Способ производства дегареликса и его промежуточных соединений - RU2602042C2
Код документа: RU2602042C2
Чертежи












Описание
Область техники
Настоящее изобретение относится к способу производства в жидкой фазе (или растворе) для получения декапептида дегареликса, его защищенного предшественника и других полезных промежуточных соединений. Изобретение дополнительно относится к полипептидам, полезным при жидкофазном способе производства, и к очистке самого дегареликса.
Предшествующий уровень техники
Рак предстательной железы является основной причиной заболеваемости и смертности мужчин в промышленно развитых странах. Дегареликс, также известный как FE200486, является антагонистом рецепторов гонадотропин-рилизинг-гормона (ГнРГ) третьего поколения (блокатор ГнРГ), который был разработан и недавно разрешен к применению страдающими раком предстательной железы пациентами, нуждающимися в андроген-депривационной терапии (Doehn et al., Drugs 2006, vol.9, No.8, pp.565-571; WO 09846634). Дегареликс действует путем непосредственной и конкурентной блокады рецепторов ГнРГ в гипофизе и, как другие антагонисты ГнРГ, не вызывает начальной стимуляции выработки лютеинизирующего гормона посредством гипоталамо-гипофизарно-гонадной оси и, поэтому не вызывает всплеск тестостерона или клиническую вспышку (Van Poppel, Cancer Management and Research, 2010:2 39-52; Van Poppel et al., Urology, 2008, 71(6), 1001-1006); James, E.F. et al., Drugs, 2009, 69(14), 1967-1976).
Дегареликс является синтетическим линейным декапептидом, содержащим семь неприродных аминокислот, пять из которых представляют собой D-аминокислоты. Он имеет десять хиральных центров в остове молекулы декапептида. Аминокислотный остаток в положении 5 в последовательности имеет дополнительный хиральный центр при замещении в боковой цепи, давая в целом одиннадцать хиральных центров. Он имеет регистрационный номер CAS 214766-78-6 (в виде свободного основания) и имеется в продаже под товарным знаком Firmagon™. Лекарственное вещество имеет химическое название N-ацетил-3-(2-нафталинил)-D-аланил-4-хлор-D-фенилаланил-3-(3-пиридинил)-D-аланил-L-серил-4-[[[(4S)-гексагидро-2,6-диоксо-4-пиримидинил]карбонил]амино]-L-фенилаланил-4-[(аминокарбонил)амино]-D-фенилаланил-L-лейцил-N6-(1-метилэтил)-L-лизил-L-пролил-D-аланинамид и представлено приведенной ниже химической формулой (в дальнейшем также именуемой как формула I):

Структура дегареликса может также быть представлена как:
Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-Hor)-D-4Aph(Cbm)-Leu-Lys(iPr)-Pro-D-Ala-NH2,
где Ac представляет собой ацетил, 2Nal представляет собой 2-нафтилаланин, 4Cpa представляет собой 4-хлорфенилаланин, 3Pal представляет собой 3-пиридилаланин, Ser представляет собой серии, 4Aph представляет собой 4-аминофенилаланин, Hor представляет собой гидрооротил, Cbm представляет собой карбамоил, Leu представляет собой лейцин, Lys(iPr) представляет собой N6-изопропиллизин, Pro представляет собой пролин, и Ala представляет собой аланин.
В целях описания этого изобретения каждой аминокислоте в дегареликсе будет дано сокращенное обозначение, как изложено ниже:
AA1 соответствует D-2Nal, AA2 соответствует D-4Cpa, AA3 соответствует D-3Pal, AA4 соответствует Ser, AA5 соответствует 4Aph(L-Hor), AA6 соответствует D-Aph(Cbm), AA7 соответствует Leu, AA8 соответствует Lys(iPr), AA9 соответствует Pro, и AA10 соответствует D-Ala.
Таким образом, например, дегареликс может быть представлен как Ас-AA1-AA10-NH2, тетрапептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser может быть представлен как AC-AA1-AA4, а гексапептид 4Aph(L-Hor)-D-4Aph(Cbm)-Leu-Lys(iPr)-Pro-D-Ala-NH2 как AA5-AA10-NH2.
Дегареликс ранее получали, используя методику твердофазного синтеза пептидов (SPPS) с Boc-защитой, как сообщалось в WO 98/46634 и Jiang et al., J. Med. Chem. 2001, 44, 453-467. Как правило, Boc-защищенный D-Ala первоначально присоединяют к МВНА (4-метилбензилгидриламиновой) смоле в диметилформамиде (ДМФА)/CH2Cl2 при использовании диизопропилкарбодиимида (DIC) и 1-гидроксибензотриазола (HOBt) в качестве активирующих агентов или агентов сочетания. Как только D-Ala присоединяют к смоле, синтез продолжают посредством промывания, деблокирования и затем присоединения следующего аминокислотного остатка, до тех пор пока построение декапептида не будет завершено. Первичные аминогруппы боковых цепей 4Aph, находящегося в положении 5, и D-4Aph, находящегося в положении 6, защищают с помощью Fmoc, когда к ним присоединяют и модифицируют их при помощи L-Hor и Cbm соответственно, перед присоединением следующей аминогруппы в цепи. Это требует дополнительных стадий сначала для снятия защиты с боковой цепи с помощью пиперидина, взаимодействия вновь освобожденной аминогруппы на пептидосмоле с трет-бутилизоцианатом или L-гидрооротовой кислотой при контроле окончания взаимодействия с помощью нингидриновой реакции, а затем промывки пептидосмолы перед добавлением следующего аминокислотного остатка (см. также Sorbera et al., Drugs of the Future 2006, Vol.31, No.9, pp 755-766).
Хотя методика Boc-SPPS до сих пор обеспечивает достаточные количества дегареликса, растущая потребность в этом полипептиде означает, что требуются все возрастающие количества. Методика Boc-SPPS, при которой требуется HF расщепление (с помощью плавиковой кислоты), не подходит для крупномасштабного промышленного синтеза. Действительно, в WO 98/46634 упомянуто, что SPPS подходит только для ограниченных количеств до 1 кг, тогда как классический синтез пептидов в растворе или жидкофазный пептидный синтез (LPPS) является предпочтительным для получения больших количеств продукта. В WO 98/46634 не указано, как следует проводить такой синтез. Несмотря на то, что сообщалось о существовании жидкофазного пептидного синтеза дегареликса [EMEA Report: Assessment Report for Firmagon™ (Дегареликс): Doc. Ref. EMEA/CHMP/635761/2008], на данный момент никаких подробностей такого способа не было публично раскрыто.
WO 97/34923 и WO 99/26964 являются документами, посвященными жидкофазным способам получения биологически активных пептидов. WO 99/26964, в частности, посвящен жидкофазному синтезу декапептидов, обладающих активностью в качестве антагонистов ГнРГ. В WO 99/26964 перечислен ряд ограничений, присущих SPPS методике получения антагонистов ГнРГ, включая ограниченную емкость смолы, большой избыток необходимых реагентов и аминокислот, а также необходимость защиты всех реакционноспособных боковых цепей, таких как гидроксигруппа в Ser, ароматические аминогруппы в Aph и D-Aph, ε-i-пропиламиногруппа в Lys(i-Pr).
В WO 99/26964 предложен жидкофазный способ, который включает сначала получение центральных фрагментов пептидов 5 и 6 положений декапептида с полностью законченными боковыми цепями, а затем сборку пептида по схеме сборки фрагментов "4-2-4", "3-3-4" или "3-4-3". Например, при получении антагониста ГнРГ, азалина B, тетрапептид соединяют с гексапептидом с образованием требуемого декапептида. Когда такую же схему сборки фрагментов пытаются применить для дегареликса, происходит рацемизация аминокислоты Ser (AA4), что в результате приводит к примерно 20% примеси L-Ser. Эта примесь переходит в конечный декапептид и, ее трудно удалить. Кроме того, при получении тетрапептида AA1-AA4 посредством добавления молекулы Ser к трипептиду AA1-AA3 согласно методу, описанному в WO 99/26964, ионы тетрабутиламммония, образовавшиеся при гидролизе бензильной сложноэфирной группы, не могли быть полностью удалены во время последующих операций и переходили в конечный продукт. В дальнейшем было установлено, что при синтезе дегареликса L-гидрооротильная группа превращается в свой гидантоинацетильный аналог, когда L-дигидрооротиловую кислоту соединяют с 4Amp с получением AA5. Эти и другие проблемы при жидкофазном синтезе дегареликса теперь преодолены, и новый жидкофазный полипептидный синтез этого декапептида раскрыт здесь впервые.
Краткое описание сущности изобретения
Проблемы SPPS методов получения дегареликса и недостатки LPPS методов, которые описаны в WO 97/34923 и WO 99/26964, теперь преодолены и являются объектом этого изобретения.
В основном, это изобретение относится к жидкофазному синтезу декапептида дегареликса.
В одном аспекте изобретение относится к жидкофазному способу получения дегареликса, имеющего формулу Ac-AA1-AA10-NH2, или его фармацевтически приемлемой соли или сольвата, включающему стадию, при которой предшественник дегареликса формулы II или его соль или сольват подвергаются обработке расщепляющим агентом:

