Способ получения гетерологичного полипептида в дрожжах saccharomyces cerevisiae - RU2194758C2
Код документа: RU2194758C2
Чертежи















Описание
Изобретение относится к полипептидам, экспрессированным и процессированным в дрожжах, к векторной ДНК, включающей ДНК последовательность, кодирующую такие полипептиды, к векторам несущим такие ДНК фрагменты и к дрожжевым клеткам, трансформируемым векторами, а также к способу продуцирования гетерологических протеинов в дрожжах.
Дрожжевые организмы продуцируют ряд протеинов, синтезируемых внутриклеточно, но функционирующих вне клетки. Такие внеклеточные протеины относятся к секретируемым /выделяемым/ протеинам. Эти секретируемые протеины первоначально экспрессируются внутри клетки на предшественнике или пре-протеиновой структуре, содержащей предпоследовательность, определяющей эффективное направление экспрессируемого продукта через мембрану эндоплазматического ретикулума /ЭР/. Пред-последовательность, обычно называемая сигнальным пептидом, как правило отщепляется от желаемого продукта в процессе транслокации. Вступив на секреторный путь, протеин транспортируется к аппарату Гольджи. Из аппарата Гольджи протеин может далее следовать различными путями, попадая либо в клеточную вакуоль, либо - клеточную мембрану или выйти за пределы клетки, выделившись во внешнюю среду /Pfefer, S.R., Rothman, J. Ann. Rev. Biochem, 56/1987/, 829-852/.
Были предложены некоторые подходы для объяснения экспрессии и секреции в дрожжах протеинов, гетерологических дрожжам. Европейская опубликованная патентная заявка 008632 А содержит описание способа, согласно которому происходит экспрессирование протеинов, гетерологических дрожжам, их процессирование и секретирование путем трансформирования организма дрожжей с экспрессией оболочки, несущей ДНК, кодирующую желаемый протеин и сигнальный пептид, подготовка культуры трансформируемого организма, выращивание культуры и выделение протеина из среды клеточной культуры. Сигнальный пептид может быть как сигнальным пептидом самих желаемых протеинов, гетерологическим сигнальным петидом или гибридом нативного и гетерологического сигнального пептида.
Проблема, которая может неожиданно возникнуть в связи с использованием сигнальных пептидов, гетерологических дрожжам, возможно состоит в том, что гетерологический сигнальный пептид не гарантирует эффективной транслокации и/или расщепления после сигнального пептида. MFα I/α - фактор S. cerevisiae синтезируют в предпроформе из 165 аминокислот, включающих 19 аминокислот длинного сигнального или пред-пептида, за которым следуют 64 аминокислоты длинного "лидирующего" или пропептида, окруженных тремя N-связанными гликозилированными сайтами, за которым следует (лизарг(асп/глу,ала)2-3 α - фактор /4/Kurjah J., Herzkowitz, J. Cell 30 /1982/, 933-943. Участок сигнал-лидер предпро-MFα-1 широко использовался для осуществления синтеза и секреции гетерологических протеинов в S. cerevisiae.
Использование сигнальных и лидирующих пептидов, гомологических дрожжам, известно из спецификации патента США 4546082, Европейской опубликованной патентной заявке 0116201 А, 0123294 А, 0123544 А, 0163529 А и 0123289 А и ДК патентные описания 2484/84 и 3614/83.
В ЕП 0123289А описано использование предшественника α - фактора S. cerevisiae, в то время как в ДК 2484/84 описано использование сигнального пептида инвертазы Saccharomyсes cerevisiae, а ДК 3614/83 описывает использование сигнального пептида РН05 Saccharomyсes cerevisiae для секреции чужеродных протеинов.
В спецификации патента США 4564082, ЕП 0016201 А, 0123294 А, 0123544 А и 0163529 А описаны способы, согласно которым α - фактор сигнал-лидера из Saccharomyсes cerevisiae /MFα1 или МFα2/ используется в процессе секреции экспрессированного гетерологического протеина в дрожжах. При включении ДНК последовательности, кодирующей S. cerevisiae MFα1 сигнал/лидер последовательность у 5 конца гена желаемого протеина, было продемонстрировано процессирование и секреция желаемого протеина.
В ЕП 206783 раскрывается система секреции полипептидов из S. cerevisiae, в которой α - фактор лидирующей последовательности усечен таким образом, чтобы устранить четыре α - факторных пептида, присутствующих в нативной лидирующей последовательности таким образом, чтобы оставить лидирующий пептид слитым с гетерологическим полипептидом через α - фактор процессингового сайта лиз-арг-глу-ала-глу-ала. Эта конструкция приводит к эффективному процессированию небольших пептидов/содержащих менее 50 аминокислот/. Для секреции и процессирования больших полипептидов нативный α - фактор усекают так, чтобы оставить один или два α - факторных пептида между лидирующим пептидом и полипептидом.
Ряд секретируемых протеинов направляется к протеолитической процессирующей системе, которая может расщеплять пептидную связь у карбоксильного конца двух последовательных основных аминокислот. Эта ферментативная активность, присущая S. cerevisiae, закодирована КЕХ2 геном /Julius, D, А и др. Cell 37/ 1984b/, 1075/. Процессинг продукта геном КЕХ2 необходим для секреции скрещивающего фактора α1 (MFα1 или α - фактора), активным и S. cerevisiae, но не включен в секрецию скрещивающего фактора а, активными S. cerevisiae.
Секреция и корректный процессинг полипептида, предназначенного к выделению, достигается в некоторых случаях при культивировании дрожжей нового организма, который трансформируют вектором, сконструированным так, как это сказано в ссылках, приведенных выше. Во многих случаях, однако, уровень секреции очень низок или секреция вообще не имеет места, или протеолитический процессинг не верен или незавершен. Настоящие изобретатели полагают, что это может быть связано в какой-то степени с недостаточной доступностью процессингового сайта, расположенного между С-терминальным концом лидирующего пептида и N-терминальным концом гетерологического протеина, так, что он оказывается бесполезным, или, по крайней мере, менее восприимчивым к протеолитическому расщеплению.
Неожиданно было обнаружено, что при осуществлении некоторых модификаций вблизи процессингового сайта в С-терминальном конце лидирующего пептида и/или N-терминального конца гетерологического полипептида, слитого с лидирующим пептидом, возможно получить более высокий выход правильно процессированного протеина, чем в случае с немодифицированными конструкциями, лидирующий пептид - гетерологический полипептид.
Итак, настоящее изобретение относится к полипептиду, включающему слияние сигнального пептида, лидирующего пептида и гетерологического протеина или полипептида, полипептида, который модифицирован в его аминокислотной последовательности, примыкающей к процессинговому сайту дрожжевой клетки, расположенному между С-терминальным концом лидирующего пептида и N-терминальным концом гетерологического протеина таким образом, чтобы обеспечить представление процессингового сайта, в результате которого становится возможным протеолитическое расщепление, к полипептиду, имеющему следующее строение: сигнальный пептид-лидирующий пептид-Х1-X2-X3-Х4 - гетерологический протеин, в котором Х1 - это пептидная связь или Х1 представляет одну или более аминокислот, которые могут быть одинаковыми или различными, Х2 и Х3 одинаковы или отличаются друг от друга и представляют основную аминокислоту, выбранную из группы, состоящей из лизина и аргинина, Х2 и Х3 вместе определяют процессинговый сайт дрожжевой клетки, и X4 - это пептидная связь или Х4 представляет одну или более аминокислот, которые могут быть одинаковы или различны, при условии, что Х1 и Х4 представляют одну или более аминокислот, и, что, по крайней мере, одна из аминокислот, представленных Х1 и/или Х4 - это отрицательно заряженная аминокислота, выбранная из группы, состоящей из глутаминовой и аспарагиновой кислот. В настоящем контексте термин "сигнальный пептид" означает предпоследовательность, преимущественно гидрофобную по природе и присутствующую в виде N-терминальной последовательности на форме предшественнике внеклеточного протеина, экспрессированного в дрожжах. Функция сигнального пептида состоит в том, чтобы обеспечить вход секретируемого гетерологического протеина в эндоплазматический ретикулум. В течение этого процесса сигнальный пептид обычно отщепляется. Сигнальный пептид может быть гетерологическим или гомологическим по отношению к дрожжевому организму, продуцирующему протеин, но, как объяснялось выше, более эффективно отщепление сигнального пептида происходит в том случае, когда он гомологичен к дрожжевому организму, о котором идет речь.
Под выражением "лидирующий пептид" подразумевается преимущественно гидрофильный пептид, функция которого состоит в том, чтобы обеспечить направление секретируемого гетерологического протеина от эндоплазматического ретикулума к аппарату Гольджи и далее в секреторную везикулу для секреции в среду /т.е. в том, чтобы обеспечить прохождение экспрессированного протеина или полипептида через стенку клетки или, по крайней мере, через клеточную мембрану в околоплазменное пространство клетки/.
Выражение "гетерологический протеин или полипептид" выбрано для обозначения протеина или полипептида, который не продуцируется в природе организмом дрожжевой клетки - хозяина. Термин "протеин" и "полипептид" в последующем описании по существу взаимозаменяемы.
Модификация полипептида в процессинговом сайте/сайт-место отделения лидирующего пептида от гетерологического протеина при протеолитическом отщеплении под действием протеолитических ферментов дрожжей/ представлена Х1 и/или Х4 и может быть осуществлена как за счет удлинения, замещения или утраты одной или более аминокислот в С-терминальном конце лидирующего пептида, так и/или в N-терминальном конце гетерологического протеина.
В данном изобретении было обнаружено, что более эффективное и корректное процессирование гетерологического протеина может быть достигнуто в том случае, когда, по крайней мере, одна из аминокислот, участвующих в удлинении последовательности полипептида или которой замещают одну или более из нативных аминокислот последовательности - это отрицательно заряженная аминокислота, например, одна из указанных выше. Аналогичный эффект наблюдается, когда отрицательно заряженную аминокислоту располагают в близости с процессинговым сайтом за счет того, что одну или более аминокислот убирают от C-терминального кольца лидирующего пептида или от N-терминального конца последовательности протеина до тех пор, пока отрицательно заряженная аминокислота не окажется примыкающей к процессинговому сайту.
Не желая ограничиться какой-либо частной теорией, предположили, что этот эффект может быть объяснен возросшей гидрофильностью вызванной отрицательно заряженными аминокислотами, примыкающими к процессинговому сайту, что приводит к увеличению экспонирования третичной структуры полипептида в процессинговом сайте в водном внутриклеточном окружении и вследствие этого становится более восприимчивой к протеолитическому отщеплению под действием процессингового фермента. Преимущественное влияние отрицательно заряженных аминокислот в отличие от положительно заряженных или нейтральных аминокислот может быть отнесено за счет того, что отрицательно заряженная сторона цепочек этих аминокислот увеличивает гидрофильность третичной структуры в процессинговом сайте, никоим образом не увеличивая потенциал ингибирования процессингового фермента.
Возможно также, что отрицательно заряженные аминокислоты содействуют созданию и поддержанию третичной структуры в процессинговом сайте /например, оборотных, петлевых или "шпилечных" структур/ тем или иным способом, например, таким как взаимодействие с другими аминокислотными остатками в полипептиде. Далее, может иметь место также непосредственное взаимодействие отрицательно заряженных аминокислот и процессингового фермента. Таким образом полагают, что отрицательно заряженные аминокислоты, позиционно примыкающие к двум основным аминокислотам процессингового сайта, могут направлять процессинговый фермент таким образом, чтобы осуществить корректное и эффективное расщепление при взаимодействиях заряда протеинов и/или протеина и растворителя.
С другой стороны, настоящее изобретение относится к векторной ДНК, включающей ДНК последовательность, кодирующую полипептид, определенный выше.
Кроме того, данное изобретение относится к рекомбинантному вектору экспрессии, способному реплицироваться в дрожжах и несущему векторную ДНК, кодирующую вышеупомянутый полипептид, а также к дрожжевому штамму, способному экспрессировать гетерологический протеин или полипептид и трансформируемому этим вектором.
Помимо всего, данное изобретение относится к способу получения /продуцирования/ гетерологического протеина или полипептида в дрожжах, включающему культивацию трансформированного дрожжевого штамма в подходящей среде для достижения экспрессии и секреции гетерологического протеина или полипептида, с последующим выделением протеина или полипептида из среды.
Согласно объяснению, данному выше, в тех случаях когда Х1 представляет единственную аминокислоту, то ею является глутаминовая или аспарагиновая кислота. Х1 и/или Х4 соответственно представляют 1-6 аминокислот.
Если Х1 представляет последовательность двух аминокислот, то она может иметь структуру ВА, в которой А - это глу или асп, а В - это глу, асп, вал, гли или лей, например лей,глу.
Если Х1 представляет последовательность трех аминокислот, то она может иметь структуру СВА, в которой А и В определены выше, а С - это асп, глу, про, вал, лей, арг, или лиз, например, асп, лей, глу.
Если Х1 представляет последовательность, состоящую из более чем двух аминокислот, то одна из дополнительных аминокислот может соответственно быть про или гли, поскольку известно, что эти аминокислоты представляют и/или образуют часть витков, "шпилечных" или петлевых структур, возможно облегчающих доступ процессингового сайта к протеолитическому ферменту.
Если Х1 представляет последовательность четырех аминокислот, то она может иметь структуру ДСВА, в которой А, В и С определены выше, а Д имеет те же значения, что и С, например, Х1 - это Глу, Арг, Лей, Глу или Лей, Асп, Лей, Глу или Лиз, Глу, Лей, Глу.
Если Х1 представляет последовательность пяти аминокислот, то она может иметь структуру ЕДСВА, в которой А, В, С и Д определены выше, а Е имеет те же значения, что и С, например, Х1 - это Лей, Глу, Арг, Лей, Глу.
Если X1 представляет последовательность шести аминокислот, то она может иметь структуру ФЕСДВА, в которой А,В, С,Д и Е определены выше, а Ф имеет те же значения, что и С, например, Х1 - это Вал, Лей, Глу, Арг, Лей, Глу.
Нужно иметь ввиду, что возможны и другие последовательности аминокислот, определяющих Х1, в рамках данного изобретения, при условии, что, по крайней мере, одна из аминокислот в последовательности - это отрицательно заряженная аминокислота, как это объяснялось выше.
Соответствующие значения Х4 могут быть теми же, что значения, приведенные выше для Х1, хотя порядок аминокислот в последовательности будет, как правило, обратным /т.е. вместо АВС-СВА и т.д./.
