Способ получения гетерологичного полипептида в эукариотических микроорганизмов - RU2091490C1
Код документа: RU2091490C1
Чертежи























Описание
Изобретение относится к генетической инженерии, в частности к получению рекомбинантных ДНК, содержащих транслируемую часть сигнального пептида гена Saccharomyces ceremisiae BARI и один структурный ген, являющийся чужеродным для клетки, трансфомированной указанной ДНК. Трансформация организмов хозяев такими конструкциями ДНК приводит в результате к экспрессии первичного продукта трансляции, включающего структурный белок, кодируемый чужеродным геном, слитым с сигнальным пептидом BARI. В результате данный белок секретируется из клетки хозяина и может выделяться в питательную среду или в периплазменное пространство.
В качестве хозяев для получения гетерологичных полипептидов были использованы различные прокариотические и зукариотические микроорганизмы. Особый интерес вызывало использование различных видов зукариотических грибковых организмов, включая Saccharomyces cerevisiae, Schizosaccharomyces pombe, Aspergillus и Neurospora. В частности, была проделана большая работа по культивированию дрожжей S. cerevisiae. Дрожжевые клетки при трансформации их соответствующих конструкций ДНК, например, плазмидой, экспрессировали полипептид, закодированный в гетерологичном гене, содержащемся в данной плазмиде.
Однако данная технология ограничена тем, что белковые продукты не выделяются из клеток-хозяев в среду и в связи с этим необходимо разрушение клеток и очистка белка от различных загрязняющих клеточных компонентов без его денатурации или инактивации.
Таким образом, желательно создать трансформированные клетки, секретирующие гетерологичный продукт, что позволит упростить очистку данного продукта.
Кроме того, желательно, чтобы некоторые белки прошли путь секреции клетки-хозяина для образования, например, бисульфидной связи.
Известно, что S. cerevisiae секретирует некоторые из своих белков в питательную среду, хотя сведения о данном процессе очень ограничены по сравнению с тем, что известно о секреции белков из бактерий и клеток млекопитающих. Как известно, большинство секретируемых дрожжевых белков представляют собой ферменты, которые остаются в периплазменном пространстве, хотя такие ферменты как инвертаза и кислотная фосфатаза, могут входит в стенку клетки. Белки, которые, как известно, выделяются S. ceremisiae в питательную среду, включают спаривающие феромоны (альфа-фактор и a-фактор), уничтожающий токсин и белок, который отвечает за активность Баррьер.
Клетки S. ceremisiae α-спаривающего типа продуцируют альфа-фактор, в то время как a-клетки продуцируют два полипептида: альфа-фактор и Баррьер, все секретируемые в питательную среду; ген альфа-фактора был клонирован, секвенирован и проанализирован (Kurjan and Herskowitz, Cell, 30, 933 943, 1982). Сигнальный пептид (короткая пептидная последовательность, обеспечивающая секрецию связанного с ней белка), главная последовательность (включающая полипептид-предшественник, который отщепляется при созревании альфа-фактора) и нетранслируемые нуклеотидные последовательности (включающие области стимулятора и регулятора) гена альфа-фактора могут использоваться для секреции чужеродных белков в дрожжах (Brake et al. Proc. Natl. Acad. Sci, USA, 81, 4642 4646, 1984). Экспрессия гена альфа-фактора регулируется продуктом гена МАТα1 и переработка предшественника альфа-фактора в созревший белок требует осуществления двух этапов под контролем генов STE13 и KEX2.
В противоположность альфа-фактору Баррьер гликолизируется его способностью связывать конканавалин A. Баррьер продуцируется a-клетками и экспрессируется под контролем гена МАТa 2. За исключением отщепления сигнального пептида никаких обработок предшественника Баррьер ранее не осуществлялось и гены STE13 и KEX2 не рассматривались как гены, участвовавшие в выражении Баррьер.
Учитывая указанные выше различия между контролем экспрессии и преобразования альфа- фактора и Баррьер, можно определить, какой из этих дрожжевых генов подходит лучше для направления секреции специфического инородного белка. Ввиду того, что эти два белка проходят через различные пути секреции, желательно использовать свойства системы секреции Баррьер.
Известен способ получения гетерологичного полипептида в эукариотических микроорганизмах, предусматривающий конструирование рекомбинантной плазмидной ДНК, содержащий промотор, последовательность, кодирующую сигнальный пептид и структурный ген, трансформацию полученной рекомбинантной ДНК штаммов-реципиетов (EPO, N 0121884, кл. C 12 N 15/00, 1983).
Объектом предлагаемого изобретения является способ получения гетерологичных полипептидов путем экспрессии чужеродных генов в микробном хозяине.
Изобретение касается способов использования конструкций ДНК, содержащих по меньшей мере кодирующую последовательность сигнального пептида гена BARI Saccharomyces cerevisiae, по меньшей мере один структурный ген, являющийся чужеродным геном для штамма-реципиента, и стимулятор, который контролирует экспрессию в штамме слитого белка, включающего сигнальный пептид BARI и чужеродный белок.
Изобретение поясняется чертежами, на которых:
фиг.
1 3 -нуоклеотидная последовательность гена BARI и выведенная
аминокислотная последовательность первичного трансляционного продукта, где связывающая точка МАТa2 подчеркнута, предполагаемая точка
расщепления сигнального пептида показана стрелкой, а точки
возможного гликозилирования отмечены звездочкой;
фиг. 4 физическая карта плазмиды pZV9;
фиг. 5 конструирование плазмиды
p254;
фиг. 6 конструирование плазмид pZV30, pZV31,
pZV32 и pZV33;
фиг. 7 и 8 конструирование плазмиды pZV50;
фиг. 9 конструирование плазмиды m115;
фиг. 10
конструирование плазмиды pZV49;
фиг. 11 конструирование
плазмиды pZV134, включающей стимулятор TPI1;
фиг. 12 -субклонирование части гена MFa1;
фиг. 13 конструирование
плазмиды pZV75;
фиг. 14 конструирование плазмид, включающих
стимулятор TPI1 и слияние BARI-MFa1;
фиг. 15 -конструирование плазмиды pSW22;
фиг. 16 конструирование плазмид,
включающих слияние BAR1-MFa1;
фиг. 17 конструирование плазмиды
pZV100;
фиг. 18 конструирование плазмиды pZV102;
фиг. 19 конструирование плазмиды pSW96;
фиг. 20
конструирование плазмиды pSW97;
фиг. 21 конструирование плазмид
pSW98 и pSW99; D указывает мутацию у кодона 25.
Под термином "конструкция ДНК" имеется в виду молекула ДНК, включающая плазмиду, которая была модифицирована при участии человека таким образом, чтобы нуклеотидная последовательность данной молекулы не совпадала с последовательностью, которая создана естественным путем. Понятие "конструкция ДНК" охватывает также клоны молекул ДНК, модифицированные таким образом. Термины "вектор экспрессии" и "плазмида экспрессии" определены как конструкция ДНК, которая включает точку инициирования транскрипции, и по меньшей мере один структурный ген, кодирующий желаемый белок, который экспрессируется в организме хозяина. Вектор экспрессии обычно включает также соответствующие области, такие как стимулятор и концевую часть, которые направляют экспрессию желаемого белка в организме хозяина, а также источник репликации. Векторы экспрессии обычно включают селективный маркер, например, геноустойчивости к антибиотикам или маркер зависимости от питательного вещества.
Понятие "конструкция ДНК" включает также части вектора экспрессии, входящего в хромосому хозяина.
Термин "плазмида" имеет его общепринятое значение, т. е. конструкция ДНК с автономной репликацией, обычно с замкнутой петлей.
Термин "сигнальный пептид" относится к той части первичного продукта трансляции, которая определяет секрецию полипептида из клетки. Сигнальный пептид обычно отщепляется от остальной части полипептида сигнальной пептидазой. Сигнальный пептид характеризуется наличием гидрофобных аминокислот, находящихся у концевой аминогруппы первичного продукта трансляции и обычно состоит из 17 25 аминокислот. Точки расщепления сигнальной пептидазой были охарактеризованы von Heinye (Eur. J. Bijchem. 133, 17, 1983). Под термином "сигнальный пептид" могут также иметься в виду функциональные части сигнального пептида естественного происхождения.
Предлагаемое изобретение предусматривает способ получения гетерологичных полипептидов с помощью трансформированных клеток, обеспечивающих секреций гетерологичных белков через клеточную стенку. Эти клетки содержат конструкцию ДНК с чужеродным геном, связанным с дрожжевым BARI геном или его частью, кодирующей по меньшей мере сигнальный пептид BARI. Гетерологичные белки выделяются в периплазменное пространство или в питательную среду. Дрожжевой ген BARI кодирует гликозилированный белок, который секретируется клетками a типа S. cerevisiae и обладает активностью Баррьер. Секретированный белок Баррьер позволяет созревшим клеткам a-типа преодолевать задерживающее действие G1, вызванное альфа-фактором. При этом предлагается, что Баррьер может представлять собой протеазу (Manney, J. Bacteriol, 155, 291 301, 1983). Транскрипция гена BARI стимулируется альфа-фактором. Баррьер или аналогичная активность не обнаруживается в a- или a/α-клетках и ген BARI не транскрибируется в этих типах клеток.
На фиг. 1 3 показана нуклеотидная последовательность размером 2750 пар оснований гена BARI вместе с выведенной аминокислотной последовательностью первичного продукта трансляции. ATG точка-инициации трансляции гена BARI находится в положении 681 фрагмента, составляющего примерно 2,75 kb, показано на фиг. 1, который субклонирован от фрагмента, полученного из библиотеки дрожжевых геномов (Nasmyth and Tatchell, Cell, 19, 753 764, 1980). Открытая рамка считывания начинается с кодона ATG в +1 и простирается до 1761 bp в 3'-направлении. Последовательность первых 24 аминокислот первичного продукта трансляции BARI аналогична известным последовательностям сигнальных пептидов дрожжей и млекопитающих. Так, аланин в положении 24 может быть использован как точка расщепления как дрожжевой инвертазой, так и кислотной фосфатазой. Расщепление может происходить также после 23 аминокислоты. В первичном продукте трансляции имеется не менее девяти возможных, связанных с аспарагином точек гликозилирования, хотя степень гликозилирования созревшего выделенного Баррьер пока еще неизвестна. Области стимулятора и регулятора гена BARI располагаются в пределах зоны примерно 680 bp на 5'-стороне кодона инициации трансляции. Полная функция стимулятора и реакция на стимулирование альфа-фактора были локализованы у ближайшего ATG, примерно 680 bp нетранслируемой области 5'.
Конструкция ДНК предлагаемого изобретения содержит точку расщепления в месте соединения Баррьер и частей чужеродного белка. Такой предпочтительной точкой является точка расщепления KEX2, последовательность аминокислот, которая распознается и расщепляется продуктом гена KEX2 S. cerevisiae (Julius et al. Cell, 37, 1075-1080, 1984). Точка KEX2 характеризуется парой основных аминокислот, таких как лизин и аргинин. Желательно, чтобы последовательность точки KEX2 представляла собой Lys-Arg или Arg-Arg. Первичный продукт трансляции BARI содержит две такие пары, локализованные в структурной области: Arg-Arg в положениях 177-178 и Lys-Lys в положениях 404 405.
Как отмечалось выше, ген KEX2 не участвует в обработке исходного белка Баррьер и это говорит о том, что возможные точки обработки блокируются белковой конформацией или гликозилированием. Кроме того, это говорит также о том, что Баррьер обычно при его обработке может проходить путь, отличный от того, который используется такими KEX2-обработанными белками, как альфа- фактор.
Однако авторы изобретения обнаружили, что при включении точки KEX2-обработки в белок слияния, содержащий часть первичного продукта трансляции гена BARI с сигнальным пептидом и желаемый белок, белок слияния расщепляется в этой точке, что приводит в результате к секреции желаемого белка.
Кроме того, было установлено, что снизив эффективность отщепления сигнальной пептидазой части Баррьер в белке, включающем точку расщепления KEX2, из клетки выделяются повышенные количества желаемого белка. Точка расщепления KEX2 может находиться в последовательности BARI желаемого гена, или же она может быть включена с помощью сайт-специфического мутагенеза, т.д.
Согласно изобретению часть гена BARI, включающая кодон инициации ATG и кодирующую последовательность сигнального пептида, может быть присоединена к желаемому чужеродному гену и трансформирована в эукариотическую клетку хозяина. Образуемый в результате ген слияния будет включать точку обработки, предпочтительно точку расщепления KEX2, расположенную в месте соединения последовательностей гена BARI и чужеродных последовательностей. Такая конструкция может включать также области регулятора и промотора от 5'-некодирующей зоны гена BARI или же может включать области регулятора и/или промотора от других генов.
Кроме стимулятора от гена BARI могут использоваться другие промоторы от генов алкогольдегидрогеназы I или II S.cerevisiae и генов гликолитической обработки S.cerevisiae, таких как промотор TPII и соответствующих генов других видов, включая дрожжи Sxhizosaccharomyces pombe (Russell and Hall, J. Biol. Chem. 258, 143 149, 1983 and Russell, Nature, 301, 167 169, 1983).
Ген алкогольдегидрогеназы I S. cerevisiae был описан в работе Ammerer (Method in Enzymology, 101, 192 201, 1983).
Ген алкогольдегидрогеназы II был описан в работе Рассела (Russell et al. J. Biol. Chem. 258, 2674 2682, 1983).
Гликолитические гены S. crevisiae были описаны в ряде работ (Kawasaki, Ph. D.Thesis, University of Washington, 1979; Hitzeman et al. J. Biol. Chem. 225, 12073 12080, 1980; Kawasaki and Fraenkel, Biochem. Biophys. Res. Comm. 108, 1107 1112, 1982 and Alber и Kawasaki, J. Mol. Appl. Genet. 1, 419 - 434, 1982).
Предпочтительно изобретение предусматривает преобразование сигнального пептида, кодируемого последовательностью гена BARI, которое снижает эффективность отщепления сигнальной пептидазой части Баррьер в белке слияния, включающем точку расщепления KEX2. Это может осуществляться путем сайт-специфического мутагенеза точек возможного расщепления, предпочтительно точек у места соединения аминокислоты 23 24 (последовательности белка Баррьер) или места соединения аминокислоты 24 25.
