Способ высокоэффективного получения полипептидного фрагмента, подходящего для ncl - RU2605411C2
Код документа: RU2605411C2
Чертежи













Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу эффективного получения полипептидного фрагмента, подходящего для NCL.
Уровень техники
Известны разные способы синтеза белка: биосинтез, химический синтез, бесклеточный синтез. Для биосинтеза используют Е. coli, в клетки которой встраивают ДНК, кодирующую нужный белок, и добиваются экспрессии этого белка. При химическом синтезе целевой белок синтезируют путем последовательного связывания аминокислот методами органической химии. А при бесклеточном синтезе белок синтезируют без клеток с помощью ферментов, присутствующих в различных клетках, например в Е. coli. Перечисленные способы можно использовать изолированно или комбинируя друг с другом в зависимости от цели применения белка, его размера и свойств.
Чтобы синтезировать белок с модифицированной структурой, включающей сахарную цепочку или липид в средней части аминокислотной последовательности, использован способ отдельного синтеза пептидного фрагмента, содержащего сахарную цепочку, и пептидного фрагмента без сахарной цепочки и последующего лигирования этих фрагментов в молекулу целевого белка. Пептидный фрагмент, содержащий сахарную цепочку можно синтезировать химическим путем, используя аминокислоту, предварительно модифицированную сахарной цепочкой, липидом и т.д. Кроме того, пептидный фрагмент, не содержащий сахарной цепочки, можно получить путем химического синтеза или биосинтеза.
Твердофазный синтез в основном применяют в качестве способа химического синтеза пептидной цепи. Однако пептидная цепь, полученная с помощью твердофазного синтеза, обычно короткая и содержит около 50 аминокислотных остатков.
Имеются сообщения о различных способах связывания пептидных цепей, и одним их таких способов, имеющих широкое применение, является способ нативного химического лигирования (NCL - Native Chemical Ligation). Способом NCL можно соединить незащищенные пептидные цепи; этот способ применим для получения естественной амидной (пептидной) связи в месте лигирования (см. ссылку 1 в патентной литературе). Способ NCL представляет собой избирательную химическую реакцию между первым пептидом, имеющим сложную альфа-карбокситиоэфирную часть на C-конце, и вторым пептидом, имеющим цистеиновый остаток на N-конце, в котором тиоловая группа в боковой цепи цистеина (SH-группа называется также сульфгидрильной группой) избирательно реагирует с карбонильным атомом углерода сложной тиоэфирной группы и в реакции обмена тиоловой группы образуется ранний промежуточный продукт со сложной тиоэфирной связью. В этом промежуточном продукте происходит спонтанная внутримолекулярная перестройка, приводящая к смещению амидной связи к месту лигирования и восстановлению тиоловой группы боковой цепи цистеина.
NCL является способом, позволяющим связать две пептидные цепи с помощью пептидной связи путем обычного перемешивания в буферном растворе. С помощью способа NCL избирательно связывают С-конец одного пептида с N-концом другого пептида, даже в случае если реакция происходит между соединениями, имеющими многочисленные функциональные группы, например между пептидами. В связи с этим в синтезе белка важно, как применяется способ NCL.
Однако при применении способа NCL стоит проблема получения сложной тиоэфирной формы пептида, имеющего сложную альфа-карбокситиоэфирную часть на C-конце, которая необходима как исходный материал. Хотя сообщается о существовании различных способов получения пептидных сложных тиоэфиров (источник 1 в непатентной литературе и источник 2 в патентной литературе), в основе всех способов лежит твердофазный синтез, поэтому все они имеют присущие ему ограничения, и размер пептидного сложного тиоэфира, который можно синтезировать с помощью этих способов, ограничен. Кроме того, при применении способа, в котором используется линкер, необходимо отдельно путем химического синтеза получить неприродное или особое производное аминокислоты, в связи с чем способ NCL не всегда прост.
Между тем, в литературе сообщалось также о способе получения полипептидного фрагмента в форме сложного тиоэфира (интеиновый способ) путем биосинтеза (источник 2 в непатентной литературе). Однако при применении данного способа экспрессии полипептида, а также для белкового сплайсинга понадобится целевая пептидная последовательность, и экспрессированный интеин-белковый комплекс всегда должен быть упакован в присущую ему трехмерную структуру. В связи с этим получение пептидного сложного тиоэфира не всегда зависит от полипептидной последовательности, которую предстоит экспрессировать, необходимо всегда оптимизировать условия биосинтеза, а также учитывать его сложность.
Кроме того, в литературе, помимо интеинового способа, сообщается также о способе получения сложного тиоэфира, который можно применить к полипептиду, полученному путем биосинтеза (источник 3 в патентной литературе). Это способ активирования цистеина, содержащегося в синтезированном полипептиде, и расщепления с его помощью сложного тиоэфира в положении активированного остатка цистеина. Иными словами, на N-конце активированного остатка цистеина можно получить пептидный фрагмент в форме сложного тиоэфира. Однако при данном способе полипептид, расщепленный на C-конце активированного остатка цистеина, после расщепления образует на своем N-конце стабильную циклическую структуру и поэтому не может быть использован в дальнейшем при синтезе белка. В связи с этим возникла необходимость в отдельном получении пептидного фрагмента на N-конце пептидного фрагмента, подлежащего лигированию.
Кроме того, несмотря на то что при биосинтезе экспрессируются полноразмерные молекулы белков, экспрессия пептидных фрагментов иногда оказывается неполноценной, например, из-за того, что они воспринимаются клеткой как ошибочные, и экспрессированный пептид разрушается. Соответственно, когда при упомянутом выше способе необходимый пептидный фрагмент после биосинтеза полноразмерного белка отщепляется в виде сложного тиоэфира, полипептид с длинной цепью на C-конце отщепленного пептидного фрагмента не может быть использован.
Список литературы
[Патентная литература 1] International Publication No. 96/34878
[Патентная литература 2] International Publication No. 2007/114454
[Патентная литература 3] International Publication No. 2010/150730
[Непатентная литература]
[Непатентная литература 1] Ingenito et al., J. Am. Chem. Soc. 1999, 121, 11369-11374
[Непатентная литература 2] Schwartz et al., CHEM. COMMUN., 2003 2087-2090
Сущность изобретения
Задачи, которые призвано решить данное изобретение
Цель настоящего изобретения состоит в том, чтобы представить эффективный способ получения первого полипептидного фрагмента, имеющего цистеиновый остаток на N-конце, и второго полипептидного фрагмента, имеющего модифицированный С-конец, которые подходят для лигирования способом NCL.
Средства для решения поставленных задач
Авторы настоящего изобретения в результате многократных экспериментов обнаружили, что первый полипептидный фрагмент, имеющий цистеиновый остаток на N-конце, и второй полипептидный фрагмент, имеющий модифицированный С-конец, которые подходят для соединения способом NCL, можно получить в виде одного полипептида с особой вставленной последовательностью.
Иными словами, настоящее изобретение относится к способу эффективного получения первого полипептидного фрагмента, имеющего на N-конце цистеиновый остаток, и второго полипептидного фрагмента, имеющего модифицированный С-конец, которые можно соединить способом NCL, включающим:
1) Стадию взаимодействия полипептида, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met-первый полипептидный фрагмент (C-концевой),
где n является целым числом от 0 до 10, Cys означает цистеин, W означает 1, 2 или 3 аминокислоты, Z означает 0, 1 или 2 аминокислоты, His означает гистидин, Met означает метионин, а N-концом первого полипептидного фрагмента является цистеиновый остаток
и для получения приведенных ниже полипептидных фрагментов используется цианид брома CNBr:
(A) первый полипептидный фрагмент, имеющий на N-конце цистеиновый остаток и
(B) третий полипептидный фрагмент, имеющий следующую структуру:
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met′ (C-концевой) [где Met′ означает производное Met]; и
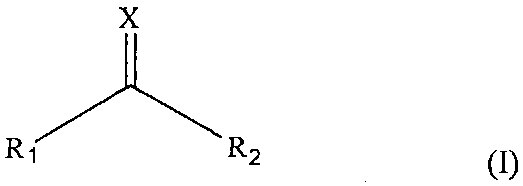
(2) Стадию взаимодействия упомянутого третьего полипептидного фрагмента с соединением, имеющим следующую формулу (I):
[Химическая формула 1]

где
X является атомом серы или кислорода и
R1 и R2 являются удаляемыми группами,
и затем с соединением в органическом растворителе, имеющим следующую формулу (II):
Химическая формула 2

где
Y является атомом кислорода, атомом серы или =NH и
R3 является атомом водорода, ацильной группой или алкоксикарбонильной группой,
для получения второго полипептидного фрагмента, имеющего модифицированный С-конец следующей структуры:
(N-концевой) второй полипептидный фрагмент-C(=O)-NH-С(=Y)NHR3 (C-концевой).
Один вариант осуществления способа по изобретению отличается тем, что включает в себя еще и стадию взаимодействия второго полипептидного фрагмента, имеющего модифицированный С-конец и полученного на упомянутой стадии (2), с тиолом, представленным формулой:
R4-SH,
где R4 является любой из групп, выбираемых из группы, состоящей из замещенной или незамещенной бензильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной алкильной группы
и обмена -NH-C(=Y)NHR3 группы на C-конце с тиоловой группой для получения второго полипептидного фрагмента, имеющего модифицированный С-конец следующей структуры:
(N-концевой) второй полипептидный фрагмент-C(=O)-SR4 (C-концевой).
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что X в соединении, представленном упомянутой формулой (I) является атомом серы.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что R1 в соединении, представленном упомянутой формулой (I), является -O-C6 арильной группой.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что R2 в соединении, представленном упомянутой формулой (I), является атомом галогена или замещенной или незамещенной -S-C6-10 арильной группой.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что Y в соединении, представленном упомянутой формулой (II), является =NH.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что R3 в соединении, представленном упомянутой формулой (II), является ацетильной группой.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что упомянутый полипептид, имеющий следующую структуру:
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met-первый полипептидный фрагмент (C-концевой),
является рекомбинантным полипептидным фрагментом, экспрессируемым клеткой.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что упомянутой клеткой является Е. coli.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что W является аминокислотой, выбираемой из группы, состоящей из Val, Ile, Leu и Trp.
Кроме того, в одном варианте осуществления способ по изобретению отличается тем, что n является целым числом от 6 до 10.
Кроме того, другой объект настоящего изобретения относится к способу получения первого полипептидного фрагмента, имеющего на N-конце цистеиновый остаток, подходящего для применения способа NCL, и отличается тем, что полипептид, имеющий следующую структуру:
(N-концевой) второй полипептидный фрагмент-P-Met-первый полипептидный фрагмент (C-концевой),
где P является любым количеством аминокислот от 0 до 10, Met означает метионин, а N-концевым остатком полипептидного фрагмента является цистеиновый остаток,
взаимодействует с CNBr.
Кроме того, другой объект настоящего изобретения относится к способу получения гликозилированного полипептида, включающему:
(1) Стадию классификации и конструирования пептидной последовательности целевого гликозилированного полипептида, получаемого в виде по меньшей мере:
- полипептидного фрагмента, содержащего сахарную цепочку, который состоит из полипептида, содержащего гликозилированную аминокислоту,
- второго полипептидного фрагмента на N-концевой части полипептидного фрагмента, содержащего сахарную цепочку и состоящего из полипептида, содержащего N-концевую часть целевого гликозилированного пептида,
- первого полипептидного фрагмента на C-концевой части полипептидного фрагмента, содержащего сахарную цепочку и состоящего из полипептида, содержащего C-концевую часть целевого гликозилированного пептида.
- полипептидного фрагмента между полипептидным фрагментом, содержащим сахарную цепочку, и вторым полипептидным фрагментом, если присутствие такого фрагмента возможно, и
- полипептидного фрагмента, если присутствие такого фрагмента возможно, между полипептидным фрагментом, содержащим сахарную цепочку, и первым полипептидным фрагментом, в котором первый полипептидный фрагмент сконструирован так, чтобы на N-конце его был цистеиновый остаток.
(2) Стадию получения полипептидного фрагмента, имеющего упомянутую структуру, с помощью вектора экспрессии, включающего нуклеотидную последовательность, кодирующую полипептид следующей структуры:
(N-конец) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met-первый полипептидный фрагмент (С-конец),
где n равно целому числу от 0 до 10, Cys означает цистеин, W означает 1, 2 или 3 аминокислоты, Z означает 0, 1 или 2 аминокислоты, His означает гистидин, a Met означает метионин и N-концевым аминокислотным остатком первого полипептидного фрагмента является цистеин
и обеспечивающую возможность экспрессии клетками Е. coli;
(3) Стадию взаимодействия полипептида со стадии (2) с CNBr для получения следующих полипептидных фрагментов:
(A) первого полипептидного фрагмента с цистеиновым аминокислотным остатком на N-конце и
(B) третьего полипептидного фрагмента следующей структуры:
(N-конец) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met′ (С-конец),
где Met′ означает производное Met;
(4) Стадию взаимодействия упомянутого третьего полипептидного фрагмента с соединением, представленным следующей формулой (I):
Химическая формула 3
где X является атомом серы или атомом кислорода, a R1 и R2 являются удаляемыми группами,
и затем взаимодействия в органическом растворителе с соединением, представленным следующей формулой (II):
Химическая формула 4