В тех вариантах полипептида настоящего изобретения, в которых гетерологический протеин инициирован одной или более положительно заряженными аминокислотами или гидрофобными аминокислотами, Х4 преимущественно представляет одной или более аминокислот, а не пептидную связь, так как полагают, что в этом случае достигается большая доступность процессингового сайта для протеолитичесиих ферментов.
В тех случаях, когда Х4 представляет N-терминальное удлинение из 1-6 аминокислот, он может соответственно обеспечить дополнительный процессинговый сайт между аминокислотой или кислотами, представленными X4 и N-терминальным концом гетерологического протеина. Это, в частности, важно тогда, когда протеин должен использоваться для целей, в которых необходимо присутствие протеина, не имеющего N-терминального удлинения /удлиняющего сегмента/. Дополнительные N-терминальные аминокислоты могут быть удалены in vitro при противолитическом отщеплении в присутствии соответствующего протеолитического фермента, например трипсин-подобной протеазы, или путем химической обработки, например, бромистым цианом. Возможно также осуществление отщепления у дополнительного процессингового сайта организма дрожжевой клетки хозяина за счет выбора процессингового сайта, специфичного для другого протеолитического фермента дрожжей.
В некоторых случаях, однако, возможно лучше сформировать N-терминальный удлиненный сегмент для решения специфической задачи. Так, удлинение может служить маркером, определяющим протеин, может способствовать очистке протеина или может служить средством, позволяющим осуществлять контроль за действием in vivo фармацевтического препарата, например пролонгировать полупериод существования лекарства в организме или направлять лекарственное вещество в конкретное место в теле.
В предпочтительном варианте полипептида настоящего изобретения Х1 и/или Х4 представляют аминокислотную последовательность их 1-4 аминокислот. В таком варианте аминокислота, непосредственно примыкающая к X2 - это предпочтительно глутаминовая или аспарагиновая кислота, аминокислота, непосредственно примыкающая к Х3 - это предпочтительно глу или асп, или обе вместе, так как в этом случае обеспечивается предпочтительное представление процессингового сайта, обусловленное гидрофильной природой этих аминокислот, как уже детально объяснялось выше. Аминокислотная последовательность, представленная Х1 и Х4, может соответственно включать более одной Глу или Асп.
В другом, представляющем интерес, варианте полипептида настоящего изобретения Х1 и Х4 обе представляют одну или более аминокислот, другими словами, полипептид модифицирован как у С-терминального конца лидирующего пептида, так и у N-терминального конца гетерологического протеина. В этом варианте Х1 и Х4 могут быть симметрично идентичны, это значит, что аминокислота или кислоты, представленные Х1 и Х4, одинаковы и удлиняются в направлении от Х2 и Х3 соответственно.
Последовательность сигнального пептида полипептида настоящего изобретения может быть любым сигнальным пептидом, который обеспечивает эффективное направление экспрессированного полипептида в секреторный канал /путь/ клетки. Сигнальный пептид может представлять сигнальный пептид естественного происхождения или быть его частью, или он может быть синтетическим пептидом. Обнаружено, что соответствующие сигнальные пептиды представляют α - фактор сигнального пептида, например сигнальный пептидамилазы слюны мыши, модифицированный сигнальный пептид карбоксипептидазы или сигнальный пептид дрожжей BARI. Сигнальная последовательность амилазы слюны мыши описана в работе O. Hagenbuchee и др. Nature 2891981, 643-646. Сигнальная последовательность карбоксипептазы описана в работе L.A. Valls и др., Cell 48, 1987, стр.887-897. Сигнальный пептид BARI раскрыт в WO 87/02670.
Лидирующим пептидом полипептида настоящего изобретения может быть любой лидирующий пептид, функцией которого
является направление экспрессированного полипептида в эндоплазматический ретикулум и далее
по секреторному пути. Возможные лидирующие последовательности, которые подходят для этой цели, это природные
лидирующие пептиды, выделяемые из дрожжей или других организмов, например, лидер α
- фактора или его функционального аналога. Лидирующий пептид может быть также синтетическим лидирующим
пептидом, например, один из синтетических лидирующих пептидов раскрыт в Международной патентной
заявке, публикация WO 89/02463. Ниже приведены аминокислотные последовательности:
А.
Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Gly-Phe-Leu-Asp-Leu-Ala-Gly-Glu-Glu-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala
В.
Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala
С.
Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala
D.
Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala-Asn-Val-Ala-Met-Ala and
Е.
Gln-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala-Asn-Val-Ala-Met-Ala
и производные на их основе.
Гетерологический протеин, продуцированный по методу настоящего изобретения, может быть любым протеином, преимущественно продуцируемым в дрожжах. Примерами таких протеинов являются апротинин или любые другие ингибиторы протеазы, инсулин/ включая предшественники инсулина/, гормоны роста человека или бычьи, интерлейкин, глюкагон, плазминогенный активатор ткани, фактор УП, фактор VIII, фактор ХIII, фактор роста из тромбоцитов, ферменты и т.д. или их функциональные аналоги. В настоящем контексте термин" функциональный аналог" означает полипетид с такой же функцией, как у нативного протеида /под функцией следует понимать природу действия, а не уровень биологической активности нативного протеина/. Такой полипептид может иметь такой же аминокислотный состав, что и у нативного протеина, а может быть получен из нативного протеина путем добавления одной или более аминокислот как к одному концу, так и к обоим к С-терминальному и N-терминальному концам нативного протеина, замещением одной или более аминокислоты в одном или ряде различных мест/сайтов/ в нативной аминокислотной последовательности, путем делении /вычитания, утраты/ одной или более аминокислоты с одного или с обоих концов нативного протеина или из одного или различных сайтов в аминокислотной последовательности, или путем включения в один или более сайтов в нативной аминокислотной последовательности одной или более аминокислот. Также модификации хорошо известны для различных протеинов, которые были упомянуты выше.
В соответствии с настоящим изобретением было установлено, что модификации у С-терминального конца лидирующего пептида или у N-терминального конца гетерологического протеина согласно вышеприведенным описаниям позволяют получить с высокими выходами корректно процессированный апротинин/ или его функциональный аналог, в соответствии с вышеприведенным определением/. Апротинин - это протеин, ингибирующий протеазу, свойства которого дают возможность использовать его при лечении ряда заболеваний /например, при лечении панкреатитов, синдрома септического шока: гиперфибринолитического кровотечения и инфарктов миокарда/. Введение апротинина в больших дозах резко снижает потери крови при операциях на сердце или других крупных операциях. Апротинин также полезен в качестве добавки, вводимой в культурную среду, поскольку он ингибирует протеазы клеток-хозяев, которые, в свою очередь, могут вызывать нежелательное отщепление экспрессированных протеинов. При использовании в качестве лидирующего пептида при синтезе в дрожжах корректно процессированного протинина имелись трудности, связанные с получением высокого выхода продукта, известных природных лидирующих последовательностей, например, α - факторного лидера. Нативный апротинин зарождается у N-терминала основной аминокислотой - аргинином /арг/, и, в связи с этим, предшествующий процессинговый сайт может быть менее доступен /подвержен/ для протеолитического расщепления /как объяснено выше/, когда один из известных лидирующих пептидов /например, α - факторный лидер/ используют без того, чтобы модифицировать согласно настоящему изобретению, что приводит к низким выходам корректно процессированного апротинина.
Согласно настоящему изобретению особенно хорошие результаты при продуцировании апротинина достигаются тогда, когда Х1 представляет Глу, лей или Глу, лей, асп, лей /см. нижеследующие примеры/, хотя можно ожидать приличных выходов апротинина и при других значениях Х1 и Х4, при условии, что, по крайней мере, одна аминокислота, а наиболее предпочтителен вариант, когда ближайшая к процессинговому сайту аминокислота является отрицательно заряженной аминокислотой, как объяснено выше.
Также в настоящем изобретении установлено, что модификации у С-терминального конца лидирующего пептида или у N-терминального конца гетерологического протеина в соответствии с вышеприведенным описанием позволяют получать с высоким выходом корректно процессированный предшественник инсулина /или функциональный аналог на его основе/. В этом варианте осуществления изобретения, если Х1 представляет глу-арг-лей-глу или лиз-глу-лей-глу, то Х4 представляет пептидную связь Альтернативно, когда Х4 представляет глу, то Х1 обычно представляет пептидную связь. В частности, полезные результаты были получены согласно настоящему изобретению при экспрессии предшественника аналога инсулина В/1-29/ала-ала-лиз-А/1-29/, когда Х1 представляет лиз-глу-лей-глу, или, когда Х4 представляет предшественник инсулина, в котором первая аминокислота-фенилаланин /фен/ замещена на глутаминовую кислоту /глу/ /см. следующие далее примеры/. Вполне приличные выходы предшественника аналога инсулина В/1-29/ сер-асп-асп-ала-лиз -А/1-29/ были получены, когда Х1 представлял лиз-глу лей-глу. Кроме того, ожидают, что высокие выходы предшественника инсулина могут быть получены и при других значениях Х1 и Х4 при условии, что, по крайней мере, одна аминокислота, а наиболее предпочтителен вариант, когда ближайшая к процессинговому сайту аминокислота является отрицательно заряженной аминокислотой.
Векторная кислота настоящего изобретения, кодирующая полипептид настоящего изобретения, может быть получена синтетическим путем с использованием традиционных методов синтеза, например, фосфоамидитного метода, описанного в работе S. L. Beaucage и M.N. Caruthers, Tetrahedron Letters 22, 1981, стр. 1859-1869 или согласно методу, описанному Matthes и др., ЕМВО Journal 3,1984, стр. 801-805. Согласно фосфоамидитному способу синтезируют олигонулкеотиды, например, в автоматическом ДНК синтезаторе, очищают, удваивают и лигируют, получая синтетическую векторную ДНК.
Векторная ДНК настоящего изобретения может быть также геномного или кДНК происхождения, например, получена при создании банка /библиотеки/ геномных или кольцевых ДНК и скрининга последовательностей ДНК, кодирующих весь или часть полипептида данного изобретения, путем гибридизации с использованием проб синтетического олигонуклеотида в соответствии со стандартной техникой /см. Т. Maniatis и др., Molecular Cloning; Alobarotary Manual, Cold Spring Harbor, 1982/. В этом случае последовательность геномной или кДНК, кодирующая сигнальный и лидирующий пептид, может быть соединена с последовательностью геномной или кДНК, кодирующей гетерологический протеин, после чего ДНК последовательность может быть модифицирована у сайта, соответствующего аминокислотной последовательности Х1-Х2-X3-X4 полипептида, например сайт-направленного мутагенеза, использующего синтетические олигонуклеотиды, кодирующие нужную аминокислотную последовательность при гомологической рекомбинации в соответствии с хорошо известными способами.
И, наконец, векторная ДНК может представлять смесь синтетической и геномной, смесь синтетической и кДНК или смесь геномной и кДНК, полученной при ренатурации фрагментов синтетической, геномной или кДНК, фрагментов, соответствующих различным частям полной векторной ДНК в соответствии со стандартными методами. Следовательно, можно представить, что ДНК последовательность, кодирующая гетерологический протеин может быть геномного происхождения, в то время как последовательность, кодирующая лидирующий пептид, может быть получена синтетическим путем.
Предпочтительными векторными ДНК, кодирующими апротинин, являются ДНК, представленные на фиг.4,7,9,11 и 12, или соответствующие из модификации, кодирующие апротинин или его функциональные аналоги. Примерами подходящих модификаций ДНК служат нуклеотидные замены, которые не дают возможности возникнуть другой аминокислотной последовательности протеина, но которые могут соответствовать использованию кодона организма дрожжей, в который проведена вставка векторной ДНК или нуклеотидных замен, которые могут привести к возникновению отличной аминокислотной последовательности и, следовательно, возможно, к отличной структуре протеина, однако, без ослабления антипротеазных свойств нативного протеина. Другими примерами возможных модификаций являются вставка одного или более нуклеотидов в последовательность, дополнение одного или более нуклеотидов у одного и другого конца последовательности и деления одного или более нуклеотидов у концов последовательности или внутри последовательности. Примерами конкретных аналогов апротинина являются те, что приведены в Европейской патентной заявке, публикация 339942.
Предпочтительные векторные ДНК, кодирующие предшественников инсулина, приведены на фиг. 16 и 17, здесь же даны соответствующие им модификации.
Рекомбинантный вектор экспрессии, несущий ДНК последовательность, кодирующую полипептид настоящего изобретения, может представлять любой вектор, который способен реплицироваться в организмах дрожжей. В векторе ДНК последовательность, кодирующая полипептид настоящего изобретения, должна быть связана с подходящим промотором /стимулятором/ последовательности. Стимулятор может быть любой последовательностью ДНК, проявляющей транскрипционную активность в дрожжах и может быть выделен из генов, кодирующих протеины, как гомологических, так и гетерологических дрожжам. Предпочтительно выделение стимулятора из гена, кодирующего протеин, гомологический дрожжам. Примерами соответствующих стимуляторов служат Mα 1, TPI, АДН или PGК стимуляторы Saccharomyces cerevisiae.
ДНК последовательность настоящего изобретения, кодирующая полипептид настоящего изобретения, должна быть также оперативно связана с соответствующим терминатором, например ТР1 терминатором /см. T. Alber и Kawasaki, J. Moll Appl. Genet 1, 1982, стр.419-434/.
Рекомбинантный вектор экспрессии данного изобретения кроме того, включает ДНК последовательность, дающую возможность вектору реплицировать в дрожжах. Примеры таких последовательностей - это дрожжевая плазмида 2μ, репликация генов REP 1-3 и источник репликации. Вектор может также включать селективный маркер, например ТРI ген, Schizosaccharomyces pombe, как это описано P.R. Russel Gene 40, 1985, стр.125-130.
Процедуры, используемые для легирования /сшивания/ ДНК последовательностей, кодирующих полипептид данного изобретения, стимулятора и терминатора соответственно и для того, чтобы осуществить их вставки в подходящие дрожжевые векторы, содержащие информацию, необходимую для репликации дрожжей, хорошо известны специалистам в данной области /см., например, Maniatis и др. /. Следует понять, что вектор может быть сконструирован либо путем получения вначале векторной ДНК, содержащей полную ДНК последовательность, кодирующую полипептид настоящего изобретения и последующей вставкой этого фрагмента в соответствующий экспрессионный вектор, либо путем последовательной вставки ДНК фрагментов, содержащих генетическую информацию для индивидуальных элементов /таких как сигнальный, лидирующий или гетерологический протеин/ с последующим их лигированием.