Соединение формулы II, таким образом, соответствует

где обозначения от AA1 до AA10 являются такими, как в формуле (I),
P1 представляет собой защитные группы для амино или ацетил;
P4 представляет собой водород или защитную группу для гидроксила, предпочтительно защитную группу для гидроксила;
P6 представляет собой водород или защитные группы для амино; предпочтительно защитные группы для амино; и
P8 представляет собой защитную группу для амино.
Предпочтительно, защитная группа P1, если присутствует, является ортогональной относительно P8, то есть обе защитные группы можно отщеплять независимо.
В предпочтительном воплощении, представляет собой ацетил и как P4, так и P8 представляют собой защитные группы, которые можно отщеплять за одну стадию, и P6 представляет собой водород или защитную группу, которую можно отщеплять вместе с защитными группами P4 и P8. Отщепление защитных групп предпочтительно проводят посредством обработки предшественника формулы II трифторуксусной кислотой (ТФУК). Особенно предпочтительно, когда P4, P6 и P8 представляют собой защитные группы, выбранные из трет-бутила (tBu) и трет-бутилоксикарбонила (Boc), и наиболее предпочтительным является предшественник формулы II, где P4 представляет собой tBu, P6 представляет собой водород или tBu, и P8 представляет собой Boc.
Настоящее изобретение также относится к способу жидкофазного производства промежуточного соединения, представленного формулой (II), или его фармацевтически приемлемой соли или сольвата, включающему стадию сочетания первого полипептида, представленного формулой (III)

или его соли со вторым полипептидом, представленным формулой (IV)

или его солью в жидкой реакционной среде в присутствии агента сочетания пептидов возможно вместе с органическим амином в форме основания с образованием декапептида, представленного формулой (II). В этом случае обозначения от AA1 до AA10, P4, P6 и P8 являются такими, как в формуле (II).
Соли включают кислые соли, такие как гидрохлориды, и основные соли, такие как соли щелочных металлов, соли щелочноземельных металлов и соли аммония.
Согласно изобретению второй полипептид, представленный формулой (IV), может быть получен посредством отщепления защитной группы PN от следующего соединения (IVa):

PN предпочтительно представляет собой защитную группу N-концевой аминогруппы и более предпочтительно защитную группу, которую можно отщепить посредством гидрирования, такую как бензилоксикарбонил.
Соединение формулы (IVa) может быть получено посредством сочетания полипептида, представленного формулой (V), с полипептидом, представленным формулой (VI):