где
Y является атомом кислорода, атомом серы или NH группой, и
R3 является атомом водорода, ацильной группой или алкоксикарбонильной группой,
для получения второго полипептидного фрагмента с модифицированным С-концом, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент-C(=O)-NH-C(=Y)NHR3 (C-концевой);
(5) Условно стадию взаимодействия второго полипептидного фрагмента, имеющего модифицированный С-конец и полученного на стадии (4), с тиолом, представленным следующей формулой
R4-SH,
в которой R4 является одной какой-либо группой, выбираемой из группы, состоящей из замещенной или незамещенной бензильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной алкильной группы,
и обмена -NH-C(=Y)NHR3 группы на C-конце с тиоловой группой для получения второго полипептидного фрагмента с модифицированным С-концом, имеющим следующую структуру:
(N-концевой) второй полипептидный фрагмент-C(=O)-SR4 (C-концевой); и
(6) Стадию связывания:
- упомянутого полипептидного фрагмента, содержащего сахарную цепочку,
- упомянутого полипептидного фрагмента, если присутствие его возможно, между полипептидным фрагментом, содержащим сахарную цепочку, и вторым полипептидным фрагментом и
- упомянутого полипептидного фрагмента, если присутствие его возможно, между полипептидным фрагментом, содержащим сахарную цепочку, и первым полипептидным фрагментом, полученным отдельно химическим синтезом с:
первым полипептидным фрагментом, имеющим на N-конце цистеиновый аминокислотный остаток и полученным на стадии (3) и
вторым полипептидным фрагментом, имеющим модифицированный С-конец и полученным на стадиях (4) и (5)
в порядке, который позволит получить целевой гликозилированный полипептид с помощью лигирования.
Кроме того, в одном варианте осуществления изобретения способ получения гликозилированного полипептида отличается тем, что X в соединении, представленном формулой (I), является атомом серы.
Кроме того, в одном варианте осуществления изобретения способ получения гликозилированного полипептида отличается тем, что R1 в соединении, представленном формулой (I), является -O-C6 арильной группой.
Кроме того, в одном варианте осуществления изобретения способ получения гликозилированного полипептида отличается тем, что R2 в соединении, представленном в формуле (I), является атомом галогена или замещенной либо незамещенной -S-C6-10 арильной группой.
Кроме того, в одном варианте осуществления изобретения способ получения гликозилированного полипептида отличается тем, что Y в соединении, представленном формулой (II), является NH группой.
Кроме того, в одном варианте осуществления изобретения способ получения гликозилированного полипептида отличается тем, что R3 в соединении, представленном формулой (II), является ацетильной группой.
Кроме того, в одном варианте осуществления изобретения способ получения гликозилированного полипептида отличается тем, что W является какой-либо аминокислотой, которую выбирают из группы, состоящей из Val, Ile, Leu и Trp.
Кроме того, в одном варианте осуществления изобретения способ получения гликозилированного полипептида отличается тем, что n соответствует любому числу из 6-10.
Эффект от изобретения
В настоящем изобретении предложен способ эффективного получения первого полипептидного фрагмента, имеющего на N-конце цистеиновый остаток, и второго полипептидного фрагмента, имеющего модифицированный С-конец, подходящих для соединения способом NCL.
Кроме того, в соответствии с настоящим изобретением доступный для лигирования пептидный фрагмент, даже если он не может экспрессироваться, можно экспрессировать связанным с другим пептидным фрагментом; способ по изобретению эффективен также в получении множественных пептидных фрагментов.
Кроме того, комбинируя способ по изобретению с традиционными способами синтеза пептидов, можно легко получить длинные цепи пептидов, имеющих модифицированные участки, которые до этого трудно было синтезировать, например, можно использовать способ по изобретению для получения относительно длинной полипептидной цепи из немодифицированных фрагментов, получаемых путем биосинтеза, и твердофазный синтез модифицированного фрагмента, и затем соединить их.
В частности, если модификация состоит в присоединении сахарной цепочки, можно легко получить более длинный пептид с сахарной цепочкой, конструируя путем химического синтеза только фрагмент, содержащий аминокислоту с нативной сахарной цепочкой, и другие пептидные участки путем биосинтеза и затем соединять подходящие пептидные фрагменты с модифицированным фрагментом, лигируя их способом по изобретению.
Кроме того, хорошо известен способ последующего добавления сахарной цепочки и т.д. в пептидную цепь с помощью линкера и возможно также последующее добавление сахарной цепочки к длинной пептидной цепи, полученной путем биосинтеза. Тем не менее, при этом способе связывания с помощью линкера сахарную цепочку и т.д. связывают, использую конкретную аминокислоту или другую структуру. И соответственно, если, например, в пептиде имеется много сайтов, в которых к пептиду можно присоединить сахарную цепочку, то способом по изобретению легче, чем при традиционном способе, выполнить сайт-специфическое гликозилирование. Для этого получают пептид с длинной цепью с помощью биосинтеза, отщепляют от него пептидный фрагмент, содержащий только целевую связывающую область длинной цепи пептида, добавляют в нее сахарную цепочку сахара и затем вновь связывают фрагмент с остальной частью пептида.
Как было показано выше, способ тиоэстерификации пептида, представленный в изобретении, применим для синтеза белка вообще.
Краткое описание фигур
На фиг. 1 показана последовательность генетической информации вектора pET32a для экспрессии полипептида в одном варианте осуществления изобретения.
На фиг. 2 приведена схема биосинтеза полипептидного фрагмента, имеющего вставочную последовательность по изобретению, в клетках Е. coli в одном варианте осуществления изобретения.
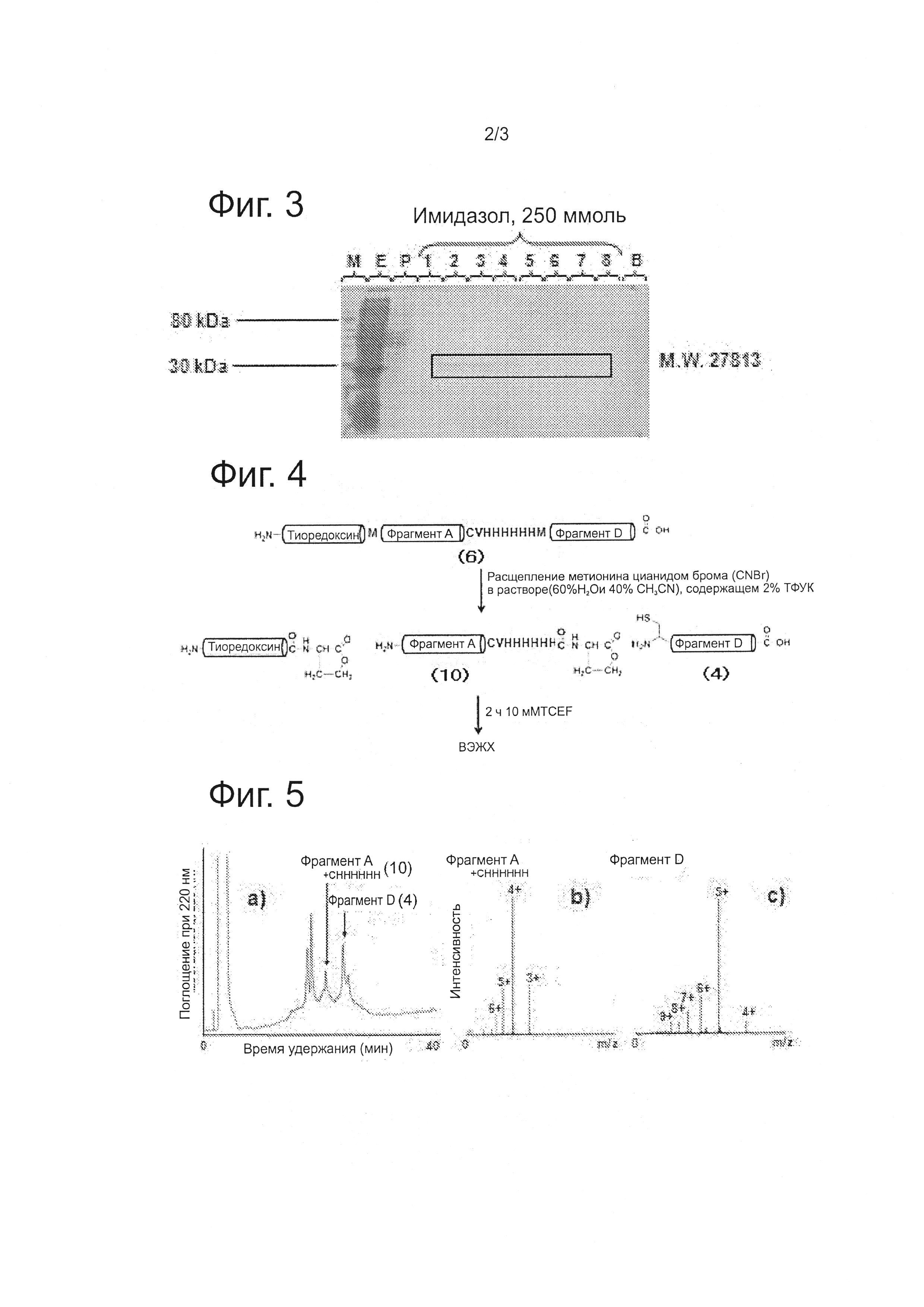
На фиг. 3 приведена фотография экспрессированного белка, очищенного на никелевой колонке. Обозначения на фиг. 3: М означает молекулярную массу, Е означает экспрессированную смесь, P означает фракцию, которая течет через колонку, 1-8 - номера фракций, элюированных с помощью 250 мМ имидазола, и В (blank) означает холостой образец.
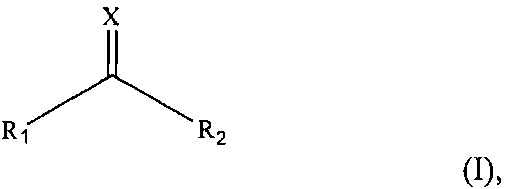
На фиг. 4 показана схема стадии расщепления полипептидного фрагмента, имеющего вставочную последовательность, экспрессируемого Е. coli, на два полипептидных фрагмента с помощь CNBr в одном варианте осуществления изобретения. Тиоредоксин, добавляемый через метионин к N-концу полипептидного фрагмента, экспрессируемого E. coli, также расщепляется при обработке CNBr.
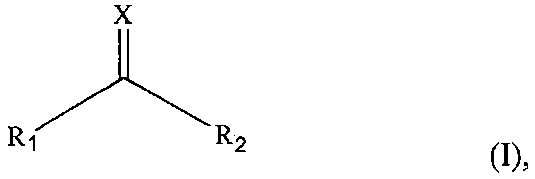
На фиг. 5a показан результат анализа с помощью ВЭЖХ продукта деградации после обработки CNBr. На фиг. 5b показан результат масс-спектрометрии пептидного фрагмента A, который является производным интерлейкина-13 и имеет его аминокислотную последовательность 1-27. На фиг. 5c показан результат масс-спектрометрии пептидного фрагмента D, который является производным ИЛ-13 и имеет его аминокислотную последовательность 56-112.
На фиг. 6 схематически представлен вариант осуществления изобретения, при котором способ по изобретению применен для получения полипептидного фрагмента, подходящего для лигирования способом NCL; показано разделение производного интерлейкина-13 на четыре полипептидных фрагмента и биосинтез фрагментов A и D в виде слитых белков, имеющих вставочную последовательность.
Описание вариантов осуществления изобретения
Далее будут описаны предпочтительные варианты осуществления изобретения.
Настоящее изобретение включает в себя стадию взаимодействия полипептидом, имеющего следующую структуру (i)
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met-первый полипептидный фрагмент (C-концевой)…(i),
где n означает любое целое число от 0 до 10, Cys означает цистеин, W означает 1, 2 или 3 аминокислоты, Z означает 0, 1 или 2 аминокислоты, His означает гистидин, Met означает метионин, а N-концевым остатком первого полипептидного фрагмента является цистеиновый остаток,
с CNBr для получения следующих полипептидных фрагментов:
(A) первого полипептидного фрагмента, в котором N-концевым аминокислотным остатком является цистеиновый остаток, и
(B) третьего полипептидного фрагмента, имеющего следующую структуру (ii):
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met′ (C-концевой)…(ii),
где Met′ означает производное Met.
Понятие «пептид» в описании не имеет каких-либо ограничений и подразумевает две или более аминокислоты, связанные между собой амидной связью; оно включает в себя хорошо известные и новые пептиды, а также измененные пептиды. Молекулы, которые упоминаются в описании как белки, также включены в пептиды. Кроме того, «полипептид» аналогичным образом включен в понятие «пептиды». Пептидная цепь, использованная при применении способа по изобретению, может быть натуральным белком или может быть пептидной цепью, полученной с помощью биосинтеза, химического синтеза или бесклеточного синтеза.
В понятие «измененный пептид» в описании включен натуральный вариант, посттрансляционная модифицированная форма или искусственно измененное соединение пептида. Примеры получения измененных пептидов включают в себя, например, алкилирование, ацилирование (например, ацетилирование), амидирование (амидирование С-конца пептида), карбоксилирование, получение сложного эфира, образование дисульфидной связи, гликозилирование, липидирование, фосфорилирование, гидроксилирование и связывание маркирующего компонента с одним или более аминокислотным остатком или присоединением к одному или более аминокислотному остатку пептида.
Понятие «аминокислота» в описании применяется в самом широком смысле и включает не только натуральные аминокислоты, например серии (Ser), аспарагин (Asn), валин (Val), лейцин (Leu), изолейцин (Ile), аланин (Ala), тирозин (Tyr), глицин (Gly), лизин (Lys), аргинин (Arg), гистидин (His), аспарагиновую кислоту (Asp), глутаминовую кислоту (Glu), глутамин (Gln), треонин (Thr), цистеин (Cys), метионин (Met), фенилаланин (Phe), триптофан (Trp), и пролин (Pro), но и неприродные аминокислоты, в частности варианты и производные аминокислот. Специалистам в данной области понятно, что столь широкое определение аминокислот охватывает также L-аминокислоты, D-аминокислоты, химически модифицированные аминокислоты, в частности их варианты и производные; аминокислоты, не используемые in vivo в синтезе белка, такие, как норлейцин, бета-аланин и орнитин; и синтезированные химическим путем производные, обладающие свойствами аминокислот, которые известны специалистам.
Под выражением «полипептидный фрагмент» в описании подразумевается полипептид, включающий часть аминокислотной последовательности целевого белка, получаемую в процессе синтеза целевого белка.
В описании под «первым полипептидным фрагментом» и «вторым полипептидным фрагментом» подразумеваются фрагменты, каждый из которых включает аминокислотную последовательность целевого белка. «Первый полипептидный фрагмент» и «второй полипептидный фрагмент» синтезированы в связанном через вставочную последовательность виде. Кроме того, в способе по изобретению как «первый полипептидный фрагмент», так и «второй полипептидный фрагмент» можно получить в виде полипептидного фрагмента, подходящего для лигирования.
Первый полипептид является сконструированным полипептидом, в котором аминокислотным остатком на N-конце является цистеиновый остаток. Он при отщеплении первого полипептидного фрагмента обеспечивает возможность его лигирования на N-конце с другим пептидным фрагментом. Кроме того, второй полипептидный фрагмент после отщепления в качестве второго полипептида имеет модифицированный С-конец, подходящий для лигирования, и это обеспечивает возможность лигирования его с другим полипептидным фрагментом на C-конце.
Кроме того, полипептид (i) по изобретению имеет вставочную последовательность «(N-концевой-Cys-W-(His)n-Z-Met-(C-концевой» между первым и вторым полипептидными фрагментами.
В описании n означает целое число от 0 до 10, Cys означает цистеин, W означает 1, 2 или 3 аминокислоты, Z означает 0, 1 или 2 аминокислоты, His означает гистидин, Met означает метионин и аминокислотным остатком на N-конце первого полипептидного фрагмента является цистеиновый остаток.
Кроме того, полипептид (i) можно связать и с другим полипептидным фрагментом на N-конце и/или C-конце через упомянутую выше вставочную последовательность.
Во вставочной последовательности аминокислота, обозначенная буквой «W», предпочтительно включает любую аминокислоту, выбираемую из группы, состоящей из Val, Ile, Leu и Trp, и в частности, предпочтительно, чтобы аминокислота, расположенная рядом с цистеином на N-конце, была выбрана из перечисленных аминокислот. В таком случае, встраивая массивную аминокислоту рядом с Cys, можно подавить побочную реакцию (b), применяя соединение, представленное формулой (II), приведенной ниже, и таким образом увеличить выход второго целевого полипептида с модифицированным С-концом.
Кроме того, во вставочной последовательности (His)n означает гистидиновую метку, которая облегчает очистку полипептида, полученного путем биосинтеза. Способ, с помощью которого можно очистить полипептид, в который добавлена гистидиновая метка, известен специалистам. Например, полипептид можно очистить с помощью поступающего в продажу Ni-NTA агарозного геля.
Если в упомянутую выше вставочную последовательности не включить гистидиновую метку (n=0), полипептид (i) также можно экспрессировать с добавленной на N- или C-конце гистидиновой меткой. Когда вставочная последовательности имеет гистидиновую метку, N предпочтительно является целым числом от 6 до 10.
Кроме того, во вставочной последовательности Met является аминокислотой, используемой для отщепления первого полипептида на C-конце Met.
При отщеплении полипептида, полученного путем биосинтеза (i) в положении Met, его можно обработать, например, CNBr и 70% водным раствором муравьиной кислоты HCOOH при комнатной температуре. Если в пептидной последовательности, которую предстоит обработать содержится количестве Ser или Thr, у которых имеются гидроксильная группа в боковой цепи, возможно нежелательное образование сложного эфира муравьиной кислоты с боковой цепью. В таком случае предпочтительно предпринять меры, позволяющие избежать этерификации, например, уменьшить содержание муравьиной кислоты в реакционном растворе, использовать неорганическую кислоту или ТФУК, чтобы образующийся сложный эфир трифторуксусной кислоты можно было легко удалить.
Примеры предпочтительной кислоты, используемой в упомянутой выше реакции, включают, но не ограничиваются ими, муравьиную кислоту, фосфорную кислоту, трифторуксусную кислоту (ТФУК), трибромуксусную кислоту и метансульфоновую кислоту. Эти кислоты предпочтительно добавляют в концентрации 0,1-50%, более предпочтительно 1-20% и еще более предпочтительно 1-5%.
Кроме того, в описанной выше реакции можно использовать также смешивающийся с водой растворитель. Смешивающийся с водой растворитель не ограничен конкретным количеством растворителей, поскольку может смешиваться с водой, и включает, например, ацетонитрил, трифторэтанол, диметилформамид, диметилсульфоксид (ДМСО) и метиленхлорид, из которых предпочтительны ацетонитрил, трифторэтанол и диметилформамид. Такой растворитель предпочтительно добавляют в концентрации 1%-70%, более предпочтительно 20%-60%, еще предпочтительнее 35%-50%.
В таком случае скорость образования целевого полипептида можно увеличить расщеплением в присутствии кислоты и смешивающегося с водой растворителя.
При отщеплении полипептида (i) в положении Met в вышеупомянутой реакции можно получить полипептидный фрагмент, имеющий аминокислотную последовательность второй полипептидный фрагмент-Cys-W-(His)n-Z-Met′ на N-конце и первый полипептидный фрагмент на C-конце.
Met′ означает производное Met и указывает на образовавшееся производное Met в реакции расщепления, упомянутой выше.
Что касается выражения «первый полипептидный фрагмент» и «второй полипептидный фрагмент», которые содержатся в полипептиде (i) по изобретению, можно сконструировать подходящий предпочтительный фрагмент с учетом длины аминокислотной последовательности или типа аминокислот. Конструируют полипептидный фрагмент по изобретению таким образом, чтобы полученный при биосинтезе полипептидный фрагмент в качестве первого полипептидного фрагмента имел на N-конце цистеиновый остаток.
Полипептидом (i) по изобретению может быть натуральный белок или пептидная цепь, полученная путем биосинтеза, химического синтеза или бесклеточного синтеза, но предпочтительно, чтобы он был рекомбинантным белом, экспрессированным бактериями или клетками. Рекомбинантным белком могут быть белки, имеющие такую же пептидную последовательность, как и натуральный белок, или пептидная последовательность, модифицированная, например, путем мутации или введения метки для очистки, поскольку экспрессируется искусственно в бактериях или других клетках.
Полипептид (i) по изобретению можно получить способом, известным специалистам в данной области. Например, его можно экспрессировать введя целевой ген в рекомбинантньш вектор и создавая условия для экспрессии. В качестве рекомбинантного вектора можно использовать любой вектор, который может трансформировать клетку-хозяина; авторы в зависимости от клетки-хозяина использовали вирусные векторы, такие как плазмиду для E. coli, плазмиду для Bacillus subtilis, плазмиду для дрожжевых грибов, векторы на основе ретровируса и вируса осповакцины, бакуловируса и др. Предпочтительными являются векторы, содержащие регуляторную последовательность, например промотор, который может вызвать экспрессию. Кроме того, клеткой-хозяином может быть любая клетка, которая может экспрессировать чужой ген, встроенный в рекомбинантный вектор; часто в качестве клетки-хозяина используют E. coli, Bacillus subtilis, дрожжевые грибы, клетки насекомых и животных и др.
Для трансфекции клетки-хозяина рекомбинантным вектором можно использовать рутинные способы, например для E. coli - способ теплового шока, способ, основанный на использовании хлорида кальция, способ электропорации, для дрожжевых грибов - способ, основанный на использовании хлорида лития, или элетропорацию. Кроме того, добиться трансформации животной клетки можно с помощью физических методов, таких, как электропорация, или химических методов, таких, как липосомный способ или способ, основанный на применении фосфата кальция, либо с помощью вирусного вектора, например ретровируса. После встраивания в клетку вектора предпочтительно с помощью способа, известного специалистам в данной области, подтвердить, что целевая последовательность ДНК надлежащим образом встроена. Что касается культивирования трансформированной клетки-хозяина условия культивирования можно подобрать исходя из особенностей нутриентов, используемых клеткой-хозяином для питания.
Использованный пептид по изобретению предпочтительно должен быть очищенным. Для этого можно воспользоваться обычно способом общей очистки. Например, в случае рекомбинантного белка после культивирования бактерий или клеток, экспрессирующих используемый здесь рекомбинантный белок, готовят необработанный экстракт пептидов путем удаления бактерий или клеток по способу, известному специалистам, суспендируя их в подходящем буферном растворе, разрушая бактерии или клетки, например ультразвуком, лизоцимом и/или чередованием замораживания и оттаивания, после чего экстракт центрифугируют или фильтруют. Буферный раствор может включать агент, денатурирующий белок, например мочевину или гидрохлорид гуанидина либо поверхностно-активное вещество, например Triton X-100™. Способ очистки полученного экстракта или пептида, содержащегося в культуральной надосадочной жидкости, хорошо известен специалистам. Например, для выделения и очистки выбирают и комбинируют такие методы, как аффинная хроматография, ионообменная хроматография, фильтрация, ультрафильтрация, гель-фильтрация, электрофорез, осаждение в растворе соли, диализ и др.
Кроме того, для облегчения очистки рекомбинантного белка в вектор экспрессии также можно встроить различные метки. Примерами меток, которые можно встроить в вектор экспрессии, могут быть метки, известные специалистам в данной области, например метка, которая повышает эффективность экспрессии, или метка, которая повышает степень очистки, например тиоредоксин, GST-метка, Myc-метка, FLAG-метка и мальтоза-связывающий белок (МВР).
Кроме того, указанные метки также можно связать с полипептидом (i) через Met (метионин). Приготовляя метку, присоединенную, как описано, к полипептиду (i) через метионин, метку можно отщепить при расщеплении на первый и второй полипептидные фрагменты с использованием метионина в качестве мишени.
Кроме того, любой известный в настоящее время способ химического синтеза можно применить для синтеза других полипептидных фрагментов, необходимых для синтеза белка, отличного от полипептида (i). В частности, синтез части полипептидной цепи, связывающей сахарную цепочку, - это предпочтительно химический синтез, чтобы получить пептидный фрагмент, имеющий единообразную структуру сахарной цепочки. Этот способ не ограничен и включает, например, жидкофазный синтез, твердофазный синтез, способ защиты Boc-группой и Fmoc-группой. Выражаясь более конкретно, можно, как и при обычном белковом синтезе, применять способ получения гликозилированного пептидного фрагмента, используя гликозилированный аспарагин (Asn) в качестве гликозилированной аминокислоты и хорошо известный способ пептидного синтеза, например твердо- и жидкофазный синтез. Такой способ описан в публикации №2004/005330 (US 2005222382 (A1)), открытие которого полностью включено в описание путем ссылки.
В аминокислотной последовательности целевого белка при получении пептидного фрагмента, который необходимо лигировать на N- и С-концах, во избежание лигирования побочного продукта N-конец связывают с тиазолидиновым производным цистеина или цистеин защищают, например S-ацетамидометильной группой (Acm группа). Иными словами, на стадии лигирования связывание происходит от пептидного фрагмента на C-конце целевого белка, и N-концевой цистеиновый остаток пептидного фрагмента после связывания вновь переходит в состояние готовности к последовательному присоединению пептидных фрагментов к N-концу белка. Например, при использовании тиазолидинового производного цистеина его можно ввести во время твердофазного синтеза или цистеин можно также избирательно тиазолидинировать после синтеза полипептидного фрагмента способом, известным специалистам.
Кроме того, сахарная цепочка может быть также добавлена к цепи синтезированного полипептида через функциональную группу. Для такого связывания сахарной цепочки можно воспользоваться любым известным способом. Такой способ не ограничен. Примерами его являются способ конденсации производного с сахарной цепочкой, в котором имеется группа, выбираемая из группы, состоящей из -NH-(CO)-CH2X, -NH-(CO)-(CH2)b-CH2X, изотиоцианатная группа, -NH-(CO)a-(CH2)b-CO2H и -NH-(CO)a-(CH2)b-CHO (где X является атомом галогена, «а» означает 0 или 1, «b» означает целое число от 1 до 4), на восстанавливающем конце, на котором имеется сульфгидрильная группа цистеина (см. WO 2005/010053), или способ применения связующего реагента, описанный в заявке WO 2005/095331.
Кроме того, перед стадией связывания полученного пептидного фрагмента в качестве фрагмента части целевого пептида в вышеупомянутом способе необходима стадия тиоэстерификации С-конца полипептида для связывания с N-концом. Тиоэстерификацию можно выполнить способом, известным специалистам, например ее можно выполнить, активируя C-концевую карбоксильную кислоту с помощью РуВОР и ДИЭА и затем добавляя в избытке алкилтиол. При применении данного способа, добавлять алкилтиол предпочтительно при низкой температуре, более предпочтительно при 10-80°C и еще более предпочтительно при 0-40°C, чтобы контролировать конфигурацию альфа-углерода аминокислоты на конце фрагмента. Кроме того, тиоэстерификацию можно также выполнить способом защиты Fmoc-группой или Вос-группой, описанным в статье Yamamoto et al., J. Am. Chem. Soc. 2008, 130(2), 501-510.
«Сложная тиоэфирная форма пептида» (далее в тексте она называется просто сложным тиоэфиром) называется здесь пептид, имеющий карбокситиоэфирную часть (-C=O-SR) на C-конце. Сложный тиоэфир пептида, примененный здесь, не ограничен, если это сложный тиоэфир, который может вступить в реакцию обмена с другими тиоловыми группами. Примеры R-группы включают группы, представленные далее в качестве примеров R4.
Из описанного здесь полипептида (i) по изобретению можно получить следующие полипептидные фрагменты путем расщепления в положении Met во вставочной последовательности:
(A) первый полипептидный фрагмент, имеющий цистеиновый остаток на N-конце, и
(B) третий полипептидный фрагмент, имеющий следующее строение (ii):
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met′ (С-конец)…(ii),
где Met′ означает производное Met.
Если к N- и/или C-концевой группе полипептида (i) через упомянутую выше вставочную последовательность присоединяют еще и другой полипептидный фрагмент, например можно получить приведенный ниже полипептидный фрагмент.
(B′) (N-концевой) полипептидный фрагмент-Cys-W-(His)n-Z-Met′ (C-концевой)
Первый полипептидный фрагмент (A), имеющий на N-конце цистеиновый остаток и полученный по способу, упомянутому выше, можно лигировать с пептидом, имеющим на C-конце сложный тиоэфир. Соответственно в настоящем изобретении предлагается также способ получения полипептида, включающий стадию лигирования первого полипептидного фрагмента, имеющего на N-конце цистеиновый остаток и полученного способом по изобретению, с пептидом, имеющим на C-конце тиоэфир путем лигирования.
Итак, способ по изобретению включает в себя:
стадию взаимодействия третьего полипептидного фрагмента (ii), полученного путем отщепления полипептида (i), с соединением, представленным следующей формулой (I):
Химическая формула 5
где X является атомом серы или атомом кислорода, R1 и R2 являются удаляемыми группами
и затем с соединением в органическом растворителе, представленным следующей формулой (II):
Химическая формула 6

