Новые способы получения барусибана и его промежуточных соединений - RU2726414C2
Код документа: RU2726414C2
Чертежи

Описание
Область изобретения
Настоящее изобретение относится к новым твердофазным способам пептидного синтеза аналогов, которые проявляют активность антагонизма окситоцина, в частности, барусибана и его промежуточных соединений, и которые являются пригодными, среди прочего, для снижения и блокирования сокращения мышц матки.
Уровень техники, к которому относится изобретение
Окситоцин представляет собой пептидный гормон, который стимулирует сокращение мышц матки, и полагают, что он вовлечен в этиологию преждевременных родов и дисменореи. Антагонисты окситоцина оказались полезными для контроля этих состояний, и пептиды-антагонисты окситоцина с высокой эффективностью и селективностью для терапевтического применения описаны в WO 95/02609, опубликованной 26 января 1995 года. Они часто предназначены для введения в водном растворе, и производство готовых для применения доз таких антагонистов может потребовать, чтобы такие растворы были стабильными в течение длительных периодов времени; что не всегда соответствует истине. Потенциальная необходимость в приготовлении такого лекарственного средства непосредственно перед применением была сочтена неудобной и было осуществлено улучшение.
Барусибан представляет собой синтетический циклический гептапептид, содержащий пять неприродных аминокислот, одна из которых представляет собой D-аминокислоту. Его регистрационный номер CAS представляет собой 285571-64-4 (для свободного основания). Лекарственное вещество химически называется C4,6,S1-цикло(N-(3-сульфанилпропаноил)-D-триптофил-L-изолейцил-L-аллоизолейцил-L-аспарагинил-L-2-аминобутаноил-N-метил-L-орнитинол), и оно соответствует химической структуре, представленной ниже:
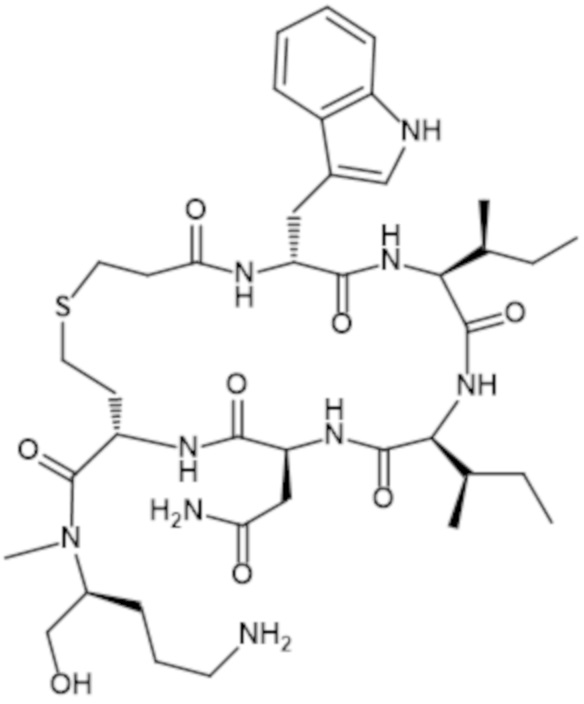
Структура барусибана также может быть представлена как:
c[Pra-D-Trp-Ile-AlloIle-Asn-hCy]-N-Me-Orn-ол,
где Pra представляет собой пропионовую кислоту, Trp представляет собой триптофан, Ile представляет собой изолейцин, AlloIle представляет собой аллоизолейцин, Asn представляет собой аспарагин, hCy представляет собой гомоцистеин и Orn представляет собой орнитин. "c" означает, что последовательность в скобках ([Pra-D-Trp-Ile-AlloIle-Asn-hCy]) имеет циклическую форму.
Для целей описания настоящего изобретения каждая аминокислота в барусибане приводится под кратким обозначением следующим образом:
c[AA1-AA6]-AA7-ол,
где AA1 представляет собой пропионовую кислоту (pra), AA2 представляет собой D-Trp, AA3 представляет собой Ile, AA4 представляет собой AlloIle, AA5 представляет собой Asn, AA6 представляет собой hCy и AA7 представляет собой N-Me-Orn-ол.
В патенте США № 6143722 (EP 938496; WO 98/23636) описаны эквивалентные гептапептидные аналоги, которые демонстрируют активность антагониста окситоцина, которые сходны с аналогами, описанными ранее в заявке WO 95/02609, но где C-конец пептида восстановлен до спирта.
Такие пептиды-антагонисты окситоцина можно синтезировать способом синтеза, описанным в патенте США № 6143722. Это требует приблизительно 7 отдельных стадий, считая синтез пептида в качестве одной и не учитывая синтез модифицированного остатка гомоцистеина (hCy).
Попытка осуществить полный способ SPPS была описана в WO2003072597. В этом документе описан способ получения гептапептидного аналога или его фармацевтически приемлемой соли, обладающих активностью антагониста окситоцина и состоящих из гексапептидной части A и C-концевого остатка β-аминоспирта B, связанного с частью A амидной связью, где (1) β-аминоспирт B представляет собой:
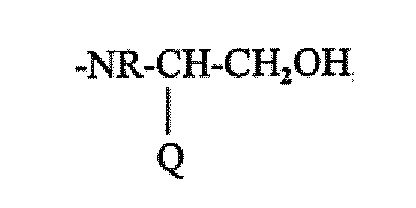
причем Q представляет собой (CH2)n-NH2, n равен 2, 3 или 4, и R представляет собой CH3 или C2H5; и (2) часть A представляет собой:
Mpa-AAb-Ile-AAd-Asn-Abu-, и причем AAb представляет собой D-ароматическую α-аминокислоту, боковая цепь которой необязательно может быть защищена; и AAd представляет собой алифатическую α-аминокислоту.
В этом синтезе обычно используется такой алкилированный диаминоспирт и защищенная аминокислота, обозначаемая как Fmoc-carba-6, т.е.
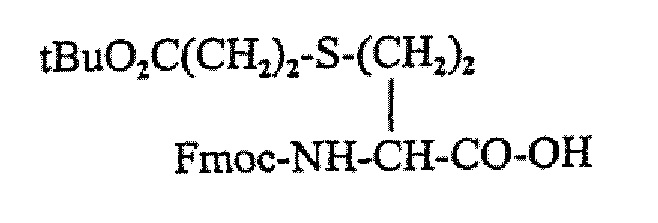
В этом способе циклизацию проводят в растворе через амидную связь (связывание Fmoc-hCy((CH2)2-COOBu)-OH) с AAb, где AAb может представлять собой D-Trp-OH). Таким образом, этот подход требует синтеза ортогонально защищенных производных гомоцистеина (например, Fmoc-hCy((CH2)2-COOBu)-OH)), что может быть трудоемким при производстве в крупном масштабе.
В документе CN 2012-1036484 описан способ получения барусибана с использованием SPPS. Способ включает стадии (1) проведения реакции Fmoc-N-Me-Orn(Boc)-ола с карбокси-смолой с получением Fmoc-N-Me-Orn(Boc)-смолы; (2) затем присоединения Fmoc-hCy(mmt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-AlloIle-OH, Fmoc-Ile-OH, Fmoc-D-Trp(Boc)-OH и 3-галогенированной пропионовой кислоты с получением X-CH2CH2CO-D-Trp(Boc)-Ile-AlloIle-Asn(Trt)-hCy(mmt)-N-Me-Orn(Boc)-смолы; и (3) удаления защитной группы mmt боковой цепи для проведения циклизации полученного линейного пептида на смоле с последующим отщеплением от смолы с получением барусибана.
Хотя такие пептиды-антагонисты окситоцина можно синтезировать способом синтеза, описанным, например, в патенте США № 6143722, WO2003072597, CN102875650 и CN 2012-1036484, для химических соединений, потенциально представляющих коммерческий интерес, часто пытаются найти более экономичный синтез, применимый в крупном масштабе, с улучшенным выходом, лучшим профилем чистоты и/или более надежными процессами. Настоящее изобретение удовлетворяет эту потребность.
Сущность изобретения
Как правило, настоящее изобретение относится к твердофазному синтезу гептапептида барусибана. Его регистрационный номер CAS представляет собой 285571-64-4 (для свободного основания). Его химическим названием является C4,6,S1-цикло(N-(3-сульфанилпропаноил)-D-триптофил-L-изолейцил-L-аллоизолейцил-L-аспарагинил-L-2-аминобутаноил-N-метил-L-орнитинол).
Настоящее изобретение также относится к гептапептидным аналогам или к их фармацевтически приемлемым солям, обладающим активностью антагониста окситоцина, и к новым способам твердофазного пептидного синтеза для их получения.
Настоящее изобретение относится к твердофазному способу получения соединения, имеющего формулу c[AA1-AA6]-AA7-ол, или его фармацевтически приемлемой соли или сольвата, где AA1 представляет собой пропионовую кислоту, AA2 представляет собой AAb, AA3 представляет собой Ile, AA4 представляет собой AAd, AA5 представляет собой Asn, AA6 представляет собой hCy, и AA7 представляет собой -NR-CHQ-CH2OH, где R представляет собой CH3 или C2H5, предпочтительно CH3, Q представляет собой (CH2)n-NH2, где n равен 2, 3 или 4, предпочтительно 3, причем способ включает стадии:
a) реакция защищенной (P5;P3)AA7 со смолой с получением (P5;P3)AA7-СМОЛЫ,
где AA7, присоединяемая к смоле в процессе синтеза на стадии a), представляет собой (P5)NR-CHQ'-CH2OH, где R представляет собой CH3 или C2H5, Q' представляет собой (CH2)n-N P3P4;
b) пошаговое удлинение (P5;P3)AA7-смолы с получением (P7)AA2AA3AA4(P1)AA5(P2)AA6-(P3)AA7-СМОЛЫ;
c) реакция X-AA1 (где X представляет собой атом галогена, выбранный из F, Cl, Br и I) с (P7)AA2AA3AA4(P1)AA5(P2)AA6-(P3)AA7-СМОЛОЙ с получением X-AA1(P7)AA2AA3AA4(P1)AA5(P2)AA6-(P3)AA7-СМОЛЫ;
d) проведение стадии отщепления и удаления защитной группы с получением X-AA1AA2AA3AA4AA5AA6-AA7-ола; и
e) циклизация X-AA1AA2AA3AA4AA5AA6-AA7-ола с получением циклического пептида c[AA1-AA6]-AA7-ола, где AA1 и AA6связаны через тиол из гомоцистеина AA6,
где:
P1, P5 и P7 представляют собой защитные группы,
AAb представляет собой D-ароматическую α-аминокислоту;
AAd представляет собой алифатическую α-аминокислоту;
X представляет собой галоген;
P2представляет собой Trt
R представляет собой CH3 или C2H5;
Q' представляет собой (CH2)n-NP3P4;
n равен 2, 3 или 4;
P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга и которые могут быть такими же или отличными от P1и/или P2.
В частности, настоящее изобретение относится к твердофазному процессу получения соединения, имеющего формулу c[AA1-AA6]-AA7-ол, или его фармацевтически приемлемой соли или сольвата, где AA1 представляет собой пропионовую кислоту, AA2 представляет собой AAb, предпочтительно D-Trp, AA3 представляет собой Ile, AA4 представляет собой AAd, предпочтительно AlloIle, AA5 представляет собой Asn, AA6 представляет собой hCy; AA7 представляет собой NR-CHQ-CH2OH, где R представляет собой CH3 или C2H5, Q представляет собой (CH2)n-NH2, где n равен 2, 3 или 4, где предпочтительно R представляет собой CH3 и n равен 3, где AA7 предпочтительно представляет собой N-Me-Orn-ол, причем способ включает стадии:
Стадия 1a
a) реакция защищенной (P5;P3)AA7 со смолой с получением (P5;P3)AA7-СМОЛЫ;
где AA7, присоединяемая к смоле в ходе синтеза, представляет собой (P5)NR-CHQ'-CH2OH, где R представляет собой CH3 или C2H5, Q' представляет собой (CH2)n-N P3P4;
b) удаление P5 из (P5;P3)AA7-СМОЛЫ с получением (P3)AA7-СМОЛЫ;
Стадия 1b
c) реакция защищенной (P6;Trt)AA6 с (P3)AA7-СМОЛОЙ с получением (P6;Trt)AA6-(P3)AA7-СМОЛЫ;
d) удаление P6 из (P6;Trt)AA6-(P3)AA7-СМОЛЫ с получением (Trt)AA6-(P3)AA7-СМОЛЫ;
e) реакция защищенной (P6;P1)AA5 с (Trt)AA6-(P3)AA7-СМОЛОЙ с получением (P6;P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
f) удаление P6 из (P6;P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ с получением (P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
g) реакция защищенной (P6)AA4 с (P1)AA5(Trt)AA6-(P3)AA7-СМОЛОЙ с получением (P6)AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
h) удаление P6 из (P6)AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ с получением AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
i) реакция защищенной (P6)AA3 с AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛОЙ с получением (P6)AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
j) удаление P6 из (P6)AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ с получением AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
k) реакция защищенной (P6;P7)AA2 c AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛОЙ с получением (P6;P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
Стадия 1c
l) удаление P6 из (P6;P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ с получением (P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
m) реакция X-AA1 (где X представляет собой атом галогена, выбранный из F, Cl, Br и I) с (P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛОЙ с получением X-AA1(P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
Стадия 2
n) проведение стадии отсщепления и удаления защитной группы с получением X-AA1AA2AA3AA4AA5AA6AA7-ола; и
Стадия 3
o)циклизация X-AA1AA2AA3AA4AA5AA6AA7-ола через простую тиоэфирную связь посредством внутримолекулярного замещения галогена X AA6(гомоцистеин)тиолом, с получением циклического пептида c[AA1-AA6]-AA7-ола,
где P1, P5, P6 и P7 представляют собой защитные группы и где P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга и которые могут быть такими же или отличающимися от P1, и где n равен 2, 3 или 4.
Кроме того, настоящее изобретение относится к промежуточному соединению, пригодному для получения пептида, имеющего фармацевтические свойства, который имеет формулу:
X-CH2CH2CO-AAb-Ile-AAd-Asn(P1)-hCy(P2)-NR-CHQ'-CH2OW
где
AAb представляет собой D-ароматическую α-аминокислоту;
AAd представляет собой алифатическую α-аминокислоту;
X представляет собой галоген (F, Cl, Br, I);
P1 представляет собой защитную группу
P2 представляет собой защитную группу (Trt)
R представляет собой CH3 или C2H5;
Q' представляет собой (CH2)n-NP3P4;
n равен 2, 3 или 4;
P3и P4независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга и которые могут быть такими же или отличными от P1 и/или P2; и
W представляет собой H (а именно, C-конец представляет собой спирт), защитную группу или смолу.
В одном варианте осуществления X не является Cl, когда P5 представляет собой Fmoc.
P5 предпочтительно представляет собой o-NBS (NBS).