Дрожжевой организм, использованный в процессе данного изобретения, может быть любым подходящим дрожжевым организмом, который, при культивировании, продуцирует большие количества гетерологического протеина или полипептида. Примерами подходящих дрожжевых организмов могут служить штаммы дрожжей вида Saccharomyces cerevisiae, Saccharomyces kluyveri, Schizosaccharomyces pombe, или Saccharomyces uvarum.
Трансформация дрожжевых клеток может быть осуществлена, например, при формировании протопласта /см. пример 1/ с последующей трансформацией per se. Среда, используемая для культивации клеток - это любая традиционная среда, подходящая для выращивания дрожжевых организмов. Секретированный гетерологический протеин значительная часть которого может присутствовать в среде в корректно процессированной форме, может быть выделен из среды стандартными методами, включая центрифугирование и фильтрацию для отделения дрожжевых клеток от среды, осаждение протеинообразных компонентов из супернатантного слоя или фильтрата добавлением соли, например сульфата аммония, последующую очистку с использованием различных хроматографических методов, например ионно-обменной хроматографии, афинной хроматографии и тому подобных методов.
Кроме вышеперечисленного, данное изобретение относится также к новому аналогу апротинина общей формулы Х4-апротинин /1-58/, в которой Х4 представляет N-терминальное удлинение одной или более аминокислотами, по крайней мере, одна из которых - это отрицательно заряженная аминокислота, выбранная из группы, состоящей из глутаминовой /глу/ и аспаргиновой /асп/ кислот. Х4 может представлять последовательность из 1-6 аминокислот, в частности 1-4 аминокислот, и может иметь значения, приведенные выше. Особенно предпочтительные значения Х4 - это глу-лей и глу-лей-асп-лей.
Далее изобретение раскрывается в следующих примерах и прилагаемых чертежах.
Фиг. 1 - ДНК и аминокислотная последовательность апротинина /1-58/. Зигзагообразные линии, изображенные на ДНК-последовательности, означают сайты, у которых осуществлялось сшивание /лигирование/ дуплексов, образованных из синтетических олигонуклеотидов.
Фиг.2 - схема конструирования плазмид pKFN-802 и pKFN-803.
Фиг.3 - схема конструирования плазмид pKFN-849 и рКFN-855.
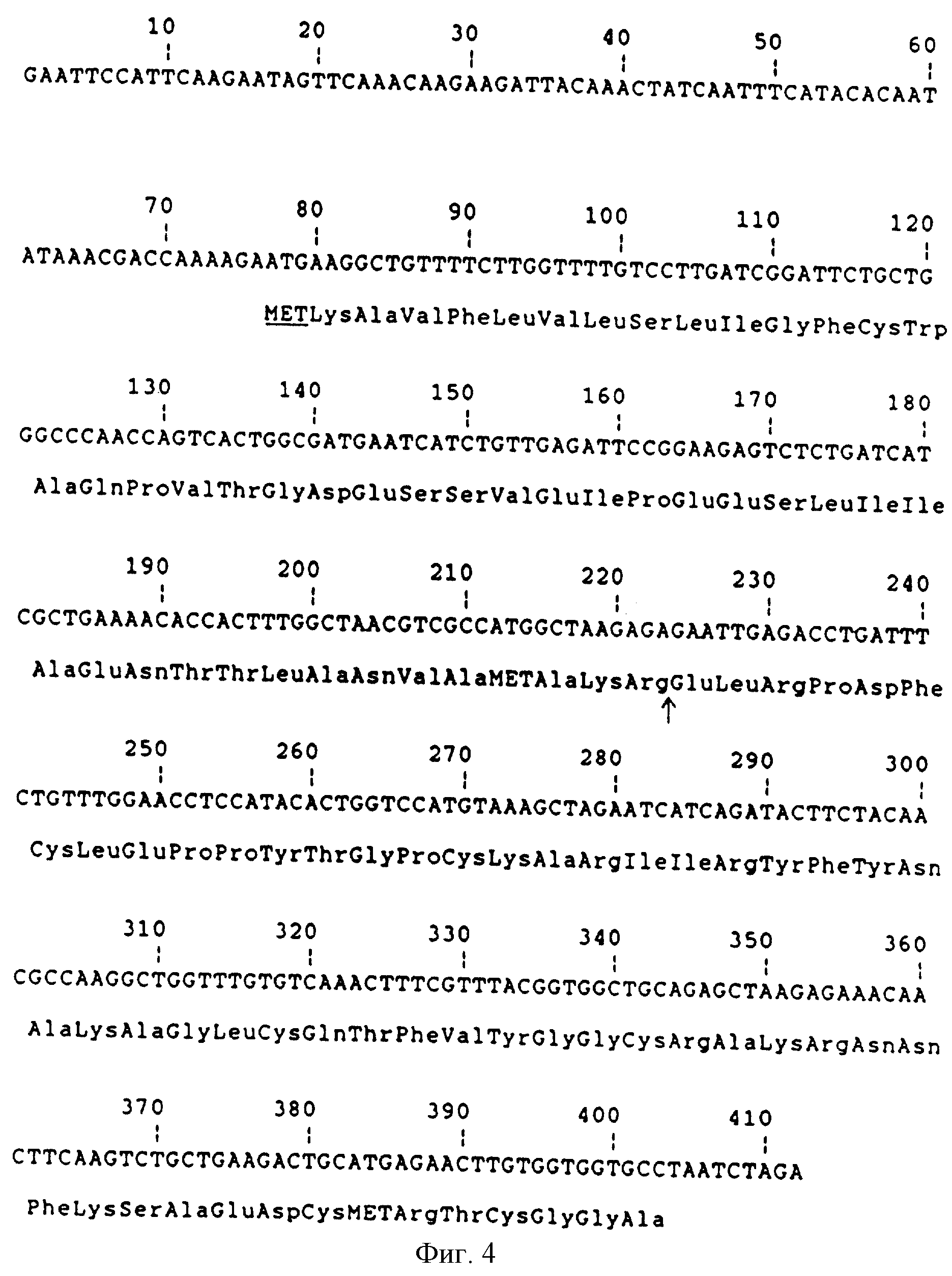
Фиг. 4 - ДНК последовательность фрагмента bpEcoRI-XbaI из pKFN-849 и pKFN-855. Стрелка отмечает сайт, у которого происходит протеолитическое отщепление во время секреции.
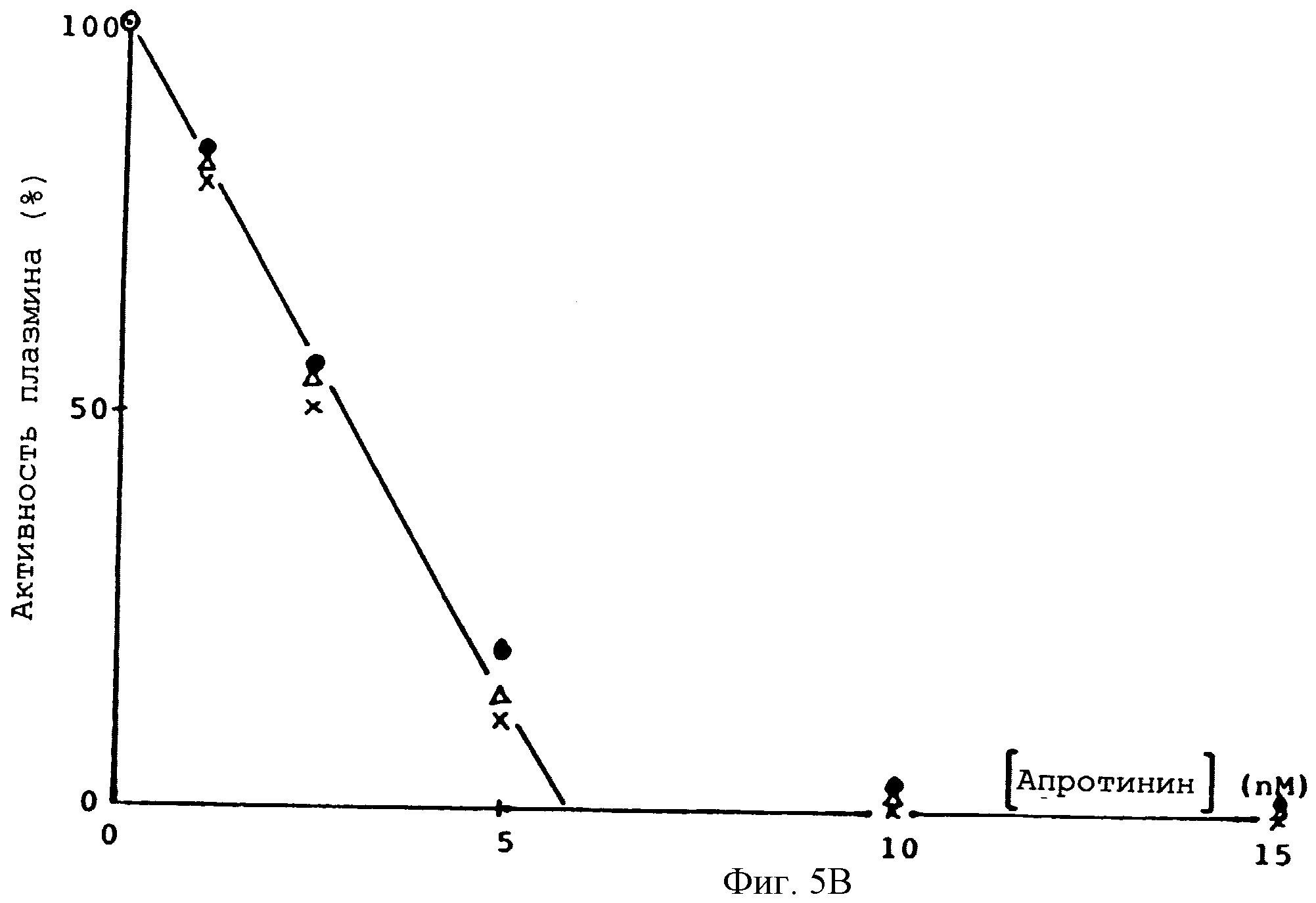
Фиг. 5А и 5В - зависимости ингибирования трипсина и плазмина, соответственно, апротинином, • - бычий апротинин из поджелудочной железы, x - глу-лей-протинин,

Фиг.6 - конструирование плазмид pKFN-852 и pKFN-858.
Фиг.7 - ДНК последовательность фрагмента 412 bр ЕсоRI - ХbаI из pKFN-852 и рKFN-858. Стрелка отмечает сайт, у которого происходит протеолитическое отщепление во время секреции.
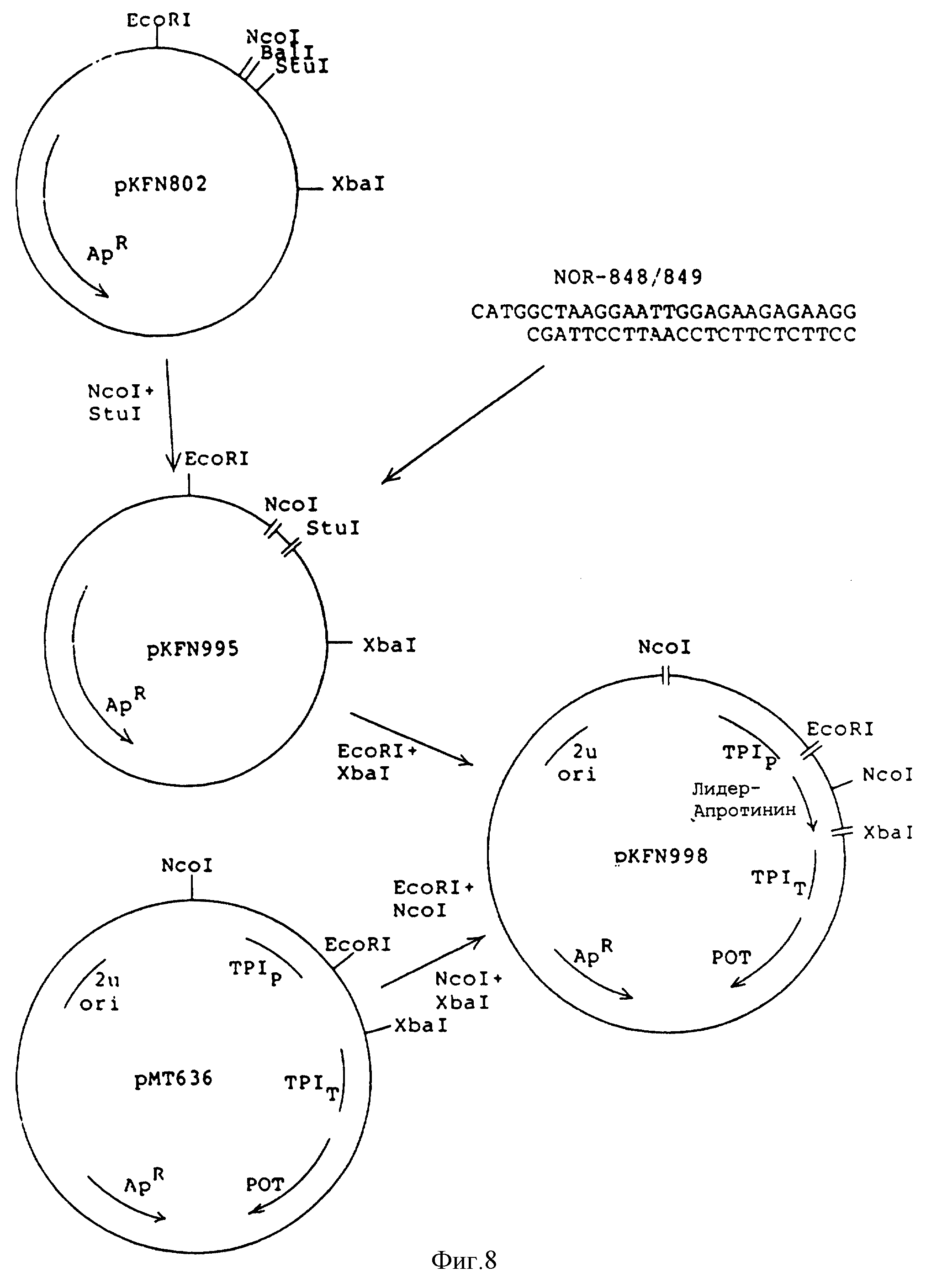
Фиг.8 - конструирование плазмид pKFN-995 и pKFN-998.