где Y является атомом кислорода, атомом серы или =NH группой, R3 является атомом водорода, ацильной или алкоксикарбонильной группой,
для получения второго полипептидного фрагмента, имеющего модифицированный С-конец следующей структуры:
(N-концевой) второй полипептидный фрагмент-C(=O)NH-C(=Y)NHR3 (C-концевой).
На указанной выше стадии сначала получают первый промежуточный продукт в результате взаимодействия тиоловой группы цистеинового остатка, который имеется в третьем полипептидном фрагменте (ii), с соединением, представленным следующей формулой (I) (реакция а).
Соединение, используемое в реакции (а), представлено следующей формулой (I).
Химическая формула 7
В приведенной формуле X является атомом серы или кислорода, но предпочтительно атомом серы. На R1 и R2 особые ограничения не накладываются при условии, что они имеют низкую нуклеофильность, чем замещенный атом или атомная группа, и имеют «отделяемую» функцию как и удаляемые группы в условиях протекания реакции (а). В частности, в качестве R1 и R2 используют атом галогена, замещенную или незамещенную -O-алкильную группу, замещенную или незамещенную -O-алкенильную группу, замещенную или незамещенную -O-алкинильную группу, замещенную или незамещенную -O-арильную группу, замещенную или незамещенную -O-гетероарильную группу, замещенную или незамещенную -S-алкильную группу, замещенную или незамещенную -S-алкенильную группу, замещенную или незамещенную -S-алкинильную группу, замещенную или незамещенную -S-арильную группу или замещенную или незамещенную -S-гетероарильную группу. Более предпочтительно, чтобы в качестве R1 и R2 была использована комбинация удаляемых групп, выбираемых из группы, состоящей для R1 из замещенной или незамещенной -O-C6-10 арильной группы и замещенной или незамещенной -S-C1-8 алкильной группы, для R2 - из атома галогена, замещенной или незамещенной -S-C1-8 алкильной группы и замещенной или незамещенной -S-C6-10 арильной группы.
Под выражением «алкильная группа» здесь имеется в виду одновалентная группа, индуцированная удалением одного какого-либо атома водорода из алифатического углеводорода и имеющая подмножество содержащих атом водорода и углерода, гидрокарбила или углеводорода. Алкильная группа бывает с прямой или разветвленной цепью. Алкильная группа по изобретению предпочтительно включает алкильную группу, имеющую от 1 до 8 атомов углерода. Алкильная группа C1-8 является алкильной группой, имеющей от 1 до 8 атомов углерода; примеры такой группы включают метальную, этильную, пропильную, бутильную, пентильную, гексильную, гептильную и октальную группы.
Под выражением «алкенильная группа» здесь имеется в виду одновалентная группа, в которой имеется по крайней мере одна двойная связь. По своей геометрической форме двойная связь может быть двух типов: E (от немецкого Entgegen - против, навстречу) и Z (от немецкого Zusammen - вместе, сообща) или иметь цис- или трансконфигурацию. Алкенильная группа бывает с прямой или разветвленной цепью. Алкенильная группа по изобретению предпочтительно включает алкенильную группу, имеющую от 2 до 8 атомов углерода. Алкенильная группа C2-8 является алкенильной группой, имеющей от 2 до 8 атомов углерода; примеры такой группы включают винильную, аллильную, пропенильную, бутенильную, пентенильную, гексенильную, гептенильную и октенильную группы.
Под выражением «алкинильная группа» здесь имеется в виду одновалентная группа, в которой имеется по меньшей мере одна тройная связь. Алкинильная группа бывает с прямой или разветвленной цепью. Алкинильная группа по изобретению включает алкинильную группу, имеющую от 2 до 8 атомов углерода. Алкинильная группа C2-8 является алкинильной группой, имеющей от 2 до 8 атомов углерода; примеры такой группы включают этинильную, 1-пропинильную, 2-пропинильную, бутинильную, пентинильную, гексинильную, гептинильную и октинильную группы.
Под «арильной группой» здесь имеется в виду циклическая группа ароматического углеводорода. Арильная группа по изобретению предпочтительно включает арильную группу, имеющую от 6 до 10 атомов углерода; примеры такой группы включают фенильную группу, 1-нафтильную группу и 2-нафтильную группу.
Под «гетероарильной группой» здесь имеется в виду одновалентная или двухвалентная группа, индуцированная удалением из гетероарильного кольца одного или двух атомов водорода в любом положении. Выражение «гетероарильная группа» означает ароматическое кольцо, содержащее один или более гетероатом среди атомов, образующих кольцо, и предпочтительно имеет 5-9-членное кольцо. Кольцо может быть моноциклическим или может быть бициклической гетероарильной группой, конденсированной с бензольным кольцом или моноциклическим гетероарильным кольцом. Примеры гетероарильной группы включают фуранильную, тиофенильную, пирролильную, бензофуранильную, бензотиофенильную, индолильную, пиридильную и хинолинильную группы.
Тип и количество замещающих групп и положение удаляемой группы не ограничены; примеры замещающих групп включают алкильную, алкенильную, алкокси группу, арильную, формильную, карбонильную, карбоксильную, алкилкарбоксильную, алкоксикарбонильную группы, галоген, сульфонильную или нитрогруппу.
В одном варианте осуществления изобретения соединение, представленное формулой (I) включает, например, следующие соединения.
Химическая формула 8