Отвечающее изобретению конструирование ДНК включает общепринятые приемы. Структурный ген BARI или его часть, а также структурный ген, который экспрессируется, находится под контролем единственного промотора.
Способы лигирования фрагментов ДНК описаны достаточно широко и специалисты в данной области в состоянии их практически осуществить. Кодируемая ДНК последовательность белка, который экспрессируется, может быть по сути последовательностью любого белка, в частности, последовательностью белков коммерческого значения, таких как интерфероны, инсулин, проинсулин, α--1-антитрипсин, факторы роста, активатор тканевого плазминогена.
После получения конструкции ДНК, включающей ген BAR1 или его часть и структурный ген, который экспрессируется, их трансформируют в штаммы хозяина. Способы трансформации прокариотов и эукариотов (включая клетки млекопитающих) уже известны из литературы.
Штамм хозяина (реципиента) представляет собой предпочтительно штамм дрожжей Saccharomyces cerevisiae, однако могут использоваться и другие грибки, включая дрожжи Schizosaccharomyces pombe и нитевидные грибки Axpergillus nidulans и Neurospora spp.
Приведенные ниже примеры даются как иллюстрация. Во всех случаях, за исключением тех, которые указаны особо, используются стандартные методы молекулярной биологии. Рестрикционные эндонуклеазы были получены из научно-исследовательских лабораторий Bethesda, New England BioLabs и Boehringer-Mannheim Biochemicals и использованы в соответствии с указанием изготовителя, обычно с добавлением панкреатической рибонуклеазы (10 мкг/мл) в продукты переваривания.
Т4 ДНК-лигаза была получена из научно-исследовательских лабораторий "Bethesda Res. Laborat. " или "Boehringer-Mannheim" и была использована в соответствии с указаниями.
Штаммы хозяина M13 и pUC и векторы были получены из научно-исследовательских лабораторий "Bethesda".
Клонирование М13 осуществлялось, как описано в работе "Messing" (Methods in Enzymology, 101, 20 77, 1983).
ДНК полимераза 1 (фрагмент Кленова) использовалась, как описано в работе Maniatis et al. "Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory", 1982. Культуры E. coli были трансформированы методом Bolivar et al. Gene, 2, 95-113 1977.
Культуры S.cerevisiae были трансформированы способом Beggs, Nature, 275, 104 108, 1978, модифицированным MacKay (Methods in Enzymology, 101, 325, 1983.
S.pombe был трансформирован, как описано Russell (Nature, 301, 167 -169, 1983).
альфа- фактор спаривающего феромона получен методом Duntze et al. Eur. j. Biochem. 35, 357-365, 1973, модифицированным Manney et fl. j. Cell Biol. 96, 1592 1600, 1983 или был приобретен у фирмы "Sigma Chemical Co".
Олигонуклеотиды были синтезированы в устройстве для синтеза ДНК (Applied Biosystems Model 38OA) и очищены путем электрофореза на полиакриламидном геле или с использованием денатурирующего геля.
Методы анализа.
Анализ на активность Баррьер. Анализ на определение продуцирования Баррьер трансформированными дрожжевыми клетками связан со способностью Баррьер изменять на обратное ингибирование роста чувствительных клеток, подвергнутых воздействию альфа- фактора. Испытываемый штамм был таким штаммом, который проявлял чрезмерную чувствительность к альфа- фактору, такой как штамм RC629 MATa bar1, поскольку он не продуцирует активность Баррьер. Культуру приготавливали с использованием такого штамма в мягком верхнем слое агара в чашке с агаровой средой. В верхний слой агара вводили альфа- фактор в количестве (0,05 0,1 ед, как определено Manney, ibid), достаточном для ингибирования роста клеток. Трансформанты, отбираемые на продуцирование Баррьер, наносились отдельными точками на культуру. Секреция Баррьер трансформированными клетками не ингибировала рост клеток альфа- фактором непосредственно вокруг нанесенной точки. Выделенные клетки наблюдались в виде бахромы роста, окаймляющей обычно гладкий край колонии трансформированных клеток. Наличие этой бахромы указывает на то, что плазмида в трансформированном штамме обеспечивает экспрессию и секрецию Баррьер.
Анализ на IRI и IRC.
Анализ на IRI и IRC осуществляли с использованием набора инструментов, полученного от фирмы "Novo Industri" (Bagsvaerd, Denmark). Вместе с данным набором поставлялись анти-свиной инсулин морских свинок и антитела античеловеческого C-пептида морских свинок.
Анализ на IRI.
Образец в количестве 50 мкл в NaFAM (0,04 моль буферный раствор фосфата pH 7,4, содержащий альбумин бычьей сыворотки).
Антитело в количестве 50 мкл (разбавленный раствор 1:30) 16 24 ч при 4oC.
50 мкл125
I-инсулина (разбавление 1:100) 2 ч при 4oC
50 мкл 1%-ного Staphylococcus aureu в NaFAM 45 мин при 0o
C.
Двукратная промывка 1-ным BSA/TNEN*
*TNEN 20 мМол Трис, pH 8,0; 100мМол. NaCl, 1 мМол этилендиаминтетрауксусной кислоты, 0,5% NP-4.
Центрифугирование и приготовление гранул для отсчета.
Различные этапы могут успешно осуществляться в микротитровальных чашках до тех пор, пока гранулы не переносятся в сцинтилляционные склянки.
Анализ на IRC.
Образец в количество 50 мкл в NaFAM.
50 мкл антитела (разбавленный раствор 1:50) 16 24 ч при 4oC.
50 мкл125I-C пептида (разбавленный раствор 1:30) 2 4 ч при 4oC
50 мкл 1% S. aureus в NaFAM 45 мин при 0oC
Двукратная промывка 1% BSA/TNEN.
* TNEN см. выше.
Центрифугирование и приготовление гранул для отсчета.
Анализ на активность альфа-фактора.
Анализ, используемый для обнаружения экспортируемого трансформированным дрожжевыми клетками альфа-фактора, основан на использовании способности альфа-фактора ингибировать рост чувствительных клеток. Испытываемый штамм [такой как штамм S.cerevisiae RC629 (MATa barl)] содержит мутацию в гене BAR1, которая предотвращает продуцирование белка Баррьер и придает клеткам сверхчувствительность к альфа-фактору. Культивирование испытываемого штамма осуществляется в верхнем слое мягкого агара на чашке со стандартной селективной, синтетической дрожжевой средой (например, средой с недостатком лейцина). Трансформанты, отбираемые на экспортирование альфа-фактора, наносились в виде точек на культуру и термостатировались при 30oC. Секреция альфа-фактора данными трансформантами вызывает ингибирование роста клеток в культуре непосредственно вокруг колонии. Ореол ингибирования роста культуры испытываемых клеток указывает на то, что данная колония выделяет активный альфа-фактор. Сопоставление размера ореола позволяет определить относительные количества альфа-фактора, выделяемого каждым трансформантом.
Пример 1. Экспрессия проинсулина в S.cerevisiae с использованием гена BARI.
Рекомбинатная плазмидная совокупность, включающая весь дрожжевой геном, была построена (Nasmyth и Tatchell, Cell, 19, 753-764, 1980) с использованием челночного вектора YEp13 (Broach et al. Gene, 8, 121-133, 179). Дрожжевые ДНК-фрагменты, полученные в результате частичной обработки Sau 3A рестриктазой, были вставлены в YEp13, гидролизованную Bam HI. Данная плазмидная совокупность была использована для трансформации штамма S. cerevisiae XP635-10C MATa leu2-3 leu2-112 barl1-1 gal12; ATCC 20679) и трансформанты были отобраны на лейциновую прототрофность и на концентрацию альфа-фактора, т. е. подавление клеток a barl. Полученные в результате колонии затем отбирались по способности выделять белок Баррьер, при этом были обнаружены две колонии, которые несут ген, обеспечивающий независимость от лейцина, так и способность выделять белок Баррьер. Эти колонии несут плазмиды, обозначенные как pBAR2 и pBAR3.
Плазмидная ДНК, выделенная из этих двух трансформантов, использовалась для трансформации штамма E.coli RRI (ATCC 31343). Трансформанты были отобраны на стойкость к ампициллину. Плазмиды pBAR3 и pBAR3 были очищены из трансформантов E.coli и были охарактеризованы путем обработки рестрикционной эндонуклеазой и путем электрофореза на агарозных или акриламидных гелях. Плазмида pBAR2, как установлено, включает вставку примерно 9,2 kb. Культура E.coli RRI, трансформированная плазмидов pBAR2, была сдана на хранение в коллекцию ATCC, где находится под номером 39410.
Субклонирование показало, что плазмидная вставка pBAR3 содержит часть вставки pBAR2, но ориентирована в данном векторе в противоположном направлении. Дальнейшее субклонирование и отбор на секрецию Баррьер позволили выявить функциональную генную последовательность BAR1 до области примерно 2,75 kb. Этот фрагмент включает кодирующую последовательность, не транслируемые, но транскрибируемые последовательности, промотор, регуляторные области, концевой терминатор транскрипции и боковые хромосомные последовательности.
Плазмида pBAR2 обрабатывалась рестрикционными эндуклеазами Hind III и Xho I и фрагмент примерно 3 kb очищался путем электрофореза на агарозном геле. Этот фрагмент был вставлен в плазмиду pUC13, которая была гидролизована Hind III и Sal I. Полученная рекомбинантная плазмида, обозначенная как pZV (фиг. 3) могла использоваться для трансформации E.coli, но в ней отсутствовал необходимый источник репликации и селективный маркерный ген для дрожжевого вектора. Данная плазмида pZV9 в штамме E.coli RRI была сдана на хранение в коллекцию ATCC, где хранится под номером 53283.
Из гена BAR1, служащего для направления секреции проинсулина, использовались фрагменты, включающие регуляторную область 5' и часть кодирующей последовательности. Слияние BAR1 фрагментов и гена проинсулина осуществлялось в соответствующей рамке считывания в точке BAR1 последовательности, в которой полученный полипептид слияния может быть расщеплен, в условиях in vivo. Некоторые точки гена являются точками возможного расщепления; Arg-Arg в положении 177 178 была выбрана как испытательная точка слияния с проинсулином. Согласно этому регуляторные 5' последовательности и кодирующей последовательности BARI размером примерно 800 bp были очищены от плазмиды pZV9, как Hind III-Sal I фрагмент размером 1,9 kb.
Фиг. 3 4 иллюстрируют способ субклонирования с ДНК человеческого препоинсулина. с ДНК человеческого препроинсулина (клон preBCA), p27, получается путем G-гидролиза плазмиды pBR327, обработанной Pst I и вставки C-разрезанной ДНК, полученной путем обратной транскрипции общей ДНК из поджелудочной железы человека. Плазмида pBR327 была описана в работе "Soberon et al.", 9, 287 305, 1980 и последовательность человеческого препроинсулина в работе "Bell et al.", Nature, 232, 525 527, 1979. Полная транслированная последовательность была выделена на Nco I-Hga I фрагменте. Выступающие концы были дополнены ДНК-полимеразой I (фрагмент Кленова) и одновременно были присоединены синтезированные связывающие звенья Eco RI (GGAATTCC), связывающие звенья Xba I (CTCTAGAG). Данный фрагмент был субклонирован в pUC13 (Vieira and Messing, Gene, 19, 259 268, 1982; and Messing, Methods in Enzymology 101, 20 -77, 1983), который был обработан Eco RI и Xba I. Поскольку ввод связывающего звена Eco RI в 5' концевом положении восстанавливает точку Nco I (CCATGG) в кодоне инициирования, то плазмиды отбирались на наличие точек Eco RI, Nco I и Xba I, граничащих с вставкой 340 bp. Плазмида, обладающая такими свойствами, носит название p47, как показано на фиг. 4. Фрагмент проинсулина (BCA) с затупленной 5' концевой частью образован в ходе синтеза предшественника (описанного в работе Lawn et al. Nuc. Acids Res. 9, 6103 6114, 1981) плазмиды p47. Последующая обработка Xba I приводит к образовано фрагмента 270 bp, который вставлен в pUC12 (Vieira and Messing, ibid and Messing, ibid). Данный вектор получен путем гидролиза Hind III, затупления концов ДНК полимеразой I (фрагмент Кленова), а также путем обработки Xba I и гелевой очистки. Полученный в результате векторный фрагмент, включающий затупленный конец и клейкий конец Xba I, был лигирован с описанным выше фрагментом BCA. Поскольку созревший BCA начинается с фенилаланина (кодон III), то лигирование затупленного конца данных двух фрагментов восстанавливает Hind III сайт в месте соединения. Плазмиды сначала отбирались на выделение сайта Hind III, затем путем секвенирования зоны соединения с использованием M13 в качестве секвенирующего праймера. Плазмида p254 имела корректную последовательность.
Как показано на фиг. 5, плазмиду p254 обрабатывали с помощью Hind III и Eco RI, а фрагмент проинсулина, составляющий примерно 270 bp, подвергали гелевой очистке. Концы данного фрагмента затуплялись с использованием ДНК-полимеразы I (фрагмент Кленова) и деоксинуклеотидтрифосфатов.
Последовательности связывающего звена Sal I (GGTCGACC) обрабатывались Т4 полинуклеотидкиназой и γ-32P-АТР и лигировались с затупленнным у конца фрагментом гена проинсулина. После обработки Sal I и Bam HI осуществлялся электрофорез на 1, 5-ном агарозном геле, в результате чего получался фрагмент проинсулина с когезивными концами Sal I и Bam HI.
Фрагмент проинсулина и фрагмент BAR1 размером 1,9 kb лигировали с pUC13, который обрабатывался с Hind III и Bam I. Эта конструкция использовалась для трансформации E.coli K12 (JM83).
Трансформированные клетки отбирались на стойкость к ампициллину и продуцированию белых колоний. Дополнительный отбор путем обработки с помощью рестрикционных эндонуклеаз с использованием Hind III, Bam HI и Sal I обнаружил плазмиду (pZV27), содержащую фрагмент Hind III-Bam HI нужного размера и единственную точку Sal I.
Для того, чтобы пришить первую аминокислоту проинсуллина к потенциальной точке обработки генного продукта BAR1 Arg-Arg, осуществлялось удаление материала, расположенного в зоне слияния BARI-проинсулина, при этом использовали синтетический олигонуклеотид для направленного ввода мутации.
