Мутагенез днк за счет неупорядоченной фрагментации и вторичной сборки - RU2157851C2
Код документа: RU2157851C2
Чертежи
























Описание
Область изобретения
Изобретение относится к способу получения полинуклеотидов, сообщающих нужный фенотип
и/или кодирующих протеин, обладающий ценными заранее определенными свойствами, причем этот протеин является селектируемым. В этом аспекте способ используют для создания и отбора фрагментов нуклеиновых
кислот, кодирующих мутантный протеин.
Описание известного уровня
Сложность активной последовательности биологической макромолекулы, например, протеина, ДНК и т.д., называют его
информационным содержанием ("UC"; 5-9). Информационное содержание протеина было определено как устойчивость активного протеина по отношению к изменению последовательности аминокислот, рассчитываемую
как число инвариабельных аминокислот (битов), необходимых для описания семейства родственных последовательностей с одинаковыми функциями (9, 10). Протеины, которые чувствительны к неупорядоченному
мутагенезу, отличаются высоким информационным содержанием. В 1974 г., когда было введено это понятие, многообразие протеинов существовало только как таксономическое многообразие.
Развитие молекулярной биологии, например, создание молекулярных библиотек, позволило идентифицировать гораздо большее число различных оснований и даже выбрать функциональные последовательности из случайных библиотек. Можно варьировать большинство остатков, хотя обычно и не в одно и то же время, в зависимости от компенсационных изменений в контексте. Так, протеин, состоящий из 100 аминокислот, может содержать только 2000 различных мутаций, но 20100 возможных комбинаций этих мутаций.
Информационной плотностью называют информационное содержание на единицу длины последовательности. Активные сайты энзимов обычно имеют высокую информационную плотность. Напротив, гибкие линкеры энзимов отличаются низкой информационной плотностью (8).
Широко распространенные современные способы, которые используют для создания мутантных протеинов в библиотечном формате, представляют собой подверженную ошибкам полимеразную цепную реакцию (11, 12, 19) и кассетный мутагенез (8, 20, 21, 22, 40, 41, 42), в которых специфический участок нужно оптимизировать, заменяют синтетически мутагенизированным олигонуклеотидом. В обоих случаях "облако мутаций" (4) создается вокруг определенных сайтов в исходной последовательности.
В подверженной ошибкам ПЦР (полимеразиоцельевая реакция) используют нестрогие условия полимеризации для введения в длинную последовательность небольшого количества неупорядоченных точечных мутаций. Подверженную ошибкам ПЦР можно использовать для мутагенизации смеси фрагментов неизвестной последовательности. Однако, компьютерная симуляция позволяет предположить, что один только точечный мутагенез часто оказывается слишком длительным, для того, чтобы обеспечить блок изменений, которые необходимы для непрерывной эволюции последовательности. Опубликованные протоколы подверженных ошибкам ПЦР не позволяют амплицировать ДНК фрагменты более чем от 0,5 до 1,0 к.п.о., что ограничивает их применение. Кроме того, повторяющиеся циклы подверженных ошибкам ПЦР приводят к накоплению нейтральных мутаций, которые могут, например, сделать протеин иммуногенным.
В сайт-направленном мутагенезе с использованием олигонуклеотидов короткую последовательность заменяют синтетическим мутагенизированным олигонуклеотидом. Такой подход не приводит к образованию комбинаций отдаленных мутаций, и поэтому не является комбинаторным. Ограниченный размер библиотеки по сравнению с остальной длиной последовательности означает, что для оптимизации протеина неизбежно придется провести множество циклов отбора. Мутагенез с использованием синтетических олигонуклеотидов требует секвенирования отдельных клонов после каждого цикла селекции с последующей группировкой в семействе, с произвольным выбором одного семейства и сведением его к консенсусному фрагменту, который вновь синтезируют и вторично встраивают в отдельный ген с последующим дополнительным отбором. Этот процесс представляет статистическое узкое место, так как он очень трудоемкий и непрактичный из-за необходимости осуществлять множество циклов мутагенеза.
Подверженная ошибкам ПЦР и сайт-направленный мутагенез с использованием олигонуклеотидов пригодны, таким образом, для отдельных циклов тонкого изменения последовательности, но быстро становятся ограничением, если их использовать для множества циклов.
Подверженную ошибкам ПЦР можно использовать для мутагенизации смеси фрагментов неизвестной последовательности (11, 12). Однако опубликованные протоколы подверженных ошибкам ПЦР (11, 12) страдают низкой процессивностью полимеразы. Поэтому такие схемы не могут привести к неупорядоченному мутагенезу гены среднего размера. Такая их неспособность ограничивает практическое применение подверженных ошибкам ПЦР.
Другим серьезным ограничением подверженных ошибкам ПЦР является то, что скорость супрессорных мутаций растет с ростом информационного содержания последовательности. При определенном информационном содержании, размере библиотеки и скорости мутагенеза баланс супрессорных мутаций и активаторных мутаций статистически предотвращает отбор или дальнейшие усовершенствования (статистический потолок).
И, наконец, повторные циклы подверженных ошибкам ПЦР также приведут к накоплению нейтральных мутаций, что может повлиять, например, на мутагенность, но не на афинность связывания.
Таким образом, было обнаружено, что подверженные ошибкам ПЦР слишком медленны для изменений блоков, что необходимо для непрерывной эволюции последовательности (1, 2).
При кассетном мутагенезе блок последовательности отдельной матрицы обычно заменяют (частично) рандомизированной последовательностью. Поэтому максимум информационного содержания, который можно получить, статистически ограничен числом неупорядоченных последовательностей (т.е. размером библиотеки). Это представляет статистически узкое место, исключая другие семейства последовательностей, которые в данный момент не являются лучшими, но которые могли бы обладать гораздо более длительным потенциалом.
Далее, мутагенез с синтетическими олигонуклеотидами требует секвенирования отдельных клонов после каждого цикла селекции (20). Поэтому такой подход является утомительным и непрактичным для многих циклов мутагенеза.
Подверженная ошибкам ПЦР и кассетный мутагенезы, таким образом, наиболее пригодны и широко используются для участков тонкой модификации с относительно низким информационным содержанием. Одним очевидным исключением является выбор рибозима РНК лигазы из неупорядоченной библиотеки, с использованием множества циклов амплификации с помощью подверженных ошибкам ПЦР и отбора (13).
Становится все более очевидно, что возможности конструирования рекомбинантных линейных биологических последовательностей, таких, как протеин, РНК и ДНК, не столь могущественны, как те возможности, которые создала природа. Обнаружение все более подходящих мутантов зависит от поиска все более подходящих последовательностей внутри все увеличивающегося числа библиотек и требует увеличения числа циклов мутагенной амплификации и отбора. Однако, как обсуждалось ранее, существующие способы мутагенеза, которые в настоящее время находят широкое применение, обладают четкими ограничениями, если их использовать для повторяющихся циклов.
Эволюция большинства организмов происходит за счет природного отбора и полового размножения. Половое размножение обеспечивает смешение и комбинирование генов потомства отдельных индивидуумов. Во время мейоза гомологичные хромосомы из родительских линий подходят друг к другу и происходит кроссинговер по части их длины, в результате чего происходит обмен генетическим материалом. Такой обмен или перетасовка ДНК дает возможность организмам эволюционировать более быстро (1, 2). При половой рекомбинации, так как встраиваемые последовательности отличаются доказанной полезностью в гомологичном окружении, эти встроенные последовательности, по-видимому, все еще обладают существенным информационным содержанием, будучи встроены в новую последовательность.
Мартон с сотр. (Marton, 27) описывает использование PCR ин витро для контроля за рекомбинацией в плазмиде, содержащей непосредственно повторяющиеся последовательности. Мартон указывает, что рекомбинация происходит во время PCR в результате образования разрывов или "ников" ДНК. В результате образуются рекомбинантные молекулы. Мейерханс (Meyerhans) с сотр. (23) также раскрывает существование ДНК рекомбинации в процессе PCR ин витро.
Термин "Искусственная молекулярная эволюция" ("Applied Molecular Evolution ["AME"J) означает осуществление алгоритма эволюционной конструкции для конкретной, полезной цели. Хотя было опубликовано множество различных библиотечных форматов для AME для полинуклеотидов (3, 11-14), пептидов и протеинов (фаг 15-17), lacI (18) и полисом, ни один из этих форматов не отличается использованием неупорядоченного кроссоверинга для преднамеренного создания комбинаторной библиотеки.
Теоретически могут существовать 2000 различных отдельных мутантов для протеина из 100 аминокислот. Протеин из 100 аминокислот предполагает 201000 возможных комбинаций мутаций, - число, которое слишком велико, чтобы его исчерпывающе осуществить обычными методами. Поэтому выгодно разработать систему, которая позволила бы создать и скринировать все эти возможные комбинационные мутации.
Уинтер (Winter) с сотрудниками (43, 44) использует систему ин виво сайт-специфической рекомбинации для объединения генов антител легких цепей с генами антител тяжелых цепей для экспрессии в фаговую систему. Однако эти системы основаны на специфических сайтах рекомбинации и, таким образом, ограничены. Хайаши (Hayashi) с сотр. (48) сообщает об одновременном мутагенезе участков CDR антител в одноцепочечных антителах (ScFv) за счет перекрывания удлиняющих сегментов и ПЦР.
Карен (Caren) с сотр. (45) описывает способ создания крупной популяции множественных мутантов с использованием неупорядоченной рекомбинации ин виво. Однако, их способ требует рекомбинации двух различных библиотек плазмид, причем каждая из библиотек содержит различные селектируемые маркеры. Таким образом, этот способ ограничен конечным числом рекомбинаций, равным числу существующих селектируемых маркеров, и приводит к линейному увеличению числа маркерных генов, связанных с выбранной последовательностью (выбранными последовательностями), Калогеро (Calogero) с сотр. (46) и Galtzzi с сотр. (47) сообщают, что ин виво рекомбинация между двумя гомологичными, но усеченными генами насекомых на плазмиде может привести к образованию гибридного гена.
Радман (Radman) с сотр. (49) сообщает об ин виво рекомбинации практически несовместимых ДНК последовательностей в клетках хозяина, содержащих дефективные ошибочно репарированные энзимы, приводящей к образованию гибридной молекулы.
Было бы выгодно разработать способ получения мутантных протеинов, который позволил бы создавать крупные библиотеки мутантных последовательностей нуклеиновых кислот, которые легко разыскиваются. Раскрываемое здесь изобретение направлено на использование повторяющихся циклов точечного мутагенеза, перетасовки нуклеиновых кислот и отбора, которые позволили бы управлять молекулярной эволюцией ин витро весьма сложных линейных последовательностей, таких, как протеины, за счет неупорядоченной рекомбинации.
Соответственно, было бы выгодно разработать способ, который позволил бы получать крупные библиотеки мутантных ДНК, РНК или протеинов и отбирать конкретные мутанты для нужных целей.
Описываемое здесь изобретение направлено на использование повторяющихся циклов мутагенеза, ин виво рекомбинацию и отбор, которые обеспечивали бы направленную молекулярную эволюцию ин виво, позволили бы управлять молекулярной эволюцией очень сложных линейных последовательностей, таких, как ДНК, РНК или протеины, за счет рекомбинации.
Дальнейшие преимущества настоящего изобретения станут очевидны из описания изобретения со ссылками на прилагаемые чертежи.
Краткое
содержание изобретения
Настоящее изобретение направлено на способ создания выбранной полинуклеотидной последовательности, или популяции выбранных полинуклеотидных последовательностей, обычно
в форме амплифицированных и/или клонированных полинуклеотидов, где выбранные полинуклеотидные последовательности (или последовательность) обладают нужными фенотипическими характеристиками (например,
кодируют полипептид, промотируют транскрипцию связанных полинуклеотидов, связывают протеин и т.п.), по которым может быть произведен отбор. Один из способов, идентифицирующих полипептиды, которые
обладают нужной структурой или функциональными свойствами, например, связываются с заранее определенной биологической макромолекулой (например, рецептором), включает скринирование большой библиотеки
полипептидов для поиска отдельных членов этой библиотеки, которые обладают структурой или функциональными свойствами, сообщаемыми аминокислотной последовательностью полипептида.
Настоящее изобретение включает способ создания библиотек представленных полипептидов или представленных антител, подходящих для скринирования за счет афинного взаимодействия или фенотипического скринирования. Этот способ включает (1) получение первого множества членов выбранной библиотеки, содержащего представленный полипептид или представленное антитело, и ассоциированного полинуклеотида, кодирующего представленный полипептид или представленное антитело, и получение указанных ассоциированных полинуклеотидов или их копий, причем указанный ассоциированный полинуклеотид включает участок практически идентичной последовательности, необязательно введение мутаций в указанные полинуклеотиды или копии и (2) сбор и фрагментацию, обычно неупорядоченную, указанных ассоциированных полинуклеотидов или копий для получения их фрагментов в условиях, подходящих для PCR амплификации, осуществления PCR амплификации и необязательного мутагенеза, и за счет этого гомологическую рекомбинацию указанных фрагментов для получения перетасованного пула рекомбинированных полинуклеотидов, в результате чего существенная фракция (например, более 10%) рекомбинированных полинуклеотидов указанного перетасованного пула отсутствует в первом множестве членов выбранной библиотеки, причем указанный перетасованный пул содержит библиотеку представленных полипептидов или представленных антител, пригодных для скринирования за счет афинного взаимодействия. При желании этот способ включает дополнительную стадию скринирования членов библиотеки перетасованного пула идентификации отдельных членов перетасованной библиотеки, способных связываться или взаимодействовать каким-либо другим образом (например, каталитические антитела) с заранее определенной макромолекулой, например, с протеиновым рецептором, пептидом, олигосахаридом, вирионом или другими заранее определенными соединениями или структурами. Представленные полипептиды, антитела, имитирующие пептиды антитела и последовательности вариабельных участков, которые идентифицированы из таких библиотек, можно использовать для терапевтических, диагностических, исследовательских и аналогичных целей (например, в качестве катализаторов, в качестве растворяемых веществ для изменения осмотических свойств водных растворов и т.п.) и/или отбора по афинности. Способ можно модифицировать таким образом, чтобы стадия отбора осуществлялась по фенотипическим характеристикам, отличающимся от связывающей афинности для заранее определенной молекулы (например, для каталитической активности, стабильности, устойчивости к окислению, устойчивости к лекарствам, или детектируемому фенотипу, возникающему на клетках хозяина).
В одном из вариантов первое множество членов выбранной библиотеки фрагментируют и гомологически рекомбинируют за счет PCR ин витро.
В одном из вариантов первое множество членов выбранной библиотеки фрагментируют ин витро, полученные фрагменты переносят в клетки или организм хозяина и гомологически рекомбинируют для получения членов перетасованной библиотеки ин виво.
В одном из вариантов первое множество членов выбранной библиотеки клонируют или амплифицируют на эписомально реплицируемых векторах, причем множество указанных векторов переносят в клетку и гомологически рекомбинируют для образования ин виво членов перетасованной библиотеки.
В одном из вариантов первое множество членов выбранной библиотеки не фрагментируют, но клонируют или амплифицируют на эписомально реплицируемом векторе как прямой повтор, причем каждый повтор содержит отличающийся вид последовательности члена выбранной библиотеки, а указанный вектор переносят в клетку и гомологически рекомбинируют за счет внутривекторной рекомбинации для образования членов перетасованной библиотеки ин виво.
В еще одном варианте предложены комбинации ин витро и ин виво перетасовки для усиления комбинаторного разнообразия.
В настоящем изобретении предложен способ создания библиотеки представленных антител, пригодных для скринирования за счет афинных взаимодействий. Этот способ включает (1) получение первого множества членов выбранной библиотеки, содержащих представленное антитело, и ассоциированного полинуклеотида, кодирующего указанное представленное антитело, и получение указанных ассоциированных полинуклеотидов или их копий, где указанные ассоциированные полинуклеотиды содержат участок, практически идентичный каркасной последовательности вариабельного участка, и (2) объединение и фрагментирование указанных ассоциированных полинуклеотидов или их копий для получения их фрагментов в условиях, подходящих для PCR амплификации, и за счет этого осуществление гомологической рекомбинации указанных фрагментов для получения перетасованного пула рекомбинированных полинуклеотидов, содержащих новые комбинации CDR, где значительная часть (например, более 10%) рекомбинированных полинуклеотидов указанного перетасованного пула содержит CDR комбинации, которые отсутствуют в первом множестве членов выбранной библиотеки, причем указанный перетасованный пул состоит из библиотеки представленных антител, содержащих CDR пермутации, и подходящий для скринирования за счет афинного взаимодействия. При желании перетасованный пул подвергают афинному скринированию для выбора членов перетасованной библиотеки, которые связываются с определенным заранее эпитопом (антигеном), и за счет этого осуществляют выбор из множества членов перетасованной библиотеки. При желании множество членов выбранной перетасованной библиотеки можно перетасовать и скринировать итерационно, в результате 1 - до около 1000 циклов или, при желании, до получения членов библиотеки, обладающих нужной связывающей афинностью.
Соответственно, в одном аспекте настоящего изобретения предложен способ введения одной или более из мутаций в матричный двухцепочечный полинуклеотид, где матричный двухцепочечный полинуклеотид был расщеплен на неупорядоченные фрагменты нужного размера, за счет добавления к полученной популяции двухцепочечных фрагментов одного или более одно- или двухцепочечных нуклеотидов, где указанные олигонуклеотиды содержат участок идентичности и участок гетерологичности к матричному полинуклеотиду; денатурирование полученной смеси двухцепочечных неупорядоченных фрагментов и олигонуклеотидов в одноцепочечные фрагменты с помощью полимеразы в условиях, которые приводят к отжигу указанных одноцепочечных фрагментов по участку идентичности между одноцепочечными фрагментами, и образования мутагенизированных двухцепочечных полинуклеотидов; и при желании повторения вышеуказанных стадий.
В другом аспекте настоящее изобретение направлено на способ получения рекомбинантных протеинов, обладающих биологической активностью, за счет обработки образца, содержащего двухцепочечные матричные полинуклеотиды, кодирующие дикого типа протеин, в условиях, которые обеспечивают расщепление указанных матричных полинуклеотидов на беспорядочные двухцепочечные фрагменты, обладающие нужным размером; добавления к полученной популяции неупорядоченных фрагментов одного или более из одно- или двухцепочечных олигонуклеотидов, причем указанные олигонуклеотиды содержат участки идентичности и участки гетерологичности с матричным полинуклеотидом; денатурации полученной смеси двухцепочечных фрагментов и олигонуклеотидов до одноцепочечных фрагментов; инкубирования полученной популяции одноцепочечных фрагментов с полимеразой в условиях, которые приводят к отжигу указанных одноцепочечных фрагментов по участкам идентичности и образованию мутагенизированных двухцепочечных полинуклеотидов; при желании повторения указанных стадий; и последующей экспрессии рекомбинантного протеина из мутагенизированного двухцепочечного полинуклеотида.
Третий аспект настоящего изобретения направлен на способ получения химерического полинуклеотида за счет обработки образца, содержащего различные двухцепочечные матричные полинуклеотиды, где указанные различные матричные полинуклеотиды содержат участки идентичности и участки гетерологичности, в условиях, которые обеспечивают расщепление указанных матричных полинуклеотидов на неупорядоченные двухцепочечные фрагменты нужного размера; денатурирования полученных неупорядоченных двухцепочечных фрагментов, содержащихся в обработанном образце, до одноцепочечных фрагментов; инкубирования полученных одноцепочечных фрагментов с полимеразой в условиях, которые обеспечивают отжиг одноцепочечных фрагментов по участкам идентичности, и образования химерических двухцепочечных полинуклеотидных последовательностей, содержащих матричные полинуклеотидные последовательности; и повторения при желании вышеуказанных стадий.
Четвертый аспект настоящего изобретения направлен на способ репликации матричного полинуклеотида за счет объединения ин витро одноцепочечных матричных полинуклеотидов с мелкими неупорядоченными одноцепочечными фрагментами, полученными при расщеплении и денатурировании матричного полинуклеотида; и инкубирования указанной смеси фрагментов нуклеиновых кислот в присутствии полимеразы нуклеиновых кислот в условиях, где образуется популяция двухцепочечных матричных полинуклеотидов.
В настоящем изобретении предложено также использование перетасовки полинуклеотидов ин витро и/или ин виво, для перетасовки кодирующих полинуклеотиды полипептидов и/или полинуклеотидов, содержащих транскрипционные регуляторные последовательности.
В настоящем изобретении предложен также способ использования полинуклеотидной перетасовки для перетасовки популяции вирусных генов (например, капсидных протеинов, спайк-гликопротеинов, полимераз, протеаз и т.д.) или вирусных геномов (например, paramyxoviridae, orthomyxoviridae, герпес-вирусов, ретровирусов, реовирусов, риновирусов и т.д.).
В этом варианте в настоящем изобретении предложен способ перетасовки последовательностей, кодирующих части или целые иммуногенные вирусные протеины для создания новых комбинаций эпитопов, а также новых эпитопов, созданных за счет рекомбинации; такие перетасованные вирусные протеины могут содержать эпитопы или комбинации эпитопов, которые, по-видимому, возникают в нейтральном окружении как следствие консенсуса вирусной эволюции (например, за счет рекомбинации штаммов вирусов инфлюэнцы).
В настоящем изобретении предложен также способ перетасовки полинуклеотидных последовательностей для создания генных терапевтических векторов и конструкций репликационно-дефективной генной терапии, таких, которые могут быть использованы для генной терапии человека, включая (но не ограничиваясь ими) векторы вакцинации для вакцинации на основе ДНК, а также антинеопластической генной терапии и других форм генной терапии.
Краткое описание чертежей
Фиг. 1 представляет схематическую
диаграмму сравнения мутагенного перетасовывания и подверженного ошибкам ПЦР: (a) исходная библиотека, (b) пул выбранных последовательностей в первом цикле афинного отбора; (d) ин витро рекомбинация
выбранных последовательностей ("перетасовка"); (f) пул выбранных последовательностей во втором цикле афинного отбора после перетасовки; (c) подверженная ошибкам ПЦР; (e) пул выбранных
последовательностей во втором цикле афинного отбора после подверженной ошибкам ПЦР.
Фиг. 2 иллюстрирует новую сборку 1,0 к.п.о. LacZ-альфа генного фрагмента из 10-50 п.о. неупорядоченных фрагментов. (a) Фотография геля ПЦР амплифицированных ДНК фрагментов, содержащих LacZ-альфа ген. (b) Фотография геля ДНК фрагментов после переваривания ДНКазой. (c) Фотография геля ДНК фрагментов 10-50 п.о., выделенных из переваренного ДНК фрагмента LacZ-альфа гена; (d) Фотография геля 10-50 п.о. ДНК фрагментов после указанного числа циклов ДНК повторной сборки; (e) Фотография геля рекомбинационной смеси после амплификации за счет ПЦР с праймерами.
Фиг. 3 представляет схематическую иллюстрацию мутантов стоп кодона LacZ-альфа гена и их ДНК последовательностей. Заключенные в прямоугольники участки представляют гетерологические зоны, служащие маркерами. Стоп кодоны заключены в маленькие прямоугольники или подчеркнуты. Знак "+" обозначает ген дикого типа, а "-" указывает мутированную зону гена.
Фиг. 4 представляет схематическую иллюстрацию встраивания или создания шпильки синтетического олигонуклеотида в процессе вторичной сборки LacZ-альфа гена.
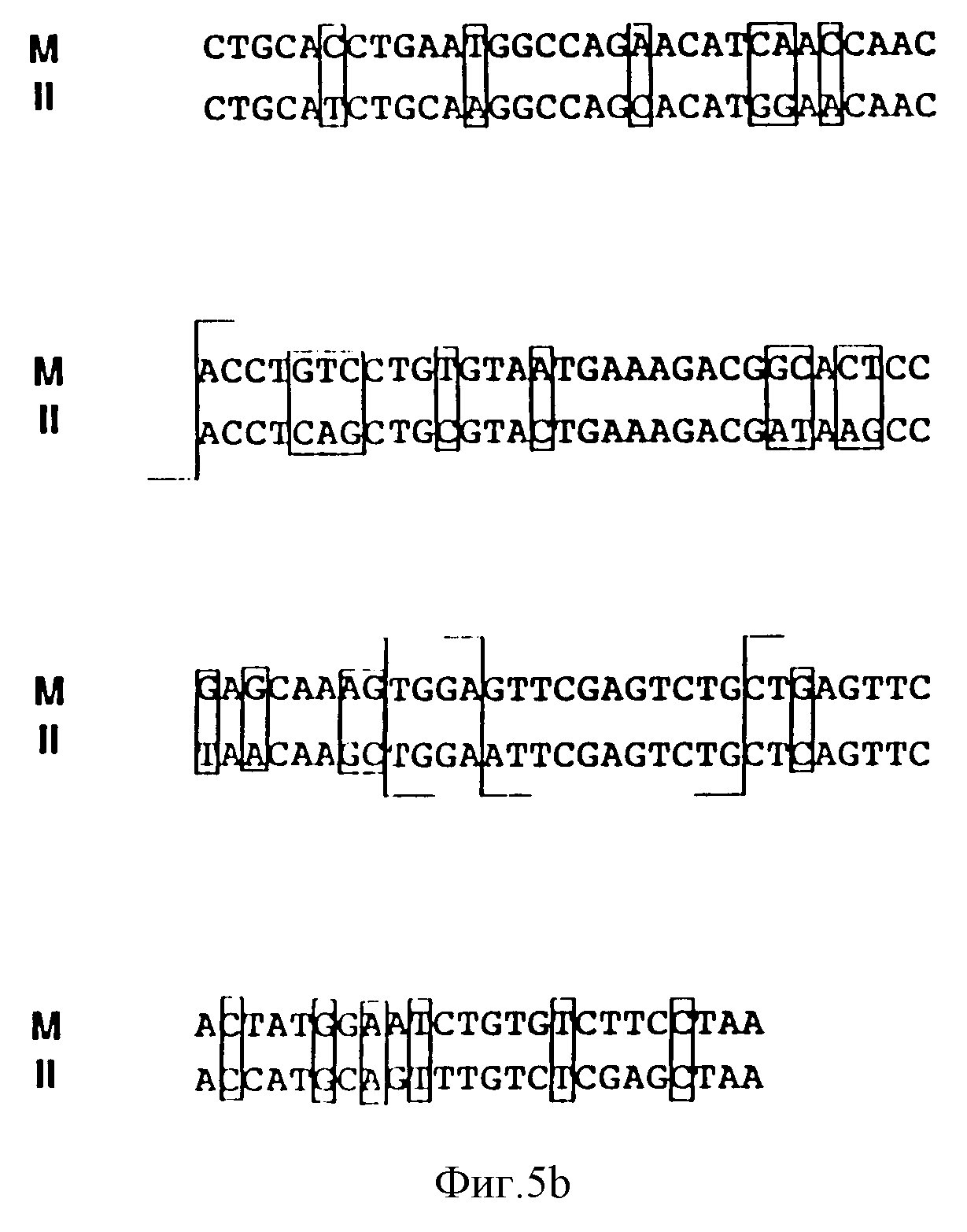
Фиг. 5 иллюстрирует участки гомологичности между мышиным IL1-B геном (М) и человеческим IL1-B геном (4) при использовании кодона E.coli. Участки гетерологичности заключены в прямоугольники. Знак

Фиг. 6 представляет схематическую диаграмму модельной системы перетасовки СDR антитела, с использованием антител антикроличьего IgG-(A10B).
