Способ синтеза пептидов - RU2777327C1
Код документа: RU2777327C1
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к области органической химии и более предпочтительно к синтезу пептидов или белков исходя из аминокислот. Изобретение относится к способу синтеза пептидов in vitro в жидкой среде, в котором не используют твердую подложку или нерастворимую смолу. Этот способ основан на применении новой группы линкеров, а именно производных соединений полиолефинов или олигомеров полиолефинов или полиалкенов, которые, будучи связанными с аминокислотой или с производным соединением аминокислоты, обладают хорошей растворимостью в неполярных растворителях и низкой растворимостью в воде. Этот способ позволяет получать более чистые и/или легче очищаемые пептиды, чем известные способы с твердой подложкой или жидким носителем. Способ можно легко автоматизировать.
Предшествующий уровень техники
Фармакология пептидов представляет огромный интерес в медицинском и экономическом аспектах, но в научном плане она остается тенью биохимии белков и генетики. Несмотря на громадные успехи инсулина, природного белка, образованного 51 аминокислотой, число пептидов, которые были разработаны в качестве фармацевтически активных веществ, оставалось небольшим до конца 20-го века. Однако, между 2000 и 2016 годами были получены лицензии для коммерческой реализации на три десятка новых активных веществ, образованных пептидами, а значительное число пептидов было объектом клинических и доклинических испытаний (см. публикацию "A. Henninot et al., "The Current State of Peptide Drug Discovery: Back to the Future?", напечатанную в J. Med. Chem., 2018, 61, 4, 1382-1414). Большая часть этих кандидатов в лекарственные средства представляет собой природные пептиды, известные своей специфической функцией, другая часть представляет собой производные соединения природных последовательностей, а еще одна часть веществ представляет собой полностью синтетические вещества.
В случае инсулина, назначаемого миллионам пациентов, синтез генетически модифицированными организмами представляет собой прекрасный путь для получения достаточно чистого инсулина в промышленном масштабе. В случае многих других пептидов терапевтического назначения синтез ab initio (то есть исходя из отдельных аминокислот) представляет собой основной способ, используемый для получения пептидов в необходимых количествах. В этом контексте чистота пептида представляет собой большую технологическую и экономическую цель для выбранного способа синтеза. В то же время экологический фактор становится все более и более сильной общественной целью. Именно поэтому химики находятся в поисках способа с учетом этого критерия (см. публикацию "A. Isidro-Llobet et al., "Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production", напечатанную в J. Org. Chem., 2019, 84, 4615-4628). Очистка пептида (которую осуществляют в общем случае высокоэффективной жидкостной хроматографией ВЭЖХ) может определять значительную долю себестоимости конечного продукта, а также быть источником отходов.
Существует две основные методики синтеза пептидов, которые отличаются физическим состоянием среды, в которой протекают химические реакции: жидкая среда (этот синтез называют также "синтезом в растворе") и твердая среда (этот синтез называют также "синтезом на твердой подложке").
Все аминокислоты обладают по меньшей мере двумя реакционноспособными функциональными химическими группами: аминогруппой (Nα-центром) и карбоксигруппой (C-концом). Некоторые аминокислоты обладают к тому же боковыми цепями, способными взаимодействовать с Nα-центрами или C-концами. Ключевой аспект стратегии синтеза пептидов состоит в выборе защитных групп (называемых также группами защиты), которыми защищают реакционноспособные центры во время некоторых стадий синтеза. Таким образом, каждое добавление аминокислоты требует осуществления цикла стадий: защита - активация/присоединение - снятие защиты. В конце синтеза защитные группы отщепляют для получения требуемого пептида. Таким образом, соответственно физическому состоянию среды, в которой происходит синтез пептидов, общий базовый цикл "защита - активация/присоединение - снятие защиты" является неизменным, а отличается только используемой защитной группой. Более точно в такой цикл входят несколько стадий:
- защита аминогруппы (Nα) аминокислоты защитной группой, отщепляемой после реакции присоединения аминокислоты;
- осуществляемая в случае необходимости защита боковой цепи защитной группой, отщепляемой в конце синтеза пептида;
- активация карбоксигруппы Nα-защищенной аминокислоты и последующее связывание ее с производным соединением аминокислоты или пептидом, Nα-аминогруппа которых является свободной, а карбоксигруппа защищена;
- в конце итераций "активация/присоединение - снятие защиты" для всех аминокислот последовательности требуемый пептид получают удалением всех защитных групп.
Цикл "защита - активация/присоединение - снятие защиты" в данном случае поясняется для примера синтеза трипептида.
Согласно стратегии, в случае раствора называемой "Boc" и показанной на схеме реакций 1, осуществляют подготовку первой, второй и третьей аминокислот (называемых в данном случае AA1, AA2 и AA3). Аминогруппу (Nα) аминокислоты AA2 временно защищают трет-бутоксикарбонильной группой (обычно называемой "Boc"). Затем активируют карбоксигруппу (C-конец) этой защищенной аминокислоты, которую соединяют со свободной аминогруппой (Nα) (т.е. незащищенной) сложного метилового эфира аминокислоты AA1, в результате чего образуется дипептид. Далее снимают защиту аминогруппы (Nα) аминокислоты AA2 образованного дипептида (т.е. отщепляют защитную группу Boc кислотой, такой как трифторуксусная кислота). Затем защищают аминогруппу (Nα) аминокислоты AA3. Далее активируют карбоксигруппу (C-конец) аминокислоты AA3, которую связывают с аминогруппой (Nα) аминокислоты AA2 (незащищенного дипептида) для образования защищенного трипептида. После удаления защитной группы Boc, внесенной аминокислотой AA3, получают трипептид. Этот процесс включает в себя не менее пяти стадий реакций, из которых две стадии (активация/присоединение - снятие защиты) осуществляют для каждой дополнительной аминокислоты, добавленной к пептиду.
Схема реакций 1

Эта реакция может происходить с прикреплением аминокислоты AA1 к твердой подложке или к солюбилизирующей органической молекуле в жидкой среде.
Флуоренилметоксикарбонильная группа может быть использована в качестве группы, защищающей аминогруппу, согласно стратегии "Fmoc", показанной на схеме реакций 2. Эта стратегия является особенно приемлемой для синтеза на твердой подложке. Осуществляют подготовку первой, второй и третьей аминокислот (AA1, AA2 и AA3). Синтез начинают с защиты аминогруппы (Nα) аминокислоты AA1 флуоренилметоксикарбонильной группой (обычно называемой "Fmoc") и последующим ее закреплением на твердой смоле (обозначенной на схеме реакций 2 черным кружком) посредством ее карбоксигруппы (C-конца), образующей ковалентную связь типа сложного эфира или амида с функциональной химической группой функционализованной смолы (спиртовой группой или аминогруппой). Затем снимают защиту аминогруппы (Nα) аминокислоты, закрепленной на смоле. Далее аминогруппу (Nα) аминокислоты AA2 защищают защитной группой Fmoc. Полученное производное соединение присоединяют посредством активации его карбоксигруппы (C-конца) к аминогруппе (Nα) аминокислоты AA1, закрепленной на смоле, в результате чего образуется N-защищенный дипептид, закрепленный на смоле. Затем отщепляют защитную группу Fmoc от дипептида (на AA2). После защиты аминогруппы (Nα) аминокислоты AA3 группой Fmoc осуществляют активацию ее карбоксигруппы (C-конца) и затем осуществляют связывание полученного соединения с закрепленным дипептидом, содержащим свободную аминогруппу, образуя таким образом N-защищенный трипептид, который остается зафиксированным на твердой подложке благодаря кислотной группе (C-концу) его аминокислоты AA1. Этот процесс включает в себя не менее семи стадий реакций, из которых две стадии (активация/присоединение - снятие защиты) осуществляют для каждой дополнительной аминокислоты, добавляемой к пептиду.
