Мутанты стрептокиназы и их ковалентно модифицированные формы - RU2542392C2
Код документа: RU2542392C2
Чертежи

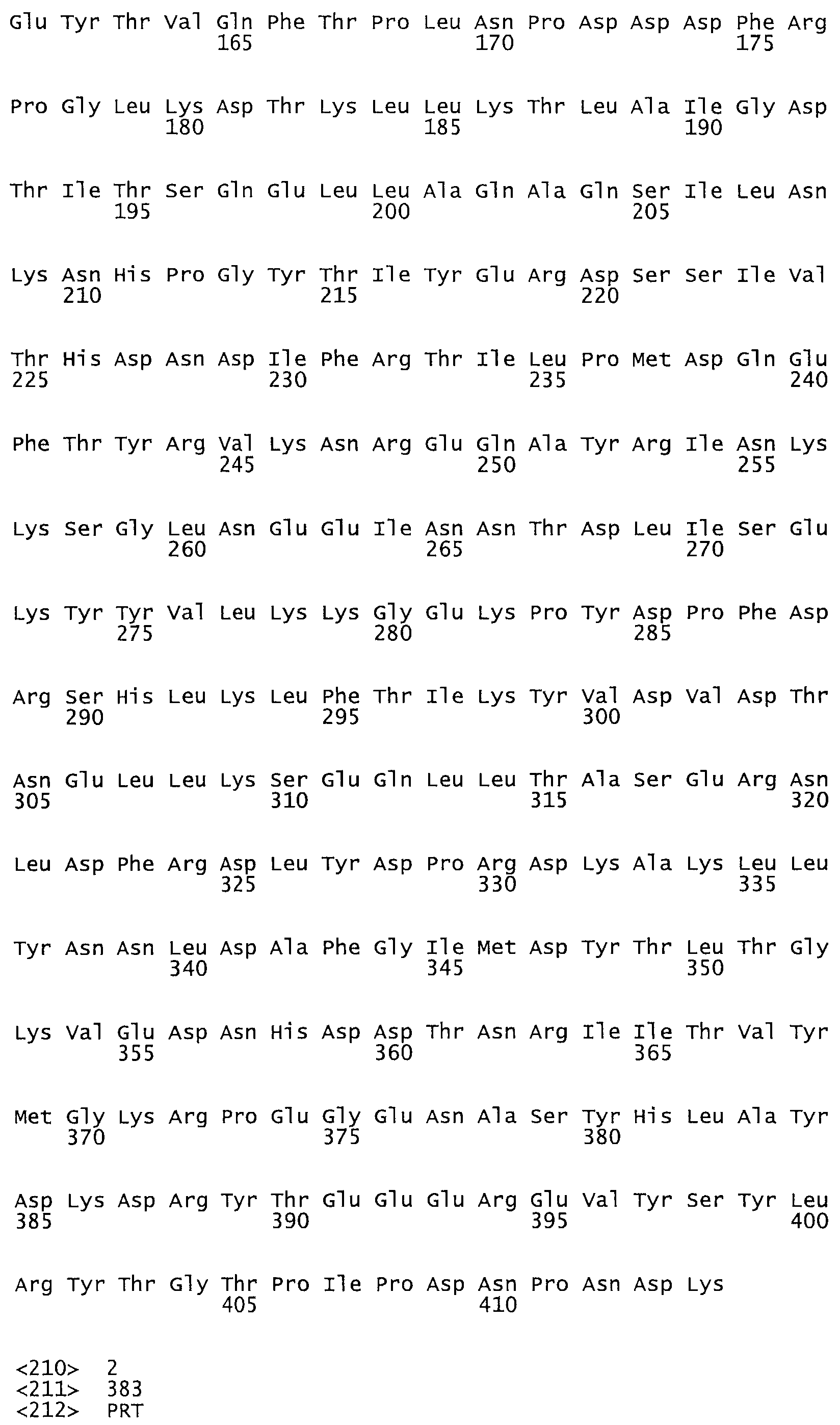
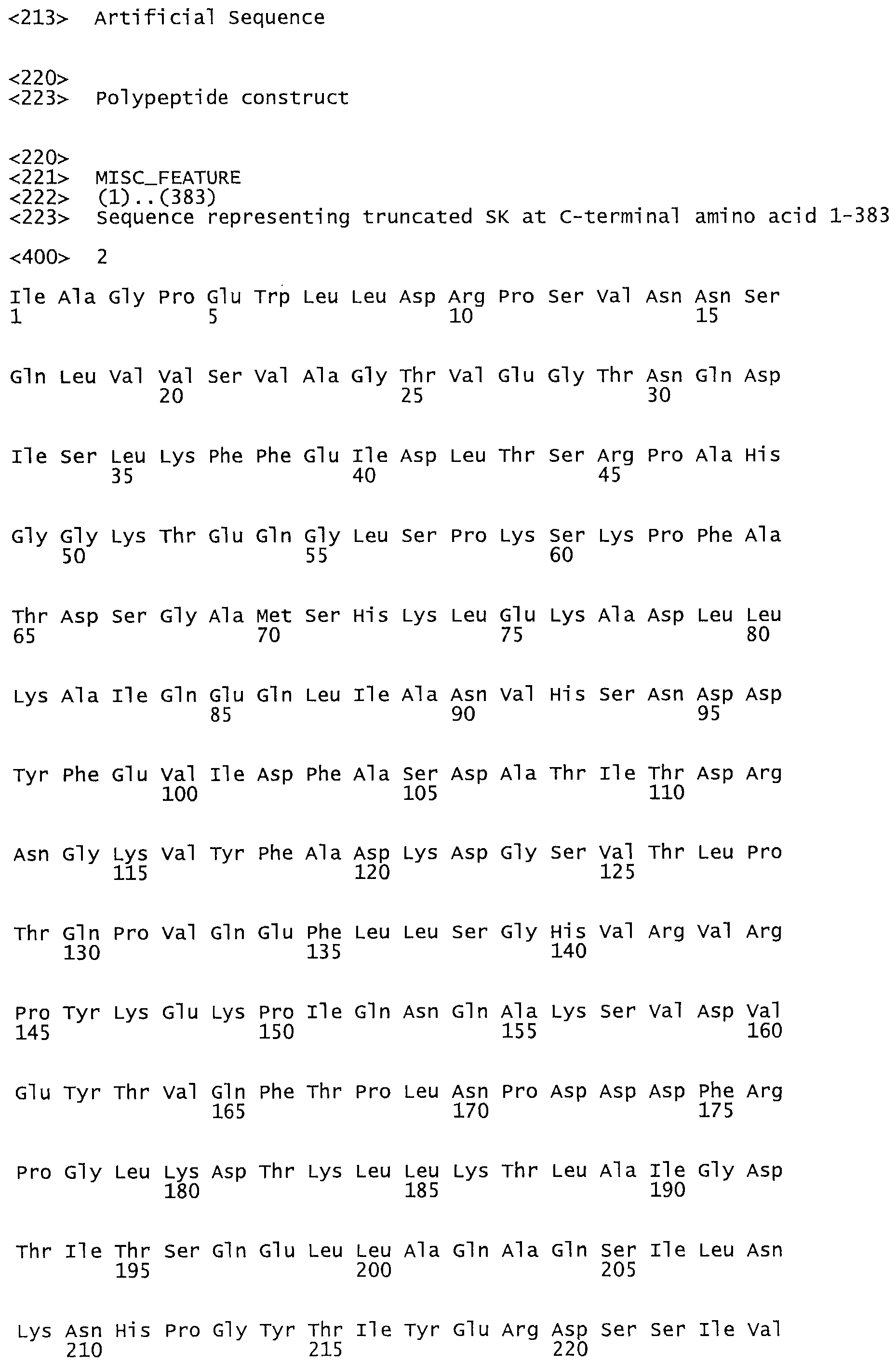

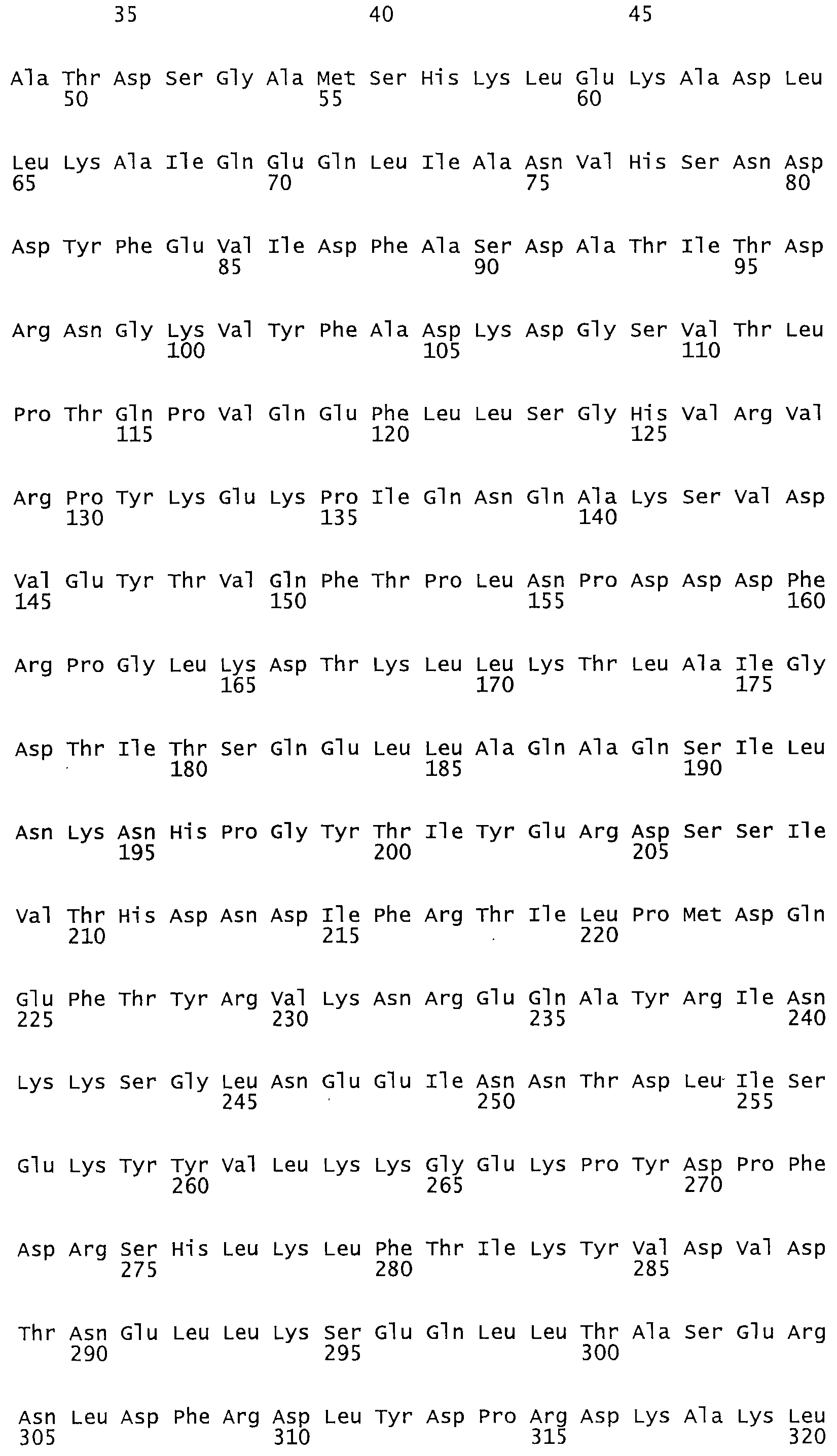

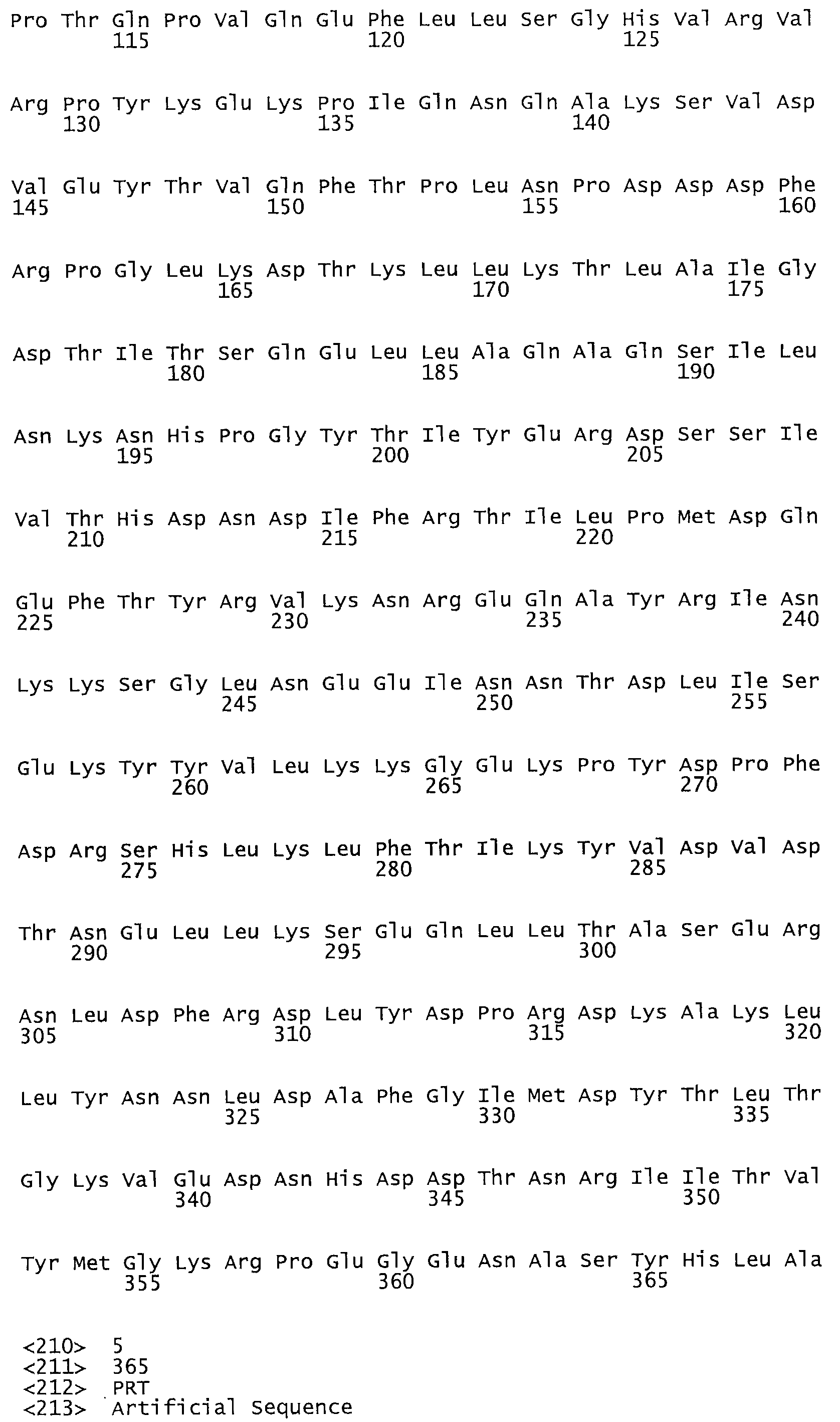
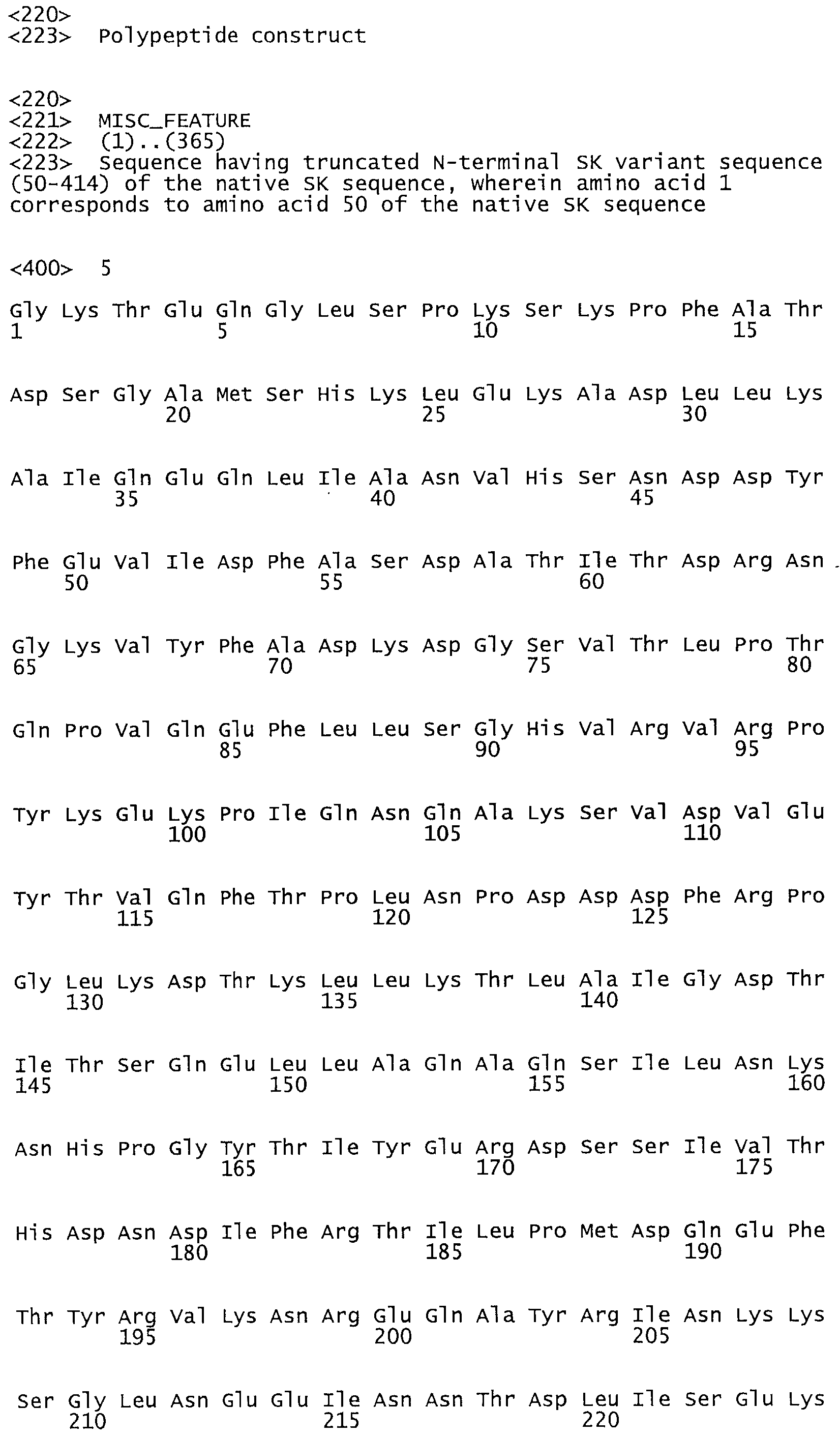

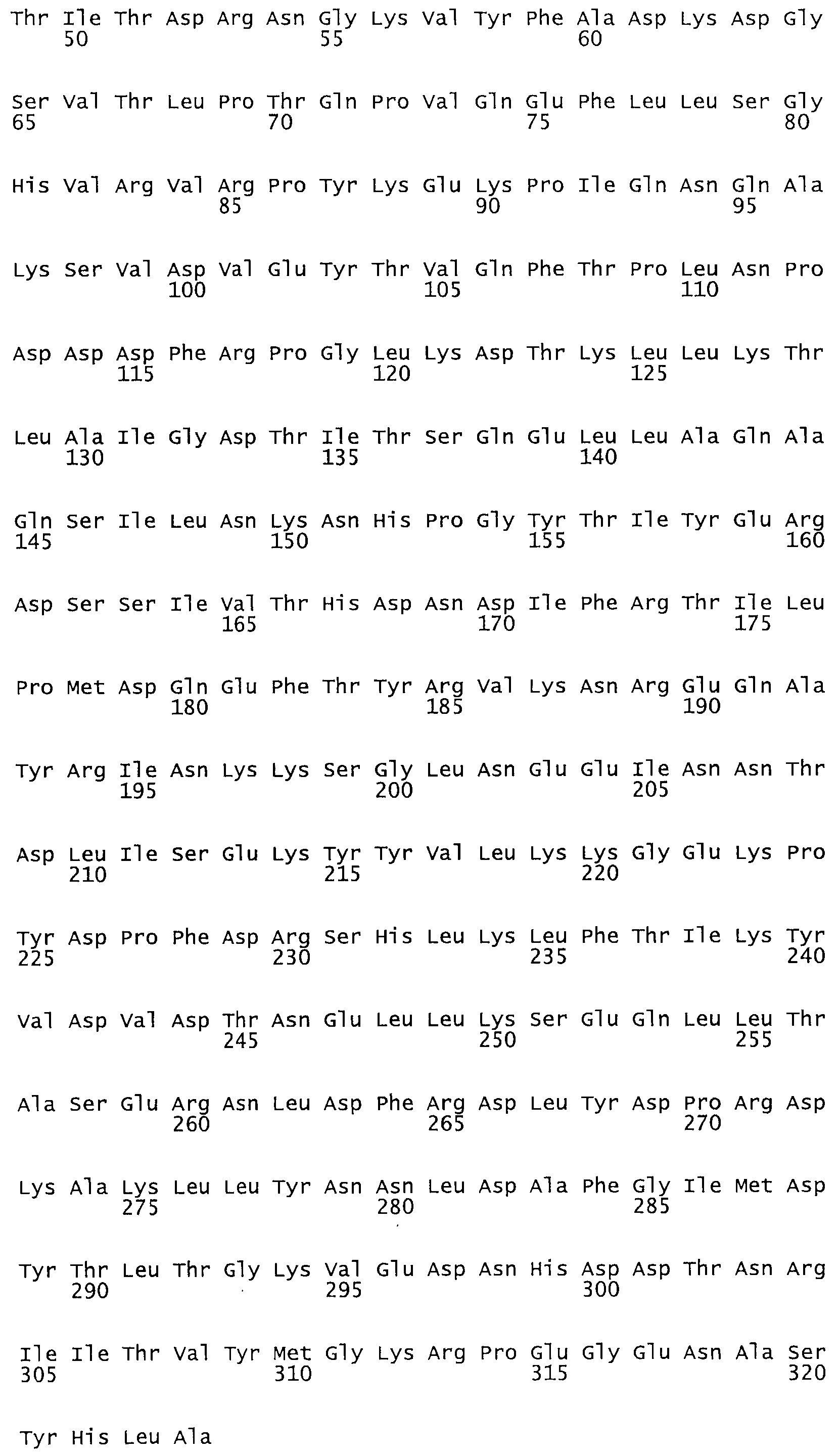

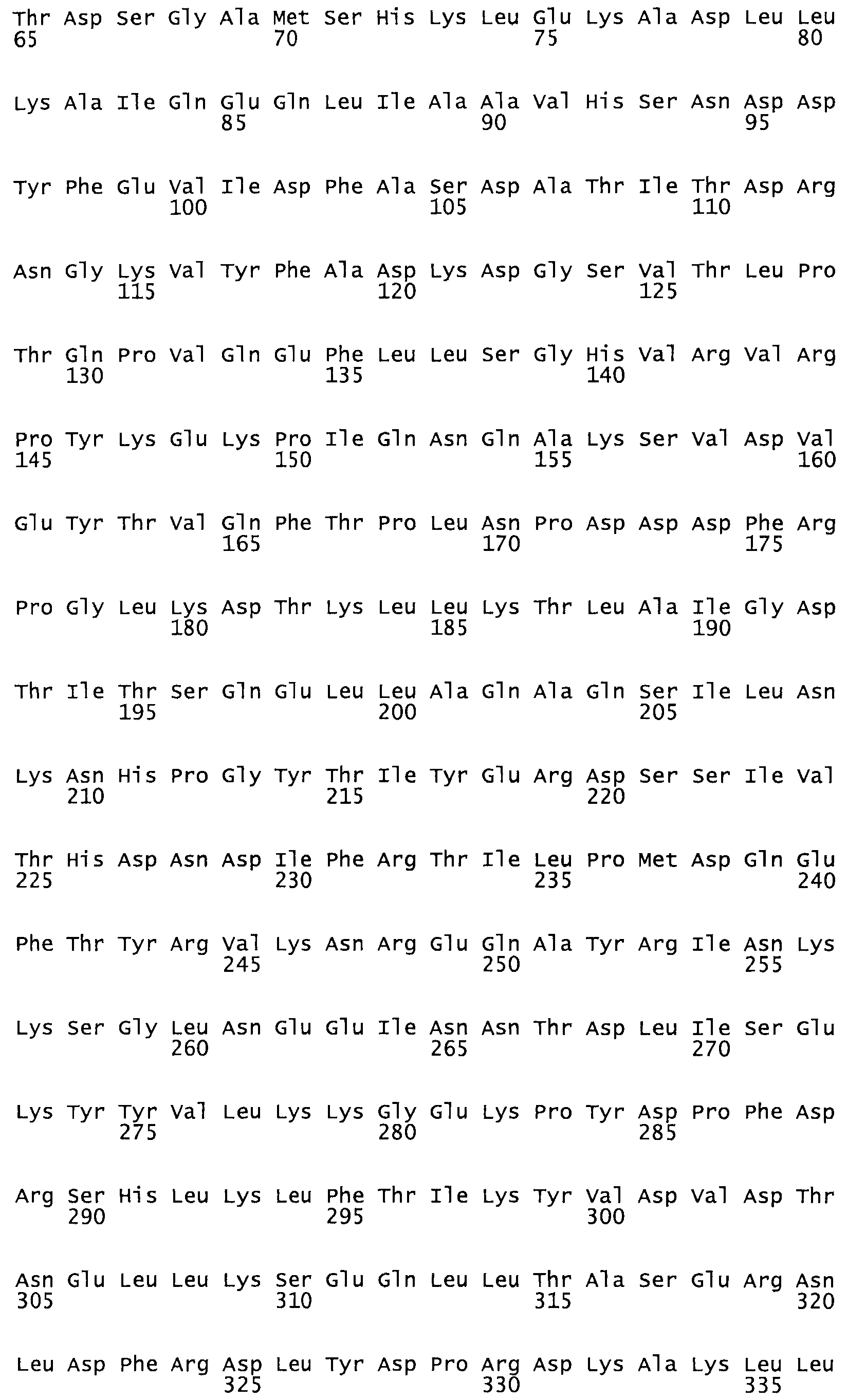

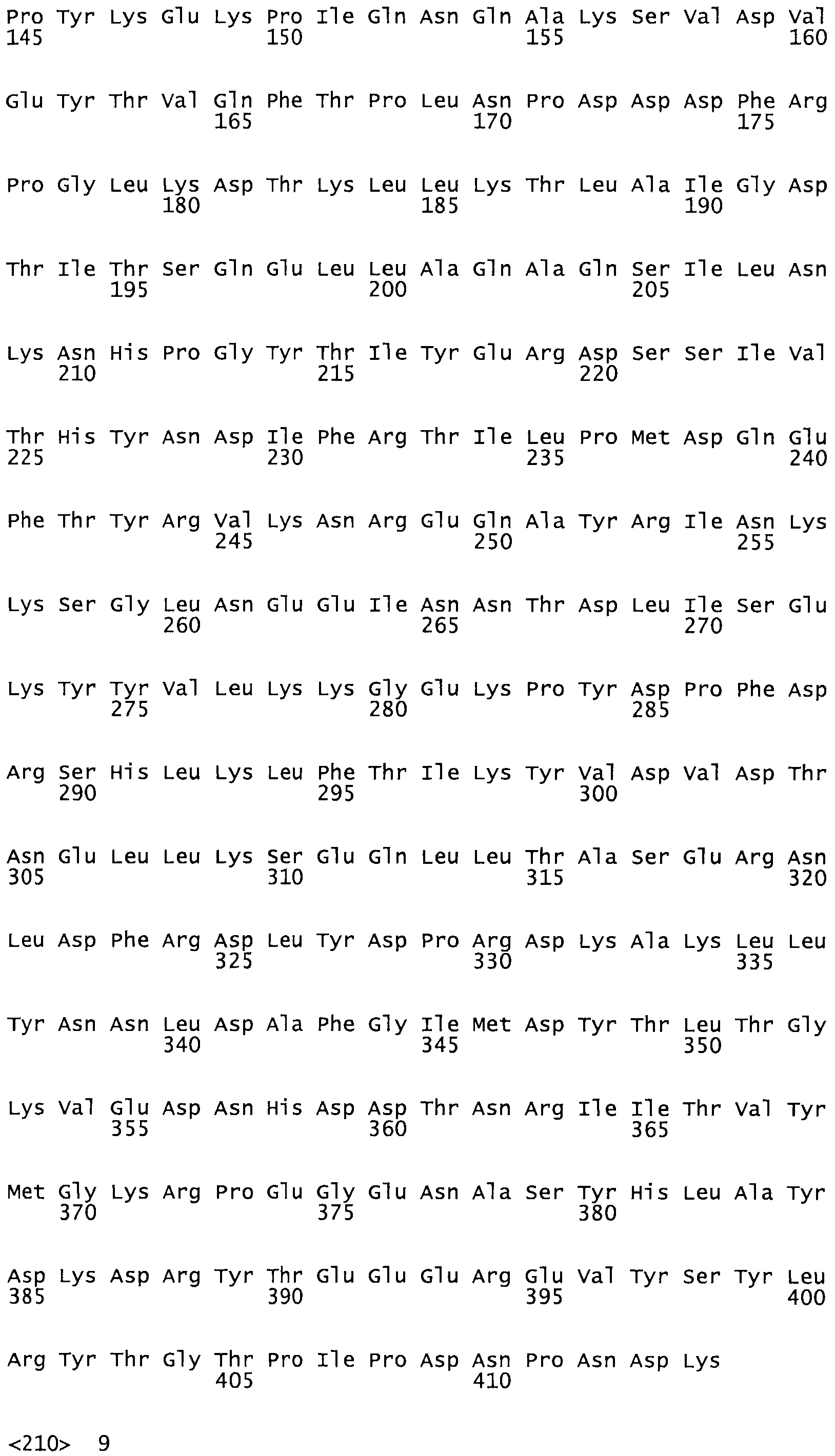
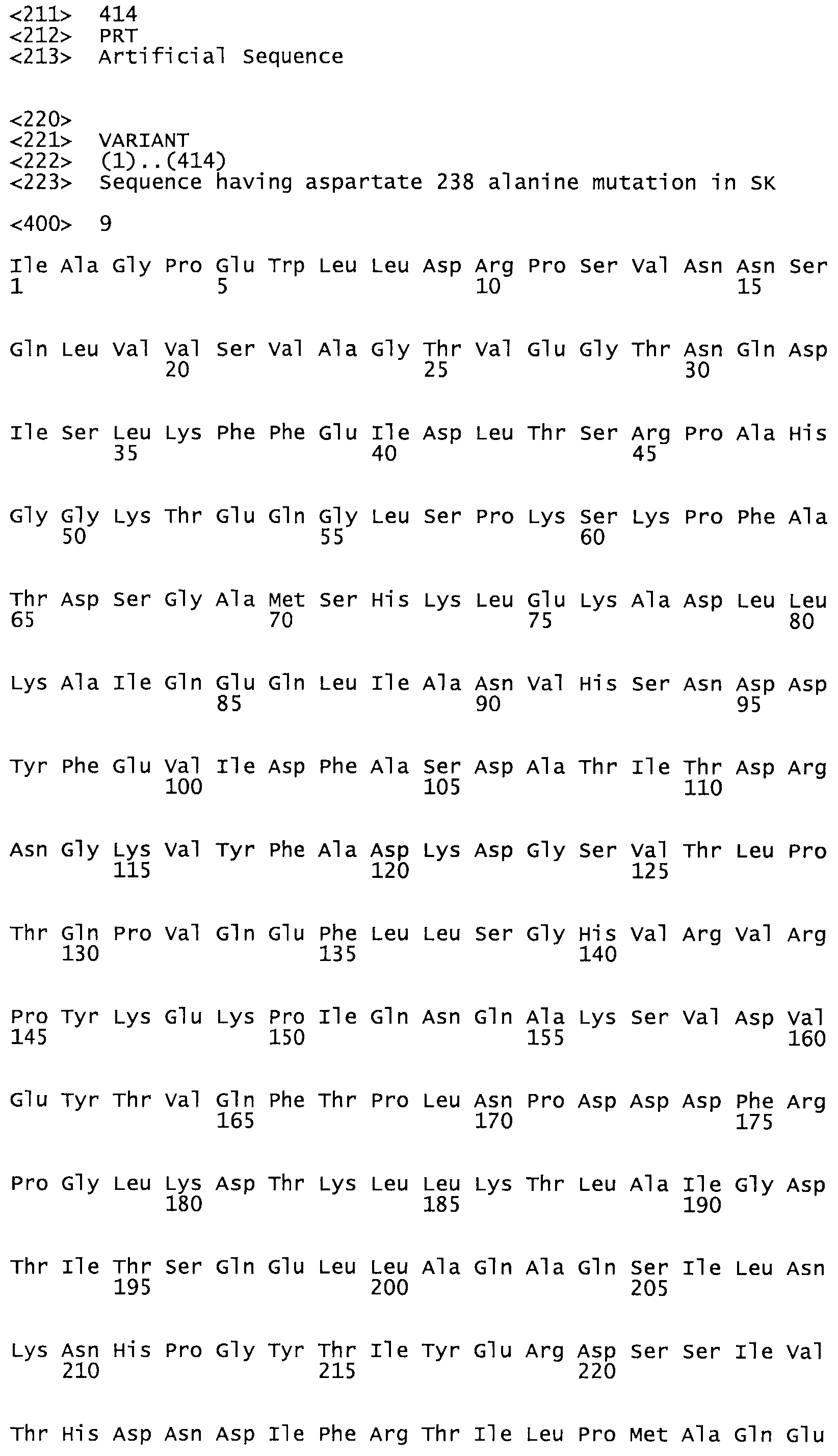

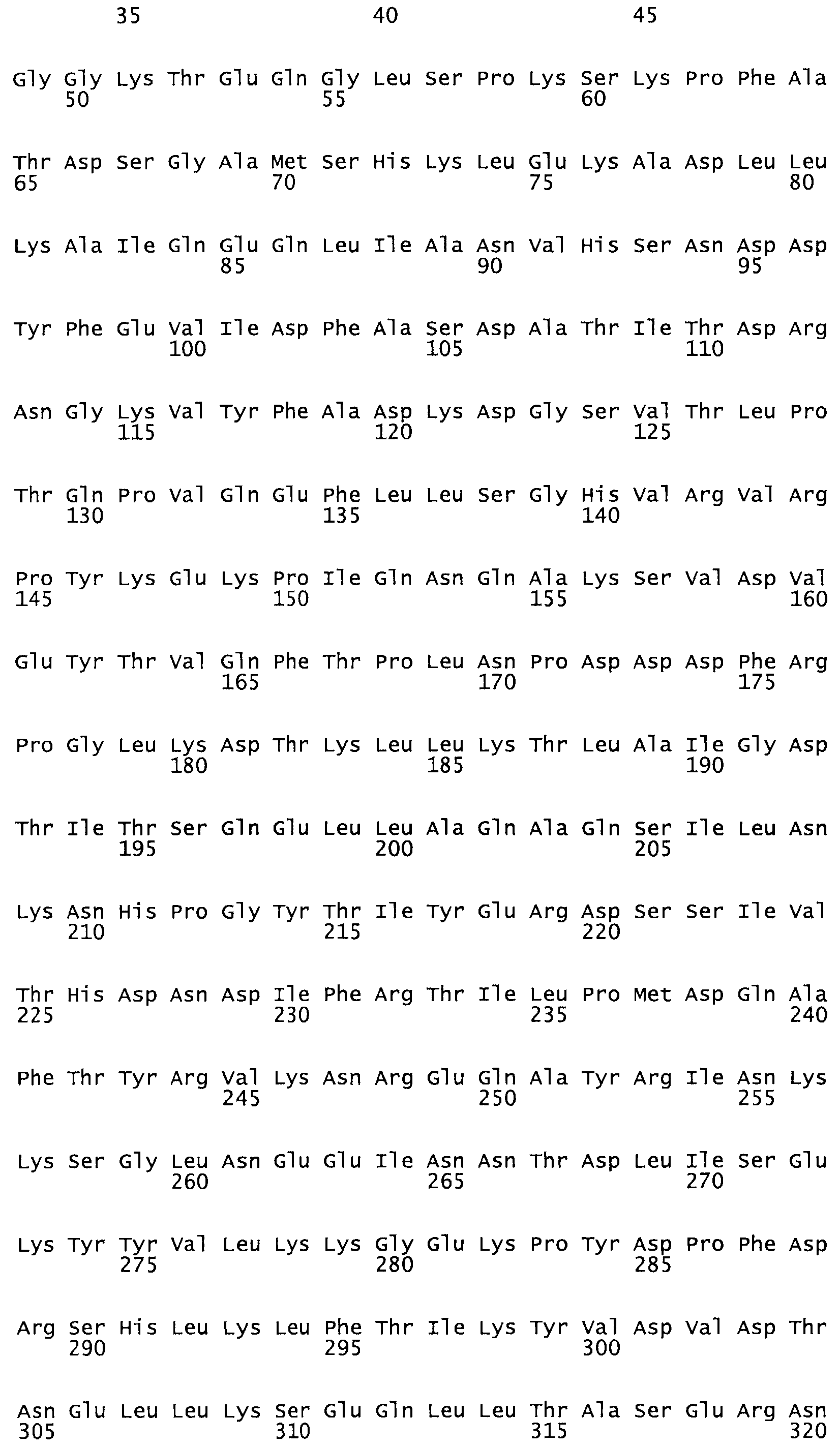


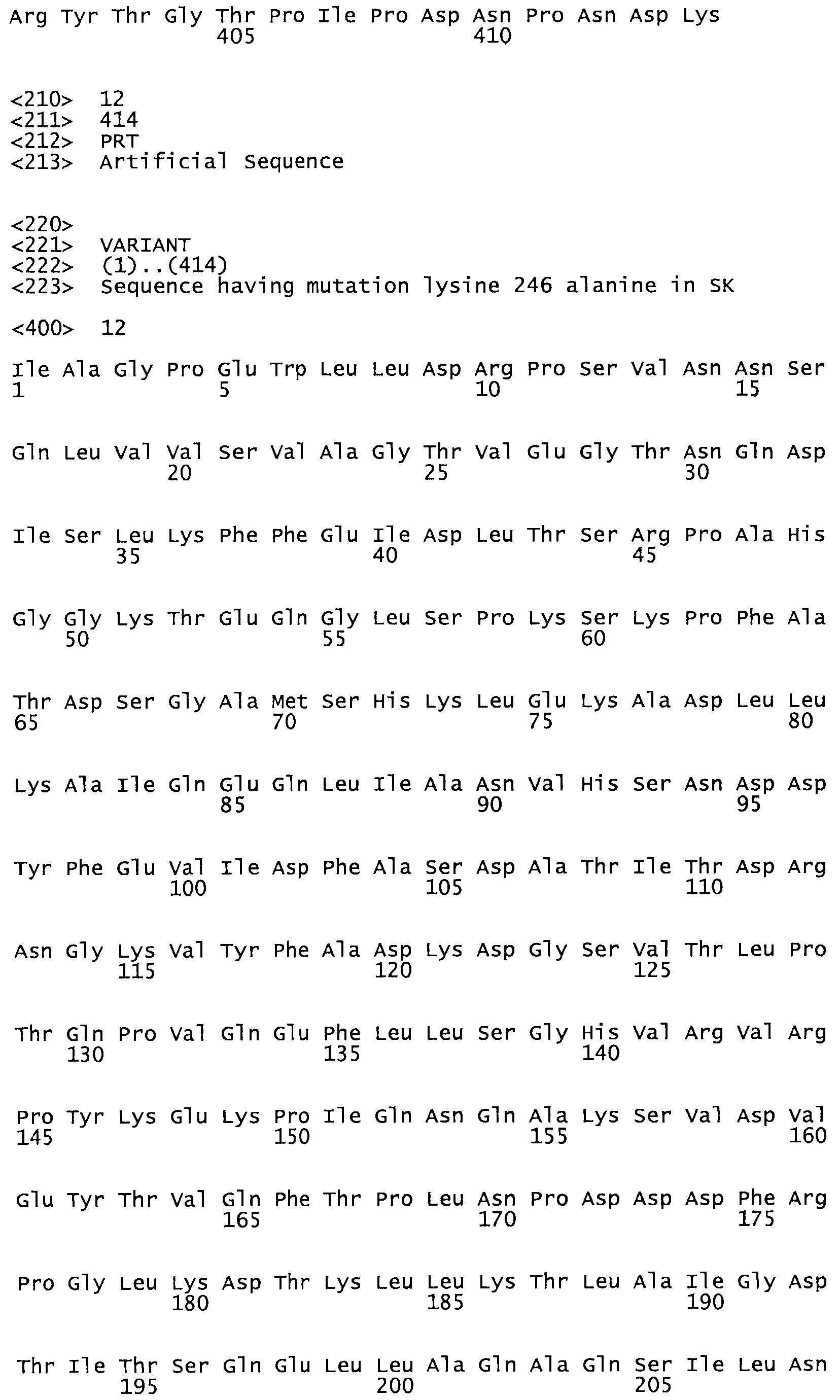
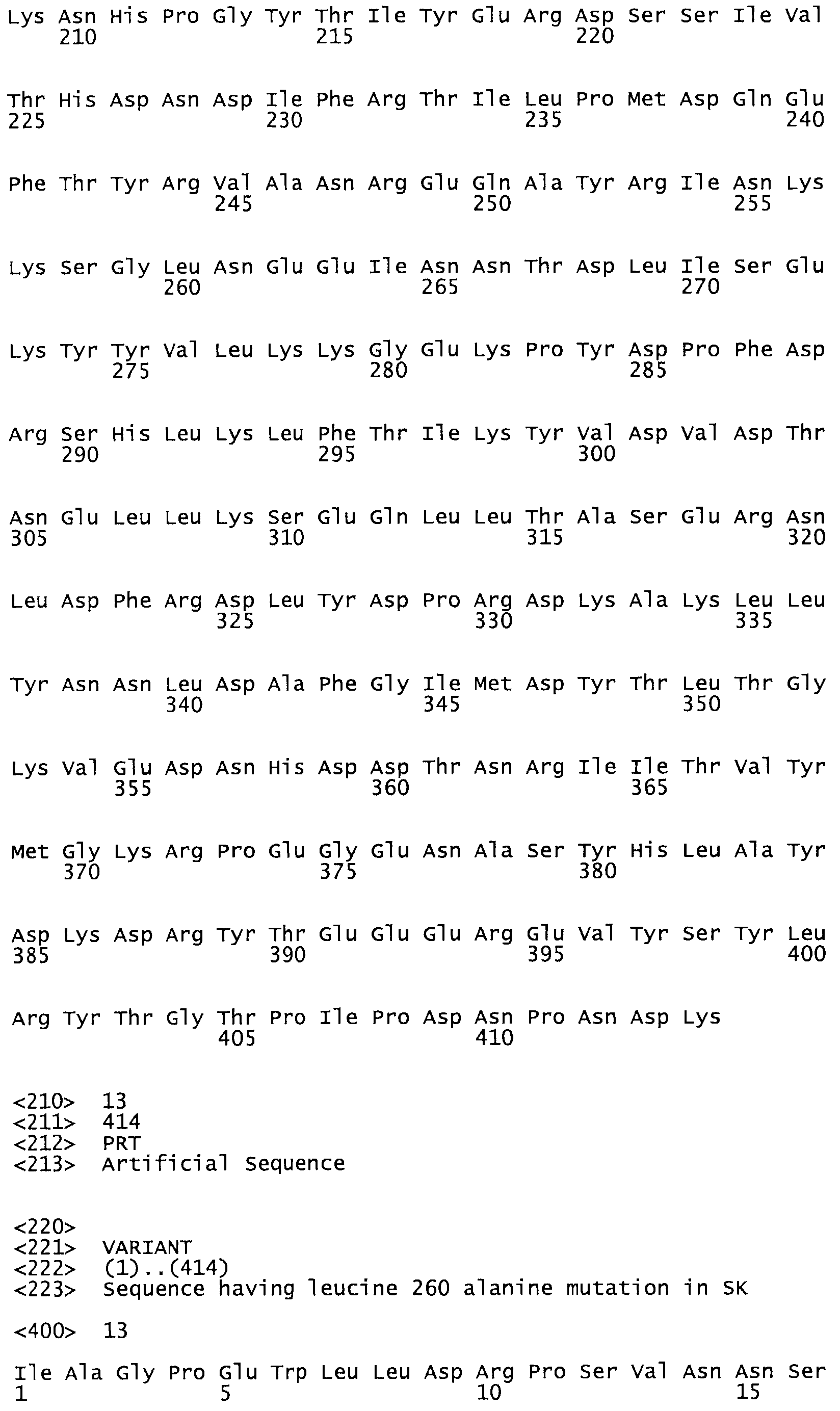
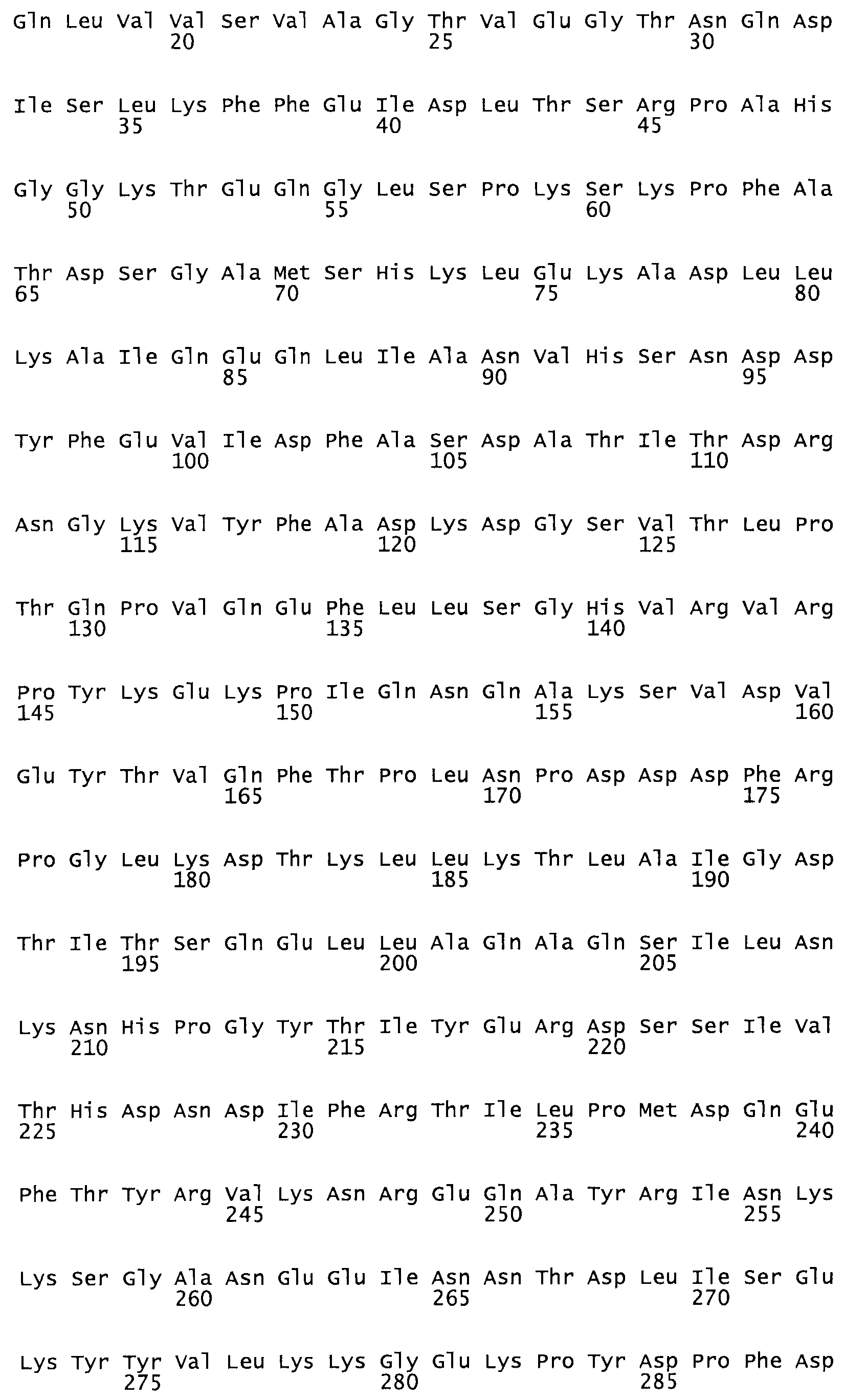



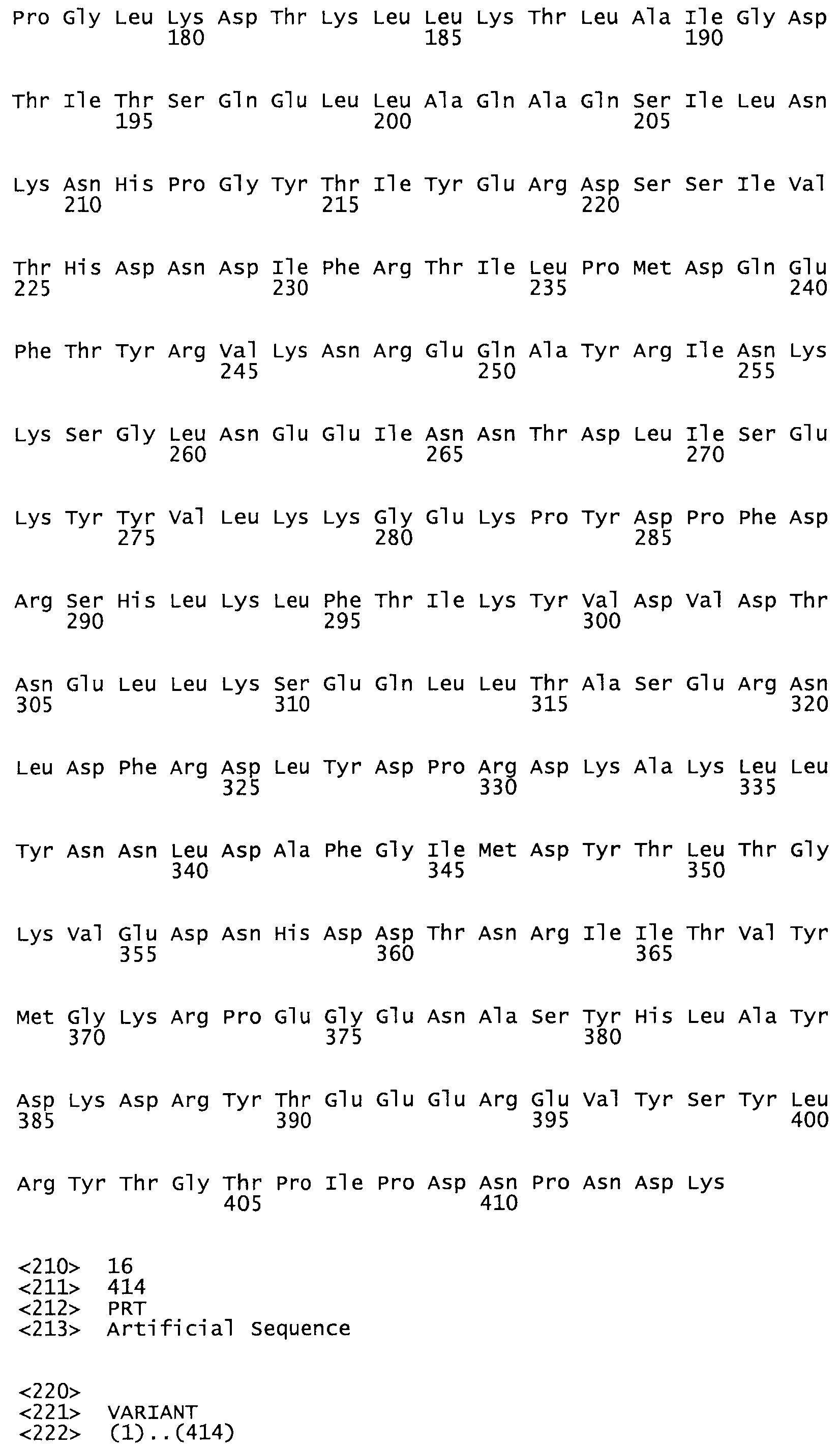
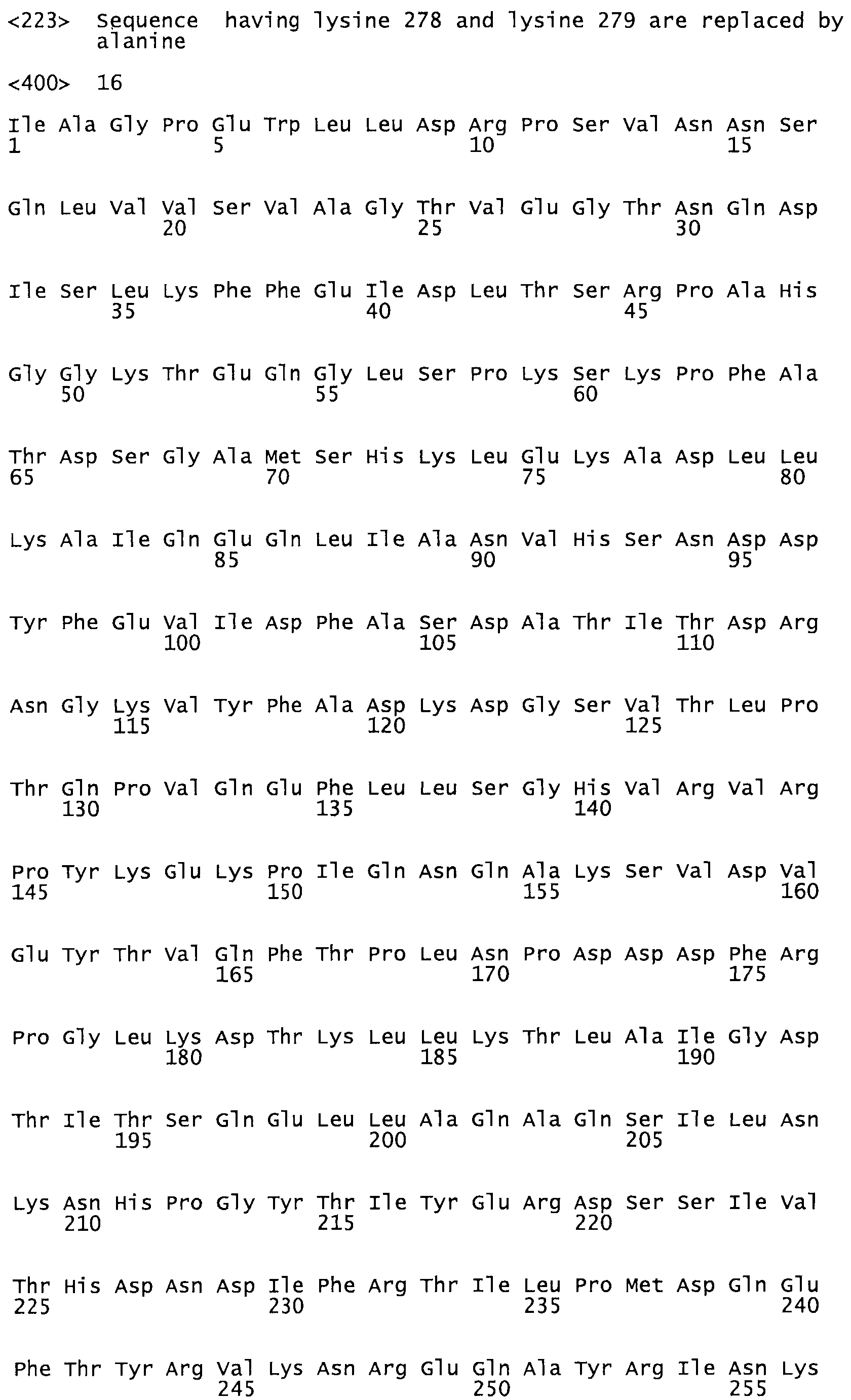

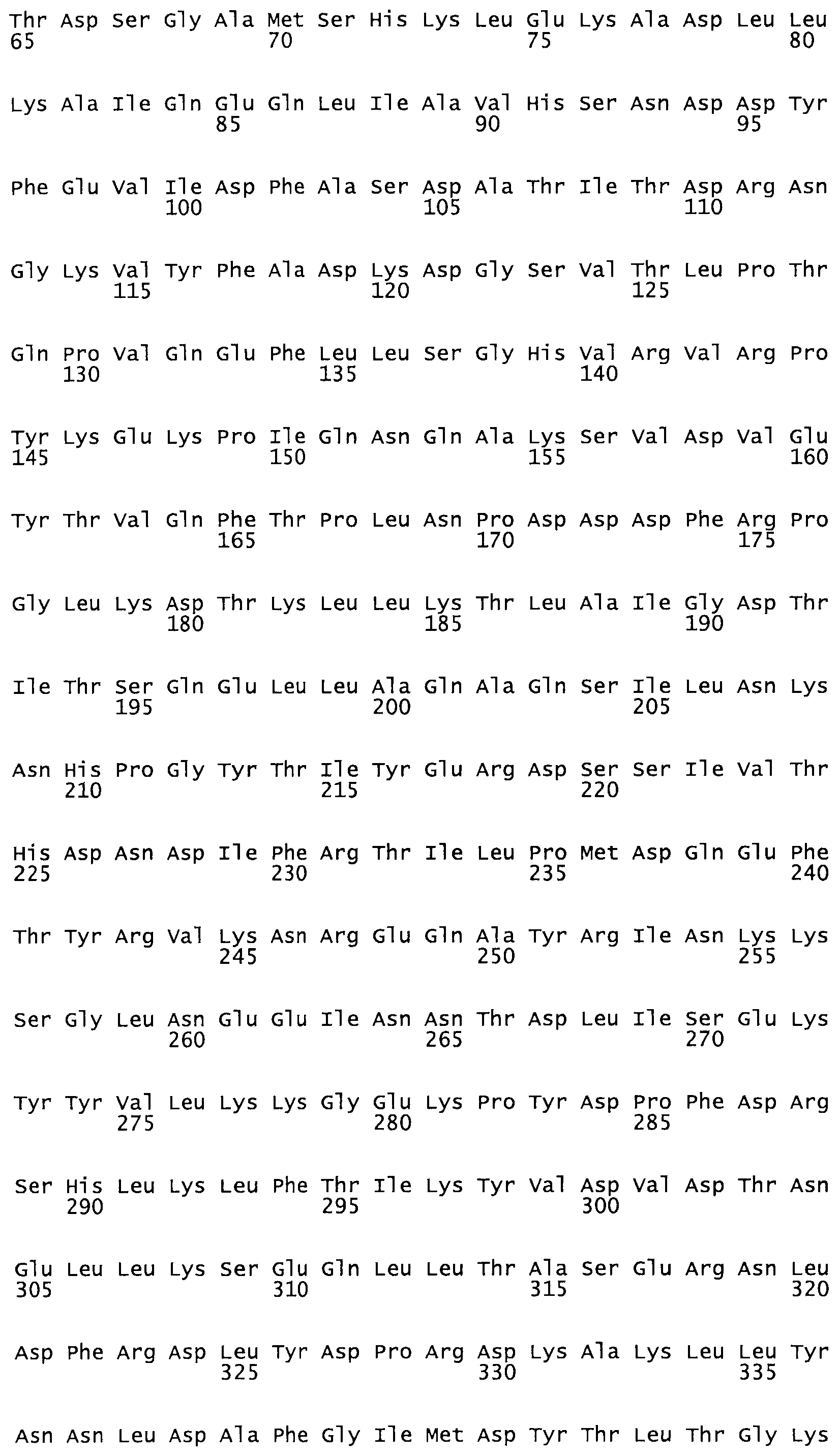

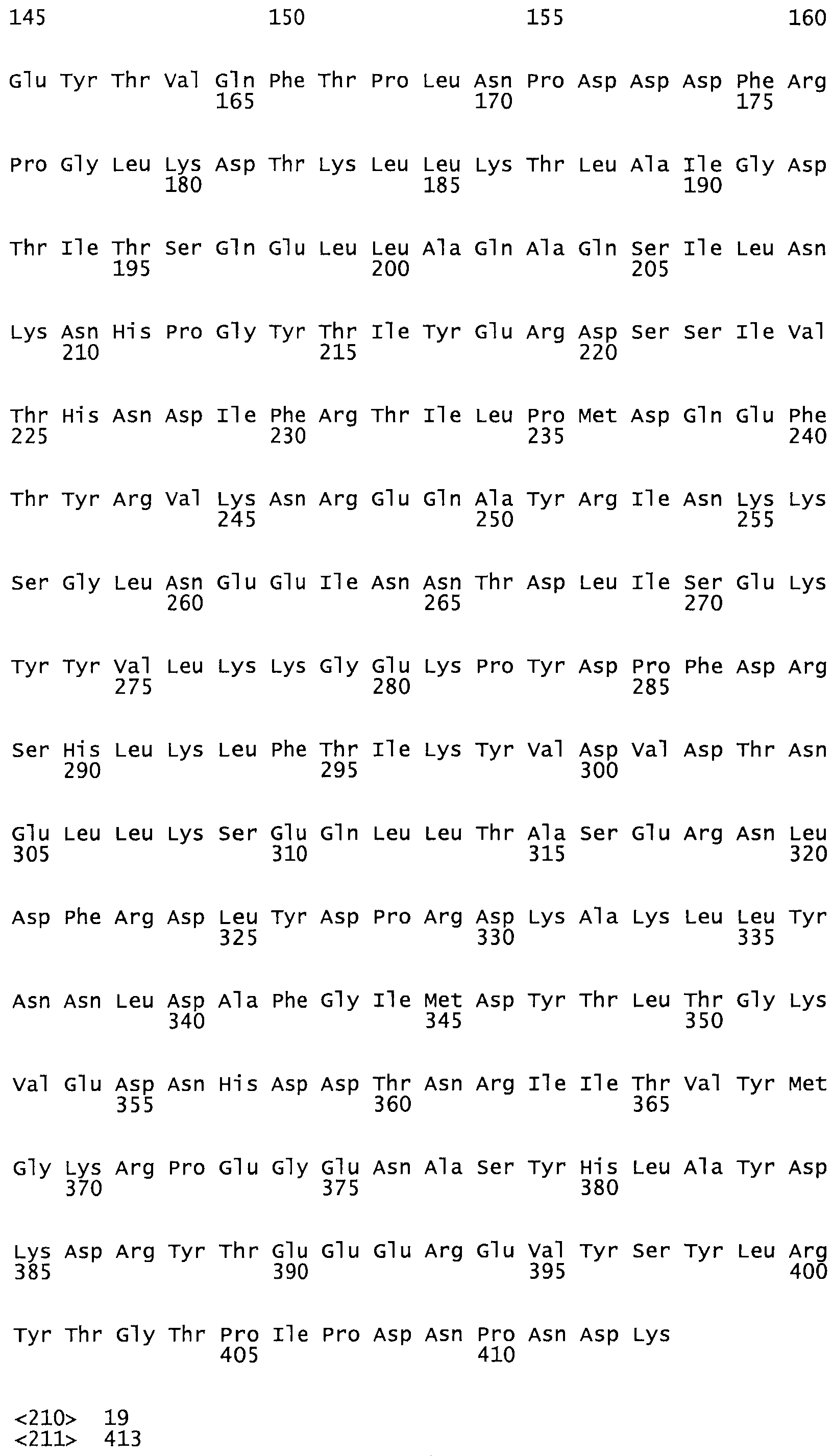
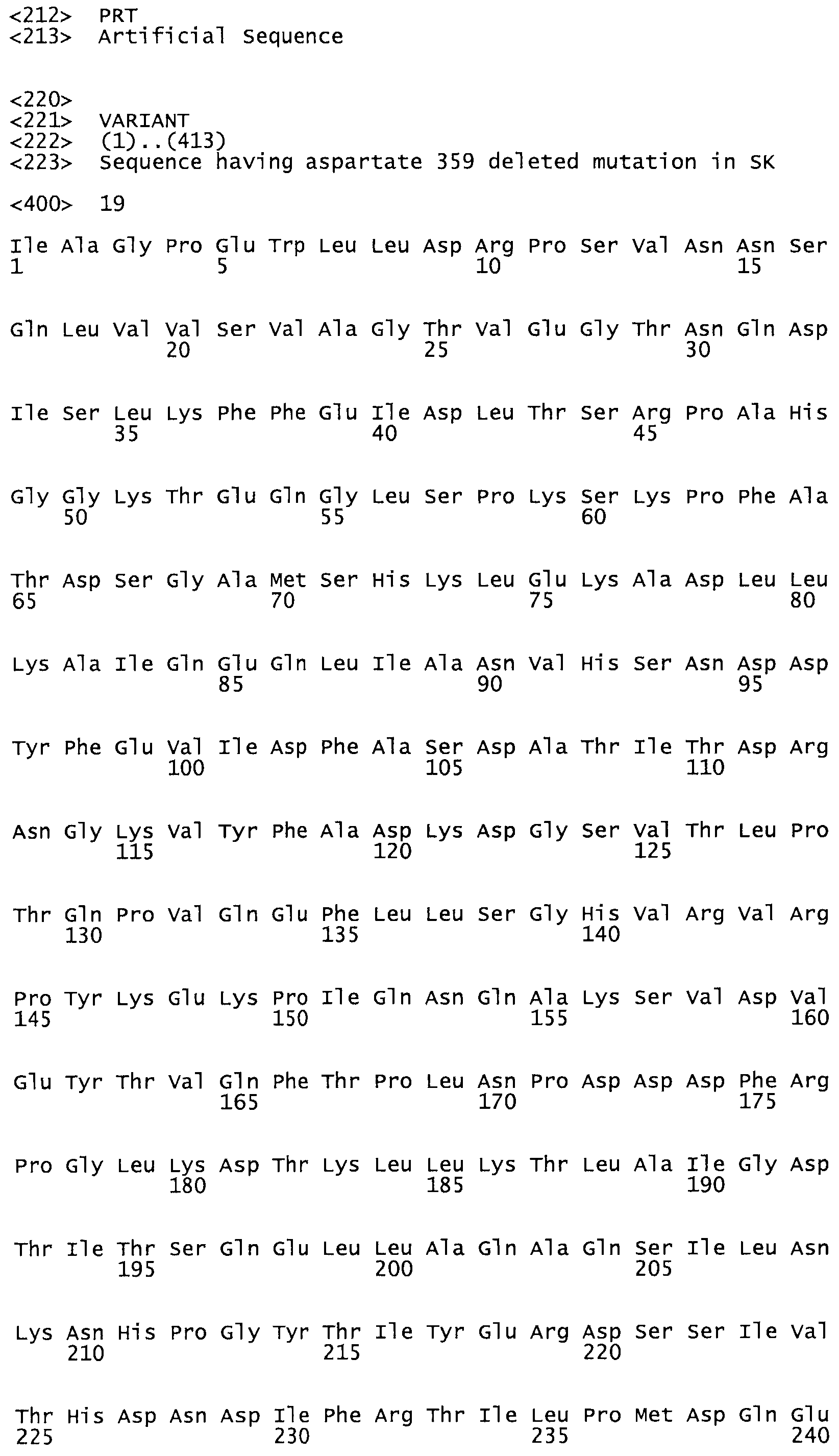



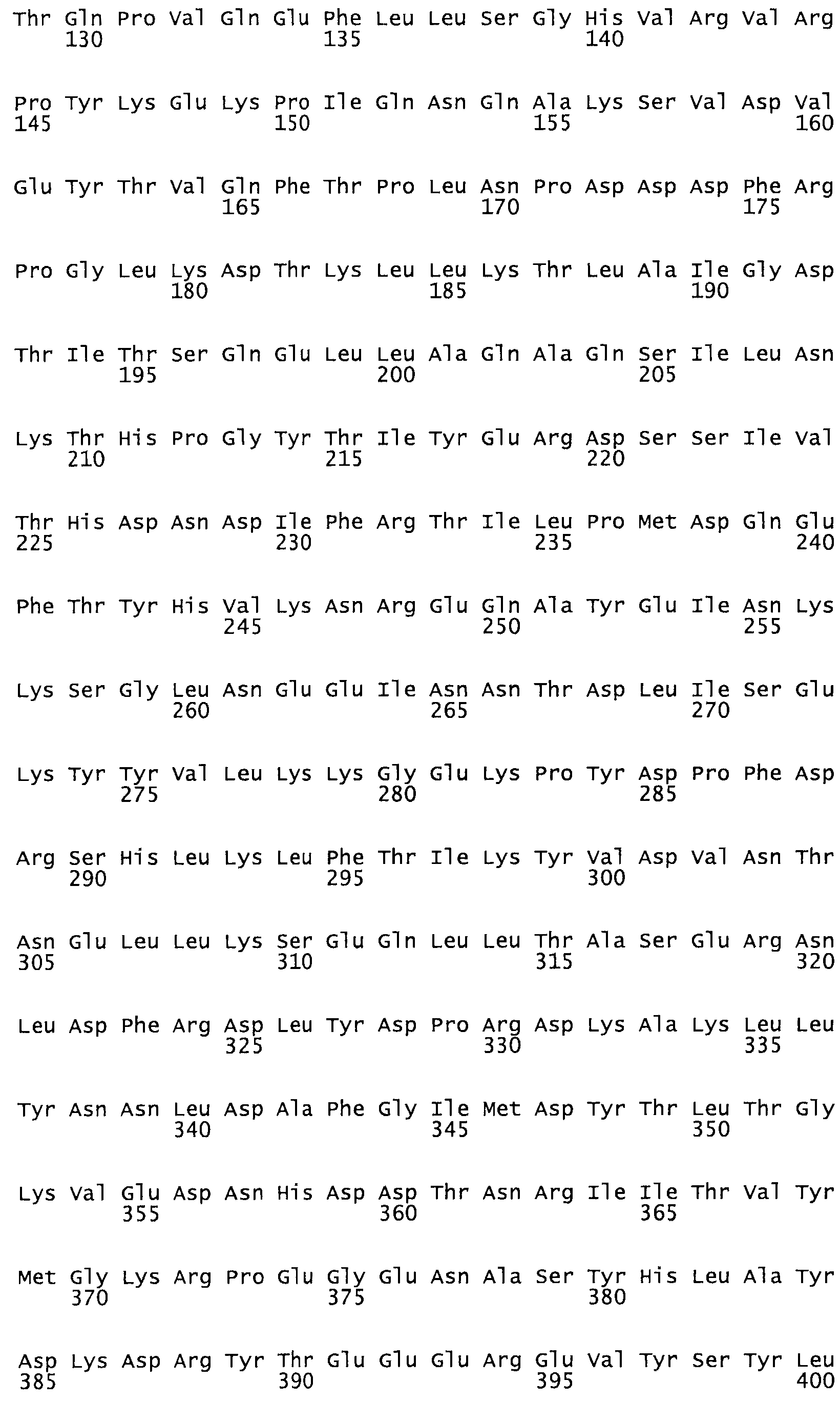

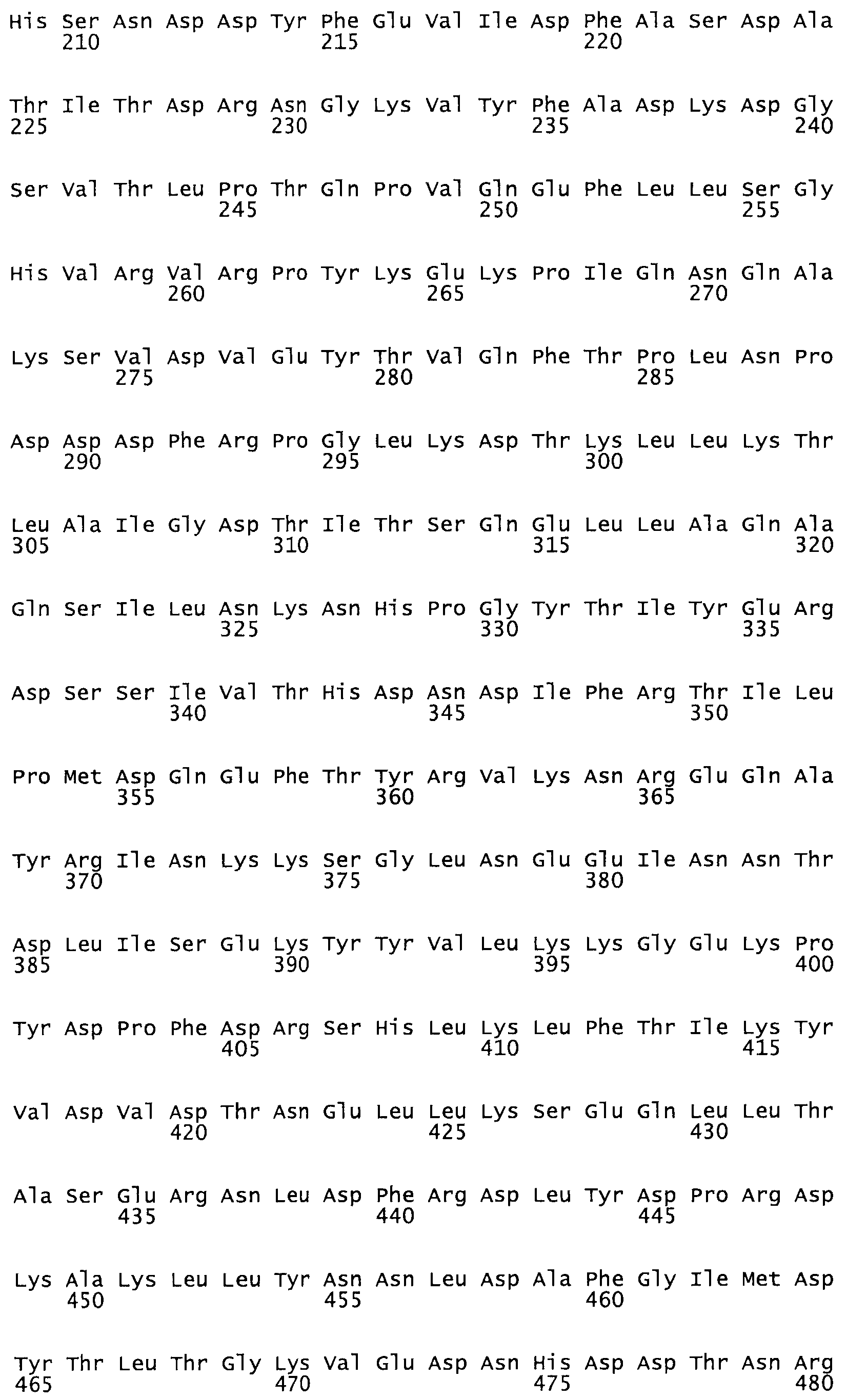

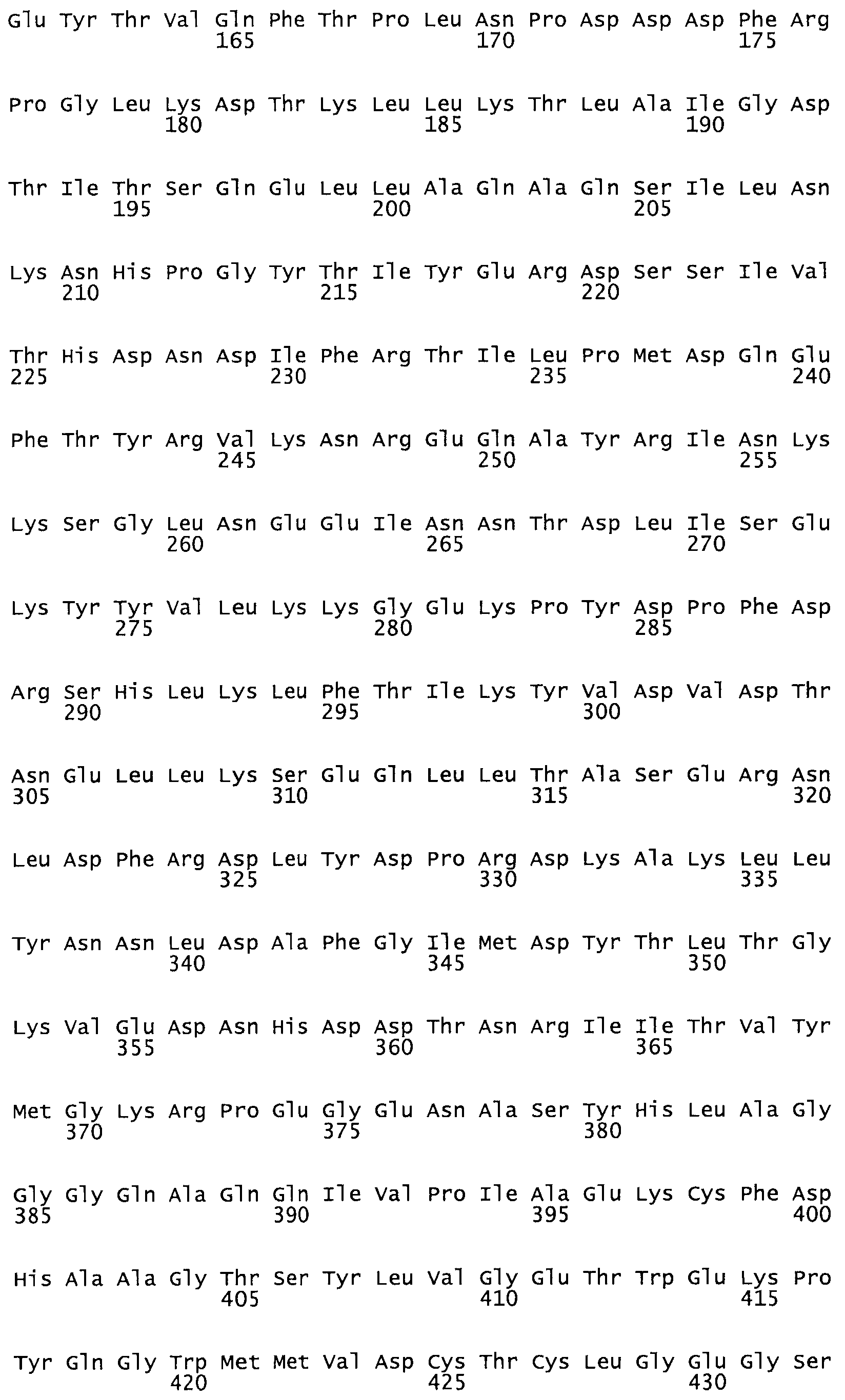

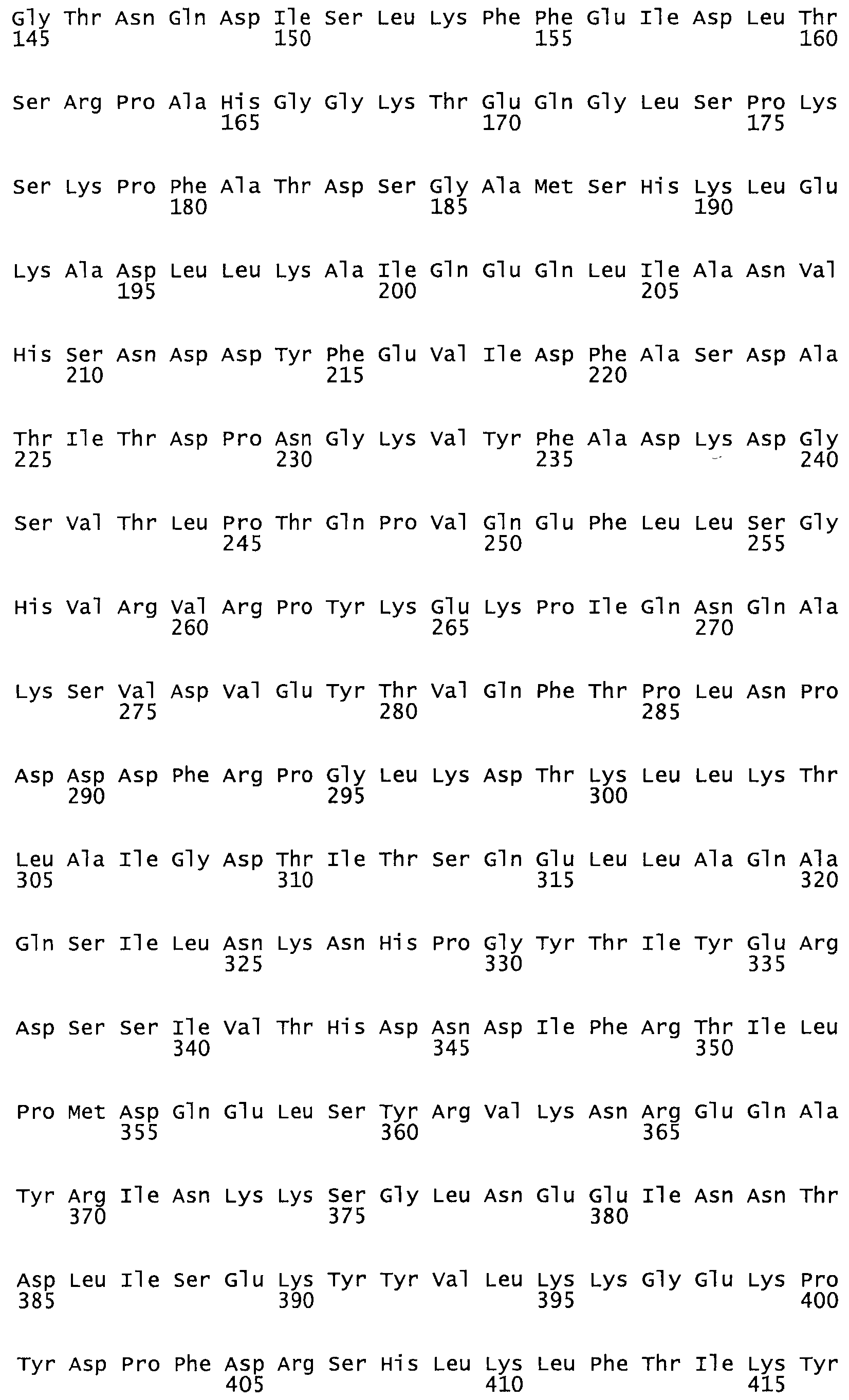

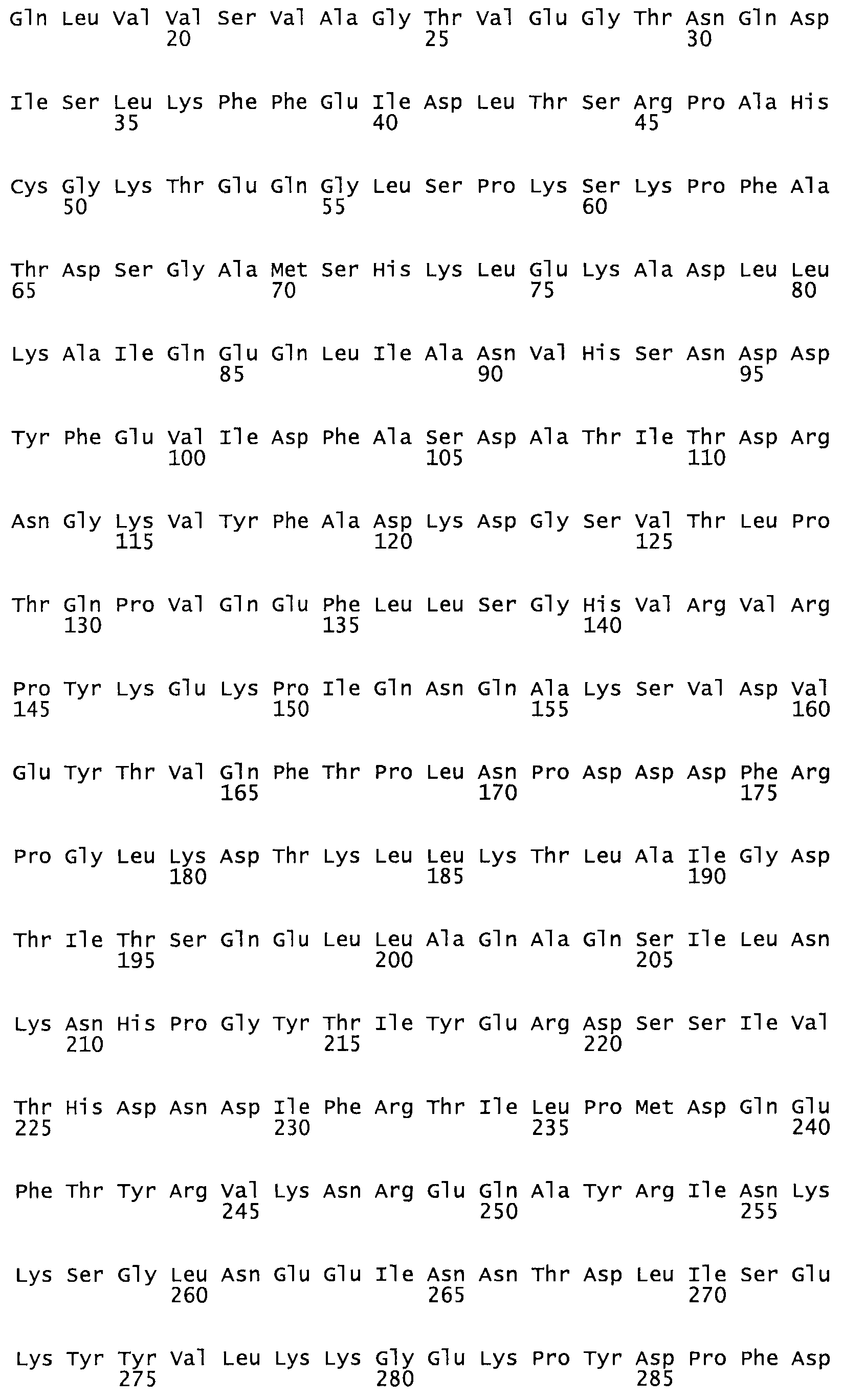

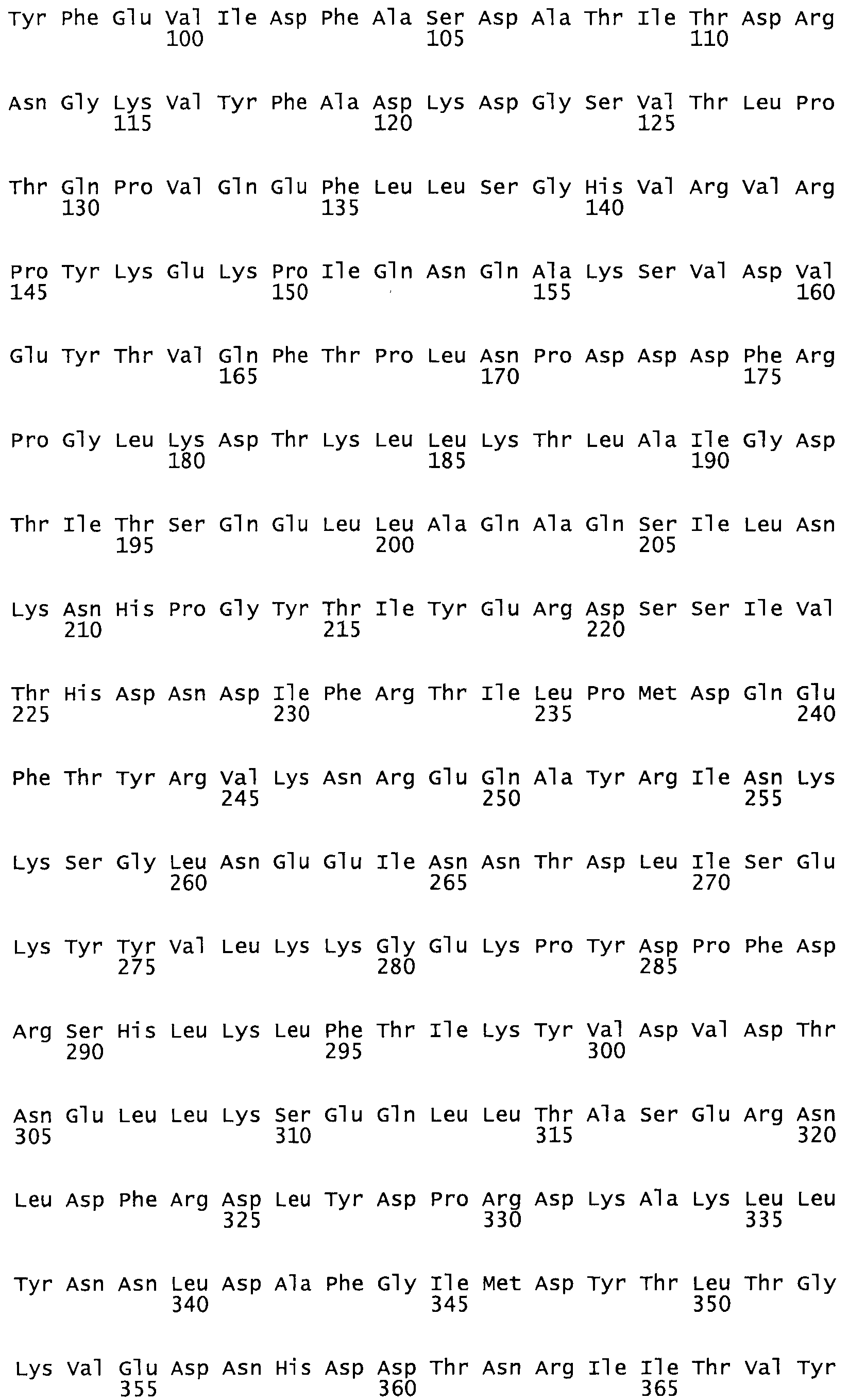

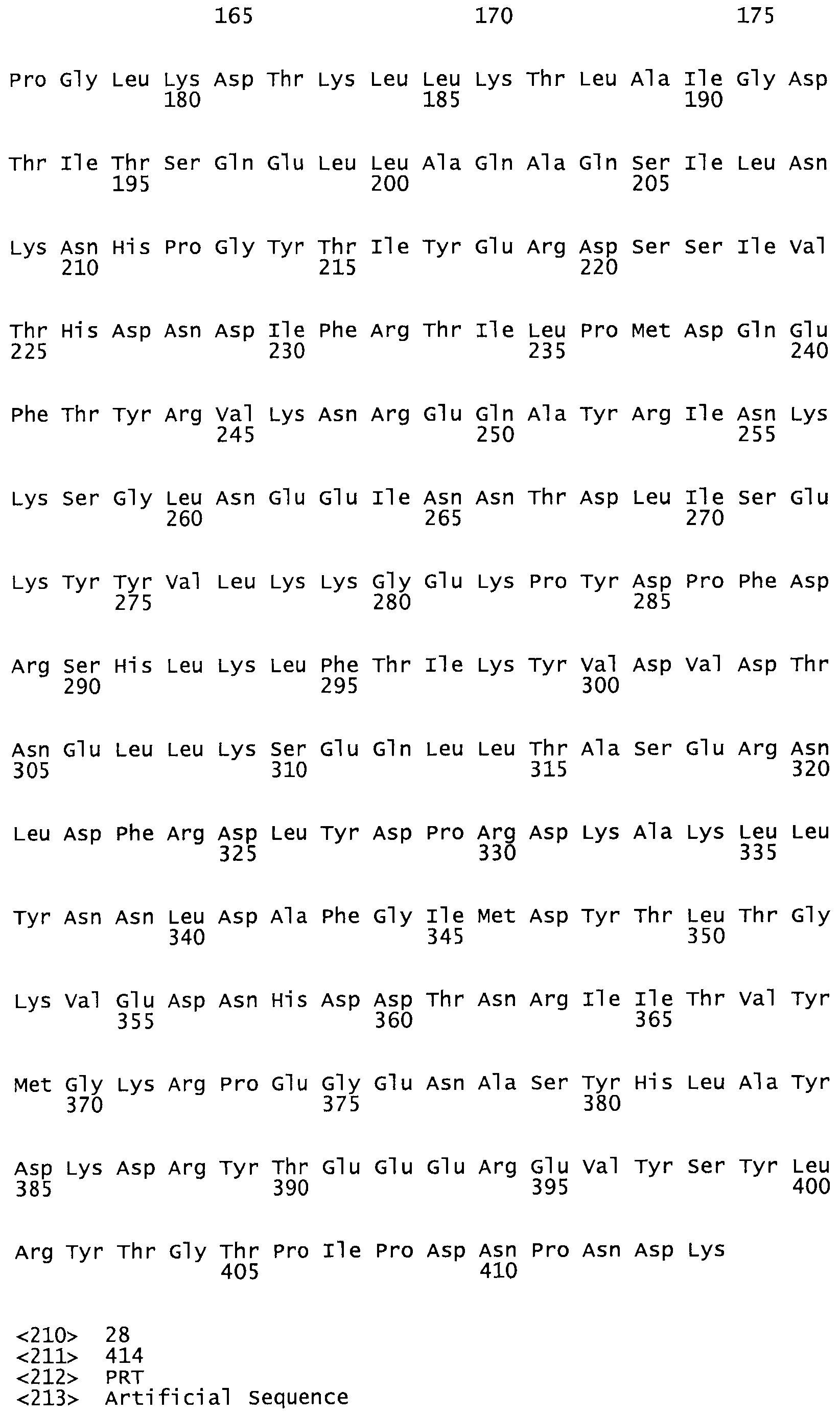
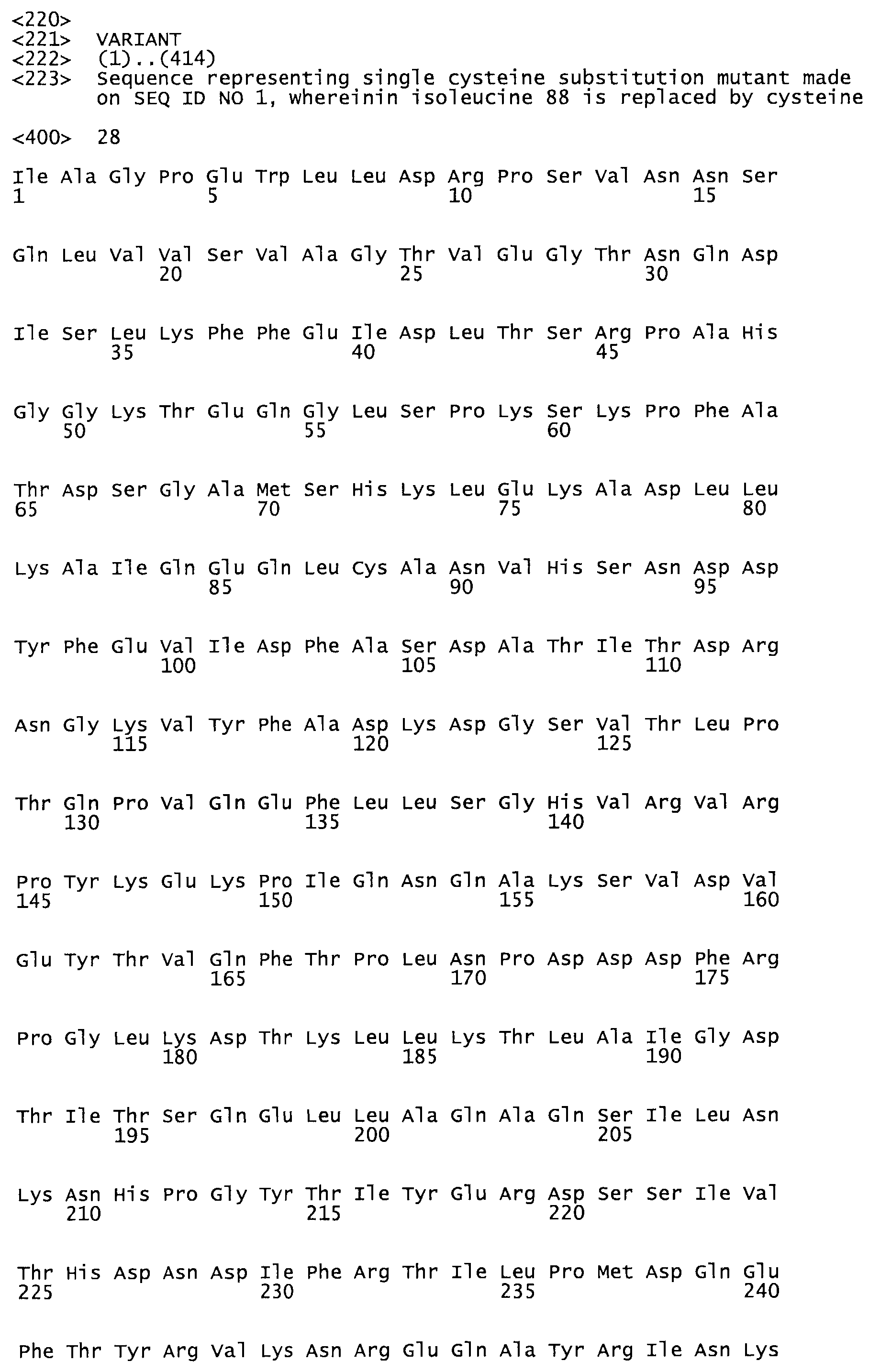

