный и фенилглиоксильный ионы. Ионы переходного металла включают в себя, не ограничиваясь ими, ионы меди, никеля, кобальта или цинка. Льюисовские основания включают в себя, но не ограничиваются ими, ацетат и пиридин.
Составляющие, которые реагируют с N-концевой α-карбонильной группой трансаминированного полипептида с образованием гидразоновой связи, включают, но не ограничиваются ими, гидразинкарбоксилат, гидразин, семикарбазид, гидразид, тиосемикарбазид, дигидразид угольной кислоты, карбазид, тиокарбазид и арилгидразид. Водорастворимые полимеры с этими составляющими, ковалентно связанными с ними, коммерчески доступны. Кроме того, составляющие, которые реагируют с N-концевой α-карбонильной группой трансаминированного полипептида с образованием оксимовой связи, включают, но не ограничиваются, оксиламин. Водорастворимые полимеры с оксиламином (а также с другими оксимобразующими составляющими), ковалентно связанные с ним, коммерчески доступны.
В предпочтительном варианте способов согласно изобретению рН на стадии (а) составляет примерно от 5,0 до 7,0, рН на стадии (b) составляет примерно от 3,0 до 5,0, а белок представляет собой ЕРО или его производное.
В одном варианте способов согласно изобретению полимер представляет собой PEG или его производное. В другом варианте PEG или его производное обладает молекулярным весом примерно от 700 до 20000 дальтон. В предпочтительном варианте PEG или его производное обладает молекулярным весом порядка 5000 дальтон.
В одном варианте осуществления первого способа составляющая, связанная с полимером, которая реагирует с данным трансаминированным полипептидом, представляет собой гидразинкарбоксилат. В предпочтительном варианте полипептид представляет собой ЕРО, полимер представляет собой mPEG, обладающий молекулярным весом около 5000 дальтон, а составляющая, ковалентно связанная с полимером, представляет собой гидразинкарбоксилат.
В одном варианте осуществления второго способа составляющая, связанная с полимером, который реагирует с трансаминированным полипептидом, представляет собой оксиламин.
В каждом способе настоящего изобретения время предпочтительного контактирования на стадии (а) составляет от 20 минут до 2-х часов, а на стадии (b) время предпочтительного контактирования и температура составляют соответственно от 10 минут до 50 часов и от 4oС до комнатной температуры.
У некоторых полипептидов N-конец полипептида "утоплен", т.е. не подвергается действию растворителей или реагентов, когда полипептид представлен в своей нативной конформации. Реагенты, такие как тетраметилмочевина или мочевина, могут применяться для разворачивания такого полипептида с тем, чтобы дать возможность подвергнуть его N-концевой остаток нужным реакциям способов настоящего изобретения.
В настоящем изобретении предложена также фармацевтическая композиция, которая включает эффективное количество первой или второй композиции и фармацевтически приемлемый носитель. Для примера, фармацевтическая композиция согласно изобретению может включать с себя количество mPEG-EPO настоящего изобретения, эффективного для лечения субъекта, страдающего анемией.
Фармацевтически приемлемые носители также хорошо известны специалистам в данной области и включают в себя, но не ограничиваются ими, 0,01-0,1 М и предпочтительно 0,05 М фосфатного буфера или 0,8% физиологического раствора. Кроме того, такие фармацевтически приемлемые носители могут быть водными или неводными растворами, суспензиями и эмульсиями. Примеры неводных растворителей представляют собой пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и инъецируемые органические эфиры, такие как этилолеат. Водные носители включают в себя воду, спиртово-водные растворы, эмульсии или суспензии, включающие в себя физиологический раствор и забуференную среду. Парентеральные носители включают в себя раствор хлорида натрия, декстрозу Рингера, декстрозу или хлорид натрия, молочнокислый раствор Рингера/или нелетучие масла. Внутривенные носители включают в себя жидкие и питательные подкрепляющие добавки, электролитные подкрепляющие добавки, такие которые основаны на декстрозе Рингера, и тому подобное. Могут также присутствовать консерванты и другие добавки, такие, например, как противомикробные, антиоксиданты, хелатирующие агенты, инертные газы и тому подобное.
Наконец, в настоящем изобретении созданы наборы для применения в получении композиций согласно изобретению. Первый набор для получения первой композиции настоящего
изобретения включает в себя нижеследующее:
a) глиоксилатный ион или его производное;
b) ион переходного металла;
c) основание Льюиса;
d) водорастворимый полимер,
обладающий молекулярным весом примерно от 200 до 200000 дальтон и обладающий составляющей, ковалентно связанной с ним, которая реагирует с N-концевой α-карбонильной группой трансаминированного
полипептида с образованием гидразоновой связи, ковалентно связывающейся таким образом с полимером по N-концевому α-углеродному атому полипептида.
