Карбамиды для разделения урана (vi) и плутония (iv) без восстановления плутония (iv) - RU2782220C2
Код документа: RU2782220C2
Чертежи

Описание
Область техники
Изобретение относится к области обработки отработавшего ядерного топлива.
Точнее изобретение относится к применению карбамидов (т.е. производных мочевины) в качестве экстрагентов для полного или частичного отделения урана (VI) от плутония (IV) на основании водного раствора, полученного растворением отработавшего ядерного топлива в азотной кислоте, посредством жидкофазной экстракции без применения какого-либо восстановления плутония (IV) в плутоний (III). Оно также относится к новым карбамидам с чрезвычайно полезными экстрагирующими свойствами.
В частности, изобретение находит применение при обработке отработавшего ядерного топлива на основе урана (в частности, оксидов урана – UOX) или урана и плутония (в частности, смешанных оксидов урана и плутония - MOX).
Предшествующий уровень техники
В способе PUREX, применяемом в мире на всех заводах по обработке отработавшего ядерного топлива (Аг во Франции, Роккашо в Японии, Селлафилд в Англии и пр.), используется три-n-бутил-фосфат (или TBP) в качестве экстрагента для извлечения урана (VI) и плутония (IV) жидкофазной экстракцией из водных растворов, полученных растворением этих видов ядерного топлива в азотной кислоте.
В данном способе ТВР применяется в виде 30%-ного раствора (в объёмном отношении) в алифатическом растворителе (гидрогенизированном тетрапропилене (ТРН), n-додекане или керосине). В данной области этот органический раствор называют обычно растворителем.
Регенерация урана и плутония способом PIREX проводится в несколько циклов:
- первый цикл: очистка урана (VI) и плутония IV) (называется «1CUPu») с целью дезактивировать эти элементы по отношению к америцию, кюрию и продуктам деления с разделением урана (VI) и плутония (IV) на два водных потока, начиная с этого первого цикла;
- второй цикл: очистка урана (VI) (обозначаемого как ZCU) с целью завершения дезактивации этого элемента для соблюдения технических условий, оговоренных в стандартах ASTM по урану, как целевому продукту, и
- второй цикл, а на некоторых заводах это третий цикл: очистка плутония (IV) (обозначаемого как соответственно «2CPu» и «3CPu») с целью завершения дезактивации этого элемента для соблюдения технических условий, оговоренных в стандартах ASTM по плутонию, как целевому продукту, и его обогащения до превращения в оксид PuO2.
Параметры процесса PUREX являются удовлетворительными и обращение к опыту, накопленному с момента пуска заводов, на которых этот способ применяется, положителен.
Однако применение три-n-бутил-фосфата (TBP) характеризуется ограничениями, лишающими возможности достичь с помощью этого экстрагента таких целей, как простота, компактность и повышенная безопасность, предназначенных для будущих заводов по обработке отработавшего ядерного топлива.
Главное из этих ограничений состоит в том, что разделение урана и плутония на два водных потока делает необходимым восстановление плутония (IV) в плутоний (III) (поскольку при использовании ТВР коэффициент разделения урана (VI) и плутония (IV) недостаточен независимо от кислотности водного раствора, применяемого для такого разделения) и, следовательно, применение восстанавливающих и противоазотистых агентов в значительных количествах, которые образуют вследствие разложения неустойчивые и реакционноспособные продукты, требующие соответствующие меры безопасности.
Снабжение экстрагентами для количественной соэкстракции урана и плутония из водного раствора, полученного растворением отработавшего ядерного топлива в азотной кислоте, с последующим полным или частичным разделением этих обоих элементов без необходимости восстановление плутония (IV) в плутоний (III), послужило поводом для проведения некоторого количества работ.
Таким образом было предложено применять симметричные N,N-диалкиламиды, такие, как N,N-ди(2-этилгексил)-3,3-диметилбутанамид (или DEHDMBA) в международной заявке PCT WO 2017/017207, далее [Д1], а также асимметричные N,N-диалкиламиды, такие, как N-метил-N-октил-2-этилгексанамид (или MOEHA) в международной заявке PCT WO 2017/017193, далее отсылка [Д2]. В обоих случаях N,N-диалкиламиды показали весьма обнадёживающие свойства.
Между тем в литературе было опубликовано три исследования об извлечении урана и плутония из азотного водного раствора посредством карбамидов. Речь идёт об исследовании, проведённом Е.K. Dukes и T.H. Sidall, с применением N,N,N’,N’-тетра-n-бутилмочевины, о котором сообщалось в Journal of Inorganic and Nuclear Chemistry 1966, 28(10), стр. 2307 – 2312, далее ссылка [3], а также об исследовании Г.М. Чумаковой и др. также с N,N,N’,N’-тетра-n-бутилмочевиной, опубликованном в «Радиохимии» 1980, 22(2), стр. 213–217, далее отсылка [4], и о более раннем исследовании, проведённом B.G. Vats et al. с N,N-диэтил-N’,N’-диизобутилмочевиной (или DEDiBU) и N,N-диэтил-N,N’-ди-n-октилмочевиной (или DEDOU), опубликованном в Dalton Transactions 2016, 45(25), стр. 10319 – 10325, далее [5].
Этими исследованиями был выявлен потенциал тетраалкильных карбамидов в качестве экстрагентов урана (VI) и плутония (IV). Однако ни одно из них не допускало возможности увидеть применение соединений такого типа для соэкстракции урана (VI) и плутония (IV) из водного раствора, полученного растворением отработавшего ядерного топлива, и для полного или частичного разделения урана (VI) и плутония (IV) без восстановления плутония (IV) в плутоний (III).
Однако оказалось, что в рамках своих работ авторами изобретения было установлено, что некоторые карбамиды обладают экстрагирующими свойствами, а именно:
- в присутствии водной фазы с сильной кислотностью, такой, какую имеют водные растворы после растворения отработавшего ядерного топлива в азотной кислоте, они обеспечивают коэффициенты распределения урана (VI) и плутония (IV), позволяющие проводить количественную соэкстракцию этих обоих элементов из данной водной фазы, и
- в присутствии водной фазы с умеренной кислотностью они обеспечивают коэффициенты разделения U(VI) / Pu(IV), при которых происходит отделение урана (VI) от плутония (IV) без необходимости восстановления плутония (IV) в плутоний (III), причём это разделение может быть полным или частичным по желанию.
И именно на этих выводах основано настоящее изобретение.
Описание изобретения
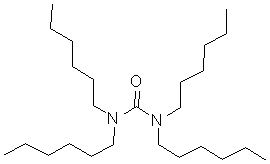
Предметом изобретения является в первую очередь применение карбамида общей формулы (I):

где R1, R2, R3, одинаковые или разные, означают линейную или разветвлённую алкильную группу с 1–12 атомами углерода, циклоалкильную группу с 3–12 атомами углерода или циклоалкильную группу с 4–13 атомами углерода;
R4 означает атом водорода, линейную или разветвлённую алкильную группу с 1–12 атомами углерода, циклоалкильную группу с 3–12 атомами углерода или циклоалкильную группу с 4–13 атомами углерода;
в качестве экстрагента для полного или частичного отделения урана (VI) от плутония (IV) без восстановления плутония (IV) в плутоний (III) на основе водного раствора А1, полученного растворением отработавшего ядерного топлива в азотной кислоте.