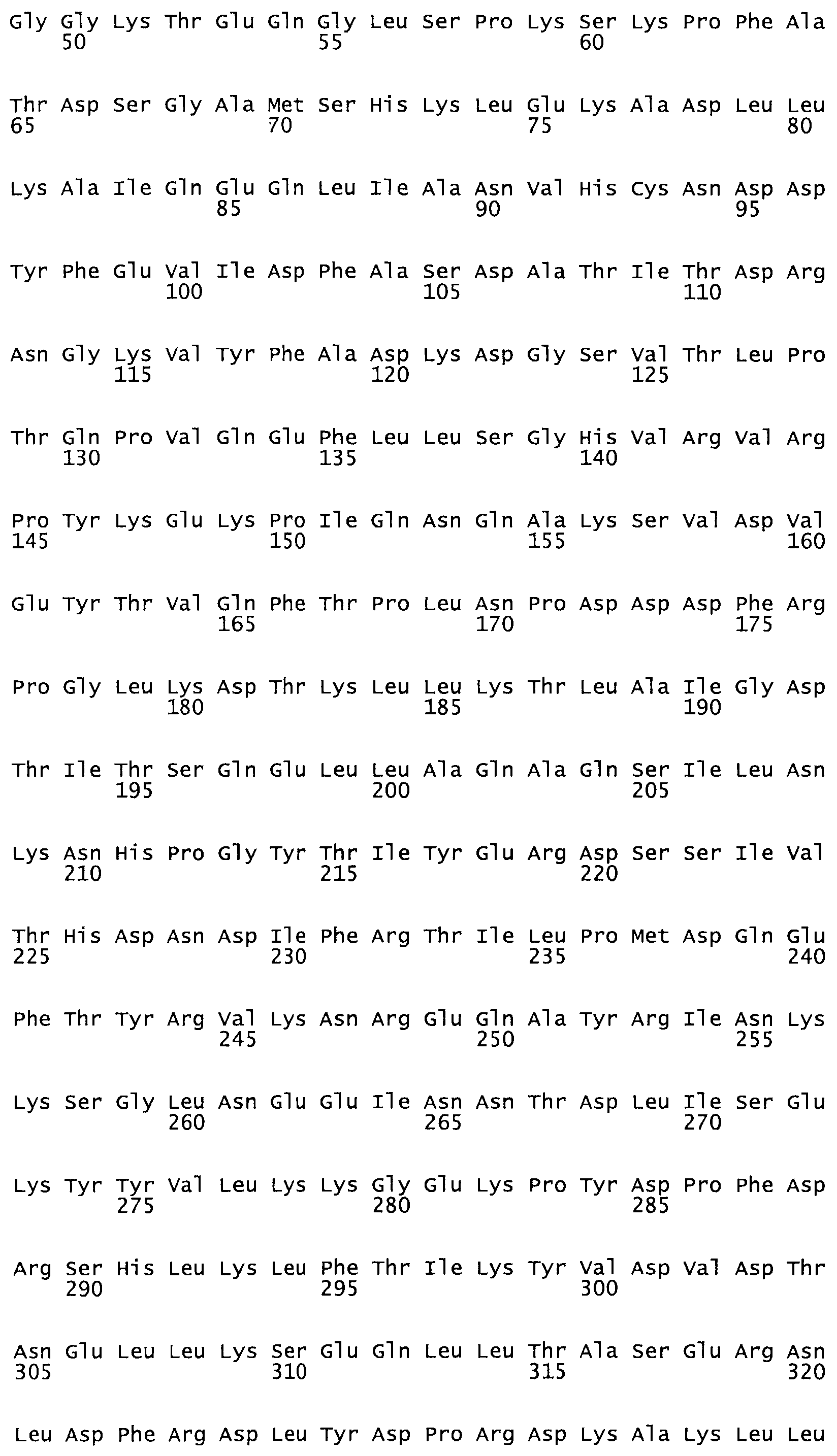

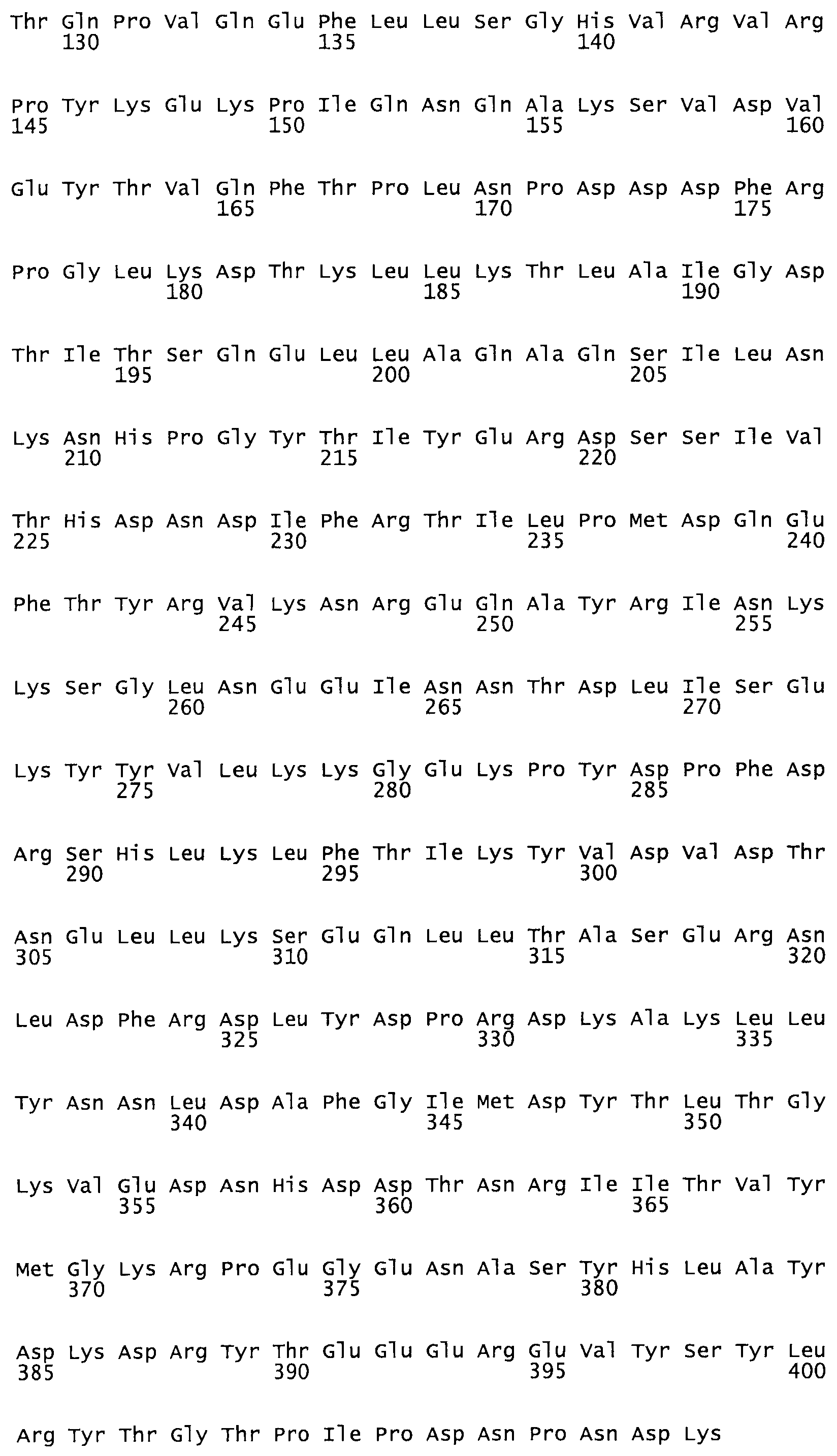


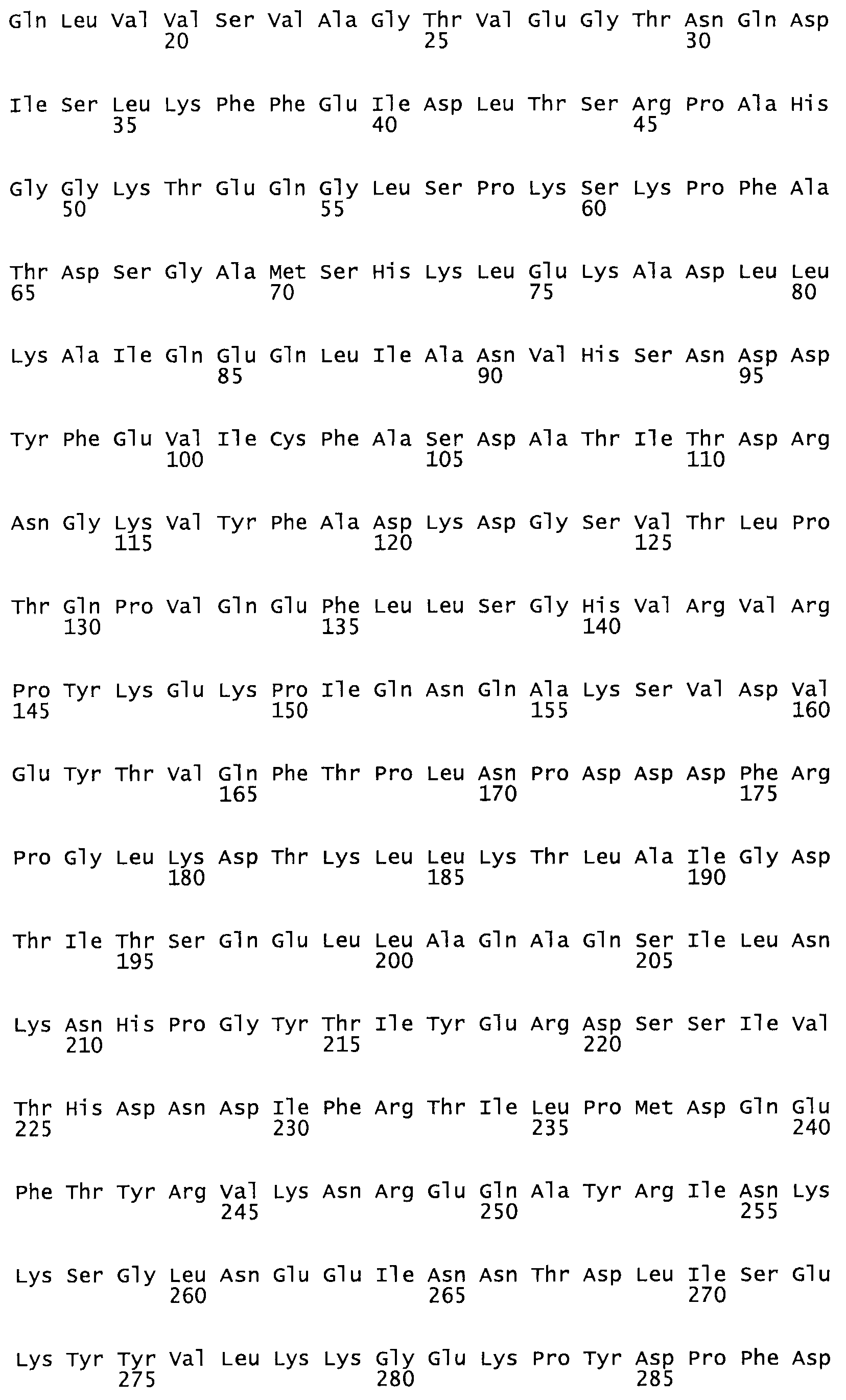

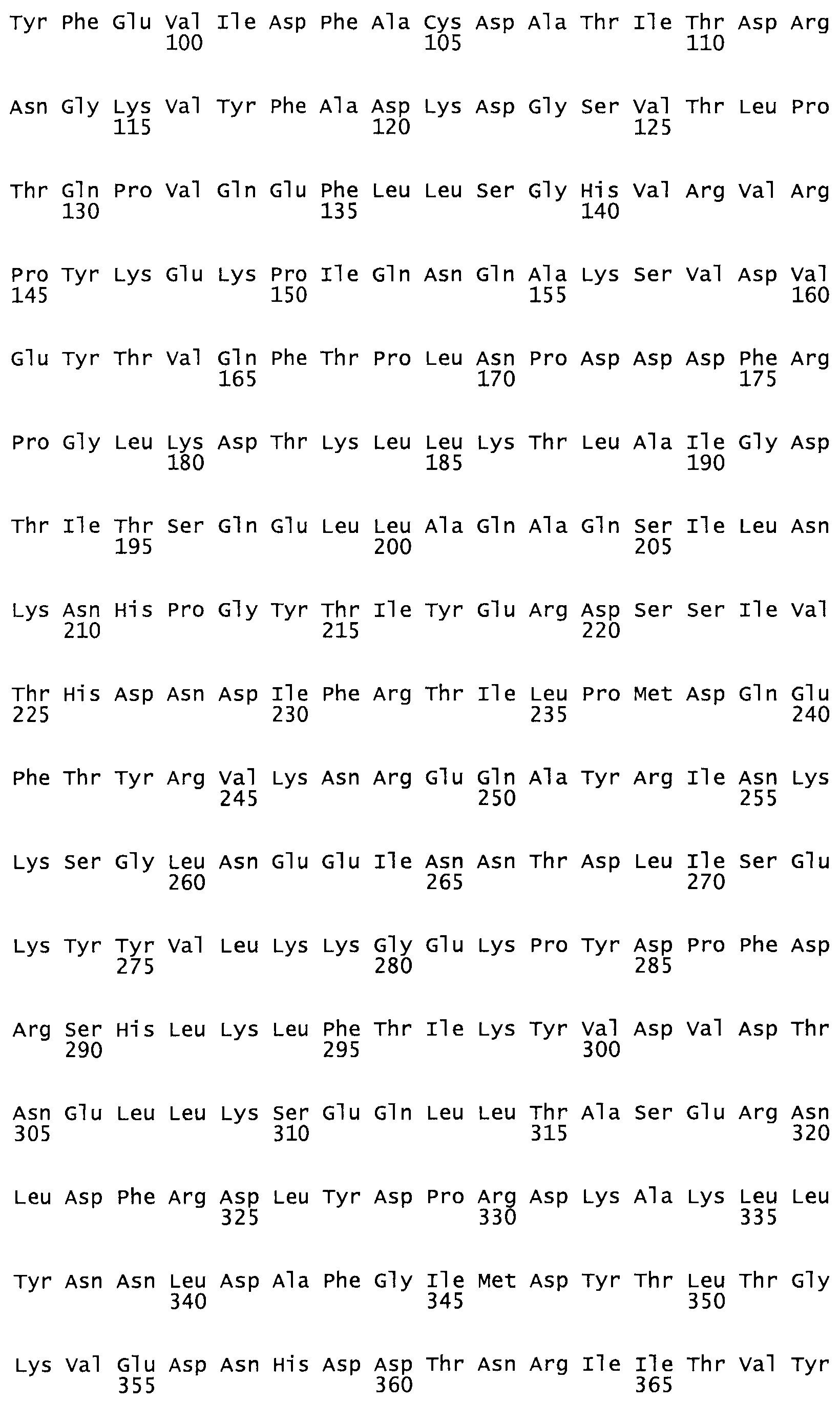

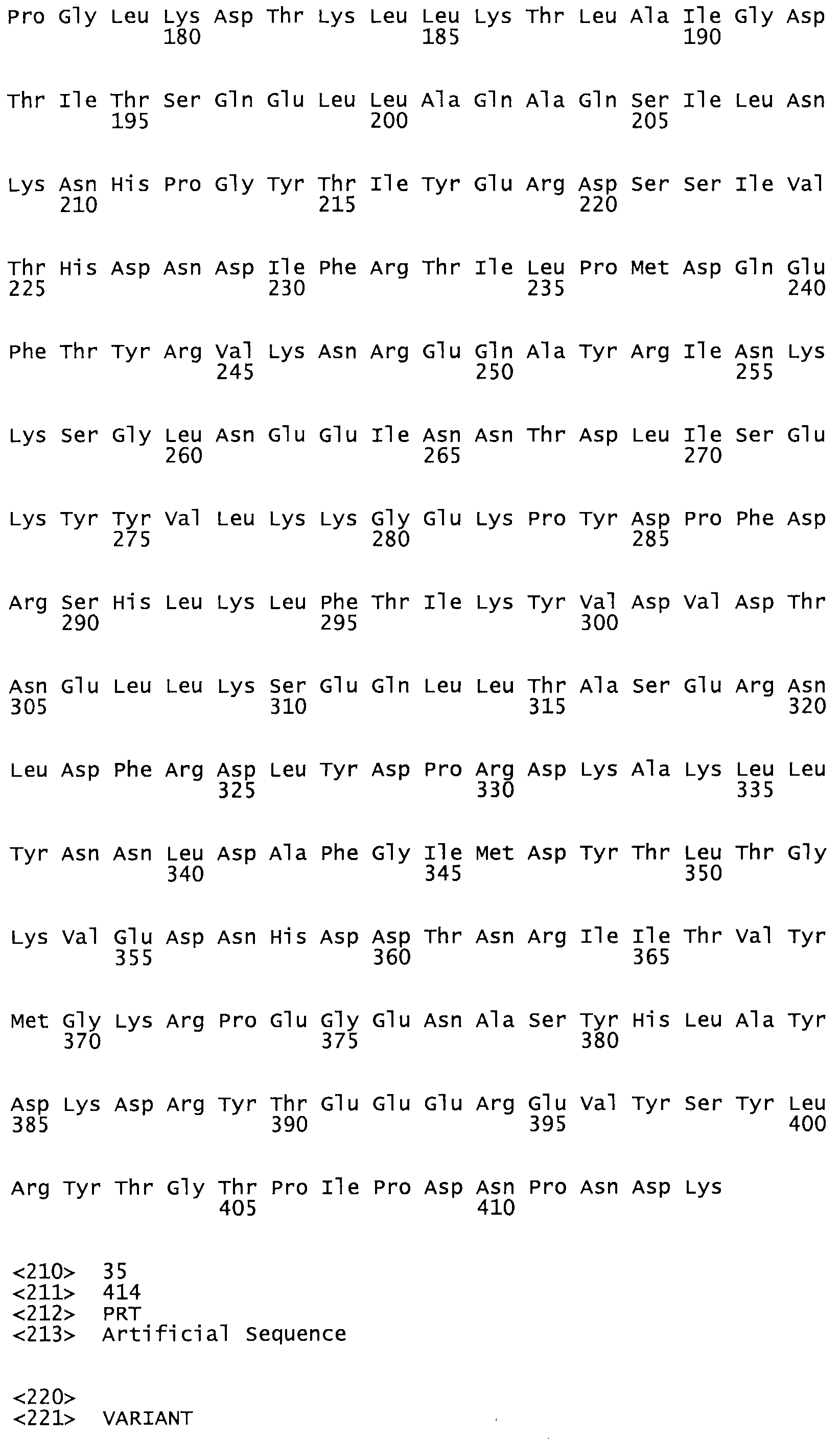
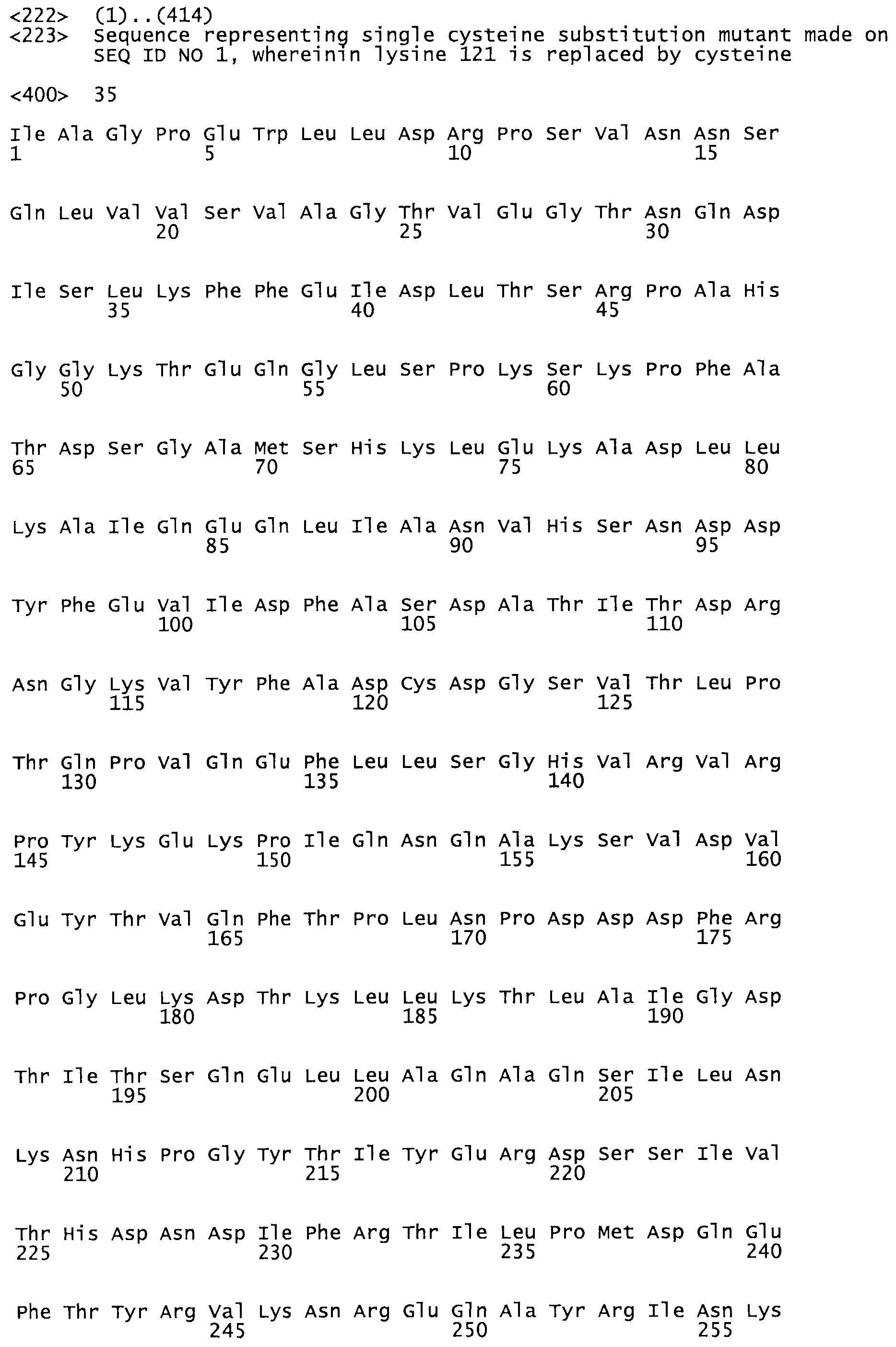

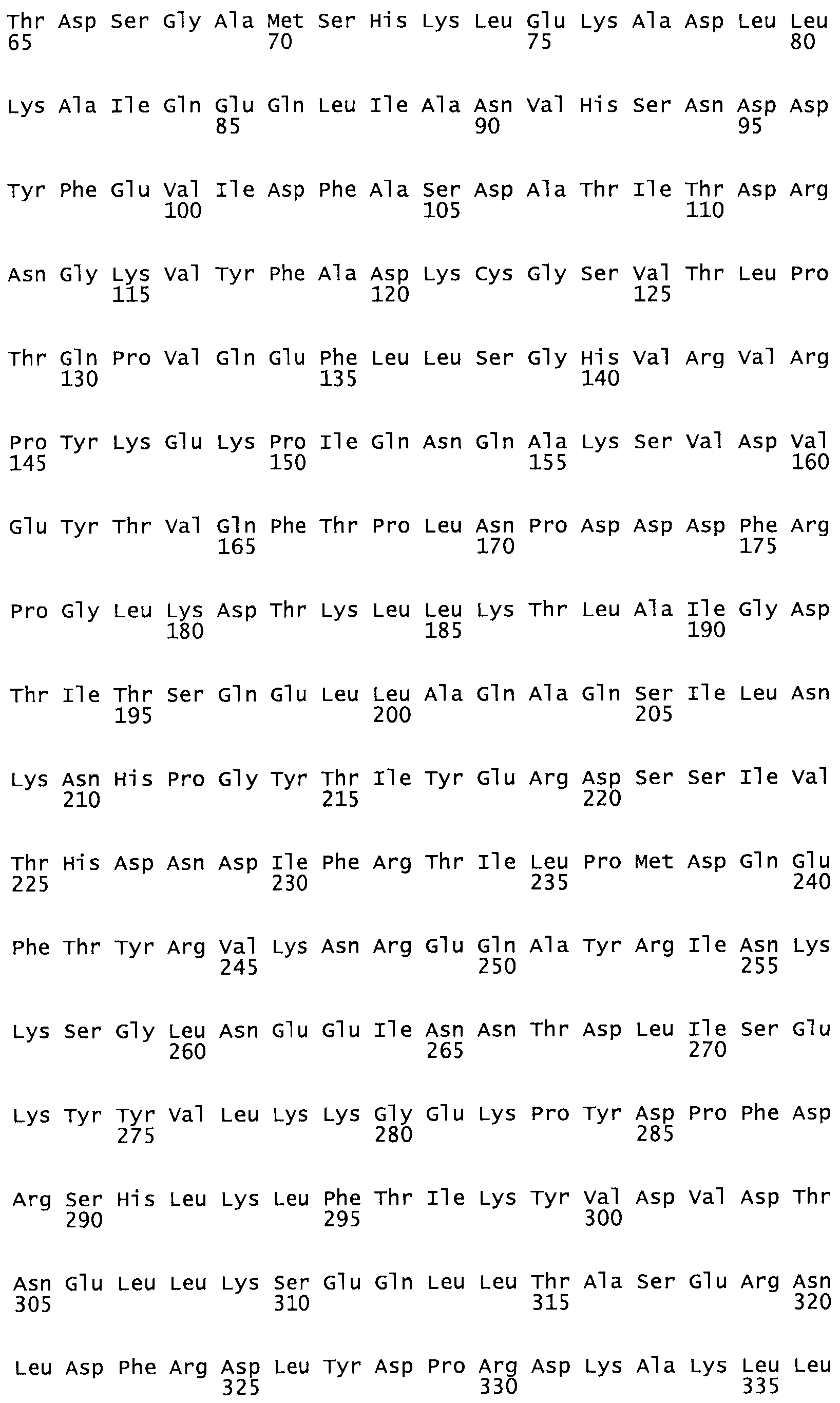

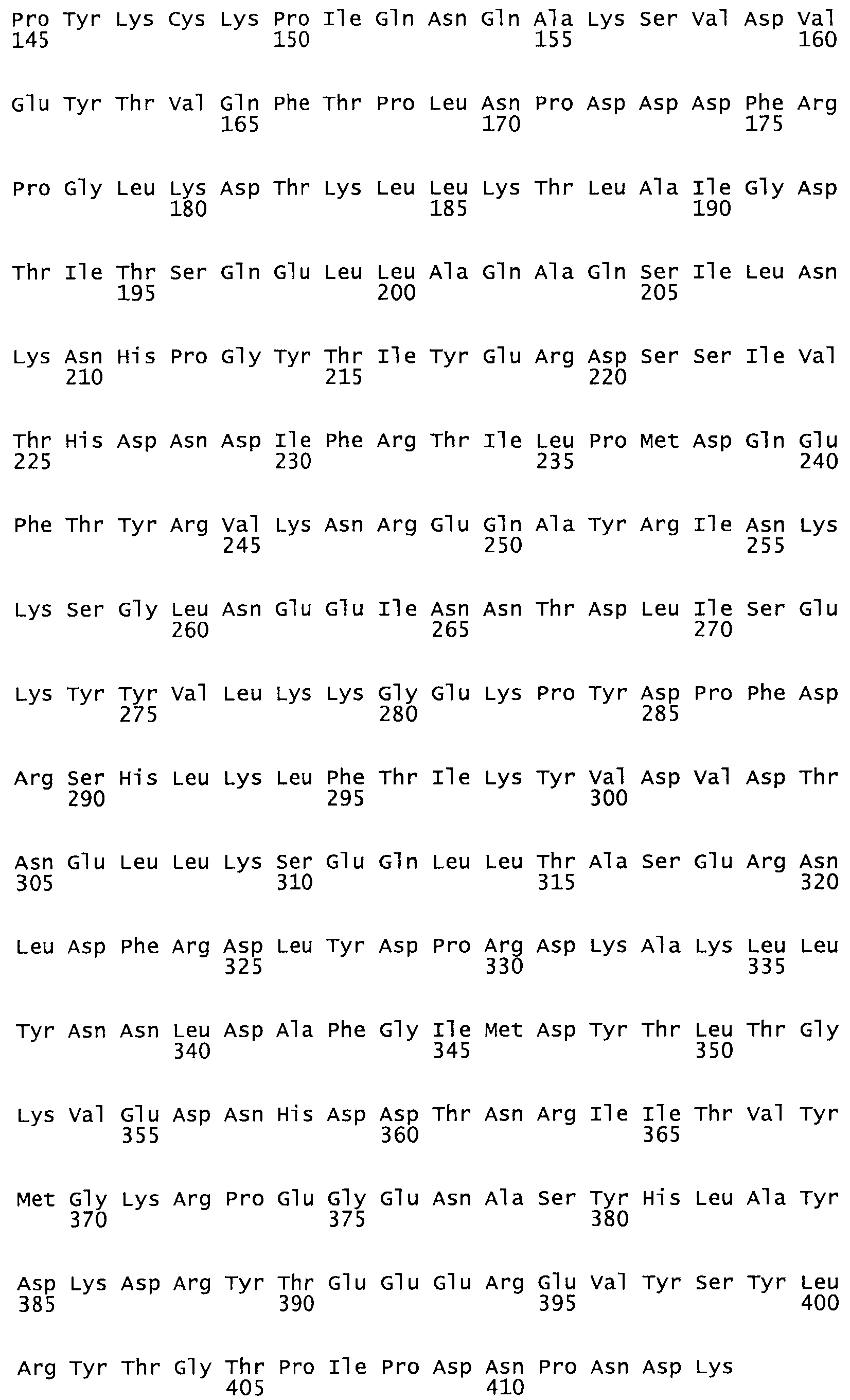
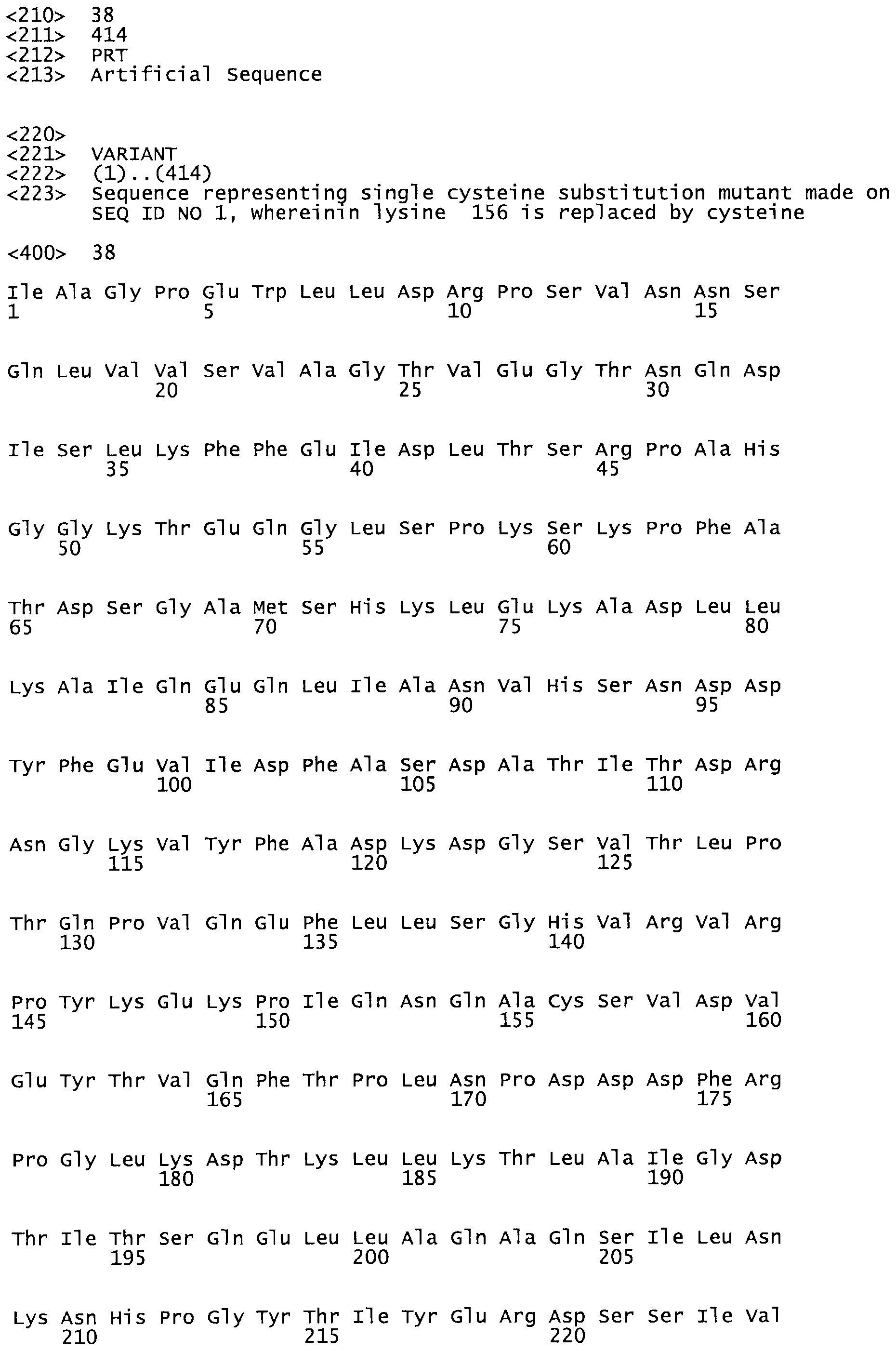

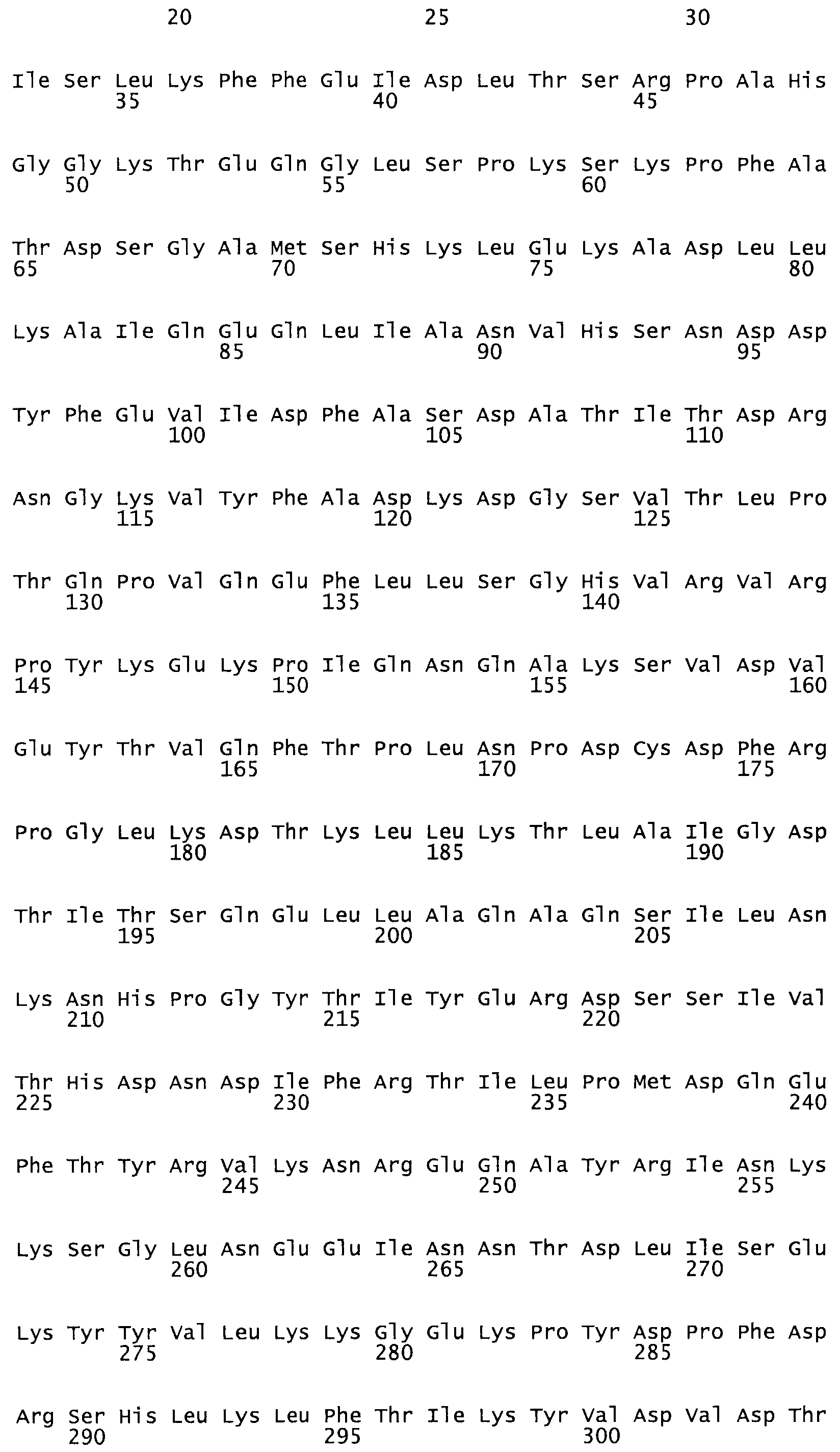

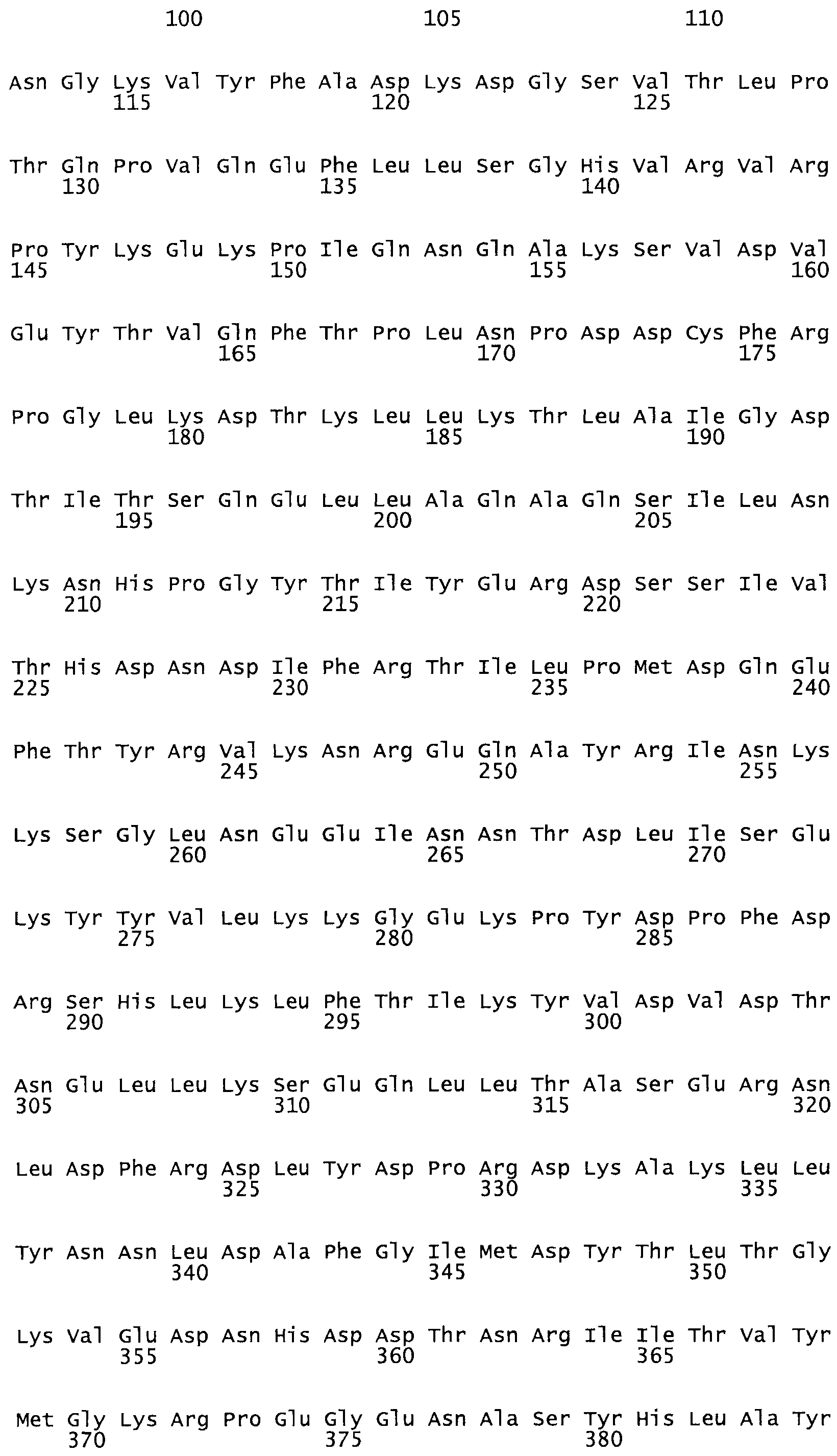

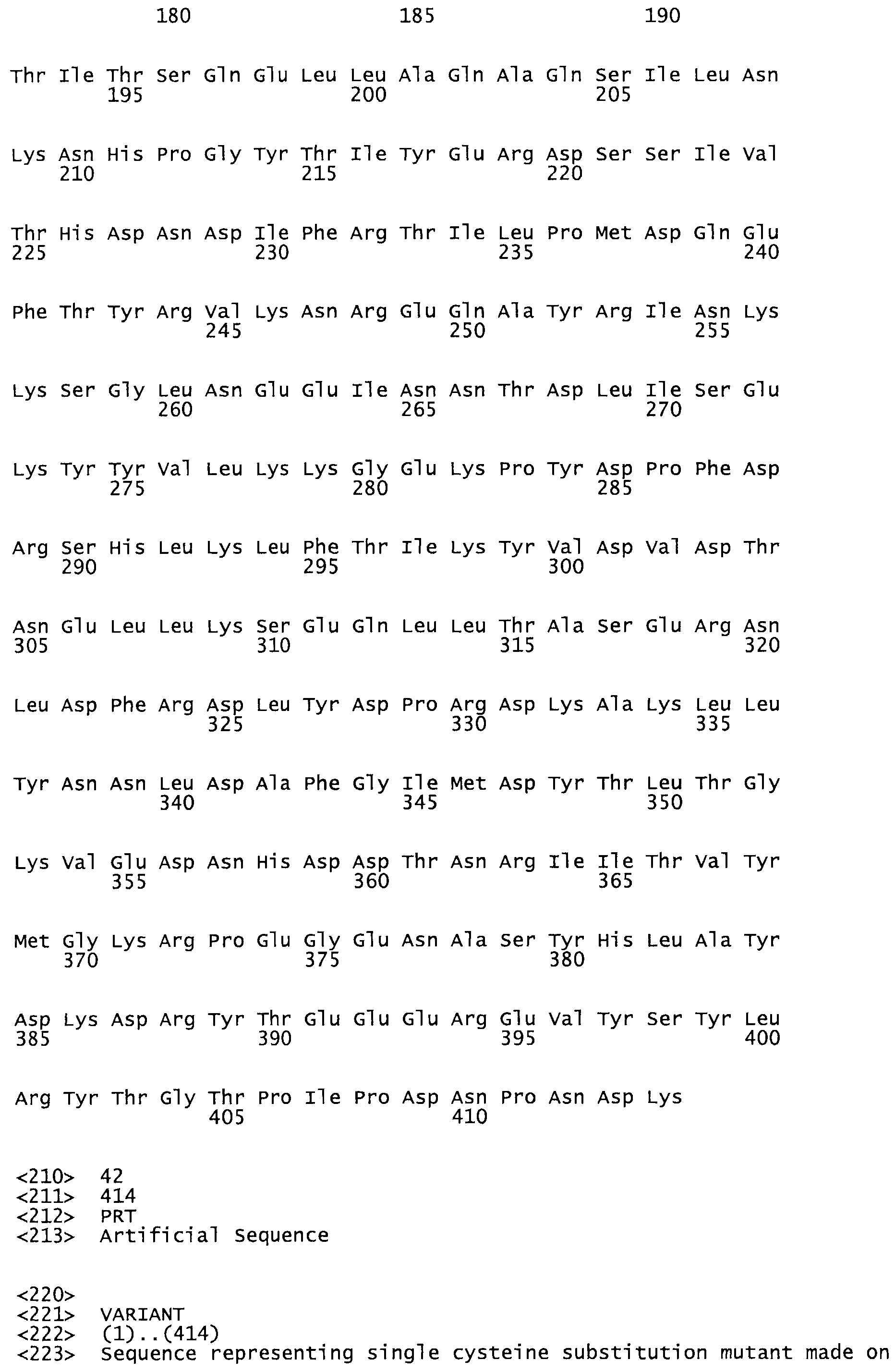
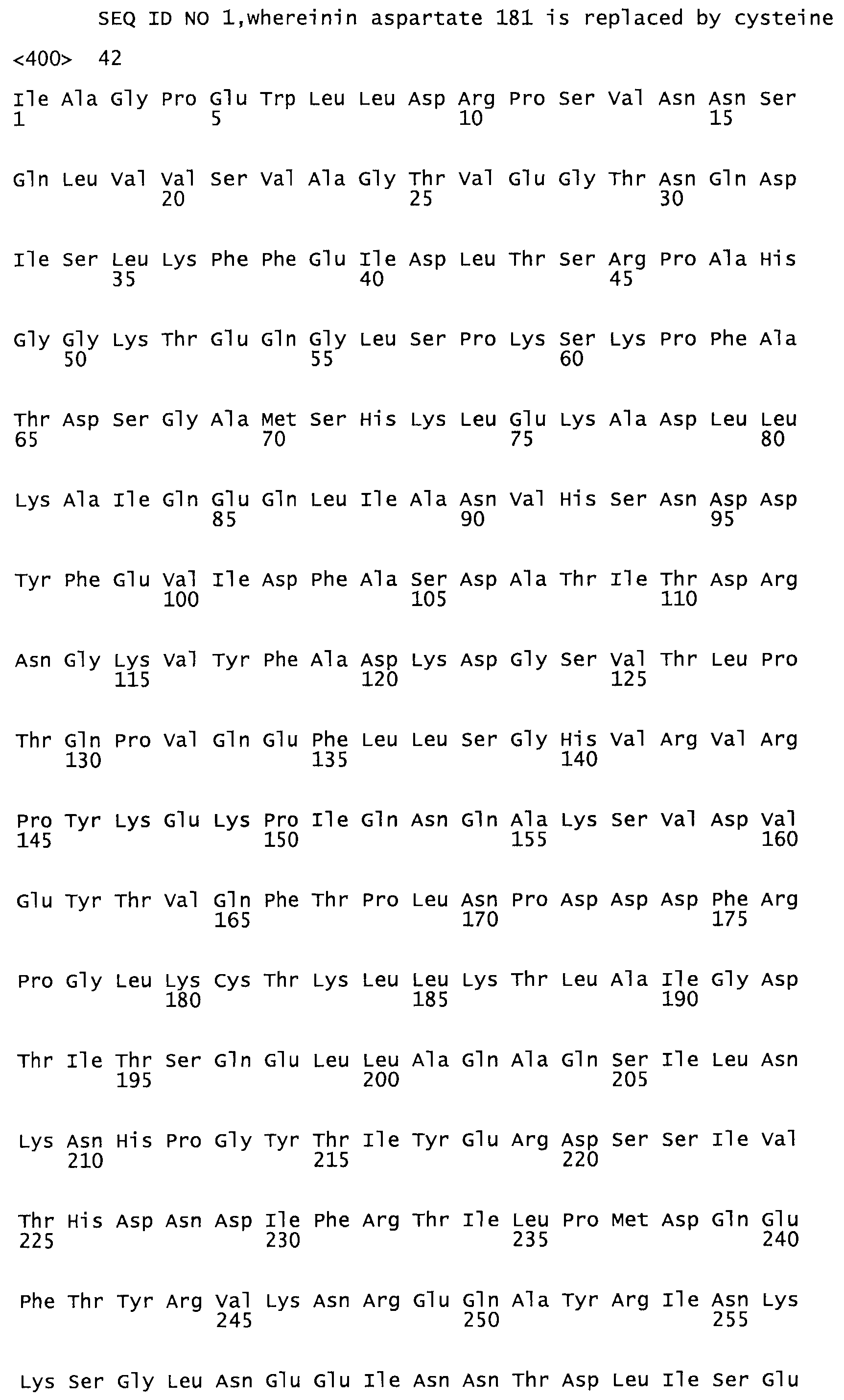

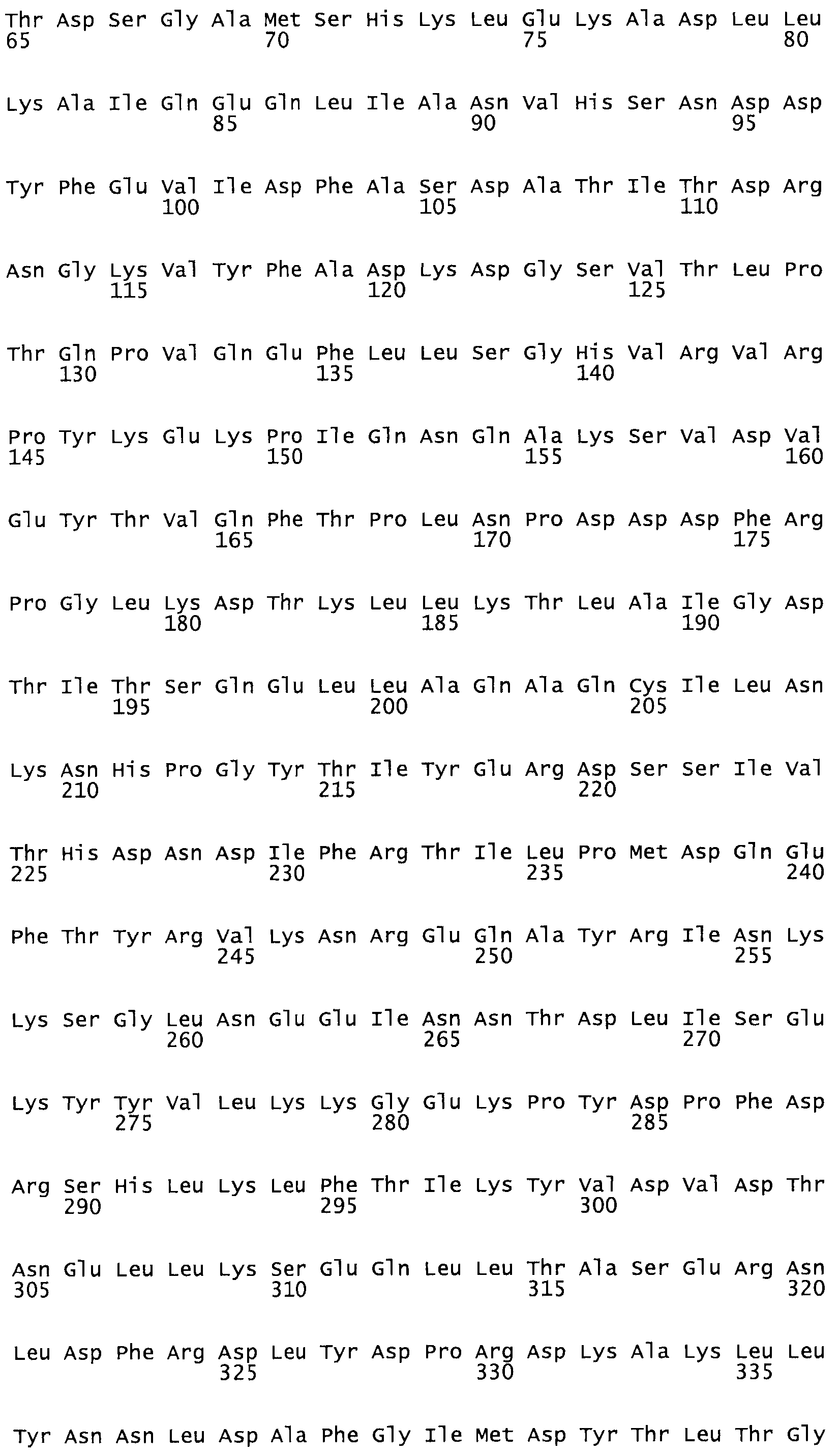

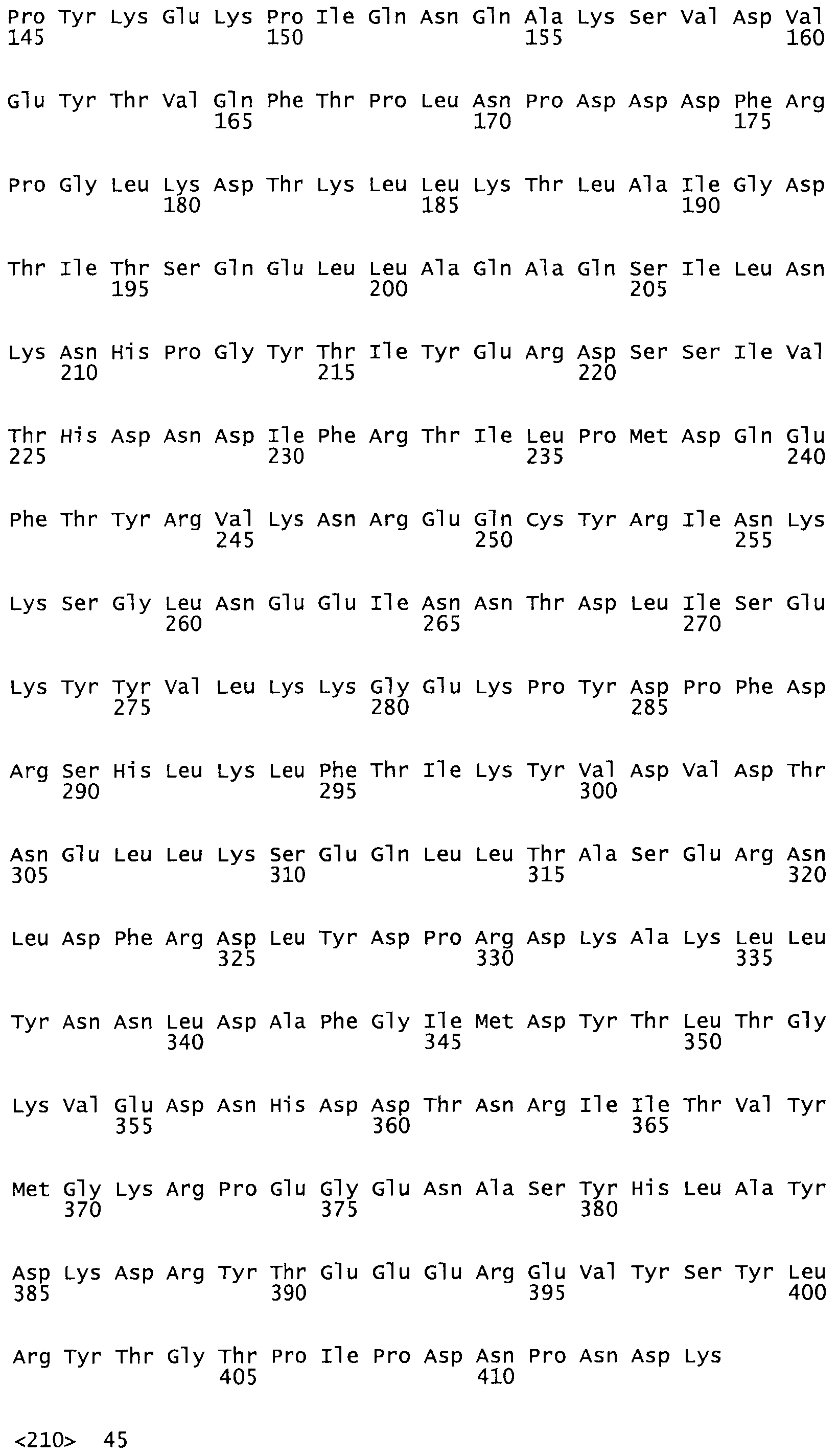
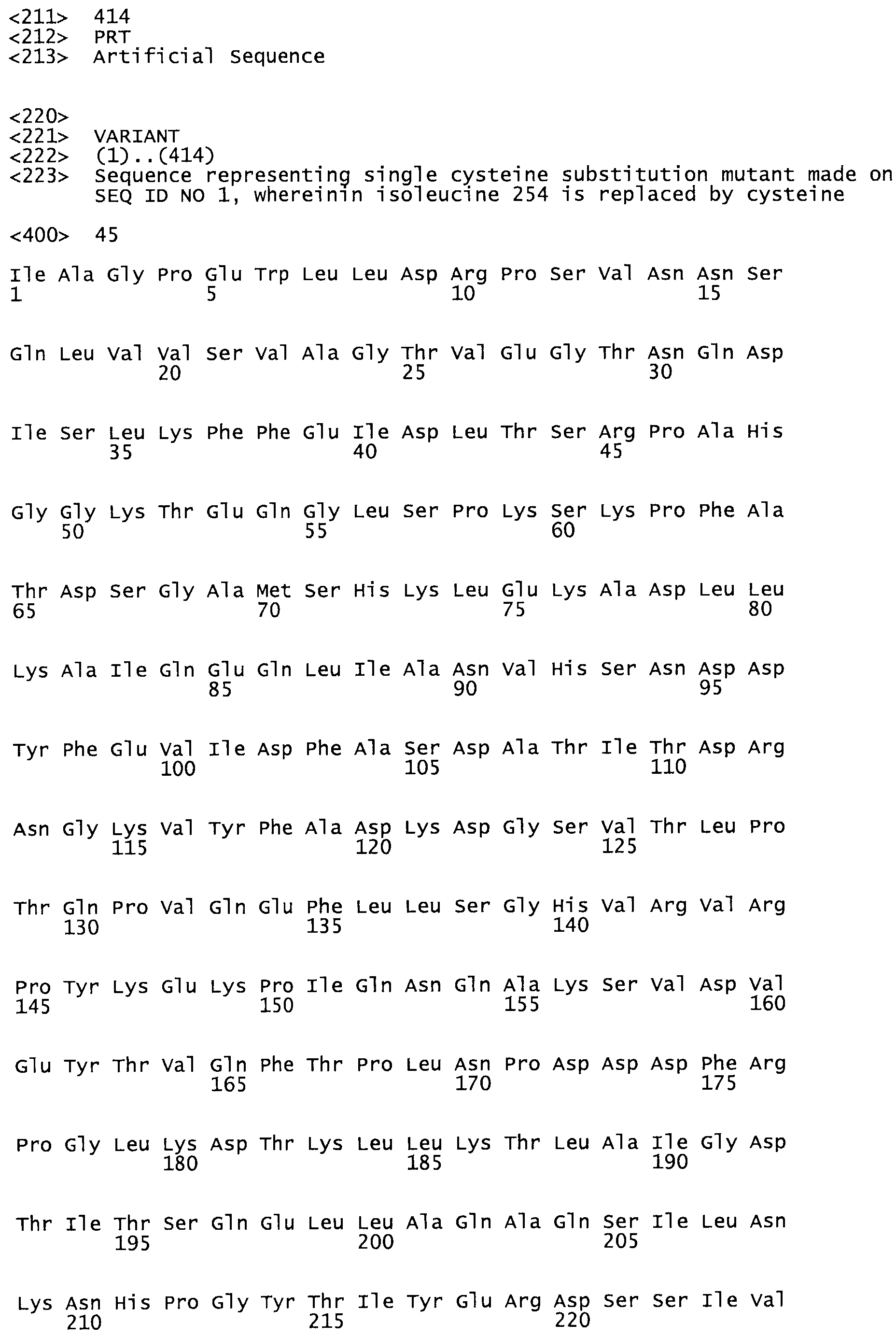



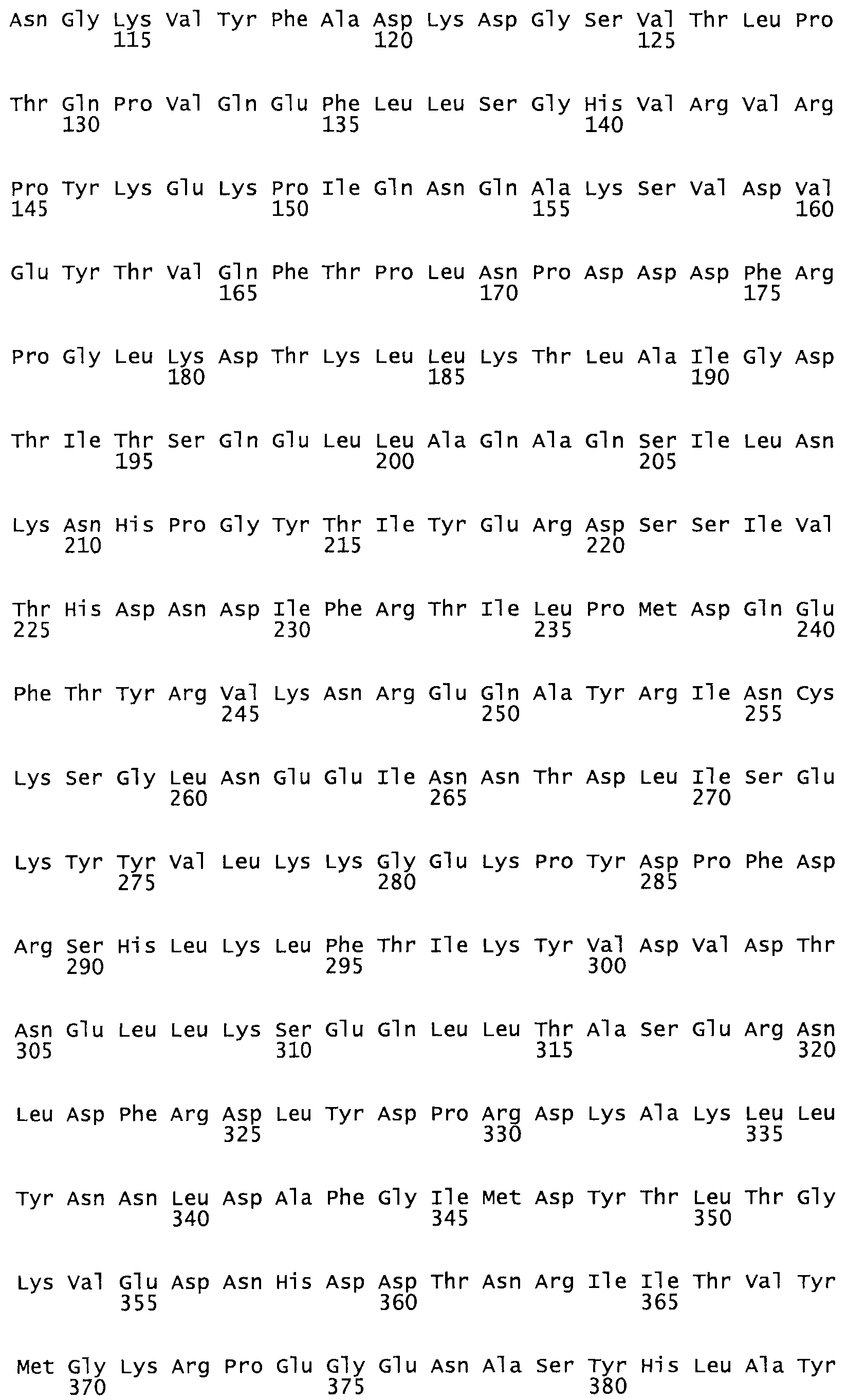

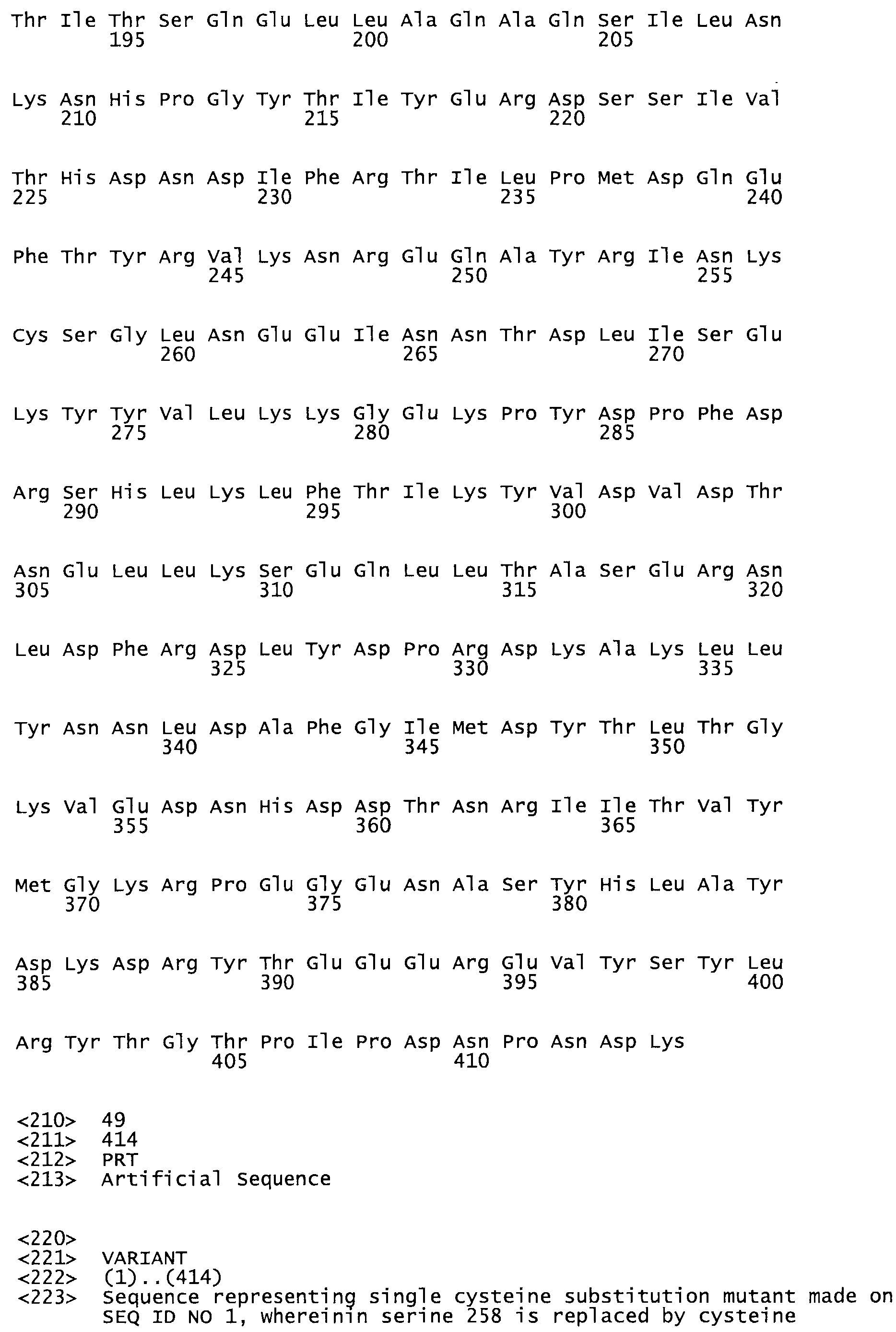
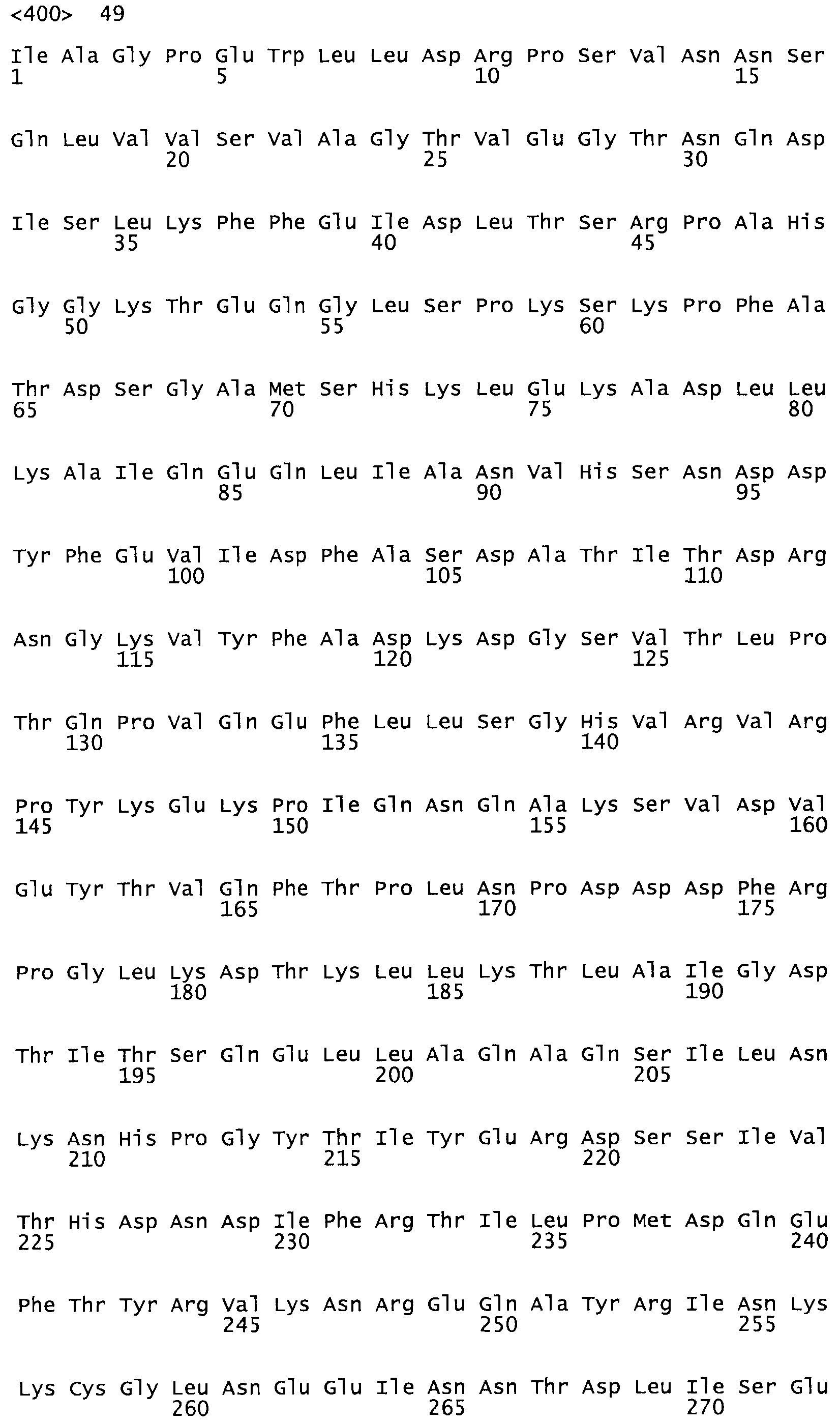