или солей этих соединений, где обозначения от AA4 до AA10, PN, P4, P6 и P8 являются такими, как в формуле (IVa).
Изобретение также относится к полипептидам, представленным формулами со (II) по (VI), которые полезны в жидкофазном способе производства по изобретению.
Описание графических материалов
На фиг.1 показана блок-схема синтеза производных AA1 и AA2 для синтеза пептидов.
На фиг.2 показана блок-схема синтеза производного AA2-AA3 для синтеза пептидов.
На фиг.3 показана блок-схема синтеза Ac-AA1-AA3.
На фиг.4 показана блок-схема синтеза производного AA6-AA7.
На фиг.5 показана блок-схема синтеза производного АА5-АА7.
На фиг.6 показана блок-схема синтеза Z-AA4-AA7.
На фиг.7 показана блок-схема синтеза AA9-AA10.
На фиг.8 и 9 показана блок-схема синтеза Z-AA8-AA10.
На фиг.10 показана блок-схема синтеза Z-AA4-AA10.
На фиг.11 показана блок-схема синтеза предшественника формулы (II).
На фиг.12 показана блок-схема синтеза дегареликса из предшественника формулы (II) и очистки и лиофилизации.
Подробное описание сущности изобретения
Настоящее изобретение теперь будет описано более подробно.
Стадия снятия защиты
В первом аспекте настоящее изобретение относится к жидкофазному способу получения дегареликса, имеющего формулу Ac-AA1-AA10-NH2, или его фармацевтически приемлемой соли или сольвата. Способ включает стадию отщепления защитных групп (P4), (P6) и (P8), если они присутствуют, от предшественника дегареликса согласно формуле (II) ((P1)AA1-AA2-AA3-AA4(P4)-AA5-AA6(P6)-AA7-AA8(P8)-AA9-AA10-NH2) или его соли или сольвата в органическом растворе, содержащем предшественник и расщепляющий агент, растворенные в нем.
Обозначения от AA1 до AA10 в формуле (II) имеют такое же значение, как в формуле (I), и P1 представляет собой защитные группы для амино или ацетил, предпочтительно ацетил.
P8 представляет собой защитную группу для амино. Предпочтительно, P8 представляет собой любую защитную группу боковой цепи, известную в данной области техники, такую как группы, описанные в E. Gross & J. Meienhofer, The Peptides: Analysis, Structure, Biology, Vol.3: Protection of Functional Groups in Peptide Synthesis (Academic Press, N.Y., 1981). Подходящие примеры включают 9-флуоренилметилоксикарбонил (Fmoc), бензилоксикарбонил (Cbz или Z) и замещенные Cbz, такие как, например 2-бром-бензилоксикарбонил (2-Br-Z), 2-хлор-бензилоксикарбонил (2-Cl-Z), пара-хлорбензилоксикарбонил, пара-6-нитробензилоксикарбонил, пара-бромбензилоксикарбонил и пара-метоксибензилоксикарбонил, орто-хлорбензилоксикарбонил, 2,4-дихлорбензилоксикарбонил, 2,6-дихлорбензилоксикарбонил и тому подобное; алифатические защитные группы уретанового типа, такие как трет-бутилоксикарбонил (Boc), трет-амилоксикарбонил, изопропилоксикарбонил, 2-(пара-бифенилил)-изопропилоксикарбонил и тому подобное; циклоалкильные защитные группы уретанового типа, такие как циклопентилоксикарбонил, адамантилоксикарбонил и циклогексилоксикарбонил; аллилоксикарбонил (Alloc), ацетил (Ac), бензоил (Bz), трифторацетил (Tfa), толуолсульфонил (Tos), бензил (Bn), трифенилметил (Trt), орто-нитрофенил-сульфенил (Nps), трет-бутил-диметилсилилоксикарбонил, [2-(3,5-диметоксифенил)-пропил-2-оксикарбонил] (Ddz), 2,2,2-трихлорэтилоксикарбонил (Troc), бифенилилизопропилоксикарбонил (Bpoc) и орто-нитробензилоксикарбонил. Предпочтительными защитными группами являются Fmoc, Boc и Alloc, причем Boc является наиболее предпочтительной.
P4 представляет собой водород или защитную группу гидроксила, предпочтительно защитную группу гидроксила. Защитная группа гидроксила Ser (P4) представляет собой предпочтительно C4-С6алкил (например, трет-бутил, циклогексил), ацетил (Ac), тритил, бензил, бензиловый эфир, такой как пара-метоксибензил, или другие замещенные бензилы, такие как пара-нитробензиловый, пара-хлорбензиловый, орто-хлорбензиловый и 2,6-дихлорбензиловый, тетрагидропираниловый, три(C1-C6)алкилсилиловый, 2-метоксиэтоксиметиловый (MEM), 4-диметилкарбамоилбензиловый и O-феноксиацетиловый эфиры.
Особо предпочтительными являются трет-бутиловый, бензиловый и 9-флуоренилметиловый эфиры, причем трет-бутиловый является наиболее предпочтительным.
P6 представляет собой водород или защитные группы для амино, предпочтительно защитные группы для амино. Предпочтительные защитные группы включают C4-С6алкил (например, трет-бутил, циклогексил), ацетил (Ac), тритил, бензил, бензиловый эфир, такой как пара-метоксибензил, или другие замещенные бензилы, такие как пара-нитробензиловый, пара-хлорбензиловый, орто-хлорбензиловый и 2,6-дихлорбензиловый, тетрагидропираниловый, три(C1-C6)алкилсилиловый, 2-метоксиэтоксиметиловый (MEM), 4-диметилкарбамоилбензиловый и O-феноксиацетиловый эфиры. Особо предпочтительными являются трет-бутиловый, бензиловый и 9-флуоренилметиловый эфиры, причем трет-бутиловый является наиболее предпочтительным.
Расщепляющий агент, используемый для удаления защитных групп, зависит от природы защитной группы, и эти агенты хорошо известны в данной области техники.
Предпочтительными расщепляющими агентами для защитной группы гидроксила Ser, P4, являются:
- трифторуксусная кислота (ТФУК), соляная кислота (HCl) или метансульфоновая кислота, в частности для трет-бутилового эфира в качестве защитной группы,
- H2/Pd-C, фтороводородная кислота (HF) или трифторметансульфоновая кислота, в частности для бензилового эфира в качестве защитной группы, и
- SiCl4/анизол, в частности для 2-(метилсульфинил)бензилового эфира в качестве защитной группы.
Предпочтительными расщепляющими агентами для защитной группы для амино, P8, являются:
- трифторуксусная кислота (ТФУК), HCl или метансульфоновая кислота, в частности для трет-бутилкарбаматов в качестве защитной группы,
- H2/Pd-C, (HF) или трифторметансульфоновая кислота, в частности для бензилкарбаматов в качестве защитной группы, и
- пиперидин, 1,8-диаза-бицикло[5.4.0]-ундец-7-цен (DBU) и диэтиламин (DEA), в частности для Fmoc в качестве защитной группы.
Предпочтительные растворители включают ДХМ (дихлорметан), ДМФА (диметилформамид), N-метилпирролидон (NMP), диоксан, этанол (EtOH), неразбавленную HF и ТФУК.
Особо предпочтительными являются различные условия отщепления, указанные в следующей таблице 1:
Ссылка: Chem. Rev. 2009, 109, 2465-2504 (Albert Isidro-Llobet)
Типично, предшественник формулы (II) растворяют в расщепляющем агенте, предпочтительно ТФУК, с добавлением или без добавления растворителя, при комнатной температуре (от 20 до 25°C) в течение периода от 20 до 30 часов, предпочтительно 24 часов. Когда защитная группа удалена (предпочтительно степень превращения составляет более 95%, наиболее предпочтительно более 99%), неочищенный дегареликс затем предпочтительно вливают в забуференную смесь вода-этанол с получением забуференного раствора неочищенного дегареликса для последующей очистки. Предпочтительное значение pH находится предпочтительно в пределах от 2 до 4, более предпочтительно в пределах от 2,5 до 3,5 и наиболее предпочтительно приблизительно 3.
Конкретные воплощения стадии снятия защиты показаны на фиг.12 и в примере 4 (см. стадию 12).
Очистка и лиофилизация
Раствор неочищенного дегареликса предпочтительно очищают при использовании хроматографических методов, таких как препаративная обращенно-фазовая хроматография (ОФХ).
Полученный продукт затем предпочтительно лиофилизируют.
Сочетание 3+7
В дополнительном аспекте настоящее изобретение также относится к способу жидкофазного производства промежуточного соединения, представленного формулой (II), или его фармацевтически приемлемой соли или сольвата, включающему стадию сочетания первого полипептида, представленного формулой (III)
или его соли со вторым полипептидом, представленным формулой (IV)
или его солью в жидкой реакционной среде в присутствии агента сочетания пептидов и возможно органического амина в форме основания с образованием декапептида, представленного формулой (II). В этом случае обозначения от AA1 до AA10, P1, P4, P6 и P8 являются такими, как в формуле (II). Соли включают кислые соли, такие как гидрохлориды, и основные соли, такие как соли щелочных металлов, соли щелочноземельных металлов и соли аммония.