Такое применение включает в себя:
а) соэкстракцию урана (VI) и плутония (IV) из водного раствора, при этом соэкстракция предусматривает, по меньшей мере, однократное приведение в контакт в экстракторе водного раствора А1 с органическим раствором S1, содержащим карбамид в органическом растворителе, и разделение водного и органического растворов;
б) реэкстракцию плутония (IV) и фракции урана (VI) из органического раствора, полученного на этапе а), при этом реэкстракция содержит, по меньшей мере, одно приведение в контакт органического раствора S1 с водным раствором А2 в экстракторе, содержащим 0,1–0,5 моля/л азотной кислоты, и разделение органического и водного растворов;
в) полную или частичную экстракцию фракции урана (VI), присутствующей в водном растворе, полученном на этапе б), при этом экстракция содержит, по меньшей мере, одно приведение в контакт водного раствора с органическим раствором S2, содержащим карбамид в органическом растворе, в экстракторе и разделение водного и органического растворов;
в результате получают водный раствор с содержанием плутония (IV) и без содержания урана (VI) или смесь из плутония (IV) и урана (VI) и органический раствор с содержанием урана (VI), но без плутония (IV).
В приведённом выше, а также ниже изложении выражения «при содержании от … до …», « … от … до …», «содержащийся(аяся) от … до …» эквивалентны и означают предельные значения.
Под выражением «линейная или разветвлённая алкильная группа с 1–12 атомами углерода» подразумевается любая алкильная группа с линейной или разветвлённой цепочкой, содержащая 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 атомов углерода, например, метиловая, этиловая, n-пропиловая, изопропиловая, n-бутиловая, втор-бутиловая, изобутиловая, трет-бутиловая, n-амиловая, втор-амиловая, изоамиловая, n-гексильная, изогексильная, n-гептильная, изогептильная, n-октильная, изооктильная, n-нониловая, изонониловая, n-дециловая, изодециловая, n-ундециловая, n-додециловая, 2-метилбутиловая, 2-метилбутиловая, 3-метилпентиловая, 2-метилгексильная, 3-метилгексильная, 2-метилгептильная, 2-метиоктиловая, 2-этилгексильная, 1,5-диметилгексильная, 2,4,4-триметиламиловая, 2-этилгептильная, 1,2-диметилгептильная, 2,6-диметилгептильная, 3,5,5-триметилгексильная, 2-метилнониловая, 3,7-диметилоктиловая, 2,4,6-триметилгептильная, 2-бутилгексильная и др. группы.
Под «циклоалькильной группой с 1–12 атомами углерода» имеется в виду любая циклоалькильная группа с одним или несколькими слившимися или соединёнными мостиковой связью циклами, причём эти циклы могут быть замещены одной или несколькими алкильными группами, если общее количество атомов углерода, содержащихся в этой циклоалкильной группе (вместе с заместителями, если они имеются) составляет 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12; такой циклоалкильной группой являются, например, циклопропиловая, циклобутиловая, циклоамиловая, циклогексильная, циклогептильная, циклооктильная, декагидронафталениловая, бициклопропиловая, бициклогексильная группы, циклоамиловая или циклогексильная группа, замещённая одной или несколькими метиловыми, этиловыми, n-пропиловыми, изопропиловыми, n-бутиловыми, изобутиловыми, трет-бутиловыми, втор-бутиловыми и др. группами.
Под «циклоалкильной группой с 4–13 атомами углерода» подразумевается любая алкильная группа, замещённая циклоалкильной группой, при условии, что общее количество атомов углерода в алкильной группе с её заместителем составляет 4, 5, 6, 7, 8, 9, 10, 11, 12 или 13; такой циклоалкильной группой являются, например, 3-циклогексилпропиловая, 3-циклогексилбутиловая, 3-циклогексил-2-метилбутиловая, 4-циклогексил-1-метилбутиловая, 4-циклогексил-2-метилбутиловая, 4-циклогексил-3-метилбутиловая, 4-циклогексил-1-этилбутиловая, 4-циклогексил-2-этилбутидловая, 4-циклогексил-1-пропилбутиловая, 4-циклогексил-2-пропилбутиловая, 4-циклогексил-амиловая, 4-циклогексил-2-метиламиловая и др. группы.
Вместе с тем выражения «водный раствор» и «водная фаза» равнозначны и взаимозаменяемы, точно так же, как выражения «органический раствор» и «органическая фаза» равнозначны и взаимозаменяемы.
По мере того как циклоалкильные группы могут рассматриваться как алкильные группы, у которых единственная углеродная цепочка или основная углеродная цепочка является цикличной, то целесообразный карбамид по изобретению может быть охарактеризован как тетра-алкильный карбамид, если R4 отличается от атома водорода или же как три-алкильный карбамид, если R4означает атом водорода.Такая характеристика будет применяться ниже.
Согласно изобретению, общее количество атомов углерода в карбамиде составляет предпочтительно от 17 до 25.
Если карбамид является тетраалкильным, то предпочтительно, чтобы R1, R2, R3 и R4 означали линейную или разветвлённую алкильную группу с 1–12 атомами углерода.
Вместе с тем предпочтительно, чтобы:
R1 и R2 были одинаковыми, R3 и R4 были одинаковыми, но отличались от R1 и R2 или
R1и R4 были одинаковыми, R2, R3 были одинаковыми, но отличались от R1,и R4 или
R1, R2, R3, R4 были все одинаковыми.
Если R1 и R2 одинаковые, R2 и R4 одинаковые и отличаются от R1 и R2, то R1 и R2означают предпочтительно линейную или разветвлённую алкильную группу с 1–5 атомами углерода, а R3 и R4 означают предпочтительно линейную или разветвлённую алкильную группу с 6 – 10 атомами углерода, при этом общее количество атомов углерода, содержащихся в карбамиде, составляет предпочтительно 19, 21 или 23.
Аналогично в том случае, когда R1 и R4 одинаковые, R2и R3 одинаковые, но отличаются от R1 и R4, а R1 и R4 означают предпочтительно линейную или разветвлённую алкильную группу с 1–5 атомами углерода, при этом R2 и R3 означают предпочтительно линейную или разветвлённую алкильную группу с 6–10 атомами углерода, то общее количество атомов углерода в карбамиде составляет предпочтительно 19, 21 или 23.
Если R1, R2, R3, R4 являются одинаковыми, то они означают предпочтительно линейную или разветвлённую алкильную группу с 4–8 атомами углерода, при этом особо предпочтительны n-бутиловая, n-амиловая, n-гексильная, n-гептильная и n-октильная группы.
Если карбамид является три-алкильным, то предпочтительно, чтобы R1, R2 и R3 означали линейную или разветвлённую алкильную группу с 1–12 атомами углерода, предпочтительно с 2–10 атомами углерода, более предпочтительно с 4–8 атомами углерода.
Вместе с тем предпочтительно, чтобы R1 было равно R2, более того, чтобы R1, R2 и R3 были одинаковыми, в этом случае R1, R2 и R3 означают предпочтительно линейную или разветвлённую алкильную группу с 6–8 атомами углерода, причём особо предпочтительны n-гексильная, n-гептильная, n-октильная и 2-этилгексильная группы.