Химическая формула 9

Химическая формула 10
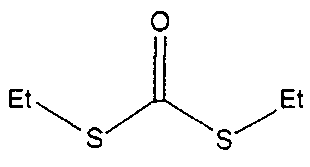
Химическая формула 11

В одном варианте осуществления изобретения можно использовать:
Химическая формула 12
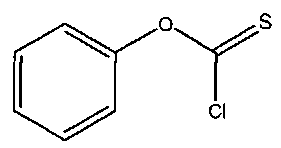
Химическая формула 13
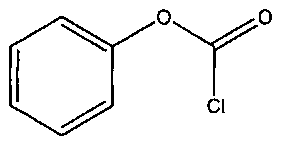
Приведенные выше соединения взаимодействуют с MPAA ((4-карбоксиметил)тиофенолом), то есть с тионоформиатными реагентами, приведенными ниже.
Химическая формула 14

Химическая формула 15

При взаимодействии соединения, представленного формулой (I), с цистеиновым остатком третьего полипептидного фрагмента (ii) можно получить первый промежуточный продукт реакции, в котором -C(=X)-R1 группа связана с SH группой цистеинового остатка, как показано на фигуре ниже.
Химическая формула 16

В настоящем изобретении реакцию (a) осуществляют предпочтительно в кислой среде, в частности предпочтительно при pH 3-5. Реакцию осуществляют в смешанном растворителе из буферного раствора и ацетонитрила при 0-50°C, предпочтительно при 15-25°C в течение 0,1-3 часов, предпочтительно в течение 10-60 мин, но длительность реакции не ограничивается указанными интервалами.
Далее в способе по изобретению упомянутый первый промежуточный продукт реакции взаимодействует в органическом растворителе с соединением, представленным формулой (II), к карбоксильной группе добавляется -NH-C(=Y)NHR3 группа, что приводит к образованию пептидной связи с аминокислотой, расположенной рядом с N-концом цистеинового остатка, и упомянутая пептидная связь расщепляется с образованием пептидного фрагмента на N-конце упомянутой расщепленной пептидной связи, являющегося вторым промежуточным продуктом (реакция (b)).
Соединение, которое было использовано в реакции (b), имеет следующую формулу (II):
Химическая формула 17
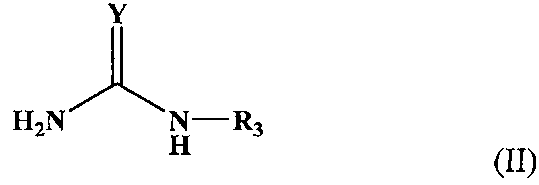
В приведенной формуле Y является атомом кислорода, NH группой или атомом серы, a R3 является атомом водорода, ацильной группой или алкоксикарбонильной группой.
Под «ацильной группой» здесь имеется в виду атомная группа, имеющая OH группу, удаленную из карбоксильной группы карбоновой кислоты. Ацильная группа в настоящем изобретении включает ацильную группу, имеющую от 1 до 5 атомов углерода. Примеры ацильной группы включают ацетильную, пивалоильную, пропионильную и бутилоильную группы.
Под «алкоксигруппой» здесь имеется в виду окси группа со связанной с ней «алкильной группой». Алкоксигруппа по изобретению может быть с прямой и разветвленной цепью. Алкоксигруппа по изобретению предпочтительно включает линейную алкоксигруппу, в которой имеется от 1 до 14 атомов углерода, или разветвленную алкоксигруппу, в которой имеется от 3 до 14 атомов углерода. Примеры алкоксигруппы включают метоксигруппу, этоксигруппу, n-пентилоксигруппу и n-гексилоксигруппу.
Под «C2-n алкоксигруппой» имеется в виду карбонильная группа, в которой имеются C1-(n-1) алкоксигруппы. Алкокси карбонильная группа по изобретению предпочтительно включает в себя алкокси карбонильную группу, содержащую от 2 до 15 атомов углерода. Примеры такой группы включают метоксикарбонильную группу, этоксикарбонильную группу, n-пропилоксикарбонильную группу, изопропоксикарбонильную группу, n-бутоксикарбонильную группу, 2-метил-2-пропилоксикарбонильную группу, n-пентилоксикарбонильную группу и n-гексилоксикарбонильную группу.
Ацильная группа предпочтительно включает ацетильную группу. Кроме того, алкоксикарбонильная группа предпочтительно включает трет-бутоксикарбонильную группу (Boc-группа).
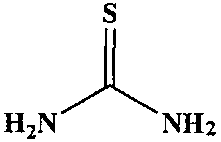
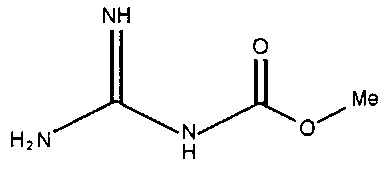
В частности, примеры соединения, представленного формулой (II) в настоящем изобретении, включают
Химическая формула 18
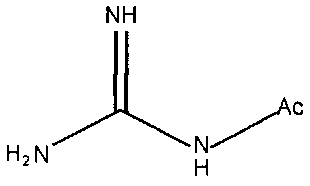
Химическая формула 19
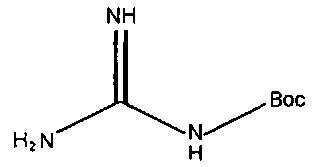
Химическая формула 20
Химическая формула 21
В настоящем изобретении предпочтительно, чтобы реакция (b) протекала в присутствии органического растворителя. Предпочтительно, чтобы органический растворитель имел высокую растворимость и низкую нуклеофильность. Примеры таких органических растворителей включают ДМСО, диметилформамид (ДМФА) и диоксан. Реакцию осуществляют при 0-50°C, предпочтительно при 15-25°C в течение 1-24 часов, предпочтительно в течение 5-10 часов, однако длительность реакции не ограничивается указанными интервалами.
Добавляя -NH-C(=Y)NHR3 группу к карбоксильной группе, участвующей в образовании пептидной связи с аминокислотой, расположенной на N-конце рядом с цистеиновым остатком, пептидную цепь расщепляют на уровне остатка цистеина на N-конце, как показано на фигуре, приведенной ниже.
Химическая формула 22
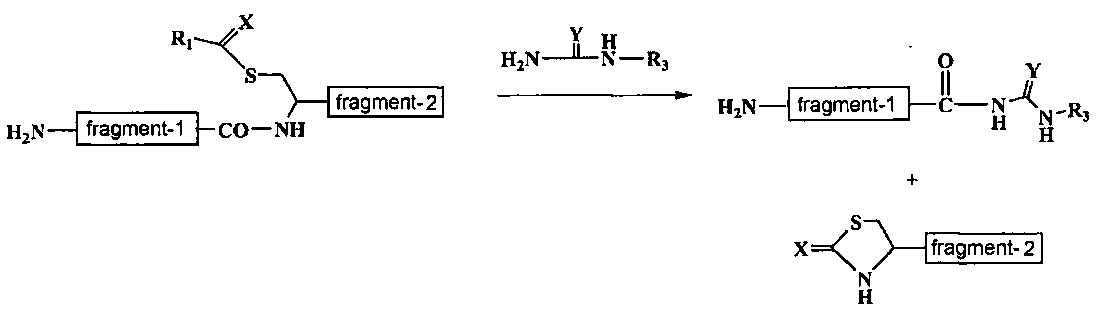
Если боковая пептидная ветвь имеет аминогруппу, то перед постановкой реакции (b) по изобретению в аминогруппу боковой цепи можно внести липофильную защитную группу. Примеры липофильных защитных групп включают, но не исчерпываются ими, защитные группы, такие, как карбонилсодержащая группа, например 9-флуоренилметоксикарбонильная группа (Fmoc), трет-бутилоксикарбонильная группа (Boc) и аллилоксикарбонильная группа (Alloc), ацильная группа, например ацетильная группа (Ac), аллильная группа и бензильная группа.
При введении липофильной защитной группы, например Fmoc группы, реакцию ставят, добавляя в реакционную среду 9-флуоренилметил-N-сукцинимидилкарбонат и гидрокарбонат натрия. Реакцию можно ставить при 0-50°C, предпочтительно при комнатной температуре в течение 1-5 часов, длительность проведения реакции не ограничена только указанным интервалом времени.
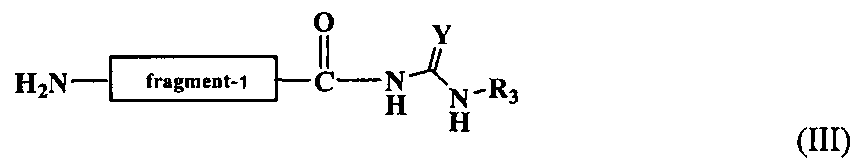
В реакции (b) можно получить второй промежуточный продукт в виде пептидного фрагмента на N-конце щепящего сайта отщепленной пептидной цепи, имеющего следующую формулу (III).
Химическая формула 23
Способ получения пептидного сложного тиоэфира по изобретению включает также стадию тиоэстерификации С-конца второго промежуточного продукта при взаимодействии упомянутого второго промежуточного продукта с тиолом для обмена C-концевой -NH-C(=Y)NHR3 группы на тиоловую (реакция (c)).
Второй промежуточный продукт, используемый для реакции (c), после реакции (b) можно изолировать, а можно и не изолировать.
В предпочтительном объекте изобретения для упомянутой реакции (с) используют тиол, представленный следующей формулой (IV).
R4-SH (формула IV)
Для R4 группы нет особых ограничений, если она не подавляет реакцию обмена тиола и становится удаляемой группой в реакции замещения на атоме углерода карбонильной группы. Предпочтительно, чтобы R4 группа был любой из групп, выбираемых из замещенной или незамещенной бензильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной алкильной группы, более предпочтительно - любой из групп, выбираемых из замещенной или незамещенной бензильной группы, замещенной или незамещенной C6-10 арильной группы и замещенной или незамещенной C1-8 алкильной группы. В частности, R4 можно выбрать из удаляемых групп бензильного типа, таких, как бензилмеркаптан, удаляемых групп арильного типа, например тиофенола и 4-(карбоксиметил)-тиофенола, и удаляемых групп алкильного типа, например 2-меркаптоэтансуфонатной группы, амида 3-меркаптопропионовой кислоты. Особых ограничений, налагаемых на тип и количество замещающих групп и положения, в которых происходит замещение удаляемых групп, нет.
В результате реакции (c) второй промежуточный продукт полностью превращается в сложный тиоэфир, как показано на фигуре ниже.
Химическая формула 24