Реакцию сочетания проводят в органическом растворе, в котором растворены два пептида и агент сочетания пептидов и возможно органический амин в форме основания. Вспомогательное вещество для сочетания пептидов и/или органический амин также могут присутствовать.
Органический растворитель, агент сочетания пептидов, вспомогательное вещество для сочетания пептидов и органический амин в форме основания могут быть любыми из тех, которые известны в области LPPS.
Типичными органическими растворителями являются ТГФ, NMP (N-метилпирролидон), ДХМ, ДМФА, ДМСО (диметилсульфоксид) и их смеси. Наиболее предпочтительным растворителем является ДМСО.
Типичными агентами сочетания пептидов являются один или более чем один из гексафторфосфата o-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU), гексафторфосфата o-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU), тетрафторбората o-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (TBTU), гексафторфосфата бензотриазол-1-ил-окси-трис(диметиламино)фосфония (BOP), гексафторфосфата бензотриазол-1-ил-окси-трис-пирролидинфосфония (PyBOP), дихлорида N,N-бис-(2-оксо-3-оксазолидинил)фосфоновой кислоты (BOP-Cl), гексафторфосфата бром-трис-пирролидин-фосфония (PyBroP), изо-бутилхлорформиата (IBCF), 1,3-дициклогексилкарбодиимида (DCC), 1,3-диизопропил-карбодиимида (DIC), гидрохлорида 1-(диметиламинопропил)-3-этилкарбодиимида (WSCDI), N-этоксикарбонил-2-этокси-1,2-дигидрохинолина (EEDQ), изопропилхлорформиата (IPCF), тетрафторбората 2-(5-норборнен-2,3-дикарбоксимидо)-1,1,3,3-тетраметилурония (TNTU), ангидрида пропанфосфоновой кислоты (PPAA) и тетрафторбората 2-сукцинимидо-1,1,3,3-тетраметилурония (TSTU).
Предпочтительным агентом сочетания является DCC. DCC предпочтительно используют без органического амина. В особо предпочтительном воплощении DCC используют в комбинации с ДМСО. DCC предпочтительно используют в количестве от 1,3 до 2, наиболее предпочтительно от 1,4 до 1,6 эквивалента относительно трипептида.
Другим предпочтительным агентом сочетания является DIC, который предпочтительно используют в комбинации с 6-хлор-HOBt, возможно совместно с солями меди.
Типичными вспомогательными веществами для сочетания пептидов являются 1-гидрокси-1H-бензотриазол (HOBt), 6-хлор-HOBt и 1-гидрокси-7-азабензотриазол (HOAt).
Типичными органическими аминами в форме оснований являются NMM (N-метилморфолин), DIPEA (N,N-диизопропилэтиламин), TEA (триэтиламин) и коллидин.
Особенно предпочтительным является проведение реакции сочетания при использовании ДМСО в качестве растворителя и DCC в качестве агента сочетания.
Конкретные воплощения стадии сочетания показаны на фиг.11 и в примере 4 (см. стадию 11).
Синтез гептапептида
Соединение формулы IV, то есть АА4(Р4)-АА5-AA6(P6)-АА7-АА8(Р8)-АА9-AA10-NH2, предпочтительно получают посредством отщепления защитной группы Pn следующего соединения IVa:
Наряду с тем, что обозначения от AA1 до AA10, P4, P6 и P8 имеют такое же значение, как и ранее, PN представляет собой защитную группу N-концевой аминогруппы. В предпочтительном воплощении PN представляет собой защитную группу, которую можно отщепить посредством гидрирования, например при использовании водорода и палладиевого катализатора (такого, как Pd/C). Наиболее предпочтительная защитная группа PN представляет собой бензилоксикарбонил (Z).
Конкретные воплощения стадии отщепления PN показаны на фиг.11 и в примере 4 (см. стадию 11).
Соединение формулы (IVa) может быть получено посредством сочетания полипептида, представленного формулой (V), с полипептидом, представленным формулой (VI),
или солей одного или обоих этих соединений, где обозначения от AA4 до AA10, P4, P6 и P8 являются такими, как в формуле (IVa). Реакцию сочетания проводят в органическом растворе, в котором растворены два пептида и агент сочетания пептидов. Вспомогательное вещество для сочетания пептидов и/или органический амин также могут присутствовать. Могут быть использованы такие же агенты сочетания пептидов, органические растворители, вспомогательные вещества для сочетания пептидов и органические амины, как описано выше. В предпочтительном воплощении DCC используют в качестве агента сочетания, возможно с этил ацетатом в качестве растворителя.
Синтез тетрапептида формулы V
В предпочтительном воплощении тетрапептид формулы V, то есть (PN)AA4(P4)-AA5-AA6(P6)-A7, получают способом, включающим следующие стадии:
a) получение (PN2)АА5-АА6(P6)-АА7(PC), где P6 имеет такое же значение, как выше в формуле IVa, (PN2) представляет собой защитную группу N-концевой аминогруппы или водород, и (PC) представляет собой защитную группу C-концевого карбоксила, которую можно отщепить посредством гидрирования;
b) удаление защитной группы для амино (PN2), если она присутствует;
c) гидрирование H-AA5-AA6(P6)-AA7(PC) с получением H-AA5-AA6(P6)-AA7; и
d) взаимодействие H-AA5-AA6(P6)-AA7 с активированным сложным эфиром (PN)AA4(P4) с получением (PN)AA4(P4)-AA5-AA6(P6)-AA7, где (P4) представляет собой защитную группу гидроксила или водород, P6 представляет собой водород или защитную группу для амино, и PN представляет собой защитную группу, которую можно предпочтительно отщепить посредством гидрирования, например при использовании водорода и палладиевого катализатора (такого, как Pd/C).
Наиболее предпочтительной защитной группой PN является бензилоксикарбонил (Z).
Предпочтительным активированным эфиром (PN)AA4(P4) является 4-нитрофениловый эфир (ONp).
Поэтому, предпочтительно, бензиловый эфир удаляли в промежуточном соединении AA5-AA7 с получением как N-, так и C-незащищенного AA5-AA7. Этот трипептид затем подвергали взаимодействию с предварительно активированным серином (например, Z-Ser(tBu)-ONp).
Производство дегареликса
Настоящее изобретение, таким образом, относится к производству дегареликса, при этом рассмотренная выше стадия снятия защиты является существенной стадией этого производства. В предпочтительном воплощении эта стадия снятия защиты следует за сочетанием 3+7, рассмотренным выше. В еще более предпочтительном воплощении сочетание 3+7 следует за синтезом гептапептида, рассмотренным выше, то есть способ включает следующие стадии:
a) синтез AA4(P4)-AA5-AA6(P6)-AA7-AA8(P8)-AA9-AA10-NH2 или его соли, или сольвата;
b) сочетание АА4(Р4)-AA5-AA6(P6)-АА7-АА8(P8)-AA9-АА10-NH2 и (P1)AA1-AA2-АА3 с получением (P1)АА1-АА2-AA3-АА4(Р4)-АА5-AA6(P6)-AA7-AA8(P8)-AA9-АА10-NH2 или его соли, или сольвата;
c) снятие защиты с (P1)АА1-АА2-AA3-АА4(Р4)-АА5-AA6(P6)-AA7-AA8(P8)-АА9-AA10-NH2 или его соли, или сольвата с получением дегареликса или его сольвата, или соли.
Обозначения от AA1 до AA10 и P1, P4, P6 и P8 имеют такие же значения, как определено ранее.
В особо предпочтительном воплощении:
- P1 представляет собой ацетил;
- P4 представляет собой защитную группу, которая расщепляется ТФУК, предпочтительно tBu;
- P6 представляет собой водород или защитную группу, которая расщепляется ТФУК, предпочтительно tBu;
- P8 представляет собой защитную группу, которая расщепляется ТФУК, предпочтительно Boc.
В наиболее предпочтительном воплощении гептапептид получают при использовании синтеза гептапептидов, описанного выше. Кроме того, за стадией снятия защиты предпочтительно следуют методы очистки и лиофилизации, описанные выше.
При синтезе дегареликса или его предшественников и особенно на всех стадиях, содержащих пептид с гидрооротильной группировкой, значение pH предпочтительно поддерживают ниже 9, предпочтительно ниже 8,5, еще более предпочтительно ниже 8. Предпочтительно использовать слабое основание, такое как NaHCO3, для регулирования pH. Особенно предпочтительно, чтобы все процессы экстракции после стадий сочетания проводились в пределах значений pH от 2 до 9, предпочтительно от 2,5 до 8 (см. стадии 6, 7, 10 и 11 в экспериментальной части). Кроме того, предпочтительным является и добавление C4-5алифатического спирта, такого как н-бутанол или 2-бутанол, перед стадией экстракции или промывки.
Промежуточные соединения
Изобретение также относится к полипептидам, представленным формулами от (II) до (VI), которые являются полезными при жидкофазном способе производства по изобретению.
Предпочтительные воплощения формулы (II)
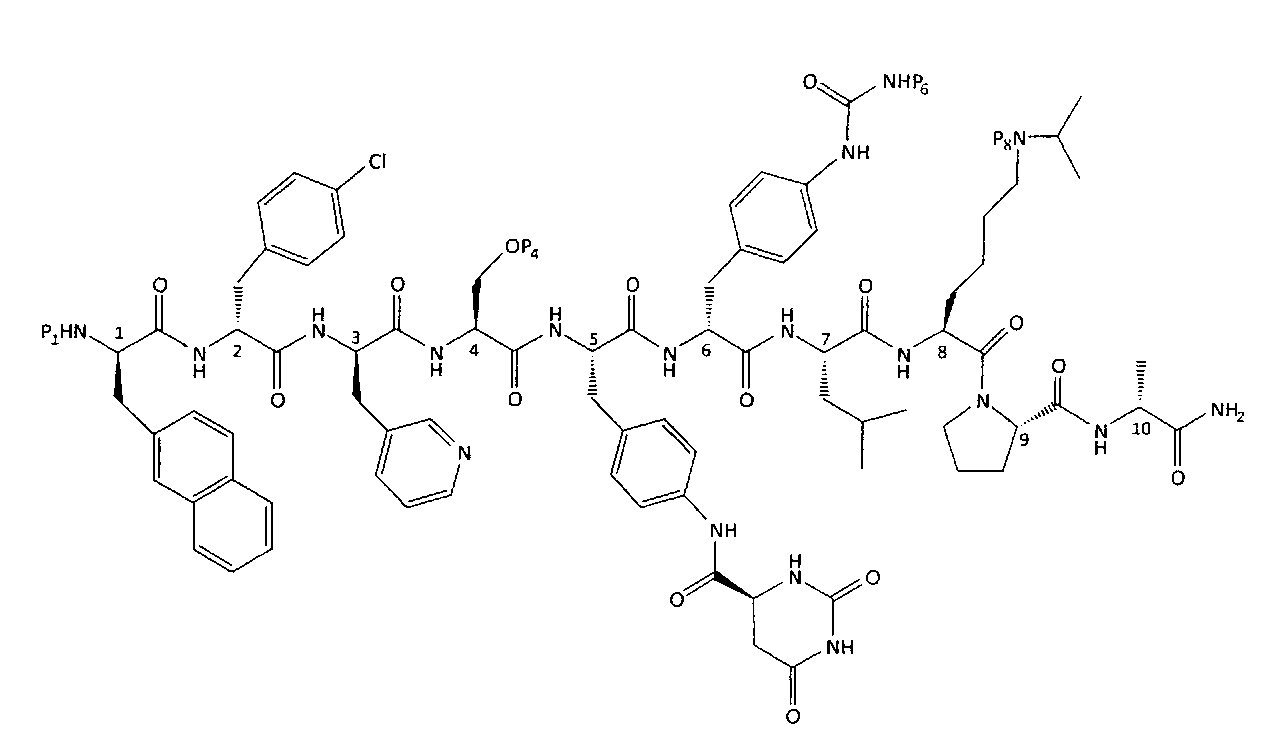