Схема реакций 2
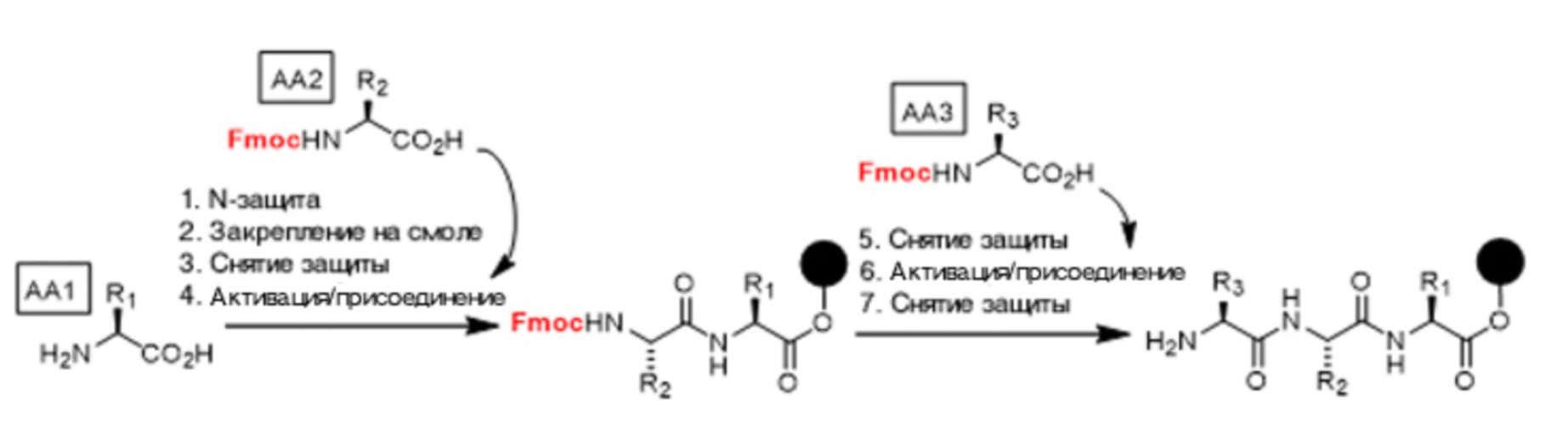
Эти два пути синтеза хорошо известны специалистам в данной области техники (см. разделы 7-5 справочника "D. Voet et J.G. Voet, "Biochimie", 2eme edition, Bruxelles 2005). Они могут быть осуществлены в жидкой среде или на твердой подложке (в этом случае аминокислоту закрепляют на твердой подложке, т.е. осуществляют синтез Меррифилда). На практике аминокислоты переводят в защищенное состояние группами Fmoc или Boc и используют непосредственно в реакциях активации/присоединения в этом защищенном состоянии.
Во время синтеза пептидов в жидкой среде (SPPL) все реакции происходят в гомогенном растворе. Этот способ был описан Бодански и Дю Виньо (J. Am. Chem. Soc., 1959, 51, 5688-5691). Карбоксигруппу (C-конец) исходной аминокислоты защищают в форме сложного метилового эфира, а последующие аминокислоты последовательно присоединяют после защиты их аминогрупп (Nα) бензилоксикарбонильной группой и активации их карбоксигрупп (C-концов) сложным нитрофениловым эфиром. Все синтезированные промежуточные соединения очищают осаждением или промывкой водой (экстракцией). Этот способ синтеза пептидов является долгим и утомительным, а пептиды в его случае образуются с низким выходом. В качестве примера можно назвать синтез ACTH с общим выходом около 7%, описанный Швицером и Зибером (Helv. Chim. Acta 1966, 49, 134-158).
О модификации этого способа было сообщено Байерманом и соавт. (Rec. Trav. Chim. Pays Bas 1973, 92, 481-492). Она состоит в том, чтобы защитить карбоксигруппу (C-конец) аминокислоты или пептида в форме сложного бензилового эфира и осуществить реакцию присоединения (или конденсации) в присутствии избытка ангидрида N-защищенной (Nα) аминокислоты с целью улучшения выхода. В конечном счете, хотя выход реакций присоединения был увеличен, было отмечено ухудшение растворимости в органической среде, когда образованный пептид содержит порядка пяти остатков аминокислот.
Для улучшения растворимости аминокислота или белок могут быть связаны с защитной группой, называемой солюбилизирующей, такой как фенилазобензилсульфонилэтилоксигруппа (OPSE), описанная в EP 0017536 (CM Industries). Действительно, эта защитная группа обеспечивает солюбилизацию синтезированной аминокислоты или пептида в N, N-диметилформамиде. После каждой итерации (активация/присоединение - снятие защиты) осуществляют очистку осаждением и отделением твердых веществ фильтрованием, что порождает технологические затруднения.
Подход, вводящий применение несшитого линейного полимера, такого как полиэтиленгликоль (PEG), для поддержания аминокислоты или пептида в растворе, был описан Муттером и Байером (Nature 1972, 237, 512-513). В этом случае также побочные продукты удаляют осаждением или ультрацентрифугированием.
В более недавнее время в EP 2612845 A1 и US 2014/0296483 (Ajinmoto Co., Inc.) были описаны новые защитные группы (или линкеры), обеспечивающие солюбилизацию аминокислоты и пептида. Таким образом, в зависимости от использованного линкера очистку осуществляют как простой промывкой водой, так и осаждением и фильтрованием. Благодаря этим сильно липофильным группам, защищающим карбоксигруппы, бивалирудин, представляющий собой антикоагулянт, образованный 20 остатками аминокислот, получали с общим выходом 73% и чистотой 84% (см. "D. Takahashi et al., Angew. Chem. Int. Ed., 2017, 56, 7803-7807"). Несмотря на преимущества этого способа, ее предел, а именно число остатков аминокислот, которые могут быть присоединены, остается неизвестным. К тому же, себестоимость и затраты на экологию в случае этих линкеров представляют собой неблагоприятные факторы.
Известны и другие стратегии присоединения аминокислот. Например, можно использовать дифункциональные группы, которые одновременно активируют карбоксигруппы (C-концы) и защищают аминогруппы (Nα) аминокислот, образуя промежуточные циклические структуры, являющиеся сильно реакционноспособными. В общем случае они легко реагируют с аминогруппой (Nα) второго производного соединения аминокислоты с образованием дипептида. К сожалению, полученные соединения являются очень реакционноспособными, при этом часто происходит нежелательная реакция полимеризации. Среди описанных дифункциональных групп можно назвать фосген (см. "R. B. Woodward et C. H. Schramm, J. Am. Chem. Soc., 1947, 69, 1551-1552"), дихлордиметилсилан (см. "S. H. van Leeuwen et al., Tetrahedron Letters 2002, 43, 9203-9207"), гексафторацетон (см. "J. Spengler et al., Chem. Rev., 2006, 106, 4728-4746") и формальдегид (см. "J. M. Scholtz, P. A. Bartlett, Synthesis 1989, 542-544").