Как показано на фиг. 5 плазмиду pZV27 обрабатывали с помощью Hind III и Bam HI и фрагмент слияния BARI-проинсулин подвергали гелевой очистке. Этот фрагмент затем был вставлен в репликационную форму вектора фага M13mp11 (Messing, Methods in Enzymology, 101, 20 77, 1983), который обрабатывался Hind III и Bam HI.
Данная рекомбинантная ДНК использовалась для трансфекции E.coli K12 (JM103) (Messing, ibid). Были выбраны белые пятна и рестрикционные формы рекомбинантного фага отбирались на основании правильных рестрикционных структур путем двукратной обработки ферментов с использование Hind III + Sal и Sal I + Bam HI. Конструкция, показывающая желаемую структуру, представляет собой mp11-ZV29. Олигонуклеотидный праймер (последовательность: 3' GGATCTTCTAAACACTTG 5') метился с использованием γ-32P--АТР и Т4- полинуклеотидкиназы. 7,5 пмолей праймера киназы смешивали с 80 мг секвенированного праймера М13 (Bethesda Research Laboratories, Inc. ). Эта смесь аннелировалась с 2 мкг однониточной mp11-ZV29 и вторая нить строилась с использованием Т4 ДНК-лигазы и ДНК-полимеразы I (фрагмент Кленова), как описано для направленного олигонуклеотидом мутагенеза (двухпраймерного метода) Zoller et al (Manual for Advanced Techniques in Molecular Cloning Course, Cold Spring Harbor Laboratory, 1983).
ДНК, полученная таким образом, использовалась для трансфекции E.coli K12 (JM103) и отбирались колонии, используемые в качестве пробы содержащего киназу олигомера (Zoller et al. ibid). Идентифицированные таким образом колонии использовались для приготовления ДНК репликационной формы фага (RF) (Messing, ibid).
Обработка с помощью рестрикционного фермента (RF) ДНК обнаруживала два клона, имеющих подходящую рестрикционную структуру Xba I (фрагменты 7,5 kb, 0,81 kb и 0,65 kb), отсутствие сайта рестрикции Sal I (который присутствует в зоне делеции слияния BAR1-проинсулин).
RF ДНК от этих двух клонов обрабатывались Hind III и Bam HI и фрагмент слияния 1,9 kb от каждого был подвергнут гелевой очистке. Эти фрагменты пришивались к векторам pUC13 и YEp13 (Broach et al. Gene, 8, 121 133, 1979), которые были обработаны с Hind III и Bam HI. Гибридные плазмиды pUC/BARU-проинсулин для секвенирования трансформировались в E.coli K12 (JM83). Обе эти плазмиды были обозначены pZV31 и pZV33. Рекомбинанты YEp13 используют для трансформации E.coli RRI (Nasmyth and Reed, Proc. Nat. Acad. Sci. USA, 77, 2119 2123, 1980). Две из этих плазмид были обозначены, как pZV30 и pZV31 (фиг. 4).
Секвенирование pZV32 и pZV33 осуществлялось способом Maxam и Gilbert (Methids in Ebzymology, 65, 57, 1980). Последовательность слияния BAR1-проинсулина начиналась от сайта Bgl II, расположенного примерно у 190 bp, и продолжалась до 5' конца места соединения; и начиналась от сайта Sau 961, расположенного примерно у 140 bp и продолжалась до 3'конца места соединения (в проинсулиновом гене). Данные этих экспериментов подтвердили, что получена желаемая конструкция путем слияния между BAR1 и проинсулиновыми генами.
Штамм S.cerevisiae XP635-10C трансформировался плазмидами pZV30 и pZV31. Один литр культуры выращивался в стандартной дрожжевой синтетической среде, в которой отсутствовал лейцин. По прошествии 34 ч в 10-мл аликвоту каждой культуры вводили альфа-фактор. Спустя дополнительных 11 ч культуры собирали путем центрифугирования. Клеточные гранулы и насадочная жидкость (после центрифугирования) подвергались анализу на присутствие инсулина или аналогичного инсулину материала. Результаты двух таких анализов надосадочной жидкости культуры, трансформированной плазмидой pZV31, показали наличие 3 пмоль IRI на мл питательной среды разведения и 5,8 пмоль IRC на мл питательной среды разведения. IRI содержал инсулин, проинсулин или продукты их разложения. IRC был свободен от С-пептида и содержал проинсулин или продукты его разложения.
Пример 2. Экспрессия проинсулина в S.cerevisiae с использованием промотора алкогольдегидрогеназы I, гена BAR1 и терминатора. Триозофосфатизомеразы.
Промотор алкогольдегидрогеназы I S. cerevisiae (далее будет называться промотор ADHI; известен также как промотор ADCI) анализировался на возможность использования для экспрессии чужеродных полипептидов в сочетании с последовательностями BAR1. При этом была построена плазмида, включающая эти последовательности. Плазмида pZV50 (фиг. 8) включает промотор S. ctrtvisiae ADH1, описанное выше слияние BAR1 проинсулин и область терминатора гена S. cerevisiae триизофосфатизомеразы (TPII) (Alber and Kawasaki, J. Molec. Appl. Genet. 1, 419 -434, 1982). Она была построена следующим образом. Как показано на фиг. 7, плазмида pAH5 (Ammerer, ibid) обрабатывалась с помощью Hind III и Bam HI и фрагмент промотора ADHI 1,5 kb подвергался гелевой очистке. Этот фрагмент вместе с полилинкерным фрагментом Hind III Eco RI от pUC13 вставлялся в плазмиду pBR327, обработанную Eco RI, Bam HI с использованием лигазы ДНК Т4. Полученная плазмида, обозначенная как pAM5, гидролизовалась с Sph I и Xba I, и фрагмент промотора ADHI примерно 0,5 kb очищался на 2-ном агарозном геле. Плазмида pZV9 обрабатывалась с Xba I и фрагмент BARI примерно 2 kb, содержащий всю кодирующую область BAR1, подвергался аналогичной гелевой очистке.
Эти два фрагмента, ADHI промотор и последовательность BAR1 сшивались с YEp13, обработанной Xba I, Sph I, в результате чего получалась плазмида pZV24. Обработка pZV24 с Sph I и BqI II с последующей гелевой очисткой приводила к образованию слияния промотора FDHI-BAR1 примерно 800 bp, которое содержало кодон начала трансляции ATG, но в котором отсутствовали кодоны для Arq-Arg сайт возможного процессинга. Плазмида pZV33, содержащая слияние BAR1-проинсулин, обрабатывалась BqI II и Xbf I, и фрагмент слияния (примерно 500 bp), который включал кодоны для Arq-Arq, подвергался очистке.
Как показано на фиг. 9, терминатор TPI1 был получен из плазмиды pFGI (Alber and Kawasaki, ibid). Плазмиду pFG1 обрабатывали Eco RI, концы линеаризованной плазмиды затуплялись с использованием ДНК полимеразы 1 (фрагмента Кленова) и вводились последовательности линкера Bav HI (CGGATCCA). Данный фрагмент обрабатывался Bav HI, и снова сшивался с образованием плазмиды p136. Терминатор TPII 700 bp очищался от p136 как фрагмент Xba I-Bam HI. Данный фрагмент вставлялся в YEp13, гидролизованную с Xba I и Bam HI, который затем обрабатывался Hind III, края затуплялись с использованием ДНК полимеразы I (фрагмента Кленова), и снова сшивались, образуя плазмиду p270. Терминатор TPI1 очищался от p270 как фрагмент Хba I-Bam HI и вставлялся в pUC13, обработанную Xba I, Bam HI, в результате чего получалась плазмида m115.
Как показано на фиг. 8, терминатор TPI1 удалялся из плазмиды m115 путем обработки с Xba I и Sst I с последующей гелевой очисткой. Три фрагмента - фрагмент слияния ADHI-BAR1, фрагмент слияния BAR1-проинсулин и концевая части TP11 вставлялись в плазмиду pUC18 (Norrander et al. Gene 26: 101 106, 1983), которая была обработана Sph I и Sst I. Данная ДНК была использована для трансформации E. coli K12 (JM83). Отбор на устойчивость к ампициллину и отбор на продуцирование белых колоний обнаружили плазмиду pZV45, содержащую желаемые вставки. Плазмида pZV45 далее гидролизовалась Sph I т Bam HI, и последовательность ADHI-BAR1-проинсулин-терминатор TPI подвергалась гелевой очистке. Этот фрагмент вставлялся в YEp13, которая обрабатывалась Sph I и Bam HI, в результате чего получался вектор экспрессии S.cerevisiae pZV50.
Штамм S.cerevisiae XP635-10C трансформировался плазмидой pZV50 и культивировался, а затем анализировался, как описано в примере 1. В среде разведения не было обнаружено никакого материала IRI и было обнаружено менее чем 0,5 пикомоля IRC на мл. Клетки, экстрагированные 0,1% Nonidet P-40, показали присутствие IRC материала с концентрацией 1 пикомоль на мл клеточного экстракта.
Пример 3. Экспрессия проинсулина в Schizosaccharjmyces pombe и использованием гена BAR1 и промотора алкогольдегидрогеназы S.pombe.
На данном примере демонстрируется использование фрагментов гена BAR1 для направления секреции чужеродных полипептидов, экспрессированных в трансформированном хозяине Schizosaccharomyces pombe. При этом была построена плазмида, которая сочетает промотор (ADH) алкогольдегидрогеназы S. pombe с геном слияния BAR1-проинсулин.
Промотор ADH S.pombe был получен из библиотеки фрагментов ДНК, образованных от штамма S.pombe 972h- (АТСС 24843), которые были клонированы в YEp13, как описано в работе Russel and hall (J. Biol. Chem. 258, 143-149, 1983). Данная промоторная последовательность была выделена из библиотеки как фрагмент SphI -Eco RI 0,7 5 kb. Этот фрагмент и фрагмент многозвенного линкера Eco RI-Hind III плазмиды pUC12 были вшиты в YEp13, которая обрабатывалась Shp I и Hind III. Полученная плазмида известна как pEVP-11.
Как показано на фиг. 10, иллюстрирующей построение вектора экспрессии S. pombe, промотор ADH очищали от pEVP-11 как фрагмент Sph I-Xba I.
Плазмида pZV33 вываривалась с Xba I и Bgl II, и фрагмент BAR1 примерно 340 bp, который включал инициирующий кодон ATG, подвергался очистке, pZV33 обрабатывался с Bgl II и Sst I, и последовательность слияния BARI-пронсулин подвергалась очистке. Эти три фрагмента комбинировались с pUC18, гидролизованной Sph I, Sst I, в результате чего получалась плазмида pZV46. Поскольку pUC18 неэффективна для трансформации S.pombe, то данная плазмида подвергалась двум обработкам ферментами. Фрагмент слияния ADH промотор-BAR1 очищался от Hind III-Bgl II-фрагмента и последовательность слияния BAR1-пронсулин очищалась от Bgl II-Xba I фрагмента. Эти фрагменты вставлялись в YEp13, обработанную Hind III, Xba I, в результате чего получался вектор экспрессии pZV49 в S. pombe.
Один литр культуры трансформированного штамма S. pombe 118-4h- (АТСС 20680) выращивали в течение 36 ч при 30oC в стандартной синтетической дрожжевой среде (-leu D), содержащей 200 мг/л аспаргиновой кислоты и по 100 мг/л гистидина, аденина и урацила. Культуры собирали путем центрифугирования и надосадочную жидкость анализировали на IRI и IRC. Две пробы трансформированных pZV49 клеток содержали 1, 6 пмоль/мл материала IRI и 0,5 пмоль/мл IRC-активного материала соответственно. Контрольная проба от культуры, трансформированной YEp13, не показала наличия IRC-активного материала.
Пример 4. Выделение альфа-фактора с использованием сигнального пептида BAR1.
Сигнальный пептид BAR1 подвергался анализу на определение способности выделять альфа-фактор из дрожжевого трансформанта. При этом было построено несколько плазмид, содержащих фрагменты ДНК, кодирующие белки слияния с различной длиной белка BAR1, и 1 или 4 копии созревшего альфа- фактора. Эти плазмиды трансформировались в штамм а/α-диплоидного хозяина, а трансформанты анализировались на продуцирование альфа- фактора по наличию ореола.
Плазмиды pSW94, pSW95, pSW96 и pSW97 включают промотор триозофосфатизомеразы (TPI1) S.cerevisiae, фрагмент 355 bp или 767 bp гена BARI (содержащего 114 или 251 кодонов 5' конца кодирующей последовательности BARI1 соответственно) и либо одну, либо четыре копии кодирующей последовательности альфа-фактора (MF a). Эти конструкции приводятся в табл. 1.
Плазмида pM220 (известная также как pM210) использовалась как источник фрагмента стимулятора TPI1. E.coli RRI, трансформированная плазмидой pM220, была депонирована в коллекции АТСС, где находится под номером 39853. Плазмида pM220 обрабатывалась Eco RI, и фрагмент 0,9 kb, включающий промотор TPI1, был выделен путем электрофореза на агарозном геле, и края его были затуплены ДНК полимеразой I (фрагментом Кленова). Содержащие киназу линкеры Xba I встраивались в данный фрагмент, который затем обрабатывался с Bgl и Xba I. Этот фрагмент модифицированного промотора TPI1 затем встраивался в векторный фрагмент 3,4 kb Bgl II-Xba I плазмиды pDR1107, в результате чего получалась pZV118. Плазмида pDR1107 была построена путем субклонирования фрагмента промотора TPI1 900 bp. Bgl II-Eco RI плазмиды pM220 в pIC7 (Marsh, Erfle and Wykes, Gene, 32, 481 485, 1984), в результате чего образуется плазмида pDR1101. Плазмида pDR1101 была обработана Hind III и Sph I для выделения фрагмента промотора TPI1 700 bp. Плазмида pDR1100, включающая фрагмент терминатора размером 800 bp Xba I-Bam HI, плазмиды pM220, субклонированный в pUC18, гидролизовалась Hind III и Sph I. Частичный фрагмент промотора TPI1 700 bp встраивался в линеаризованную pDR1100, и в результате получалась pDR1107. Затем сайт Eco RI на 3' конце промотора TPI1 в плазмиде pZV118 застраивают. Данную плазмиду обрабатывают Hind III и Eco RI, и фрагмент 0,9 kb выделяют и сшивают с синтетическим линкером, полученным путем слияния олигонуклеотидов ZC708 (5' AATTGCTCGAGT3' и ZC709 (3' CGAGCTCAGATC5'). Ввод этого линкера убирает сайт Eco RI на 3' концевой части фрагмента промотора TPI1 и вводит сайты XhoI и Xba I. Затем данный фрагмент присоединяется к pUC13, а Hind II-Xba I фрагмент вырезается. Образованная плазмида обозначается как pZV134 (фиг. 11).