Полученный пептидный сложный тиоэфир, как описано выше, можно способом лигирования связать с пептидом, который содержит аминокислотный остаток, имеющий -SH группу на C-конце (или модифицированный пептид), выбирая из пептидов или модифицированных пептидов. Соответственно в настоящем изобретении также предлагается способ получения полипептида, включающий в себя стадию связывания пептидного сложного тиоэфира, полученного способом по изобретению, с пептидной цепью, в которой на N-концевым аминокислотным остатком является цистеиновый остаток.
В другом варианте осуществления изобретения в способе лигирования можно также вместо вышеупомянутого пептидного сложного тиоэфира использовать второй промежуточный продукт, полученный на стадии (b). Такой вариант предпочтителен тем, что можно пропустить стадию превращения в сложный тиоэфир.
Под «способом лигирования» здесь имеется в виду не только способ нативного химического лигирования (способ NCL), описанный в источнике 1 патентной литературы, но и случаи применения упомянутого способа нативного химического лигирования к пептидам, содержащим неприродные аминокислотные остатки и производные аминокислот (например, треониновое производное A, защищенный метионин и гликозилированную аминокислоту). Способом лигирования можно получить пептид, в котором имеется естественная амидная связь (пептидная связь) в месте лигирования.
Связывание с помощью способа лигирования можно осуществить между любыми пептидами в следующих вариантах: пептид-пептид, пептид-модифицированный пептид и модифицированный пептид-модифицированный пептид.
Используемые здесь термины применимы для описания вариантов осуществления изобретения, но их применение не ограничивается этими вариантами.
Кроме того, слово «содержащий», используемое в описании, если не очевидно иное, отражает наличие описанных признаков (например, компонентов, стадий, элементов и чисел) и не исключает наличие других признаков (например, компонентов, стадий, элементов и чисел).
Если не оговорено иначе, все термины в описании (включая технические и научные) имеют тот же смысл, который известен широкому кругу специалистов, занятых в области технологии, к которой относится настоящее изобретение. Использованные здесь термины, если на них не налагаются явные ограничения, следует толковать в соответствии с их значением в данном изобретении и в смежных областях техники, и они не должны быть идеализированы и чрезмерно формализованы.
Варианты осуществления изобретения можно описать, прибегая к использованию схем. В этом случае схемы можно увеличить для внесения большей ясности в описание.
Слова «первый» и «второй» используются для выражения различных элементов и понятно, что эти элементы не ограничиваются этими словами, которые использованы лишь для того, чтобы подчеркнуть отличие одного элемента от другого, и можно, например, описать первый элемент как второй элемент и, наоборот, второй как первый, не отклоняясь от сути изобретения.
Далее изобретение описывается более детально с привлечением примеров. Тем не менее, оно имеет различные варианты осуществления и не ограничено приведенными здесь примерами.
Примеры
Синтез производного интерлейкина-13 (ИЛ-13) (SEQ ID NO. 1) (это вещество названо «производным», так как сахарная цепочка отлична от цепочки в натуральном ИЛ-13) способом по изобретению показан на приведенных ниже примерах.
В частности, производное ИЛ-13, являющееся предпочтительным целевым гликозилированным полипептидом, классифицировано и сконструировано так, как описано ниже:
- полипептидный фрагмент A, соответствующий аминокислотной последовательности 1-27 в молекуле ИЛ-13 (она соответствует «второму полипептидному фрагменту, присутствующему на N-конце полипептидного фрагмента, имеющего сахарную цепочку и состоящего из полипептида, который содержит на N-концевую часть целевого гликозилированного пептида»).
- полипептидный фрагмент B, соответствующий аминокислотной последовательности 28-43 в молекуле ИЛ-13 (эта последовательность соответствует «полипептидному фрагменту, расположенному между полипептидным фрагментом, имеющим сахарную цепочку, и вторым полипептидным фрагментом»).
- полипептидный фрагмент С, соответствующий аминокислотной последовательности 44-55 в молекуле ИЛ-13 (она содержит гликозилированную аминокислоту. Этот фрагмент соответствует «полипептидному фрагменту, имеющему цепочку сахара и состоящему из полипептида, который содержит гликозилированную аминокислоту»).
- полипептидный фрагмент D, соответствующий аминокислотной последовательности 56-112 в молекуле ИЛ-13 (эта последовательность соответствует «первому полипептидному фрагменту, присутствующему на C-конце полипептидного фрагмента, имеющего сахарную цепочку и состоящему из полипептида, который содержит на C-концевую часть целевого гликозилированного пептида»).
На фиг. 6 представлена схема эксперимента, в котором получают указанные полипептидные фрагменты.
Как описано далее, вышеупомянутые полипептидные фрагменты A и D получают методом экспрессии в клетках E. coli, который имеет следующие стадии.
Полипептидные фрагменты B и C, упомянутые выше, получают путем химического синтеза.
Затем получают производное ИЛ-13, связывая все четыре полипептидных фрагмента путем лигирования.
Например, обозначение C·D полипептидных фрагментов означает, что полипептидный фрагмент C и полипептидный фрагмент D находятся в связанной форме.
1. Получение полипептидного фрагмента A (аминокислотная последовательность 1-27) и полипептидного фрагмента D (аминокислотная последовательность 56-112)
Два негликозилированных участка (полипептидный фрагмент A (соединение 1) (SEQ ID NO. 2) и полипептидный фрагмент D (соединение 4) (SEQ ID NO. 3)) были получены в виде слитого белка (соединение 6) (SEQ ID NO. 4) с помощью экспрессионной системы на основе клеток E. coli.
1-1. Встраивание молекулы нуклеиновой кислоты, кодирующей негликозилированную часть, в клетку E. coli
(1) В пробирку, содержащую 5 мл среды LB A (содержит 10 г бакто-триптона, 5 г дрожжевого экстракта, 10 г NaCl и 15 г агара в 1 л H2O), добавляли 10 мкл суспензии E. coli (штамм BL21) и после перемешивания с помощью вихревой мешалки инкубировали в течение ночи при 37°C.
(2) Готовили две пробирки, содержащие 5 мл среды LB A, в которые добавляли 200 мкл культуральной среды со стадии (1), и инкубировали их при 37°C. Измеряли мутность (OD600), и через 50 мин, когда OD600 достигала 0,4, пробирки помещали в лед.
(3) Содержимое пробирки со стадии (2) переносили в коническую пробирку и центрифугировали при 0°C, 2000 об/мин в течение 20 мин.
(4) Надосадочную жидкость сливали, осадок растирали, добавляли в него 3 мл 0,1 М раствора CaCl2 и оставляли на льду на 20 мин.
(5) Пробирку со стадии (4) центрифугировали при 0°C, 2000 об/мин в течение 20 мин.
(6) Надосадочную жидкость сливали, осадок растирали, добавляли в него 0,5 мл 0,1 М раствора CaCl2 и после перемешивания вихревой мешалкой, переносили в отдельную пробирку.
(7) В пробирку со стадии (6) добавляли 2 мл вектора рЕТ32а, содержащего молекулу нуклеиновой кислоты, кодирующую аминокислотную последовательность, соответствующую аминокислотам 1-27 ИЛ-13, вставочную последовательность (Cys-Val-His-His-His-His-His-His-Met) и аминокислотную последовательность, соответствующую аминокислотам 56-112, соединенным друг с другом (SEQ ID NO. 5), содержимое пробирки слегка перемешивали и охлаждали на льду в течение 1 часа. Вектор авторами изобретения был безвозмездно предоставлен третьим лицам. Упомянутую молекулу нуклеиновой кислоты встраивали в вектор рЕТ32а (Novagen, Inc.) на уровне сайта NcoI/BamHI кзади по ходу от гена тиоредоксина (см. фиг. 1).
(8) Образец со стадии (7) флотировали на водяной бане при 42°C в течение 3 мин (метод теплового шока), после чего охлаждали на льду в течение 1 мин.
(9) К образцу со стадии (8) добавляли 5 мл среды LB A и после инкубации при 37°C в течение 45 мин центрифугировали при 3000 об/мин и комнатной температуре в течение 10 мин.
(10) Надосадочную жидкость после фильтрации сливали, осадок растирали, добавляли к нему 1 мл среды LB A, перемешивали вихревой мешалкой и общий объем делили на две части, равные 100 мкл и 900 мкл. Каждую из частей высевали шпателем на чашку Петри, содержащую 30 мл среды LB B (Для приготовления среды 10 г бакто-триптона, 5 г дрожжевого экстракта, 5 г NaCl и 5 г глюкозы растворяли в 1 л H2O, добавляли 1,5% агара (15 г/л), автоклавировали в течение 20 мин, давали остыть до 50-60°C и добавляли 100 мг ампициллина).
(11) Чашки Петри со стадии (10) оставляли на 10 ч при 37°C.
(12) Колонии, образовавшиеся на стадии (11), собирали в пробирку (содержащую 3 мл среду LBamp, представляющую собой среду LB A, в которую добавлен 10% ампициллин в соотношении 1000:1) с помощью простерилизованной зубочистки и после перемешивания вихревой мешалкой инкубировали в течение ночи при 37°C.
2. Подтверждение присутствия гена
2-1. Экстракция плазмидной ДНК
(1) 1,5 мл образца со стадии (12) пункта 1-1 переносили в пробирку типа эппендорф.
(2) После центрифугирования при 7000 об/мин и комнатной температуре в течение 3 мин надосадочную жидкость сливали и осадок растирали.
(3) К осадку добавляли 100 мл GTE (50 мМ глюкозы, 25 мМ трис-HCl и 10 мМ ЭДТА) и после перемешивания готовили суспензию на шейкере в течение 5 мин, затем добавляли 200 мл щелочной раствор SDS (0,2N NaOH, 1% SDS) и перемешивали, переворачивая пробирку, после чего охлаждали на льду в течение 5 мин.
(4) Добавляли 150 мл 5 М раствора ацетата калия и перемешивали, переворачивая пробирку, затем охлаждали на льду в течение 5 мин.
(5) К смеси добавляли 15 мл хлороформа и перемешивали с помощью вихревой мешалки, центрифугировали при 13000 об/мин и 4°C в течение 15 мин, после чего надосадочную жидкость переносили в другую пробирку типа эппендорф, к надосадочной жидкости добавляли 3 мл РНКазы и оставляли при 37°C на 1 час.
(6) К образцу со стадии (5) добавляли 500 мл смеси из фенола, хлороформа и изоамилового спирта в соотношении 25:24:1 и центрифугировали при 13000 об/мин и комнатной температуре в течение 10 мин.
(7) Надосадочную жидкость переливали в другую пробирку типа эппендорф, добавляли в нее 40 мл 3 М раствора NaOAc и 1 мл EtOH и перемешивали, переворачивая пробирку, после чего перемешивали с помощью вихревой мешалки и оставляли при -80°C на 30 мин.
(8) Пробирку типа эппендорф со стадии 7 центрифугировали при 13000 об/мин и 4°C в течение 30 мин.
(9) Надосадочную жидкость сливали, к осадку добавляли 1 мл 70% EtOH и после перемешивания с помощью вихревой мешалки центрифугировали при 13000 об/мин и комнатной температуре в течение 5 мин.
(10) Надосадочную жидкость сливали и осадок высушивали в сушильном шкафу.
(11) Затем к нему добавляли 15 мл автоклавированной воды (aH2O) и перемешивали на шейкере в течение 10 мин.
2-2. Обработка рестрикционными ферментами и электрофорез плазмидной ДНК
(1) К 5 мл образца со стадии (11) пункта 2-1 добавляли 15 мл раствора А, содержащего BamHI и NcoI (2,5 мл буферного раствора 10×K (200 мМ трис-HCl pH 8,5, 100 мМ MgCl2, 10 мМ DTT и 1 М KCl), 2,5 мл 0,1% BSA, 0,85 мл NcoI, 0,85 мл BamHI и 10,76 мл aH2O), перемешивали и оставляли на 2 часа при 37°C.
(2) Смесь охлаждали до комнатной температуры и добавляли в нее 5 мл гасящего раствора (50% глицерол, 0,5% SDS, 2 мМ ЭДТА, 0,25% бромфеноловый синий в H2O).
(3) Образец со стадии (2) наносили на 4% полиакриламидный гель (1,995 мл 30% акрилового раствора, 1,5 мл 10×ТВЕ (108 г Trisma base, 55 г борной кислоты и 80 мл 0,25 М раствор ЭДТА в 1 л H2O), 75 мл 10% APS (0,1 г персульфата аммония в 1 л H2O), 10 мл TEMED и 15 мл H2O) и проводили электрофорез при постоянном напряжении 50 В. (В качестве подвижного буфера использовали 1×TBE (10×ТВЕ, разведенный в 10 раз), а в качестве маркера молекулярной массы использовали Ф×174).
(4) Выделенное вещество в течение 15 мин окрашивали бромидом этидия, после чего исследовали при 300 нм для оценки полосы, соответствующей около 759 п.о. Поскольку полипептидная цепь белка, который надо экспрессировать, содержит 253 аминокислоты, было подтверждено отщепление ДНК соответствующей длины.
3. Экспрессия белка
(1) 5 мл 2×YTamp (16 г бакто-триптона, 10 г бактодрожжевого экстракта, 5 г NaCl и 100 мг ампициллина в 1 л aH2O) переносили в каждую из двух пробирок, добавляли в каждую из них 30 мл раствора со стадии (12) пункта 1-1 (образец хранили при 4°C в течение 2 нед) и затем инкубировали в течение ночи при 37°C.
(2) К 1 л 2×YTamp добавляли 10 мл раствора, приготовленного на стадии (1) и инкубировали по достижении температуры 37°C.
(3) Измеряли мутность (OD600) и через 2 ч, когда OD600 достигала 0,6, добавляли 1 мл 1 М раствора изопропил-бета-D-тиогалактопиранозида (IPTG) и осуществляли индукцию в течение 3 ч.
(4) Образец со стадии (3) погружали в лед, затем центрифугировали при 3500 об/мин и 4°C в течение 10 мин.
(5) Надосадочную жидкость сливали и собирали бактерии.
(6) Собранные бактерии хранили при -20°C.
4. Очистка экспрессированного белка
(1) Образец, полученный на стадии (6) пункта 3. оттаивали при комнатной температуре, растворяли в 100 мл буферного раствора A (100 мМ NaH2PO4 10 мМ трис-HCl, 6 М гуанидина гидрохлорида и 5 мМ имидазола, pH 8,0) и обрабатывали на льду ультразвуком для разрушения клеточной стенки.
(2) 10 мл Ni-NTA агарозы (50% EtOH) переносили во флакон (колонку) для твердофазного синтеза и замещали буферным раствором A. Загружали образец со стадии (1), смешивали с Ni-NTA агарозой и затем элюировали.
(3) Элюат собирали и вновь загружали в колонку. Эту операцию выполняли 10 раз, и окончательный элюат собирали (пермеат (1)).
(4) Ni-NTA агарозу со стадии (3) промывали в 100 мл буферного раствора В (100 мМ NaH2PO4, 10 мМ трис-HCl, 6 М гуанидина гидрохлорида и 250 мМ имидазола, pH 8,0) и промывную воду собирали (пермеат (2)).
(5) Элюирование осуществляли с 10 мл буферного раствора С (100 мМ NaH2PO4, 10 мМ трис-HCl, 6 М гуанидина гидрохлорида и 400 мМ имидазола, pH 8,0) (элюат). Элюирование белка подтверждали с помощью раствора CBBG (Coomassie Brilliant Blue G250 - бриллиантовый голубой Кумасси G250).
(6) Элюат со стадии (5) загружали в диализную пробирку, диализную трубку флотировали в 5-литровом сосуде, содержащем чистую воду, и перемешивали при 4°C в течение ночи для осуществления диализа.
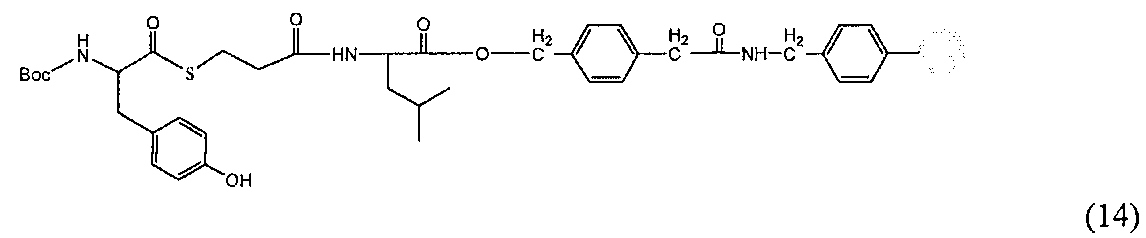
(7) Содержимое диализной пробирки собирали в центрифужную пробирку, центрифугировали при 3500 об/мин и 4°C в течение 10 мин; после сливания надосадочной жидкости оставалось около 25 мг слитого полипептида, имеющего следующее строение: полипептидный фрагмент A-Cys-Val-His метка-Met-полипептидный фрагмент D, и связанного с белком тиоредоксина (соединение 6) (см. фиг. 2 и 3).
Химическая формула 25