Предпочтительные воплощения включают соли этих соединений.
Предпочтительные воплощения формулы (III)

Предпочтительные воплощения включают соли этих соединений.
Предпочтительные воплощения формулы (IV)/(IVA)
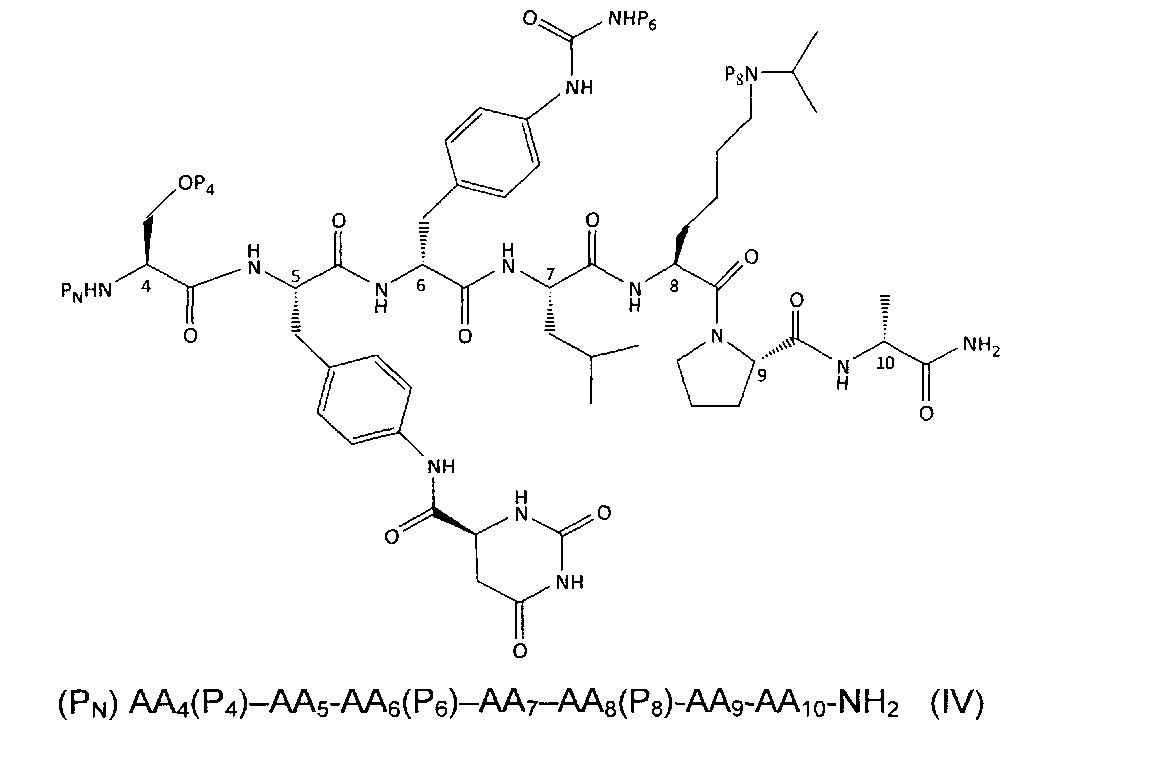
Предпочтительные воплощения включают соли этих соединений.
Предпочтительные воплощения формулы (V)


Предпочтительные воплощения включают соли и сольваты этих соединений.
Предпочтительные воплощения формулы (VI)