Используют следующие сокращения:
-Abu: 2-аминомасляная кислота
-AcOH: уксусная кислота
-Ac2O: уксусный ангидрид
-AlloIle/aIle: аллоизолейцин
-Asn: аспарагин
-BH3THF: комплекс боран-тетрагидрофуран
-Boc: трет-бутилоксикарбонил
-Br: бром
-Br-(CH2)2-COOH: 3-бромпропионовая кислота
-CH3CN: ацетонитрил
-Cl: хлор
-Cl-(CH2)2-COOH: 3-хлорпропионовая кислота
-CTC: хлортритилхлорид
-DBU: 1,8-диазабицикло[5.4.0]ундец-7-ен
- MeCN: ацетонитрил
-DCM: дихлорметан
-DIAD: диизопропилазодикарбоксилат
-DIC: 1,3-диизопропилкарбодиимид
-DIEA: N,N-диизопропилэтиламин
-DIPEA: N,N-диизопропилэтиламин
-DME: 1,2 диметоксиэтан
-DMF: N,N-диметилформамид
-DMAP: 4-диметиламинопиридин
-DTT: дитиотреитол
-EDT: этандитиол
-Et2O: этиловый эфир
-EtOAc: этилацетат
-Fmoc: 9-флуоренилметилоксикарбонил
-Fmoc-N-MeOrn(Boc)-ол: метил-N-α-(9-флуоренилметоксикарбонил)-N-δ-Boc-L-орнитол
-Fmoc-hCy(Trt)-OH: N-α-(9-флуоренилметоксикарбонил)-S-тритил-L-гомоцистеин
-Fmoc-hCy(mmt)-OH: N-α-(9-флуоренилметоксикарбонил)-S-метокситритил-L-гомоцистеин
-Fmoc-Asn(Trt)-OH: N-α-(9-флуоренилметоксикарбонил)-N-β-тртил-L-аспарагин
-Fmoc-aIle-OH: N-α-(9-флуоренилметоксикарбонил)-N-L-аллоизолейцин
-Fmoc-Ile-OH: N-α-(9-флуоренилметоксикарбонил)-N-L-изолейцин
-Fmoc-D-Trp(Boc)-OH: N-α-(9-флуоренилметоксикарбонил)-N(in)-Boc-D-триптофан
-FmocONSu: N-(9-флуоренилметоксикарбонилокси)сукцинимид
-hCy/hCys/Hcy: гомоцистеин
-HOBt: 1-гидроксибензотриазол
-ВЭЖХ: высокоэффективная жидкостная хроматография
-H2O: деионизированная вода
-IPA: изопропиловый спирт
-LC/MS: жидкостная хроматография-масс-спектрометрия
-MeOH: метанол
-Mpa: 3-меркаптопропионовая кислота
-MTBD: 7-метил-1,5,7-триазабицикло[4.4.0]дец-5-ен
-MTBE: метил трет-бутиловый эфир
-Na2CO3: карбонат натрия
-NMM: N-метилморфолин
-NMP: 1-метилпирролидин-2-он
-Orn: орнитин
-o-NBS (NBS): o-нитробензолсульфонил
-o-NBS-Cl: o-нитробензолсульфонилхлорид
-PhOH: фенол
-PyBOP: гексафторфосфат бензотриазол-1-илокси-трис-пирролидинoфосфония
-RRT: относительное время удержания
-SPPS: твердофазный пептидный синтез
-TBTU: тетрафторборат 2-(1-H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония
-TEAP: фосфат триэтиламмония
-TFE: трифторэтанол
-TFA: трифторуксусная кислота
-THF: тетрагидрофуран
-TIS: триизопропилсилан
-TNBS: 2,4,6-тринитробензолсульфоновая кислота
-TMSBr: триметилсилилбромид
-TPP: трифенилфосфин
-Trp: триптофан
-Trt: тритил
-Z: бензилоксикарбонил
Краткое описание чертежей
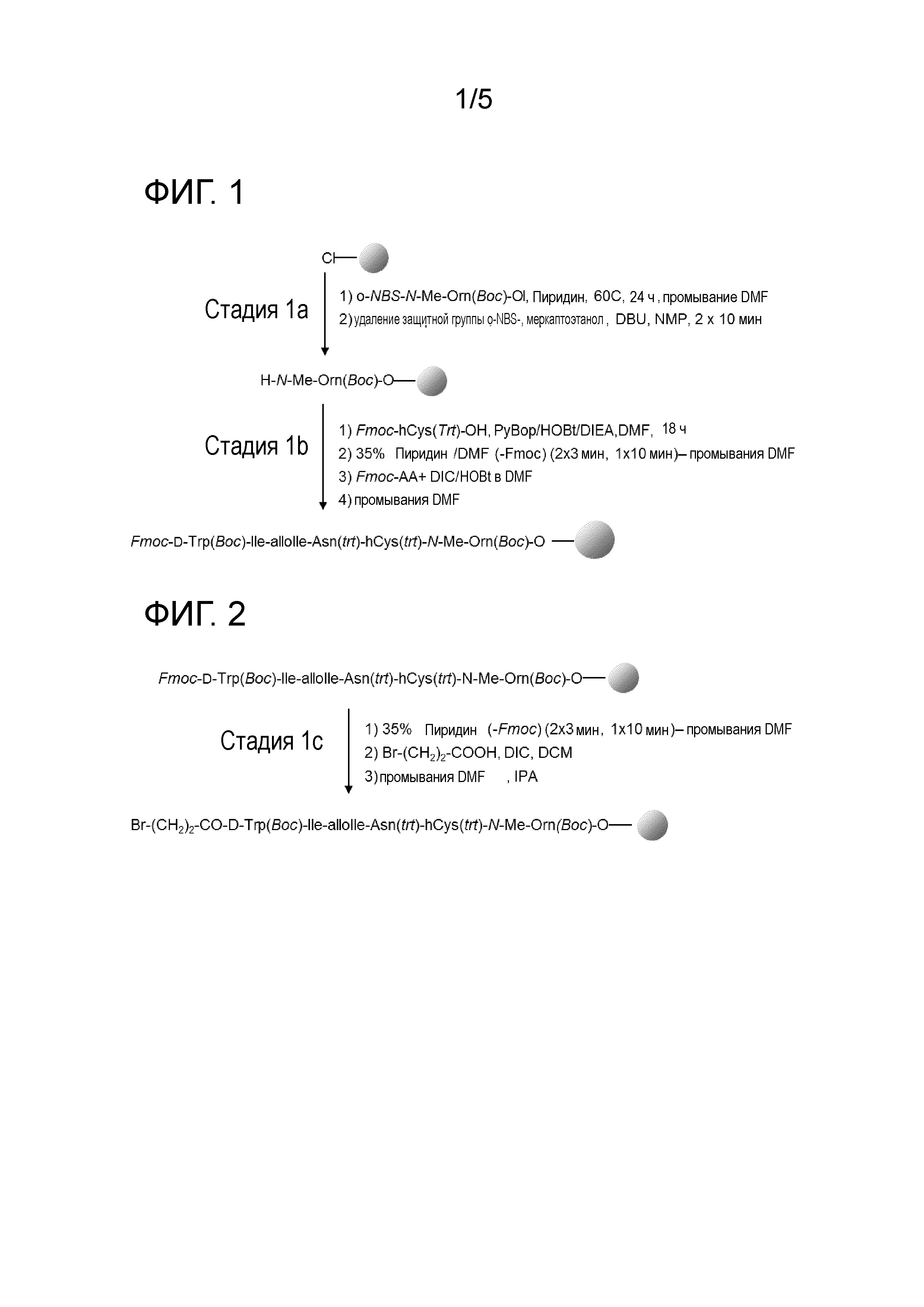
Фиг. 1: Присоединение аминокислот 2 и 7.
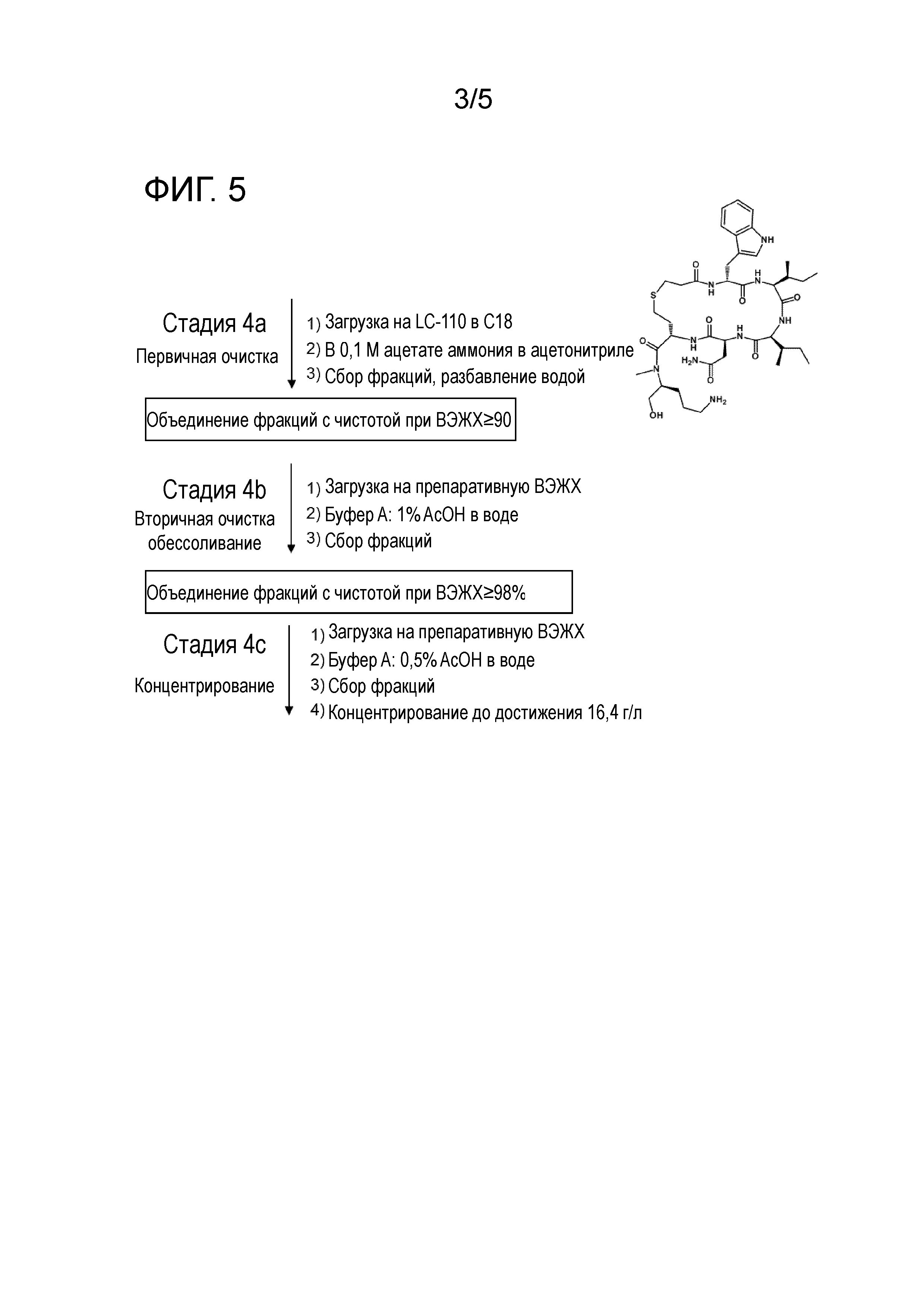
Фиг. 2: Присоединение аминокислоты 1.
Фиг.3: Удаление защитной группы и отщепление гептапептида от смолы с получением линейного пептида.
Фиг.4: Циклизация пептида в растворе с получением барусибана.
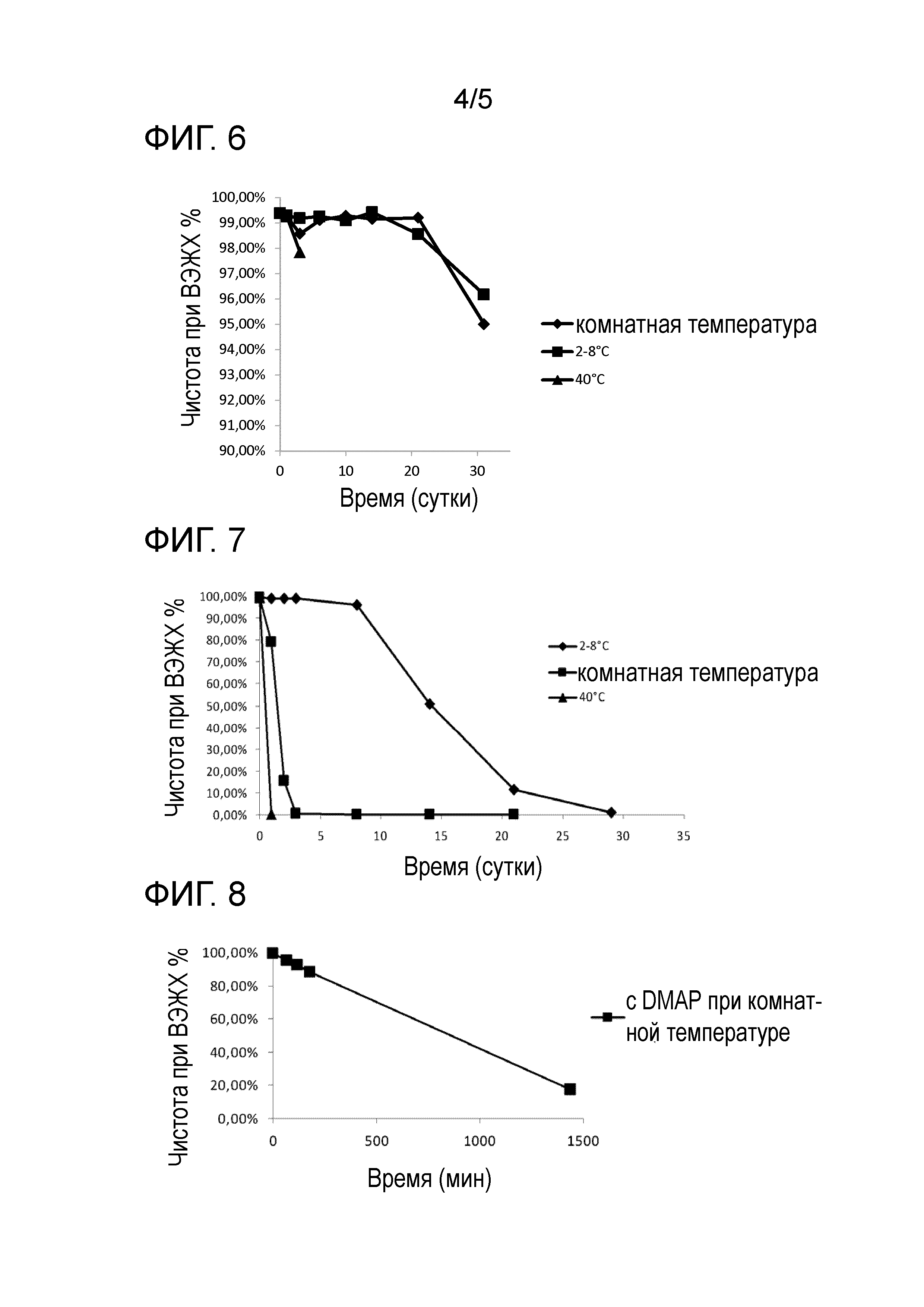
Фиг.5: Очистка циклического пептида.
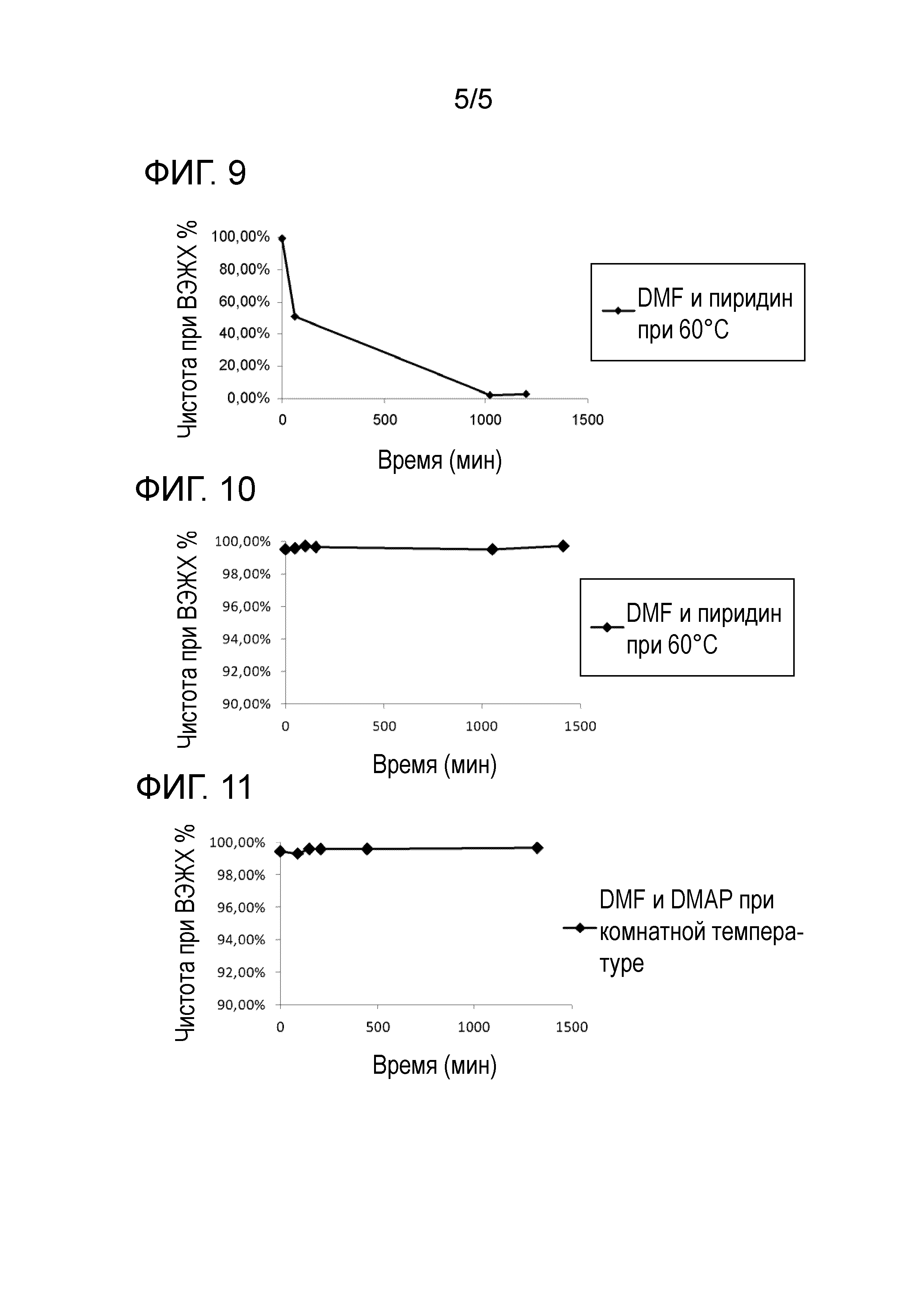
Фиг.6: Исследование стабильности Fmoc-N-Me-L-Orn(Boc)-ола в виде порошка при различных температурах хранения.