Второй набор для применения в
получении второй композиции настоящего изобретения включает в себя нижеследующее:
a) глиоксилатный ион или его производное;
b) ион переходного металла;
c) основание Льюиса;
d) водорастворимый полимер, обладающий молекулярным весом примерно от 200 до 200000 дальтон и обладающий составляющей, ковалентно связанной с ним, которая реагирует с N-концевой α
-карбонильной группой трансаминированного полипептида с образованием оксимовой связи, ковалентно связывающейся таким образом с полимером по N-концевому α-углеродному атому полипептида.
Реагенты в этих наборах могут быть упакованы в заранее предопределенном количестве и могут находиться в отдельных ячейках. С другой стороны, некоторые реагенты могут содержаться в одной и той же ячейке в соответствии со способами настоящего изобретения. Наконец, наборы могут дополнительно включать в себя реагенты для образования восстановленной гидразоновой и оксимовой связей в соответствии со способами настоящего изобретения, а также подходящие буферы и реакционные сосуды.
Настоящее изобретение будет лучше всего понято при ссылке на нижеследующие экспериментальные примеры, однако специалистам в данной области необходимо иметь ввиду, что конкретные эксперименты детализированы лишь для иллюстрации настоящего изобретения, которое наиболее полно описано в следующей за ними формуле изобретения.
Экспериментальные примеры
1. Получение трансаминированного ЕРО
5 мг ЕРО в 20 мМ цитрате натрия (рН 6,9) и 100 мМ NaCl
меняли на 100 мМ натрийацетатного буфера (рН 7,0) с использованием Centricon-10 (Amicon, Beverly, МА). Конечные концентрации доводили до 1 мг/мл ЕРО 2 М ацетатом натрия, 0,4 М уксусной кислоты, 0,1 М
глиоксалевой кислоты и 10 мМ сульфатом меди (рН 5,5) (фигура 1). Данную реакцию осуществляли в течение 2-х часов при комнатной температуре и останавливали добавлением 100 мл 0,5 М ЭДТА.
Трансаминированный ЕРО выделяли очисткой на колонке с Сефадексом G-25 (Pharmacia, Piscataway, NJ) с использованием 100 мМ натрийацетатного (рН 4,5) буфера.
Продолжительность трансаминирования определяли с помощью 2,4-динитрофенилгидразина, как описано в литературе (Fields R. с соавт., Biochem. J., 1971, 121, 587). Экстинкцию при 37 нм измеряли после первых нескольких минут и через один час. Разница в абсорбции пропорциональна количеству карбонильных групп, представленных в молекуле ЕРО. Полученный трансаминированный ЕРО подвергали также аминокислотному анализу в системе ABI 420Н (Applied Biosystems, Foster City, CA) с использованием предваряющей PITC-химии на колонке. Полученные результаты свидетельствуют, что лизиновые остатки, т.е. не N-концевые остатки, не трансаминировались.
2. Получение mPEG-EPO с mPEG-гидразинкарбоксилатом
Трансаминированный ЕРО (1 мг) в 100 мМ ацетате натрия (рН 4,5) доводили 0,5 М хлоридом натрия до
конечного объема 1 мл, к которому добавляли 10 мг mРЕG5000-гидразинкарбоксилата. Данную реакционную смесь перемешивали в течение 40 часов при комнатной температуре и выделяли очисткой на колонке с
Сефакрилом S-200 (Pharmacia, Piscataway, NJ) с использованием 20 мМ натрийцитратного (7,0) буфера, содержащего 100 мМ NaCl. Кроме того, к данной реакционной смеси может быть добавлен 0,1% SDS для
увеличения конъюгатного выхода.
При гель-проникающей хроматографии конъюгат выявил значительно увеличенный молекулярный вес в сравнении с ЕРО и mРЕG5000-гидразинкарбоксилатом (фигура 2).
Матрично-ассоциированную лазерно-десорбционную масс-спектрометрию (Finnigan-MAT LaserMAT 2000, линейный времяпролетный) использовали для характеристики mPEG-EPO для определения молекулярного веса (фигура 3). mPEG5000-гидразинкарбоксилат показывает ион при m/z 5157,4. ЕРО показывает двухзарядный мономер (m/z 14604), однозарядный мономер (m/z 28569), димер (m/z 57208) и тример (m/z 85284). Аналогично, mPEG-EPO показывает двухзарядный мономер (m/z 17092), однозарядный мономер (m/z 34279), димер (m/z 69071) и тример (m/z 102955).
