Способ синтеза терапевтических пептидов - RU2625793C2
Код документа: RU2625793C2
Чертежи


Описание
Настоящее изобретение относится к новому способу крупномасштабного синтеза терапевтических пептидов, содержащих неприродные или рукотворные аминокислоты. Указанный способ является масштабируемым до крупных объемов способом, и позволяет осуществлять эффективное по себестоимости производство пептидов высокой степени чистоты.
Твердофазный пептидный синтез (SPPS) представляет собой чрезвычайно успешный способ, впервые предложенный Меррифильдом в 1963 г. (Merrifield, R. B., J. Amer. Chem. Soc, 1963, 85:2149-54). С тех пор таким способом было синтезировано множество пептидов. Обзор способов, которые использовали до химического синтеза пептидов и белков, представлен у Кента, Kent, S. B. H., Ann. Rev. Biochem., 1988, 57:957-89. Твердофазный синтез позволяет синтезировать природные пептиды, которые трудно экспрессировать в бактериях, встраивать неприродные или синтетические аминокислоты, модифицировать пептидный остов и синтезировать D-белки, содержащие D-аминокислоты.
Используют две стратегии для сборки пептидных цепей способом твердофазного синтеза: 1) поэтапный твердофазный синтез и 2) способ твердофазной конденсации фрагментов. При поэтапном SPPS, C-концевая аминокислота в N-α-защищенной форме, и при необходимости, реакционноспособное производное с защищенными боковыми цепями, ковалентно связывают или непосредственно или с использованием подходящего линкера с ʺтвердымʺ носителем, например, полимерной смолой, обычно набухшей в органическом растворителе. N-α-защитную группу удаляют, и последующие защищенные аминокислоты добавляют поэтапно. По достижении необходимой длины пептидной цепи, защитные группы боковых цепей удаляют, и пептид отщепляют от смолы. Процесс отщепления/удаления защитных групп можно осуществить на отдельных этапах или одновременно. В способе твердофазной конденсации фрагментов, целевую последовательность собирают последовательной конденсацией фрагментов на твердом носителе, используя защищенные фрагменты, полученные поэтапным SPPS.
Одна из форм SPPS основана на использовании флуоренилметоксикарбонила (или ʺFmocʺ) для временной защиты α-аминогрупп. Используя такой способ, Fmoc группу ковалентно связывают с аминогруппой для подавления ее нуклеофильности. C-концевую аминокислоту ковалентно связывают со смолой, используя линкер. Затем Fmoc группу удаляют основанием, таким как пиперидин. Это высвобождает аминогруппу, которая затем оказывается доступной для реакции с активированной аминокислотой. Реакции ведут до завершения, используя избыток (обычно двух-четырех-кратный) активированной аминокислоты. После каждого этапа удаления защитных групп и присоединения, осуществляют одну или более из промывок для удаления избытка реагентов. Отщепление пептидов от смолы с удалением защитных групп боковых цепей можно осуществить, используя ацидолиз, используя раствор кислоты, такой как трифторуксусная кислота (TFA). Обычной практикой является добавление дополнительных химических соединений, называемых ʺакцепторамиʺ, таких как триизопропилсилан (TIPS), триэтилсилан (TES), фенол, анизол, тиоанизол, вода, 1,2-этандитиол (EDT), 1-додекантиол, дитиотреитол (DTT) и индол, с кислотой в отщепляющей смеси для взаимодействия с высвобожденными защитными группами боковых цепей, тем самым, препятствуя повторному присоединению указанных высвобожденных групп к отщепленным пептидам.
Аминокислоты содержат реакционноспособные фрагменты на N- и C-концах, что облегчает присоединение аминокислот во время синтеза. Кроме того, реакционноспособные функциональные группы боковых цепей, существующие у большинства аминокислот, могут взаимодействовать со свободными концами или группами других боковых цепей в процессе синтеза и удлинения пептидов и негативно влиять на выход и степень чистоты. Для облегчения синтеза соответствующих аминокислот с минимальной реакционной способностью боковых цепей, используют химические группы, именуемые как ʺзащитные группыʺ, для связывания со специфическими функциональными группами аминокислот для ʺблокированияʺ или ʺзащитыʺ указанных функциональных групп от неспецифических реакций. Защитные группы боковых цепей известны как постоянные или полупостоянные защитные группы, так как они могут выдержать множество циклов химических обработок в процессе синтеза, и обычно их удаляют сильными кислотами только после завершения синтеза пептида.
Вышеописанные существующие в настоящее время стратегии нежелательны для производства терапевтических пептидов в промышленных масштабах, так как используемые в них смолы требуют для удаления пептидов высоких концентраций кислоты для отщепления пептидов от полимерной смолы. Помимо проблем безопасности, связанных с использованием больших количеств крайне коррозивных материалов, для возможности их использования может потребоваться специальное оборудование. Кроме того, использование высоких концентраций сильных кислот для отщепления пептидов и удаления защитных групп может привести к значительной деградации целевых пептидов, приводя к низким выходам и/или к образованию новых примесей в результате экспонирования пептидов сильной кислоте в течение промежутка времени, необходимого для отщепления и обработки в нужном количестве. Такие примеси могут включать продукты дегидратации или окисления или примеси, образовавшиеся в результате присоединения всех или части смолы-линкеров к пептиду - так как указанные примеси затем будет трудно удалить. Таким образом, существует необходимость в создании эффективного крупномасштабного способа получения терапевтических пептидов.
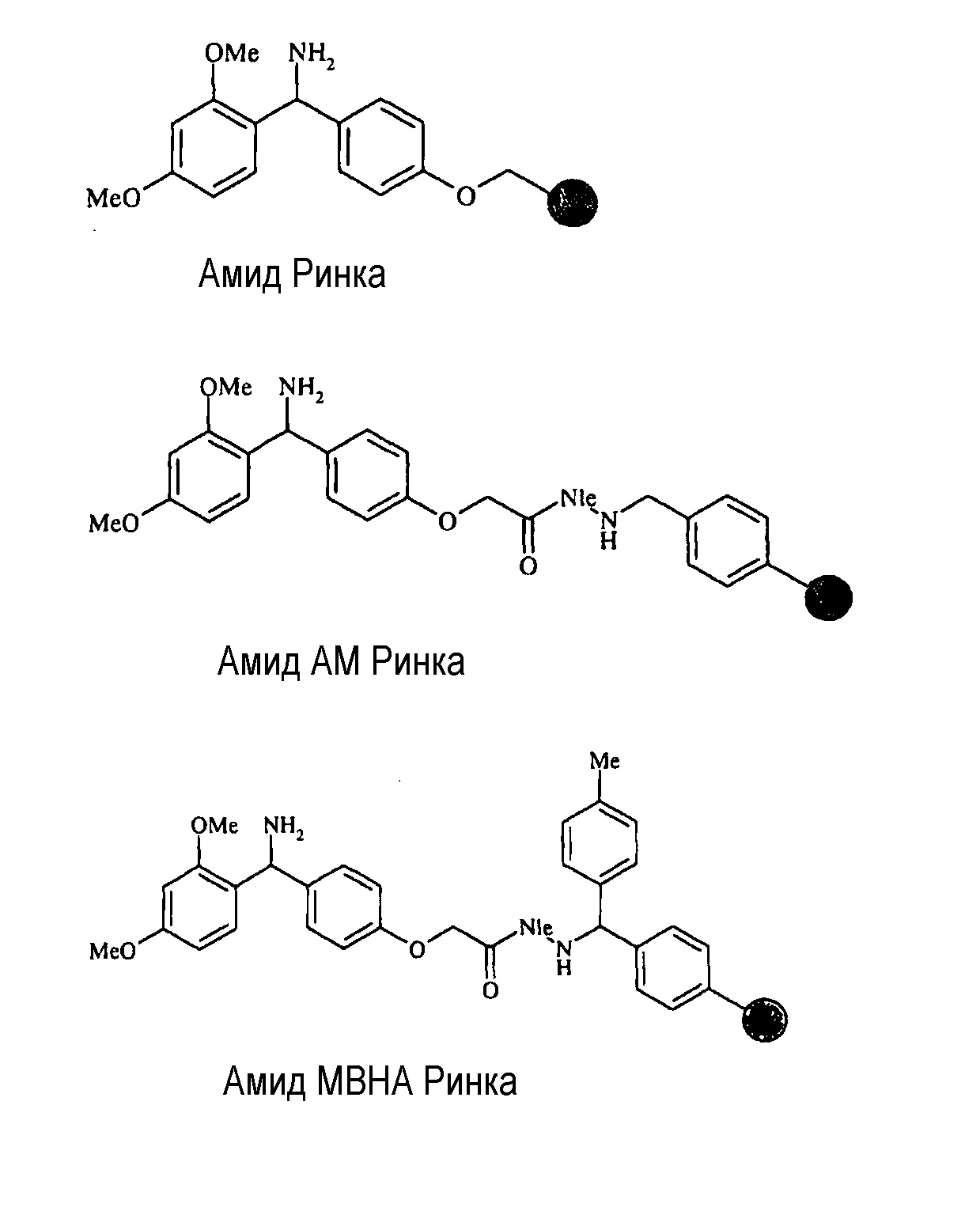
Как было указано ранее, твердофазный пептидный синтез инициируют на ʺтвердомʺ носителе или ʺякореʺ. Такие ʺносителиʺ в промышленности называют ʺсмоламиʺ. Смолы могут быть получены из полистирола или других полимерных материалов, таких как полимеры этиленоксида, например, смолы на основе ПЕГ, или смеси обоих, например, ʺгибридныеʺ или ПЕГ-полистирольные смолы. Обычно используемые смолы для получения пептидных амидов способом Fmoc SPPS включают смолы на основе полистирола, комбинированные с линкерами, подходящими для высвобождения пептидных амидов с полностью удаленными защитными группами после обработки высококонцентрированными кислотами. Обычно используемые смолы включают амидные смолы Ринка, например, амидную смолу Ринка, амидную MBHA смолу Ринка и амидную AM смолу Ринка. Амидные смолы Ринка высвобождают пептидные амиды с полностью удаленными защитными группами после обработки отщепляющим коктейлем с высоким процентом об/об кислоты - например, обычно используют 80-95% об/об трифторуксусной кислоты (TFA).
В 1987, новую кислотно-лабильную смолу для твердофазного синтеза C-концевых амидов предложил Зибер (Tetrahedron Lett., 1987, 28(19):2107-10). В этой смоле используется 9-ксантенильная группа с -OCH2- группой, введенной между указанной ксантенильной группой и полистиролом, для повышения кислотной лабильности. Отщепление пептидных амидов от такой смолы осуществляют очень мягким ацидолизом. В статье раскрыт синтез двух пептидов на указанной смоле - первого без защитных групп боковых цепей (Z-Val-Gly-Ala-Pro-NH2), где отщепление от смолы в масштабе 0,5 г осуществляют, прокачивая кислотную отщепляющую смесь (TFA:l,2-дихлорэтан 2:98 об/об) через смолу в стеклянной колонке; второй пептид (α-MSH), пептид из 13 аминокислот, содержащий защитные группы боковых цепей (трет-бутил, Trt, Mtr и Boc), отщепляют от смолы, прокачивая кислотную отщепляющую смесь (TFA/l,2-дихлорэтан -/l,2-этандитиол 2:98:0,1) через колонку со смолой. Два следующих этапа с использованием высоких концентраций кислоты и нагревания (TFA/вода 9:1 при 30°C, затем 95% TFA с акцепторами при 50°C) требуют удаления всех защитных групп боковых цепей.
Хотя в вышеприведенной статье это и не указано непосредственно, основное преимущество амидной смолы Зибера (по мере того, как стала известна ксантенильная смола) состояло в получении пептидов с полностью защищенными боковыми цепями для последующего использования в реакциях конденсации фрагментов. Этого можно достичь, используя низкие проценты об/об кислоты в отщепляющем коктейле - обычно 1-5% об/об. По данным коммерческих поставщиков (Novabiochem®, Merck KGaA), смола Зибера представляет собой ʺ[a] гипер кислотно-лабильный линкер (смола) для FMOC SPPS защищенных пептидных амидов способом мягкого отщепления 1% TFA.ʺ
Было обнаружено, что используя амидную смолу Зибера скомбинированную с Fmoc химией и a отщепляющим раствором, используя определенные концентрации трифторуксусной кислоты (TFA) (например, выше 10% об/об), можно синтезировать пептидные амиды с полностью удаленными защитными группами практично и в крупном масштабе (масштаб кг). Этот способ получения пептидных амидов с полностью удаленными защитными группами превосходит способ с использованием амидных смол Ринка, так как:
(i) используя указанный способ, можно достигать больших выходов продукта
(ii) используя указанный способ, можно получать пептиды с более высокой степенью чистоты, что позволяет облегчить последующую очистку
(iii) способ позволяет снизить расход исходных материалов и растворителей, и поэтому является более стоимостно-эффективным способом производства
(iv) способ является надежным и воспроизводимым способом, как в малых, так и в крупных масштабах, что позволяет облегчить масштабирование процесса.
В настоящем изобретении предложен новый способ промышленного синтеза терапевтических пептидов, который включает поэтапную твердофазную Fmoc-химию. В одном аспекте в настоящем изобретении предложен способ синтеза терапевтических пептидов, включающий последовательные стадии:
(a) набухания Fmoc-смолы Зибера (называемой также амидной смолой Зибера или Fmoc амидной смолой Зибера) в диполярном апротонном растворителе;
(b) удаления защитных Fmoc групп, используя раствор пиперидина в диполярном апротонном растворителе;
(c) промывки указанной смолы после удаления Fmoc защитных групп диполярным апротонным растворителем;
(d) активации Fmoc-аминокислоты для присоединения к смоле, у которой удалены защитные группы, путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в диполярном апротонном растворителе и последующего добавления основания и перемешивания;
(e) добавления раствора активированной Fmoc-аминокислоты к смоле в реакторе;
(f) присоединения активированной Fmoc-аминокислоты, используя (2-(6-хлор-1H-бензотриазол-l-ил)-l,l,3,3-тетраметиламиний гексафторфосфат) (HCTU) или 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторкорат (TBTU)/1-гидроксибензотриазол (HOBt) с основанием в диполярном апротонном растворителе в качестве связующего реагента;
(g) промывки смолы после каждого присоединения Fmoc-аминокислоты;
(h) повторения стадий (b)-(g) до образования пептида;
(i) отщепления целевого пептида от смолы при одновременном удалении защитных групп боковых цепей аминокислоты, используя отщепляющий коктейль;
(j) отфильтровывания отщепляющей смеси от смолы; и
(k) выпаривания фильтратов и осаждения и частичной очистки сырого продукта от концентрированного раствора органическим растворителем до получения частично очищенного пептида.
В соответствии со стадиями (a), (b), (c) и (f) способа настоящего изобретения, как раскрыто выше, используют диполярный апротонный растворитель. Такой диполярный апротонный растворитель может быть выбран из диметилформамида (ДМФ), диметилацетамида (DMA) или N-метилпирролидона (NMP), или их комбинаций. В предпочтительном варианте в качестве диполярного апротонного растворителя используют ДМФ.
В другом аспекте в настоящем изобретении предложен способ синтеза терапевтических пептидов, включающий последовательные стадии:
(a) набухания Fmoc-смолы Зибера (также именуемой как амидная смола Зибера или Fmoc амидная смола Зибера) в диметилформамиде (ДМФ);
(b) удаления защитных Fmoc групп, используя раствор пиперидина в ДМФ;
(c) промывки смолы после удаления защитных Fmoc групп с использованием ДМФ;
(d) активирования Fmoc-аминокислоты для присоединения к смоле, у которой удалены защитные группы, путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в ДМФ с последующим добавлением основания и перемешиванием;