Фиг.7: Исследование стабильности раствора Fmoc-N-Me-L-Orn(Boc)-ола в DMF при различных температурах.
Фиг.8: Исследование стабильности раствора Fmoc-N-Me-L-Orn(Boc)-ола+DMAP в DMF при комнатной температуре.
Фиг.9: Исследование стабильности раствора Fmoc-N-Me-L-Orn(Boc)-ола+пиридин в DMF при 60°C.
Фиг.10: Исследование стабильности раствора o-NBS-N-Me-L-Orn(Boc)-ола+пиридин в DMF при 60°C.
Фиг.11: Исследование стабильности раствора o-NBS-N-Me-L-Orn(Boc)-ола+DMAP в DMF при комнатной температуре.
Подробное описание изобретения
Настоящее изобретение относится к твердофазному способу получения циклического гептапептидного аналога, предпочтительно барусибана или его фармацевтически приемлемой соли, обладающего активностью антагониста, включающему стадии:
Стадия 1a
1. Присоединение P5NR-CHQ'-CH2OH (первая аминокислота, AA7), где P5представляет собой защитную группу аминокислоты, предпочтительно NBS, к смоле. R представляет собой CH3 или C2H5. Q' представляет собой (CH2)n-NP3P4, где n равен 2, 3 или 4, и P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга. Предпочтительно R представляет собой CH3, n равен 3, P4 представляет собой H, P3 представляет собой Boc и P5 представляет собой o-NBS (NBS).
Твердая подложка
Твердая подложка, которую можно использовать в способе по настоящему изобретению, конкретно не ограничена, и для этого процесса можно использовать по существу все твердые подложки, которые используются для твердофазного синтеза пептидов. В настоящей заявке твердую подложку также обозначают как "-СМОЛА". Иллюстративными твердыми положками являются хлорметилированные смолы, гидроксиметилированные смолы, смола MBHA, смола BHA, смола NAMM, амидная cмола AM Ринка, амидная смола MBHA Ринка, амидная смола MBHA Ринка, смолы SASRIN, смолы Зибера, смолы Ванга, сверхчувствительные к действию кислот смолы, такие как хлортритильные смолы. Особенно предпочтительными являются смолы, способные образовывать простую эфирную связь с алифатическим спиртом, например, такие как тритильная смола, хлортритильная смола (CTC) и/или смола 2-CTC. Предпочтительно смола представляет собой хлортритильную смолу, еще более предпочтительно смолу 2-CTC.
Предпочтительно, первой аминокислотой, подлежащей связыванию со смолой (AA7), является NBS-N-Me-Orn(P3)-ол. Предпочтительно P3 представляет собой Boc. Выбор этой аминокислоты из других аминокислот, которые можно использовать (например, Fmoc-N-Me-Orn(Boc)-ол) является следствием ее высокостабильного профиля, как можно видеть в экспериментальной части настоящего писания.
Перед присоединением первой аминокислоты из смолы удаляют защитную группу, если это необходимо.
Первую аминокислоту предпочтительно добавляют к смоле (предпочтительно c удаленной защитной группой) в количестве приблизительно от 0,5 до 1,5 эквивалента, например, 0,5 эквивалента, 0,6 эквивалента, 0,7 эквивалента, 1 эквивалента, 1,3 эквивалента или 1,5 эквивалента. Предпочтительно, первую аминокислоту (которая предпочтительно представляет собой NBS-N-Me-Orn(Boc)-ол) добавляют к смоле (предпочтительно с удаленной защитной группой) в количестве приблизительно 0,7 эквивалента. Эквиваленты представляют собой молярные эквиваленты.
Предпочтительно первую аминокислоту (предпочтительно NBS-N-Me-Orn(Boc)-ол) добавляют к смоле (предпочтительно с удаленной защитной группой) в DMF. Предпочтительно, далее к реакционной смеси (смола+аминокислота+DMF) добавляют пиридин. Например, реакцию можно проводить при высоких температурах, таких как от 30 до 80°C, предпочтительно приблизительно 60°C. Время реакции может варьироваться, однако оно может составлять от 2 до 30 ч, как например, от 10 до 25 ч, как например, от 15 до 18 ч. Предпочтительно, время реакции приблизительно составляет 17 ч (например, 17 ч ± 15 мин) или приблизительно 18 ч. Время реакции может составлять приблизительно 24 ч.
После добавления первой аминокислоты к смоле оставшиеся активные центры можно кэппировать, например, с использованием DIEA/MeOH, при комнатной температуре приблизительно в течение одного часа. Специалисту в данной области известны другие способы кэппирования.
2. Удаление защитной группы (P5, которая предпочтительно представляет собой o-NBS (NBS)) из α-аминогруппы первой аминокислоты, связанной со смолой. Специалисту в данной области известны условия удаления каждой из защитных групп. Например, в Chem. Rev. 2009, 109, 2465-2504 (Albert Isidro-Llobet et al.) представлен обзор защиты аминокислот. В предпочтительном варианте осуществления, когда первая аминокислота имеет NBS в качестве α-аминозащитной группы, удаление NBS проводят с использованием β-меркаптоэтанола и DBU в NMP (меркаптоэтанол, DBU, NMP).
Стадия 1b
3. Присоединение следующих аминокислот с использованием обычных протоколов твердофазного синтеза пептидов с Fmoc в этом порядке:
P6-hCy(Trt) (AA6), P6-Asn (AA5), P6-AAd (AA4), P6-Ile (AA3) и, наконец, P6-AAb (AA2)
AAb представляет собой D-ароматическую α-аминокислоту, в которой необязательно боковая цепь может быть защищена P7.P7 представляет собой защитную группу (предпочтительно Boc). AAd представляет собой алифатическую α-аминокислоту. Предпочтительно, AAb представляет собой D-Trp и AAd представляет собой AlloIle.
Эти аминокислоты (с AA6 по AA2) присоединяют к смоле со свободной карбоксильной группой и защищенной α-аминогруппой (P6), которые могут быть одинаковыми или могут различаться, причем предпочтительной защитной группой P6 является Fmoc. Свободную карбоксильную группу предпочтительно предварительно активируют, подвергая α-аминозащищенную кислоту воздействию связующего реагента для пептидов и добавки для пептидного присоединения в органическом растворителе. Эти защищенные аминокислоты предпочтительно добавляют в количестве приблизительно 1-3 эквивалента, таком как приблизительно 1, приблизительно 1,5, приблизительно 2 или приблизительно 3 эквивалента. Например, защищенные аминокислоты AA6, AA5, AA3 и AA2 добавляют к смоле в количестве приблизительно 2 эквивалента. Например, защищенную аминокислоту AA4 добавляют к смоле в количестве приблизительно 1,5 эквивалента. Например, связующие реагенты (например, DIC/HOBt) добавляют к смоле в тех же количествах эквивалентов, что и аминокислоты (например, приблизительно 2 эквивалента для присоединения AA6, AA5, AA3 и AA2, и, например, приблизительно 1,5 эквивалента для присоединения AA4).
Как известно специалисту в данной области, после присоединения каждой аминокислоты и/или удаления защитной группы из нее можно проводить тест присоединения для подтверждения того, что присоединение/удаление защитной группы произошло. Если присоединение/удаление защитной группы не произошло, реакцию можно повторять (либо присоединение, либо удаление защитной группы). Примерами этих тестов являются "тест Кайзера" (также известный как "тест с нингидрином") (E. Kaiser, R. L. Colescott, C. D. Bossinger, P. I. Cook, Analytical Biochemistry 34 595 (1970)) или "тест с хлоранилом" (для тестирования на свободные вторичные амины, T Christensen, Acts Chemica Scandinavica B 33 (1979) 763-766).
Органический растворитель, связующий реагент для пептидов и добавка для пептидного присоединения могут представлять собой любые агенты, известные в области твердофазного пептидного синтеза.
Типичными органическими растворителями являются THF, NMP, DCM, DMF, DMSO, IPA и их смеси. Предпочтительными растворителями являются NMP, DCM и DMF или их смеси. Например, предпочтительным для выбора растворителем для реакции присоединения является DMF. Например, предпочтительным для выбора растворителем для реакции удаления защитной группы является DMF.
Типичными связующими реагентами для пептидов являются один или несколько из гексафторфосфата o-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU), гексафосфата o-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU), тетрафторбората o-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (TBTU), гексафторфосфата бензотриазол-1-илокситрис(диметиламино)фосфония (BOP), гексафторфосфата бензотриазол-1-илокситриспирролидинoфосфония (PyBOP), дихлорида N,N-бис-(2-оксо-3-оксазолидинил)фосфония (BOP-Cl), гексафторфосфата бром-трис-пирролидинoфосфония (PyBroP), изобутилхлорформиата (IBCF), 1,3-дициклогексилкарбодиимида (DCC), 1,3-диизопропилкарбодиимида (DIC), гидрохлорида 1-(диметиламинопропил)-3-этилкарбодиимида (WSCDI), N-этоксикарбонил-2-этокси-1,2-дигидрохинолина (EEDQ), изопропилхлорформиата (IPCF), тетрафторбората 2-(5-норборнен-2,3-дикарбоксимидо)-1,1,3,3-тетраметилурония (TNTU), ангидрида пропанфосфоновой кислоты (PPAA), пиридина и тетрафторбората 2-сукцинимидо-1,1,3,3-тетраметилурония (TSTU). Предпочтительными связующими реагентами являются DIC, PyBOP и HBTU.
Типичными добавками для пептидного присоединения являются 1-гидрокси-1H-бензотриазол (HOBt) и 1-гидрокси-7-азабензотриазол (HOAt), предпочтительно HOBt. В особенно предпочтительном варианте осуществления DIC и HOBt используют в комбинации. В другом варианте осуществления PyBOP и HOBt можно использовать в комбинации.
Предпочтительно, вторую аминокислоту (P6-hCy(P2)-OH, предпочтительно P6-hCy(trt)-OH, более предпочтительно Fmoc-hCy(Trt)-OH) добавляют в количестве приблизительно 1,5 эквивалента или приблизительно 2 эквивалентов. Предпочтительно это присоединение проводят в присутствии PyBOP. Например, это присоединение проводят в присутствии PyBOP/HOBt/DIEA в DMF.
Предпочтительно, третью присоединяемую кислоту (P6-Asn(P1)-OH, предпочтительно P6-Asn(Trt)-OH, еще более предпочтительно Fmoc-Asn(Trt)-OH) добавляют в количестве приблизительно 2 эквивалентов. Предпочтительно это присоединение проводят в присутствии DIC/HOBt, предпочтительно в DMF, предпочтительно в количестве приблизительно 2 эквивалентов относительно добавляемой аминокислоты.
Предпочтительно четвертую присоединяемую аминокислоту (P6-AAd-OH, предпочтительно P6-AlloIle-OH, еще более предпочтительно Fmoc-AlloIle-OH) добавляют в количестве приблизительно 1,5 эквивалента. Предпочтительно это присоединение проводят в присутствии DIC/HOBt, предпочтительно в DMF, предпочтительно в количестве приблизительно 1,5 эквивалента относительно добавляемой аминокислоты.
Предпочтительно, пятую присоединяемую аминокислоту (P6-Ile-OH, предпочтительно Fmoc-Ile-OH) добавляют в количестве приблизительно 2 эквивалентов. Предпочтительно, это присоединение проводят в присутствии DIC/HOBt, предпочтительно в DMF, предпочтительно в количестве приблизительно 2 эквивалентов.
Предпочтительно шестую присоединяемую аминокислоту (P6-AAb(P7)-OH, предпочтительно P6-D-Trp(Boc)-OH, еще более предпочтительно Fmoc-D-Trp(Boc)-OH), добавляют в количестве приблизительно 2 эквивалентов. Предпочтительно, это присоединение проводят в присутствии DIC/HOBt, предпочтительно в DMF, предпочтительно в количестве приблизительно 2 эквивалентов относительно добавляемой аминокислоты.
Связующий реагент для пептидов обычно добавляют в количестве приблизительно от 1,5 до 4, таком как приблизительно 1,5, приблизительно 2, приблизительно 3 или приблизительно 4 эквивалента, предпочтительно приблизительно 1,5 или приблизительно 2 эквивалента относительно добавляемой аминокислоты (эквиваленты представляют собой молярные эквиваленты).
После каждой стадии присоединения α-аминозащитную группу (P6 для от второго до шестого присоединения, которая предпочтительно представляет собой Fmoc) необходимо удалять перед последующей стадией присоединения. Такая стадия известна в данной области. Если α-аминозащитная группа P6 представляет собой Fmoc, что является предпочтительным, как утверждалось выше, удаление предпочтительно проводят посредством реакции с пиперидином, DBU (1,8-диазабицикло[5.4.0]-ундец-7-ен) или DEA (диэтиламин). Предпочтительным растворителем является DMF. Предпочтительно удаление группы Fmoc проводят с использованием смеси пиперидин/DMF, предпочтительно с приблизительно 35% пиперидина в DMF (пиперидин/DMF 35/65).
Получают следующую пептид-смолу:
P6-AAb-Ile-AAd-Asn(P1)-hCy(Trt)-NR-CHQ'-CH2-O-смола
AAb представляет собой D-ароматическую α-аминокислоту, предпочтительно триптофан (D-Trp).
AAd представляет собой алифатическую α-аминокислоту, предпочтительно аллоизолейцин (alloIle).
R представляет собой CH3 или C2H5. Q' представляет собой (CH2)n-NP3P4, где n равен 2, 3 или 4, и P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга. Предпочтительно, P4 представляет собой H. Предпочтительно, R представляет собой CH3, n равен 3, P4 представляет собой H и P3 представляет собой Boc. P1, P3 и P7 могут быть одинаковыми или могут различаться. Предпочтительно, P3и P7представляют собой одинаковые защитные группы, предпочтительно Boc. Предпочтительно, P1 представляет собой Trt. Предпочтительно, P5 представляет собой NBS. P6предпочтительно является ортогональным относительно P1, P3 и P7. В Barany et al. (Barany, G.; Merrifield, R. B. J. Am. Chem. Soc. 1977, 99, 7363; Barany, G.; Albericio, F. J. Am. Chem. Soc. 1985, 107, 4936) описана концепция ортогональности в том смысле, что две или более защитных группы принадлежат к независимым классам и удаляются посредством различных механизмов. Таким образом, эти защитные группы (P1, P3и P7) выбирают так, чтобы они не удалялись на стадии удаления α-аминозащитной группы (P5 и P6) и предпочтительно так, чтобы они могли быть удалены вместе на одной стадии реакции, наиболее предпочтительно на стадии отщепления пептида от смолы. Наиболее предпочтительным для P1 является Trt, и для P3 и P7 - Boc. P6 предпочтительно представляет собой Fmoc.
В следующей таблице приведены предпочтительные защитные группы для -NH2 и -OH вместе с предпочтительными расщепляющими реагентами.
Таблица 1: Защитные группы боковых цепей и условия расщепления (Isidro-Llobet et al., Chem. Rev. 2009, 109, 2465-2504)
Стадия 1c
4. Удаление защитной группы аминокислоты P6 и присоединение галогенпропионовой кислоты (X-pra) (X-CH2CH2-COOH). X представляет собой галоген, выбранный из группы, состоящей из F, Cl, Br и I. Например, X может представлять собой Cl или Br. Предпочтительно X представляет собой Br.