Фиг. 7 иллюстрирует наблюдаемую частоту осуществления определенных комбинаций CDR в перетасованных ДНК ScFv антител антикроличьего IgG (A10B).
Фиг. 8 иллюстрирует повышенную активность ScFv антикроличьих антител после перетасовки ДНК и каждого цикла отбора.
Фиг. 9 представляет схематическое изображение pBR322-Sfi -BL-LA-Sfi и ин виво внутриплазмидную рекомбинацию за счет прямых повторов, а также скорость образования ампициллин-устойчивых колоний за счет внутриплазмидных рекомбинаций, воссоздающих функциональный бета-лактамазный ген.
Фиг. 10 представляет схематическое изображение pBR322-Sfi -2Bla-Sfi и ин виво внутриплазмидную рекомбинацию за счет прямых повторов, а также скорость образования ампициллин-устойчивых колоний за счет внутриплазмидной рекомбинации, восстанавливающей функциональный бета-лактамазный ген.
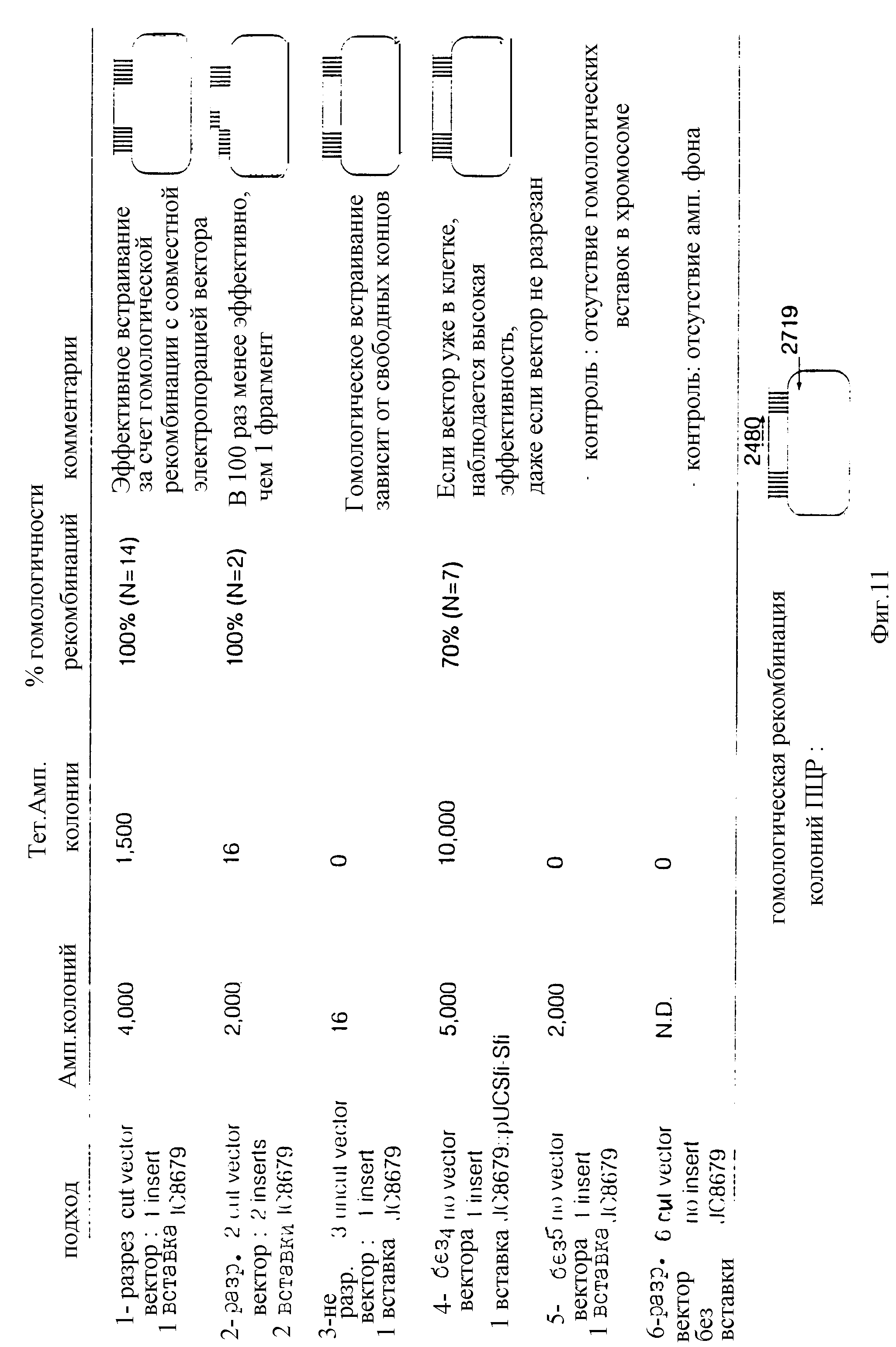
Фиг. 11 иллюстрирует способ тестирования эффективности множества циклов гомологических рекомбинаций после введения полинуклеотидных фрагментов в клетки для образования рекомбинантных протеинов.
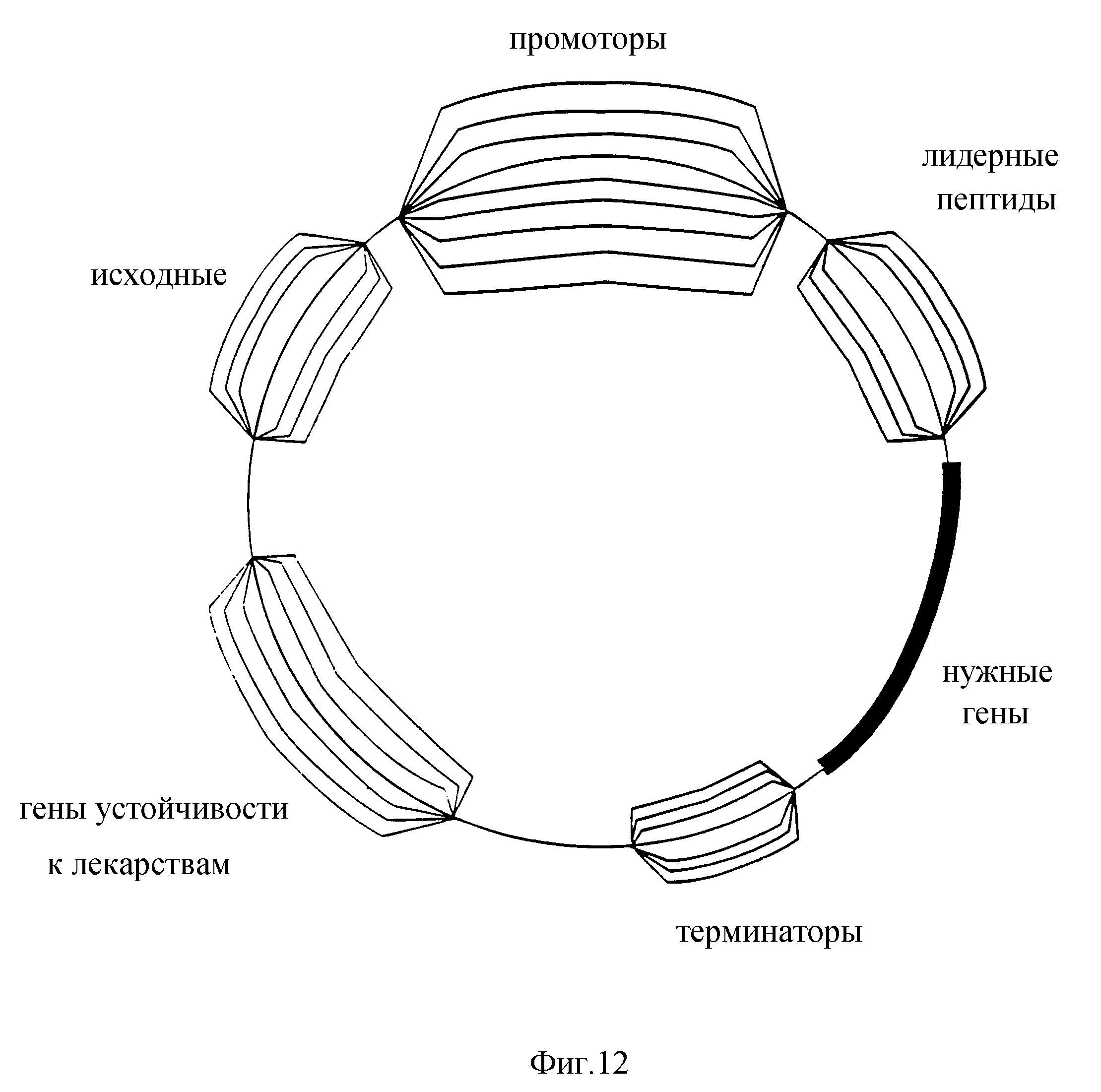
Фиг. 12 схематически изображает создание библиотеки векторов за счет перетасовки кассет по следующим локусам: промотор, лидерный пептид, терминатор, селектируемый по устойчивости к лекарствам ген, и источник репликации. Множественные параллельные линии в каждом локусе представляют множество кассет для указанной кассеты.
Фиг. 13 схематически представляет некоторые примеры кассет, пригодных по различным локусам для конструирования библиотек прокариотных векторов за счет перетасовки.
Описание предпочтительного варианта
Настоящее изобретение относится к способу вторичной сборки молекул нуклеиновых кислот после неупорядоченной фрагментации и к
его применению для мутагенеза ДНК последовательностей. Описан также способ получения фрагментов нуклеиновых кислот, кодирующих мутантные протеины, обладающие повышенной биологической активностью. В
частности, настоящее изобретение относится также к способу повторяющихся циклов мутагенеза, перетасовки нуклеиновых кислот и отбору, которые позволяют создавать мутантные протеины, обладающие
повышенной биологической активностью.
Настоящее изобретение направлено на способ создания очень большой библиотеки ДНК, РНК или мутантов протеинов. Этот способ имеет особенные преимущества при создании родственных ДНК фрагментов, из которых можно осуществить отбор нужного (нужных) фрагмента (фрагментов) нуклеиновой кислоты. В частности, настоящее изобретение относится также к способу повторяющихся циклов мутагенеза, гомологической рекомбинации и отбора, которые позволяют создавать мутантные протеины, обладающие повышенной биологической активностью.
Однако прежде, чем обсуждать настоящее изобретение более подробно, следует дать определения следующих терминов.
Определения
В том смысле, как здесь использованы, следующие термины
имеют значения:
Термин "ДНК вторичная сборка" используется в тех случаях, когда рекомбинация происходит между идентичными последовательностями. Напротив, термин "перетасовка ДНК" используют в
тех случаях, когда необходимо указать, что рекомбинация происходит между существенно гомологичными, но не идентичными последовательностями, причем в некоторых случаях перетасовка ДНК может включать
кроссовер за счет негомологичной рекомбинации, как,
например, за счет cre/lox и/или flp/frt систем и т.п.
Термин "амплификация" означает, что количество копий фрагмента нуклеиновой кислоты увеличивается.
Термин "идентичны" или "идентичность" означает, что две последовательности нуклеиновой кислоты имеют одинаковую или комплементарную последовательность. Так, "участки идентичности" означают, что участки или зоны фрагмента нуклеиновой кислоты или полинуклеотида идентичны или комплементарны другому полинуклеотиду или фрагменту нуклеиновой кислоты.
Термин "соответствует" используют для обозначения того, что последовательность полинуклеотида гомологична (то есть, идентична, но не строго эволюционно родственна) всей или части сравнительной полинуклеотидной последовательности, или что последовательность полинуклеотида идентична сравнительной последовательности полинуклеотида. Напротив, термин "комплементарна" используют здесь для обозначения того, что комплементарная последовательность гомологична всей или части сравнительной полинуклеотидной последовательности. Для иллюстрации, нуклеотидная последовательность "TATAC" соответствует сравнительной последовательности "TATAC" и комплементарна сравнительной последовательности GTATA".
Следующие термины используют для описания связей между двумя и более полинуклеотидами: "сравнительная последовательность", "окно сравнения", "идентичность последовательности", "процент идентичности последовательности" и "практически идентична". Термин "сравнительная последовательность" определяет последовательность, используемую как основу для сравнения последовательностей; "сравнительной последовательностью" может быть часть более крупной последовательности, например, сегмент полной длины кДНК или генной последовательности, приведенной в списке последовательностей, такая, как полинуклеотидная последовательность на фиг. 1 или фиг. 2(b), или может представлять собой полную кДНК или генную последовательность. Обычно сравнительная последовательность состоит из, по крайней мере, 20 нуклеотидов, часто из, по крайней мере, 25 нуклеотидов, и часто из, по крайней мере, 50 нуклеотидов в длину. Так как возможно, что два нуклеотида (1) могут каждый содержать последовательность (т. е. часть полной нуклеотидной последовательности), которая аналогична для двух полинуклеотидов, и (2) могут дополнительно содержать последовательность, которая отличается для двух полинуклеотидов, сравнение последовательностей двух (или более) полинуклеотидов обычно осуществляют, сравнивая последовательности двух полинуклеотидов в "окне сравнения" для определения и сравнения локальных участков аналогичности последовательностей.
Термин "окно сравнения" в том смысле, как здесь использован, относится к концептуальному сегменту из, по крайней мере, 20 непрерывных положений нуклеотидов, в котором полинуклеотидную последовательность можно сравнивать со сравнительной последовательностью, состоящей из, по крайней мере, 20 непрерывных нуклеотидов, и где часть полинуклеотидной последовательности в окне сравнения может содержать дополнения или делеции (т.е. разрывы или бреши) порядка 20% или менее по сравнению со сравнительной последовательностью (которая не содержит дополнений или делеций) для оптимального сравнения этих двух последовательностей. Оптимальное выравнивание последовательностей для соответствия окну сравнения можно осуществить за счет алгоритма локальной гомологичности Смита и Уотермана (Smith and Waterman (1981) Adv. Appl. Math. 2:482 за счет алгоритма сравнения гомологичности Needleman and Wunsch (1970) J. Mol.Biol. 48:443, за счет способа поиска аналогичности Pearson and Lipman (1988) Proc.Natl.Acad.Sci. (USA) 85: 2444, за счет компьютерного осуществления этих алгоритмов (GAP, BESTFLT, FASTA и TFASTA в Wisconsin Genetics Software Rackage Release 7.0, Genetics Computer Group, 575 Science Dr. Madison, WI) или за счет изучения и наилучшего соответствия (т.е. достижения наивысшего процента гомологичности в окне сравнения), создаваемого за счет различных методов.
Термин "идентичность последовательности" означает, что две полинуклеотидные последовательности идентичны (т.е., на основании нуклеотид за нуклеотидом) в окне сравнения. Термин "процент идентичности последовательности" рассчитывают для сравнения двух оптимально соответствующих последовательностей в окне сравнения, определения числа положений, в которых находятся идентичные основания нуклеиновых кислот (например. A, T, C, G, U или I) в обоих последовательностях для получения числа соответствующих положений, деления числа соответствующих положений на полное число положений в окне сравнения (т.е. на размер окна) и умножения результата на 100 для получения процента идентичности последовательности.
Термин "практически идентична" в том смысле, как здесь использован, обозначает характеристику полинуклеотидной последовательности, в которой полинуклеотид содержит последовательность, которая содержит, по крайней мере, 80% идентичности последовательности, предпочтительно, по крайней мере, 85% идентичности, и часто от 90 до 95% идентичности последовательности, и чаще, по крайней мере, 99% идентичности последовательности по сравнению со сравнительной последовательностью в окне сравнения, состоящем из, по крайней мере, 20 нуклеотидных положений, часто в окне, состоящем из, по крайней мере, 25-50 нуклеотидов, где процент идентичности последовательности рассчитывают из сравнения сравнительной последовательности с полинуклеотидной последовательностью, которая может содержать делеции или дополнения, составляющие всего 20% или менее от сравнительной последовательности в окне сравнения.
Замещения консервативных аминокислот относятся к взаимозаменяемости остатков, содержащих аналогичные боковые цепи. Так, например, группа аминокислот с алифатическими боковыми цепями является глицином, аланином, валином, лейцином и изолейцином; группа аминокислот с алифатически-гидроксильными боковыми цепями представлена серином и треонином; а группа аминокислот с амид-содержащими боковыми цепями представлена аспарагином и глутамином; группа аминокислот с ароматическими боковыми цепями представлена фенилаланином, тирозином и триптофаном; группа аминокислот, содержащих основные боковые цепи, представлена лизином, аргинином и гистидином; группа аминокислот с серусодержащими боковыми цепями, представлена цистеином и метионином. Предпочтительными группами замещения консервативных аминокислот являются: валин-лейцин-изолейцин, фенилаланин-тирозин, лизин-аргинин, аланин-валин и аспарагин-глютамин.
Термин "гомологичный" или "гомолог" означает, что одна одноцепочечная последовательность нуклеиновых кислот может гибридизоваться с комплементарной одноцепочечной последовательностью нуклеиновых кислот. Степень гибридизации может зависеть от ряда факторов, включая степень идентичности между последовательностями и условия гибридизации, такие, как температура и концентрация солей, что будет обсуждаться далее. Предпочтительно, чтобы участок идентичности был более чем около 5 пар оснований, более предпочтительно, чтобы участок идентичности составлял более 10 пар оснований.
Термин "гетерологичны" означает, что одна одноцепочечная последовательность нуклеиновых кислот не может гибридизоваться с другой одноцепочечной последовательностью нуклеиновых кислот или с ее комплементом. Так, участок гетерологичности может означать, что фрагменты нуклеиновых кислот или полинуклеотидов имеют участки или зоны в последовательности, которые не способны гибридизоваться с другой нуклеиновой кислотой или полинуклеотидом. Такие участки или зоны являются, например, участками мутаций.
Термин "родственный" (cognate) в том смысле, как здесь использован, относится к генной последовательности, которая эволюционно и функционально является родственной для образцов. Так, например (но не ограничиваясь этим), в геноме человека человеческий ген CD4 является родственным геном с мышиным CD4 геном, так как последовательности и структуры этих двух генов показывают, что они высоко гомологичны и оба гена кодируют протеин, функции которого состоят в подаче сигнала активации T клеток за счет MHC класса 11-ограниченного распознавания антигена.
Термин "дикого типа" означает, что фрагмент нуклеиновой кислоты не содержит никаких мутаций. Протеин "дикого типа" означает, что этот протеин будет активным на уровне активности, соответствующей природной, и будет содержать последовательность аминокислот, находящуюся в природе.
Термин "родственные полинуклеотиды" означает, что участки или зоны полинуклеотидов идентичны и что участки или зоны полинуклеотидов гетерологичны.
Термин "химерный полинуклеотид" означает, что этот полинуклеотид включает участки дикого типа и участки, которые подвергались мутациям. Он также может означать, что полинуклеотид содержит дикого типа участки из одного полинуклеотида и дикого типа участки из другого родственного полинуклеотида.
Термин "расщепление" означает переваривание полинуклеотида энзимами или разрушение полинуклеотида.
Термин "популяция" в том смысле, как здесь использован, означает набор таких компонентов, как полинуклеотиды, фрагменты нуклеиновых кислот или протеинов. Термин "смешаная популяция" означает набор компонентов, которые принадлежат к одному и тому же семейству нуклеиновых кислот или протеинов (т. е. являются родственными), но которые отличаются своими последовательностями (т. е. не являются идентичными) и, следовательно, отличаются своей биологической активностью.
Термин "специфический фрагмент нуклеиновой кислоты" относится к фрагменту нуклеиновой кислоты, который содержит определенные концевые точки и определенную последовательность нуклеиновых кислот. Два фрагмента нуклеиновых кислот, в которых один фрагмент нуклеиновых кислот имеет идентичную последовательность как часть второго фрагмента нуклеиновых кислот, но различные концы, содержат два различных специфических фрагмента нуклеиновых кислот.
Термин "мутации" означает изменения в последовательности дикого типа последовательности нуклеиновых кислот или изменения в последовательности пептида. Такие мутации могут быть такими точечными мутациями, как транзиция или трансверсия. Мутации могут быть делециями, вставками или дупликациями.
В используемых здесь обозначениях полипептидов левое направление является аминотерминальным направлением, а правое направление является карбокси-терминальным направлением, в соответствии со стандартным принятым обозначением. Аналогично, если нет других указаний, левый конец одноцепочечной полинуклеотидной последовательности является 5' концом; левое направление двухцепочечных полинуклеотидных последовательностей обозначается как 5' направление. Направление 5' к 3' добавления образующихся РНК транскриптов определяется как направление транскрипции; участки ДНК нитей, имеющие ту же последовательность, что и РНК, и которые являются 5' на 5'-конец РНК транскрипта, определяются как "последовательности в обратном направлении", участки последовательности на ДНК цепочке, содержащие ту же последовательность, что и РНК, и которые являются 3' на 3'- конец кодирующего РНК транскрипта, определяются как "последовательности в прямом направлении".
Термин "встречающийся в природе" в том смысле, как здесь использован, применяют к объекту для обозначения того факта, что этот объект может быть найден в природе. Так, например, полипептид или полинуклеотидная последовательность, которая присутствует в организме (включая вирусы), которая может быть выделена из природного источника и которая не была намеренно модифицирована человеком в лаборатории, является природной (или нативной). Обычно термин "встречающийся в природе" (или нативный) относится к объекту, который присутствует у непатологических (здоровых) индивидуумов, которые могут быть определены как типичные для данных видов.
Термин "агент" используют здесь для обозначения химического соединения, смеси химических соединений, ряда пространственно локализованных соединений (например, VLSIPS пептидного ряда, полинуклеотидного ряда, и/или ряда комбинаторных небольших молекул), биологических макромолекул, библиотек пептидов бактериофагов, библиотеки антител бактериофагов (например, ScFv), библиотеки полисомных пептидов, или экстрактов, полученных из таких биологических материалов, как бактерии, растения, грибки, или клетки или ткани животных (особенно млекопитающих).
Агенты оценивают по их потенциальной активности в качестве антинеопластов, противовоспалительных агентов или модуляторов апоптоза путем включения их в анализы скринирования, описываемые далее. Агенты оценивают по их потенциальной активности в качестве специфических протеиновых ингибиторов взаимодействия (т. е. , агент, который селективно ингибирует взаимодействие связывания двух заранее определенных полипептидов, но который практически не препятствует жизнеспособности клеток), за счет включения их в анализы скринирования, описываемые далее.
В том смысле, как здесь использован, термин "практически чистый" относится к видам объектов, в которых присутствует преимущественно этот вид (т.е. на молярной основе в этой композиции его больше, чем любых других видов отдельных макромолекул), и предпочтительно практически чистая фракция представляет собой композицию, в которой вид составляет, по крайней мере, около 50% (на молярной основе) от всех присутствующих видов макромолекул. Обычно практически чистая композиция составляет более чем около 80-90% от всех видов присутствующих макромолекул в композиции. Наиболее предпочтительно, чтобы целевой вид был очищен до существенной гомогенности (загрязненные образцы не могут быть детектированы в композиции с помощью обычных способов детектирования), где композиция состоит, главным образом, из одного вида макромолекул. Различные растворители, мелкие молекулы (менее 500 Дальтон) и различные элементные ионы не рассматриваются в качестве макромолекулярных образцов.
В том смысле, как здесь использован, термин "физиологические условия" относится к температуре, pH, ионной силе, вязкости и тому подобным биохимическим параметрам, которые совместимы с живым организмом и/или которые обычно существуют внутриклеточно в жизнеспособных культивируемых дрожжевых клетках или клетках млекопитающих. Так, например, внутриклеточные условия для роста дрожжевых клеток в условиях выращивания типичных лабораторных культур являются физиологическими условиями. Подходящие условия реакции ин витро для ин витро транскрипционных коктейлей обычно являются физиологическими условиями. Обычно ин витро физиологические условия составляют 50-200 мМ NaCl или KCl, pH 6,5-8,5, 20-45oC и 0,001-10 мМ двухвалентных катионов (например, Mg++, Ca++); предпочтительно около 150 мМ NaCl или KCl, pH 7,2-7,6, 5 мМ двухвалентных катионов, и часто включают 0,01-1,0% неспецифического протеина (например, BSA). Часто может присутствовать неионный детергент (Tween, NP-40, Triton Х-100) обычно от около 0,001 до 2%, обычно 0,05-0,2% (об/об). Конкретные водные условия можно выбрать на основе практики в соответствии с обычными методиками. Для общего сведения, могут быть использованы следующие условия буферированных водных растворов: 10-250 мМ NaCl, 5-50 мМ Tris-HCl, pH 5-8, с добавлением при желании двухвалентного катиона (катионов) и/или хелатообразующих металлов, и/или неионных детергентов, и/или мембранных фракций, и/или противовспенивающих агентов и/или сцинтиллирующих агентов.
Специфическую гибридизацию определяют как образование гибридов между первым полинуклеотидом и вторым полинуклеотидом (например, полинуклеотидом, имеющим определенную, но практически идентичную последовательность с первым нуклеотидом), где первый полинуклеотид, предпочтительно, гибридизуется со вторым полинуклеотидом в жестких условиях гибридизации, когда существенно не родственные полинуклеотидные последовательности в этой смеси не образуют гибридов.