Твердофазный синтез пептидов был описан Меррифилдом (J. Am. Chem. Soc., 1963, 85, 2149-2154). Синтез состоит в том, чтобы закрепить карбоксигруппу (C-конец) первой аминокислоты или пептида на нерастворимой смоле (подложке). Вследствие этого реагенты используют в избытке для того, чтобы обеспечивать полную конверсию на стадиях активации/присоединения. Очистку осуществляют простым фильтрованием и промывкой смолы. Хотя этот способ является автоматизируемым и более простым, существует множество недостатков, таких как стоимость и потеря реагентов, используемых в избытке, и отсутствие однородности синтезированных пептидов, поскольку почти невозможно получать однородные пептиды: в данном случае говорят, что система является вырожденной. К тому же очистка этих разнородных пептидов препаративной высокоэффективной жидкостной хроматографией является дорогостоящей, поскольку требует большого расхода растворителей и экологически мало приемлема.
С учетом того, что каждая стадия реакции редко проходит количественно и, следовательно, сопровождается потерями, накапливающимися по ходу всего синтеза, и с учетом того, что каждая стадия требует применения растворов реагентов (связывающих агентов и побочных продуктов), удаление которых является проблематичным, было бы интересно располагать более простым способом, который обеспечивал бы одновременно получение пептидов высокой чистоты, меньшую стоимость производства и более высокую экологическую совместимость. Этот способ должен быть приемлемым для всех видов природных аминокислот и для широкого спектра аминокислот неприродного происхождения. Это является целью настоящего изобретения.
Следует подчеркнуть, что в случае пептида терапевтического назначения выход меньше 100% обуславливает не только потерю реагентов, но и образование вторичных продуктов, которые могут быть трудно отделяемыми от требуемого пептида; желательность получения как можно более чистых пептидов для четкого определения их биологических эффектов означает выяснение того, насколько приемлемо удорожание, определяемое очисткой, насколько приемлемо присутствие примесей, способных обуславливать риск ошибки в оценке исследованных биологических эффектов.
Проблема, на решение которой направлено настоящее изобретение, состоит в разработке способа синтеза как можно более чистых пептидов или белков с более высоким выходом, который был бы более экологически приемлемым, менее затратным и был бы автоматизируемым. этот способ должен быть приемлемым по меньшей мере в случае всех природных аминокислот и предпочтительно в случае широкого спектра аминокислот неприродного происхождения. В этом способе не должны применяться дорогостоящие или трудно синтезируемые линкеры или подложки.
Цель изобретения
В настоящем изобретении предлагается решить проблемы, существующие на предшествующем уровне техники, за счет применения полиолефинов или олигомеров полиолефинов или полиалкенов для получения пептидов или белков высокой чистоты в жидкой среде. Действительно, авторами было найдено, что применение полиолефинов и предпочтительно производных соединений полиизобутенов (PIB) в качестве линкеров или жидкой подложки обеспечивает солюбилизацию аминокислот и синтез пептидов в органическом растворе (галогенированные и негалогенированные растворители), облегчая их очистку простой экстракцией или промывкой, в данном случае водой или смесью "вода/этанол" или "вода/ацетонитрил", или простым фильтрованием. Кроме того, некоторые из этих линкеров (предпочтительно некоторые производные соединения полиизобутенов (PIB)) представляют собой вещества, поставляемые в торговую сеть, или же их синтез является простым, прямым и мало затратным.
Следовательно, цель изобретения представляет собой способ синтеза пептидов или белков последовательным удлинением второй концевой части (Nα) пептидной цепи, первая концевая часть которой прикреплена посредством ее карбоксигруппы (C-конца) или ее аминогруппы (Nα), к линкеру, растворимому в неполярном растворителе, причем способ отличается тем, что линкер содержит полиолефиновую цепь, содержащую по меньшей мере 10 мономерных звеньев и предпочтительно от 15 до 50 звеньев. Предпочтительно линкер в одной из своих концевых частей содержит функциональную группу с целью обеспечения связывания с первой аминокислотой.
Указанный линкер предпочтительно представляет собой полиолефин.
Указанный линкер предпочтительно содержит только полиолефиновую цепь; таким образом, эта полиолефиновая цепь может быть получена простой реакцией полимеризации (изобутена).
Указанный линкер может содержать в каждом из своих звеньев одинаковые или разные алкилы, предпочтительно выбранные из группы, которую образуют метил и этил. Указанная полиолефиновая цепь предпочтительно имеет среднемассовую молекулярную массу в интервале от 600 до 20 000 и предпочтительно от 700 до 15 000. Указанная полиолефиновая цепь может содержать ненасыщенные связи "углерод-углерод" в количестве не более 5% и предпочтительно не более 3%. Предпочтительно речь идет о полиизобутеновой цепи.
В предпочтительном варианте осуществления способа по настоящему изобретению линкер содержит полиолефиновую цепь (или представляет собой полиолефиновую цепь), заканчивающуюся группировкой, выбранной из группы, которую образуют:
- функциональная группа -X, где X выбрана из группы, которую образуют -OH, -NH2, -SH;
- функциональная группа -Z-C6H4X1, где:
- Z представляет собой O или отсутствует;
- X1 выбрана из группы, которую образуют -OH, -NH2, -SH, -NH-NH2, -CXRR1, -C6H3R'(CRX), где X выбрана из группы, которую образуют -OH, -NH2, -SH, а R выбрана из группы, которую образуют -H, арил, гетероарил, и R' выбрана из группы, которую образуют -H, -алкил, -O-алкил, -арил, -O-арил, гетероарил, -O-гетероарил;
- функциональная группа -CR"=CH-CHX или функциональная группа -CR''H-CH=CH-CHX, где X выбрана из группы, которую образуют -OH, -NH2, -SH, а R'' означает метил или этил.
При этом эта группировка означает функционализацию полиолефиновой цепи.
Первая концевая часть пептидной цепи представляет собой первое аминокислотное звено AA1; именно эта первая аминокислота AA1 связана с линкером как с его карбоксигруппой (с C-концом), так и с его аминогруппой (Nα). Указанная пептидная цепь образована n аминокислотными звеньями; ее вторая концевая часть представляет собой другое аминокислотное звено AAn. Во время осуществления способа пептидная цепь увеличивается за счет последовательного удлинения, а в ходе каждой из этих стадий удлинения добавляют другое аминокислотное звено AA(n+1) к указанной второй концевой части.
Аминогруппа (Nα) аминокислот, используемая в способе по настоящему изобретению, может быть защищена группировкой Boc или Fmoc или любыми другими приемлемыми защитными группами.
В пептидной цепи можно использовать природные аминокислоты и/или аминокислоты неприродного и/или синтетического происхождения.
Способ по настоящему изобретению включает в себя по меньшей мере одну стадию, на которой пептидную цепь связывают с линкером и отделяют от реакционной смеси экстракцией в неполярном растворителе.
Способ по настоящему изобретению позволяет получать очень чистые пептиды или белки, которые отщепляют от линкера после последней стадии удлинения пептидной цепи, для применения согласно их назначению, например в качестве активного вещества для доклинических или клинических испытаний.