C означает Cys, V означает Val, Н означает His и М означает Met. Обозначения в нижеприведенной формуле такие же.
5. Обработка цианидом брома (CNBr)
(1) К 100 мг образца слитого белка (соединение 6), полученного на стадии (7) пункта 4., после очистки ВЭЖХ и лиофилизации добавляли 20 мл водного раствора, содержащего 2% ТФУК и 40% ацетонитрила в атмосфере аргона, далее добавляли 76 мг CNBr, защищая от света, и перемешивали в течение ночи. При такой обработке происходит гидролиз амидной связи на C-концевом метионине между слитым белком, который является тиоредоксиновой частью + гистидиновая метка, и негликозилированной частью, которая является полипептидным фрагментом D, и можно выделить полипептидный фрагмент A с добавленной вставочной последовательностью и полипептидный фрагмент D.
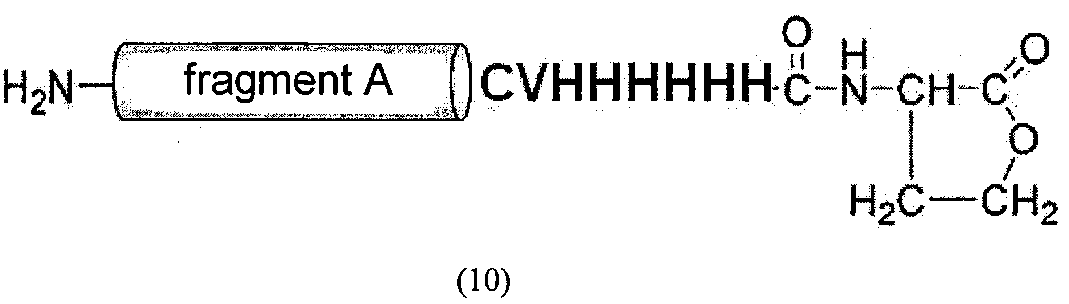
(2) Анализ с помощью ВЭЖХ и последующая масс-спектрометрия подтвердили получение 2,3 мг производного целевого полипептидного фрагмента A-C-Val-His-His-His-His-His-His-Met (соединение 10) (SEQ ID NO. 6) и 4,2 мг полипептидного фрагмента D (соединение 4) при времени удержания 12 мин (см. фиг. 4 и 5).
Химическая формула 26
Химическая формула 27

6. Тиоэстерификация
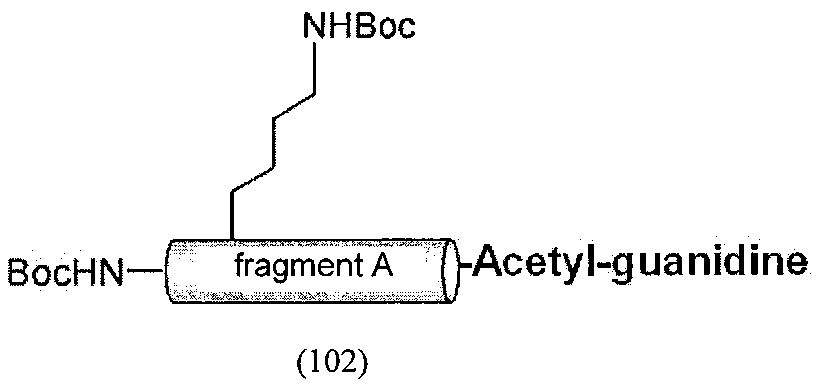
После растворения 6 мг производного полипептидного фрагмента A-C-Val-His-His-His-His-His-His-Met (соединение 10), полученного в пункте 5., в 680 мкл диметилформамида (ДМФА), к раствору добавляли 2,6 мкл (10 экв) N,N-диизопропилэтиламин (DIEtN) и 3,2 мг (10 экв) Boc-OSu и после взаимодействия в течение 1 часа образовывался частично защищенный полипептидный фрагмент из 37 остатков, имеющий Вос-группу на двух сайтах на N-концевой аминогруппе производного полипептидного фрагмента A-C-Val-His-His-His-His-His-His-Met (соединение 10) и аминогруппе боковой цепи лизина в последовательности (соединение 100) (SEQ ID NO. 7). Полученный пептид из 37 остатков растворяли в фосфатном буфере (pH=5,0, 100 нМ), добавляли хлортионоформиат (5 экв), тиоловую группу цистеина модифицировали по тиокарбонатному типу и очищали ВЭЖХ. Полученный полипептидный фрагмент (соединение 101) (SEQ ID NO. 8) лиофилизировали и получали 1,7 мг вещества. Это общее количество растворяли в ДМСО, добавляли ацетилгуанидин (0,5 М в ДМСО) и реакционную среду оставляли при температуре окружающей среды в течение 8 ч. После очистки продукта реакции ВЭЖХ получали 0,3 мг полипептидного фрагмента, имеющего C-концевую карбоксильную группу полипептидного фрагмента A, аминокислотная последовательность которого соответствует последовательности аминокислот 1-27 ИЛ-13, модифицированного гуанидиновой группой (соединение 102) (SEQ ID NO. 9).
Гуанидиновое производное пептида можно обработать алкилтиолом, например меркаптоэтилсерной кислотой, для превращения его в сложное тиоэфирное производное, но его можно использовать и для нативного химического лигирования непосредственно в виде гуанидиновой группы.
Химическая формула 28

M′ обозначает производное Met, такое же обозначение сохраняется и приведенной ниже формуле.
Химическая формула 29