В том смысле, как здесь использован, термин "одноцепочечное антитело" относится к полипептиду, содержащему VH домен и VL домен в полипептидной связи, обычно связанные пространственным (спейсерным) пептидом (например, [Gly-Gly-Gly-Gly- Ser]x), и которые могут содержать дополнительные аминокислотные последовательности по амино- и/или карбоксиконцу. Так, например, ScFv представляет собой одноцепочечное антитело. Одноцепочечными антителами обычно являются протеины, состоящие из одного или более полипептидных сегментов, из, по крайней мере, 10 непрерывных аминокислот, практически кодируемые генами суперсемейства иммуноглобулинов (например, см. The Immunoglobulin Gene Superfamily, A.F. Williams and A.N. Barclay, in Immunoglobulin Genes, T. Honjo F.W: Alt and T.H. Rabbitts eds. (1989) Academic Press: San Diego, CA, pp 361-387, включено сюда по ссылке), более часто кодируемые генными последовательностями тяжелых или легких цепей грызунов, приматов (не людей), птиц, свиней, быков, овец, коз или людей. Функциональное одноцепочечное антитело обычно содержит достаточный участок генного продукта суперсемейства иммуноглобулинов, так что сохраняет свойство связывания со специфической мишеневой молекулой, обычно рецептором или антигеном (эпитопом).
В том смысле, как здесь использован, термин "определяющий комплементарность участок" (complementarity-determining region) и "CDR" относится к известному специалистам термину, определенному, например, Kabat and Chothia, известному так же, как гипервариабельные участки или гипервариабельные петли (Chothia and Lesk (1987) J.Mol. Biol. 196:901; Chothia et.al. (1989) Nature 342: 877; E. A.Kabat et al. Sequences of Proteins of Immunological Interest (National Institute of Health, Bethesda, MD) (1987), и Tramontano et al. (1990), J. Mol.Biol. 215:175). Домены вариабельных участков обычно содержат примерно 105-115 амино-терминальных аминокислот из нативной иммуноглобулиновой цепи (например, аминокислоты 1-110), хотя несколько более короткие или несколько более длинные вариабельные домены также годятся для образования одноцепочечных антител.
Вариабельный участок легкой или тяжелой цепи иммуноглобулина состоит из каркасного участка, прерываемого тремя гипервариабельными участками, также называемыми CDR. Протяженности каркасного участка CDR были точно определены (см. "Sequences of Proteins of Immunological Interest", E.Kabat et al. 4-th Ed. U.S. Departament of Health and Human Services, Bethesda, MD (1987).
Последовательности каркасных участков различных легких и тяжелых цепей относительно консервативны внутри одного вида. В том смысле, как здесь использован, термин "каркасный участок человека" представляет собой каркасный участок, который практически идентичен (примерно на 85% или более, обычно на 90-95% или более) каркасному участку, нативно встречающемуся в иммуноглобулине человека. Каркасный участок антитела, то есть, объединенные каркасные участки, составляющие легкую и тяжелую цепи, служат для определения положения и сравнения CDR. Именно CDR, главным образом, ответственны за связывание с эпитопом антигена.
В том смысле, как здесь использован, термин "вариабельный сегмент" относится к части образующегося пептида, который содержит неупорядоченную, псевдонеупорядоченную или определенную основную последовательность. Вариабельный сегмент может содержать как вариантные, так и инвариантные положения остатков и степень вариации остатков в вариантном положении остатков может быть ограничена; обе возможности можно селектировать. Обычно вариабельные сегменты имеют в длину от около 5 до 20 аминокислотных остатков (например, 8-10), хотя вариабельные участки могут быть длиннее и могут содержать участки антител или рецепторных протеинов, рецепторные протеины и т.п.
В том смысле, как здесь использован, термин "неупорядоченная пептидная последовательность" относится к аминокислотной последовательности, состоящей из двух или более аминокислотных мономеров и сконструированная за счет стохастического или неупорядоченного процесса. Неупорядоченный пептид может включать каркасные или опорные участки, которые могут содержать инвариантные последовательности.
В том смысле, в каком здесь использован, термин "библиотека неупорядоченных пептидов" относится к набору полинуклеотидных последовательностей, который кодирует набор неупорядоченных пептидов, и к набору неупорядоченных пептидов, кодируемых этими полинуклеотидными последовательностями, а также к протеинам слияния, содержащим эти неупорядоченные пептиды.
В том смысле, как здесь использован, термин "псевдонеупорядоченный" относится к набору последовательностей, которые отличаются ограниченной вариабельностью, так что, например, степень вариабельности остатков в одном положении отличается от степени вариабельности в другом положении, но в любом из псевдонеупорядоченных положений разрешена некоторая степень вариации остатков, однако в определенных пределах.
В том смысле, как здесь использован, термин "определенная каркасная последовательность" относится к набору определенных последовательностей, которые отбирают на не неупорядоченной основе, обычно на основе экспериментальных результатов или структурных данных; например, определенная последовательность каркаса может содержать набор определенных аминокислотных последовательностей, которые, как известно, образуют β -листовую структуру, или может содержать лейциновый семичленный повторяющийся фрагмент типа "застежки-молнии", цинк-фингер домен, наряду с другими вариациями. "Определенное ядро последовательности" представляет набор последовательностей, который охватывает ограниченный объем вариабельности. Поскольку (1) полностью неупорядоченная 10- мерная последовательность из 20 обычных аминокислот может быть любой из (20)10 последовательностей, и (2) псевдонеупорядоченная 10-мерная последовательность из 20 обычных аминокислот может быть любой из (20)10 последовательностей, но должна демонстрировать предпочтение для определенных остатков в определенных положениях и/или (3) определенное ядро последовательности является субнабором последовательностей, которые представляют менее чем максимальное число потенциальных последовательностей, если положение каждого остатка может быть любым из допустимых 20 обычных аминокислот (и/или допустимых необычных амино/имино-кислот). Определенное ядро последовательности обычно содержит положения вариантных и инвариантных остатков и/или включает положения вариантных остатков, которые могут включать остатки, выбранные из определенного субнабора аминокислотных остатков, и т. п., либо сегментно, либо по полной длине отдельно выбранной последовательности - члена библиотеки. Определенные ядра последовательностей могут относиться либо к аминокислотным последовательностям, либо к полинуклеотидным последовательностям. Для иллюстрации (но не с целью ограничений) последовательности (NNK)10 и (NNM)10, где N представляет A, T, G или C; K представляет G или T, а M представляет A или C, являются определенными ядерными последовательностями.
В том смысле, как здесь использован, термин "эпитоп" относится к части антигена или другой макромолекулы, способной к образованию связывающего взаимодействия, которое взаимодействует с вариабельным участком связывающего "кармана" антитела. Обычно такое связывающее взаимодействие проявляется как межмолекулярный контакт одного или более из аминокислотных остатков CDR.
В том смысле, как здесь использован, термин "рецептор" относится к молекуле, которая отличается афинностью для данного лиганда. Рецепторы могут быть нативными или синтетическими молекулами. Рецепторы можно использовать в неизмененном состоянии или в виде агрегатов с другими видами. Рецепторы можно присоединить, ковалентно или нековалентно, к связывающему члену, либо непосредственно, либо за счет специфического связывающего вещества. Примеры рецепторов включают (но не ограничиваются ими) антитела, включая моноклональные антитела и антисыворотки, реагирующие со специфическими антигенными детерминантами (такими, как на вирусах, клетках или других материалах), рецепторы клеточных мембран, комплексы углеводов и гликопротеинов, энзимы и рецепторы гормонов.
В том смысле, как здесь использован, термин "лиганд" относится к молекуле, такой, как неупорядоченный пептид или последовательность вариабельного сегмента, которая распознается конкретным рецептором. Как понятно специалистам, молекула (или макромолекулярный комплекс) может быть как рецептором, так и лигандом. Обычно, участвующий в связывании партнер с меньшим молекулярным весом называют лигандом, а партнер с большим молекулярным весом называют рецептором.
В том смысле, как здесь использован, термин "линкер" или "спейсер" относится к молекуле или к группе молекул, которые соединяет две молекулы, такие, как ДНК связывающий протеин и неупорядоченный пептид, и служит для размещения двух молекул в предпочтительной конфигурации, например, так, чтобы неупорядоченный пептид мог связываться с рецептором при минимальных стерических затруднениях со стороны ДНК связывающего протеина.
В том смысле, как здесь использован, термин "операбельно связанный относится к связи полинуклеотидных элементов в функциональном соотношении. Говорят, что нуклеиновая кислота "операбельно связана", если она находится в функциональном отношении с другой последовательностью нуклеиновой кислоты. Так, например, промотор или энхансер операбельно связаны с кодирующей последовательностью, если они осуществляют транскрипцию кодирующей последовательности. Термин операбельно связанный означает, что ДНК последовательности, будучи связаны, обычно непрерывны и там, где необходимо соединить кодирующие участки двух протеинов, непрерывны и находятся в считывающей рамке.
Методика
Перетасовка нуклеиновых кислот представляет собой способ для ин витро или ин виво
гомологической рекомбинации пулов фрагментов нуклеиновых кислот или полинуклеотидов. Смеси последовательностей родственных нуклеиновых кислот или полинуклиотидов неупорядоченно фрагментируют и
осуществляют вторичную сборку для получения библиотеки смешаной популяции молекул нуклеиновых кислот или полинуклеотидов.
В отличие от кассетного мутагенеза, только перетасовка и подверженные ошибкам ПЦР позволяют осуществить мутацию пула последовательностей вслепую (без какой-либо информации о последовательностях помимо праймеров).
Преимущество мутагенной перетасовки настоящего изобретения по сравнению с одной только подверженной ошибкам ПЦР для повторяющегося отбора можно наилучшим образом продемонстрировать на примере конструирования антитела. На фиг. 1 представлена схематическая диаграмма ДНК перетасовки в соответствии с настоящим описанием. Исходная библиотека может состоять из родственных последовательностей различного происхождения (т.е. антител из нативной мРНК) или может быть получена за счет любого типа мутагенеза (включая перетасовку) гена отдельного антитела. Набор выбранных определяющих комплементарность участков ("CDR") получают после первого цикла афинного отбора (фиг. 1). На диаграмме выделенные жирно CDR придают молекуле антитела повышенную афинность по отношению к антигену. Перетасовка позволяет обеспечить свободную комбинаторную ассоциацию всех CDR1 со всеми CDR2, со всеми CDR3 и т.д. (фиг. 1).
Этот способ отличается от ПЦР тем, что представляет обратную цепную реакцию. В ПЦР количество полимеразных стартовых сайтов и количество молекул растет экспоненциально. Однако последовательности полимеразных стартовых сайтов и последовательности молекул остаются практически теми же самыми. Напротив, вторичная сборка или перетасовка неупорядоченных фрагментов нуклеиновых кислот приводит к снижению с течением времени количества стартовых сайтов и количества (но не размера) неупорядоченных фрагментов. Для фрагментов, полученных из целых плазмид, теоретической конечной точкой является отдельная, крупная конкатемерная молекула.
Так как кроссоверы происходят в участках гомологичности, рекомбинации будут происходить, главным образом, между членами одного семейства последовательностей. Это исключает комбинации CDR, которые существенно несовместимы (например, направленные против различных эпитопов одного и того же антигена). Считается, что множество семейств последовательностей может быть перетасовано в одной и той же реакции. Далее, перетасовка сохраняет относительный порядок, так что, например, CDR1 не может оказаться в положении CDR2.
Редкие члены после перетасовки будут содержать большое число лучших (например, с наивысшей афинностью) CDR, и эти редкие члены могут быть отобраны на основании их превосходной афинности (фиг. 1).
Для CDR из пула, состоящего из 100 различных выбранных последовательностей антитела, можно осуществить вплоть до 1006 способов пермутаций. Столь большое число пермутаций невозможно представить в одной библиотеке ДНК последовательностей. Соответственно, ожидается, что может понадобиться множество циклов ДНК перетасовки и отбора в зависимости от длины последовательности и различий в последовательности, которую желательно получить.
Напротив, подверженная ошибкам ПЦР сохраняет все выбранные CDR в одной и той же родственной последовательности (фиг. 1), создавая гораздо меньшее облако мутаций.
Матричный полинуклеотид, который можно использовать в способах настоящего изобретения, может быть ДНК или РНК. Они могут быть различной длины в зависимости от размеров гена или ДНК фрагмента, которые нужно подвергнуть рекомбинации или вторичной сборке. Предпочтительно, чтобы матричный полинуклеотид был от 50 п.о. до 50 к.п.о. Предполагается, что полные векторы, содержащие нуклеиновую кислоту, кодирующую представляющий интерес протеин, можно использовать в способах настоящего изобретения, и в действительности они были успешно использованы.
Матричный полинуклеотид можно получить за счет амплификации, используя ПЦР реакцию (патент США 4683202 и 4683195) или с помощью других способов амплификации или клонирования. Однако наиболее эффективные результаты обеспечивает удаление свободных праймеров из PCR продукта перед фрагментацией. Если удалить соответствующим образом праймеры не удается, это может привести к низкой частоте кроссоверных клонов.
Матричный полинуклеотид часто должен быть двухцепочечным. Двухцепочечная молекула нуклеиновой кислоты необходима для обеспечения того, чтобы участки полученных фрагментов одноцепочечных нуклеиновых кислот были комплементарны друг другу, и поэтому могли бы гибридизоваться с образованием двухцепочечной молекулы.
Предполагается, что одноцепочечные или двухцепочечные фрагменты нуклеиновых кислот, содержащие участки идентичности с матричным полинуклеотидом и участки гетерологичности с матричным полинуклеотидом, можно добавлять к матричному полинуклеотиду на этой стадии. Предполагается также, что две различные, но родственные полинуклеотидные матрицы можно смешивать на этой стадии.
Двухцепочечную полинуклеотидную матрицу и любые добавляемые двух- или одноцепочечные фрагменты можно неупорядоченно переваривать до фрагментов с длиной от около 5 п.о. до около 5 к.п.о. или более. Предпочтительно, чтобы размер неупорядоченных фрагментов составлял от около 10 п.о. до 1000 п.о., более предпочтительно, чтобы размер ДНК фрагментов составлял от около 20 п. о. до 500 п.о.
В другом варианте, можно предположить, что двухцепочечную нуклеиновую кислоту с множеством ников можно использовать в способах настоящего изобретения. Ник представляет собой разрыв в одноцепочечной или двухцепочечной нуклеиновой кислоте. Расстояние между такими никами предпочтительно составляет от 5 п.о. до 5 к.п.о., более предпочтительно от 10 п.о. до 1000 п.о.
Фрагмент нуклеиновой кислоты можно переварить многими различными способами. Фрагмент нуклеиновой кислоты можно переварить такими нуклеазами, как ДНКаза1 или РНКаза. Нуклеиновую кислоту можно неупорядоченно разрезать с помощью ультразвука или пропуская ее через трубку с маленькими отверстиями.
Ожидается, что нуклеиновую кислоту можно частично переварить с помощью одного или более из рестрикционных энзимов, так, чтобы некоторые точки кроссовера могли бы сохраниться статистически.
Концентрация любого из фрагментов специфических нуклеиновых кислот должна быть не более чем 1 вес.% от всей нуклеиновой кислоты, более предпочтительно, чтобы концентрация любой из последовательностей специфических нуклеиновых кислот была бы не более чем 0,1 вес.% в расчет на всю нуклеиновую кислоту.
Количество различных фрагментов специфических нуклеиновых кислот в смеси должно быть, по крайней мере, около 100, предпочтительно, по крайней мере, около 500 и более предпочтительно, по крайней мере, около 1000.
На этой стадии одноцепочечные или двухцепочечные фрагменты нуклеиновых кислот, как синтетических, так и нативных, можно добавлять к неупорядоченным фрагментам двухцепочечных нуклеиновых кислот для повышения гетерогенности смеси фрагментов нуклеиновых кислот.
Предполагается также, что популяции двухцепочечных фрагментов неупорядоченно разрушенных нуклеиновых кислот можно смешать или скомбинировать на этой стадии.
Если необходимо осуществить вставку или мутации в матричном полинуклеотиде, фрагменты одноцепочечной или двухцепочечной нуклеиновых кислот с участками идентичности с матричным полинуклеотидом и участками гетерологичности с матричным полинуклеотидом можно добавлять в 20-кратном избытке по весу по сравнению со всей нуклеиновой кислотой, более предпочтительно, добавлять фрагменты одноцепочечной нуклеиновой кислоты в 10-кратном избытке по отношению ко всей нуклеиновой кислоте.
Если необходима смесь различных, но родственных матричных полинуклеотидов, популяции фрагментов нуклеиновых кислот из каждой из матриц можно объединить в отношении менее чем около 1:100, более предпочтительно, чтобы это отношение было менее чем около 1:40. Так, например, при бэккроссе дикого типа нуклеотида с популяцией мутированного полинуклеотида может понадобиться исключить нейтральные мутации (например, мутации за счет несущественных изменений фенотипических свойств, по которым идет отбор). В таком примере отношение неупорядоченно переваренных фрагментов дикого типа полинуклеотида, которые можно добавлять к неупорядоченно переваренным фрагментам мутантного полинуклеотида, составляет примерно от 1:1 до около 100:1, предпочтительно от 1:1 до 40:1.
Смешанную популяцию неупорядоченных фрагментов нуклеиновой кислоты денатурируют для получения фрагментов одноцепочечной нуклеиновой кислоты, а затем снова отжигают. При этом снова отожгутся только те фрагменты одноцепочечных нуклеиновых кислот, которые содержат участки гомологичности с другими фрагментами одноцепочечных нуклеиновых кислот.
Неупорядоченные фрагменты нуклеиновых кислот можно денатурировать за счет нагревания. Специалист может определить условия, необходимые для полной денатурации двухцепочечной нуклеиновой кислоты. Предпочтительно, чтобы температура составляла от 80oC до 100oC, более предпочтительно, чтобы температура составляла от 90oC до 96oC. Другими способами, которые можно использовать для денатурации фрагментов нуклеиновых кислот, являются соответствующие величины давления (36) и pH.
Фрагменты нуклеиновых кислот можно повторно отжечь за счет охлаждения. Предпочтительная температура составляет от 20oC до 75oC, более предпочтительная температура составляет от 40oC до 65oC. Если необходима высокая частота кроссоверов на основании в среднем только 4 последовательных оснований гомологичности, рекомбинацию можно усилить, используя низкие температуры отжига, хотя процесс при этом становится более трудным. Степень ренатурации, которая осуществляется, зависит от степени гомологичности между популяцией одноцепочечных фрагментов нуклеиновых кислот.
Ренатурацию можно ускорить, добавляя полиэтиленгликоль ("PEG") или соль. Концентрация соли составляет, предпочтительно, от 0 мМ до 200 мМ, более предпочтительно, чтобы концентрация соли составляла от 10 мМ до 100 мМ. Соль может быть KCl или NaCl. Концентрация PEG предпочтительно составляет от 0% до 20%, более предпочтительно, от 5% до 10%.
Фрагменты отожженных нуклеиновых кислот затем инкубируют в присутствии полимеразы нуклеиновых кислот и dNTP (т.е., dATP, dCTP, dGTP и dTTP). Полимеразой нуклеиновых кислот может быть фрагмент Кленова, Taq полимераза или любая другая известная ДНК полимераза.
Подход, который следует использовать для сборки, зависит от минимальной степени гомологичности, при которой все еще происходит кроссовер. Если участки идентичности достаточно велики, можно использовать Taq полимеразу при температуре отжига от 45 до 65oC. Если участки идентичности малы, можно использовать полимеразу Кленова при температуре отжига от 20 до 30oC. Специалисты могут варьировать температуру отжига для увеличения числа осуществляемых кроссоверов.
Полимеразу можно добавлять к неупорядоченным фрагментам нуклеиновой кислоты перед отжигом, одновременно с отжигом или после отжига.
Цикл, включающий денатурацию, ренатурацию и инкубирование в присутствии полимеразы, здесь называют перетасовкой или вторичной сборкой нуклеиновой кислоты. Этот цикл повторяют нужное число раз. Предпочтительно, повторять этот цикл от 2 до 50 раз, более предпочтительно повторять эту последовательность от 10 до 40 раз.
Полученная нуклеиновая кислота представляет собой более крупный двухцепочечный полинуклеотид, содержащий от около 50 п.о. до около 100 к.п.о., предпочтительно, чтобы этот более крупный полинуклеотид содержал от 500 п.о. до 50 к.п.о.
Такой более крупный полинуклеотидный фрагмент может содержать ряд копий фрагмента нуклеиновой кислоты того же размера, что и матричный полинуклеотид в тандеме. Такой конкатемерный фрагмент переваривают затем до отдельных копий матричного полинуклеотида. В результате образуется популяция фрагментов нуклеиновых кислот примерно того же размера, что и матричный полинуклеотид. Такая популяция будет смесью популяций, где одноцепочечные и двухцепочечные фрагменты нуклеиновых кислот, содержащие участок идентичности и участок гетерологичности, были добавлены к матричному полинуклеотиду перед перетасовкой.
Затем эти фрагменты клонируют в соответствующий вектор, и эту смесь для легирования используют для трансформации бактерий.
Ожидается, что отдельные фрагменты нуклеиновых кислот можно получить из более крупных конкатемерных фрагментов нуклеиновых кислот за счет амплификаций отдельных фрагментов нуклеиновых кислот перед клонированием с помощью различных способов, включая PCR (патенты США N 4683195 и 4683202) скорее, нежели за счет переваривания конкатемера.
Вектор, который используют для клонирования, не является критическим, при условии, что он примет ДНК фрагмент нужного размера. Если необходима экспрессия ДНК фрагмента, вектор клонирования должен содержать далее сигналы транскрипции и трансляции после сайта вставки ДНК фрагмента для обеспечения экспрессии фрагмента ДНК в клетку хозяина. Предпочтительные векторы включают серии pUC и серии pBR плазмид.
Полученная бактериальная популяция будет включать ряд фрагментов рекомбинантных ДНК с неупорядоченными мутациями. Эта смешанная популяция может быть протестирована для идентификации нужного фрагмента рекомбинантной нуклеиновой кислоты.
Так, например, если нужен фрагмент ДНК, который кодирует протеин, который повышает эффективность связывания с лигандом, протеины, экспрессируемые каждым из ДНК фрагментов в популяции или библиотеке, можно тестировать по их способности связываться с лигандом способами, известными специалистам (т.е., за счет пэннинга или афинной хроматографии). Если нужен ДНК фрагмент, который кодирует протеин, который повышает устойчивость к лекарствам, протеины, экспрессируемые каждым из ДНК фрагментов в популяции или библиотеке, можно тестировать по их способности придавать устойчивость к лекарствам организму хозяина. Специалист, которому известна информация о нужном протеине, может легко тестировать популяцию для идентификации ДНК фрагментов, которые придают нужные свойства протеину.
Считают, что специалист может использовать фаговую систему, в которой фрагменты протеина экспрессируются как протеины слияния на поверхности фага (Pharmacia, Milwaukee WI). Рекомбинантные ДНК молекулы клонируют в фаговую ДНК по сайту, который приводит к транскрипции протеина слияния, часть которого кодируется рекомбинантной ДНК молекулой. Фаг, содержащий молекулу нуклеиновой кислоты, претерпевает репликацию и транскрипцию в клетке. Лидерная последовательность протеина слияния управляет транспортом протеина слияния к верхней части фаговой частицы. Так, протеин слияния, который частично кодируется рекомбинантной ДНК молекулой, располагается на фаговой частице для определения и отбора описанными ранее способами.
Предполагается далее, что ряд циклов перетасовки нуклеиновых кислот можно осуществить с фрагментами нуклеиновых кислот из субпопуляции первой популяции, причем эта субпопуляция содержит ДНК, кодирующую нужный рекомбинантный протеин. Таким образом можно получить протеины, обладающие даже более высокими связывающими афинностями или энзиматическими активностями.
Предполагают также, что ряд циклов перетасовки нуклеиновых кислот можно проводить со смесью фрагментов дикого типа нуклеиновой кислоты и субпопуляции нуклеиновых кислот из первого или последующих раундов перетасовки нуклеиновых кислот для удаления всех молчащих мутаций из субпопуляции.
Любой источник нуклеиновой кислоты в очищенной форме можно использовать в качестве исходной нуклеиновой кислоты. Так, в процессе можно использовать ДНК или РНК, включая информационную РНК, причем эти ДНК или РНК могут быть одно- или двухцепочечными. Кроме того, гибрид ДНК-РНК, который содержит одну цепь из каждой, может быть использован. Последовательность нуклеиновой кислоты может быть различной длины, в зависимости от размера нуклеиновой кислоты, которая подлежит мутации. Предпочтительно, чтобы специфическая последовательность нуклеиновой кислоты содержала от 50 до 50000 пар оснований. Ожидают, что полный вектор, содержащий нуклеиновую кислоту, кодирующую представляющий интерес протеин, можно использовать в способах настоящего изобретения.
Нуклеиновую кислоту можно получить из любого источника, например, из такой плазмиды, как pBR322, из клонированных ДНК или РНК, или из нативных ДНК или РНК из любого источника, включая бактерии, дрожжи, вирусы и такие высшие организмы, как растения и животные. ДНК или РНК можно экстрагировать из крови или материалов тканей. Матричный полинуклеотид можно получить за счет амплификации, используя полинуклеотидную цепную реакцию (PCR) (патенты США N 4683202 и 4683195). В другом варианте полинуклеотид может присутствовать в векторе, присутствующем в клетке, и подходящую нуклеиновую кислоту можно получить, культивируя клетку и экстрагируя нуклеиновую кислоту из клетки известными специалистам способами.
Любую специфическую последовательность нуклеиновой кислоты можно использовать для получения популяции мутантов в способе настоящего изобретения. Необходимо только, чтобы небольшая популяция мутантных последовательностей специфической последовательности нуклеиновых кислот существовала или была создана до этого процесса.
Исходная небольшая популяция специфических последовательностей нуклеиновых кислот, содержащих мутации, может быть создана рядом различных способов. Мутации можно создавать за счет подверженной ошибкам ПЦР. Подверженная ошибкам ПЦР использует произвольные условия полимеризации для введения небольшого числа точечных мутаций неупорядоченно по всей длине последовательности. В другом варианте мутации можно ввести в матричный полинуклеотид за счет олигонуклеотид-направленного мутагенеза. В олигонуклеотид-направленном мутагенезе короткую последовательность полинуклеотида удаляют из полинуклеотида, используя переваривание рестрикционным энзимом и заменяя синтетическим полинуклеотидом, в котором различные основания были изменены по сравнению с исходной последовательностью. Полинуклеотидную последовательность можно также изменить за счет химического мутагенеза. Химический мутагенез включает (но не ограничивается этим), например, обработку бисульфитом натрия, азотистой кислотой, гидроксиламином, гидразином или муравьиной кислотой. Другие агенты, которые являются аналогами предшественников нуклеотидов, включают нитрозогуанидин, 5-бром-урацил, 2-аминопурин или акридин. Обычно эти агенты добавляют в реакцию PCR вместо нуклеотидного предшественника, осуществляя тем самым мутацию последовательности. Можно также использовать такие агенты интеркаляции, как профлавир, акрифлавин, хинакрин и т.п. Неупорядоченный мутагенез полинуклеотидной последовательности можно также осуществить за счет облучения рентгеновскими лучами или ультрафиолетовым светом. Обычно фрагменты ДНК или плазмидные ДНК, подвергнутые такому мутагенезу, вводят в Е.Coli и культивируют как пул или библиотеку мутантных плазмид.