Следовательно, в качестве примера можно привести следующие предпочтительные карбамиды:
- N,N,N’,N’-тетра-n-бутилмочевина (или TBU), отвечающая общей формуле (I), где: R1, R2, R3 и R4 означают n-бутиловую группу;
- N,N,N’,N’-тетра-n-амилмочевина (или TPU), отвечающая общей формуле (I), где: R1, R2, R3 и R4 означают n-амиловую группу;
- N,N,N’,N’-тетра-n-гексилмочевина (или THU), отвечающая общей формуле (I), где: R1, R2, R3 и R4 означают n-гексильную группу;
- N,N,N’,N’-тетра-n-октилмочевина (TOU), отвечающая общей формуле (I), где: R1, R2, R3 и R4 означают n-октильную группу;
- N,N’-ди-n-бутил-N,N’-ди-n-гексилмочевина (или DBDHU), отвечающая общей формуле (I), где: R1 и R4 означают n-бутиловую группу, R2 и R3 означают n-гексильную группу;
- N,N’-ди-n-гептил-N,N’-ди-n-пропилмочевина (или DHDPU), отвечающая общей формуле (I), где R1 и R4 означают n-пропиловую группу, R2 R3 означают n-гептильную группу;
- N,N’-диэтил-N,N’-ди-n-октилмочевина (или sym-DEDOU), отвечающая общей формуле (I), где R1 и R4 означают этиловую группу, R2 и R3 означают n-октильную группу;
- N,N’-диметил-N,N’-ди-n-нонилмочевина (или DMDNU), отвечающая общей формуле (I), где: R1 и R4 означают метиловую группу, R2 и R3 означают n-нониловую группу;
- N,N,N’-три-n-октилмочевина (или TrOU), отвечающая общей формуле (I) , где: R1, R2 и R3 означают n-октильную группу, R4 означает атом водорода;
- N,N,N’-три(2-этилгексил)мочевина (или TrEHU), отвечающая общей формуле (I), где: R1, R2 и R3 означают 2-этилгексильную группу, R4 означает атом водорода, и
- N,N-ди(2-этилгексил)-N’-n-октилмочевина (или DEHOU), отвечающая общей формуле (I), где: R1 и R2 означают 2-этилгексильную группу, R3 означает n-октиловую группу, R4 означает атом водорода.
Из этих карбамидов особо предпочтительны N,N,N’,N’-тетра-n-бутилмочевина, N,N,N’,N’-тетра-n-гексилмочевина, N,N,N’,N’-тетра-n-пентилмочевина, N,N’-ди-n-бутил-N,N’-ди-n-гексилмочевина, N,N’-ди-n-гептил-N,N’-ди-n-пропилмочевина, N,N’-диэтил-N,N’-ди-n-октилмочевина и N,N’-диметил-N,N’-ди-n-нонилмочевина ввиду того, что они обладают особо повышенной избирательностью к урану (VI) по сравнению с плутонием (IV) при умеренной кислотности, т.е. в присутствии водной фазы, содержащей от 0,1 до 0,5 моля/л азотной кислоты.
Согласно изобретению, органические растворы S1 и S2 содержат в себе преимущественно 0,5 - 2 моля/л, предпочтительно 1–1,4 моля/л, карбамида.
Органическим растворителем служит преимущественно ациклический углеводород или смесь ациклических углеводородов, например, n-додекан, гидрогенизированный тетрапропилен (или ТРН), керосин, IsaneТМ IP-185Т или IsaneТМ IP-175T, предпочтительно ТРН.
Что касается водного раствора, полученного растворением отработавшего ядерного топлива, то в нём обычно содержится азотная кислота в количестве от 3 до 6 молей/л.
Предпочтительно этап а) включает в себя дополнительно дезактивацию органического раствора, полученного соэкстракцией урана (VI) и плутония (IV), по отношению к америцию, кюрию и продуктам деления, при этом такая дезактивация включает в себя, по меньшей мере, приведение в контакт в экстракторе органического раствора с водным раствором А3, содержащим 1–6 молей/л азотной кислоты, и разделение органического и водного растворов. При необходимости эта дезактивация дополняется раскислением органического, таким образом дезактивированного раствора, при этом такое раскисление включает в себя, по меньшей мере, приведение в контакт в экстракторе органического раствора, полученного дезактивацией, с водным раствором А4, содержащим 0,1–1 моль/л, предпочтительно 0,5 моля/л азотной кислоты, и разделение органического и водного растворов.
Предпочтительно также, чтобы приведение в контакт в экстракторе, в котором протекает этап б), органического раствора, полученного на этапе а), с водным раствором А2 предусматривало противоточную циркуляцию органического и водного растворов при соотношении расходов О/А (органический раствор/водный раствор), превышающим преимущественно 1, предпочтительно равным или превышающим 3, более предпочтительно равным или превышающим 5, таким образом, чтобы обеспечивалась обогащающая плутоний (IV) реэкстракция, т.е. реэкстракция плутония (IV) с образованием водного раствора, в котором концентрация плутония (IV) превышает его концентрацию в органическом растворе, из которого его повторно экстрагировали.
Согласно изобретению уран (VI), присутствующий в органическом растворе, полученном на этапе в), может быть повторно экстрагирован из этого раствора, по меньшей мере, приведением в контакт в экстракторе органического раствора с водным раствором А5, содержащим не более 0,05 моля/л, предпочтительно не более 0,01 моля/л азотной кислоты, затем разделяют органический и водный растворы. Такая реэкстракция может проводиться при комнатной температуре или при температуре от 40 до 50°С, что способствует реэкстракции урана (VI). Вместе с тем она может проводиться при соотношении расходов О/А более 1, с тем чтобы уран (VI) повторно экстрагировался, сопровождаясь обогащением.
Из пригодных карбамидов согласно изобретению некоторые, такие, как тетра-алкильные карбамиды N,N,N’,N’-тетра-n-бутилмочевина, N,N,N’,N’-тетра-n-гексилмочевина, N,N,N’,N’-тетра-n-октилмочевина и N,N,N’,N’-тетра-n-пентилмочевина известны из уровня техники.
Другие же, такие как, с одной стороны, тетра-алкильные карбамиды N,N’-ди-n-бутил-N,N’-ди-n-гексилмочевина, N,N’-ди-n-гептил-N,N’-ди-n-пропилмочевина, N,N’-диэтил-N,N’-ди-n-октилмочевина, N,N’-диметил-N,N’-ди-n-нонилмочевина (или DMDNU), и, с другой стороны, три-алкильные карбамиды N,N,N’-три-n-октилмочевина, N,N,N’-три(2-этилгексил)мочевина и N,N-ди(2-этилгексил)-N’-n-октилмочевина не были известны авторам изобретения и никогда не были описаны в уровне техники.
Поэтому также предметом изобретения является тетра-алкильный карбамид приведенной выше общей формулы (I), в которой R1 и R4 являются одинаковыми и означают линейную алкильную группу с 1–4 атомами углерода, R2 и R3 являются одинаковыми и означают линейную алкильную группу с 6–9 атомами углерода, при этом общее количество атомов углерода в карбамиде составляет 21.
Этим тетра-алкильным карбамидом могут быть карбамиды DBDHU, sym-DEDOU, или карбамид DMDNU, предпочтительны карбамиды sym-DEDOU и DMDNU.
Как известно само по себе, этот тетра-алкильный карбамид может быть получен, в частности, посредством реакции N,N’-диалкилмочевины формулы RNHC(O)NHR, где: R имеет то же значение, что и R2, R3 в карбамиде с содержанием галогеноалкана HalR’, например, йодалкан IR’, где R’ имеет то же значение, что и R1 и R4 в карбамиде в органической среде, например, в тетрагидрофуране и в присутствии металлического гидрида, такого, как гидрид натрия.
Также предметом изобретения является триалкильный карбамид отдельной формулы (II):

где: R1, R2 и R3, одинаковые или разные, означают линейную или разветвлённую алкильную группу с 8-12 атомами углерода или циклоалкильную группу с 3–12 атомами углерода при условии, что, по меньшей мере, одно из значений R1, R2 или R3 будет отличаться от циклоалкильной группы.
Кроме того, общее количество атомов углерода, содержащихся в карбамиде отдельной формулы (II), составляет предпочтительно от 17 до 25.