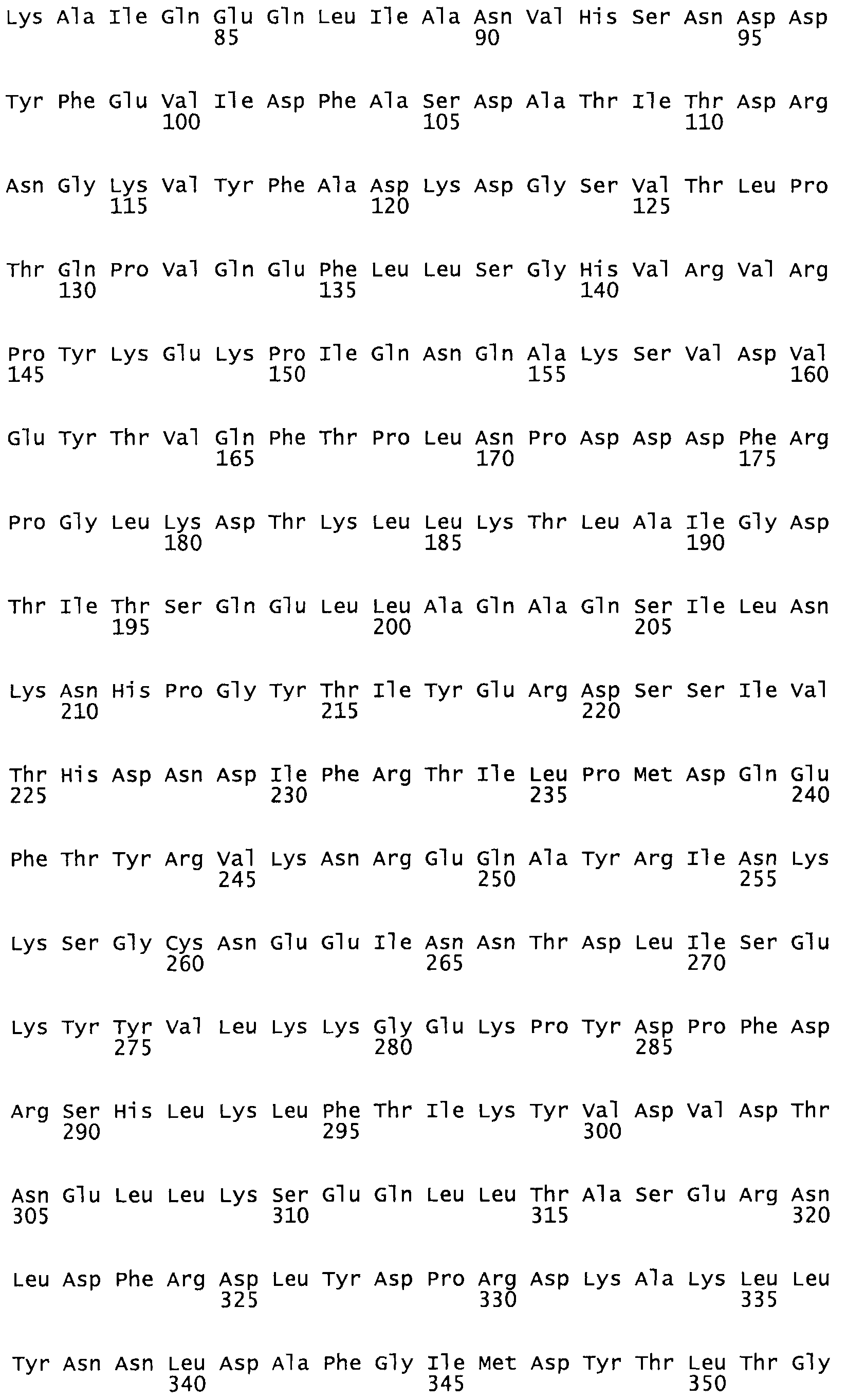

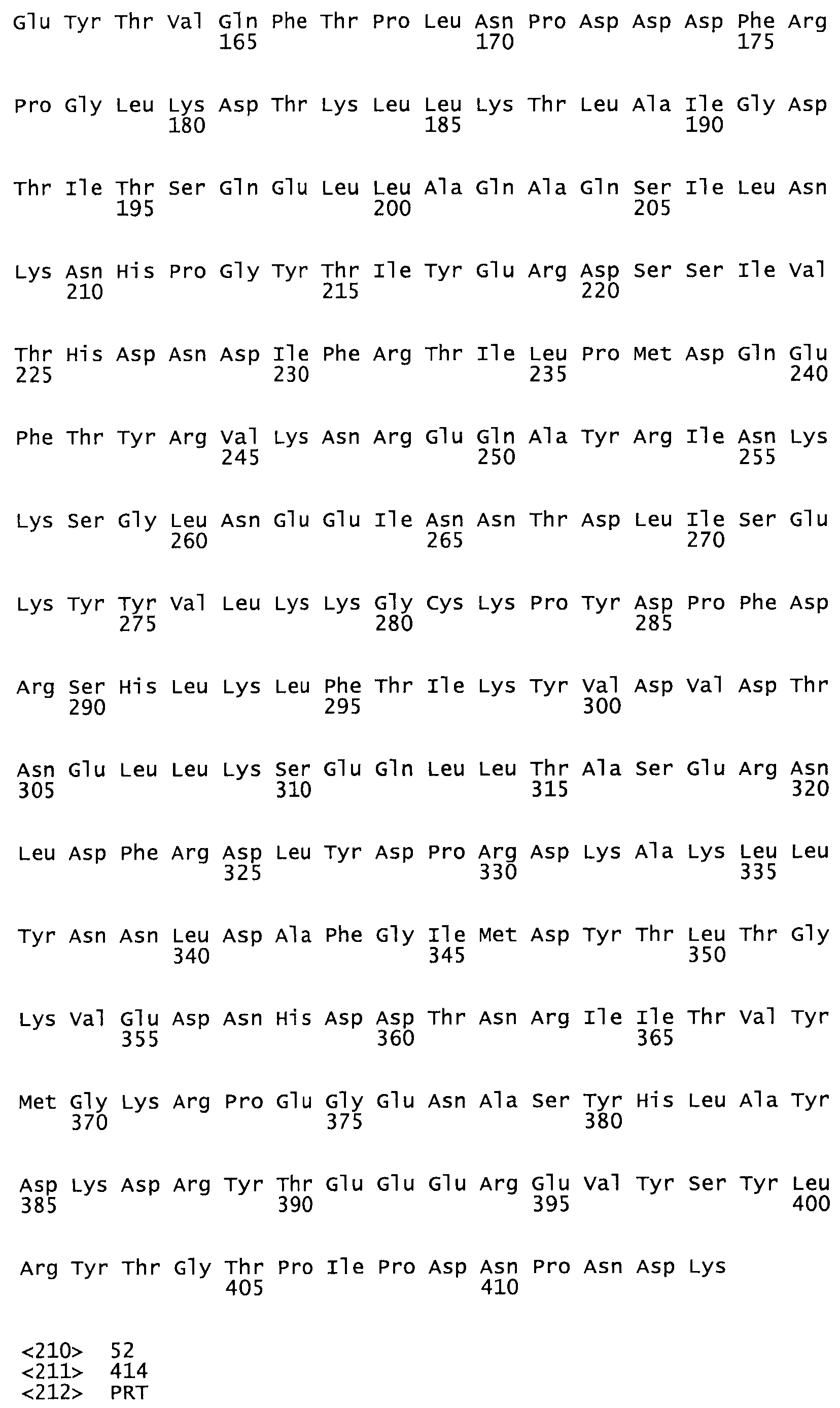


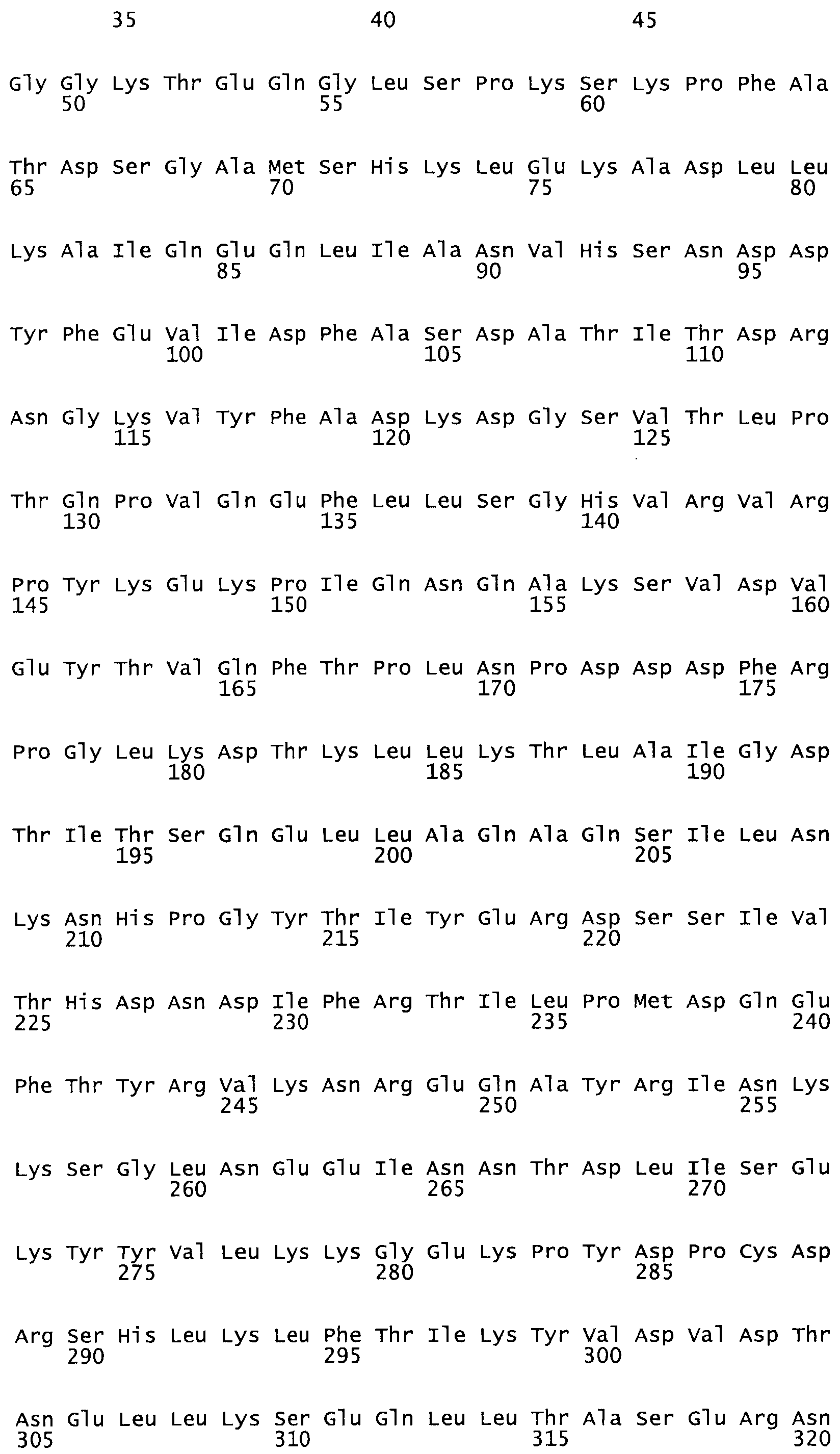



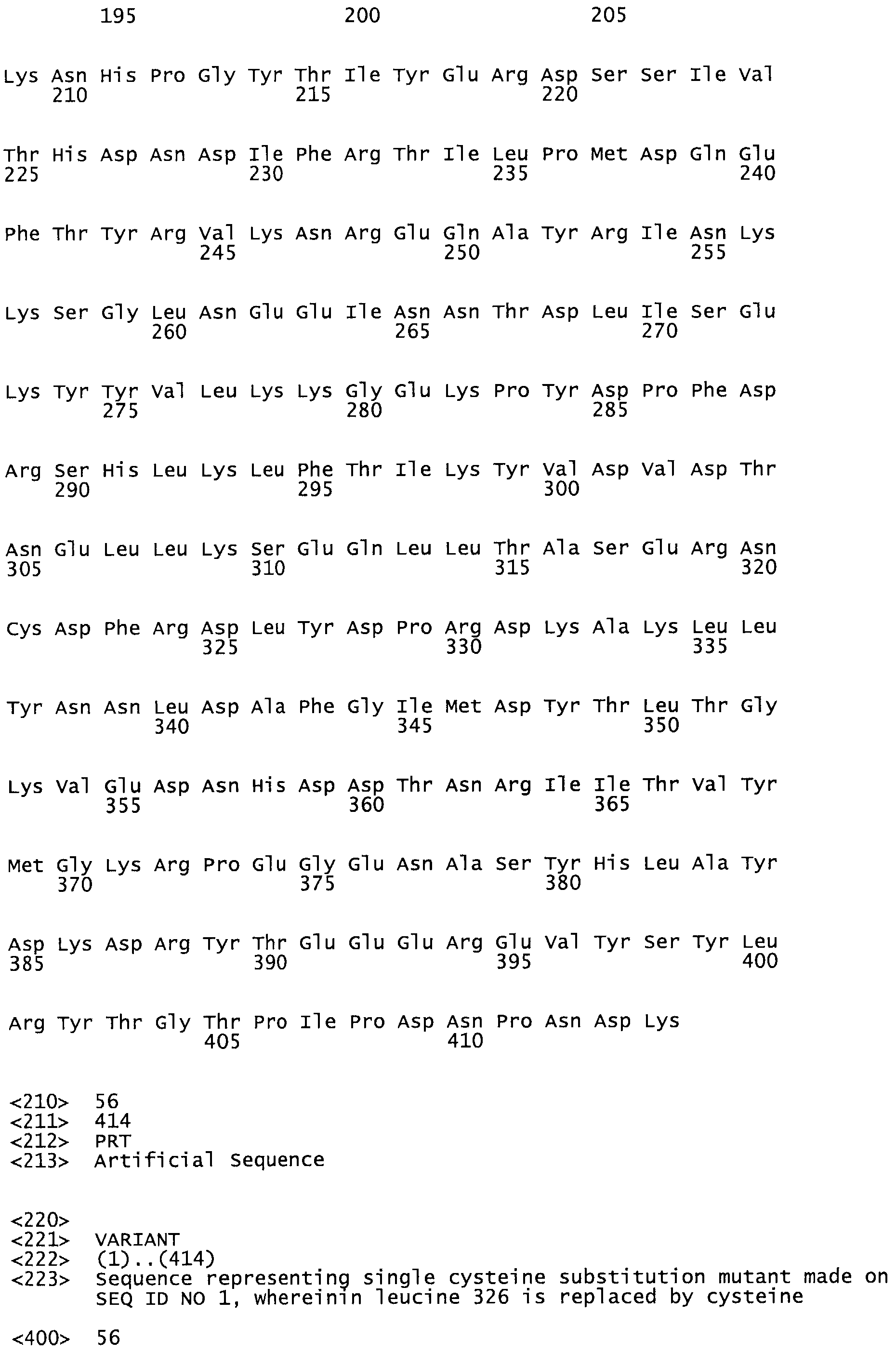


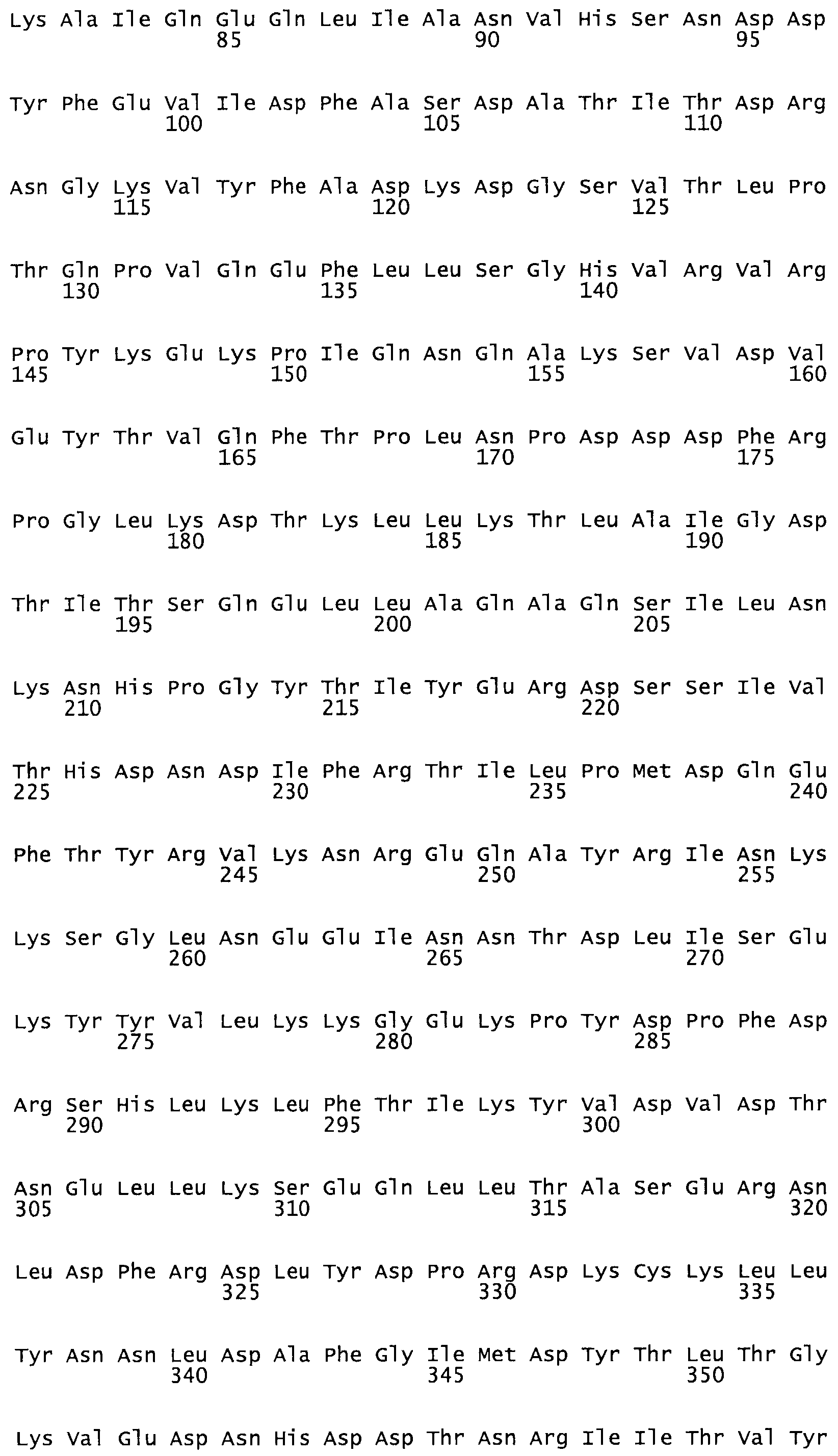

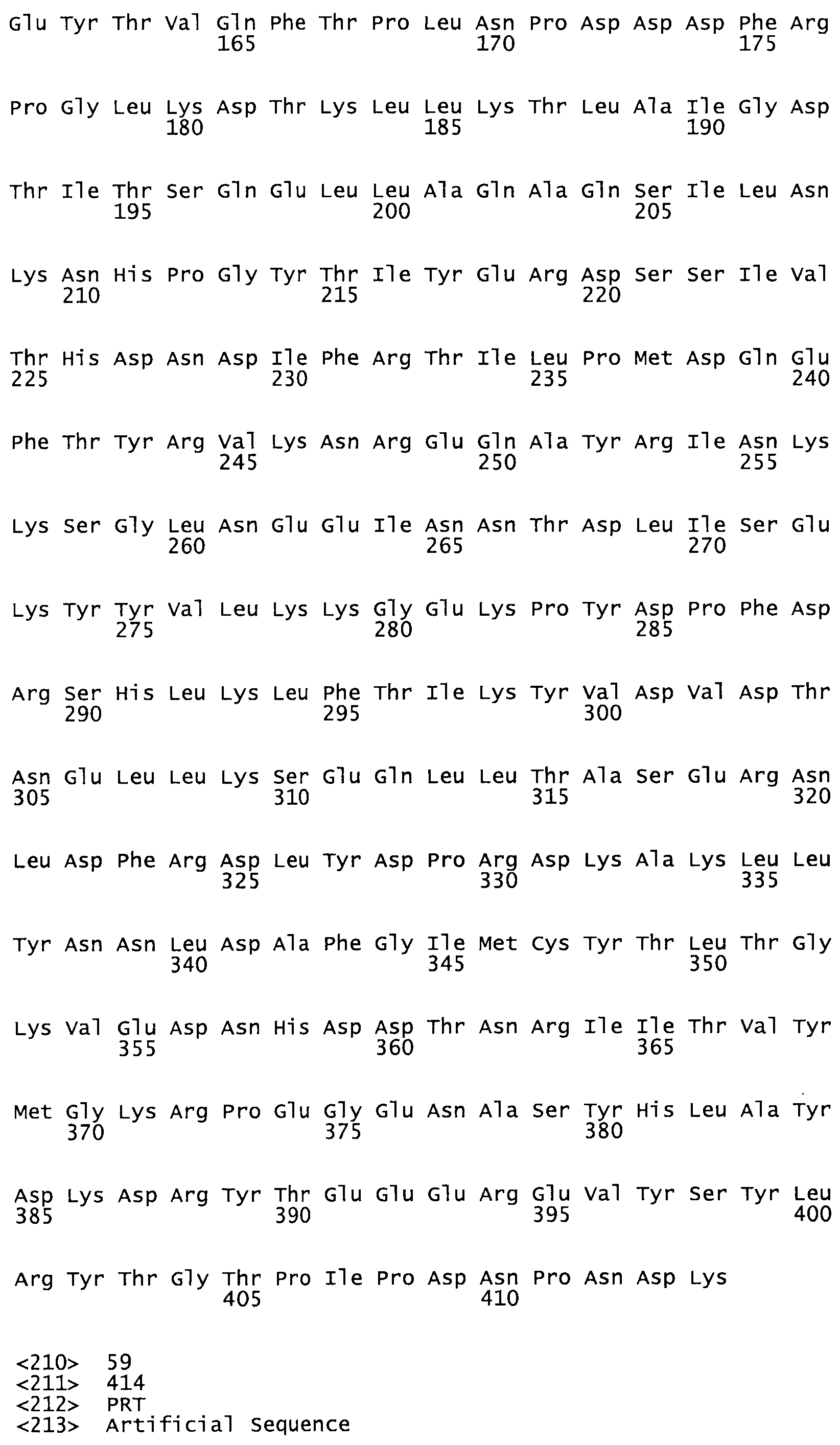


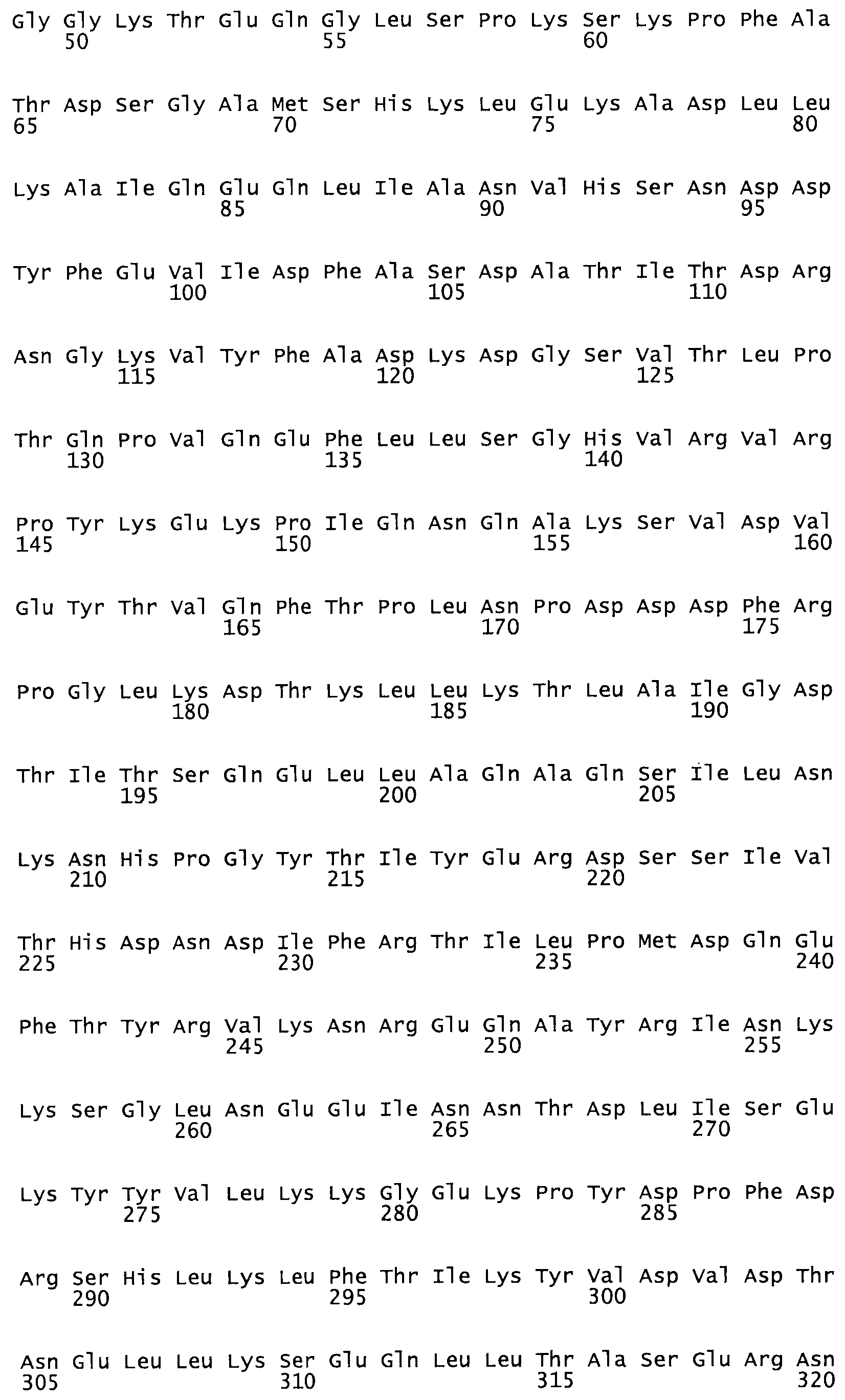

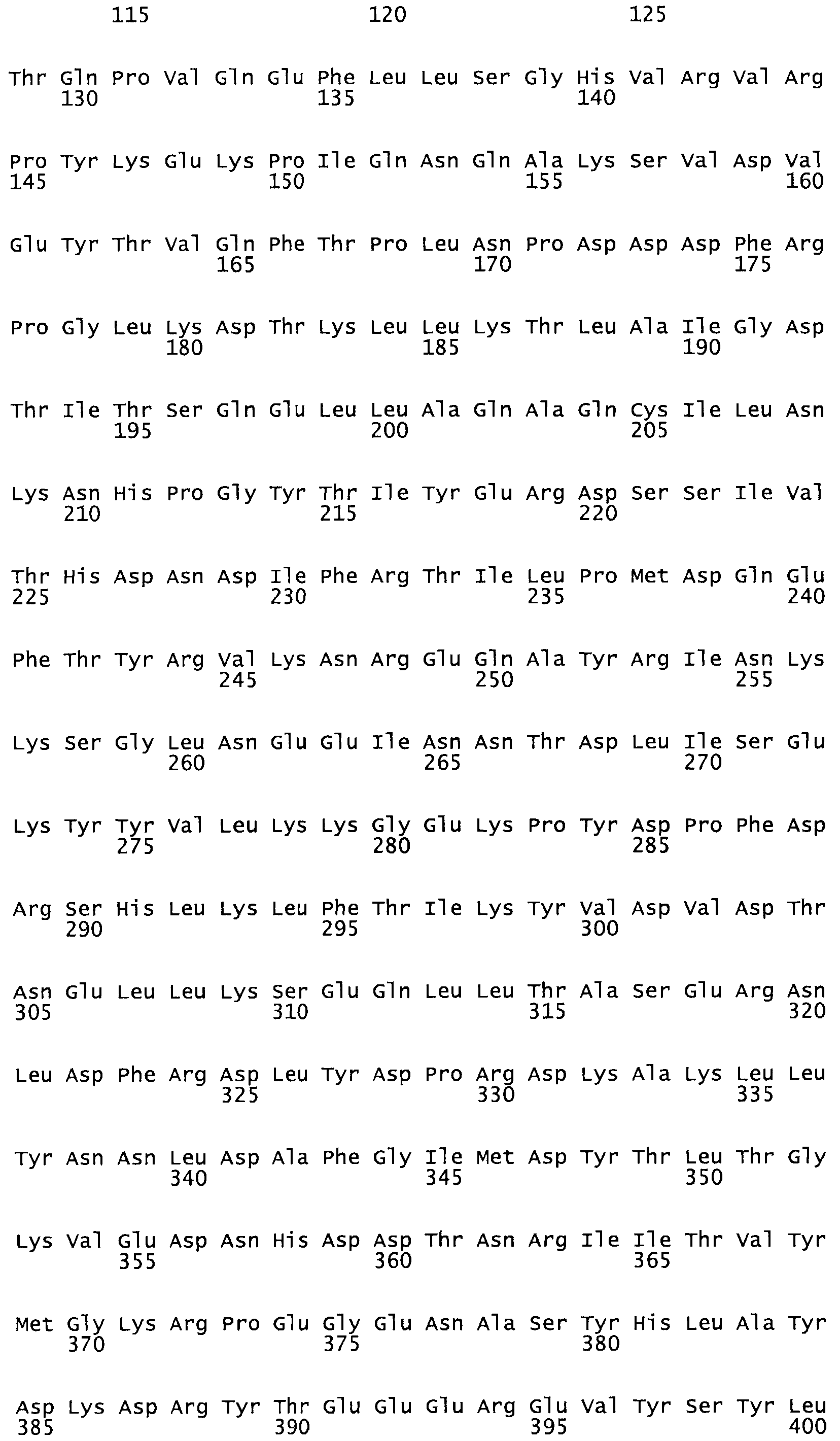

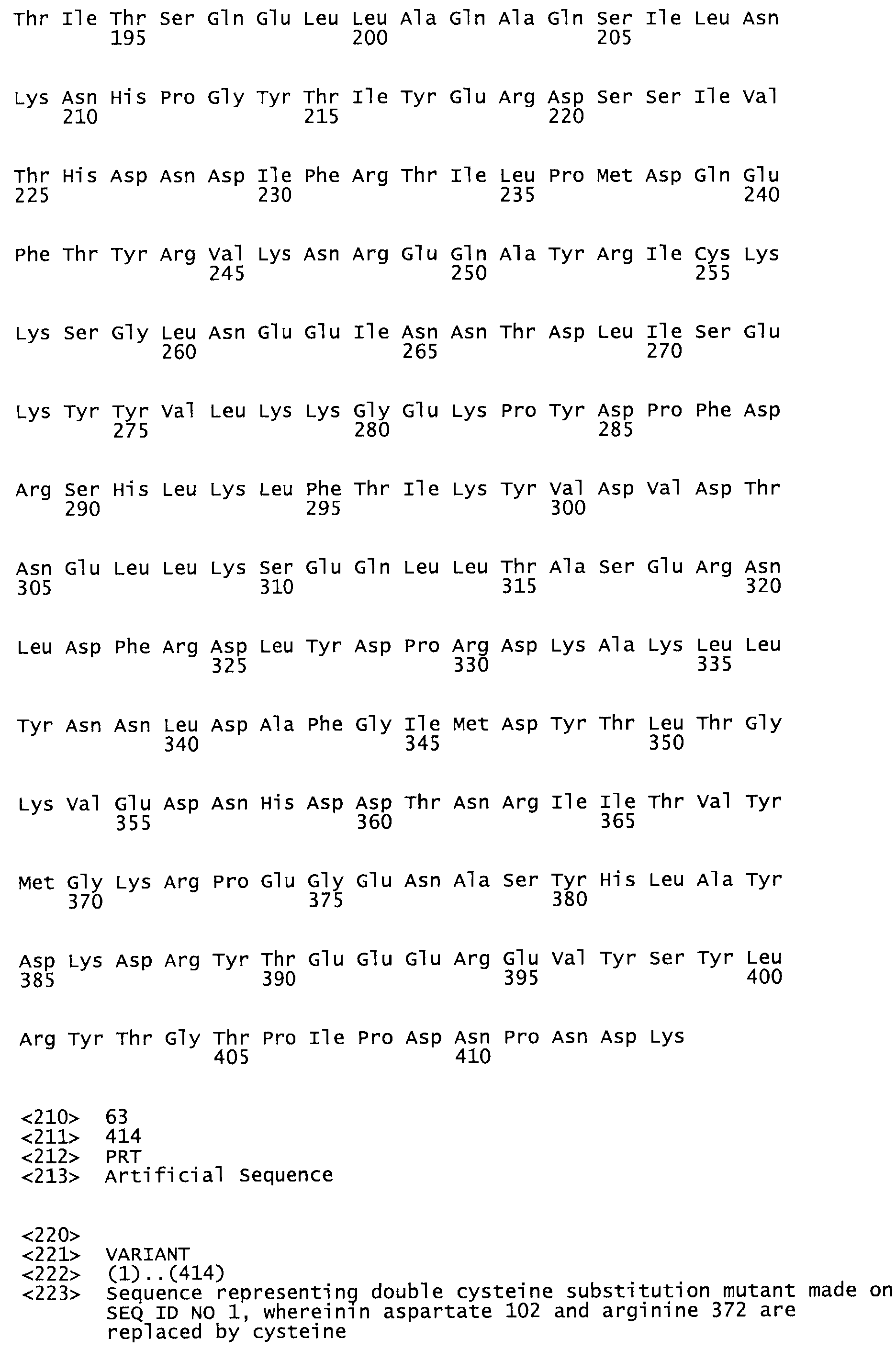
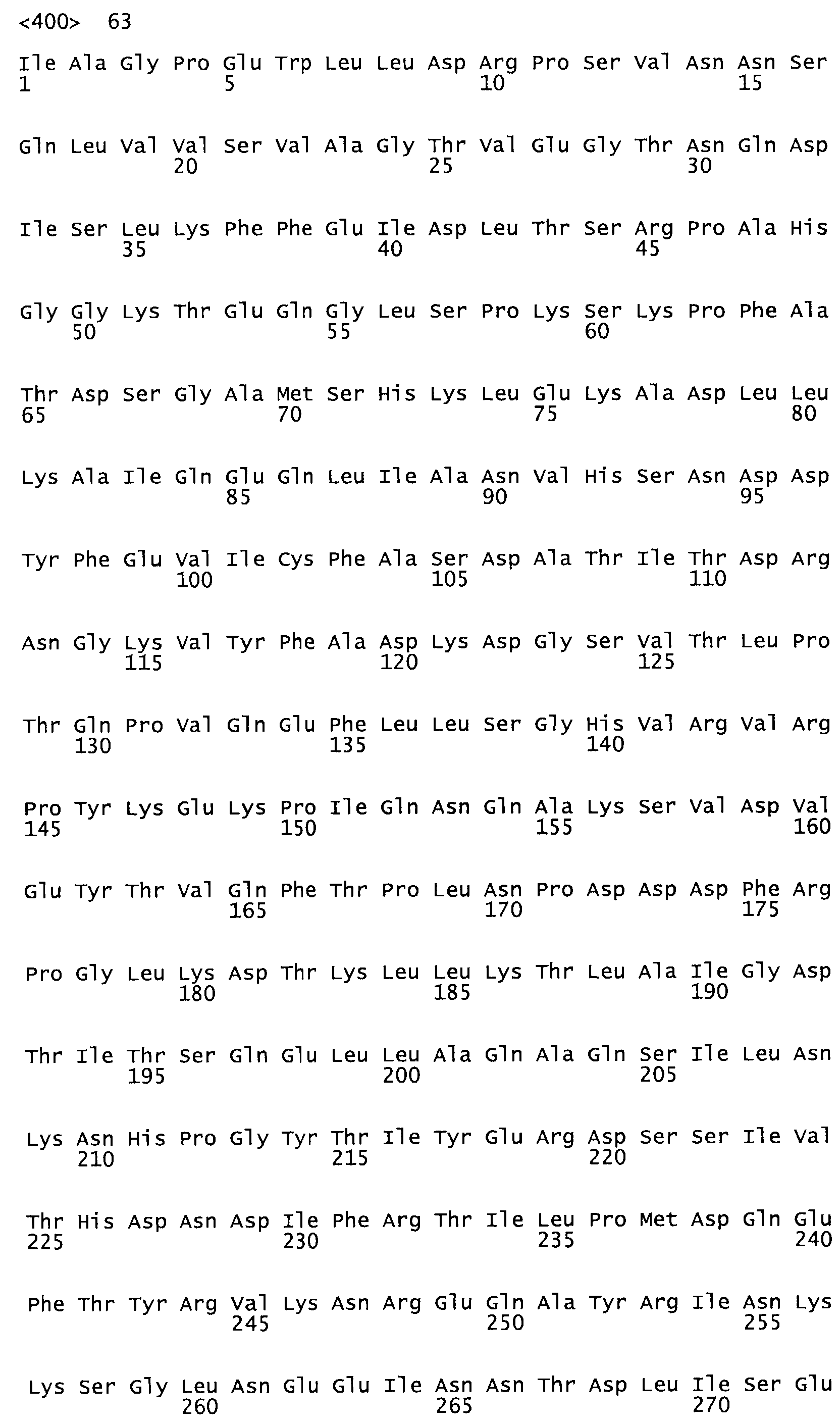

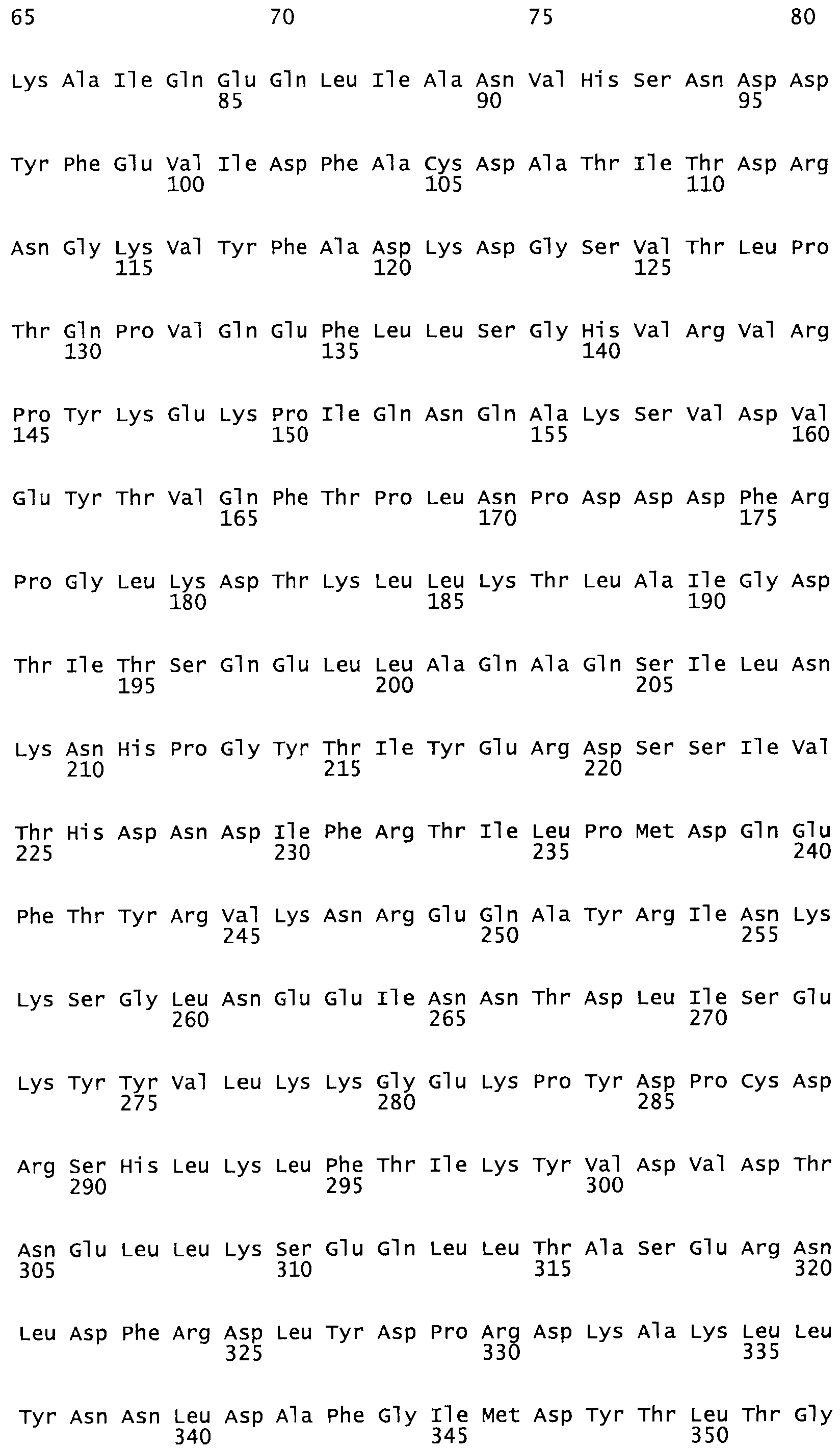

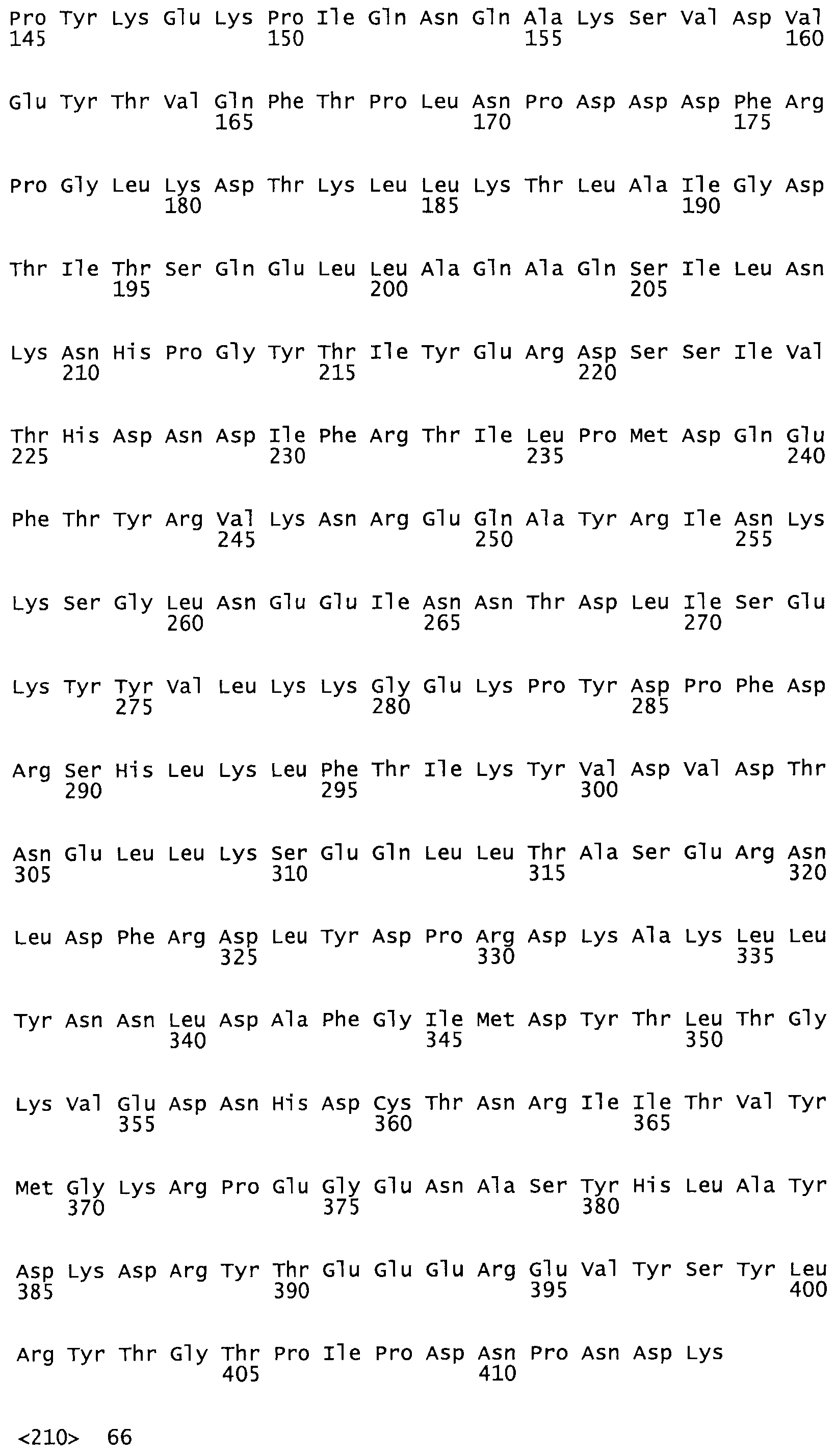
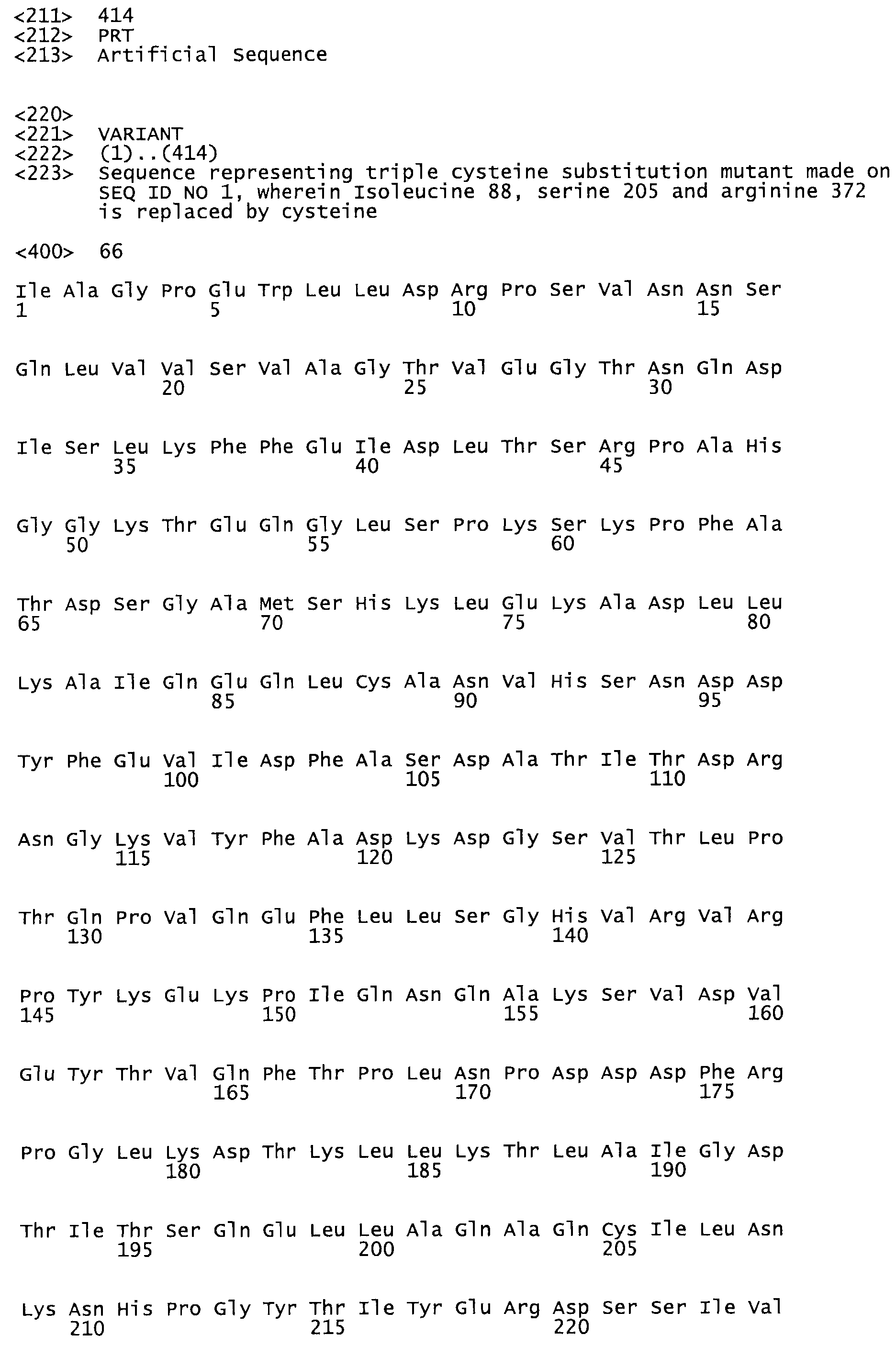

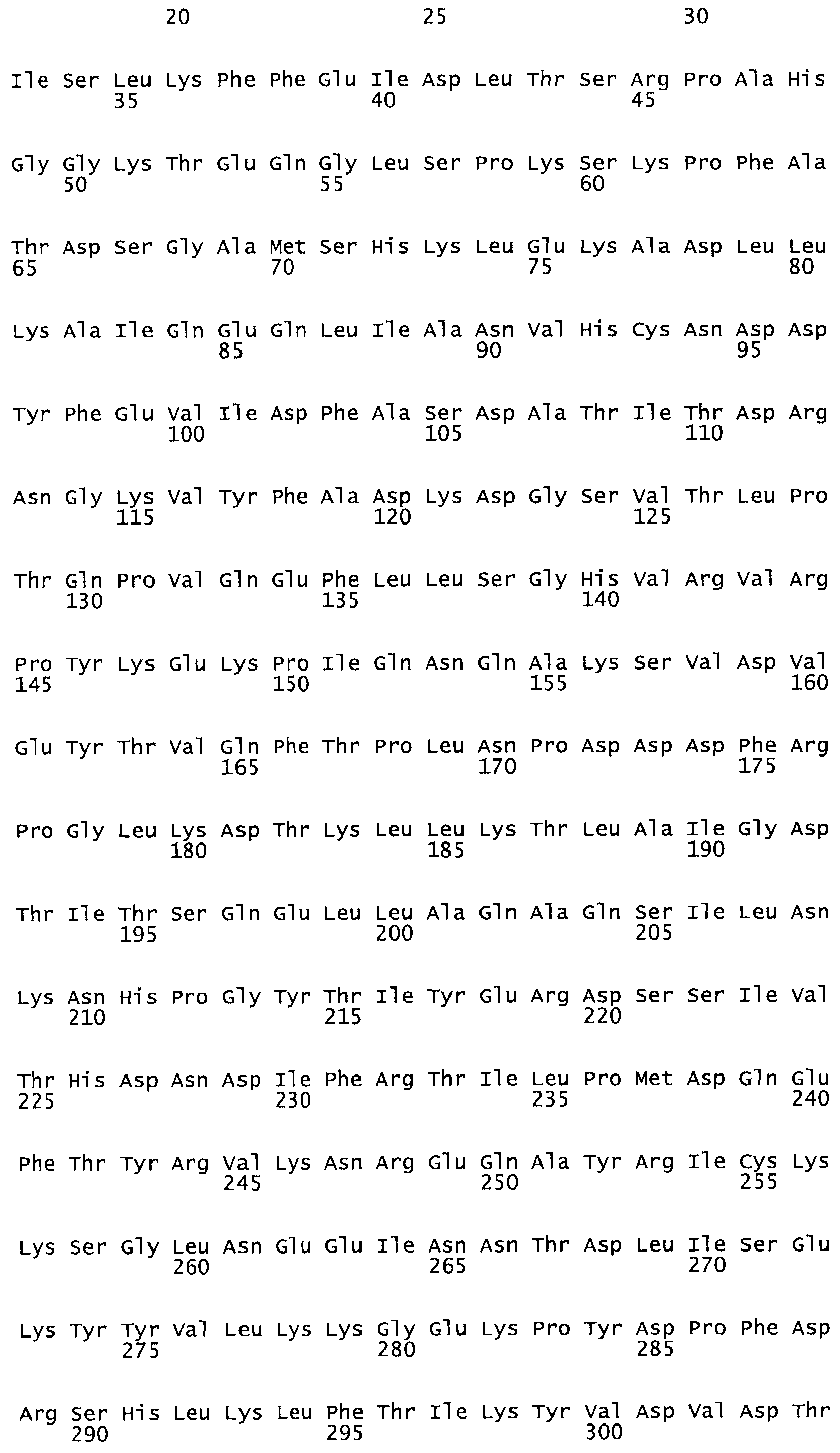

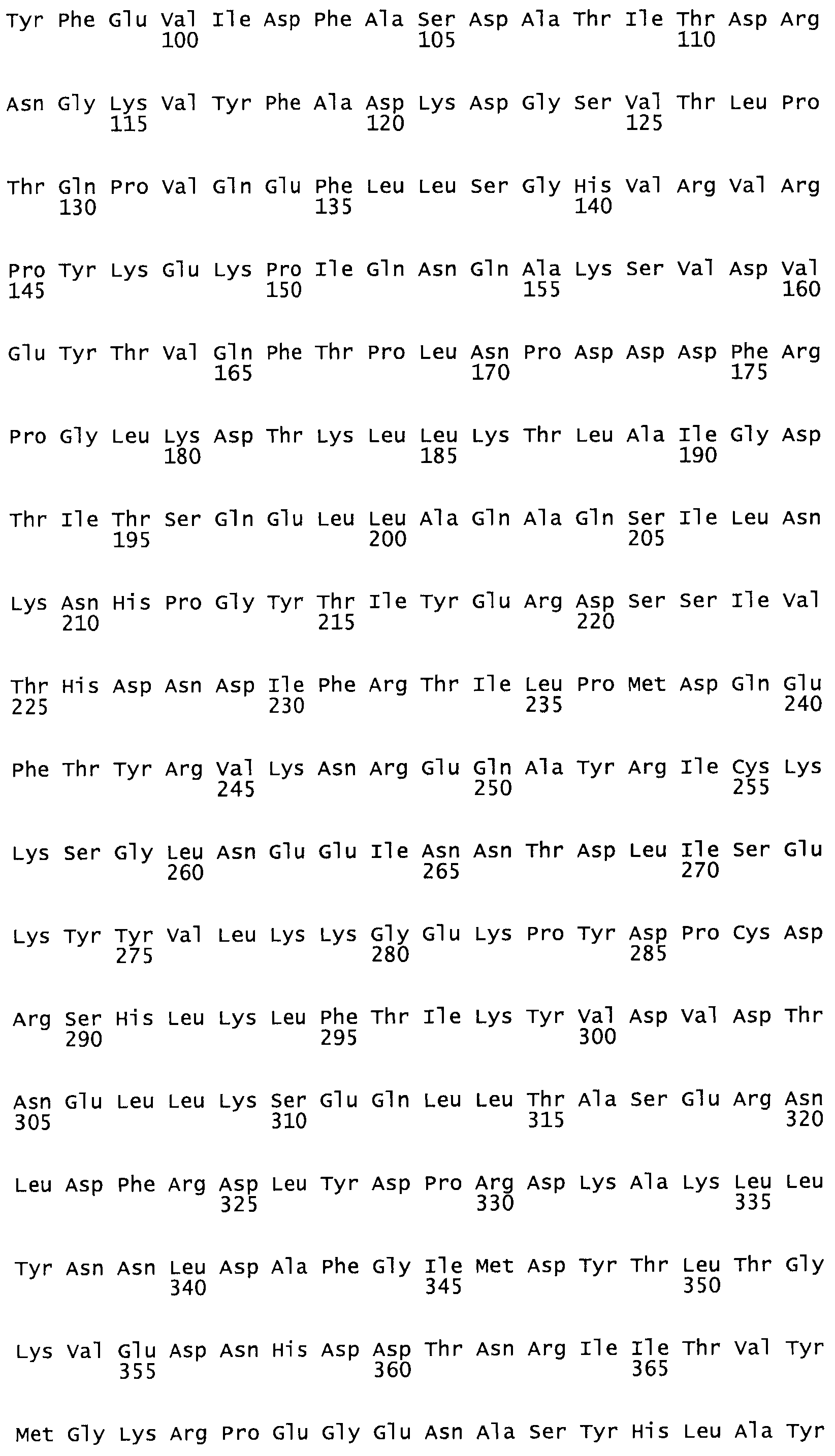

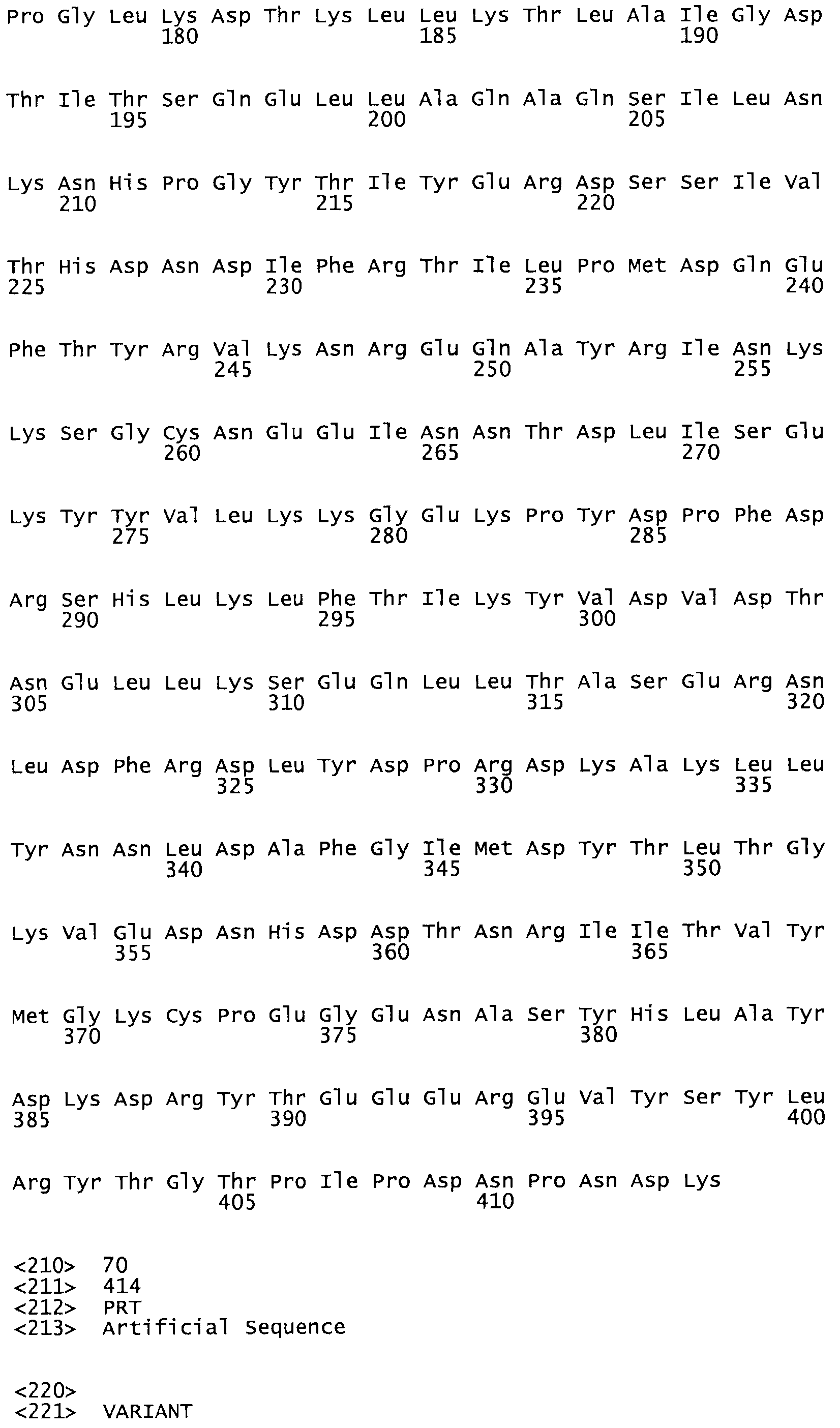
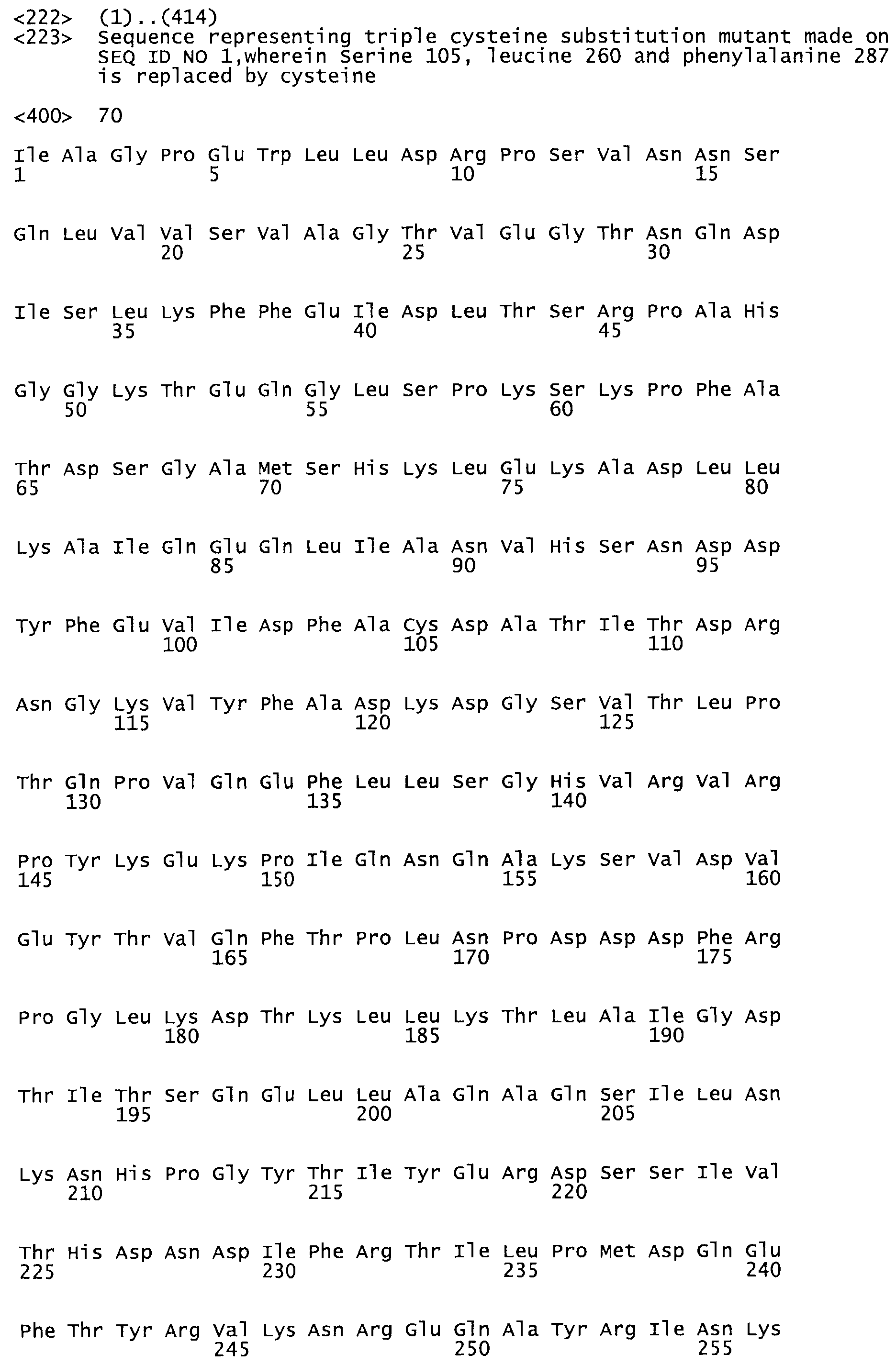

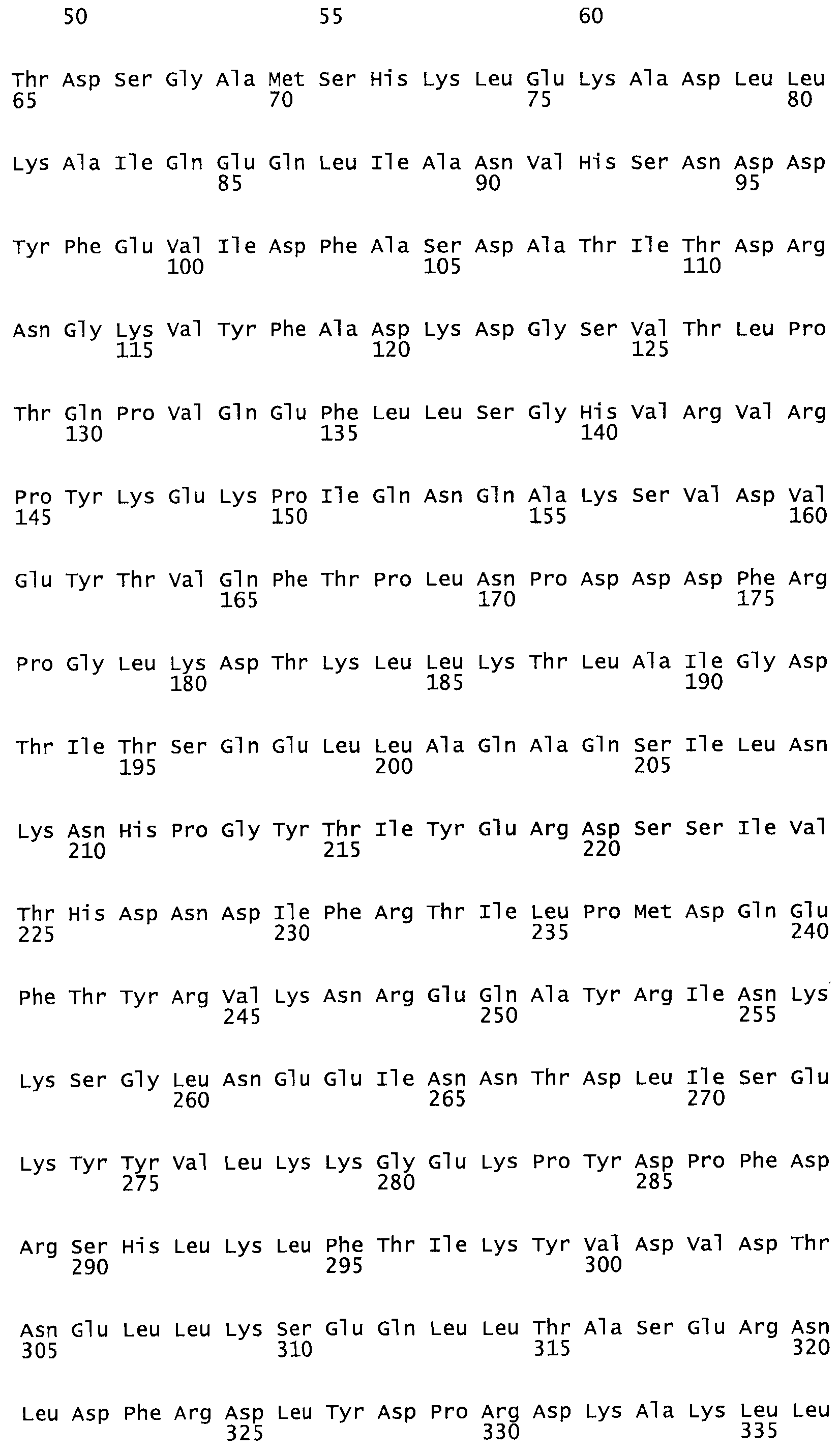

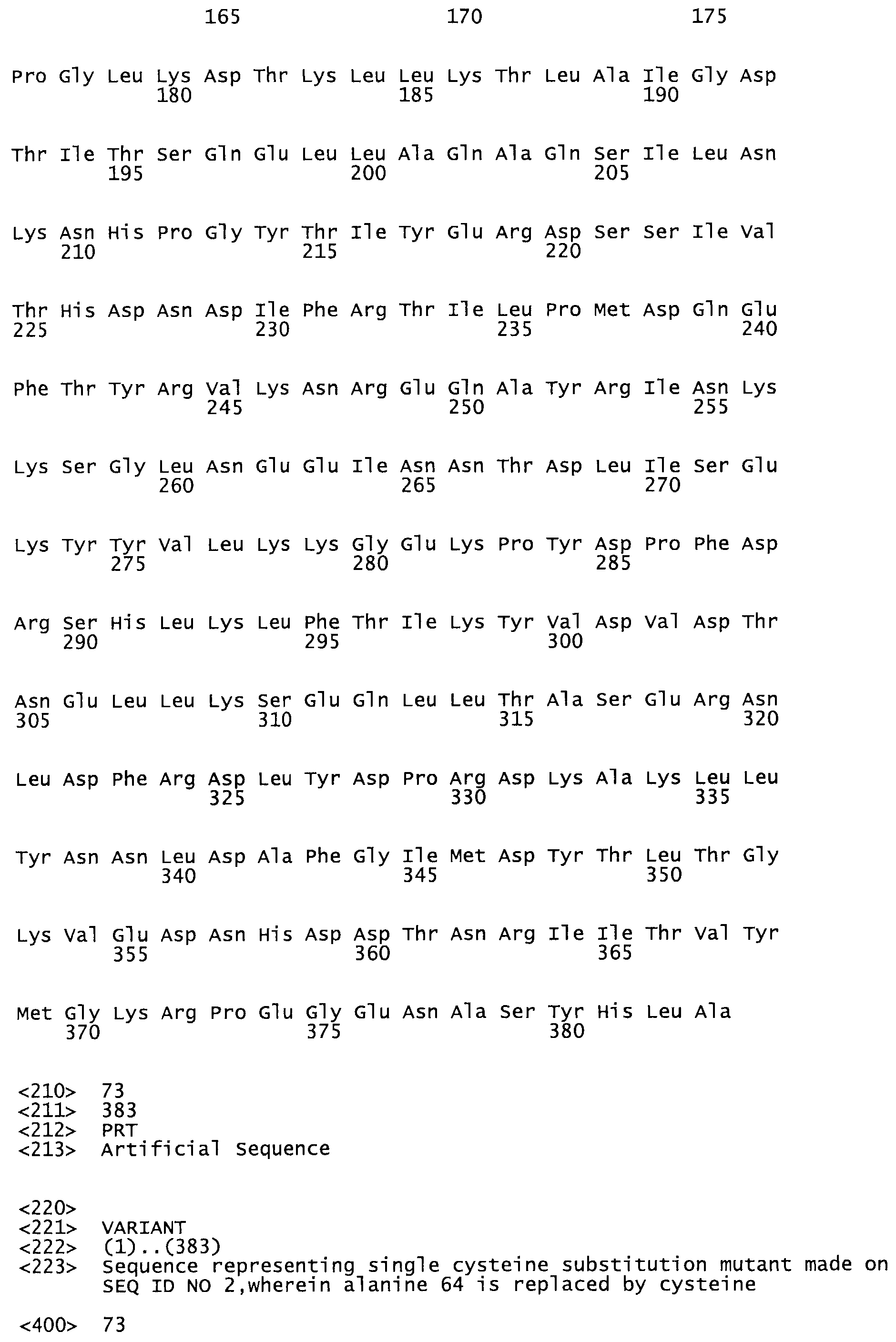
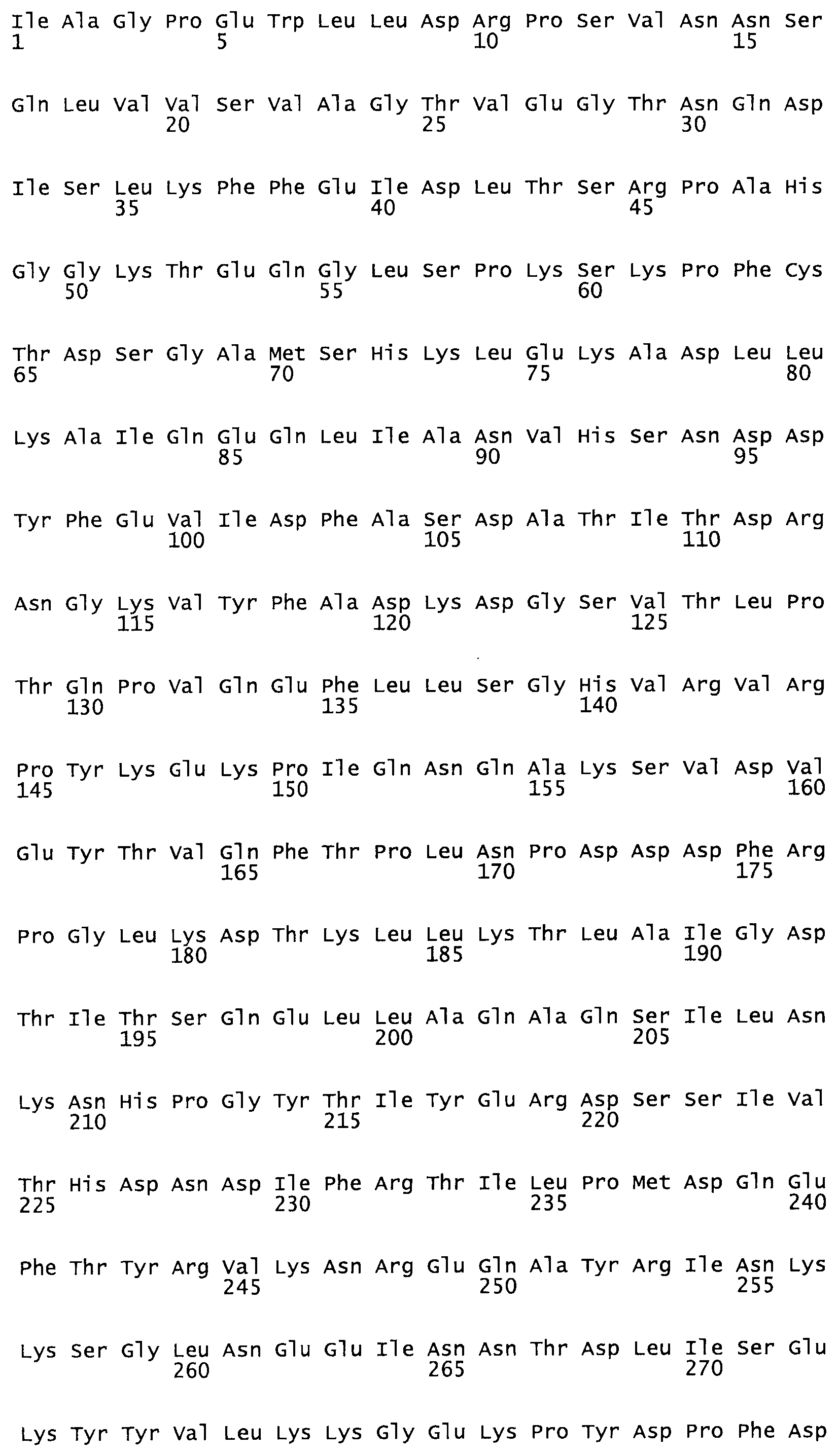

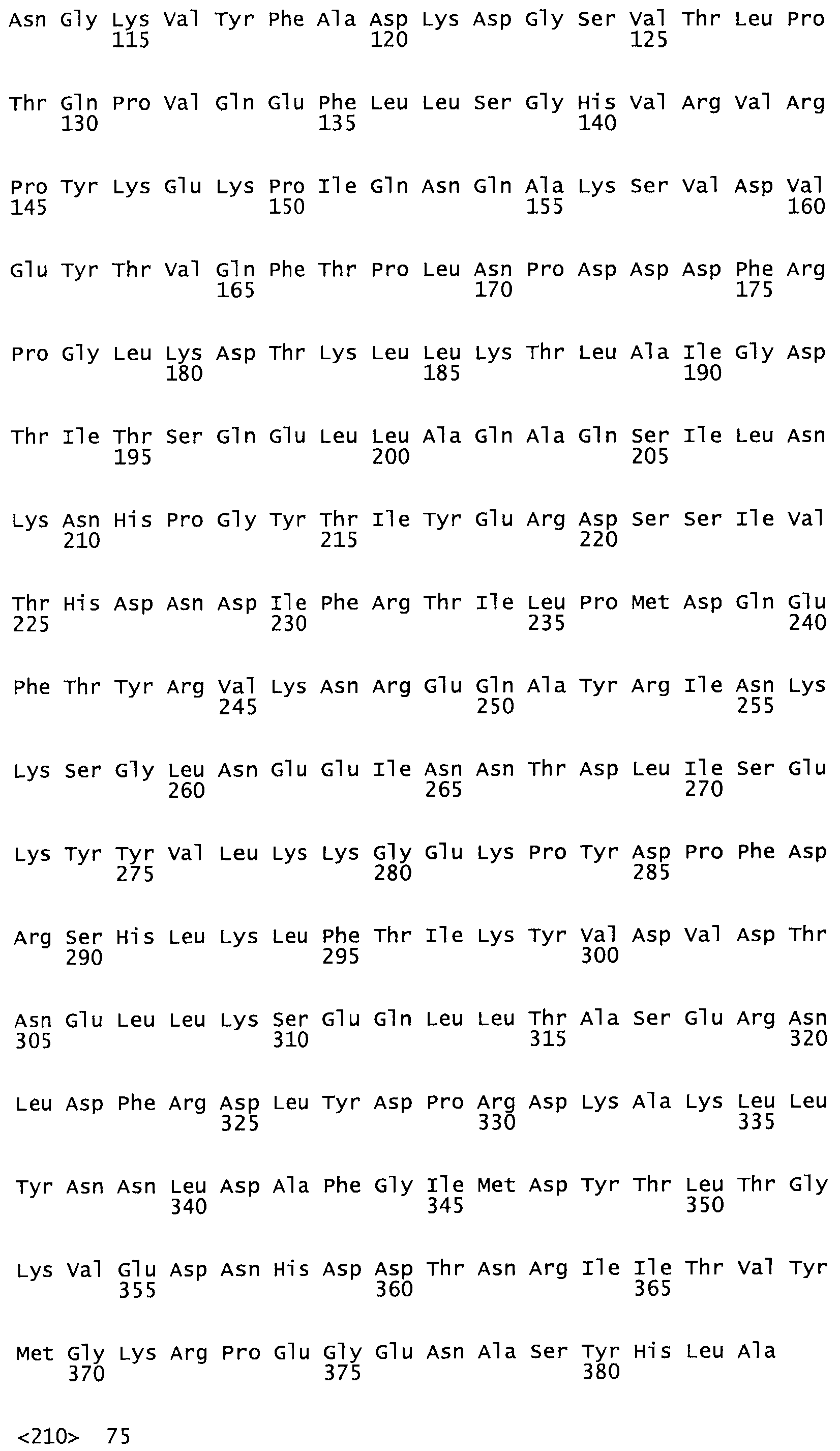
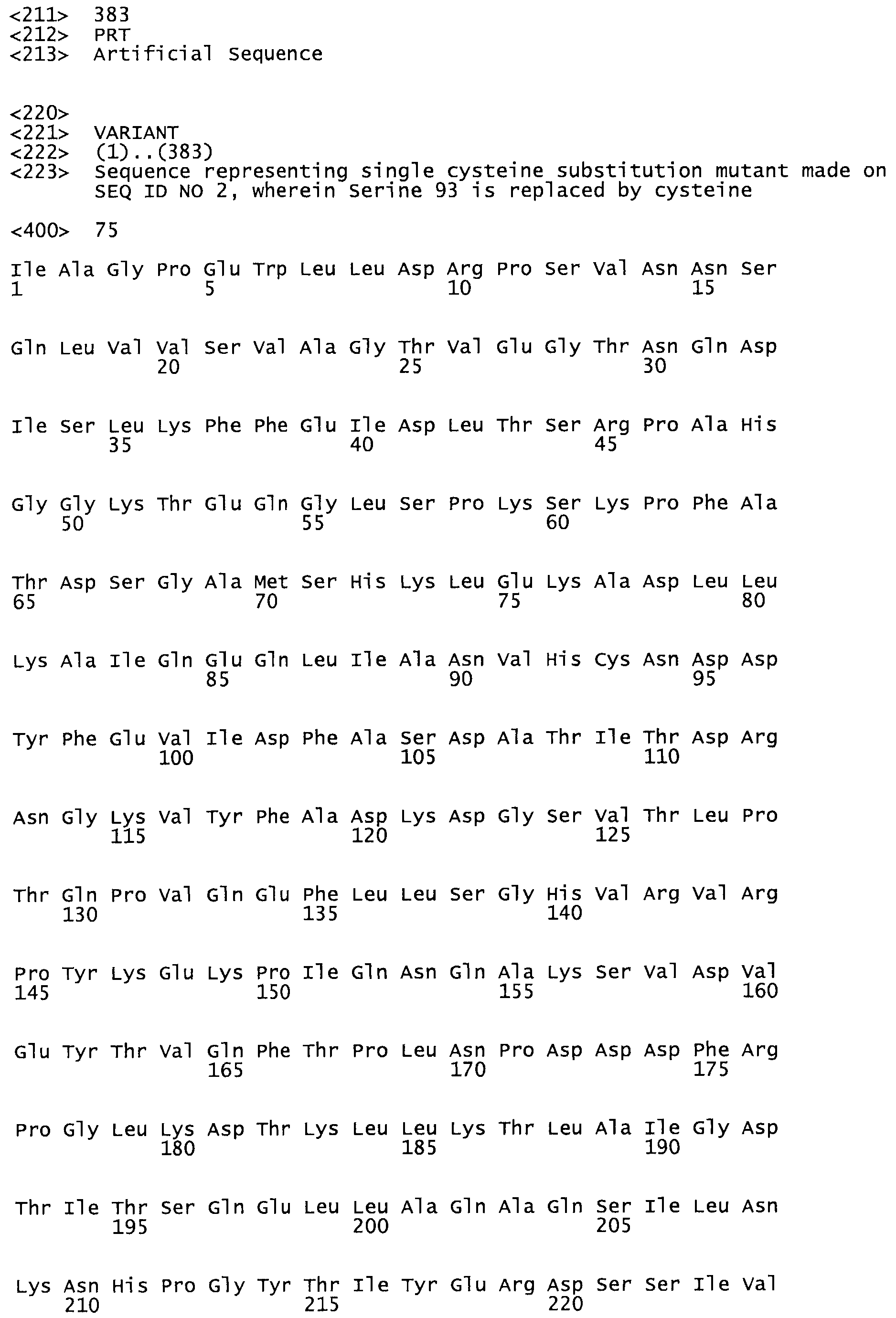

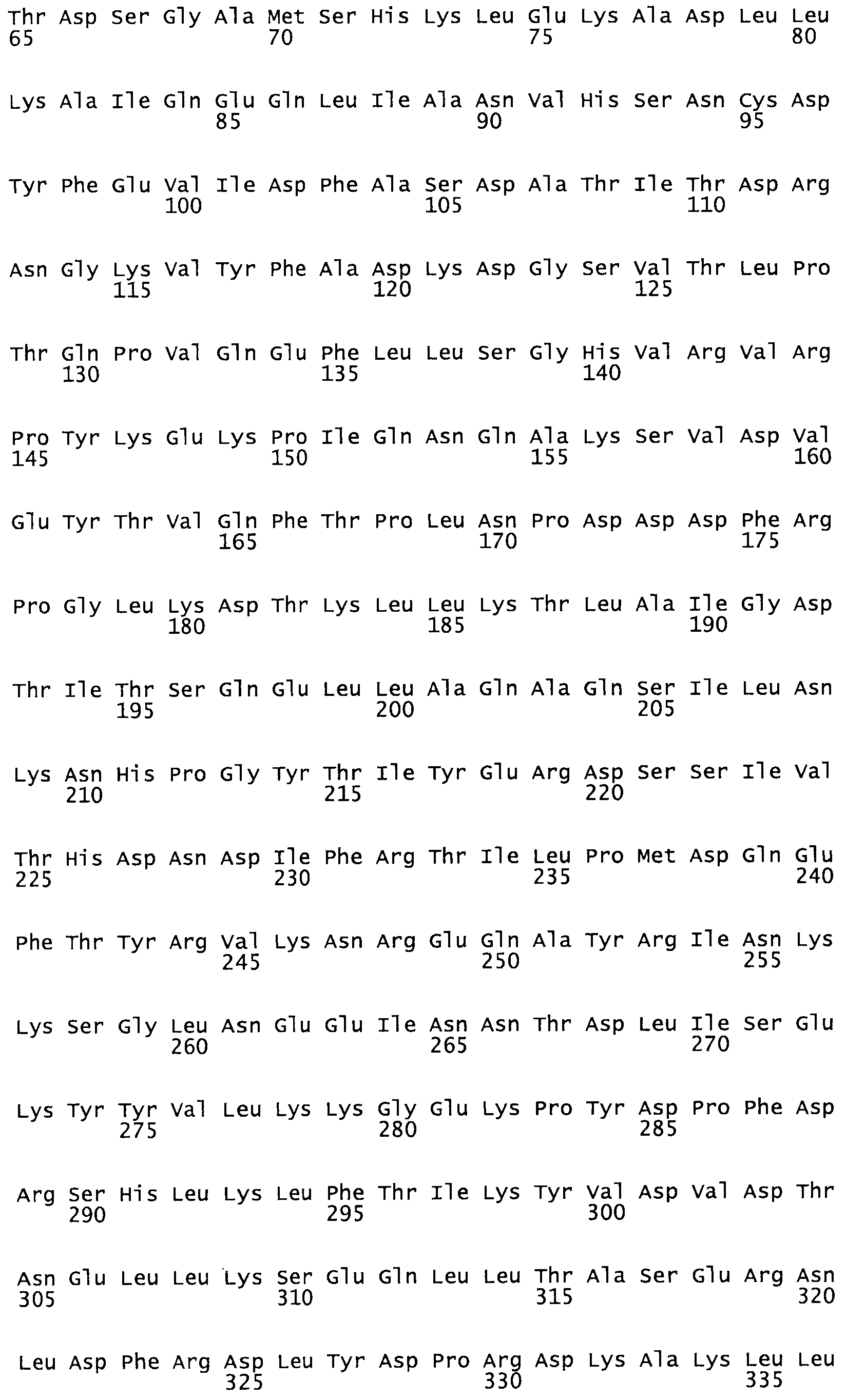


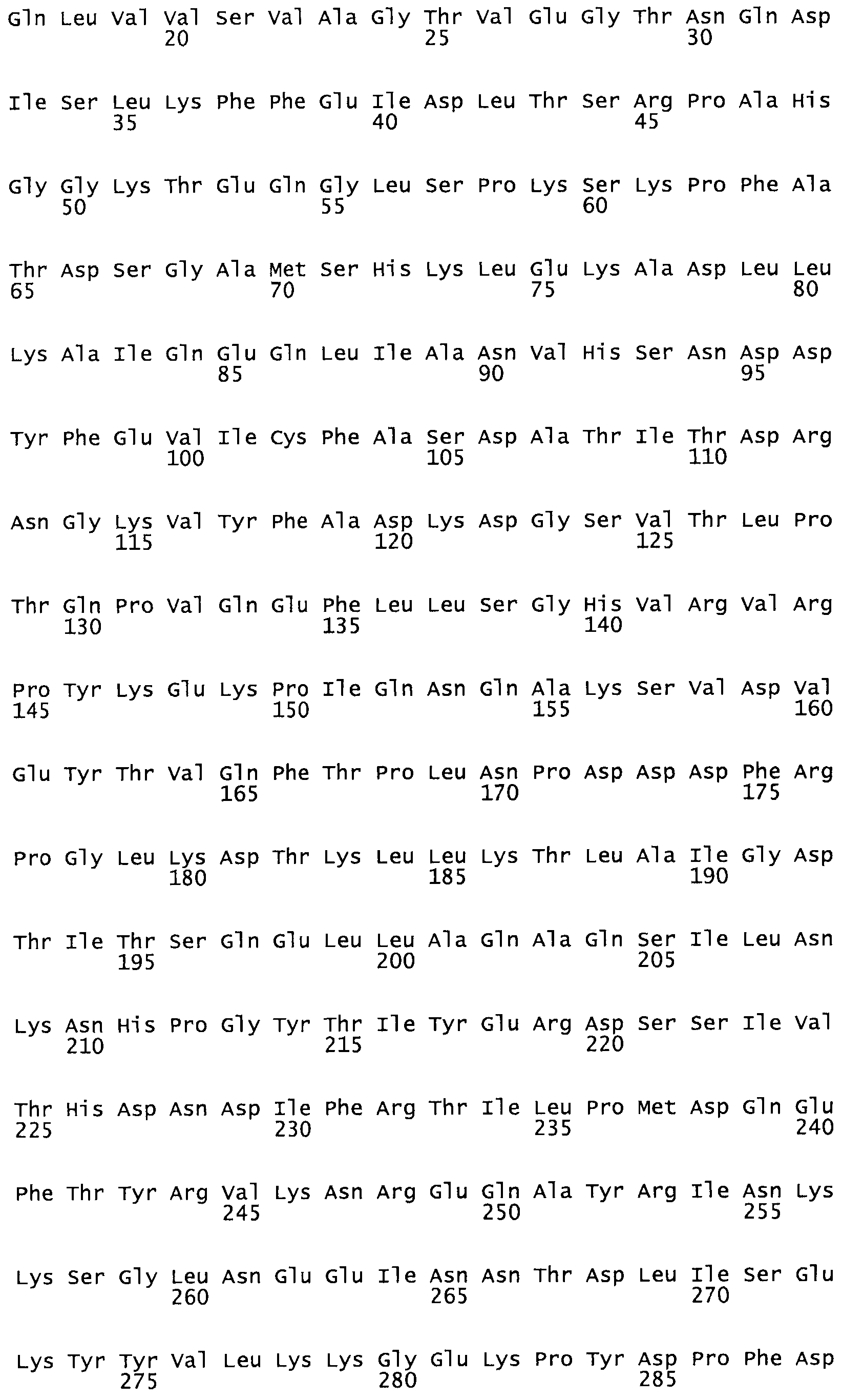

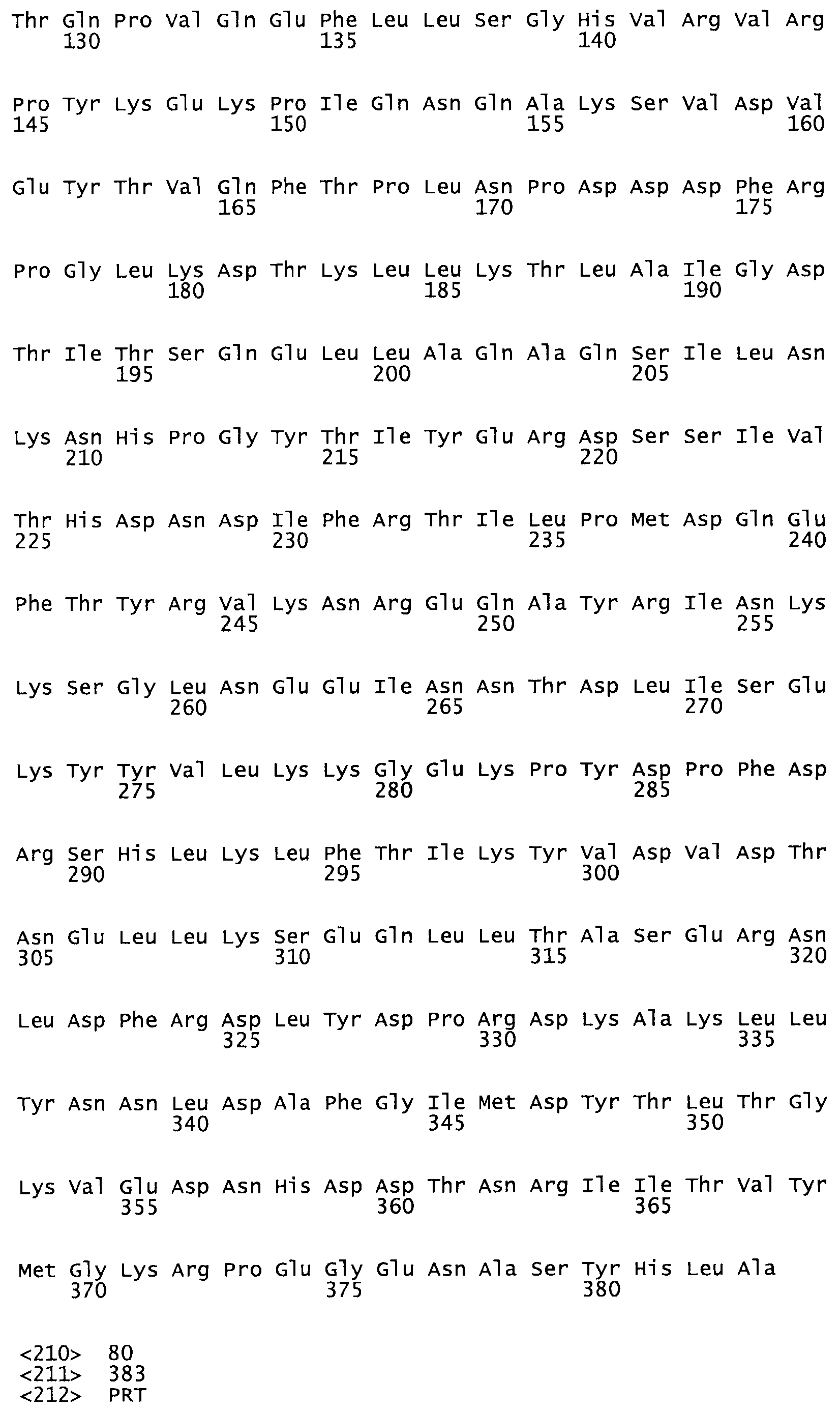
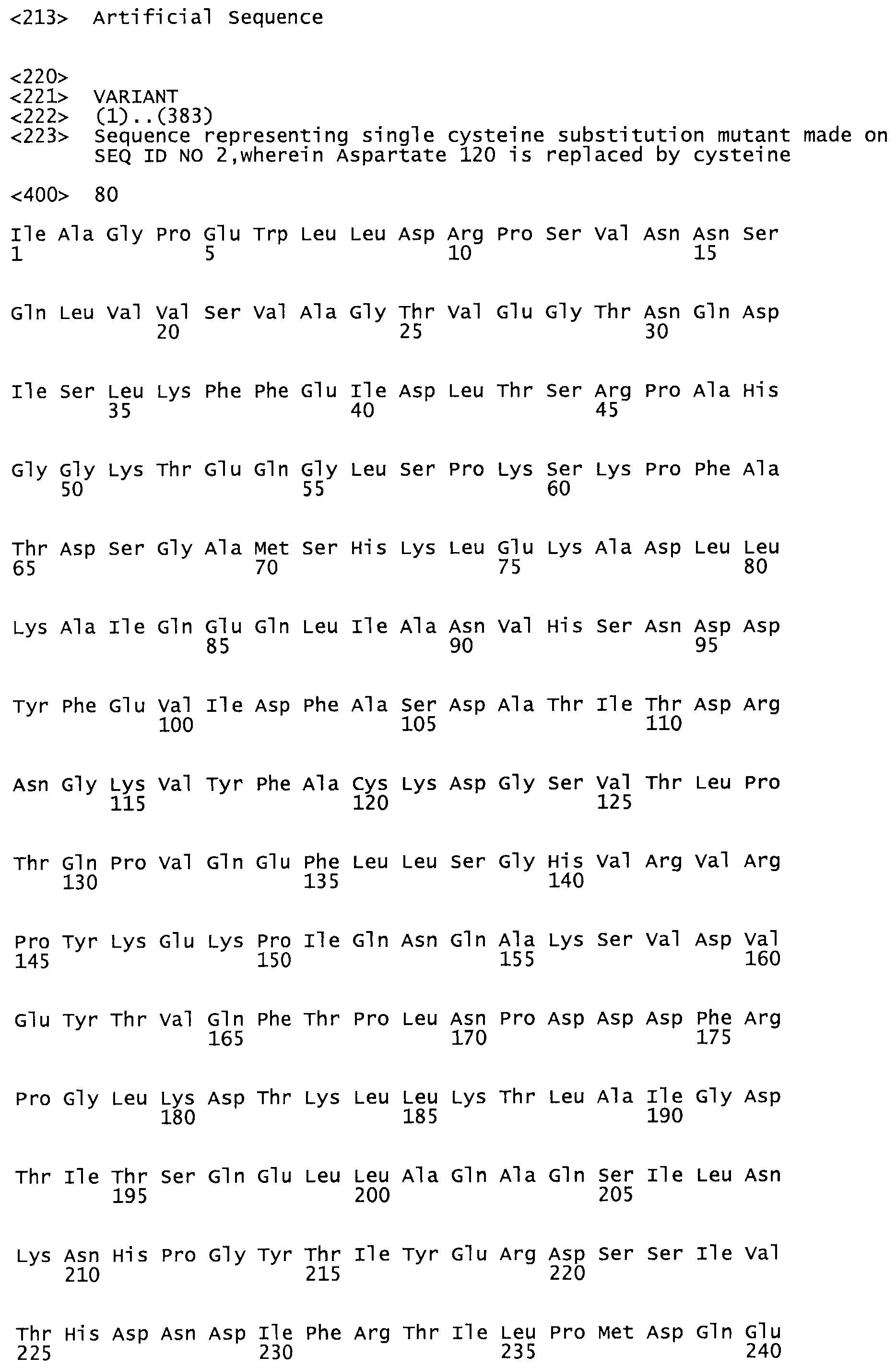

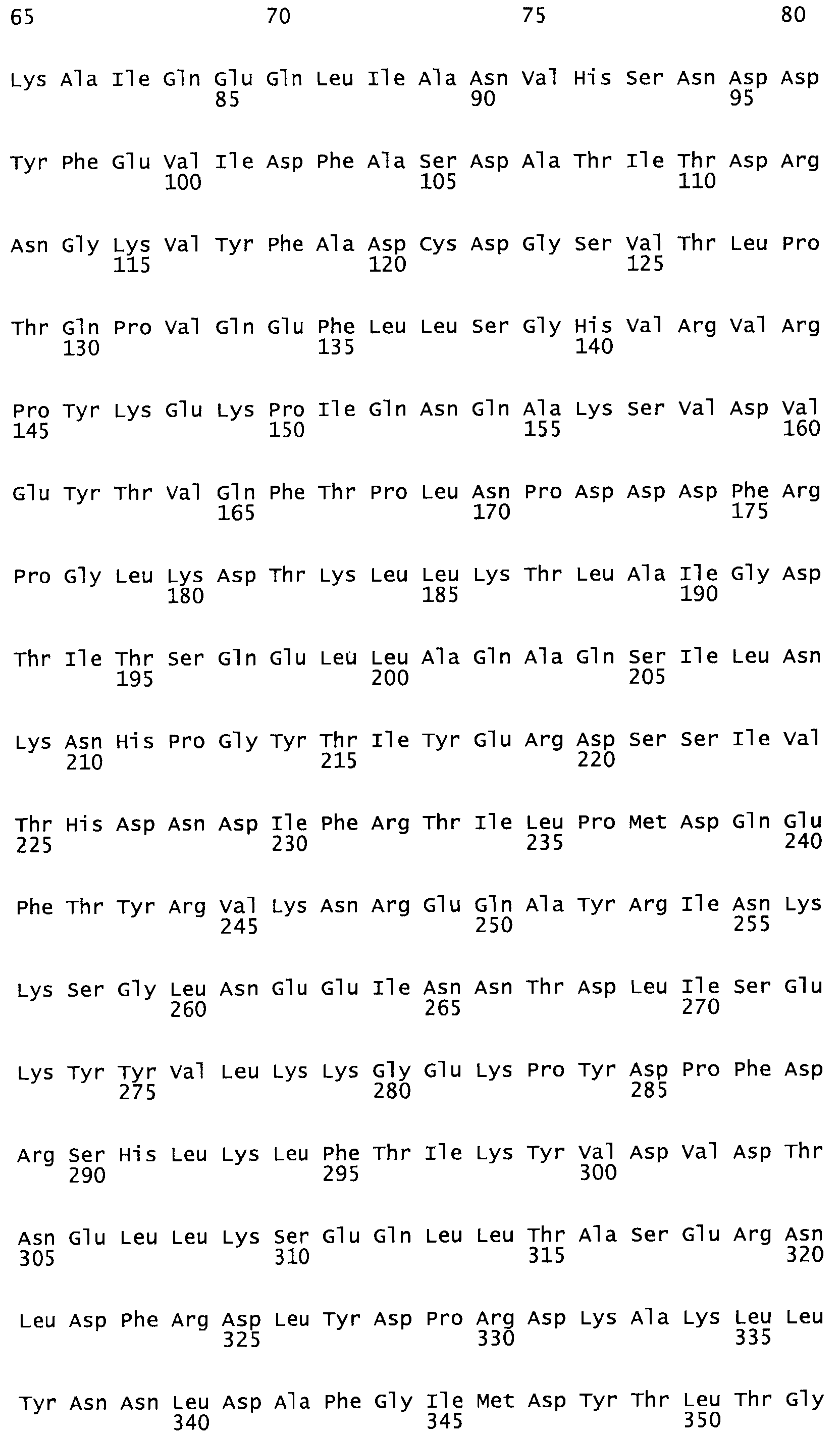
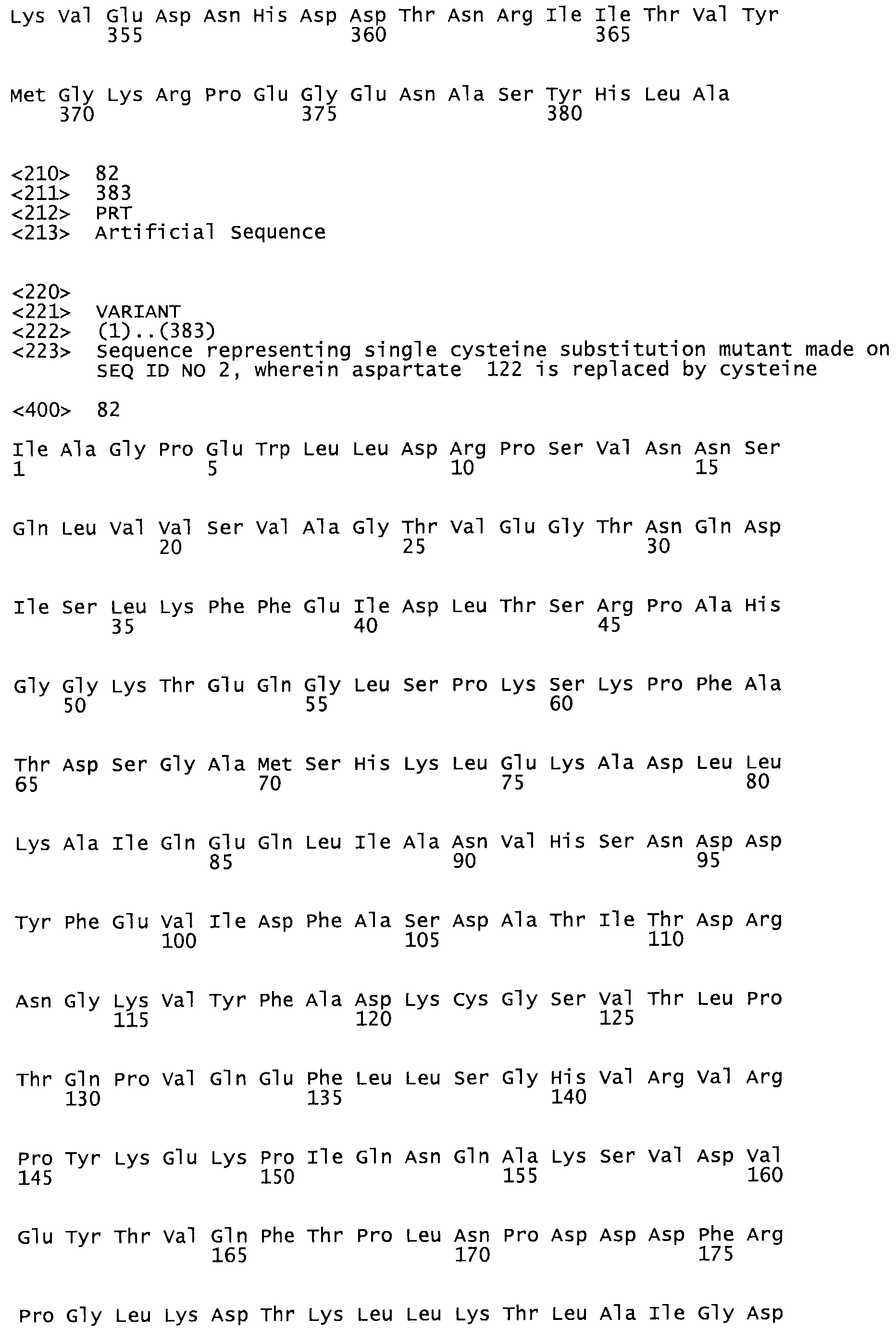

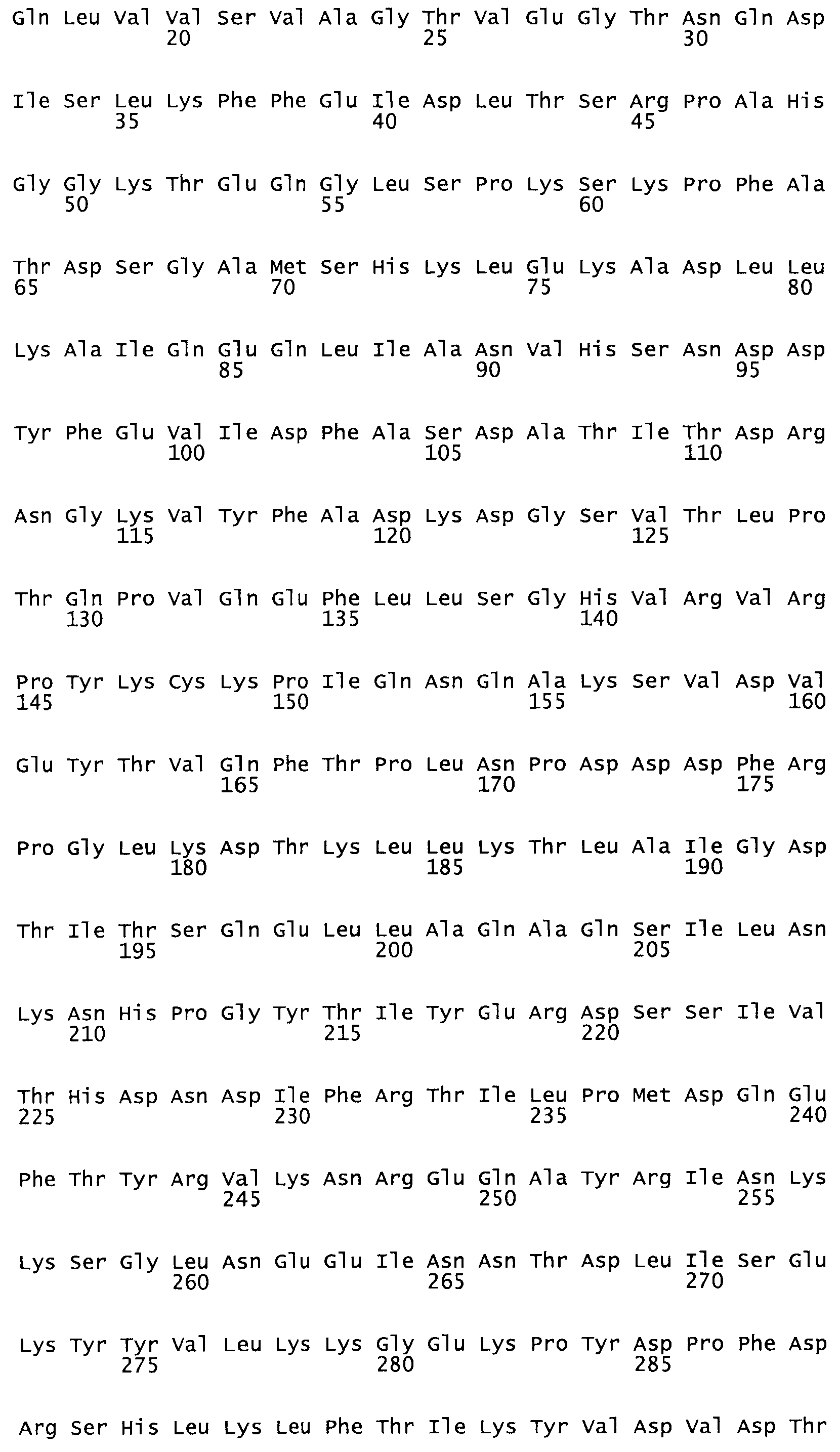



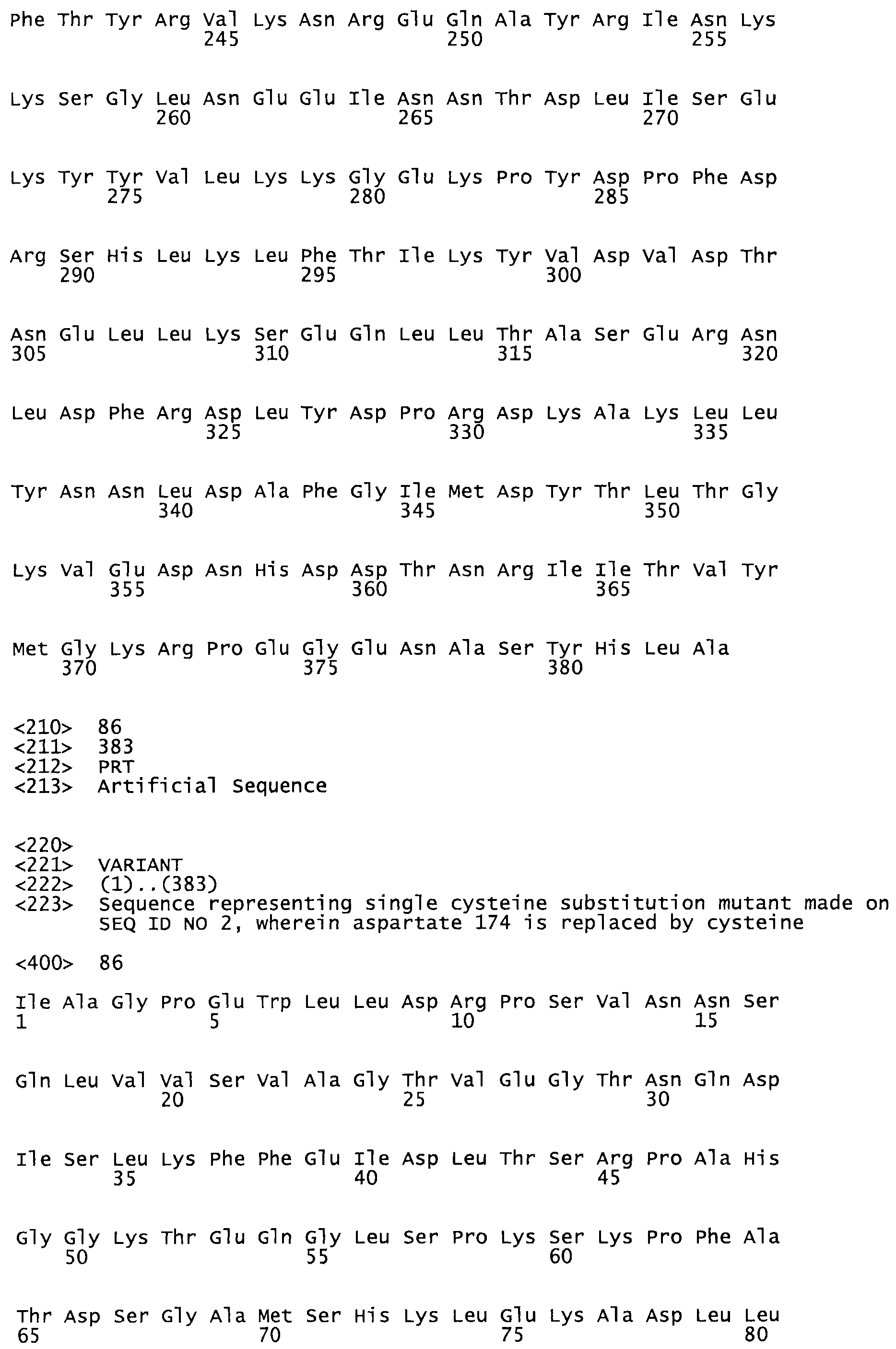
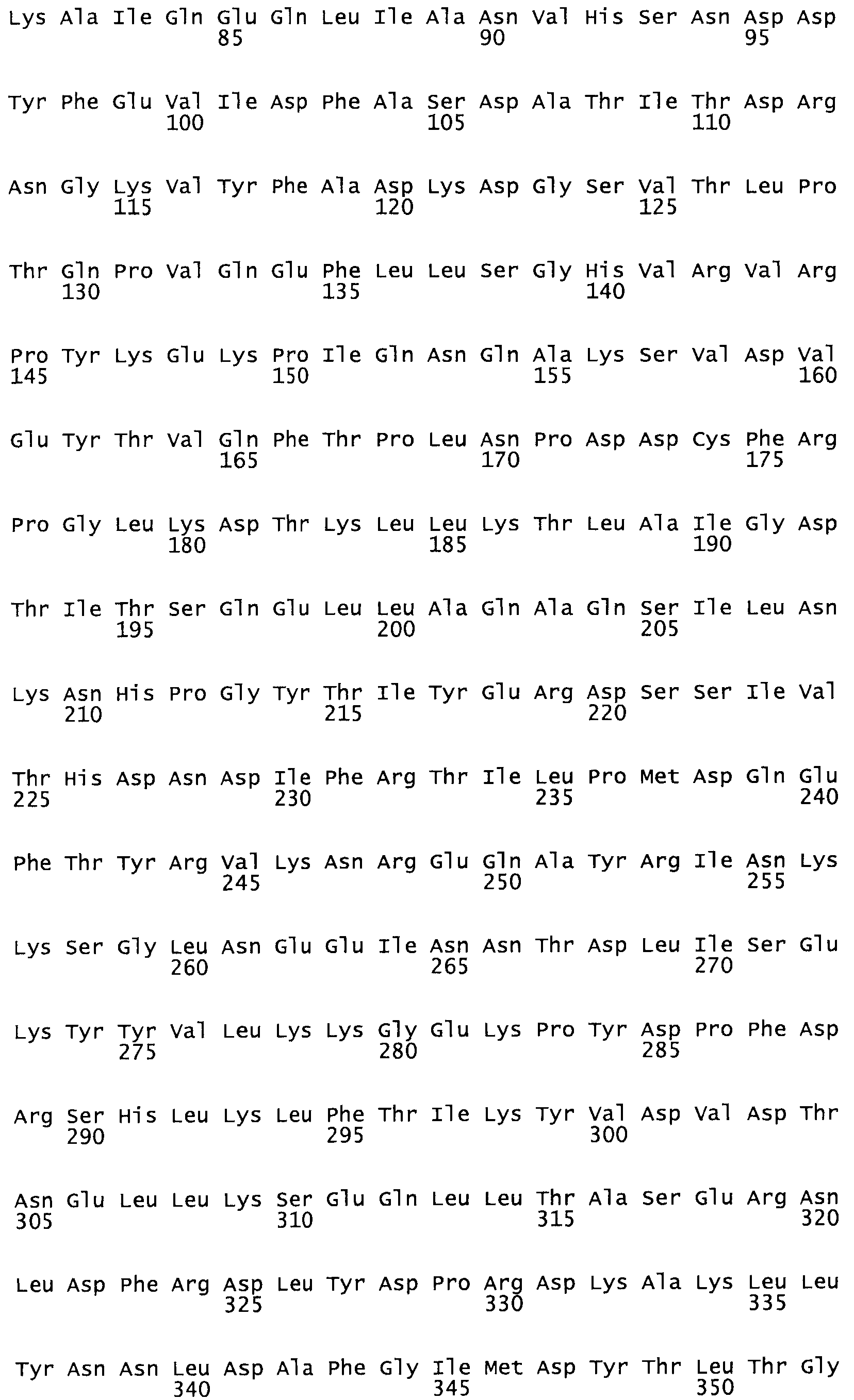