Подробное описание
Определения
В рамках настоящего изобретения под "аминокислотой" понимают природные аминокислоты и аминокислоты неприродного происхождения. "Природные" аминокислоты имеют L-форму аминокислот, которые могут быть найдены в белках природного происхождения, то есть аланин (Ala), аргинин (Arg), аспарагин (Asn), аспарагиновая кислота (Asp), цистеин (Cys), глутамин (Gln), глутаминовая кислота (Glu), глицин (Gly), гистидин (His), изолейцин (ILe), лейцин (Leu), лизин (Lys), метионин (Met), фенилаланин (Phe), пролин (Pro), серин (Ser), треонин (Thr), триптофан (Trp), тирозин (Tyr) и валин (Val).
Аминокислоты "неприродного происхождения" имеют D-форму определенных ранее природных аминокислот, гомоформы некоторых природных аминокислот (таких как аргинин, лизин, фенилаланин и серин) и нор-формы лейцина и валина. К ним относятся также аминокислоты неприродного происхождения, такие как:
Abu=аминобутировая кислота CH3-CH2-CH(COOH)(NH2);
iPr=изопропиллизин (CH3)2C-NH-(CH2)4-CH(COOH)(NH2);
Aib=2-аминоизобутировая кислота;
F-trp=N-формилтриптофан;
Orn=орнитин;
Nal2=2-нафтилаланин.
К аминокислотам "неприродного происхождения" относятся также все синтетические аминокислоты.
Термин "защищенная аминокислота" в данном случае применяют также для обозначения временно защищенных аминокислот, описанных, в частности, ранее; например, аминогруппа (Nα) может быть защищена группами Fmoc, Boc, бензильной группой или любыми другими приемлемыми защитными группами.
В способе получения пептидов по настоящему изобретению используют полиолефины, или более точно олигомеры полиолефинов (полиолефинов, называемых также полиалкенами), и их производные соединения в качестве линкеров или защитных групп в случае карбоксигруппы (C-конца) аминокислоты или пептида или аминогруппы (Nα) аминокислоты или пептида, или боковой цепи указанных аминокислоты или пептида (в форме связей сложных эфиров, амидов, простых эфиров, тиоэфиров или любых других функциональных групп) в жидкой среде. Молекулы полиолефинов содержат цепь атомов углерода, соединенных простыми связями. Они могут содержать ответвления, образованные одинаковыми или разными алкильными группами, но предпочтительно одинаковыми. Предпочтительно применяют полимеры с числом мономерных звеньев, которое составляет по меньшей мере 10 и предпочтительно находится в интервале от 15 до 50. Предпочтительным является гомополимеры, но также можно использовать сополимеры; в последнем случае число ненасыщенных связей в цепи атомов углерода преимущественно не превышает 5% и предпочтительно не превышает 3%.
Предпочтительно речь идет о производных соединениях полиизобутенов (PIB), представляющих собой класс полимеров, известный с 30-х годов прошлого века, но также можно использовать производные соединения полипропиленов.
Эти линкеры предпочтительно применяют в случае способа по настоящему изобретению в форме производных соединений, содержащих функциональные группы, что будет пояснено дальше более подробно.
Согласно настоящему изобретению эти линкеры связываются с карбоксигруппой аминокислоты (с C-концом) или с аминогруппой (Nα) посредством ковалентной связи типа связи амидной, сложноэфирной, бензильной, аллильной группы или любых других функциональных групп. Это предполагает, чтобы линкеры были использованы в форме, которая содержит надлежащие функциональные группы и которую в настоящем описании называют "производные соединения PIB" с учетом того, что этот термин включает в себя в данном случае также производные соединения линкеров, которые не являются производными соединениями полиизобутена и которые являются производными соединениями других полиолефинов согласно определению, приведенному ранее. Эта функционализация линкера, как правило, представляет собой введение функциональной группы в концевую часть, предпочтительно в одну из концевых частей цепи атомов углерода, что будет описано далее.
Олигомеры полиолефинов, применяемые в качестве линкера, как правило, характеризуются среднемассовой молекулярной массой, но также можно использовать "чистые" олигомеры, которые содержат одинаковые молекулы с заданной длиной цепи.
Такая реакция между производным соединением PIB и аминокислотой ведет к получению соединения, отличающегося тем, что когда производное соединение PIB связано с аминокислотой или производным соединением аминокислоты, содержащим в случае необходимости защищенную боковую цепь, получают соединение, растворимость которого в воде является низкой (< 30 мг/мл).
Более точно, способ синтеза пептидов, защищенных в случае необходимости, в жидкой среде (в растворе) согласно настоящему изобретению отличается тем, что аминокислоты или пептиды солюбилизируют в органической среде производным соединением PIB, связанным с карбоксигруппой (с C-концом) или с аминогруппой (Nα) аминокислоты или пептида. Производное соединение PIB действует в качестве линкера или жидкой подложки аминокислоты или пептида, которые синтезируют последовательным прикреплением аминокислот к последней аминокислоте, связанной с этой линкером. Таким образом, линкер служит также в качестве защитной группы в ходе последовательных итераций во время синтеза пептида.
Аминокислота или пептид, защищенные в случае необходимости, связанные с молекулой PIB, отличаются тем, что карбоксигруппа (C-конец) или аминогруппа (Nα) указанных аминокислоты или пептида связаны ковалентной связью типа связи сложного эфира, амидной, бензильной, аллильной группы или любых других функциональных химических групп липофильного производного соединения PIB, определяющего очень низкую растворимость в воде (< 30 мг/мл). В этом смысле в способе по настоящему изобретению линкер, который предпочтительно представляет собой производное соединение PIB, действует в качестве жидкой подложки для синтеза пептидов или белков.
Получение производного соединения аминокислоты (Nα-защищенной или Nα-незащищенной) или пептида (Nα-защищенного или Nα-незащищенного) с производным соединением PIB в значительной степени увеличивает растворимость указанных аминокислоты или пептида в органической неполярной жидкой среде. Более точно, эти аминокислоты и эти пептиды становятся растворимыми в органических растворителях, таких как галогенированные растворители (метиленхлорид, хлороформ), этилацетат, тетрагидрофуран, циклогексан, гексаны или ароматические растворители, такие как бензол или толуол. Следовательно, аминокислоты и пептиды, связанные с производным соединением PIB, имеют высокий коэффициент распределения относительно органической среды во время экстракции/декантации в присутствии воды или смеси "вода/этанол" или же "вода/ацетонитрил", что позволяет, таким образом, осуществлять их простую и быструю очистку.
В настоящем изобретении предлагается также способ синтеза пептидов (защищенных или незащищенных) в жидкой среде, отличающийся тем, что начинают с аминокислоты или пептида в растворе (или производного соединения аминокислоты или пептида в растворе), которые будут связаны с одним из линкеров, определенных ранее, посредством карбоксигруппы (C-конца) или аминогруппы (Nα) исходного производного соединения аминокислоты или пептида, и тем, что добавляют или присоединяют следующие аминокислоты или пептиды, содержащие защищенные аминогруппы (Nα) и в случае необходимости защищенные боковые цепи, после активации их карбоксигрупп (C-концов).
Активация карбоксигруппы (C-конца) Nα-защищенных аминокислоты или пептида может быть осуществлена любыми известными способами синтеза путем получения ангидрида разными реагентами, такими как карбодиимид, хлорангидрид кислоты, алкилхлорформиат, или любым другим способом активации Nα-защищенной аминокислоты.