Предпочтительно, чтобы в этом карбамиде R1, R2 и R3 означали каждый линейную или разветвлённую алкильную группу, при этом особо предпочтительны n-октильные и 2-этилгексильные группы.
Вместе с тем предпочтительно, чтобы R1 было одинаково с R2 и чтобы R1, R2 и R3 были одинаковы между собой.
Согласно изобретению карбамидом отдельной формулы (II) предпочтительно являются карбамиды TrOU, TrEHU или карбамид DEHOU, при этом карбамиды TrOU и TrEHU особо предпочтительны.
Как известно само по себе, данный карбамид может быть получен в результате реакции вторичного амина формулы R1R2NH, где R1 и R2 имеют приведённые выше значения, с изоцианатом формулы R3NCO, где R3 имеет приведённое выше значение, в органической среде, например, безводном дихлорметане.
Помимо обладания очень полезными экстрагирующими свойствами карбамиды имеют также то преимущество, что в них, как и в их продуктах распада, содержатся только атомы углерода, водорода, кислорода и азота, вследствие чего они полностью озоляемы и не создают вторичных вредных отходов.
Другие признаки и преимущества изобретения станут понятны из последующего описания.
Во всяком случае, само собой разумеется, что приводимое ниже описание служит для пояснения предмета изобретения и совершенно не должно восприниматься как ограничение предмета изобретения.
Краткое описание фигуры
На фиг. 1 изображена принципиальная схема предпочтительного варианта выполнения изобретения, при этом прямоугольниками 1–4 обозначены многоуровневые экстракторы, которые традиционно применяются при обработке отработавшего ядерного топлива (смесители-отстойники, пульсационные колонны или центробежные экстракторы); органические фазы обозначены сплошной линией, водные фазы - пунктирной линией.
Подробное описание частных вариантов выполнения
Пример 1. Синтез тетра-алкильных и три-алкильных карбамидов
1. 1. N,N’-ди-n-бутил-N,N’-n-гексилмочевина или DBDHU
Карбамид DBDHU синтезируют посредством:
- реакции n-гексилизоцианата (5 мл, 34,3 ммоля) с n-гексиламином (0,3 моля/л, 1,2 грамм-эквивалент) в безводном дихлорметане с получением N,N’-ди-гексилмочевины,
- реакции N,N’-ди-n-гексилмочевины (7,6 г, 33,4 моль) с йодобутаном (5 грамм-эквивалентов) в тетрагидрофуране (THF).
При этом раствор n-гексилизоцианата добавляли по капле при 0°С в раствор n-гексиламина в безводном дихлорметане. Реакционную смесь встряхивали в течение 1 ч при 0°С, затем довели до комнатной температуры и встряхивали в течение ночи. Растворитель испарился при пониженном давлении и продукт очистили на силикагели смесью дихлорметана с метанолом (95/5 в объёмном отношении), чтобы получить 7,6 г N,N’-ди-n-гексилмочевины (выход: >97%).
Затем суспензию NaH при 60% по массе (8 грамм-эквивалентов) в THF порционно добавляли в раствор N,N’-ди-n-гексилмочевины в ТHF при 0°С. Реакционную смесь встряхивали в течение 2 часов при 0°С, затем по капле добавляли йодобутан. Смесь довели до комнатной температуры, затем до флегмы. Через ночь смесь охладили до 0°С и добавили в неё воду. Водную фазу экстрагировали диэтиловым эфиром. Затем органическую фазу промыли насыщенным хлоридом натрия (NaCl) раствором и просушили на сульфате натрия (Na2SO4). Растворитель испарился при пониженном давлении, полученное твёрдое вещество очистили на силикагели смесью циклогексана с ацетатом этила (99/1 – 90/10 в объёмном отношении) для получения 10,5 г карбамида DBDHU (выход: >95%). Описание свойств этого карбамида приведено ниже.
ЯМР1H (CDCl3, 400 МГц) δ (ч./млн): 3,12 (m, 8H); 1,50 (m, 8H); 1,28 (m, 16H); 0,92 (m, 12H)
ЯМР13C (CDCl3, 101 МГц) δ (ч./млн): 165; 48,5; 48,1; 31,6; 30,1; 27,9; 26,7; 22,6; 20,2; 14,0; 13,9
MS (ионизированное электронапыление в положительном режиме (ESI)): 341 (MH+); 682 (2MH+); 703 (2MNa+)
GC-HRMS (положительный режим EI): чистота >99%; точная расчётная масса для C21H44N2O: 340,3448; получено: 340,3412
I.2 – N,N’-ди-n-гептил-N,N’-ди-n-пропиловая мочевина или DHDPU:
Карбамид DHDPU синтезировали посредством:
- реакции n-гептилизоцианата (5 г, 35,4 ммоля) с n-гептиламином (0,3 моля/л, 1,2 грамма-эквивалента) в безводном дихлорметане для получения N,N’-ди-n-гептилмочевины, и
- реакции N,N’-ди-n-гептилмочевины (8,8 г, 34,3 ммоля) с йодопропаном (5 грамм-эквивалентов) в THF;
при соблюдении того же протокола, что и описанный выше в п. 1.1 при синтезе DBDHU.
Таким образом получили 11,2 г карбамида DHDPU (выход: >96%), описание свойств которого приведено ниже:
ЯМР1H (CDCl3. 400 МГц) δ (ч./млн): 3,07 (m, 8H); 1,50 (m, 8H); 1,25 (m, 16H); 0,86 (m, 12H)
ЯМР13C (CDCl3. 101 МГц) δ (ч./млн): 163,7; 48,5; 46,7; 30,2; 27,5; 26,4; 25,4; 21,0; 19,6; 12,4; 9,8
MS (ESI в положительном режиме): 341 (MH+); 363 (MNa+); 682 (2MH+); 703 (2MNa+)
GC-HRMS (EI в положительном режиме): чистота: 99,1%; точная расчётная масса для C21H44N2O: 340,3448; получено: 340,3438
I.3 – N,N’-диэтил-N,N’-ди-n-октилмочевина или sym-DEDOU:
Карбамид sym-DEDOU синтезировали посредством:
- реакции n-октилизоцианата (5 г, 32,2 ммоля) с n-октиламином (0,3 моля/л, 1,2 грамм-эквивалента) в безводном дихлорметане для получения N,N’-ди-n-октилмочевины и
- реакции N,N’-ди-n-октилмочевины (8.9 г, 31.3 ммоль) с йодоэтаном (5 грамм-эквивалентов.) в THF;
при соблюдении того же протокола, что и описанный выше в п. 1.1 при синтезе DBDHU.
Получено 10,2 г карбамида sym-DEDOU (выход: >96%), описание свойств которого приведено ниже.
ЯМР1H (CDCl3, 400 МГц) δ (ч./млн.): 3,14 (q, J = 7 Гц, 4HEt); 3,07 (m, 4HOct); 1,48 (m, 4HOct); 1,25 (m, 20HOct); 1,08 (t, J = 7 Гц, 6HEt); 0,86 (m, 6HOct)
ЯМР13C (CDCl3, 101 МГц) δ (ч./млн.): 164,9; 47,8; 42,9; 31,8; 29,4; 29,3; 28,0; 27,1; 22,6; 14,1; 13,2
MS (ESI в положительном режиме): 341 (MH+); 363 (MNa+); 703 (2MNa+)
GC-HRMS (EI в положительном режиме): чистота: 99,5%; точная расчётная масса для C21H44N2O: 340,3448; получено: 340,3437
I.4 – N,N’-диметил-N,N’-ди-n-нонилмочевина или DMDNU:
Карбамид DMDNU синтезировали посредством реакции N,N’-ди-n-метилмочевины (2 г, 22,7 ммоля) с йодононаном (5,3 мл, 40,2 ммоля, 2 грамм-эквивалента) в THF.