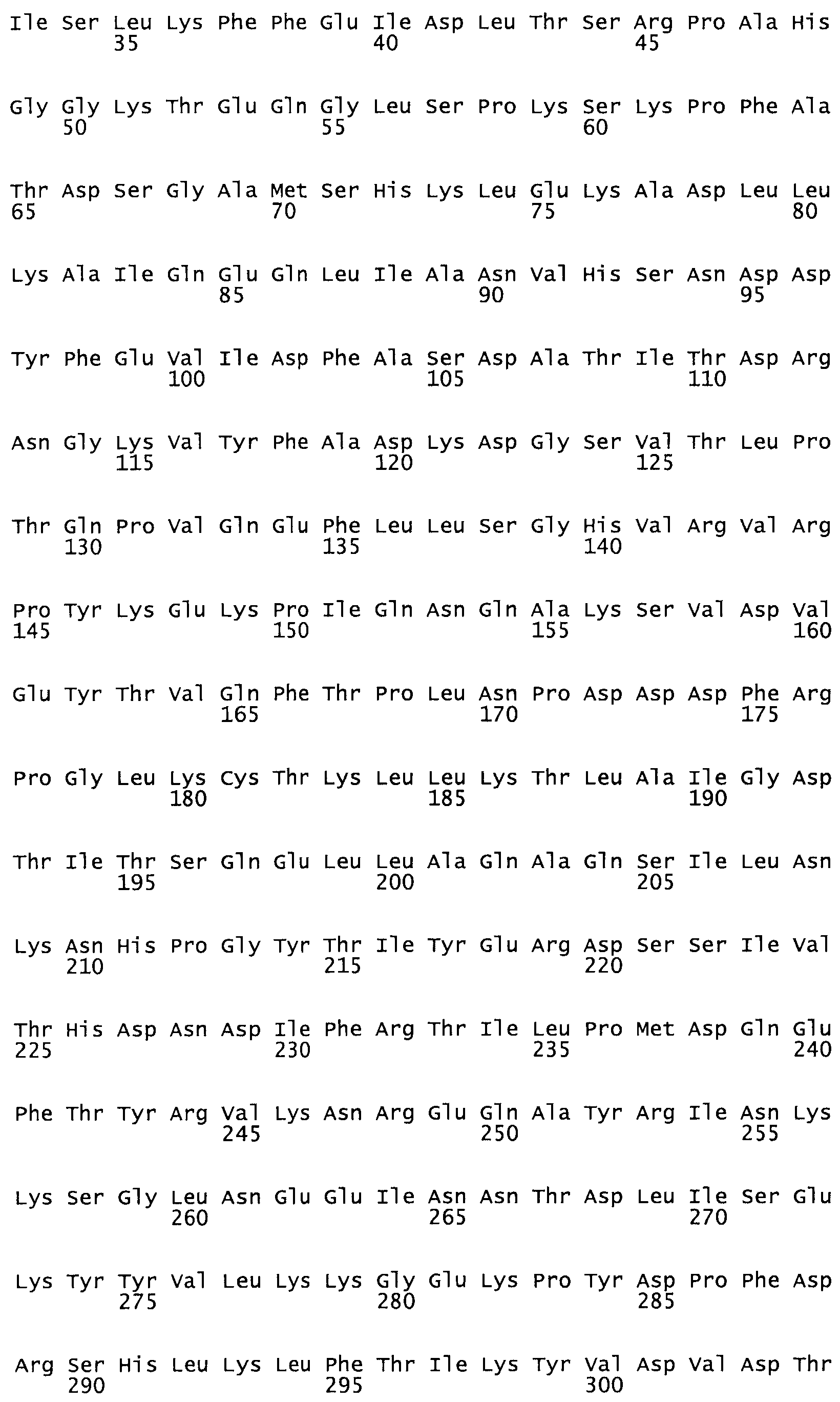
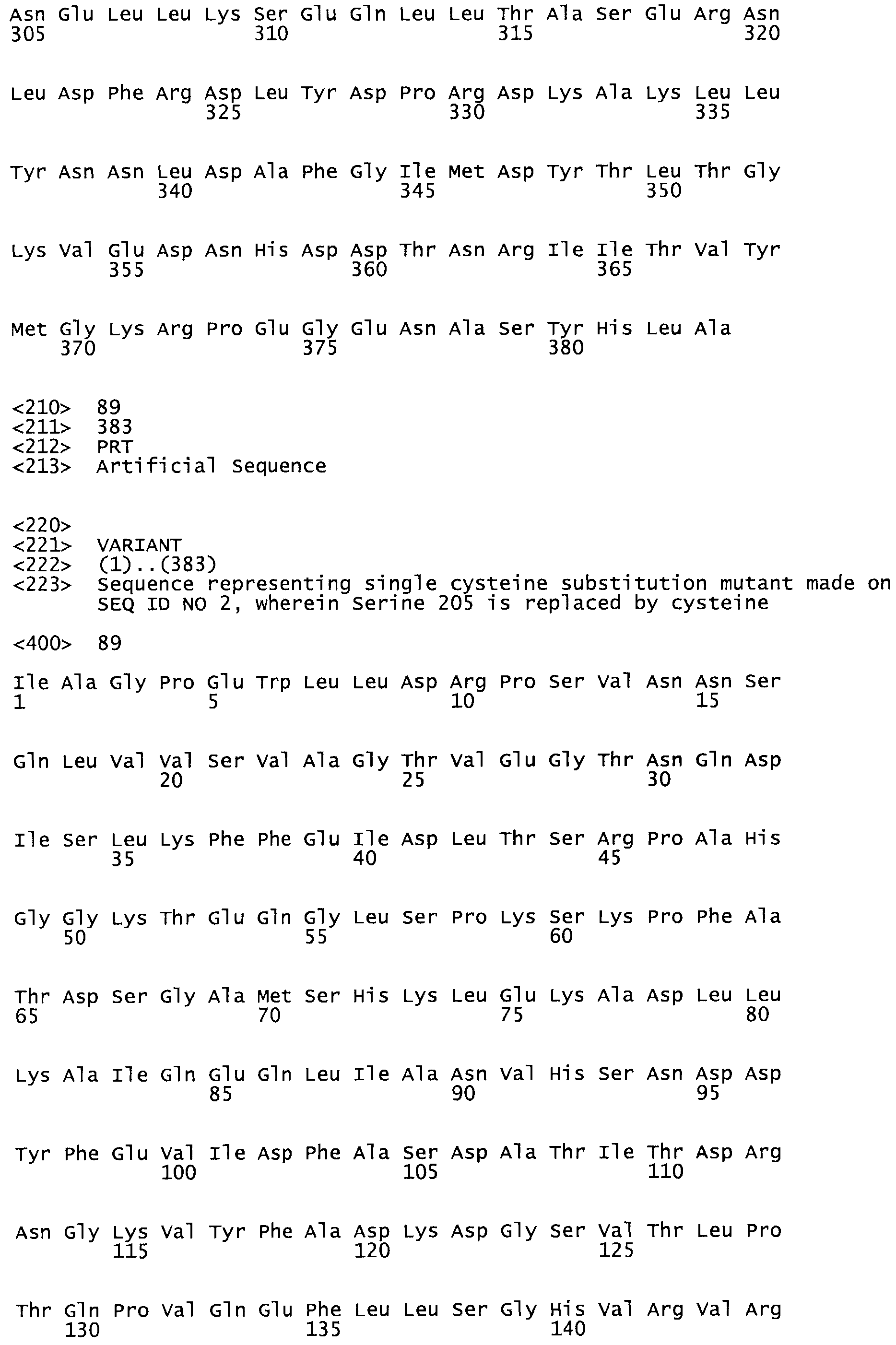
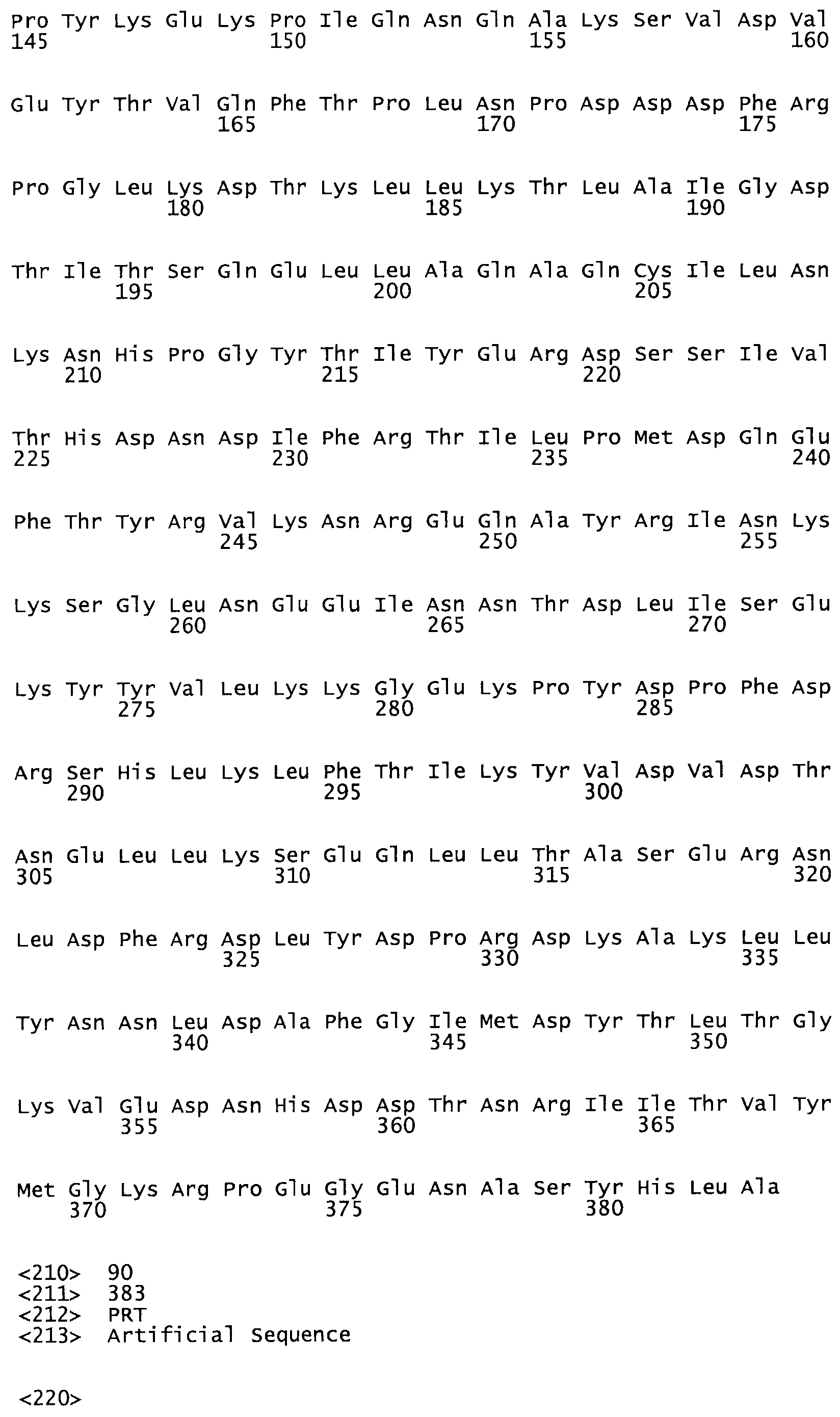





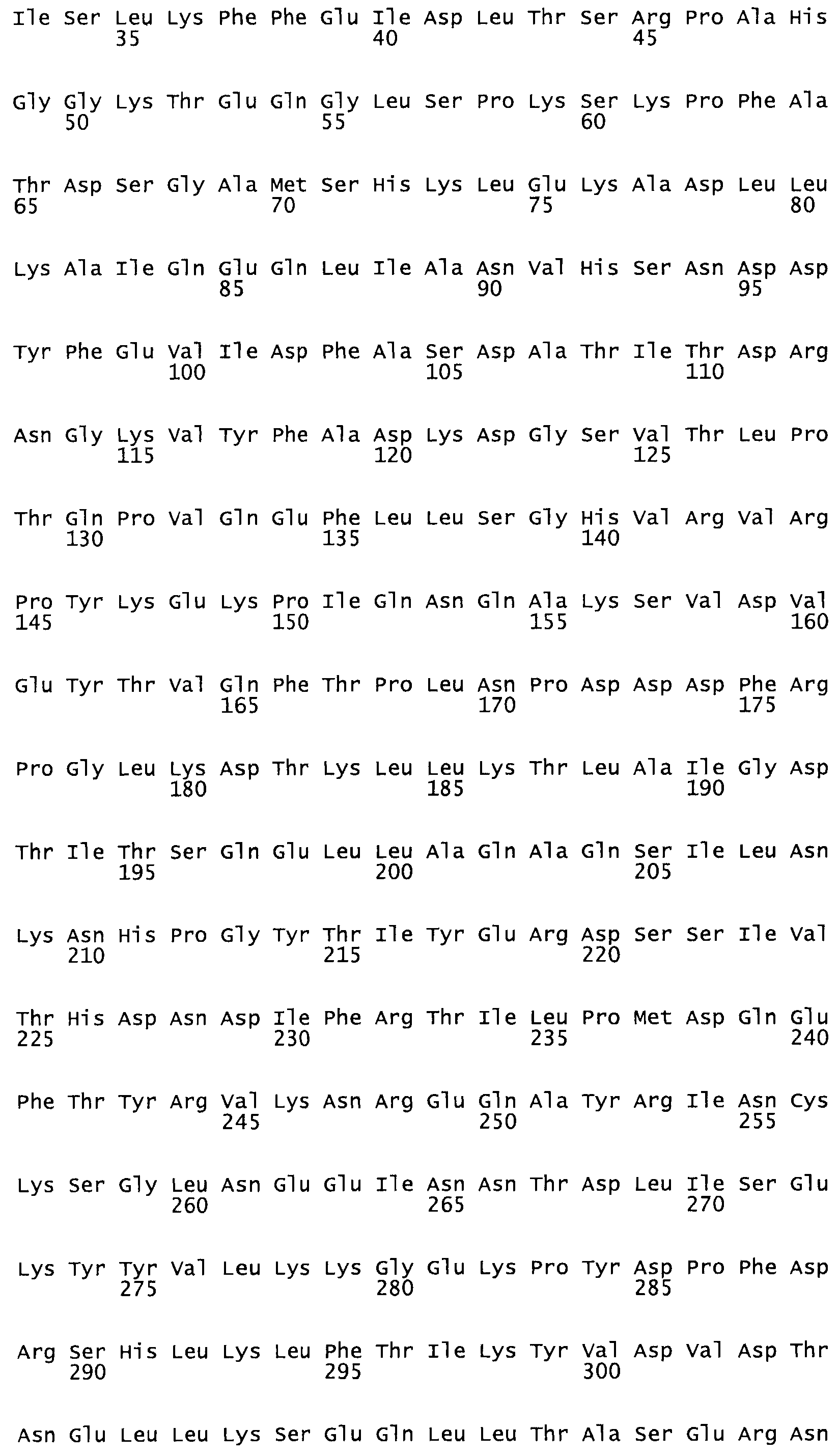

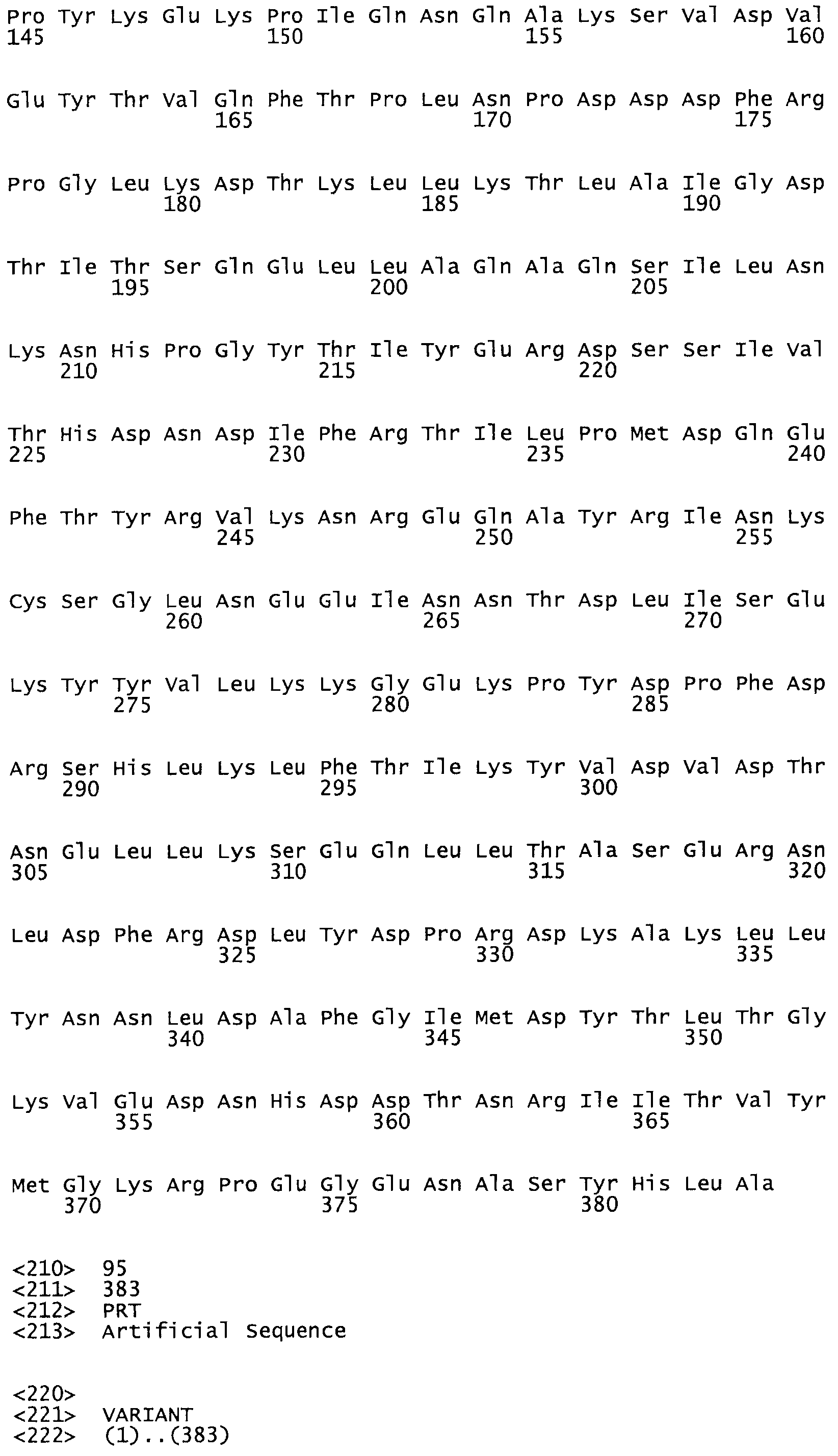
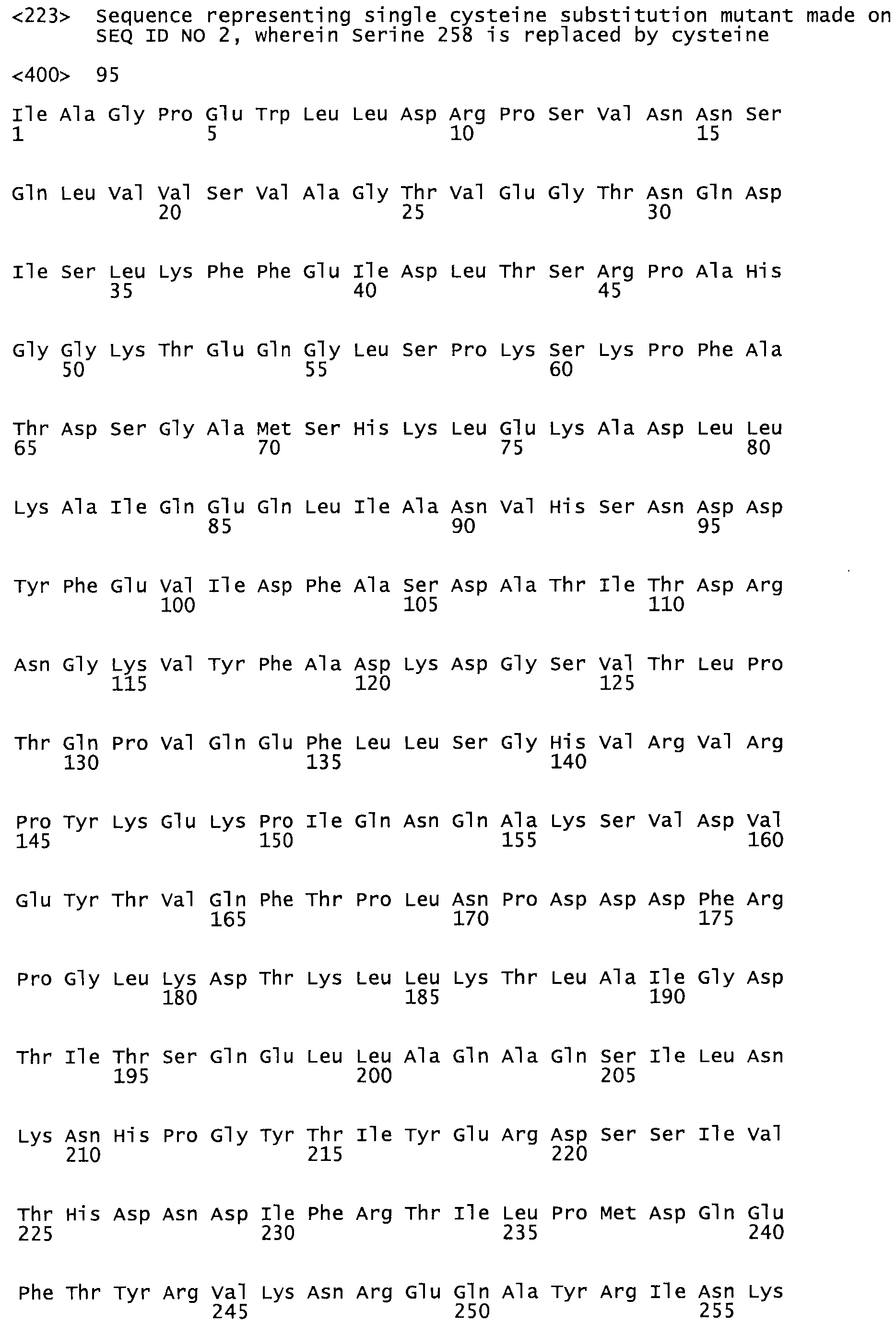

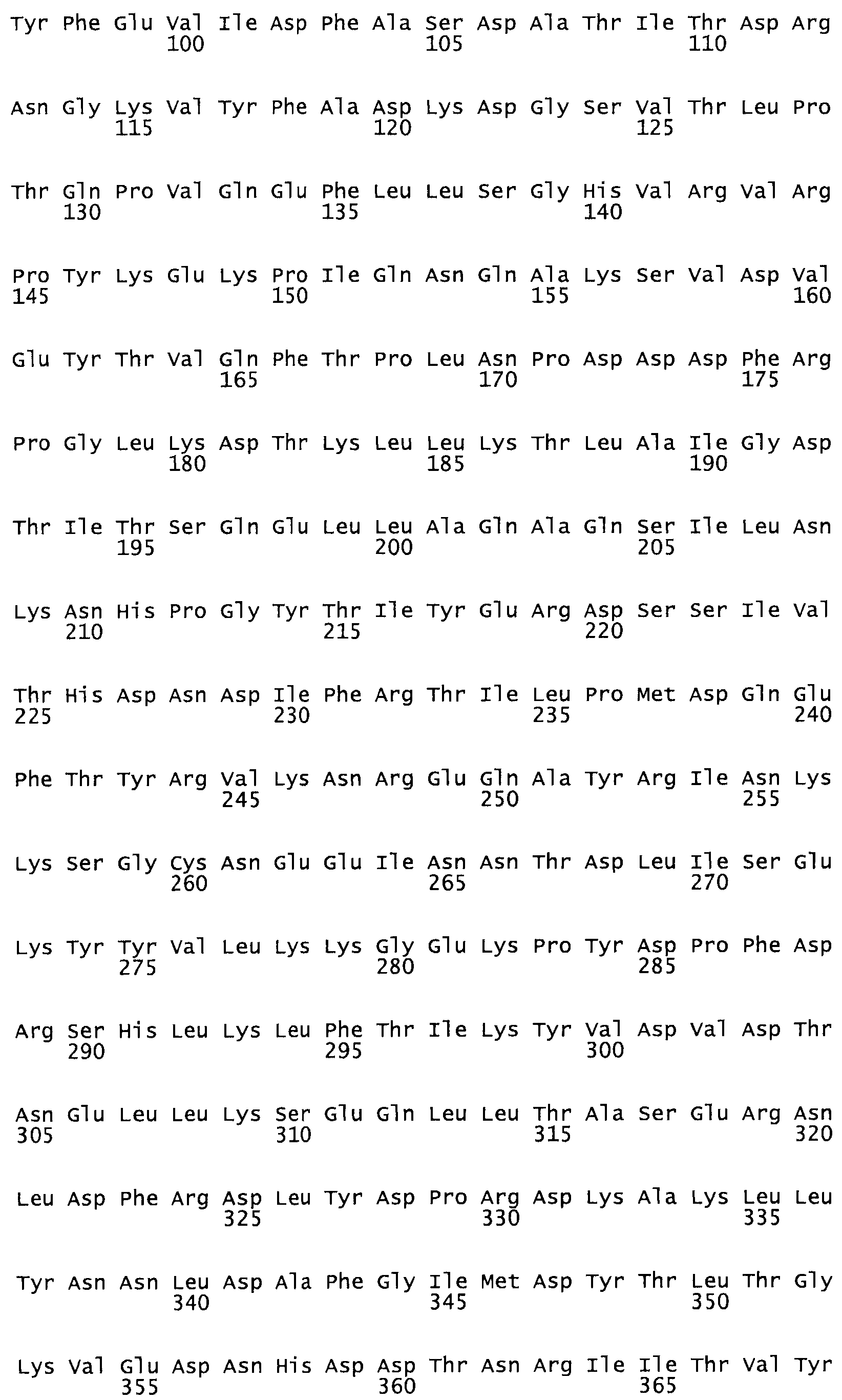


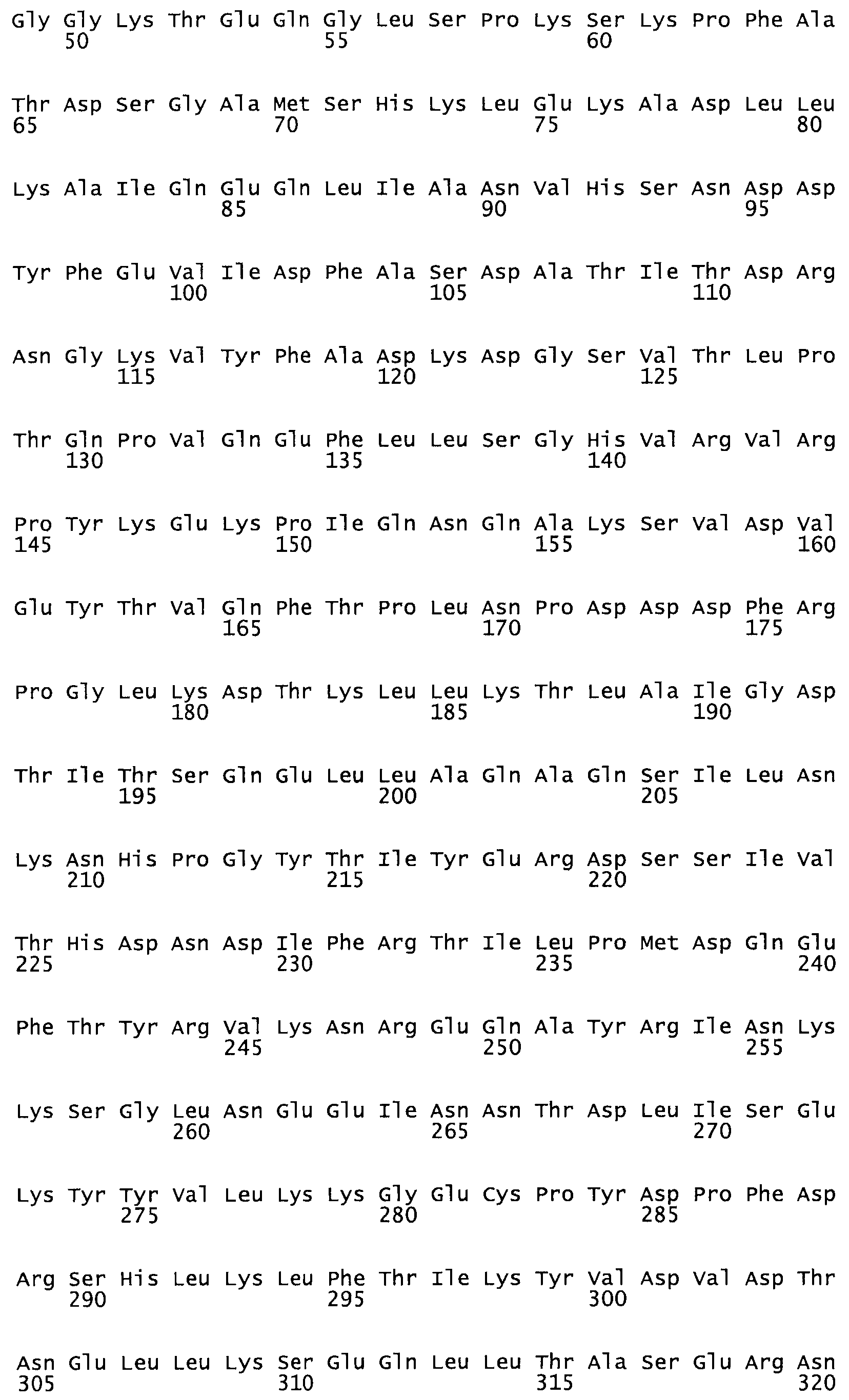

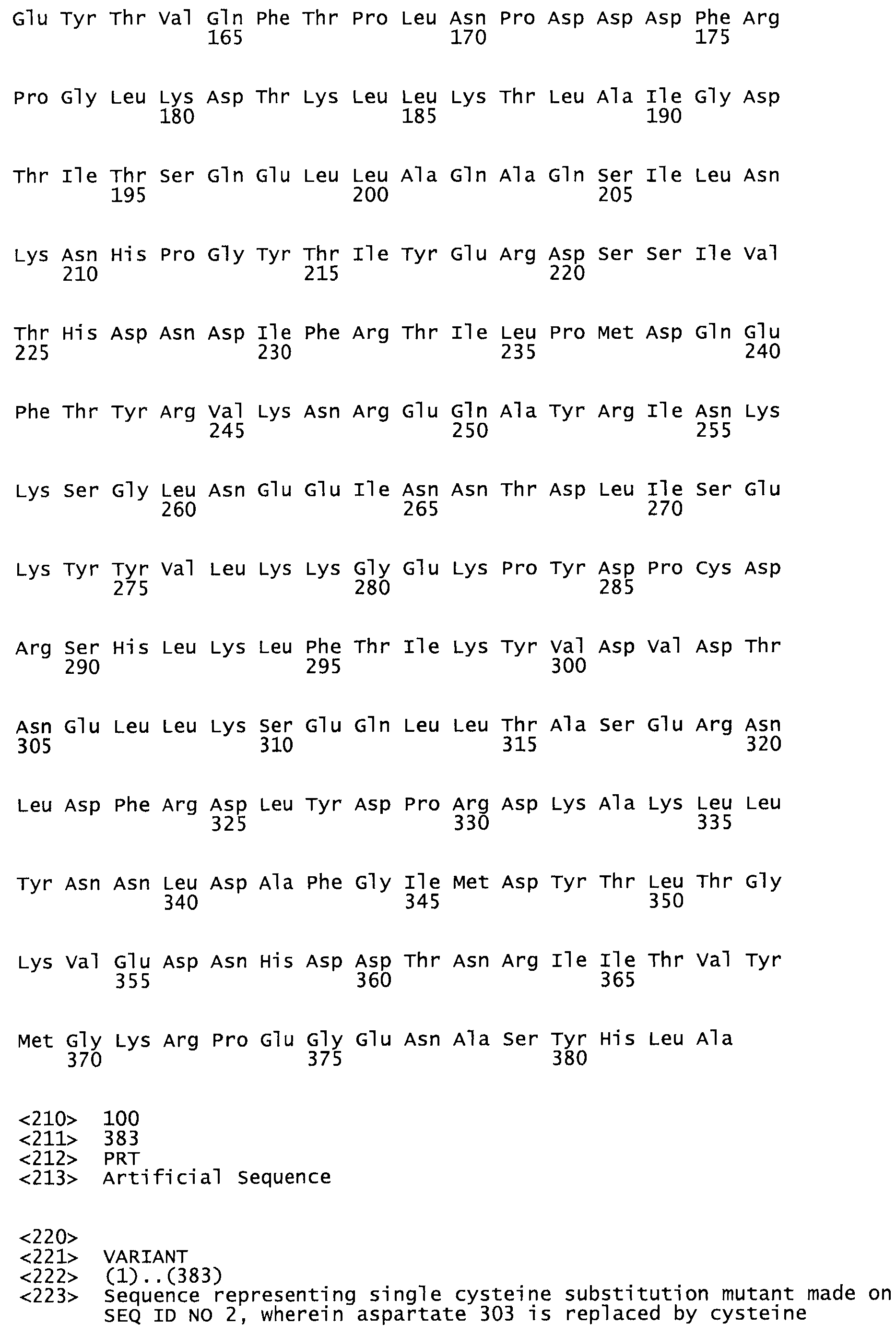
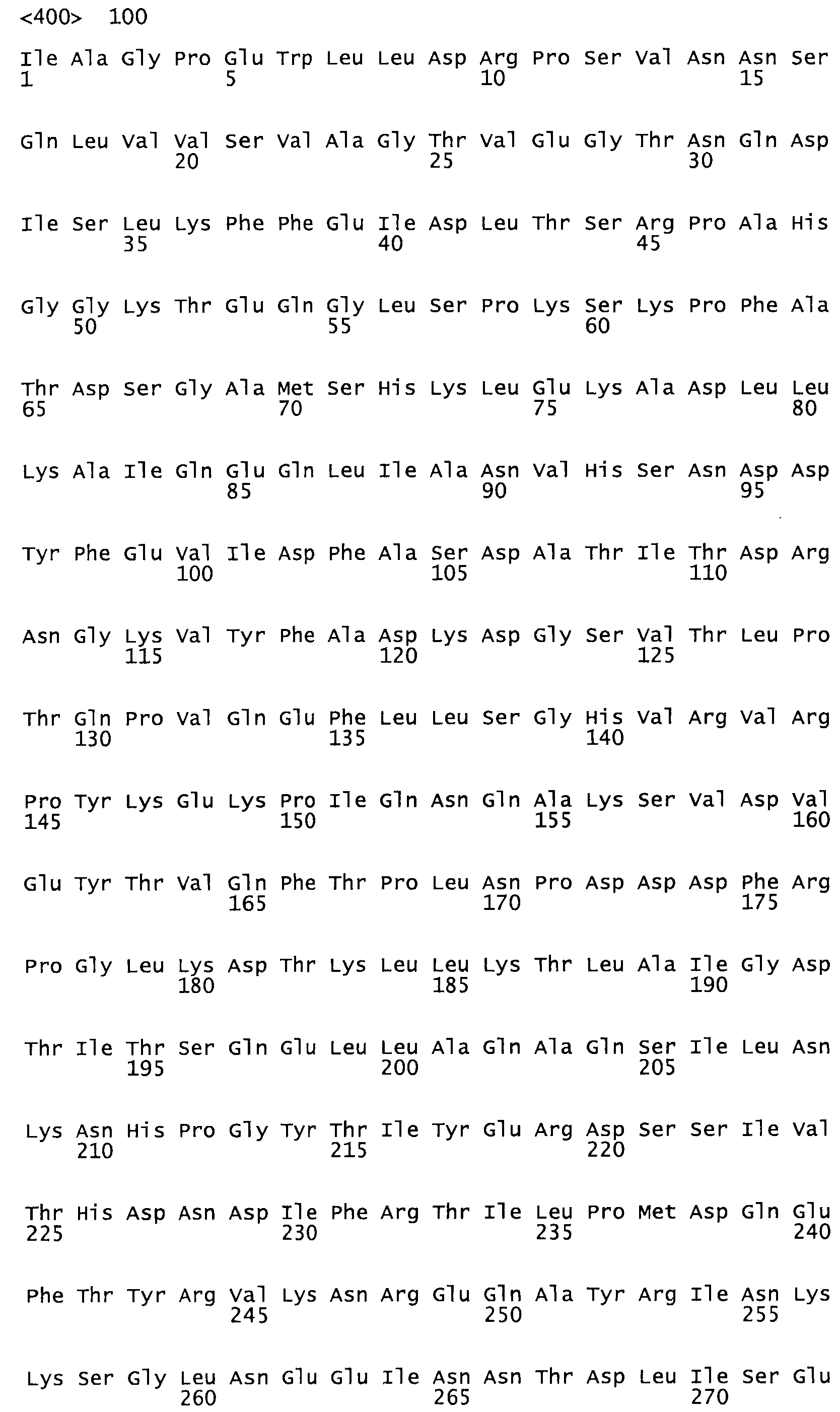

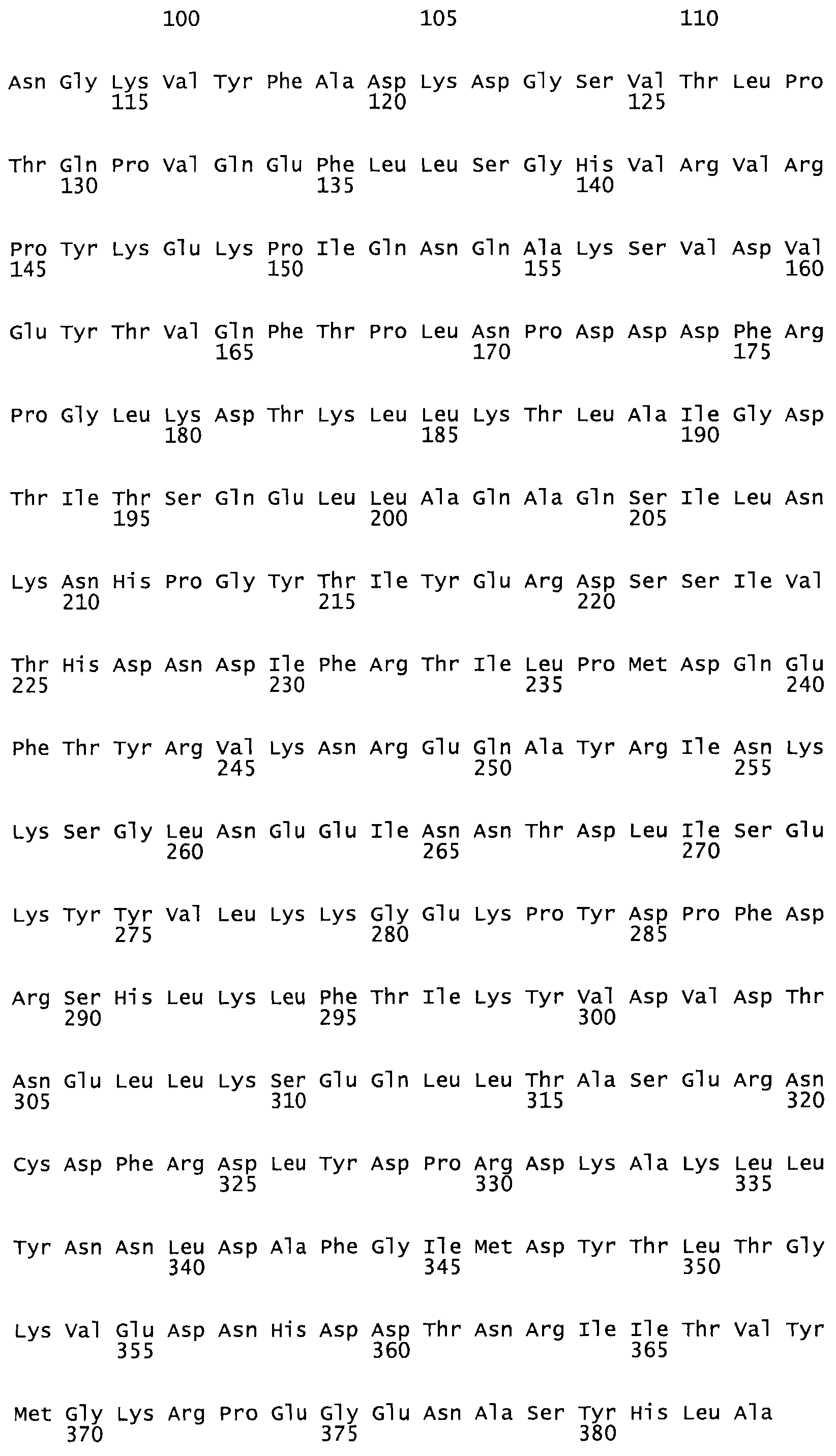
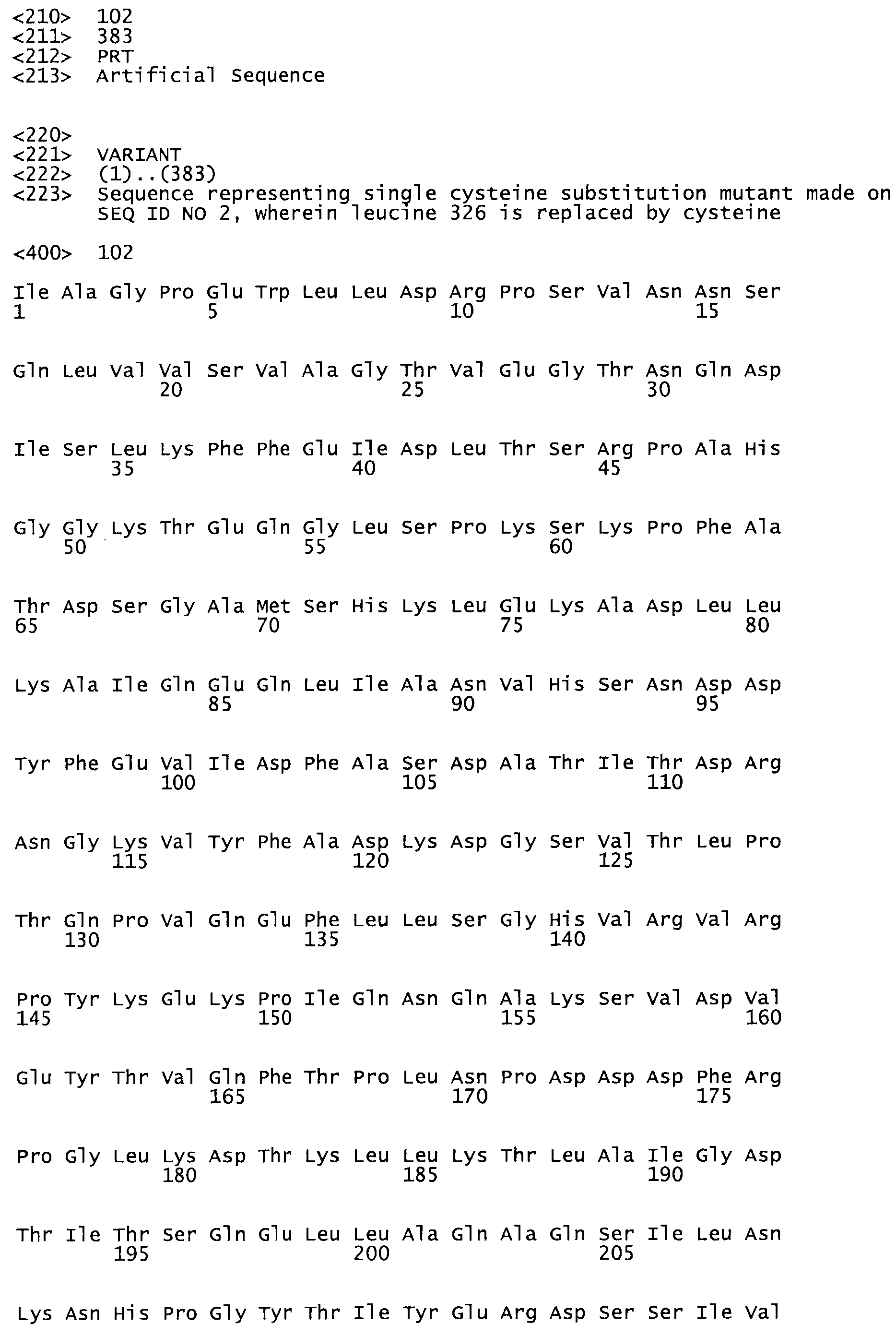

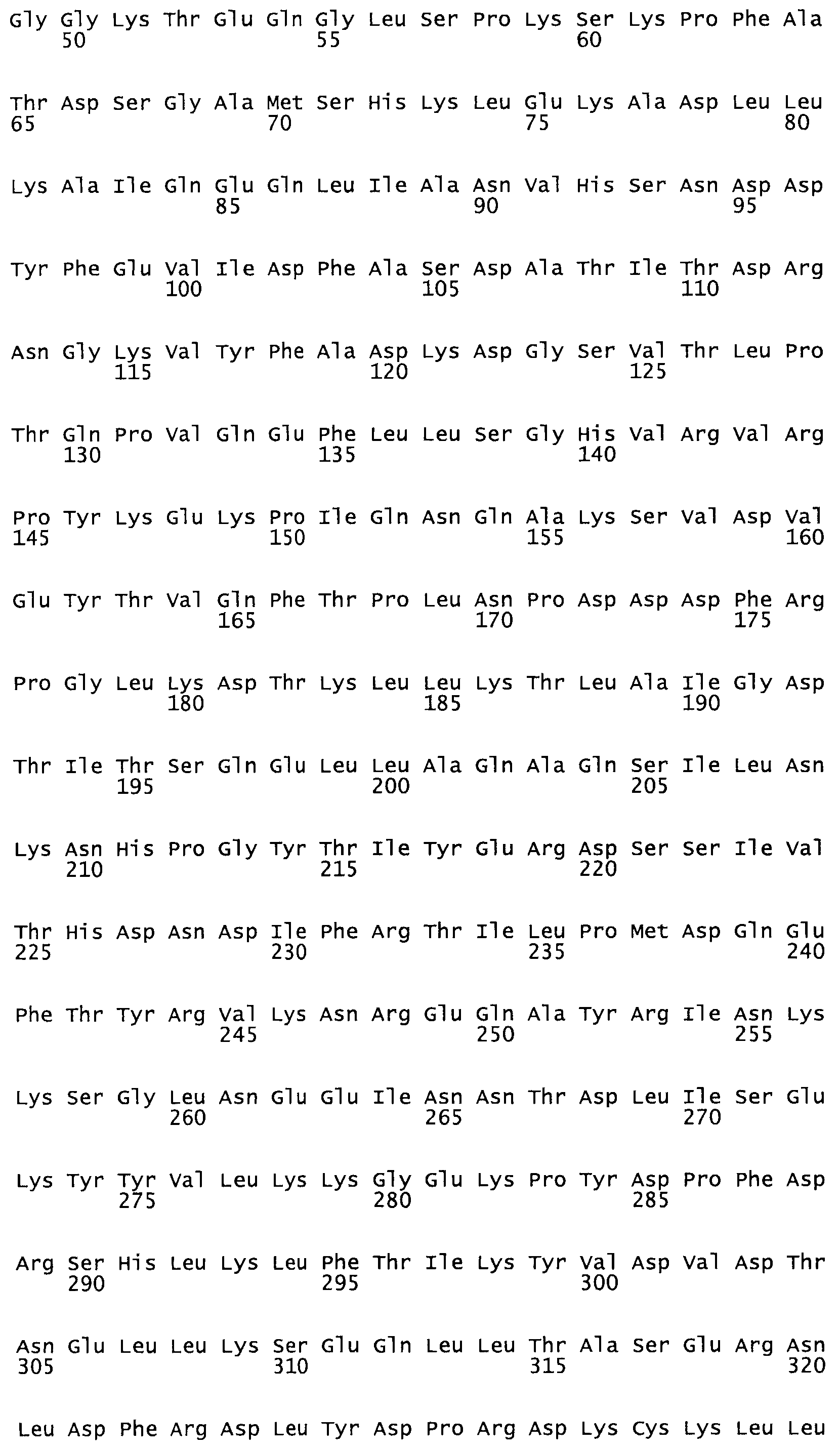

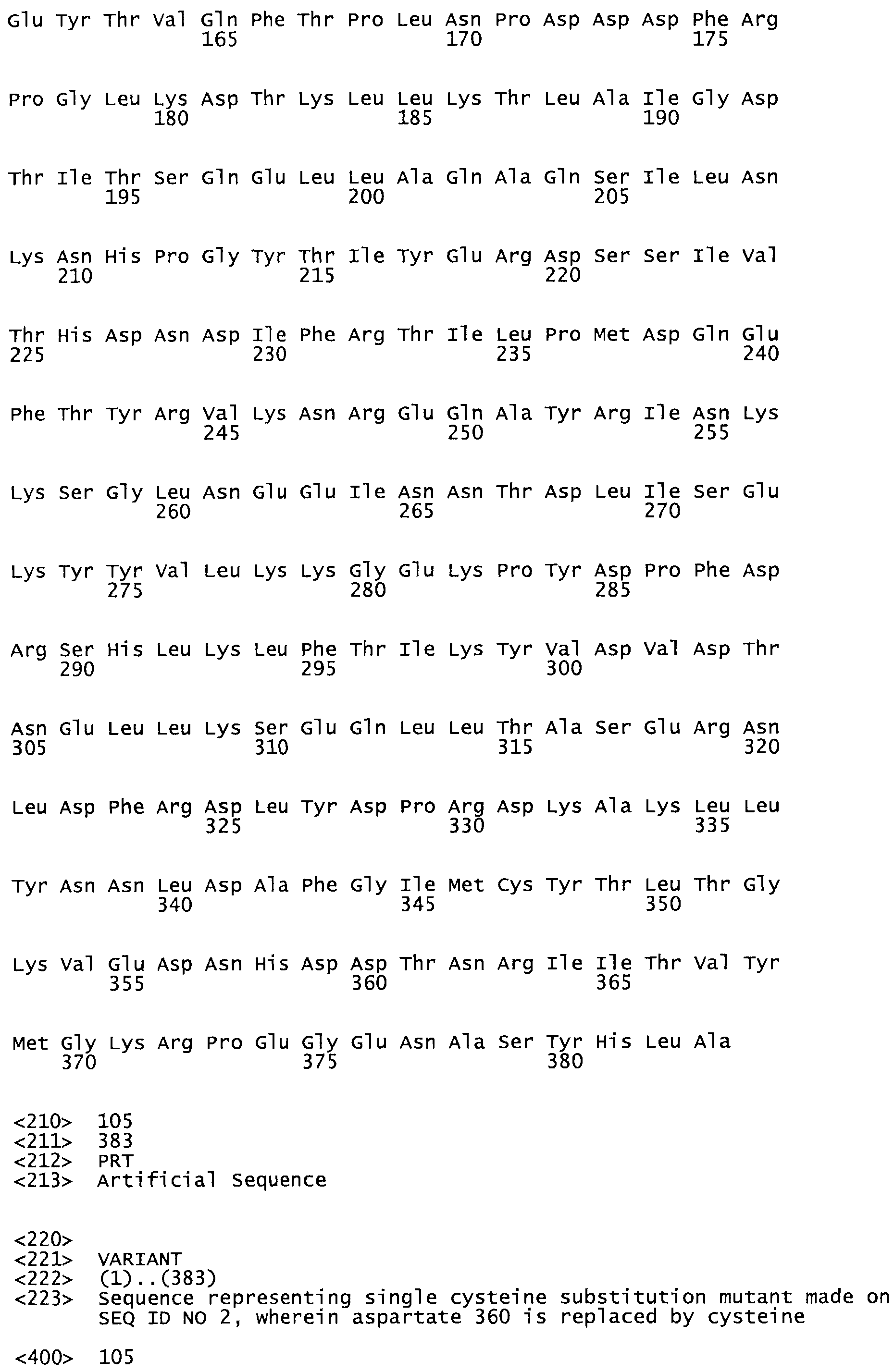
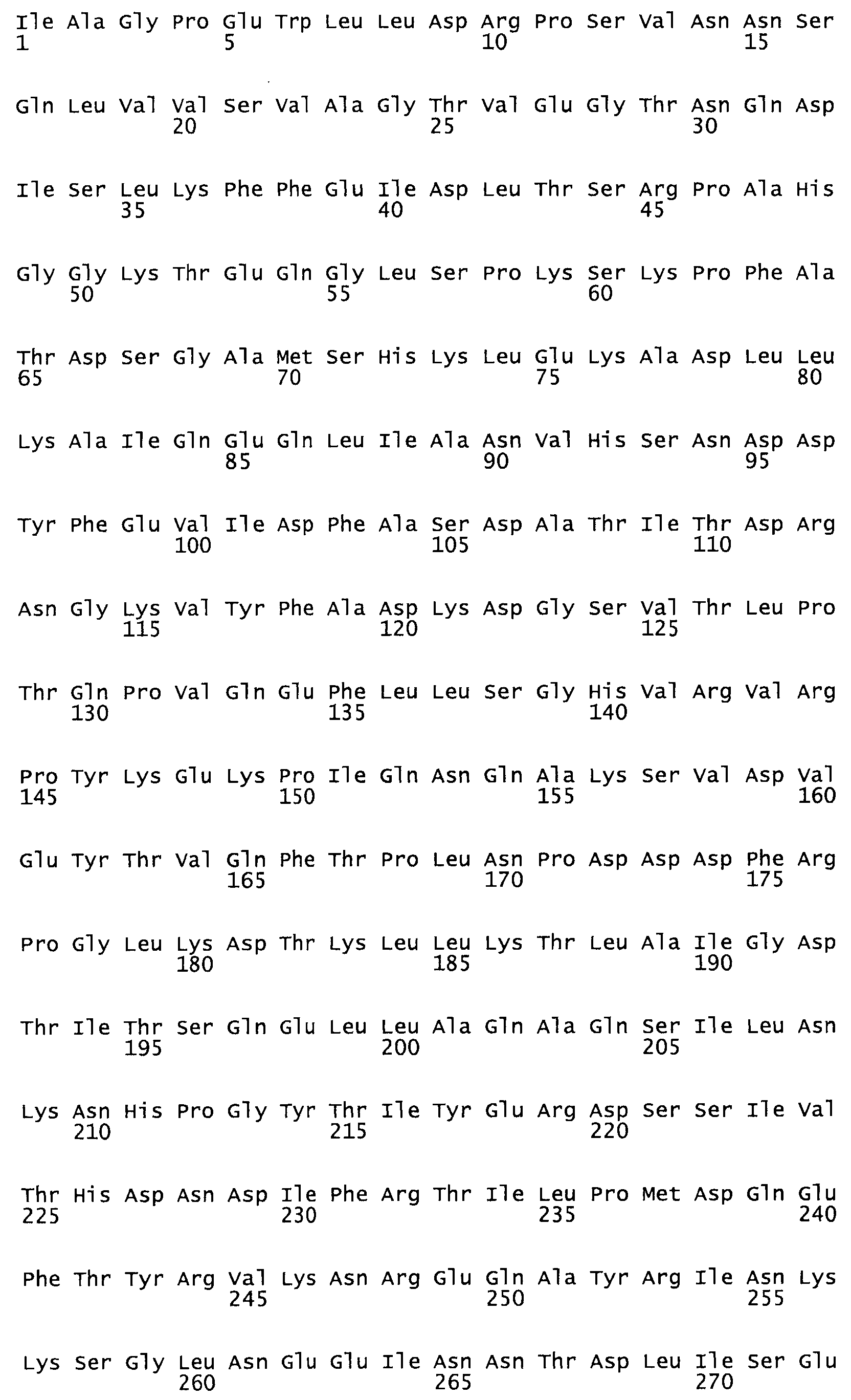

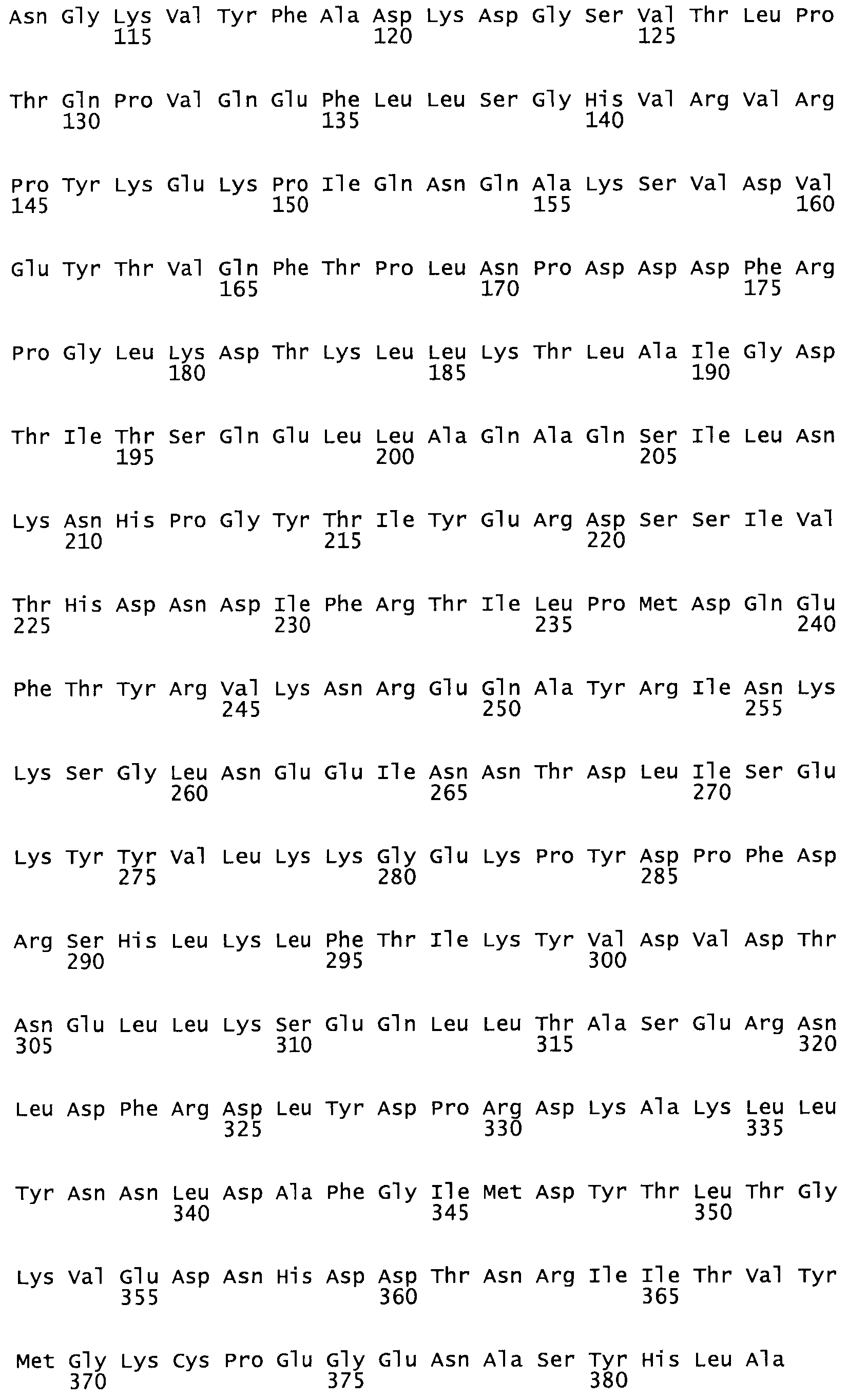
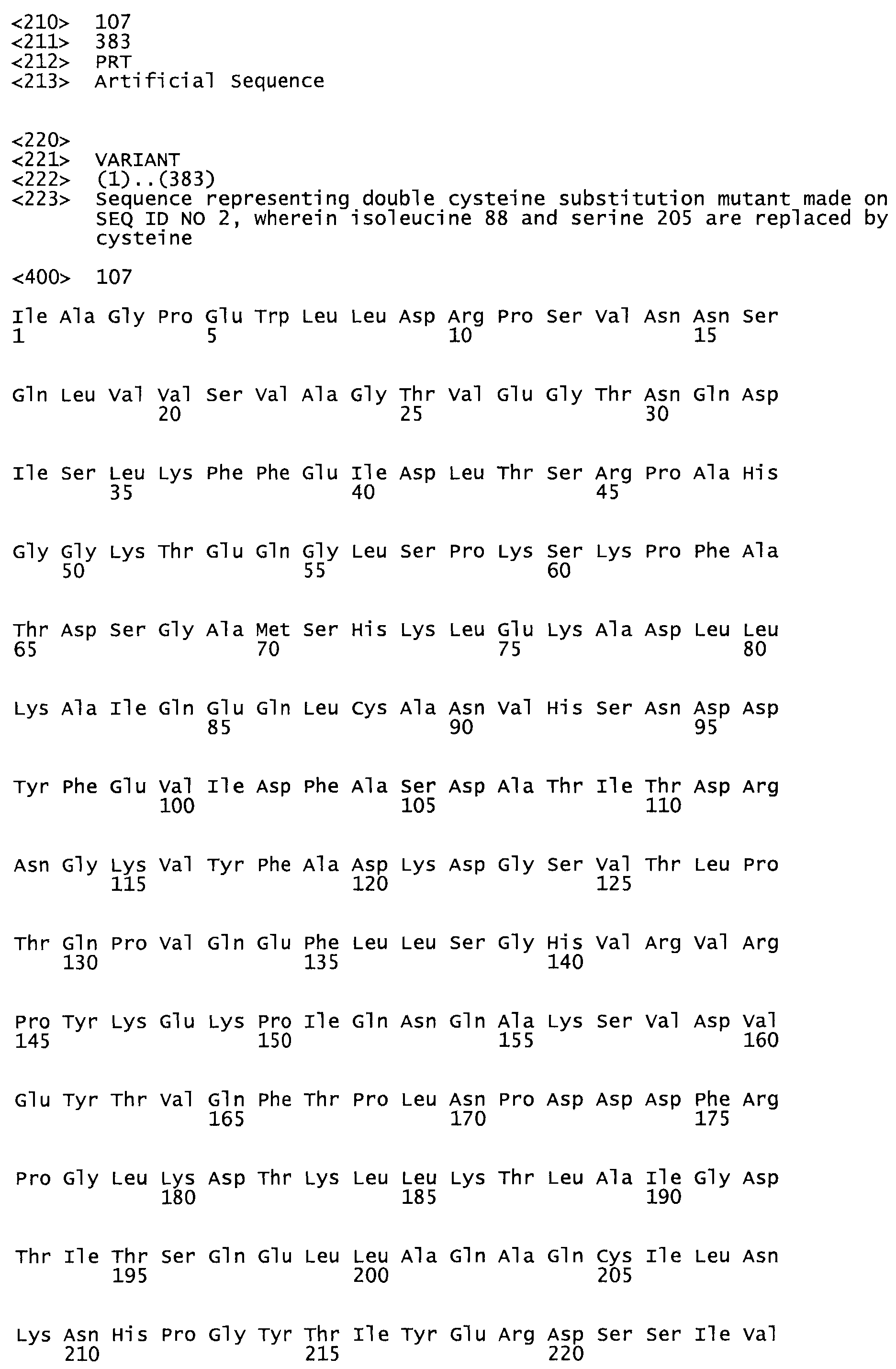

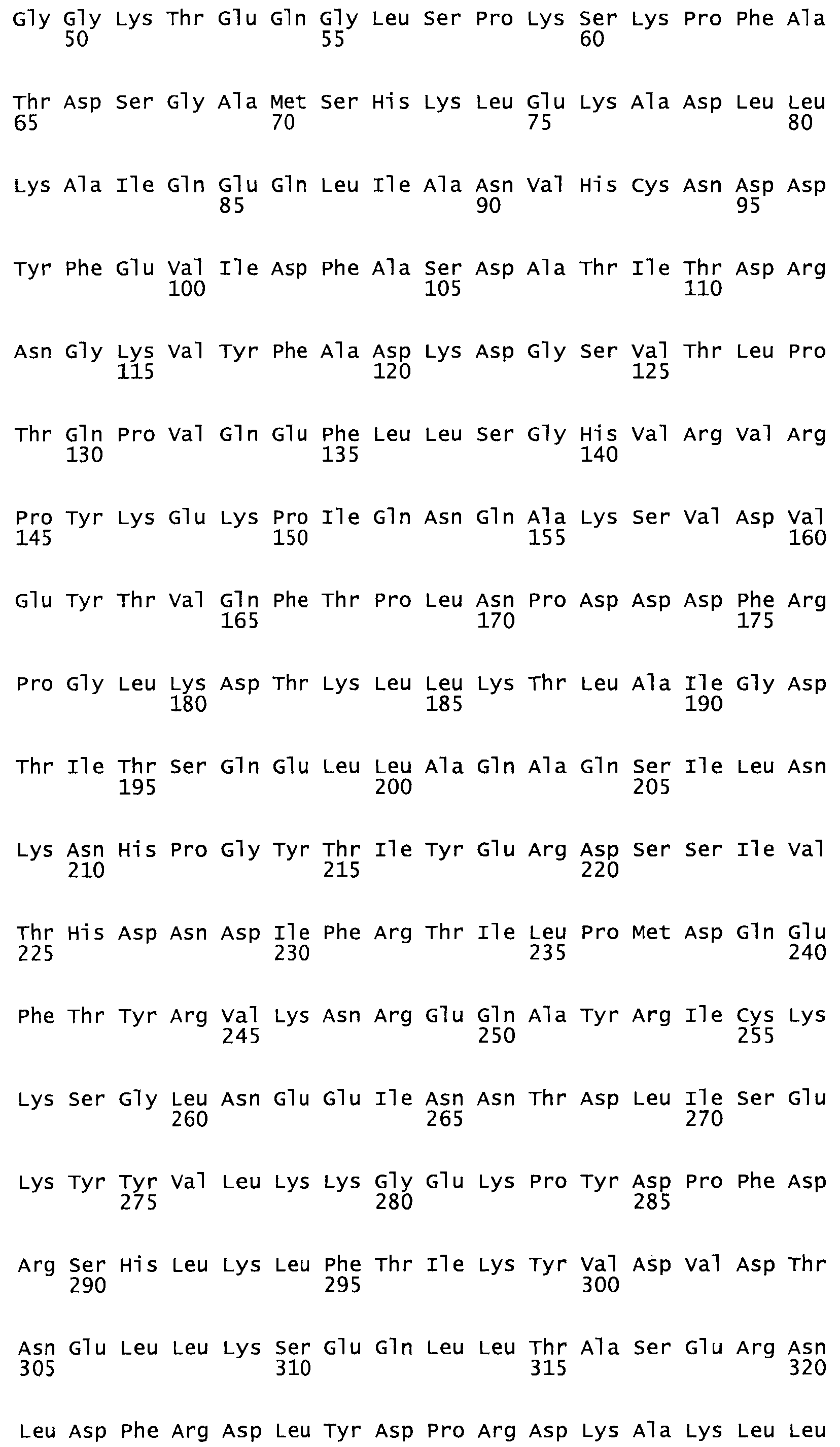

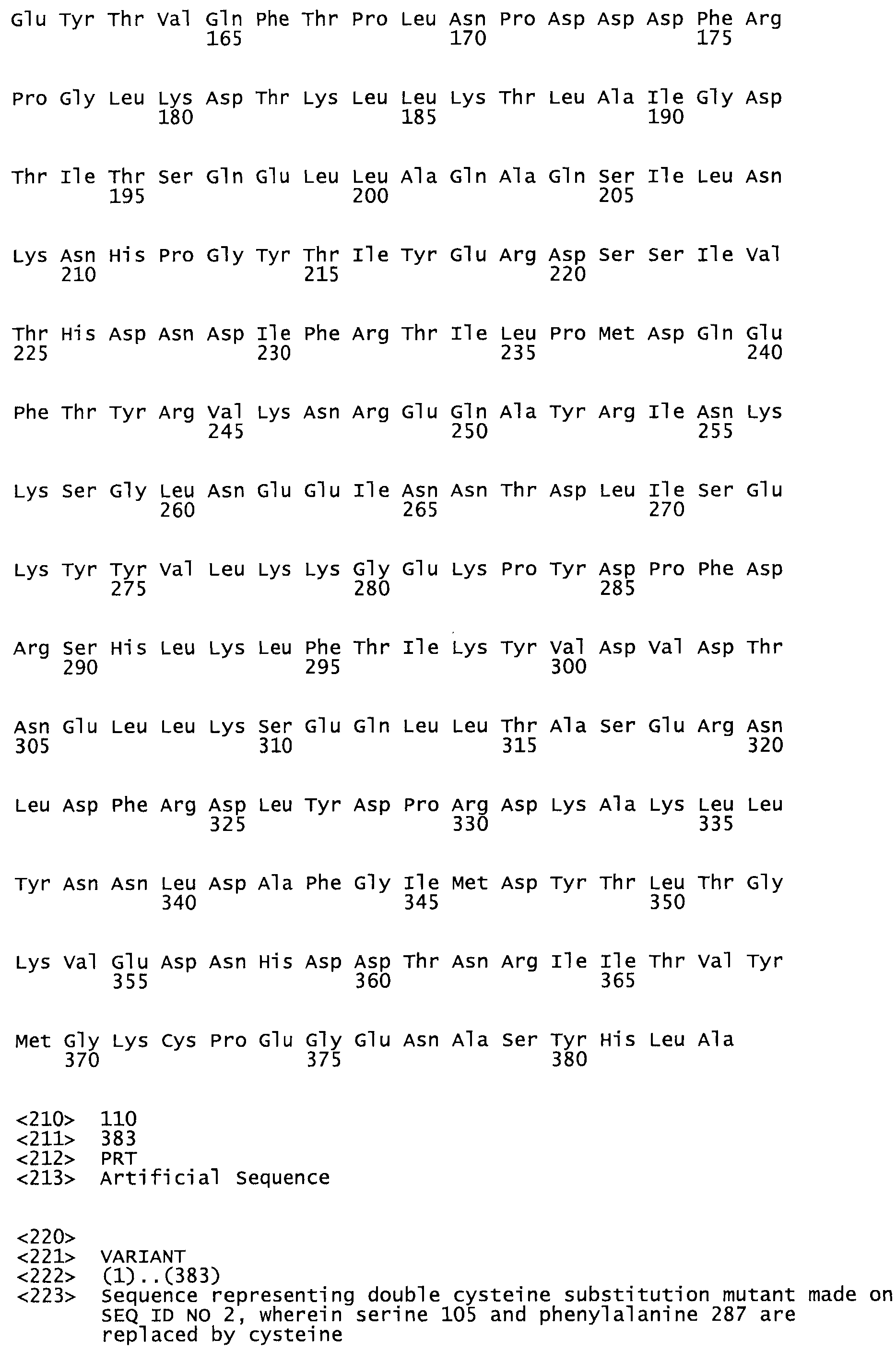


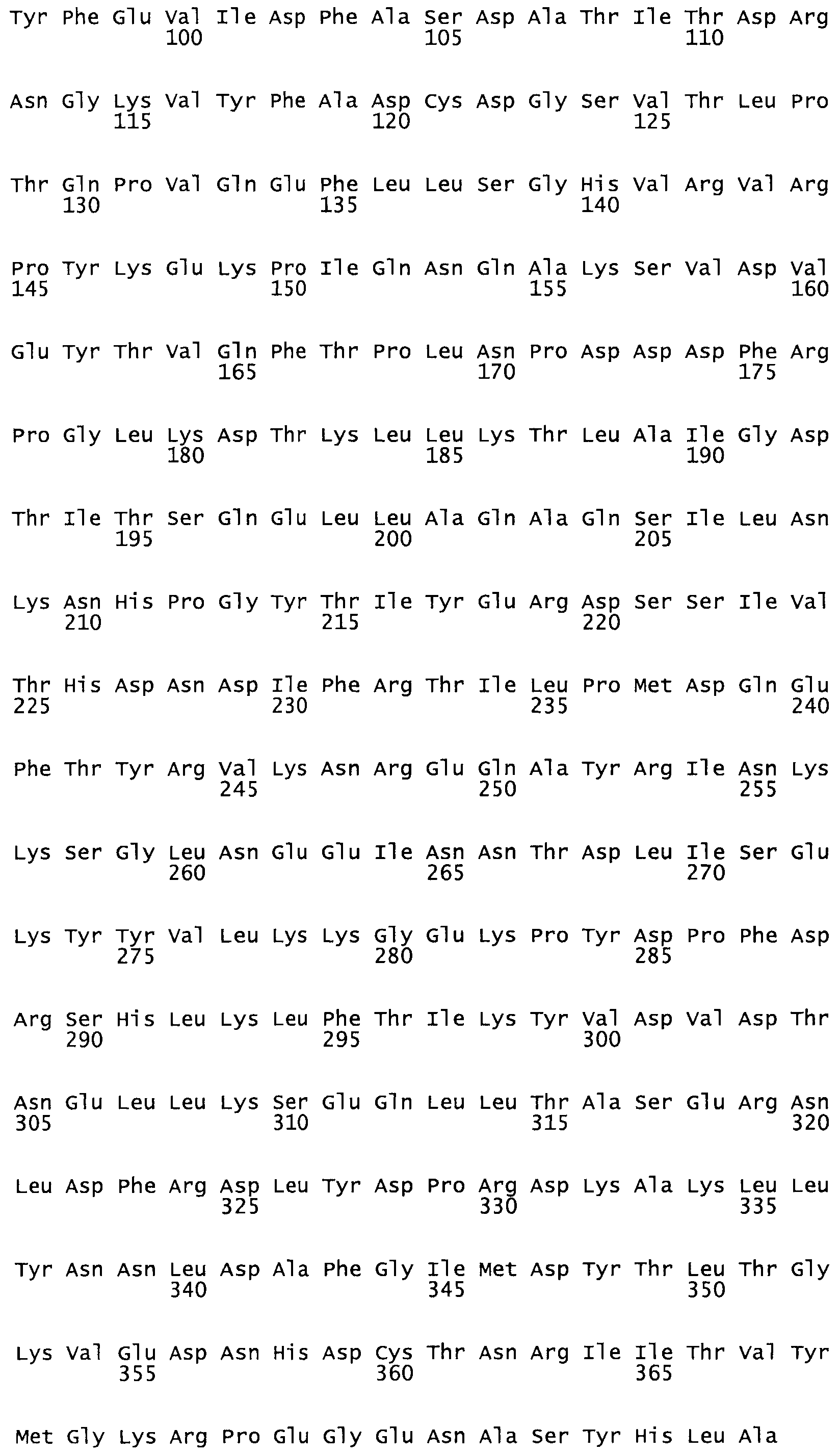




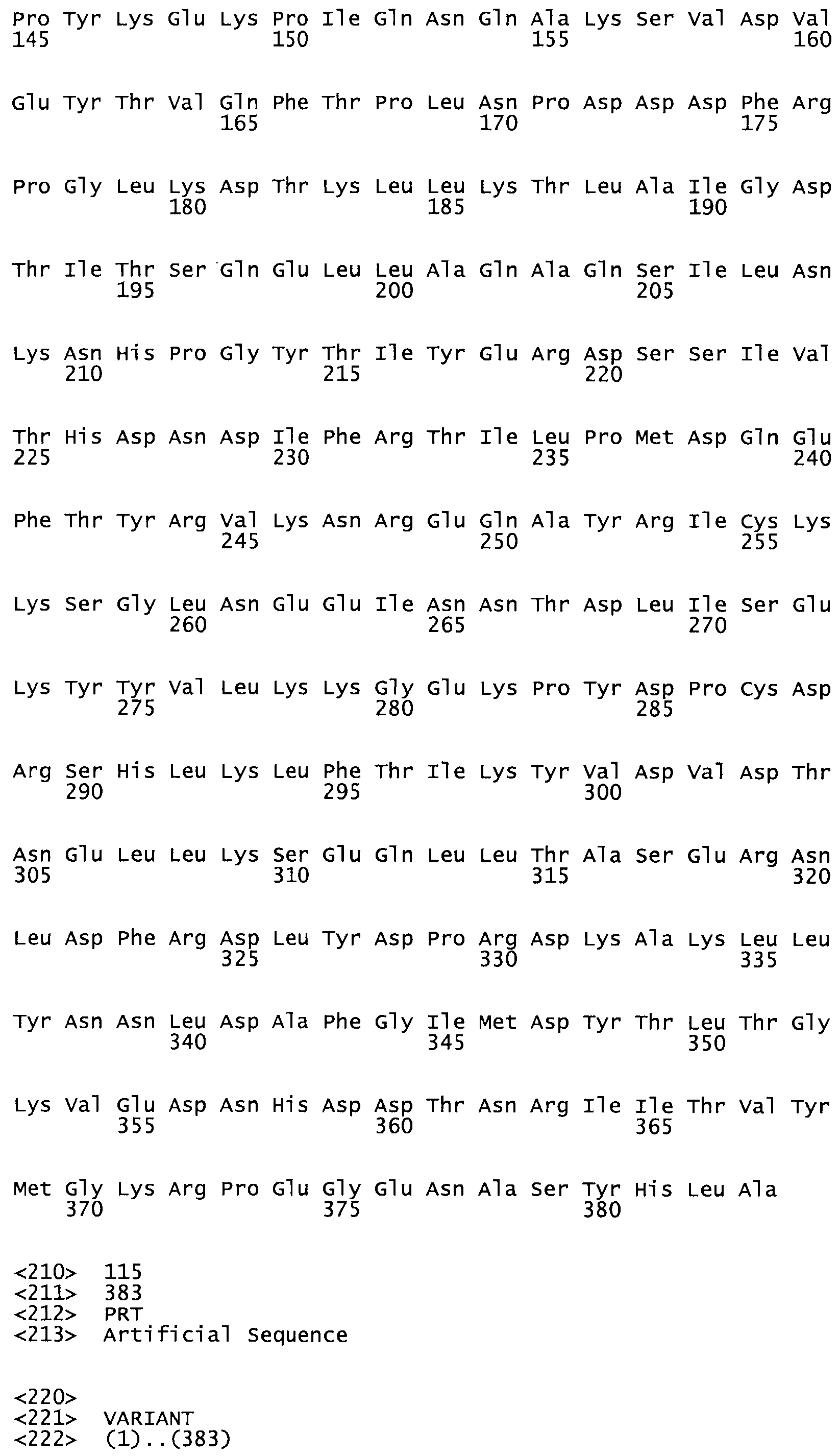


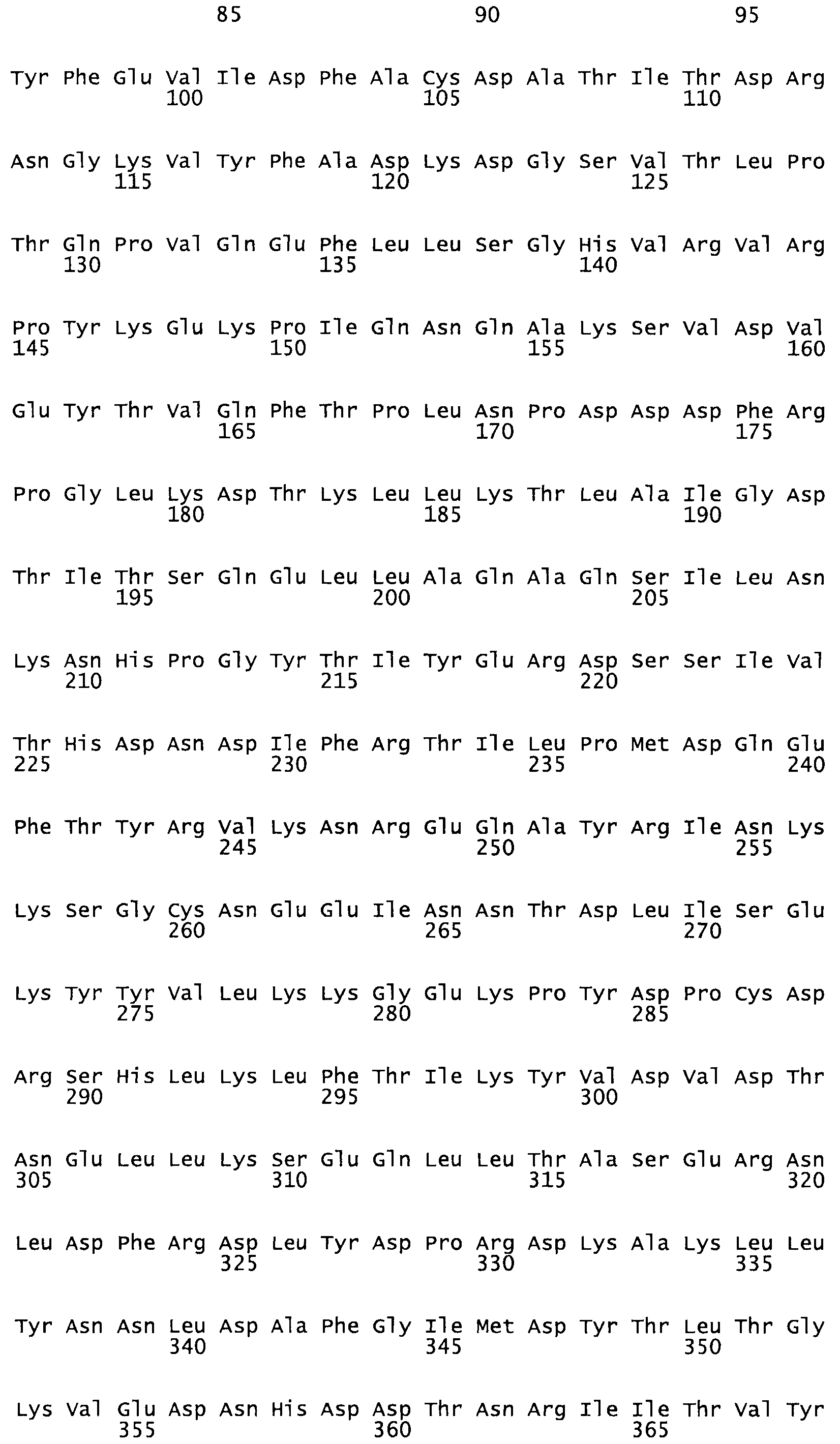

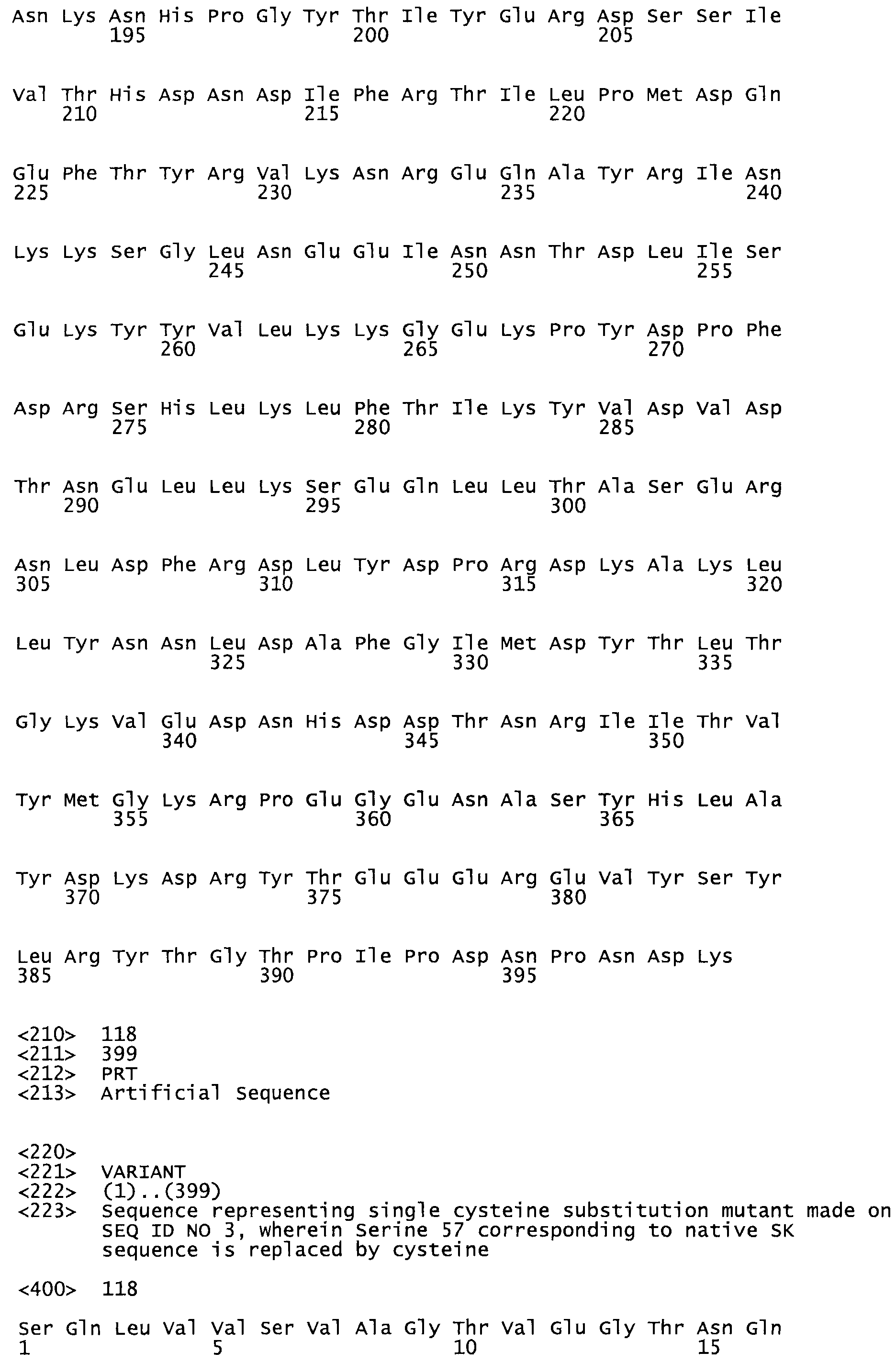
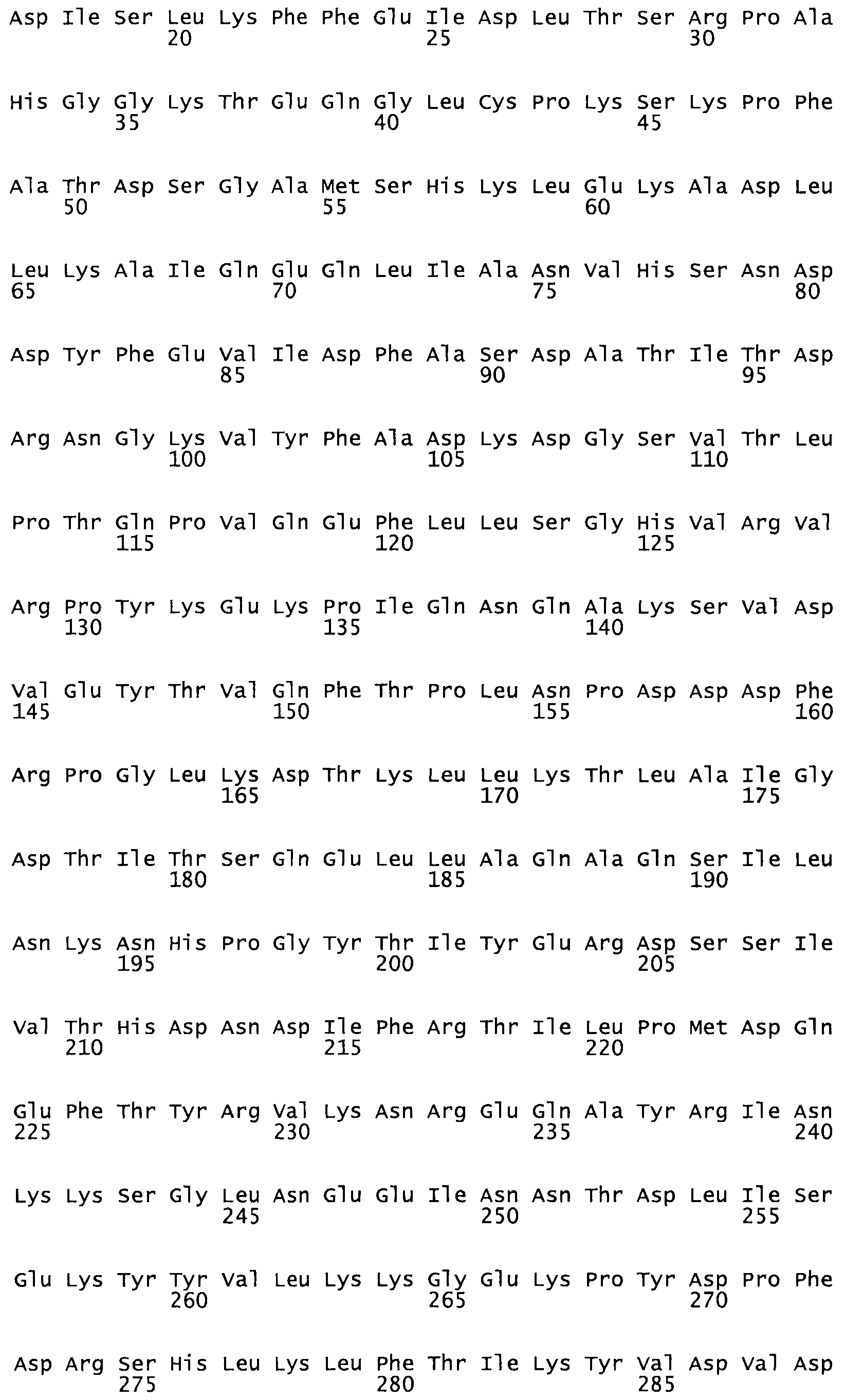