(e) добавления раствора активированной Fmoc-аминокислоты к смоле в реакторе;
(f) присоединения активированной Fmoc-аминокислоты, используя (2-(6-хлор-1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламиний гексафторфосфат) (HCTU) или 2-(lH-бензотриазол-l-ил)-l,l,3,3-тетраметилуроний тетрафторкорат (TBTU)/1-гидроксибензотриазол (HOBt) с основанием в ДМФ в качестве связующего реагента;
(g) промывки смолы после каждого присоединения Fmoc-аминокислоты;
(h) повторения стадий (b)-(g) до образования пептида;
(i) отщепления целевого пептида от смолы, при одновременном удалении защитных групп боковых цепей аминокислоты, используя отщепляющий коктейль;
(j) отфильтровывания отщепляющей смеси от смолы; и
(k) выпаривания фильтратов и осаждения и частичной очистки сырого продукта из концентрированного раствора органическим растворителем до получения частично очищенного пептида.
В соответствии со стадией (d) способа настоящего изобретения, как раскрыто выше, используют основание. Указанное основание может быть основанием третичного амина или их смесью, и может быть выбрано из N,N-диизопропилэтиламина (DIEA), триэтиламина (TEA), N-метилморфолина (NMM), 2,4,6-триметилпиридина (TMP, известного также как коллидин), 2,3,5,6-тетраметилпиридина (TEMP), 2,6-ди-трет-бутил-4-диметиламинопиридина (DBDMAP) или 4-диметиламинопиридина (DMAP). Предпочтительный вариант непосредственно вышеизложенного аспекта настоящего изобретения отличается тем, что основанием, используемым на стадии (d) является третичный амин, и что в более предпочтительном варианте, указанным основанием является N,N-диизопропилэтиламин (DIEA).
В другом аспекте в настоящем изобретении предложен способ синтеза терапевтических пептидов, включающий стадии:
(a) набухания Fmoc-смолы Зибера (именуемой также как амидная смола Зибера или Fmoc амидная смола Зибера) в диметилформамиде (ДМФ);
(b) удаления защитных Fmoc групп, используя раствор пиперидина в ДМФ;
(c) промывки смолы ДМФ после удаления защитных групп Fmoc;
(d) активации Fmoc-аминокислоты для присоединения к смоле, у которой удалены защитные группы, путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в ДМФ с последующим добавлением основания и перемешиванием;
(e) добавления раствора активированной Fmoc-аминокислоты к смоле в реакторе;
(f) присоединения активированной Fmoc-аминокислоты, используя (2-(6-хлор-1H- бензотриазол-l-ил)-l,l,3,3-тетраметиламиний гексафторфосфат) (HCTU) или 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторкорат (TBTU)/1-гидроксибензотриазол (HOBt) с N,N-диизопропилэтиламином (DIEA) в ДМФ в качестве связующего реагента;
(g) промывки смолы после каждого присоединения Fmoc-аминокислоты;
(h) повторения стадий (b)-(g) до образования пептида;
(i) отделения целевого пептида от смолы при одновременном удалении защитных групп боковых цепей аминокислоты, используя отщепляющий коктейль;
(j) отфильтровывания отщепляющей смеси от смолы; и
(k) выпаривания фильтратов и осаждения и частичной очистки сырого продукта от концентрированного раствора органическим антирастворителем до получения частично очищенного пептида.
В другом аспекте в настоящем изобретении предложен способ синтеза терапевтических пептидов, включающий последовательные стадии:
(a) набухания Fmoc-смолы Зибера (также именуемой как амидная смола Зибера или Fmoc амидная смола Зибера) в диметилформамиде (ДМФ);
(b) удаления защитных Fmoc групп, используя раствор пиперидина в ДМФ;
(c) промывки смолы ДМФ после удаления защитных Fmoc групп;
(d) активирования Fmoc-аминокислоты для присоединения к смоле, у которой удалены защитные группы, путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в ДМФ, с последующим добавлением N,N-диизопропилэтиламина (DIEA) и перемешиванием;
(e) добавления раствора активированной Fmoc-аминокислоты к смоле в реакторе;
(f) присоединения активированной Fmoc-аминокислоты, используя (2-(6-хлор-1H-бензотриазол-l-ил)-l,l,3,3-тетраметиламиний гексафторфосфат) (HCTU) или 2-(1H-бензотриазол- 1-ил)-1,1,3,3-тетраметилуроний тетрафторкорат (TBTU)/1-гидроксибензотриазол (HOBt) с N,N-диизопропилэтиламином (DIEA) в ДМФ в качестве связующего реагента;
(g) промывки смолы после каждого присоединения Fmoc-аминокислоты;
(h) повторения стадий (b)-(g) до образования пептида;
(i) отщепления целевого пептида от смолы при одновременном удалении защитных групп боковых цепей аминокислоты, используя отщепляющий коктейль;
(j) отфильтровывания отщепляющей смеси от смолы; и
(k) выпаривания фильтратов и осаждения и частичной очистки сырого продукта от концентрированного раствора органическим растворителем до получения частично очищенного пептида.
Другой предпочтительный вариант настоящего изобретения отличается тем, что указанный отщепляющий коктейль, использованный на стадии (i) способа настоящего изобретения, как раскрыто выше, состоит из TFA, одного или более из акцепторов и DCM, где указанный акцептор выбирают из группы, состоящей из триизопропилсилана (TIPS), триэтилсилана (TES), фенола, анизола, тиоанизола, воды, 1,2-этандитиола (EDT), 1-додекантиола, дитиотреитола (DTT) и индола, при условии, что процент TFA в указанном отщепляющем коктейле не превышает 25%.
Предпочтительный вариант непосредственно вышеизложенного аспекта настоящего изобретения отличается тем, что указанный акцептор выбирают из группы, состоящей из TIPS, TES, анизола и воды.
Другой предпочтительный вариант настоящего изобретения отличается тем, что указанный отщепляющий коктейль, использованный на стадии (i), как раскрыто выше, состоит из TFA, одного или более из акцепторов и DCM, где указанный акцептор выбирают из группы, состоящей из TIPS, TES, анизола и воды, при условии, что процент TFA в указанном отщепляющем коктейле не превышает 25%.
Для пептидов, у которых требуется удалить только Boc и tBu защитные группы боковых цепей, предпочтительный вариант настоящего изобретения отличается тем, что указанный отщепляющий коктейль состоит из 15-25% об/об TFA с 2,5-12% об/об TIPS и 62,5-82,5% об/об DCM; и еще более предпочтительно, если указанный отщепляющий коктейль состоит из 20% об/об TFA с 10% об/об TIPS и 70% об/об DCM.
Для пептидов, у которых требуется удалить только Boc и tBu защитные группы боковых цепей, предпочтительный вариант настоящего изобретения, как раскрыто выше, отличается тем, что: указанный отщепляющий коктейль, использованный на стадии (i), как раскрыто выше, состоит из 15-25% об/об TFA с 2,5-12% об/об TIPS и остальную часть отщепляющего коктейля составляет до 100% DCM; и еще более предпочтительно, если указанный отщепляющий коктейль, использованный на стадии (i), как раскрыто выше, состоит из приблизительно 20% об/об TFA с приблизительно 10% б/об TIPS и 70% об/об DCM.
Следующий предпочтительный вариант способа настоящего изобретения, как раскрыто выше, на стадиях (a)-(k) состоит в том, что полученный пептид отщепляют от амидной смолы Зибера одновременно с удалением защитных групп боковых цепей.
Другой предпочтительный вариант настоящего изобретения отличается тем, что Fmoc группы вначале удаляют со смолы, используя пиперидин в ДМФ. В более предпочтительном варианте Fmoc группу вначале удаляют со смолы, используя пиперидин в ДМФ, где концентрация указанного пиперидина в ДМФ меньше чем 20% (об/об) и более предпочтительно, около 15% (об/об).
В соответствии со стадией (f) способа настоящего изобретения, как раскрыто выше, присоединение активированной Fmoc-аминокислоты осуществляют, используя (2-(6-хлор-1H- бензотриазол-l-ил)-l,l,3,3-тетраметиламиний гексафторфосфат) (HCTU) или 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторкорат (TBTU)/1-гидроксибензотриазол (HOBt) с основанием, таким как N,N-диизопропилэтиламин (DIEA) в диполярном апротонном растворителе, таком как ДМФ, отдельно или в комбинации. В предпочтительном варианте любого одного из предшествующих аспектов настоящего изобретения, остатки аминокислоты присоединяют, используя “комбинацию связующих реагентов”, выбранных из группы, состоящей из TBTU/HOBt/DIEA, HBTU/HOBt/DIEA, HATU DIEA, HCTU DIEA, DIC/HOBt, DIC/HOAt, HATU HOBt/DIEA и HCTU/HOBt/DEEA, более предпочтительно, выбранных из группы, состоящей из HCTU/DIEA и TBTU/HOBt/DIEA.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (a) (Fmoc-амидную смолу Зибера вначале набухают, используя от 1 до 3 обработок 7-12 объемами ДМФ в течение вплоть до 1 часа, еще более предпочтительно, 3 обработки 10 объемами ДМФ длительностью от 10 до 30 минут за обработку.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями от (a) до (j), как раскрыто выше, отличается тем, что на стадии (b), Fmoc группы у смолы Зибера удаляют, используя 1-2 обработки раствором пиперидина в ДМФ (10-20% об/об) длительностью от 5 до 20 минут, еще более предпочтительно 2 обработки 15% об/об пиперидина в ДМФ длительностью 10 минут.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (c), смолу с удаленными защитными группами промывают от 3 до 5 раз 7-12 объемами ДМФ, причем каждая промывка длится до 5 минут, еще более предпочтительны 3 промывки 10 объемами ДМФ, причем каждая промывка длится вплоть до 5 минут.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (d) от 1,2 до 2,0 молярных эквивалентов (относительно масштаба загрузки смолы) Fmoc-аминокислоты активируют для присоединения путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в ДМФ, добавления DIEA, перемешивания в течение вплоть до 5 минут; и более предпочтительно 1,5 молярных эквивалентов (относительно масштаба загрузки смолы) Fmoc-аминокислоты активируют для присоединения путем растворения Fmoc-аминокислоты и связующего реагент (реагентов) в ДМФ, добавления DIEA, перемешивания в течение 1-2 минут.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (f), от 0,5 до 1,5 молярных эквивалентов (относительно Fmoc-аминокислоты) связующего реагента (реагентов) используют с 1,5-2,5 молярными эквивалентами (относительно Fmoc-аминокислоты) DIEA в 4-10 объемах ДМФ в течение 30-120 минут при комнатной температуре; и более предпочтительно, от 0,5 до 1,5 молярных эквивалентов (относительно Fmoc-аминокислоты) связующего реагента (реагентов) используют с 1,5-2,5 молярными эквивалентами (относительно Fmoc-аминокислоты) DIEA в 5-7 объемах ДМФ в течение 60 минут при температуре от 15 до 30°C.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (g), смолу промывают после каждого присоединения, от 2 до 4 раз в 7-12 объемах ДМФ в течение вплоть до 5 минут; и в более предпочтительном варианте, смолу после каждого присоединения промывают 2 раза в 10 объемах ДМФ в течение вплоть до 5 минут.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (i):
- указанную смолу погружают в отщепляющий коктейль и перемешивают в течение 2-3 часов при комнатной температуре, и более предпочтительно, указанную смолу погружают и перемешивают в течение 2,5 часа, и
- раствор смола/отщепляющий коктейль периодически барботируют газообразным азотом.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (j):
- отработанную смолу промывают небольшим объемом или свежего отщепляющего коктейля или смесью TFA/DCM (20:80 об/об) 1-2 раза;
- отработанную смолу необязательно промывают небольшим объемом MeOH.
Предпочтительный вариант способа настоящего изобретения с последовательными стадиями (a)-(j), как раскрыто выше, отличается тем, что на стадии (k), после комбинации объединения фильтратов и выпаривания:
- сырой пептид осаждают, используя от 5 до 15 объемов MtBE;
- выпавший в осадок пептид сушат до необходимого уровня сухости;
- выпавший в осадок пептид растворяют разбавленной кислотой, или разбавленной кислотой с органическим модификатором, чтобы использовать в последующем процессе хроматографической очистки
- пептид очищают и осуществляют стадию солевого обмена, используя препаративную хроматографию с обращенной фазой.
Другой предпочтительный вариант способа настоящего изобретения отличается тем, что включает следующие последовательные стадии (a)-(j):
(a) набухания Fmoc-амидной смолы Зибера, используя от 1 до 3 обработок в 7-12 объемах ДМФ в течение вплоть до 1 часа, еще более предпочтительно, 3 обработки 10 объемами ДМФ длительностью от 10 до 30 минут на обработку;