Фиг.9 - ДНК последовательность фрагмента 412 bр EcoRI-XbaI из pKFN-995 и рKFN-998. Стрелка отмечает сайт протеолитического отщепления во время секреции.
Фиг.10 - конструирование плазмид рKFN-1000 и pKFN-1003.
Фиг. 11 - ДНК последовательность фрагмента 412 bpEcoRI - XbaI из pKFN-1000 и рKFN-1003. Стрелка отмечает сайт, у которого происходит протеолитическое отщепление во время секреции.
Фиг. 12 - ДНК последовательность гена синтетического апротинина /3-58/. Зигзагообразные линии внутри последовательности указывают сайты, у которых лигируются пять дуплексов, сформированных 10 синтезированными олигонуклеотидами.
Фиг.13 - конструирование плазмид pKFN-305 и рKFN-374/375.
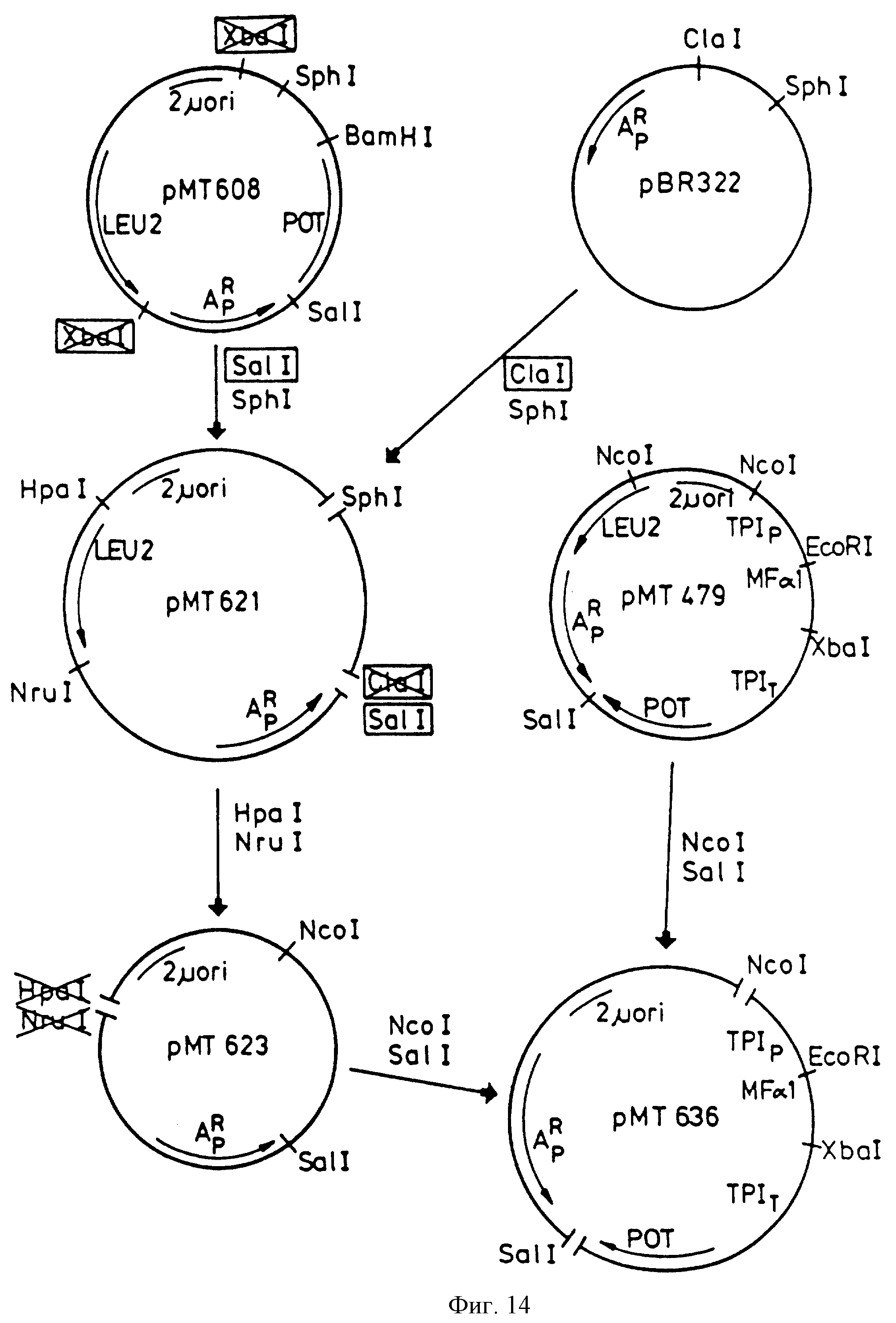
Фиг.14 - конструирование плазмиды рМТ-636.
Фиг.15 - конструирование плазмиды рLаС-240.
Фиг. 16 - ДНК последовательность гена модифицированного предшественника лидер-инсулина из рLаС-240.
Фиг.17 - ДНК последовательность фрагмента 508 bpEcoRI- XbaI из pKFN-458, кодирующая MFα1-сигнал-лидер /1-85/ и предшественник аналога инсулина В/1-29, 1Глу+27 Глу/ -Ала-Ала-Лиз -А/1-21/.
Пример 1.
Продуцирование Глу-лей-апротинин
/1-58/ из дрожжевого штамма KFN-837
а) Конструирование плазмида pKFN-802.
Легированием /сшиванием/ 10 олигонуклеотидов сконструировали синтетический ген, кодирующий апротинин /1-58/.
Олигонуклеотиды синтезировали в автоматическом ДНК синтезаторе на стеклянной пористой подложке с применением фосфоамидитной химии /Beaucage S.L., Caruthers, M. N., Tetrahedron Letters 22/1981/1859-1869. Были синтезированы следующие 10 олигонуклеотидов.
NOR-760: CATGGCCAAAAGAAGGCCTGATTTCTGTTTGGAACCTCCATACACTGGTCC
NOR-754:
TTACATGGACCAGTGTATGGAGGTTCCAAACAGAAATCAGGCCTTCTTTTGGC
NOR-354: ATGTAAAGCTAGAATCATCAGATACTTCTACAACG
NOR-355: CTTGGCGTTGTAGAAGTATCTGATGATTCTAGCT
NOR-356:
CCAAGGCTGGTTTGTGTCAAACTTTCGTTTACGGTGGCT
NOR-357: CTCTGCAGCCACCGTAAACGAAAGTTTGACACAAACCAGC
NOR-358:
GCAGAGCTAAGAGAAACAACTTCAAGT
NOR-359: AGCAGACTTGAAGTTGTTTCTCTTAG
NOR-360: CTGCTGAAGACTGCATGAGAACTTGTGGTGGTGCCTAAT
NOR-361: CTAGATTAGGCACCACCACAAGTTCTCATGCAGTCTTC
5
дуплексов были сформированы из 10 вышеприведенных олигонуклеотидов в соответствии с
фиг.1.
20 пикомолей каждого дуплекса А-Е были сформированы из соответствующих пар 5'-фосфорилированных олигонуклеотидов при нагревании в течение 5 мин при 90oС, с последующим охлаждением до комнатной температуры в течение 75 мин. 5 дуплексов смешали и обработали Т4 ДНК лигазой. Синтезированный ген изолировали после электрофореза сшитой смеси на геле 2% агарозы /окрашенная полоса 191bp/. Полученный синтезированный ген представлен на фиг.1.
Синтезированный ген сшили с фрагментом 209 bp ЕсоRI-NсоI из pLaC212spx3 и с 2.7kb EcoRI - XbaI фрагментом плазмиды рИ с19 /Yanish Perren, C., Vieira, J., Messing J., Gene 33 (1985) 103-119/. Плазмид pLaC212spx3 описан в примере 3 международной патентной заявки, публикация WO/89/02483.
Фрагмент 209 bp EcoRI-NcoI из pLaC212spx3 кодирует синтетический лидирующий пептид дрожжей.
Сшитая смесь была использована для трансформации компетента Е. соli штамма r-, m+, выбранного по устойчивости к ампициллину. Определение первичной структуры /секвенирование/ фрагмента 32p-XbaI-EcoRI /Maxam. Gilbert, W. , Methods Enzymel 65/1980/, 499-560/, показало, что плазмиды из полученных колоний содержали корректную ДНК последовательность для апротинина /1-58/.
Один плазмид рKFN-802 был отобран для дальнейшего использования. Конструирование плазмида pKFN-802 приведено на фиг.2.
б) Конструирование плазмидов pKFN-849, рKFN-855 и дрожжевого штамма KFN-837.
3.0 kbNcoI-StuI
фрагмента рKFN-802 сшили с синтезированным фрагментом NOR-790/791, использовав Т4 ДНК лигазу:

Сшитую смесь гидролизовали с помощью рестрикционного фермента StuI для того, чтобы снизить фон pKFN-802, и полученную смесь использовали для трансформации компетента штамма (r-, m+) E.соli выбранного по устойчивости к амцициллину. Согласно /Sanger, F., Micklen, Coulson, A.R., Proc. Natl. Acad. Sci. USA 74(1977) 5463-5467/ была определена первичная структура ДНК плазмида рKFN-849 из одной из полученных колоний для того, чтобы осуществить корректную вставку ДНК последовательности для Глу-лей-апротинина /1-58/ в синтезированный лидерный ген дрожжей. Конструирование плазмида рKFN-849 проиллюстрировано на фиг.3.
pKFN-849 был разрезан EcoRI и ХbaI и фрагмент 406bp был сшит с 9,5 kbNcoI-XbaI фрагментом из рМТ636 и 1,4 KbNcoI-EcoRI фрагментом из рМТ636, в результате получили плазмид pKFN-855, см. фиг.3. Плазмид рМТ636 описан в международной патентной заявке РСТ/ ДК88/00138.
рМТ636 - это Е.соli - S.cerevisiae челночный вектор, содержащий Schizosacharomycer pombe ТРI ген/РОТ/ Russel, P.R., Gene 40 (1985 ) 125-130/, стимулятор и терминатор S. cerevisiae триосефосфатизомеразы, ТРIP и ТРIT /Alber T., Kawasaki J. Mol. Appl. Gen. 1 /1982/, 419-434/. Плазмид pKFN-855 содержит следующую последовательность: ТРIp - LаС212sрх3 сигнал-лидер-глу-лей-апротинин/1-58/-ТРIT, в которой LaC212spx3 сигнал-лидер - это синтетический дрожжевой лидер, описанный в международной патентной заявке, публикация WO 89/02463. Последовательность ДНК фрагмента 406 bрEcoRI-XbaI из pKFN-849 и pKFN-855 приведена на фиг.4.
Штамм МТ663 S. cerevisiae /E2-7B XEII-36 a/α, Δtpi Δtpi, pep 4-3/ рер4-3/ вырастили на УРУaL /1% Bacto дрожжевой экстракт, 2% Bacto пептон, 2% галактоза, 1% лактоза/, оптическая плотность при 600 нм равна 0,6. 100 мл культуры собрали путем центрифугирования, промыли 10 мл воды, рецентрифугировали и ресуспендировали в 10 мл раствора, содержащего 1,2М сорбитол, 25 мМ Na2 ЭДТА рН=8,0 и 6,7 мг/мл дитиотрейтола. Суспензию инкубировали при 30o в течение 15 мин, центрифугировали и клетки ресуспендировали в 10 мл раствора, содержащего 1,2 М сорбитола, 10 мМ Na2 ЭДТА, 0,1 М цитрата натрия, рН= 5,8 и 2 мг Vovozym®234. Суспензию инкубировали при 30oС в течение 30 мин, клетки собрали путем центрифугирования, промыли в 10 мл 1,2 М сорбитола и 10 мл САS /1,2 М сорбитол, 10 мМ СаСl2, 10 мМ ТрисНСl/ Трис=трис /гидроксиметил/ аминометан/, рН= 7,5/ и ресуспендировали в 2 мл СаS. Для трансформации 0,1 мл ресуспендированных в САS клеток смешали с примерно 1 мкг плазмида pKFN-855 и оставили при комнатной температуре на 15 мин. 1 мл /20% полиэтиленгликоль 4000, 20 мМ СаСl2 , 10 мМ СаСl2, 10 мМ Трис НСl, рН= 7,5/ добавили и смесь оставили при комнатной температуре еще на 30 мин. Смесь центрифугировали и образовавшиеся катышки ресуспендировали в 0,1 мл SOS /1,2 М сорбитол, 33% об./об. УPD 6,7 мМ СаСl2, 14 мг/мл лейкина/ и инкубировали в течение 2 часов при 30oС. Суспензию затем центрифугировали и катышки ресуспендировали в 0,5 мл 1,2 М сорбитола. Затем при 52oС добавили 6 мл top агара /SC среда Sherman'a/ Methods in Yeast Genetics, Cold Spring Harbor Laboratory /1981//, содержащего 1,2 М сорбитола + 2,5% агара/ и суспензию вылили на пластинки, заполненные тем же затвердевшим агаром с сорбитолсодержащей средой. Трансформированные колонии были отобраны после выдержки в течение 3 дней при 30oС, реизолированы и использованы для выращивания суспензионных культур. Один такой трансформант KFN-837 был выбран для последующего охарактеризования.