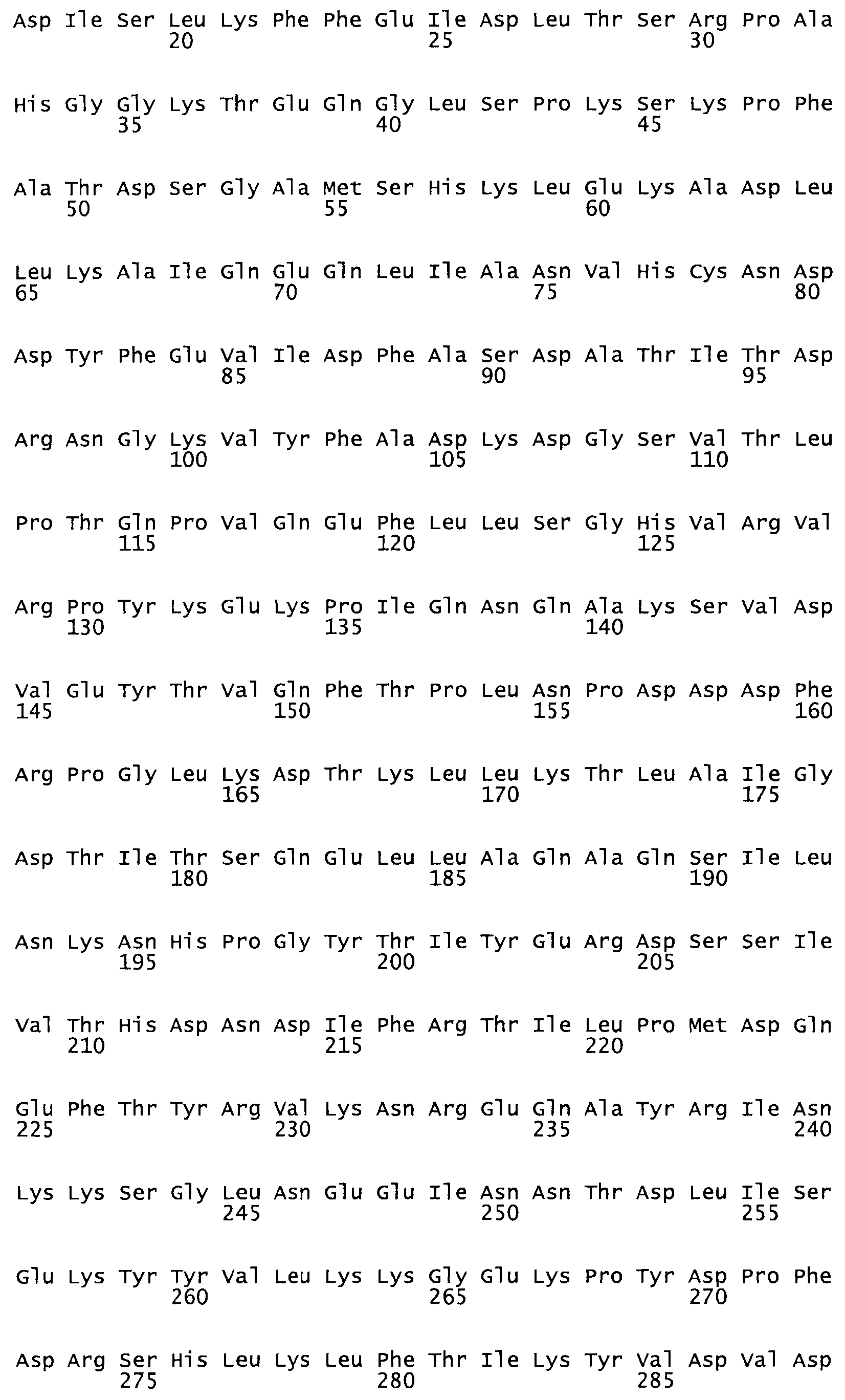

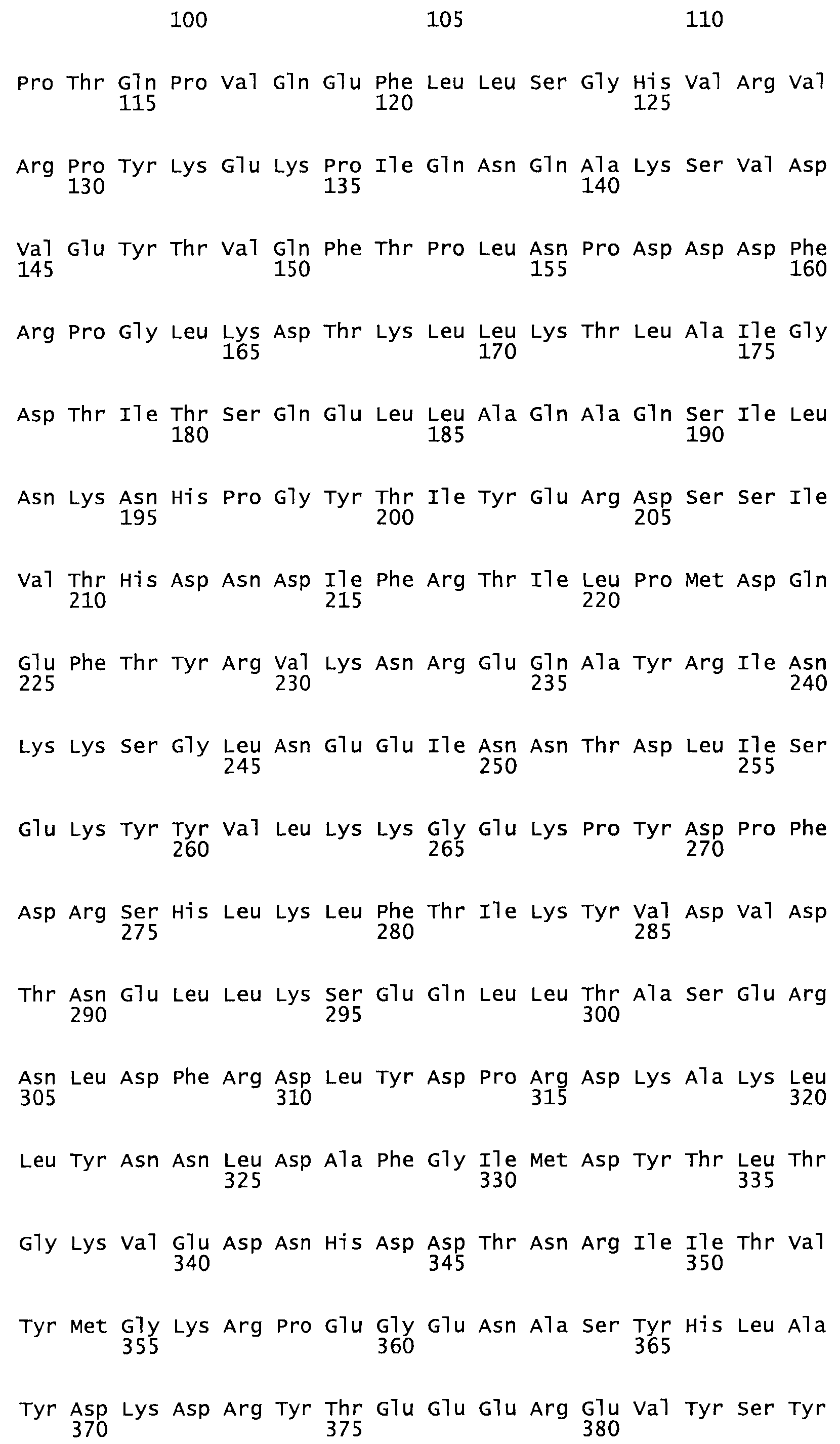


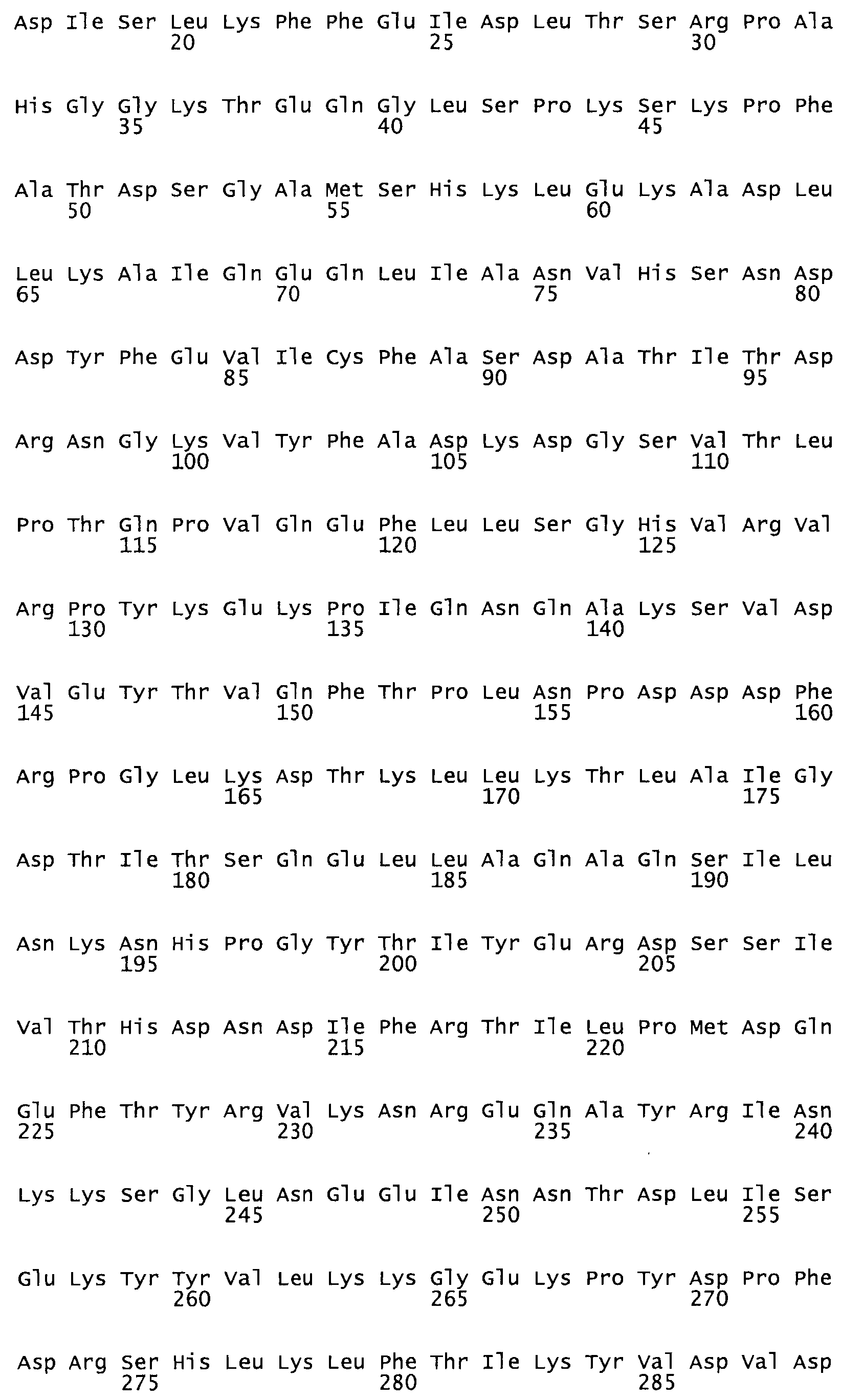

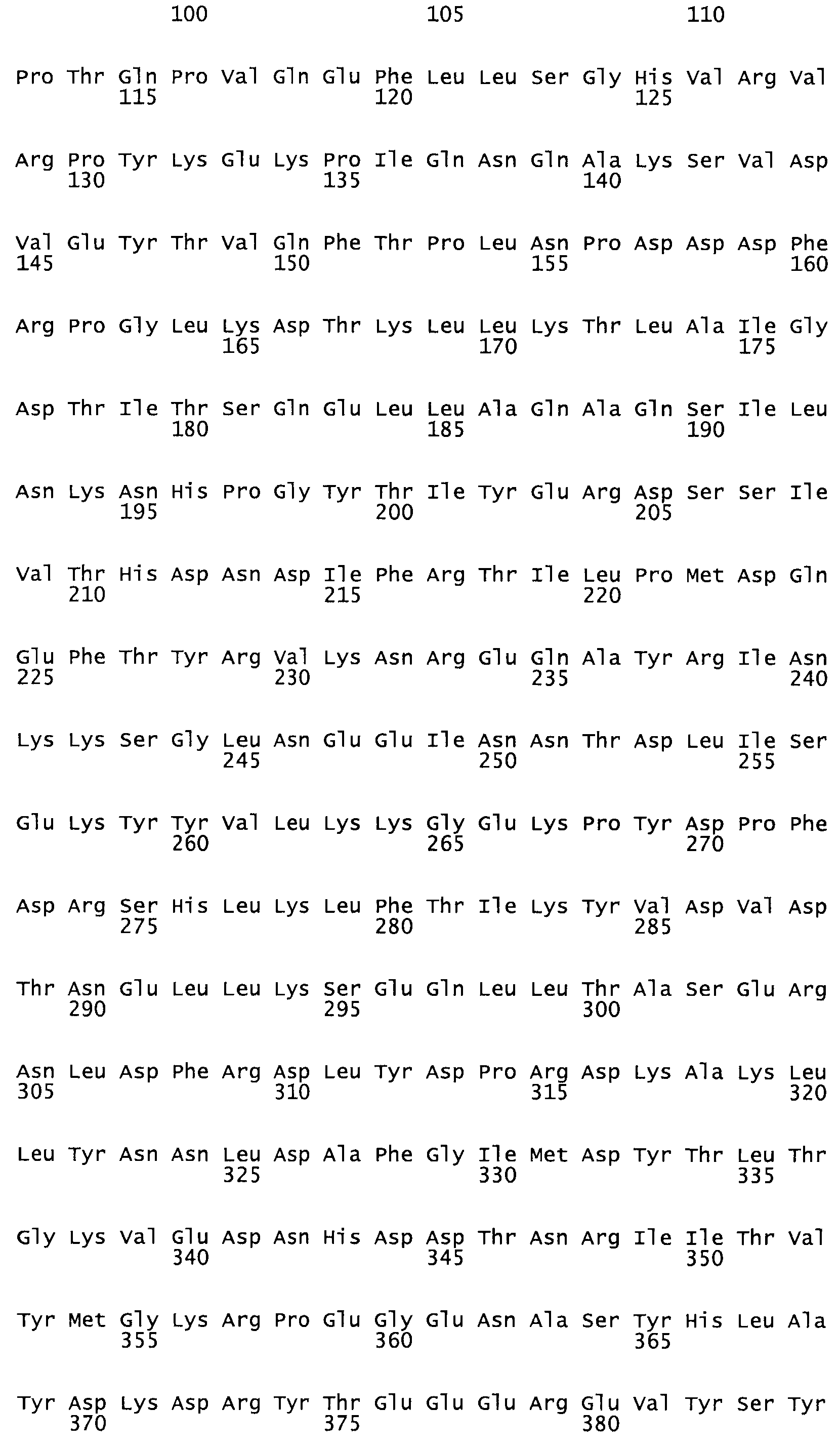

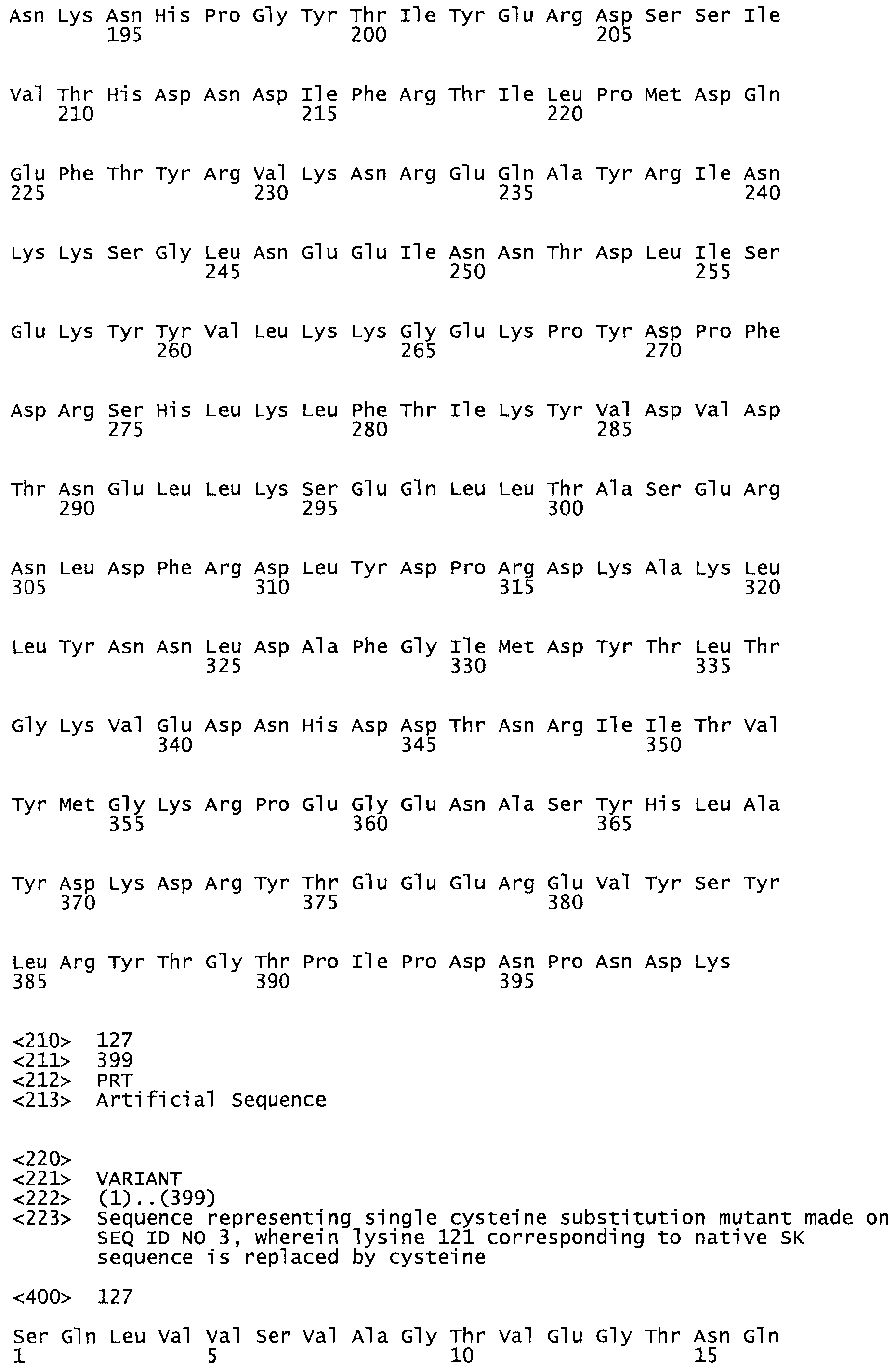
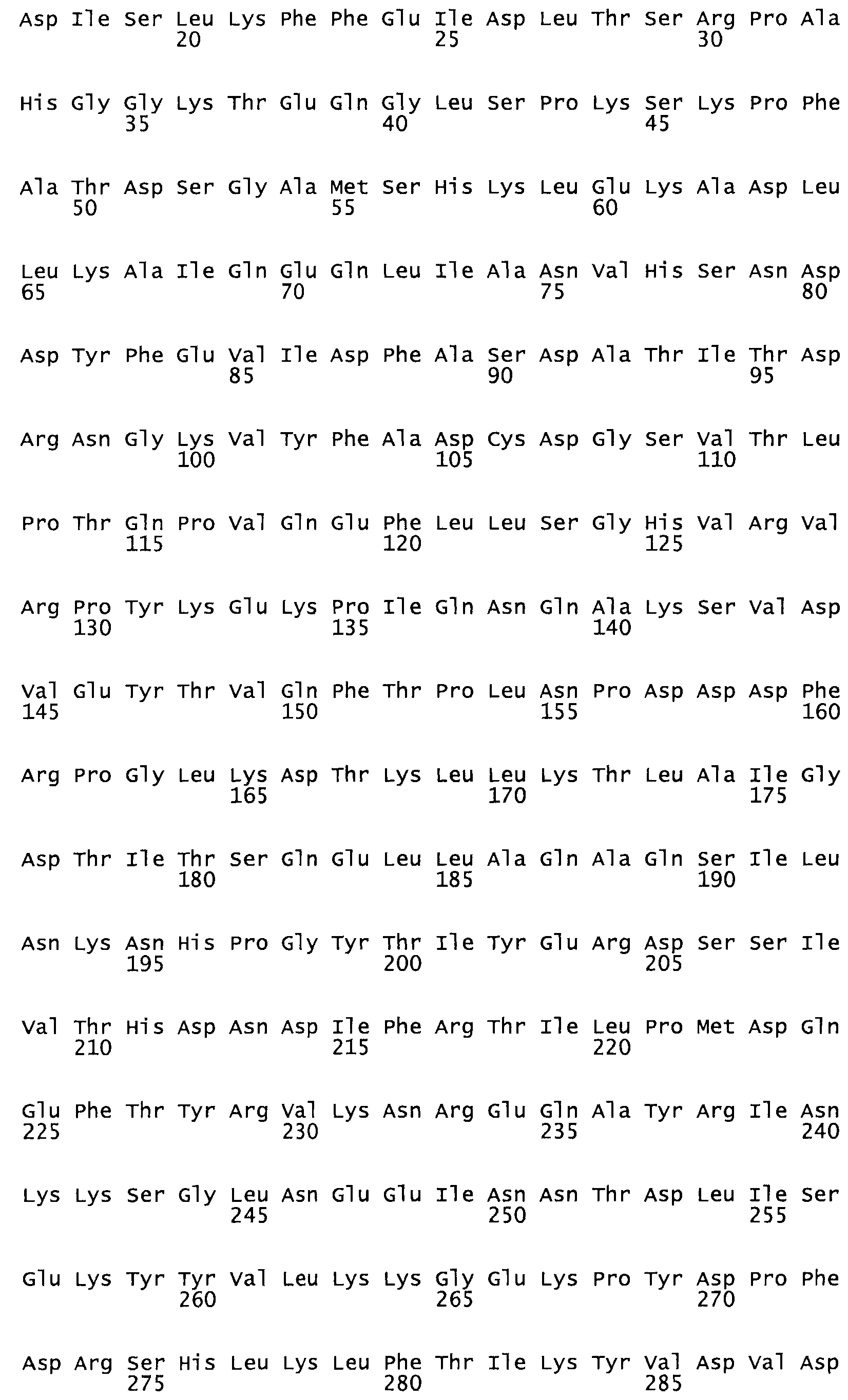

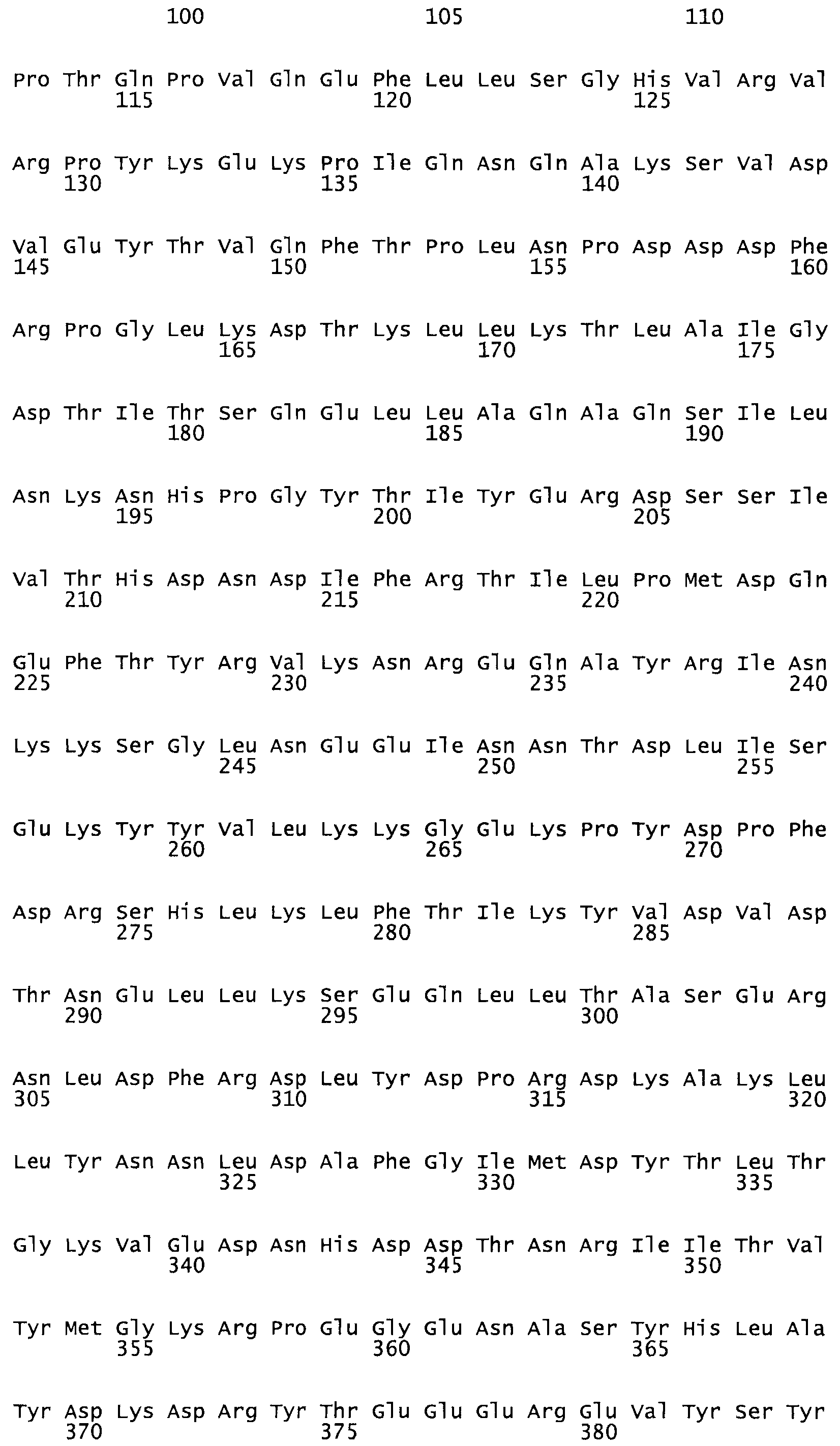


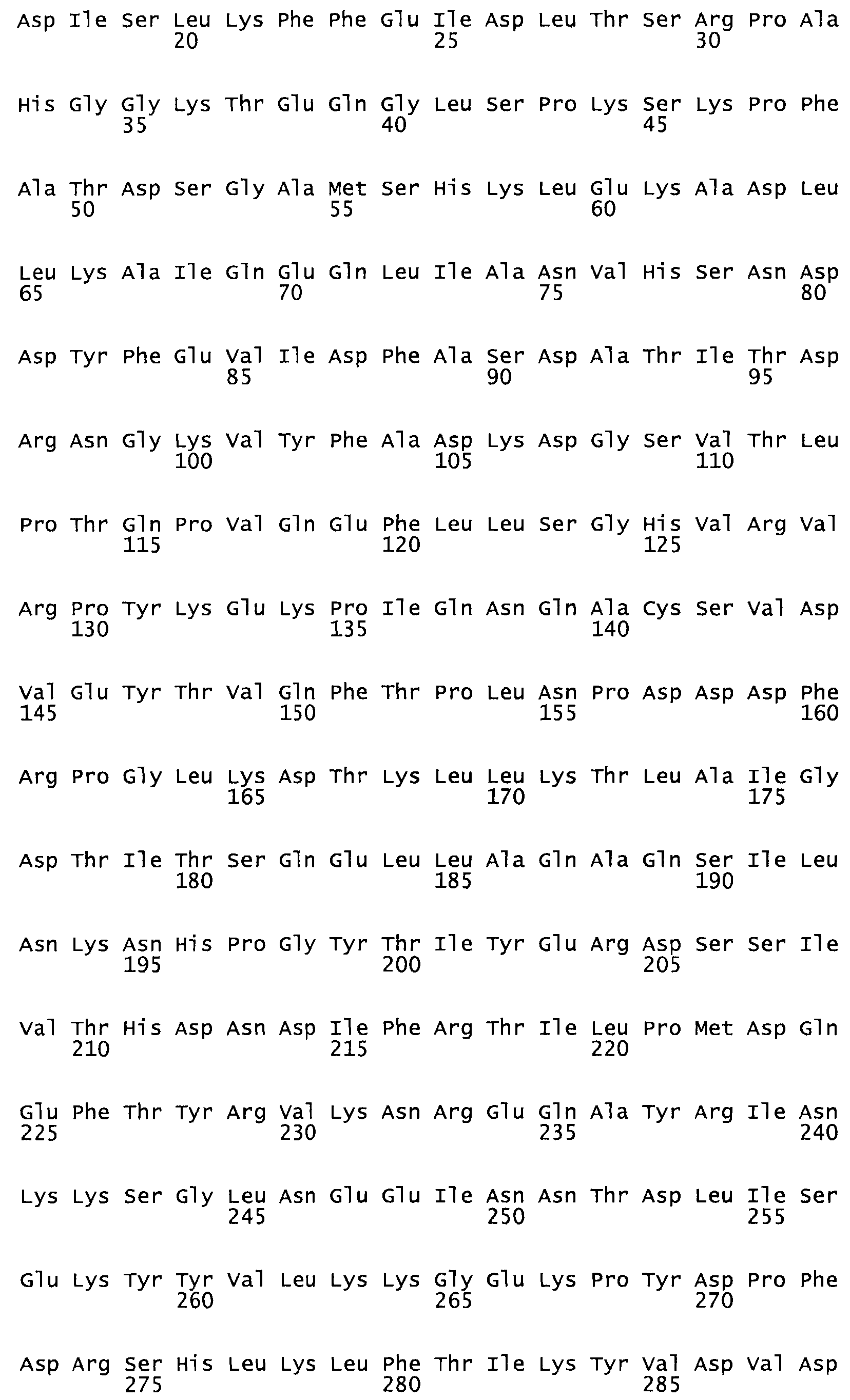

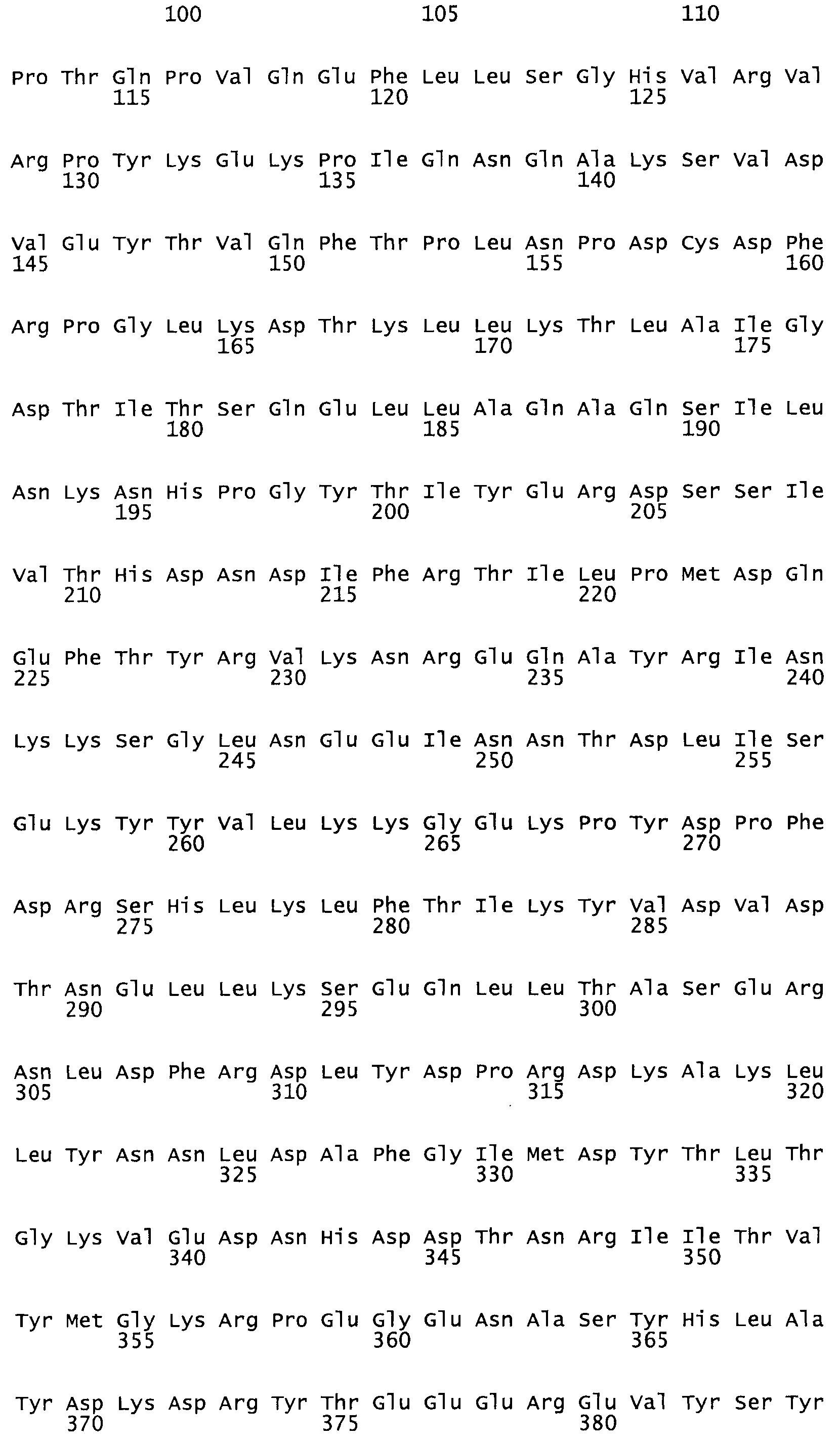


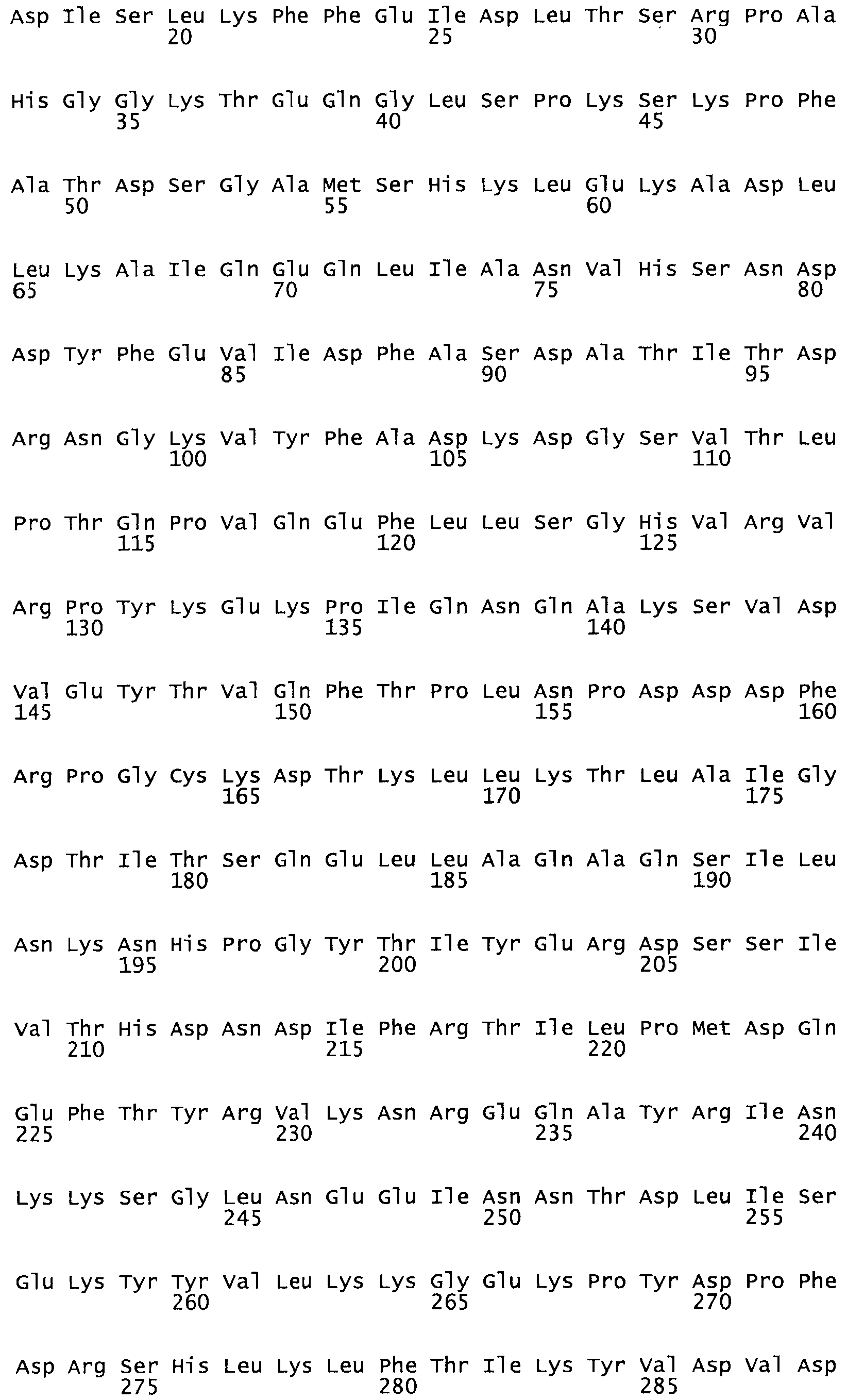

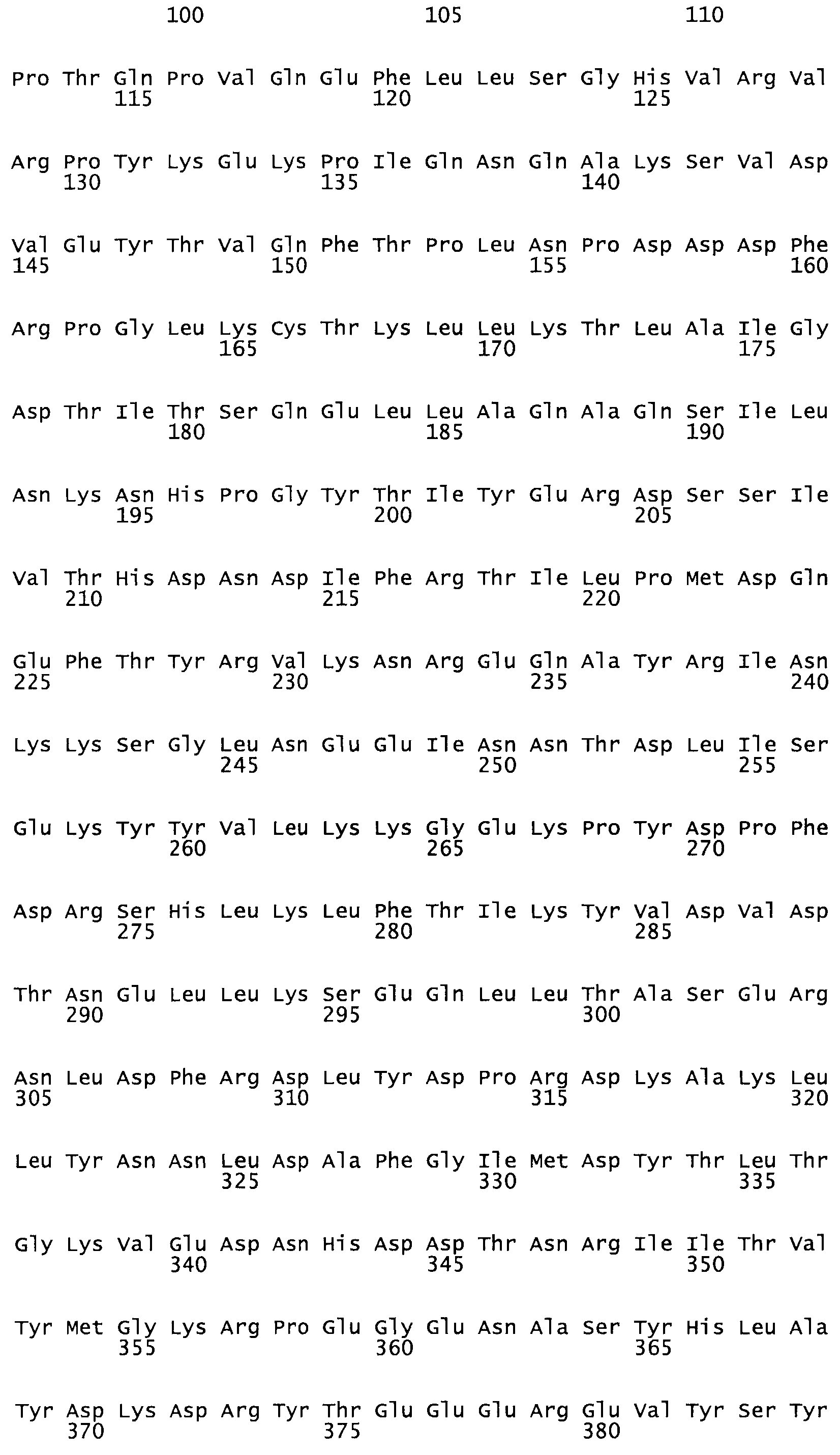

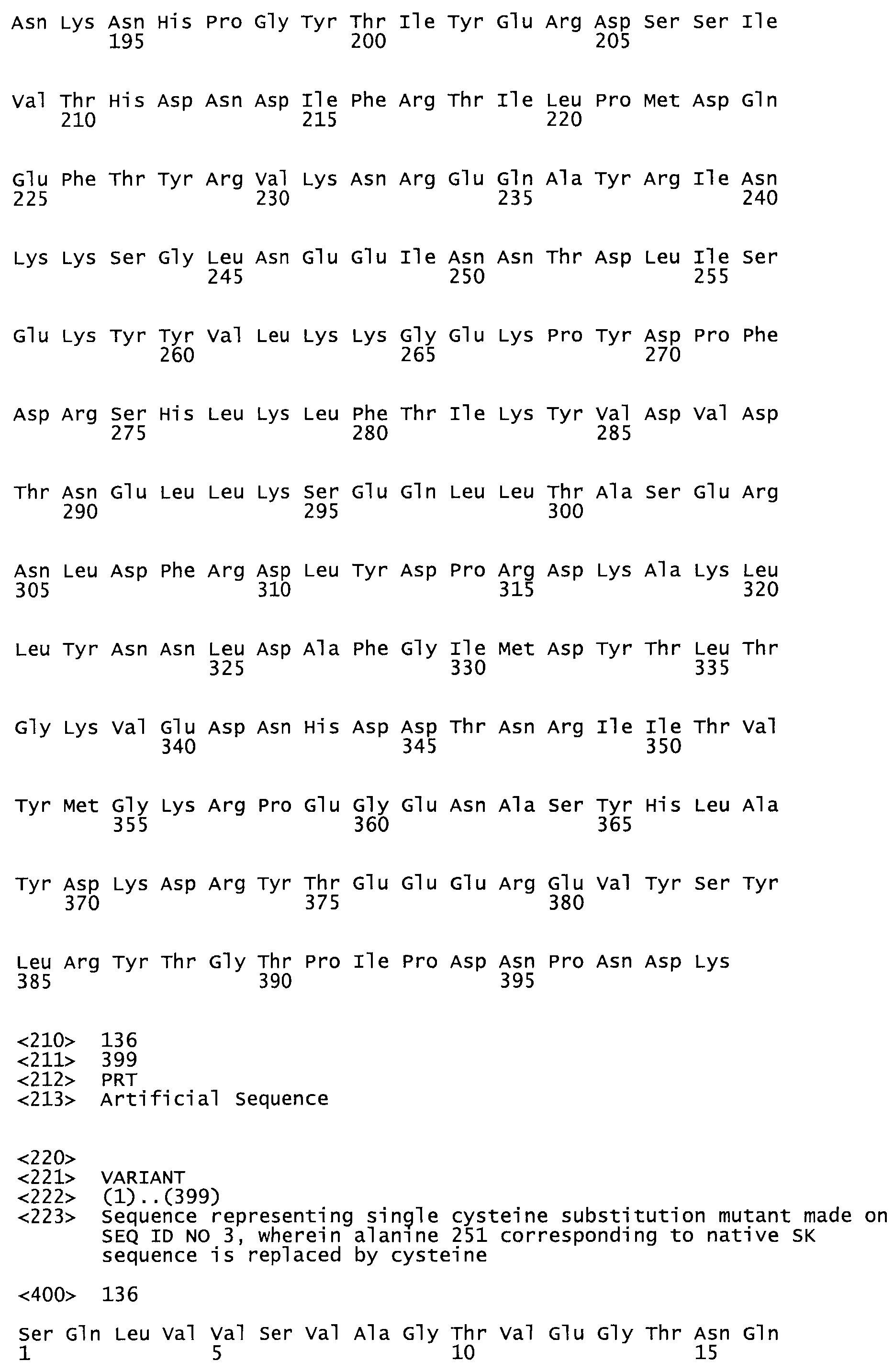


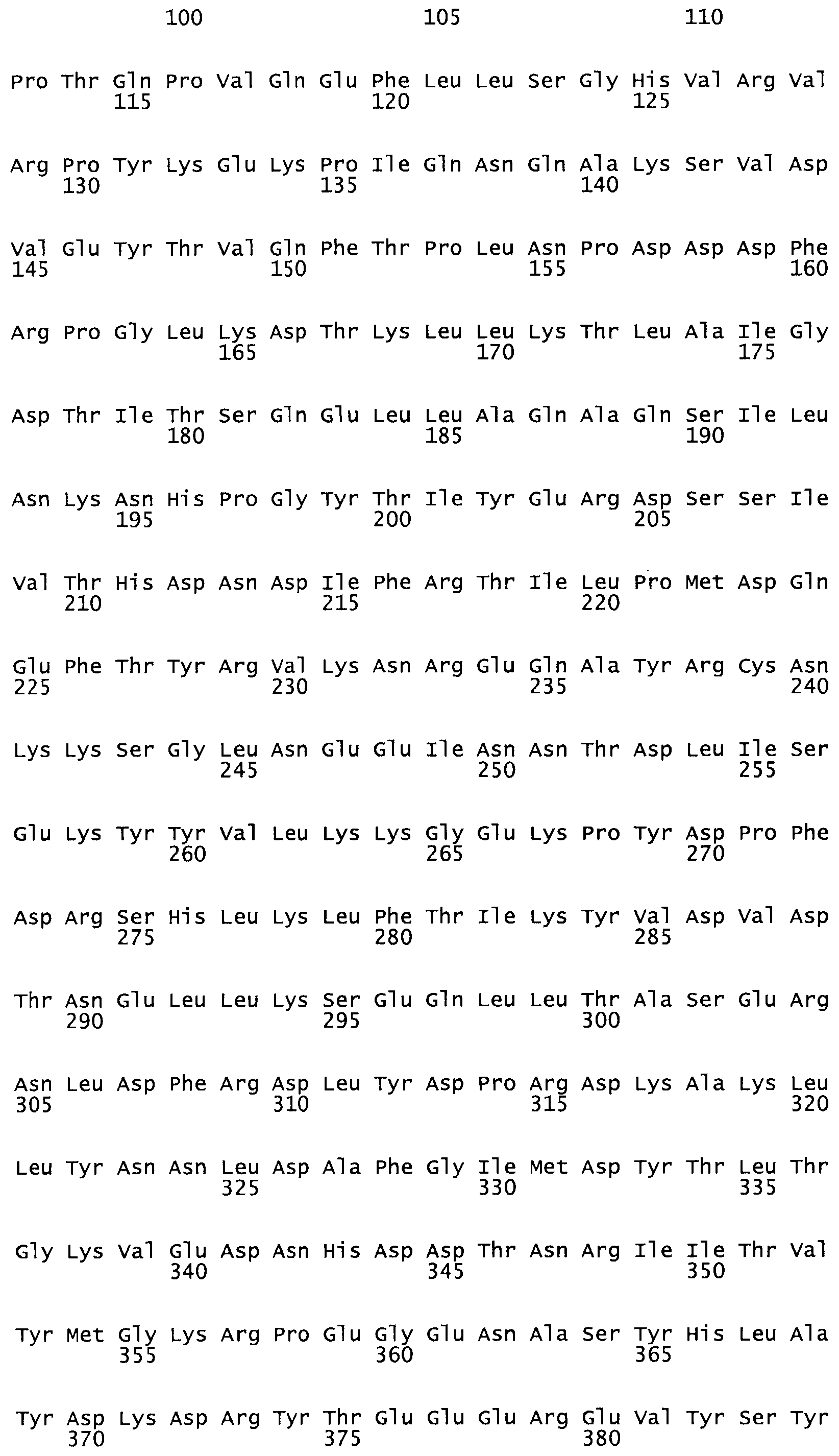

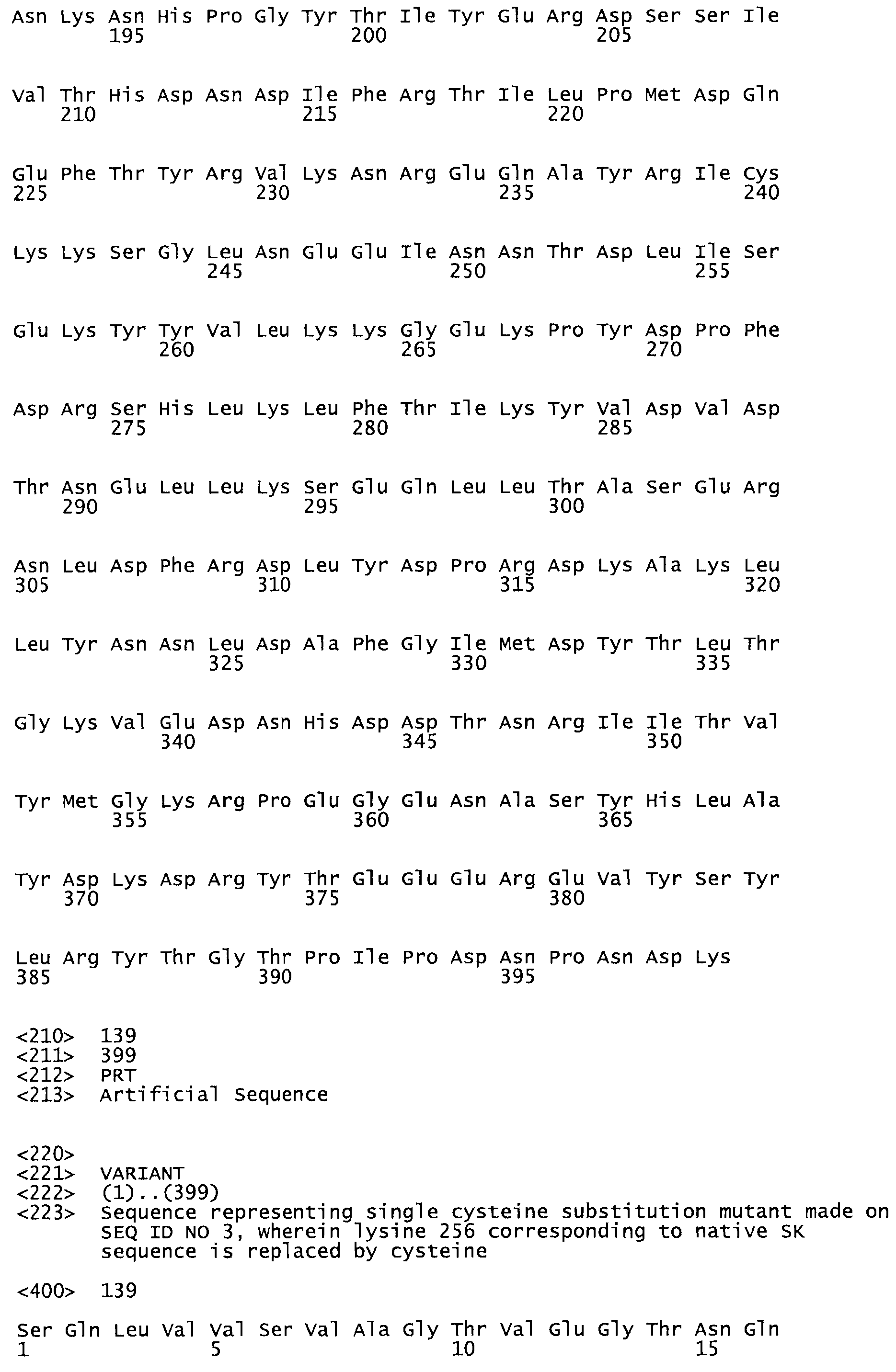
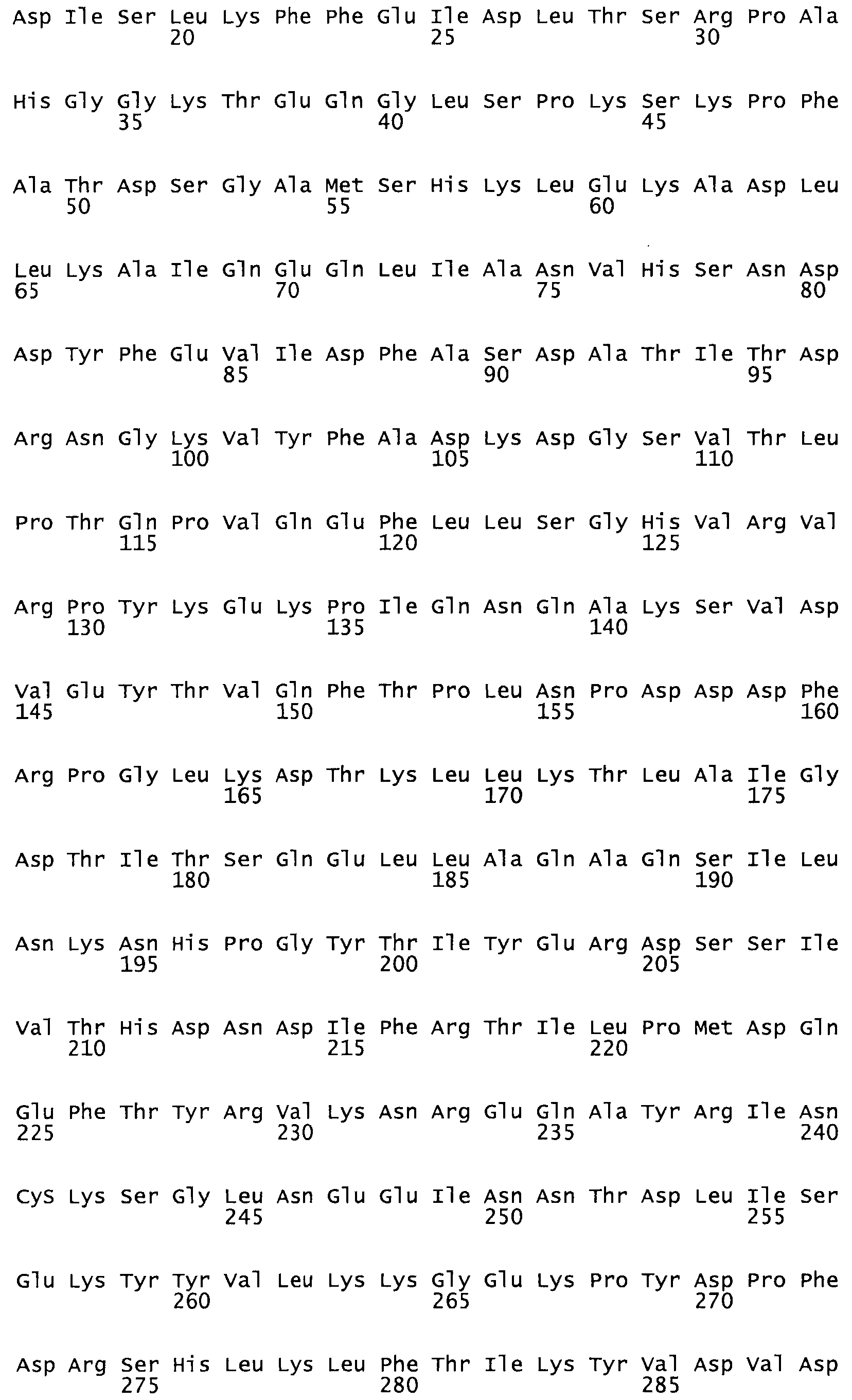

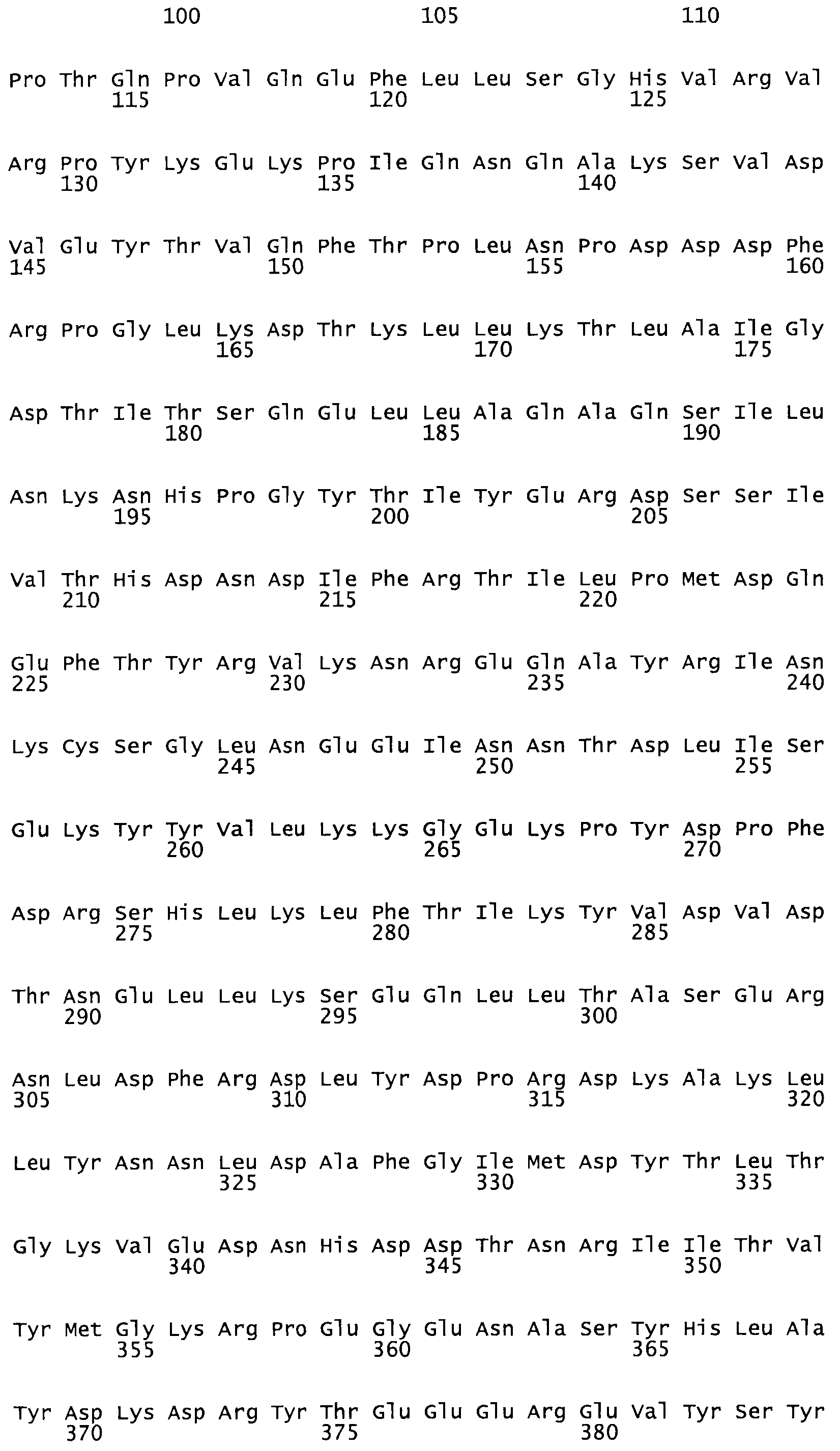


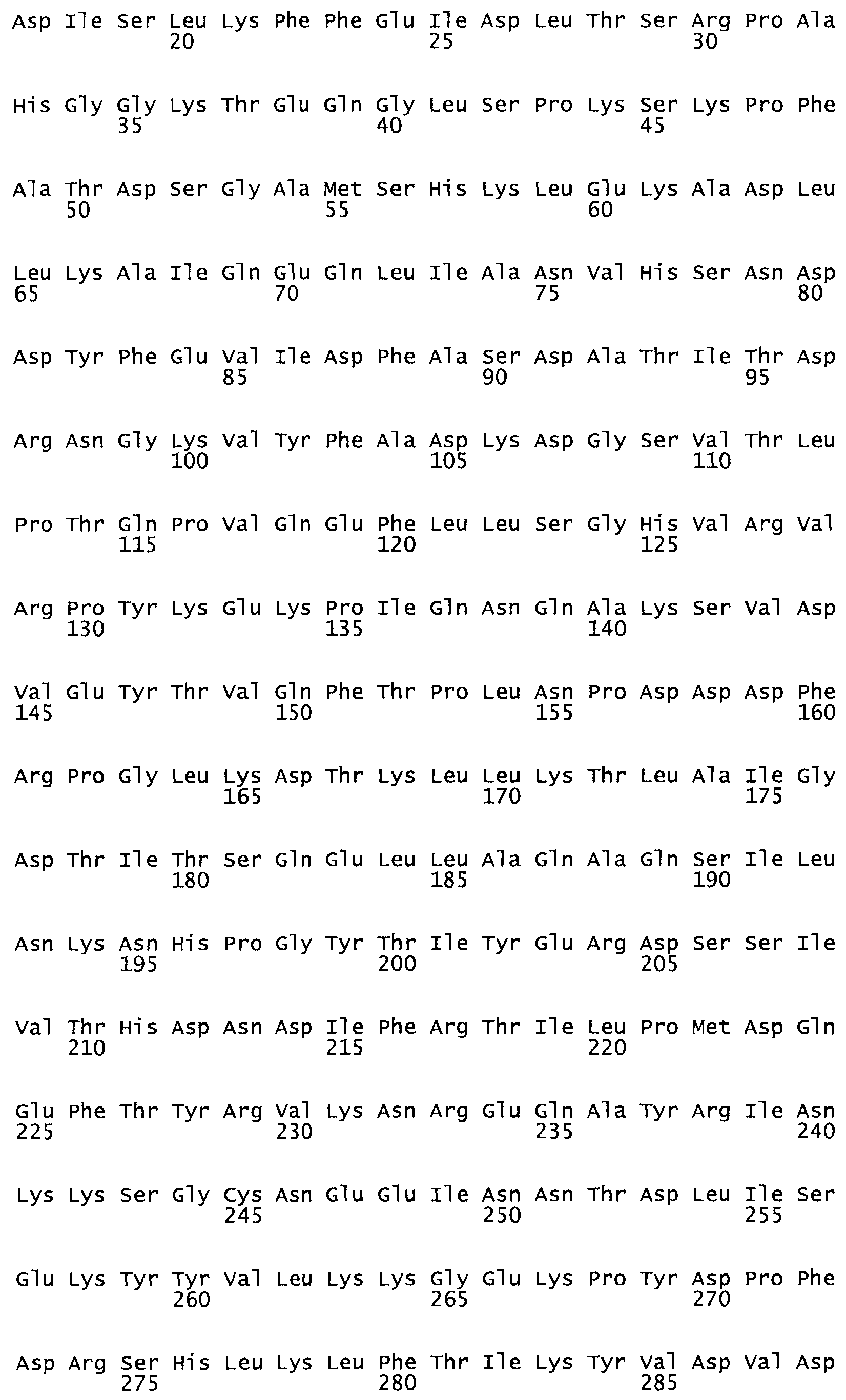

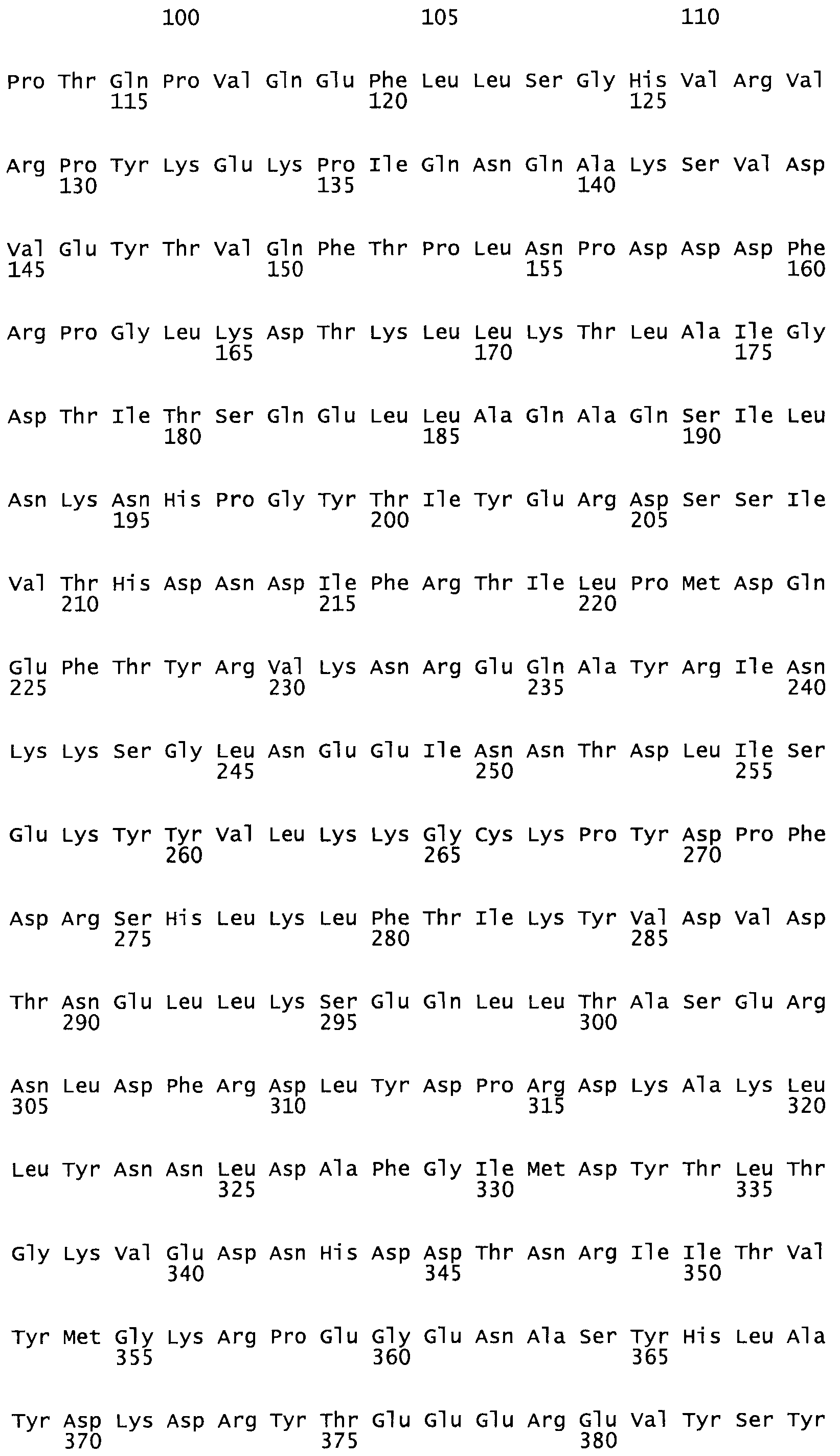

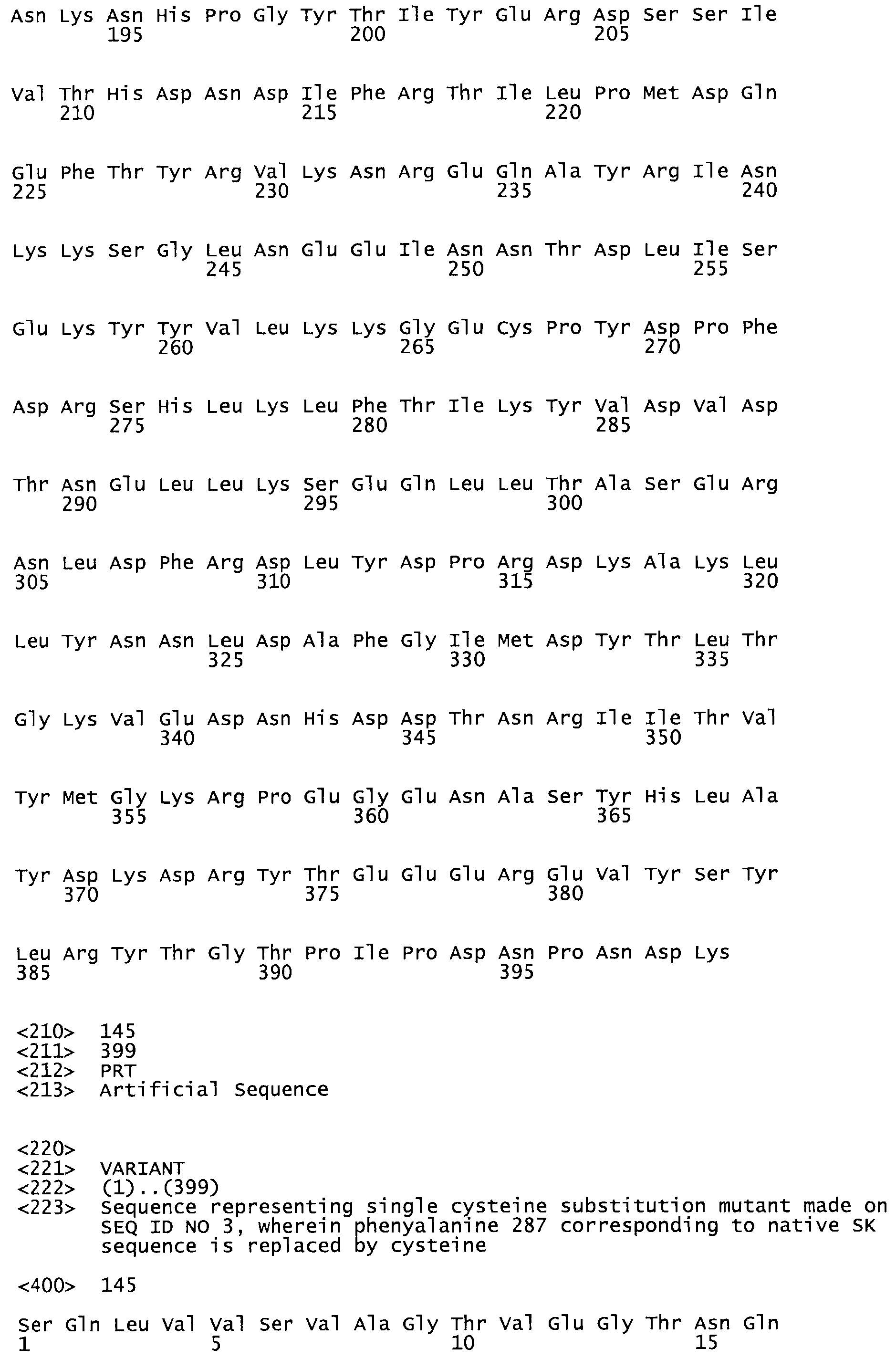
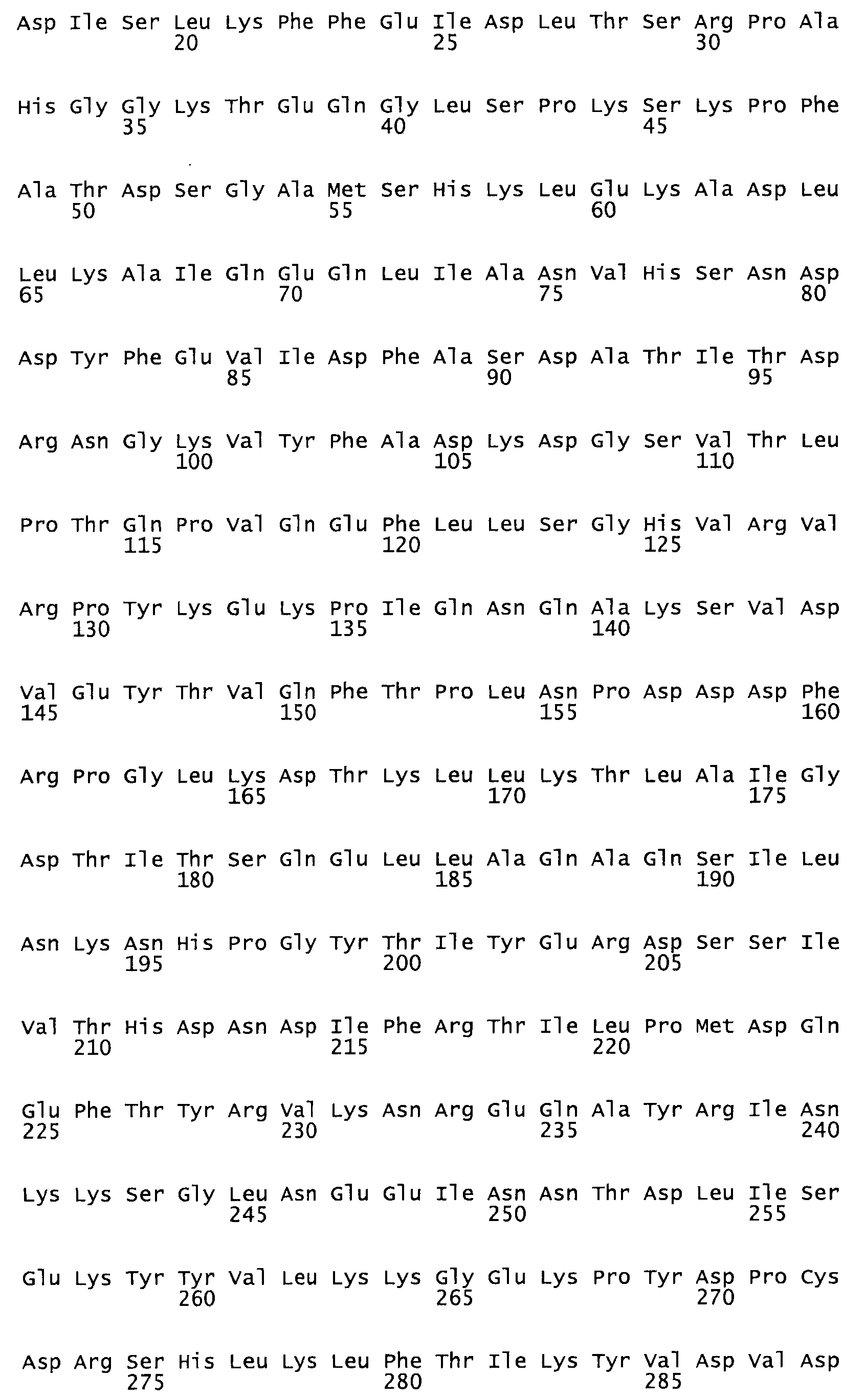

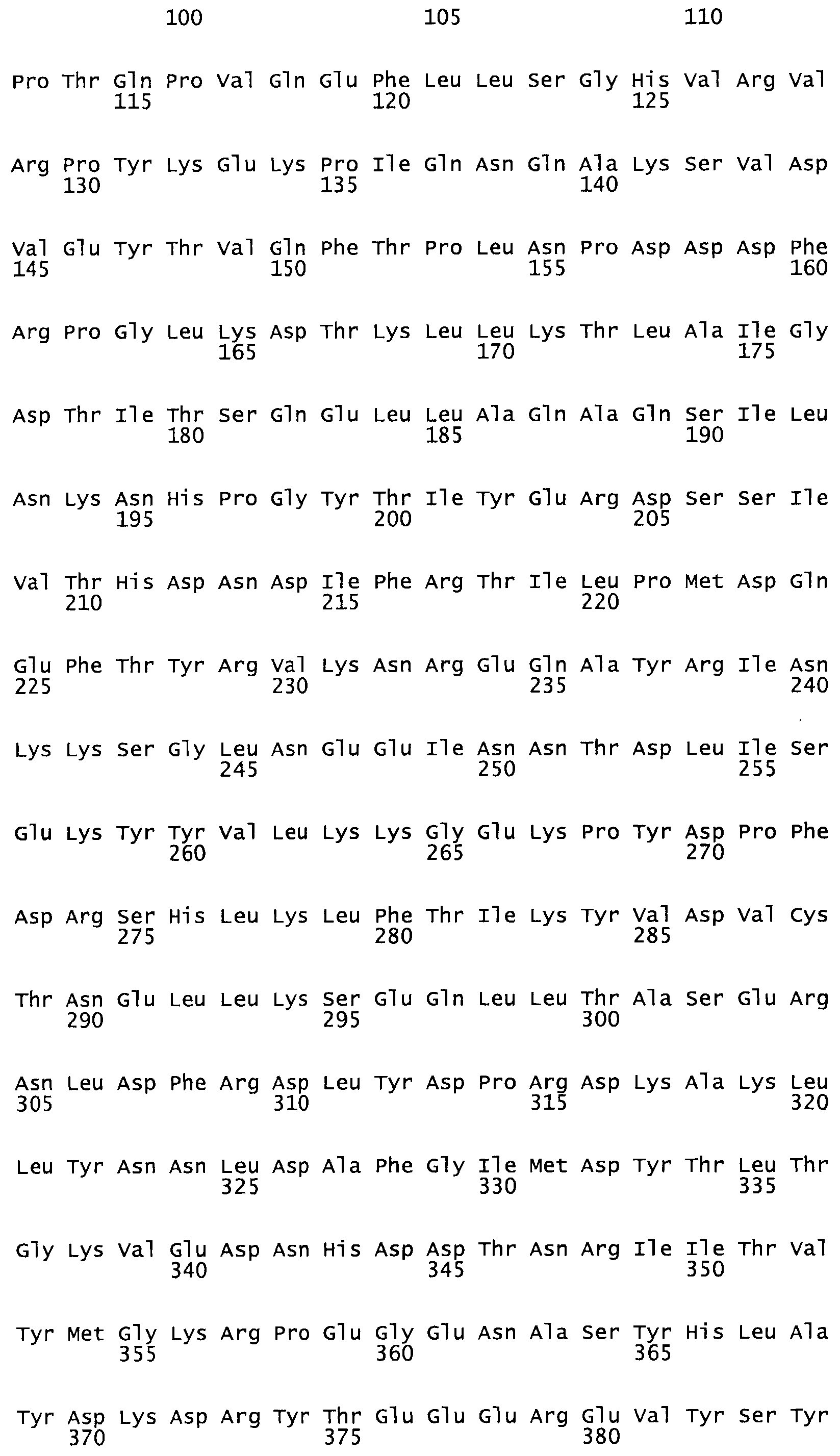




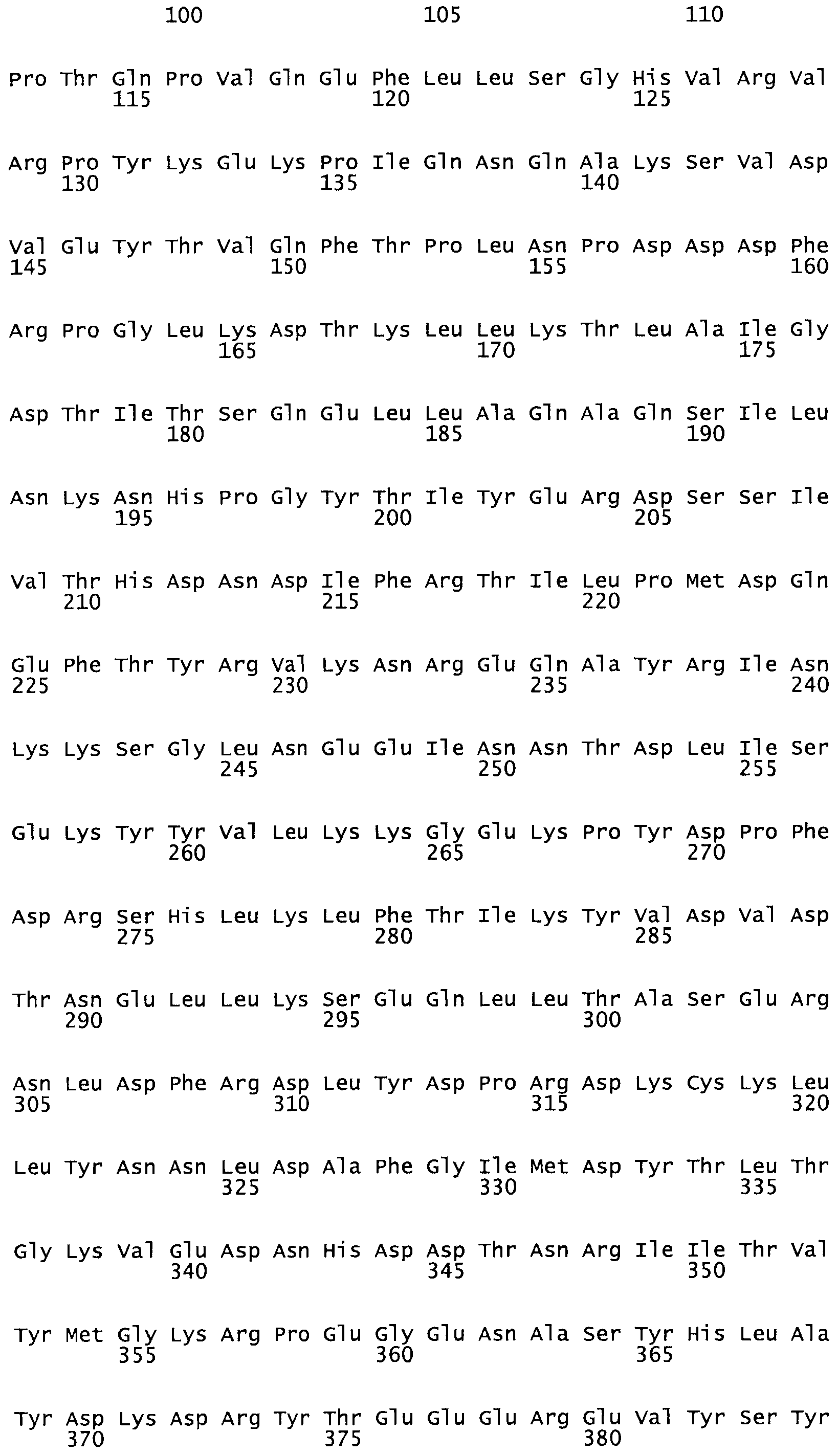

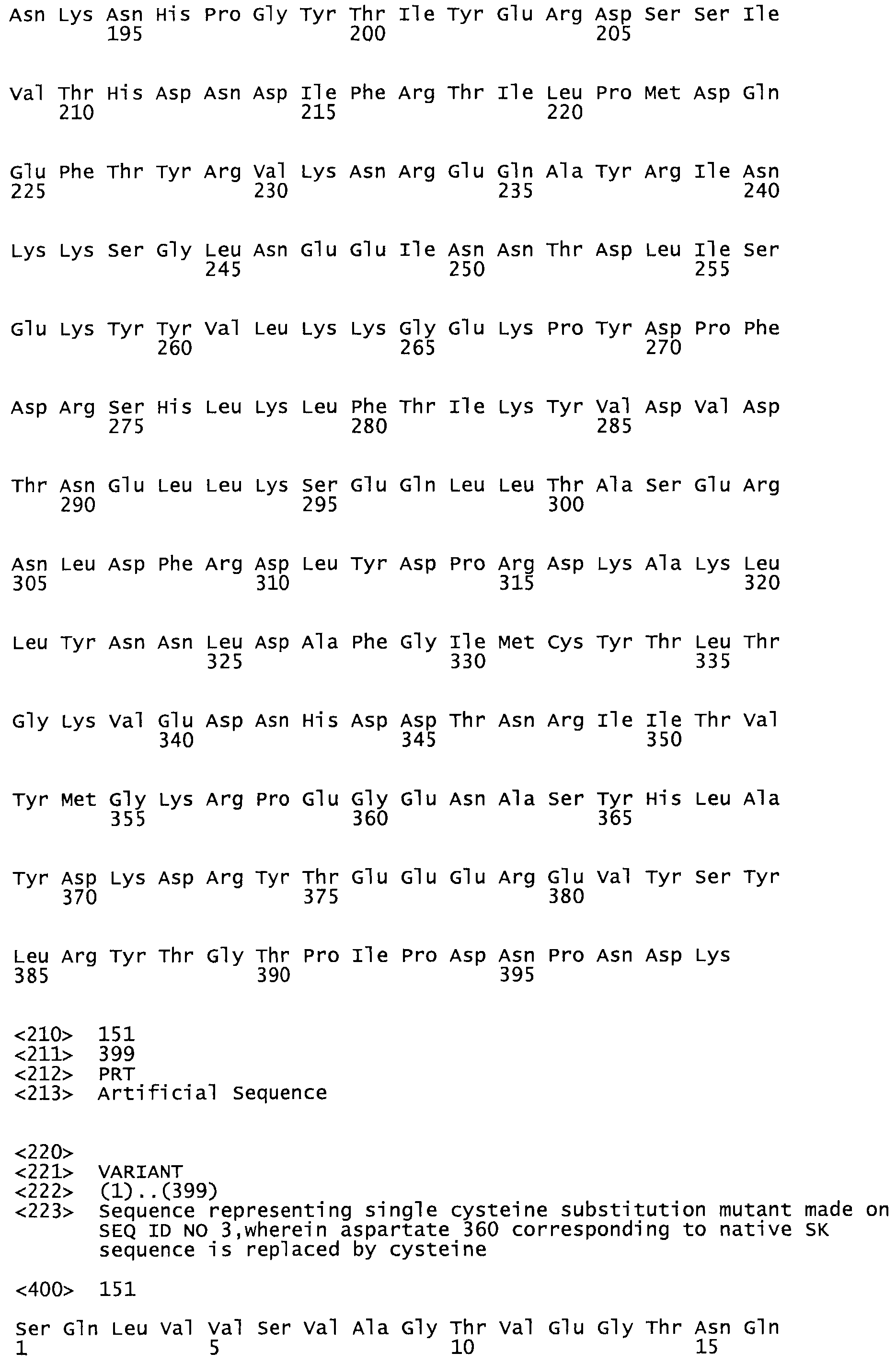
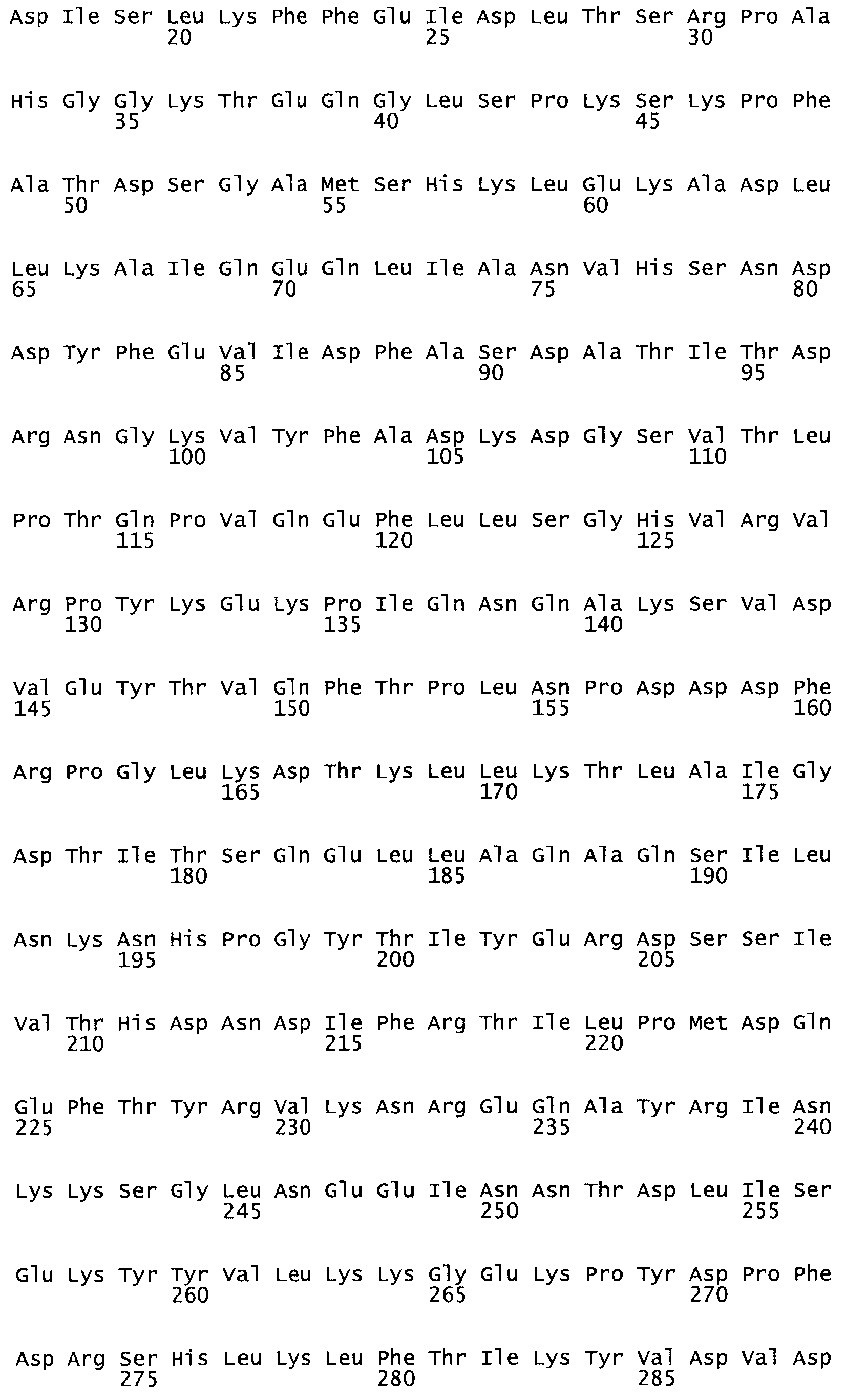