Клонирование гена альфа-фактора (MF a1) дрожжевого спаривающего феромона было описано Kurjan и Herskowitz (ibid). Этот ген был выделен в данной лаборатории из дрожжевой геномной библиотеки частичной обработкой фрагментов Sau 3A, клонированных в точку Bam HI вектора YEp13 (Nasmyth and Tatchell, Cell, 19, 753 764, 1980). Из этой библиотеки была выделена плазмида, которая экспрессировала альфа-фактор в диплоидном штамме гомозиготных дрожжей с мутацией mata2-34 (Manney et al. J. Cell. Biol. 96, 1592, 1983). Клон, содержащий вставку, охватываемую геном MFa1, был охарактеризован Kurjan и Herskowitz. Данную плазмиду, известную как pZA2, обрабатывали EcoRI, и фрагмент 1,7 kb, содержащий MFa1, был выделен и встроен в Eco RI сайт плазмиды pUC13. Полученная плазмида, обозначенная как p192, расщеплялась Eco RI, и образующийся фрагмент MFa1 1,7 kb выделен и обработан Mbo II. Фрагмент Mbo II-Eco RI размером 550 bp был выделен и сшит с содержащими киназу линкерами Sal I. Этот сшитый фрагмент был обработан Sal I. Полученный фрагмент Sal I размером 0,3 kb встроен в сайт Sal I плазмиды pUC4 (Viera and Messing, Gene, 19, 259-268, 1982) и в результате получалась плазмида, обозначенная как p489 (фиг. 12).
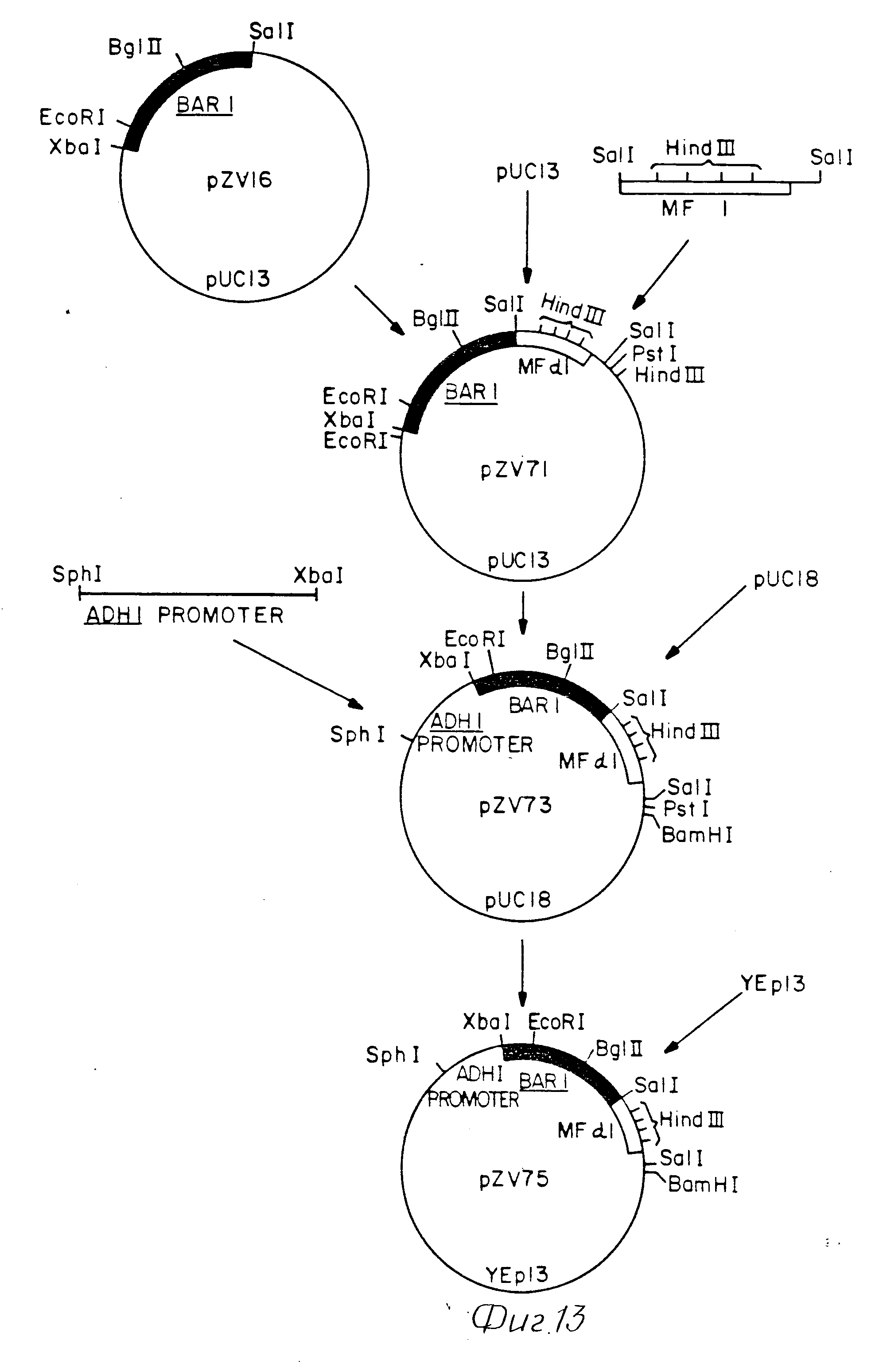
Затем было построено слияние гена, включающее части BARI (114 кодонов) и кодирующей последовательности MFa1. Плазмида pSV24 (пример 2) обрабатывалась с Sph I и Bgl II, был выделен фрагмент промотор ADHI-BAR1 размером 0,8 kb. Плазмида p489 расщеплялась Bam HI и выделялся фрагмент MFa1 размером 0,3 kb. Эти два фрагмента сшиты Sph I-Bam HI, фрагментом YEp13. Полученная плазмида обозначена как pZV69 (фиг. 14).
Затем был построен второй ген слияния, кодирующий 251 аминокислоту Баррьер, связанный с частью предшественника альфа- фактора. Плазмида pZV16, включающая фрагмент Xba I-Sal I с BAR1 размером 767 bp плазмиды pZV9 (пример 1), сшитый с вырезом Xba I-Sal I, фрагментом pUC13 линеаризовалась с Sal I. Этот фрагмент размером 4,0 kb соединялся с Sal I фрагментом p489 размером 0,3 kb, кодирующим четыре копии альфа- фактора. Плазмида, включающая слияние BAR1-MFa1 в правильной ориентации, обозначена как pZV71. Слияние BAR1-MFa1 от pZ71 затем было соединено с промотором ADH1. Плазмиду pZV71 обрабатывали Xba I и Pst I и выделяли фрагмент размером 1,07 kb. Промотор ADH1 был выделен на фрагменте Sph I-Xba I размером 0,42 kb из pZV24. Эти два фрагмента были соединены с фрагментом Sph I-Pst I pUC18. Полученная в результате плазмида pZV73 обрабатывалась Sph I и Bam HI и фрагмент 1,5 kb, включающий данную единицу экспрессии, был выделен и лигирован с фрагментом Sph I-Bam HI, плазмиды YEp13, образуя pZV75 (фиг. 13).
Для облегчения фрагменты слияния BAR1-MFa1 из pZV69 и pZV75 были субклонированы с промотором TPI1 в pUC18 (фиг. 11). Плазмида pZV69 обрабатывалась Eco RI и Bam HI и был выделен фрагмент размером 0,55 kb, содержащий данное слияние. Фрагмент промотора 0,9 kb TPI1 был выделен из pZV118 путем обработки Hind III и Eco RI. Сшитый из трех частей фрагмент был получен с использованием BAR1-MFa1 фрагмента размером 0,55 kb, TPI1 фрагмента размером 0,9 kb и pUC19, обработанной Hind III и Bam HI. Полученная в результате плазмида была обозначена как pSW59. Плазмида pZV75 обрабатывалась Eco RI и Bam HI, в результате чего выделялся фрагмент слияния BAR1-MFa1 размером 954 bp. Этот фрагмент BAR1-MFa1 был лигирован с фрагментом промотора TPI1 Hind III+Eco RI размером 0,9 kb с образованием лигированного фрагмента, состоящего из трех частей, и pUC18 обрабатывалась Hind III и Bam HI, образуя плазмиду pSW60.
При построении плазмид экспрессии источником 5' 116 bp кодирующей последовательности BAR1 являлась pSW11, которая была построена следующим образом (фиг. 12). Кодирующая область BAR1 в pSW22 происходит от pZV9. Плазмида pZV9 (пример 1) обрабатывалась Sal I и Bam HI, в результате чего был выделен BARI фрагмент размером 1,3 kb. Этот фрагмент лигировали SaI и Bam HI pUC13, в результате чего образовалась плазмида, обозначенная как pZV17. Плазмида pZV17 обрабатывалась Eco RI, в результате чего удалялась небольшая 3' часть в кодирующей последовательности BARI размером 0,5 kb. Векторный BAR1 фрагмент снова сшивался, образуя плазмиду, обозначенную как pJH66. Плазмида pJH66 линеаризовалась Eco RI и затуплялась фрагментом Кленова. Затем вводили содержащие киназу линкеры Bam Hi (5' CCGGATCCGG3') и избыточные линкеры удалялись путем обработки Bam HI. Полученная плазмида, pSW8, обрабатывалась Sal I и Bam HI, и в результате был выделен фрагмент размером 824 bp, кодирующий аминокислоты 252 525 BAR1. Данный фрагмент BAR1 сшивался с фрагментом, кодирующим C-концевую часть вещества P (Munro and Pelham, EMBO J. 3, 3087 3093, 1984). Плазмида pPM2, содержащая синтетическую олигонуклеотидную последовательность, кодирующую димерную форму вещества P в M13mp8, была получена от Munro и Pelham.
Плазмида pPM2 была обработана Bam HI и Sal I и сшивалась с фрагментом BAR1 размером 824 bp. Полученная в результате плазмида pSW14 обрабатывалась Sal I и Sma I, в результате чего выделялся фрагмент BAR1 вещество P размером 871 bp. Плазмида pZV16 (фиг. 10) обрабатывалась Xba I и Sal I и в результате выделялась 5' кодирующая последовательность фрагмента BARI размером 767 bp. Этот фрагмент был сшит вместе с фрагментом BAR1 вещество P размером 871 bp плазмиды pUC18, обработанной Xba I и Sma I, образуя лигированный фрагмент из трех частей. Полученная плазмида была обозначена как pSW15. Плазмида pSW15 обрабатывалась Xba I и Sma I, в результате чего выделялся фрагмент BAR1-вещество P размером 1,64 kb. Был получен промотор ADH1 pRL029, включающий фрагмент Sph I, Eco RI размером 0,54 kb, содержащий промотор ADH1 и 5' кодирующую зону BAR1 pZV24 размером 116 bp в pUC18. Плазмида pRL029 обрабатывалась Sph I и Xba I, в результате чего был выделен фрагмент промотора 0,42 kb ADH1. Терминатор TPI1 (Alber and Kawasaki, J. Mol. Appl. Gen. 1, 419 434, 1982) выделялся как фрагмент Xba I Eco RI размером 0,7 kb из pUC18. Данный линеаризованный фрагмент, содержащий терминатор TPI1 и Xba I-Sph I pUC18 и фрагмент Кленова, сшивался с фрагментом промотора ADH1 размером 0,42 kb и с фрагментом BARI-вещество P размером 1,64 kb рекомбинантного фрагмента, состоящего из трех частей, образуя плазмиду PSW22.
Затем была построена плазмида pSW94 (фиг. 13). Фрагмент размером 2,3 kb, содержащий слияние BARI-вещество P и терминатор TPI1, был выделен из плазмиды pSW22 как фрагмент Xba I-Sst I.
Hind III-Xba I фрагмент промотора TPI1, выделенный из pZV134, соединялся с фрагментом BAR1-вещество P концевая часть TPI1 путем сшивки трех частей pUC18, Hind III-Sst I фрагментом pUC18. Полученная плазмида pSW81 разщеплялась Hind III и Eco RI и в результате выделялся фрагмент размером 1,02 kb, содержащий промотор TPI1 и 5' фрагмент BARI размером 116 bp. Плазмида pSW59 обрабатывалась Eco RI и Bam HI, в результате чего выделялся фрагмент слияния BAR1-MFa1 размером 0,55 kb. Данный фрагмент затем сшивался тремя фрагментами: TPI1 промотор BARI из pSW81 и YEp13, которую предварительно обработали Hind III и Bam HI, образуя в результате плазмиду pSW94.
Конструкция плазмиды pSW95 иллюстрируется на фиг. 16. Плазмида pSW60 обрабатывалась Eco RI и Bam HI, в результате чего выделен фрагмент слияния BAR1-MFa1 размером 954 kb. Плазмида pSW81 обрабатывалась Hind III и Eco RI, и в результате выделялся фрагмент промотора TPI1-BAR1 размером 1,02 kb, который соединялся с фрагментом BAR1-MFa1 в результате сшивки трех частей с фрагментом Hind III-Bam HI плазмиды YEp13. Полученная плазмида была названа pSW95.