При этом раствор N,N’-диметилмочевины в THF добавляли по капле при 0°C в суспензию NaH при 60% в весовом отношении (7,2 г, 45 ммолей, 2 грамм-эквивалента) в THF. Реакционную смесь встряхивали в течение 1 часа при 0°C, затем добавляли по капле йодононан. Смесь довели до комнатной температуры и затем до рефлюкса. Через ночь среду охладили до 0°С и добавили воду. Водную фазу экстрагировали диэтиловым эфиром. Затем органическую фазу промыли насыщенным NaCl раствором и сушили на Na2SO4.Растворитель испарился при низком давлении, полученное твёрдое вещество очистили на силикагеле смесью циклогексана с этилацетатом (99/1–90/10 в объёмном отношении) и получили 7,8 г карбамида DMDNU (выход: >98%). Описание свойств приведено ниже.
ЯМР1H (CDCl3, 400 МГц) δ (ч./млн): 3,08 (t, J = 7.5 Гц, 4H); 2,76 (s, 6H); 1,52 (m, 4H); 1,25 (m, 24H); 0,87 (m, 6H)
ЯМР13C (CDCl3, 101 МГц) δ (ч./млн): 165,4; 50,6; 36,5; 31,8; 29,5; 29,4; 29,3; 27,6; 26,9; 22,6; 14,1
MS (ESI в положительном режиме): 341 (MH+); 682 (2MH+); 703 (2MNa+)
GC-HRMS (EI в положительном режиме): чистота: 99.2 %; точная расчётная масса для C21H44N2O: 340,3448; получено: 340,3454
I.5 – N,N,N’-три-n-октилмочевина или TrOU:
Карбамид TrOU синтезировали посредством реакции ди-n-октиламина (1,5 мл, 4,9 ммоль, 1 грамм-эквивалент) с n-октилизоцианатом (1,7 мл, 9,7 ммоль, 2 грамм-эквивалента) в безводном дихлорметане (0,1 моля/л).
При этом раствор, содержавший ди-n-октиламин и n-октилизоцианат в безводном дихлорметане, встряхивали в течение 4,5 часов при температуре окружающей среды. Затем реакционную смесь дважды промыли раствором соляной кислоты 1 М и дважды насыщенным раствором бикарбоната натрия (NaHCO3). Органическую фазу просушили на сульфате магния (MgSO4), концентрировали при пониженном давлении, остаток очистили мгновенной хроматографией на силикагеле посредством смеси дихлорметана с этилацетатом (95/5 в объёмном отношении) и получили 1,9 г TrOU в виде бесцветного масла (выход: 100%). Описание свойств приводится ниже.
ЯМР1H (CDCl3, 400 МГц) δ (ч./млн): 4,25 (s, 1H); 3,20 (q, J = 5,4 Гц, 2H); 3,14 (q, J = 7,7 Hz, 4H); 1,50 (m, 6H); 1,46 (m, 30H); 0,87 (t, J = 7,2 Гц, 9H)
ЯМР13C (CDCl3, 101 МГц) δ (ч./млн): 157,8; 47,5; 41,0; 32,0; 30,6; 29,6; 29,5; 29,4; 28,8; 27,2; 22,8; 14,2
IR (KBr, νmax/cm-1): 3345; 2955; 2922; 2853; 1622; 1533; 1465; 1376; 1274; 767; 722
MS (ESI в положительном режиме): 397 (MH+), 816 (2MNa+)
HRMS (ESI в положительном режиме): рассчитано для C25H53N2O: 397,4158; получено: 397,4156.
Элементный анализ (%) (C25H52N2O + 0.18AcOEt): рассчитано: C 74,88; H 13,06; N 6,79; получено: C 75,17; H 12,74; N 6,88
I.6 – N,N,N’-три(2-этилгексил)мочевина или TrEHU:
TrEHU синтезировали на основе ди(2-.этилгексил)амина (1,49 мл, 4,92 ммоля, 1 грамм-эквивалент) и 2-этилгексилизоцианата (1,76 мл, 9,84 ммоля, 2 грамм-эквивалента) в безводном дихлорметане (0,1 моля/л).
При этом раствор, содержавший ди(2-этилгексил)амин и 2-этилгексилизоцианат в безводном дихлорметане, встряхивали в течение 5 часов при комнатной температуре. Затем реакционную смесь дважды промыли раствором соляной кислоты 1М и дважды насыщенным раствором NaHCO3. Органическую фазу просушили на MgSO4, концентрировали при пониженном давлении и остаток очистили мгновенной хроматографией на силикагеле при градиенте дихлорметан/этилацетата (99/1 – 90/10 в объёмном отношении) и получили 1,94 г TrEHU в виде густого бесцветного масла (выход: 99%). Описание свойств приводится ниже.
ЯМР1H (CDCl3, 400 МГц) δ (ч./млн): 4,26 (s, 1H); 3,17 (t, J = 5,7 Гц, 2H); 3,09 (m, 4H); 1,62 (m, 2H); 1,46 (m, 25H); 0,88 (m, 18H)
ЯМК13C (CDCl3, 101 МГц) δ (ч./млн): 158,2; 51,6; 43,6; 39,8; 38,4; 31,2; 30,7; 28,9; 24,4; 24,0; 23,1; 14,1; 11,0; 10,8.
IR (KBr, νmax/cм-1): 3348; 2957; 2925; 2859; 1617; 1534; 1459; 1378; 1240; 765; 727
MS (ESI в положительном режиме): 397 (MH+); 816 (2MNa+)
HRMS (ESI в положительном режиме): рассчитано для C25H53N2O: 397,4158; получено: 397,4156
Элементный анализ (%) (C25H52N2O): рассчитано C 75,69; H 13,21; N 7,06; получено: C 75,49; H 13,45; N 6,91
I.7 – N,N-ди(2-этилгексил)-N’-n-октилмочевина или DEHOU:
Карбамид DEHOU синтезировали на основе ди(2-этилгексил)амина (1,49 мл, 4,92ммоля, 1 грамм-эквиалент) и n-октилизоцианата (1,75 мл, 9,84 ммоля, 2 грамма-эквивалента) в безводном дихлорметане (0,1 моля/л) при соблюдении того же протокола, что и протокол, описанный выше в п. 1.6 при синтезе карбамина TrEHU. Получили 1,84 г карбамида DEHOU в виде бесцветного масла (выход 94%). Описание свойств приводится ниже.