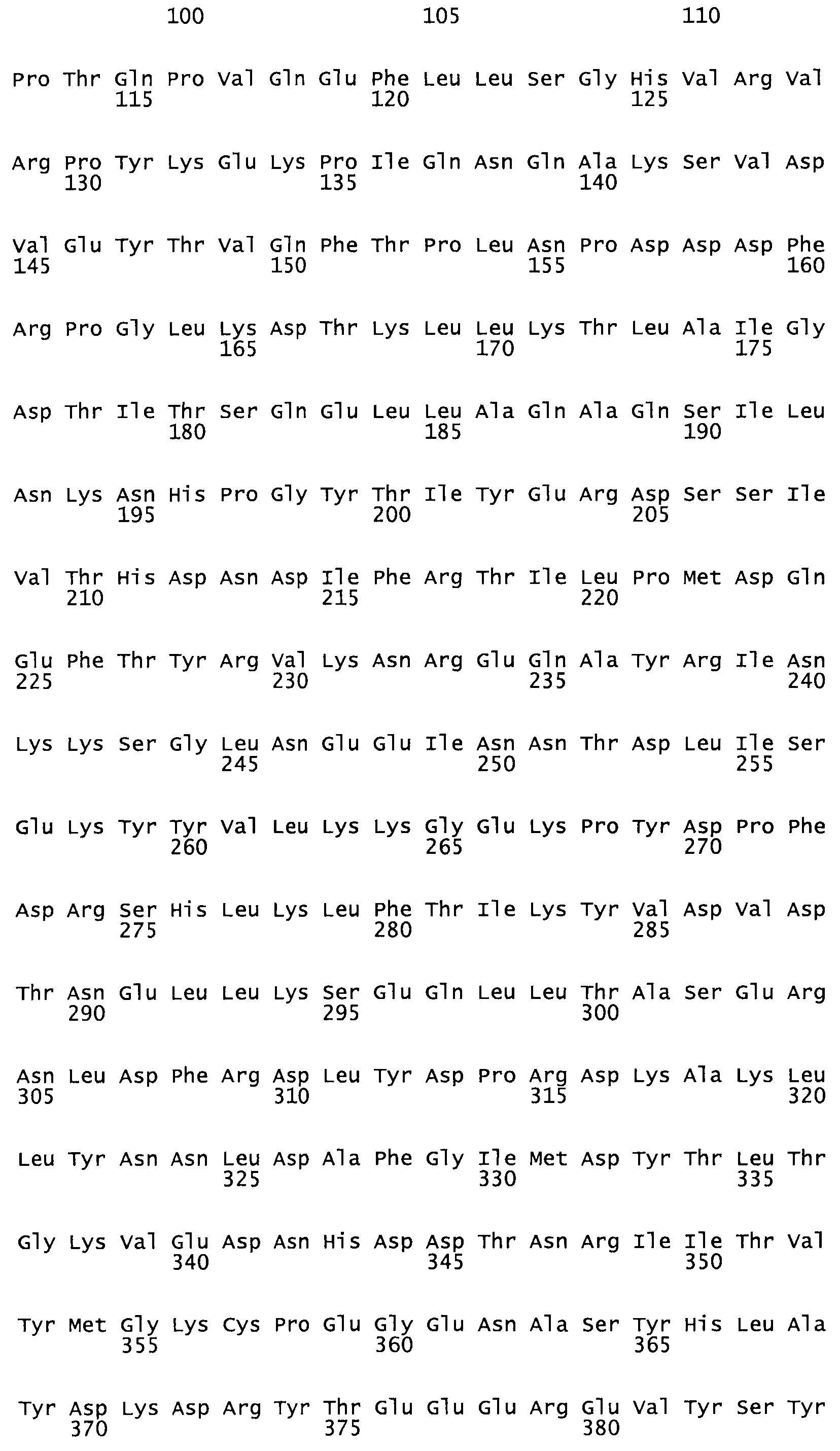






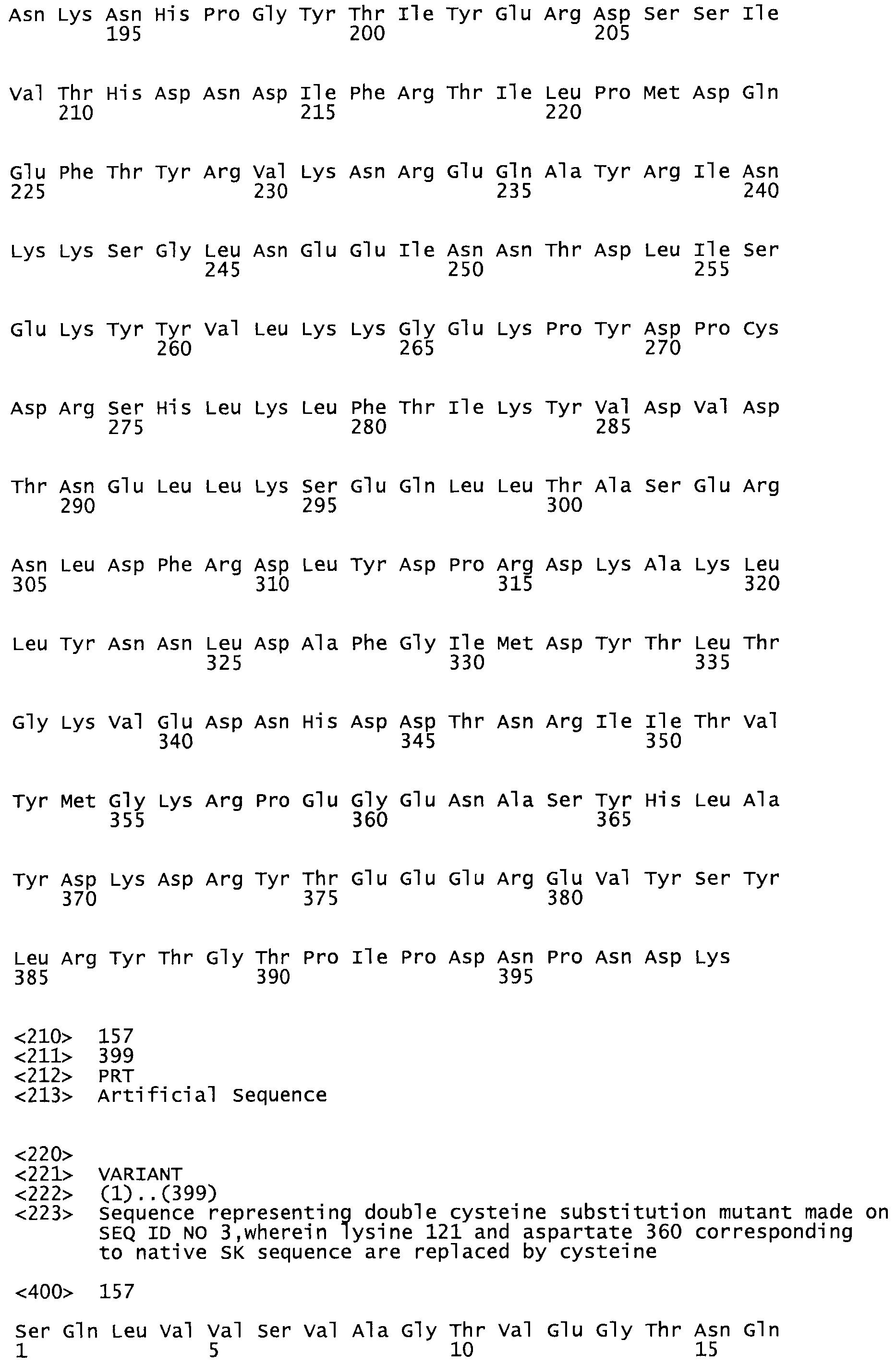
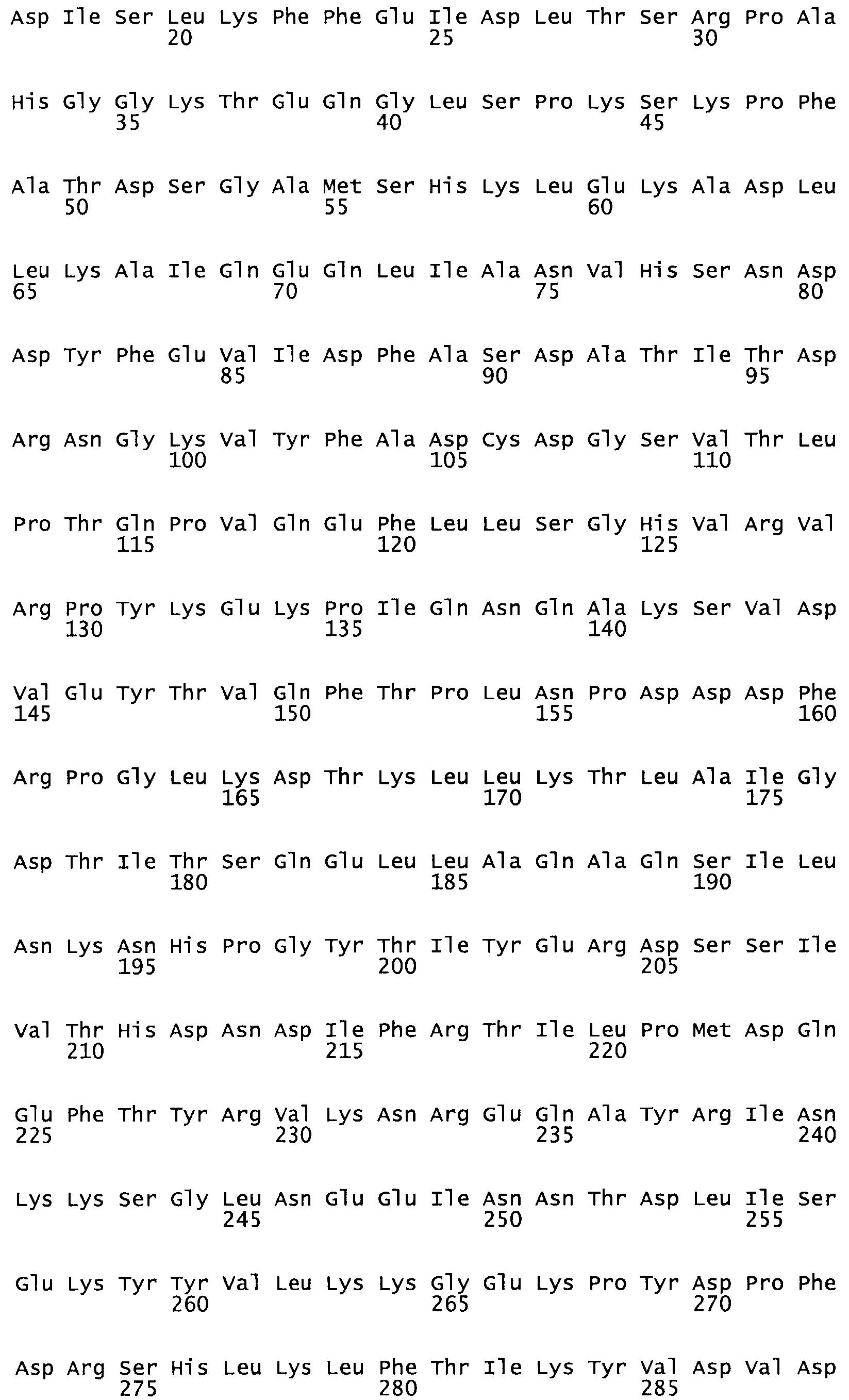

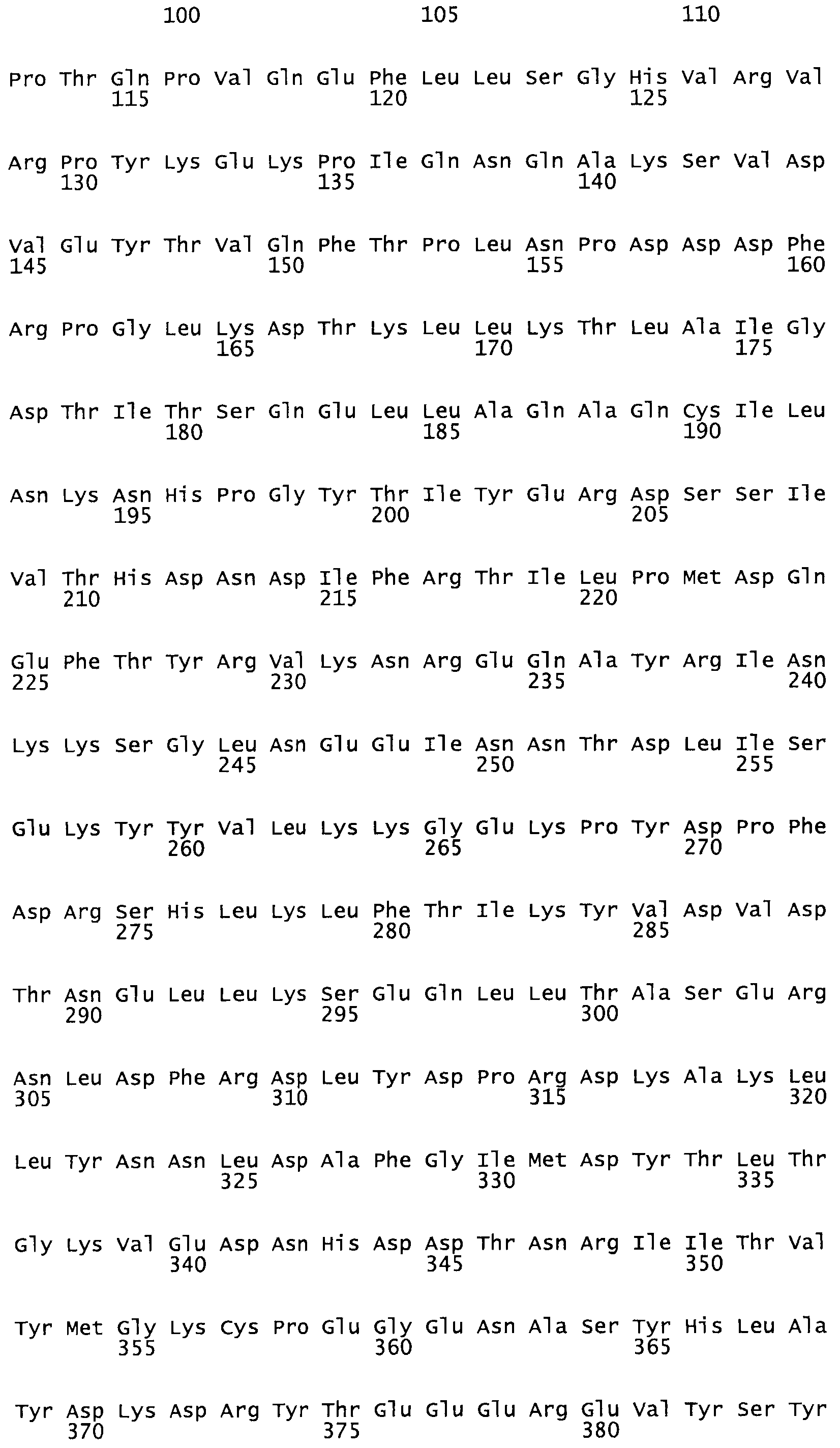


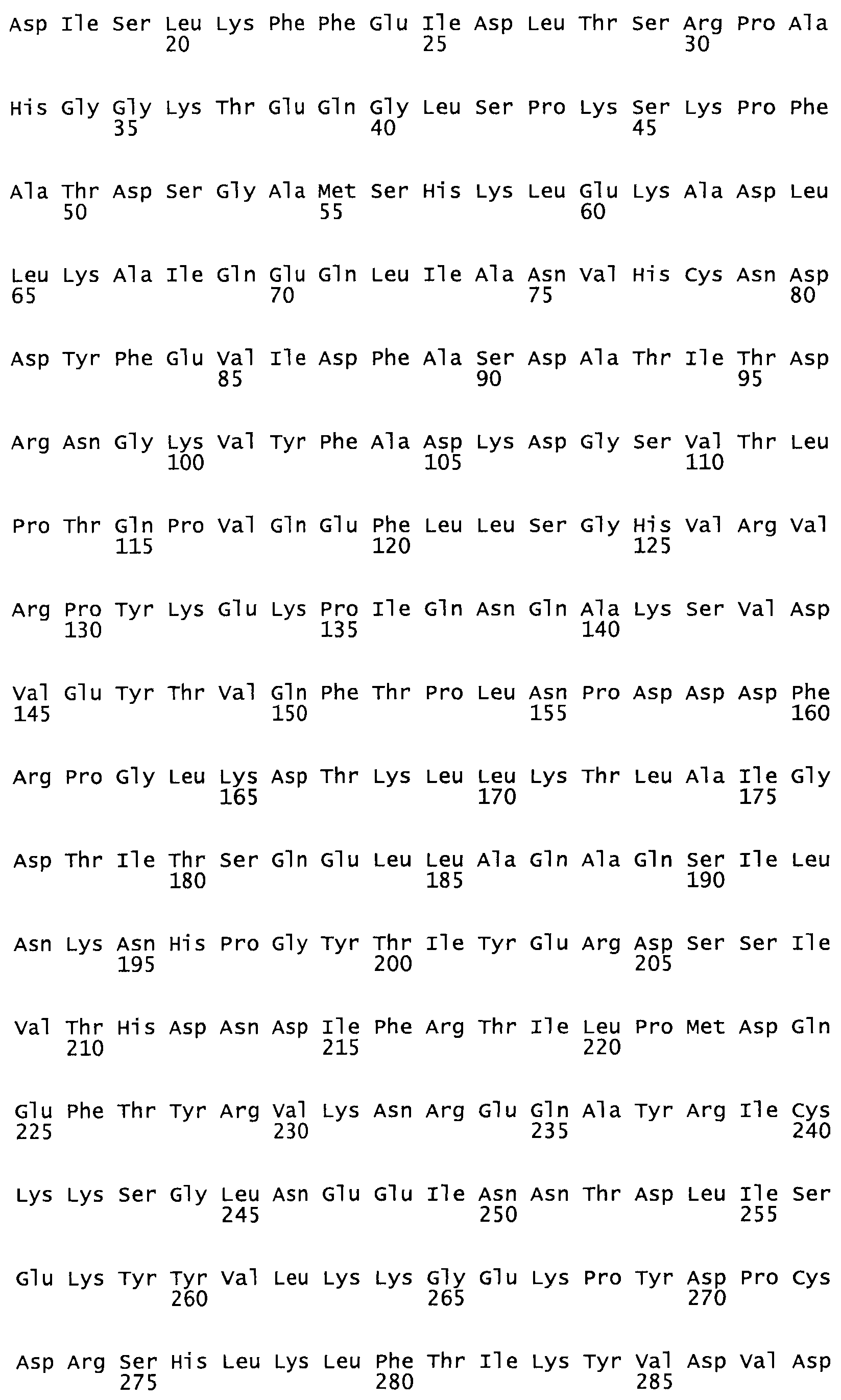

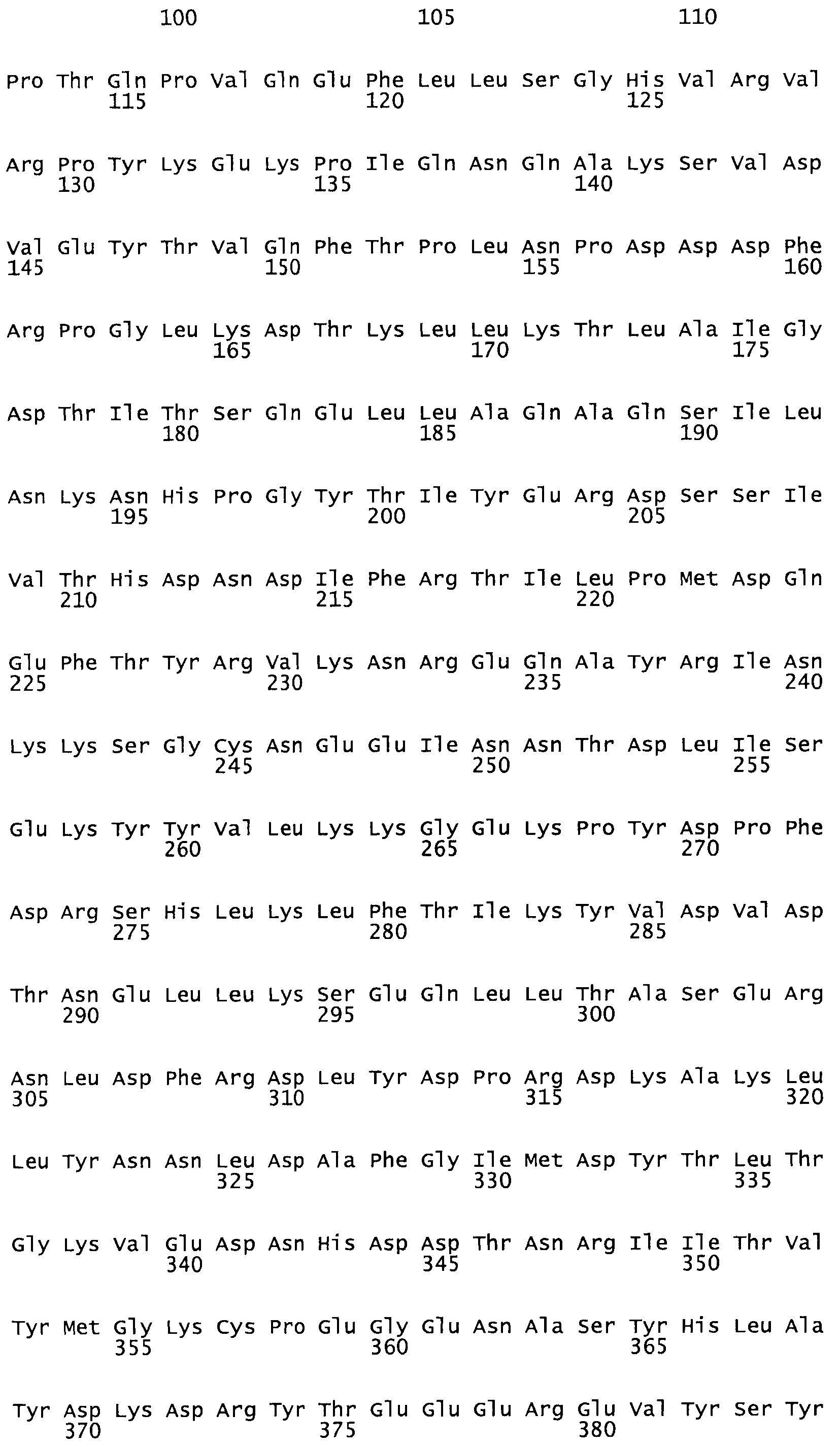

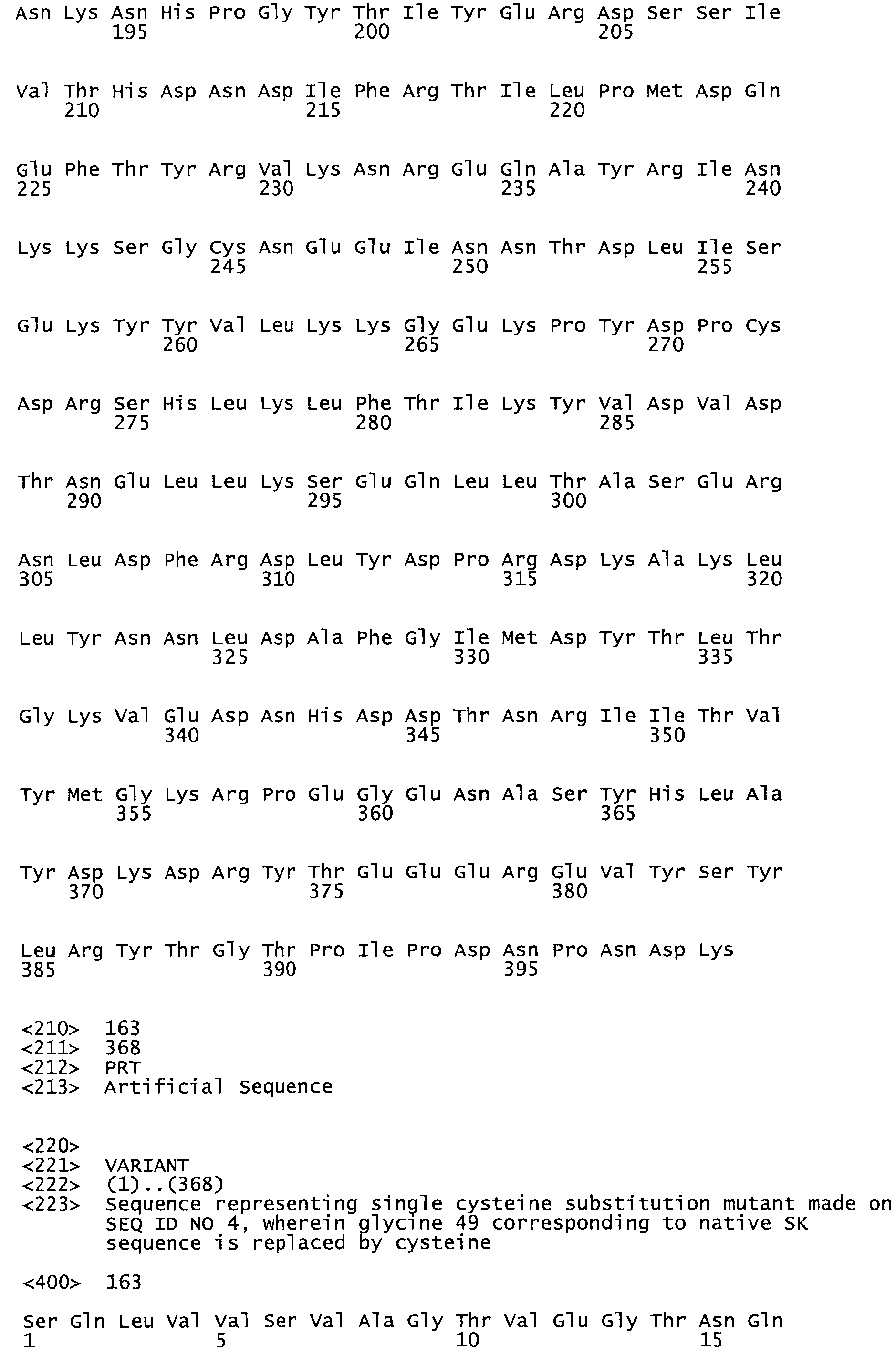
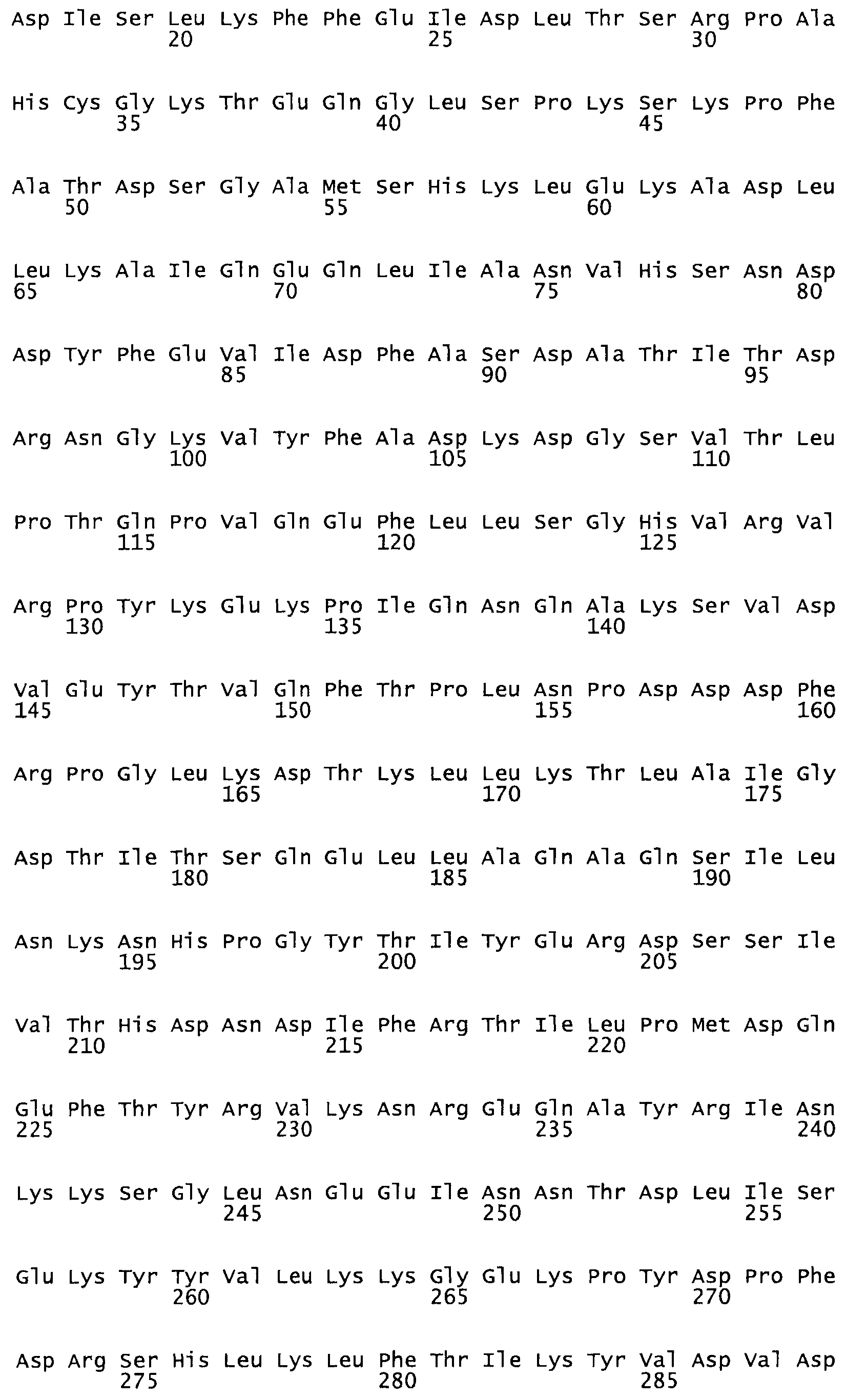
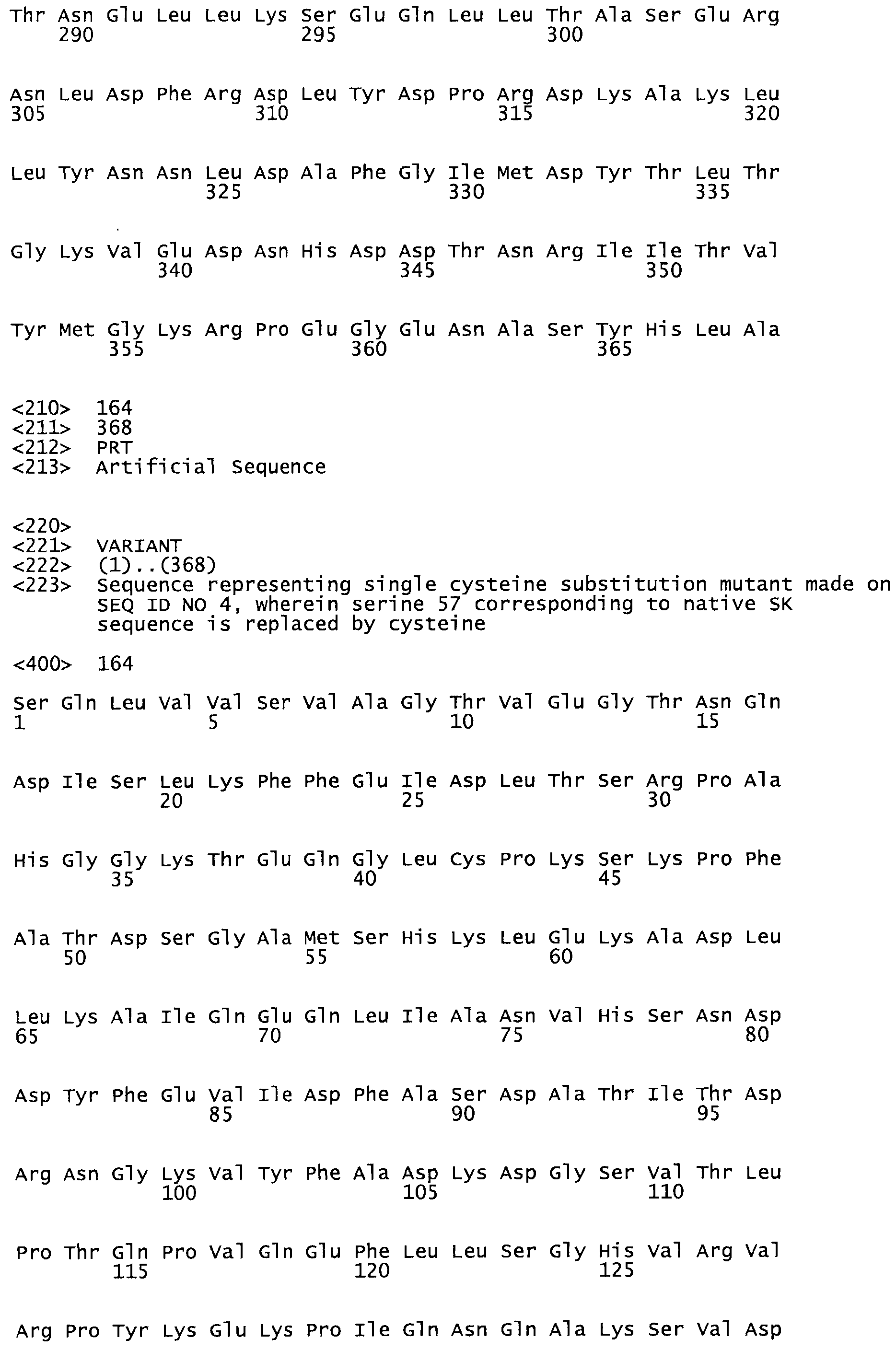
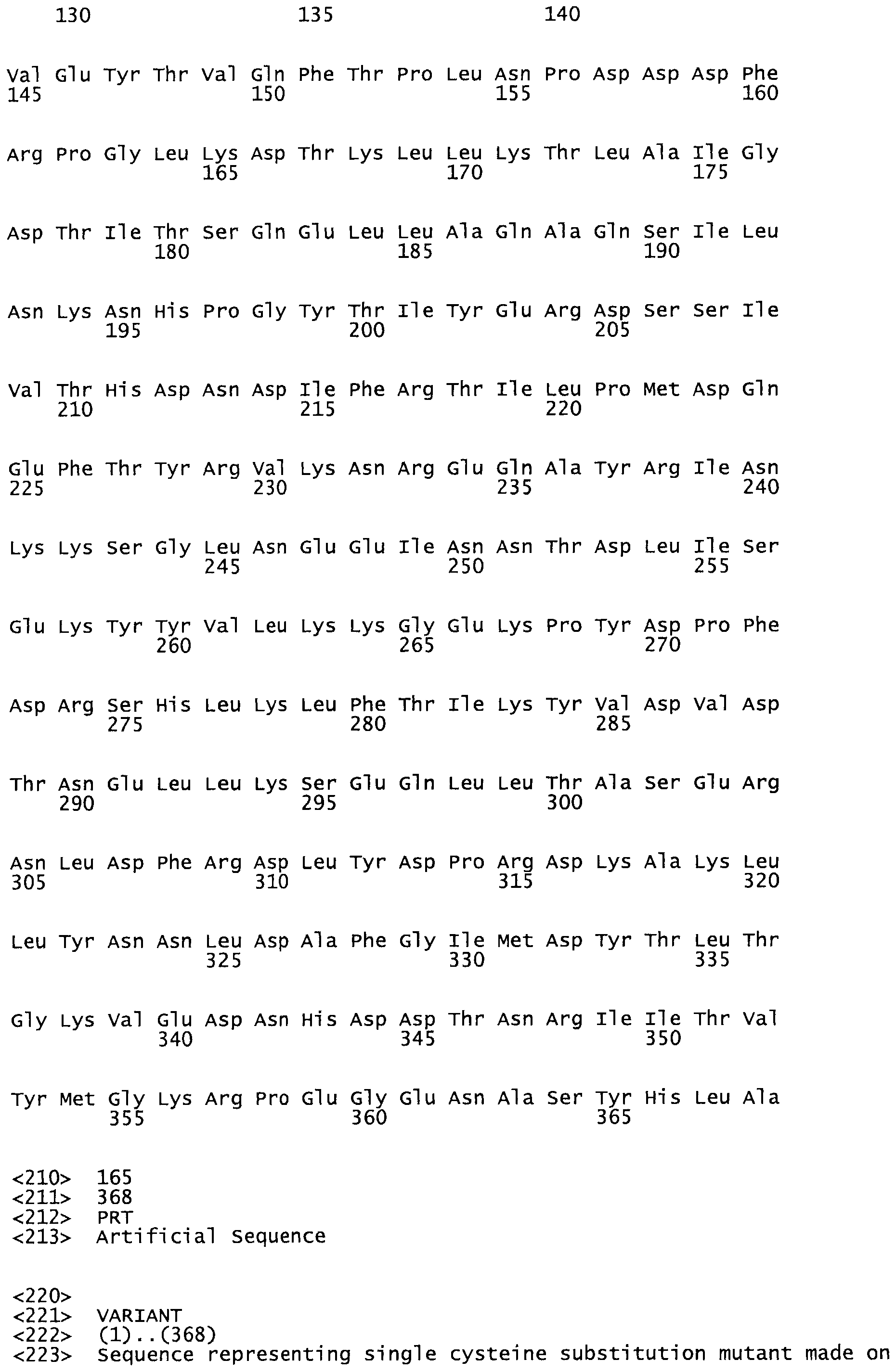
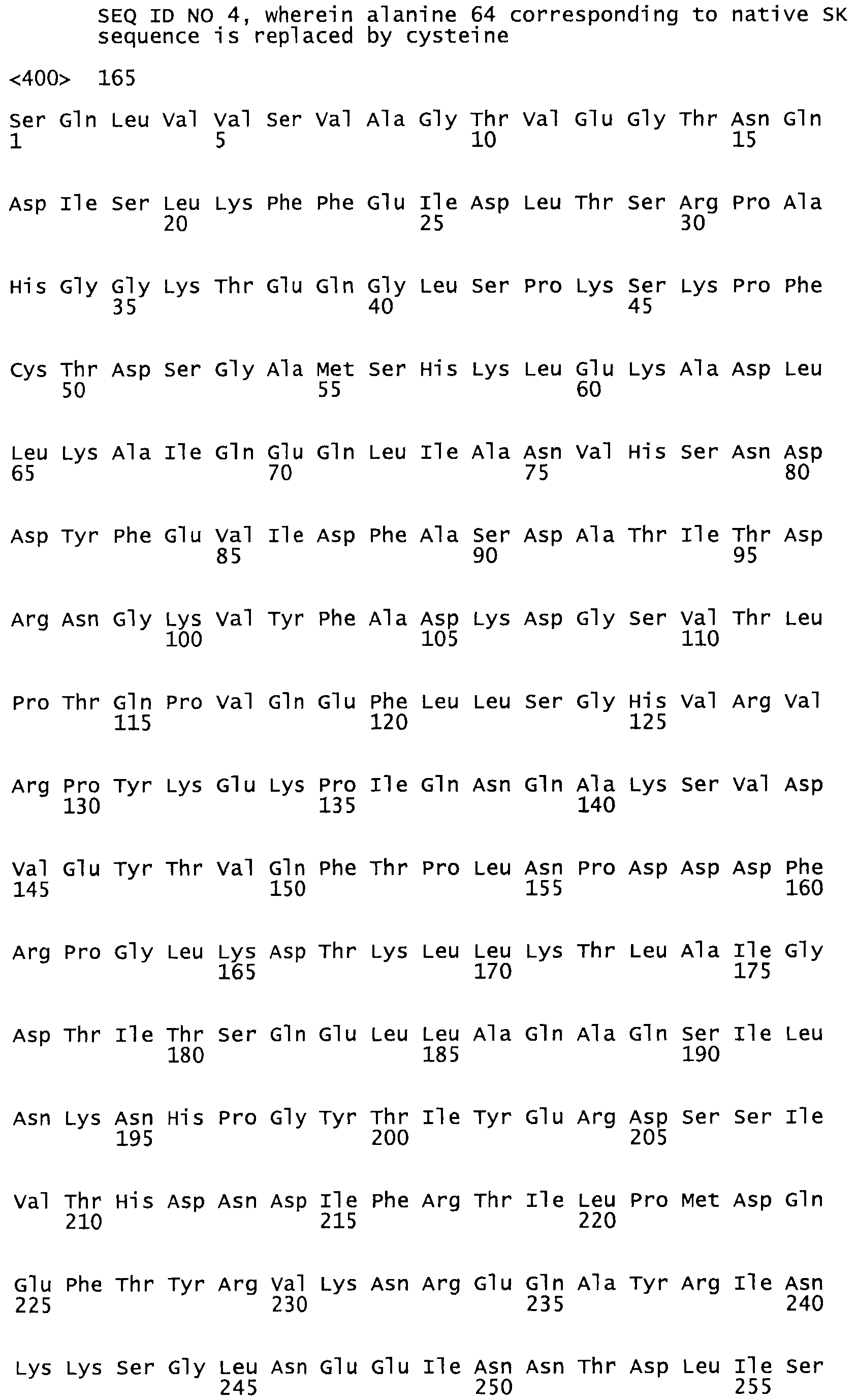




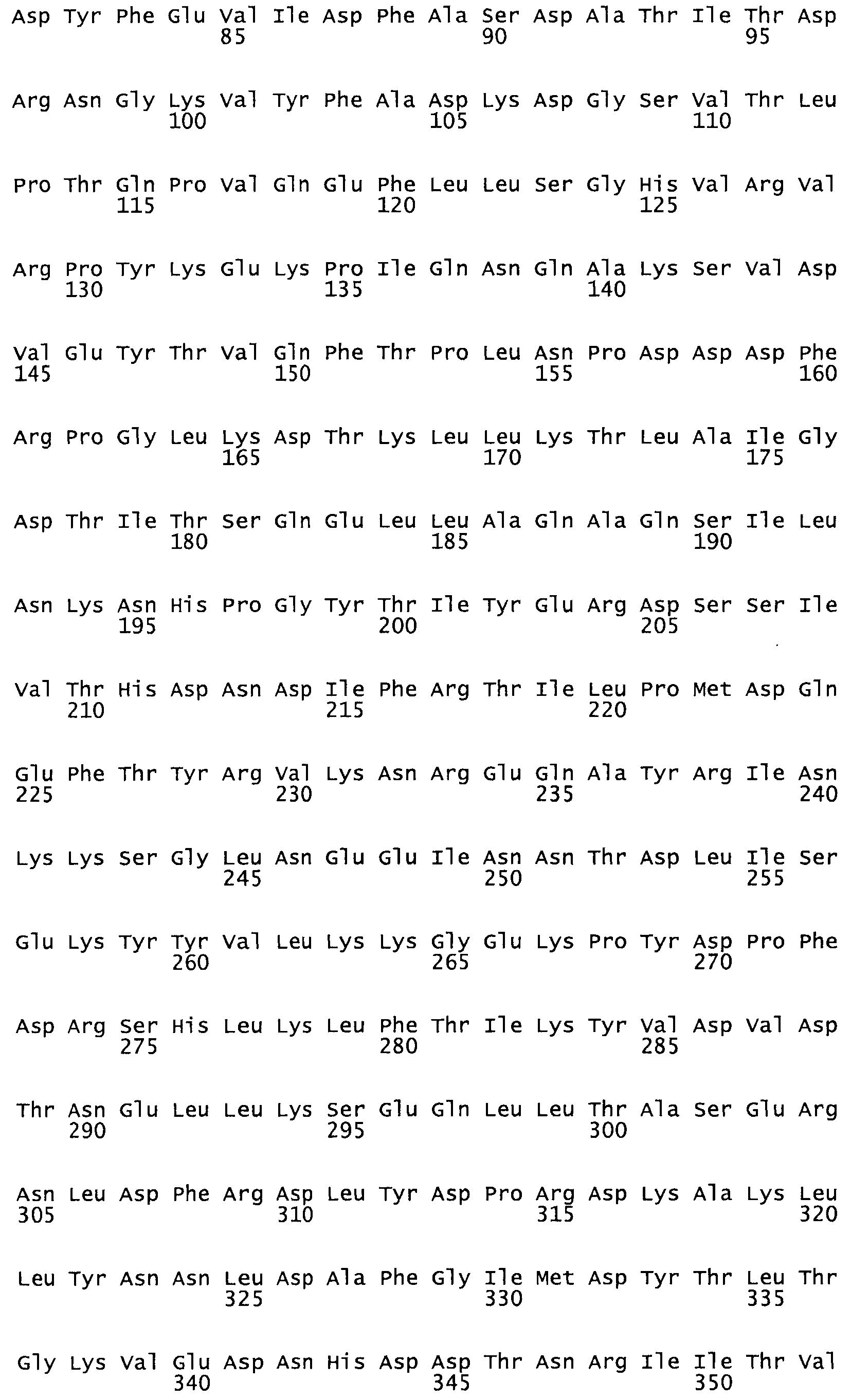


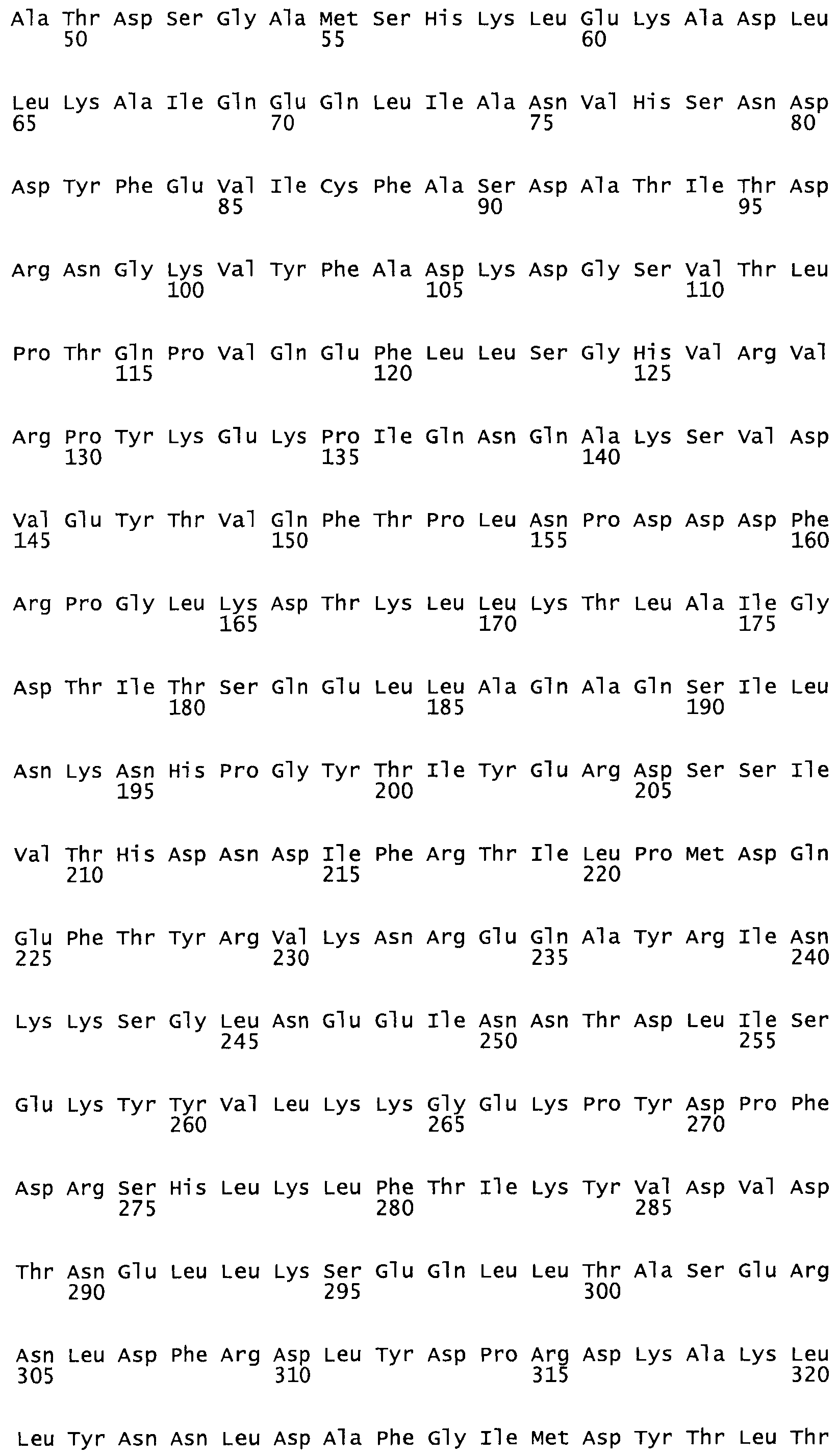


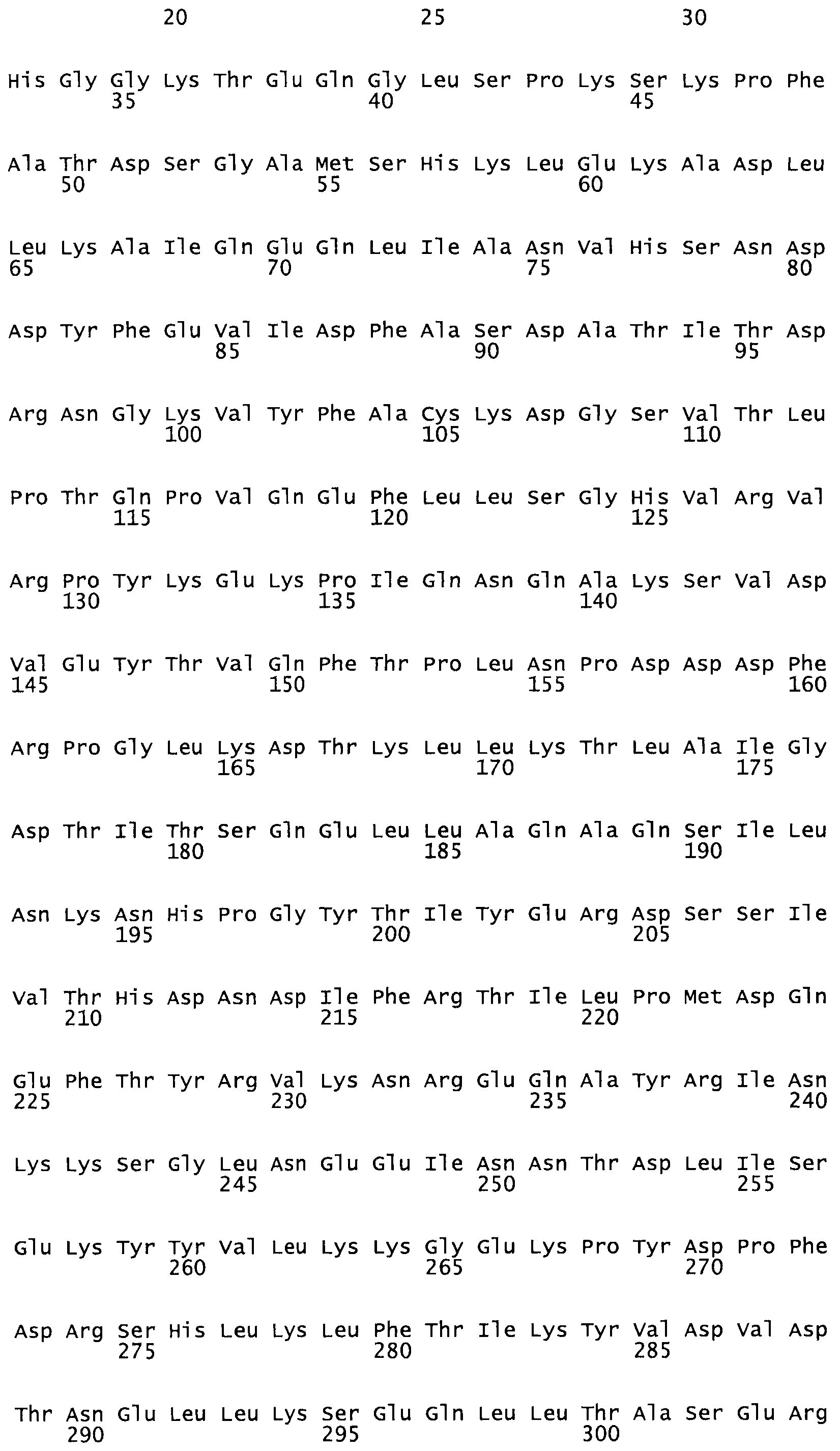

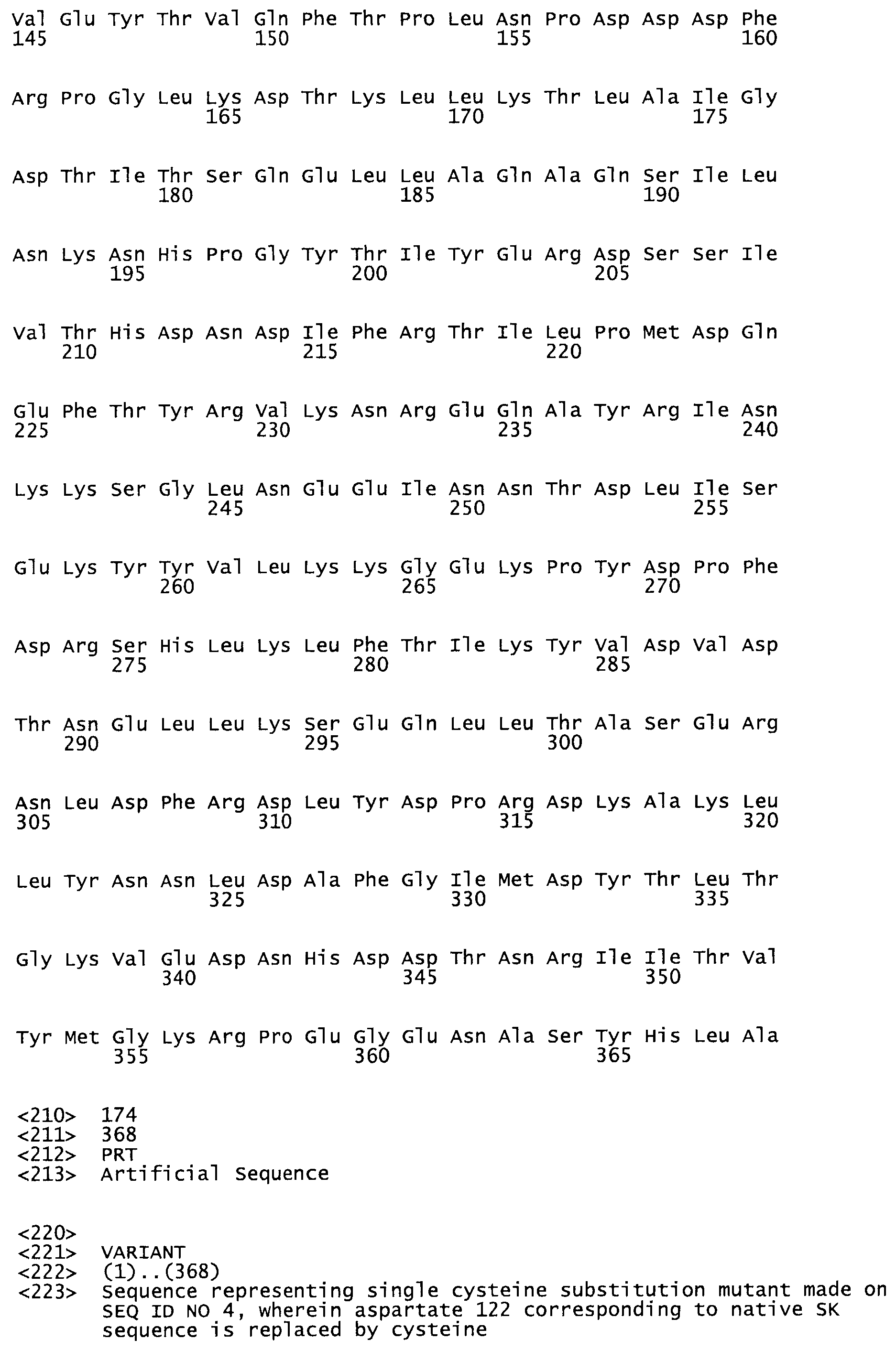
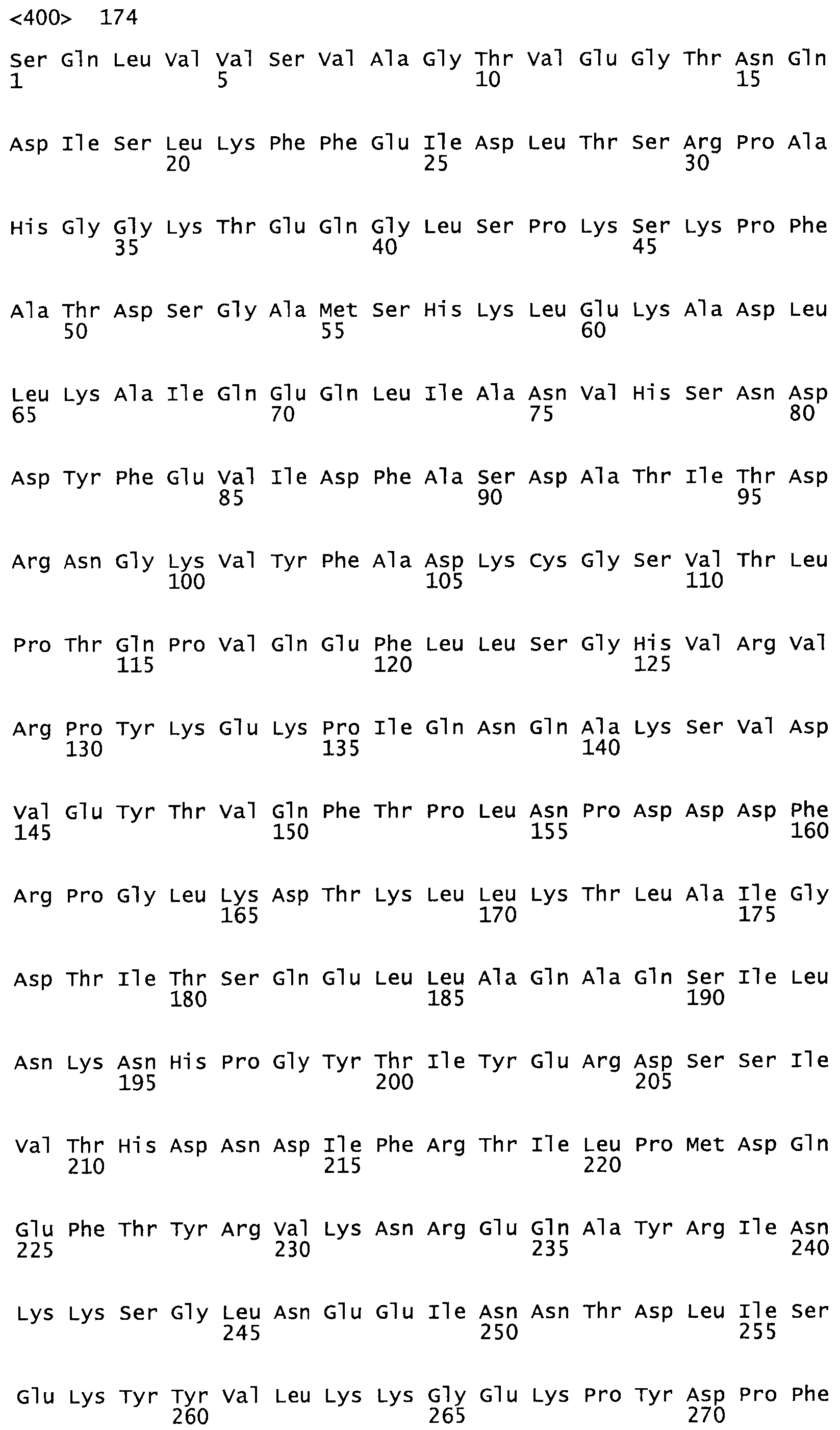

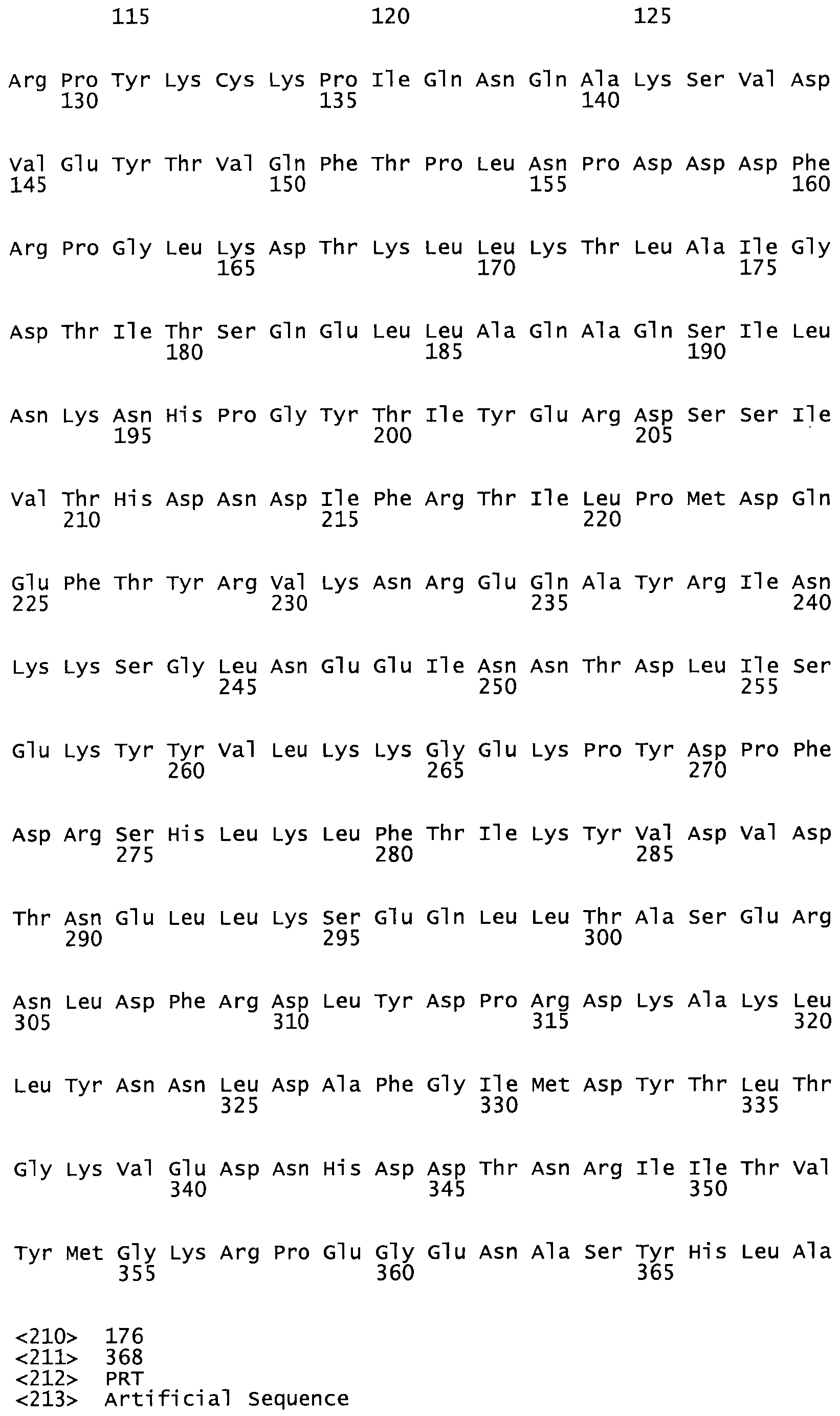





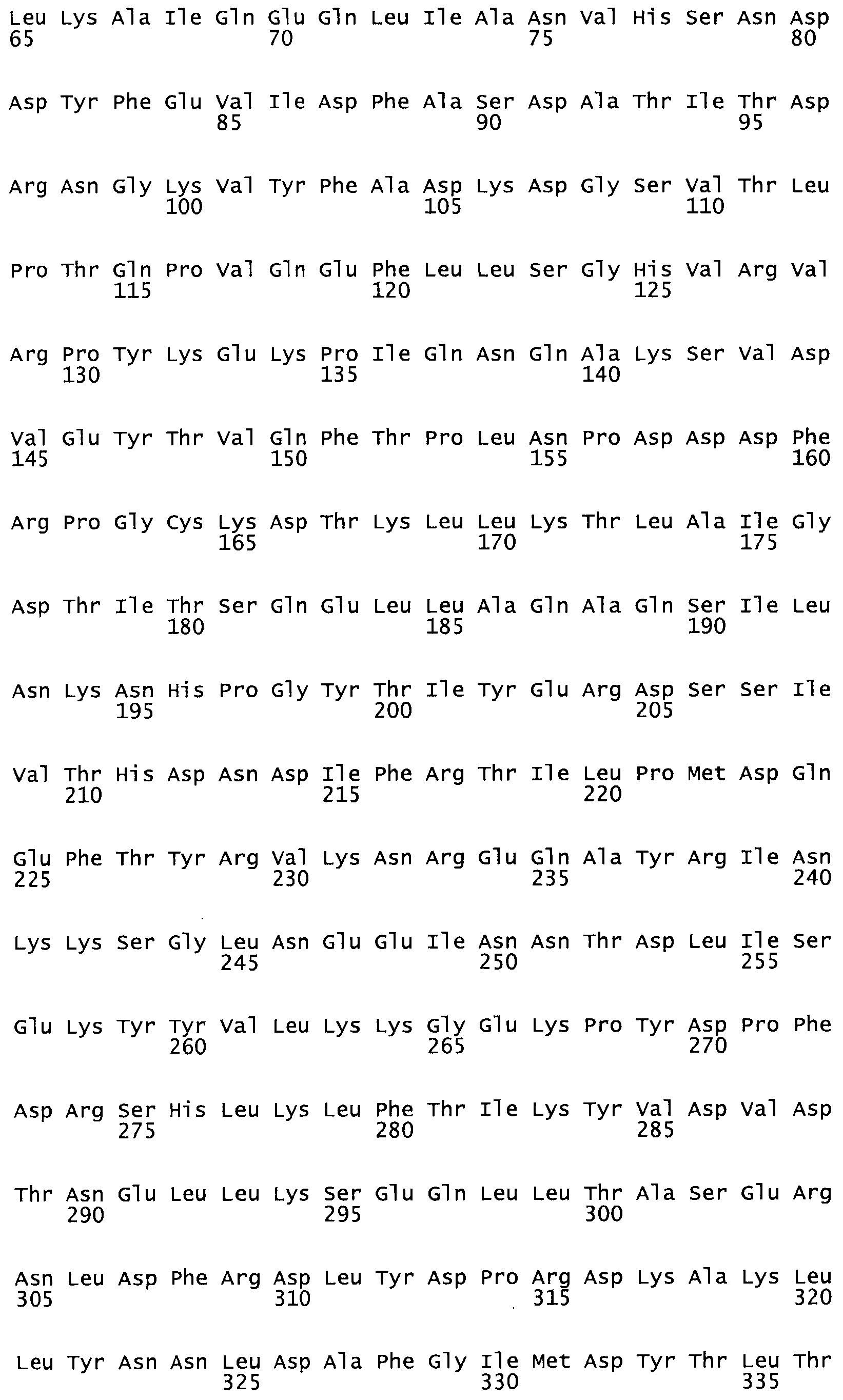


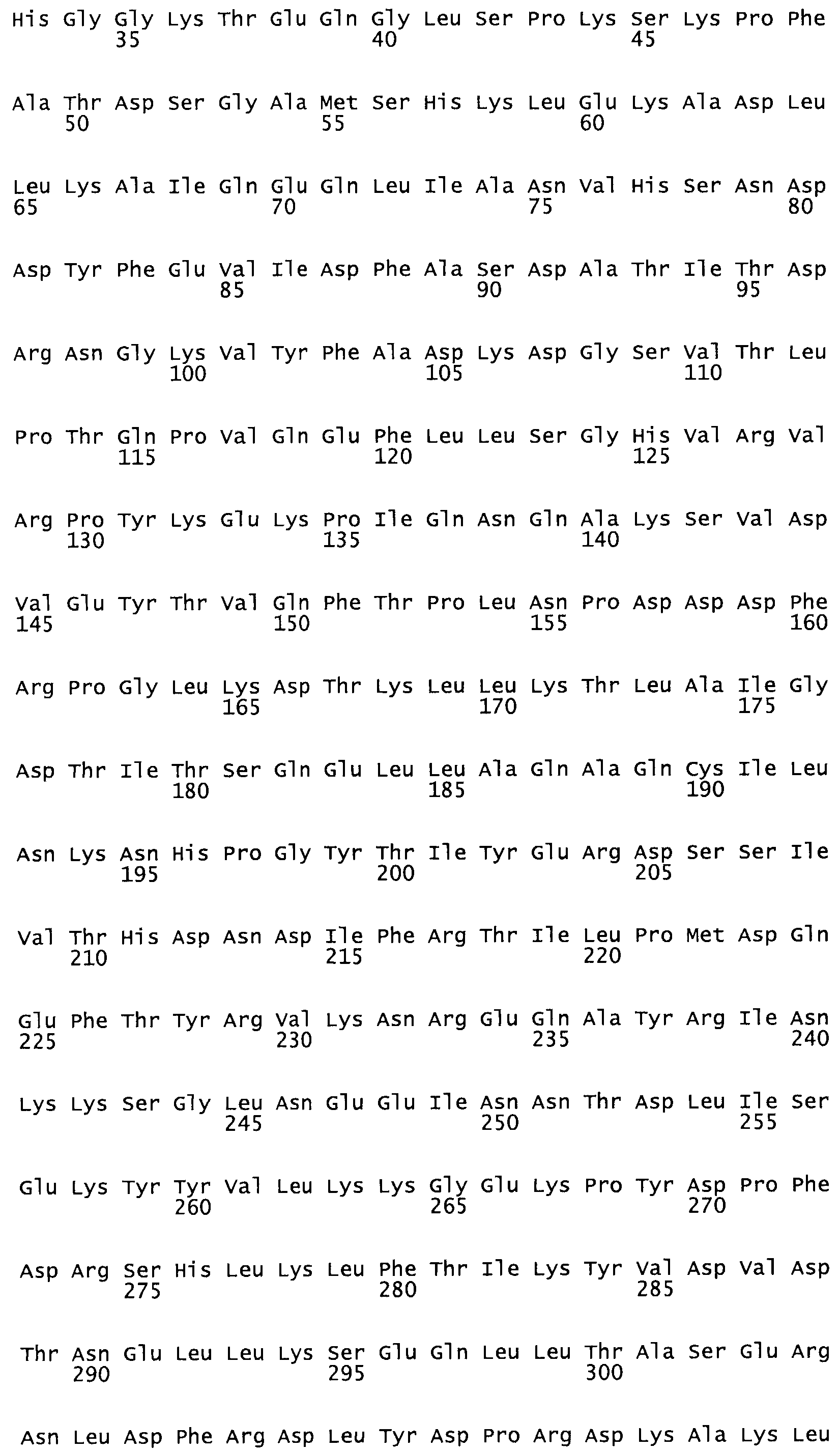

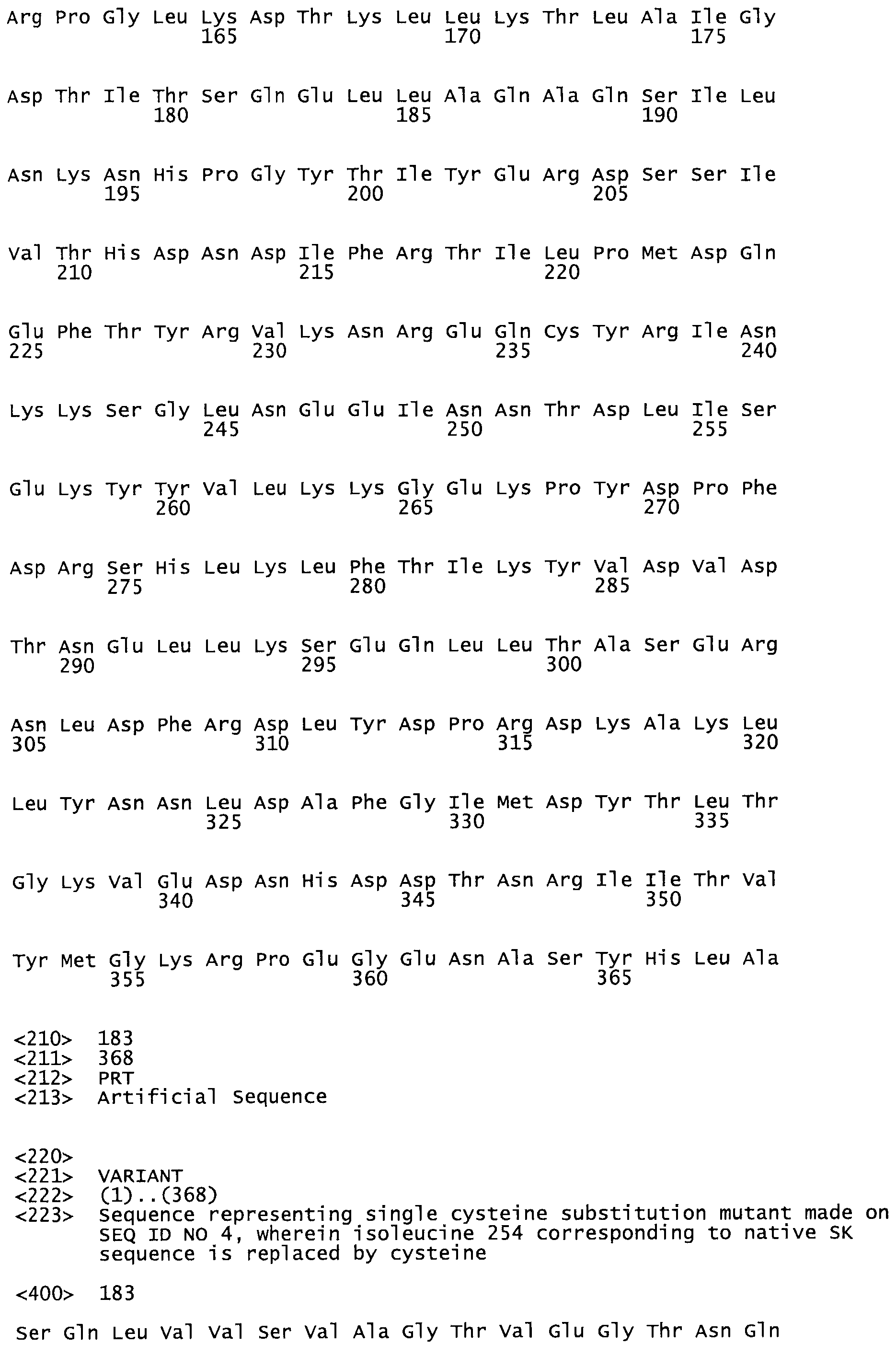
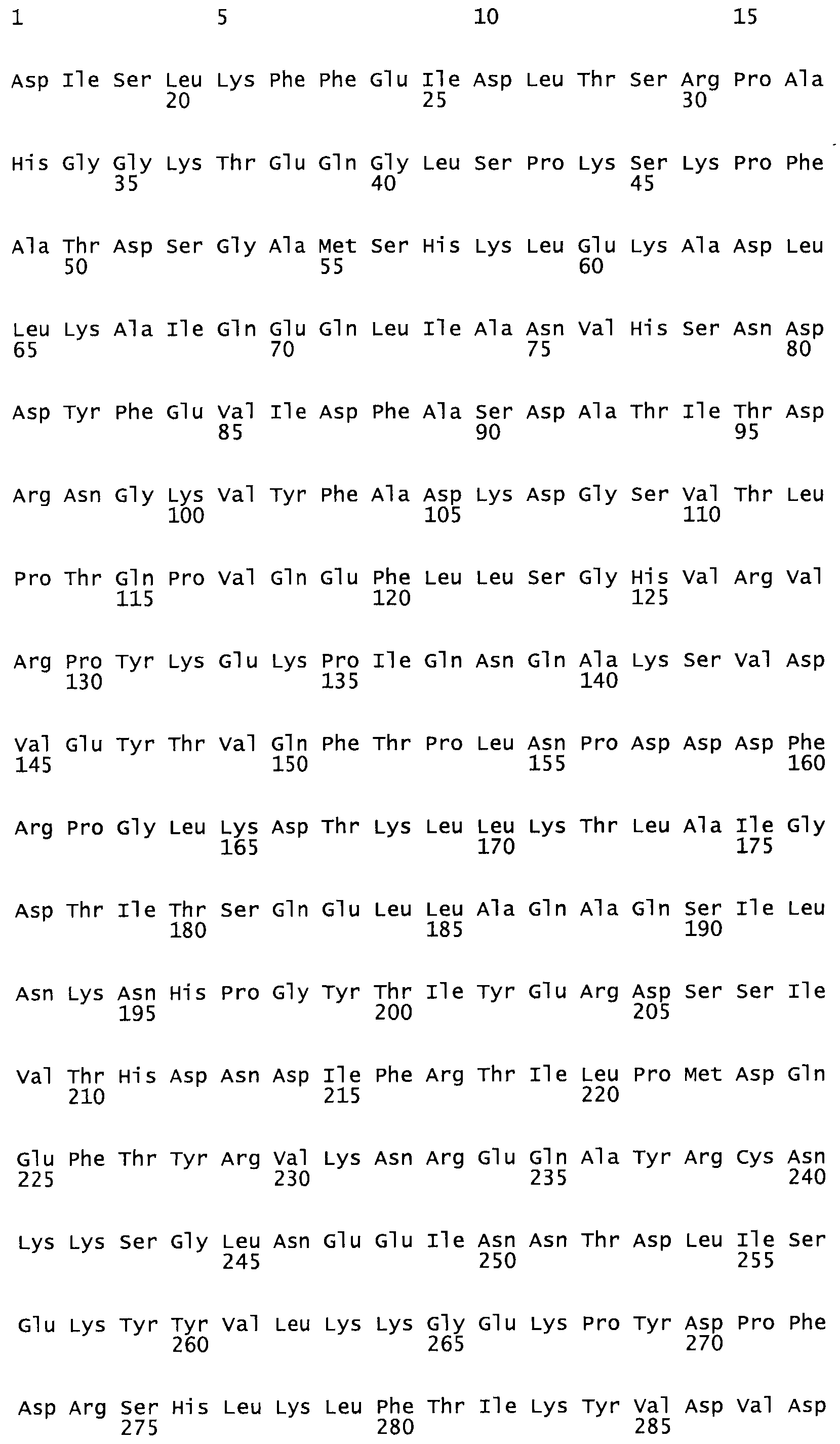

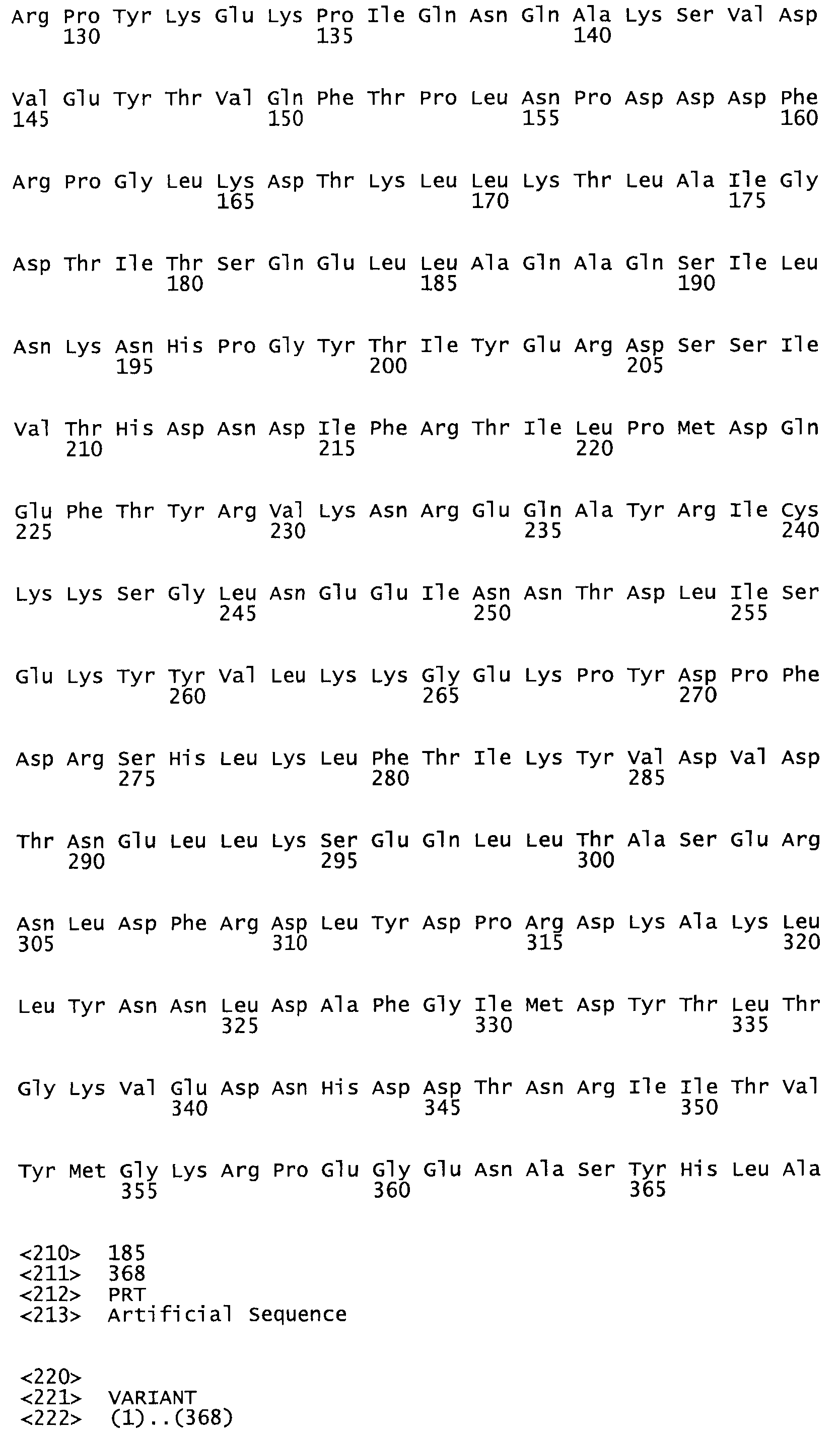
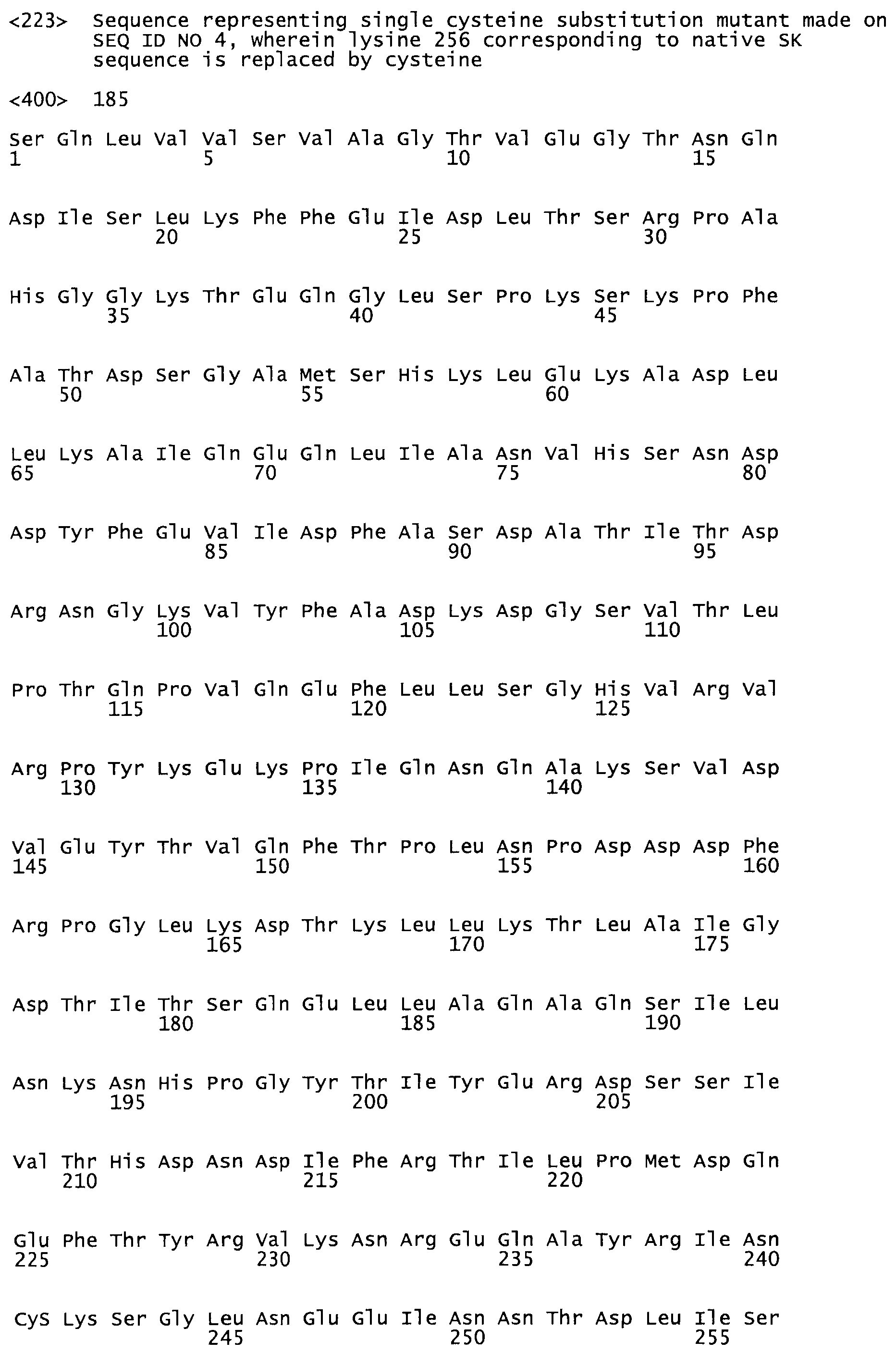

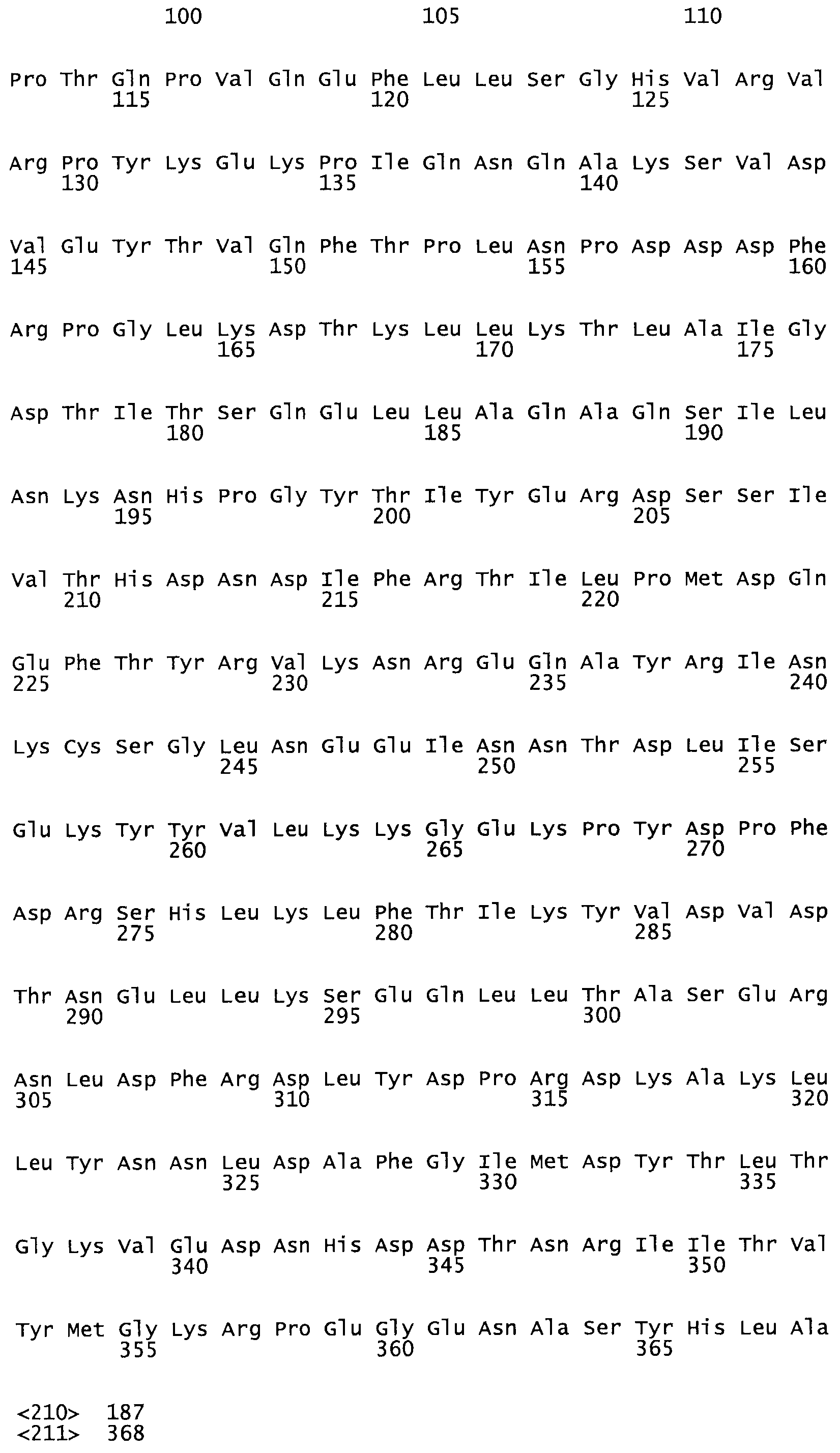
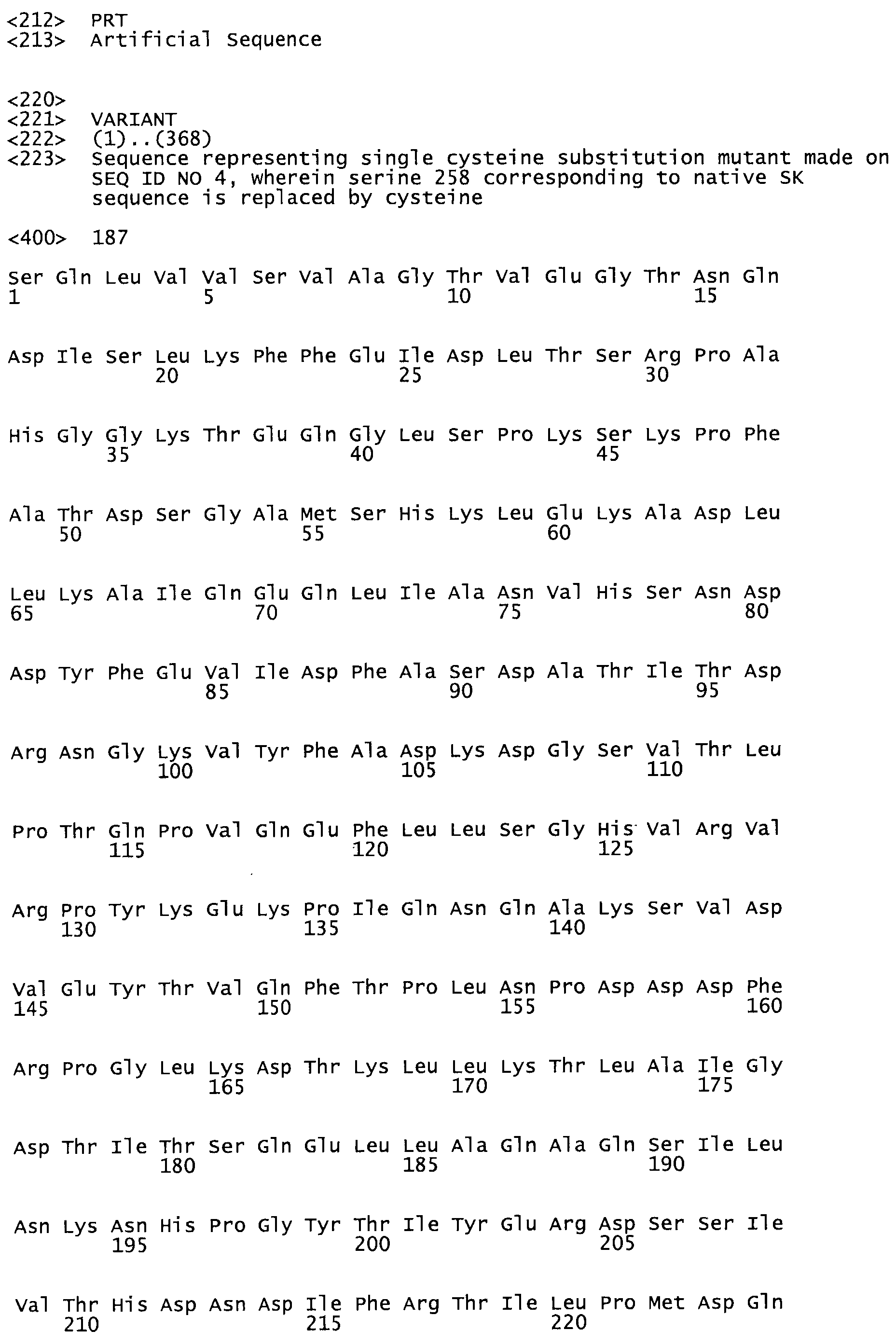