Предпочтительно, эту (седьмую) присоединяемую аминокислоту (галогенпропионовая кислота (X-pra), предпочтительно 3-Br-пропионовая кислота) добавляют в количестве приблизительно 3 эквивалента. Предпочтительно это присоединение проводят в присутствии DIC/DCM, предпочтительно в количестве приблизительно 3 эквивалента относительно добавляемой аминокислоты.
Как утверждалось выше, после присоединения галогенпропионовой кислоты (X-pra) (X-CH2CH2-COOH) (предпочтительно Br-CH2CH2-COOH), можно проводить тест присоединения для подтверждения того, что присоединение произошло. Если присоединение не произошло, реакцию можно повторять (либо присоединение, либо удаление защитной группы). Примерами этих тестов являются "тест Кайзера" (также известный как "тест с нингидрином") или "тест с хлоранилом". Например, после присоединения галогенпропионовой кислоты (X-pra) (X-CH2CH2-COOH) (предпочтительно Br-CH2CH2-COOH) можно проводить тест с нингидрином для подтверждения завершения реакции.
После присоединения галогенпропионовой кислоты (X-pra) (X-CH2CH2-COOH) (предпочтительно Br-CH2CH2-COOH), пептид-смолу можно промывать DCM и IPA.
Агент пептидного присоединения обычно добавляют в количестве приблизительно от 1,5 до 4 эквивалентов (например, приблизительно 1,5, приблизительно 2 эквивалента, или приблизительно 3 эквивалента, или приблизительно 4 эквивалента, предпочтительно приблизительно 3 эквивалента в случае седьмого присоединения, как описано выше) относительно добавляемой аминокислоты (эквиваленты представляют собой молярные эквиваленты).
После присоединения галогенпропионовой кислоты (X-pra) (X-CH2CH2-COOH) (предпочтительно Br-CH2CH2-COOH) со смолой связывают следующий защищенный пептид:
X-AA1(P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛА
предпочтительно:
X-(CH2)2-CO-AAb-Ile-AAd-Asn(P1)-hCy(Trt)-NR-CHQ'-CH2O-СМОЛА,
еще более предпочтительно
Br-(CH2)2-CO-D-Trp(Boc)-Ile-alloIle-Asn(trt)-hCy(Trt)-N-Me-Orn(Boc)-O-СМОЛА
R представляет собой CH3 или C2H5, Q' представляет собой (CH2)n-NP3P4, где n равен 2, 3 или 4 и P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга. X представляет собой остаток галогена, такой как F, Cl, Br или I, предпочтительно Br. AAb представляет собой D-ароматическую α-аминокислоту, предпочтительно триптофан (D-trp). AAd представляет собой алифатическую α-аминокислоту, предпочтительно аллоизолейцин (alloIle). Предпочтительно R представляет собой CH3, n равен 3, P4 представляет собой H и P3 представляет собой Boc. Предпочтительно, P7 представляет собой Boc. Предпочтительно, P1представляет собой Trt.
В одном варианте осуществления X не является Cl, когда P5 представляет собой Fmoc.
Стадия 2
5. Удаление защитных групп боковых цепей (P1, P3, P4 и P7, если присутствуют все из них) из пептида и отщепление пептида от смолы с получением линейного пептида с удаленной защитной группой:
X-AA1AA2AA3AA4AA5AA6AA7-ол, предпочтительно
X-(CH2)2-CO-AAb-Ile-AAd-Asn-hCy-NR-CHQ-CH2OH, еще более предпочтительно
Br-(CH2)2-CO-D-Trp-Ile-alloIle-Asn-hCy-N-Me-Orn-ол
где R представляет собой CH3 или C2H5, Q представляет собой (CH2)n-NH2, и n равен 2, 3 или 4. Предпочтительно, R представляет собой CH3 и n равен 3.
X представляет собой остаток галогена, такой как Cl или Br, предпочтительно Br.
AAb представляет собой D-ароматическую α-аминокислоту, предпочтительно триптофан (D-trp).
AAd представляет собой алифатическую α-аминокислоту, предпочтительно аллоизолейцин (alloIle).
Условия отщепления зависят, в частности, от типа смолы и используемых защитных групп. Однако в соответствии с настоящим изобретением отщепление предпочтительно проводят в кислых условиях с использованием TFA.
В соответствии с настоящим изобретением эти две стадии (расщепление и удаление защитных групп боковых цепей (P1, P3, P4 и P7, если все из них присутствуют) предпочтительно объединяют в одну стадию удаления защитной группы и отщепления.
Предпочтительно отщепление и удаление защитной группы проводят на одной стадии в присутствии расщепляющего коктейля, содержащего (или альтернативно состоящего из) TFA, H2O и DTT, предпочтительно в соотношении приблизительно 86/5/9 (TFA/H2O/DTT), предпочтительно в количестве приблизительно 5 мл (расщепляющего коктейля/г пептидной смолы), в течение приблизительно 1-5 ч, предпочтительно в течение приблизительно 3 ч.
Например, отщепление можно проводить при комнатной температуре, такой как приблизительно 25°C. Отщепление можно проводить при температуре 15-30°C, такой как приблизительно 15°C, приблизительно 20°C, или приблизительно 25°C, или приблизительно 30°C. Предпочтительно отщепление проводят при комнатной температуре (к.т.).
После отщепления пептид с удаленной защитной группой и смолу можно осаждать, например, путем охлаждения посредством TFA (например, от -5 до 8°C, как например, от 2 до 8°C, предпочтительно при температуре 0°C±5°C) и добавления, например, MBTE/н-гептана, предпочтительно в соотношении 80:20, или предпочтительно в соотношении 70:30 (MBTE:н-гептан). Предпочтительно, температуру контролируют так, что она остается ниже приблизительно 20°C.
После расщепления пептид с удаленной защитной группой можно экстрагировать из пептид-смолы, например, с использованием, например, смеси вода/CH3CN: 7/3, 1,6 г/л. После расщепления неочищенный линейный пептид и смолу можно отфильтровывать и промывать, например, посредством MTBE (например, три раза в количестве приблизительно 3 мл на грамм пептид-смолы) и сушить в вакууме.
6. Необязательно очистка линейного пептида. Очистку можно проводить хроматографическими способами, такими как препаративная обращено-фазовая хроматография (RPC).
Таким образом, получают линейный пептид с удаленной защитной группой (необязательно очищенный):
X-AA1AA2AA3AA4AA5AA6AA7-ол,
предпочтительно
X-(CH2)2-CO-AAb-Ile-AAd-Asn-hCy-NR-CHQ-CH2-OH,
более предпочтительно
Br-(CH2)2-CO-D-Trp-Ile-alloIle-Asn-hCy-N-Me-Orn-ол.
R представляет собой CH3 или C2H5, Q представляет собой (CH2)n-NH2, и n равен 2, 3 или 4. Предпочтительно, R представляет собой CH3 и предпочтительно n равен 3. X представляет собой галоген, такой как F, Cl, Br или I, предпочтительно Br. AAb представляет собой D-ароматическую α-аминокислоту, предпочтительно триптофан (D-Trp). AAd представляет собой алифатическую α-аминокислоту, предпочтительно аллоизолейцин (alloIle).
Стадия 3
7. Проведение циклизации пептида в растворе посредством внутримолекулярного замещения галогена X (предпочтительно Br) тиолом гомоцистеина (hCy). Предпочтительно циклизация происходит в присутствии подходящего основания, такого как карбонат (например, карбонат натрия, карбонат калия) или органические основания, такие как DIEA, с последующим подкислением (например, pH 5,5 или pH 6) подходящей кислотой (например, уксусная кислота). Предпочтительно, циклизация происходит в присутствии карбоната натрия (Na2CO3).
К 1 и 2 г пептида/л раствора (предпочтительно к концентрации приблизительно 1 г пептида/л раствора), добавляют Na2CO3 (например, в количестве приблизительно 10 г/л). Мониторинг реакции циклизации можно проводить посредством ВЭЖХ, как известно специалисту в данной области.
После завершения циклизации pH раствора можно доводить, например, до 5,5 или pH 6, добавлением подходящего основания, например, AcOH. Затем циклический пептид можно отфильтровывать и промывать смесью вода/CH3CN (7/3).
Таким образом, получают циклический гептапептидный аналог, предпочтительно барусибан или его фармацевтически приемлемую соль: c[(CH2)2-CO-AAb-Ile-AAd-Asn-hCy]-NR-CHQ-CH2OH, предпочтительно c[(CH2)2-CO-D-Trp-Ile-AlloIle-Asn-hCy]-N-MeOrn-ол, также изображаемый как
CO-AAb-Ile-AAd-Asn-hCy-NR-CHQ-CH2OH, предпочтительно

CO-D-Trp-Ile-AlloIle-Asn-hCy-N-MeOrn-ол:

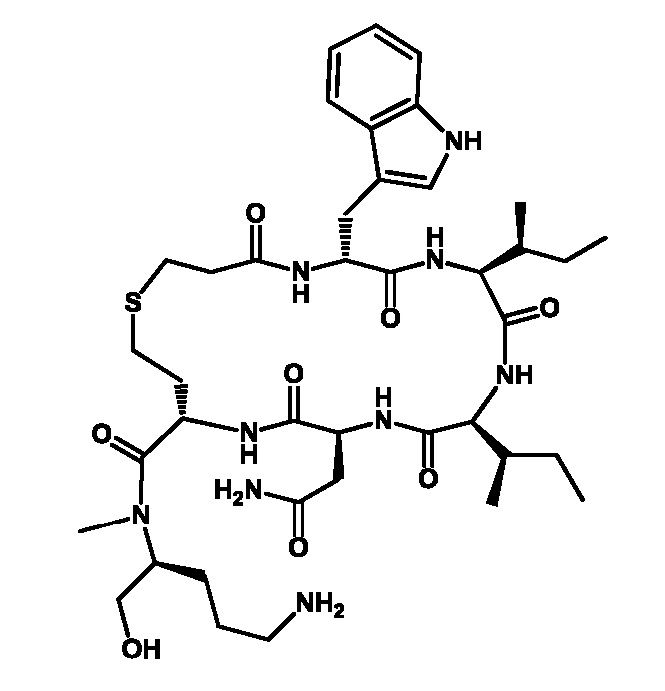
R представляет собой CH3 или C2H5, Q представляет собой (CH2)n-NH2, и n равен 2, 3 или 4. Предпочтительно, R представляет собой CH3 и предпочтительно n равен 3. X представляет собой остаток галогена, такой как F, Cl, Br или I, предпочтительно Br. AAb представляет собой D-ароматическую α-аминокислоту, предпочтительно триптофан (D-Trp). AAd представляет собой алифатическую α-аминокислоту, предпочтительно аллоизолейцин (alloIle).
Необязательно, циклический пептид можно очищать. Очистку можно проводить хроматографическими способами, такими как препаративная обращено-фазовая хроматография (RPC). После очистки пептид можно лиофилизировать.
Кроме того, изобретение относится к промежуточному пептиду, который имеет формулу:
X-(CH2)2-CO-AAb-Ile-AAd-Asn(P1)-hCy(P2)-NR-CHQ'-CH2OW
где AAb представляет собой D-ароматическую α-аминокислоту (предпочтительно D-Trp), боковая цепь которой необязательно может быть защищенной, AAd представляет собой алифатическую α-аминокислоту (предпочтительно alloIle), X представляет собой остаток галогена, выбранный из группы, состоящей из F, Cl, Br и I, предпочтительно Br, P1представляет собой защитную группу (предпочтительно Trt), P2 представляет собой Trt, R представляет собой CH3 или C2H5, Q' представляет собой (CH2)n-NP3P4, где n равен 2, 3 или 4, и P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга и которые могут иметь быть такими же или отличающимися от P1 и/или P2, и где W представляет собой H (а именно, C-конец представляет собой спирт), защитную группу или смолу. Предпочтительно P4 представляет собой H и P3представляет собой Boc. Предпочтительно, R представляет собой CH3, n равен 3, P4 представляет собой H и P3 представляет собой Boc.
Предпочтительно AAb представляет собой D-Trp, предпочтительно с боковой цепью, защищенной защитной группой P7. Предпочтительно, P7 представляет собой Boc. Предпочтительно, AAd представляет собой AlloIle.
Кроме того, изобретение относится к промежуточному пептиду, который имеет формулу:
X-(CH2)2-CO-D-Trp(Boc)-Ile-AlloIle-Asn(Trt)-hCy(Trt)-N-Me-Orn(Boc)-OW,
где X представляет собой галоген, выбранный из группы, состоящей из F, Cl, Br и I, предпочтительно Br, и где W представляет собой H (а именно, C-конец представляет собой спирт), защитную группу или смолу. Предпочтительно, изобретение относится к промежуточному пептиду, который имеет формулу:
X-(CH2)2-CO-D-Trp-Ile-AlloIle-Asn-hCy-N-Me-Orn-ол,
где X представляет собой галоген, выбранный из группы, состоящей из F, Cl, Br и I, предпочтительно Br.
Кроме того, изобретение относится к промежуточному пептиду, который имеет формулу:
Br-(CH2)2-CO-D-Trp-Ile-AlloIle-Asn-hCy-N-Me-Orn-ол.
Как используют в рамках изобретения, термины "приблизительно" и "приближенно" означают указанную величину ±1% от этой величины, или термины "приблизительно" и "приближенно" означают указанную величину ±2% от этой величины, или термины "приблизительно" и "приближенно" означают указанную величину ±5% от этой величины, термины "приблизительно" и "приближенно" означают указанную величину ±10% от этой величины, и термины "приблизительно" и "приближенно" означают указанную величину ±20% от этой величины, или термины "приблизительно" и "приближенно" означают указанную величину ±30% от этой величины; предпочтительно термины "приблизительно" и "приближенно" означают в точности указанную величину (± 0%).