ЯМР1H (CDCl3, 400 МГц) δ (ч./млн.): 4,25 (t, J = 4,9 Гц, 1H); 3,20 (q, J = 6,1 Hz, 2H); 3,07 (m, 4H); 1,61 (hept, J = 5,6 Гц, 2H); 1,46 (quint, J = 6,4 Hz, 2H); 1,25 (m, 26H); 0,86 (m, 15H)
ЯМР13C (CDCl3, 101 МГц) δ (ч./млн.): 158,3; 51,5; 41,0; 38,5; 31,9; 30,8; 30,5; 29,5; 29,4; 29,0; 27,2; 24,1; 23,2; 22,8; 14,2; 10,9
IR (KBr, νmax/cм-1): 3344; 2957; 2924; 2857; 1618; 1533; 1459; 1408; 1378; 1241; 765; 725
MS (ESI в положительном режиме): 397 (MH+); 816 (2MNa+)
HRMS (ESI в положительном режиме): рассчитано для C25H53N2O: 397,4158; получено: 397,4154
Элементный анализ (%) (C25H52N2O): рассчитано: C 75,69; H 13,21; N 7,06; получено: C 75,87; H 13,49; N 6,89
I.8 – N-циклогексил-N’,N’-ди-n-октилмочевина или CyDOU:
Карбамид CyDOU, отвечающий приведенной выше формуле (I), в которой R1= R2 = n-октил, R3 = циклогексил и R4 = водород, синтезировали на основе ди-n-октиламина (1,50 мл, 4,87 ммоля, 1 грамм-эквивалент) и циклогексилизоцианата (1,27 мл, 9,74 ммоля, 2 грамм-эквивалента) в безводном дихлорметане (0,1 моля/л) при соблюдении того же протокола, что и описанный выше в п. 1,6 протокол синтеза карбамида TrEHU за исключением того, что градиент дихлорметана/этилацетата, применявшийся для мгновенной хроматографии на силикагеле с некоторым градиентом, составлял 96/4 – 90/10. При этом получено 1,48 г карбамида CyDOU в виде бесцветного масла (выход: 83%).
Описание свойств приводится ниже.
ЯМР1H (CDCl3, 400 МГц) δ (ч./млн): 4,11 (s, 1H); 3,64 (m, 1H); 3,13 (t, J = 7,6 Гц, 4H); 1,94 (m, 2H); 1,59 (m, 8H); 1,28 (m, 24H); 0,87 (t, 6H)
ЯМР13C (CDCl3, 101 МГц) δ (ч./млн): 157,1; 49,3; 47,5; 34,3; 32,0; 29,6; 29,4; 28,8; 27,2; 25,9; 25,2; 22,8; 14,2
IR (KBr, νmax/cм-1): 3330; 2922; 2853; 1617; 1528; 1451; 1407; 1314; 1251; 1214; 890; 767; 722
MS (ESI в положительном режиме): 367 (MH+); 756 (2MNa+)
HRMS (ESI в положительном режиме): рассчитано для C23H47N2O: 367,3688; получено: 367,3686
Элементный анализ (%) (C23H46N2O): рассчитано: C 75,35; H 12,65; N 7,64; получено: C 75,33; H 12,80; N 7,47
II. Экстрагирующие свойства карбамидов
II.1. Тетраалкильные карбамиды
Тесты по экстрагированию урана (IV) и плутония (VI)
Тесты по экстрагированию проводились с применением:
- в качестве органических фаз: растворов с содержанием либо ≈ 0,5 моля/л, либо ≈ 1,2 моля/л одного из карбамидов: TBU, THU, TOU, TPU, DBDPU, DHDPU, sym-DEDOU и DMDNU в гидрогенезированном тетрапропилене (TPH) и
- в качестве водных фаз: водных растворов с содержанием 10 г/л урана (VI), ≈ 200 кБк/мл плутония (IV) и азотной кислоты при концентрации либо 4 моля/л (для симуляции кислотности, присущей водному раствору, полученному растворением отработавшего ядерного топлива в азотной кислоте), либо 0,5 моля/л (для симуляции кислотности, присущей водному раствору для повторного экстрагирования плутония согласно изобретению).
Каждый из этих опытов проводился при контакте в трубе при встряхивании органической фазы с водной фазой в течение 30 минут при 25°С. Объёмное соотношение О/А составило 1.
После центрифугирования и разделения фаз проводили измерение концентрации урана в водных фазах атомным эмиссионным спектрометром с наведённым плазменным факелом (или ICP-AES), а концентрация урана в органических фазах определялась путём реэкстракции этого элемента в растворе азотной кислоты при концентрации 0,01 моля/л и путём измерения с помощью ICP-AES в водной фазе, образовавшейся при этой реэкстракции. Показатели концентрации плутония замеряли в водных и органических фазах посредством спектрометрии α.
В приводимой ниже таблице 1 показаны по каждой концентрации, при которой тестировали карбамид, коэффициенты распределения урана, обозначенные как DU,,и плутония, обозначенные как DPt, полученные для водных фаз при 4 молях/л HNO3, и 0,5 моля/л HNO3,а также коэффициенты разделения,полученные для водных фаз при 0,5 моля/л HNO3.
Для сравнения в таблицу внесены также результаты тестов по экстрагированию при тех же условиях испытания, но с применением в качестве органических фаз растворов с содержанием известных из уровня техники N,N-диалкиламидов, а именно:
- раствора с содержанием 0,5 моля/л карбамида DEHDMBА (N,N-ди(2-этилгексил)-3,3-диметибутирамид) в гидрогенезированном тетрапропилене (ТРН), предложенного в источнике [1],
- раствора с содержанием либо 0,5 моля/л, либо 1,2 моля/л MOEHA (N-метил-N-октил-2-этилгексанамидв в ТРН, предложенного в источнике [2].
Таблица 1









[C] = концентрация тестированного соединения в органической фазе;
[HNO3] = концентрация азотной кислоты в водной фазе.
Из этой таблицы видно, что будь то сильная кислотность ([HNO3] = 4 моля/л) или умеренная кислотность ([HNO3] = 0,5 моль/л), коэффициенты распределения урана (IV), полученные для восьми тестированных тетраалкильных карбамидов, все превышают коэффициенты, полученные для N,N-диалкиламидов DEHDMBA и MOEHA при схожей концентрации органической фазы, что подтверждает выраженную способность тетраалкильных карбамидов к экстрагированию урана (VI) из сильнораскисленной водной фазы.
В частности, будь то при сильной кислотности ([NHO3] = 4 моля/л) или при умеренной кислотности (HNO3] = 0,5 моля/л), коэффициенты распределения урана (VI), полученные для карбамидов TPU, DBDHU, DHDPU, sym-DEDOU, DMDNU при концентрации около 1,2 моля/л в органической фазе, превышают более, чем в два раза коэффициенты, полученные при схожей концентрации MOEHA в органической фазе.
Также эта таблица указывает на то, что при сильной кислотности ([HNO3] = 4 моля/л) и при концентрации 0,5 моля/л органической фазы карбамиды TBU и TOU также создают коэффициенты распределения плутония (IV), которые больше коэффициентов, полученных при схожей концентрации органической фазе в DEHDMBA и MOEHA.
Напротив, при концентрации 1,2 моля/л органической фазы карбамиды TBU, THU, TPU, DBDHU, DHDPU и sym-DEDOU образуют коэффициенты распределения плутония (IV), которые меньше коэффициента, полученного при схожей концентрации органической фазы в MOHE, однако эти коэффициенты распределения остаются, тем не менее, весьма удовлетворительными, поскольку коэффициенты карбамидов TBU, THU, TPU, DBDPU, DHDPU больше 2, коэффициент карбамида TOU около 2 (DPu+= 1,9), коэффициент карбамида sym-DEDOU составляет более 1,3.
Из таблицы также видно, что при умеренной кислотности ([HNO3]) = 0,5 моля/л) коэффициенты разделения U(VI) / Pu (IV), полученные для карбамидов TBU, THU, TPU, DBDHU, DHDPU, sym-DEDOU и DMDNU, превышают коэффициенты для N,N-диалкиламидов DEHDMBA и MOEHA при схожей концентрации органической фазы.