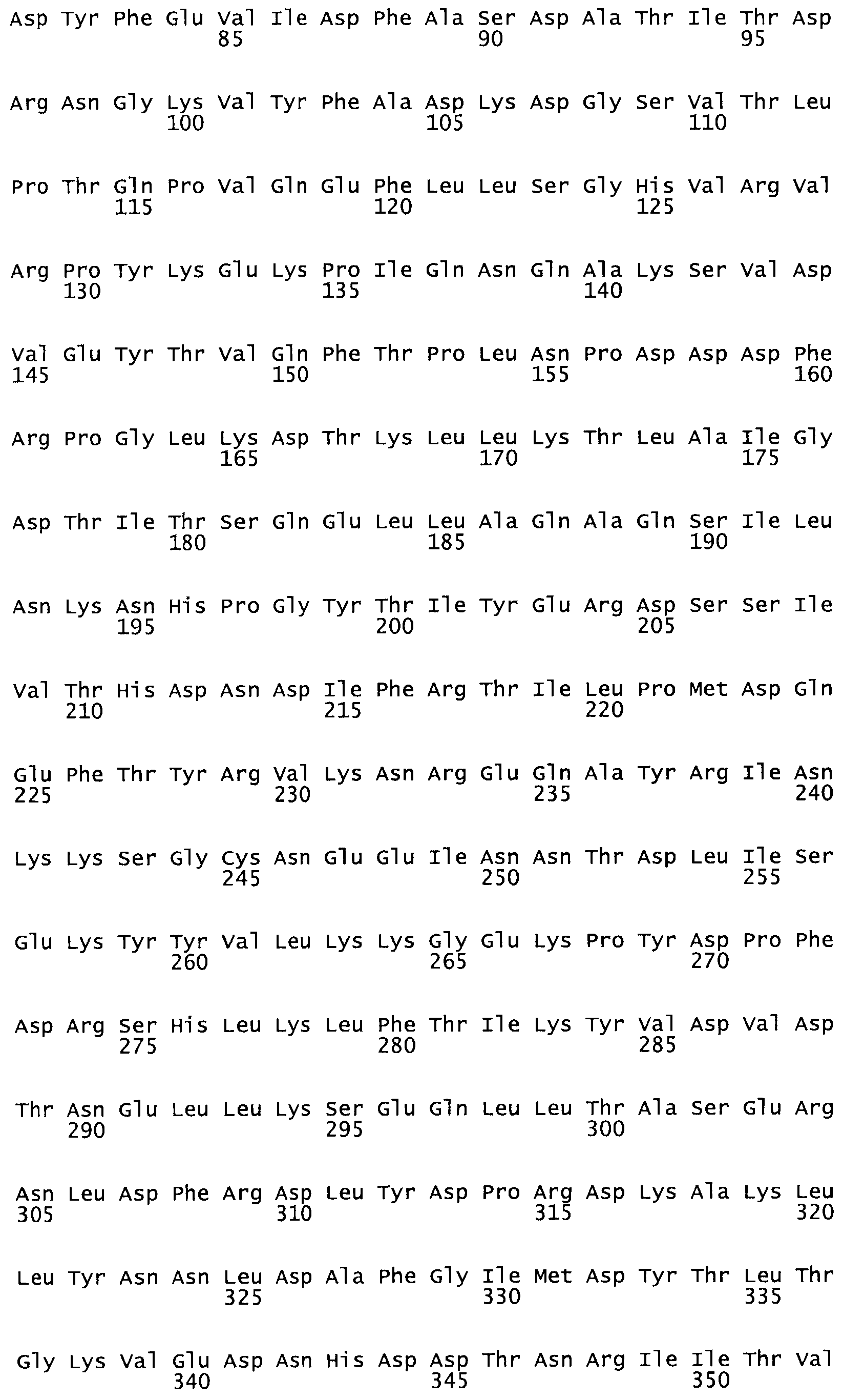





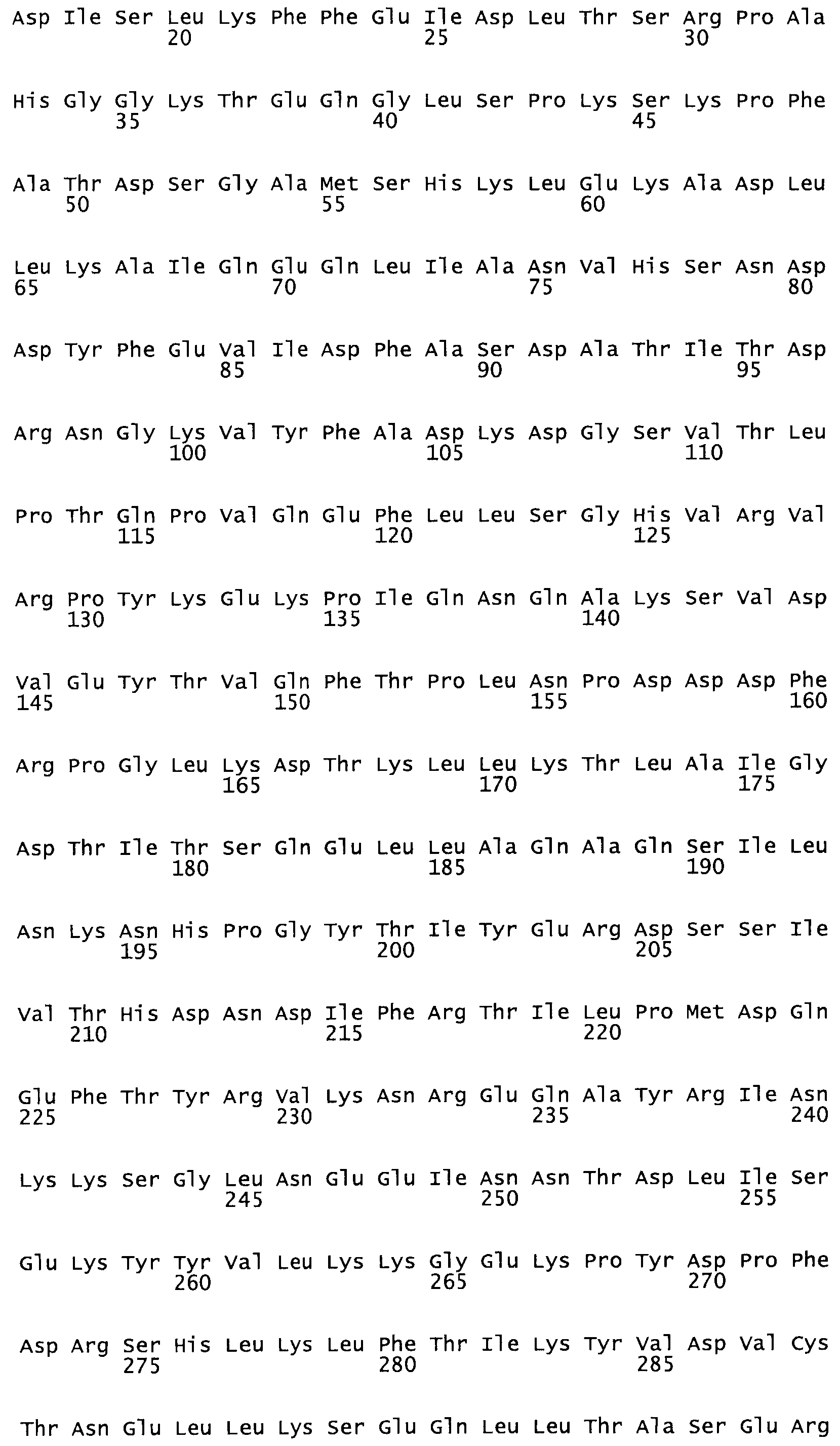

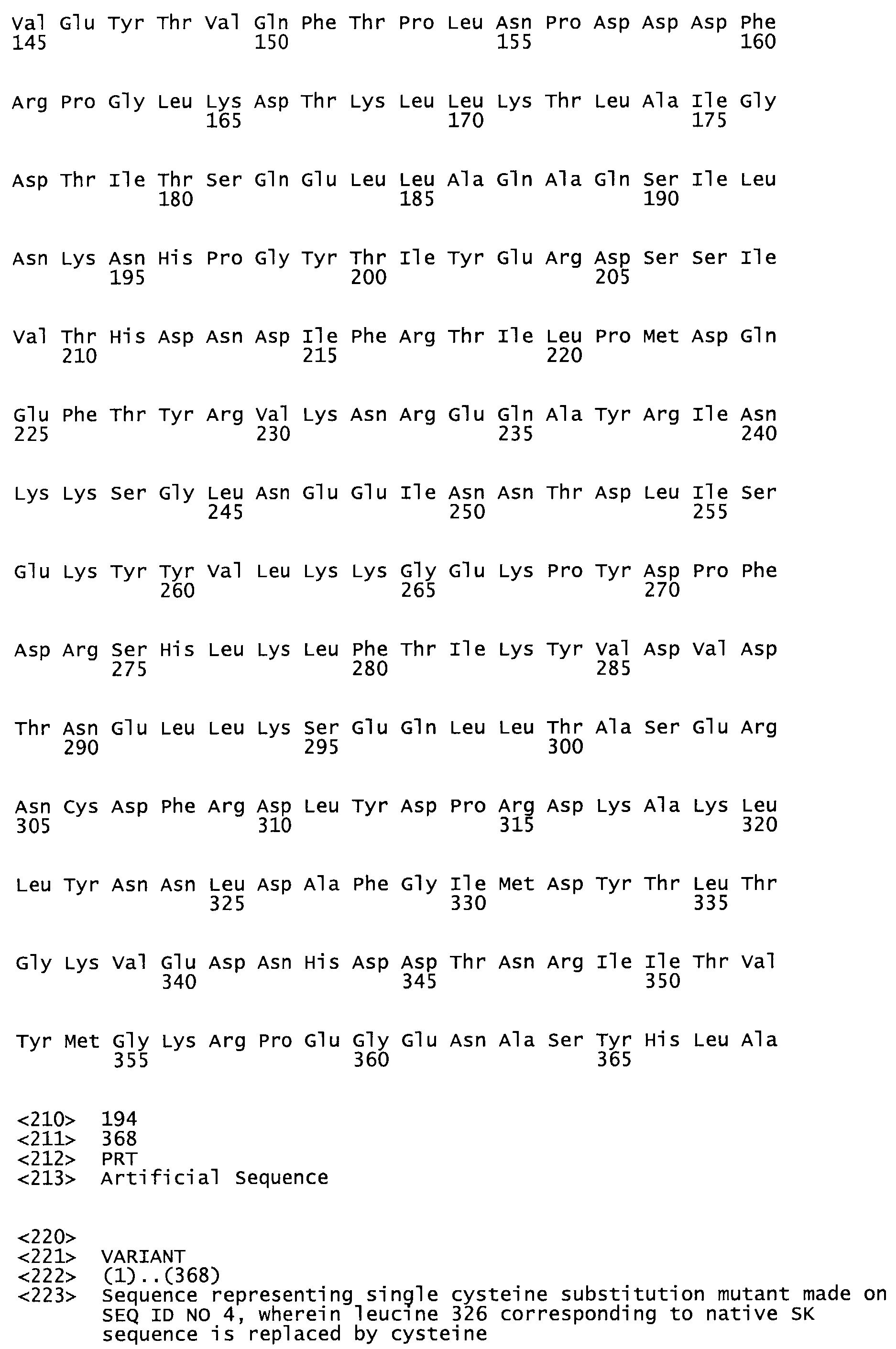


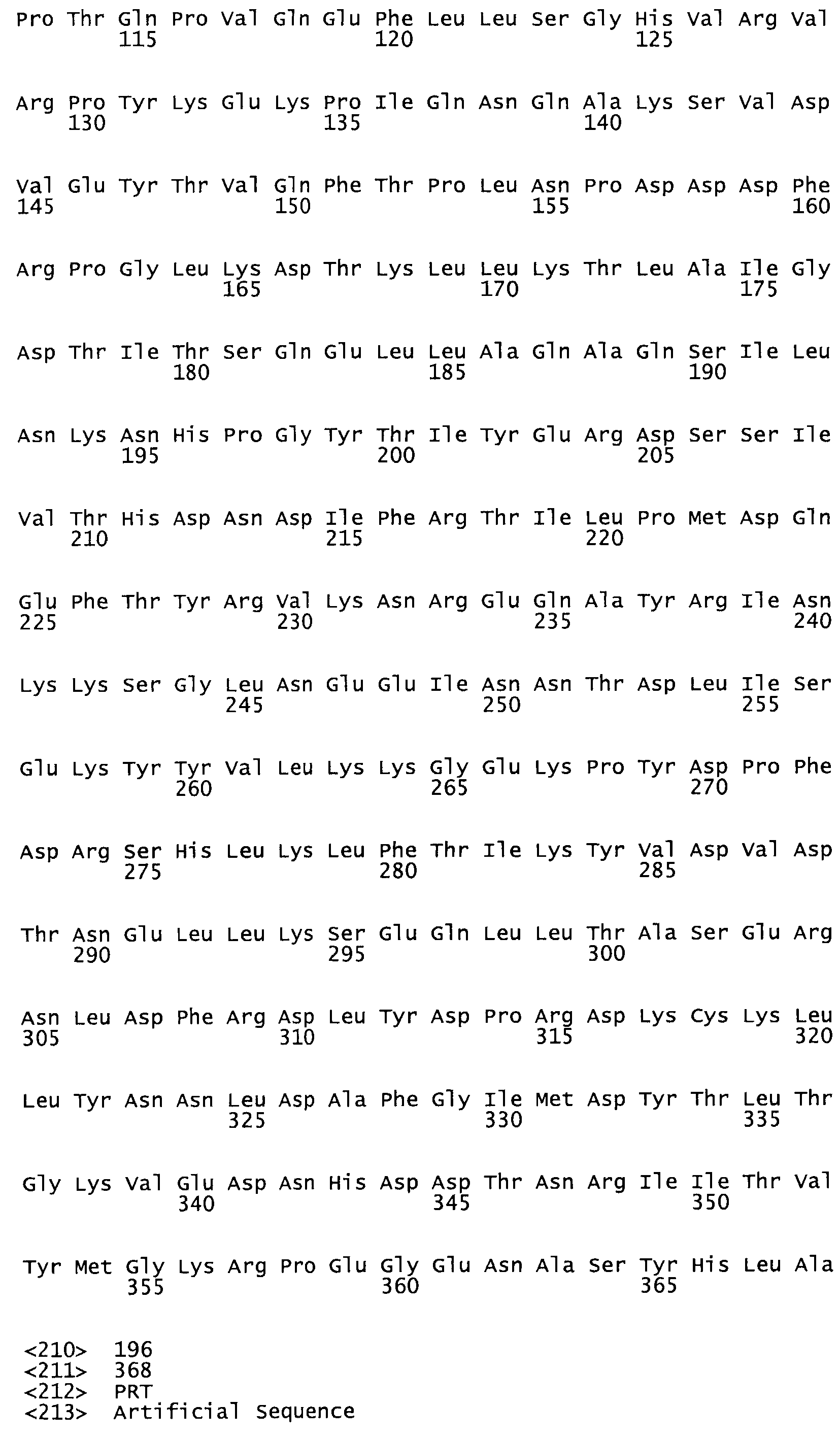
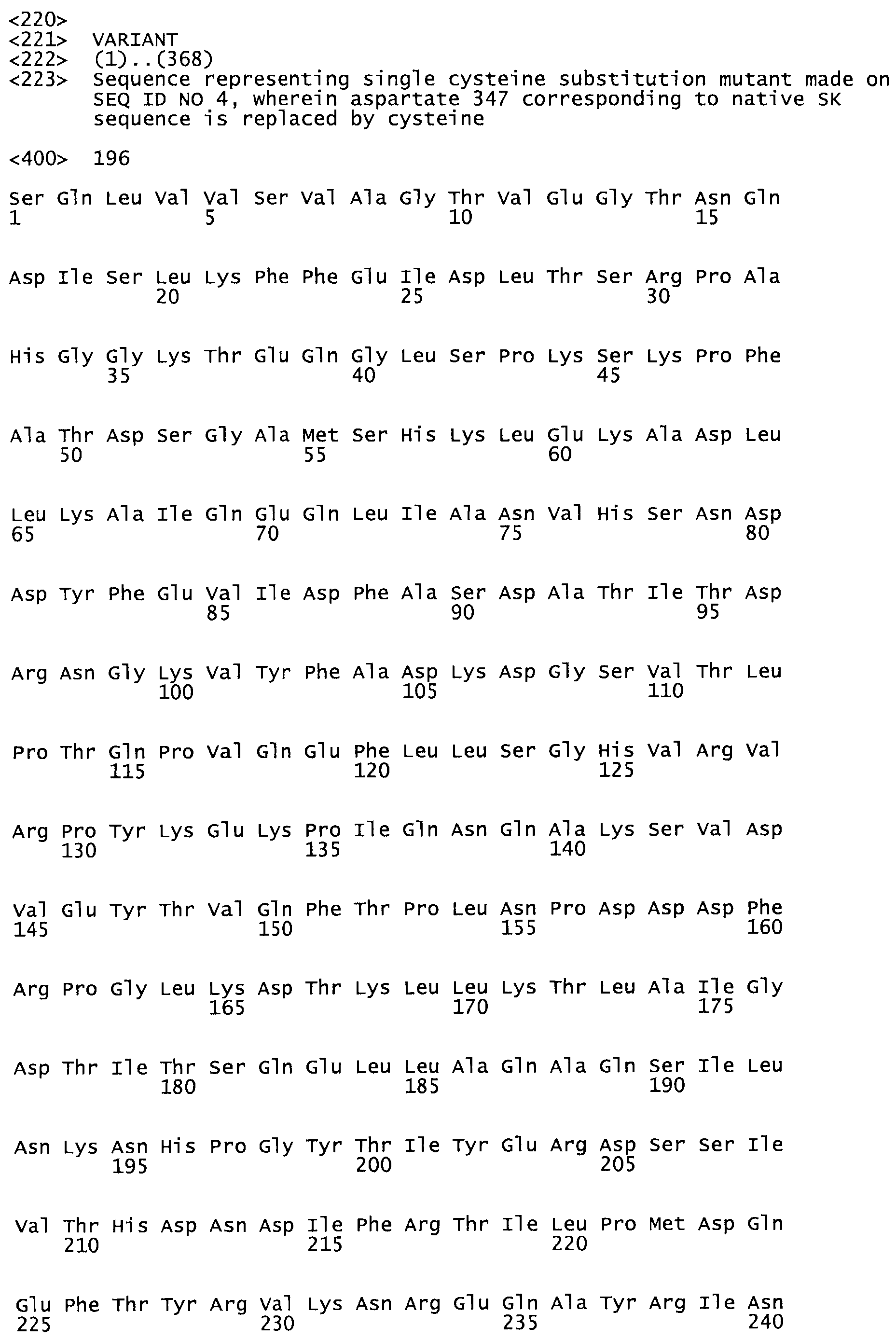

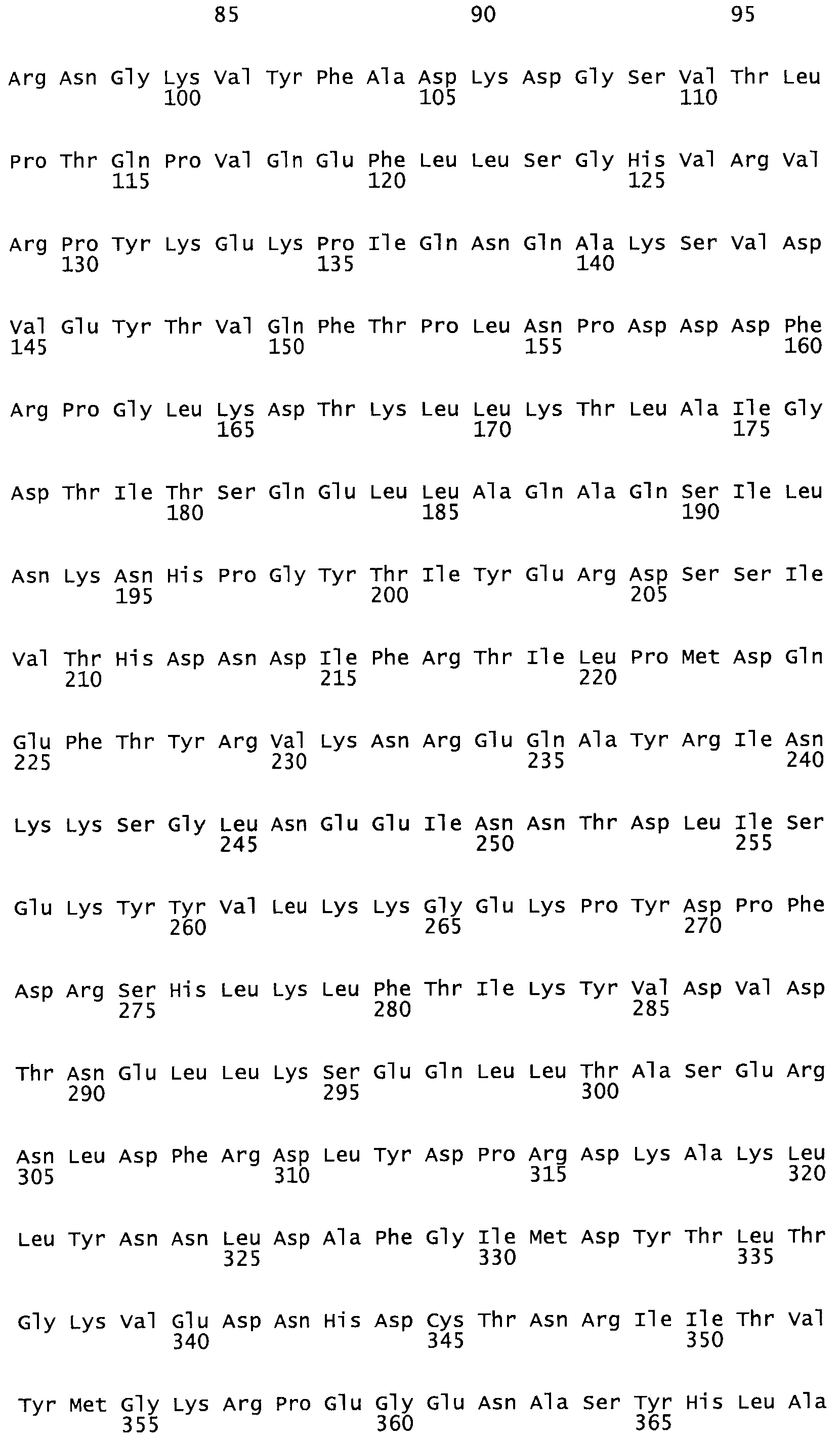
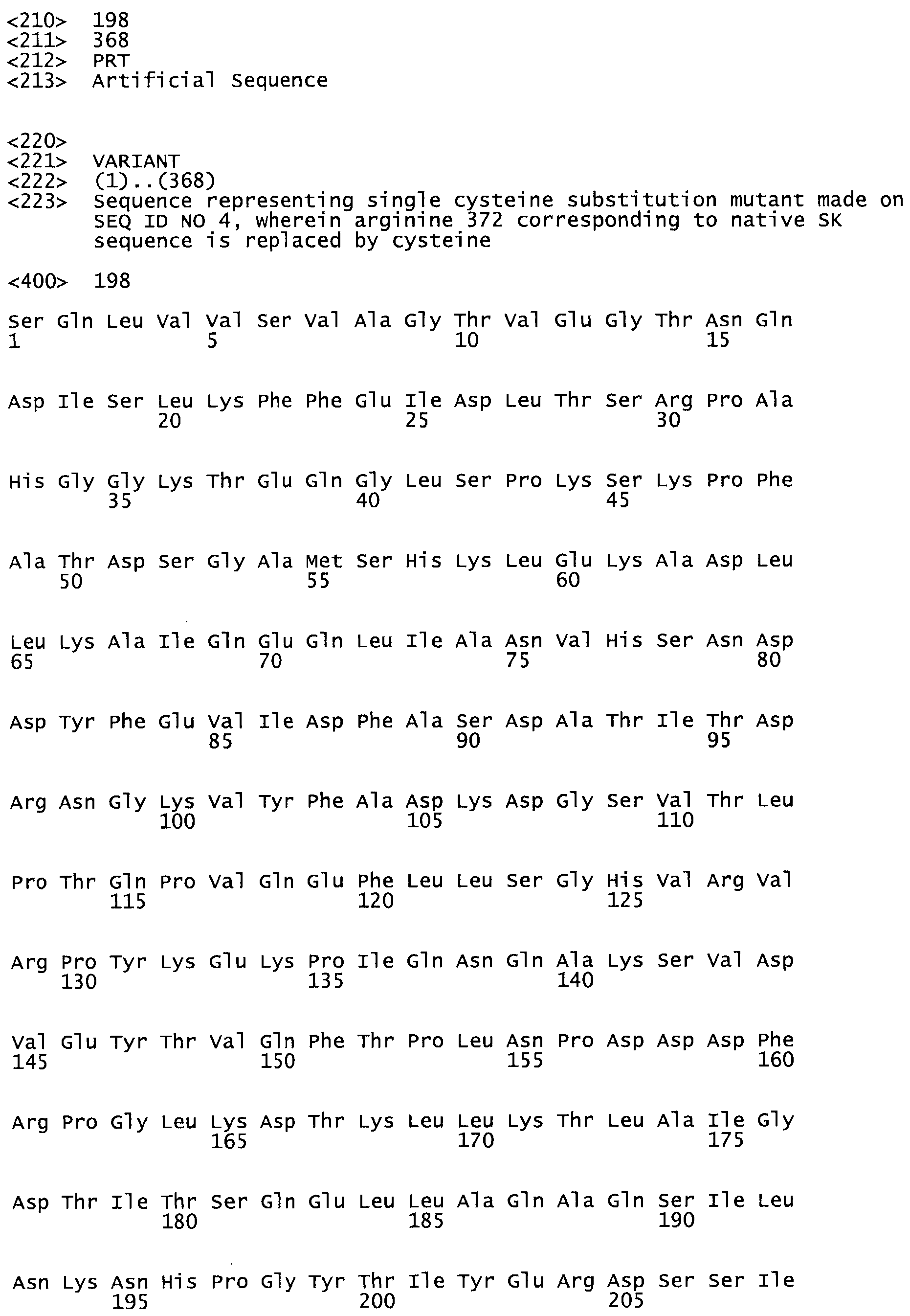



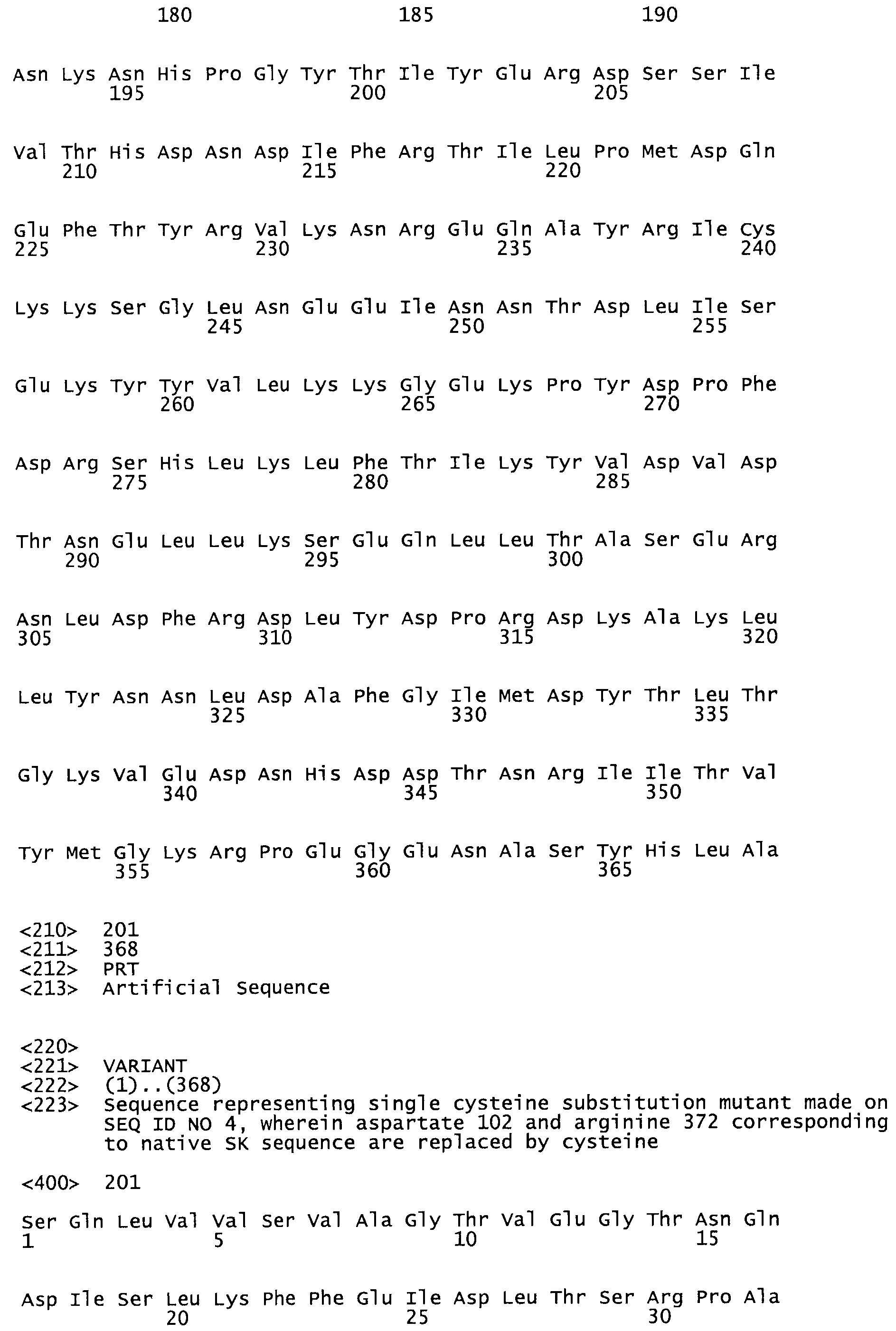
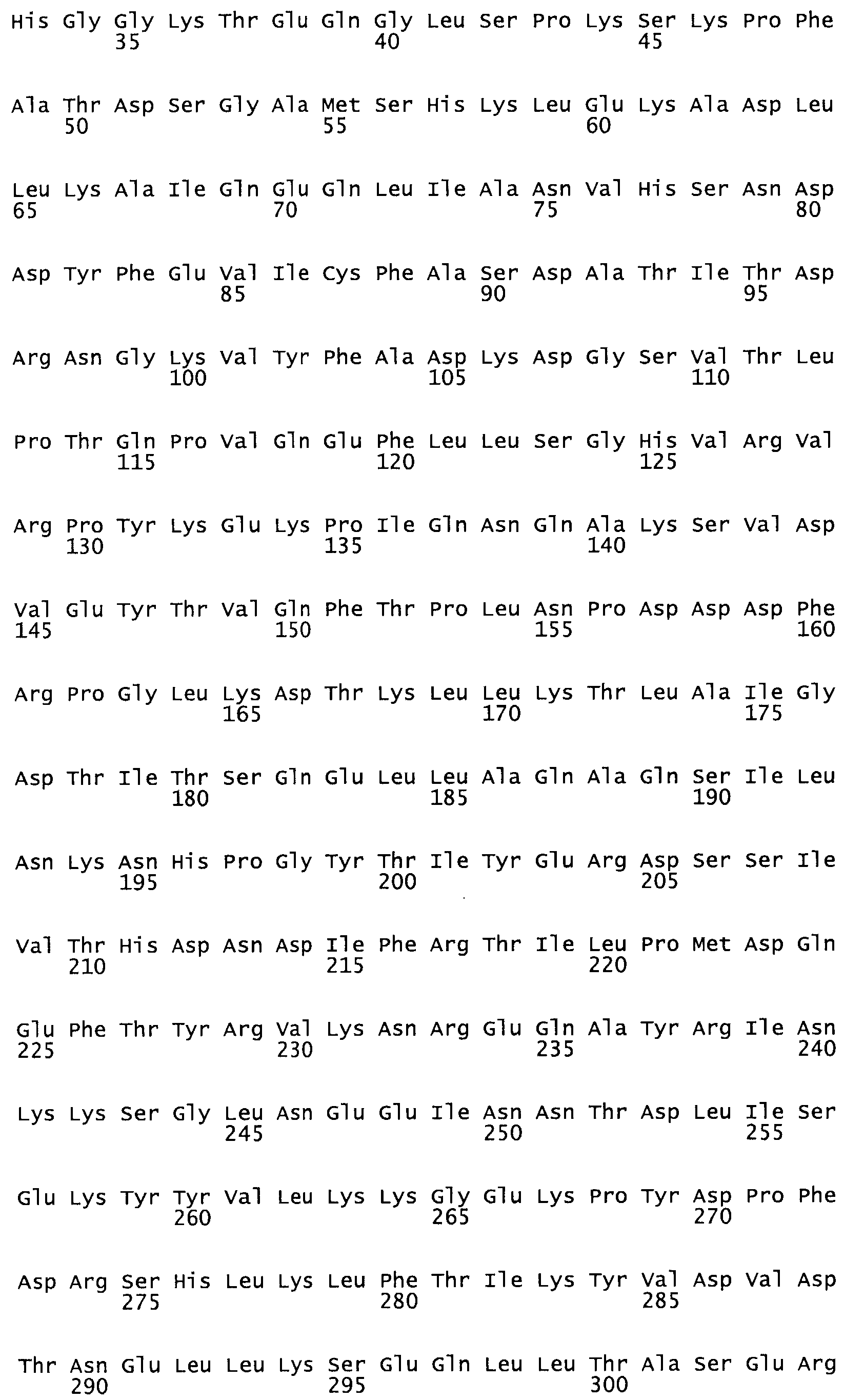

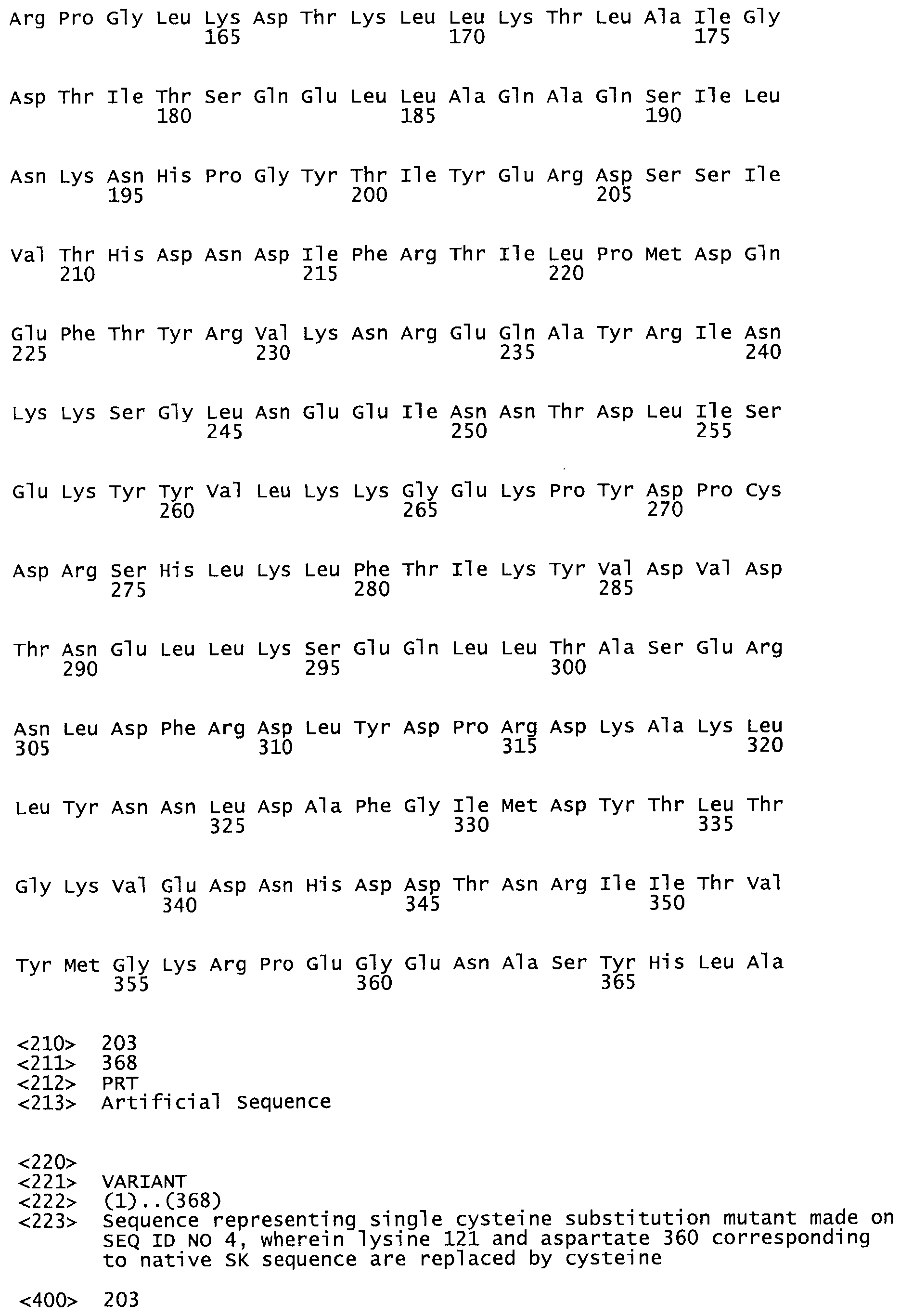
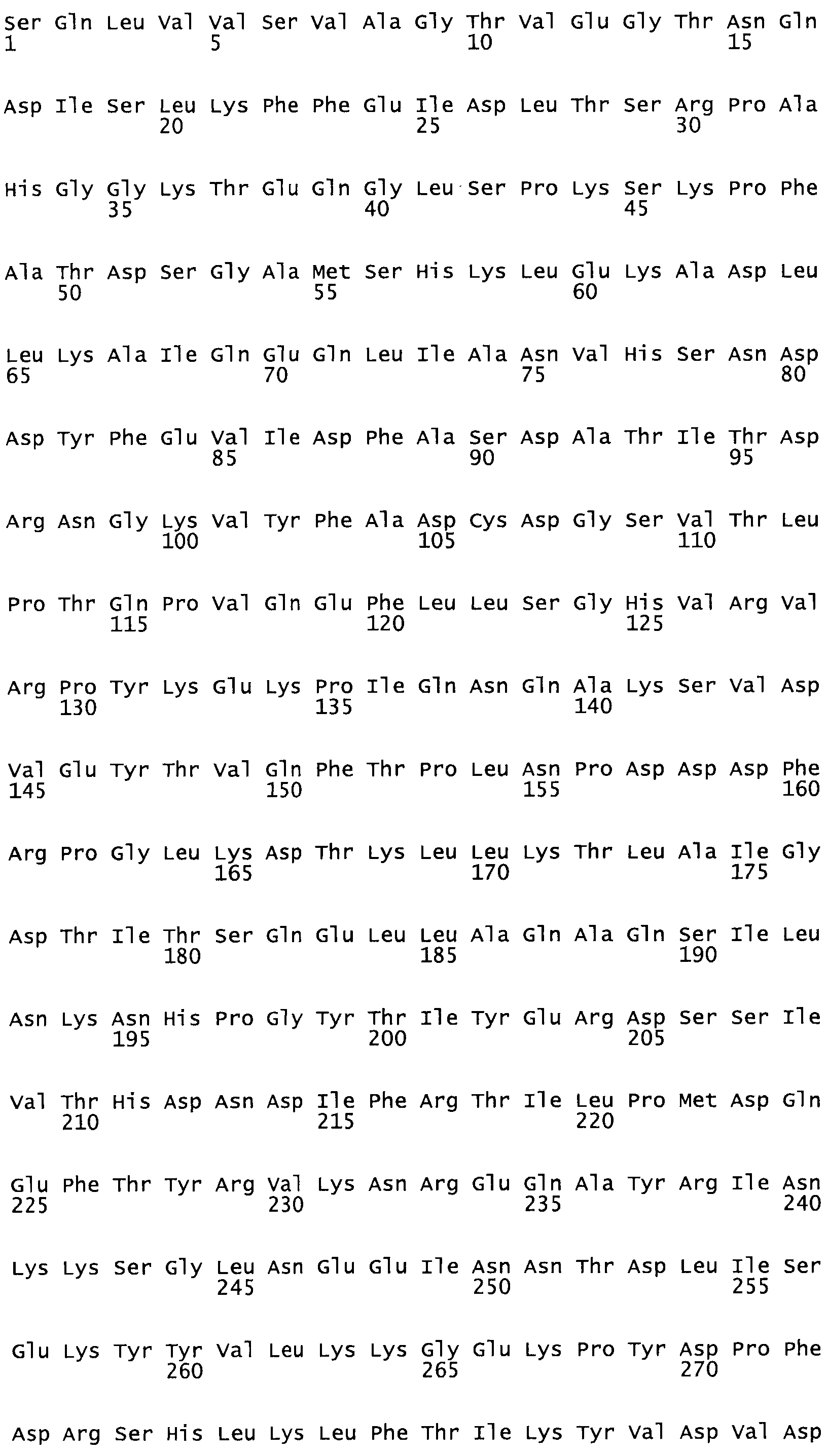

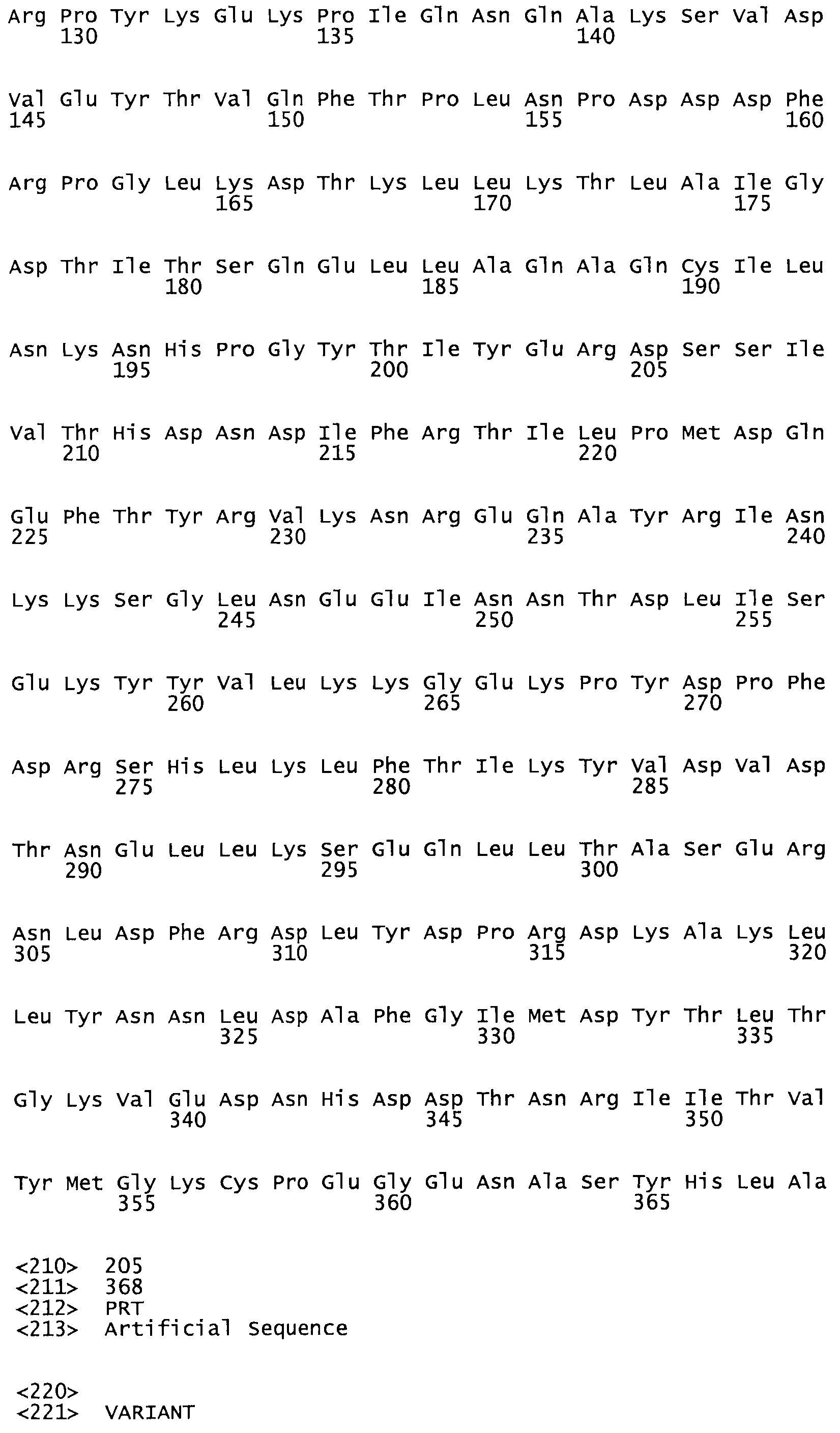
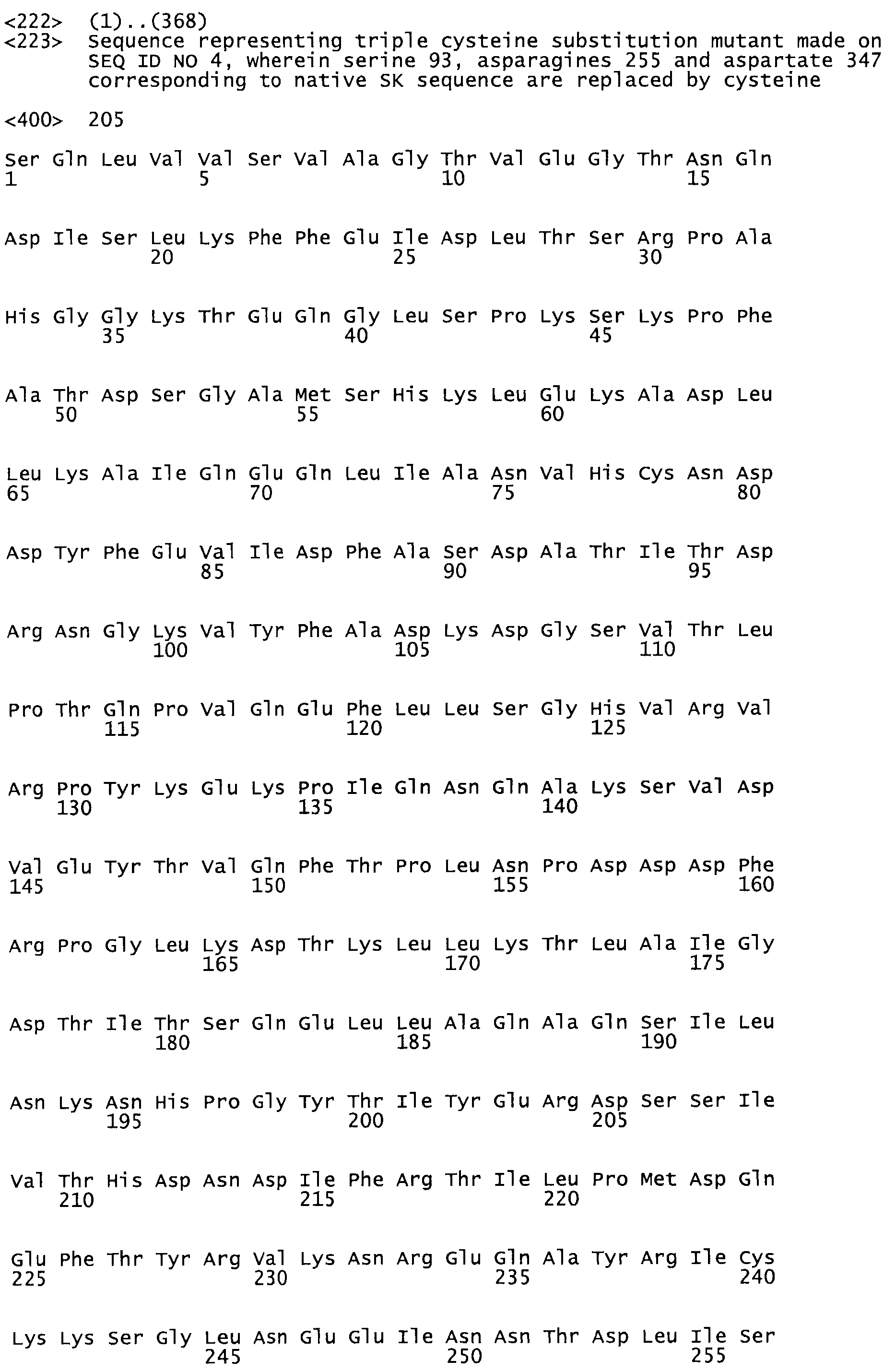

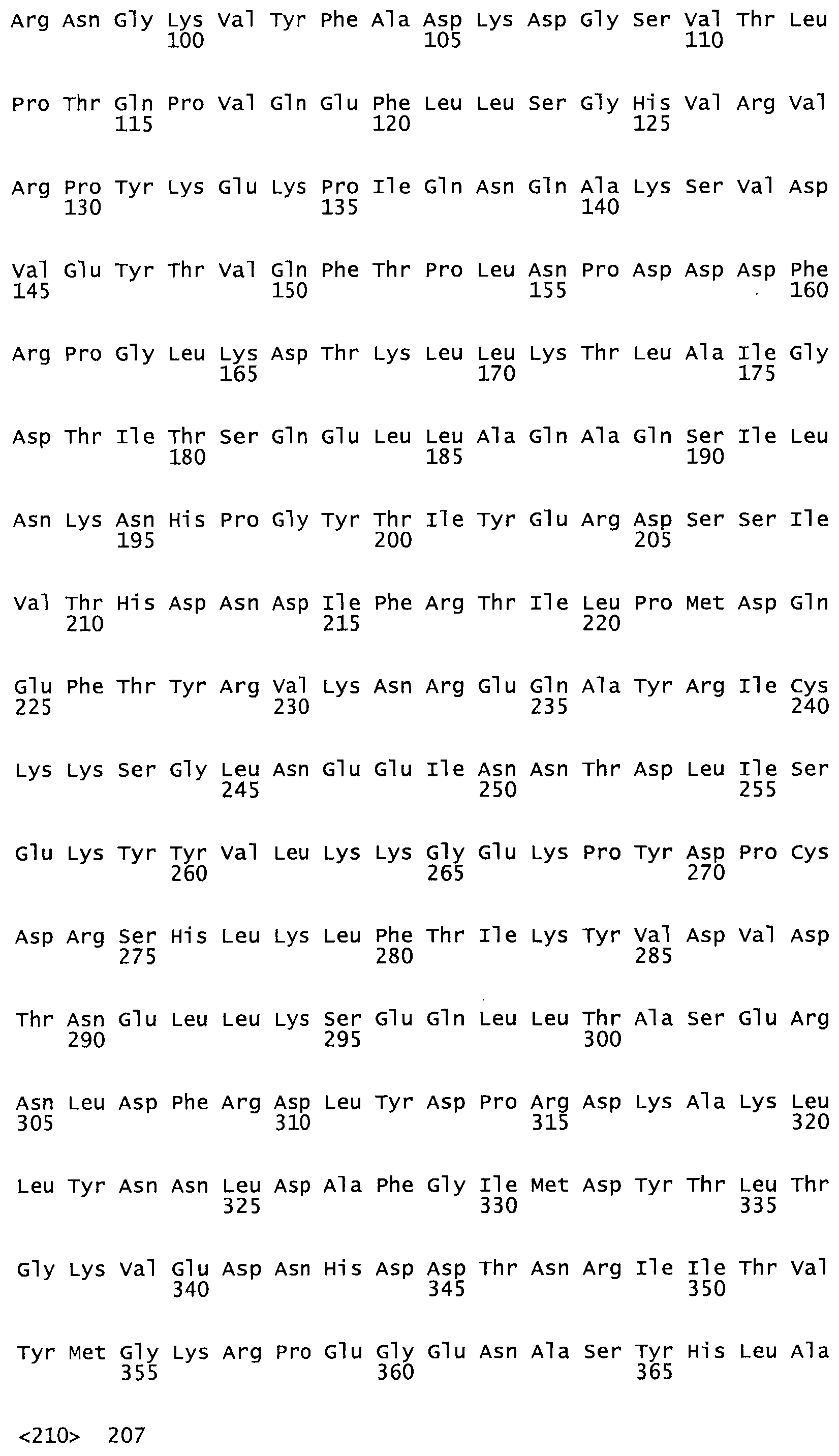
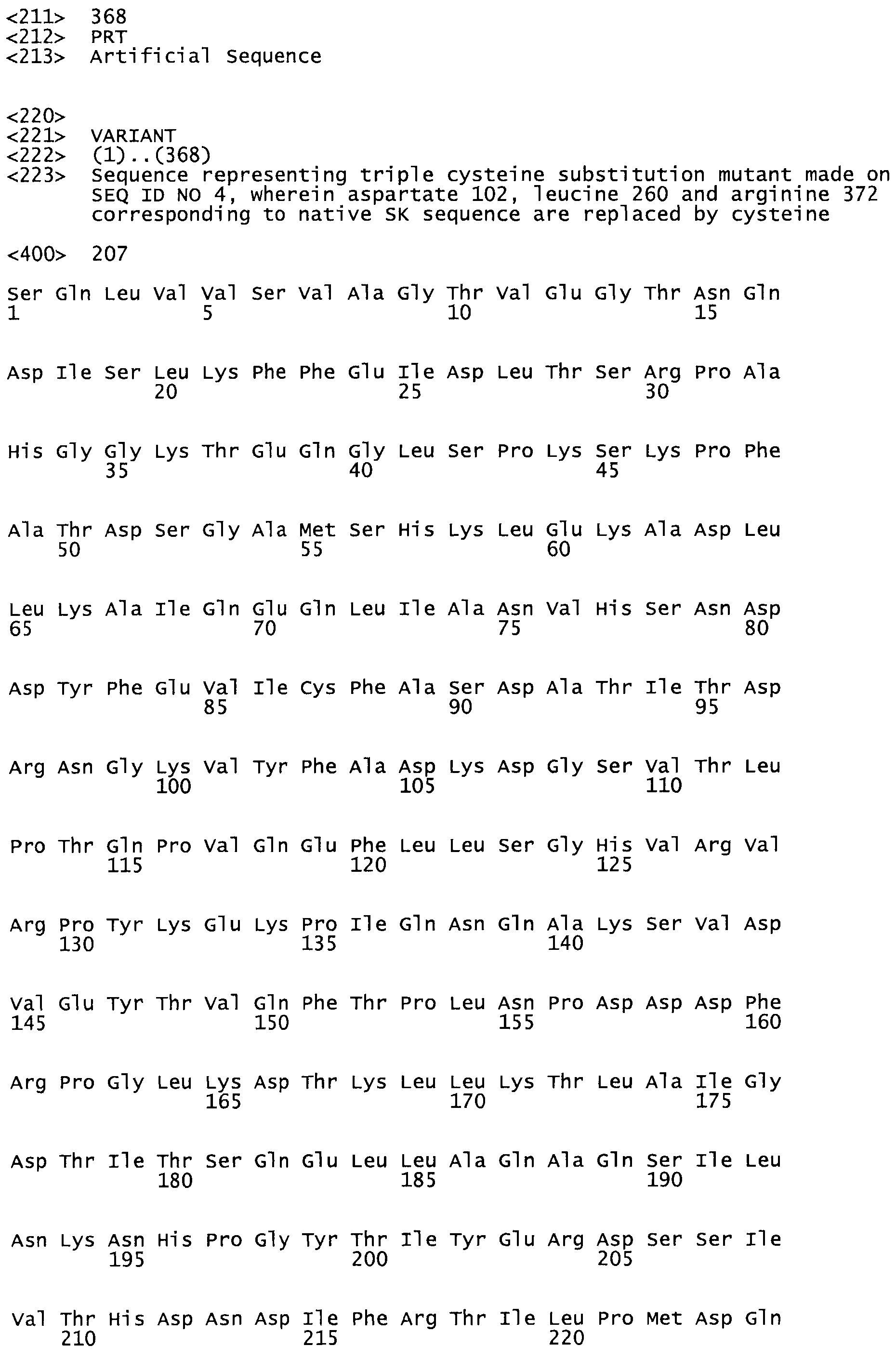

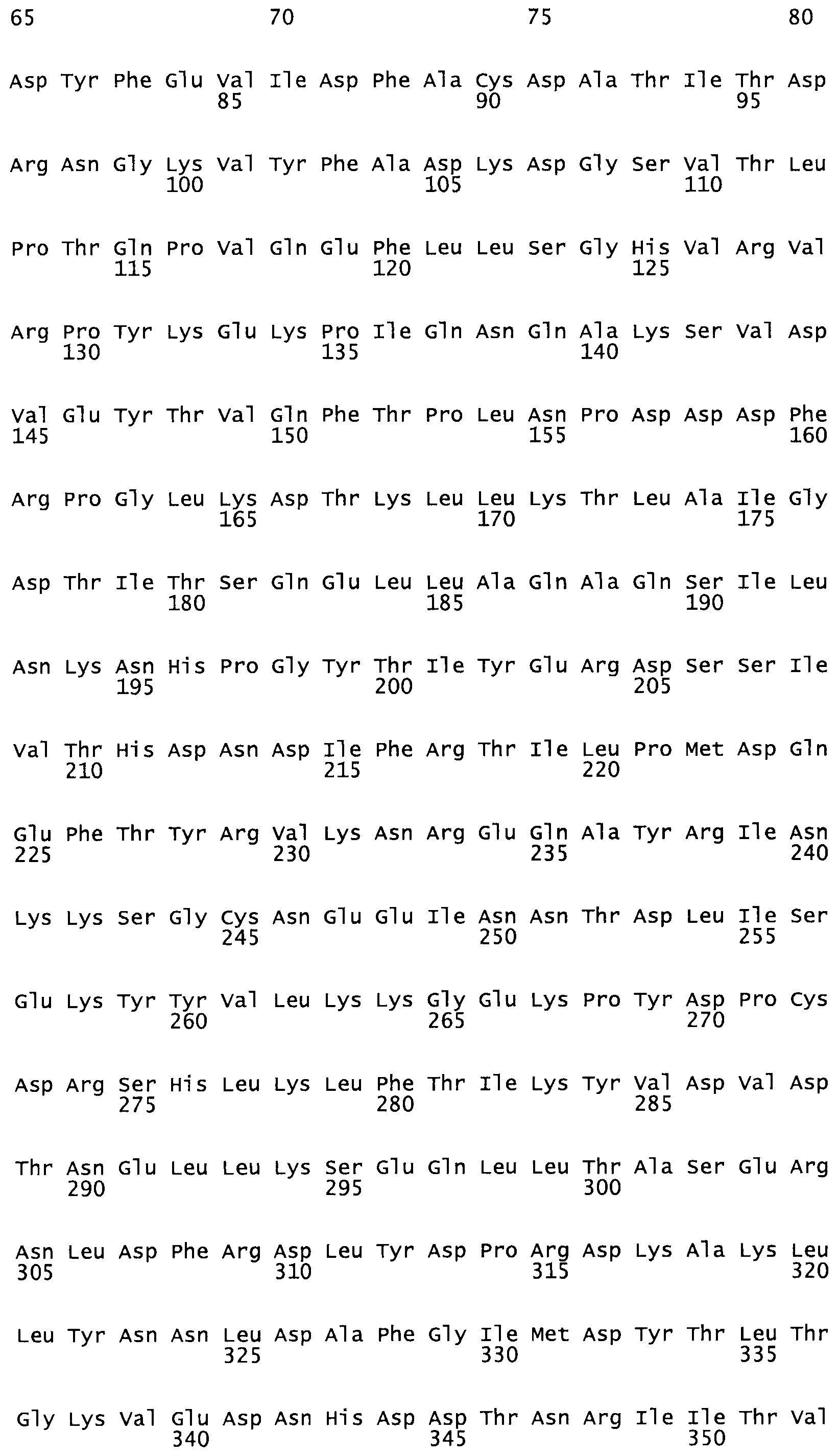




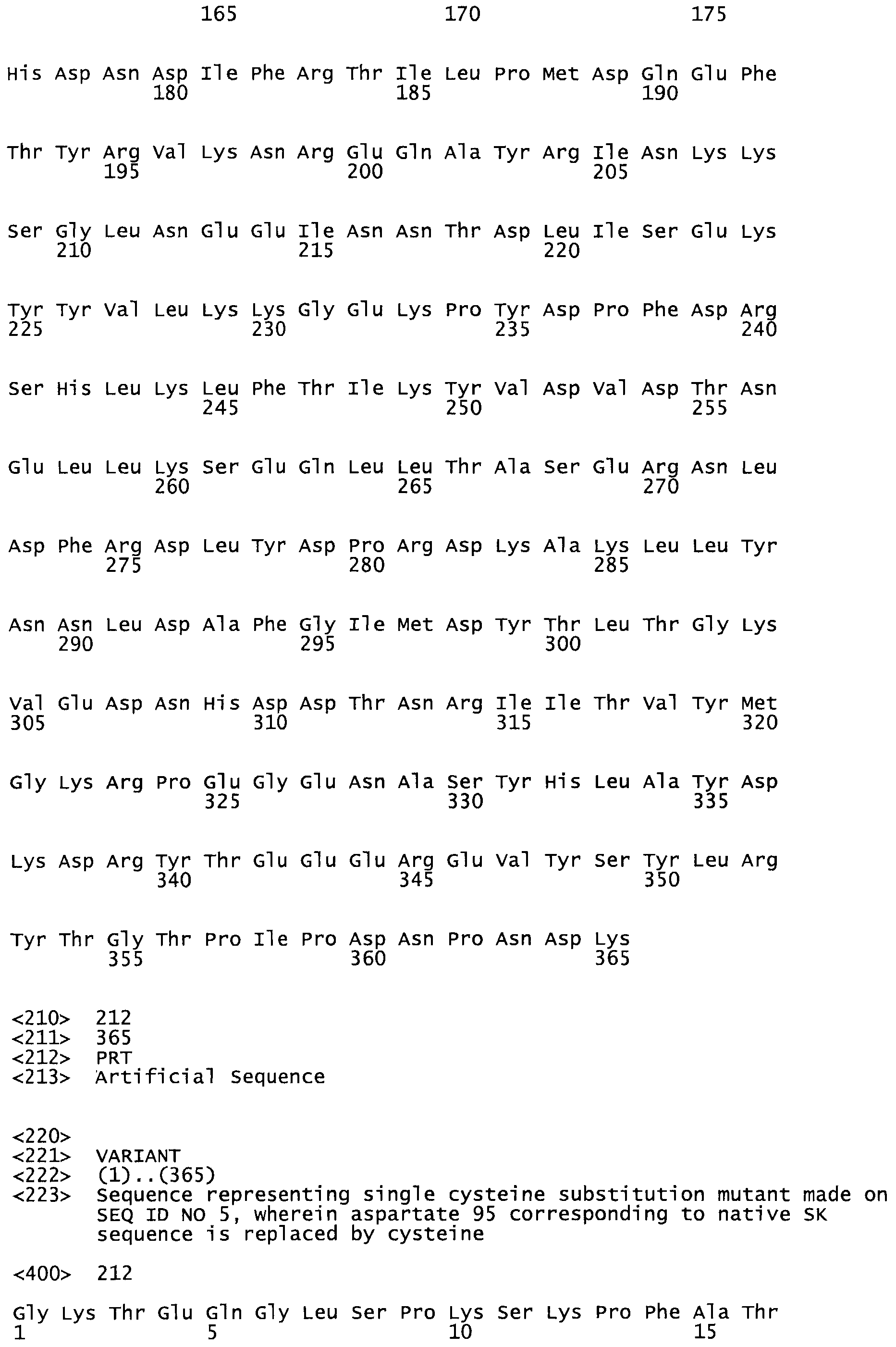
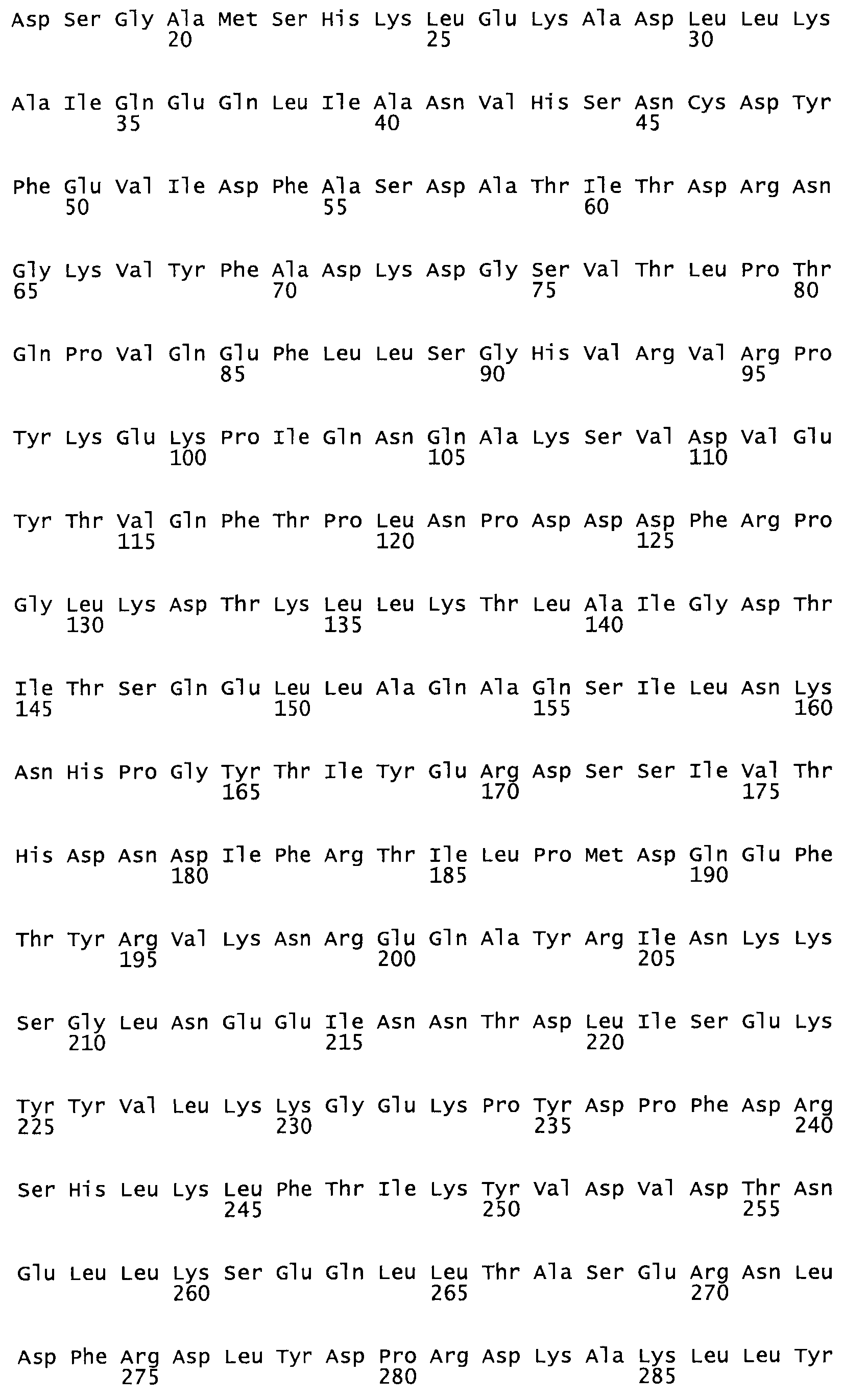

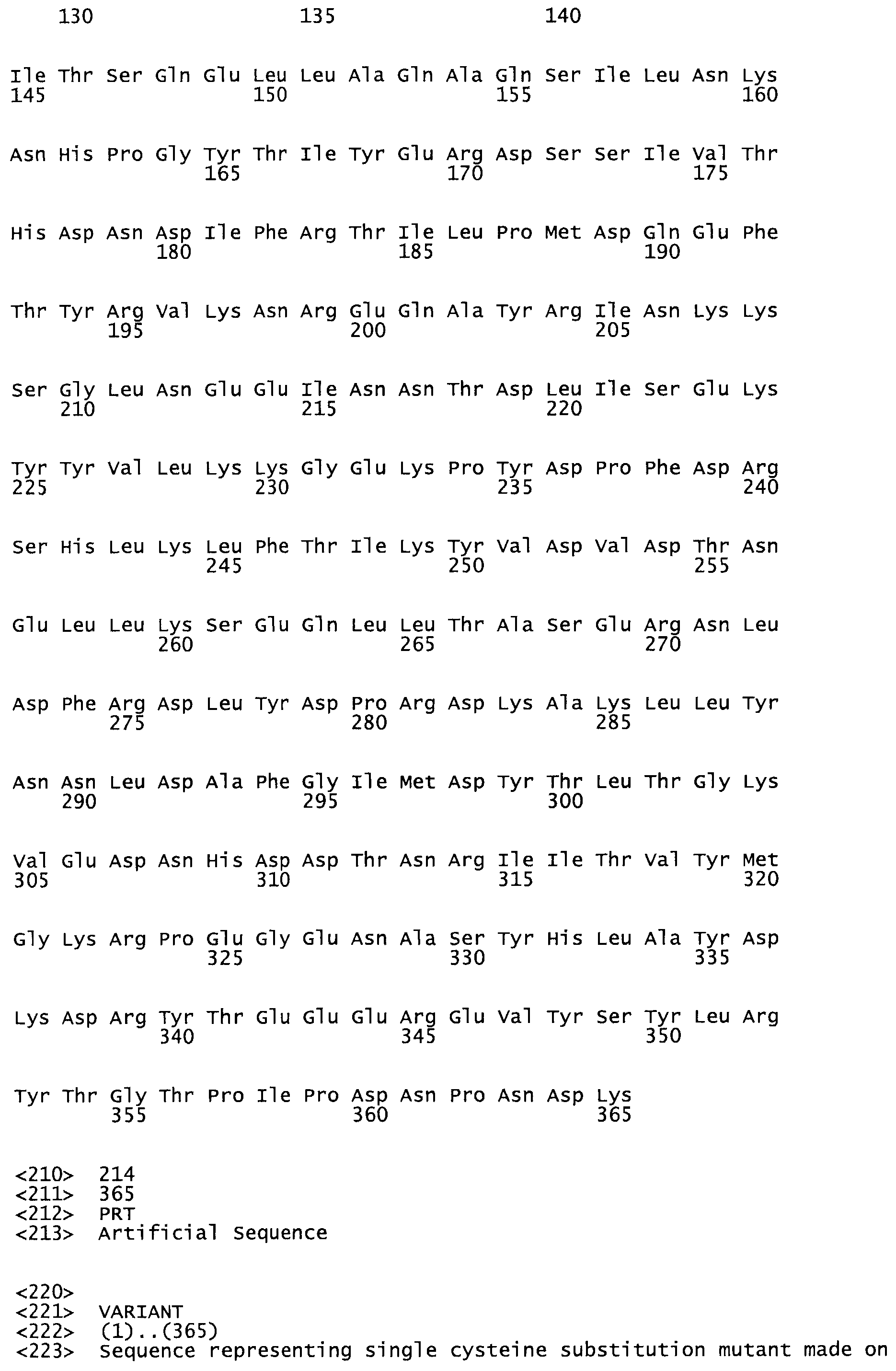


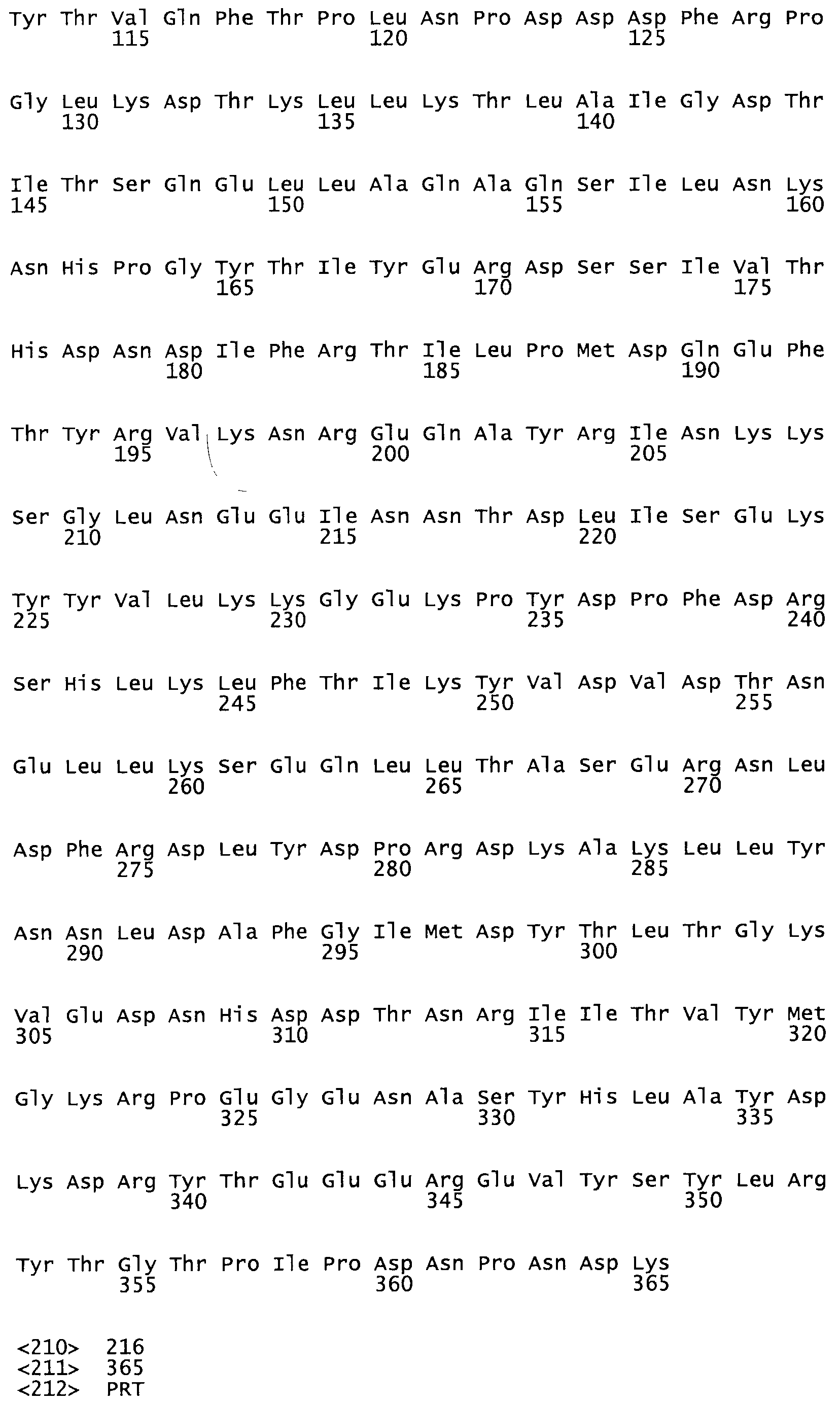
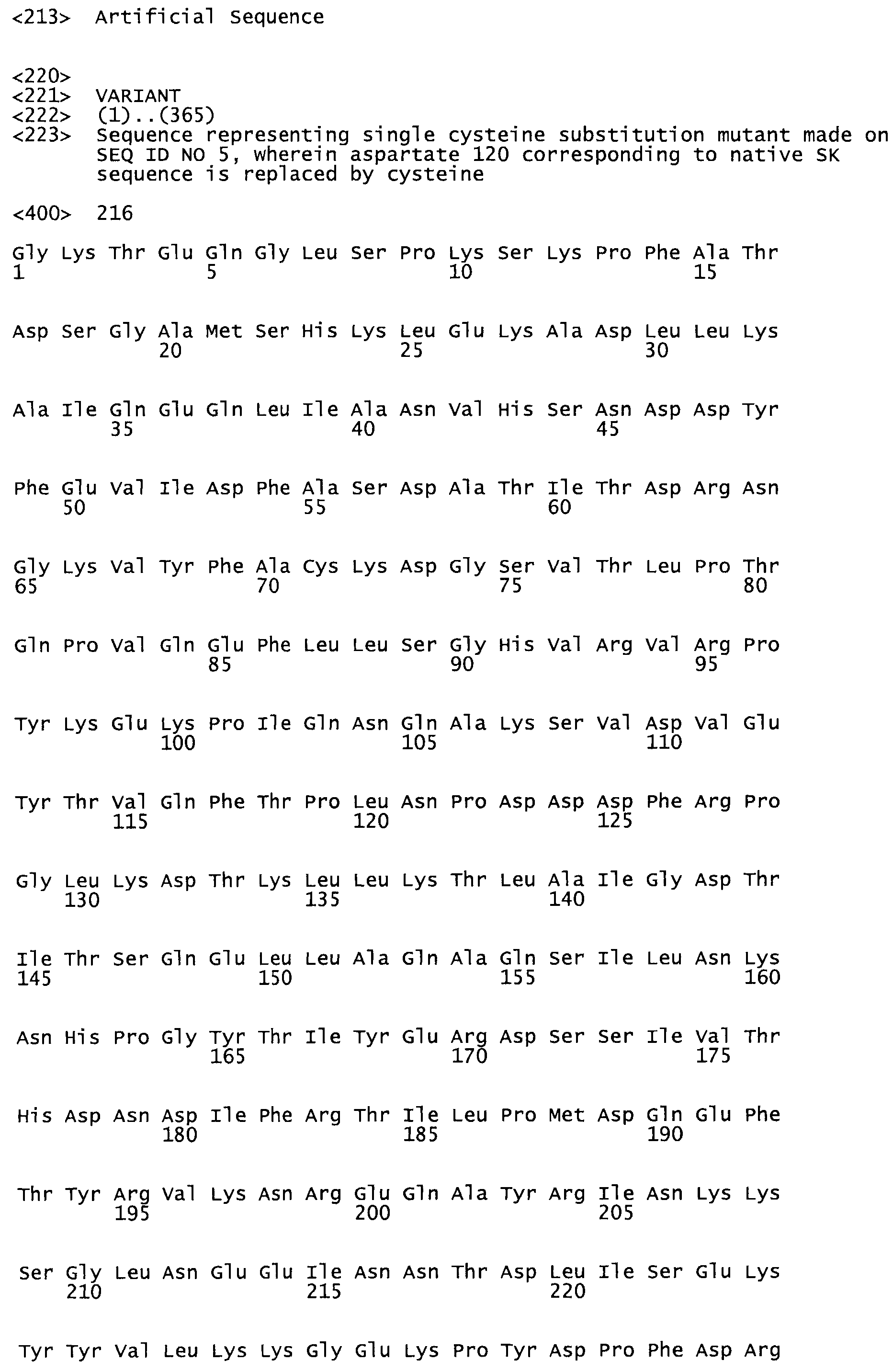

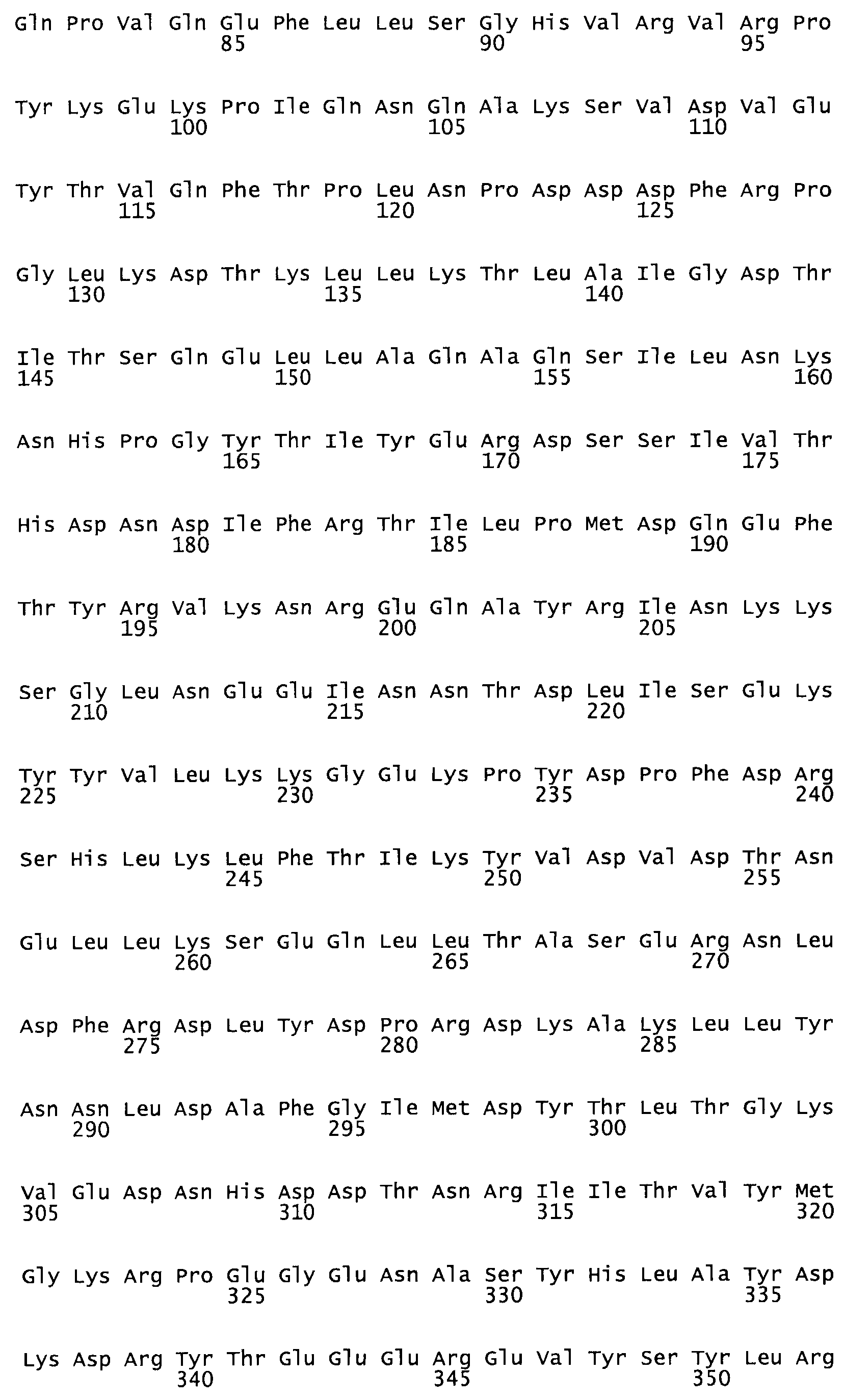





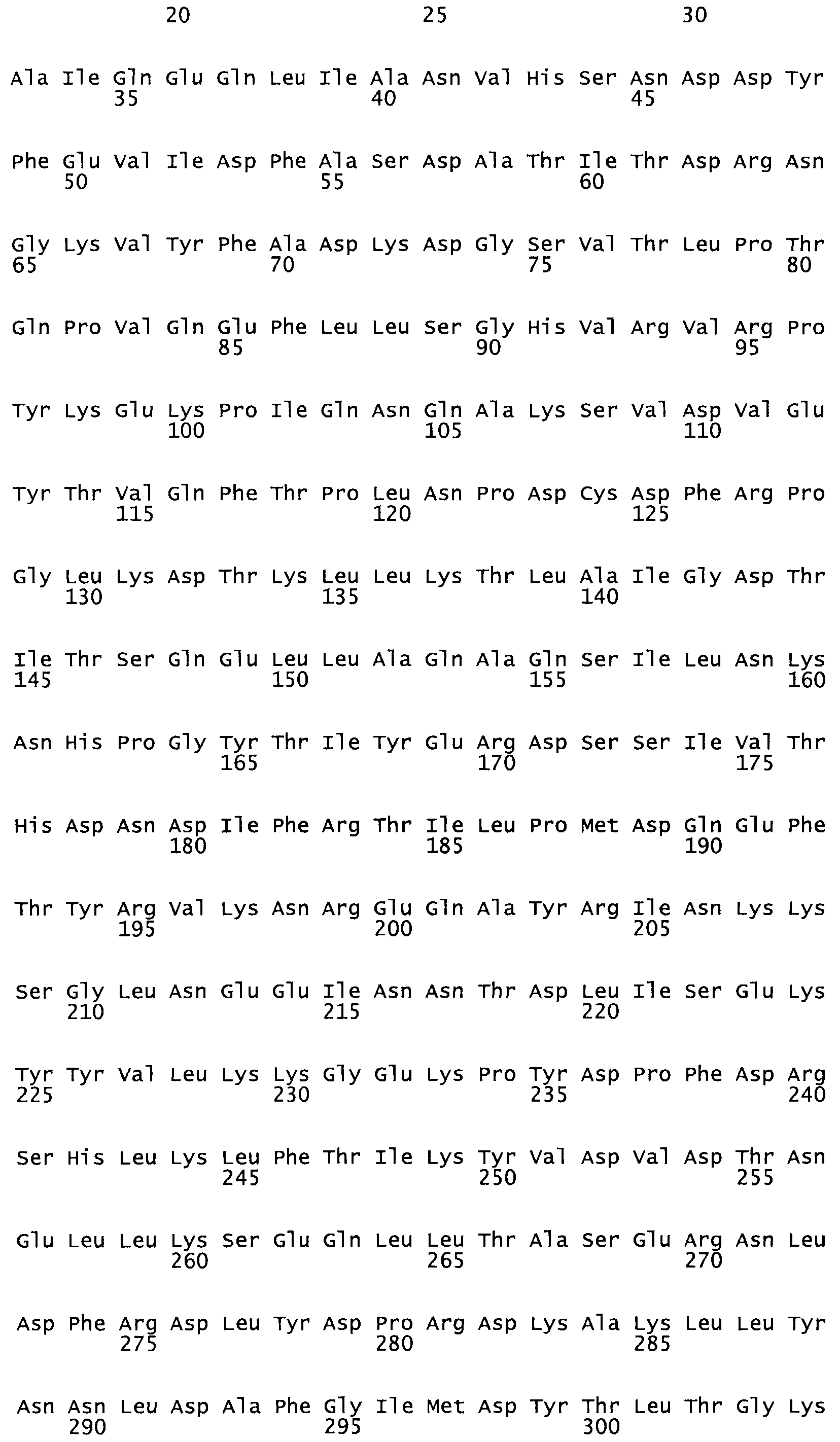

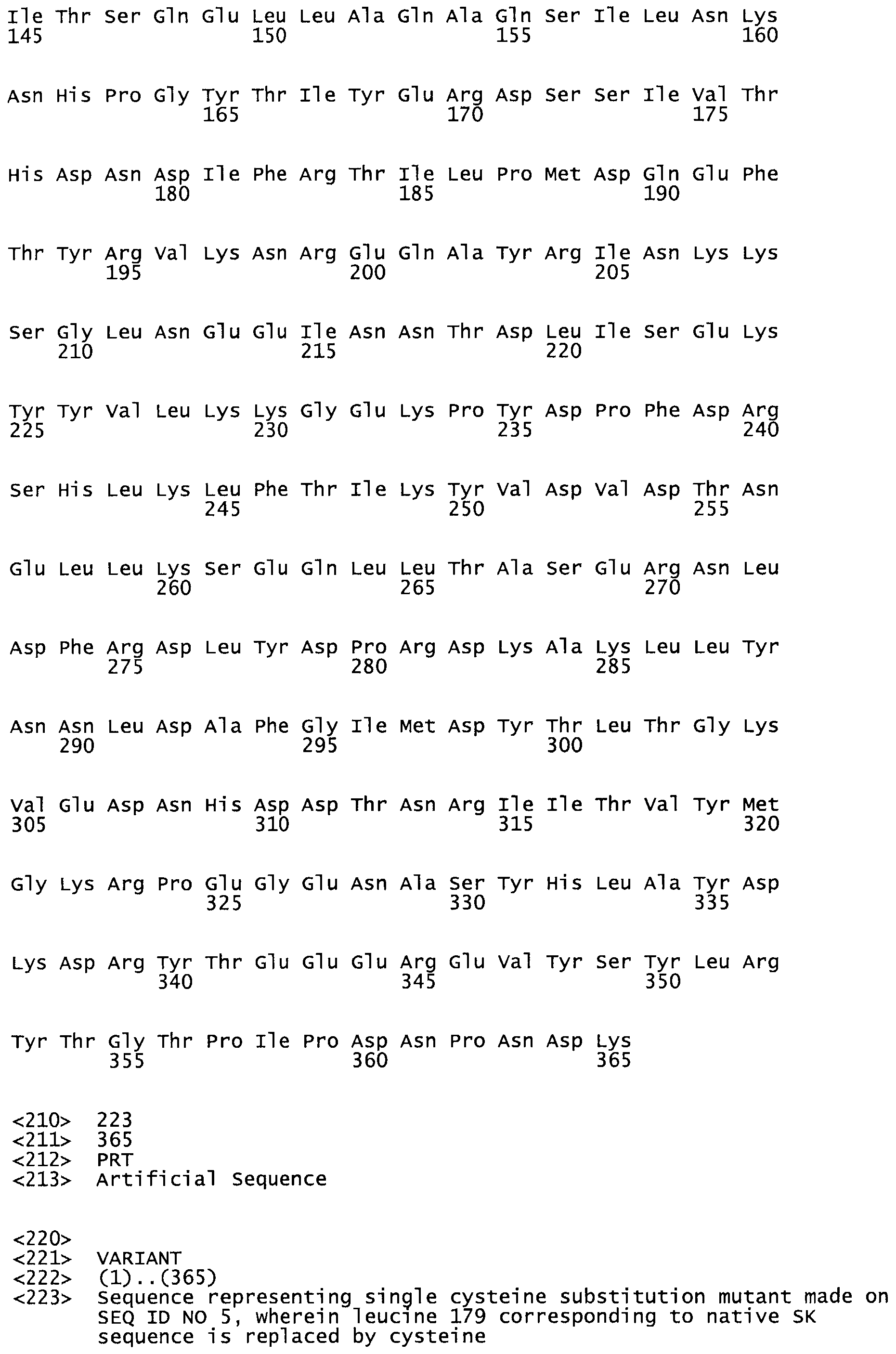
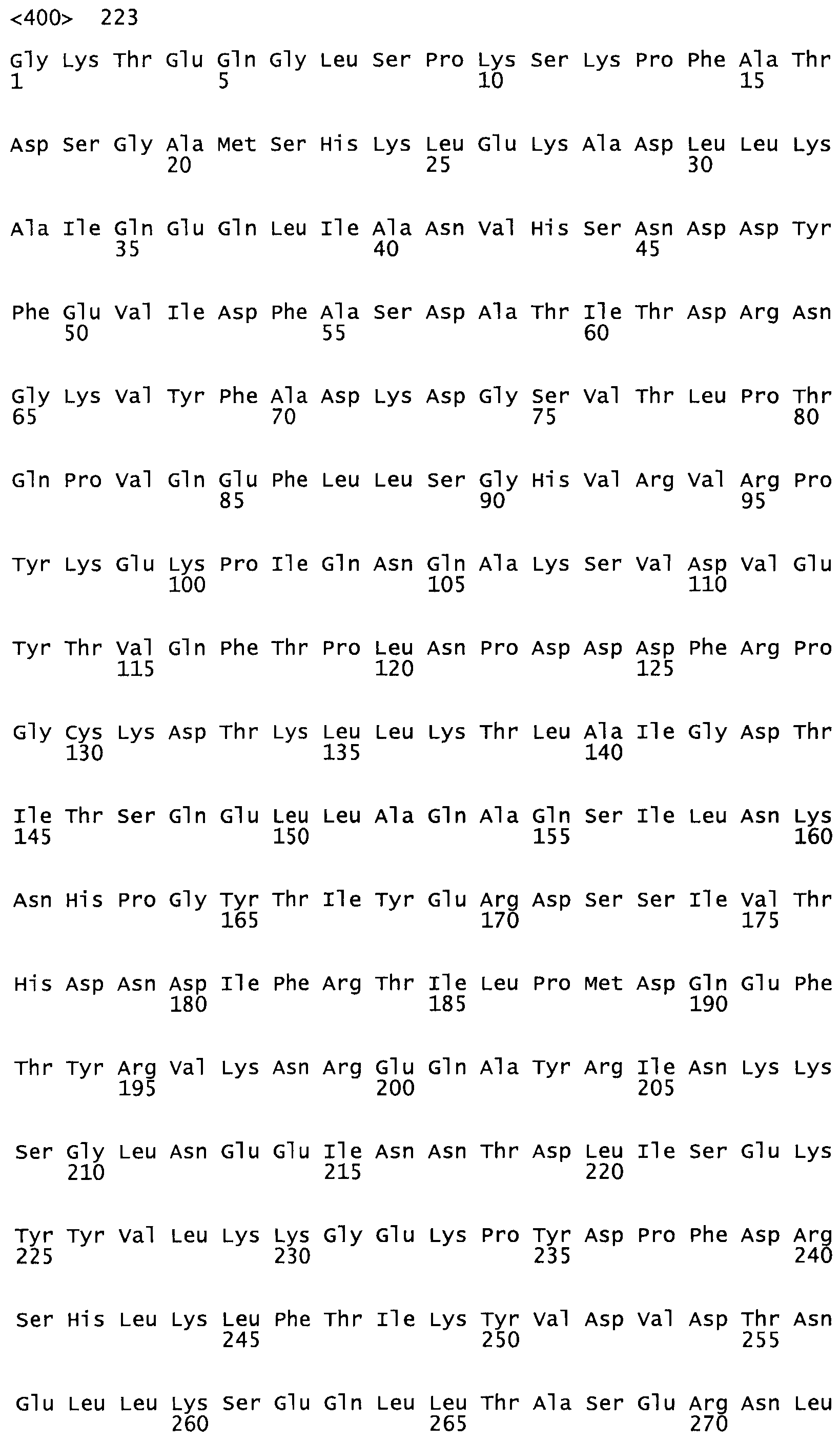