1. ПРИМЕРЫ
Пример 1: Синтез барусибана
1.1. Схема-алгоритм синтеза

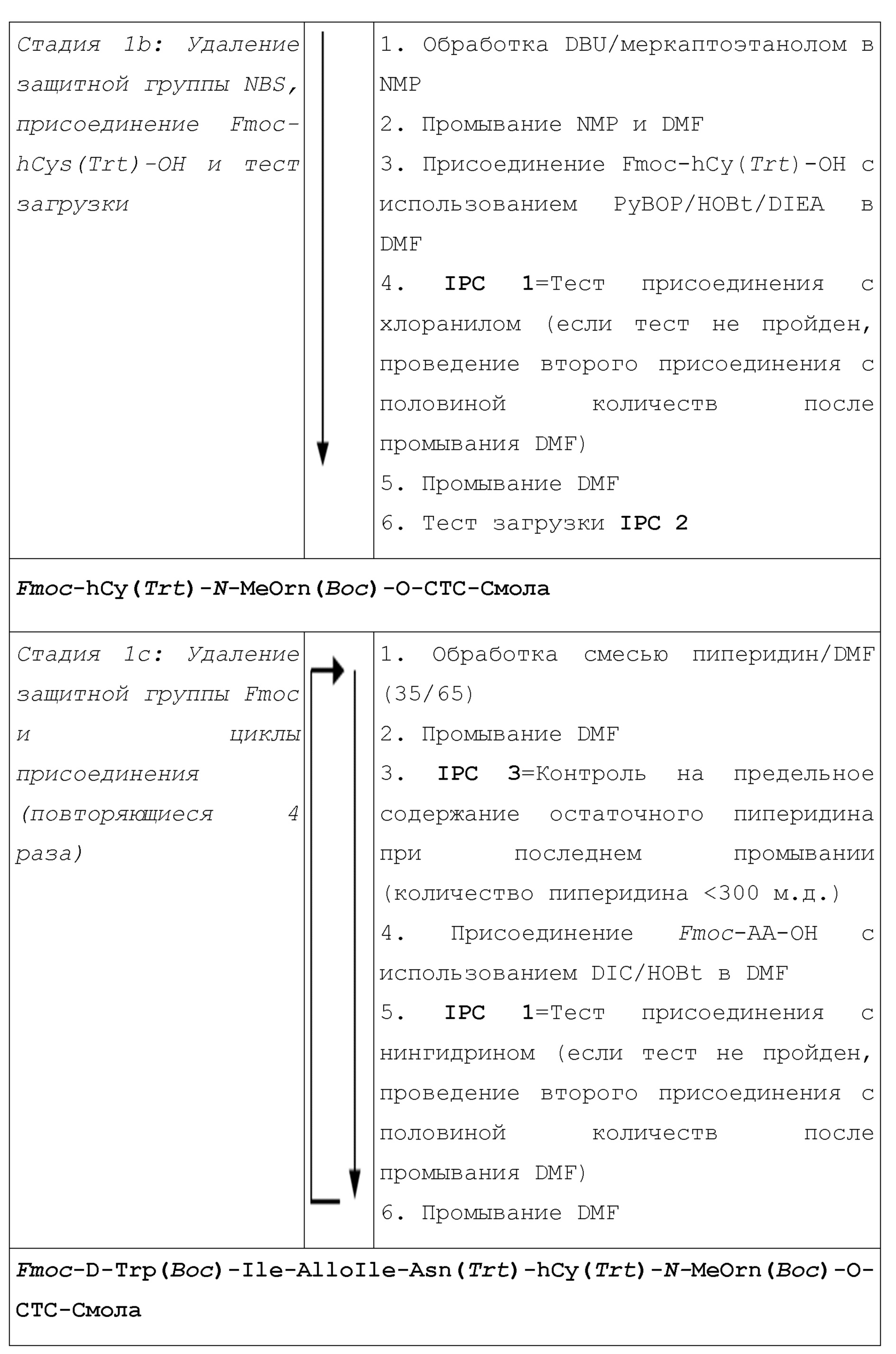


1.2. Описание процесса производства
Сборка
Защищенный o-NBS-N-MeOrn(Boc)-ол присоединяют прямо к хлор-2-хлортритил-смоле (CTC-смола) в DMF при 60°C в присутствии пиридина в течение 17 часов. После кэппирования смолы защитную группу NBS удаляют с использованием смеси DBU/ меркаптоэтанол/NMP. После удаления защитной группы NBS присоединяют вторую защищенную аминокислоту (Fmoc-hCy(Trt)-OH) с PyBOP/HOBt/DIEA в DMF. Завершение реакции проверяют посредством теста присоединения (тест с хлоранилом). Четыре других остатка (Fmoc-Asn(Trt)-OH, Fmoc-AlloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH) включают путем последовательных циклов удаления защитной группы Fmoc и присоединения аминокислот:
1. Удаление Fmoc
2. Промывание DMF
3. Присоединение Fmoc-AA-OH с использованием DIC/HOBt в DMF
4. Тест присоединения (тест с нингидрином или тест с хлоранилом)
5. Промывание DMF
Объемы реакции вычисляют, исходя из 5 мл/г пептид-смолы. Удаление защитной группы Fmoc происходит в пиперидине (35% в DMF) посредством трех повторяющихся циклов (3 мин, 3 мин и 10 мин). Промывание проводят посредством DMF посредством семи повторяющихся циклов после удаления защитной группы Fmoc и посредством трех повторяющихся циклов после стадии присоединения. Все реакции присоединения проводят с 2 экв. защищенной Fmoc аминокислоты и 2 экв. DIC/HOBt в DMF за исключением присоединения Fmoc-alloIle-OH (1,5 экв. вместо 2 экв.). После каждой реакции присоединения аминокислоты завершенность контролируют полуколичественным цветным тестом на основе выявления не вступивших в реакцию аминов. Первичные амины тестируют с использованием теста с нингидрином (тест Кайзера) (Kaiser et al., Analytical Biochem., 39, 305, (1975); E. Kaiser, R.L. Colescott, C.D. Bossinger, P.I. Cook, Analytical Biochem., 34 (Issue 2), 595-598, (1970)) и вторичные амины тестируют с использованием теста с хлоранилом (Kaiser et al., Analytical Biochem., 39, 305, (1975)). Последнюю реакцию присоединения проводят с 3 экв. 3-бромпропионовой кислоты и 3 экв. DIC в растворе в DCM с получением защищенного линейного пептида.
Отщепление:
После стадии сборки проводят отщепление от смолы и сопутствующее удаление защитной группы боковой цепи в смеси с TFA с получением неочищенного линейного пептида. Пептид отщепляют от смолы и из него удаляют защитную группу с использованием смеси TFA/H2O/DTT (86/5/9 об./об./масс.) в концентрации 5 мл на грамм пептид-смолы в течение 3,00 ч при комнатной температуре. Порционно добавляют пептид-смолу, контролируя температуру (T≤20°C). Реакционную смесь охлаждают при 0°C±5°C, и в реакционную смесь выливают смесь MTBE/н-гептан (7/3; 50 мл на грамм пептид-смолы) для осаждения расщепленного пептида в смоле, контролируя температуру (T<20°C). Неочищенный линейный пептид+смола отфильтровывают, промывают MTBE (3×3 мл на грамм пептид-смолы) и сушат в вакууме.
Циклизация:
Линейный пептид растворяют в растворе вода/CH3CN (7/3) в концентрации 1,6 г/л. Затем раствор доводят до концентрации 1 г/л и перемешивают в течение ночи для завершения декарбоксилирования пептида. Перед циклизацией количество линейного пептида в растворе оценивают посредством ВЭЖХ с помощью эталонного образца линейного пептида в концентрации 1 г/л. Пептид циклизуют в концентрации 1 г/л пептида со смолой и в присутствии карбоната натрия (10 г/л). Мониторинг завершенности циклизации проводят посредством ВЭЖХ. После завершения циклизации (менее 1% линейного пептида), pH реакционной смеси снижают приблизительно до 5,5 добавлением уксусной кислоты. Смолу отфильтровывают и промывают смесью вода/CH3CN (7/3 об./об.). Затем раствор фильтрата разбавляют наполовину водой и хранят при 2-8°C.
2. Пример 2: синтез неочищенного барусибана с использованием 3-хлорпропионовой кислоты вместо 3-бромпропионовой кислоты
Эксперимент проводили в соответствии со способом, описанным выше (см. пример 1), где 3-бромпропионовую кислоту заменяют 3-хлорпропионовой кислотой.
2.1. Сборка
Загрузка смолы
Сборку проводили в масштабе 5 ммоль (7,14 г хлор-2-хлортритилсмолы (CTC-смола) с замещением 0,7 мэкв./г). После набухания смолы в DMF (7 мл) в течение 15 минут o-NBS-N-MeOrn(Boc)-ол (1 экв., 2,92 г) растворяли в 9 мл DMF и добавляли к смоле. Добавляли пиридин (2 экв., 0,81 мл) и реакционную смесь нагревали до 60°C и перемешивали в течение 17 часов. Через 1 ч добавляли 6 мл DMF для гомогенизации реакционной смеси. Концентрация o-NBS-N-MeOrn(Boc)-ола в ходе реакции загрузки составляла 0,22 ммоль/л. Через 17 ч к смоле добавляли 15 мл DMF для гомогенизации реакционной смеси перед кэппированием. Проверяли стабильность o-NBS-NMeOrn(Boc)-ола в этих условиях; исследование описано в примере 5. Оказалось, что материал является стабильным без какой-либо деградации в этих условиях загрузки.
Кэппирование смолы
DIEA (5,7 экв., 4,96 мл) и MeOH (4,96 мл, об./об.) добавляли к смоле и перемешивали в течение 1 часа при комнатной температуре для нейтрализации не вступивших в реакцию участков смолы. Затем смолу осушали и промывали DMF (3×45 мл).
Удаление защитной группы NBS и присоединение Fmoc-hCy(Trt)-OH
Защитную группу NBS удаляли посредством двух повторяющихся циклов (2×10 мин) с использованием смеси DBU/меркаптоэтанол (5 экв./10 экв., 3,74 мл/ 3,51 мл) в NMP (30 мл). Вторую аминокислоту (Fmoc-hCy(Trt)-OH) удаляли с использованием PyBOP/HOBt/DIEA (1,5 экв./1,5 экв./3,75 экв.; 1,15 г/3,90 г/ 3,26 мл) в DMF (23 мл). Через 16 часов завершение реакции проверяли посредством теста с хлоранилом и смолу промывали посредством DMF (3×51 мл).
Удаление защитной группы Fmoc:
Следующие аминокислоты (Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH) включали путем последовательного удаления защитной группы Fmoc и циклов присоединения аминокислот в соответствии со способом получения, описанным выше (пример 1). Защитную группу Fmoc удаляли посредством трех повторяющихся циклов (3, 3 и 10 мин) с использованием смеси 35% пиперидина в DMF. После удаления защитной группы Fmoc проводили промывание DMF посредством семи повторяющихся циклов и после стадии присоединения с DMF посредством трех повторяющихся циклов. Объемы удаления защитной группы Fmoc вычисляют, исходя из 5 мл/г пептид-смолы.
Присоединение Fmoc-AA-OH:
Для Fmoc-Asn(Trt)-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH), 2 экв. аминокислоты присоединяли в присутствии 2 экв. DIC/HOBt в DMF (25 мл). Для Fmoc-alloIle-OH, 1,5 экв. аминокислоты присоединяли в присутствии 1,5 экв. DIC/HOBt в DMF (25 мл). Для всех реакций присоединения завершение реакции проверяли с использованием теста с нингидрином и смолу промывали с использованием DMF посредством трех повторяющихся циклов.
Присоединение 3-хлорпропионовой кислоты
Последнее присоединение проводили путем растворения 1,63 г 3-хлорпропионовой кислоты (3 экв.) и 2,34 мл DIC в DCM (42,5 мл). Завершение реакции проверяли с использованием теста с нингидрином (время присоединения=2 часа). В конце сборки смолу промывали DCM (3×77 мл), IPA (5×77 мл) и сушили в вакууме. Получали 12 г защищенной пептид-смолы, которые соответствуют выходу 92,4%.
Отщепление и удаление защитной группы
12 г защищенной пептид-смолы добавляли порционно к смеси TFA/H2O/DTT (86/5/9 об./об./масс.) (60 мл), контролируя температуру (T≤20°C). Реакционную смесь перемешивали при комнатной температуре в течение 3,00 ч. Затем реакционную смесь охлаждали до 2,7°C и в реактор выливали смесь MTBE/н-гептан (7/3 об./об.) (600 мл) посредством поддержания температуры ниже 20°C. Неочищенный линейный пептид+смола отфильтровывали, промывали MTBE (3×36 мл) и сушили в вакууме в течение ночи. Получали 13 г неочищенного хлорсодержащего линейного пептида+смола. Чистота ВЭЖХ составляет 76,6%.
2.2. Циклизация
Общее количество хлорсодержащего линейного пептида+смола делили на две порции и циклизацию проводили два раза в количестве приблизительно 6,5 г.
2.2.1. Первая циклизация
Первую циклизацию проводили на 6,5 г в соответствии со способом, описанным в примере 1 выше. Циклизация не завершилась согласно анализу посредством ВЭЖХ. Затем начинали вторую циклизацию оставшихся 6,5 г хлорсодержащего линейного пептида.
2.2.2. Вторая циклизация
Вторую циклизацию проводили на 6,44 г хлорсодержащего линейного пептида+смола. Хлорсодержащий линейный пептид+смола растворяли в 595 мл раствора вода/CH3CN (7/3 об./об.) с получением пептида в растворе в концентрации 1 г/л. Добавляли 5,95 г твердого карбоната натрия (pH 11,3). Регулярно проводили мониторинг реакции посредством ВЭЖХ). Через 25 ч мониторинг посредством ВЭЖХ продемонстрировал менее 1% линейного пептида. Добавляли 8,35 мл уксусной кислоты для нейтрализации реакционной смеси; полученное значение pH раствора составляло 5,6. После подкисления смолу удаляли фильтрацией и промывали два раза 30 мл смеси вода/CH3CN (7/3). Затем раствор фильтрата разбавляли 660 мл воды и хранили при 2-8°C. Конечная концентрация неочищенного пептида в растворе составляла 0,45 г/л с чистотой 86,4% согласно ВЭЖХ. Результаты, полученные в результате обеих сборок, сравнивали для оценки различий между использованием 3-бромпропионовой кислоты (пример 1) и 3-хлорпропионовой кислоты (пример 2) в процессе синтеза барусибана, как описано в примерах 1 и 2.
Результаты сборки
В следующей таблице (таблица 2) обобщенно представлены и сравниваются данные сборки, полученные в эксперименте примера 2 и при получении согласно примеру 1:
Таблица 2: Сравнительная таблица данных сборки для эксперимента согласно примеру 1 и получения согласно примеру 2.
Вплоть до присоединения 3-галогенпропионовой кислоты обе сборки проводили в одинаковых условиях; следовательно, не является неожиданным наблюдение сходного времени присоединения. Более того, не наблюдали значительных отличий во времени присоединения посредством замены последнего реагента; 3-хлорпропионовая кислота (эксперимент согласно примеру 2) демонстрирует сходное время присоединения с 3-бромпропионовой кислотой (пример 1), обе реакции завершались в течение менее чем 2 часов. Учитывая загрузку смолы, обе сборки демонстрируют сравнимый выход (приблизительно 90%). Основное отличие лежит во времени реакции, требуемом для циклизации; действительно, циклизация завершается после 1 часа процесса с использованием бромсодержащего производного (пример 1), в то время как при использовании хлорсодержащего производного требуется 25 часов (пример 2). Бромсодержащий линейный пептид демонстрирует лучшую реактивность по сравнению с хлорсодержащим линейным пептидом.
Чистота циклических неочищенных соединений согласно ВЭЖХ является сходной; 86,36% для 3-хлорпропионовой кислоты и 85,24% для 3-бромпропионовой кислоты.
Эти результаты сборки показывают, что барусибан можно синтезировать из 3-хлорпропионовой кислоты, однако время циклизации является более длительным, чем при использовании 3-бромпропионовой кислоты, как показано в примере 1 и 2.
Профиль примесей при ВЭЖХ для неочищенного барусибан
Неочищенные растворы барусибана, синтезированные как из 3-хлорпропионовой кислоты, так и из 3-бромпропионовой кислоты, анализировали посредством ВЭЖХ и LC/MS для идентификации примесей и для сравнения профилей примесей.
Основные примеси приведены в таблице 3 ниже:
Таблица 3: Сравнительная таблица примесей для экспериментов согласно примеру 1 и примеру 2.
Способ получения по изобретению (например, пример 1) с использованием 3-бромпропионовой кислоты является более адаптируемым, поскольку реакция циклизации является более быстрой и приводит к меньшим побочным реакциям.
Заключение по примерам 1 и 2
Целью эксперимента согласно примеру 2 была оценка замены 3-бромпропионовой кислоты 3-хлорпропионовой кислотой в процессе синтеза барусибана, как описано в примере 1. Исходя из всех результатов, полученных после этих испытаний (см. таблицу 2 и таблицу 3), применение 3-хлорпропионовой кислоты является возможным; тем не менее реакция циклизации является более длительной вследствие более низкой реактивности хлорсодержащего линейного пептида. Увеличенное время реакции вовлекает частичное дезамидирование пептида в водном основном растворе. Образовавшиеся примеси может быть трудно отделить на стадии очистки.
3. Пример 3. Синтез барусибана с помощью другого подхода синтеза
3.1. Схема алгоритма синтеза
Ниже представлена схема алгоритма синтеза барусибана согласно процессу примера 3:
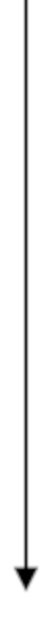



CO-D-Trp-Ile-alloIle-Asn-hCy-N-MeOrn-ол

Неочищенный барусибан
3.2. Описание процесса производства
3.2.1. Сборка
Защищенный Fmoc-N-MeOrn(Boc)-ол присоединяют прямо к карбоновой смоле в DMF при комнатной температуре в присутствии DMPA в течение 2 часов. После кэппирования смолы защитную группу Fmoc удаляют посредством промывания раствором пиперидина (20% в DMF). Пять других остатков (Fmoc-hCy(mmt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH) включают путем последовательного удаления защитной группы Fmoc и циклов присоединения аминокислоты:
1. Удаление Fmoc
2. Промывание DMF
3. Присоединение Fmoc-AA-OH с использованием DIC/HOBt в DMF посредством предварительной активации аминокислоты
4. Тест присоединения (тест с нингидрином или тест с хлоранилом)
5. Промывание DMF
Объемы реакции вычисляют, исходя из 5 мл/г пептид-смолы. Удаление защитной группы Fmoc проводят в пиперидине (20% в DMF) посредством двух повторяющихся циклов (10 мин и 20 мин). Проводят промывание DMF посредством семи повторяющихся циклов после удаления защитной группы Fmoc и посредством трех повторяющихся циклов после стадии присоединения. Предварительную активацию проводят с использованием 3 экв. Fmoc-AA-OH и 3,3 экв. DIC/HOBt в DMF путем перемешивания в течение 5 минут при 0°C. После предварительной активации все реакции присоединения проводят в течение 2 часов при комнатной температуре. Поскольку эта стадия является критической, после каждой реакции присоединения аминокислоты контролируют завершенность посредством полуколичественного цветного теста на основе обнаружения не вступивших в реакцию аминов. Первичные амины тестируют с использованием теста с нингидрином (тест Кайзера), и вторичные амины тестируют с использованием теста с хлоранилом. Последнее присоединение проводят с 3-галогенпропионовой кислотой в растворе в DMF с получением защищенного линейного пептида.