Химическая формула 30
7. Синтез полипептидного фрагмента В (аминокислоты 28-43)
Удлинение полипептидного фрагмента В (соединение 2) осуществляли путем
твердофазного синтеза способом, основанным на использовании защитных групп Fmoc или Boc.
94,34 мг (50 мкмоль) смолы Boc-Leu-PAM загружали во флакон для твердофазного синтеза, промывали дистиллированным ДМФА и дистиллированным ДХМ, после чего высушивали. Аминогруппа аминокислоты, использованной для конденсации, была аминогруппой, защищенной Boc-группой. Кроме того, реакция протекала во флаконе для твердофазного синтеза, если не указано иное.
Смолу тщательно промывали ДМФА и ДХМ, затем к ней добавляли 10% раствор серной кислоты/диоксана (1,0 мл) и перемешивали в течение 30 мин для удаления защитной Boc-группы, затем смолу промывали ДХМ и ДМФА.
Брали 5 эквивалентов S-тритиол-3-меркаптопропионовой кислоты (87,1 мг, 250 мкмоль) в качестве линкера, 10 эквивалентов ДИЭА (87,3 мкл, 500 мкмоль) и 5 эквивалентов HBTU (94,8 мг, 250 мкмоль) растворяли в ДМФА (1,0 мл), заполняли этим раствором флакон для твердофазного синтеза, содержащий смолу, приготовленную, как описано выше, и перемешивали при комнатной температуре в течение 0,5 часа. После перемешивания смолу отмывали несколько раз ДХМ и ДМФА и затем тщательно промывали ДХМ и ДМФА. К смоле добавляли ТФУК и перемешивали в течение 30 мин для удаления защиты с тритильной группы, после чего смолу промывали ДХМ и ДМФА.
После промывания смолы ДМФА 5 эквивалентов первого остатка Boc-Tyr(Br-Z)-COOH (123,6 мг, 250 мкмоль), 5 эквивалентов HBTU (94,8 мг, 50 мкмоль) и 10 эквивалентов ДИЭА (87,3 мкл, 500 мкмоль) смешивали в ДМФА растворителе (1,0 мл), последний добавляли к смоле и перемешивали при комнатной температуре в течение 0,5 ч для получения промежуточного продукта (соединение 14).
Химическая формула 31
После конденсации смолу отмывали ДМФА. С помощью теста Кайзера контролировали конденсацию аминокислоты на смоле, и последовательно снимали Boc-защиту с аминокислот, как в случае Boc-Leu-PAM-смола, конденсируя аналогичным образом аминокислотный остаток 2 и следующие аминокислотные остатки. Каждый такой цикл соответствовал конденсации одного аминокислотного остатка (одиночное сопряжение) до 16-го аминокислотного остатка.
Аминокислоты с защитной группой тиазолидинового типа использовали для Cys, соответствующего аминокислотному остатку 28 в аминокислотной последовательности ИЛ-13.
Смолу после завершения конденсации 28 аминокислот промывали ДМФА и ДХМ, добавляли к ней 1 мл коктейля, содержащего ТФУК, ДМС, m-крезол, ЭДТ, TfOH в соотношении соответственно 5:3:0,8:0,2:1, и перемешивали на льду в течение 1 часа для деблокирования боковой цепи от смолы. Смолу тщательно промывали диэтиловым эфиром и высушивали.
8. Отщепление от смолы
Чтобы отщепить пептид, полученный на стадии 7 от смолы, к ней добавляли 1 мл коктейля, содержащего ТФУК : тиоанизол : ЭДТ : TfOH в соотношении 8:0,8:0,2:0,8 и перешивали на льду в течение 1 часа. После выделения сырого пептида путем осаждения диэтиловым эфиром его высушивали при комнатной температуре и проводили ВЭЖХ (колонка: Synmetory300™ C4, 3,5 мкм, размеры колонки 4,6×150 мм, скорость потока 1,0 мл/мин, подвижная фаза 0,09% ТФУК, линейный градиент: ацетонитрил 18 мин от 18% до 54%). При времени удержания 13 мин образуется сложная тиоэфирная форма пептида (соединение 103) с аминокислотной последовательностью, соответствующей аминокислотам 28-43 молекулы ИЛ-13, которая отщеплялась в форме сложного тиоэфира вследствие добавления линкера. Этот сложный тиоэфирный пептид (соединение 103) (SEQ ID NO. 10) очищали ВЭЖХ и после лиофилизации фракционированного раствора получали 5 мг сложной тиоэфирной формы ИЛ-13 (28-3) (соединение 103); Электроспрей-МС: m/z C85H130N20O25S4; расчетное значение: [M+H]+ 1959,8 [+2H]2+ 980,4, измеренное значение: 1960,8, 981,0.
9. Синтез полипептидного фрагмента С гликозилированной части (аминокислоты 44-55)
Удлинение полипептидного фрагмента осуществляли путем твердофазного синтеза способом защиты Fmoc-группой (соединение 3).
Навеску смолы массой 1,67 мг (50 мкмоль), представляющую собой сополимер 4-(4-гидроксиметил-3-метоксифенокси)масляную кислоту (НМРВ)-поли(этиленгликоль)-поли(диметилакриламид) (PEGA), переносили во флакон для твердофазного синтеза, промывали дистиллированным ДМФА и дистиллированным ДХМ и затем высушивали. Аминогруппа аминокислоты, взятой для конденсации, была аминокислотой, защищенной Fmoc-группой. Реакцию осуществляли в пробирке для твердофазного синтеза, если не указано иное.
5 эквивалентов Fmoc-Gly-OH (74,3 мг, 250 мкмоль) и 3,75 эквивалентов N-метилимидазола (14,9 мкл, 187,5 мкмоль) растворяли в ДХМ (1,0 мл), переносили во флакон для твердофазного синтеза, подготовленный, как было описано выше, перемешивали при комнатной температуре в течение 2 часов. После перемешивания смолу несколько раз промывали раствором, содержащим ДХМ : MeOH : ДИЭА в соотношении 17:2:1 и затем тщательно промывали ДХМ и ДМФА. Затем к смоле добавляли 20% раствор пиперидин/ДМФА (1,0 мл) и перемешивали в течение 30 мин для снятия защиты Fmoc-группой и смолу промывали ДМФА.
После промывания смолы ДМФА 5 эквивалентов аминокислотного остатка 2 Fmoc-Ser(tButyl)-COOH (95,9 мг, 50 мкмоль), 5 эквивалентов гидроксибензотриазола (HOBtH2O) (33,8 мг, 50 мкмоль) и 5 эквивалентов диизопропилкарбодиимида (DIPCDI) (38,5 мкл, 50 мкмоль) перемешивали в растворителе ДМФА (1,0 мл), добавляли к смоле и перемешивали при комнатной температуре в течение 1 часа. После конденсации аминокислоты смолу промывали ДМФА. С помощью теста Кайзера подтверждали полноту конденсации аминокислоты на смоле и деблокировали аминокислоту от защитной Fmoc-группы, как в случае аминокислотного остатка 1. Аналогичным образом конденсировали аминокислотный остаток 3. Затем конденсировали аминокислотный остаток 4 Fmoc-Asn-асиало-сахарная цепочку, представленный следующей формулой (20).
Химическая формула 32

Иными словами, после снятия у валина, являющегося аминокислотным остатком 3 защиты Fmoc-группой смолу промывали ДМФА и добавляли в растворитель, содержащий ДМСО : ДМФА в соотношении 4:1 (2,5 мл) и Fmoc-Asn-асиало-сахарную цепочку (от Otsuka Chemical Co., Ltd., 198 мг, 100 мкмоль), 3 эквивалента DEPBT (45 мг, 150 мкмоль) и 2 эквивалента ДИЭА (18,5 мкл, 100 мкмоль) для введения гликозилированного аспарагина в положение аминокислотного остатка 4. Затем его конденсировали аналогично тому, как конденсировали аминокислотные остатки 1-3, с той лишь разницей, что количество ДМФА подбирали так, чтобы концентрации аминокислот, защищенных Fmoc-группами, которые вводили в реакционный раствор, составила около 50 мМ. После добавления гликозилированного аспарагина аминокислоты, завершали конденсацию защищенных Fmoc-группой аминокислот, при этом каждый цикл соответствовал конденсации одного аминокислотного остатка.
Цистеиновый остаток на N-конце (Cys, соответствующий аминокислотному остатку 45 в аминокислотной последовательности ИЛ-13), который был использован в синтезе, был остатком, имеющим защиту тиазолидинового типа.
Смолу после завершения конденсации тщательно промывали ДМФА и ДХМ, добавляли к ней 2 мл коктейля, содержащего AcOH : ДХМ : MeOH в соотношении 5:4:1, и перемешивали в течение 3 часов, затем полипептидный фрагмент, содержащий сахарную цепочку и имеющий защищенную боковую цепь аминокислоты (соединение 104) (SEQ ID NO. 11), отщепляли от смолы. Последнюю высушивали под вакуумом при комнатной температуре, и уксусную кислоту азеотропно удаляли бензолом. К части полученного вещества добавляли раствор, состоящий из ТФУК (95%), ТИС (2,5%) и H2O (2,5%) и удаляли защитную группу с боковой цепи аминокислоты, чем Cys, соответствующий 44-й аминокислоты в аминокислотной последовательности ИЛ-13 и после упаривания при пониженном давлении и комнатной температуре, очищали с помощью ВЭЖХ. При времени удерживания 11 мин подтверждали масс-спектрометрии образование целевого полипептидного фрагмента С, содержащего сахарную цепочку (соединение 105) (SEQ ID NO. 12).
Соединение 105: Электроспрей-МС: m/z C112H187N17O63S; расчетное значение: [M+2H]2+ 1406,1, [M+3H]3+ 937,7, измеренное значение: 1406,8, 938,3.
10. Тиоэстерификация полипептидного фрагмента C, имеющего сахарную цепочку
Полипептидный фрагмент, содержащий защищенную сахарную цепочку, который был получен на стадии 9. (соединение 104), подвергали азеотропной обработке бензолом, после чего высушивали в сушильном шкафу. 10,1 мг (3,3 мкмоль) этого полипептидного фрагмента (соединение 104) растворяли в 0,4 мл ДМФА, добавляли сухие молекулярные сита 4 Å (6 мг) и 30 эквивалентов бензилтиола (11,6 мкл, 99 мкмоль), перемешивали при -20°C в течение 1 часа, добавляли 5 эквивалентов РуВОР (8,58 мг, 16,5 мкмоль) и 5 эквивалентов ДИЭА (2,9 мкл, 16,5 мкмоль) и в результате 2-часовой реакции происходила тиоэстерификация С-конца. После завершения реакции реакционный раствор по каплям добавляли к 6 мл диэтилового эфира, центрифугировали и собирали полученный белый осадок. Осадок промывали несколько раз диэтиловым эфиром, высушивали и получали белое твердое вещество в виде сложного тиоэфира полипептида, содержащего сахарную цепочку (соединение 106) (SEQ ID NO. 13). К этому веществу добавляли 1 мл коктейля, содержащего ТФУК : H2O : ТИС : ЭДТ в соотношении 90:5:2,5:2,5 и перемешивали в течение 3 ч для удаления защитной группы с боковой цепи пептида. После упаривания при пониженном давлении и комнатной температуре образец анализировали с помощью ВЭЖХ (колонка: Synmetory300™ C4, 3,5 мкм, размеры колонки 4,6×150 мм, скорость потока 1,0 мл/мин, подвижная фаза 0,09% ТФУК, линейный градиент: ацетонитрил 18 мин от 18%) до 54%). При времени удерживания 9,5 мин получали сложный тиоэфир полипептида, содержащего сахарную цепочку и имеющего аминокислотную последовательность, которая соответствует аминокислотам 44-55 в пептидной цепи ИЛ-13 (соединение 107) (SEQ ID NO. 14). Этот сложный тиоэфир полипептида, содержащего сахарную цепочку (соединение 107), очищали с помощью ВЭЖХ и после лиофилизации фракционированного раствора получали 7,2 мг.
Сложная тиоэфирная форма ИЛ-13 (44-55) (соединение 107); электроспрей-MC: m/z C119H193N17O62S2; расчетное значение: [M+2H]2+ 1459,1, [M+3H]3+ 973,1, измеренное значение: 1459,7 и 973,5.
11. Связывание полипептидного фрагмента С, содержащего сахарную цепочку, и полипептидного фрагмента D
Тиоэстерифицированный полипептидный фрагмент, имеющий сахарную цепочку и полученный на стадии 10. (соединение 107) (1,2 мг, 425 нмоль), и негликозилированную часть, которая является полипептидным фрагментом D, полученным на стадии 5. (соединение 4) (2,3 мг, 355 нмоль), добавляли к 180 мкл буферного раствора (6 М гуанидина гидрохлорид, 0,2 М PBS, 20 ммоль ТКЭФ (трис(2-карбоксиэтил)фосфин - ТСЕР), и 0,04 М МРАА (4-меркаптофенилуксусная кислота), pH 6,8) и в реакции при комнатной температуре за 3 часа получали связанные друг с другом полипептидный фрагмент, содержащий сахарную цепочку, полипептидного фрагмента С, имеющего сахарную цепочку, и полипептидный фрагмент D (соединение 108) (SEQ ID NO. 15). После завершения лигирования способом NCL в реакционный раствор непосредственно добавляли метоксиамина гидрохлорид (3,0 мг 35,9 мкмоль), pH доводили до 4,0, N-концевой тиазолидин превращался в цистеин. Через 12 ч реакционную смесь анализировали с помощью ВЭЖХ и определяли пик вещества. Кроме того, вещество, соответствующее данному пику, очищали и выполняли масс-спектрометрию, которая подтвердила получение 2,3 мг полипептидного фрагмента, содержащего сахарную цепочку (соединение 109) (SEQ ID NO. 16).
Соединение 109: Электроспрей-MC: m/z C402H654N102O141S3; расчетное значение: [M+4H]4+ 2316,7, [M+5H]5+ 1853,5, [M+6H]6+ 1544,8, [M+7H]7+ 1324,2, [M+8H]8+ 1158,8, [M+9H]9+ 1030,2, измеренные значения: 2317,7, 1854,5, 1545,7, 1325,2, 1030,9
12. Связывание полипептидного фрагмента В и полипептидного фрагмента C·D, содержащего сахарную цепочку
Полипептидный фрагмент, содержащий сахарную цепочку, который был получен на стадии 11. (соединение 109) (1,3 мг, 140 нмоль), и тиоэстерифицированный полипептидный фрагмент В, полученный ранее на стадии 8. (соединение 103) (0,40 мг, 209 нмоль), добавляли к 70 мкл буферного раствора (6 М гуанидина хлорид, 0,2 М PBS, 40 мМ ТКЭФ (трис(2-карбоксиэтилэтил)фосфин) и 0,02 М MPAA (4-меркаптофенилуксусная кислота), pH 7,1)) и в результате реакции при комнатной температуре в течение 8 ч образовался полипептидный фрагмент, содержащий сахарную цепочку, полипептидного фрагмента В и полипептидный фрагмент C·D, содержащий сахарную цепочку, связанные вместе (соединение 110) (SEQ ID NO. 17). После завершения лигирования по способу NCL в реакционный раствор непосредственно добавляли метоксиамина гидрохлорид (1,2 мг, 14 мкмоль), pH доводили до 4,0 и N-концевой тиазолидин превращался в цистеин. Через 12 часов реакционную смесь анализировали с помощью ВЭЖХ и определяли пик соответствующего вещества. Кроме того, соединение, соответствующее данному пику очищали и выполняли масс-спектрометрию, которая подтверждала получение 0,6 мг полипептидного фрагмента, содержащего сахарную цепочку (соединение 111) (SEQ ID NO. 18).
Соединение 111: Электроспрей-MC: m/z C477H767N121O163S6; расчетные значения: [M+5H]5+ 2199,1, [M+6H]6+ 1832,7, [M+7H]7+ 1571,1, [M+8H]8+ 1374,8, [M+9H]9+ 1222,2, [M+10H]10+ 1100,0, [M+11H]11+ 1000,1, измеренные значения: 2200,4, 1834,1, 1572,3, 1375,9, 1223,3, 1100,9, 1000,9.
13. Связывание полипептидного фрагмента А и полипептидного фрагмента B·C·D
Полипептидный фрагмент, содержащий сахарную цепочку (соединение 111) (0,6 мг, 54 нмоль), полученный на стадии 12., и гуанидиновое производное полипептидного фрагмента (соединение 102) (0,2 мг, 62 нмоль), полученное ранее на стадии 6., добавляли к 50 мкл буферного раствора (6 М гуанидина хлорида, 0,2 М PBS, 20 мМ ТКЭФ (трис(2-карбоксиэтилэтил)фосфин - ТСЕР), и 0,1 М МРАА (4-меркаптофенилуксусная кислота), pH 7,2) и через 24 часа, в течение которых протекала реакция при комнатной температуре, реакционную смесь анализировали с помощью ВЭЖХ и определяли пик, соответствующий полученному веществу. Выполняли масс-спектрометрию полученного вещества, которая подтверждала получение 0,1 мг полипептида, содержащего сахарную цепочку (соединение 112) (SEQ ID NO. 19).
Соединение 112: Электроспрей-MC: m/z C617H1000N156O207S6; расчетные значения: [M+8H]8+ 1763,4, [M+9H]9+ 1567,6, [M+10H]10+ 1410,9, [M+11H]11+ 1282,7, [M+12H]12+ 1175,9, [M+13H]13+ 1085,5, [M+14H]14+ 1008,1, измеренные значения: 1764,3, 1568,5, 1411,8, 1283,6, 1176,8, 1086,3, 1009,0.
14. Удаление Вос-группы
0,1 мг полипептида, содержащего 112 аминокислотных остатков сахарную цепочку, который был получен на стадии 13. (соединение 112), растворяли в 200 мкл 5% водного раствора трифторуксусной кислоты и реакционную среду выдерживали при комнатной температуре в течение 1 ч. Раствор по завершении реакции концентрировали продувкой аргоном, затем разбавляли 20 мкл буферного раствора (6 М гуанидина гидрохлорид и 0,2 М PBS) и полученный раствор очищали с помощью ВЭЖХ. Собирали вещество, соответствующее основному пику, количество полученного производного интерлейкина-13, имеющего единообразную целевую сахарную цепочку, составило 0,1 мг, (соединение 113) (SEQ ID NO. 1).
15. Оптимизация вставочной последовательности
Для оптимизации W во вставочной последовательности (-Cys-W-(His)n-Z-Met-) сравнивали практический выход гуанидиновой формы полипептида, образовавшейся при гуанидинировании тионоформиата пептида, имеющего гистидиновую метку на C-конце (формула реакции приведена ниже).
Химическая формула 33
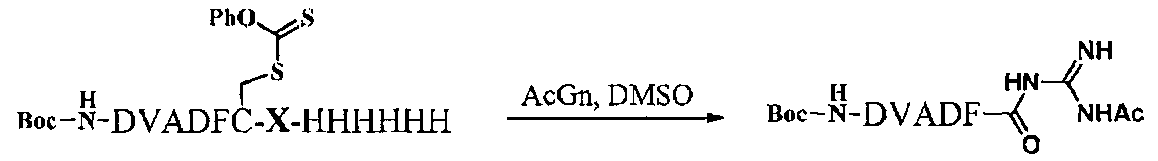
где X соответствует G, V, I, P, F, W, L,
D означает Asp, V означает Val, А означает Ala, F означает Phe, C означает Cys, X может быть любой из следующих аминокислот: Gly, Val, Ile, Pro, Phe, Trp и Leu.
Для реакции гуанидинирования были использованы шесть тионоформиатных пептидов, различающиеся по аминокислоте Xaa в последовательности Boc-Asp-Val-Ala-Asp-Phe-Cys(C(S)OPh)-Xaa-His-His-His-His-His-His-OH (Xaa означает Gly, Val, Ile, Phe, Pro и Trp) (SEQ ID NO. 20). Каждый из тионоформиатных пептидов растворяли в ДМСО (2 мМ), добавляли к раствору такой же объем раствора 1-ацетилгуанидина в ДМСО (1 М) и перемешивали с помощью вихревой мешалки. Полученный раствор оставляли на водяной бане при 37°C на 1 день и затем осаждали с диэтиловым эфиром и осадок отмывали. Затем для получения целевого соединения (SEQ ID NO. 21) осадок очищали полупрепаративной ВЭЖХ. Выход каждого из пептидов приведен в таблице 1. Электроспрей-МС: расчетное значение [M+H]+ 748,3, измеренное значение 748,3.
Как видно из таблицы 1, чрезвычайно большое количество гуанидиновой формы можно получить, если ввести в N-концевую часть гистидиновой метки аминокислоту Val, Trp или Ile. В частности, аминокислоты Val и Ile могут подавить образование побочного продукта.