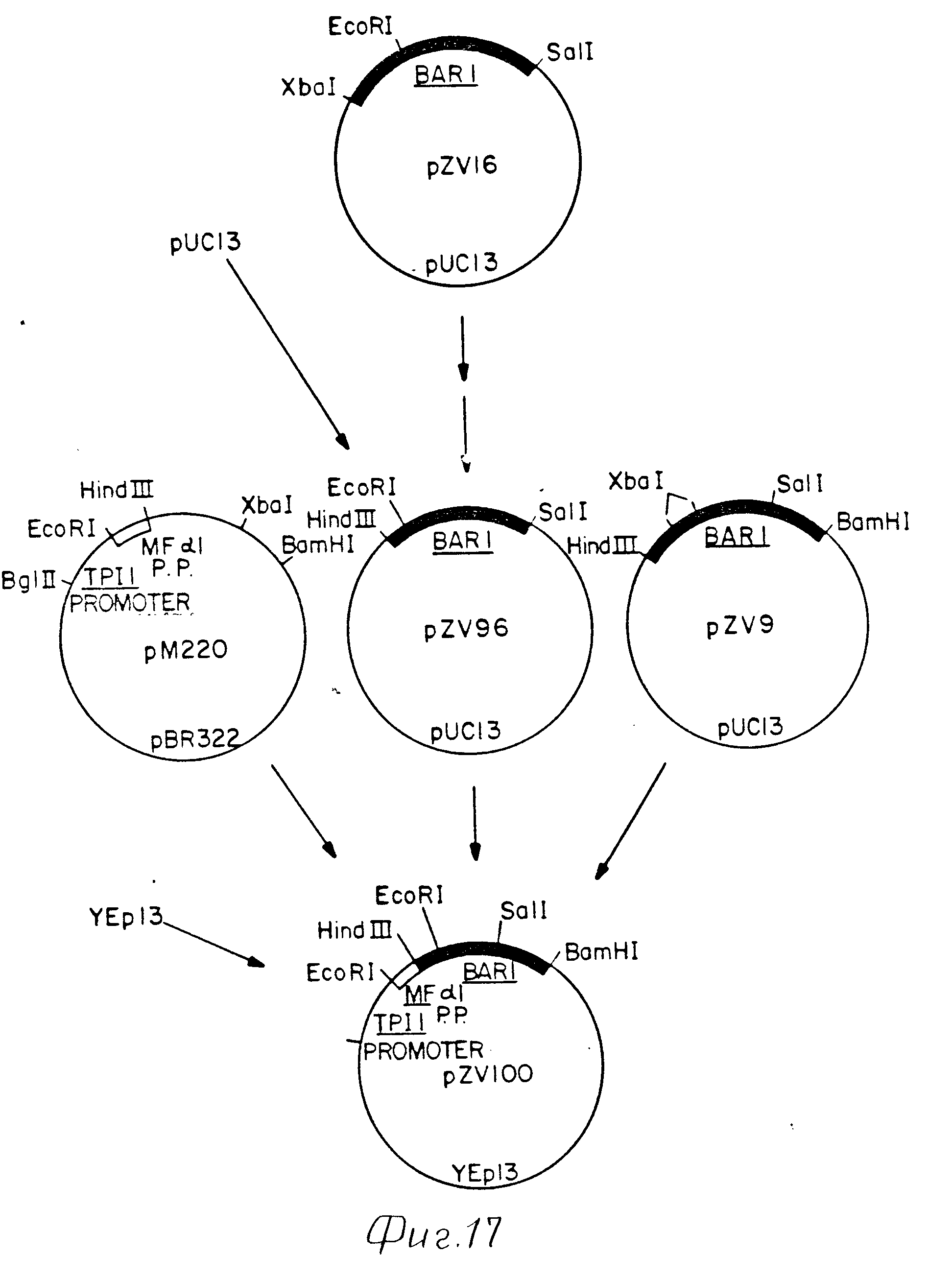
Конструкция фрагментов слияния стимулятор TPI1-BAR1-MFa1, содержащая лишь одну копию альфа-фактора, была образована от слияний BAR1-MFa1 (кодирующих четыре копии альфа- фактора), которые содержали промотор TPI1 и препоследовательность MFa1 (фиг. 17 19). Плазмида pZV16 обрабатывалась Eco RI и Sal I. Выделенный фрагмент BARI размером 651 bp встраивался вместе с содержащим киназу специфическим адаптором Hind III-Eco RI BAR1 (получен путем слияния олигонуклеотида ZC566:5'AGCTTTAACAAACGATGGCACTGGTCACTTAG3' и олигонуклеотида ZC567:5' AATTCTAAGTGACCAGTGCCATCGTTTGTTAA3') с Hind III-Sal I фрагментом pUC13. Полученная плазмида pZV96 обрабатывалась Hind III и Sal I, в результате чего выделялся фрагмент BAR1 размером 684 bp. Плазмида pM220 содержала промотор TPI1 слитый с препоследовательностью MFa1. Плазмида pM220 обрабатывались Bgl II и Hind III, в результате чего получали фрагмент промотора TPT1 препро MFa1 размером 1, 2 kb.3' часть зоны кодирующей BAR1 получалась в результате обработки Sal I и Bam HI плазмиды pZV9, в результате чего был выделен фрагмент BAR1 размером 1,3 kb. Hind III-Sal I BAR1 фрагмент размером 684 bp промотор TPI1 [Bgl II-Hind] препро MFa1 размером 1,2 kb и фрагмент Sal I-Bam HI- BAR1 (1,3 kb) встраивались в YEp13 на линеаризованной Bam HI путем лигирования четырех частей. Данная конструкция с желаемой ориентацией промотора и слияниям MG a 1-BAR1 была обозначена как pZV100 (фиг. 17).
Для облегчения обработки усеченный фрагмент слияния препропоследовательность MFa1-BAR1 от плазмиды pZV100 субклонировался в pUC13 как фрагмент Pst I-Bam HI (1,6 kb). Полученная плазмида pZV101 обрабатывалась Pst I и Eco RI, в результате чего был выделен фрагмент препро MFa1-BAR1 размером 270 bp. Плазмиду pZV69 обрабатывали Eco RI и Bam HI, в результате чего был выделен фрагмент слияния BAR1-MFa (кодирующий четыре копии альфа- фактора) (0,55 kb). Этот фрагмент и фрагмент препро MFa1-BAR1 (270 kb) лигировался с Pst I Bam HI фрагментом pUC13. Образующаяся плазмида была обозначена как pZV102 (фиг. 18).
Затем было построено звено экспрессии, включающее промотор TPI1, часть BAR1 и единственную копию кодирующей последовательности альфа-фактора (фиг. 16). Плазмида pZV102 была обработана Pst I и Bam HI, в результате чего был получен фрагмент препро MFa1-BAR1 (0,82 kb). Hind III-Pst I фрагмент (1,0 kb), включающий промотор TPI1 и усеченную препропоследовательность MFa1 от pM220, был соединен вместе с фрагментом препро MF a1-BAR1 (0,82 kb), выделенным из pZV102 путем лигирования с YEp13, обработанной Hind III и Bam HI. Образованная плазмида была обозначена как pZV105. Плазмиду pZV105 гидролизовали Hind III с выделением фрагмента промотор TPI1-препро MFa1 (1,2 kb). Плазмида pZV102 обрабатывалась Hind III, в результате чего выделялся векторный фрагмент, содержащий концевую копию альфа- фактора. Этот фрагмент вектор -MFa1 (2,8 kb) был сшит с фрагментом промотор TPI1-препро MFa1 (1,2 kb). Данная плазмида с правильной ориентацией и единственной копией кодирующей последовательности MFa1 была обозначена как pSW61. Плазмида pSW61 линеаризировалась путем частичного гидролиза Hind III. Плазмида pZV102 была обработана Hind III, в результате чего был выделен фрагмент BAR1-MFa1 (0,3 kb). Этот фрагмент был встроен в pSW61. Данная плазмида с указанной вставкой в Hind III (284 bp) в правильной ориентации в 3' к начальному кодону MF a 1 была обозначена как pSW70. Плазмида pSW70 обрабатывалась Eco RI и Bam HI, и в результате был выделен фрагмент BAR1-MFa1 размером 361 bp. Плазмида pSW81 (фиг. 16) обрабатывалась Hind III и Eco RI, и в результате был выделен фрагмент промотор TPI1-BAR1 размером 1,02 kb. Этот фрагмент был слит с фрагментом BAR1-MFa1 путем лигирования трех частей с YEp13, обработанной Hind III и Bam HI. Полученная плазмида pSW96 содержит промотор TPI1 и 5' кодирующую последовательность BAR1 (356 bp), слитой с одной копией кодирующей последовательности альфа- фактора.
Вторая конструкция BAR1-MFa1, содержащая фрагмент BAR1 (767 bp), слитый с одной копией кодирующей последовательности MFa1, была получена с использованием pZV75 в качестве источника фрагмента BAR1 (фиг. 20). Плазмида pZV75 обрабатывалась с Eco RI и Bam HI, в результате чего был выделен фрагмент BAR1-MFa1 (размером 954 bp). Плазмида pZV101, содержащая препропоследовательность MFa1, слитую с BAR1, обрабатывалась Pst I и Eco RI, в результате чего был выделен фрагмент препро MFa1-BAR1 (0,27 kb). Этот фрагмент был соединен с фрагментом BAR1-MFa1 (954 bp), в результате лигирования в виде сшивки, состоящей из трех частей с pUC13, обработанной с Pst I и Bam HI. Полученная плазмида pZV104 была обработана Hind III, в результате чего был выделен фрагмент BAR1-MFa1 (0,70 kb). Этот фрагмент был встроен в pSW61, которая была линеаризована в результате обработки Hind III. Данная плазмида со вставкой в Hind III в правильной ориентации (264 bp) к 3'- начальному кодону MFa1 была обозначена как pSW74. Плазмида pSW74 была обработана Eco RI и Bam HI, в результате чего был получен фрагмент BAR1-MFa1 (738 bp). Плазмида pSW81 была обработана Nind III и Eco RI и в результате был выделен фрагмент промотор TPI1-BAR1 размером 1,02 kb. Этот фрагмент был лигирован с фрагментом BAR1-MFa1 (738 bp) в результате сшивки трех частей с фрагментом Hind III Bam YEp13. Полученная плазмида pSW97 включает промотор TPI1 и 5' конец BAR1 (767 bp), слитый с единственной копией кодирующей последовательности альфа- фактора.
Диплоидный штамм a/a S. cerevisiae XP733 (MATa leu2-3 leu2-112 barl-1 gal2/Mataleu2-3 leu2-112 bar1-1 gal2) был трансформирован плазмидами pSW73, pSW94 и pSW95. Плазмида pSW73 включает промотор TPI1, сигнальный пептид и препропоследовательность MFa и кодирующую зону четырех копий альфа- фактора в YEp13. Данные трансформанты наносились как пятна на культуру клеток S. cerevisiae RC629 в мягком поверхностном агаровом слое на чашке с селективной средой и инкубировались в течение ночи при 30oC. Сопоставление размеров ореолов с использованием pSW73 и контрольной пробы показывает, что pSW94 выделяет не менее примерно 15% количества альфа- фактора кодируемого pSW73.
Конструкции, содержащие BAR1, слитый с одной копией кодирующей последовательности MFa1, анализировали на выделение альфа-фактора таким же образом. Плазмида pSW67, включающая промотор TPI1, сигнальный пептид MFa, препро и кодирующую зону одной копии альфа- фактора в YEp13, использовалась как контрольная для плазмид pSW96 и pSW97. Сопоставление размеров ореола показывает, что pSW96 направляет секрецию примерно до 30 40% количества альфа- фактора pSW67, и pSW97 направляет секрецию примерно до 10 15% количества альфа- фактора pSW67.
Пример 5. Мутация точки расщепления сигнального пептида BAR1.
Как описывалось выше, установлено, что изменение точки расщепления сигнального пептида предшественника Баррьер может упростить процесс и выделение содержащих Баррьер белков слияния по пути KEX2.
Возможные точки расщепления находятся между аминокислотами 23 и 24 и между аминокислотами 24 и 25. Так, последовательность ДНК, кодирующая первичный продукт трансляции BAR1, мутировали в положении 25, кодирующем пролиновый остаток. Плазмиды pSW98 и pSW99 представляют собой основанные на YEp13 плазмиды, включающие промотор S. cerevisiae, фрагменты гена BAR1 (355 bp или 767 bp), включающие точку расщепление точку расщепления сигнального пептида, и одну копию кодирующей последовательности альфа- фактора.
Мутация последовательности, кодирующей сигнальный пептид была осуществлена стандартными способами мутагенеза в условиях in vitro (Zoller et al. Manual for Advanced Techniques in Molecular Cloning Course. Cold Spring Harbor Laboratory, 1983) с использованием модели фага M13 и синтетического мутагенного олигонуклеотида (последовательность5'ATTACTGCTCCTACAAACGAT3'). Модель фага pSW54 была построена путем сшивки Sph I-Eco RI фрагмента плазмиды pSW22 (0,54 kb) с M13mp19, обработанном Sph I-Eco RI. После мутагенеза, в условиях in vitro, потенциально содержащие мутации фаги отбирались путем гибридизации фага с32P-меченым олигонуклеотидом, содержащим мутацию, и выстраивались в последовательность, которая подтверждала наличие мутации. Репликативная форма одного из фагов mZC634-7, содержащая мутацию, была обработана Sph I и Eco RI и был выделен фрагмент (0, 54 kb), который был лигирован с Sph + Eco RI фрагментом. Образованная плазмида, pSW66 (фиг. 21) была обработана Hind III и Xba I, в результате чего удалялся промотор ADH1, и фрагмент, включающий последовательности данного вектора и BAR1, был сшит с Hind III-Xba I фрагментом промотора TPI1 (0,9 kb) плазмиды pSV134. Эта плазмида с промотором TPI1 и 5'-концом фрагмента BAR1 (119 bp), включающая мутацию в точке отщепления сигнального пептида, была обозначена как pSW82.
Как показано на фиг. 21, плазмида pSW82 обрабатывалась Hind III и Eco RI и с Bgl II и Eco RI, и были выделены фрагменты размером 1,02 kb. Фрагмент Hind III-Eco RI плазмиды pSW82 был лигирован с фрагментом Eco RI-Bam HI плазмиды pSW74 с Hind-Bam HI фрагментом YEp13, образуя pSW99. Фрагмент Bgl II-Eco RI плазмиды pSW82 был лигирован с фрагментом Eco RI-Bam HI (0,30 kb) плазмиды pSW70 и YEp13, обработанной Bam HI, образуя pSW98. Плазмида pSW98 включает промотор TPI1, 5'-конец последовательности BAR1, содержащий мутацию (355 bp) и единственную копию кодирующей последовательности альфа- фактора. Плазмида pSW99 включает идентичное звено экспрессии, за исключением того, что имеет последовательность BAR1, содержащую мутацию, размером 767 bp.
Анализ путем измерения ореола показал, что мутация точки расщепления усиливает выделение альфа- фактора при использовании плазмид, кодирующих одну копию альфа- фактора. Трансформанты, содержащие pSW98, выводят примерно на 50% больше альфа- фактора, чем трансформанты, содержащие pSW96 (контроль естественного типа).