3.2.2. Удаление защитной группы гомоцистеина и циклизация
После стадии сборки защитную группу mmt гомоцистеина (hCy) удаляют и пептид-смолу подвергают циклизации на смоле. Объемы реакции вычисляют на основе 5 мл/г пептид-смолы. Пептид-смолу промывают DCM посредством трех повторяющихся циклов и группу mmt удаляют посредством пяти повторяющихся циклов со смесью TFA/EDT/DCM (2/3/95 об./об./об.). После удаления защитной группы проводят промывание DCM и DMF посредством трех повторяющихся циклов. Пептид подвергают циклизации на смоле с раствором тетраметилгуанидина (1% в DMF) в течение 8 часов при комнатной температуре. После циклизации циклический пептид-смолу промывают DMF, DCM и MeOH перед сушкой в вакууме.
3.2.3. Отщепление
После циклизации проводят отщепление от смолы и сопутствующее удаление защитной группы боковой цепи в смеси в TFA с получением неочищенного барусибана. Пептид отщепляют от смолы и из него удаляют защитную группу с использованием смеси TFA/TIS/EDT/PhOH/H2O (90/3/2/1/4 об./об./об./об./об.) в концентрации 10,6 мл на грамм пептид-смолы в течение 2 часов при комнатной температуре. Реакционную смесь фильтруют и смолу промывают TFA. Реакционную смесь выливают на сухой Et2O (106 мл/г пептид-смолы) для осаждения неочищенного циклического пептида. Осадок отфильтровывают, промывают сухим Et2O (3×3,3 мл на грамм пептид-смолы) и сушат в вакууме.
3.3. Схема эксперимента
Для сравнения синтеза барусибана после различного времени присоединения, было решено разделить эксперименты на два пути:
- первый путь, в котором присоединение останавливают через 2 часа даже если тест на присоединение является положительным (пример 3.1).
- Второй путь, в котором присоединение проводят в течение минимум 2 часов и останавливают только после завершения согласно тесту Кайзера; в этом случае время реакции может отличаться (IPC) (пример 3.2).
Кроме того, для каждого эксперимента (3.1 и 3.2), вносили дополнительную переменную, а именно последнюю присоединенную аминокислоту, представляющую собой 3-бромпропионовую кислоту или 3-хлорпропионовую кислоту (см. схему ниже). Всего проводили 4 различных синтеза:
Эксперимент 3.1.1: присоединение останавливали через 2 часа; 3-хлорпропионовая кислота
Эксперимент 3.1.2: присоединение останавливали через 2 часа; 3-бромпропионовая кислота
Эксперимент 3.2.1: присоединение проводили в течение минимум 2 часов и останавливали только после завершения согласно тесту Кайзера; 3-хлорпропионовая кислота
Эксперимент 3.2.2: присоединение проводили в течение минимум 2 часов и останавливали только после завершения согласно тесту Кайзера; 3-бромпропионовая кислота
На следующей схеме (схема 1) обобщенно представлен план эксперимента, которому следовали для синтеза неочищенного барусибана согласно примеру 3 (3.1.1, 3.1.2, 3.2.1 и 3.2.2):
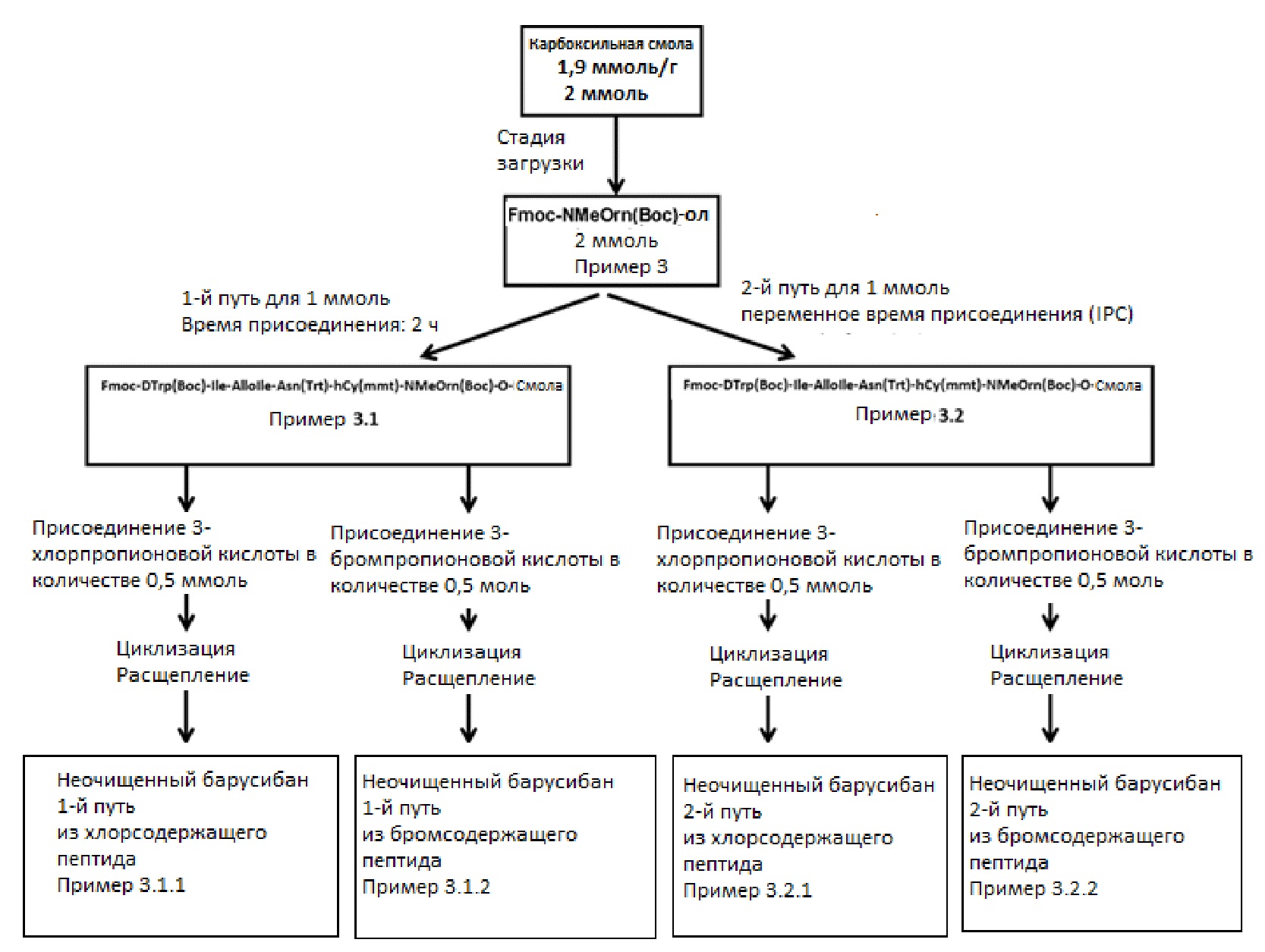
Схема 1: План эксперимента согласно примеру 3 (способы синтеза 3.1.1, 3.1.2, 3.2.1 и 3.2.2)
Загрузку смолы проводят в количестве 2 ммоль карбоновой смолы с замещением 1,9 ммоль/г. После стадии загрузки количество разделяют на 2 равные части (по 1 ммоль каждая) и сборку продолжают с использованием либо первого пути (эксперимент 3.1), либо второго пути (эксперимент 3.2). После присоединения Fmoc-D-Trp(Boc)-OH количество разделяют на 2 равных части (по 0,5 ммоль каждая) и сборку продолжают для каждого пути с использованием либо 3-хлорпропионовой кислоты (эксперименты 3.1.1 и 3.2.1), либо 3-бромпропионовой кислоты (эксперименты 3.1.2 и 3.2.2).
3.4. Синтез Fmoc-N-MeOrn(Boc)-ола
Fmoc-N-MeOrn(Boc)-ол используют в качестве исходного материала в способах, описанных в примере 3. Это производное не является таким же, как производное, использованное в способе, описанном в примере 1 и 2, таким образом, оно было синтезировано до сборки. Производное Fmoc получали из материала o-NBS-N-MeOrn(Boc)-ола, использованного в примерах 1 и 2, по методике из двух стадий, описанной ниже.
3.4.1. Схема реакции
Fmoc-N-MeOrn(Boc)-ол синтезируют из o-NBS-N-MeOrn(Boc)-ола за две стадии по схеме реакции, описанной ниже (схема 2).
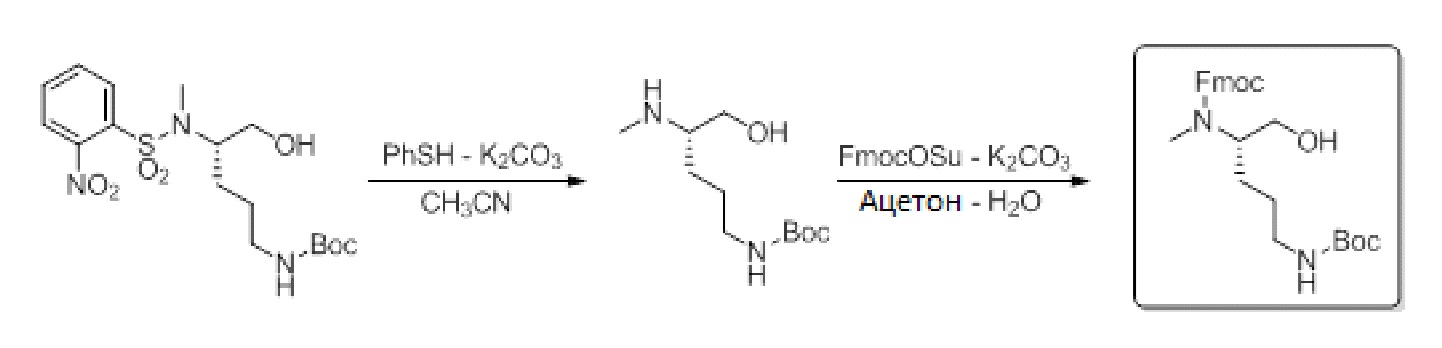
Схема 2: Синтез Fmoc-N-MeOrn(Boc)-ола из o-NBS-N-MeOrn(Boc)-ола за две стадии
3.4.2. Стадия 1: Удаление защитной группы o-NBS с использованием PhSH
Из группы o-NBS удаляют защитную группу посредством обработки тиофенолом в основном растворе. После концентрирования и добавления подкисленной воды основные органические примеси удаляют промыванием IPE.
Экспериментальная часть:
o-NBS-N-Me-L-Orn(Boc)-ол (50 г, 1 экв., 119,8 ммоль) растворяли в CH3CN (16 об.) в колбе с 3 горлышками. К реакционной смеси добавляли K2CO3 (54,6 г, 3,5 экв., 395,3 ммоль) и PhSH (30,5 мл/33g, 2,5 экв., 299,4 ммоль) и перемешивали при комнатной температуре. Завершение реакции проверяли посредством TLC (AcOEt/циклогексан/AcOH, 5/5/0,5 и CHCl3/метанол/AcOH, 60/10/5). Через 19 ч растворитель удаляли концентрированием на роторном испарителе и добавляли воду (800 мл). Водную фазу промывали два раза IPE (2×300 мл) и раствор использовали в таком виде на следующей стадии.
3.4.3. Стадия 2: Защита посредством Fmoc с использованием Fmoc-OSu
Водную фазу используют как есть и защиту посредством Fmoc проводят с использованием Fmoc-OSu в смеси ацетон/вода. После подкисления реакционной смеси раствор экстрагируют посредством AcOEt и промывают основным и кислотным растворами.
Экспериментальная часть:
Fmoc-OSu (36,4 г, 0,9 экв., 107,8 ммоль) в ацетоне (800 мл) добавляли к реакционной смеси, содержавшей H-N-Me-L-Orn(Boc)-ол и перемешивали при комнатной температуре в течение ночи. Завершение реакции проверяли посредством TLC (AcOEt/циклогексан/AcOH, 5/5/0,5 и CHCl3/метанол/AcOH, 60/10/5). После ночи добавляли AcOEt (800 мл) и водную фазу снимали. Органическую фазу промывали два раза раствором NaHCO3 (5% в воде) (2×400 мл), два раза 1 Н раствором KHSO4 (2×400 мл) и один раз водой (400 мл) и рассолом (400 мл). Затем органическую фазу сушили над Na2SO4. Соединение очищали флэш-хроматографией с использованием силикагеля и AcOEt/циклогексана в качестве элюента. После концентрирования очищенных фракций Fmoc-N-MeOrn(Boc)-ол выделяли в виде белой пены.
Экспериментальная часть:
После фильтрации и концентрирования на роторном испарителе неочищенный продукт очищали флэш-хроматографией на силикагеле (элюент: AcOEt/циклогексан 3/7 (5 л), а затем 7/3 (10 л)). Очищенные фракции концентрировали, совместно упаривали с циклогексаном (300 мл) и сушили в вакууме (≤100 мм. рт. ст.) в течение 20 ч с получением белого воска. Добавляли холодный пентан (-20°C) и взвесь перемешивали в течение 1 ч при -20°C. Фильтрация была невозможной, поскольку Fmoc-N-MeOrn(Boc)-ол не образовывал твердое вещество. Пентан удаляли концентрированием на роторном испарителе и белую пену сушили в вакууме (≤100 мм рт. ст.) в течение 20 ч. Получали 34,1 г Fmoc-N-MeOrn(Boc)-ола, что соответствовало выходу 70% за две стадии. Чистота производного Fmoc при ВЭЖХ составила 99,10%. Стабильность Fmoc-N-MeOrn(Boc)-ола проверяли в различных условиях, и она описана в примере 5.
3.5. Синтез неочищенного барусибана в соответствии со способом согласно примеру 3
3.5.1. Загрузка и кэппирование
Загрузка смолы:
Загрузку проводили в количестве 2 ммоль карбоновой смолы с замещением 1,9 мэкв./г.
Карбоновой смоле (1 экв., 1,05 г) позволяли набухать в DMF (5,26 мл) в течение 30 минут и осушали. Параллельно Fmoc-N-MeOrn(Boc)-ол (2 экв., 1,82 г) растворяли в DMF (3,4 мл) и предварительно активировали DIC (1 экв., 0,31 мл) в течение 5 минут при 0°C. После предварительной активации к смоле добавляли Fmoc-N-MeOrn(Boc)-ол. Добавляли DMAP (0,22 экв., 54 мг) и смесь перемешивали в течение 2 часов при комнатной температуре. Через 2 ч смолу осушали и промывали DMF (3×7,6 мл). Стабильность Fmoc-N-MeOrn(Boc)-ола в этих условиях описана в примере 5, как указано выше. Оказалось, что частичная деградация уже наблюдалась через 2 часов при комнатной температуре; материал потенциально мог разлагаться в ходе реакции загрузки.
Кэппирование смолы:
В реактор добавляли DMF (3,4 мл), а затем добавляли DIEA (0,83 экв., 290 мкл) и MeOH (1,7 мл). Реакционную смесь перемешивали в течение 1 ч 30 мин при комнатной температуре для нейтрализации не вступивших в реакцию участков смолы. Затем смолу осушали и промывали DMF (3×7,6 мл). Согласно схеме эксперимента, описанной выше (схема I), общее количество пептид-смолы разделяли на две порции. Первую порцию использовали для синтеза через путь 1 (первый путь, время присоединения 2 ч, пример 3.1), а вторую часть использовали для синтеза через путь 2 (второй путь, переменное время присоединения (минимум 2 ч) в зависимости от завершенности согласно тесту Кайзера, пример 3.2).
3.5.2. Первый путь: эксперимент согласно примеру 3.1
Для первого пути следующие пять аминокислот собирали в масштабе 1 ммоль вплоть до пептида Fmoc-D-Trp(Boc)-Ile-alloIle-Asn(Trt)-hCy(mmt)-N-MeOrn(Boc)-O-смола
Удаление защитной группы Fmoc:
Следующие аминокислоты (Fmoc-hCy(mmt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH) включали путем последовательного удаления защитной группы Fmoc и циклов присоединения аминокислот в соответствии со способом получения согласно примеру 3, как описано выше (см. раздел 3.2). Защитную группу Fmoc удаляли посредством двух повторяющихся циклов (10 и 20 мин) с использованием смеси пиперидина 20% в DMF. После удаления защитной группы проводили промывание DMF посредством семи повторяющихся циклов и после стадии присоединения с использованием DMF посредством трех повторяющихся циклов. Объемы реакций удаления защитной группы Fmoc вычисляли, исходя из 5 мл/г пептид-смолы.