Спектр кругового дихроизма (CD) (Jobin-YVON CD6, Dichrograph Spectrometer Instruments, SA, Edison, NJ) mPEG-EPO показывает, что данный белок сохраняет свернутую α-спиральную структуру, присутствующую в нативном ЕРО (данные не приведены). Данный результат означает, что молекула PEG на N-конце ЕРО не нарушает его вторичную структуру.
3. Получение mPEG-EPO с mPEG-гидразидом, mPEG-семикарбазидом и
оксиламином
Трансаминированный ЕРО (1 мг) в 100 мМ ацетате натрия (рН 4,5) доводили до 0,5 М хлоридом натрия, 0,1% SDS до конечного объема 1 мл и добавляли 10-20 мг mРЕG5000-гидразида,
семикарбазида или оксиламина. Реакционную смесь перемешивали в течение 40 часов при комнатной температуре и выделяли очисткой на колонке с Сефакрилом S-200 (Pharmacia, Piscataway, NJ) с использованием
20 мМ натрийцитратного (7,0) буфера, содержащего 100 мМ NaCl.
Конъюгаты mPEG-EPO анализировали с помощью SDS-PAGE (Bio-RAD, Herculus, СА) различными методами: окраской Кумасси (специфичной для белков), вестерн-блоттинг (специфичным для ЕРО) и окраской йодом (специфичной для PEG). Миграционное расстояние конъюгатов mPEG-EPO с большим молекулярным весом в SDS-PAGE меньше, чем у нативного ЕРО. Полученный образец изоэлектрического фокусирования свидетельствует, что изоэлектрическая точка (рI) ЕРО существенно не меняется при модификации. Однако трансаминированный ЕРО несколько более закислен, чем нативный ЕРО и mPEG-EPO.
Так как полосы ЕРО и mPEG-EPO хорошо разделялись в SDS-PAGE, данную методику можно было использовать для эффективного мониторинга реакции конъюгации. Наблюдали, что реакция конъюгации достигала >95% полноты при использовании mРЕG5000-гидразидкарбоксилата, тогда как данная реакция составляла лишь около 20% полноты при использовании mРЕG5000-гидразида, mРЕG5000-семикарбазида или mPEG5000-оксиламина. Следовательно, гидразидкарбоксилатная составляющая является более реакционноспособной в отношении карбонильных групп, чем гидразидная, семикарбазидная или оксиламинная составляющая.
4. Реакционная способность mPEG-EPO с антителом анти-ЕРО
Антигенность mPEG-EPO изучали с использованием набора
QuantikineTM IVDTM ЕРО ELISA (R& D systems, Minneapolis, MN). Данный анализ состоит из плашки для микротитрования, покрытой моноклональным антителом к ЕРО. ЕРО или
mPEG-EPO давали возможность реагировать с покрытой плашкой. После отмывания плашки добавляли конъюгат поликлонального антитела анти-ЕРО и пероксидазы хрена. После удаления избытка конъюгата в лунки
добавляли хромоген и окисляли с помощью ферментативной реакции с образованием комплекса, окрашенного в голубой цвет. Абсорбцию данного комплекса измеряли при 450 нм.
Результаты, полученные в анализе ELISA, для mPEG-EPO с гидразоновой связью, образованной из гидразинкарбоксилата (HZC) и гидразида (HZ), представлены на фигуре 5. Эти данные свидетельствуют о том, что даже одна молекула PEG, присоединенная по N-концу ЕРО, существенно уменьшает сродство моноклональных антител к связыванию с ЕРО, что, вероятно, обусловлено стерическим препятствием.
5.
Активность mPEG-EPO in vitro
Биологическую активность mPEG-EPO in vitro оценивали на основании анализа клеточной пролиферации с использованием клеток FDC-P1/HER, мышиной гемопоэтической
клеточной линии. Эта линия клеток экпрессирует рецептор ЕРО, и ее рост зависит от ЕРО. После того как клетки растили в отсутствие ЕРО в течение 24 часов, к этим клеткам добавляли ЕРО или mPEG-EPO.
Клетки инкубировали в течение 42 часов, а затем к клеткам добавляли меченный по тритию тимидин. Через 6 часов определяли клеточный рост по включению тимидина.