В другом варианте небольшая смешанная популяция специфических нуклеиновых кислот может обнаружиться в природе, в том, что они могут состоять из различных аллелей одного и того же гена, или одного гена из различных родственных видов (т. е. родственных генов). В другом варианте они могут быть родственными ДНК последовательностями, обнаруживаемыми в одном образце, например, генами иммуноглобулина.
После того, как создана смешанная популяция специфических последовательностей нуклеиновых кислот, полинуклеотиды можно использовать непосредственно или встроить в соответствующий вектор клонирования, используя хорошо известные специалистам методики.
Выбор вектора зависит от размера полинуклеотидной последовательности и клетки хозяина, которую намерены использовать в способе настоящего изобретения. Матрицами настоящего изобретения могут быть плазмиды, фаги, фагемиды, вирусы (например, ретровирусы, вирусы парагриппа, герпесвирусы, реовирусы, парамиксовирусы и т.п.), или их выбранные части (например, протеин оболочки, спайк-гликопротеин, капсидный протеин). Так, например, космиды и фагэмиды предпочтительны в тех случаях, когда специфическая последовательность нуклеиновой кислоты, которая подлежит мутации, большего размера, так как эти векторы способны стабильно размножать фрагменты нуклеиновых кислот.
Если смешанная популяция специфической последовательности нуклеиновой кислоты клонирована в вектор, она может быть клонально амплифицирована за счет встраивания каждого вектора в клетку хозяина и обеспечения возможности для клетки хозяина амплифицировать этот вектор. Это называют клональной амплификацией, так как, хотя абсолютное число последовательностей нуклеиновых кислот возрастает, количество мутантов не увеличивается.
Применение
Способ перетасовки ДНК
настоящего изобретения можно осуществить вслепую на пуле неизвестных последовательностей. Добавляя для вторичной сборки смесь олигонуклеотидов (с концами, которые гомологичны последовательности,
которые подвергают вторичной сборке), любую смесь последовательностей можно встроить по любому специфическому положению в другую смесь последовательностей. Так, ожидается, что смеси синтетических
олигонуклеотидов, PCR фрагментов или даже целых генов можно смешать в другую библиотеку последовательностей в определенном положении. Встраивание одной последовательности (смеси) не зависит от
встраивания последовательности в другую часть матрицы. Так, степень рекомбинации, необходимая гомологичность и разнообразие библиотеки можно независимо и одновременно варьировать по всей длине
вторично собранной ДНК.
Такой подход к смешиванию двух генов может быть полезен для гуманизации антител, полученных из мышиных гибридом. Подход смешивания двух генов или встраивания мутантных последовательностей в гены может оказаться полезным для любого терапевтически используемого протеина, например, для интерлейкина 1, антител, tPA, гормона роста и т.д. Этот подход может быть также полезен для любой нуклеиновой кислоты, например, для промоторов или интронов или 3' нетранслируемых участков или 5' нетранслируемых участков генов для усиления экспрессии или изменения специфичности экспрессии протеинов. Этот подход можно также использовать для осуществления мутаций рибозимов или аптамеров.
Перетасовка требует присутствия гомологичных участков, разделяющих участки разнообразия. Протеины со структурой, похожей на виселицу (Scaffold-like), могут быть особенно подходящими для перетасовки. Консервативная "виселица" определяет полную складчатость за счет самоассоциации, хотя и демонстрирует относительно неограниченные петли, за счет которых происходит специфическое связывание. Примерами таких "виселиц" являются бета-цилиндр и четырехспиральный узел (24). Такую перетасовку можно использовать для создания протеинов в форме виселицы с различными комбинациями мутированных последовательностей для связывания.
Ин витро
перетасовка
За счет перетасовки ин витро можно также осуществить эквиваленты некоторых стандартных генетических скрещиваний. Так, например, молекулярный "бэккросс" можно осуществить, повторяя
смешивание мутантной нуклеиновой кислоты с дикого типа нуклеиновой кислотой, отбирая при этом представляющие интерес мутанты. В качестве традиционного бридинга, такой подход можно использовать для
объединения фенотипов из различных источников в выбранный фон. Это, например, полезно для удаления нейтральных мутаций, которые влияют на неселективность характеристики (т.е. на иммуногенность). Таким
образом, это можно использовать для определения того, какая из мутаций в протеине вовлечена в усиление биологической активности, а какая - нет; преимущество, которого нельзя достичь в результате
подверженного ошибкам мутагенеза или кассетного мутагенеза.
Крупные функциональные гены можно правильно собрать из смеси мелких неупорядоченных фрагментов. Такая реакция может быть использована для вторичной сборки генов из сильно фрагментированных ДНК ископаемых (25). Кроме того, неупорядоченные фрагменты нуклеиновых кислот ископаемых можно объединить с фрагментами нуклеиновых кислот из аналогичных генов родственных видов.
Предполагается также, что способ настоящего изобретения можно использовать для ин витро амплификации полного генома из отдельной клетки, что бывает необходимо для различных исследований и диагностических подходов. ДНК амплификация за счет PCR на практике ограничена длиной около 40 к.п.о. Амплификация полного генома, такого, как геном Е.coli, (5000 к.п.о.) за счет ПЦР потребовала бы 250 праймеров, что привело бы к 125 фрагментам по 49 к.п. о. Такой подход на практике невозможен из-за недоступности данных о последовательностях. С другой стороны, неупорядоченное переваривание генома с ДНКазой 1 с последующей очисткой на геле мелких фрагментов позволяет получить множество возможных праймеров. Использование этой смеси неупорядоченных мелких фрагментов в качестве праймеров в PCR реакции отдельно или с полным геномом в качестве матрицы должно привести к обратной цепной реакции с теоретической конечной точкой отдельного конкатемера, содержащего множество копий генома.
100-кратная амплификация числа копий и средний размер фрагмента более чем 50 к.п.о. можно получить, если использовать только неупорядоченные фрагменты (см. фиг. 2). Считают, что более крупный конкатемер образуется за счет перекрывания множества более мелких фрагментов. Качество полученных продуктов специфической ПЦР с использованием синтетических праймеров будет неотличимым от продуктов, полученных из неамплифицированной ДНК. Ожидается, что такой подход может оказаться полезным для картирования геномов.
Подлежащий перетасовке полинуклеотид можно фрагментировать неупорядоченно или упорядоченно, по выбору практикующего специалиста.
Перетасовка ин виво
В варианте
перетасовки ин виво смешанную популяцию специфической последовательности нуклеиновых кислот вводят в бактериальные или эукариотные клетки в таких условиях, что, по крайней мере, две различные
последовательности нуклеиновых кислот присутствуют в каждой клетке хозяина. Фрагменты можно вводить в клетки хозяина различными способами. Клетки хозяина можно трансформировать фрагментами, используя
способы, известные специалистам, например, обрабатывая их хлоридом кальция. Если фрагменты встроены в геном фага, клетки хозяина можно транфектировать рекомбинантным фаговым геномом, содержащим
специфические последовательности нуклеиновых кислот. В другом варианте последовательности нуклеиновых кислот можно вводить в клетки хозяина, используя электропорацию, трансфекцию, липофекцию,
biolistics, конъюгацию и т.п.
Вообще в этом варианте последовательности нуклеиновых кислот будут присутствовать в векторах, которые способны к стабильной репликации последовательности в клетках хозяина. Кроме того, предполагается, что векторы кодируют маркерный ген таким образом, что клетки хозяина, содержащие такие векторы, можно селектировать. Это обеспечивает возможность того, что мутированную специфическую последовательность нуклеиновой кислоты можно выделить после введения в клетку хозяина. Однако предполагается, что полная смешанная популяция специфических последовательностей нуклеиновых кислот не должна присутствовать в векторной последовательности. Скорее лишь достаточное число последовательностей необходимо клонировать в векторы для того, чтобы быть уверенными в том, что после введения фрагментов в клетки хозяина каждая из клеток хозяина содержит один вектор, содержащий, по крайней мере, одну специфическую последовательность нуклеиновой кислоты, в нем присутствующую. Предполагается также, что скорее, чем наличие субнабора популяции специфических последовательностей нуклеиновых кислот, клонированных в векторы, такой субнабор может быть уже стабильно интегрирован в клетку хозяина.
Было обнаружено, что если в клетку хозяина вводят два фрагмента, которые имеют участки идентичности, между этими двумя фрагментами происходит гомологическая рекомбинация. Такая рекомбинация между двумя мутированными специфическими последовательностями нуклеиновых кислот приводит к получению в некоторых ситуациях двойных или тройных мутантов.
Было также обнаружено, что частота рекомбинаций возрастает, если некоторые из мутированных специфических последовательностей нуклеиновых кислот присутствуют в линейных молекулах нуклеиновых кислот. Поэтому в предпочтительном варианте некоторые специфические последовательности нуклеиновых кислот присутствуют в линейных фрагментах нуклеиновых кислот.
После трансформации трансформанты клеток хозяина помещают в условия отбора для определения тех клеточных трансформантов хозяина, которые содержат мутированные последовательности специфических нуклеиновых кислот, отличающиеся нужными свойствами. Так, например, если необходима повышенная устойчивость к конкретному лекарству, тогда трансформированные клетки хозяина можно обрабатывать повышенными концентрациями конкретного лекарства, и отобрать те трансформанты, которые продуцируют мутированные протеины, способные придавать повышенную устойчивость к лекарству. Если нужна повышенная способность конкретного протеина связывать рецептор, тогда экспрессию этого протеина можно индуцировать из трансформантов, а полученный протеин проанализировать в анализе со связыванием лиганда способами, известными специалистам, для определения того субнабора мутированной популяции, которая демонстрирует более эффективное связывание с лигандом. В другом варианте протеин можно экспрессировать в другой системе для обеспечения соответствующего процессинга.
После того, как субнабор первых рекомбинированных специфических последовательностей нуклеиновых кислот (дочерние последовательности), обладающих нужными характеристиками, определен, его подвергают второму циклу рекомбинации.
Во втором цикле рекомбинации специфические последовательности нуклеиновых кислот можно смешать с исходными мутированными специфическими последовательностями нуклеиновых кислот (родительскими последовательностями) и цикл повторяют, как указано ранее. Таким способом набор вторично рекомбинированных специфических последовательностей нуклеиновых кислот можно идентифицировать, причем этот набор обладает улучшенными характеристиками или кодирует протеины, обладающие улучшенными свойствами. Этот цикл можно повторять столько раз, сколько нужно.
Предполагается также, что во втором или последующих циклах рекомбинации можно осуществить бэккросс. Молекулярный бэккросс можно осуществить за счет смешивания нужных специфических последовательностей нуклеиновых кислот с большим числом дикого типа последовательностей, так, что, по крайней мере, одна дикого типа последовательность нуклеиновой кислоты и мутированная последовательность нуклеиновой кислоты присутствуют в одной и той же клетке хозяина после трансформации. Рекомбинация с дикого типа специфической последовательностью нуклеиновой кислоты исключит те нейтральные мутации, которые могут повлиять на неселектируемые характеристики, такие, как иммуногенность, а не селектируемые свойства.
В другом варианте настоящего изобретения предполагается, что во время первого цикла субнабор специфических последовательностей нуклеиновых кислот можно фрагментировать до введения в клетку хозяина. Размеры фрагментов должны быть достаточно велики, чтобы содержать некоторые участки идентичности с другими последовательностями, с тем, чтобы была возможна гомологическая рекомбинация с другими последовательностями. Размеры фрагментов составляют величину от 0,03 к.п.о. до 100 к.п.о., более предпочтительно, от 0,2 к.п.о. до 10 к.п.о. Предполагают также, что в последующих циклах все специфические последовательности нуклеиновых кислот, отличающиеся от последовательностей, выбранных в предыдущих циклах, можно расщепить на фрагменты до введения в клетки хозяина.
Фрагментацию последовательностей можно осуществить различными способами, известными специалистам. Эти последовательности можно фрагментировать неупорядоченно или фрагментировать по специфическим сайтам в последовательности нуклеиновой кислоты. Неупорядоченные фрагменты можно получить за счет разрушения нуклеиновой кислоты или экспонирования ее жесткой физической обработке (например, обработке с высоким сдвигом или облучению), или обработке ее жесткими химическими агентами (например, свободными радикалами, ионами металлов; кислотной обработке для расщепления). Неупорядоченные фрагменты можно также получить в случае ДНК, используя ДНКазу или подобные нуклеазы. Последовательности можно расщепить по специфическим сайтам, используя рестрикционные энзимы. Фрагментированные последовательности могут быть одноцепочечными или двухцепочечными. Если исходные последовательности одноцепочечные, их можно денатурировать за счет нагревания, химически или с помощью энзимов перед тем, как вводить в клетку хозяина. Условия реакции, подходящие для разделения цепей нуклеиновых кислот, хорошо известны специалистам.
Стадии этого процесса можно повторять неопределенно долго, причем ограничением является только количество возможных мутантов, которые можно получить. После определенного числа циклов, все возможные мутанты получены и дальнейшие циклы не целесообразны.
В этом варианте ту же самую мутированную матричную нуклеиновую кислоту повторно рекомбинируют, и полученные рекомбинанты отбирают по целевым характеристикам.
Поэтому исходный пул или популяцию мутированных матричных нуклеиновых кислот клонируют в вектор, способный к репликации в такие бактерии, как E. Coli. Конкретный вектор несущественен до тех пор, пока он способен к автономной репликации в Е.coli. В предпочтительном варианте конструируют вектор, который обеспечивает экспрессию и продуцирование любого протеина, кодируемого мутированной специфической нуклеиновой кислотой, связанной с вектором. Предпочтительно также, чтобы вектор содержал ген, кодирующий селектируемый маркер.
Популяцию векторов, содержащих пул мутированных последовательностей нуклеиновой кислоты, вводят в клетки хозяина Е. coli. Векторные последовательности нуклеиновой кислоты можно вводить за счет трансформации, трансфекции или информирования в случае фага. Концентрации векторов, используемые для трансформации бактерий, должны быть такими, чтобы несколько векторов оказались введенными в каждую клетку. Если они присутствуют в клетке, эффективность гомологической рекомбинации такова, что гомологическая рекомбинация происходит между различными векторами. Это приводит к созданию мутантов (дочерних), содержащих комбинации мутаций, которые отличаются от исходных родительских мутированных последовательностей.
Затем клетки хозяина клонально реплицируют и отбирают по маркерному гену, присутствующему на векторе. В условиях отбора растут только клетки, содержащие плазмиду.
Клетки хозяина, которые содержат вектор, тестируют затем на наличие благоприятных мутаций. Такое тестирование может сводиться к помещению клеток в условия отбора, например, если ген, который нужно отобрать, является геном повышенной устойчивости к лекарству. Если вектор обеспечивает экспрессию протеина, кодируемого мутированной последовательностью нуклеиновой кислоты, тогда такой отбор может включать обеспечение экспрессии кодируемого таким образом протеина, выделение протеина и тестирование протеина для определения, например, того, связывается ли он с повышенной эффективностью с представляющим интерес лигандом.
После того, как конкретная дочерняя мутированная последовательность нуклеиновой кислоты была идентифицирована (определено, что она придает нужные свойства), нуклеиновую кислоту выделяют либо в виде, уже связанном с вектором, либо отдельно от вектора. Затем нуклеиновую кислоту смешивают с первой или родительской популяцией нуклеиновых кислот, и цикл повторяют.
Было показано, что таким способом можно селектировать последовательности нуклеиновых кислот, обладающие улучшенными целевыми свойствами.
В альтернативном варианте первое поколение мутантов сохраняют в клетках, и родительские мутированные последовательности снова добавляют в клетки. Соответственно, первый цикл варианта 1 осуществляют, как указано ранее. Однако после того, как идентифицируют дочерние последовательности нуклеиновых кислот, сохраняют клетки хозяина, содержащие эти последовательности.
Популяцию родительских мутированных специфических нуклеиновых кислот либо в виде фрагментов, либо клонированную в тот же вектор, вводят в клетки хозяина, уже содержащие дочерние нуклеиновые кислоты. Дают возможность осуществиться рекомбинации в клетках, и следующее поколение рекомбинантов или "внучатых" последовательностей селектируют описанным ранее способом.
Такой цикл можно повторять много раз до тех пор, пока не получают нуклеиновую кислоту или пептид, обладающие нужными характеристиками. Предполагается, что в последующих циклах популяция мутированных последовательностей, которую добавляют к предпочтительным мутантам, может происходить из родительских мутантов или из мутантов любых последующих поколений.
В альтернативном варианте в изобретении предложен способ осуществления "молекулярного" бэккросса полученной рекомбинантной специфической нуклеиновой кислоты для исключения любых нейтральных мутаций. Нейтральными мутациями являются такие мутации, которые не придают нуклеиновой кислоте или пептиду целевых свойств. Однако, такие мутации могут придавать нуклеиновой кислоте или пептиду нежелательные характеристики. Соответственно, желательно исключить нейтральные мутации. И способ настоящего изобретения предлагает средства для осуществления этой задачи.
В этом варианте после того, как способом этого варианта получают мутантную нуклеиновую кислоту, обладающую нужными характеристиками, выделяют эту нуклеиновую кислоту, вектор, содержащий эту нуклеиновую кислоту, или клетку хозяина, содержащую этот вектор и нуклеиновую кислоту.
Затем нуклеиновую кислоту и вектор вводят в клетку хозяина с большим избытком нуклеиновой кислоты дикого типа. Предоставляют возможность осуществления рекомбинации для последовательностей нуклеиновой кислоты мутанта и нуклеиновой кислоты дикого типа. Полученные рекомбинанты помещают в те же самые условия отбора, что и мутантную нуклеиновую кислоту. Отбирают только те мутанты, которые сохраняют нужные характеристики. Любые молчащие мутации, которые не обеспечивают нужные характеристики, будут утрачены во время рекомбинации с дикого типа ДНК. Этот цикл можно повторять несколько раз до тех пор, пока не будут исключены все молчащие мутации.
Таким образом, способ настоящего изобретения можно использовать в молекулярных бэккроссах для исключения ненужных молчащих мутаций.
Применение
Способ ин виво рекомбинации настоящего изобретения можно осуществлять вслепую на пуле неизвестных мутантов или аллелей фрагментов или последовательностей специфических нуклеиновых кислот. Однако,
нет необходимости знать реальную ДНК или РНК последовательность фрагмента специфической нуклеиновой кислоты.
Подход использования рекомбинации без смешивания популяции генов может быть использован для создания любых нужных протеинов, например, интерлейкена I, антител, tPA, гормонов роста и т.п. Такой подход может быть использован для создания протеинов, обладающих измененной специфичностью или активностью. Такой подход может быть также полезен для создания последовательностей мутантных нуклеиновых кислот, например, промоторных участков, интронов, эксонов, энхансерных последовательностей, 3' нетранслируемых участков или 5' нетранслируемых участков генов. Этот подход можно использовать для создания генов, обладающих повышенной экспрессией. Этот подход может быть также полезен для изучения повторяющихся ДНК последовательностей. И, наконец, этот подход может быть полезен для мутации рибозим или аптамеров.
Похожие на "виселицы" участки, разделяющие участки разнообразия в протеинах, могут быть особенно полезны для осуществления способов настоящего изобретения. Консервативные "виселицы" определяют общую складчатость за счет самоассоциации, хотя и демонстрируют относительно неограниченные петли, за счет которых происходит специфическое связывание. Примерами таких "виселиц" служат бета-цилиндры иммуноглобулина и четырехспиральные узлы. Способы настоящего изобретения можно использовать для создания "виселицеподобных" протеинов с различными комбинациями мутированных последовательностей для связывания.
Эквивалентами некоторых стандартных генетических спариваний могут также быть кодирующие информацию пептидные последовательности; так, представленная пептидная последовательность может быть определена за счет детерминации нуклеотидной последовательности члена выделенной библиотеки.
Хорошо известный способ представления пептидов включает представление пептидной последовательности на поверхности нитеподобного бактериофага, обычно в виде слияния с протеином оболочки бактериофага. Библиотеку бактериофага можно инкубировать с иммобилизованной, заранее определенной молекулой или мелкой молекулой (например, рецептором) с тем, чтобы частицы бактериофага, которые представляют пептидную последовательность, которая связывается с иммобилизованной макромолекулой, могла быть дифференциально отделена от тех, которые не представляют пептидные последовательности, которые связывают заранее определенную макромолекулу. Частицы бактериофага (т.е. члены библиотеки), которые связаны с иммобилизованной макромолекулой, затем выделяют и реплицируют для амплификации выбранной субпопуляции бактериофага для последующего цикла афинного обогащения и фаговой репликации. После нескольких циклов афинного обогащения и фаговой репликации члены библиотеки бактериофага, которые были таким образом отобраны, выделяют, и определяют нуклеотидную последовательность, кодирующую выставленную пептидную последовательность, идентифицируя, таким образом, последовательность (последовательности) пептидов, которые связываются с заранее определенной макромолекулой (например, с рецептором). Такие способы более подробно описаны в PCT патентной публикации N 91/17271, 91/18980, 91/19818 и 93/08278.
В последней PCT публикации раскрыт метод рекомбинантных ДНК для выставления пептидных лигандов, который включает получение библиотеки протеинов слияния, причем каждый протеин слияния состоит из первой полипептидной части, обычно содержащей вариабельную последовательность, то есть, способной к потенциальному связыванию с заранее определенной макромолекулой, и второй полипептидной части, которая связывается с ДНК, например, векторной ДНК, кодирующей индивидуальный протеин слияния. Когда трансформированные клетки хозяина культивируют в условиях, которые позволяют обеспечить экспрессию протеина слияния, этот протеин слияния связывается с векторной ДНК, кодирующей его. После лизиса клетки хозяина, комплексы протеин слияния/векторная ДНК можно скринировать в отношении заранее определенной макромолекулы в значительной степени так же, как скринируют частицы бактериофага в системе представления на основе фага, причем репликация и секвенирование ДНК векторов в отобранных комплексах протеин слияния/векторная ДНК служат в качестве основания для идентификации выбранной библиотеки пептидной последовательности (последовательностей).
Другие системы создания библиотек пептидов и аналогичных полимеров содержат аспекты обоих - рекомбинантного и ин витро химического синтеза - способов. В этих способах гибридизации бесклеточный энзиматический механизм используют для осуществления ин витро синтеза членов библиотеки (т.е. пептидов или полинуклеотидов). В этом способе одного типа РНК молекулы со способностью связываться с заранее определенным протеином или молекулой определенного красителя отбирают в процессе чередующихся циклов селекции и PCR амплификации (Tuerk and Gold (1990) Science 249:505; Ellington and Szostak (1990) Nature 346: 818). Аналогичную методику используют для идентификации ДНК последовательностей, которые связываются с заранее определенным фактором транскрипции человека (Thiesen and Bach (1990) Nucleic Acids Res. 18:3203; Beaudry and Joyce (1992) Science 257; 635; патент PCT N 92/05258 и 92/14843. Аналогичным образом методику ин витро трансляции использовали для синтеза протеинов, представляющих интерес, и она была предложена как способ создания крупных библиотек пептидов. Эти способы, которые основаны на ин витро трансляции, обычно содержат стабилизированные полисомные комплексы, и они были описаны более подробно в PCT патентных публикациях N 88/08453, 90/05785, 90/07003, 91/02076, 91/05058 и 92/02536. Заявители описали способы, в которых библиотека членов содержит протеин слияния, содержащий часть первого полипептида с ДНК связывающей активностью и часть второго полипептида, содержащего уникальную пептидную последовательность члена библиотеки; такие способы пригодны для использования, наряду с другими, в бесклеточных ин витро селекционных форматах.
Выставленные пептидные последовательности могут иметь различную длину, обычно от 3 до 5000 аминокислот или более, чаще от 5 до 100 аминокислот и часто от около 8 до 15 аминокислот. Библиотека может содержать библиотеку членов, отличающихся различной длиной выставленной пептидной последовательности, или может содержать библиотеку членов, отличающихся фиксированной длиной выставленной пептидной последовательности. Части или вся выставленная пептидная последовательность (последовательности) может быть неупорядоченной, псевдонеупорядоченной, фиксированной или т.п. Настоящий способ представления включает способы для ин витро и ин виво представления одноцепочечных антител, таких, как образующиеся ScFv на полисомах или ScFv представленные на фаге, которые обеспечивают крупномасштабное скринирование ScFv библиотек, обладающих широким разнообразием последовательностей вариабельных участков и специфичностями связывания.
В настоящем изобретении предложены также библиотеки неупорядоченных, псевдоупорядоченных и каркасных пептидов, а также способы создания и скринирования этих библиотек для идентификации полезных соединений (например, пептидов, включая одноцепочечные антитела), которые связываются с рецепторными молекулами или эпитопами, представляющими интерес, или с генными продуктами, которые модифицируют пептиды или РНК нужным образом. Неупорядоченных, псевдоупорядоченных и определенных каркасных последовательностей пептиды получают из библиотек членов пептидных библиотек, которые включают представленные пептиды или представленные одноцепочечные антитела, присоединенные к полинуклеотидной матрице, из которой эти представленные пептиды были синтезированы. Способ присоединения может варьироваться в соответствии со специфическим вариантом изобретения и может включать инкапсуляцию в фаговые частицы или включение в клетку.
Способ афинного обогащения позволяет скринировать очень крупные библиотеки пептидов и одноцепочечных антител, подлежащие скринированию, а также полинуклеотидные последовательности, кодирующие целевой пептид (пептиды) или одноцепочечные антитела. Затем полинуклеотид можно выделить и перетасовать для комбинаторной рекомбинации аминокислотной последовательности выбранного пептида (пептидов) (или предварительно определенных их частей) или одноцепочечных антител (или только VH, VL или CDR их фрагментов). Используя эти способы, можно идентифицировать пептид или одноцепочечное антитело, как обладающее нужной связывающей афинностью для молекулы, и можно использовать этот процесс перетасовки для быстрой сходимости к целевому с высокой афинностью пептиду или ScFv. Затем этот пептид или антитело можно синтезировать в большем количестве, используя обычные способы для любого подходящего применения (например, в качестве терапевтического или диагностического агента).