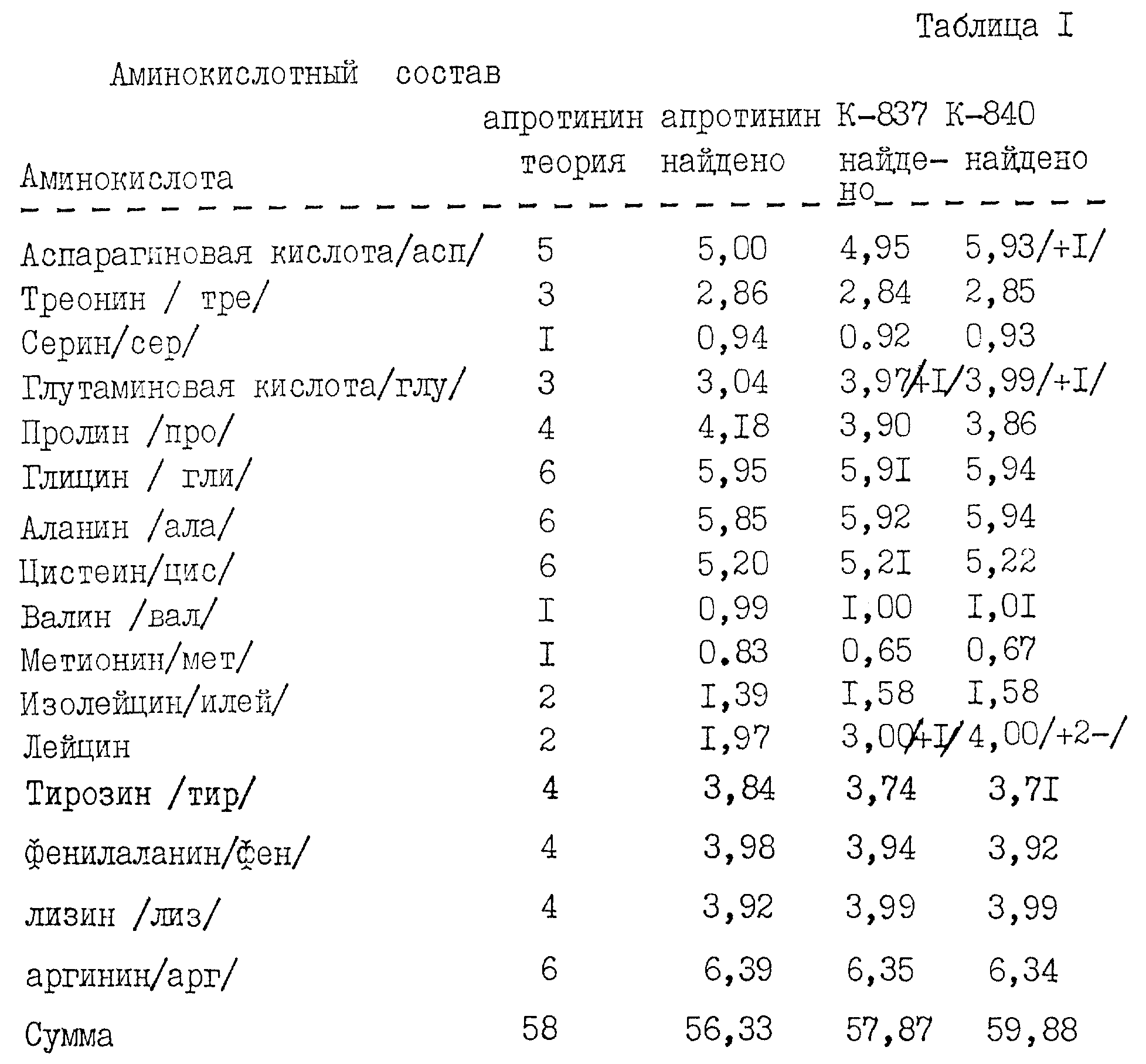
Дрожжевой штамм KFN-837 вырастили на YPD-среде /1% дрожжевой экстракт, 2% пептон /Difco Laboratories/ и 6% глюкоза/, 200 мл культуры штамма встряхивали при 30oС при 250 об/мин в течение 3 дней до оптической плотности при 600 нм равной 20 (сухая биомасса дрожжей, 18,8 г/л). После центрифугирования супернатантный слой проанализировали методом ионно-обменной хроматографии FPLC /жидкостная экспресс-хроматография белков/. Дрожжевой супернатант профильтровали через Millex GV фильтр с размером пор 0, 22 мкм, и 1 мл перенесли в ионно-обменную колонку Monos/ 0,5х5 см/, уравновешенную 20 мМ Bicine, рН= 8,7. После промывки равновесным буффером колонку проэлюировали раствором с линейным градиентом концентрации NaCl /0-1 М/ в равновесном буфере. Ингибирующая активность элюированных фракций была определена по отношению к трипсину спектрофотометрическим методом /Kassel, В., Меthods Enzymol 19, 1970 /844-852/ и интегрированием поглощения при 280 нм Е2801% /апротинин/=8,3.
Выход составил 120 мг /л глу-лей-апротинина /1-58/.
Для проведения аминокислотного анализа и определения первичной структуры N-терминала провели концентрирование и последующую очистку градиента элюата /глу-лей-апротинин/ 1-58 // методом ВЭЖХ на колонке с реверсивной фазой /Vydac C4, 4,6 х 250 мм/. Элюирование проводили в градиенте CH3CN в 0,1% ТFA. Собранные фракции сконцентрировали примерно до 100 мкл методом вакуумного центрифугирования, и далее были отобраны образцы для определения первичной структуры N-терминала и аминокислотного анализа.
Секвенирование N-терминала позволило установить следующую
последовательность с
N-конца: глу-лей-арг-про-асп-фен-Х-лей-глу-про-про-тир-тре-гли-про-Х-лиз-ала-арг-илей-илей-арг-тир-фен-тир-асп-N-ала-лиз-ала, подтверждающую корректность N-терминального конца.
Половина цистеиновых
остатков не определяется этим методом, что и отмечено Х, обозначающим остатки 7 и 16. Данные аминокислотного анализа приведены в табл. 1. Из этих данных очевидно, что продукт
имеет ожидаемый
аминокислотный состав, т.е. больше глу и лей. Слегка заниженное содержание илей может в большинстве случаев отнесено за счет неполного
гидролиза илей/18/-илей/19// это хорошо
известно
специалистам/. Также следует отметить слегка завышенное содержание, по сравнению с ожидаемым, аргинина. Однако, как видно из табл. 1, колонка 3, подобное явление характерно и для нативного
апротинина.
При сравнении специфической активности глу-лей-апротинина /1-58/ и специфической активности нативного апротинина, проведенном с использованием упомянутого выше метода Kassel, было установлено, что в пределах ошибки эксперимента они совпадают. Кривые ингибирующего титрования трипсина и плазмина глу-лей-апротинином /1-58/ неотличимы от кривых титрования апротинином из поджелудочной железы быка /Апротинин Novo/. После инкубирования фермента с апротинином или его аналогами в течение 30 мин добавили 0,6 мМ 2251 /Kabivitrum/, измерил активность по скорости образования нитроанилина. Активность, как функция концентрации апротинина, представлена на фиг.5А и 5В. Отмечено полное ингибирование обоих ферментов всеми тремя ингибиторами.
Пример
2
Продуцирование глу-лей-асп-лей-апротинина/1-58/ из штамма дрожжей KFN-840
Синтетический ген, кодирующий глу-лей-асп-лей-апротинин/1-58/ был сконструирован так, как это описано в
примере 1. Синтезированный фрагмент NOR-793/ 794 был использован вместо NОR-790/791:

Плазмид pKFN-852 был сконструирован аналогично плазмиду pKFN-849. Согласно процедуре, описанной в примере 1, был получен плазмид pKFN-858, содержащий следующую конструкцию: ТР1р-LAC212sрх3 сигнал-лидер-глу-лей-асп-лей-апротинин/1-58/-ТРIT.
Конструирование плазмидов pKFN-852 и pKFN-858 проиллюстрировано на фиг. 6, ДНК последовательность фрагмента 412bp ЕcoRI - ХFаI из pKFN-852 и из pKFN-858 приведена на фиг.7.
Плазмид pKFN-858 был трансформирован в дрожжевой штамм МТ663 согласно методике, описанной выше, результатом явился дрожжевой штамм KFN-840.
200 мл культуры KFN-840 в среде УРD встряхивали при 30oС в течение 3 дней со скоростью 250 об/мин до оптической плотности при 600 нм, равной 18 /сухая биомасса 16, 7 мг/л/. После FРLС ионной хроматографии супернатанта получили глу-лей-асп-лей-апротинин /1-58/ с выходом 90 мг/л.
Данные аминокислотного анализа, приведенные в табл. 1, подтверждают ожидаемый аминокислотный состав.
Кривые ингибирующего титрования трипсина и плазмина глу-лей-асп-лей-апротинином /1-58/ неотличимы от кривой титрования апротинином из поджелудочной железы быка. /Апротинин Novo/ см. фиг.5А и 5В.
Пример 3
Продуцирование апротинина/1-58/ из штамма дрожжей KFN-1006
Плазмид pKFN-995 сконструирован из pKFN-802 путем
сшивания 3,0 кb NcoI-StuI фрагмента с синтезированным фрагментом NOR-848/849

Сшитая смесь была гидролизована с помощью рестрикционного фермента BaII с целью снижения фона pKFN-802.
Дрожжевой экспрессионный плазмид pKFN-998 сконструирован в основном как описано в примере 1 и показано на фиг.8: ТРIP-LaС212sрх3 сигнал-лидер/1-47/ лиз-глу-лей-глу-/лиз-арг/-апротинин /1-58/-TPIT.
ДНК последовательность фрагмента 412 bp EcoRI-XbaI- из pKFN-995 и из pKFN-998 приведена на фиг.9.
Плазмид pKFN-998 был трансформирован в дрожжевой штамм МТ663 в соответствии с описанием примера 1, в результате получили штамм KFN-1006. Культивирование штамма KFN 1006 в УРD-среде и анализ супернатанта на апротинин был проведен в соответствии с приведенным выше описанием.
Выход апротинина /1-58/ составил 30 мг/л.
Аминокислотный анализ очищенного материала подтвердил ожидаемый аминокислотный состав.
Пример 4
Продуцирование апротинина /1-58/ из дрожжевого штамма
KFN-1008.
PUС - полученный плазмид pKFN-1000 был сконструирован в соответствии с описанием примера 1 путем сшивания 3.0 KbNcoI-StuI фрагмента pKFN-802 и синтетического фрагмента
NOR-850/851:

Следуя процедуре, описанной в примерах 1 и 3, был получен дрожжевой экспрессионный плазмид pKFN-1003, содержащий следующую конструкцию TPIP-LAC212spx3 сигнал-лидер/1-47/ - глу-арг-лей-глу/Лиз-Арг/ апротинин /1-58/-TPIT.
Конструирование плазмид pKFN-1000 и pKFN-1003 проиллюстрировано на фиг. 10. ДНК последовательность 412bp EcoRI -Xbal фрагмента из pKFN-1000 и pKFN-1003 приведено на фиг.11.
Плазмид pKFN-1003 был трансформирован в дрожжевой штамм МТ663, как описано выше, в результате получили дрожжевой штамм KFN-1008.
Выращивание трансформированного штамма KFN-1008 в УРD-среде и анализ на апротинин /1-58/ супернатанта проведен в соответствии с проведенным выше описанием. Выход апротинина /1-58/составил 120 мг/л. Аминокислотный анализ подтвердил ожидаемый аминокислотный состав. Кроме того, подтверждение полной первичной структуры было получено путем газофазного секвенирования редуцированного пиридилэтилированного полипептида.
Далее, специфическая ингибирующая активность рекомбинантного апротинина /1-58/ в отношении трипсина совпадает с активностью апротинина, выделенного из поджелудочной железы быка.
Пример 5
Продуцирование апротинина /1-58/ из дрожжевого штамма KFN-783. Плазмид pKFN-802 /см. пример 1/ разрезали с помощью ЕсоRI и Xb aI и фрагмент 400 bр сшили с фрагментом 9,5 кb NcoI-ХbaI из рМТ636 и
фрагментом 1,4 kbNcoI-ЕсoRI из рMT636, в результате получили плазмид pKFN-803, см. фиг.2. pKFN-803 содержит следующую конструкцию: TPIp-LaC212sрх3 сигнал-лидер-апротинин/1-58/-ТРIT.
Плазмид pKFN-803 трансформировали в дрожжевой штамм МТ663 согласно описанию, приведенному выше. Выращивание трансформированного штамма KFN-783 в УРD-среде и FPLС ионная хроматография супернатанта были проведены согласно описанию, приведенному выше. Апротинин /1-58/ количественно охарактеризовали путем сравнения с хроматограммой стандартного раствора аутентичного апротинина, выделенного из бычьей поджелудочной железы. Выход апротинина /1-58/ был ниже 1 мг/л.
Пример 6
Продуцирование апротинина /3-58/
Синтетический ген для
апротинина /3-58/ был сконструирован из ряда нуклеотидов путем их сшивания /лигирования/.
Следующие 10 нуклеотидов были синтезированы в соответствии с описанием, приведенном в
примере
1:
I: AAAGAGATTTCTGTTTGGAACCTCCATACACTGGTCC 37-mer
II: TTACATGGACCAGTGTATGGAGGTTCCAAACAGAAACT 38-mer
III: ATGTAAAGCTAGAATCATCAGATACTTCTACAACG 35-mer
IV:
CTTGGCGTTGTAGAAGTATCTGATGATTCTAGCT 34-mer
V: CCAAGGCTGGTTTGTGTCAAACTTTCGTTTACGGTGGCT 39-mer
VI: CTCTGCAGCCACCGTAAACGAAAGTTTGACACAAACCAGC 40-mer
VII:
GCAGAGCTAAGAGAAACAACTTCAAGT 27-mer
VIII: AGCAGACTTGAAGTTGTTTCTCTTAG 26-mer
IX: CTGCTGAAGACTGCATGAGAACTTGTGGTGGTGCCTAAT 39-mer
X: CTAGATTAGGCACCACCACAAGTTCTCATGCAGTCTTC
38-mer
Пять дуплексов А-Е были сформированы из 10 вышеприведенных олигонуклеотидов, см. фиг.12.
20 пкмоль каждого из дуплексов А-Е было сформировано из соответствующих пар 5'-фосфорилированных олигонуклеотидов I-Х при нагревании в течение 5 мин при 90oС с последующим охлаждением до комнатной температуры в течение 75 мин. Пять дуплексов смешали и обработали Т4 лигазой. После электрофореза сшитой смеси на геле 2% агарозы выделили синтезированный ген /окрашенная полоса 176bp/. Полученный синтетический ген представлен на фиг.12.
Синтетический ген 176bp сшили с 330bp EcoRI-HgaI фрарментом из плазмида pKFN-9, кодирующего последовательность-скрещивающий фактор αI-сигнал-лидер (1-85) S. cerevisiae) /Mar Kussen, J. и др., Protein-Engineering 1/1987/, 215-223/, и с2.7kb EcoRI-XbaI фрагментом из рUC19/ /Janish-Perron, C., Vieira, J. , Messing, J., Gene 33/1985/ 103-119/. Конструкция pKFN-9, у которой HgaI сайт находится сразу после последовательности MFαI лидер описана в ЕП 214826.
Сшитую смесь использовали для трансформирования компетента штамма /r-, m+/ E. coli, отобранного по устойчивостии к ампициллину. Секвенирование32P-XbaI-EcoRI фрагмента /Maxam, A. , Gilbert, W. , Methods Enzymol 65, /1980/499-560/ показало, что плазмиды из выращенных колоний содержали корректную ДНК последовательность для кодирования апротинина /3-58/.