Пример. Экспрессия pJH39 в дрожжах.
A. Конструирование плазмиды pJH39.
Последовательности ДНК, кодирующие BAR1, использовали для секреции активатора плазминогена ткани (tPA). Кодирующую BAR1 последовательность (от -17 до 342), показанную на фиг.1 настоящей заявки, лигировали последовательностью ДНК, кодирующей зрелую форму tPA, как описано ниже.
кДНК, кодирующую активатор плазминогена ткани (tPA), получали из плазмиды pDR1296, депонированной в качестве трансформанта E.coli в коллекции American Type Culture Collection (Rockville, MD) под номером 53347. Плазмида pDR1296 включает промотор TPI1, tPA кДНК и векторные последовательности pUC19. Терминатор TPI1 получали в качестве фрагмента Xba 1-Bam (0,8 kb) и субклонировали в pDR1296, расщепленную Xba I-Bam HI, с образованием конструкции плазмиды pJH33. Плазмиду pJH33 расщепляли Sph I и Bgl II с выделением фрагмента (5,1 kb), содержащего вектор. Плазмиду pZV24 (пример 2 заявки) расщепляли Sph I и Bgl II с выделением фрагмента (0,76 kb), включающего промотор ADH1 и кодированную BAR1 (от -17 bp до 342 bp), показанную на фиг. 1 3. Фрагмент Sph I-Bgl II pZV24 и фрагмент (5,1 kb) pJH33 лигировали. Полученную плазмиду обозначили pJH35.
Единицу экспрессии, присутствующую в плазмиде pJH35, субклонировали в дрожжевой челночный вектор YEp13. Плазмиду YEp13 расщепляла Bam HI и Sph I с выделением фрагмента, содержащего вектор. Плазмиду pJH35 расщепляли Sph I и Bam HI с выделением единицы экспрессии (3,1 kb). Sph I-Bam HI фрагменты YEp13 и Sph I-Bam, pJH35 лигировали. Полученную плазмиду обозначили pJH39.
Плазмидой pJH39 трансформировали штамм Saccharomyces cerevisiae E8-11C (MATaleu2-3, 112 pep4-3) по методу, описанному по существу автором Hinnen et at (Proc. Natl. Acad. Sci. USA, 57, 1929 1933, 1978). Трансформанты отбирали в зависимости от их способности расти с синтетической полной среде, не содержащей аминокислоту лейцин.
Трансформаторы подвергали фибринолитическому анализу на определение их способности tPA. Было найдено, что трансформанты секретировали 22 мкг/л (фибринолитическая активность).
Пример VII.
Пример 1. Трансформация клеток-хозяина и экспрессия аналога инсулина MI-3,
Векторы
экспрессии pSW167 и pSW200, включающие единицы экспрессии в векторе YEp13, и векторы экспрессии pSW210, pSW219 и pZV187, включающие единицы экспрессии в векторе pSW197, трансформированы стандартными
способами в приемлемые дрожжевые хозяева. Штаммы-хозяева S. cerevisiae содержали мутации, которые дополняют селектируемые маркерные гены, присутствующие в плазмидах.
Плазмиду pSW167, включающую кодирующую последовательность для первых 526 аминокислот кодирующего участка BAR1, слитую с кодирующей последовательностью MI-3 в YEp13, и плазмиду pSW200, включающую кодирующие последовательности для сигнального пептида BAR1 и третий участок BAR1, слитый с кодирующей последовательностью MI-3 в Yp13, трансформировали в штамм S. cerevisiae (MATa leu2-3 leu2-112 ura3 pep4:URA3 barl gal2). Трансформанты отбирали в соответствии с их способностью расти в синтетической среде выращивания, не включающей лейцина.
Трансформаторы выращивали в течение ночи при 30oC в 5 мл -LeuD (Wickerham, L. J. Bact. 52, 293 301, 1946; содержащие Difco Yeast Nitrogen Base в качестве источника азота). Трансформанты разбавляли в соотношении 1: 100 в 20 или 50 мл -LeuD) и выращивали при 30oC в течение 24 или 48 ч. Клетки гранулировали и промывали до замораживания при -70oC. Истощенную среду дважды центрифугировали и декантировали ее от клеточного материала перед замораживанием при -70oC. Содержание MI-3, определенное с помощью радиоиммуноанализа (RIA, см. пример II), показало, что трансформанты pSW167 продуцировали 38 пиког/мл иммунореактивного материала MI-3, в то время как трансформанты pSW200 продуцировали 113 мкг/мл иммунореактивного материала MI-3 в течение 54 ч.
Плазмиду pSW210, включающую последовательность, кодирующую первые 526 аминокислот BAR1, слитую с кодирующей последовательностью MI-3 в pSW197, и плазмиду pSW219, включающую кодирующие последовательности сигнального пептида BAR1 и третий участок BAR1, слитые с кодирующей последовательностью MI-3 в pSW197, трансформировали в штаммы S. cerevisiae GA18-1C (Mata leu2-3 leu2-112 ura 3Dtpil:LEU2 [ciro]) и ZM114 (MATa leu2-3, 112, ura3-52 ade2-101 pep4:TPI промотор-CATDtpil:URA3 vpt3 suc2-D[ciro]). Трансформанты отбирали в соответствии с их способностью расти в присутствии глюкозы.
Экспрессию и секрецию MI-3 из штамма GA18-1C, трансформированного плазмидами pSW210 и pSW219, осуществляли путем выращивания трансформантов в течение ночи при 30oC в 5 мл MED 1 (2% "Bacto" Yeasr Extract, 0,5% сульфата аммония, 6% глюкозы). Трансформанты разбавляли в соотношении 1:100 в 20 или 50 мл MED 1 и выращивали при 30oC в течение 24 или 48 ч. Клетки гранулировали, промывали и затем замораживали при -70oC. Истощенную среду дважды центрифугировали и декантировали ее от клеточного материала, а потом замораживали ее при 70oC. Содержание MI-3, определенное радиоиммуноанализом (RIA), показывает, что трансформанты pSW210 продуцировали 0,3 мкг/мл иммунореактивного материала MI-3, что и трансформанты pSW219 продуцировали 0,15 мкг/мл иммунореактивного материала MI-3 в течение 24 ч.
Степень экспрессии и секреции MI-3 из трансформантов pSW219 штамма ZM114 также измеряли с помощью высокоэффективной жидкостной хроматографии (HPLC). Трансформанты выращивали в течение ночи при 30oC в 5 мл пополненном YEPD (YEPD + 40 мг/л Ade + 80 мг/L Ley + 10 мМ CaCl2, доведенном до 6% глюкозы). Культуру, выращенную в течение ночи, разбавляли в соотношении 1:100 в 50 мл пополненного YEPD и выращивали при 30oC. По два образца объемом 4 мл брали в час 30, 48 и 75 соответственно. Центрифугировали образцы и отделяли поверхностные слои. Аликвоты 5 мл поверхностных слоев смешивали с 0,5 мл ферментативного бульона (552 г 96% EtOH + 349 г H2O + 5 мл конц, H2 SO4) и смеси дают охладиться при комнатной температуре в течение 30 мин. Смеси затем фильтровали через Acrodiscs (Gelman Sciences, Ann Arbor, Mich.) размером 2 мкм и их замораживали при -20oC. Содержание MI-3, определенное высокоэффективной жидкостной хроматографии, показывало, что трансформанты pSW219 продуцируют 14 мкг/мл MI-3 в течение 75 ч.
Плазмиду pZV187, содержащую сайт расщепления тромбина между третьим участком BAR1 и кодирующей MI-3 последовательностью, трансформировали в штаммы S.cerevisiae GA18-1C и ZM114. Отбирали трансформанты в соответствии с их способностью расти в присутствии глюкозы. Трансформанты выращивали в течение ночи в 5 мл YEP + 6% глюкозы (1% Bacto Yeast Extract, 2% Bacto Yeast Peptone, причем добавляли 6% декстрозы после обработки в автоклаве). Культуры, выращенные в течение ночи, разбавляли в соотношении 1:100 в 10 мл YEP + 6% глюкозы и их выращивали при 30oC. Образцы брали через 26 и 48 ч. Образцы центрифугировали для того, чтобы гранулировать клетки, и поверхностные слои декантировали и замораживали при -70oC. Содержание M1-3 определяли радиоиммуноанализом. Было установлено, что трансформанты GA18-1C продуцировали 0,9 нг/мг иммунореактивного материала MI-3 в течение 48 часов, а трансформанты ZM114 продуцировали 0,52 нг/мл иммунореактивного материала MI-3 в течение 48 ч.
Пример VIII. Трансформация клеток-хозяина и экспрессия урокиназы.
Плазмиду pSW163, включающую промотор TPI1, сигнальную последовательность PHO5, uPA кДНК, третий участок BAR1 и терминатор TPI1 в дрожжевом векторе YEp13, трансформировали в дрожжевые штаммы ZY100 MATa, ade2-101 leu2-3, 112 ura3-52 suc2Dgal2 pep4:CAT) и ZY200 (MFTf ade2-101 leu2-3, 112 ura3-52 suc2-D gal2 pep4:CAT vpt3). Трансформанты отбирали в соответствии с их способностью расти в синтетической среде выращивания, не содержащей лейцина.
Экспрессию и секрецию свиной урокиназы из трансформаторов pSWWW163 осуществляли выращиванием трансформантов в течении ночи при 30oC в 5 мл -Leu6%D + 0,1 М сукцината при pH 5,5 (причем -Leu содержала 6% глюкозы и 0,1 М сукцината, pH доводили до 5,5 добавлением NaOH перед осуществлением обработки в автоклаве). Культуры, выращенные в течении ночи, разбавляли в соотношении 1: 1000 в 5 мл -Leu6%D + 0,1 M сукцината при pH 5,5 и выращивали в течении 37 ч при 30oC. Клетки гранулировали и поверхностный слой декантировали и отделяли. Активность UPA измеряли фибринолитическим анализом. При использовании этого способа обнаруживали, что содержание uPA составляет 7,2 мкг.л в клеточном экстракте, а 38 мкг/л в поверхностном слое трансформаторов pSW163 ZY200.
Пример IX. Экспрессия и секреция PDGF BB при использовании сигнала секреции BAR1.
А. Клонирование последовательной PDGF. Конструирование последовательности, кодирующей В-цепь PDGF, описано авторами Murae et al (патент США NN 4,766,073 и 4,769,328). В патенте США N 4,766,073 вектор экспрессии pB12 включает последовательность ДНК, кодирующую В-цепь PDGF человека, которая оперативно связана с промотором ТРI1 S. ctrevisiae, с препро-последовательностью MFa1 и с терминатором TPI1. В патенте США N 4,766,073 также описано, что вектор pSB1 включает единицу экспрессии, содержащую промотор TPI1, препро-последовательность MFa1, последовательность, кодирующую v-sis, и терминатор TPI1.
Последовательность MFa1/v-sis замещали MFa1/B-цепью последовательности в векторе pSB1. Единицу экспрессии PSB1 вставляли в модифицированную плазмиду pBR322, не содержащую сайт Eco RI. Полученный вектор, обозначенный как pKP 10, обрабатывали с Eco RI и Xba I для того, чтобы удалить фрагмент MFa1/v-sis. Фрагмент MFa1/B-цепь из pB12 затем вставляли в единицу экспрессии pKP10 с тем, чтобы сконструировать pKP26.
Последовательность альфа- фактора с оптимизированными кодонами вставляли затем в единицу экспрессии. Фрагмент Eco RI-Xba 1, включающий последовательность препро альфа- фактора и последовательность инсулина, клонировали в pUC118, обработанную Eco RI и Xba 1 (Vieira and Messing, Methods in Enzymology, 153, 3 -11, 1987), получали однонитевую ДНК. Эту ДНК затем подвергали мутагенезу в соответствии с двухпраймерным методом (Zoller and Smith, DNA, 3, 479 488, 1984), применяя олигонуклеотид ZC862 с мутацией (5' CGA ATC TTT TGA GCT CAG AAA CAC C3'). В результате образовывался Sst i-сайт в конечном В' положении лидера альфа- фактора. Отбирали плазмиду, измененную правильным образом, и обозначали ее как pKP23. Расщепляли главную (лидерную) последовательность из рКР23 путем обработки Есо RI и Sst I и лидерный фрагмент субклонировали в pIC19H, расщепленную Есо RI и Sac I (Marsh et al. Gene, 32, 48111-486, 1984). Полученную плазмиду обозначали как зКЗ24. Плазмиду зКЗ26 расщепляли ЕсоRI и Sst I для того, чтобы удалить последовательность альфа- фактора. Последовательность альфа- фактора с оптимизированными кодонами затем удаляли из рКР24 в качестве фрагмента Есо RI-SSt I и присоединяли ее к линеаризованной рКР26. Полученный вектор обозначали как рКР28.
Сайт Sst I, вставленный в лидер альфа- фактора с тем, чтобы облегчить конструирование вектора, затем удаляли, в результате чего восстанавливали кодирующую последовательность дикого типа. Плазмиду рКР28 обрабатывали Eco RI и Xbf I и извлекали последовательность слияния В-цепи альфа- фактора. Этот фрагмент клонировали в плазмиду pUC118 и выделяли однонитевую ДНК. ДНК подвергали мутагенезу двухпраймерным методом, применяя олигонуклеотид с мутацией ZC1019 (5' ACC CAA GGA TCT CTT GTC CAA AGA AAC ACC TTC TTC 3'). Плазмиду, содержащую нужную мутацию, обозначали как рКР32.
Полную единицу экспрессии затем реконструировали, Плазмиду рКР32 обрабатывали Eco RI и Xba I и извлекали фрагмент В-цепи альфа- фактора. Этот фрагмент вставляли в плазмиду рКР10, расщепленную ECO RI и Xba I, для того, чтобы сконструировать плазмиду рКР34. Плазмиду рКР34 обрабатывали Cla I и Bfm HI и выделяли единицу экспрессии. Этот фрагмент вставляли в плазмиду pMPON2, отработанную Cla I и Bam Hl, с тем, чтобы сконструировать плазмиду рКР36 (дрожжевая плазмида на основе 2 мкм, содержащая источники репликации дрожжей и бактерий, ген устойчивости к ампициллину и селектируемый маркер POT1, сдана на хранение в коллекцию АТСС, где хранится под номером 67788).