Когда защитная группа отсутствует, функциональная группа является незащищенной группой (например, >NH).
Предпочтительные воплощения включают соли этих соединений.
Экспериментальная часть
Материалы, используемые в экспериментальной части
Материалы, используемые в экспериментальной части, перечислены ниже.
Химические вещества:
Водный аммиак NH3 (водн.)
Ацетонитрил C2H3N
н-Бутанол C4H10O
2-Бутанол C4H10O
Изопропанол (изопропанол) C3H8O
Бутилацетат C5H12O2
Этанол, 99,9% C2H6O
Метанол CH4O
Гептан C7H16
Очищенная вода H2O
Этилацетат C3H8O2
Уксусная кислота C2H4O2
Ацетат аммония C2H7NO2
цетилимидазол C5H6N2O
Триэтиламин C6H15N
N-Метилморфолин C5H11NO
N-Метилпирролидон C5H9NO
N,N'-Дициклогексилкарбодиимид C13H22N2
Дициклогексиламин C12H23N
N,N'-Диизопропилкарбодиимид C7H14N2
N,N-Диметилэтилендиамин C4H12N
N,N-Диметилформамид C3H7NO
Диметилсульфоксид C2H6OS
1-Гидроксибензотриазол C6H5N3O
пара-Нитрофенол C6H5NO3
N-Гидроксисукцинимид C4H5NO3
Изобутилхлорформиат C5H9ClO2
Хлорид натрия NaCl
Гидроксид натрия, водный NaOH (водн.)
Соляная кислота, водная HCl (водн.)
Фосфорная кислота H3PO4
Гидросульфат натрия NaHSO4
Гидрокарбонат натрия NaHCO3
Метансульфоновая кислота CH4SO3
Трифторуксусная кислота C2HF3O2
Палладий на угле, 5% Pd-C
Водород H2
Толуол C7H8
Исходные вещества:
Пример 1: синтез промежуточного соединения Ac(1-3)ONa: Ac-D-2Nal-D-4Cpa-D-3Pal-ONa [7]
Активация Boc-D-4Cpa-OH и выделение
Стадия 1 (стадия реакции)
Boc-D-4Cpa-OH (299,75 г) растворяли в изопропиловом спирте (3,53 кг), смесь перемешивали и добавляли HOSu (0,184 кг) и DIC (0,164 кг) и перемешивали при 0°C в течение 1 часа. Осадок отфильтровывали и промывали изопропиловым спиртом. Твердое вещество сушили при пониженном давлении с получением Boc-D-4Cpa-OSu [1].
Активация Boc-D-2Nal-OH и выделение
Стадия 2 (стадия реакции)
Boc-D-2Nal-OH (315,38 г) растворяли в изопропиловом спирте (5,35 кг) и добавляли IBC (157,07 г) и NMM (116,7 г). Смесь воды (42 мл), изопропилового спирта (1,1 кг) и HOSu (230,14 г) добавляли после охлаждения до -10°C вместе с дополнительным количеством NMM (10,11 г) и смесь перемешивали в течение 30 минут. Добавляли воду (0,82 л), осадок отфильтровывали и промывали изопропиловым спиртом и сушили при пониженном давлении с получением Boc-D-2Nal-OSu [2].
Синтез Boc(2-3)ОН: Boc-D-4Cpa-D-3Pal-OH
Стадия 3 (стадия реакции)
H-D-3Pal-OH × 2 HCl (0,251 кг) и Boc-D-4Cpa-OSu [1] (0,397 кг) со стадии (1) растворяли в ДМСО (3,33 л) и добавляли NMM (318,8 г). Смесь перемешивали при 20°C в течение 6 часов.
Добавляли воду (17 л), и pH регулировали посредством добавления HCl до pH 4,25. Осадок отфильтровывали и диспергировали в воде. Полученную суспензию затем фильтровали и промывали водой. Твердое вещество сушили при пониженном давлении с получением Boc-D-4Cpa-D-3Pal-OH [3].
Синтез промежуточного соединения Ac(1-3)ONa: Ac-D-2Nal-D-4Cpa-D-3Pal-ONa [7] (соединение формулы IIIa)
Стадия 4 (стадия реакции)
Boc-D-4Cpa-D-3Pal-OH [3] (447,93 г) со стадии (3) растворяли в смеси AcOEt (3,4 л) и АсОН (675 мл), смесь охлаждали при 5°C, после в нее добавляли MSA (672,77 г). Взаимодействие продолжается при 10°C в течение 2 часов и в раствор добавляли TEA (1214,28 г) с получением H-D-4Cpa-D-3Pal-OH [4].
Boc-D-2Nal-OSu [2] (412,44 г) со стадии (2) добавляли к H-D-4Cpa-D-3Pal-ОН [4], перемешивали в течение 24 часов при 20°C. Добавляли 25%-ный водный NH3 (0,154 л) и н-бутанол (4,5 л) и смесь перемешивали при 45°C в течение 1 часа.
Раствор промывали:
- водой,
- водой при pH 9,5 (pH регулировали при смешивании с водным NaOH),
- водой.
AcOH (4,5 л) добавляли в органическую фазу, и раствор концентрировали до получения масла при пониженном давлении. Масло повторно растворяли в AcOH (4,5 л) и повторно концентрировали при пониженном давлении с получением Boc-D-2Nal-D-4Cpa-D-3Pal-OH [5] в виде масла.
Boc-D-2Nal-D-4Cpa-D-3Pal-OH [5] растворяли в воде (0,09 л) и уксусной кислоте (AcOH) (1,8 л). Добавляли MSA (672,77 г), и смесь перемешивали при температуре ниже 35°C в течение 2 часов. Раствор нейтрализовали TEA (779,16 г). Раствор концентрировали при пониженном давлении до получения масла. Масло повторно растворяли в толуоле (2,5 л) и повторно концентрировали при пониженном давлении до получения масла. Последнюю стадию повторяли с получением H-D-2Nal-D-4Cpa-D-3Pal-OH [6].
H-D-2Nal-D-4Cpa-D-3Pal-OH [6] растворяли в толуоле (2,0 л) и добавляли раствор ацетилимидазола (132,14 г) в толуоле (0,25 л). Раствор перемешивали при 20°C в течение 2 часов и добавляли воду (0,1 л).
Добавляли н-бутанол (4,5 л), и органическую смесь промывали при 35°C:
- 5% водным NaCl,
- метанолом и водой при кислых значениях pH 5,5 (pH регулировали при смешивании с водным NaOH),
- метанолом и водой при pH 11 (рН регулировали при смешивании с водн. NaOH),
- метанолом и 10% водным NaCl.
К перемешиваемой органической фазе после процессов экстракции добавляли гептан (15 л) при 20°C в течение 1 часа, полученную суспензию оставляли при перемешивании при 20°C в течение 1 часа. Осадок выделяли посредством фильтрации и суспендировали в гептане (3,5 л). Суспензию снова фильтровали. Последнюю стадию промывки гептаном и фильтрацию повторяли. Твердое вещество затем сушили при пониженном давлении с получением Ac-D-2Nal-D-4Cpa-D-3Pal-ONa [7].
Характеристики ключевых промежуточных соединений
Промежуточное соединение Ac(1-3)ONa [7] стадии 4
Пример 2: синтез промежуточного соединения Z(4-7)OH × DCHA: Z-SerftBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OH × DCHA [15]
Синтез промежуточного соединения Boc(6-7)OBzl: Boc-D-4Aph(tBuCbm)-Leu-OBzl
Стадия 5 (стадия реакции)
Boc-D-4Aph(tBuCbm)-OH (379,45 г) растворяли в NMP (0,76 л) и AcOEt (4,4 кг). После охлаждения при -4°C добавляли IBC (150,2 г) и NMM (101,1 г), и раствор перемешивали при -7°C в течение 0,5 часа с получением Boc-D-4Aph(tBuCbm)-OAct [8].
H-Leu-OBzl×TOS (491,88 г) растворяли в NMP (1,5 л) и добавляли AcOEt (2,7 кг) с последующим добавлением NMM (126,4 г). Этот раствор затем переносили в Boc-D-4Aph(tBuCbm)-OAct [8] и перемешивали при -10°C в течение 1 часа. Затем добавляли воду (0,5 л).
Реакционную смесь промывали при 20°С:
- водой при pH 8,5 (рН регулировали при смешивании с водным NaOH),
- водой при pH 2,0 (рН регулировали при смешивании с водным HCl),
- водой.
Органическую фазу концентрировали при пониженном давлении до получения масла. Масло повторно растворяли в AcOEt (0,6 кг) и повторно концентрировали при пониженном давлении до получения масла. Оставшееся масло растворяли в AcOEt (0,6 кг). Добавляли гептан (15,5 л) при перемешивании при 20°C. Осадок выделяли посредством фильтрации и промывали гептаном и затем сушили при пониженном давлении с получением Boc-D-4Aph(tBuCbm)-Leu-OBzl [9].
Синтез Boc-(5-7)-OBzl: Boc-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OBzl
Стадия 6 (стадия реакции)
Boc-D-4Aph(tBuCbm)-Leu-OBzl [9] (582,7 г) со стадии 5 растворяли в AcOEt (3,15 кг). Добавляли MSA (481 г) и перемешивали при температуре ниже 15°C в течение 5 часов и добавляли TEA (406 г). Добавляли ДМФА (0,333 кг) с последующим добавлением TEA (101 г) и NMM (51 г) с получением H-D-4Aph(tBuCbm)-Leu-OBzl [10].
Boc-4Aph(L-Hor)-OH (462,46 г) растворяли в ДМФА (2,09 кг) и AcOEt (1,44 кг). Добавляли IBC (150,24 г) и NMM (111,27 г) и перемешивали при -10°C в течение 0,5 часа с получением Boc-4Aph(L-Hor)-OAct [11].
H-D-4Aph(tBuCbm)-Leu-OBzl [10] добавляли к Boc-4Aph(L-Hor)-OAct [11] и перемешивали при -10°C в течение 1,5 часа. Затем добавляли AcOEt (5,4 кг) и л-бутанол (6,0 л).
Органическую фазу промывали при 20°C:
- 5% водным NaHCO3 при значении pH примерно 8 (pH регулировали при смешивании с водным NaHCO3),
- 10% водным NaCl при pH 2,5 (pH регулировали при смешивании с водным H3PO4).
ДМФА (0,9 л) добавляли в органическую фазу, которую затем концентрировали при пониженном давлении до получения масла. Раствор вливали в воду (14 л) при перемешивании. Осадок выделяли на фильтре и промывали водой. Твердое вещество сушили при пониженном давлении с получением Boc-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OBzl [12].
Синтез промежуточного соединения Z(4-7)OH × DCHA: Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OH × DCHA (соединение формулы Va)
Стадия 7 (стадия реакции)
Boc-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OBzl [12] (885,02 г) со стадии 6 добавляли в смесь MSA (961,1 г) и AcOEt (7,2 кг) и добавляли 2-бутанол (2 л), полученную смесь перемешивали при 0°C в течение 6 часов. Затем MSA нейтрализовали с помощью TEA (909,0 г).
Добавляли диспергированный в 2-бутаноле (1 л) 5% Pd/C (88,5 г), смесь гидрировали под давлением при 20°C в течение 3 часов. Затем Pd/C отфильтровывали и промывали 2-бутанолом с получением раствора, содержащего H-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OH [13].
Z-Ser(tBu)-OH (413,5 г) растворяли в MeCN (2,5 л), раствор охлаждали до -5°C. Добавляли HONp (195 г) с последующим добавлением DCC (278,5 г), смесь перемешивали при 20°C в течение 24 часов. Смесь затем фильтровали и промывали MeCN с получением Z-Ser(tBu)-ONp [14].
NMM (354,2 г), ДМФА (4,75 кг) и Z-Ser(tBu)-ONp [14] добавляли к раствору H-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OH [13], смесь оставляли при перемешивании при 20°C в течение 3 суток.
Полученную смесь промывали:
- 10% водным NaCl при pH 2,5 (pH регулировали при смешивании с водным HCl),
- водой при кислых значениях pH (pH 2,5) (pH регулировали при смешивании с водным HCl),
- 7,5% водным NaHCO3,
- 5% водным NaCl при (pH 2,5) (pH регулировали при смешивании с водным HCl),
-10% водным NaCl.
В конечную органическую фазу добавляли дициклогексиламин (DCHA) (181 г), органическую фазу концентрировали при пониженном давлении до получения масла. Масло повторно растворяли в iPrOH (3,14 кг) и повторно концентрировали при пониженном давлении до получения масла. Оставшееся масло повторно растворяли в iPrOH (3,14 кг) и при перемешивании раствор вливали в AcOEt (31,5 кг). Перемешивание продолжали при 20°C в течение 1 часа до осаждения, осадок затем выделяли посредством фильтрации и промывали AcOEt. Твердое вещество сушили при пониженном давлении при 30°C в течение 30 часов с получением Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OH × DCHA [15]. Чистота промежуточного соединения Z(4-7)OH × DCHA [15] составляет не менее 80% (ВЭЖХ (высокоэффективная жидкостная хроматография).
Промежуточное соединение Z(4-7)OH [15] стадии 7