Реферат
Изобретение относится к способу эффективного получения полипептидного фрагмента, подходящего для связывания способом NCL. Способ получения включает стадию взаимодействия полипептида, состоящего из первого полипептидного фрагмента, имеющего на N-конце цистеиновый аминокислотный остаток, и второго полипептидного фрагмента, связанного через вставочную последовательность -Cys-W-(His)n-Z-Met- с CNBr для получения первого полипептидного фрагмента, имеющего на N-конце цистеиновый аминокислотный остаток, и третьего полипептидного фрагмента, стадию последующего взаимодействия с третьим полипептидным фрагментом с соединением, представленным формулой (I), и соединением, представленным формулой (II), для получения второго полипептидного фрагмента, имеющего модифицированный С-конец. Способ эффективен для получения множественных пептидных фрагментов. Также способ позволяет легче, чем при традиционном способе, выполнить сайт-специфическое гликозирование. 2 н. и 17 з.п. ф-лы, 6 ил., 1 пр.Химическая формула 1Химическая формула 2
Формула
(1) Стадию взаимодействия полипептида, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met-первый полипептидный фрагмент (С-концевой),
где n представляет собой целое число от 0 до 10, Cys означает цистеин, W означает любые 1, 2 или 3 аминокислоты, Z означает любые 0, 1 или 2 аминокислоты, His означает гистидин, Met означает метионин, и N-концевым аминокислотным остатком первого полипептидного фрагмента является цистеин с CNBr для получения следующих полипептидных фрагментов:
(A) первого полипептидного фрагмента с N-концевым цистеиновым остатком и
(B) третьего полипептидного фрагмента, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент- Cys-W-(His)n-Z-Met′ (С-концевой),
где Met′ означает производное Met;
(2) Стадию взаимодействия упомянутого третьего полипептидного фрагмента с соединением, представленным следующей формулой (I):
Химическая формула 1

где
X является атомом серы или кислорода, и
R1 и R2 являются удаляемыми группами,
а в дальнейшем взаимодействия в органическом растворителе с соединением, представленным формулой (II):
Химическая формула 2
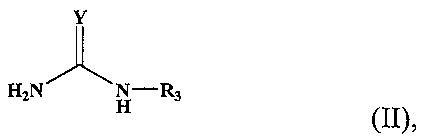
в которой
Y является атомом кислорода, атомом серы или =NH, а
R3 является атомом водорода, ацильной группой или алкоксикарбонильной группой
для получения второго полипептидного фрагмента с модифицированным С-концом, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент-C(=O)-NH-C(=Y)NHR3 (С-концевой).
стадию взаимодействия второго полипептидного фрагмента, имеющего
модифицированный С-конец и полученного на упомянутой стадии (2), с тиолом, представленным формулой
R4-SH,
где R4 является любой из групп, выбираемых из группы, состоящей из замещенной или незамещенной бензильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной алкильной группы
и обмена группы -NH-C(=Y)NHR3 на С-конце с тиоловой группой с образованием второго полипептидного фрагмента с модифицированным С-концом, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент-С(=O)-SR4 (С-концевой).
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met-первый полипептидный фрагмент (С-концевой) является рекомбинантным полипептидным фрагментом, экспрессируемым клеткой.
(1) Стадию классификации и конструирования пептидной последовательности целевого гликозилированного полипептида, получаемого в виде по меньшей мере:
- полипептидного фрагмента, содержащего сахарную цепочку, который состоит из полипептида, содержащего гликозилированную аминокислоту,
- второго полипептидного фрагмента на N-концевой части полипептидного фрагмента, содержащего сахарную цепочку и состоящего из полипептида, содержащего N-концевую часть целевого гликозилированного пептида,
- первого полипептидного фрагмента на С-концевой части полипептидного фрагмента, содержащего сахарную цепочку и состоящего из полипептида, содержащего С-концевую часть целевого гликозилированного пептида,
- полипептидного фрагмента между полипептидным фрагментом, содержащим сахарную цепочку, и вторым полипептидным фрагментом, если присутствие такого фрагмента возможно, и
- полипептидного фрагмента между полипептидным фрагментом, содержащим сахарную цепочку, и первым полипептидным фрагментом, если присутствие такого фрагмента возможно, в которых первый полипептидный фрагмент сконструирован так, чтобы на N-конце его был аминокислотный остаток цистеина;
(2) Стадию получения полипептидного фрагмента упомянутой выше структуры с помощью вектора экспрессии, включающего нуклеотидную последовательность, кодирующую полипептид следующей структуры:
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met-первый полипептидный фрагмент (С-концевой),
где n равно целому числу от 0 до 10, Cys означает цистеин, W означает 1, 2 или 3 аминокислоты, Z означает 0,1 или 2 аминокислоты, His означает гистидин, a Met означает метионин, и N-концевым аминокислотным остатком первого полипептидного фрагмента является цистеин,
и обеспечивающего возможность экспрессии клетками Е. coli;
(3) Стадию взаимодействия полипептида со стадии (2) с CNBr для получения следующих полипептидных фрагментов:
(A) первого полипептидного фрагмента с цистеиновым аминокислотным остатком на N-конце и
(B) третьего полипептидного фрагмента следующей структуры:
(N-концевой) второй полипептидный фрагмент-Cys-W-(His)n-Z-Met′ (С-концевой),
где Met′ означает производное Met;
(4) Стадию взаимодействия упомянутого третьего полипептидного фрагмента с соединением, представленным следующей формулой (I):
Химическая формула 1

в которой X является атомом серы или атомом кислорода, R1 и R2 являются удаляемыми группами,
и затем взаимодействия в органическом растворителе с соединением, представленным следующей формулой (II):
Химическая формула 2

в которой
Y является атомом кислорода, атомом серы или NH-группой, и
R3 является атомом водорода, ацильной группой или алкоксикарбонильной группой,
для получения второго полипептидного фрагмента с модифицированным С-концом, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент-C(=O)-NH-C(=Y)NHR3 (С-концевой);
(5) Условно стадию взаимодействия второго полипептидного фрагмента, имеющего модифицированный С-конец и полученного на стадии
(4), с тиолом, представленным следующей формулой
R4-SH,
в которой R4 является одной какой-либо группой, выбираемой из группы, состоящей из замещенной или незамещенной бензильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной алкильной группы,
и обмена -NH-C(=Y)NHR3 группы на С-конце с тиоловой группой для получения второго полипептидного фрагмента с модифицированным С-концом, имеющего следующую структуру:
(N-концевой) второй полипептидный фрагмент-С(=O)-SR4 (С-концевой); и
(6) Стадию связывания:
- упомянутого полипептидного фрагмента, содержащего сахарную цепочку,
- упомянутого полипептидного фрагмента, если присутствие его возможно, между полипептидным фрагментом, содержащим сахарную цепочку, и вторым полипептидным фрагментом и
- упомянутого полипептидного фрагмента, если присутствие его возможно, между полипептидным фрагментом, содержащим сахарную цепочку, и первым полипептидным фрагментом, полученным отдельно химическим синтезом с:
первым полипептидным фрагментом, имеющим на N-конце цистеиновый аминокислотный остаток и полученным на стадии (3) и
вторым полипептидным фрагментом, имеющим модифицированный С-конец и полученным на стадиях (4) и (5)
в порядке, который позволит получить целевой гликозилированный полипептид с помощью лигирования.
Документы, цитированные в отчёте о поиске
Способ получения тиоэфира пептида
Комментарии