Последовательность А-цепи PDGF с оптимизированными кодонами из плазмиды рА7 (Murray et al. патент США N 4,766,073) объединяли с лидерной последовательностью альфа- фактора с оптимизированными кодонами в течении разных стадий конструирования, аналогичных стадиям, описанных выше для В-цепи. Последовательность А-цепи из рА7 выделяли как Sst I-Xba I фрагмент и вставляли ее в плазмиду рК28, для того, чтобы сконструировать плазмиду рК27. Плазмиду рКР27 обрабатывали Eco RI и Xba I и фрагмент А-цепи альфа- фактора клонировали в плазмиду pUC118.
Затем осуществляли мутагенез, применяя олигонуклеотид ZC1018 (5' TTC GAT AGA TCT CTT GTC CAA AGA AAC ACC TTC TTC 3') в соответствии с вышеизложенным с тем, чтобы удалить сайт Sst I и восстановить альфа- факторную последовательность дикого типа. Исправленную плазмиду обозначали как рКР31.
Затем конструировали вектор оптимизированными кодонами. Плазмиду рКР31 обрабатывали Eco RI и Xba I и фрагмент А цепи альфа- фактора присоединяли к плазмиде рКР10, расщепленной с Eco RI и Xba I. Полученный вектор, обозначенный как рК33, содержал единицу экспрессии. Плазмиду рКР33 обрабатывали Cla I и Bam HI и выделяли фрагмент единицы экспрессии. Этот фрагмент вставляли в рМРОТ2, расщепленную Cla I и Bam Hl, для того, чтобы сконструировать вектор экспрессии рКР35.
Кодирующую последовательность В-цепи PDGF получали из плазмиды рКР51. Плазмиду рКР32 трансформировали в штамм E. coli MV1193. Получали однонитевую ДНК. ДНК подвергали мутагенезу, применяя олигонуклеотид с мутацией ZC1078 (табл. 3). Мутагенез ДНК с ZC1078 приводили к вставке сайта рестрикции Bam HI в 5'конце кодирующей последовательности В-цепи PDGF. Положительные клоны обнаруживали путем гибридизации фагов, рестрикционного анализа и дидезоксисеквенирования. Положительный клон обозначали как рКР47.
Сигнальную последовательность MFa1, присутствующую в плазмиде рКР47б замещали синтетической сигнальной последовательностью. Плазмиду рКР47 обрабатывали Eco RI и Bam YI для того, чтобы выделить фрагмент, включающий последовательность В-цепи человека и последовательности вектора pUC118. Конструировали олигонуклеотиды ZC1157, ZC1158, ZC1076 и ZC1077 (табл. 1) таким образом, чтобы они кодировали при отжиге Eco RI-Bam HI-адаптер, кодирующий синтетическую сигнальную последовательность. Олигонуклеотиды ZC1158 и ZC1076 обрабатывали киназой. Отжигали олигонуклеотиды ZC1158 и ZC1157, а ZC1076 и ZC1077 отжигали в отдельных реакциях. Фрагмент Eco RI-Bam HI из рКР47 присоединяли к ZC1158/ZZC1157 и ZC1076/ZC1077 в результате лигирования трех частей. Полученная плазмида, обозначенная как рКР49, включала синтетическую сигнальную последовательность В-цепи PDGF и последовательность вектора pUC118.
Единицу экспрессии, включающую промотор ТР11, синтетическую сигнальную последовательность, последовательность В-цепи PDGF и терминатор TPI1, конструировали из плазмиды рКР49 с последующим субклонированием в дрожжевой вектор экспрессии. Плазмиду рКР34 обрабатывали Cla Iи Bam НI, в результате чего выделяли фрагмент 2,3 kb, включающий промотор TPI1, сигнальную последовательность MFa1, последовательность В-цепи PDGF и Eco RI. Олигонуклеотиды ZC1016 и ZC1017 (табл. 2) подвергали обработке киназой и отжигу, в результате чего образуется полинкер-адаптер, включающий сайты рестрикции Cak I, Hind III, Xho I, Acc I, Xbf I и Bam HI. Литейный вектор лигировали с адаптером ZC1016/ZC1017, подвергнутыми обработке киназой и отжигу. В результате этого теряли сайты Hind III и Eco RI плазмиды pUC12. Полученный вектор pHC12 обрабатывали путем гидролиза Асс I и Bam HI. Фрагмент единицы экспрессии (2,3 kb) присоединяли к линеаризованной pUC12. Полученную плазмиду обозначали как рКР38. плазмиду обрабатывали Eco RI и Xba для того, чтобы выделить фрагмент, включающий промотор TPI1, терминатор TPI1 и последовательности вектора pUC12 (4,3 kd). Плазмиду рКР49 обрабатывали Eco RI и Xbf I для того, чтобы выделить фрагмент 0,8 кb, включающий синтетическую сигнальную последовательность и последовательность В-цепи PDGF. Присоединяли фрагмент 0,8 kb к фрагменту 4,3 kb из рКР37. Полученную плазмиду обозначали как рКР51.
В. Конструирование вектора экспрессии.
Последовательность В-цепи PDGF затем присоединяли к сигналу секреции, включающему лидерную последовательность и последовательность, кодирующую третий участок гена BARI S.cerevisiae. Сигнал секреции BAR I присоединяли к кодирующей В-цепь последовательности, в результате чего конструировали векторы экспрессии pSW304 и pZY76.
Сначала конструировали плазмиду pSW255, включающую промотор TPI1 и сигнал секреции BAR1. Последовательность BAR1, кодирующую третий участок и присутствующую в плазмиде pSW195, сливали с синтетическим адаптером, кодирующим аминокислоты 81 85 альфа- фактора, сайт расщепления Lys-Arg, 5' Eco R1, адгезивный конец 3' Bgl II и первую аминокислоту B-цепи PDGF. Олигонуклетиды ZC1135 и ZC1136 (таблица 1) подвергали обработке киназой и отжигу, как описано автором Maniatis et al (ibid). Плазмиду pSW195 обрабатывали Hind III и Eco R1, в результате чего выделяли фрагмент 1, 4 kb, включающий промотор TPI1 и кодирующие последовательности BAR1. Фрагмент 1,4 kb присоединяли к ZC1135/ZC1136-адаптером и pIC19R, которая была обработана Hind III и Bgl II, в результате слияния трех частей. Полученную плазмиду обозначали как pSW255.
Последовательность B-цепи PDGF, присутствующую pKP51, присоединяли к промотору TPI1, сигнальной последовательности BAR1, третьему участку BAR1 и ZC1135/ZC1136-адаптеру (кодирующему сайт расщепления Lys-Arg) для того, чтобы сконструировать плазмиду pSW262. Плазмиду pKP51 обрабатывали Bam HI, в результате чего выделялся фрагмент 1,09 kb, включающий кодирующую B-цепь PDGF последовательность и терминатор TPI1. Плазмиду pSW255 вываривали Sph и Bgl II для того, чтобы выделить фрагмент 0,75 kb, включающий частичный промотор TPI1, сигнальную последовательность BAR1 и ZC1135/ZC1136-адаптер. Два фрагмента присоединяли путем лигирования трех частей с pUC18, которую гидролизовали Sph и HI. Обнаружили плазмиду, включающую составные фрагменты в правильной ориентации. Плазмиду обозначили как pSW262.
Дрожжевой вектор экспрессии pSW304, включающий промотор TPI1, сигнальную последовательность BAR1, третий участок BAR1, B-цепь PDGF и терминатор TPI1 в векторе pMOT2, конструировали. Плазмиду pKP36 обрабатывали Cla I и Sph I для того, чтобы выделить 5' участок промотора TPI1 (0,76 kb). Плазмиду pKP36 также обработали Cla I и Bgl II для того, чтобы выделить фрагмент 11 kb, содержащий вектор. Этот фрагмент включал последовательность B-цепи PDGF, терминатор TPI1 и последовательности вектора PMPOT2. Плазмиду pSW262 вываривали Sph I и Bgl II для того, чтобы выделить частичный промотор TPI1 (0,75 kb), сигнальную последовательность BAR1, третий участок BAR1 и ZC1135/ZC1136. Три фрагмента соединяли в результате сшивки трех частей и полученную плазмиду обозначили как pSW304.
Вектор экспрессии pZY76 конструировали путем встраивания единицы экспрессии B-цепи в вектор pRPOT. Вектор pRPOT получен из pCOT (750 bp) фрагментом Sph I-Bam HI плазмиды pBR322 (186 bp). Полученную плазмиду, pDPOT, обрабатывали Sph I и Bam HI, в результате чего выделяли фрагмент 10,8 kb. Олигонуклеотиды ZC1551 и ZC1552 подвергали обработке киназой и отжигу для образования адаптера, имеющего адгезивный конец Dfm HI и адгезивный конец Sph I, расположенные сбоку сайтов рестрикции Sma I, Sst I и Xho I. Фрагмент 10,8 kb pDPOT рециркулирован путем сшивки с ZC1551/ZC1552-адаптером. Полученную плазмиду обозначают как pRPOT.
Терминатор TPI1 субклонировали следующим образом.
Плазмиду pSW195 обрабатывали Bgl II и Sma I для того, чтобы выделить фрагмент 2,38 kb, включающий промотор TPI1, концевую аминогруппу BAR1 и третий участок BAR1, последовательность, кодирующую MI-3, и терминатор TPI1. Фрагмент 2,38 kb сшивали с плазмидой PRPOT, которую обрабатывали Sma I и Bgl II. Полученную плазмиду, обозначенную как pSW313, обработали Xba I и Sph I для того, чтобы выделить фрагмент терминатор TPI1 0,76 kb. Фрагмент 0,76 kb присоединяли к плазмиде pUC18, обработанной Sph I и Xba I. Полученную плазмиду обозначают как pZY75.
Затем конструировали плазмиду pZY76. Плазмиду pSW195 обрабатывали Bgl II и Eco RI для того, чтобы выделить фрагмент 1,4 kb, включающий промотор TPI1 и концевую аминогруппу BAR1 и третий участок BAR1. Плазмиду pSW262 обработали Eco RI и Xba I для того, чтобы выделить фрагмент 0,35 kb, включающий ZC1135/ZC1136-адаптер и последовательность, кодирующую B-цепи PDGF. Плазмиду pZY75 обработали Xba I и Sph I для того, чтобы выделить фрагмент 0,75 kg, включающий терминатор TPI1. Три фрагмента присоединяли к pRPOT, гидролизованной с Bam HI и Sph I, в результате сшивки четырех частей. Полученную плазмиду, включающую промотор TPI1, концевую аминогруппу BAR1 и третий участок BAR1, кодирующую PDGF последовательность, терминатор TPI1 и последовательности вектора pRPOT, обозначали как pZY76.
C. Экспрессия гомодимера BB.
Экспрессию PDGF BB в дрожжевых штаммах, трансформированных плазмидами pSW304 и pZY76, сравнивают с экспрессией PDGF из контрольных плазмид pB1 70m (Murray en al. USSN 896, 485) и pKP57 (включающая единицу экспрессии pKP34 в pRPOT). Плазмиды pSW304, pZY76, pB1 70m и pKP57 трансформировали в дрожжевые штаммы E18# 9 (MATa leu2-3,112 his4-580 pep4-3 Dtpil:LEU2/MATa leu2-3,112 pep4-3 Dtpil: LEU2) XB13-5B (MAT a leu2-3,112 ura 3 barl gal12 D tpil:URA3 [ciro] ) и ZM114 (MATa ade2-101 leu2-3,112 ura3-52 D tpil:URA3 vpt3 suc2-D 9 gal2 pep4: TPI промотор-CAT [ciro]) согласно описанию автора Beggs (Nature 275: 104 108, 1978).
Трансформанты из отдельных колоний инокулировали в среду ферментации и выращивали в течение 24 ч при 30oC. В течение 24 ч добавляли глюкозу к культурам, обеспечивая при этом конечную концентрацию 2% Культуры выращивали в течение 24 ч при 30oC.
Среда ферментации, г:
NZ амина типа A 20
KH2PO4 7
NH4SO4 6
MgSO4 2
Твердые частицы
растворяли в воде и объем доводили до 1 л. Раствор обрабатывали в автоклаве
в течение 25 мин. После обработки в автоклаве добавляли 2 мл/л раствора микроэлементов (состав приведен ниже), 4 мл раствора
витаминов (состав приведен ниже), 2 M сукцината натрия, pH 5,5, до
конечной концентрации 0,1 M, и 50% глюкозы до конечной концентрации 2%
Раствор микроэлементов
9,45 мг ZnSO4
284,8 мг Fe2(SO4)3
48 мг CuSO4•5H2O
Твердые частицы растворяли в дистиллированной воде и доводили
конечный объем до 100 мл. Осуществляли стерилизацию фильтрованием.
Раствор витаминов:
420 мг рибофлавина
5,4 г пантотеновой кислоты
6,1 г ниацина
1,
4 г пиридоксина
60 мг биотина
40 мг фолиевой
кислоты
6,6 г инозита
1,3 г тиамина
При этом растворяли в дистиллированной воде и доводили до конечного объема
1 л, затем осуществляли стерилизацию фильтрованием.
Клетки удаляли из среды путем центрифугирования. Поверхностные слои затем фильтровали через фильтр с диаметром отверстий 0,45 мкм, в результате чего удалялись всякие клетки или отходы от клеток. Профильтрованные поверхностные слои культур подвергали анализу на митогенез, как описано авторами Raines and Ross (Methods in Enzymology, 109, 749 773, 1985). Результаты, выраженные в нг активности PDGF на мл культуральной среды, приведены в табл. 3.
PDGF-аналоги, продуцированные трансформированными дрожжевыми клетками, выделяли из концентрированных поверхностных слоев культур очисткой с помощью разных стадий хроматографии и концентрации.
Поверхностные слои культур концентрировали, применяя Millipore Pellican Cassettes (Millipore, Bedford, Mass.), и концентраты подвергали гранулированию путем центрифугирования в центрифуге типа Beckman J-6B (Beckman Instruments, Inc. Brea, Calif.) при 4200 оборотов/мин в течение 30 мин, в результате чего устранено помутнение. Затем добавляли EDTA, вследствие чего конечную концентрацию доводили до 10 mM, и добавлением 5 M NaOH pH смеси доводили до pH 5,5. После этого концентраты разбавляли водой для того, чтобы проводимость составляла примерно 10 миллимхоз.