Пример 3: синтез промежуточного соединения H(8-10)NH2: H-Lys(iPr, Boc)-Pro-D-Ala-NH2 [21]
Синтез Boc(9-10)NH2: Boc-Pro-D-Ala-NH2
Стадия 8 (стадия реакции)
Boc-Pro-OH (226,02 г) растворяли в iPrOH (1,73 кг). Реакционную смесь охлаждали до -5°C. Добавляют IBC (143,4 г) и NMM (106,2 г), смесь перемешивали при 5°C в течение 0,5 часа с получением Boc-Pro-OAct [16].
H-D-Ala-NH2× HCl (124,57 г) суспендировали в смеси iPrOH (1,57 кг) и NMM (106,2 г). Суспензию добавляли к Boc-Pro-OAct [16]. Реакционную смесь оставляли при перемешивании при 10°C в течение 3 часов. Затем добавляли DMEDA (10,6 мл). Смесь фильтровали, фильтрат концентрировали при пониженном давлении до получения масла. Масло повторно растворяли и повторно концентрировали с AcOEt (1,125 кг).
Остаточное масло растворяли в смеси AcOEt (1,8 кг) и н-бутанола (0,6 л). Органическую фазу промывали:
- 15% водным NaCl при pH 2,5 (pH регулировали при смешивании с водным HCl),
- 15% водным NaCl при pH 9,5 (pH регулировали при смешивании с водным NaOH).
Органическую фазу концентрировали при пониженном давлении, повторно растворяли в AcOEt (1,08 кг) и повторно концентрировали до получения масла.
Добавляли смесь AcOEt (0,33 кг) и гептана (0,75 л) при 20°C и перемешивали в течение 16 часов. Полученный осадок фильтровали и промывали смесью AcOEt и гептана на фильтре. Твердое вещество затем сушили при пониженном давлении с получением Boc-Pro-D-Ala-NH2 [17]
Синтез промежуточного соединения H(8-10)NH2: H-Lys(iPr, Boc)-Pro-D-Ala-NH2 (соединение формулы VIa)
Стадия 9 (стадия реакции)
Boc-Pro-D-Ala-NH2 [17] (313,89 г) со стадии 8 растворяли в смеси MSA (528,61 г) и iPrOH (0,785 кг), раствор перемешивали при 45°C в течение 1 часа. Смесь затем нейтрализовали TEA (607,14 г) с получением H-Pro-D-Ala-NH2[18].
Z-Lys(iPr, Boc)-OH × DCHA (603,83 г) суспендировали в AcOEt (1,17 кг) и промывали:
- 12% водным NaHSO4,
- водой,
- 15% водным NaCl.
Органическую фазу Z-Lys(iPr, Boc)-OH [19] после процессов экстракции добавляли к H-Pro-D-Ala-NH2 [18]. Добавляли растворенные в AcOEt (0,135 кг) HOBt (183,79 г) и DCC (227,0 г), смесь перемешивали при 2°C в течение 0,5 часа. Затем добавляли воду (0,2 л). Смесь фильтровали и промывали AcOEt. Объединенные фильтраты концентрировали при пониженном давлении до получения масла. Масло растворяли в AcOEt (0,9 кг), фильтровали и раствор промывали:
- водой при pH 2,5 (pH регулировали при смешивании с водным HCl),
- водой при pH 9 (pH регулировали при смешивании с водным NaOH),
- 10% водным NaCl при pH 7 (pH регулировали при смешивании с водным HCl или водным NaOH).
Органическую фазу концентрировали при пониженном давлении с получением Z-Lys(iPro,Boc)-Pro-D-Ala-NH2 [20].
Z-Lys(iPro, Boc)-Pro-D-Ala-NH2 [20] растворяли в этаноле (0,04 кг) и воде (0,5 л) и добавляли 5% Pd/C (50 г). Суспензию подкисляли до значения pH 2,5 посредством добавления 6 М HCl и гидрировали при 20°C. После завершения взаимодействия катализатор удаляли посредством фильтрации и pH повышали до значения pH 7,0 посредством добавления 32% NaOH. Затем этанол удаляли посредством выпаривания при пониженном давлении. н-Бутанол (1 л) добавляли к полученной водной фазе, и уровень pH регулировали до щелочных значений pH 9 водным NaOH, и экстракция начинается. Эту стадию повторяли. Объединенные органические фазы концентрировали при пониженном давлении до получения масла.
Масло растворяли в AcOBu (0,5 л), концентрировали при пониженном давлении при 20°C и повторно растворяли в AcOBu (0,5 л). Затем добавляли гептан (2 л) при 50°C в течение 1 часа. Суспензию оставляли при перемешивании при 0°C в течение 16 часов. Осадок выделяли посредством фильтрации и промывали гептаном. В конце твердое вещество сушили при пониженном давлении с получением H-Lys(iPr, Boc)-Pro-D-Ala-NH2 [21]. Чистота промежуточного соединения H(8-10)NH2 [21] составляла не менее 95% (ВЭЖХ).
Промежуточное соединение H(8-10)NH2 [21] стадии 9
Пример 4: процессы сегментной конденсации конечного промежуточного соединения (соединение формулы II)
Промежуточное соединение Z(4-10)NH2: Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [22] (соединение формулы IVg)
Стадия 10 (стадия реакции)
Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OH×DCHA [15] (1153,41 г) со стадии 7 растворяли в ДМФА (2,1 кг). Затем добавляли HOBt (153,2 г) совместно с AcOEt (6,9 кг) и H-Lys(iPr,Boc)-Pro-D-Ala-NH2 [21] (569,5 г) со стадии 9. Когда все твердые вещества растворились, добавляли MSA (96,1 г). Раствор охлаждали до температуры ниже 5°C и добавляли DCC (309,5 г), растворенный в AcOEt (0,810 кг). Температуру повышали до 20°C, реакция продолжалась в течение 24 часов. Превращение Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-OH × DCHA [15] составляет не менее 96% (ВЭЖХ). Добавляли AcOEt (4,95 кг) и воду (5,5 л), и смесь перемешивали и фильтровали. 7,5% NaHCO3 (водн.) (35 л) добавляли к фильтрату при перемешивании. Фазы разделяли, и органический слой дополнительно промывали:
- 7,5% NaHCO3,
- водой при pH 3 (pH регулировали при смешивании с водным HCl),
- водой.
Конечную органическую фазу концентрировали при пониженном давлении до получения масла. Масло повторно концентрировали ЕЮН (0,405 кг) и затем AcOEt (0,45 кг). Оставшееся масло растворяли в ЕЮН (0,405 кг) и добавляли AcOEt (0,45 кг) и AcOBu (4,6 л). Раствор добавляли к гептану (27,6 л) при 20°C в течение 1 часа. Затем осадок фильтровали и промывали гептаном. Твердое вещество сушили при пониженном давлении при максимуме с получением Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [22]. Чистота Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr, Boc)-Pro-D-Ala-NH2 [22] составляет не менее 70% (ВЭЖХ).
Конечное промежуточное соединение Ac(1-10)NH2: Ac-D-2Nal-D-4Cpa-D-3Pal-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [24]
Стадия 11 (стадия реакции)
Z-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [22] (1409,67 г) со стадии 10 добавляли к смеси EtOH (10,98 кг) и воды (3,2 л) и перемешивали до тех пор, пока раствор не станет гомогенным. Добавляли 5% Pd/C (211 г). Смесь гидрировали при 20°C с контролем pH при pH 2,5 водным HCl.
Катализатор удаляли посредством фильтрации и pH регулировали с помощью водного NaOH до pH 3,8. Фильтрат концентрировали при пониженном давлении до получения масла. EtOH (4,7 кг) добавляли к маслу и повторно концентрировали. Затем к маслу добавляли AcOEt (5,4 кг) и повторно концентрировали, этот процесс повторяли снова с получением H-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [23].
H-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [23] диспергировали в AcOEt (1,125 кг), затем добавляли HOBt (153,16 г) и смесь охлаждали до 0°C. Ac-D-2Nal-D-4Cpa-D-3Pal-ONa [7] (609 до 05 г) со стадии 4 растворяли в ДМСО (2,5 л), этот раствор смешивали с суспензией, содержащей H-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [23], и добавляли DCC (309,5 г), растворенный в AcOEt (0,45 кг). Смесь перемешивали при °C в течение 24 часов. Превращение соединения [23] составляет не менее 96% (ВЭЖХ).
Добавляли воду (150 мл) и ДМСО (0,5 л), и перемешивание продолжали при 20°C в течение более чем 3 часов. Осадок фильтровали и промывали смесью AcOEt и ДМСО. Фильтраты объединяли и добавляли н-бутанол (17 л). Органический раствор промывали:
- водой при pH 2,5 (pH регулировали при смешивании с водным HCl),
- 7% NaHCO3 (водным),
- 10% водным NaCl (pH смеси нейтрализовали до значения pH 7,0, если это необходимо, при смешивании с водным HCl).
Добавляли ДМФА (4,75 кг), и органическую фазу концентрировали при пониженном давлении до получения масла. Масло медленно добавляли в воду (50 л) при 20°C в течение 1 часа при интенсивном перемешивании. Осадок выделяли на фильтре и дважды промывали водой. Твердое вещество затем сушили при пониженном давлении с получением Ac-D-2Nal-D-4Cpa-D-3Pal-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [24] [Конечное промежуточное соединение]. Чистота соединения [24] составляет не менее 70% (ВЭЖХ).
Пример 5: Снятие защиты с конечного промежуточного соединения Ас(1-10)NH2 с получением неочищенного дегареликса [25]
Стадия 12 (стадия реакции)
Ac-D-2Nal-D-4Cpa-D-3Pal-Ser(tBu)-4Aph(L-Hor)-D-4Aph(tBuCbm)-Leu-Lys(iPr,Boc)-Pro-D-Ala-NH2 [24] (соединение формулы IIb)(1844,59 г) со стадии 11 растворяли в ТФУК (28,3 кг) при 20°C. Раствор перемешивали при 20°C (удаление 3 защитных групп) в течение 24 часов. Превращение соединения [24] составляет не менее 99% (ВЭЖХ).
Реакционную смесь затем смешивали с холодным раствором (ниже 10°C) AcONH4 (19,1 кг), AcOH (18,4 л) и EtOH (14,52 кг) в воде (74 л). Во время смешивания двух растворов температуру поддерживали ниже 25°C. pH конечного раствора регулировали до значения pH 3 с помощью ТФУК или AcONH4, если это необходимо, с получением раствора Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-Hor)-D-4Aph(Cbm)-Leu-Lys(iPr)-Pro-D-Ala-NH2 [25] [Неочищенный дегареликс].
Стадия 13 (очистка и лиофилизация)
Раствор неочищенного дегареликса прокачивали через колонку с обращенной фазой. Дегареликс элюировали с колонки градиентом EtOH/0,12% ТФУК в воде. Фракции с чистотой не менее 95% повторно очищали на колонке с обращенной фазой при использовании градиента EtOH/1% АсОН в воде. Фракции высокой чистоты лиофилизировали.
Реферат
Изобретение относится к жидкофазному способу получения декапептида - дегареликса и промежуточных соединений для получения декапептида. Изобретение дополнительно относится к промежуточным полипептидам для жидкофазного способа получения дегареликса. Способ осуществляют путем сочетания первого полипептида, представленного формулой (III): (P)AA-AA-AA(III) или его соли со вторым полипептидом, представленным формулой (IV): AA(P)-AA-AA(P)-AA-AA(P)-AA-AA-NH(IV), или его солью в жидкой реакционной среде в присутствии агента сочетания пептидов c получением декапептида формулы (II): (P)AA-AA-AA-AA(P)-AA-AA(P)-AA-AA(P)-AA-AA-NH(II), где Р, Ркаждый независимо представляет собой водород или защитную группу; Р, Р, каждый независимо представляет собой защитную группу; AAпредставляет собой D-2Nal; ААпредставляет собой D-4Cpa; ААпредставляет собой D-3Pal; ААпредставляет собой Ser; ААпредставляет собой 4Aph(L-Hor); ААпредставляет собой D-Aph(Cbm); ААпредставляет собой Leu; ААпредставляет собой Lys(iPr); ААпредставляет собой Pro; ААпредставляет собой D-Ala. Заявленный способ обеспечивает возможность крупномасштабного производства дегареликса. 8 н. и 4 з.п. ф-лы, 12 ил., 6 табл., 5 пр.
Формула