Плазмид pKFN-305 отобрали для дальнейшего использования. Конструкция плазмида pKFN-305 проиллюстрирована на фиг.13.
pKFN-305 разрезали ЕсоRI и XbаI и 0,5kb фрагмент сшили с 9,5kb NcoI-XbaI фрагментом из рМТ636 и 1,4kb NcoI-EcoRI фрагментом из рМТ636, в результате получили плазмид pKFN-734, см. фиг.13. Плазмид рМТ636 сконструировали из рМТ608 после делеции LEU-2 гена и из рМТ479, см. фиг.14. рМТ608 описан в ЕП 195691, рМТ479 описан в ЕП 163529. рМТ479 содержит Schizosaccharomyces pombe ТРI ген/ рот/, промотор/стимулятор/ и терминатор S. cerevisiae триозефосфат изомеразы, TPIP и ТРIT /Alber, T. и Kawasaki, G.J. Mol Appl. Gen 1/1982/, 419-434/.
Плазмид pKFN-734 содержит следующую последовательность:
TPIP-MFαI сигнал лидер-/1-85/-апротинин/3-58/-ТРIT, в которой MFαI
- это
скрещивающий фактор αI S. cerevisiae кодирующий последовательность /Kurjian, J. и Hers-Kowits, J., Cell 30/1982/, 933-943/, сигнал-лидер/1-85/ означает, что последовательность содержит
первые
85 аминокислотных остатка MFαI сигнал-лидер последовательности, и апротинин /3-58/ - это синтетическая последовательность, кодирующая производное апротинина, в котором отсутствуют два
первых
аминокислотных остатка.
Штамм МТ663/Е2-7В ХЕII-36 а) α Δtpi Δtpi pep 4-3/рер 4-3/ S. cerevisiae вырастили на УРGal. /1% Bacto дрожжевой экстракт, 2% Bacto пептон, 2% галактоза, 1% лактат/ до оптической плотности 0,6 при длине волны 600 нм.
Путем центрифугирования отобрали 100 мл выращенной культуры, промыли 10 мл воды, рецентрифугировали и ресуспендировали в 10 мл раствора, содержащего 1,2 М сорбитола, 25 мМ Na2 ЭДТА, рН=8,0 и 6,7 мг/м л дитиотрейтола. Суспензию инкубировали при 30oС в течение 15 мин, центрифугировали и клетки ресуспендировали в 10 мл раствора, содержащего 1,2 М сорбитола, 10 мМ Na2 ЭДТА, 0,1 М цитрата натрия, рН=5,8 и 2 мг Novozym® 234. Суспензию инкубировали при 30oС в течение 30 мин клетки собрали путем центрифугирования, промыли в 10 мл 1,2 М сорбитола и 10 мл САS /1,2 М сорбитол, 10 мл СаСl2 10 мМ трис НСl/Тристри/ гидроксиметил/аминометан, рН=7,5/ и ресуспендировали в 2 мл САS. Для трансформации 0,1 мл клеток, ресуспендированных в САS, смешали с примерно 1 мкг плазмида pKFN-374 и оставили при комнатной температуре на 15 мин. 1 мл /20% полиэтиленгликоль 4000, 10 мМ СаСl2 , 10 мМ трис HCl, рН= 7,5/ добавили, и смесь оставили еще на 30 мин при комнатной температуре. Смесь центрифугировали, и катышки ресуспендировали в 0,1 мл SOS /1,2 М сорбитол, 33% об/об УPD 6,7 мМ СаСl2 14 г/мл лейкина/ и инкубировали при 30oС в течение 2 ч. Суспензию вновь центрифугировали, и катышки ресуспендировали в 0,5 мл 1,2 М сорбитола. Затем 6 мл top агара/SС среда Sherman и др./, содержащая 1,2 М сорбитола плюс 2,5% агар/ при 52oС добавили, и суспензию вылили на пластины, содержащие тот же затвердевший агар с сорбитолсодержащей средой. После выдержки в течение 3 дней при 30oС колонии трансформированных клеток были отобраны, реизолированы и использованы для выращивания суспензионных культур. Один такой трансформант KFN322 был отобран для дальнейшего охарактеризования.
Дрожжевой штамм KFN332 вырастили на УРD-среде /1% дрожжевой экстракт, 2% пептон/, полученный из Difco Laboratories/, и 2% глюкоза/. 10 мл культуры штамма встряхивали при 30oС до достижения оптической плотности 32 при длине волны 600 нм. После центрифугирования супернатант проанализировали методом FLPС ионно-обменной хроматографии. Дрожжевой супернатант профильтровали через фильтр Millex® с размером пор 0,22 мкм, и 1 мл внесли в колонку, заполненную катионообменной смолой Monos /0,5 х 5 см/, уравновешенную 20 мМ Bicine, рН= 8,7. После промывания равновесным буферным раствором содержимое колонки проэлюировали в линейном градиенте NaCl /0-1 М/ в равновесном буфере. Ингибирующую активность элюированных фракций в отношении трипсина определяли спектрофотометрическим методом и интегрированием поглощения при 280 нм от Е2801% /апротинин/=8,3.
Выход апротинина /3-58/ составил примерно 3 мг/л.
Пример 7
Продуцирование
инсулина предшественника В/1-29/-ала-ала лиз-А/1-21/ из дрожжевого штамма LаС1667
589 bp SphI-NcoI и 172 bp HpaI-XbаI фрагменты были выделены из рLаС212spx3 /описание дано в WO 89/02463/.
Эти
фрагменты были соединены с синтетическим адаптером

И С 10 kb XbaI-SphI фрагментом из рМТ743/ описано в WO 89/02463, в результате получили плазмид рLАС240/ см.фиг.15/. ДНК последовательность гена из рLаС240 для модифицированного предшественника лидер-инсулина представлена на фиг.16.
Трансформирование дрожжевого штамма МТ633 плазмидсм рLаС240 привело к образованию штамма LаС1667 (МТ663/ pLaС240), секретирующего предшественник инсулина, продуктивность которого составляет 165%, производительность штамма LАС1414 (МТ663/ рLаC212spx3, содержащего ген для немодифицированного предшественника лидер-инсулина, описанного в WO 9/02463.
Пример 8
Продуцирование предшественника аналога инсулина В/1-29,1 глу + 27 глу/-ала-ала-лиз -А/1-21/ из дрожжевого штамма KFN-470
Синтетический ген, кодирующий предшественник аналога
инсулина
В/1-29, 1 глу + 27 глу / -ала-ала-лиз - А/1-21/ сконструировали путем сшивания 10 олигонуклеотидов. В /1-29/ 1 глу + 27 глу/ означает полипептид, содержащий первые 29 аминокислотных остатков
В-цепи
человеческого инсулина, в которой глу-остатки были замещены на BIPne и В27Тnr остатки. А/1-21/ - это А-цепь человеческого инсулина. Трипептид-ала-ала-лиз -связывает В29 лиз остаток с остатком
АIглу.
Были синтезированы следующие 10 олигонуклеотидов:
NOR-542: AAAGAGAAGTTAACCAACACTTGTGCGGTTCCCAC
NOR-543: AACCAAGTGGGAACCGCACAAGTGTTGGTTAACTTC
NOR-69:
TTGGTTGAAGCTTTGTACTTGGTTTGCGGTGAAAGAGGTTTCT
NOR-73: GTAGAAGAAACCTCTTTCACCGCAAACCAAGTACAAAGCTTC
NOR-315: TCTACGAACCTAAGGCTGCTAAGGGTATTGCT
NOR-316:
ATTGTTCGACAATACCCTTAGCAGCCTTACGTTC
NOR-70: GAACAATGCTGTACCTCCATCTGCTCCTTGTACCAAT
NOR-71: TTTTCCAATTGGTACAAGGAGCAGATGGAGGTACAGC
NOR-78:
TGGAAAACTACTGCAACTAGACGCAGCCCGCAGGCT
NOR-72: CTAGAGCCTGCGGGCTGCGTCTAGTTGCAGTAG
5 дуплексов было сформировано из приведенных выше 10 олигонуклеотидов и эти дуплексы были сшиты таким
же путем, как это было сделано в примере 1.
Синтетический l78bp ген был сшит с 330 bp ЕсоRI-НgаI фрагментом из pKFN-9, кодирующим последовательность скрещивающего фактора альфа 1-сигнал-лидер /1-85/ S. cerevisiae /Markussen, J. и др., Protein Engineering 1/1987/, 215-223/ и с 2.7kb EcoRI-XbаI фрагментом плазмида рUC 19 /Yanish-Perron, C., Vieira, и Messing, J., Gene 33/ 1985/,103-119/.
Сшивание смеси было использовано для трансформирования компетента штамма/r-, m+/ E.coli, отобранного по устойчивости к ампициллину. Секвенирование32P-XbaI-EcoRI фрагмента /Maxam, A. и Gilbert, W. Methods Enzymol 65 /1980/, 499-560/ показало, что плазмиды из полученных колоний содержали корректную ДНК последовательность для кодирования предшественника аналога инсулина. Плазмид pKFN-456 был выбран для дальнейшего использования.
Согласно процедуре примера 1 был получен дрожжевой экспрессионный плазмид, содержащий следующий полигенный экспрессирующий кластер: ТРIP МFαI-сигнал-лидер /1-85/-В /1-29, 1 глу + 27 глу/ ала-ала-лиз-А-/1-21/ - TPIT.
ДНК последовательность 508bp EcoRI-XbaI фрагмента из рKFN-456 приведена на фиг.17.
Плазмид pKFN-458 был трансформирован в дрожжевой штамм МТ-633 в соответствии с описанием, приведенным выше, в результате получили дрожжевой штамм KFN-470.
Выращивание трансформированного штамма провели на УРD среде в соответствии с вышеприведенной методикой. Содержание предшественника аналога инсулина в супернатанте было определено методом ВЭЖХ в соответствии с описанием, приведенным в работе L. Shel и др., Chromatographia 24/1987/, 329-332.
В табл. 2 сравниваются уровни экспрессии предшественников аналога инсулина В/1-29/ 1 глу + 27 глу /-ала-ала-лиз-А/1-21/, В/1-29/ 27 глу/-ала-ала-лиз-А/1-21/ и предшественника инсулина В/1-29/-алаалализ-А/1-21/. Все три предшественника были экспрессированы в одном и том же штамме хозяине МТ-663, трансформированном S. pombe ТРI геном, содержащим плазмиды с полигенным экспрессирующим кластером. TPIP -MFαI сигнал-лидер/1-85/-предшественник-ТРIT.
Уровни экспрессии представлены как процентное отношение к уровню экспрессии предшественника инсулина В/1-29/-ала-ала-лиз -А/1-21/, который принят за 100%.
Пример 9
Получение ЕАЕАЕАK-предшественника инсулина в дрожжевом штамме уАК344.
Синтетическая последовательность ДНК N-концевого удлинения предшественника инсулина B(1-29)SDDAK-A(1-21) (далее называемого Ml5) был сконструирован из двух олигонуклеотидов путем лигирования.
Следующие два олигонуклеотида были синтезированы как описано в примере 1 настоящей заявки.
АК#78 5'С ATG GCT AAG AGA GAA GCT GAA GCT GAA GCT AAG ТТС GTT-3'
АК#79 5' ААС GAA
СТТ AGC ТТС AGC ТТС AGC ТТС ТСТ СТТ AGC-3'
Из АК#78 и АК#79 был получен дуплекс как описано в примере 1.
Фрагмент ДНК был залигирован в плазмиду рАК188, разрезанную рестрикционными эндонуклеазами NcoI и НраI. Плазмида рАК188 состоит из последовательности ДНК 412 п.о., кодирующей сигнальный/лидерный ген дрожжей LaC212spx3 (описан в примере 3 WO 89/02463) и гена инсулинового предшественника M15 (последовательность см. на фиг. 2), вставленного в вектор (фагмиду) pBLUESCRIPT IIsk (+/-) (Stratagene, USA). Плазмида рФК188 представлена на фиг.1.
Смесь для лигирования использовалась для трансформирования компетентного штамма E. соli (r-, m+), который селектировался по устойчивости к ампициллину. Плазмиды изолировали из получившихся колоний E.соli стандартным методом минипрепа [Sambrook J, Fritsch EL Maniatis T, Molecular cloning. A laboratory Manual, Cold Spring Harbor Laboratory Press, 1989]. Секвенирование ДНК, проведенное с использованием энзиматического терминирования цепи (Sequenase, United States Biochemicals) в соответствии с инструкциями производителя, показало, что плазмида содержит правильные последовательности ДНК, кодирующие удлиненный с N-конца ЕАЕАЕАK предшественник инсулина, точно соединенный с LAC212spx3 и инсулиновым предшественником M15. Конечная плазмида названа рАK342. рАK34 была разрезана рестрикционными эндонуклеазами EcoRI и ХbаI и фрагмент 427 п.о. был лигирован в экспрессирующий вектор сРОТ как описано в примере 11 ниже и трансформирован в компетентные Е coli как описано в примере 1 настоящей заявки. Полученная плазмида рАK344 была трансформирована в дрожжевой штамм МТ663 как описано в примере 1. Последовательность ДНК, кодирующая предшественник инсулина М15, удлиненный на N-конце ЕАЕАЕАK, показана на фиг.3. Полученный дрожжевой штамм назван уАK344. Конструирование рАK344 показано на фиг.1.
Пример 10.
Получение ЕАЕАЕАK-инсулинового предшественника M15 в дрожжевом штамме уАK387.