Способ синтеза пептидов согласно настоящему изобретению отличается также тем, что затем добавляют аминокислоты или пептиды, подлежащие присоединению. Эти аминокислоты или пептиды, содержащие защищенные аминогруппы (Nα) и в случае необходимости также защищенные боковые цепи, активируют по их кислотным группам.
Авторами постадийно представлен способ по настоящему изобретению на примере, в котором получают трипептид H-Tyr-Gly-Phe-OH, используя в качестве защитных групп Boc или Fmoc.
На схеме реакций 3 показана первая стадия связывания карбоксигруппы первой аминокислоты (AA1), в данном случае фенилаланина (Phe), с производным соединением PIB, содержащим фенольную функциональную группу. Аминокислоту AA1 используют в форме, защищенной группой Fmoc.
Схема реакций 3
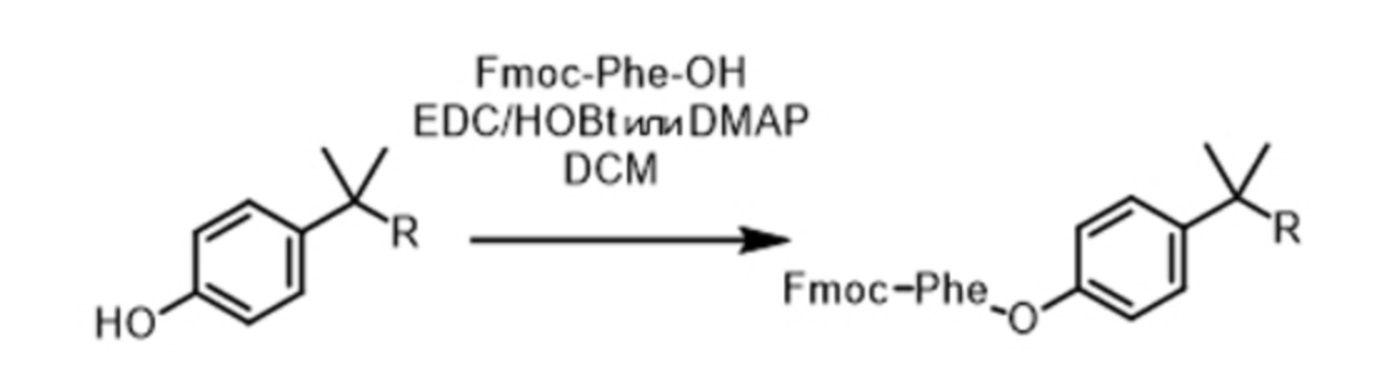
В этой схеме реакций DCM означает дихлорметан, EDC означает 1-этил-3-(3-диметиламинопропил)карбодиимид, HOBt означает гидроксибензотриазол, а DMAP означает 4-диметиламинопиридин.
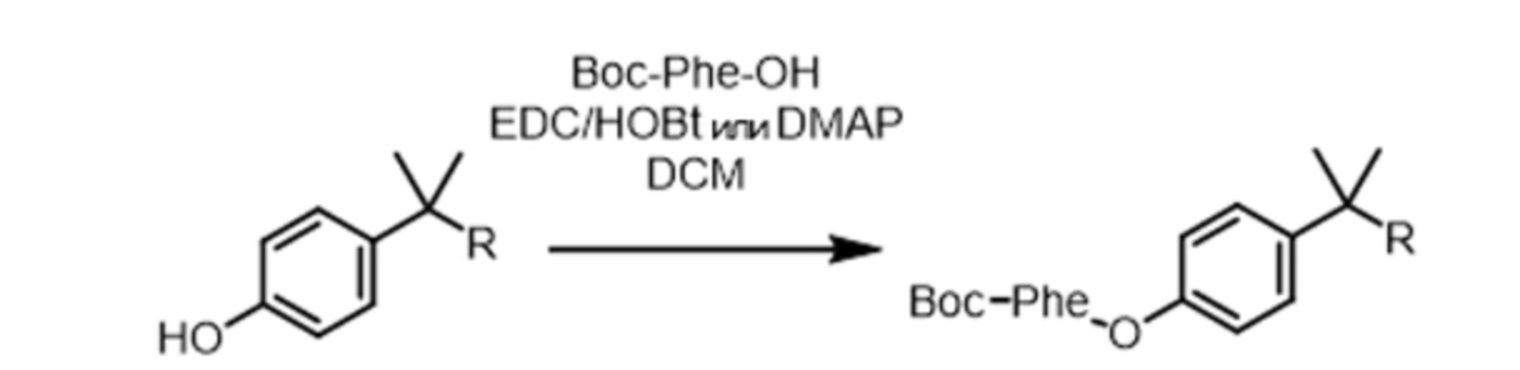
На схеме реакций 4 показан вариант, в котором AA1 используют в форме, защищенной группой Boc.
Схема реакций 4
На второй стадии защитную группу (Fmoc или Boc) отщепляют для высвобождения аминогруппы (Nα) AA1. Эта стадия, называемая стадией снятия защиты, поясняется схемой реакций 5 для случая Fmoc и 6 для случая Boc. Она ведет к получению соединения, называемого в данном случае PIB-AA1.
Схема реакций 5
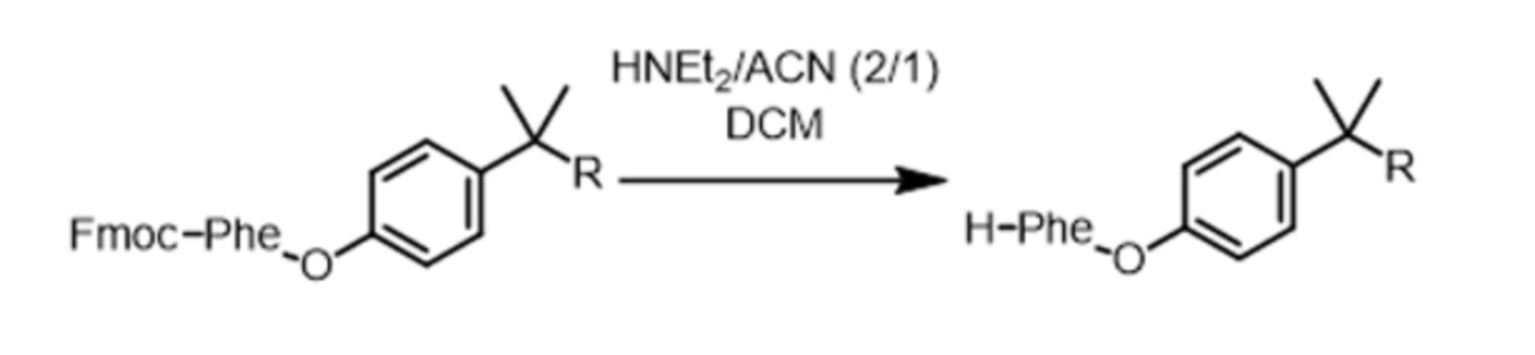
Схема реакций 6
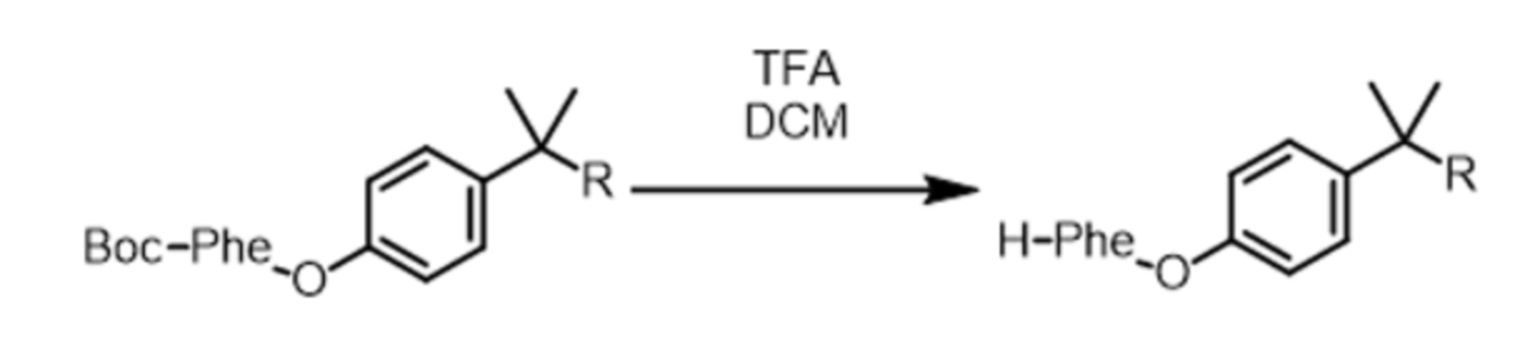
ACN означает ацетонитрил, HNEt2 означает диэтиламин, а TFA означает трифторуксусную кислоту.