(b) удаления защитных Fmoc групп на смоле Зибера, используя от 1 до 2 обработок раствором пиперидина в ДМФ (10-20% об/об) длительностью от 5 до 20 минут, еще более предпочтительно, 2 обработки 15% об/об пиперидина в ДМФ длительностью 10 минут;
(c) промывки смолы, у которой удалены защитные группы, от 3 до 5 раз в 7-12 объемах ДМФ, причем каждая промывка длительностью вплоть до 5 минут, еще более предпочтительно, 3 промывки 10 объемами ДМФ, причем каждая промывка длительностью вплоть до 5 минут.
(d) активирования 1,2-2,0 молярных эквивалентов (относительно масштаба загрузки смолы) Fmoc-аминокислоты для присоединения к смоле, у которой удалены защитные группы, путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в ДМФ, добавления DIEA, перемешивания в течение 1-2 минут; и более предпочтительно, активирования 1,5 молярных эквивалентов (относительно масштаба загрузки смолы) Fmoc-аминокислоты для присоединения путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в ДМФ, добавления DIEA, перемешивания в течение 1-2 минут.
(e) добавления раствора активированной Fmoc-аминокислоты к смоле в реакторе;
(f) присоединения Fmoc-аминокислоты к смоле, у которой удалены защитные группы, путем погружения и перемешивания смолы, у которой удалены защитные группы, с раствором активированной Fmoc-аминокислоты в течение 30-120 минут при комнатной температуре; причем указанный раствор активированной Fmoc-аминокислоты состоит из Fmoc-аминокислоты, как раскрыто выше в (d), вместе с 0,5-1,5 молярными эквивалентами (относительно Fmoc-аминокислоты) связующего реагента (реагентов) с от 1,5 до 2,5 молярных эквивалентов (относительно Fmoc-аминокислоты) DIEA в 4-10 объемах ДМФ при комнатной температуре; и более предпочтительно, от 0,5 до 1,5 молярных эквивалентов (относительно Fmoc-аминокислоты) связующего реагента (реагентов) с 1,5 до 2,5 молярными эквивалентами (относительно Fmoc-аминокислоты) DIEA в 5-7 объемах ДМФ в течение 60 минут при температуре от 15 до 30°C.
(g) промывки смолы после каждого присоединения, от 2 до 4 раз в 7-12 объемах ДМФ в течение вплоть до 5 минут; и более предпочтительно 2 раза в 10 объемах ДМФ в течение вплоть до 5 минут;
(h) повторения стадий (b)-(g) до образования пептида;
(i) отщепления целевого пептида от смолы при одновременном удалении защитных групп боковых цепей аминокислоты, используя отщепляющий коктейль, путем:
- погружения смолы в отщепляющий коктейль и перемешивания в течение 2-3 часов при комнатной температуре, и более предпочтительно, путем погружения и перемешивания в течение 2,5 часа, и
- периодического барботирования смеси смола/отщепляющий коктейль газообразным азотом.
(j) отфильтровывания отщепляющей смеси от смолы, затем
- промывки отработанной смолы небольшим объемом или свежего отщепляющего коктейля или смеси TFA/DCM (20:80 об/об) 1-2 раза; и
- необязательной промывки отработанной смолы небольшим объемом MeOH.
(k) комбинации комбинирования фильтрата и промывок и выпаривания, затем
- осаждения сырого пептида из объединенных выпаренного фильтрата и промывок, используя от 5 до 15 объемов MtBE;
- сушки выпавшего в осадок пептида до необходимого уровня сухости;
- растворения выпавшего в осадок пептида разбавленной кислотой, или разбавленной кислотой с органическим модификатором для использования в последующей хроматографической очистке;
- очищения пептидов и осуществления стадии солевого обмена, используя препаративную хроматографию с обращенной фазой.
Предпочтительный вариант по любому одному из непосредственно вышеприведенных аспектов настоящего изобретения, который отличается тем, что стадии (a)-(j) (новые подстадии обозначают как -№) далее определяют следующим образом:
(a) набухания Fmoc-амидной смолы Зибера, вначале используя от 1 до 3 обработок 7-12 объемами ДМФ в течение вплоть до 1 часа, еще более предпочтительно, 3 обработки в 10 объемах ДМФ длительностью от 10 до 30 минут на обработку;
(b) удаления защитных Fmoc групп на смоле Зибера, используя от 1 до 2 обработок раствором пиперидина в ДМФ (10-20% об/об) длительностью от 5 до 20 минут, еще более предпочтительно, 2 обработки 15% об/об пиперидина в ДМФ длительностью 10 минут;
(c) промывки смолы, у которой удалены защитные группы, от 3 до 5 раз в 7-12 объемах ДМФ, причем каждая промывка длительностью вплоть до 5 минут, еще более предпочтительно, 3 промывки 10 объемами ДМФ, причем каждая промывка длительностью вплоть до 5 минут;
(d) активирования Fmoc-аминокислоты (1,2-2,0 молярных эквивалента, или более предпочтительно, 1,5 молярных эквивалента относительно масштаба загрузки смолы) для присоединения путем растворения Fmoc-аминокислоты и связующего реагента (реагентов) в ДМФ, добавления DIEA, перемешивания в течение вплоть до 5 минут, еще более предпочтительно, перемешивания в течение от 1 до 2 минут;
(e) добавления связующего раствора к смоле в реакторе;
(f) использования от 0,5 до 1,5 молярных эквивалентов связующего реагента (реагентов) относительно Fmoc-аминокислоты с 1,5-2,5 молярными эквивалентами DIEA относительно Fmoc-аминокислоты в 4-10 объемах ДМФ в течение 30-120 минут при комнатной температуре, еще более предпочтительно, используя от 5 до 7 объемов ДМФ в течение 60 минут при температуре от 15 до 30°C;
(g) промывки смолы после каждого присоединения от 2 до 4 раз в 7-12 объемах ДМФ в течение вплоть до 5 минут, еще более предпочтительно, 2 промывки 10 объемами ДМФ в течение вплоть до 5 минут;
(h) повторения стадий (b)-(g) до образования пептида;
(i) погружения смолы в отщепляющий коктейль и перемешивания в течение от 2 до 3 часов при комнатной температуре, более предпочтительно, погружения и перемешивания смолы в течение 2,5 часа;
(i-1) периодического барботирования раствора смола/отщепляющий коктейль газообразным азотом;
(j) отфильтровывания отщепляющей смеси от смолы;
(j-1) промывания отработанной смолы небольшим объемом или свежего отщепляющего коктейля, или смеси TFA/DCM (20:80 об/об) от 1 до 2 раз;
(j-2) промывки отработанной смолы небольшим объемом MeOH;
(k) комбинации комбинирования фильтратов и выпаривания
(k-1) осаждения сырого пептида, используя от 5 до 15 объемов MtBE;
(k-2) сушки выпавшего в осадок пептида до необходимого уровня сухости
(k-3) растворения выпавшего в осадок пептида разбавленной кислотой, или разбавленной кислотой с органическим модификатором для использования в последующей хроматографической очистке
(k-4) очистки пептида и осуществления стадии солевого обмена, используя препаративную хроматографию с обращенной фазой.
Известно, что некоторые защитные группы (например, 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонил (Pbf), защитная группа боковой цепи Arg) требуют более высокого процента кислоты, обычно 50-80%, для удаления защитных групп боковых цепей в разумных для практики рамках времени, однако, все остальные аспекты настоящего изобретения остаются теми же самыми. Другие предполагаемые защитные группы боковых цепей, включают, но ими не ограничиваются, метокситриметилбензолсульфонил (Mtr), 2,2,5,7,8-пентаметилхроман-6-сульфонилхлорид (Pmc), 4,4-диметилоксибензгидрил (Mbh) и 2,4,6-триметоксибензил (Tmob).
В настоящем изобретении также предложено решение для таких ситуаций, когда желательно сохранить некоторые защитные группы боковых цепей во время и после отщепления. Так, например, важно сохранить защитные группы боковых цепей ацетамидометила (Acm) на Cys с тем, чтобы полный пептид можно было очистить в его линейно форме, и после этого осуществить циклизацию, когда защитные группы удаляют и дисульфидный мостик образуется между двумя Cys остатками.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1: график, изображающий голова к голове сравнение пептидов, синтезированных с использованием амидной смолы Ринка с пептидами, синтезированными с использованием амидной смолы Зибера в аналогичных процедурах синтеза. Получены пептиды различной длины, от 5 аминокислот до 30 аминокислот, в соответствии с раскрытыми в описании процедурами, и определен выход продуктов синтеза. Для пептидов каждой длины (отложена по y-оси графика), процентный выход (определяется по x-осям) с использованием амидной смолы Ринка представлен верхними полосками, и процентный выход с использованием амидной смолы Зибера представлен нижними полосками. Для каждого синтезированного пептида, то есть, для последовательности из 5 аминокислот, двух последовательностей из 8 аминокислот, аминокислотной последовательности из 8 аминокислот, модифицированной одним или более из фрагментов дофамина, и 30 аминокислотной последовательности, использование амидной смолы Зибера привело к более высоким % выходов.
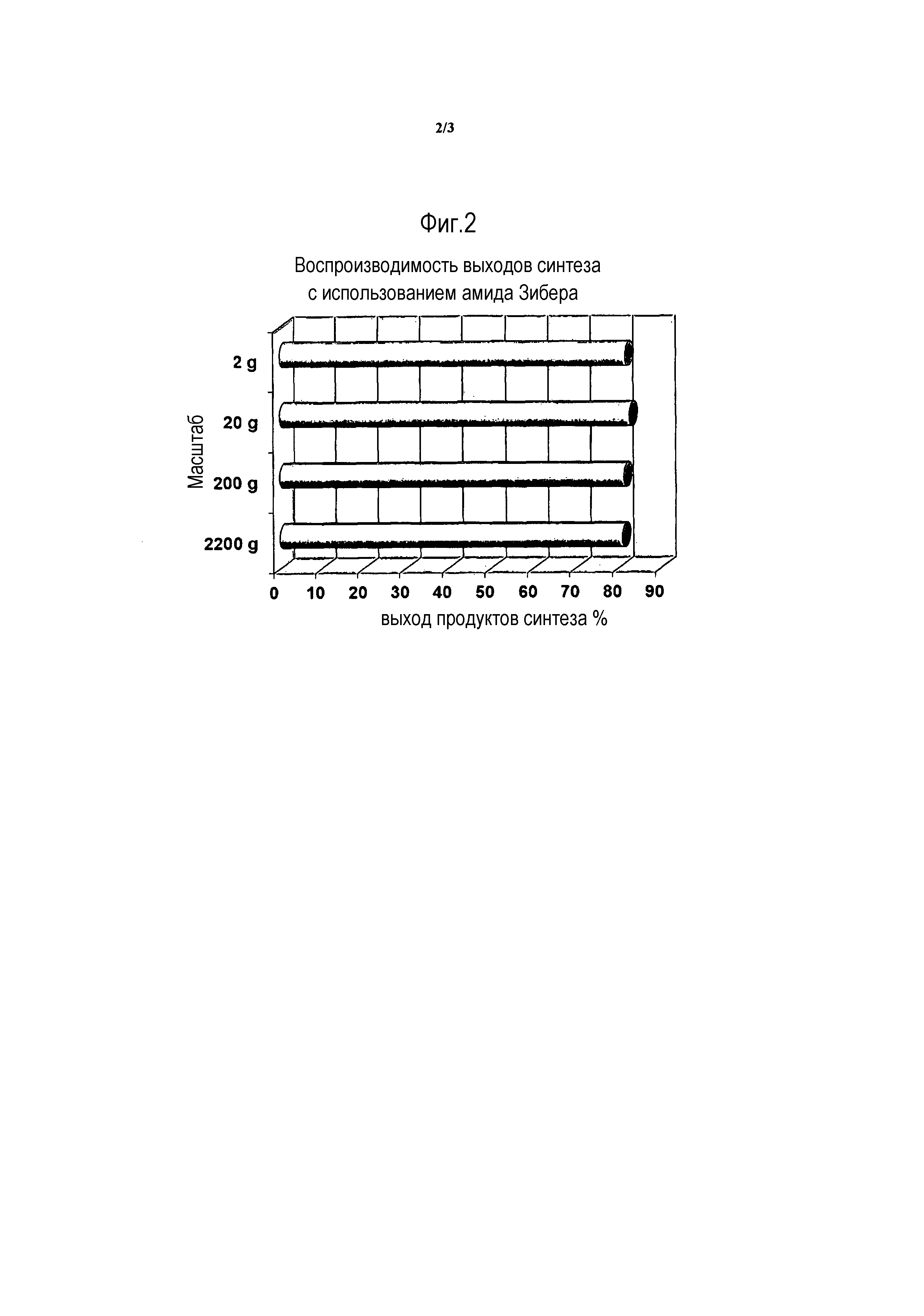
Фиг. 2: график, представляющий воспроизводимость выходов продуктов синтеза для масштабов от 2 г до 2200 г при использовании амидной смолы Зибера для синтеза 5 пептидов из 5 аминокислот. Сообщают, что выход синтеза, составляющий около 80%, неизменно достигается для каждого масштаба.
Фиг. 3: график, представляющий относительную стоимость в расчете на материал, используемый для синтеза пептида из 8 аминокислот и пептида из 30 аминокислот, с использованием амидной смолы Ринка по сравнению с амидной смолой Зибера. Относительные стоимости в расчете на материал, используемый для синтеза пептидов, синтезированных с использованием амидной смолы Ринка для пептида из 8 аминокислот и пептида из 30 аминокислот, представлены верхними полосками, тогда как относительная стоимость для тех же самых пептидов, но полученных с использованием амидной смолы Зибера, представлены нижними полосками для каждого пептида. Как сообщалось, относительная стоимость производства с использованием амидной смолы Зибера меньше, чем стоимость с использованием амидной смолы Ринка.
Большинство поэтапных твердофазных способов синтеза требует использования полистирольной смолы для синтеза пептидных амидов. Амидные смолы Ринка используют в твердофазном синтезе пептидов для получения пептидных амидов с использованием Fmoc-защищенных аминокислот. Присоединение первой аминокислоты можно обеспечить, используя типичные способы образования амидной связи. Пептидные последовательности собирают в основных или в нейтральных условиях на амидной смоле Ринка, затем законченный пептид отщепляют от смолы в кислотных условиях. Обычно пептид отщепляют от амидной смолы Ринка, используя более 80% TFA об/об. (Stathopoulos, P.; Papas, S.; и Tsikaris, V., J. Pept. Sci., 2006, 12:227-37). Более сильные кислоты или более высокие концентрации TFA иногда отщепляют часть линкеров Ринка от полистирольного носителя, и привносят окрашенные примеси в отщепленный продукт. Поэтому для некоторых пептидов выход продуктов синтеза при использовании амидной смолы Ринка традиционно низок. Примерами смол Ринка являются:
Лабильность линкеров ʺсупер кислотно-чувствительнойʺ или ʺгипер кислотно-чувствительнойʺ смолы к низкой концентрации кислоты позволяет отщеплять от смолы полностью защищенные пептиды. Обычно 1-5% об/об TFA требуется для отщепления пептида. За исключением более низкой силы кислоты, необходимой для отщепления, такие смолы аналогичны амидным смолами Ринка, а именно, они представляют собой полистирольную матрицу близкого размера шариков с аналогичной емкостью загрузки. Как таковые, указанные смолы пригодны для конвергентного синтеза, использующего ту же самую Fmoc-химию в отношении введения первого и присоединения последующих остатков.
Амидную смолу Зибера, как пример ʺсупер восприимчивой к кислотеʺ смолы (Sieber, P., Tetrahedron Lett., 1987, 28(19):2107-10), используют главным образом для синтеза пептидных амидов, сохраняющих защитные группы боковых цепей, включая, но ими не ограничиваясь, трет-бутилоксикарбонил (Boc) и трет-бутиловый эфир (tBu), если используют низкие концентрации трифторуксусной кислоты (TFA) (1-5% об/об) в отщепляющем коктейле.
Так как амидную смолу Зибера традиционно используют для Fmoc твердофазного синтеза, необходимо, чтобы аминокислоты были Fmoc-защищены. Соответственно, защитные группы, которые остаются после завершения пептидного синтеза, необходимо отщепить. Отщепление оставшихся защитных групп требует жестких ацидолитических условий, таких как вплоть до 95% TFA, содержащей вплоть до 5% этандиола и вплоть до 5% 4-(метилмеркапто)фенола (Sieber, P., Tetrahedron Lett., 1987, 28(19):2107-10).