Значительное преимущество настоящего изобретения состоит в том, что до сих пор не было информации относительно того, какая ожидаемая структура лиганда необходима для выделения представляющих интерес пептидных лигандов или антител. Идентифицированный пептид может обладать биологической активностью, что подразумевает, что он обладает, по крайней мере, специфической связывающей афинностью для выбранной рецепторной молекулы, и в некоторых случаях обладает также способностью блокировать связывание других соединений для стимуляции или ингибирования метаболических процессов, для того, чтобы действовать в качестве сигнала или мессенджера, для стимуляции или ингибирования клеточной активности и т.п.
В настоящем изобретении предложен также способ перетасовки пула полинуклеотидных последовательностей, отобранных в результате афинного скринирования библиотеки полисом, представляющих образующиеся пептиды (включая одноцепочечные антитела) для членов библиотеки, которые связываются с заранее определенным рецептором (например, такими протеиновыми рецепторами млекопитающих, как, например, рецептор пептидергического гормона, рецептор клеточной поверхности, внутриклеточный протеин, который связывается с другим протеином (протеинами) с образованием внутриклеточных протеиновых комплексов, таких как гетеродимеры и т.п.) или эпитопы (например, иммобилизованный протеин, гликопротеин, олигосахарид и т.п.).
Полинуклеотидные последовательности, выбранные в первом цикле селекции (обычно афинной селекции по связыванию с рецептором (например, лигандом) за счет любого из этих способов собирают в пул и этот пул (пулы) перетасовывают за счет ин витро и/или ин виво рекомбинации до получения перетасованного пула, содержащего популяцию рекомбинированных отобранных полинуклеотидных последовательностей. Эти рекомбинированные отобранные полинуклеотидные последовательности подвергают, по крайней мере, одному последующему циклу селекции. Полинуклеотидные последовательности, селектированные в последующем цикле (циклах) селекции, можно использовать непосредственно, секвенировать и/или подвергнуть еще одному или более циклу перетасовки и последующей селекции. Селектированные последовательности можно подвергнуть бэккроссу с полинуклеотидными последовательностями, кодирующими нейтральные последовательности (например, оказывающими несущественный функциональный эффект на связывание), осуществляя, например, бэккросс с дикого типа или нативными последовательностями, практически идентичными выбранной последовательности, для получения похожего на нативный функционального пептида, который может оказаться менее иммуногенным. Обычно, во время бэккросса используют последующую селекцию для того, чтобы сохранить свойство связывания с заранее определенным рецептором (лигандом).
Перед перетасовкой выбранных последовательностей или одновременно с ней, последовательности можно подвергнуть мутагенезу. В одном из вариантов члены выбранной библиотеки клонируют в прокариотный вектор (например, плазмиду, фагемид или бактериофаг), где получают коллекцию отдельных колоний (или бляшек), представляющих дискретные члены библиотеки. Отдельные отобранные члены библиотеки можно затем подвергнуть дальнейшей обработке (например, за счет сайт-направленного мутагенеза, кассетного мутагенеза, химического мутагенеза, ПЦР мутагенеза и т.п.) для создания коллекции членов библиотеки, представляющих основу разнообразных последовательностей на основе последовательности выбранного члена библиотеки. Последовательность отдельно выбранного члена библиотеки или пула может быть подвергнута дальнейшей обработке для включения неупорядоченной мутации, псевдоупорядоченной мутации определенной ядерной мутации (т.е. включающей положения вариантных и инвариантных остатков и/или содержащей положения вариантных остатков, которые могут содержать остаток, выбранный из определенного субнабора аминокислотных остатков), мутаций на основе кодонов и т.п., либо сегментно либо по всей длине последовательности отдельного выбранного члена библиотеки. Мутированные селектированные члены библиотеки затем перетасовывают за счет ин витро и/или ин виво рекомбинационной перетасовки, как здесь раскрыто.
В настоящем изобретении предложены также пептидные библиотеки, содержащие множество членов отдельных библиотек настоящего изобретения, где (1) каждый отдельный член библиотеки указанного множества содержит последовательность, полученную при перетасовке пула селектированных последовательностей, и (2) каждый отдельный член библиотеки содержит последовательность сегмента вариабельного пептида или последовательность сегмента одноцепочечного антитела, которая отличается от последовательностей вариабельного пептидного сегмента или последовательностей одноцепочечного антитела других индивидуальных членов библиотеки в указанном множестве (хотя некоторые члены библиотеки могут присутствовать в более чем одном экземпляре на библиотеку за счет неоднородной амплификации, стохастической вероятности или т.п.).
В изобретении также предложен продукт побочного процесса, в котором селектированные полинуклеотидные последовательности, содержащие (или кодирующие пептид) заранее определенную специфичность связывания, образуются в процессе: (1) скринирования представленной пептидной или одноцепочечного антитела библиотеки против заранее определенного рецептора (т.е. лиганда) или эпитопа (т.е. макромолекулы антигена) и идентификации и/или обогащения членов библиотеки, которые связываются с заранее определенным рецептором или эпитопом до получения пула членов выбранной библиотеки, (2) перетасовки за счет рекомбинации членов выбранной библиотеки (или амплифицированных или клонированных ее копий), которые связываются с заранее определенным эпитопом и были за счет этого выделены и/или обогащены из библиотеки для создания перетасованной библиотеки, и (3) скринирования перетасованной библиотеки против заранее определенного рецептора (например, лиганда) или эпитопа (например, макромолекулы антигена) и идентифицируя и/или обогащая членов перетасованной библиотеки, которые связываются с заранее определенным рецептором или эпитопом для получения пула членов селектированной перетасованной библиотеки.
Методы и представления антител и
скринирования
Настоящий способ можно использовать для перетасовки за счет ин витро и/или ин виво рекомбинации любым из раскрытых способов и в любых комбинациях полинуклеотидных
последовательностей, селектированных методами представления антител, где ассоциированный полинуклеотид кодирует представленное антитело, которое скринируют по фенотипу (например, по афинности для
связывания заранее определенного антигена (лиганда).
Были сконструированы различные молекулярные генетические подходы для выделения обширного иммунологического материала, представленного чрезвычайно большим числом различных вариабельных участков, которые могут присутствовать в цепях иммуноглобулина. Локус тяжелой цепи нативного иммуноглобулина состоит из отдельных тандемных рядов генов вариабельного (V) сегмента, расположенного в обратном направлении от тандемного ряда генов сегмента разнообразия (Д), которые, в свою очередь, расположены в обратном направлении от тандемного ряда генов, соединяющего (I) участки, которые расположены в обратном направлении от генов постоянного (CH) участка. В процессе развития лимфоцитов происходит K-D-I перегруппировка, при которой ген вариабельного участка тяжелой цепи (VH) образуется за счет перегруппировки с образованием слитого D-I сегмента с последующей перегруппировкой с V сегментом до образования V-D-I гена продукта присоединения, который, если продуктивно перестроен, кодирует функциональный вариабельный участок (VH) тяжелой цепи. Аналогично, локус перестройки легкой цепи одного из нескольких V сегментов с одним из нескольких I сегментов до образования гена, кодирующего вариабельный участок (VL) легкой цепи.
Обширный репертуар вариабельных участков, возможный для иммуноглобулинов, получают частично из множества комбинаторных возможностей соединения V и I сегментов (и, в случае локус тяжелой цепи, D сегментов) во время перегруппировки в развитии В клеток. Дополнительное разнообразие последовательностей в вариабельных участках тяжелых цепей возникает из неоднородных перегруппировок D сегментов во время соединения V-D-I и от добавления N участков. Далее, антиген-селекция специфических клонов В клеток отбирает по наивысшей афинности варианты, содержащие нонгермлайн (nongermline) мутации в одном или в обоих вариабельных участках тяжелой и легкой цепи; явление, которое носит название "созревание афинности" или "обострение афинности". Обычно такие мутации "обострения афинности" приводят к образованию кластеров в специфических зонах вариабельных участков, чаще всего в участках, определяющих комплементарность (CDR).
Для того чтобы преодолеть многие из этих ограничений при получении и идентификации высокоафинных иммуноглобулинов за счет антиген-стимулированного развития В клеток (т. е. иммунизации), были разработаны различные прокариотные системы экспрессии, с помощью которых можно получать комбинаторные библиотеки антител, которые можно скринировать на высокоафинные антитела к специфическим антигенам. Последние достижения в экспрессии антител в системах Escherichia coli и бактериофагов (см "Alternative Peptide Display Methods") выдвинули возможность того, что виртуально любую специфичность можно получить, либо клонируя гены антител из охарактеризованных гибридом, либо осуществляя заново селекцию, используя библиотеки генов антител (например, из Ig кДНК).
Комбинаторные библиотеки антител были созданы в системах экспрессии бактериофага лямбда, который можно скринировать как бляшки бактериофага или как колонии лизогенов (Huse et al. (1989) Science 246:1275; Caton and Koprowski (1990) Proc. Natl. Acad. Sci. (USA) 87:6459; Mullinax et al. (1990) Proc. Natl. Acad. Sci. (USA) 87:8095; Persson et al. (1991) Proc. Natl.Acad. Sci (USA) 88:2432).
Были описаны различные варианты библиотек, представляющих антитела
бактериофагов и библиотеки экспрессии лямбда фага. (Kang et al. (1991) Proc. Natl. Acad. Sci. (USA) 88: 4363; Clackson et al. (1991) Nature 352: 624; McCafferty et al. (1990) Nature 348: 552; Burton
et al. (1991) Proc.NatI. Acad. Sci. (USA) 88: 10134; Hoogenboom et al. (1991) Nucleic Acids Res. 19: 4133; Chang et al. (1991) J.linmunol. 147: 3610; Breitling et al. (1991) Gene 104: 147; Marks et
al. (1991) J.Mol.Biol. 222: 581; Barbas et al. (1992) Proc. Natl. Acad. Sci. (USA) 89: 4457; Hawkins and Winter (1992) J. Immunol. 22: 867; Marks et al. (1992) Biotechnology 10: 779;
Marks et
al. (1992) J. Biol.Chem. 267: 16007; Lownan et al. (1991) Biochemistry 30: 10831; Lemer et al. (1992) Science 258: 1313, включены сюда по ссылкам). Обычно библиотеку представления антител бактериофага
скринируют за счет рецептора (например, полипептида, углевода, гликопротеина, нуклеиновой кислоты), которой иммобилизован (например, за счет ковалентной связи с хроматографической смолой для
обогащения для реактивного фага за счет афинной хроматографии) и/или метят (например, для скринирования реплик бляшек или колоний).
Одним из наиболее выгодных подходов является использование так называемых библиотек одноцепочечных вариабельных фрагментов (ScFv) (Marks et al. (1992) Biotechnology 10: 779; Winter G and Milstein C (1991) Nature 349:293; Clackson et al. (1991) op cit; Marks et al. (1991) J. Mol. Biol, 222:581; Chaudhary et al. (1990) Proc. Natl. Acad. Sci. (USA) 87:1066; Chiswell et al. (1992) TJBTECH 10:80; McCafferty et al. (1990) op cit.; Huston et al. (1988) Proc. Natl. Acad. Sci. (USA) 85:5879).
Были описаны различные варианты ScFv библиотек, представленных на протеинах оболочки бактериофага.
Начиная с 1988 г., одноцепочечные аналоги Fv фрагментов и их протеинов слияния стало легко получать методами генной инженерии. Первая стадия обычно включает получение генов, кодирующих VH и VL домены с нужными свойствами связывания: эти V гены можно выделить из специфической гибридомной клеточной линии, выбранной из комбинаторной V-генной библиотеки или полученной за счет синтеза V гена. Одноцепочечные Fv получают, соединяя компонент V генов с олигонуклеотидом, который кодирует соответствующим образом сконструированный линкерный пептид, например, (Gly-Gly-Gly-Gly-Ser)3 или эквивалентный линкерный пептид (пептиды). Линкерные мостики C-концов первого V участка и N-конца второго имеют вид либо VH-линкер-VL либо VL-линкер-VH. В принципе, сайт связывания ScFv может успешно реплицировать как афинность, так и специфичность его сайта, объединяющего родительские антитела.
Таким образом, ScFv фрагменты содержат VH и VL домены, связанные в единую полипептидную цепь за счет гибкого линкерного пептида. После того, как ScFv гены собраны, их клонируют в фагемид и экспрессируют у конца М13 фага (или аналогичного нитеобразного бактериофага), как протеины слияния с протеином оболочки бактериофага pIII (ген 3). Обогащение для экспрессии фага, представляющего интерес антитела, сопровождается пэннингом рекомбинатного фага, представляющего популяцию ScFv для связывания с заранее определенным эпитопом (например, с мишеневым антигеном, рецептором).
Связанный олигонуклеотид из членов библиотеки предоставляет основу для репликации члена библиотеки после процедур скринирования или селекции, а также предоставляет основу для детерминации за счет нуклеотидного секвенирования, идентичности представленной пептидной последовательности или VH и VL аминокислотных последовательностей. Представленный пептид (пептиды) или одноцепочечные антитела (например, ScFv) и/или его VH и VL домены или их CDR можно клонировать и экспрессировать в подходящую экспрессионную систему. Часто полинуклеотиды, кодирующие выделенные VH и VL домены, можно легировать с полинуклеотидами, кодирующими постоянные участки (CH и CL) до получения полинуклеотидов, кодирующих полные антитела (например, химерические или полностью человеческие), фрагменты антител и т.п. Часто полинуклеотиды, кодирующие выделенные CDR, можно прививать на полинуклеотиды, кодирующие каркас подходящего вариабельного участка (и необязательно постоянные участки) до получения полинуклеотида, кодирующего полное антитело (например, гуманизированное или полностью человеческое), фрагменты антител и т.п. Антитела можно использовать для выделения препаративных количеств антигена за счет иммуноафинной хроматографии. Различные другие применения таких антител относятся к диагностике и/или определению стадии заболевания (например, неоплазии), и для терапевтических применений для лечения таких заболеваний, как, например, неоплазия, автоиммунные заболевания, AIDS, сердечно-сосудистые заболевания, инфекция и т.п.
Имеются сообщения о различных способах для повышения комбинаторного разнообразия ScFv библиотеки для расширения репертуара типов связывания (спектр идиотипов). Применение PCR позволяет быстро клонировать вариабельные участки либо из специфических гибридомных источников, либо в виде генной библиотеки из неиммунизированных клеток, предоставляя комбинаторное разнообразие в ассортименте VH и VL кассет, которые можно объединить. Кроме того, VH и VL кассеты можно сами диверсифицировать, например, за счет неупорядоченного, псевдоупорядоченного или направленного мутагенеза. Обычно VH и VL кассеты диверсифицируют в/или вблизи участков, определяющих коплементарность (CDR), часто в третьем CDR, CDR3. Энзиматически инверсный ПЦР мутагенез, как было показано, является простым и надежным способом конструирования относительно крупных библиотек ScFv сайт-направленных мутантов (Stemmer et al. (1993). Biotechniques 14: 256), каким является подверженный ошибкам PCR и химический мутагенез (Deng et al. (1994) J. Biol. Chem. 269:9533). Riechmann et al. (1993) Biochemistry 32:8848) представил полурациональную конструкцию ScFv фрагмента антитела с использованием сайт-направленной рандомизации за счет дегенеративной олигонуклеотидной ПЦР и последующего представления на фаге полученных StFv мутантов. Barbas et al. (1992) op. cit. попытался обойти проблему ограниченного размера репертуара, возникающую из-за использования ошибочных последовательностей вариабельных участков за счет рандомизии последовательности на участке синтетического CDR Fab, связывающего токсоид столбняка человека.
CDR рандомизация имеет потенциал создавать примерно 1• 1020 CDR только для CDR3 тяжелой цепи и примерно аналогичное число вариантов CDR1 и CDR2 тяжелой цепи, а также вариантов CDR1-3 легкой цепи. Взятые отдельно или вместе, комбинации CDR рандомизации тяжелой и/или легкой цепей требуют создания запрещающего числа клонов бактериофага для получения библиотеки клонов, представляющей все возможные комбинации, основное число которых окажется несвязывающими. Создание столь большого числа первичных трансформантов несложно при существующей в настоящее время технике трансформации и систем представления бактериофагов. Так, например, только Barbas et al. (1992) создал 5•107 трансформантов, что представляет только малую часть потенциального разнообразия библиотеки тщательно рандомизованных CDR.
Несмотря на эти существенные ограничения, представление бактериофага ScFv уже приводит к разнообразию полезных антител и протеинов слияния антител. Биспецифическое одноцепочечное антитело, как было показано, осуществляет эффективный лизис опухолевых клеток (Gruber et. al. (1994) J. Immunol. 152: 5368). Как было показано, внутриклеточная экспрессия анти-Rev-ScFv ингибирует репликацию вируса H1V-1 ин витро (Duan et al. (1994) Proc. Natl.Acad. Sci (USA) 91: 5075) и было показано, что внутриклеточная экспрессия анти-р21ras ScFv ингибирует меиотическое созревание Xenopus ооцитов (Biocca et al. (1993) Biochem. Biophys. Res.Commun. 197:422. Было также сообщение о рекомбинантных ScFv, которые можно использовать для диагностики H1V инфекции, что демонстрирует диагностические возможности ScFv (Lilley et al. (1994) J.Immunol. Meth. 171: 211). Протеины слияния, в которых ScFv связан со вторым полипептидом, такие, как токсин или фибринолитический активаторный протеин, также были описаны (Holvost. et al. (1992) Eur. J. Biochem. 210:945; Nicholls et al. (1993) J. Biol.Chem. 268:5302).
Если бы можно было создать библиотеки ScFv, содержащие большее разнообразие антител, и преодолеть многие ограничения CDR мутагенеза и методов рандомизации, которые могут охватывать только очень маленькую часть потенциальных комбинаций последовательностей, число и качество ScFv антител, пригодных для терапевтического и диагностического применения, можно было бы существенно расширить. Для этого методы перетасовки ин витро и ин виво настоящего изобретения используют для рекомбинации CDR, которые были получены (обычно за счет PCR амплификации или клонирования) из нуклеиновых кислот, полученных из селектированных представленных антител. Такие представленные антитела можно представить на клетках, на частицах бактериофагов, на полисомах, или на любой системе представления антител, где антитело ассоциировано с его кодирующей нуклеиновой кислотой (кислотами). В одном из вариантов вначале получают CDR из мРНК (или кДНК) из продуцирующих антитела клеток (например, из клеток плазмы) спленоцитов из иммунизованных дикого типа мышей, человека или трансгенных мышей, способных вырабатывать человеческие антитела, как указано в WO92/03918, WO93/12227 и WO94/25585, включая полученные из них гибридомы.
Полинуклеотидные последовательности, отобранные в первом цикле селекции (обычно за счет афинной селекции для связывания представленных антител с антигеном (например, лигандом), любым из этих способов объединяют, и этот пул (пулы) перетасовывают за счет ин витро и/или ин виво рекомбинации, особенно за счет перетасовки CDR (обычно перетасовывания CDR тяжелых цепей с CDR других тяжелых цепей и CDR легких цепей с CDR других легких цепей) до получения перетасованного пула, содержащего популяцию рекомбинированных последовательностей селектированных полинуклеотидов. Рекомбинированные селектированные полинуклеотидные последовательности экспрессируют в формат селекции как представленное антитело и подвергают, по крайней мере, одному последующему циклу селекции. Полинуклеотидные последовательности, отобранные в последующем цикле (циклах) селекции, можно использовать непосредственно, секвенировать и/или подвергнуть одному или более из дополнительных циклов перетасовки и последующего отбора до тех пор, пока не получат антитело с нужной связывающей афинностью. Селектированные последовательности можно также подвергнуть бэккроссу с полинуклеотидными последовательностями, кодирующими последовательности каркаса нейтрального антитела (например, оказывающими несущественный функциональный эффект на связывание антигена), такими, как, например, бэккросс с каркасом вариабельного участка человека до получения человекоподобной последовательности антител. Обычно, после бэккроссинга применяют последующую селекцию для сохранения свойств связывания с заранее определенным антигеном.
В другом варианте или в сочетании с указанными вариантами, валентность мишеневого эпитопа можно варьировать для контроля средней связывающей афинности членов селектированной ScFv библиотеки. Затем мишеневый эпитоп можно связать с поверхностью или субстратом при различных плотностях, как, например, включая конкурирующий эпитоп, за счет разбавления или за счет других способов, известных специалистам. Высокая плотность (валентность) заранее указанного эпитопа может быть использована для обогащения членов ScFv библиотеки, которые обладают относительно низкой афинностью, тогда как низкую плотность (валентность) можно предпочтительно обогатить для более высокой афинности членов ScFv библиотеки.
Для создания разнообразных вариабельных сегментов коллекцию синтетических олигонуклеотидов, кодирующих неупорядоченные, псевдоупорядоченные, определенный каркасный набор пептидных последовательностей можно встроить за счет легирования в заранее определенный сайт (например, в CDR).
Аналогично, разнообразие последовательностей одного или более из CDR кассеты (кассет) одноцепочечных антител можно расширить за счет введения мутации в CDR за счет сайт-направленного мутагенеза, CDR-замещения и т.п. Полученные молекулы ДНК можно размножить в хозяине для клонирования и амплификации перед перетасовкой или можно использовать непосредственно (т.е. можно избежать потери разнообразия, что могло бы произойти при размножении в клетке хозяина) и существенно перетасовать члены селектированной библиотеки.
Представленные комплексы пептид/полинуклеотид/ члены библиотеки/, которые кодируют последовательность вариабельного сегмента пептида, представляющую интерес, или представляющее интерес одноцепочечное антитело, отбирают из библиотеки с помощью методики афинного обогащения. Это осуществляют с помощью иммобилизованной макромолекулы или эпитопа, специфического для пептидной последовательности, представляющей интерес, например, рецептора, другой макромолекулы или другого типа эпитопа. Повторение процедуры афинного отбора обеспечивает обогащение членов библиотеки, кодирующих целевые последовательности, которые можно затем выделить для создания пула и перетасовки, для секвенирования и/или для дальнейшего размножения и афинного обогащения.
Члены библиотеки без нужной специфичности удаляют промывкой. Степень жесткости условий необходимой промывки определяется для каждой пептидной последовательности или одноцепочечного антитела, представляющих интерес, и иммобилизованной заранее определенной макромолекулы или эпитопа. Определенную степень контроля можно обеспечить за характеристиками связывания выделенных образующихся комплексов пептид/ДНК, выделенными за счет регулирования условий связывания, инкубирования и последующей промывки. Температура, pH, ионная сила, концентрация двухвалентных катионов и объем и длительность промывки позволяют отобрать образовавшиеся комплексы пептид/ДНК в конкретных интервалах афинности для иммобилизованных макромолекул. Селекция на основании медленной диссоциации, которая обычно является предвестником высокой афинности, часто является наиболее удобным способом. Это можно осуществить либо за счет непрерывного инкубирования в присутствии насыщающего количества свободных заранее определенных макромолекул, либо за счет увеличения объема, количества и длительности промывок. В каждом случае предотвращается вторичное связывание диссоциированных комплексов образовавшихся комплексов пептид/ДНК или пептид/РНК, и при увеличении времени выделяют образовавшиеся комплексы пептид/ДНК или пептид/РНК все более и более высокой афинности.
Дополнительные модификации процедур связывания и промывки можно использовать для того, чтобы найти пептиды со специальными характеристиками. Афинности некоторых пептидов зависят от ионной силы или концентрации катионов. Это является удобной характеристикой для пептидов, которую можно использовать при афинной очистке различных протеинов, если необходимы мягкие условия для удаления протеинов из пептидов.
Один из вариантов включает использование множества связывающих мишеней (множества типов эпитопов, множества типов рецепторов), например, так, чтобы ScFv библиотеку можно было одновременно скринировать для множества ScFv, которые отличаются различными специфичностями связывания. Учитывая, что размер ScFv библиотеки часто ограничивает разнообразие потенциальных ScFv последовательностей, обычно желательно использовать ScFv библиотеки насколько это возможно крупного размера. Время и экономические соображения относительно создания ряда очень крупных полисомных ScFv-представляющих библиотек могут стать ограничивающими факторами. Чтобы избежать этой существенной проблемы, можно одновременно скринировать множество заранее определенных видов эпитопов (видов рецепторов) в одной библиотеке или последовательно скринировать в отношении ряда видов эпитопов. В одном из вариантов множество видов мишеневых эпитопов, каждый из которых кодирует отдельную гранулу (или субнабор гранул), можно смешать и инкубировать с полисом-представляющей ScFv библиотекой в подходящих условиях связывания. Набор гранул, содержащих множество видов эпитопов, можно затем использовать для выделения за счет афинной селекции членов ScFv библиотеки. Обычно последующие циклы афинного скринирования можно включать в ту же смесь гранул, их субнабор, или в гранулы, содержащие только один или два отдельных вида эпитопов. Такой подход обеспечивает эффективное скринирование и совместим с автоматикой в лаборатории, порционными обработками и высокими пропускными способностями методов скринирования.
В настоящем изобретении можно использовать различные методики для диверсификации пептидной библиотеки или библиотеки одноцепочечных антител, или для диверсификации, перед или одновременно с перетасовкой, пептидов вариабельных сегментов или VH, VL или CDR, обнаруженных на ранних циклах пэннинга, для достижения достаточной активности связывания с заранее определенной макромолекулой или эпитопом. В одном подходе позитивные селекционированные пептид/нуклеотидные комплексы (те, которые идентифицированы в ранних циклах афинного обогащения), секвенируют для определения идентичности активных пептидов. Затем синтезируют олигонуклеотиды на основании этих активных пептидных последовательностей, используя низкие уровни всех оснований, включаемых на каждой стадии, для получения небольших вариаций первичных олигонуклеотидных последовательностей. Эту смесь (незначительно) дегенеративных олигонуклеотидов затем клонируют в вариабельные сегментные последовательности в соответствующих положениях. Этот способ приводит к получению систематических контролируемых вариаций исходных пептидных последовательностей, которые можно затем перетасовать. Однако это требует, чтобы отдельные позитивные образовавшиеся пептид/полинуклеотидные комплексы были секвенированы перед мутагенезом и, таким образом, полезны для расширения разнообразия небольшого числа выделенных комплексов и отбора вариантов, обладающих более высокой связывающей афинностью и/или более высокой специфичностью связывания. При вариациях, мутагенной PCR амплификация позитивных селектированных пептид/полинуклеотидных комплексов / особенно последовательностей вариабельных участков, продукты амплификации которых перетасовывают ин витро и/или ин виво, и один или более из дополнительных циклов скринирования осуществляют перед секвенированием. Тот же самый общий подход можно использовать для одноцепочечных антител для того, чтобы расширить разнообразие и повысить связывающую афинность/специфичность, обычно за счет диверсификации CDR или соседних каркасных участков перед или одновременно с перетасовкой. При желании реакции перетасовки можно совместить с мутагенными олигонуклеотидами, способными к ин витро рекомбинации с членами выбранной библиотеки, которые могут быть включены. Таким образом, смеси синтетических олигонуклеотидов и ПЦР фрагментов (синтезированных за счет подверженных ошибкам или в высшей степени произвольных методов) можно добавить к ин витро перетасованной смеси, и они могут быть включены в число полученных членов перетасованной библиотеки.