В частности, когда они применяются в количестве 1,2 моля/л в органической фазе , TBU позволяет получить коэффициент разделения U(VI) / Pu (IV), равный 36, т. е. почти в два раза больший, чем коэффициент для MOEHA при схожей концентрации органической фазы; карбамиды TPU и DBDHU позволяют получить коэффициенты разделения U (VI) / Pu (IV) соответственно 73 и 74, т.е. почти в 4 раза больший, чем коэффициент для MOEHA; карбамид DHDPU позволяет получить коэффициент разделения U(VI) / Pu (IV), равный 94, т.е. почти в 5 раз больший, чем коэффициент для MOEHA; что касается коэффициента разделения U(VI) / Pu (IV) , полученного для sym-DEDOU, то он в 8,5 раза больше коэффициента для MOHEA (170 против 20).
Следовательно? данная таблица подтверждает, что тетра-алкильные карбамиды могут успешно применяться в качестве экстрагентов в способе обработки водного раствора, полученного растворением отработавшего ядерного топлива в азотной кислоте, включающем в себя соэкстракцию урана (VI) и плутония (IV) из такого раствора, являющегося сильно кислым, и обеспечивать частичное или полное разделение плутония (IV) и урана (VI), присутствующих в органической фазе, полученной при такой реэкстракции плутония (IV) из этой органической фазы посредством водного раствора умеренной кислотности.
Из тетраалкильных карбамидов карбамид DMDNU представляется особо полезным для разработки такого способа, так как, с одной стороны, он обладает при сильной кислотности значительно большей способностью к экстракции урана (DU и DPu > 30), чем способность карбамида MOEHA (DU = 7,5 и DPu = 4,3) и, с другой стороны, он обеспечивает умеренную кислотность при коэффициенте разделения уран (VI) / плутоний (IV) (FSU/Pu = 30¸ который, в свою очередь, значительно более высокий, чем коэффициент, полученный при использовании MOEHA.
Даже если он при сильной кислотности обладает более слабой способностью к экстрагированию плутония, чем MOEHA, то представляется, что карбамид syn-DEBOU выступает столь же положительным претендентом, так как он позволяет получить при умеренной кислотности особо высокий коэффициент разделения урана ((VI) и плутония (IV) (FSU/Pu= 170).
Тесты на способность к наполнению ураном (VI)
Проводились тесты по определению способности к наполнению ураном (VI) при 4-кратном приведении в контакт в трубах при встряхивании органических фаз с содержанием 1,2 моли/л одного из карбамидов TPU, DBDHU, DHDPU, sym-DEDOU и DMDNU в ТРН с аликвотными частями водной фазы, содержавшей 200 г/л урана (VI) и 3,4 моля/л азотной кислоты.
Каждый контакт проводился при 25°С в течение 30 минут при соотношении О/А, равным 2.
После центрифугирования и разделения фаз замерили концентрацию урана в органических фазах после реэкстракции этого элемента из раствора азотной кислоты при концентрации 0,01 моля/л; прибором ICP-AES измеряли концентрацию в водной фазе после реэкстракции.
Ниже в таблице II приведены по каждому тестированному карбамиду показатели концентрации урана (VI) в г/л, которые получили в органических фазах после каждого из 4 контактов.
Для сравнения в эту таблицу также внесены результаты тестов на способность к наполнению ураном (VI), проведённых при тех же условиях опыта, но с использованием в качестве органических фаз растворов с содержанием 1,2 моль/л MOEHA в ТРН.
Таблица II
- После растворения 3-й фазы.
После второго контакта органическая фаза, содержавшая MOEHA, образовала третью фазу. Добавка 300 мл органической фазы (предварительно уравновешенной контактом с азотной кислотой) позволила устранить эту третью фазу: насыщение органической фазы ураном (VI) составило, следовательно, 124 г/л, т.е. приблизительно 87% от теоретической способности органической фазы к наполнению (при учёте стехиометрии уран/экстрагент = ½).
Зато при тех же условиях карбамиды не расслаивались и было возможно наполнить содержащую карбамид органическую фазу ураном (VI) в количестве от 130 до 143 г/л, что составляет более 88% от теоретической способности к наполнению органической фазы (при учёте стехиометрии уран/экстрагент = ½).
Эти результаты показывают, что карбамиды обладают сильно выраженной способностью к наполнению ураном (VI), что совместимо с разработкой способа обработки ядерного топлива и что они даже позволяют исключить проблемы с расслоением органической фазы, наблюдавшиеся в случае с MOEНA.
II.2 Три-алкильные карбамиды
Проводились тесты по экстрагированию, аналогичные описанным выше в разделе II.1, но применялись в качестве органических фаз растворы с содержанием 0,4 – 0,5 моля/л одного из карбамидов TrOU, TrEHU и DEHOU в гидрогенизированном тетрапропилене (TPH).
Результаты этих тестов представлены ниже в таблице III.
Для сравнения в эту таблицу внесены также результаты, внесённые ранее в таблицу I для MOEHA при 0,5 моля/л в ТРН.
Таблица III



[C] - концентрация тестированного соединения в органической фазе;
[HNO3] - концентрация азотной кислоты в водной фазе.
Из этой таблицы видно, что при сильной кислотности ([HNO3] = 4 моля/л) три тестированных три-алкильных карбамида экстрагировали в большей степени уран (VI) и плутоний ((IV), чем N,N-диалкиламид MOEHA, поскольку они образовали коэффициенты распределения урана (VI) и плутония (IV), которые все были больше коэффициентов, полученных при этом N,N-диалкиламиде.
Из неё также видно, что при умеренной кислотности ([HNO3] = 0,5 моля/л) три тестированных триалкильных карбамида позволяют удержать уран (VI) в органической фазе одновременно очень эффективно (DU>0,5) и избирательно по отношению к плутонию (IV), поскольку коэффициент распределения полученного плутония (IV) при этих карбамидах составляет менее 0,04.
Карбамид TrEHU является особо избирательным, поскольку он обеспечивает коэффициент разделения U(VI) / Pu (IV), равный 121, т.е. в 10 раз больший, чем коэффициент при N,N-диалкиламид MOEHA при сопоставимой концентрации.
Кроме того этой таблицей подтверждается, что три-алкильные карбамиды могут эффективно применяться в качестве экстрагентов в способе обработки водного раствора, полученного растворением отработавшего ядерного топлива в азотной кислоте, включающем в себя соэкстракцию урана (VI) и плутония (IV) из этого водного раствора, являющегося сильно кислым, и частичное или полное разделение плутония (IV) и урана (VI), присутствующих в этом органическом растворе, полученном соэкстракцией путём реэкстракции плутония (IV) из этой органической фазы посредством водного раствора с умеренной кислотностью.
III. Принципиальная схема предпочтительного варианта применения изобретения
Обратимся к фиг. 1, на которой изображена принципиальная схема предпочтительного варианта применения изобретения.
Как следует из этой фигуры, применение предусматривает четыре этапа.
На первом этапе, обозначенном как «Соэкстракция урана и плутония» на фиг. 1, ставится целью совместная экстракция урана (VI) и плутония (IV) из водного азотного раствора, полученного растворением отработавшего ядерного топлива, обозначенного как «А1» на фиг. 1.
Такой раствор содержит, как правило, 3 – 6 молей/л HNO3, уран, плутоний, второстепенные актиниды (в частности, америций и кюрий), продукты деления (La, Ce, Pr, Nd, Sm, Eu, Gd, Mo, Zr, Ru, Tc, Rh, Pd, Y, Cs, Ba …), а также некоторые продукты коррозии, например, железо.
Этап «Соэкстракция U + Pu» проводится при циркуляции в экстракторе 1 водного раствора А1 в противотоке органической фазе, обозначенной S1 на фиг. 1, которая содержит 0,5 – 2 моля/л, предпочтительно 1,0 - 1,4 моля/л, карбамида общей формулы (I) в виде раствора с органическим растворителем.