Полученные концентраты подвергали хроматографии на колонке типа S-Sepharose Fast Flow (Pharmacia, Piscataway, N.J.). Колонку промывали 20 мМ фосфата натрия, 0,1 M хлорида натрия, причем pH составляет 7,3. Затем колонку элюировали 20 мМ фосфата натрия, 1М хлорида натрия, причем pH составляла 7,3. При этом наблюдали кривую спектрального поглощения элюата при 280 нм и пиковые фракции собирали и объединяли.
Элюаты замораживали при -20o C и потом их оттаивали.
Материалы в виде частиц удаляли из элюатов путем центрифугирования. Собирали поверхностные слои и добавлением 0,87 M уксусной кислоты доводили до pH 3,0. Затем элюаты концентрировали, причем применяли фильтр типа Amicon YM10 (Amicon, Danvers, Mass.) Разбавляли концентрированные элюаты 5 объемами 1 М уксусной кислоты для того, чтобы снизить концентрацию хлорида натрия до примерно 0,2 М.
Элюаты подвергали хроматографии на второй колонке типа S-Sepharose. Затем промывали колонку 1 М уксусной кислотой и наблюдали кривую спектрального поглощения элюатов при 280 нм, пока кривая не возвратилась к основной линии. Затем элюировали колонку 1 М уксусной кислоты, 1,5 М хлорида аммония, причем pH составляла 4,8 5,0. После этого наблюдали кривую А280 элюатов и собирали PDGF как последний пик А280. Пиковые фракции собирали и концентрировали с помощью фильтра типа Amicon YM10.
Концентрированные элюты затем пропускали через колонку типа Sephadex G-50 Superfine (Pharmacia, Piscataway, N.J.), причем использовали образец в объеме, составляющем примерно 1% объема колонки. Колонка работала при скорости потока 5 см/ч в 1 М ацетата аммония, причем pH равен 9,0. При этом собирали самые чистые фракции, установленные путем электрофореза на SDS-геле, и добавлением уксусной кислоты доводили pH до 4,0.
Пример Х. Использование лидера BAR1 для секреции эпидермального фактора роста и трансформирующего альфа- фактора роста. Кодирующие последовательности для эпидермального фактора роста (EGF) и трансформирующего альфа- фактора роста (TGFa) получали из синтетических олигонуклеотидов. Затем последовательности использовали для получения единиц экспрессии, включающих промотор и терминатор TPI1 и лидер BAR1 и последовательности третьего участка.
Кодирующая EGF-последовательность сконструирована из олигонуклеотидов ZC1734, ZC1735, ZC1534 и ZC1535. Пары олигонуклеотидов (ZC173-4+ZC1735 и ZC1534+ZC1535) отдельно подвергали отжигу путем инкубации при 100oC в водяной ванне в течение 5 мин с последующим медленным охлаждением в водяной ванне в течение 60 мин. Подвергнутые отжигу пары затем соединяли и сшивали. Собранный кодирующий фрагмент клонировали в M13mp19 для подтверждения последовательности. Полученный клон обозначали как pZY77.
Последовательность TFGa сконструировали из пар олигонуклеотидов ZC1732+ZC1733 и ZC1198+ZC1200 по способу, в основном описанному выше для EGF. Собранная кодирующая последовательность клонировалась в M13mp19, в результате чего образовалась pZY78.
Последовательности EGF и TGFa присоединяли к терминатору TPI1. Клоны фагов pZY77 и pZY78 обрабатывали Eco RI и Xba I и очищали соответствующие фрагменты факторов роста. Плазмиду pB170SW, включающую фрагмент Sal I-Bam HI, который в свою очередь включает 3' участок последовательности B-цепи PDGF и терминатор TPI1, клонированный в pIC19R, обрабатывали Xba I и Bam HI, и выделяли фрагмент терминатора TPI1. Каждый из фрагментов факторов роста присоединяли к терминатору TPI1 и к pUC18, обработанной Eco RI и Bam HI, в результате сшивки трех частей. Полученные плазмиды обозначали как pZY80 (EGF) и pZY81 (TGFa).
Последовательности факторов к промотору TPI1 и к сигналу секреции BAR1. Каждую плазмиду pZY80 и pZY81 обрабатывали Eco RI и Bam HI и очищали фрагменты терминаторов факторов роста. Плазмиду pSW195 обрабатывали Hind III и Eco PI, и выделяли фрагмент промотора TPI1-BAR1 (около 1,4 kb). Фрагменты терминаторов факторов роста присоединяли к фрагменту промотора BAR1 в плазмиде pUC18, обработанной Bam HI и Hind III.
Полученные плазмиды обозначали как pZY83 (EGF) и pZY84 (TGF a).
Для конструирования векторов экспрессии каждую из pZY83 и pZY84 обрабатывали c Bg1 I и Sst I, и выделяли фрагменты единиц экспрессии. Эти единицы вставляли в плазмиду pRPOT (пример III), обработанную Sst I Bam I, для того, чтобы сконструировать pZY85 (EGF) pZY86 (TGFa). Вторую совокупность векторов экспрессии конструировали путем вставки единиц экспрессии Bam HI-Sst I плазмид pZY83 и pZY84 в плазмиду pMPOT2 (пример III), обработанную Sst I и Bam I. Полученные векторы экспрессии обозначают как pZY89 (EGF) pZY90(TGF a).
Для экспрессии EGF TGFa плазмиды pZY85, pZY86, pZY89 и pZY90 трансформировали в штаммы S.cerevisiae ZM120 и ZM122 (оба ciro, диплоиды а/ a, leu2 ura3 tpi:LEU2 pep4:URA3 bar1). Трансформанты культивировали по существу, как описано в примере III. Поверхностные слои культур подвергали анализу по митогенетической активности EGF и TGFa8 согласно описанию авторов Raines and Ross (Vethods in Enzymology, 109, 749-773, 1985) при использовании EGF-стандарта.
Пример XI. Очистка проинсулина из трансформированных дрожжей.
Клетки S. cerevisiae XP635-10C, трансформированные плазмидой pZV30 или pZV31 (см. пример 1), выращивали при 30oC в культурах объемом 1 л с использованием среды, в которой отсутствует лейцин (-leu среда). Эта среда содержала, в расчете на 1 л, 20 г глюкозы, 6,7 г "Bacto" азотного основания для дрожжей, не содержащего аминокислот (Difco), 40 мг аденина, 30 мг L-аргинина HCl, 50 мг L-аспарагиновой кислоты, 20 мг L-гистидина, 60 мг L-изолейцина, 40 мг L-лизина HCl, 20 мг L-метионина, 60 мг L-фенилаланина, 50 мг L-серина, 50 мг L-тирозина, 40 мг урацила, 60 мг L-валина, 150 мг L-треонина и 40 мг L-триптофана. Культуры инкубировали в течение 34 ч, после чего добавляли альфа- фактор. Культуры инкубировали еще 11 ч и затем собирали путем центрифугирования.
Проинсулин очищали от лизатов дрожжевых клеток и надосадочных жидкостей культуральной среды с помощью афинной хроматографии с использованием моноклонального антитела против инсулина.
В целях получения антитела мышей иммунизировали свиным проинсулином с проведением иммунизации каждые две недели в течение 6 нед (50 мкг на 1 иммунизацию). Через 5 дн после проведения последней иммунизации удаляли селезенку и клетки селезенки сливали с линией клеток NS-1 миеломы (АТСС TIB-18) с полиэтиленглоколем. После слияния выделяли одну гибридому, продуцирующую антитела, которые вступали в реакцию со свиным инсулином, свиным проинсулином и человеческим инсулином, что подтверждается осаждением125J-меченных инсулинов. Оказывается, что моноклональное антитело представляет собой IgG1, который, по-видимому, не содержит NS-lai-легкой цепи. Эту гибридому (5.4.4.1) клонировали и выращивали в асците. Атитело 5.4.4.1 выделяли из асцитической жидкости путем афинной хроматографии на белок-А-сефарозе и получали антиинсулин-афинную матрицу за счет присоединения антитела к GNBr-активированной сефарозе.
В целях очистки проинсулина образцы концентрировали на колонке типа Lichrorep RP-18. Элюат из колонки лиофилизировали и опять растворяли в 1 мл фосфатбуферного буффера (PBS), содержащего 0,5M NaCI) и затем 5 мл H2O. Образец элюировали 2 мл 3М уксусной кислоты, содержащей 0,5 мг BSA на 1 мл. Элюат лиофилизировали и опять растворяли в 150 мкл 45-ного раствора CH3CN с 0,1% TFA. Элюат из колонки подвергали анализу путем высокоэффективной жидкостной хроматографии (HPLC) на колонке, содержащей Varian Micro Pak SP-C18-белок. В качестве растворителей использовали 0,1% TFA в H2O и 0,07% TFA в CH3CN(A и B, соответственно). Образец элюировали линейным градиентом от 10% B до 45% B в течение 30 мин при скорости протока 1,0 мл/мин.
При этом собирали фракции и подвергали их анализам на IRI и IRC описанным ранее способом.
Пример XII. Использование BARI для секреции t-PA.
В целях использования последовательностей BAR1 для экспрессии тканевого активатора плазминогена (t-PA) участок гена BAR1 сначала присоединяют к промотору ADHI дрожжей в плазмиду pZV24 в соответствии с примером 2. Затем конструируют плазмиду, включающую последовательность BAR1, присоединенную к последовательности, кодирующей Ser-t-PA, (сайт BgI II гена BAR1 (фиг. 19. pDR1107 (включающую терминатор TPI S. cerevisie, клонированный в pUC18, расщепленную с Xbai + Bam HI) и pDR1296 (включающую BgI II-Xba кодирующую t-PA последовательность в pUC18; она депонирована в коллекции АТСС как трансформ E. coli JM 83 и имеет номер депонирования 53347) трансформировали в E.coli G-48 (dam-). Плазмиды получали и расщепляли с помощью Xba I и Bam HI в буфере Xba I. Фрагмент терминатора TPI (600 bp) из pDR1107 и фрагмент t-PA + вектор (4,3 kb) плазмиды pDR1296 подвергали гелевой очистке. Эти два фрагмента лигировали при комнатной температуре в течение ночи и использовали для трансформации E. COli RR1. Плазмидную ДНК получали из трансформантов и расщепляли с помощью Xba i и Bam HI в буфере Xba I для определения положительных клонов. Один из этих клонов обозначали pJH33.
Плазмиду pJH33 расщепляли с помощью Bgl II и SPh I в "uni" буфере (0, 05 Трис pH 7,4, 0,006M MgCl2, 0,05M NaCI). t-PA-TPI-терминаторный + секторный фрагмент (5,1 kb) подвергали гелевой очистке. pZV24 расщепляли с помощью Sph I + Bgl II в "uni" буфере и фрагмент промотор ADHI-BARI (800 bp) подвергали гелевой очистке. Фрагмент Bgl II-Sph (5,1 kb) лигировали с фрагментом промотор ADHI-BARI (800 bp) из pZV24 при комнатной температуре в течение ночи. Смесь, полученную в результате лигации трасформировали в E.coli RRI. Плазмидную ДНК отбирали путем обработки Sph I+ Bgl II и Sph I + Bam HI. Выбирали одну положительную плазмиду и обозначали ее pJH35.
Затем конструировали дрожжевую плазмиду pJH39, включающую кодирующую t-PA последовательность в слиянии с последовательностью BAR1 в положении 1025 (аминокислота N 114) плазмиды BAR1. В результате этого слияния получали первичный продукт трансляции, состоящий из участка Баррьер полипептида, присоединенного к Ser-t-PA, причем Arg остаток одной аминокислоты 5' заменяли на Ser в t-PA. В целях конструирования плазмиды pJH39, плазмиды pJH35 и YEpI3расщепляли Sph I и BamHI в "uni" буфере. Фрагмент YEp13 и вставку (3 kb) из pJH35 (включающей промотор ADHI, фрагмент слияния BAR1-t-PA и терминатор TPI) подвергали гелевой очистке и затем их подвергали лигированию в течение ночи. Полученную в результате плазмиду обозначали pJH39 (фиг. 19).
Вектор экспрессии дрожжей pJH39 использовали для трансформации штамма S. cerevisiae E8-11c. Клетки выращивали в среде без leu (пример 6) + 6% глюкозы, содержащей 0,1 M гидрогенфталата калия, pH 5,5, при 30oC в течение 26 ч. Сырые экстракты подвергали анализу ELISA и фибринолитическому анализу, получали следующие результаты (табл. 5).
t-PA выделяли из экстрактов дрожжевых клеток путем афинной хроматографии с использованием моноклонального антитела против участка Крингл 1 t-PA, присоединенного к CNBr-активированной сефарозе. Образцы доводили до 1M NaCI и пропускали через колонку для иммуно-афинной хроматографии. Колонку затем промывали калий-фосфатным буфером (25 mM фосфата калия, 0,2M NaCl, 2% Tween-80 и 0,5% азида натрия) при pH 8,5, пока спектральная поглощательная способность элюата при A280 не достигла стабильной основной линии. Затем колонку подвергали предварительному элюированию калийфосфатным буфером при pH 10,0. После достижения стабильной основной линии для A280 в промывной жидкости t-PA элюировали с колонки калийфосфатным буфером при pH 11,5. Собирали фракции, содержащие t-PA, очищенный афинным способом, и добавлением уксусной кислоты pH доводили до 7,0. После очистки образцы хранили при -80oC.
Реферат
Использование: биотехнология, в частности генная инженерия при получении гетерологичного полипептида в эукариотических микроорганизмах. Сущность изобретения: способ предусматривает конструирование рекомбинантной плазмидной ДНК, содержащей промотор BAP1 Saccharomyces cerevisial или промотор алкогольдегидрогеназы IS cerevisial часть последовательности гена BARI, кодирующей сигнальный пептид, и место отщепления сигнального пептида KEX2 от гетерологичного полипептида, которое представляет собой Arg-Arg или Lys-Lys, а также структурный ген. Затем проводят трансформацию полученной ДНК штаммов - реципиентов, культивирование трансформатов, выделение и очистку целевого продукта. В качестве штаммов - реципиентов целесообразно использовать штамм дрожжей Saccharomyces cerevisial или Schizosauharomyces pombe или Aspergillus или Neurospora. 8 з. п. ф-лы, 21 ил., 5 табл.

Комментарии