Настоящее изобретение перетасовки позволяет создавать обширную библиотеку CDR-вариантов одноцепочечных антител. Одним из путей создания таких антител является встраивание синтетических CDR в одноцепочечное антитело и/или CDR рандомизация перед или одновременно с перетасовкой. Последовательности синтетических CDR кассет выбирают, обращаясь к данным об известных последовательностях, и отбирают в соответствии со следующей схемой: синтетические CDR должны содержать, по крайней мере, 40% идентичности положений последовательности с известными CDR последовательностями и, предпочтительно, должны содержать, по крайней мере, от 50 до 70% идентичности положений последовательности с известными CDR последовательностями. Так, например, коллекцию синтетических CDR последовательностей можно создать, синтезируя коллекцию олигонуклеотидных последовательностей на основании нативных CDR последовательностей человека, перечисленных у Kabat et al. (1991) op. cit; пул/пулы синтетических CDR последовательностей рассчитывают так, чтобы он кодировал CDR пептидные последовательности, содержащие, по крайней мере, 40% идентичности последовательностей с, по крайней мере, одной известной нативной CDR последовательностью человека. В другом варианте коллекцию нативных CDR последовательностей можно сравнить с созданными консенсусными последовательностями так, что аминокислотные последовательности, используемые в оставшихся положениях, часто (т.е. по крайней мере, на 5% от известных CDR последовательностей) были бы включены в синтетические CDR в соответствующем положении (положениях). Обычно, несколько (например, от 3 до около 50) известных CDR последовательностей сравнивают, и наблюдаемые вариации нативных последовательностей по сравнению с известными CDR сводят в таблицу и синтезируют коллекцию олигонуклеотидов, кодирующих CDR пептидные последовательности, охватывающие все или большинство пермутаций наблюдаемых вариантов нативных последовательностей. Так, например (но не для ограничения), если коллекция VH CDR последовательностей человека имеет карбокситерминальные аминокислоты, которые являются Tyr, Val, Phe, либо Asp, тогда пул (пулы) синтетических CDR олигонуклеотидных последовательностей конструируют таким образом, чтобы позволить карбокси-терминальному CDR остатку быть любой из аминокислот. В некоторых вариантах остатки, отличающиеся от тех, которые нативно встречаются в положении остатка в коллекции CDR последовательностей, оказываются встроенными. Замещения консервативных аминокислот часто включают, и вплоть до 5 положений остатков можно варьировать для встраивания неконсервативных замещений аминокислот по сравнению с известными нативно встречающимися CDR последовательностями. Такие CDR последовательности можно использовать в членах первичных библиотек (перед первым циклом скринирования) и/или их можно использовать в ин витро реакциях перетасовки последовательностей членов выбранных библиотек. Конструирование таких пулов определенных и/или дегенеративных последовательностей можно легко осуществить специалистам.
Коллекция синтетических CDR последовательностей содержит, по крайней мере, один член, который, как известно, не является нативной CDR последовательностью. От решения практика зависит, включать или нет часть неупорядоченной или псевдоупорядоченной последовательности, соответствующей N участку, дополнительно в CDR тяжелой цепи; последовательность N участков состоит из от 1 нуклеотида до около 4 нуклеотидов, находящихся в V-D и D-I соединениях. Коллекция синтетических CDR последовательностей тяжелой цепи содержит, по крайней мере, около 100 уникальных CDR последовательностей, обычно, по крайней мере, около 1000 уникальных CDR последовательностей, предпочтительно, по крайней мере, около 10000 уникальных CDR последовательностей, часто более чем около 50000 уникальных CDR последовательностей; однако обычно не более чем около 1•106 уникальных CDR последовательностей включены в коллекцию, хотя иногда присутствует от 1•107 до 1•108 уникальных CDR последовательностей, особенно, если замещения консервативных аминокислот разрешены в положениях, где заместитель консервативных аминокислот отсутствует или присутствует редко (т. е. менее 0,1%) в этом положении в нативно-существующих CDR человека. Вообще, число уникальных CDR последовательностей, включенных в библиотеку, не должно превышать ожидаемое число первичных трансформантов в библиотеке более чем в 10 раз. Такие одноцепочечные антитела обычно связываются с заранее определенным антигеном (например, иммуногеном) с афинностью примерно, по крайней мере, 1•107 М-1, предпочтительно с афинностью около, по крайней мере, 5•107 М-1, более предпочтительно с афинностью, по крайней мере, 1•108 М-1 или более, иногда вплоть до 1•1010 М-1 или более. Часто заранее определенным антигеном является человеческий протеин, например, такой как антиген клеточной поверхности человека (например, CD4, CD8, IL-2 рецептор, EGF рецептор, PDGF рецептор), другие биологические макромолекулы человека (например, тромбодулин, протеин C, углеводный антиген, сиаллильный антиген Льюиса, L-селектин) или макромолекулы, связанные с нечеловеческими заболеваниями (например, бактериальные LPS, протеин капсида вириона или гликопротеин оболочки) и т.п.
Высоко афинные одноцепочечные антитела нужной специфичности можно сконструировать и экспрессировать в различных системах. Так, например, ScFv были получены в растениях (Firek et al. (1993) Plant Mol.Biol. 23:861) и можно легко получить в прокариотных системах (Owens R.I.and Young R.J (1994) J. Immunol.Meth. 168:149; Johnson S and Bird RE (1991) Methods Enzymol. 203: 88). Кроме того, одноцепочечные антитела можно использовать как основу для конструирования целых антител или их различных фрагментов (Kettleborough et al. (1994) Eur. J. Immunol. 24:952). Последовательности, кодирующие вариабельные участки, можно выделить (например, за счет PCR ампликации или субклонирования) и осуществить сплайсинг в последовательность, кодирующую нужный постоянный участок человека для того, чтобы кодировать последовательность антитела человека, более подходящую для терапевтического использования у людей, где должна быть, предпочтительно, минимальная иммуногенность. Полинуклеотид (полинуклеотиды), содержащие полученные полностью человеческие кодирующие последовательности, можно экспрессировать в клетки хозяина (например, в виде вектора экспрессии в клетки млекопитающих) и очистить для фармацевтических композиций.
Конструкции ДНК экспрессии обычно включают ДНК последовательность, контролирующую экспрессию, операбельно связанную с кодирующими последовательностями, включая нативно-связанный или гетерологически промоторные участки. Предпочтительно, чтобы последовательности, контролирующие экспрессию, были бы эукариотными промоторными системами в векторах, способных трансформировать или трансфектировать клетки эукариотного хозяина. После того, как такой вектор введен в соответствующего хозяина, этого хозяина содержат в условиях, подходящих для высокого уровня экспрессии нуклеотидных последовательностей и сбора и очистки мутантных "сконструированных" антител.
Как было указано ранее, ДНК последовательности будут экспрессироваться в хозяине после того, как последовательности будут операбельно связаны с последовательностью, контролирующей экспрессию (т. е. расположенной таким образом, чтобы обеспечить транскрипцию и трансляцию структурного гена). Эти векторы экспрессии обычно реплицируемы в организмах хозяина либо как эписомы, либо как интегральные части хромосомной ДНК хозяина. Обычно векторы экспрессии должны содержать селекционные маркеры, например, ген устойчивости к тетрациклину или неомицину, чтобы позволить осуществить детектирование тех клеток, которые трансформированы целевыми ДНК последовательностями (см. например, патент США 4704362, который включен сюда по ссылке).
В дополнение к эукариотным микроорганизмам, таким, как дрожжи, клеточные культуры тканей млекопитающих можно также использовать для получения полипептидов настоящего изобретения (см. Winnacker "From Genes to Clones" VCH Publishers N. Y. (1987), которая включена сюда по ссылке). Эукариотные клетки действительно предпочтительны, так как достаточное количество подходящих клеточных линий хозяев, способных секретировать интактные иммуноглобулины, было разработано специалистами и включает CHO клеточные линии, различные COS клеточные линии, HeLa клетки миеломные клеточные линии и т.д., но, предпочтительно, трансформированные В-клетки гибридом. Векторы экспрессии для этих клеток могут включать последовательности, контролирующие экспрессию, такие, как начало репликации, промотор, энхансер (Queen et al. (1986) Immunol. Rev. 89:49) и необходимые информационные сайты процессинга, такие, как рибосомные сайты связывания, сайты РНК сплайсинга, сайты полиаденилирования и последовательности окончания транскрипции. Предпочтительными последовательностями, контролирующими экспрессию, являются промоторы, полученные из генов иммуноглобулина, цитомегаловируса, SV40, аденовируса, вируса бычьей папилломы и т.п.
Эукариотную ДНК транскрипцию можно усилить за счет встраивания в этот вектор энхансерной последовательности. Энхансеры представляют собой цис-функционирующие последовательности от 10 до 300 п.о., которые увеличивают транскрипцию за счет промотора. Энхансеры могут эффективно усилить транскрипцию, если либо 5', либо 3'-конца связывается с транскрипционным фрагментом. Они также эффективны, если расположены внутри интрона или внутри самой кодирующей последовательности. Обычно, используют вирусные энхансеры, полиомные энхансеры и аденовирусные энхансеры. Обычно используют также энхансерные последовательности из систем млекопитающих, например, энхансер тяжелой цепи мышиного иммуноглобулина.
Системы векторов экспрессии из млекопитающих также обычно включают селектируемый маркерный ген. Примеры подходящих маркеров включают ген дигидрофолат-редуктазы (DHFR), ген тимидин киназы (TK) или прокариотные гены, придающие устойчивость к лекарствам. Первые два маркерных гена предпочитают использование мутантных клеточных линий, которые лишены способности роста без добавления к ростовой среде тимидина. Затем трансформированные клетки можно идентифицировать по их способности расти на среде без дополнительных ингредиентов. Примеры прокариотных генов устойчивости к лекарствам, которые удобно использовать в качестве маркеров, включают гены, придающие устойчивость к G-418, микофенольной кислоте и гигромицину.
Векторы, содержащие представляющие интерес ДНК сегменты, можно перенести в клетки хорошо известными способами, в зависимости от типа клеточного хозяина. Так, например, обычно используют трансфекцию кальцийхлорида для прокариотных клеток, тогда, как обработку фосфатом кальция, липофекцию или электропорацию можно использовать для других клеточных хозяев. Другие способы, используемые для трансформации клеток млекопитающих, включают использование полибрена, слияния протопластов, липосомы, электропорацию и микроинъекции (см. Sambrook et al.).
После того как экспрессированы антитела, отдельные мутированные цепи иммуноглобулинов, фрагменты мутированных антител и другие иммуноглобулиновые полипептиды настоящего изобретения, их можно очистить в соответствии со стандартными процедурами, известными специалистам, включая осаждение сульфатом аммония, фракционирование на хроматографической колонке, гель-электрофорез и т. п. (см. Scopes, R. Protein Purification, Springer-Verlag, N.Y.(1982)). После очистки (частичной или при желании до гомогенности) полипептиды можно затем использовать в терапевтических целях или для разработки и осуществления аналитических процедур, иммунофлюоресцентного окрашивания и т.п. (см. Immunological Methods, Vol. 1 and 11, Eds. Lefcovits and Pernis, Academic Press, New York. N.Y. (1979 и 1981).
Антитела, полученные по способу настоящего изобретения, можно использовать для диагностических и терапевтических целей. В качестве иллюстрации (но не ограничения! ) их можно использовать для лечения рака, автоиммунных заболеваний или вирусных инфекций. Для лечения рака антитела обычно связывают с антигеном, экспрессируемым преимущественно на раковых клетках, таких как erbB-2, CEA, CD33, и многих других антигенах и связывающих членах, хорошо известных специалистам.
Анализ со скринированием двух гибридов дрожжей
Перетасовку можно также использовать для рекомбинаторной
диверсификации пула членов выбранной библиотеки, полученного за счет скринирования в системе скринирования двух гибридов для идентификации членов библиотеки, которые связывают заранее определенную
полипептидную последовательность. Члены выбранной библиотеки объединяют в пул и перетасовывают за счет ин витро или ин виво рекомбинации. Затем перетасованный пул можно скринировать в дрожжевой
двухгибридной системе для отбора членов библиотеки, которые связываются с указанной заранее определенной полипептидной последовательностью (например, с SH2 доменом) или которые связываются
с измененной заранее указанной полипептидной последовательностью (например, SH2 доменом из другого вида протеина).
Такой подход к идентификации полипептидных последовательностей, которые связываются с заранее определенной полипептидной последовательностью, был использован для, так называемой, "двухгибридной" системы, где заранее определенная полипептидная последовательность присутствует в протеине слияния (Chien et al. (1991) Proc. Natl. Acad. Sci. (USA) 88:9578). Такой подход идентифицирует взаимодействие протеин-протеин ин виво за счет реконструирования активатора транскрипции (Fields and Song O (1989) Nature 340:245), Gal4 транскрипционного протеина дрожжей. Обычно этот способ базируется на свойствах Gal4 протеина дрожжей, который состоит из отделяемых доменов, ответственных за ДНК-связывание и активацию транскрипции. Полинуклеотиды, кодирующие два гибридных протеина, причем один состоит из дрожжевого Gal4 ДНК-связывающего домена, слитого с полипептидной последовательностью известного протеина, а другой - состоит из Gal4 активаторного домена, слитого с полипептидной последовательностью, второго протеина, конструируют и вводят в дрожжевые клетки-хозяева. Межмолекулярное связывание между двумя протеинами слияния реконструирует Gal4 ДНК-связывающий домен с Gal4 активаторным доменом, что приводит к транскрипционной активации рецепторного гена (например, lacZ, His3), который операбельно связан с Gal4 связывающим сайтом. Обычно двухгибридный метод используют для идентификации новых полипептидных последовательностей, которые взаимодействуют с известным протеином.
(Silver SC and Hunt SW (1993) Mol.Biol.Rep. 17: 155; Durfee et al. (1993) Genes Devel. 7: 555; Yang et al. (1992) Science 257: 680; Luban et al. (1993) Cell 73: 1067; Hardy et al. (1992) Genes Devel. 6; 801; Bartel et al. (1993) Biotechniques 14: 920; and Voсtek et al. (1993) Cell 74: 205).
Однако варианты двухгибридного метода были использованы для идентификации мутаций известного протеина, которые влияют на его связывание с известным протеином.
(Li В and Fields S (1993) FASEBI. 7: 957; Lalo et fl. (1993) Proc. Natl. Acad. Sci. (USA) 90: 5524; Jackson et al. (1993) Mol. Cell. Diol. 13; 2899; and Madura et al. (1993) J. Biol. Chem. 268: 12046).
Двухгибридная система была также использована для идентификации взаимодействующих структурных доменов двух известных протеинов (Bardwell et al. (1993) med. Microbiol. 8: 1177; Chakraborty et al. (1992) J.Biol.Chem. 267: 17498; Staudinger et al. (1993) J. Biol. Chem. 268:4608; Milhe GT and Weaver DT (1993) Genes.Devel. 7: 1755) или домены, ответственные за олигомеризацию отдельного протеина (Iwabuchi et al. (1993) Oncogene 8, 1693; Bogerd et al. (1993) J. Virol. 67: 5030). Варианты двухгибридных систем были использованы для изучения ин виво активности протеолитического энзима (Dasmahapatra et al. (1992) Proc. Natl. Acad. Sci. (USA) 89: 4159). В другом варианте можно использовать Е. coli (BCCP интерактивную скринирующую систему (Germino et al. (1993) Proc. Natl. Acad. Sci (USA) 90:933; Guarente L. (1993) Proc. Natl. Acad. Sci (USA) 90:1693) для идентификации взаимодействующих последовательностей протеинов (т.е. последовательностей протеинов, которые гетеродимеризуются или образуют гетеромультимеры высшего порядка). Последовательности, селектированные за счет двухгибридной системы, можно объединить в пул и перетасовать и ввести в двухгибридомную систему для одного или более последующих циклов скринирования для идентификации полипептидных последовательностей, которые связываются с гибридом, содержащим заранее определенную связывающую последовательность. Идентифицированные таким образом последовательности можно сравнить с идентифицированной консенсусной последовательностью (последовательностями) и ядрами консенсусных последовательностей.
Как можно заключить из приведенного ранее, настоящее изобретение имеет широкое применение. Соответственно, нижеследующие примеры предложены только с целью иллюстрации, и их не следует рассматривать как ограничивающие.
В приведенных далее примерах сокращения имеют следующие значения. Если они не приведены ниже, тогда, эти сокращения имеют общепринятые значения.
мл = миллилитр
мкл = микролитр
мкМ = микромолярный
нМ = наномолярный
PBS = буферированный фосфатом физиологический раствор
нг = нанограмм
мкг = микрограмм
IPTG изопропилтио- β-D-галактозид
п.о. = пара оснований
к.п.о. = килопар оснований
dNTP = деоксинуклеозидтрифосфат
ПЦР =
полимеразная цепная реакция
X-qal = 5-бром-4-хлор-3-индол-β-D-галактозид
ДНКаза1 = деоксирибонуклеаза
CDR = участок, определяющий комлементарность
MIC =
минимальная ингибирующая концентрация
ScFv = одноцепочечный Fv фрагмент антитела
Вообще, стандартные методики рекомбинационной ДНК технологии описаны в различных публикациях,
например, в книге Sambrook et al.,1989, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory; Ausubel et al. 1987, Current Protocols in Molecular Biology, v. 1 and 2 and supplements
and Berger and Kimmel, Methods in Enzymology, Y, 152, Guide to Molecular Cloning Techniques (1987), Academic Press Inc., San Diego, CA, причем все они включены сюда по ссылке. Рестрикционные энзимы и
модифицирующие полинуклеотиды энзимы используют в соответствии с рекомендациями изготовителей. Олигонуклеотиды были синтезированы на синтезаторе Applied Biosystems Inc. Model 394 HK Synthesizer,
используя химикалии ABI. При желании ПЦР амплимеры для амплификации заранее определенной ДНК последовательности можно отобрать в соответствии с усмотрением практика.
Примеры
Пример 1. Вторичная сборка LacZ альфа гена
1. Приготовление субстрата
Субстрат для реакции вторичной сборки представляет собой продукт dsДНК полимеразной цепной реакции ("ПЦР")
дикого типа LacZ альфа гена из pUC18 (фиг. 2) (28; Gene Bank N X02514). Праймерной последовательностью является последовательность 1Д N 1 = 5' AAAGCGTCGATTTTTGTGAT3', и последовательность 1Д N 2 = 5'
ATGGGGTTCCGCGCACA'. Свободные праймеры были выделены из ПЦР продукта с помощью Wizard ПЦР prep (Promega Madison, WI) в соотвествии с указаниями изготовителей. Было обнаружено, что выделение свободных
праймеров является важным.
2) Переваривание с помощью ДНКазы 1.
Около 5 мкг ДНК субстрата переваривают 0,15 единицами ДНКазы 1 (Sigma, St. Louis МО) в 100 мкл 50 мМ Tris-HCl, pH 7,4, 1 мМ MgCl2 в течение 10-20 минут при комнатной температуре. Переваривание ДНК проводят на 2% агарозном геле с низкой температурой плавления. Фрагменты из 10-70 пар оснований выделяют из 2% агарозного геля с низкой температурой плавления за счет электрофореза на ДЕ81 ионообменной бумаге (Whatman, Hillsborough OR). ДНК фрагменты элюируют из бумаги 1 М NaCl и осаждают этанолом.
3) Вторичная сборка ДНК
Очищенные фрагменты снова суспендируют в концентрации 10-30 нг/мкл в ПЦР Mix (0,2 мМ dNTP, 2,2 мМ MgCl2; 50 мМ KCl, 10
мМ Tris-HCl, pH 9,0, 0,1% Triton X-100, 0,3 мкл TagДНК полимеразы, 50 мкл полный объем). В этот момент не добавляют никаких праймеров. Программа вторичной сборки составляет: 94oC в течение
60 секунд, 30-45 циклов (94oC в течение 30 секунд, 50-55oC в течение 30 секунд, 72oC в течение 30 секунд) и 5 минут при 72oC в термоциклере MJ Research
(Watertown MA) PTC-150. ПЦР вторичная сборка мелких фрагментов в более крупные последовательности осуществляется далее за счет отбора образцов из реакции после 25, 30, 35, 40 и 45 циклов вторичной
сборки (фиг. 2).
Тогда как вторичная сборка 100-200 п.о. фрагментов может привести к отдельному ПЦР продукту нужного размера, фрагменты в 10-50 пар оснований обычно приводят к получению части продуктов нужного размера, а также к продуктам различных молекулярных весов. Большая часть таких размеров, по-видимому, связана с одноцепочечными последовательностями на концах продуктов, так как после рестрикционного энзиматического переваривания получают только одну полосу нужного размера.
4) ПЦР с праймерами
После разбавления вторичной сборки
продукта в ПЦР Mix с 0,8 мкМ каждого из вышеуказанных праймеров (последовательности 1Д N 1 и 1) и около 15 циклов ПЦР, причем каждый цикл состоит из (94oC в течение 30 секунд, 50o
C в течение 30 секунд и 72oC в течение 30 секунд) получают один продукт нужного размера (фиг. 2).
5) Клонирование и анализ
Продукт ПЦР со стадии 4 переваривают
терминальными рестрикционными энзимами BamHI и Eco0109 и очищают на геле, как указано ранее на стадии 2. Фрагменты вторичной сборки легируют в pUC18, переваривают BamHI и Есо0109. E. coli
трансформируют смесью для легирования в стандартных условиях в соответствии с рекомендациями изготовителей (Stratagene, San Diego CA) и высевают на агарные пластины, содержащие 100 мкг/мл ампициллина,
0,004%-X-qal и 2 мМ 1PTG. Полученные колонии, содержащие фрагмент Hin Д111-Nhel, который является диагностическим для ++рекомбинанты, идентифицируют, так как они становятся синими.
Этот пример иллюстрирует тот факт, что 1,0 к.п.о. последовательность, содержащая LacZ альфа ген, была переварена на 10-70 п.о. фрагменты, и что эти выделенные на геле 10-70 п.о. фрагменты можно подвергнуть вторичной сборке в отдельный продукт нужного размера, так что 84% (N = 377) полученных колоний являются LacZ+ (против 94% без перетасовки, фиг. 2).
ДНК, кодирующую LacZ ген из полученных LacZ- колоний, секвенируют с помощью набора для секвенирования (United States Biochemical Co, Cleveland OH) в соответствии с указаниями изготовителей, и было обнаружено, что гены содержат точечные мутации в результате процесса вторичной сборки (таблица 1). Было обнаружено 11/12 типов замещений и не было обнаружено сдвига рамки.
Всего было секвенировано 4437 оснований перетасованной lacZ ДНК.
Степень точечных мутаций во время вторичной сборки ДНК из отрезков 10-70 п. о. определяют по данным ДНК секвенирования, и она составляет 0,7% (N = 4,473), что аналогично результатам подверженной ошибкам PCR. Не ограничиваясь какой-либо теорией, считают, что степень точечных мутаций может быть меньше, если для вторичной сборки использовать более крупные фрагменты или если добавлять корректирующую полимеразу.
Если объединить плазмидные ДНК из 14 из этих точечно-мутированных LacZ-колоний и снова подвергнуть вторичной сборке/ перетасовке описанным выше способом, оказывается, что 34% (N = 291) из полученных колоний являются LacZ, и эти колонии, главным образом, образуются за счет рекомбинации ДНК из различных колоний.
Ожидаемая степень обращения отдельной точечной мутации за счет подверженной ошибкам ПЦР, предполагая степень мутагенеза 0,7% (10), ожидается должна быть менее 1%.
Таким образом крупные ДНК последовательности можно вторично собрать из неупорядоченной смеси мелких фрагментов за счет реакции, которая неожиданно оказывается и эффективной и простой. Одно из применений такой методики состоит в рекомбинации или перетасовке родственных последовательностей, основанных на гомологичности.
Пример 2. Перетасовка LacZ гена и
всей плазмидной ДНК
1) Перетасовка LacZ гена
Кроссовер между двумя маркерами, разделенными 75 основаниями, определяют, используя две конструкции LacZ гена. Стоп кодоны встраивают в
два отдельные участка LacZ альфа гена для того, чтобы они служили негативными маркерами. Каждый маркер представляет собой негомологичную последовательность из 25 п.о. с четырьмя стоп кодонами, из
которых два находятся в считывающей рамке LacZ гена. Негомологичная последовательность из 25 п.о. представлена на фиг. 3 крупным прямоугольником. Стоп кодоны либо заключены в прямоугольники, либо
подчеркнуты. Смесь 1:1 двух 1,0 к.п.о. LacZ матриц, содержащих + - и - + варианты LacZ альфа гена (фиг. 3), переваривают ДНКазой 1 и по способу примера 1 выделяют фрагменты в 100-200 п.о. Программу
перетасовки ведут в условиях, аналогичных тем, которые указаны для вторичной сборки в примере 1, за исключением того, что добавляют 0,5 мкл полимеразы, и полный объем составляет 100 мкл.
После клонирования число полученных голубых колоний составляет 24% (N = 386), что близко к теоретически максимальному числу голубых колоний (т.е. 25%), что указывает на то, что рекомбинация между двумя маркерами была полной. Все из 10 голубых колоний содержат ожидаемые Hind111-Nhe1 рестрикционный фрагмент.
2) Перетасовка полной плазмидной ДНК
Тестируют также
целые 2,7 к.п.о. плазмиды (pUC18-+ и pUC18+-). Смесь 1:1 из двух 2,9 к. п. о. плазмид, содержащих +- и -+ варианты LacZ альфа гена (фиг. 3), переваривают ДНКазой 1, и фрагменты 100-200 п.о. выделяют,
как указано в примере 1. Программу перетасовки осуществляют в условиях, аналогичных тем, которые указаны для вторичной сборки на стадии (1) ранее, за исключением того, что программа была для 60 циклов
(94oC в течение 30 секунд, 55oC в течение 30 секунд, 72oC в течение 30 секунд). Гель-анализ показал, что после программы перетасовки большая часть продукта оказывается
больше, чем 20 к. п. о. Таким образом, полные 2,7 к.п.о. плазмиды (pUC18 -+ и pUC18+-)эффективно подвергаются вторичной сборке из неупорядоченных 100-200 п.о. фрагментов без добавления праймеров.