На третьей стадии повторяют операции первой стадии, добавляя к молекуле PIB-AA1 вторую аминокислоту (обозначаемую как AA2), в данном случае глицин (Gly) в защищенной форме (Boc или Fmoc); кислотную группу (C-конец) аминокислоты AA2 (Nα-функциональная группа которой защищена) связывают с N-концевой функциональной группой молекулы PIB-AA1. На четвертой стадии с Nα-функциональной группы молекулы PIB-AA1-AA2 снимают защиту, получая дипептид PIB-AA1-AA2, прикрепленный к его подложке.
На пятой стадии повторяют операции первой стадии, присоединяя к молекуле PIB-AA1-AA2 третью аминокислоту (обозначаемую как AA3), в данном случае тирозин, содержащий боковую цепь, защищенную трет-бутильной группой, и находящийся в защищенной форме (Boc или Fmoc); кислотную группу (C-конец) аминокислоты AA3 (Nα-функциональная группа которой защищена) связывают с Nα-функциональной группой молекулы PIB-AA1-AA2. На шестой стадии с Nα-функциональной группы молекулы PIB-AA1-AA2-AA3 снимают защиту и получают прикрепленный трипептид, показанный на схеме 7.
Схема реакций 7
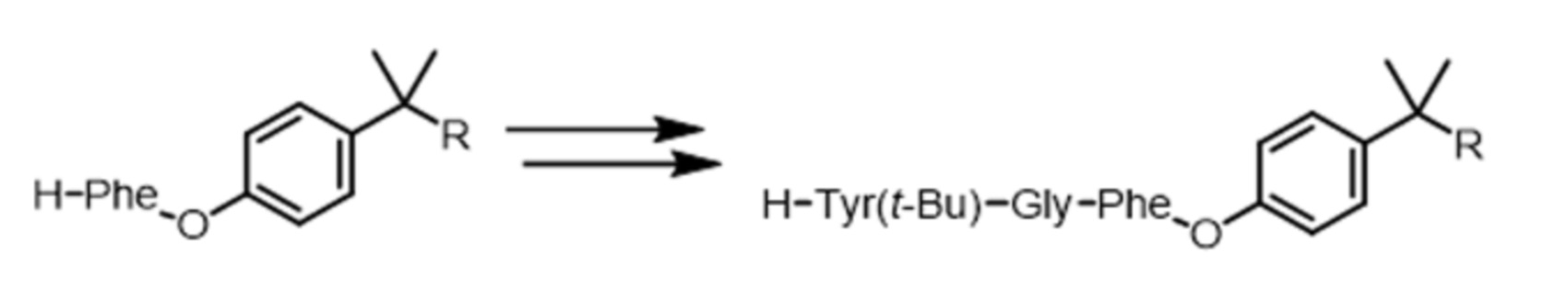
Можно легко видеть, что этот способ позволяет последовательными итерациями добавлять к аминокислоте и затем к пептиду, прикрепленным к производному соединению PIB, последующие аминокислоты для получения таким образом пептида, содержащего заданную последовательность. Пептид, прикрепленный к жидкой подложке, может быть отделен в любой момент и, в частности, после последней итерации от всех полярных соединений экстракцией неполярным растворителем. В конце этой последовательности удлинений можно отделить пептид от полимерной подложки. Таким образом, пептид утрачивает свою растворимость в неполярной среде и может быть отделен от полимерной подложки для применения соответственно своему назначению.
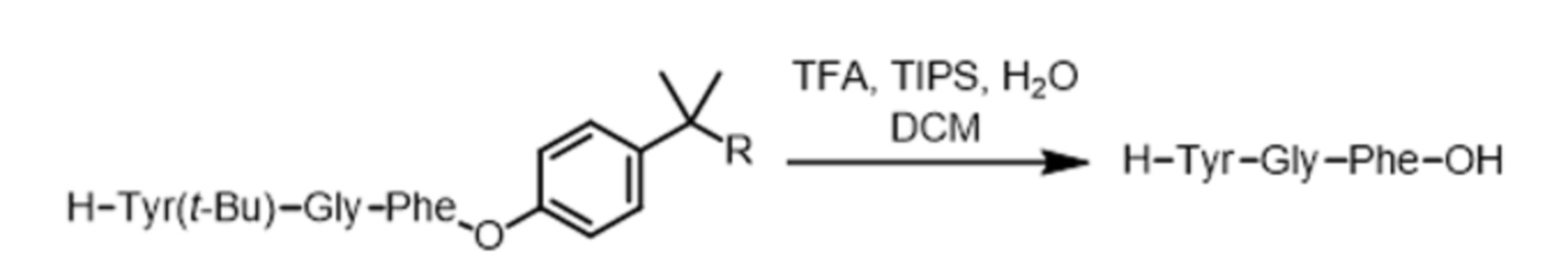
Реакция отделения пептида от жидкой подложки поясняется на схеме реакций 8 для случая трипептида со схемы реакций 7. При этом можно использовать смесь трифторуксусной кислоты (TFA), триизопропилсилана (TIPS) и воды.
Схема реакций 8
На схеме 9 показано некоторое число производных соединений PIB с введенными в них функциональными группами, являющихся приемлемыми в качестве жидкой подложки для осуществления настоящего изобретения.