Присоединение Fmoc-AA-OH:
3 экв. следующих аминокислот (Fmoc-hCy(mmt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH) предварительно активировали 3,3 экв. DIC/HOBt в DMF при 0°C в течение 5 минут, а затем добавляли к смоле в реакторе. Все присоединение проводили в течение 2 часов и проверяли с использованием теста с хлоранилом или нингидрином. Тесты присоединения были отрицательными для Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH. Несмотря на положительный тест при присоединении Fmoc-hCy(mmt)-OH, реакцию останавливали (через 2 ч). После теста присоединения смолу промывали DMF посредством трех повторяющихся циклов. После присоединения Fmoc-D-Trp(Boc)-OH общее количество пептид-смолы делили на две порции и присоединение 3-галогенпропионовой кислоты (либо Cl, пример 3.1.1 либо Br, пример 3.1.2) проводили в масштабе 0,5 ммоль.
Присоединение 3-галогенпропионовой кислоты: Эксперименты согласно примерам 3.1.1 и 3.1.2
Для последнего присоединения параллельно проводили два эксперимента. Один эксперимент проводили с 3-хлорпропионовой кислотой (пример 3.1.1), а другой эксперимент проводили с 3-бромпропионовой кислотой (пример 3.1.2). Это присоединение проводили в течение 2 часов при комнатной температуре посредством предварительной активации 3-галогенпропионовой кислоты (3 экв.) посредством DIC/HOBt (3,3 экв.) в DMF в течение 5 минут при 0°C. Через 2 часа завершение реакции проверяли посредством теста с нингидрином и TNBS:
- для бромсодержащего производного оба теста были положительными
- для хлорсодержащего производного тест с нингидрином продемонстрировал светло-синие гранулы и тест с TNBS был положительным.
Несмотря на эти положительные тесты присоединения реакции присоединения останавливали. Затем смолу промывали DMF (3×5 мл).
Удаление защитной группы из гомоцистеина и циклизация
Пептид-смолу промывали DCM (3×5 мл) и защитную группу mmt удаляли посредством пяти повторяющихся циклов (5×10 мин) с 4 мл смеси TFA/EDT/DCM (2/3/95 об./об./об.). После удаления защитной группы проводили промывание DCM (3×5 мл) и DMF (3×5 мл). Циклизацию проводили на смоле посредством добавления 5 мл раствора тетраметилгуанидина (1% в DMF) и перемешивания в течение 8 часов при комнатной температуре. После циклизации циклический пептид-смолу промывали DMF (3×6 мл), DCM (3×6 мл), MeOH (5×6 мл) и сушили в вакууме. В эксперименте согласно примеру 3.1.1 с использованием 3-хлорпропионовой кислоты получали 278 мг циклического пептида-смолы и в эксперименте согласно примеру 3.1.2 с использованием 3-бромпропионовой кислоты получали 292 мг, что соответствовало, соответственно, выходу сборки 8,3% и 13,3%, учитывая нагрузку.
Отщепление
Отщепление от смолы проводили для каждого пептида в соответствии с общими условиями, описанными в разделе 3.1.
Эксперимент с 3-хлорпропионовой кислотой: Пример 3.1.1
Защищенный циклический пептид-смолу в количестве 278 мг суспендировали в смеси TFA/TIS/EDT/PhOH/H2O(90/3/2/1/4) (об./об./об./об./об.) (2,95 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь фильтровали и смолу промывали TFA (195 мкл). Смесь в TFA переливали к сухому Et2O (29,5 мл); в противоположность ожиданиям, не достигали выпадения в осадок неочищенного пептида. Смесь, содержавшую неочищенный пептид, концентрировали досуха на роторном испарителе. Осадок после концентрирования растворяли в H2O/CH3CN (50/50 об./об.) и лиофилизировали. После лиофилизации получали очень малое количество порошка (приблизительно 1-3 мг) с чистотой 11,8%.
Эксперимент с 3-бромпропионовой кислотой: пример 3.1.2
Защищенный циклический пептид-смолу в количестве 272 мг суспендировали в смеси TFA/TIS/EDT/PhOH/H2O(90/3/2/1/4) (об./об./об./об./об.) (3,1 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь фильтровали и смолу промывали TFA (204 мкл). Смесь в TFA переливали к сухому Et2O (31 мл); как и в случае эксперимента с 3-хлорпропионовой кислотой, не наблюдали выпадения неочищенного пептида в осадок. Смесь, содержавшую неочищенный пептид, концентрировали досуха на роторном испарителе. Осадок после концентрирования растворяли в H2O/CH3CN (50/50) (50/50 об./об.) и лиофилизировали. После лиофилизации получали очень малое количество порошка (приблизительно 1-3 мг) с чистотой 1,8%.
3.5.3. Второй путь: эксперимент 3.2
Как объяснено выше в разделе 3.3, второй путь соответствует синтезу согласно способу примера 3, где все реакции присоединения контролируются посредством IPC. В этом случае присоединение проводят минимум в течение 2 часов и останавливают только после достижения отрицательного теста на присоединение. Согласно второму пути проводили два эксперимента: один эксперимент с 3-хлорпропионовой кислотой (эксперимент согласно примеру 3.2.1), а другой эксперимент проводили с 3-бромпропионовой кислотой (эксперимент согласно примеру 3.2.2).
Загрузка смолы: эксперимент 3
Загрузка смолы описана в разделе 3.5.1 (общая загрузка для двух путей). После теста загрузки общее количество пептид-смолы разделяли на две порции. Для второго пути (пример 3.2) пять следующих аминокислот собирали в масштабе 1 ммоль вплоть до пептида Fmoc-D-Trp(Boc)-Ile-alloIle-Asn(Trt)-hCy(mmt)-N-MeOrn(Boc)-O-смола (эксперимент согласно разделу 3.2).
Удаление защитной группы Fmoc:
Следующие аминокислоты (Fmoc-hCy(mmt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH) включали путем последовательного удаления защитной группы Fmoc и циклов присоединения аминокислот в соответствии со способом получения, описанным выше (см. раздел 3.2, пример 3). Защитную группу Fmoc удаляли посредством двух повторяющихся циклов (10 и 20 мин) с использованием смеси пиперидина 20% в DMF. После удаления защитной группы проводили промывание DMF посредством семи повторяющихся циклов и после стадии присоединения с использованием DMF посредством трех повторяющихся циклов. Объемы реакций удаления защитной группы Fmoc вычисляли, исходя из 5 мл/г пептид-смолы.
Присоединение Fmoc-AA-OH:
3 экв. следующих аминокислот (Fmoc-hCy(mmt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH) предварительно активировали 3,3 экв. DIC/HOBt в DMF при 0°C в течение 5 минут, а затем добавляли к смоле в реакторе. Для всех присоединений завершение реакции проверяли с использованием теста с нингидрином или хлоранилом. Тесты присоединения были отрицательными для Fmoc-Asn(Trt)-OH, Fmoc-alloIle-OH, Fmoc-Ile-OH и Fmoc-D-Trp(Boc)-OH через 1 час и перемешивание продолжали в течение дополнительного часа для достижения времени присоединения 2 часа. Присоединение Fmoc-hCy(mmt)-OH требовало 4 часов для достижения отрицательного теста присоединения. После теста присоединения смолу промывали DMF посредством трех повторяющихся циклов. После присоединения Fmoc-D-Trp(Boc)-OH общее количество пептид-смолы делили на две порции и присоединение 3-галогенпропионовой кислоты проводили в масштабе 0,5 ммоль.
Присоединение 3-галогенпропионовой кислоты:Эксперименты согласно примеру 3.2.1 и примеру 3.2.2.
Для последнего присоединения параллельно проводили два эксперимента. Один эксперимент проводили с 3-хлорпропионовой кислотой (3.2.1), а другой эксперимент проводили с 3-бромпропионовой кислотой (3.2.2). Эти присоединения проводили при комнатной температуре посредством предварительной активации 3-галогенпропионовой кислоты (3 экв.) с использованием DIC/HOBt (3,3 экв.) в DMF в течение 5 минут при 0°C. Завершение реакции регулярно проверяли посредством теста с нингидрином и теста с TNBS. Через 21 ч 15 минут тест присоединения все еще был положительным, таким образом, добавляли 1,65 экв. DIC (128 мкл) и реакционную смесь перемешивали в течение дополнительных 6 часов. После этого периода не наблюдали изменений и смолу осушали.
- Двойное присоединение проводили посредством предварительной активации 1,5 экв. 3-галогенпропионовой кислоты (3-хлорпропионовая кислота или 3-бромпропионовая кислота) в присутствии 1,65 экв. DIC/HOBt. Через 19 часов завершение реакции проверяли с использованием тестов с нингидрином. Для хлорсодержащего производного тесты были отрицательными (реакция завершилась).
- Положительные результаты тестов были получены для бромсодержащего производного (реакция не завершилась).
Несмотря на положительный тест присоединения бромсодержащего производного присоединение останавливали. Пептид-смолу промывали в обоих экспериментах посредством DMF (3×5 мл).
После положительного теста на присоединение в эксперименте с использованием 3-бромпропионовой кислоты (пример 3.2.2) проводили микрорасщепление для определения чистота линейного пептида посредством ВЭЖХ.
Микрорасщепление
Микрорасщепление проводили в соответствии с условиями, описанными для способа согласно примеру 3 (см., например, раздел 3.2.3). 40 мг защищенного пептида-смолы суспендировали в смеси TFA/TIS/EDT/PhOH/H2O(90/3/2/1/4) (об./об./об./об./об.) (424 мкл) и перемешивали в течение 2 часов при комнатной температуре. Затем реакционную смесь отфильтровывали и смолу промывали TFA. Смесь в TFA переливали к сухому Et2O (4,24 мл); не достигали выпадения неочищенного пептида в осадок. Смесь концентрировали досуха и осадок растворяли в H2O/CH3CN (50/50) в концентрации 1 мг/мл. Этот раствор анализировали посредством ВЭЖХ и LC/MS. На хроматограмме отсутствовал пик, соответствующий неочищенному линейному бромсодержащему пептиду (чистота 0%).
Основные соединения, идентифицированные посредством LC/MS, представляли собой:
-H-D-Trp-Ile-alloIle-Asn-hCy-OH, соответствующий не присоединенной бромпропионовой кислоте и делеции N-MeOrn.
-H-D-Trp-Ile-Ile-alloIle-Asn-hCy-N-MeOrn-OH или H-DTrp-Ile-alloIle-alloIle-Asn-hCy-N-MeOrn-OH, соответствующие не присоединенной бромпропионовой кислоте и присоединению либо изолейцина, либо аллоизолейцина.
-H-D-Trp-Ile-alloIle-hCy-N-MeOrn-OH, соответствующий не присоединенной бромпропионовой кислоте.
Эти результаты демонстрируют, что для этого эксперимента не достигалось присоединение 3-бромпропионовой кислоты. В соответствии с этим результатом только эксперимент с использованием 3-хлорпропионовой кислоты (эксперимент согласно примеру 3.2.1) был вовлечен в следующие стадии (удаление защитной группы цистеина и циклизация).
Удаление защитной группы из гомоцистеина и циклизация
Пептид-смолу промывали DCM (3×5 мл) и защитную группу mmt удаляли посредством пяти повторяющихся циклов (5×10 мин) с 4 мл смеси TFA/EDT/DCM (2/3/95 об./об./об.). После удаления защитной группы проводили промывание DCM (3×5 мл) и DMF (3×5 мл).
Циклизацию проводили на смоле посредством добавления 5 мл раствора тетраметилгуанидина (1% в DMF) и перемешивания в течение 8 часов при комнатной температуре. После циклизации циклический пептид-смолу промывали DMF (3×6 мл), DCM (3×6 мл), MeOH (5×6 мл) и сушили в вакууме. Выход 15,1%.
Отщепление
Отщепление от смолы проводили в соответствии с общими условиями, описанными в разделе 3.2.3. 297 мг защищенного циклического пептида-смолы суспендировали в смеси TFA/TIS/EDT/PhOH/H2O(90/3/2/1/4) (об./об./об./об./об.) (3,15 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь отфильтровывали и смолу промывали TFA (210 мкл). Смесь в TFA переливали к сухому Et2O (31,5 мл); как и в случае экспериментов по первому пути не наблюдали выпадения в осадок неочищенного пептида. Смесь, содержавшую неочищенный пептид, концентрировали досуха на роторном испарителе. Остаток после концентрирования растворяли в H2O/CH3CN (50/50 об./об.) и лиофилизировали. После лиофилизации получали очень малое количество порошка (приблизительно 1-3 мг). Чистота 10,4%.
3.5.4. Обобщение экспериментов согласно различным способам примера 3
Целью этих экспериментов была оценка различных способов получения для синтеза неочищенного барусибана с использованием двух различных путей: один путь, в котором все реакции присоединения проводят в течение 2 часов, и другой путь, в котором завершение проводили посредством выполнения теста присоединения (тест Кайзера на завершенность). В таблице ниже (таблица 4) обобщенно представлены данные о сборке, полученные для обоих путей:
*Присоединение было завешено через 1 ч, однако реакционную смесь перемешивали в течение 2 ч
**Проводили двойной присоединение (DC): 27 ч 15 мин +19 ч (DC)
Таблица 4: Обобщающая таблица данных о сборке, полученных для первого и второго путей.
Чистота неочищенных соединений посредством ВЭЖХ является очень низкой для обоих путей (примеры 3.1 и 3.2). Тем не менее, эксперименты с использованием 3-хлорпропионовой кислоты привели к чистоте неочищенных соединений приблизительно 10%, в то время как использование 3-бромпропионовой кислоты приводит к чистоте только 2%. Также следует отметить, что в эксперименте согласно примеру 3.2.2 с использованием 3-бромпропионовой кислоты не было получено неочищенного соединения даже при контроле реакций присоединения. Данные показывают, что присоединение Fmoc-hCy(mmt)-OH требует более 2 часов. Присоединение 3-галогенпропионовой кислоты также требует более 2 часов; действительно для второго пути выявлено, что присоединение не достигалось через 2 часа и для достижения отрицательного теста на присоединение в случае хлорсодержащего производного требовалось двойное присоединение. Более того бромсодержащее производное не могло включаться даже после двойного присоединения. Неочищенный барусибан мог быть получен в соответствии со способом согласно примеру 3 с низким выходом сборки (от 0 до 15%) и низкой чистотой при ВЭЖХ (от 0 до 12%).
4. Пример 4: Сравнение способов (примеры 1 и 2 против примера 3)
В разделах выше описан синтез неочищенного барусибана в соответствии со способом получения согласно примерам 1 и 2 и в соответствии со способом согласно примеру 3 (3.1 и 3.2). Для каждого способа (примеры 1 и 2 против примера 3), проводили исследование, является ли 3-хлорпропионовая кислота (примеры 2, 3.1.1 и 3.2.1) или 3-бромпропионовая кислота (примеры 1, 3.1.2 и 3.2.2) более пригодной для синтеза барусибана. Результаты, полученные для обоих способов (примеры 1 и 2 против пример 3) сравнивают для оценки того, какие их способов являются более подходящими/пригодными для синтеза барусибана.
4.1. Основные данные
В таблице ниже (таблица 5) обобщенно представлены и сравниваются данные о сборке, полученные в экспериментах согласно способам в соответствии с примерами 1 и 2 и способам в соответствии с примером 3.
* P означает "защитную группу". В зависимости от способа, P может представлять собой Fmoc или NBS, mtt или trt, см. примеры 1, 2 и 3
** 46 ч 15 мин: проводили присоединение в течение 21 ч 15 мин, с последующим добавлением 1,65 экв. DIC (6 ч)+двойное присоединение (19 ч)
Таблица 5: Сравнительная таблица данных о сборке для экспериментов в соответствии со способами примеров 1 и 2 и примера 3.
4.2. Промежуточное заключение
Как правило, не наблюдали значимых отличий для времени присоединения остатков Asn, alloIle, Ile и D-Trp. Присоединение 3-галогенпропионовой кислоты оказалось затрудненным в способах согласно примеру 3 и было необходимым двойное присоединение. Реакция завершилась для 3-хлорпропионовой кислоты, но она не завершилась для 3-бромпропионовой кислоты, даже после двойного присоединения. Время присоединения 3-галогенпропионовой кислоты занимает только менее 2 часов в случае способа согласно примеру 1 и двойное присоединение не требуется. Следуя способу получения согласно примеру 3, оказалось, что получают различные неочищенные соединения с чистотой ВЭЖХ от 0 до 12% и выходом от 0 до 15%. Эти результаты являются значительно более низкими, чем данные способа получения в соответствии с примером 1.