Авторы изобретения предприняли попытки синтеза чувствительного к кислоте пептидного амида из 8 остатков, используя амидную смолу Зибера. Они обнаружили, что линейные SPPS можно получить, используя амидную смолу Зибера вместе с Fmoc-химией. Авторы обнаружили, что регулируя условия, высокие концентрации кислоты, т.е. TFA, не требуются для отщепления конечного продукта от смолы Зибера. Кроме того, было обнаружено, что защитные группы боковых цепей можно удалить одновременно с отщеплением полученного пептида от амидной смолы Зибера, если использовать ʺумереннойʺ силы TFA/TIPS/DCM отщепляющий коктейль. В результате некоторой оптимизации авторы изобретения обнаружили, что можно синтезировать полной длины пептиды с защищенными аминокислотами, особенно такие, у которых защитные группы представляет собой Boc, tBu и/или Trt, и затем отщепить от смолы пептидные амиды с полностью удаленными защитными группами, минимизируя при этом разложение пептидов.
Авторы настоящего изобретения также предприняли попытку использовать амидную смолу Зибера для синтеза других пептидов длиной от 5 до 30 аминокислот, и пептидов, содержащих искусственные аминокислоты, также как проблематичные природные аминокислоты, такие как триптофан, цистеин и аргинин. Авторы настоящего изобретения обнаружили, что пептиды, содержащие искусственные или проблематичные аминокислоты, можно синтезировать, используя амидную смолу Зибера со средней концентрацией TFA в процессе отщепления. Было также обнаружено, что пептиды, содержащие аргинин, можно синтезировать, используя амидную смолу Зибера, хотя более высокие концентрации TFA необходимы в процессе отщепления, особенно, если присутствуют сульфонильные защитные группы боковых цепей.
Неожиданно оказалось, что, амидная смола Зибера, если ее использовать способом, противоположным тому, который раскрыт в литературе, обеспечивает более высокие выходы более чистого сырого продукта. Например, авторы изобретения обнаружили, что выход продуктов синтеза возрастает с 18-30% при использовании амидной смолы Ринка, до 78-83% при использовании амидной смолы Зибера для получения аналога грелина H-Inp-D-Bal-D-Trp-Phe-Apc-NH2, как раскрыто в WO 2004/014415. Для других пептидов, таких как гибриды дофамин-соматостатина, авторы настоящего изобретения сообщают, что выход продуктов синтеза при использовании амидной смолы Зибера составляет 72,6-80,8% по сравнению с выходами 13-71% при использовании обычно используемых смол, таких как семейство амидных смол Ринка (например, амидной MBHA смолы Ринка, амидной AM смолы Ринка, амидной смолы Ринка) в идентичных условиях. Авторы изобретения обнаружили, что при использовании амидной смолы Зибера выход возрастает в среднем на вплоть до 50% по сравнению с выходом при использовании амидных смол Ринка. Кроме того, выходы являются воспроизводимыми от загрузки к загрузке и в масштабе от 2 г до вплоть до 2,2 кг. Сравнительные выходы продуктов с использованием амидной смолы Ринка против амидной смолы Зибера в идентичных условиях синтеза пептидов различной длины, т.е. 5 аминокислот в длину до 30 аминокислот в длину, представлены на Фиг. 1. При каждом сравнении использование амидной смолы Зибера приводит к более высоким выходам продуктов синтеза, 70% против 10%. В результате, относительный процент стоимости в расчете на длину пептидов оказывается меньше при использовании амидной смолы Зибера вместо традиционной амидной смолы Ринка. Фиг. 3. Авторы настоящего изобретения также продемонстрировали воспроизводимость выходов продукта синтеза с использованием амидной смолы Зибера на Фиг. 2.
Далее, обычно используемые амидные смолы Ринка требуют высоких концентраций TFA, обычно 80-95% об/об, для отщепления конечного пептида от смолы. Авторы изобретения обнаружили, что при использовании амидной смолы Зибера требуется только 10-25% TFA. Как было указано выше, более высокие концентрации TFA смогут привести к значительному разложению пептида с течением времени, также как присутствие примесей, таких которые образуются в результате присоединения всей или части линкеров смолы к пептиду, что впоследствии может быть трудно удалить. Кроме того, обработка после отщепления происходит быстрее при использовании заявленного процесса, так как во время финального отщепления требуется меньше кислоты. Кроме того, было обнаружено, что можно уменьшить количества Fmoc-аминокислоты, связующих реагентов и растворителей, если использовать амидную смолу Зибера, без влияния на выход или степень чистоты получаемого пептида.
Ряд аминокислот, присутствующих в соединениях настоящего изобретения представлены в описании следующим образом:














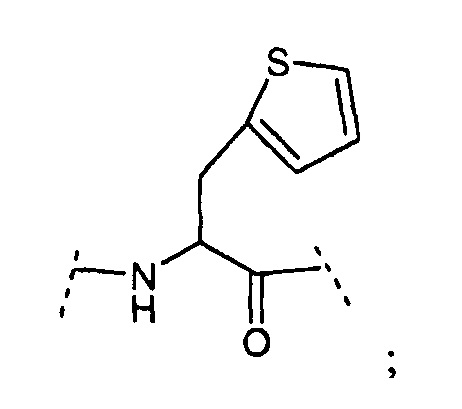

ʺDop1ʺ подразумевает соединение структурной формулы:

ʺDop2ʺ подразумевает соединение структурной формулы:

ʺDop3ʺ подразумевает соединение структурной формулы:

ʺDop4ʺ подразумевает соединение структурной формулы:
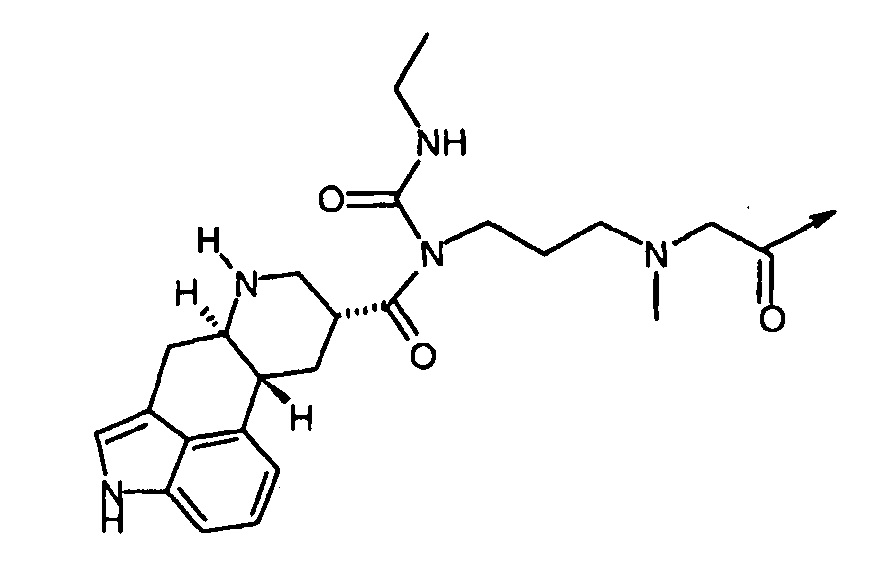
ʺDop5ʺ подразумевает соединение структурной формулы:

ʺDop6ʺ подразумевает соединение структурной формулы:
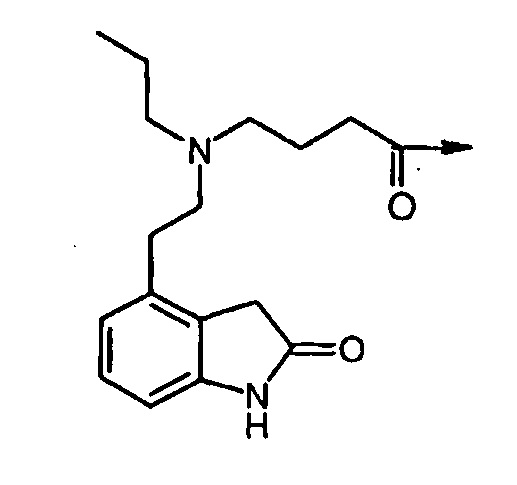
ʺDop7ʺ подразумевает соединение структурной формулы:

ʺDop8ʺ подразумевает соединение структурной формулы:

ʺDop9ʺ подразумевает соединение структурной формулы:
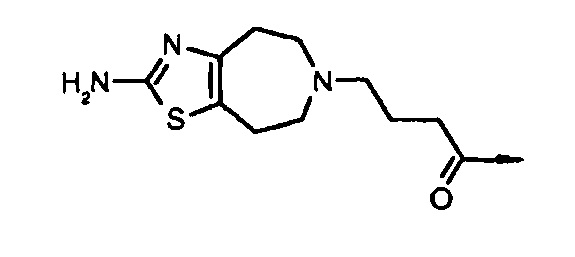
ʺDop10ʺ подразумевает соединение структурной формулы:

ʺDop11ʺ подразумевает соединение структурной формулы:
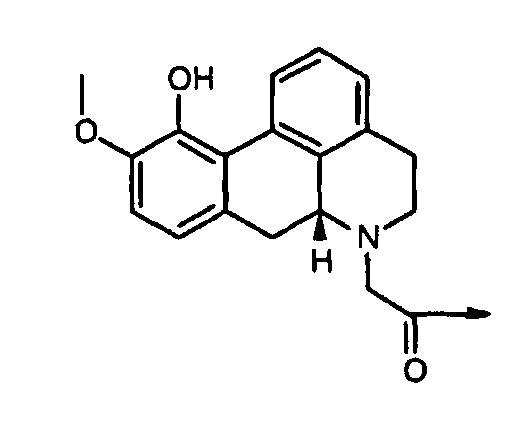
ʺDop12ʺ подразумевает соединение структурной формулы:

ʺDop13ʺ подразумевает соединение структурной формулы:
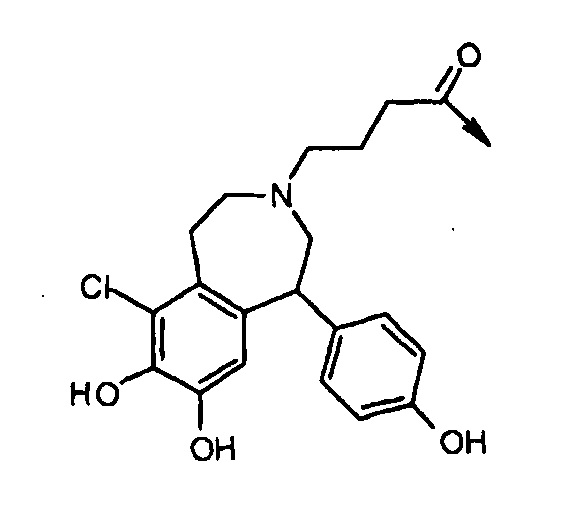
Lys(Dop2) имеет структуру:

Dop2-Lys(Dop2) имеет структуру:
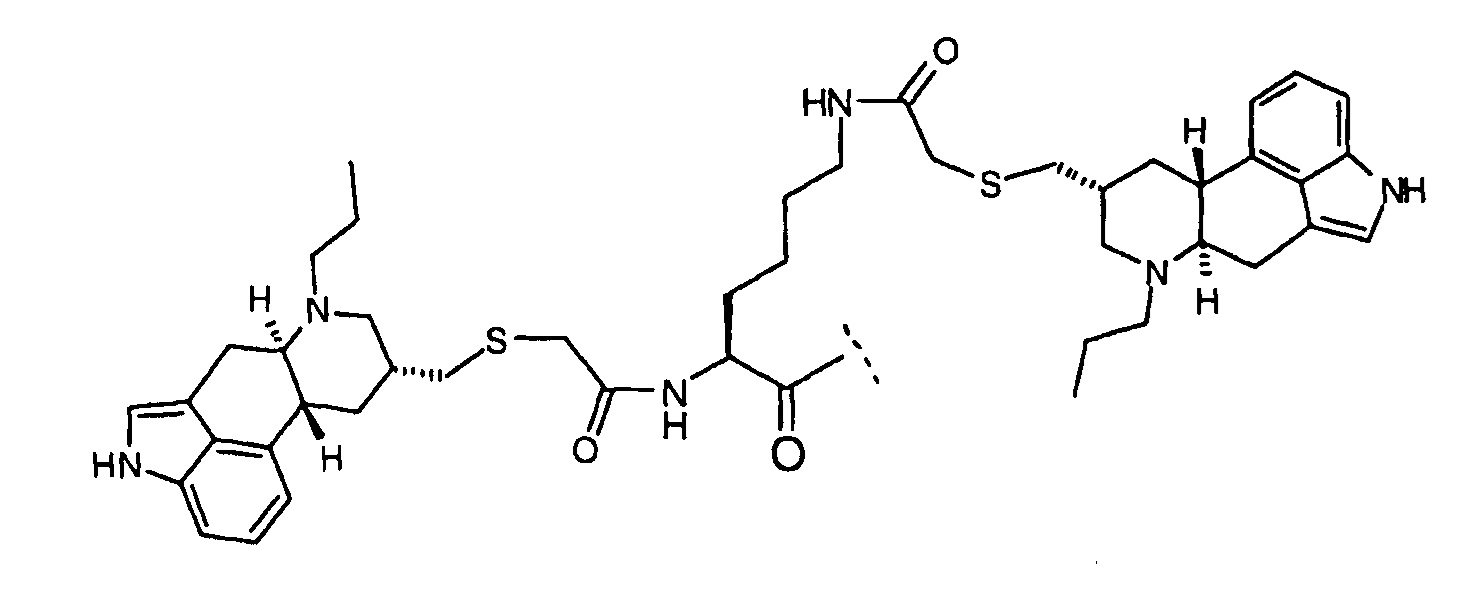
Lys(Dop5) имеет структуру:
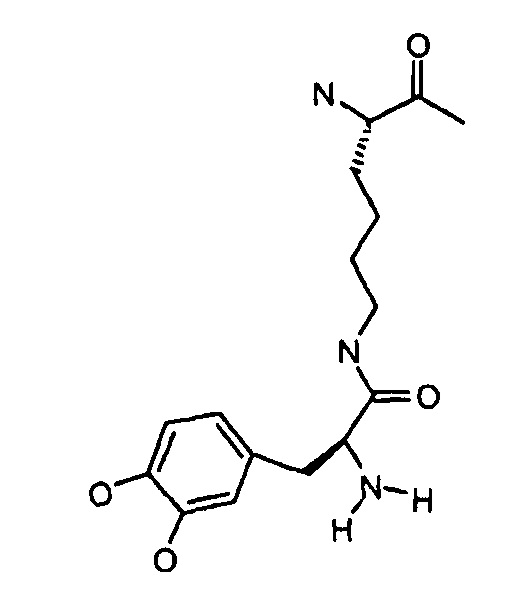
Dop5-Lys(Dop5) имеет структуру:

Греческую букву пси ʺψʺ используют в описании для указания на то, что пептидная связь была заменена псевдопептидной связью. В названии аминокислотной последовательности, формат термина Ψ представляет собой Α1-ψ-(Χ-Χ')Α2, где A1 представляет собой аминоацильный радикал, карбонильная группа которого была модифицирована до X, и A2 представляет собой аминоацильный радикал, α-аминогруппа которой была модифицирована до X'. X и X' изображены как цепочки элементарных символов элементов, разделенные связью, например, Tyr-ψ-(CH2-NH)Gly.
В заявке использованы следующие сокращения:
Если не указано иначе, следующие определения представлены далее для иллюстрации и определения смысла и объема различных терминов, используемых для описания настоящего изобретения.
Термин ʺотщепляющий коктейльʺ в том смысле, как использован в описании, относится к смеси реагентов, использованных для удаления или отщепления, собранных пептидов от смолы. Кроме того, отщепляющий коктейль также служил для удаления всех защитных групп боковых цепей и N-концевых защитных групп.
Термин ʺоколоʺ (или ʺприблизительноʺ) в том смысле, как использован в описании, в отношении параметров или количеств, означает, что указанные параметры или количества составляют величину, отличающуюся на ±5% от указанного параметра или количества. Так, например, ʺоколо 20%ʺ означает (20±20*0,05)% что равно (20±0,1)%.
Термин ʺсмолаʺ в том смысле, как использован в описании, относится или к Fmoc-амидной смоле Зибера или амидной смоле Зибера, к которым присоединена одна или более из аминокислот.
Термин ʺкомнатная температураʺ (или температура окружающей среды) означает температурный интервал от 15 до 30°C.
Следующие примеры приведены с целями иллюстрации способа настоящего изобретения и никоим образом не ограничивают настоящее изобретение.
В настоящем изобретении раскрыт новый способ синтеза пептидов, включающий постадийную твердофазную химию.
В предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтического пептида, где указанный пептид выбирают из аналогов соматостатина, бомбезина, VIP, PACAP, GHRH, глюкагена, кальцитонина, пептида YY, нейромедина B, PTH, PTHrP, PTH2, GLP-1, уротензина-II, грелина, меланокортина, MIS, LHRH, атропина, GIP, нейропептида Y, IGF-1, гибридов дофамин-соматостатина и ACTH.
В более предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанные пептиды выбирают из аналогов грелина или гибридов дофамин-соматостатина.
В более предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанный пептид выбирают из аналогов грелина.
В более предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанные пептиды выбирают из аналогов грелина формулы (I)
R1-A1-A2-A3-A4-A5-R2 (I),
где
A1 представляет собой Aib, Apc или Inp;
A2 представляет собой D-Bal, D-Bip, D-Bpa, D-Dip, D-1Nal, D-2Nal, D-Ser(Bzl) или D-Trp;
A3 представляет собой D-Bal, D-Bip, D-Bpa, D-Dip, D-1 Nal, D-2Nal, D-Ser(Bzl) или D-Trp;
A4 представляет собой 2Fua, Orn, 2Pal, 3Pal, 4Pal, Pff, Phe, Pim, Taz, 2Thi, 3Thi, Thr(Bzl);
A5 представляет собой Apc, Dab, Dap, Lys, Orn, или исключен;
R1 представляет собой водород; и
R2 представляет собой OH или NH;
при условии, что
если A5 представляет собой Dab, Dap, Lys, или Orn, тогда:
A2 представляет собой D-Bip, D-Bpa, D-Dip или D-Bal; или
A3 представляет собой D-Bip, D-Bpa, D-Dip или D-BaI; или
A4 представляет собой 2Thi, 3Thi, Taz, 2Fua, 2Pal, 3Pal, 4Pal, Orn, Thr(Bzl) или Pff;
если A5 удален, тогда:
A3 представляет собой D-Bip, D-Bpa или D-Dip; или
A4 представляет собой 2Fua, Pff, Taz, или Thr(Bzl); или
A1 представляет собой Apc и -
A2 представляет собой D-Bip, D-Bpa, D-Dip или D-Bal; или
A3 представляет собой D-Bip, D-Bpa, D-Dip или D-Bal; или
A4 представляет собой 2Thi, 3Thi, Orn, 2Pal, 3Pal или 4Pal;
и более конкретно, соединение формулы (I), где
A1 представляет собой Aib, Apc или Inp;
A2 представляет собой D-Bal, D-Bip, D-Bpa, D-Dip, D-1Nal, D-2Nal, D-Ser(Bzl) или D-Trp;
A3 представляет собой D-Bal, D-Bpa, D-Dip, D-1 Nal, D-2Nal или D-Trp;
A4 представляет собой Orn, 3Pal, 4Pal, Pff, Phe, Pim, Taz, 2Thi или Thr(Bzl); и
A5 представляет собой Apc, Lys или удален.
В более предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанные пептиды представляют собой аналог грелина формулы (I), как раскрыто выше, где
A1 представляет собой Apc или Inp;
A2 представляет собой D-Bal, D-Bip, D-1 Nal или D-2Nal;
A3 представляет собой D-Bal, D-1 Nal, D-2Nal или D-Trp;
A4 представляет собой 3Pal, 4Pal, Pff, Phe, Pim, Taz, 2Thi или Thr(Bzl); и
A5 представляет собой Apc или Lys.
В более предпочтительном варианте, настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанные пептиды представляют собой аналоги грелина, выбранные из H-Inp-D-Bal-D-Trp-Phe-Apc-NH2, H-Inp-D-2Bal-D-Trp-Phe-Apc-NH2, H-Inp-D-Bal-D-Trp-2Thi-Apc-NH2 и H-Inp-D-Bal-D-Trp-Taz-Apc-NH2, и более конкретно, H-Inp-D-Bal-D-Trp-Phe-Apc-NH2.
В более предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанные пептиды представляют собой аналоги гибридов дофамин-соматостатина, т.е. гибридные молекулы, включающие соматостатин или его аналоги и по меньшей мере один фрагмент дофамина.
В более предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанные пептиды представляют собой аналоги гибридов дофамин-соматостатина, включая структуры Dop A или DopA- Lys(DopA), где Lys представляет собой L-лизин, если только точно не указан как D-Lys, A представляет собой 1-13, например, Dop1, Dop2, Dop3, Dop4, Dop5, Dop6, Dop7, Dop8, Dop9, Dop10, Dop11, Dop12, Dop13.
В другом более предпочтительном варианте настоящее изобретение относится к способу синтеза терапевтических пептидов, где указанные пептиды представляют собой аналоги гибридов дофамин-соматостатина, включая структуру DopA-Lys(DopA), и соединение Dop2-D-Lys(Dop2)-c[Cys-Tyr-D-Trp-Lys-Abu-Cys]-Thr-NH2.
Общий способ синтеза терапевтических пептидных амидов с полностью удаленными защитными группами по способу настоящего изобретения проиллюстрирован далее.
Вначале осуществляют набухание Fmoc-амидной смолы Зибера (Merck Chemicals, Darmstadt, Germany), используя от 1 до 3 обработок 7-12 объемами, 10 объемами предпочтительно, ДМФ (Samsung, Korea), в течение вплоть до 1 часа, хотя 3 обработки длительностью около 10-30 минут каждая предпочтительны.
Удаление Fmoc с амидной смолы Зибера осуществляют, используя от 1 до 2 обработок раствором пиперидина в ДМФ (около 10-20% об/об, 15% об/об предпочтительно) длительностью от 5 до 20 минут, хотя 2 обработки длительностью 10 минут каждая предпочтительны.
Смолу с удаленными защитными группами промывают от 3 до 5 раз в 7-12 объемах ДМФ в течение вплоть до 5 минут, хотя 3 промывки 10 объемами ДМФ длительностью вплоть до 5 минут для каждой промывки предпочтительны.
Fmoc-аминокислоты активируют для присоединения к смоле путем растворения Fmoc-аминокислоты вместе со связующим реагентом (реагентами) в ДМФ, добавления основания, такого как DIEA (SAFC, Gillingham, United Kingdom), перемешивания в течение вплоть до 5 минут (1-2 минут предпочтительно), и добавления к смоле в реакторе.
Присоединение Fmoc-аминокислоты осуществляют, используя от около 1,2 до 2,0 молярных эквивалентов (1,5 молярных эквивалентов предпочтительно) Fmoc-аминокислоты относительно смолы, используя HCTU (Merck Chemicals) или TBTU/HOBt (от 0,5 до 2,0 молярных эквивалентов относительно Fmoc-аминокислоты) (как TBTU так и HOBt получены из SAFC) с основанием, предпочтительно DIEA (от около 1,5 до 3,5 молярных эквивалентов относительно Fmoc-аминокислоты, хотя специфические эквиваленты предпочтительны для конкретных аминокислот), в ДМФ (от 4 до 10 объемов, от 5 до 7 объемов предпочтительно) длительностью от 30 до 120 минут (хотя длительность варьируется в зависимости от присоединяемой аминокислоты, однако, 60 минут предпочтительны для большинства аминокислот) при комнатной температуре (предпочтительно, от 15 до 30°C). Или HCTU (1,2 эквивалента относительно Fmoc-аминокислоты) или TBTU с HOBt (0,98 молярных эквивалентов) предпочтительны в зависимости от подлежащей к присоединению аминокислоты.
После каждого присоединения Fmoc-аминокислоты, смолу промывают от 2 до 4 раз в 7-12 объемах ДМФ (2 промывки в 10 объемах ДМФ предпочтительны) причем каждая промывка длительностью вплоть до 5 минут.
Целевой пептид отщепляют от смолы и все защитные группы боковых цепей ʺудаляютʺ, используя отщепляющий коктейль, состоящей из от около 15 до 25% об/об TFA (Rhodia, Lyon, France) (хотя предпочтительно от 15 до 20% об/об, и приблизительно 20% об/об более предпочтительно) с около 2,5 до 12% об/об TIPS (SAFC, Gillingham, United Kingdom) (хотя предпочтительны 5-10% об/об, и около 10% об/об более предпочтительны) используют в качестве акцепторов, причем остальная часть отщепляющего коктейля включает от 62,5 до 82,5% об/об DCM (INEOS Chlor, Runcorn, UK) (в зависимости от процента используемых TFA и TIPS). Указанную смолу добавляют и перемешивают с отщепляющим коктейлем в течение от 2 до 3 часов (2,5 часа предпочтительны) при приблизительно комнатной температуре (от около 15 до 30°C). Периодически реакционную смесь барботируют газообразным азотом или поддерживают в атмосфере газообразного азота. Отщепляющую смесь, содержащую целевой пептид и ʺотработаннуюʺ смолу, отфильтровывают. ʺОтработаннуюʺ смолу промывают небольшим объемом или свежего отщепляющего коктейля или смеси TFA/DCM (15-20:80-85 об/об) (от 1 до 2 раз, используя от 1 до 2 объемов больше массы смолы). После этого может следовать необязательная промывка небольшим объемом MeOH (от 1 до 2 раз, используя от около 1 до 2 объемов больше массы смолы) (Univar, Dublin, Ireland).
Обогащенные пептидами фильтраты объединяют и выпаривают до <20% исходной массы фильтрата (<15% предпочтительно). Сырой пептид осаждают из концентрированного раствора, используя органический анти-растворитель, такой как MtBE (Univar, Dublin, Ireland) (от около 5 до 15 объемов, предпочтительно, от 6,5 до 10 объемов), фильтруют и промывают небольшими объемами того же самого органического анти-растворителя (вплоть до 3 раз, используя от около 1 до 2 объемов). Выпавший в осадок пептид можно высушить. Растворение сухого или полувлажного пептидного осадка для последующей очистки осуществляют, используя разбавленную кислоту, такую как уксусная кислота вместе с органическим растворителем, таким как ACN (INEOS Nitriles, Rolle, Switzerland) (около % об/об в зависимости от растворимости пептида и %, при котором он элюируется во время хроматографической очистки).
Очистку пептида до очень высокой степени чистоты (> 99%) вместе с солевым обменом (например, обменом соли TFA на ацетат) обеспечивают, используя препаративную ВЭЖХ с обращенной фазой (на C18 или C8 силикагеле, или на другой подходящей набивке) хорошо известную специалистам в данной области. Выделение очищенного пептида путем лиофилизации или другими способами выделения пептидного порошка из раствора (например, сушкой распылением, осаждением или кристаллизацией с последующей сушкой) хорошо известно специалистам в данной области.
При синтезе гибридных соединений, таких как гибриды дофамин-соматостатина, способ включает дополнительные стадии. Общую схему таких дополнительных стадий можно проиллюстрировать следующим образом: перед стадией (i),
Стадия h-1: дофамин активируют для присоединения путем растворения в HCTU и HOBt в ДМФ;
Стадия h-2: - основание добавляют к раствору со стадии (h-1);
Стадия h-3: раствор со стадии (h-2) перемешивают в течение 1 минуты, затем указанную смолу перемешивают в течение около 1,5 часа;
Стадия h-4: полученную смолу промывают ДМФ; и
Стадия h-5: указанную смолу дополнительно промывают 1-3 объемами MeOH.
Смесь TFA, TIPS и DCM используют для отщепления пептида от смолы и одновременного удаления защитных группы боковых цепей аминокислот, и предпочтительно, отношение TFA:TIPS:DCM составляет 15:5:80. Затем осадок промывают MtBE. И наконец, полученный осадок циклизуют.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пример 1: Синтез аналога грелина H-Inp-D-Bal-D-Trp-Phe-Apc-NH2 (как раскрыто в международной патентной заявке WO 2004/014415, которая включена для ссылки во всей полноте). В раскрытом далее способе все эквиваленты даны относительно масштаба загрузки смолы.
Указанный в заголовке пептид синтезируют в 50-литровом фильтр-реакторе (Buchi, Flawil, Switzerland).
Синтез осуществляют в масштабе 1,04 моля (1,4 кг загрузка смолы).
Приблизительно 1,41 кг Fmoc-амидной смолы Зибера набухают в ДМФ (3 раза по 10 объемов). Fmoc группы удаляют, используя две обработки 15% об/об раствором пиперидина (BASF, Schwarheide, Germany) в ДМФ (2×10 объемов, 10 минут каждая). Затем смолу промывают ДМФ (3×10 объемов).
Некоторые из используемых Fmoc-аминокислот (Apc, D-Trp) требуют Boc-защиты боковых цепей, другие Fmoc-аминокислоты (Phe, D-Bal, Inp) не требуют защиты боковых цепей.
Вводимый в реактор раствор 1,5 эквивалентов Fmoc-Apc(Boc)-OH, предварительно активируют 1,8 эквивалентами HCTU и 3 эквивалентами DIEA в ДМФ (6 объемов). Раствор и смолу перемешивают в течение приблизительно 90 минут. Смолу осушают и промывают ДМФ (2×10 объемов). Fmoc группы удаляют, как раскрыто выше, и присоединяют вторую аминокислоту, Fmoc-Phe-OH, используя те же самые условия, которые были указаны для Fmoc-Apc(Boc)-OH. Цикл удаления Fmoc, промывки и присоединения Fmoc-аминокислоты и промывки повторяют для Fmoc-D-Trp(Boc)-OH, Fmoc-D-Bal-OH и Fmoc-Inp-OH на стадиях присоединения Fmoc-аминокислоты, используя 1,45 эквивалентов TBTU, 1,45 эквивалента HOBt и 2,25 эквивалента DIEA в ДМФ (6-7 объемов). Время присоединения составляет 60 минут.
После завершения сборки пептида на амидной смоле Зибера, смолу промывают ДМФ и затем дополнительно дважды промывают 10 литрами метанола и сушат.
Пептид отщепляют от смолы, и защитные группы его боковых цепей удаляют, используя 10 объемов отщепляющего коктейля, включающего TFA/TIPS/DCM (20/10/70% об/об), в течение 2,5 часа. Пептид-содержащий фильтрат выпаривают при пониженном давлении, осаждают и промывают MtBE перед тем, как растворяют в разбавленной уксусной кислоте и ацетонитриле для последующей очистки. Выход продукта синтеза составляет 80,8%, степень чистоты по данным ВЭЖХ составляет 90,0%.
Пептид очищают, используя колонку препаративной ВЭЖХ с обращенной фазой (Novasep, Pompey, France), набитую C18 стационарной фазой (EKA Chemicals AB, Bohus, Sweden). Очистку и солевой обмен осуществляют, используя градиентное элюирование, используя в качестве буферов ацетат аммония и уксусную кислоту и ацетонитрил в качестве органического модификатора.
Пример 2: Синтез гибрида дофамин-соматостатина формулы