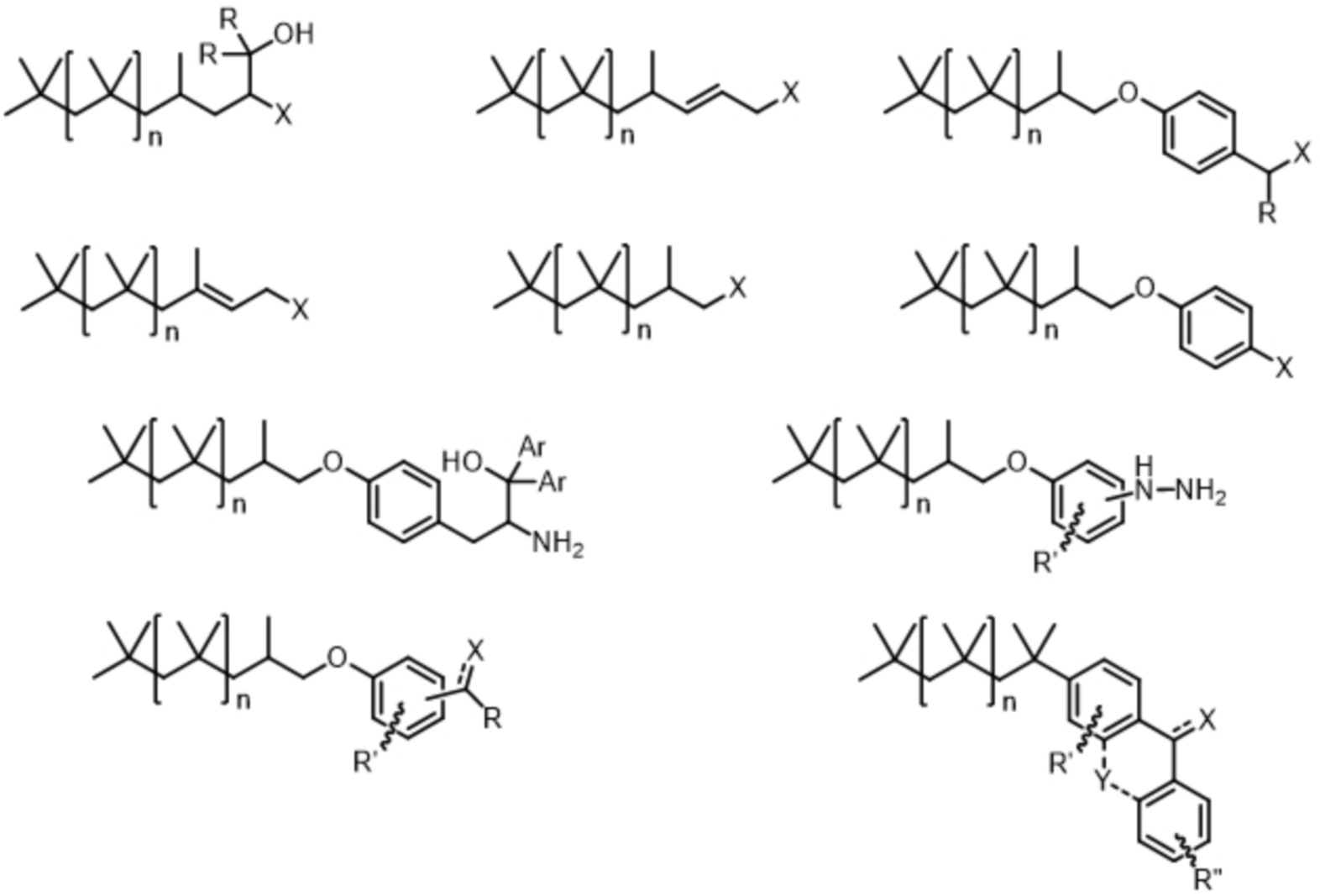
В этих формулах:
- X означает группировку, выбранную из группы, которую образуют OH, NH2, NH-NH2, SH;
- R означает группировку, выбранную из группы, которую образуют H, арил, гетероарил;
- R' означает группировку, выбранную из группы, которую образуют H, алкил, O-алкил, гетероарил, O-(гетероарил);
- R" означает группировку, выбранную из группы, которую образуют H, алкил, O-алкил, гетероарил, O-(гетероарил);
- Y означает группировку, выбранную из группы, которую образуют O, CH2CH2;
- число n означает целое число, которое, как правило, превышает 10 и предпочтительно находится в интервале от 15 до 50.
В частности, X может представлять собой свободный амин, гидразин, спирт, тиол или фенол.
В предпочтительном варианте осуществления среднемассовая молекулярная масса линкеров, за исключением функциональных групп в концевой части (например, -X, -Z-C6H4X1 или -CR"=CH-CHX, определенных ранее), находится в интервале от 600 до 20 000 и предпочтительно от 700 до 15 000. При молекулярной массе более 20 000 эти соединения обладают очень большой вязкостью, что будет создавать риск ограничения их растворимости в растворителях (галогенированных или негалогенированных), используемых для стадии активация/присоединение.
Некоторые производные соединения PIB, приемлемые для применения в рамках настоящего изобретения, имеются в продаже в качестве лигандов для гомогенного катализа. Например, можно использовать 2-метил-3-[полиизобутил(12)]пропанол (среднемассовая молекулярная масса составляет 757, включая функциональные группы в концевой части), или 4-[полиизобутил(18)]фенол (среднемассовая молекулярная масса составляет 1104, включая функциональные группы в концевой части), которые реализует компания "Strem Chemicals" под артикулами 06-1037 и 06-1048 соответственно. Эти два соединения представляют собой производные соединения полиизобутенов, в которых цепь заканчивается группой -CH2-C(CH3)(H)-CH2-OH (т.е. изопропанольной) и группой -CH2-C(CH3)2-C6H5-OH (т.е. фенольной) соответственно.
Предпочтительные линкеры, а именно производные соединения полиизобутена, могут быть получены исходя из изобутена биологического происхождения. Понятие "содержание вещества биологического происхождения" определено в стандарте ISO 16620-1:2015 "Plastiques - Teneur biosourcée - partie 1: Principes généraux", в частности, терминами "синтетический полимер биологического происхождения", "содержание синтетического полимера биологического происхождения", "содержание углерода биологического происхождения" и "содержание в массе биологического происхождения", а также в стандартах ISO 16620-2:2015 "Plastiques - Teneur biosourcée - partie 2: Détermination de la teneur en carbone biosourcé" и ISO 16620-3:2015 "Plastiques - Teneur biosourcée - partie 3: Détermination de la teneur en polymère synthétique biosourcée" для способов нахождения и количественного определения степени биологического происхождения.
Линкеры преимущественно характеризуются содержанием углерода биологического происхождения, превышающим 90%, предпочтительно превышающим 93% и более предпочтительно превышающим 95%.
Способ по настоящему изобретению предоставляет несколько преимуществ.
Первое преимущество состоит в том, что способ позволяет получать пептид в жидкой среде, в которой пептид (Nα-защищенный или Nα-незащищенный), связанный с линкером, определенным ранее, остается в органическом растворе.
Второе преимущество состоит в том, что способ позволяет получать пептиды высокой чистоты простой промывкой водой или смесью "вода/этанол" или же "вода/ацетонитрил", или фильтрованием, обеспечивая таким образом удаление побочных продуктов (солей, кислот или любых других соединений, образующихся, например, во время снятия защиты с аминогруппы), которые не связаны с производным соединением полиолефинов или олигомеров полиолефинов или полиалкенов, и реагентов, находящихся в органической фракции в избытке. Органические растворители, такие как циклогексан, гептаны, гексаны, обладающие температурой вспышки < 15°C, являются приемлемыми для солюбилизации производных соединений полиолефинов или олигомеров полиолефинов или полиалкенов во время экстракции или промывки. Способ по настоящему изобретению позволяет, следовательно, избегать все стадии очистки, необходимые в способах предшествующего уровня техники.