Этим органическим растворителем предпочтительно выступает ациклический углеводород или смесь ациклических углеводородов, например, n-додекан, гидрогенизированный тетрапропилен (ТРН), керосин, IsaneТМ IP-175T, предпочтительно гидрогенизированный тетрапропилен.
Второй этап, обозначенный на фиг. 1 как «Промывка PF (продуктов деления)», имеет своей целью вторичную экстракцию из органической фазы, образовавшейся после «соэкстракции U + Pu», фракции продуктов деления, экстрагированных из водного раствора А1 вместе с ураном (VI) и плутонием (IV).
При этом этап «Промывка PF» содержит одну или несколько операций по промывке органической фазы, полученной после «Соэкстракции U + Pu», причём каждая операция по промывке проводится при циркуляции этой органической фазы в экстракторе 2 в противотоке водному раствору, обозначенному А3 на фиг. 1, содержащему азотную кислоту, концентрация которой может составлять до 1–6 молей/л, но предпочтительно составляет от 4 до 6 молей/л.
Если этап «Промывка PF» проводится с применением водного раствора сильной кислотности, т.е. обычно равной или превышающей 3 моля/л HNO3, то этот этап содержит дополнительно раскисление органической фазы, которое проводят при циркуляции этой органической фазы в противотоке водному раствору, обозначенному А4 на фиг. 1 и являющемуся слабо кислым, т.е. содержащим от 0,1 до 1 моля/л HNO3, в качестве водного раствора с содержанием 0,5 моля/л HNO3 для исключения попадания слишком значительного количества кислоты в экстрактор, предназначенный для третьего этапа, обозначенного как «Реэкстракция Pu» на фиг. 1, и для исключения нарушения рабочих характеристик этого третьего этапа.
На этапе «Реэкстракция Pu» проводится повторное экстрагирование плутония до степени окисления +IV органической фазы, образовавшейся на этапе «Промывка PF».
Он проводится при обеспечении циркуляции этой органической фазы в экстракторе 3 в противотоке водному раствору, обозначенному как «А2» на фиг. 1, содержащему 0,1–0,5 моля/л HNO3, предпочтительно при соотношении расходов О/А свыше 1, предпочтительно равном или превышающем 3, более предпочтительно равным или превышающим 5 с тем, чтобы экстрагирование плутония (IV) сопровождалось его обогащением.
Реэкстракция плутония (IV) на этапе «Реэкстракция Pu», сопровождается реэкстракцией фракции урана (VI), который также присутствует в органической фазе, образовавшейся при «Промывке PF».
Таким образом на четвёртом этапе, обозначенном как «Промывка U» на фиг. 1, предусмотрено экстрагировать из водной фазы, образовавшейся при «Реэкстакции Pu»:
- либо всё количество присутствующего в этой водной фазе урана (VI) в том случае, когда необходимо получить в конце этого этапа водный раствор с содержанием плутония (IV), но без урана (VI),
- либо количество урана (VI), обеспечивающее получение в конце этого этапа водного раствора, содержащего уран (VI) и плутоний (IV) в заданном соотношении.
В обоих случаях «Промывка U» проводится при циркуляции в экстракторе водной фазы, полученной при «Реэкстракции Pu», в противотоке органической фазе, обозначенной как «S2» на фиг. 1, качественный и количественный состав которой предпочтительно одинаков с таким же составом органической фазы S1. Количество экстрагированного урана (VI) регулируется воздействием, с одной стороны, на соотношение О/А и, с другой стороны, на кислотность водной фазы, при этом уран (VI) в действительности экстрагируется тем лучше, чем больше соотношение между расходами органической и водной фаз и кислотность водной фазы. Добавка более или менее концентрированной азотной кислоты в водную, циркулирующую в экстракторе 4 фазу может быть предусмотрена в зависимости от кислотности, которую необходимо придать этой водной фазе.
В конце этих четырёх этапов получают:
- рафинат, соответствующий водной, выходящей из экстрактора 1 фазе и содержащий продукты деления, а также америций и кюрий,
- водную фазу, выходящую из экстрактора 4 и содержащую либо дезактивированный плутоний (IV), либо смесь дезактивированных плутония (IV) и урана (VI),
- органическую фазу, выходящую из экстрактора 3 и содержащую уран (VI) без плутония (IV).
Эта органическая фаза может быть направлена непосредственно или после других видов обработки в экстрактор, не показанный на фиг. 1, в котором уран (VI) повторно экстрагируют из этой органической фазы, например, посредством водного раствора с содержанием не более 0,05 моля/л азотной кислоты в качестве водного раствора с содержанием 0,01 моля/л HNO3 при комнатной температуре (т.е. при 20–25°С) или в горячем состоянии (т.е. обычно при температуре 40–50°С) и предпочтительно при использовании соотношения расходов О/А свыше 1, с тем чтобы уран (VI) экстрагировался с обеспечением обогащения.
Упомянутые источники
1. Международная заявка PCT WO 2017/017207.
2. Международная заявка PCT WO 2017/017193.
3. E.K. Dukes и T.H. Sidall, Journal of Inorganic and Nuclear Chemistry 1966, 28(10), стр. 2307–2312.
4. G.M. Chumakowa et al. Radiochimiya 1980, 22(2), стр. 213 – 217.
5. B.G. Vats et al., Dalton Transactions 2016, 45(25), стр. 10319 – 10325.
Реферат
Изобретение относится к применению карбамида общей формулы (I), где R1, R2 и R3, одинаковые или разные, означают линейную или разветвлённую алкильную группу с 1–12 атомами углерода, R4 означает атом водорода или линейную или разветвлённую алкильную группу с 1–12 атомами углерода, и где карбамид содержит общее количество атомов углерода от 17 до 25, в качестве экстрагента для полного или частичного разделения урана (VI) и плутония (IV), без восстановления плутония (IV) в плутоний (III), на основе водного раствора А1, полученного растворением отработавшего ядерного топлива в азотной кислоте. При этом применением предусмотрено: а) соэкстракция урана (VI) и плутония (IV) из водного раствора А1, при этом соэкстракция включает в себя, по меньшей мере, контакт в экстракторе водного раствора А1 с органическим раствором S1, содержащим карбамид в органическом растворителе, и разделение водного и органического растворов; б) реэкстракция плутония (IV) и фракции урана (VI) из органического раствора, полученного на этапе а) при реэкстракции, содержавшей, по меньшей мере, одно приведение в контакт в экстракторе органического раствора S1 с водным раствором А2, содержавшим 0,1–0,5 моля/л азотной кислоты, и разделение органического и водного растворов; в) полная или частичная экстракция фракции урана (VI), содержащегося в водном растворе, полученном на этапе б), при этом экстракция содержит, по меньшей мере, одно приведение в контакт в экстракторе водного раствора с органическим раствором S2, содержащим карбамид в органическом растворе, и разделение водного и органического растворов; в результате чего получают водный раствор с содержанием плутония (IV), но без содержания урана (VI) или смесь плутония (IV) и урана (VI), и органический раствор с содержанием урана (VI) без плутония (IV). Также изобретение относится к новым карбидам. Карбамиды формулы (I) в присутствии водной фазы с сильной кислотностью обеспечивают коэффициенты распределения урана (VI) и плутония (IV), позволяющие проводить количественную соэкстракцию этих обоих элементов из водной фазы, и в присутствии водной фазы с умеренной кислотностью обеспечивают коэффициенты разделения (VI) и плутония (IV), при которых происходит отделение урана (VI) от плутония (IV) без необходимости восстановления плутония (IV) в плутоний (III). 3 н. и 13 з.п. ф-лы, 1 ил., 3 табл., 1 пр.

Формула


Комментарии