После переваривания рестрикционным энзимом с уникальным сайтом на плазмиде (Eco0109), большая часть продукта состоит из одной полосы ожидаемого размера. Эту полосу выделяют из геля, повторно легируют, и ДНК используют для трансформации E.coli. Трансформанты высевают на 0,004% X-qal пластины по способу примера 1. 11% (N = 328) из полученных плазмид оказываются синими, и таким образом, ++ рекомбинантами.
3) Перетасовка Spiked ДНК
Олигонуклеотиды, которые смешаны с перетасованной смесью, можно включить в конечный продукт на основе гомологичности
фланкирующих последовательностей олигонуклеотидов с матричной ДНК (фиг. 4). Мутант LacZ стоп кодона (pUC18-+), описанный выше, используют в качестве ДНКазой 1 переваренной матрицы, 66-мерный
олигонуклеотид, включающий 18 оснований гомологичности с дикого типа LacZ геном с обоих концов, добавляют в реакцию в 4-кратном молярном избытке для исправления мутаций стоп кодона, присутствующих в
исходном гене. Реакцию перетасовки ведут в условиях, аналогичных условиям, указанным для стадии 2 ранее. Полученный продукт переваривают, легируют и встраивают в E. coli, как указано ранее.
оц ДНК, по-видимому, более эффективна; нежели дц ДНК, предположительно за счет конкурирующей гибридизации. Степень включения можно варьировать в широком интервале значений, регулируя молярный избыток, температуру отжига или длину гомологичности.
Пример 3. Вторичная сборка ДНК при полном отсутствии праймеров
Плазмиду pUC18 переваривают рестрикционным
энзимом EcoR1, Ecо0109, Xmn1 и AlwN1, получая фрагменты примерно в 370, 460, 770 и 1080 п.о. Эти фрагменты обрабатывают электрофоретически и отдельно выделяют из 2% агарозного геля с низкой
температурой плавления (полосы 370 и 460 п.о. не удается разделить), получая крупный фрагмент, средний фрагмент и смесь двух небольших фрагментов в 3 отдельных ампулах.
Каждый из фрагментов переваривают ДНКазой 1, как указано в примере 1, и фрагменты 50-130 п.о. выделяют из 2% агарозного геля с низкой температурой плавления для каждого из исходных фрагментов.
ПЦР смесь (как указано ранее в примере 1) добавляют к очищенным переваренным фрагментам до конечной концентрации 10 нг/мкл фрагментов. Не добавляют никаких праймеров. Реакцию вторичной сборки ведут в течение 75 циклов (94oC в течение 30 секунд, 60oC в течение 30 секунд) отдельно для каждой из трех переваренных смесей ДНК фрагментов, и полученные продукты анализируют с помощью электрофореза на агарозном геле.
Полученные результаты четко показывают, что 1080, 770 и 370 и 460 п.о. полосы, эффективно реформированные из очищенных фрагментов, демонстрируют, что перетасовка вовсе не требует использования каких-либо праймеров.
Пример 4. Перетасовка IL-1 β гена
Этот пример иллюстрирует, что кроссовер, основанный на
гомологичности менее чем 15 оснований, может быть достигнут. В качестве примера перетасовывают IL-1 β ген человека и мыши.
Используют мышиный IL1-β ген (BBG49) и IL1-β ген человека с кодоном, E. coli (BBG2; R andD System, Inc., Minneapolis MN) были использованы в качестве матрицы в реакции перетасовки. Участки полной гомологичности между человеческими и мышиными IL-1 β последовательностями составляют всего 4,1 основание в длину (фиг. 5, участки гетерологичности заключены в прямоугольники).
Получение дцДНК ПЦР продуктов для
каждого из генов, удаление праймеров, переваривание ДНКазой 1 и выделение фрагментов в 10-50 п.о. аналогично описанному в примере 1. Последовательности праймеров, используемые в ПЦР реакции, были:
5'TTAGGCACCCCAGGC' (последовательность 1Д N 3) и
5'ATGTGCTGCAAGGCGATT3' (последовательность 1Д N 4).
Первые 15 циклов реакции перетасовки осуществляют с фрагментом Кленова ДНК полимеразы 1, добавляя 1 единицу свежего энзима в каждом цикле. ДНК добавляют к ПЦР смеси примера 1, причем эта смесь не содержит полимеразы. Программа вручную составляет 94oC в течение 1 минуты, а затем 15 циклов: (95oC в течение 1 минуты, 10 секунд на смеси сухой лед/этанол (до замерзания), инкубирование около 20 секунд при 25oC, добавление 1 ед. фрагмента Кленова и инкубирование при 25oC в течение 2 минут). В каждом из циклов после стадии денатурирования ампулу быстро охлаждают в смеси сухой лед/этанол и снова нагревают до температуры отжига. Затем добавляют термолабильную полимеразу. Этот энзим необходимо добавлять в каждом из циклов. Используя такой подход, получают высокие уровни кроссоверов, основанные только на нескольких основаниях непрерывной гомологичности (фиг. 5, положения кроссоверов обозначены как
После этих 15 циклов, проведенных вручную, добавляют Tag полимеразу и проводят еще 22 цикла реакции перетасовки (94oC в течение 30 секунд, 35oC в течение 30 секунд) без праймеров.
Затем реакционную смесь разбавляют в 20 раз. Нижеследующие праймеры добавляют до конечной концентрации 0,8 мкМ:
5'AACGCCGCATGCAAGCTTGGATCCTTATT3' (послед. 1Д N
5) и
5'AAAGCCCTCTAGATGATTACGAATTCATAT3' (послед. 1Д N 6), и PCR реакцию ведут, как было указано ранее в примере 1.
Вторая пара праймеров отличается от первой пары только потому, что посчитали необходимым внести изменения в рестрикционные сайты.
После переваривания ПЦР продуктов за счет Xba1 и Sph1, фрагменты легируют в Xba1-Sph1-переваренный pUC18. Последовательности вставок из нескольких колоний определяют с помощью набора дидеокси ДНК секвенирования (United States Biochemical Co., Cleveland OH) в соответствии с рекомендациями изготовителей.
Всего 17 кроссоверов было получено в результате ДНК секвенирования девяти колоний. Некоторые из кроссоверов были основаны всего лишь на 1-2 основаниях с непрерывной гомологичностью.
Было обнаружено, что для усиления эффективности кроссоверов на основании коротких гомологичностей, необходима очень низкая эффективная температура отжига. С любой термостабильной полимеразой время охлаждения ПЦР автомата (94oC до 25oC при 1-2oC/секунду) вызывает температуру эффективного отжига выше, чем заданный набор температур отжига. Так, ни один из протоколов на основе Tag полимеразы не привел к образованию кроссовера, даже если использовать 10-кратный избыток IL1- β генов. Напротив, такая термолабильная полимераза, как фрагмент Кленова ДНК полимеразы 1, можно использовать для точного получения низкой температуры отжига.
Пример 5. Перетасовка ДНК TEM-1 беталактамазного гена
Применение мутагенной ДНК
перетасовки для управляемой молекулярной эволюции тестируют в модельной системе беталактамазы. TEM-1 беталактамаза является очень эффективным энзимом, причем скорость ее реакций ограничена, главным
образом, диффузией. Этот пример позволяет определить, можно ли изменить ее реакционную специфичность и получить устойчивость к препарату цефотаксиму, который она обычно не гидролизует.
Минимальную ингибирующую концентрацию (МИК) цефотаксима на бактериальных клетках, не содержащих плазмид, определяют, высевая 10 мкл 10-2 разбавления ночной бактериальной культуры (около 1000 кое) E.coli XL1-синих клеток (Stratagene, San Diego CA) на пластины с варьируемыми уровнями цефотаксима (Sigma, St. Louis MO), с последующим инкубированием в течение 24 часов при 37o C.
Рост на цефотаксиме чувствителен к плотности клеток, и поэтому одинаковое число клеток необходимо высеять на каждую из пластин (этого достигают, высевая на плоские LB пластины).
Обычно высевают порядка 1000 клеток.
1) Конструирование исходной плазмиды
Используют производное pUC18, содержащее бактериальный TEM-1 беталактамазный ген (28).
TEM-1 беталактамазный ген придает устойчивость к бактериям против примерно 0,02 мкг/мл цефотаксима. Sfil рестрикционные сайты добавляют в положении 5' от промотора и 3' от конца гена за счет ПЦР
векторной последовательности двух праймеров:
Праймер A (последовательность 1Д N 7)

Праймер B (последовательность 1Д N 8)

и за счет ПЦР последовательности беталактамазного гена с двумя другими праймерами:
Праймер C (последовательность 1Д N 9)
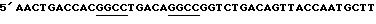
Праймер D (последовательность 1Д N 10)

Два продукта реакций переваривают за счет Sfil, смешивают, легируют и используют для трансформации бактерий.
Получают плазмиду pUC182Sfi. Эта плазмида содержит фрагмент, содержащий TEM-1 ген и P-3 промотор.
Минимальная ингибирующая концентрация цефотаксима для E.coli XL1-блю (Stratagene, San Diego CA), содержащих эту плазмиду, составляет 0,02 мкг/мл после 24 часов при 37oC.
Способность повышать устойчивость беталактамазного гена к цефотаксиму без перетасовки определяют постадийным пересевом разбавленного пула клеток (примерно 107 к. о. е. ) на двукратно повышающихся уровнях лекарства. Это предоставляет 64-кратное усиление устойчивости.
2) Переваривание ДНКазой 1
Субстратом для первой реакции перетасовки служит дцДНК длиной 0,9 к.п.о. ,
полученная в результате ПЦР pUC18Sfi с праймерами C и D, причем оба содержат Sfi1 сайт.
Свободные праймеры из ПЦР продукта удаляют с помощью препарата Wisard ПЦР prep (Promega, Madison WI) на каждом цикле.
Около 5 мкг НК субстрата (субстратов) переваривают 0,15 ед. ДНКазы 1 (Sigma, St Louis MO) в 100 мкл 50 мМ Tris-HCl, pH 7,4, 1мМ MgCl2, в течение 10 минут при комнатной температуре, фрагменты длиной 100- 300 п.о. выделяют из 2% агарозного геля с низкой температурой плавления за счет электрофореза на ДЕ81 ионообменной бумаге (Whatman Hillsborough ОР), элюируя 1 М NaCl и осаждая этанолом способами, описанными в примере 1.
3) Перетасовка гена
Очищенные фрагменты снова суспендируют в ПЦР смеси (0,2 мМ
каждого из dNTR, 2,2 мМ MgCl2, 50 мМ KCl, 10 мМ Tris-HCl, pH 9,0, 0,1 Triton X-100) в концентрации 10-30 нг/мкл. В этот момент не добавляют никаких праймеров. Программа вторичной сборки:
94oC в течение 60 секунд, затем 40 циклов (94oC в течение 30 секунд, 50-55oC в течение 30 секунд, 72oC в течение 30 секунд), а затем 72oC в
течение 5 минут; ее используют на термоциклере MJ Pesearch (Watertоwn) PTC-150.
4) Амплификация продукта вторичной сборки с праймерами
После разбавления продукта вторичной
сборки в ПЦР смеси с 0,8 мкМ каждого из праймеров (C и D) и 20 ПЦР циклов (94oC 30 секунд, 50oC 30 секунд, 72oC 30 секунд) получают один продукт длиной 900 п.о.
5) Клонирование и анализ
После переваривания продукта 900 п.о. терминальным рестрикционным энзимом Sfi1 и очистки на агарозном геле, продукт 900 п.о. легируют в вектор pUC182 Sfi
по уникальному Sfi1 сайту за счет T4 ДНК лигазы (BRL, Caithersburg VМ). Полученную смесь электропорируют в E.coli XL1-блю клетки и высевают на LB пластины с 0,32-0,64 мкг/мл цефотаксима (Sigma, St.
Louis MO). Эти клетки выращивают вплоть до 24 часов при 37oC, и полученные колонии соскребают с пластины в виде пула, который используют как матрицу в ПЦР на следующем цикле перетасовки.
6) Последующие циклы вторичной сборки
Полученные после каждого из трех циклов перетасовки трансформанты высевают на повышающиеся уровни цефотаксима. Колонии (более 100 для
сохранения разнообразия) с пластины с наивысшим уровнем цефотаксима объединяют и используют в качестве матрицы в ПЦР реакции для следующего цикла.
Смесь колоний цефотаксим-устойчивых, полученных при 0,32-0,64 мкг/мл на стадии (5), используют в качестве матрицы для следующего цикла перетасовки. 10 мкл клеток в LB бульоне используют в качестве матрицы в программе вторичной сборки в течение 10 минут при 99oC, затем следуют 35 циклов (94oC 30 секунд, 52oC 30 секунд, 72oC 30 секунд), а затем 5 минут при 72oC, как указано ранее.
Продукты вторичной сборки переваривают и легируют в pUC182Sfi, как указано на стадии (5) ранее. Полученную смесь электропорируют в Е.coli XL1-блю клетки и высевают на LB пластины, содержащие 5-10 мкг/мл цефотаксима.
Колонии, полученные при 5-10 мкг/мл, используют для третьего цикла, аналогичного первому и второму циклам за исключением того, что клетки высевают на LB пластины, содержащие 80-160 мкг/мл цефотаксима. После третьего цикла получают колонии при 80-160 мкг/мл, и после вторичного высевания на повышающиеся концентрации цефотаксима, можно получить колонии на вплоть до 320 мкг/мл после 24 часов при 37oC (M1C = 320 мкг/мл).
Рост на цефотаксиме зависит от плотности клеток, требуя, чтобы все M1C были стандартизованы (в нашем случае до около 1000 клеток на пластину). При более высоких плотностях достигают роста на вплоть до 1280 мкг/мл. 5 самых крупных колоний, выросших при 1280 мкг/мл, высевают дважды для каждой колонии, и Sfi1 вставки анализируют за счет рестрикционного картирования продуктов колоний PCR.
Получают один мутант с 16000-кратным усилением устойчивости к цефотаксиму (M1C = 0,2 мкг/мл до M1C = 320 мкг/мл).
После отбора плазмиду выбранных колоний переносят обратно в дикого типа E.coli XL1-блю клетки (Stratagene, San Diego CA) для обеспечения того, чтобы ни одна из определениях устойчивостей к лекарству не была бы связана с хромосомной мутацией.
Три цикла перетасовки и отбора позволяют получить 1,6 • 104-кратное увеличение минимальной ингибирующей концентрации широкого спектра антибиотика цефотаксима для TEM-1 беталактамазы. Напротив, повторное высевание без перетасовки приводит лишь к 16-кратному увеличению устойчивости (подверженная ошибкам PCR или кассетный мутагенез).
7) Анализ последовательности
Все 5 из наиболее крупных колоний, выросших на 1280
мкг/мл, имеет рестрикционную карту, идентичную дикого типа TEM-1 энзима, Sfi1 вставка плазмиды, полученной из одной из этих колоний, была секвенирована за счет дидеокси секвенирования (United States
Biochemical Co. Cleveland OH) в соответствии с рекомендациями изготовителей. Все номера оснований соответствуют ревизованной pBR322 последовательности (29), и номера аминокислот соответствуют ABL
стандартной схеме нумерации (30). Аминокислоты обозначены по их трехбуквенному коду, а нуклеотиды - по их однобуквенному коду. Термин G4205A означает, что нуклеотид 4205 был изменен с гуанидина на
аденин.
Было обнаружено 9 замещений отдельных оснований. G4205A расположен между -35 и -10 сайтами беталактамазного P3 промотора (31). Промоторный ап-мутант, наблюдавшийся Chen and Clowes (31), расположен вне используемого здесь Sfi1 фрагмента и, таким образом, не мог быть детектирован. Четыре мутации оказались молчащими (A3689G, G3713A, G3934A и T3959A) и четыре сводятся к изменению в аминокислотах (C3448T приводит к Gly238Ser, A3615G приводит к Met182Thr, C3850T приводит к Glu104Lys и G4107A приводит к Ala18Val).
8) Молекулярный бэккросс
Молекулярный бэккросс с избытком дикого типа ДНК используют затем для исключения несущественных мутаций.
Молекулярный бэккроссинг проводят на выбранной плазмиде из трех циклов ДНК перетасовки по способу, идентичному нормальной перетасовке, как было указано ранее, за исключением того, что переваривание ДНКазой 1 и реакцию перетасовки ведут в присутствии 40-кратного избытка дикого типа фрагмента TEM-1 гена. Для того, чтобы сделать бэккросс более эффективным, в реакции перетасовки используют очень мелкие ДНК фрагменты (от 30 до 100 п.о.). Полученные в результате бэккроссинга мутанты снова отбирают на LB пластинах с 80-160 мкг/мл цефотаксима (Sigma St Louis MO).
Такую перетасовку бэккросса повторяют с ДНК из колоний первого цикла бэккроссинга в присутствии 40-кратного избытка дикого типа TEM-1 ДНК. Для повышения эффективности бэккроссинга используют мелкие ДНКфрагменты (30-100 п. о. ). Мутанты, полученные в результате второго цикла бэккроссинга, снова отбирают на LB пластинах с 80-180 мкг/мл цефотаксима.
Полученные трансформанты высевают на 160 мкг/мл цефотаксима, и пул колоний снова высевают на повышающиеся уровни цефотаксима, вплоть до 1280 мкг/мл. Самую большую из колоний, полученных на 1280 мкг/мл, снова высевают для получения отдельных колоний.
Этот мутант бэккроссинга оказывается в 32000 раз более устойчивым нежели дикого типа (МИК = 640 мкг/мл). Полученный мутантный штамм в 64 раза более устойчив к цефотаксиму, нежели полученные ранее клинические или сконструированные TEM-1- производные штаммы. Таким образом, очевидно, что ДНК перетасовка является быстрым и эффективным инструментом для, по крайней мере, нескольких циклов направленной молекулярной эволюции.
ДНК последовательность Sfi1 вставки мутанта, подвергнутого бэккроссингу, была определена с использованием набора для дидеоксиДНК секвенирования (United States Biochemical Co., Cleveland, OH) в соответствии с указаниями изготовителей (таблица 3). Этот мутант содержит 9 отдельных мутаций пар оснований. Как и ожидалось, все четыре из идентифицированных ранее молчащих мутаций исчезли, возвращаясь в последовательность дикого типа гена. Промоторная мутация (G4205A) так же, как и три из четырех мутаций аминокислот (Glu104Lys, Met182Thr и Gly238Ser) остались в клоне, подвергнутом бэккроссингу, что дает возможность предположить, что они существенны для высокого уровня устойчивости к цефотаксиму. Однако две новые молчащие мутации (T384C и A3767G) так же, как и три новые мутации, приведшие к изменению в аминокислотах, были обнаружены (C3441T превратилась в Arg241His, C3886T превратилась в Gly926Ser и G4035C превратилась в Ala42Gly). Хотя эти две молчащие мутации не влияют на первичную последовательность протеина, они могут повлиять на уровень экспрессии протеина (например, за счет структуры мРНК) и возможно даже на складчатость протеина (за счет изменения применения кодона и поэтому сайта паузы, который может участвовать в складчатости протеина).
Как подвергнутые бэккроссингу, так и не подвергнутые бэккроссингу мутанты содержат промоторную мутацию (которая сама по себе или в сочетании приводит к 2-3-кратному повышению уровня экспрессии), а также три общих изменения аминокислот (Glu104Lys, Met182Thr и Gly238Ser). Glu104Lys и Gly238Ser представляют собой мутации, которые присутствуют в нескольких устойчивых к цефотаксиму или другим производным TEM-1 (таблица 4).
9) Сравнение уровней экспрессии
Уровни
экспрессии беталактамазного гена в плазмиде дикого типа, в мутанте, подвергнутом бэккроссингу, и в мутанте, не подвергавшемся бэккроссингу, сравнивают, осуществляя электрофорез в SDS-полиакриламидном
геле (4-20%; NoVex, San Diego CA) периплазмических экстрактов, полученных за счет осмотического шока по способу Witholt В. (32).
Очищенную TEM-1 беталактамазу (Sigma St.Louis МО) используют в качестве стандарта молекулярного веса, и клетки Е. coli. XL1-блю без плазмиды используют в качестве негативного контроля.
Мутант и подвергнутый бэккроссингу мутант, по-видимому, продуцируют уровни беталактамазного протеина, в 2-3 раза превышающие продуцирование дикого типа геном. Промоторная мутация, по-видимому, приводит к 2-3-кратному увеличению беталактамазы.
Пример 6. Конструирование мутантных комбинаций TEM-1 беталактамазного гена
Для определения устойчивости различных комбинаций мутаций и для сравнения новых
мутантов с уже опубликованными мутантами, были сконструированы несколько мутантов в идентичном плазмидном фоне. Два из этих мутантов Gly104Lys и Gly238Ser известны как мутанты цефотаксима. Все
мутантные сконструированные комбинации содержат промоторную мутацию, чтобы можно было осуществить сравнение с выбранными мутантами. Полученные результаты приведены в таблице 4.
Специфические комбинации мутаций были введены в дикого типа pUC182Sfi за счет ПЦР, с использованием двух олигонуклеотидов на мутацию.
Для получения следующих мутаций были использованы олигонуклеотиды (приведены в конце описания).
Эти отдельные ПЦР фрагменты были отделены с помощью геля от синтетических олигонуклеотидов. 10 нг каждого из фрагментов объединяют, и осуществляют реакцию вторичной сборки при 94oC в течение 1 минуты, а затем 25 циклов (94oC 30 секунд, 50oC 30 секунд и 72oC 45 секунд). ПЦР осуществляют на продукте вторичной сборки для 25 циклов в присутствии Sfi1-содержащих внешних праймеров (праймеры C и D из примера 5). ДНК переваривают за счет Sfi1 и встраивают в дикого типа pUC182Sfi вектор. Получают комбинации мутантов, приведенные в табл. 4.
Был сделан вывод, что консервативные мутации составляют 9 из 15 удвоений в МИК.
Было показано, что Glu104Lys один приводит только к удвоению МИК до 0,08 мкг/мл, а Gly238Ser (в нескольких контекстах с одним дополнительным изменением в аминокислотах) приводит только в МИК 0,16 мкг/мл (26). МИК для двойного мутанта Glu104Lys/Gly238Ser составляет 10 мкг/мл. Этот мутант соответствует TEM-15.
Те же самые Glu104Lys и Gly238Ser мутации в сочетании с Glu39Lys/TEM-3/ или Thr263Met(TEM-4) приводят к более высокому уровню устойчивости (2-32 мкг/мл для TEM-3 и 8-32 мкг/мл для TEM-4 (34, 35).
Мутант, содержащий три изменения в аминокислотах, которые остались сохраненными после бэккроссинга (Glu104Lys Met-182Thr/Gly238Ser), также имеют МИС порядка 10 мкг/мл. Это означает, что мутации, которые каждый из вновь отобранных мутантов содержит помимо трех известных мутаций, и отвечают за дальнейшее повышение (32-64-кратное) устойчивости гена к цефотаксиму.
Нативные, клинические TEM-1-полученные энзимы (TEM-1-19), каждый содержат различные комбинации только 5-7 идентичных мутаций. Так как эти мутации находятся в хорошо разделенных положениях гена, мутант с наивысшей устойчивостью к цефотаксиму не может быть получен за счет кассетного мутагенеза отдельного участка. Это может объяснить, почему максимальные значения M1C, которые были получены с помощью стандартного кассетного мутагенеза, достигали лишь величины 0,64 мкг/мл (26). Так, например, как Glu104Lys, так и Gly238Ser мутации, как было обнаружено отдельно в этом исследовании, имеют МИК ниже 0,16 мкг/мл. Использование ДНК перетасовки позволяет осуществить комбинацию, и таким образом, Glu104Lys/Gly238Ser комбинация, как было обнаружено, имеет значение МИК = 10 мкг/мл.
Важным ограничением этого примера является использование в качестве исходной точки одного гена. Считают, что наилучшие комбинации можно получить, если перетасовывать большое количество родственных нативно-встречающихся генов. Разнообразие, которое присутствует в такой смеси, имеет большее значение, чем неупорядоченные мутации, которые возникают за счет мутагенной перетасовки. Так, например, считают, что можно использовать репертуар родственных генов из одного вида, например, как существующее заранее разнообразие иммунной системы, или родственных генов, полученных из множества различных видов.
Пример 7. Усовершенствование антитела A10B за счет ДНК перетасовки библиотеки из всех шести мутантных CDR.
A10B ScFv антитело, мышиный анти-кроличий IgG было подарком от Pharmacia /Milwaukee W1/. Используют коммерчески доступную систему представления фага от Pkarmacia, в которой используется pCANTAB5 вектор представления фага.
Исходное A10B антитело имеет лишь слабую воспроизводимость, так как были получены клоны, которые только слабо связываются с иммобилизованным антигеном (кроличий IgG) (по данным ELISA фага аналитический набор Pharmacia или по титру фага). Концентрации кроличьего IgG, которые приводят к 50% ингибирования A10B связывания антитела в конкурирующем анализе, составили 13 пикомолей. Наблюдаемая низкая способность возможно также была связана с нестабильностью A10B клона.
Была секвенирована ДНК A10B ScFv (United States Biochemical Co., Cleveland OH) в соответствии с рекомендациями заявителей. Последовательность оказалась аналогичной существующим антителам, на основании сравнения с данными Kabat(33).
1) Получение фаговой ДНК
Фаговую ДНК, содержащую ген A10B дикого типа антитела (10 мкл), инкубируют при 99oC в течение 10 минут, затем 72oC в течение 2 минут. Смесь ПЦР (50 мМ KCl, 10 мМ Tris-HCl, pH 9,0, 0,1% Triton X-100, 200 мкМ каждой dNTP, 1,9 мМ MgCl2), 0,6 мкм каждого праймера и 0,5 мкл Tag полимеразы (Promega,
Madison WI) добавляют к фаговой ДНК. ПЦР программа состоит из 33 циклов (30 секунд при 94oC, 30 секунд при 45oC, 45 секунд при 72oC).
Используют
следующие праймеры:
5'ATGATTACGCCAAGCTTT 3' (послед. 1Д N 26) и
5'TTGTCGTCTTTCCAGACGTT 3' (послед. 1Д N 27).
Затем продукт PCR, из 850 п.о. обрабатывают электрофоретически и выделяют из 2% агарозного геля с низкой температурой плавления.