где AA1 представляет собой D-2Nal, АА2 представляет собой D-4Cpa, АА3 представляет собой D-3Pal, АА4 представляет собой Ser, АА5 представляет собой 4Aph(L-Hor), АА6 представляет собой D-Aph(Cbm), АА7 представляет собой Leu, АА8 представляет собой Lys(iPr), АА9 представляет собой Pro, АА10 представляет собой D-Ala;
Р1 представляет собой защитные группы для амино или ацетил;
Р4 представляет собой водород или защитную группу для гидроксила, предпочтительно защитную группу для гидроксила;
Р6 представляет собой водород или защитные группы для амино; предпочтительно защитные группы для амино; и
Р8 представляет собой защитную группу для амино,
или его фармацевтически приемлемых соли или сольвата, включающий стадию сочетания первого полипептида, представленного формулой (III)

или его соли со вторым полипептидом, представленным формулой (IV)

или его солью в жидкой реакционной среде в присутствии агента сочетания пептидов.

где Р4 представляет собой водород или защитную группу для гидроксила, предпочтительно защитную группу для гидроксила;
Р6 представляет собой водород или защитные группы для амино; предпочтительно защитные группы для амино; и
Р8 представляет собой защитную группу для амино,
посредством сочетания полипептида, представленного формулой (V), или его соли или сольвата с полипептидом, представленным формулой (VI), или его солью или сольватом,


и затем удаления защитной группы PN, где обозначения от АА4 до АА10, Р4, Р6 и Р8 являются такими, как определено в п. 1, и PN представляет собой защитную группу, которая может быть удалена посредством гидрирования.
a) получение (PN2)АА5-АА6(Р6)-АА7(РС), где Р6 имеет такое же значение, как указано выше, (PN2) представляет собой защитную группу для N-концевой аминогруппы или водород, и (РС) представляет собой защитную группу для С-концевого карбоксила, которую можно отщепить посредством гидрирования;
b) удаление защитной группы для амино (PN2), если она присутствует;
c) гидрирование Н-АА5-АА6(Р6)-АА7(РС) с получением Н-АА5-АА6(Р6)-АА7; и
d) взаимодействие Н-АА5-АА6(Р6)-АА7 с активированным сложным эфиром (PN)АА4(Р4) с получением (PN)АА4(Р4)-АА5-АА6(Р6)-АА7, где АА4 имеет такое же значение, как указано выше, (Р4) представляет собой защитную группу для гидроксила или водород, и PN представляет собой защитную группу, которую можно предпочтительно отщепить посредством гидрирования.
AA4(P4)-AA5-AA6(P6)-AA7-AA8(P8)-AA9-AA10-NH2,
(PN)AA4(P4)-AA5-AA6(P6)-AA7-AA8(P8)-AA9-AA10-NH2,
(PN)AA4(Р4)-АА5-АА6(Р6)-АА7,
или соли, или сольваты,
где АА4 представляет собой Ser, АА5 представляет собой 4Aph(L-Hor), AA6 представляет собой D-Aph(Cbm), АА7 представляет собой Leu, АА8представляет собой Lys(iPr), АА9 представляет собой Pro, АА10 представляет собой D-Ala;
Р4 представляет собой водород или защитную группу для гидроксила, предпочтительно защитную группу для гидроксила;
Р6 представляет собой водород или защитные группы для амино; предпочтительно защитные группы для амино; и
Р8 представляет собой защитную группу для амино, и PN представляет собой защитную группу.
Документы, цитированные в отчёте о поиске
Способ получения дегареликса
Комментарии