(т.e. Dop2-D-Lys(Dop2)-c[Cys-Tyr-D-Trp-Lys-Abu-Cys]-Thr-NH2, как раскрыто в международной патентной заявке WO 2004/091490, которая включена в описание по ссылке во всей своей полноте).
В раскрытом далее способе все эквиваленты приведены относительно масштаба загрузки смолы.
Указанный в заголовке пептид синтезируют в 50-литровом фильтр-реакторе.
Синтез осуществляют в масштабе 0,72 моля (1,2 кг загрузка смолы).
Используемые защищенные аминокислоты можно получить из Synthetech, Inc., Albany, Oregon, USA или Senn Chemicals, Dielsdorf, Switzerland
Около 1,2 кг Fmoc-амидной смолы Зибера набухают в ДМФ (3×10 объемов) в реакторе и Fmoc группы удаляют, используя две обработки 15% об/об раствором пиперидина в ДМФ (10 объемов на обработку длительностью 10 минут). Смолу промывают ДМФ (4×10 объемов).
Первую аминокислоту, которую необходимо присоединить к смоле, Fmoc-Thr(tBu)-OH (2,0 эквивалента, TBTU (1,96 эквивалента), HOBt (1,96 эквивалента) и DIEA (3,0 эквивалента в ДМФ (5,5 объемов) перемешивают со смолой в течение 60 минут. Смолу выделяют, и Fmoc-Thr(tBu)-OH снова присоединяют, используя Fmoc-Thr(tBu)-OH (1,0 эквивалент), TBTU (0,98 эквивалента), HOBt (0,98 эквивалента) и DIEA (1,5 эквивалента) в ДМФ (2,8 объема) в течение 60 минут.
Смолу промывают ДМФ (4×10 объемов).
Fmoc группы удаляют, как раскрыто выше, и вторую аминокислоту, Fmoc-Cys(Acm)-OH, присоединяют, используя те же самые условия, что указаны для Fmoc-Thr(tBu)-OH. Цикл удаления Fmoc, промывки, присоединения Fmoc-аминокислоты и промывки повторяют для Fmoc-Abu-OH, Fmoc-Lys(Boc)-OH, Fmoc-D-Trp(Boc)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Cys(Acm)-OH и Fmoc-D- Lys(Fmoc)-OH в указанном порядке. Стадии присоединения Fmoc-аминокислот осуществляют, используя TBTU (1,96 эквивалента), HOBt (1,96 эквивалента) и DIEA (3,0 эквивалента) в ДМФ (5,8-7 объемов) в течение 60 минут. Дофаминовая часть указанной в заголовке молекулы, т.е.

(Biomeasure, Inc., Milford, MA, USA) активируют для присоединения путем растворения ее (2,75 молярных эквивалентов относительно смолы), HCTU (2,79 эквивалента) и HOBt (3,3 эквивалента) в ДМФ (12,3 объема на грамм смолы), добавления DIEA (6,27 эквивалента) и перемешивания в течение 1 минуты перед перемешиванием со смолой в течение 1,5 часа.
После окончательной промывки пептидильной смолы, используя ДМФ, смолу дополнительно промывают MeOH (2×10 объемов) и сушат.
Отщепление гибридного пептида от смолы и удаление защитных групп боковых цепей осуществляют в одном реакторе, используя смесь TFA:TIPS:DCM (15:5:80, 12 объемов, 34,3 л) в течение 2,0 часов. Используют периодическое барботирование отщепляющей реакционной смеси газообразным азотом (длительностью 1-2 минуты каждые 30 минут). После отфильтровывания отщепляющей смеси (которая содержит целевой пептид) от смолы, ʺотработаннуюʺ смолу промывают смесью TFA/DCM (15:85) (1,3 объема от массы смолы, 3 раза). Фильтраты с большим содержанием пептидов объединяют и упаривают до 10,4% (6,2 кг) от исходной массы фильтрата. Сырой пептид осаждают из концентрированного раствора, добавляя к перемешиваемой MtBE (6,5 объемов относительно массы остатка после упаривания, 40 л), фильтруют и промывают MtBE (1 объем на массу, оставшуюся после упаривания, 6,2 л, один раз). Растворение полусухого осадка пептида для последующей циклизации осуществляют, используя 38 объемов (45 л) относительно массы смолы 0,1% об/об TFA/вода, с ACN (30% об/об).
В результате синтеза/отщепления выход составляет 72,6%, степень чистоты по данным ВЭЖХ составляет 79,1%.
Примеры терапевтических пептидов, которые можно синтезировать, используя раскрытый в описании новый процесс, включают, но ими не ограничиваются, следующие:














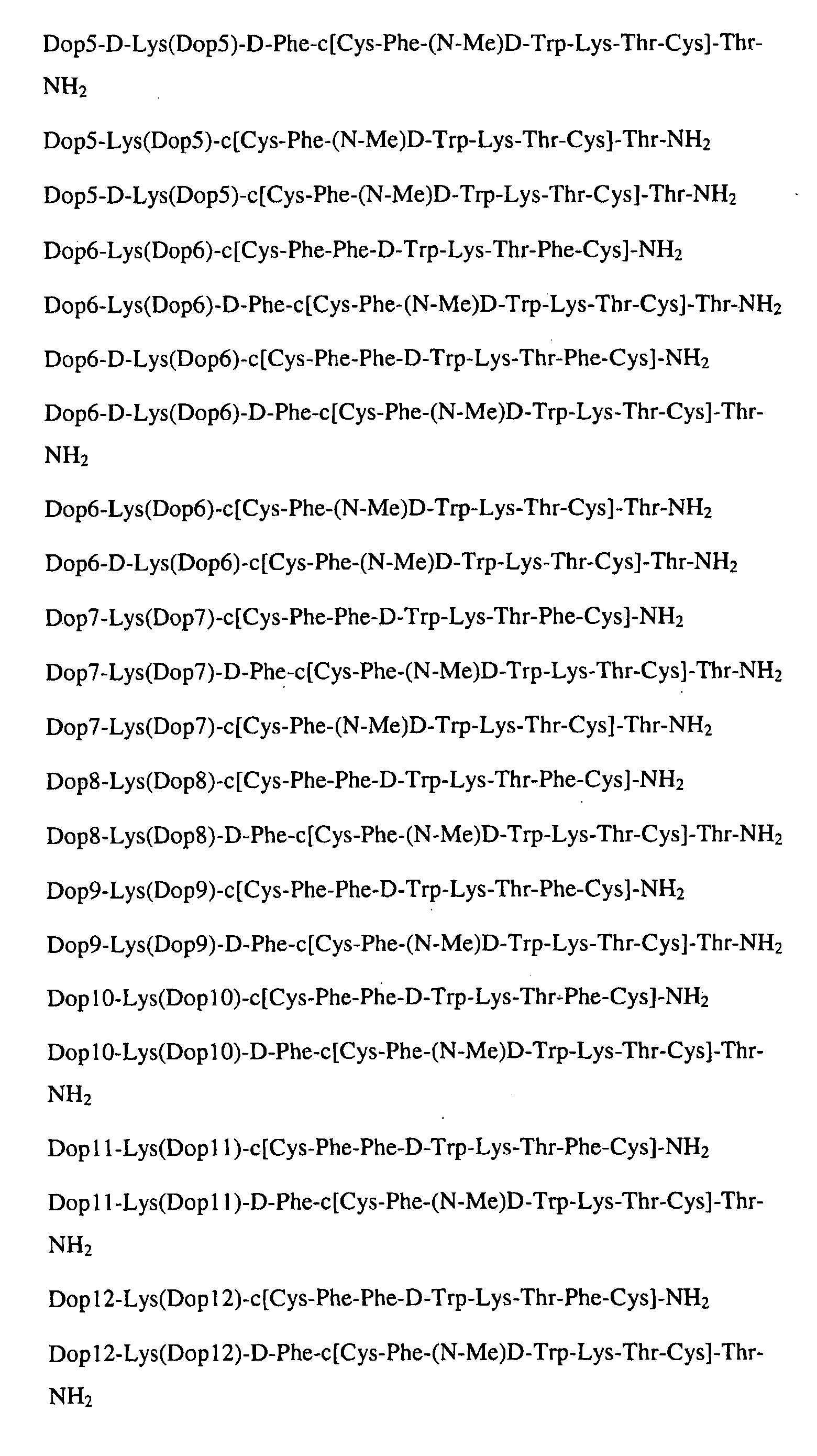
Гибриды дофамин-соматостатина, которые можно синтезировать, используя заявленный способ, включают, но ими не ограничиваются, те молекулы, которые раскрыты в WO 02/100888 и WO 04/091490, а именно:































ДРУГИЕ ВАРИАНТЫ
Из вышеприведенного описания специалисты в указанной области могут легко понять существенные характеристики настоящего изобретения, и, не выходя за рамки сути и объема изобретения, смогут внести в изобретение различные изменения и модификации, чтобы адаптировать его к различным использованиям и условиям. Таким образом, другие варианты также попадают в объем формулы изобретения.
Реферат
Изобретение относится к способу крупномасштабного синтеза терапевтических пептидов, представляющих собой аналоги грелина, методом поэтапной твердофазной Fmoc-химии с использованием амидной смолы Зибера. 13 з.п. ф-лы, 3 ил., 2 пр.
Формула

Документы, цитированные в отчёте о поиске
Новые пептиды как ингибиторы ns3-серинпротеазы вируса гепатита c
Патенты аналоги
Новые пептиды как ингибиторы ns3-серинпротеазы вируса гепатита c
Комментарии