5. Пример 5. Исследование стабильности исходных материалов
5.1. Исследование стабильности Fmoc-N-MeOrn(Boc)-ола
Нестабильность исходного материала Fmoc-N-MeOrn(Boc)-ола наблюдали в растворе в DMSO (условия для анализа посредством ЯМР). Через 24 ч Fmoc-N-MeOrn(Boc)-ол деградировал и было выявлено присутствие дибензофульвена. Вследствие этого проводили исследование стабильности Fmoc-N-MeOrn(Boc)-ола. Целью этого исследования являлась проверка стабильности Fmoc-N-Me-L-Orn(Boc)-ола в различных условиях для определения его оптимальных условий применения и хранения. Некоторые эксперименты проводили для оценки деградации Fmoc-N-Me-L-Orn(Boc)-ола в различных условиях хранения.
Стабильность исследовали следующим образом:
-Fmoc-N-Me-L-Orn(Boc)-ол, порошок, при 2-8°C, комнатной температуре и 40°C
-Fmoc-N-Me-L-Orn(Boc)-ол, раствор в DMF (545 г/л) при 2-8°C, комнатной температуре и 40°C
-Fmoc-N-Me-L-Orn(Boc)-ол+DMAP, раствор в DMF при комнатной температуре (соответствует условиям загрузки в способе синтеза согласно примеру 3)
-Fmoc-N-Me-L-Orn(Boc)-ол+пиридин, раствор в DMF при 60°C (соответствует условиям загрузки в способе согласно примерам 1 и 2)
5.1.1. Стабильность порошка Fmoc-N-Me-L-Orn(Boc)-ола
На фиг.6 представлены результаты, полученные для стабильности порошка Fmoc-N-Me-L-Orn(Boc)-ола при 2-8°C, комнатной температуре и 40°C. Fmoc-N-Me-L-Orn(Boc)-ол не является стабильным при 40°C в виде порошка. Через 3 суток наблюдают чистоту 1,55% (снижение от 99,26% до 97,84%, через 6 суток порошок превращается в клейкий материал с затрудненной солюбилизацией и невозможностью анализа посредством ВЭЖХ. Fmoc-N-Me-L-Orn(Boc)-ол является стабильным в качестве твердого вещества при 2-8°C и при комнатной температуре в течение по меньшей мере 14 суток. После этого периода наблюдают небольшую деградацию. В заключение, порошок Fmoc-N-Me-L-Orn(Boc)-ол можно хранить при 2-8°C в течение по меньшей мере 14 суток без какого-либо риска деградации.
5.1.2. Стабильность Fmoc-N-Me-L-Orn(Boc)-ола, раствор в DMF, при различных температурах
На фиг.7 представлены результаты, полученные для стабильности Fmoc-N-Me-L-Orn(Boc)-ола, раствор в DMF (545 г/л), при 2-8°C, комнатной температуре и 40°C. Чистота Fmoc-N-Me-L-Orn(Boc)-ола, хранящегося в DMF, быстро снижается с течением времени при комнатной температуре и при 40°C. Продукт полностью деградировал через 1 сутки при 40°C и через 3 суток при комнатной температуре. Хранение при 2-8°C позволяет замедление деградации. Чистоту поддерживают в течение 3 суток. 50% деградацию наблюдают через 2 недели и полное разложение через один месяц. Соединение деградирует посредством удаления защитной группы Fmoc с образованиемнезащищенного H-N-Me-L-Orn(Boc)-ола и дибензофульвена. Напротив, для хранения при 2-8°C не наблюдают значительной деградации Fmoc-N-Me-L-Orn(Boc)-ола в DMF вплоть до 3 суток. Чистота Fmoc-N-Me-L-Orn(Boc)-ола снижается на 0,3% через 3 суток и на 3% через 8 суток. Через 2 недели чистота Fmoc-N-Me-L-Orn(Boc)-ола снижается вдвое. В заключение, Fmoc-N-Me-L-Orn(Boc)-ол в DMF можно хранить при 2-8°C в течение 3 суток.
5.1.3. Стабильность Fmoc-N-Me-L-Orn(Boc)-ола+DMAP, раствор в DMF, при комнатной температуре
На фиг.8 представлены результаты, полученные для стабильности раствора Fmoc-N-Me-L-Orn(Boc)-ола+DMAP в DMF (условия загрузки в способе в соответствии с примером 3). Получали профили ВЭЖХ. Как и ожидалось, деградация Fmoc-N-Me-L-Orn(Boc)-ола возрастала в присутствии DMAP. Через 2 часа чистота Fmoc-N-Me-L-Orn(Boc)-ола при ВЭЖХ снижалась на 7,5% и через 3 часа чистота Fmoc-N-Me-L-Orn(Boc)-ола при ВЭЖХ составляла 88,24%. Через 24 часа чистота Fmoc-N-Me-L-Orn(Boc)-ола была очень низкой (17,71%). В заключение, раствор Fmoc-N-Me-L-Orn(Boc)-ола+DMAP в DMF не является стабильным в течение длительного периода в условиях загрузки в соответствии со способом примера 3. Некоторую деградацию наблюдали уже после 2 часов при комнатной температуре; материал потенциально мог частично разлагаться в ходе реакции загрузки.
5.1.4. Стабильность Fmoc-N-Me-L-Orn(Boc)-ола+пиридин, раствор в DMF, при 60°C
На фиг.9 представлены результаты, полученные для стабильности Fmoc-N-Me-L-Orn(Boc)-ола+пиридин, раствор в DMF, при 60°C (условия загрузки в соответствии со способами примеров 1 и 2).В условиях стадии загрузки, использованной в способе производства согласно примерам 1 и 2, Fmoc-N-Me-L-Orn(Boc)-ол деградирует очень быстро; 50% снижение чистоты наблюдают после 1 часа и практически полную деградацию наблюдают через 17 часов. В заключение, Fmoc-N-Me-L-Orn(Boc)-ол не является стабильным в условиях загрузки в соответствии со способами примеров 1 и 2, поскольку деградация начинается уже через 1 час и соединение не является стабильным на протяжении времени загрузки (17 часов).
5.2. Исследование стабильности o-NBS-N-Me-Orn(Boc)-ола
Проводили исследование стабильности o-NBS-N-Me-Orn(Boc)-ола. Целью этого исследования является проверка стабильности o-NBS-N-Me-L-Orn(Boc)-ола в различных условиях для определения его оптимального применения. Некоторые эксперименты проводили для оценки деградации o-NBS-N-Me-L-Orn(Boc)-ола в различных условиях загрузки.
Стабильность исследовали следующим образом:
-o-NBS-N-Me-L-Orn(Boc)-ол+DMAP, раствор в DMF при комнатной температуре (соответствующий условия нагрузки в соответствии со способом, описанным в примере 3)
-o-NBS-N-Me-L-Orn(Boc)-ол+пиридин, раствор в DMF при 60°C (соответствующий условиям загрузки в способам, как описано в примерах 1 и 2)
5.2.1. Стабильность o-NBS-N-MeOrn(Boc)-ола+пиридин, раствор в DMF при 60°C
На фиг.10 представлены результаты, полученные для стабильности раствора o-NBS-N-Me-L-Orn(Boc)-ола+пиридин в DMF (условия загрузки в способе согласно примерам 1 и 2). В условиях стадии загрузки, использованных в способе согласно примерам 1 и 2, o-NBS-N-Me-L-Orn(Boc)-ол является стабильным. Через 24 часа чистота o-NBS-N-Me-L-Orn(Boc)-ола составляет 99,68%. В заключение, o-NBS-N-Me-L-Orn(Boc)-ол является стабильным в условиях способов согласно примерам 1 и 2.
5.2.2. Стабильность раствора o-NBS-N-Me-Orn(Boc)-ола+DMAP в DMF при комнатной температуре
На фиг.11 представлены результаты, полученные для стабильности раствора o-NBS-N-Me-L-Orn(Boc)-ола+DMAP в DMF (условия загрузки способов согласно примеру 3). В условиях стадии загрузки, использованных в способах согласно примеру 3, o-NBS-N-Me-L-Orn(Boc)-ол является стабильным. Через 22 часа чистота o-NBS-N-Me-L-Orn(Boc)-ола составляет 99,62%. В заключение, o-NBS-N-Me-L-Orn(Boc)-ол является стабильным в условиях загрузки в соответствии со способами примера 3.
5.2.3. Заключение исследования стабильности
Fmoc-N-Me-Orn(Boc)-ол, использованный в способах согласно примеру 3, является чувствительным материалом, который быстро деградирует в условиях, используемых для стадии загрузки. С другой стороны, o-NBS-защищенный материал остается стабильным независимо от условий загрузки(способы согласно примерам 1, 2 или 3).
6. Основные заключения
В примерах 1 и 2 описан способ производства барусибана в соответствии с настоящим изобретением. Этот способ сравнивают с другим способом производства барусибана (пример 3).
Всего пять экспериментов проводили с использованием либо 3-хлорпропионовой кислоты, либо 3-бромпропионовой кислоты (примеры 1, 2, 3.1.1, 3.1.2, 3.2.1 и 3.2.2).
- Fmoc-N-Me-Orn(Boc)-ол, использованный в способах согласно примеру 3, является чувствительным материалом, который быстро деградирует в условиях, используемых на стадии загрузки: быстрая деградация может влиять на выход загрузки.
- С другой стороны o-NBS-защищенныйматериал остается стабильным независимо от условий загрузки (примеры 1, 2 или 3); это производное является более адаптированным, поскольку оно демонстрирует лучшую стабильность в основных условиях (пиридин или DMAP).
- Какой реагент ни использовали для последнего присоединения в способах согласно примеру 3 (3-хлор/3-бромпропионовая кислота), присоединение было затруднено и было необходимо двойное присоединение для достижения завершения. Реакция была завершенной с 3-хлорпропионовой кислотой и не завершилась с 3-бромпропионовой кислотой. Использование 3-хлорпропионовой кислоты является возможным в способе в соответствии с настоящим изобретением (пример 2).
- Неочищенные материалы в способах согласно примеру 3 получают с чистотой ВЭЖХ от 0 до 12% и выходом от 0 до 15%. Эти результаты являются значительно худшими, чем текущие данные в соответствии со способом согласно примерам 1 и 2, с чистотой при ВЭЖХ 45% и выходом 90%.
Исходя из этих заключений, наилучшие результаты определенно получены с использованием способа согласно примерам 1 и 2, в частности, с использованием способа согласно примеру 1. Процесс получения барусибана согласно примерам 1 и 2 демонстрирует лучшую надежность и хорошо адаптируется для получения надлежащей чистоты и выхода.
ПОЛОЖЕНИЯ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
1. Твердофазный способ получения соединения, имеющего формулу c[AA1-AA6]-AA7-OH, или его фармацевтически приемлемой соли или сольвата, где AA1 представляет собой пропионовую кислоту, AA2 представляет собой AAb, AA3 представляет собой Ile, AA4 представляет собой AAd, AA5 представляет собой Asn, AA6 представляет собой hCy и AA7 представляет собой -NR-CHQ-CH2OH, где R представляет собой CH3 или C2H5, Q представляет собой (CH2)n-NH2, где n равен 2, 3 или 4, причем способ включает стадии:
a) реакция защищенной (P5;P3)AA7 со смолой с получением (P5;P3)AA7-СМОЛЫ,
где AA7, добавляемая к смоле в ходе синтеза на стадии a), представляет собой (P5)NR-CHQ'-CH2OH;
b) пошаговое удлинение (P5;P3)AA7-СМОЛЫ с получением (P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
c) реакция X-AA1 (где X представляет собой атом галогена, выбранный из F, Cl, Br и I) с (P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛОЙ с получением X-AA1(P7)AA2AA3AA4(P1)AA5(Trt)AA6-(P3)AA7-СМОЛЫ;
d) проведение стадии отщепления и удаления защитной группы с получением X-AA1AA2AA3AA4AA5AA6AA7-ола; и
e) циклизация X-AA1AA2AA3AA4AA5AA6AA7-ола с получением циклического пептида:
c[AA1-AA6]-AA7-ола, где AA1 и AA6связаны через тиол из AA6 гомоцистеина,
где:
P1, P5 и P7 представляют собой защитные группы,
AAb представляет собой D-ароматическую α-аминокислоту;
AAd представляет собой алифатическую α-аминокислоту;
X представляет собой галоген;
R представляет собой CH3 или C2H5;
Q' представляет собой (CH2)n-NP3P4;
n равен 2, 3 или 4;
P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга и которые могут быть такими же или отличными от P1.
2. Твердофазный способ согласно положению 1, где смола представляет собой 2-CTC.
3. Твердофазный способ согласно одному или нескольким из предшествующих положений, где P5 представляет собой NBS, и/или где оба из P3 и P7 представляют собой Boc и/или где P1 представляет собой Trt.
4. Твердофазный способ согласно одному или нескольким из предшествующих положений, где X представляет собой Br или Cl, предпочтительно Br.
5. Твердофазный способ согласно одному или нескольким из предшествующих положений, где n равен 3 и/или R представляет собой CH3, и/или предпочтительно где P4представляет собой H и/или предпочтительно где P3 представляет собой Boc.
6. Твердофазный способ согласно одному или нескольким из предшествующих положений, где стадию реакции c) проводят с DIC в качестве связующего реагента в присутствии DCM.
7. Твердофазный способ согласно одному или нескольким из предшествующих положений, где AA2(AAb) представляет собой D-Trp, и/или AA4(AAd)представляет собой AlloIle, и/или AA7представляет собой N-Me-Orn-ол.
8. Твердофазный способ согласно одному или нескольким из предшествующих положений, где способ необязательно включает одну или несколько стадий очистки, предпочтительно одну или несколько стадий очистки после стадии i) (а именно, после стадии отщепления и удаления защитной группы) и/или одну или несколько стадий очистки после стадии j) (а именно, после стадии циклизации).
9. Промежуточное соединение, пригодное для формирования пептида, обладающего фармацевтическими свойствами, который имеет формулу:
X-(CH2)2-CO-NH-AAb-Ile-AAd-Asn(P1)-hCy(P2)-NR-CHQ'-CH2OW
где
AAb представляет собой D-ароматическую α-аминокислоту;
AAd представляет собой алифатическую α-аминокислоту;
X представляет собой галоген;
P1 представляет собой защитную группу
P2представляет собой Trt
R представляет собой CH3 или C2H5;
Q' представляет собой (CH2)n-NP3P4;
n равен 2, 3 или 4;
P3и P4 независимо представляют собой H или защитную группу аминокислоты, которые могут быть одинаковыми или могут отличаться друг от друга и которые могут быть такими же или отличающимися от P1и/или P2; и
W представляет собой H, защитную группу или смолу.
10. Промежуточное соединение согласно положению 9, где W представляет собой смолу, где предпочтительно смола представляет собой CTC.
11. Промежуточное соединение согласно положениям 9-10, где n равен 3 и/или R представляет собой CH3.
12. Промежуточное соединение согласно любому из положений 9-11, где P1 представляет собой Trt.
13. Промежуточное соединение согласно любому из положений 9-12, где P4 представляет собой H и/или P3 представляет собой Boc.
14. Промежуточное соединение согласно любому из положений 9-13, где AAb представляет собой D-Trp, и/или где AAd представляет собой alloIle.
15. Промежуточное соединение согласно любому из положений 9-14, где X представляет собой Br или Cl, предпочтительно Br.
Реферат
Изобретение относится к новым твердофазным способам пептидного синтеза аналогов, которые проявляют активность антагониста окситоцина, особенно барусибана и его промежуточных соединений. В частности, настоящее изобретение относится к твердофазному способу получения соединения, имеющего формулу[AA-AA]-AA-ол, где AAпредставляет собой пропионовую кислоту, AAпредпочтительно представляет собой D-Trp, AAпредставляет собой Ile, AAпредпочтительно представляет собой AlloIle, AAпредставляет собой Asn, AAпредставляет собой Hcy, и AAпредпочтительно представляет собой-Me-Orn-ол, или его фармацевтически приемлемой соли или сольвата. Способ демонстрирует лучшую надежность и хорошо адаптируется для получения надлежащей чистоты и выхода, что делает его приемлемым для крупномасштабного производства. 2 н. и 13 з.п. ф-лы, 11 ил., 5 табл., 5 пр.
Формула

Документы, цитированные в отчёте о поиске
Агонисты окситоциновых рецепторов
Комментарии