Результаты анализа клеточной пролиферации, полученные для трансаминированного ЕРО и mPEG-EPO с гидразоновой связью, образованной из гидразинкарбоксилата (HZC) и гидразида (HZ), представлены на фигуре 6. Трансаминированный ЕРО демонстрирует полную биологическую активность, сопоставимую с нативным ЕРО, что определено по его ED50. Образцы mPEG-EPO с гидразоновыми связями, образованными из гидразинкарбоксилата (HZC) и гидразида (HZ), сохраняют соответственно лишь 38,5 и 25% активности, как определено по их ED50. Полученные данные свидетельствуют о том, что одна молекула PEG, присоединенная по N-концу ЕРО, существенно уменьшает сродство ЕРО к своему рецептору, что, вероятно, обусловлено стерическим препятствием.
6. Активность mPEG-EPO in vivo
Активность mPEG-EPO оценивали с помощью постгипоксийного биоанализа мыши (Coates P.M. с соавт., Nature, 1961, 191, 1065). Эндогенное образование мышиных эритроцитов супрессировали с помощью
полицитемии, полученной при воздействии пониженного давления. ЕРО или конъюгат mPEG-EPO инъецировали на уровне 1 единицы/мышь. Железо-59 вводили через 48 часов после инъекции ЕРО или mPEG-EPO.
Включение железа-59, которое указывает на образование новых эритроцитов крови, измеряли через 48, 72 и 96 часов после введения ЕРО или mPEG-EPO.
Результаты постгипоксийного биоанализа мыши для mPEG-EPO с гидразоновой связью, образованной с гидразинкарбоксилатом (HZC), гидразидом (HZ) и семикарбазидом (SCZ), представлены на фигуре 7. Образцы mPEG-EPO обнаруживают высокую активность in vivo, а также более продолжительную активность в сравнении с нативным ЕРО. Результаты in vivo свидетельствуют о том, что образцы mPEG-EPO обладают более продолжительным временем кровотока in vivo и поддерживают высвобождение ЕРО в течение кровотока.
7. Получение mPEG-фибринового димера 17-29
Фибриновый димер 17-29 обладает нижеследующей структурой:

2 мг фибринового димера 17-29 растворяли в 0,5 мл 2 М ацетата натрия, 0,4 М уксусной кислоты, 0,1 М глиоксалевой кислоты и 10 мМ сульфата меди (рН 5,5). Данную реакцию осуществляли в течение 2-х часов при комнатной температуре и останавливали добавлением 20 мл 0,5 М ЭДТА. Трансаминированный фибриновый димер выделяли очисткой с помощью колонки с Сефадексом G-10 (Pharmacia, Piscataway, NJ) с использованием 100 мМ натрийацетатного (рН 4,5) буфера.
Трансаминированный фибриновый димер обнаруживает существенное уменьшение Gly в аминокислотном анализе, что свидетельствует о трансаминировании N-концевого Gly.
1 мг трансаминированного фибринового димера в 0,5 мл 100 мМ ацетата натрия (рН 4,5) добавляли к 10 мг mPEG5000-гидразинкарбоксилата. Реакционную смесь перемешивали в течение 24 часов при комнатной температуре. mPEG-фибриновый димер выделяли очисткой с помощью анионобменной хроматографии на колонке НЕМА IEC BIO CM (Alltech). Подвижная фаза А представляет собой 20 мМ ацетата натрия (рН 5,5). Подвижная фаза В представляет собой 0,2 М MOPS, 0,05 М одноосновного фосфата калия и 0,25 М двухосновного фосфата калия (рН 7,5). Градиент составлял 100% А в течение 5 минут, затем 0-100% В в течение 25 минут. Новый пик на 14,5 минутах, возникающий перед немодифицированным фибриновым димером (18 минут), собирали для дальнейшего анализа. Масс-спектр лазерной десорбции обнаруживает ион на m/z 8070,9, который доказывает, что одна молекула PEG была присоединена к фибриновому димеру (m/z 29,86,8).
Реферат
В настоящем изобретении созданы композиции, по существу состоящие из полипептида и водорастворимого полимера, ковалентно связанного с ним по N-концевому α-углеродному атому через гидразоновую или восстановленную гидразоновую связь. В настоящем изобретении созданы также способы изготовления композиций настоящего изобретения, фармацевтических композиций, включающий их же, и наборы, используемые для их получения. Изобретение позволяет точнее и проще осуществлять сложные методики получения ковалентно связанных полипептидов. 4 c. и 8 з.п. ф-лы, 7 ил., 1 табл.

Комментарии