рАK344 была разрезана рестрикционными эндонуклеазами Nco I и Eco RI и фрагмент 218 был выделен как описано выше. В дополнение, рАK405 была разрезана рестрикционными эндонуклеазами EcoRI и NcoI и фрагмент 1653 п.о. был вырезан из агарозного геля и выделен с помощью набора Gene Clean (Bio 101 inc. , PO BOX 2284, La Jolla, ca 92038, USA) согласно инструкциям производителя. Часть этого фрагмента, кодирующая модифицированный альфа-фактор (альфа-фактор был модифицирован таким образом, что в последовательности ДНК, где две С-концевые, прилегающие к Кех 2-сайту, аминокислоты лейцин и аспарагиновая кислота были заменены на метионин и аланин, был представлен сайт NcoI эндонуклазы. Далее, дрожжевой экспрессирующий вектор сРОТ был разрезан рестриктазами NcoI и ХbаI и фрагмент 9273 п.о. был выделен как описано выше. Затем три фрагмента ДНК были лигированы Т4 лигазой в стандартных условиях [Sambrook J, Fritsch EL Maniatis Т, см. выше]. Лигазую смесь использовали для трансформации компетентных клеток E.coli, как описано в примере 1 настоящей заявки. Плазмида была выделена из полученных колоний стандартными методами [Sambrook J, Fritsch EL Maniatis T, см выше], и последовательность ДНК, кодирующей удлиненный на N-конце предшественник инсулина, была проверена сиквенированием как описано в примере 2 настоящей заявки. Последовательность ДНК, кодирующая комплекс лидера фактора альфа с инсулиновым предшественником ЕАЕАЕАK-М15, показана на фиг. 4. Плазмида была названа рАK387 и была использована для трансформации компетентных клеток дрожжей МТ663 как описано в примере 1. Полученный штамм дрожжей назван уАK387.
Пример 11
Получение предшественника инсулина ЕАK-М15 в
штамме
дрожжей уАK418.
Были синтезированы следующие олигонуклеотиды как описано в примере 1:
#129 5'-САТ GGC TAA GAG AGA AGC TAA GTT CGT T-3'
#130 5'-AAC GAA CTT
AGC TTC
ТСТ СТТ AGC-3'
#94 5'-ТАА АТС ТАТ ААС ТАС ААА ААА САС АТА-3'
#16 5'-CTG CTC TAG AGC CTG CGG GCT GCG TCT-3'
Следующая полимеразная цепная реакция (PCR) была
проведена с
использованием набора реагентов Gene Amp PCR (Perkin Elmer, 761 Main AVEWALK, CT 06859, USA) согласно указаниям производителя. Сверху смесь PCR всегда покрывали 100 мкл минерального масла
(Sigma
Chemical Co., St. louis МО, USA)
PCR #1
5 мкл олигонуклеотида #129 (50 пкмоль)
5 мкл олигонуклеотида #16 (50 пкмоль)
10 мкл 10х буфера PCR
16 мкл
смеси
dNTP
0.5 мкл фермента Taq
0.5 мкл рАK387 плазмиды в качестве образца (0.2 мг ДНК)
63 мкл дистиллированной воды
PCR #2
5 мкл олигонуклеотида #130 (50
пкмоль)
5 мкл олигонуклеотида #94 (50 пкмоль)
10 мкл 10х буфера PCR
16 мкл смеси dNTP
0.5 мкл фермента Taq
0.5 мкл рАK387 плазмиды в качестве образца (0.2
мг ДНК)
63 мкл дистиллированной воды
В целом проводили 12 циклов, каждый цикл при 94oС в течение 1 мин; 37oС 2 мин; 72oС 3 мин. Затем смесь PCR
наносили на 2% агарозный гель и разделяли электрофоретически стандартным методом [Sambrook J, Fritsch EL Maniatis Т, см. выше]. Полученный фрагмент ДНК вырезали из агарозы и выделяли с использованием
набора Gene Clean (Bio 101 inc., PO BOX 2284, La Jolla, ca 92038, USA) согласно инструкциям производителя. Аналогично фрагмент ДНК длиной 371 п. о. из PCR #2 был выделен и очищен. Очищенные фрагменты
ДНК из PCR#1 и #2 были объединены и использованы в PCR как описано ниже с использованием так называемого PCR с удлинением по перекрыванию [Horton RM, Cai Z. Но SN, Pease LR, Biotechniques 8,
528-535(1990)].
PCR #3
5 мкл олигонуклеотида #16 (50 пкмоль)
5 мкл олигонуклеотида #94 (50 пкмоль)
10 мкл 10х буфера PCR
16 мкл смеси dNTP
0.5 мкл фермента Taq
10 мкл очищенного фрагмента PCR#1
10 мкл очищенного фрагмента PCR#2
43.5 мкл дистиллированной воды
В целом проводили 10 циклов, каждый цикл
при
94oС в течение 1 мин; 37oС 2мин; 72oС 3 мин. Полученный PCR фрагмент ДНК 563 п.о. был выделен и очищен как описано выше. Очищенный PCR фрагмент ДНК растворяли в
10 мкл
дистиллированной воды и буфера для рестрикционных эндонуклеаз и разрезали рестриктазами EcoRI и НраI согласно стандартной методике [Sambrook J, Fritsch EL Maniatis Т, см. выше].Фрагмент ДНК
Eco
RI/ Нра I длиной 344 п.о. был нанесен на агарозный электрофорез и очищен с использованием набора Gene Clean, как описано выше.
Плазмида рАK188 (см. фиг.1). Также была разрезана рестриктазами EcoRI и НраI и фрагмент вектора 3139 п.о. был выделен. Два фрагмента ДНК были лигированы Т4 ДНК-лигазой в стандартных условиях [Sambrook J, Fritsch EL Maniatis T, см. выше] . Лигазную смесь затем трансформировали в компетентный штамм E. coli (R-, M+) с селекцией по устойчивости к ампициллину. Плазмиды были выделены из полученных колоний E.coli с использованием стандартной техники минипрепа [Sambrook J, Fritsch EL Maniatis T, см. выше] и названы рАK413.
рАK 413 была разрезана рестрикционными эндонуклеазами NcoI и ХbаI и полученный фрагмент ДНК 206 п. о. был выделен. Дрожжевой экспрессирующий вектор сРОТ был разрезан рестрикционными эндонуклеазми ХbаI и SphI и фрагмент ДНК 9821 п.о. был выделен. рАK387 разрезали рестрикционными эндонуклеазами SphI и NcoI и фрагмент в 1413 п. о. частично деградированной SphI/NcoI ДНК был выделен. Затем фрагмент NcoI/ XbaI 206 был лигирован с другими двумя фрагментами, 9821 Sph I/XbaI фрагментом и 1413 Spb I/XbaI фрагментом с использованием стандартной методики [Sambrook J, Fritsch EL Maniatis Т, см. выше]. Лигазную смесь трасформировали в E.coli как описано выше. Плазмида из полученных E.coli была изолирована стандартным методом [Sambrook J, Fritsch EL Maniatis T. см. выше], и проверена соответствующими рестрикционными эндонуклеазами EcoRI, XbaI, SphI, NcoI, HpaI. Отобранная плазмида имела правильную последовательность для ЕАK-М15 предшественника инсулина, как показал анализ секвенированием, и была встроена за последовательностью ДНК, кодирующей фактор альфа.
Плазмида была названа рАK418. Последовательность ДНК, кодирующая комплекс лидер альфа фактора/ЕАK-М15 предшественник инсулина показана на фиг.5. Плазмиду рАK 418 трансформировали в штамм S. cereviciae МТ663 как описано в примере 1 и полученный штамм был назван уАK418.
Пример 12
Получение ЕАЕАK-М15 предшественника инсулина в штамме
дрожжей уАK419
Фрагмент ДНК, кодирующий удлиненный на N-конце, предшественник инсулина ЕАЕАK-М15 был получен как описано в примере 11, за исключением того, что олигонулеотид # 133
использовался вместо олигонуклеотида #129 и олигонуклеотид #135 использовался вместо олигонуклеотида #130.
#133 5-С ATG GCT AAG AGA GAA GCT GAA GCT AAG TTC GTT-3'
#135 5-AAC
GAA CTT CGC TTC AGC TTC TCT CTT AGC-3'
Как описано в примере 11, фрагмент ДНК, кодирующий ЕАЕАK-М15 предшественик инсулина, был сначала клонирован в рАK188 и затем субклонирован в дрожжевом
экспрессирующем векторе сРОТ. Дрожжевая экспрессирующая плазмида, кодирующая удлиненный на N-конце предшественник инсулина ЕАЕАK-М15, была названа рАK419. Последовательность ДНК, кодирующая комплекс
лидера альфа-фактора с ЕАЕА-М15 предшественником инсулина представлена на фиг.6. Наконец, рАK419 была трансформирована в дрожжевой штамм МТ663, как описано в примере 1.
Пример 13
Получение ЕАЕАЕАЕАK-М15 предшественника инсулина в штамме дрожжей уАK420
Фрагмент ДНК, кодирующий удлинненный на N-конце предшественник инсулина ЕАЕАЕАЕАK-М15, был синтезирован как
описано в примере 11, за исключением того, что олигонуклеотид #136 использовался вместо олигонуклеотида #129 и олигонуклеотид #134 использовался вместо олигонуклеотида #130.
#136
5'-CATGG CTAAG AGAGA AGCTG AAGCT GAAGC TGAAG TCTAA GTTCG TT-3'
#134 5'-AACGA ACTTA GCTTC AGCTT CAGCT TCAGC TTCTC TCTTA GC-3'
Как описано в примере 11, фрагмент ДНК, кодирующий
ЕАЕАЕАЕАK-М15 предшественник инсулина, был сначала клонирован в рАK188 и затем субклонирован в дрожжевом экспрессирующем векторе сРОТ. Дрожжевая экспрессирующая плазмида, кодирующая удлиненный на
N-конце предшественник инсулина ЕАЕАЕАЕАK-М15, была названа рАK420. Последовательность ДНК, кодирующая комплекс лидер альфа фактора - предшественник инсулина ЕАЕАЕАЕА-М15 представлена на фиг.7. Затем
рАK420 была трансформирована в дрожжевой штамм МТ663 как описано в примере 1.
Пример 14
Получение ЕАЕАЕАЕАK-М 15 предшественника инсулина в штамме дрожжей уАK421
Фрагмент ДНК, кодирующий удлиненный на N-конце предшественник инсулина ЕАЕАЕАЕАK-М15, был синтезирован как описано в примере 11, за исключением того, что олигонуклеотид #131 использовался вместо
олигонуклеотида #129 и олигонуклеотид #132 использовался вместо олигонуклеотида #130.
#131 5'-CATGG CTAAG AGAGA AGCTG AAGCT GAAGC TAAGT TCGTT-3'
#132 5'-AACGA ACTTA GCTTC
AGCTT CAGCT TCAGC TTCAG CTTCT CTCTT AG-3'
Как описано в примере 11, фрагмент ДНК, кодирующий ЕАЕАЕАЕАK-М15 предшественник инсулина, был сначала клонирован в рАK188 и затем субклонирован в
дрожжевом экспрессирующем векторе сРОТ. Последовательность ДНК, кодирующая комплекс лидер альфа-фактора - предшественник инсулина ЕАЕАЕАЕА-М15, представлена на фиг. 8. Дрожжевая экспрессирующая
плазмида, кодирующая удлиненный на N-конце предшественник инсулина ЕАЕАЕАЕАK-М15, была названа рАK421 и трансформирована в дрожжевой штамм МТ663, как описано в примере 1. Полученный штамм дрожжей
назван уАK421.
Пример 15
Получение ЕАЕАЕАK-М15 предшественника инсулина в дрожжевом штамме уАK50
pLaC246/1003 была разрезана эндонуклеазами ShpI и NcoI и был
выделен
фрагмент ДНК 649 п.о., кодирующий лидер LaC245 (лидер LaC245 описан в примере 3 WO 92/11378, где он назван LSC6315d3; см. фиг.9, прилагающийся для последовательности LaC245). рАК387 была
разрезана
эндонуклеазами NcoI и ХbаI и фрагмент ДНК 218 п.о., кодирующий предшественник инсулина M15, был выделен. Дрожжевой экспрессирующий вектор сРОТ был разрезан эндонуклеазами SphI и ХbаI и
фрагмент ДНК
9821 был выделен и лигирован с двумя упомянутыми выше фрагментами с использованием стандартной методики. [Sambrook J, Fritsch EL Maniatis T, см. выше]. После выделения полученной
плазмиды и анализа
последовательности ДНК для подтверждения точности последовательности ДНК, плазмида была трансформирована в дрожжевой штамм МТ663 как описано в примере 1. Полученный штамм был
назван уАK50.
Пример 16
Получение инсулинового предшественника в дрожжевом штамме уАK49.
pLaC246/1003 была разрезана эндонуклеазами ShpI и NcoI и выделен фрагмент ДНК 649 п.о., кодирующий лидер LaC245 как описано ранее. pLaC200M15 была разрезана эндонуклеазами NcoI и ХbаI и фрагмент ДНК 197 п.о., кодирующий предшественник инсулина M15, был выделен. Дрожжевой экспрессирующий вектор сРОТ был разрезан эндонуклеазами SphI и ХbаI и фрагмент ДНК 9821 был выделен и лигирован с двумя упомянутыми выше фрагментами с использованием стандартной методики. [Sambrook J, Fritsch EL Maniatis T, см. выше]. После выделения полученной плазмиды и плазмида для подтверждения точности последовательности ДНК была тестирована сиквенсным анализом и затем трансформирована в дрожжевой штамм МТ663 как описано в примере 1, и полученный штамм был назван уАK49.
Пример 17
Получение ЕАЕАЕАЕАK-М15 предшественника инсулина в штамме
дрожжей уАK424
рАK420 была
разрезана эндонуклеазами NcoI и ХbаI и был выделен и очищен с использованием набора Gene Clean, как описано ранее, фрагмент ДНК 236 п.о. , кодирующий ЕАЕАЕАЕАK-М15
предшественник инсулина.
pLaC246/1003 была разрезана эндонуклеазами SphI и NcoI и фрагмент ДНК 649 п.о. был выделен. Дрожжевой экспрессирующий вектор сРОТ был разрезан рестриктазами SphI и ХbаI и был
выделен фрагмент ДНК 9821
п.о. Манипуляции с рестрикционными эндонуклеазами проводились в стандартных условиях. [Sambrook J, Fritsch EL Maniatis Т, см. выше]. Затем три фрагмента ДНК были лигированы
в стандартных условиях с
использованием Т4-лигазы. [Sambrook J, Fritsch EL Maniatis T, см. выше]. Полученная экспрессирующая дрожжевая плазмида, кодирующая удлиненный на N-конце предшественник
инсулина ЕАЕАЕАЕАK-М15, была
названа рАK424. Анализ последовательности ДНК проводился как описано в примере 2. рАK424 была использована для трансформации дрожжевого штамма МТ663 и полученный штамм
назван уАK424.
Пример 18
Получение ЕАЕАЕАЕАЕАK-М15 предшественника инсулина в штамме дрожжей уАK424
рАK421 была разрезана эндонуклеазами NcoI и ХbаI и был выделен и
очищен с использованием набора
Gene Clean, как описано ранее, фрагмент ДНК 242 п.о. , кодирующий ЕАЕАЕАЕАЕАK-М15 предшственник инсулина. pLaC246/1003 была разрезана эндонуклеазами SphI и NcoI и
фрагмент ДНК 649 п.о. был выделен.