2) Фрагментация
300 нг геля, выделенного из полосы 850 п.о., переваривают с 0,18
единицами ДНКазы 1 (Sigma) в 50 мМ Tris-HCl, pH 7,5, 10 мМ MgCl2 в течение 20 минут при комнагной температуре. Переваренную ДНК выделяют на 2% агарозном геле с низкой температурой плавления,
и вырезают из геля полосы между 50 и 200 п.о.
3) Конструирование тестовой библиотеки
Целью настоящего изобретения является проверка того, является ли эффективной вставка
CDR.
Синтезируют следующие CDR последовательности, содержащие внутренние сайты рестрикционных энзимов. "CDRH" означает CDR в тяжелой цепи, a "CDRL" означает CDR в легкой цепи антитела.
CDR олигомеры с рестрикционными сайтами приведены в конце описания.
СDR олигомеры добавляют к очищенным фрагментам ДНК A10B антитела (от 50 до 200 п.о.) со стадии (2) в 10-кратном молярном избытке. ПЦР смесь (50 мМ KCl, 10 мМ Tris-HCl, pH 9,0, 0,1% Triton X-100, 1,9 мМ MgCl2, 200 мкМ каждого dNTP, 0,3 мкл Taq полимеразы (Promega), 50 мкл полный объем) добавляют, и проводят программу перетасовки: 94oC 1 минуту, 1 минуту при 72oC, а затем 75 циклов: 30 секунд при 94oC, 30 секунд при 55oC, 30 секунд при 72oC.
2 мкл перетасованной смеси добавляют к 100 мкл смеси ПЦР (50 мМ KCl, 10 мМ Tris-HCl, pH 9,0, 0,1% Triton X-100, 200 мкМ каждого из dNTR, 1,9 мМ MgCl2, 0,6 мкМ каждого из двух внешних праймеров (послед. 1Д N 26 и 27 см. далее), 0,5 мкл Tag ДНК полимеразы) и осуществляют программу PCR в течение 30 циклов (30 секунд при 94oC, 30 секунд при 45oC, 45 секунд при 72oC). Полученную смесь ДНК фрагментов размером 850 пар оснований экстрагируют смесью фенол/хлороформ и осаждают этанолом.
В
качестве внешних праймеров используют:
Внешний праймер 1: последовательность 1Д N 27
5'TTGTCGTCTTTCCAGACAGCTT 3'
Внешний праймер 2: последовательность 1Д N 26
5'ATGATTACGCCAAGCTTT 3'
Продукт ПЦР 850 п.о. переваривают рестрикционными энзимами Sfi1 и Not 1, выделяют из агарозного геля с низкой температурой плавления и легируют в вектор экспрессии
pCANTAB5, полученный от Pharmacia, Milwaukee W1. Легированный вектор подвергают электропорации по способу Invitrogen (San Diego CA) в TG1 клетки (Pharmacia) и высевают для получения отдельных
колоний.
ДНК из полученных колоний добавляют к 100 мкл ПЦР смеси (50 мМ KCl, 10 мМ Tris-HCl, pH 9,0, 0,1% Triton X-100, 200 мкМ каждой dNTR, 1,9 мМ MgCl2, 0,6 мкМ внешнего праймера ID N 27, см. далее), шесть внутренних праймеров (последоват. 1Д N 40-45, см. далее и 0,5 мкл Tag ДНК полимеразы) и ПЦР программу осуществляют в 35 циклов (30 секунд при 94oC, 30 секунд при 45oC, 45 секунд при 72oC). Размеры ПЦР продуктов определяют с помощью электрофореза на агарозном геле и используют для определения того, какой из CDR с рестрикционными сайтами был встроен.
CDR внутренних праймеров:
H1 (последовательность 1Д N 40) 5'AGAATTCATCTAGATTTG 3',
H2 (последовательность 1Д N 41) 5'GCTTATCCTTTATCTCAGGTC 3',
H3 (последовательность 1Д N 42) 5'ACTGCAGTCTTATACGAGGAT 3',
L1 (последовательность 1Д N 43) 5'GACGTCTTTAAGCGATCG 3',
L2 (последовательность 1Д N 44) 5'TAAGGGAGATCTAAACAG 3',
L3 (последовательность 1Д N 45 5'TCTGCGCGCTTAAAGGAT 3'.
Шесть синтетических CDR встраивают в ожидаемые положения дикого типа ДНК A10B антитела (фиг. 7). Эти исследования показывают, что, хотя каждый из шести CDR в специфическом клоне имеет маленький шанс быть CDR с рестрикционным сайтом, большинство из клонов содержат, по крайней мере, один CDR с рестрикционным сайтом и что были созданы все возможные комбинации CDR с рестрикционными сайтами.
4) Конструирование мутантных участков, определяющих комплементарность ("CDR")
На основании наших
данных о последовательности сконструированы шесть олигонуклеотидов, соответствующих этим шести CDR. CDR (определение Kabat) подвергают синтетическому мутагенезу в отношении 70 (существующих
оснований): 10: 10:10 и фланкируют с 5' и 3' сторон примерно 20 основаниями фланкирующих последовательностей, которые обеспечивают гомологичность для включения CDR при смешивании со смесью генных
фрагментов немутированного антитела в молярном избытке. Полученные мутантные последовательности приведены далее.
Олигомеры для CDR библиотеки даны в конце описания.
Выделенные жирным шрифтом и подчеркнутые последовательности были мутантными последовательностями, синтезированными с использованием смеси нуклеозидов 70:10:10:10, в которых 70% были дикого типа нуклеозидами.
10-кратный молярный избыток CDR мутантных олигонуклеотидов добавляют к очищенным фрагментам ДНК A10B антитела длиной от 50 до 200 п.о. со стадии (2). PCR смесь (50 мМ KCl, 10 мМ Tris-HCl, pH 9,0, 0,1% Triton X-100, 1,9 мМ MgCl2, 200 мкМ каждого из dNTP, 0,3 мкл Tag ДНК полимеразы (Promega), 50 мкл полный объем) добавляют и осуществляют программу перетасовки в течение 1 минуты при 94oC, 1 минуты при 72oC, а затем 35 циклов: (30 секунд при 94oC, 30 секунд при 55oC, 30 секунд при 72oC).
1 мкл перетасованной смеси добавляют к 100 мкл ПЦР смеси (50 мМ KCl, 10 мМ Tris-HCl, рН 9,0, 0,1% Triton X-100, 200 мкМ каждого dNTP, 1,9 мМ MgCl2, 0,6 мкМ каждого из двух внешних праймеров (последовательность 1Д N 26 и N 27, 0,5 мкл TagДНК полимеразы) и осуществляют программу ПЦР в течение 30 циклов (30 секунд при 94oC, 30 секунд при 45oC, 45 секунд при 72oC). Полученную смесь ДНК фрагментов размером в 850 пар оснований экстрагируют смесью фенол/хлороформ и осаждают этанолом.
В качестве внешних праймеров
используют:
внешний праймер 1: последовательность 1Д N 27:
5'TTGTCGTCTTTCCAGACGTT 3'
внешний праймер 2: последовательность 1Д N 26:
5'ATGATTACGCCAAGCTTT 3'
5) Клонирование ДНК ScFv антитела в pCANTAB5
Продукт ПЦР 850 п.о. переваривают рестрикционными энзимами Sfi1 и Not1, выделяют из агарозного геля с низкой температурой плавления и легируют в
вектор экспрессии pCANTAB5, полученный от Pharmacia Milwaucee WI. Легированный вектор электропорируют в соответствии с изложенным далее способом Invitrogene (San Diego CA) в TG1 клетки (Pharmacia), и
фаговую библиотеку выращивают, используя хелперный фаг в соответствии с рекомендациями изготовителя.
Затем созданную таким образом библиотеку скринируют на наличие усовершенствованных антител, используя шесть циклов селекции.
6) Селекция высокоафинных клонов
15 ячеек микротитровальной платины с 96 ячейками покрывают кроличьим IgG (Jackson Immunoresearch,
Bar Harbor ME) при 10 мкг/ячейку в течение 1 часа при 37oC, а затем блокируют 2% обезжиренным сухим молоком в PBS в течение 1 часа при 37oC.
100 мкл фаговой библиотеки (1•1010 КОЕ) блокируют 100 мкл 2% молока в течение 30 минут при комнатной температуре, а затем добавляют в каждую из 15 ячеек и инкубируют в течение 1 часа при 37oC.
Затем ячейки трижды промывают PBS, содержащим 0,5% Tween-20 при 37oC в течение 10 минут на промывку. Связанный фаг элюируют 100 мкл элюирующего буфера (Glycine-HCl, pH 2,2), с последующей немедленной нейтрализацией 2М Tris pH 7,4 и трансфекцией для получения фага. Такой цикл селекции повторяют шесть раз.
После шестого цикла отдельные клоны фага отбирают и сравнивают относительно афинности за счет ELISA фага, и специфичность к IqG кролика анализируют набором от Pharmacia (Milwakee W1), в соответствии с рекомендациями изготовителей.
Наилучший клон содержит приблизительно 100-кратно улучшенный уровень экспрессии по сравнению с дикого типа A10B при тестировании в Вестернблоттинге. Концентрацию кроличьего IgG, которые приводят к 50% ингибирования в конкурирующем анализе с наилучшим клоном, составляет 1 пикомоль. Наилучший клон воспроизводимо специфичен кроличьему антигену. Число копий антител, представляемых фагом, по-видимому, можно увеличить.
Пример 8. Ин виво рекомбинации за счет прямых повторов частичных генов
Плазмиду конструируют из двух частичных
неактивных копий одного и того же гена (беталактамазы) для того, чтобы продемонстрировать, что рекомбинация между общими участками двух прямых повторов приводит к получению полной длины активных
рекомбинантных генов.
Производное pUC18, содержащее бактериальный TEM-1 беталактамазный ген, используют в этой конструкции (Vanich-Perron et al., 1985, Gene 33:103-119). TEM-1 беталактамазный ген ("Bla") придает устойчивость бактериям против примерно 0,02 мкг/мл цефотаксима. Sfi1 рестрикционные сайты добавляют в направлении 5' от промотора и в направлении 3' конца беталактамазного гена за счет ПЦР векторной последовательности с двумя праймерами (дано в конце описания).
Эти два продукта реакции переваривают за счет Sfi1, смешивают, легируют и используют для трансформации компетентных E.coli бактерий описанным далее способом. Полученная плазмида оказалась pUC182Sfi-Bla-Sfi. Эта плазмида содержит Sfi1 фрагмент, содержащий Bla ген и P-3 промотор.
Минимальная ингибирующая концентрация цефотаксима для E.coli XLl-блю (Stratagene), содержащей pUC182Sfi-Bla-Sfi, оказалась 0,02 мкг/мл после 24 ч при 37oC.
Ген устойчивости к тетрациклину pBR322 клонируют в pUC18 Sfi-Bla-Sfi, используя гомологичные участки, в результате чего получают pBR322TetSfi-Bla-Sfi. Затем TEM-1 ген исключают за счет рестрикционного переваривания pBR322Tet Sfi-Bla-Sfi за счет SspI и FspI и легирования с тупыми концами, в результате чего получают pUC322TetSfi-Sfi.
Перекрывающиеся участки ТЕМ-1 гена
амплифицируют, используя стандартную методику ПЦР и следующие праймеры:
Праймер 2650 (последовательность 1Д N 50):
5'TTCTTAGACGTCAGGTGGCACTT 3'
Праймер 2439
(последовательность 1Д N 51):
5'TTTTAAATCAATCTAAAGTAT 3'
Праймер 2651 (последовательность 1Д N 52):
5'TGCTCATCCACGAGTGTGGAGAAGTGGTCCTGCAACTTTAT 3' и
Праймер 2652
(последовательность 1Д N 53)
ACCACTTCTCCACACTCGTGGATGAGCACTTTTAAGTT
Два полученных ДНК фрагмента переваривают за счет Sfi1 и BstX1 и легируют в Sfi сайт pBR322TetSfi-Sfi. Полученную
плазмиду называют pBR322Sfi-BL-LA-Sfi. Карта плазмиды, а также схема рекомбинаций внутри плазмиды и реконструкции функциональной беталактамазы представлена на фиг. 9.
Эту плазмиду электропорируют либо в TG1, либо в JC8679 E.coli клетки. E. coli JC8679 является RecBC sbcA (Oliher et al., 1993, NaR 21:5192). Эти клетки высевают на твердые агарные пластины, содержащие тетрациклин. Те колонии, которые растут, высевают затем на твердые агарозные пластины, содержащие 100 мкг/мл ампициллина, и подсчитывают количество жизнеспособных колоний. Беталактамазный ген встроен в те трансформанты, которые демонстрируют устойчивость к ампициллину; их амплифицируют с помощью стандартной методики ПЦР, используя праймер 2650 (последовательность 1Д N 50) 5'TTCTTAGACGTCAGGTGGCACTT3' и праймер 2493 (последовательность 1Д N 51) 5'TTTTAAATCAATCTAAAGTAT 3' и определяют длину вставки. Наличие вставки 1 к. п.о. указывает, что ген был успешно рекомбинирован, как представлено на фиг. 9 и в таблице 5.
Около 17-25% устойчивых к тетрациклину колоний оказались также устойчивыми к ампициллину, и все устойчивые к ампициллину колонии были соответствующим образом рекомбинированы при определении ПЦР колоний. Поэтому частичные гены, расположенные на одной и той же плазмиде, могут успешно рекомбинировать с созданием функционального гена.
Пример
9. Ин виво рекомбинация за счет прямых повторов генов полной длины
Конструируют плазмиду с двумя полной длины копиями различных аллелей беталактамазного гена. Гомологичная рекомбинация двух
генов приводит к отдельной рекомбинантной полной длины копии этого гена.
Конструирование pBR322TetSfi-Sfi и pBR322TetSfi-Bla-Sfi было описано ранее.
Две аллели
беталактамазного гена конструируют следующим образом. Проводят две ПЦР реакции и pUC18Sfi-Bla-Sfi в качестве матрицы. Одну реакцию ведут со следующими праймерами:
Праймер 2650
(последовательность 1Д N 50):
5'TTCTTAGACGTCAGGTGGCACTT 3'
Праймер 2649 (последовательность 1Д N 51)
5'ATGGTAGTCCACGAGTGTGGTAGTGACAGGCCGGTCTGACAGTTACCAATGCTT 3'
Вторую ПЦР реакцию ведут со следующими праймерами:
Праймер 2648 (последовательность 1Д N 54):
5'TGTCACTACCACACTCGTGGACTACCATGGCCTAAATACATTCAAATATGTAT 3'
Праймер 2493
(последовательность 1Д N 51)
5'TTTTAAATCAATCATAAGTAT 3'
В результате получают два Bla гена, один с 5' Sfi1 сайтом и с 3' BstX1 сайтом, а другой - с 5' BstX1 сайтом и 3' Sfi1
сайтом.
После переваривания этих двух генов за счет BstX1 и Sfi1 и легирования в Sfi1-переваренную плазмиду pBR322TetSfi-Sfi, получают плазмиду (pBR322-Sfi-2BLA-Sfi) с тандемным повтором Bla гена (см. фиг. 10).
Эту плазмиду электропорируют в Е.coli клетки. Эти клетки высевают на твердые агарные пластины, содержащие 15 мкг/мл тетрациклина. Затем растущие колонии высевают на твердые агарные пластины, содержащие 100 мкг/мл ампициллина, и подсчитывают количество жизнеспособных колоний. Bla вставки в тех трансформантах, которые демонстрируют устойчивость к ампициллину, амплифицируют за счет стандартной методики ПЦР, используя способ и праймеры примера 8. Наличие вставки 1 к.п.о. указывает на то, что дубликатные гены были рекомбинированы, что видно из таблицы 6.
Праймеры, которые использовали в двух различных ПЦР реакциях для создания двух плазмид Bla генов, были праймером 2650 (последовательность 1Д N 50) и праймером 2649 (последовательность 1Д N 51) для первого гена и праймерами 2648 (последовательность 1Д N 54) и праймером 2719 (последовательность 1Д N 55) для второго гена. Это приводит к смешанной популяции каждого из двух амплифицированных ДНК фрагментов: фрагмент N 89 (амплифицированный с праймерами 2648 и 2719) и фрагмент N 90 (амплифицированный с праймерами 2650 и 2649). В каждом случае число различных мутаций было введено в смешанную популяцию каждого из фрагментов.
После подверженной ошибкам ПЦР популяцию амплифицированного ДНК фрагмента N 59 переваривают за счет Sfi1, а затем клонируют в pBR322Tet Sfi-Sfi для создания смешанной популяции плазмиды pBR322Sfi-Bla-Sfi1.
После подверженной ошибкам ПЦР популяцию амплифицированных фрагментов N 90 и N89 переваривают Sfi1 и BstX1 при 50oC и легируют в pBR322Tet Sfi-Sfi для создания смешанной популяции плазмиды pBR322Tet Sfi-2Bla-Sfi1 (фиг. 10).
Плазмиды pBR322Sfi-Bla-Sfi1 и pBR322Sfi-2Bla-Sfi1 электропорируют в E. coli JC8679 и высевают на агарные пластины, содержащие различные концентрации цефотаксима для отбора устойчивых штаммов и на пластины с тетрациклином для титрования.
Равное количество колоний (в расчете на число колоний, выросших на тетрациклине) отбирают, выращивают на LB-tet, и ДНК экстрагируют из этих колоний. Это один цикл рекомбинации. Такие ДНК переваривают Ecr1 и используют для второго цикла подверженной ошибкам ПЦР, как указано ранее.
После пяти циклов МИК (минимальная ингибирующая концентрация) для цефотаксима для плазмиды из одного фрагмента составляет 0,32, тогда как МИК для плазмиды из двух фрагментов составляет 1,28. Полученный результат показывает, что после пяти циклов устойчивость, достигнутая за счет рекомбинации, в четыре раза выше в присутствии ин виво рекомбинации.
Пример 11. Ин виво рекомбинации за счет электропорации
фрагментов
Компетентные клетки Е.coli, содержащие pUC18Sfi-Bla-Sfi, получают описанным ранее способом. Плазмида pUC18Sfi-Bla-Sfi содержит стандартный TEM-1 беталактамазный ген, как указано
ранее.
TEM-1 полученный ген устойчивости к цефотаксиму из pUC18Sfi-cef-Sfi (клон ST2) (Stemmer WPC (1994) Nature 370:389-91, включено сюда по ссылке), который придает на Е.coli. содержащей плазмиду МИК в 640 мкг/мл для цефотаксима, получают в одном эксперименте полную плазмиду ДНК pUC18Sfi-cef-Sfi, электропорируют в клетки E.coli, содержащие плазмиду pUC18Sfi-Bla-Sfi.
В другом эксперименте ДНК фрагмент, содержащий ген цефотаксима из pUC18Sfi-cef-Sfi, амплифицируют за счет ПЦР, используя праймеры 2650 (последовательность 1Д N 50) и 2719 (последовательность 1Д N 55). Полученный ПЦР продукт длиной 1 к.п.о. переваривают на ДНК фрагменты менее 100 п.о. с помощью ДНКазы, и эти фрагменты электропорируют в компетентные E.coli клетки, которые уже содержат pUC18Sfi-Bla-Sfi.
Трансформированные клетки из обоих экспериментов затем анализируют по их устойчивости к цефотаксиму, высевая эти трансформированные клетки на агарные пластины с различными концетрациями цефотаксима. Полученные результаты представлены в таблице 6.
Из этих результатов становится очевидным, что полный ST-2Cef ген был выстроен либо в бактериальный геном, либо в плазмиду после электропорации. Так как большинство вставок гомологично, ожидается, что ген был встроен в плазмиду, заменяя ген дикого типа. Фрагменты Cef гена из ST-2 также эффективно встроены в дикого типа ген в плазмиде. Не наблюдается резкого повышения устойчивости к цефотаксиму при введении дикого типа гена (либо целиком, либо его фрагментов) и без ДНК. Поэтому ST-2 фрагменты, как было показано, придают гораздо более высокую устойчивость к цефотаксиму, нежели фрагменты дикого типа. Предполагается, что повторное встраивание фрагментов, полученных из генных пулов с повышенной устойчивостью, может привести к увеличению устойчивости.
Соответственно, те колонии, которые продуцируют повышенную устойчивость к цефотаксиму с ST-2 генными фрагментами, выделяют и экстрагируют плазмидные ДНК. Эти ДНК амплифицируют, используя ПЦР описанным ранее способом. Амплифицированную ДНК переваривают ДНКазой до фрагментов (менее 100 п.о.) и 2- 4 мкг фрагментов электропорируют в компетентные клетки E.coli, уже содержащие pUC322Sfi-Bla-Sfi, как указано ранее. Трансформированные клетки высевают на агар, содержащий различные концентрации цефотаксима.
В качестве контроля компетентные клетки E.coli, содержащие плазмиду pUC18Sfi-Kan-Sfi, также используют. ДНК фрагменты из переваров ПЦР продуктов pUC18Sfi-сef-Sfi электропорируют в эти клетки. Не существует гомологичности между геном канамицина и беталактамазным геном, и поэтому рекомбинация не должна происходить.
Этот эксперимент повторяют в течение 2 циклов, и полученные результаты представлены в таблице 7.
Пример 12. Определение форматов рекомбинаций
Этот эксперимент был создан для определения того, какой
формат рекомбинации создает наибольшее число рекомбинантов за цикл.
В первом подходе вектор pUC18Sfi-Bla-Sfi амплифицируют с ПЦР праймерами для создания крупного и мелкого фрагмента. Крупный фрагмент содержит плазмиду и концы, содержащие части Bla гена, а мелкий фрагмент кодирует середину Bla гена. Третий фрагмент, содержащий полный Bla ген, создают, используя PCR по способу примера 8. Более крупный плазмидный фрагмент и фрагмент, содержащий полный Bla ген, электропорируют в E.coli JC8679 клетки в то время описанным ранее способом, и полученные трансформанты высевают на пластины с различными концентрациями цефотаксима.
В подходе 2 вектор pUC18Sfi-Bla-Sfi амплифицируют для получения крупного плазмидного фрагмента, выделенного в подходе 1 ранее. Эти два фрагмента, каждый из которых содержит часть полного Bla гена, например, два фрагмента, которые вместе образуют Bla ген, которые также получают в ПЦР. Крупный плазмидный фрагмент и два Bla генных фрагмента все электропорируют в компетентные E.coli JC8679 клетки, и трансформанты высевают на пластины с различными концентрациями цефотаксима.
В третьем подходе оба вектора и плазмиду электропорируют в клетки Е. coli JC8679, а полученные трансформанты высевают на пластины с различными концентрациями цефотаксима.
В четвертом подходе полный Bla ген электропорируют в клетки E.coli JC8679, уже содержащие вектор pUCSfi-Sfi, и полученные трансформанты высевают на пластины с различными концентрациями цефотаксима. В качестве контроля клетки Е. coli JC8679 электропорируют либо полным Bla геном, либо только вектором.
Полученные результаты представлены на фиг. 11. Эффективность встраивания двух фрагментов в вектор составляет 100 X, что меньше, чем если используют один фрагмент, содержащий полный Bla ген. Подход 3 показывает, что эффективность встраивания не зависит от наличия свободных концов ДНК, так как при этом подходе не получают рекомбинантов. Однако результат подхода 3 связан также с низкой эффективностью электропорации вектора. Если вектор экспрессии уже находится в компетентных клетках, эффективность векторной электропорации уже не является фактором, оказывающим влияние, и эффективной гомологической рекомбинации можно добиться даже с неразрезанным вектором.
Пример 13. Набор для кассетной
перетасовки для оптимизации векторных характеристик
Для получения вектора, способного придавать оптимизированный фенотип (т. е. максимальную экспрессию кодирующей вектор последовательности,
например, как у клонированного гена), предложен набор, содержащий различные кассеты, которые можно перетасовывать, и оптимальные продукты перетасовки можно было бы селектировать. На фиг. 12
схематически представлен один вариант, в котором каждый локус содержит множество кассет. Так, например, в системе бактериальной экспрессии фиг. 13 демонстрирует пример кассет, которые используют в
соответствующем локусе. Каждая кассета данного локуса (например, все промоторы в этом примере) фланкированы практически идентичными последовательностями, способными перекрывать фланкирующие
последовательности кассет прилежащего локуса и предпочтительно также способными участвовать в гомологической рекомбинации или в негомологической рекомбинации (например, lox/cre или flp/frt системы), с
тем, чтобы избежать перетасовки кассет внутри локуса, но практически не между локусами.
Кассеты поставляют в наборе как ПЦР фрагменты, причем каждый тип кассеты или отдельные виды кассет упакованы в отдельные ампулы. Векторные библиотеки создают, объединяя содержимое ампул для сборки целых плазмид или их существенных частей за счет гибридизации перекрывающихся фланкирующих последовательностей кассет в каждом локусе с кассетами соседнего локуса. Собранный вектор легируют с заранее определенным геном, представляющим интерес, для получения векторной библиотеки, в которой каждый член библиотеки содержит заранее определенный ген, представляющий интерес, и комбинацию кассет, определяемую ассоциацией кассет. Эти векторы переносят в клетки подходящего хозяина, и эти клетки культивируют в условиях, подходящих для экспрессии, и отбирают нужный фенотип.
Хотя настоящее изобретение было описано со ссылкой на предпочтительный его вариант, следует понимать, что изобретение не ограничено раскрытыми примерами. Напротив, настоящее изобретение охватывает различные модификации и эквивалентные перестройки, которые включены в объем и дух прилагаемой формулы изобретения.
Все публикации, патенты и заявки на патент включены здесь ссылкой в полном объеме, как если бы каждая отдельная публикация, патент или заявка на патент была бы специально и отдельно указана с целью включения с помощью ссылки в полном их объеме.
Реферат
Изобретение относится к генной инженерии и может быть использовано для получения полинуклеотидов, обладающих нужными свойствами. Матричный двухцепочечный полинуклеотид расщепляют на двухцепочечные неупорядоченные фрагменты. К полученной популяции добавляют олигонуклеотиды, содержащие участок идентичности и гетерологичности с матричным полинуклеотидом с последующим денатурированием до одноцепочечных фрагментов и инкубированием в присутствии полимеразы в условиях, приводящих к образованию мутантных двухцепочечных полинуклеотидов. Химерный полинуклеотид получают путем неупорядоченной фрагментации различных матричных полинуклеотидов, содержащих участки идентичности и гетерологичности, с последующим денатурированием и инкубированием с полимеразой. Изобретение позволяет за счет неупорядоченной рекомбинации разработать способы управления молекулярной эволюции сложных линейных последовательностей. 2 с и 17 з.п.ф-лы, 7 табл., 13 ил.

Комментарии