Третье преимущество, являющееся особенно важным, состоит в том, что способ по настоящему изобретению позволяет синтезировать пептиды и даже белки, регулируя длину производного соединения полиолефинов или олигомеров полиолефинов или полиалкенов, делая их более липофильными.
Еще одно преимущество состоит в возможности регулировать чистоту пептида в ходе синтеза в любой момент времени отбором аликвоты с последующим анализом различными способами, известными специалистам в данной области техники (такими как масс-спектрометрия, высокоэффективная жидкостная хроматография, протонный магнитный резонанс или ядерный магнитный резонанс с углеродом-13).
Еще одно преимущество состоит в том, что предпочтительные линкеры, а именно производные соединения полиизобутена, могут быть получены исходя из изобутена биологического происхождения, что было пояснено ранее.
Другие преимущества состоят в возможности автоматизировать способ по настоящему изобретению и в возможности рециклировать линкеры (полиолефины или олигомеры полиолефинов или полиалкенов). Действительно, когда серия итераций для получения последовательности требуемого пептида завершена, с пептида снимают защиту в виде его защитных групп и в последнюю очередь отделяют линкер по одной из реакций, обычно используемых в синтезе пептидов (таких как гидролиз, омыление, гидрогенолиз), что высвобождает линкер. Таким образом, линкер может быть рециклирован.
Благодаря своей высокой чистоте пептиды или белки, получаемые этим способом, могут быть использованы в качестве фармацевтических препаратов (лекарственных средств и вакцин), косметических изделий, препаратов для борьбы с болезнями растений или продовольственных продуктов или для получения какого-либо из таких веществ.
Примеры
Приведенными примерами поясняются варианты осуществления настоящего изобретения без ограничения объема патентной охраны. Примерами 1 и 2 поясняются два варианта реакции присоединения первой аминокислоты (AA1) требуемого пептида (используемой в реакции в форме, в которой Nα защищен группами Fmoc или Boc) с жидкой подложкой (в данном случае с производным соединением PIB). Примерами 3 и 4 поясняются два варианта снятия защиты с последующей аминокислоты (AA2), используемой в реакции в форме, в которой Nα защищен группами Fmoc или Boc (называемой в данном случае "производное соединение Fmoc" или "производное соединение Boc" соответственно,). Примером 5 поясняется отделение пептида от линкера соответственно схеме реакций 8, приведенной ранее.
В этих примерах используют хлорированные растворители, а именно DCM. Подобные результаты были получены с растворителями с малой вредностью, такими как тетрагидрофуран, 2-метилтетрагидрофуран, этиленкарбонат, применяемые по отдельности или в смеси.
Пример 1. Реакция применения с применением EDC/HOBt
Nα-защищенную аминокислоту (Fmoc-AA-OH или Boc-AA-OH) (1,3 ммоль) растворяли в DCM (5 мл) при перемешивании магнитной мешалкой в атмосфере азота и затем охлаждали при 0°C в бане со льдом. Последовательно прибавляли растворенный в DCM (5 мл) 4-[полиизобутил(18)]фенол в качестве производного соединения PIB (1 ммоль) и гидроксибензотриазол (1,4 ммоль). Через 10 минут прибавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC) (1,5 ммоль) и реакционной смеси давали медленно нагреваться до комнатной температуры (в течение 3 или 16 ч). Затем реакционную смесь сгущали при пониженном давлении. К остатку прибавляли циклогексан, затем три раза промывали водой или смесью "вода/этанол" или "вода/ацетонитрил" и далее промывали насыщенным водным раствором хлорида натрия. Органическую фракцию сушили над Na2SO4, фильтровали и растворитель выпаривали при пониженном давлении.
Пример 2. Реакция применения с применением EDC/DMAP
N-защищенную аминокислоту (Fmoc-AA-OH или Boc-AA-OH) (1,3 ммоль) растворяли в DCM (5 мл) при перемешивании магнитной мешалкой в атмосфере азота и затем охлаждали до 0°C в бане со льдом. Последовательно прибавляли производное соединение PIB (свободный амин, спирт, тиол или фенол) (1 ммоль), растворенное в DCM (5 мл), и 4-диметиламинопиридин (DMAP) (0,3 ммоль). Через 10 минут прибавляли EDC (2 ммоль) и реакционной смеси давали медленно нагреваться до комнатной температуры (в течение 3 или 16 ч). Затем реакционную смесь сгущали при пониженном давлении. К остатку прибавляли циклогексан, затем три раза промывали водой или смесью "вода/этанол" или "вода/ацетонитрил" и далее промывали насыщенным водным раствором хлорида натрия. Органическую фракцию сушили над Na2SO4, фильтровали и растворитель выпаривали при пониженном давлении.
Пример 3. Удаление флуоренилметилкарбаматной защитной группы (Fmoc)
Производное соединение флуоренилметилкарбамата (1 ммоль) растворяли в DCM (5 мл) при перемешивании магнитной мешалкой и охлаждали до 0°C в бане со льдом. Прибавляли раствор ACN/HNEt2 (2/1, 5 мл) и затем реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Далее выпаривали растворители при пониженном давлении. К остатку прибавляли циклогексан, затем три раза промывали водой или смесью "вода/этанол" или "вода/ацетонитрил" и далее промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия. Органическую фракцию сушили над Na2SO4, фильтровали и растворитель выпаривали при пониженном давлении.
Пример 4. Удаление защитной трет-бутоксикарбонильной группы (Boc)
Производное соединение трет-бутилкарбамата (1 ммоль) растворяли в DCM (5 мл) при перемешивании магнитной мешалкой и охлаждали до 0°C в бане со льдом. Прибавляли смесь DCM/TFA (1/1, 35 мл) и затем реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Далее выпаривали растворители при пониженном давлении. К остатку прибавляли циклогексан, затем три раза промывали водой или смесью "вода/этанол" или "вода/ацетонитрил" и далее промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия. Органическую фракцию сушили над Na2SO4, фильтровали и растворитель выпаривали при пониженном давлении.
Пример 5. Отделение пептида от линкера
Трипептид, прикрепленный к его твердой подложке (1 ммоль), растворенной в DCM (5 мл), прибавляли к смеси "трифторуксусная кислота/триизопропилсилан/вода" (об./об./об., 95/2,5/2,5, 5 мл), предварительно охлажденной до 0°C в бане со льдом. Реакционную смесь перемешивали в течение 3 ч при комнатной температуре. К реакционной смеси прибавляли диэтиловый эфир, осадок отделяли фильтрованием и сушили в вакууме.
Реферат
Способ синтеза пептидов или белков последовательным удлинением второй концевой части пептидной цепи, карбоксигруппа (C-конец) или аминогруппа Nα которой прикреплена к линкеру, растворимому в неполярном растворителе, отличающийся тем, что линкер представляет собой полиолефиновую цепь, содержащую по меньшей мере 10 мономерных звеньев и предпочтительно от 15 до 50 звеньев. Способ позволяет получать чистые и/или легко очищаемые пептиды и может быть легко автоматизирован. 10 з.п. ф-лы, 5 пр.
Комментарии