Дрожжевой экспрессирующий вектор сРОТ был разрезан рестрикционными эндонуклеазами SphI и ХЬаI и был выделен фрагмент ДНК 9821 п.о. Манипуляции с рестрикционными
эндонуклеазами проводились в
стандартных условиях. [Sambrook J, Fritsch EL Maniatis T. см. выше]. Затем три фрагмента ДНК были лигированы в стандартных условиях с использованием Т4-лигазы. [Sambrook
J, Fritsch EL Maniatis Т, см.
выше]. Полученная экспрессирующая дрожжевая плазмида, кодирующая удлинненый на N-конце предшественник инсулина ЕАЕАЕАЕАЕАK-М15, была названа рАK425. Анализ
последовательности ДНК проводился как
описано в примере 2. рАK425 была использована для трансформации дрожжевого штамма МТ663, и полученный штамм назван уАK424.
Пример 19
Получение SEAEAEAK-M15 предшественника
инсулина в дрожжевом штамме уАK440
Фрагмент ДНК, кодирующий удлиненный на N-конце предшественник инсулина SEAEAK-M15, был синтезирован как описано в
примере 11, за исключением того, что
олигонуклеотид #160 использовался вместо олигонуклеотида #129 и олигонуклеотид #161 использовался вместо олигонуклеотида #130.
#160 5'-CATGG
CTAAG AGAGA TCTGC AGCTG AAGCT AAGTT
CGTT-3'
#161 5'-AACGA AGCTT CAGCT TCAGC TTCAG ATCTC TTAGC-3'
Как описано в примере 11, фрагмент ДНК, кодирующий SEAEAEAK-M15 предшественник
инсулина, был сначала клонирован в рАK188
и затем субклонирован в дрожжевом экспрессирующем векторе сРОТ. Дрожжевая экспрессирующая плазмида, кодирующая удлинненый на N-конце предшественник инсулина
SEAEAEAK-M15, была названа рАK440.
Последовательность ДНК, кодирующая комплекс LaC245/SEAEAEA-M15 лидера и предшественника инсулина, представлена на фиг. 10. Анализ последовательности ДНК проводился
как в примере 2. В заключение рАK440
была трансформирована в дрожжевой штамм МТ663 как описано в примере 1, и полученный штамм назван уАK440.
Пример 20
Получение
АЕАЕАЕАK-М15 предшественника инсулина в дрожжевом
штамме уАK442
Фрагмент ДНК, кодирующий удлиненный на N-конце предшественник инсулина SEAEAEAK-M15, был синтезирован как описано в примере 11,
за исключением того, что олигонуклеотид #162
использовался вместо олигонуклеотида #129 и олигонуклеотид #163 использовался вместо олигонуклеотида #130.
#160 5'-CATGG CTAAG AGAGC
TGAAG CTGAA GCTGA AGCTA AGTTC GTT-3'
#161
5'-AACGA AGCTT AGCTT CAGCT TCAGC TTCAG CTCTC TTAGC-3'
Как описано в примере 11, фрагмент ДНК, кодирующий АЕАЕАЕАK-М15 предшественик
инсулина, был сначала клонирован в рАK188 и затем
субклонирован в дрожжевом экспрессирующем векторе сРОТ. Дрожжевая экспрессирующая плазмида кодирующая удлиненный на N-конце предшественник инсулина
АЕАЕАЕАK-М15, была названа рАK442.
Последовательность ДНК, кодирующая комплекс LaC245/AEAEAEA-M15 лидерной последовательности и предшественника инсулина, представлена на фиг.11. Анализ
последовательности проводился как описано в
примере 2. Наконец, рАK442 была трансформирована в дрожжевой штамм МТ663 как описано в примере 1, и полученный штамм назван уАK442.
Пример
21
Получение ЕЕАЕАЕАK-М15
предшественника инсулина в дрожжевом штамме уАK443
Фрагмент ДНК, кодирующий удлиненный на N-конце предшественник инсулина ЕЕАЕАЕАK-М15, был синтезирован
как описано в примере 11, за
исключением того, что олигонуклеотид #166 использовался вместо олигонуклеотида #129 и олигонуклеотид #168 использовался вместо олигонуклеотида #130.
#166
5'-CATGG CTAAG AGAGA AGAAG
CTGAA GCTGA AGCTA AGTTC-3'
#168 5'-AACGA ACTTA GCTTC AGCTT CAGCT TCTCT CTTAG C-3'
Как описано в примере 11, фрагмент ДНК, кодирующий ЕЕАЕАЕАK-М15
предшественник инсулина, был сначала
клонирован в рАK188 и затем субклонирован в дрожжевом экспрессирующем векторе сРОТ. Дрожжевая экспрессирующая плазмида, кодирующая удлиненный на N-конце
предшественник инсулина АЕАЕАЕАK-М15, была
названа рАK443. Последовательность ДНК, кодирующая комплекс LaC245/AEAEAEA-M15 лидера с предшественником инсулина, представлена на фиг. 12. Анализ
последовательности ДНК проводился как в примере 2. В
заключение рАK443 была трансформирована в дрожжевой штамм МТ663 как описано в примере 1, и полученный штамм назван уАK443.
Пример
22
Получение ЕАЕАЕАЕK-М15 предшественника
инсулина в дрожжевом штамме уАK443
Фрагмент ДНК, кодирующий удлиненный на N-конце предшественник инсулина ЕАЕАЕАЕK-М15, был синтезирован
как описано в примере 11, за исключением того, что
олигонуклеотид #164 использовался вместо олигонуклеотида #129 и олигонуклеотид #165 использовался вместо олигонуклеотида #130.
#164
5'-CATGG CTAAG AGAGA AGCTG AAGCT GAAGC TGAAA AGTTC
GTT-3'
#168 5'-AACGA ACTTT TCAGC TTCAG CTTCA GCTTC TCTCT TAGC-3'
Как описано в примере 11, фрагмент ДНК, кодирующий ЕАЕАЕАЕK-М15
предшественник инсулина, был сначала клонирован в
рАK188 и затем субклонирован в дрожжевом экспрессирующем векторе сРОТ. Дрожжевая экспрессирующая плазмида, кодирующая удлиненный на N-конце
предшественник инсулина ЕАЕАЕАЕK-М15, была названа рАК444.
Последовательность ДНК, кодирующая комплекс LaC245/EAEAEAEK-M15 лидера с предшественником инсулина, представлена на фиг. 13. Анализ
последовательности ДНК проводился как в примере 2. В заключение
рАK444 была трансформирована в дрожжевой штамм МТ663 как описано в примере 1, и полученный штамм назван уАK444.
Пример
23
Получение ЕЕАЕАЕАЕK-М15 предшественника инсулина в
дрожжевм штамме уАK443
Фрагмент ДНК, кодирующий удлиненный на N-конце предшественник инсулина ЕЕАЕАЕАЕK-М15, был синтезирован
как описано в примере 11, за исключением того, что олигонуклеотид
#192 использовался вместо олигонуклеотида #129 и олигонуклеотид #193 использовался вместо олигонуклеотида #130.
#192
5'-GAAGA AGCTG AAGCT GAAGC TGAAA AGTTC GTT-3'
#193
5'-СТTTТ CAGCT TCAGC TTCAG CTTCT TCTCT-3'
Как описано в примере 11, фрагмент ДНК, кодирующий ЕЕАЕАЕАЕK-М15 предшественник инсулина,
был сначала клонирован в рАК188 и затем субклонирован в
дрожжевом экспрессирующем векторе сРОТ. Дрожжевая экспрессирующая плазмида, кодирующая удлиненный на N-конце предшественник инсулина
ЕЕАЕАЕАЕK-М15, была названа рАK448. Последовательность ДНК,
кодирующая комплекс LaC245/EEAEAEAEK-M15 лидера с предшественником инсулина, представлена на фиг. 14. Анализ последовательности ДНК
проводился как в примере 2. В заключение рАK448 была
трансформирована в дрожжевой штамм МТ663 как описано в примере 1, и полученный штамм назван уАK448.
Пример 24
Получение
EVK-инсулинового предшественника в дрожжевом штамме
уАK390
Синтетический ген, кодирующий N-концевое удлинение предшественника инсулина М15, был сконструирован из двух олигонуклеотидов путем
лигирования.
Олигонуклеотиды были синтезированы как описано в примере 1.
Были синтезированы следующие олигонуклеотиды:
АК# 61 5'-CGTCG CCATG GCTAA GAGAG
AAGTT AAGTT CGTT-3'
АK# 60 5'-TTCGT TAACG
AACTT AACTT CTCTC TTAGC CATG-3'
Дуплекс между АК#61 и АК#60 был образован как описано в примере 1.
Фрагмент ДНК был залигирован в плазмиду рАК188, которая была разрезана рестрикционными эндонуклеазами NcoI и НраI как описано в примере 2. Лигазная смесь была использована для трасформации компетентного штамма E.coli (r-, m+), который затем отбирался по устойчивости к ампициллину. Полученная плазмида была названа рАK355. рАк355 была разрезана рестрикционными эндонуклеазами NcoI и ХbаI и фрагмент 218 п.о. выделен. рАK387 была разрезана SphI и NcoI и фрагмент в 1413 п.о. частично деградированной ДНК был выделен. Экспрессирующий дрожжевой вектор сРОТ был обработан эндонуклеазами SphI и ХbаI, был выделен векторный фрагмент ДНК 9823 п.о. Три фрагмента ДНК были лигированы как описано, затем трансформированы в компетентные E.coli как описано в примере 1. Полученная плазмида рАK390 была трансформирована в дрожжевой штамм МТ663 как описано в примере 1. Анализ последовательности ДНК проводился как описано в примере 2 и показал, что плазмида из полученных колоний содержит правильные последовательности ДНК для EVK-удлиненного на N-конце предшественника инсулина M15, точно присоединенного к лидерной последовательности альфа-фактора и предшественнику инсулина M15. Последовательность ДНК, кодирующая удлиненный EVK N-конец инсулинового предшественника, представлена на фиг.15. Полученный штамм дрожжей назван уАK390.
Пример 25
Получение
KELE-KR-RPDK-инсулинового предшественника в дрожжевом штамме
уАK388.
Синтетический ген, кодирующий N-концевое удлинение предшественника инсулина M15, был сконструирован из двух олигонуклеотидов путем лигирования.
Олигонуклеотиды были синтезированы как описано в примере 1.
Были синтезированы следующие олигонуклеотиды:
АК# 80
5'-CATGG CTAAG GAATT GGAAA AGAGA AGACC AGACA AGTTC GTT-3'
AK# 81 5'-AACGA ACTTG TCTGG TCTTC TCTTT CCAAT TCCTT AGC-3'
Дуплекс между АK#61 и АK#60 был образован как описано в примере
1.
Фрагмент ДНК был залигирован в плазмиду рАK188, которая была разрезана рестрикционными эндонуклеазами NcoI и НраI как описано в примере 2. Лигазная смесь была использована для трасформации компетентного штамма E.coli (r-, m+), который затем отбирался по устойчивости к ампициллину. Полученная плазмида была названа рАK312. рАK312 была разрезана рестриктазами NcoI и ХbаI и фрагмент 221 п. о. выделен. рАK312 была разрезана SphI и NcoI и фрагмент частично деградированной ДНК в 1413 п.о. был выделен. Экспрессирующий дрожжевой вектор сРОТ был обработан эндонуклеазами SphI и ХbаI, был выделен векторный фрагмент ДНК 9823 п.о. Три фрагмента ДНК были лигированы как описано выше, затем трансформированы в компетентные E.coli как описано в примере 1. Полученная плазмида рАK390 была трансформирована в дрожжевой штамм МТ663, как описано в примере 1. Анализ последовательности ДНК проводился как описано в примере 2 и показал, что плазмида из полученных колоний содержит правильные последовательности ДНК для терминально KELE-KR-RPDK-удлиненного на N-конце предшественника инсулина M15, точно присоединенного к лидерной последовательности альфа-фактора и предшественнику инсулина M15. Последовательность ДНК, кодирующая удлиненный KELE-KR-RPDK N-конец инсулинового предшественника, представлена на фиг.16. Полученный штамм дрожжей назван уАK388.
Реферат
Изобретение относится к способу получения гетерологичного полипептида в дрожжах Saccharomyces cerevisiae. Дрожжи трансформируют рекомбинантным экспрессирующим вектором, содержащим конструкцию ДНК, кодирующую полипептид, включающий сшитые сигнальный пептид, лидерный пептид и гетерологичный полипептид. Проводят культивирование трансформированных дрожжей в подходящей для экспрессии среде и выделение конечного продукта из культуральной среды. Изобретение позволяет получать более высокий выход правильно процессированного целевого протеина. 20 з.п.ф-лы, 18 ил., 2 табл.
Формула
сигнальный пептид - лидерный пептид - X1X2Х3X4 - гетерологичный белок,
где X1 является пептидной связью или представляет собой одну или более аминокислот, которые могут быть одинаковыми или различными,
X2 и X3 являются одинаковыми или различными и представляют основную аминокислоту, выбранную из группы, состоящей из Lys и Arg, причем X2 и X3 вместе определяют сайт, обеспечивающий процессинг в дрожжах;
X4 является пептидной связью или представляет одну или более аминокислот, которые могут быть одинаковыми или различными при условии, что X1 и/или X4 представляют одну или более аминокислот и что по крайней мере одна из аминокислот, представленных X1 и/или X4 , является отрицательно заряженной аминокислотой, выбранной из группы, состоящей из Glu и Asp, и, кроме того, при условии, что когда X1 представляет пептидную связь, X4 последовательность иная, чем Glu-Ala-Glu-Ala, Glu-Ala-Glu-Ala-Ser-Leu-Asp, и что, когда X4 представляет пептидную связь, X1 последовательность иная, чем Ser-Leu-Asp.
A Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Gly-Phe-Leu-Asp-Leu-Ala-Gly-Glu-Glu-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala;
B Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala;
C Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala;
D Ala-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala-Asn-Val-Ala-Met-Ala; и
E Gln-Pro-Val-Thr-Gly-Asp-Glu-Ser-Ser-Val-Glu-Ile-Pro-Glu-Glu-Ser-Leu-Ile-Ile-Ala-Glu-Asn-Thr-Thr-Leu-Ala-Asn-Val-Ala-Met-Ala.
Комментарии