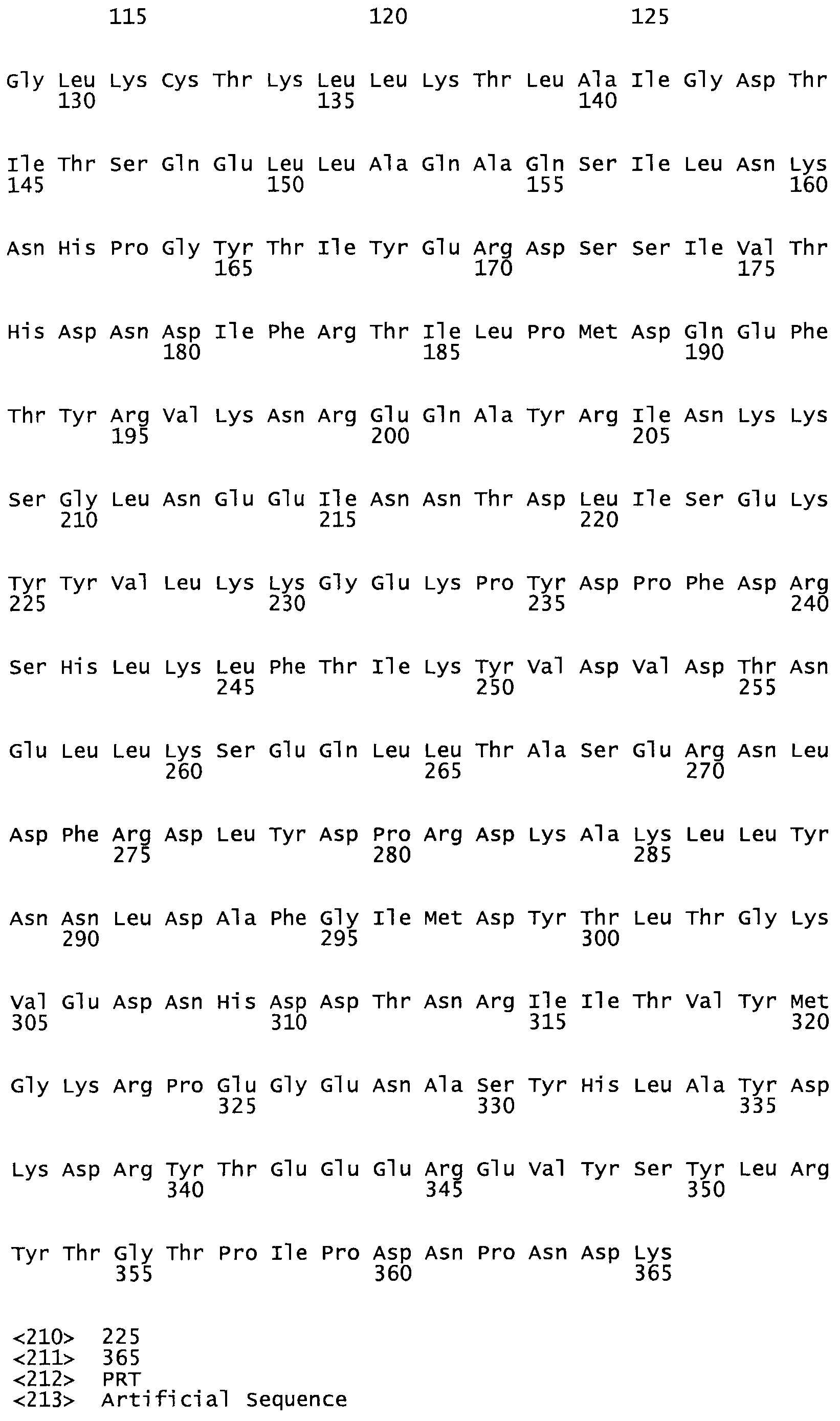
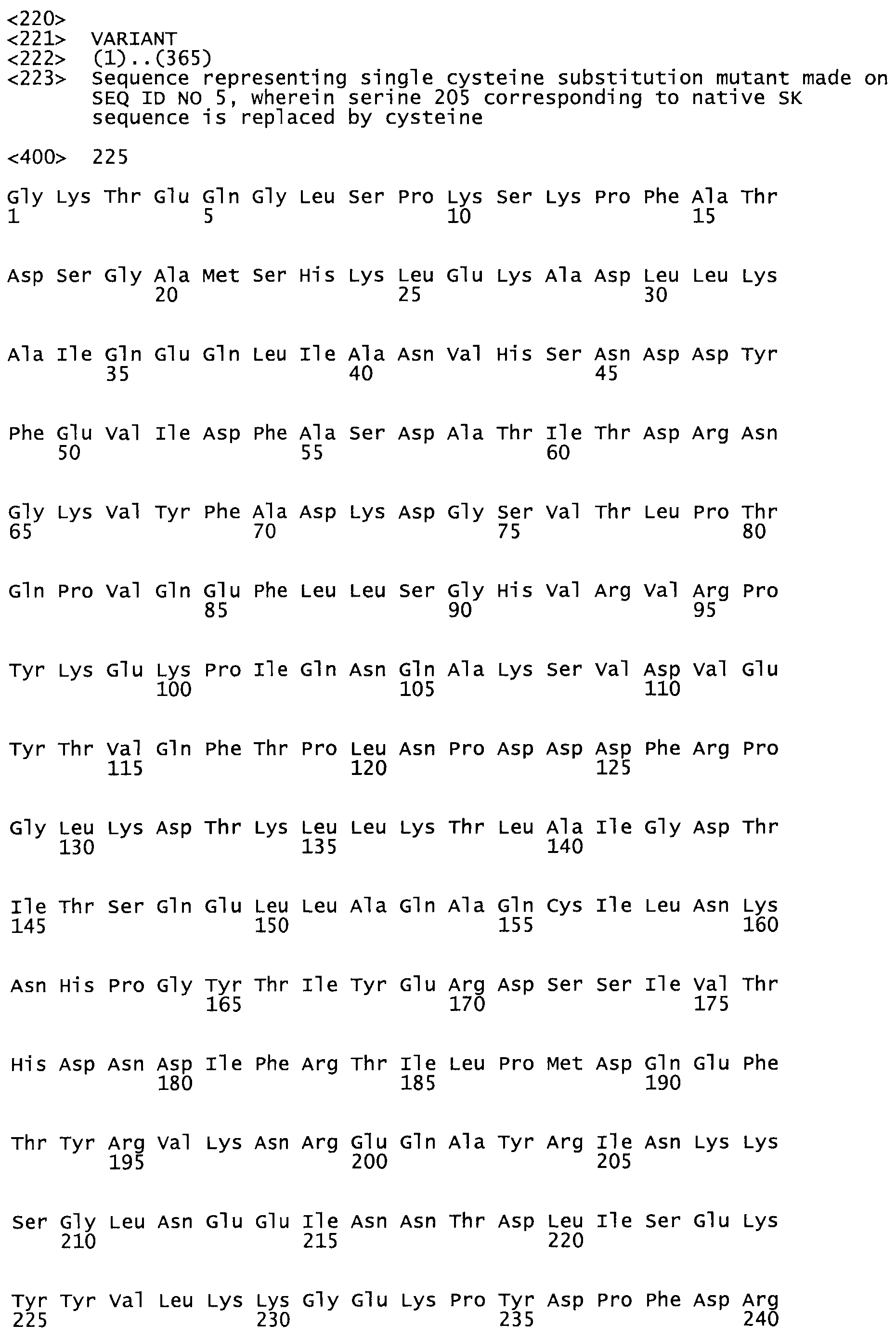

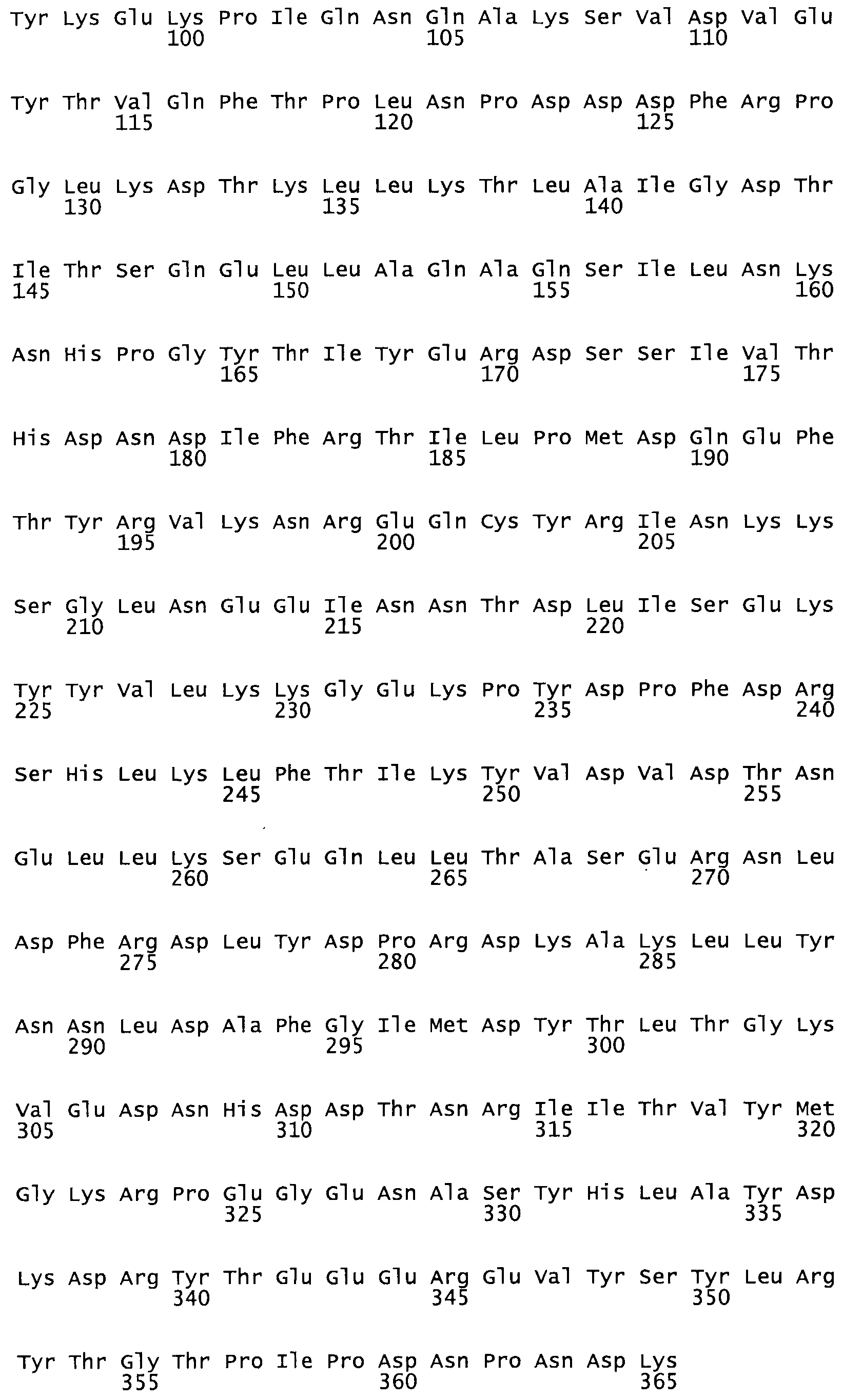


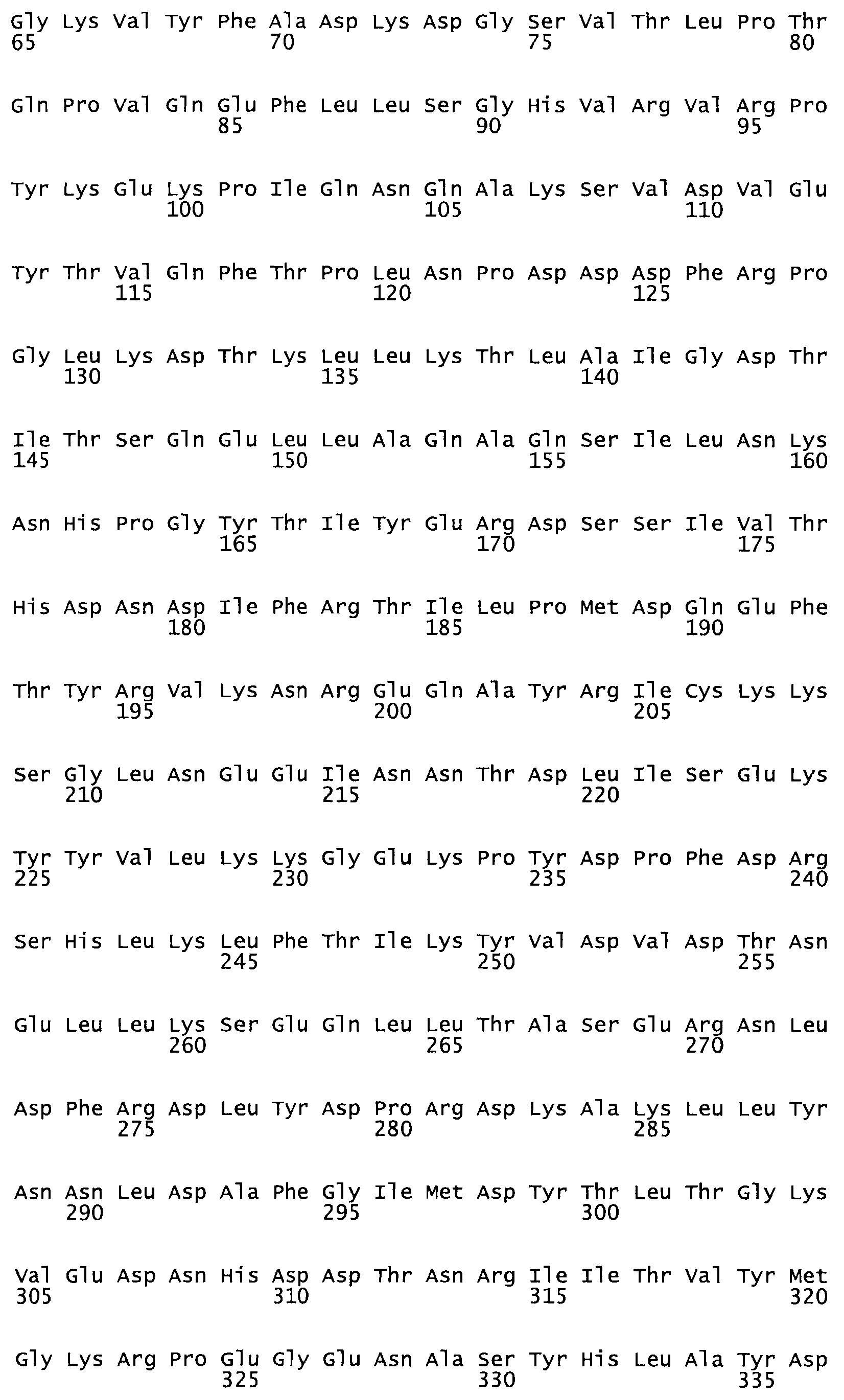


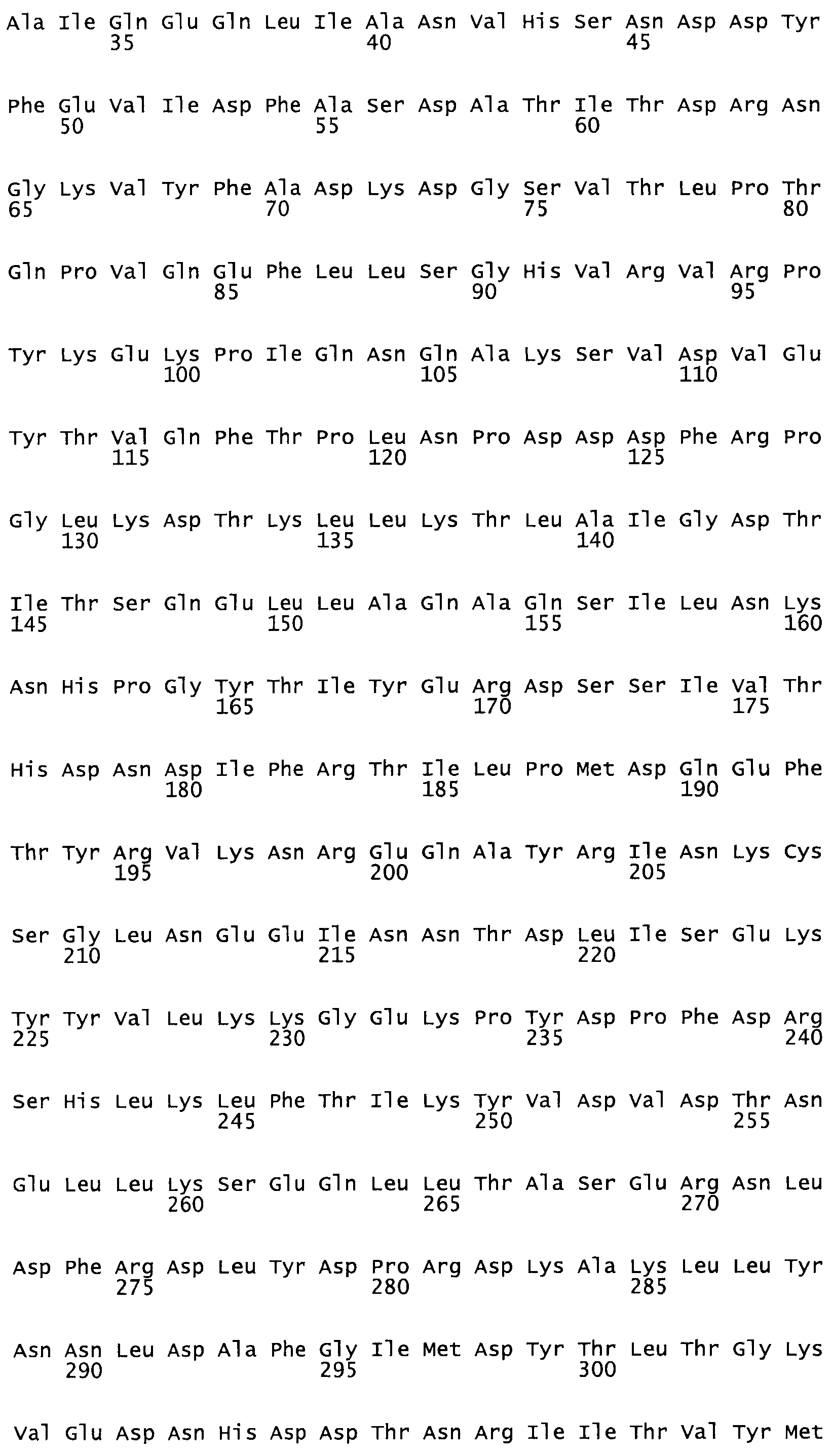

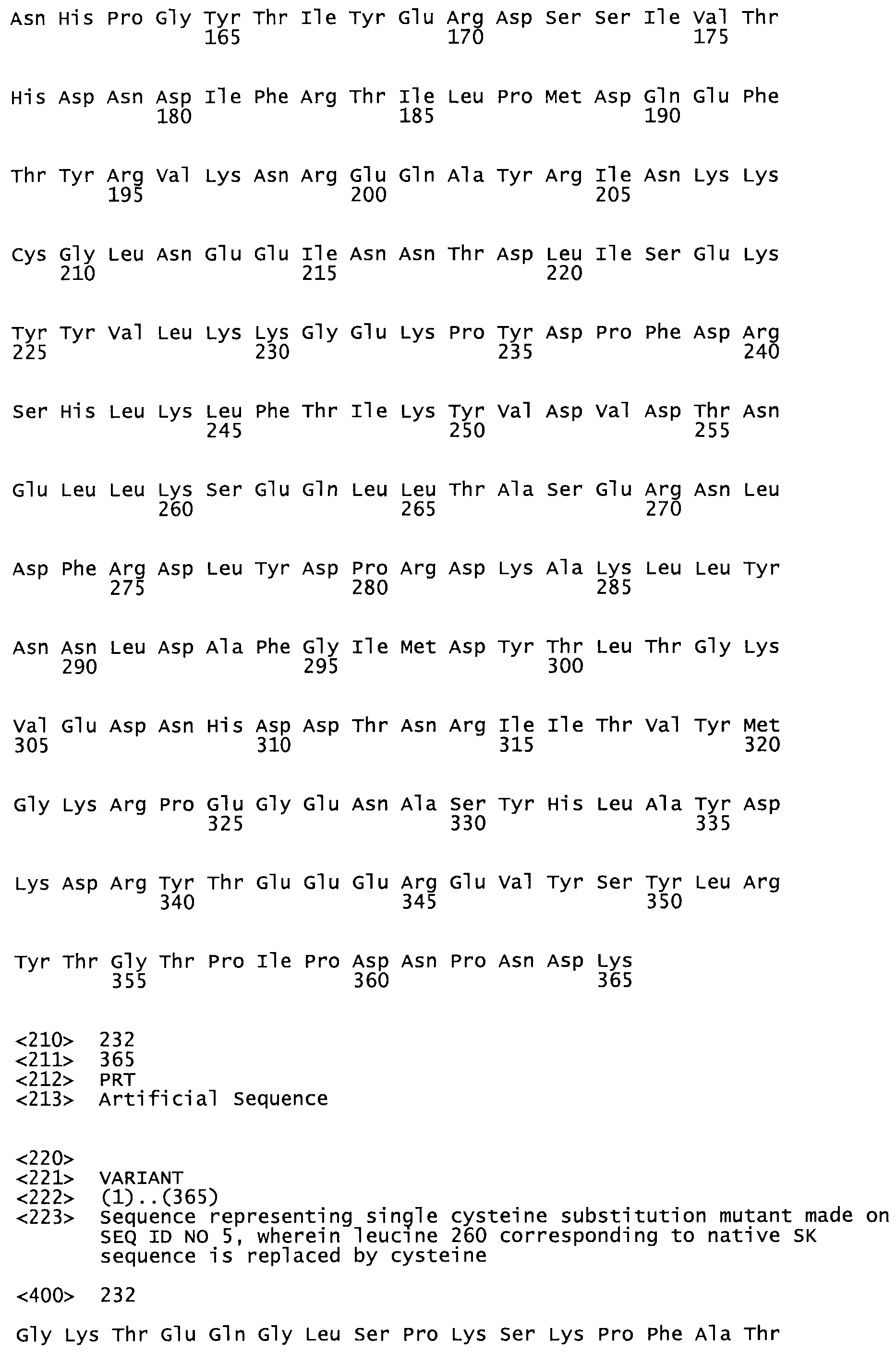


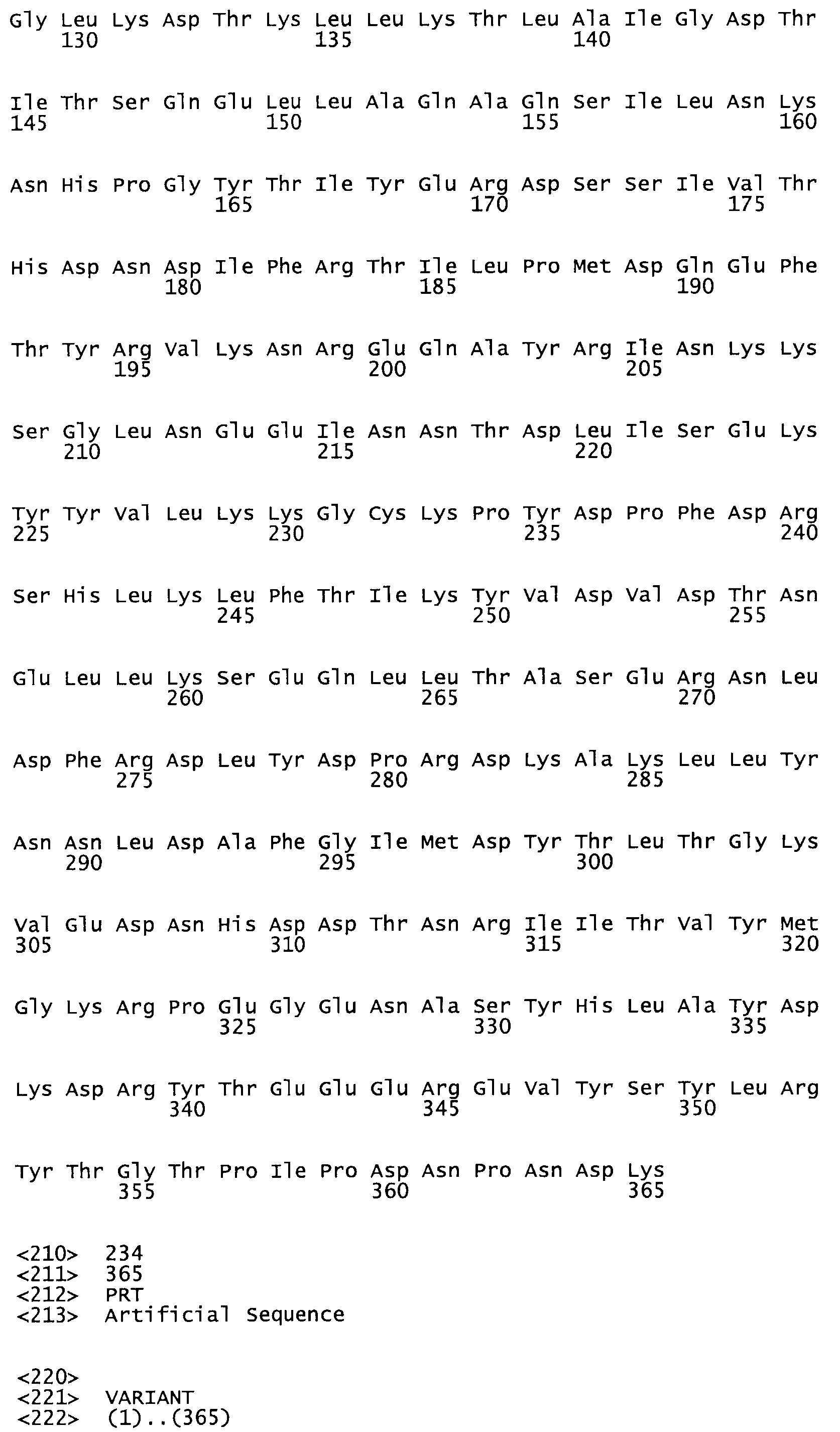
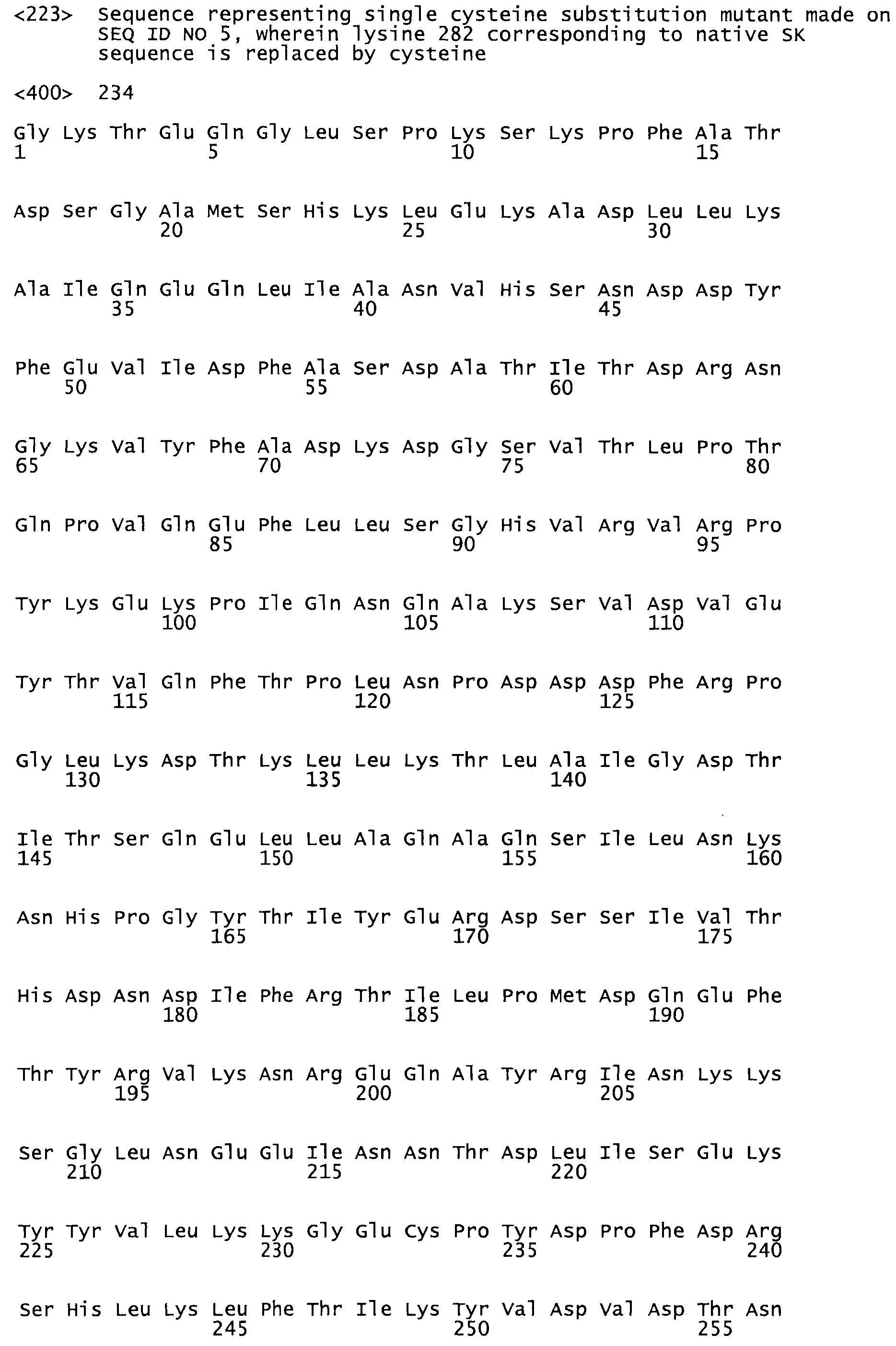

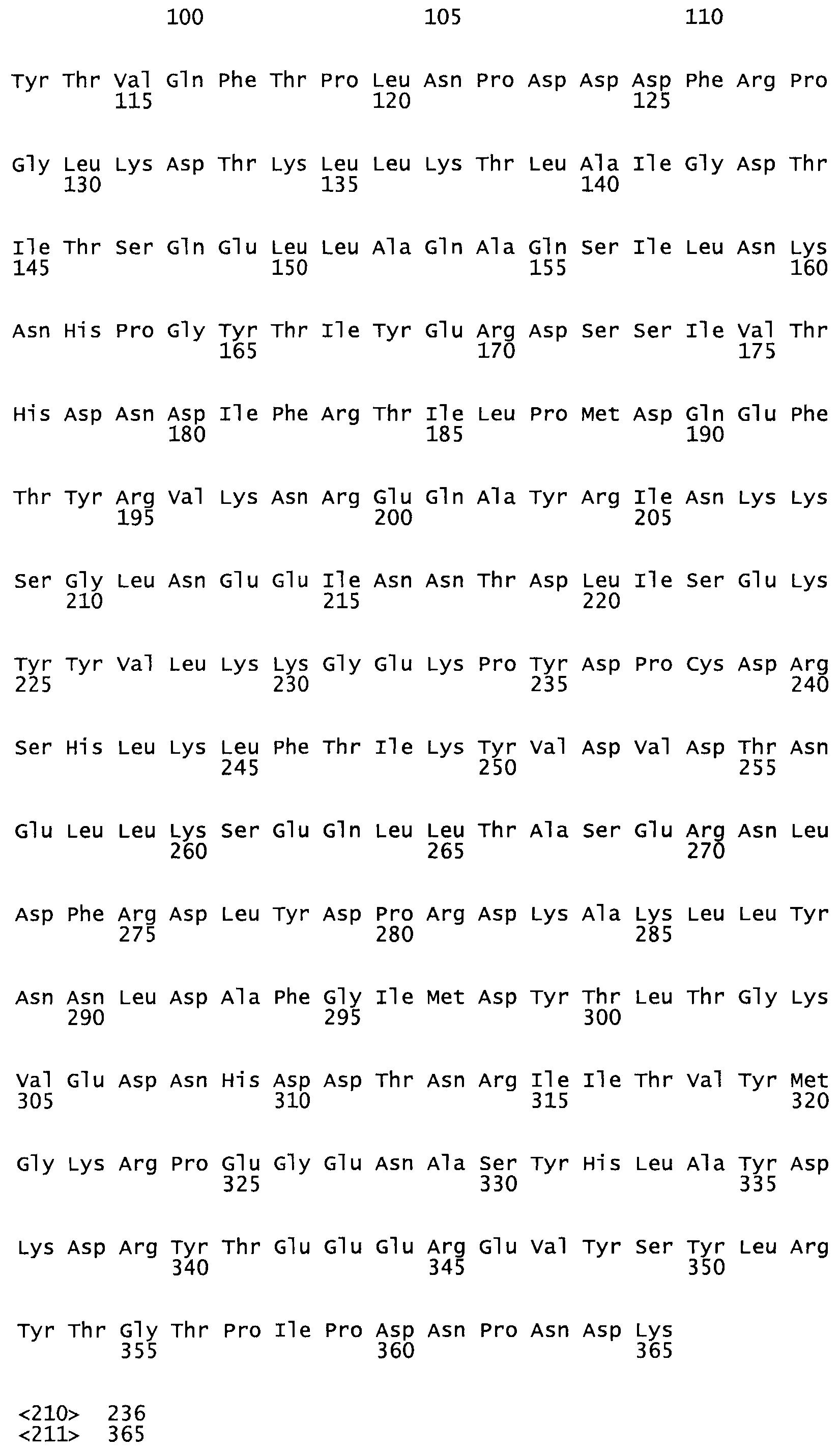
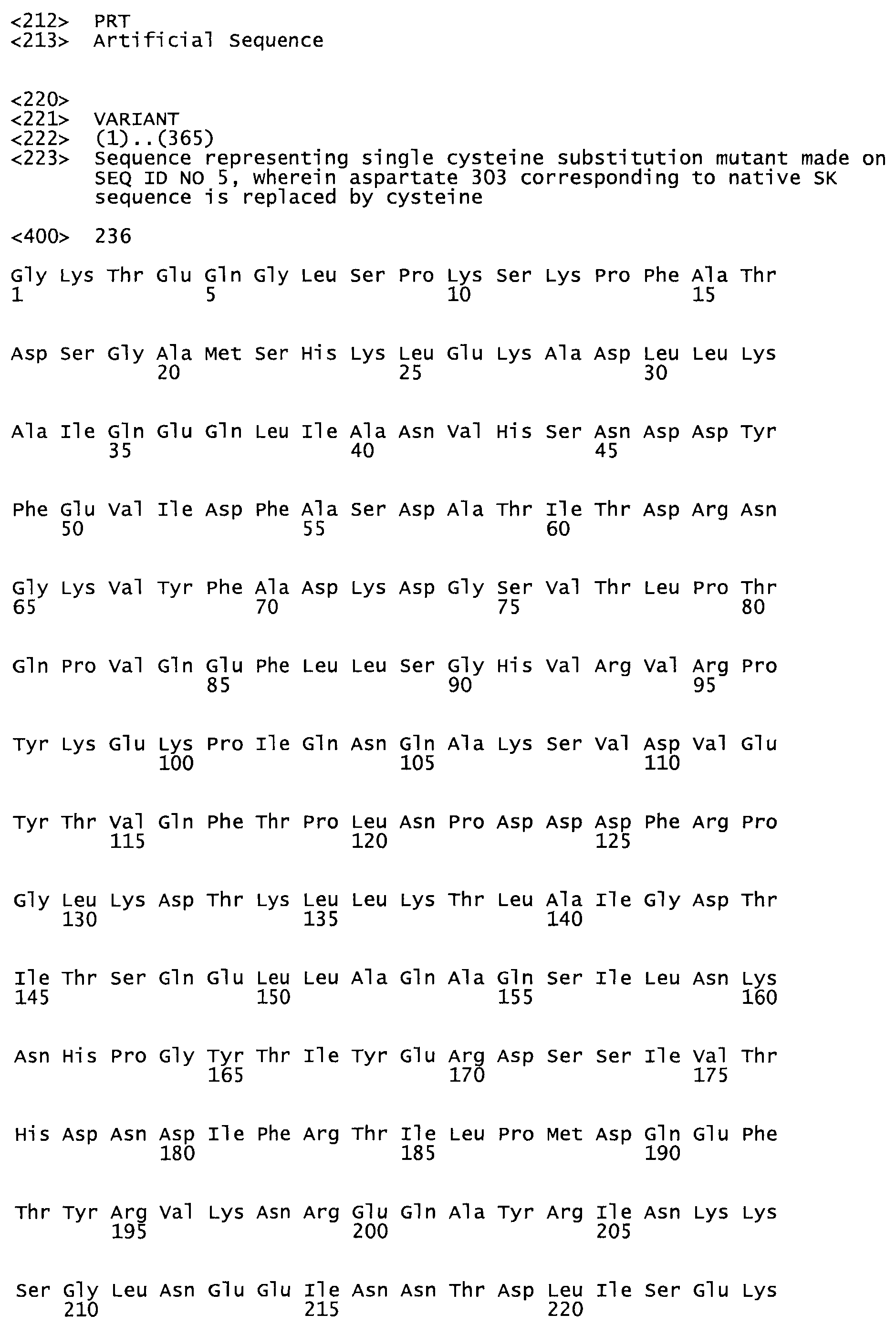

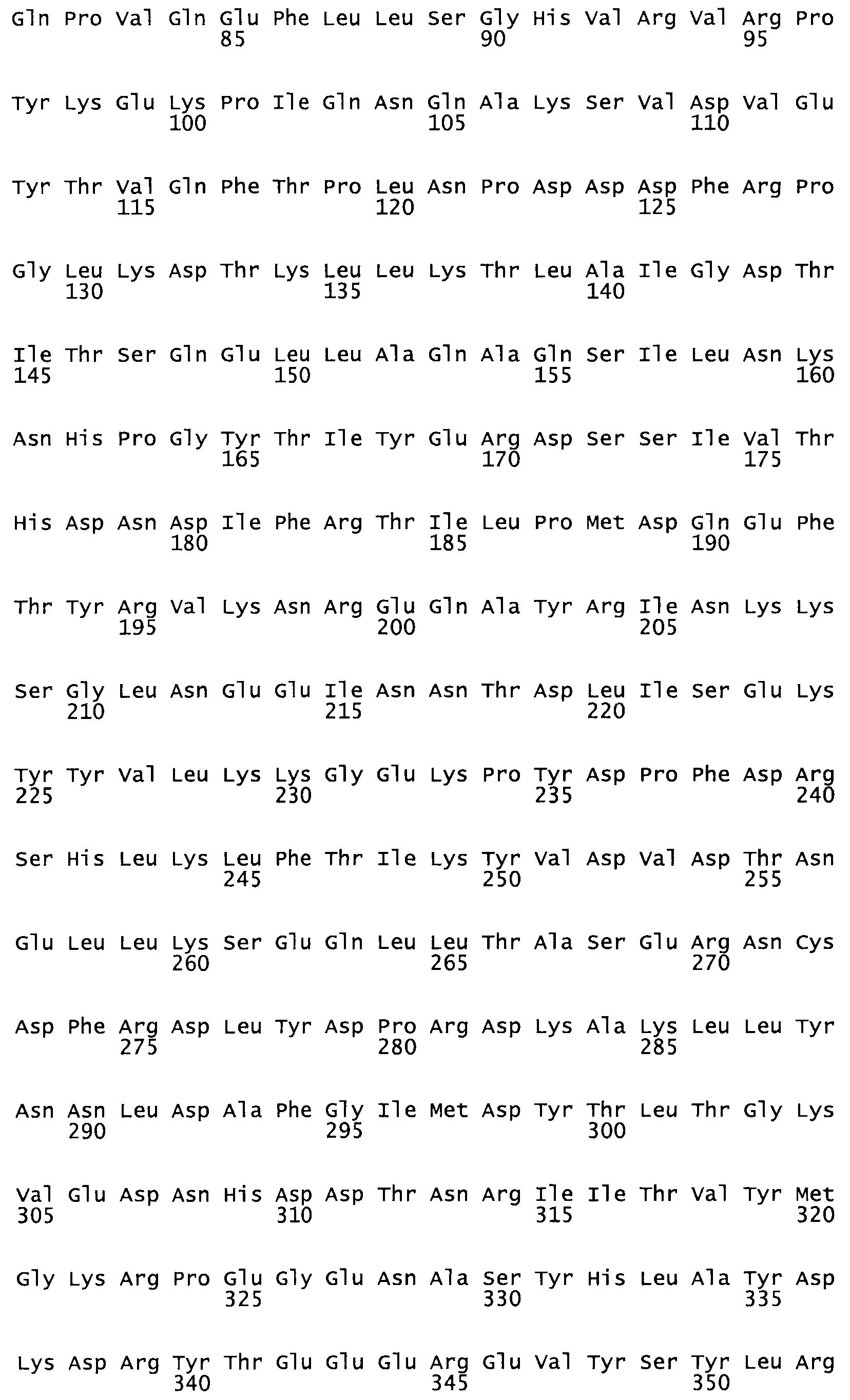







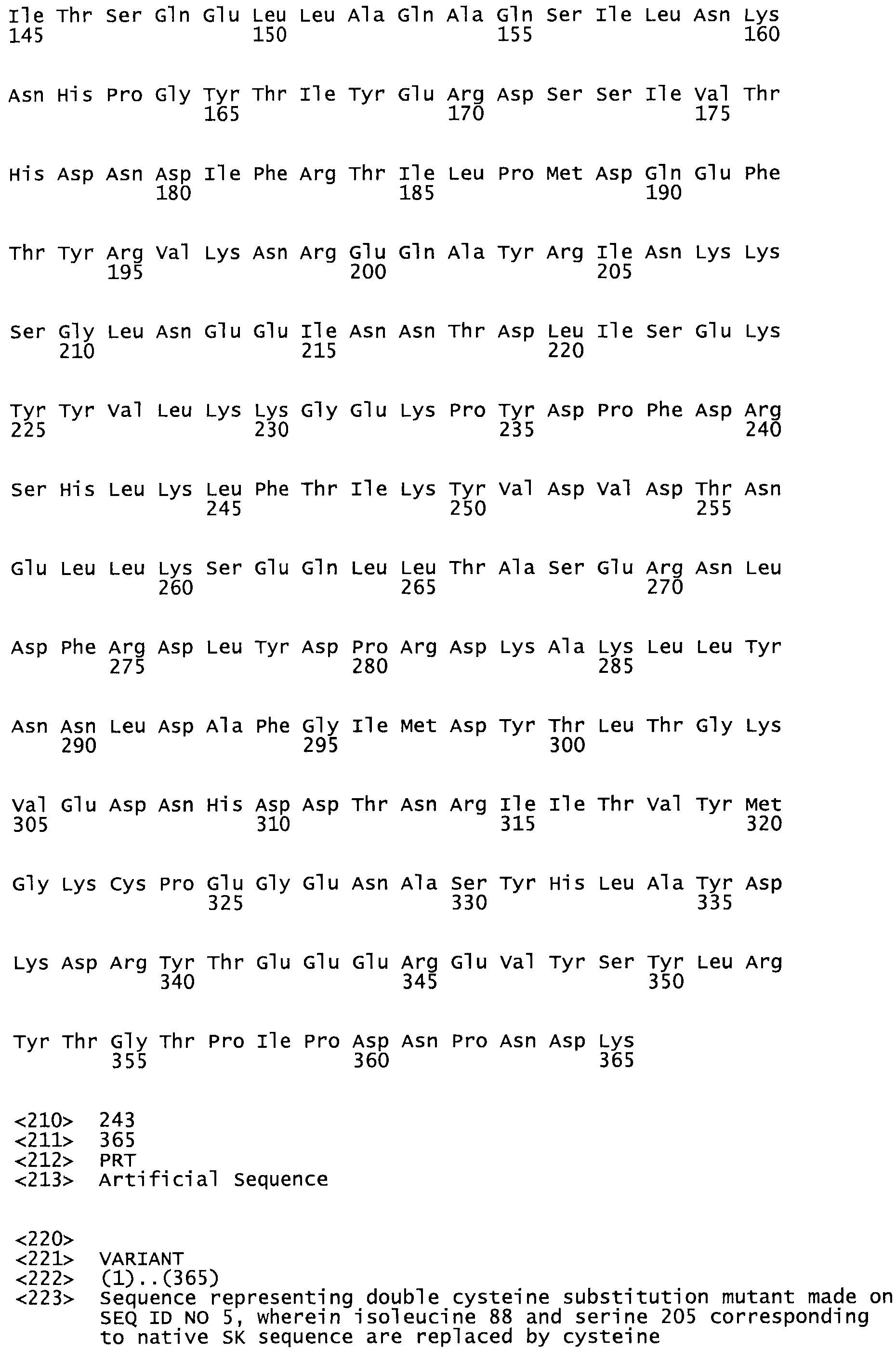
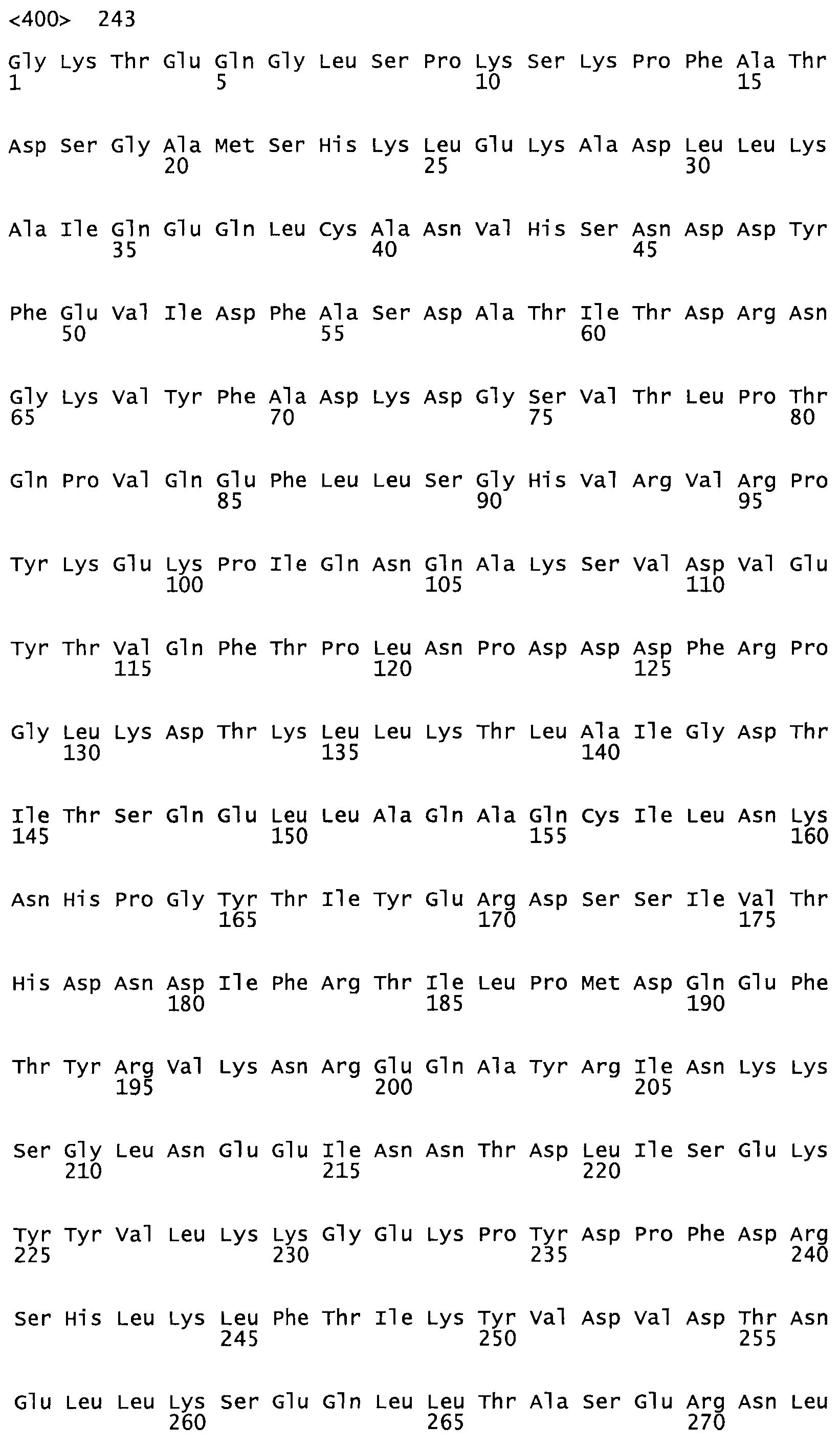

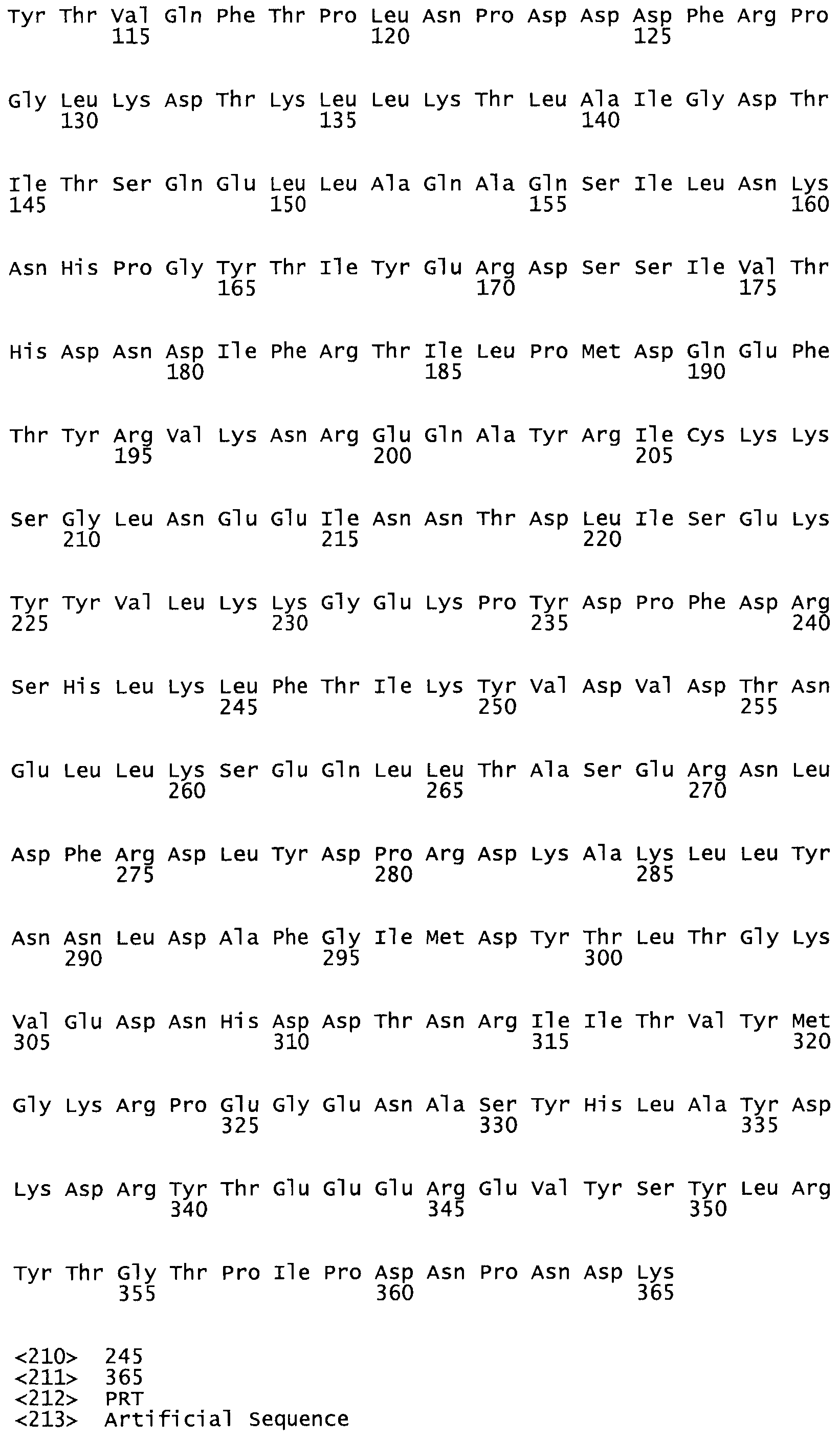
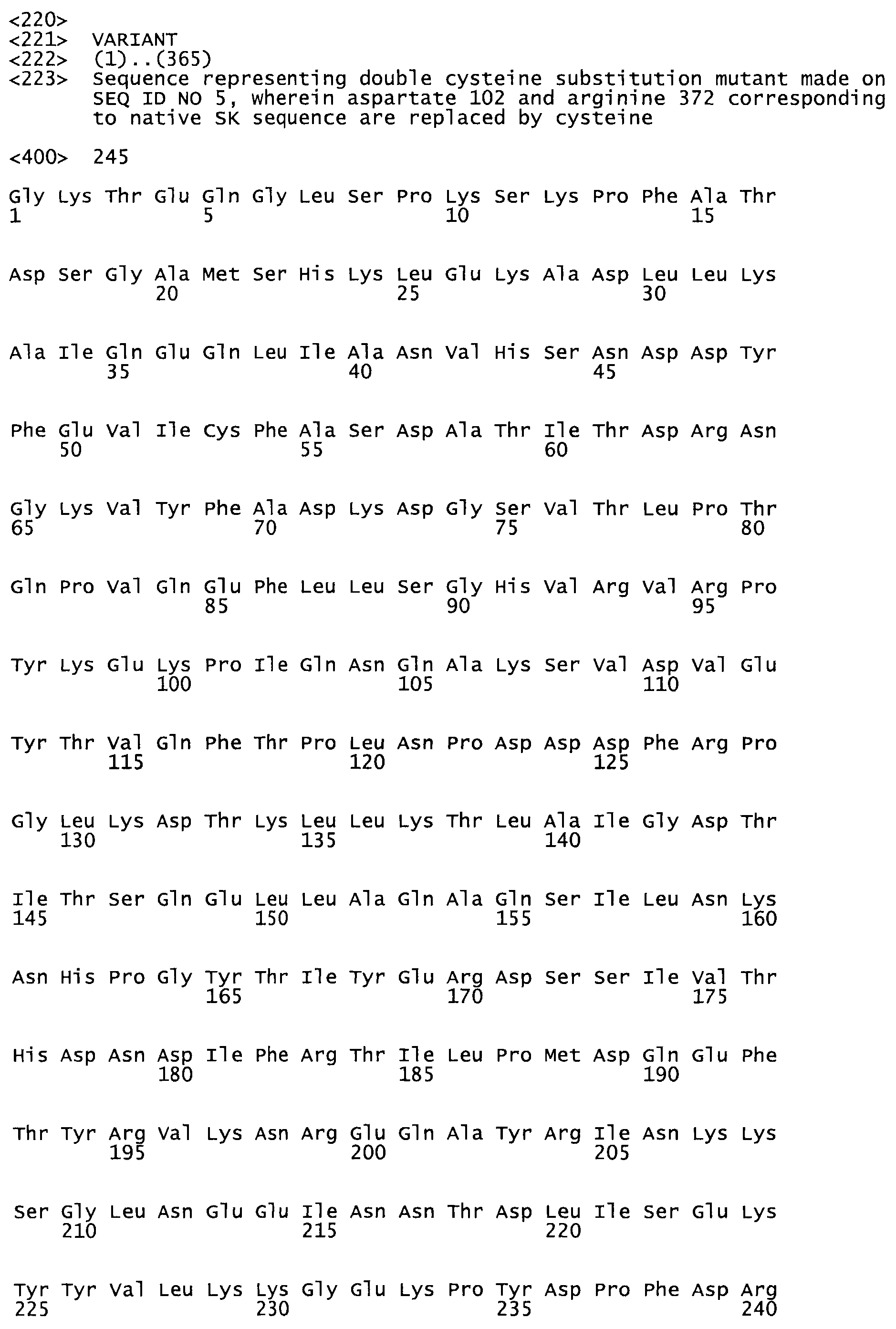

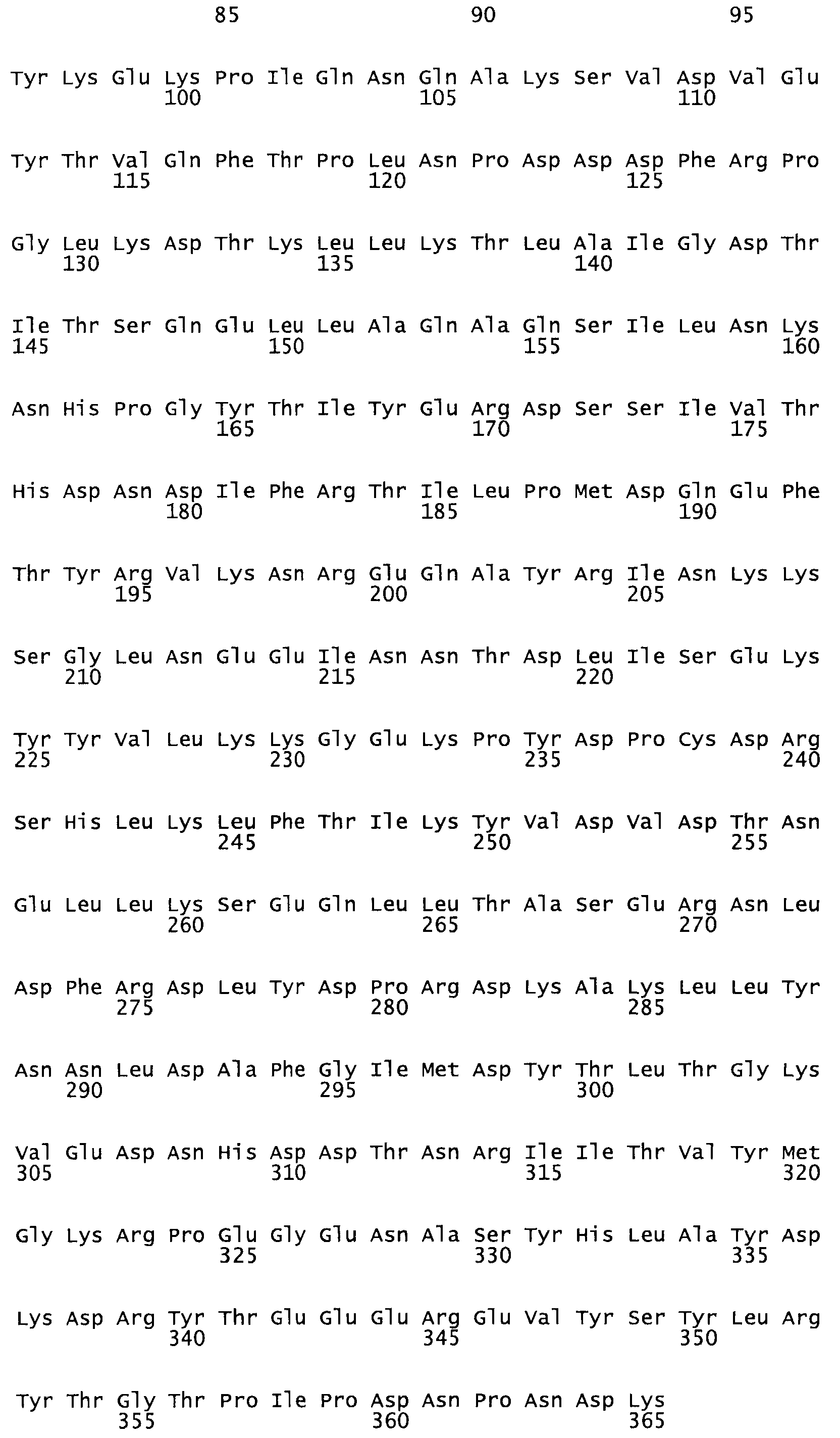


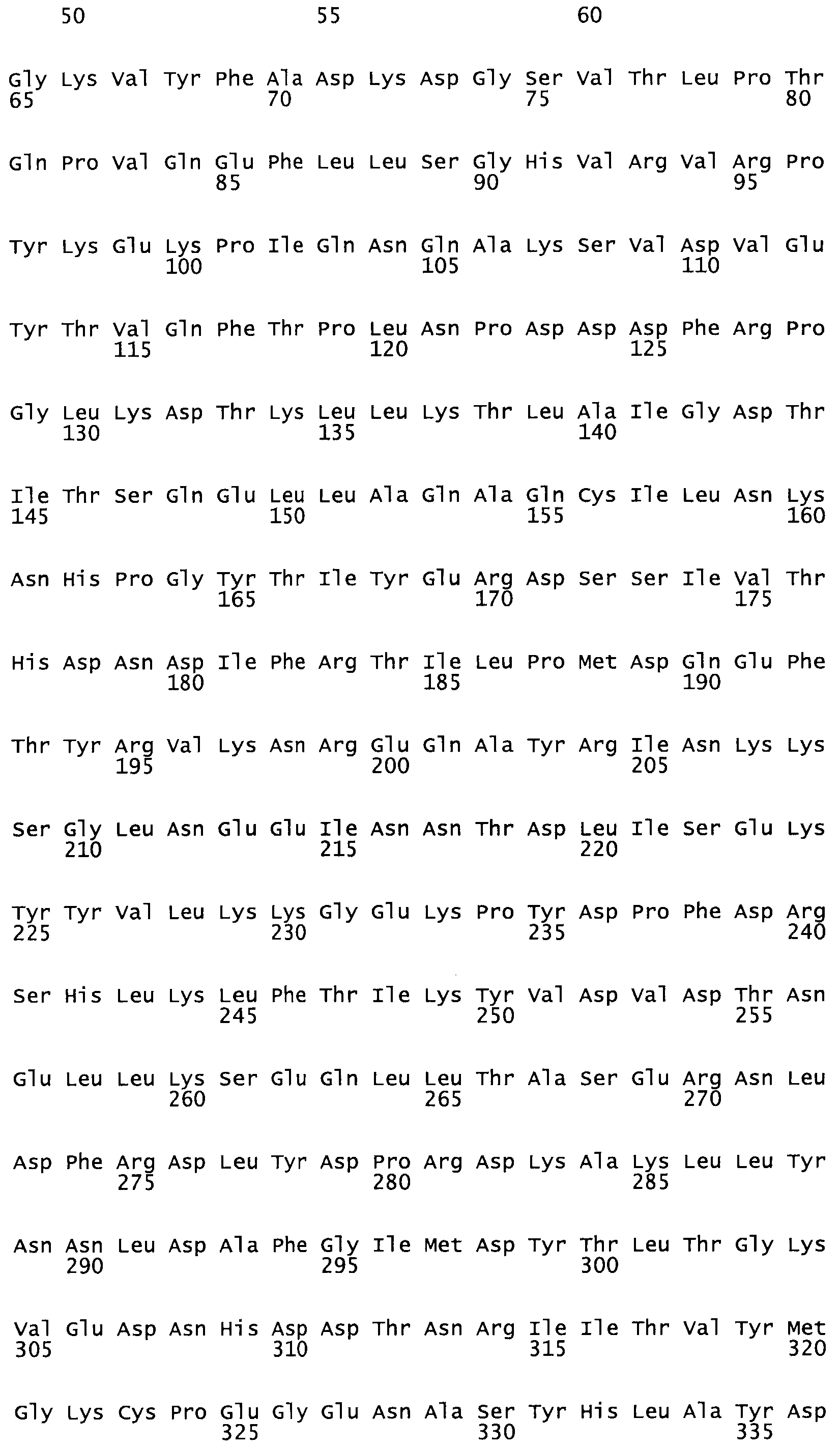




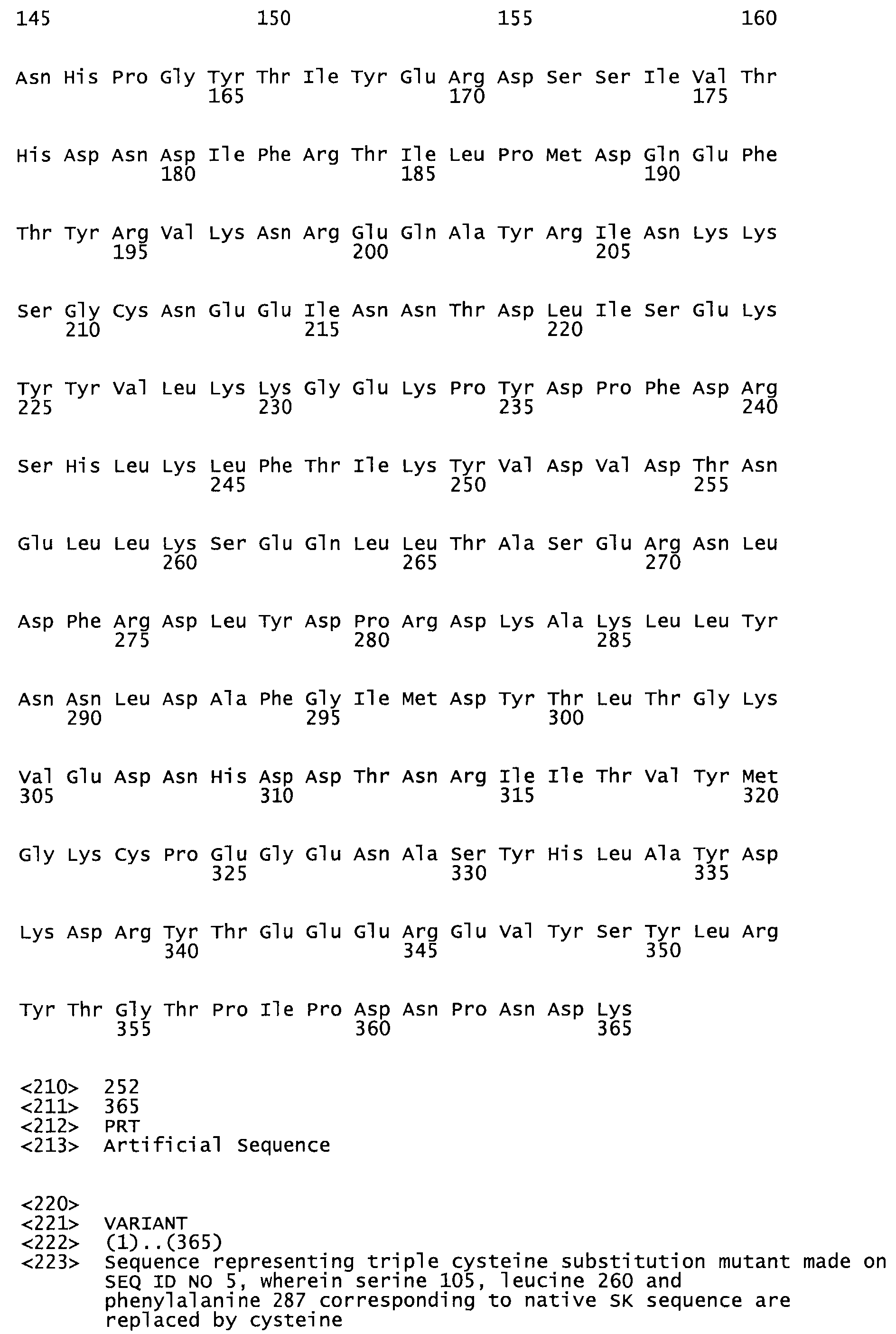


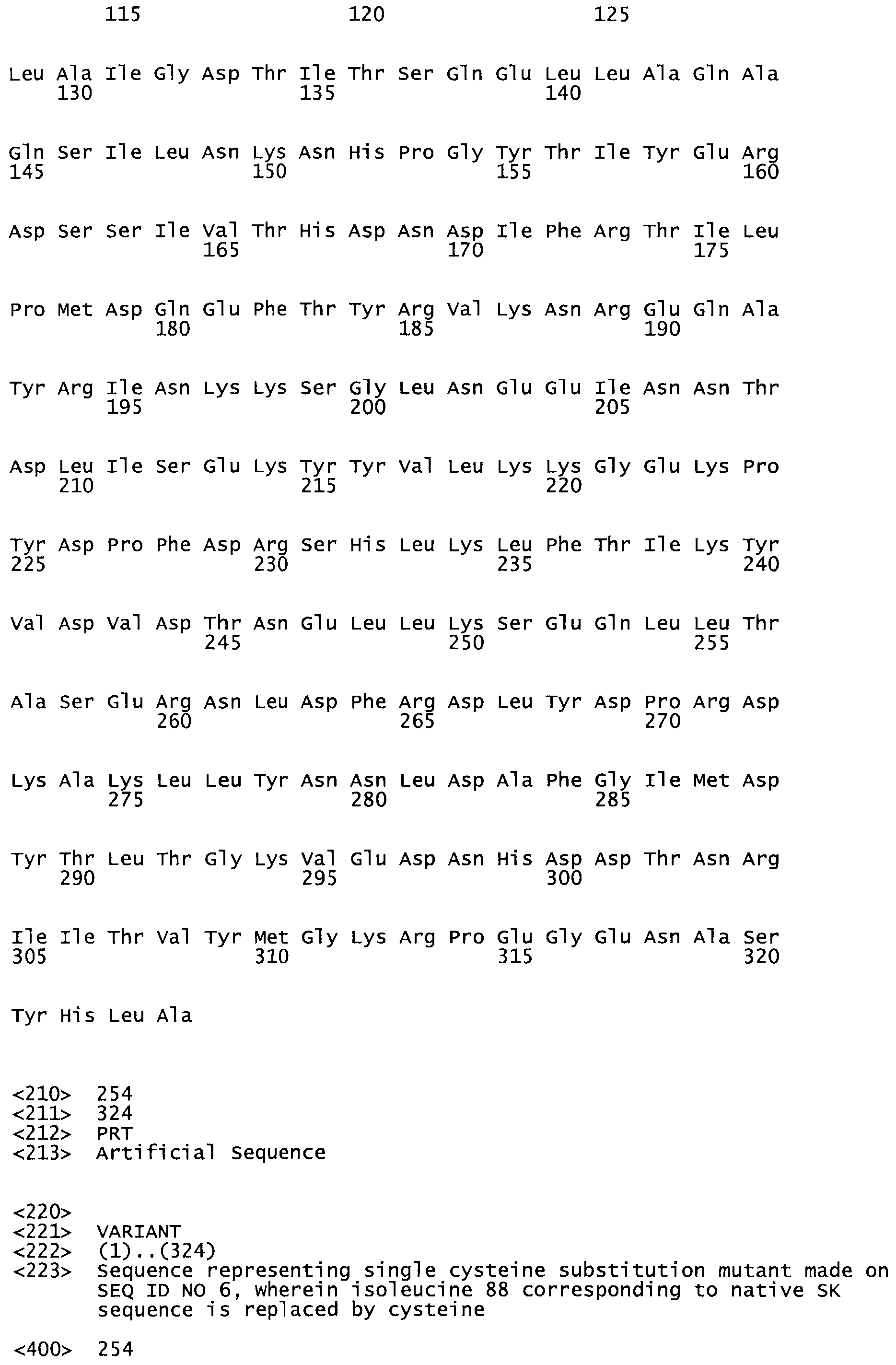
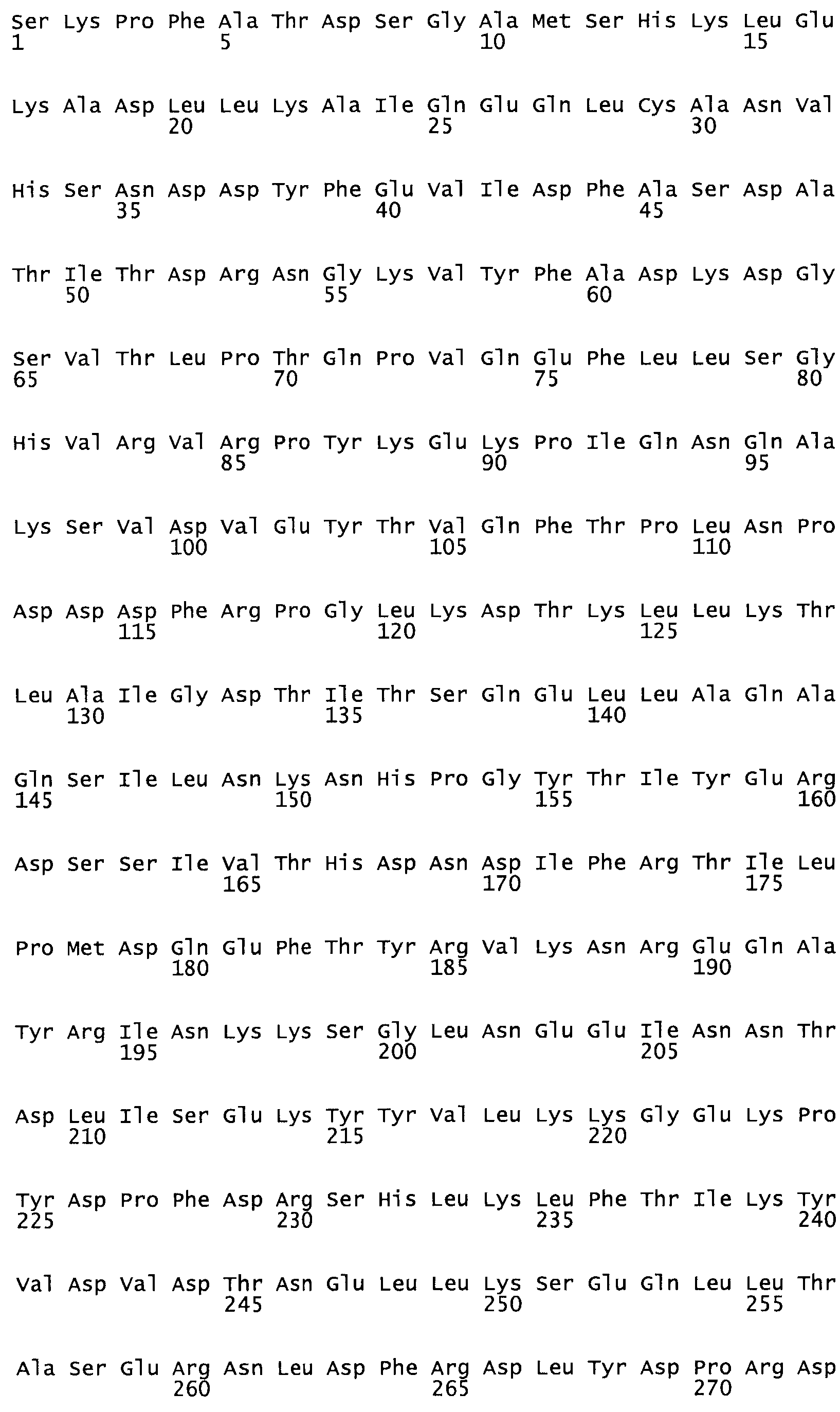


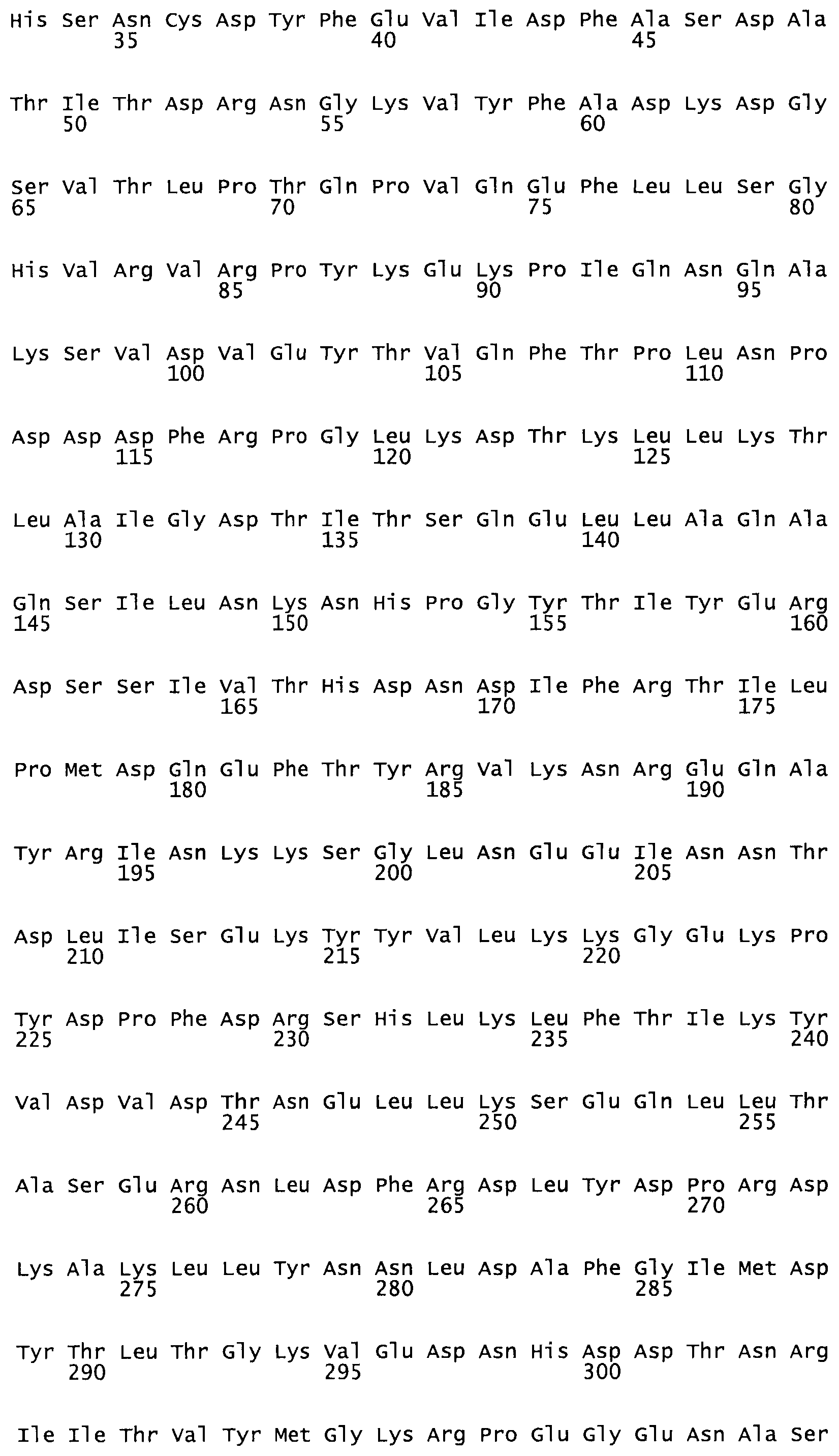



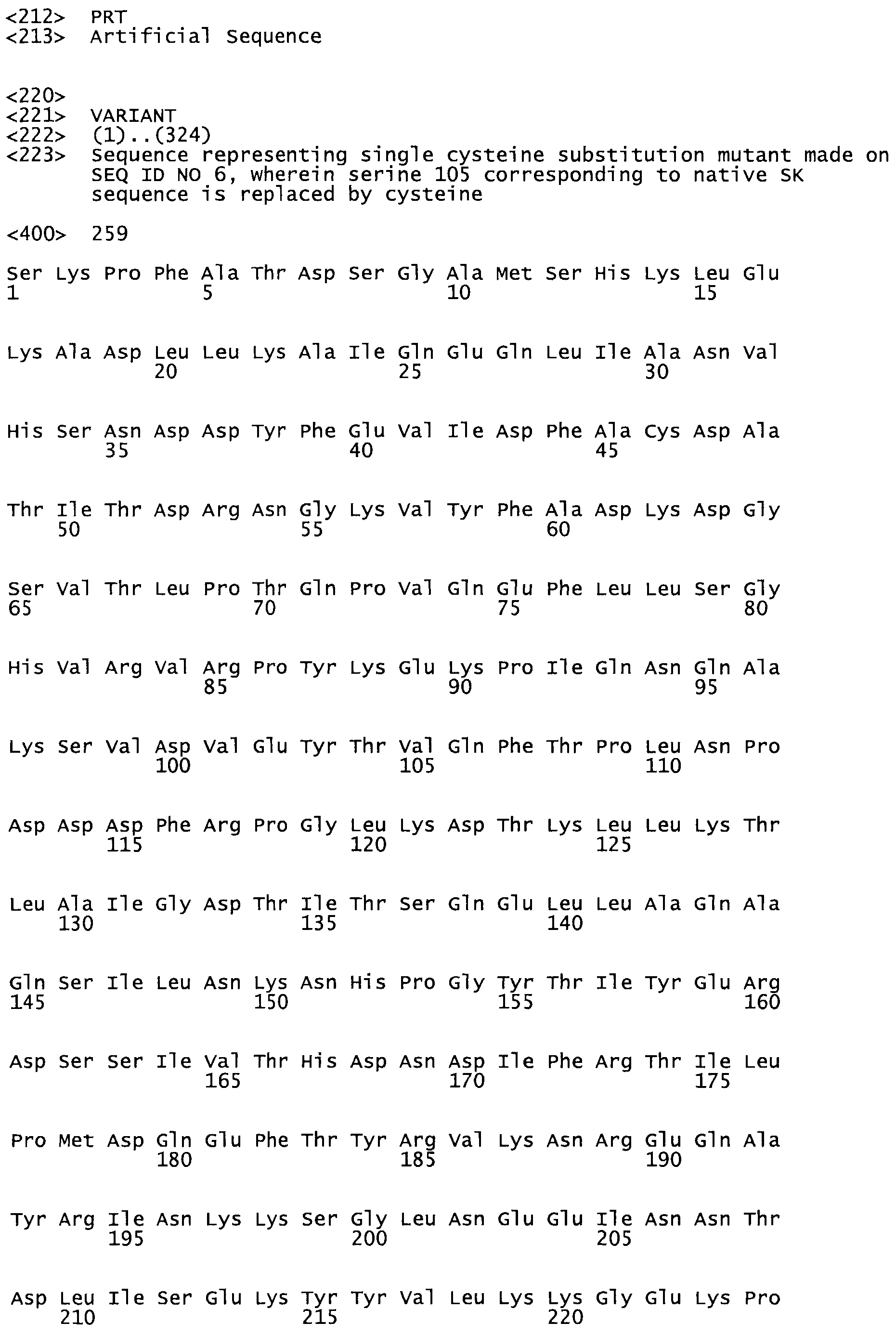

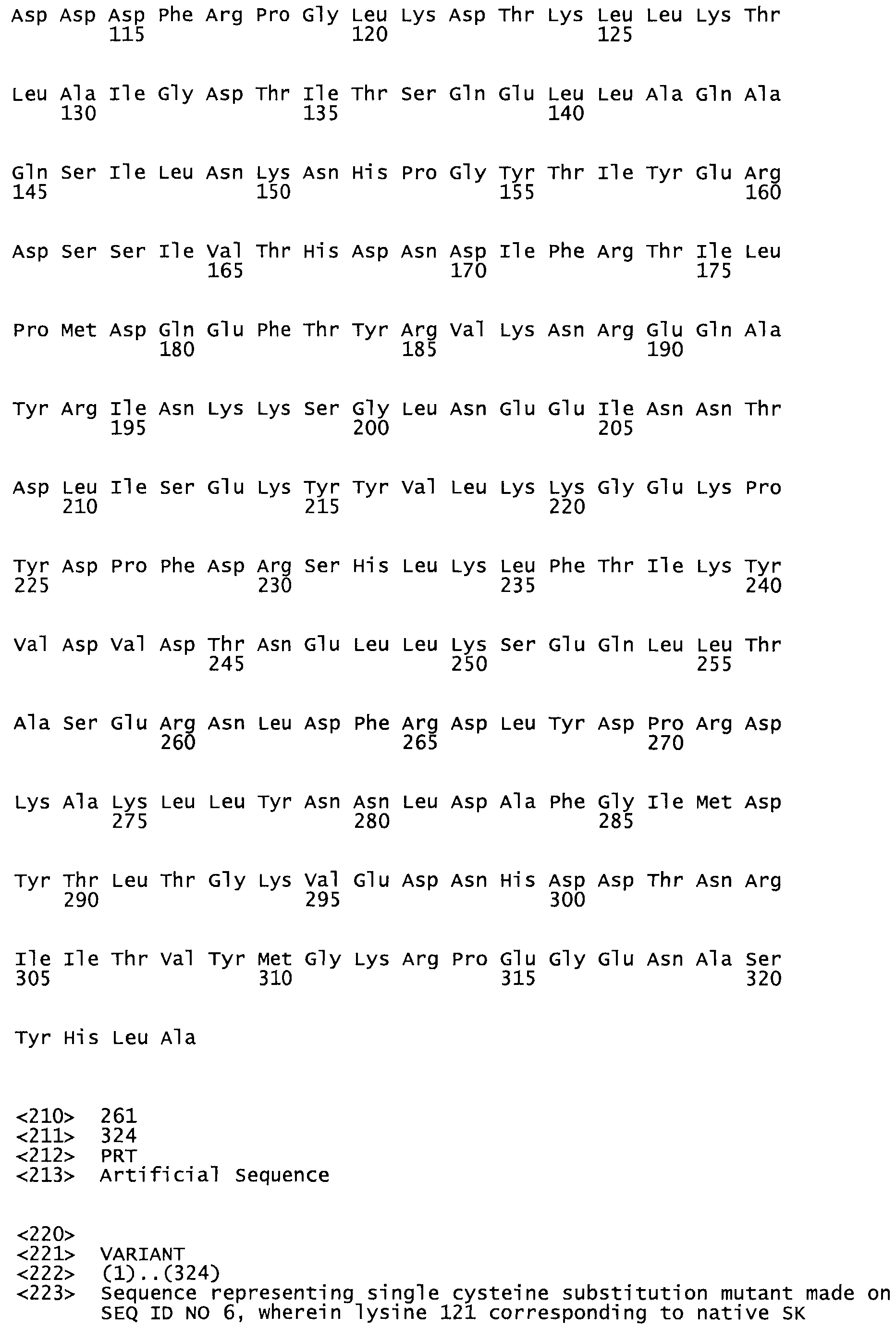



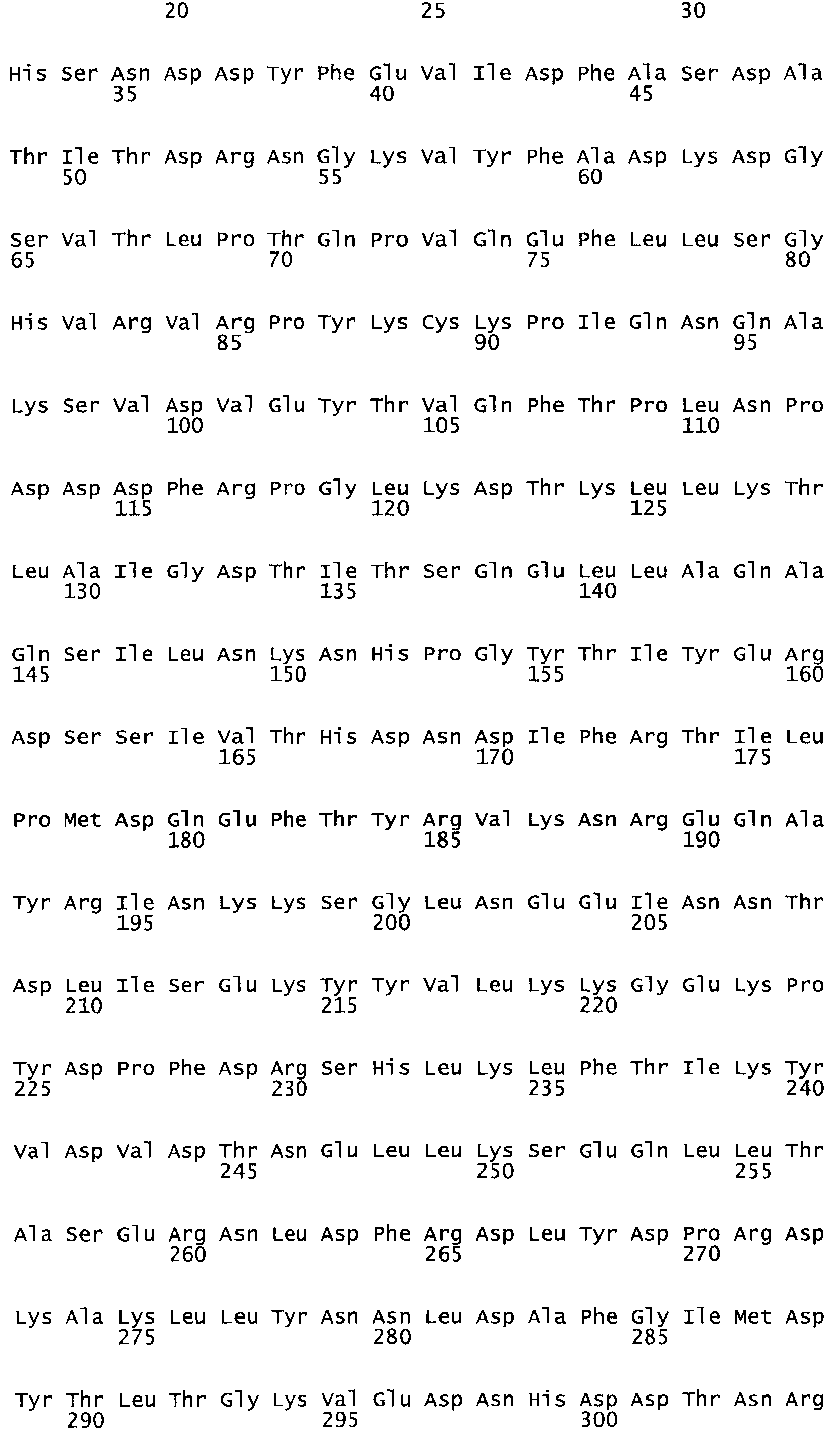





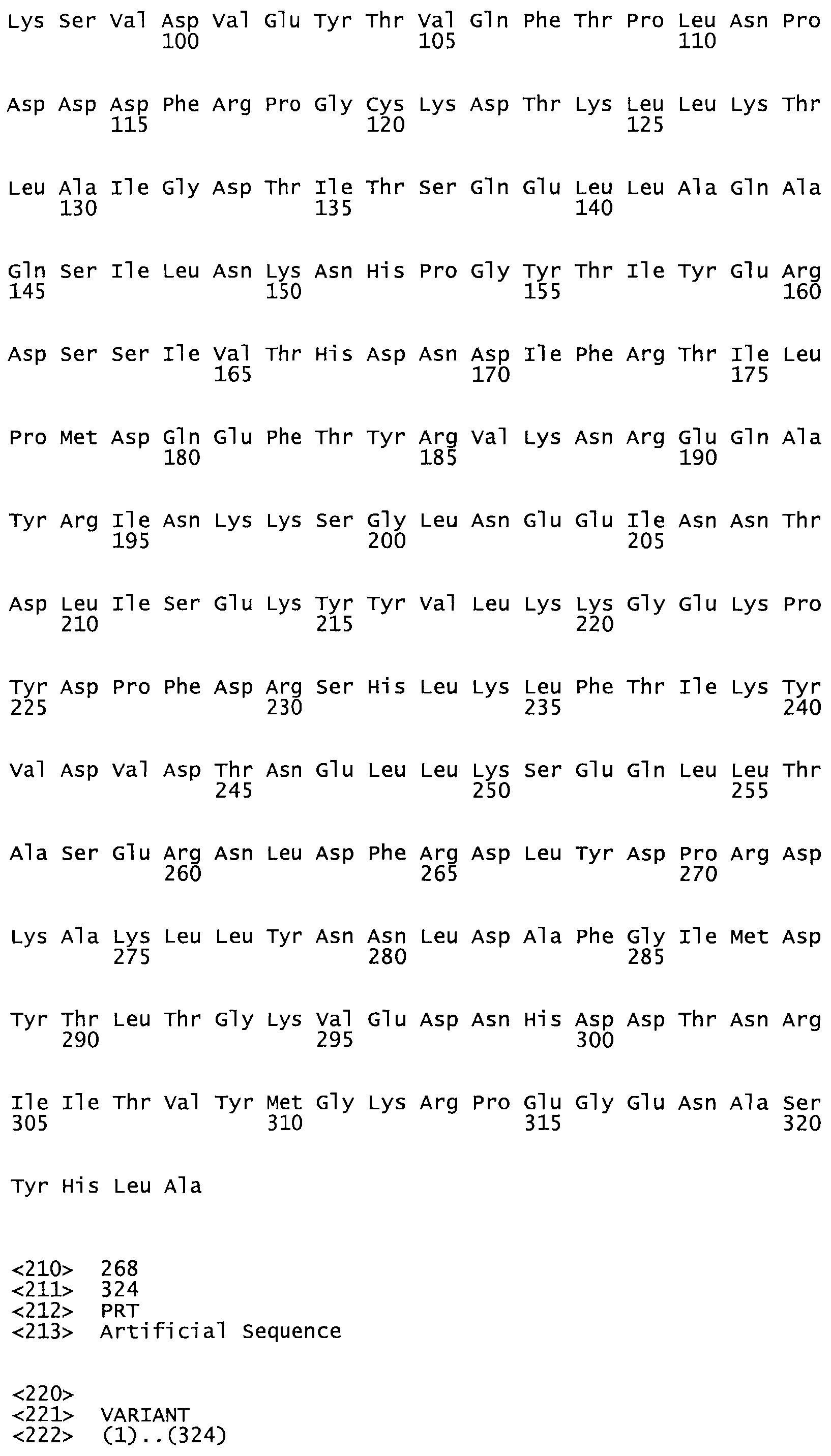
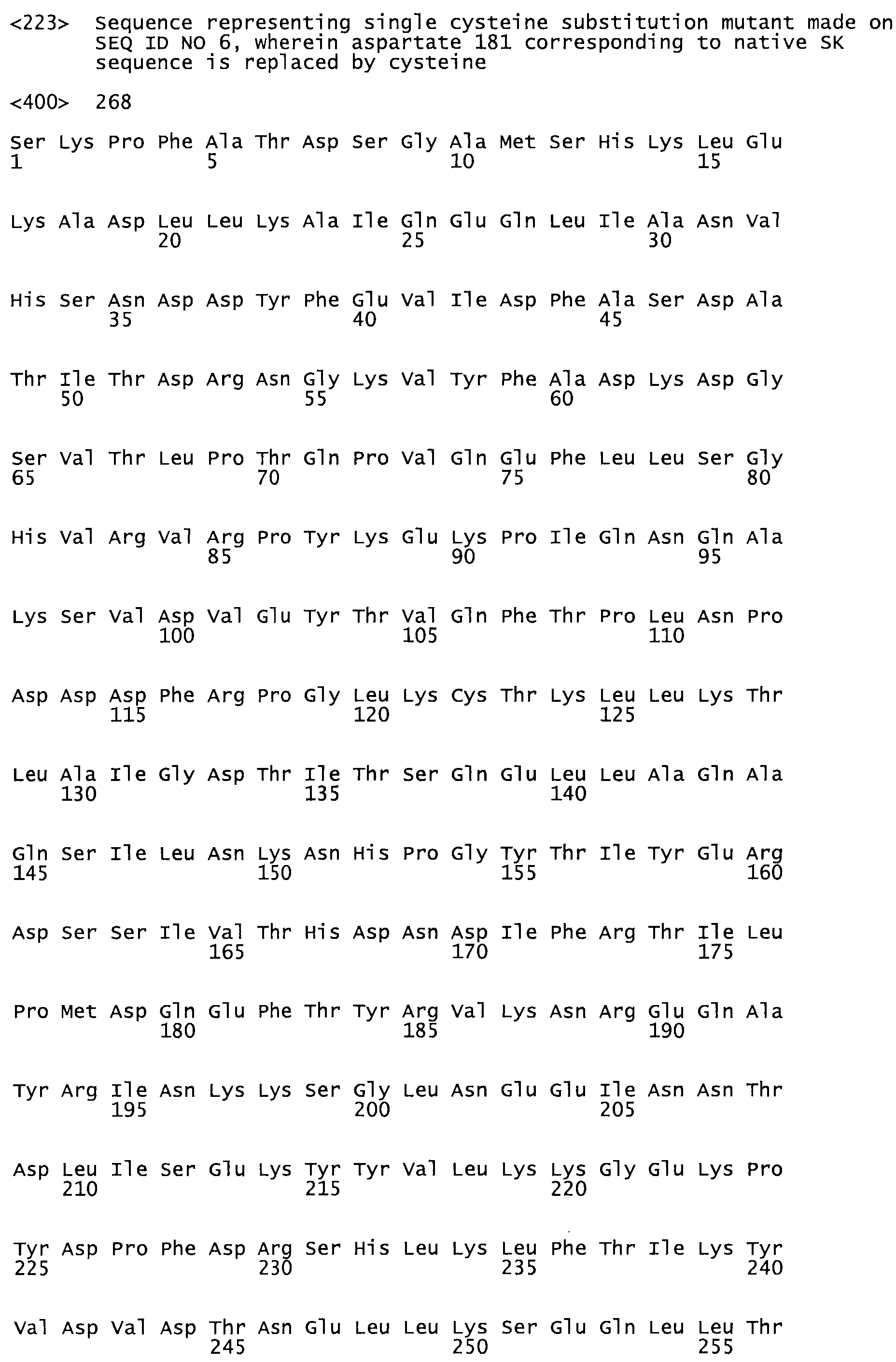


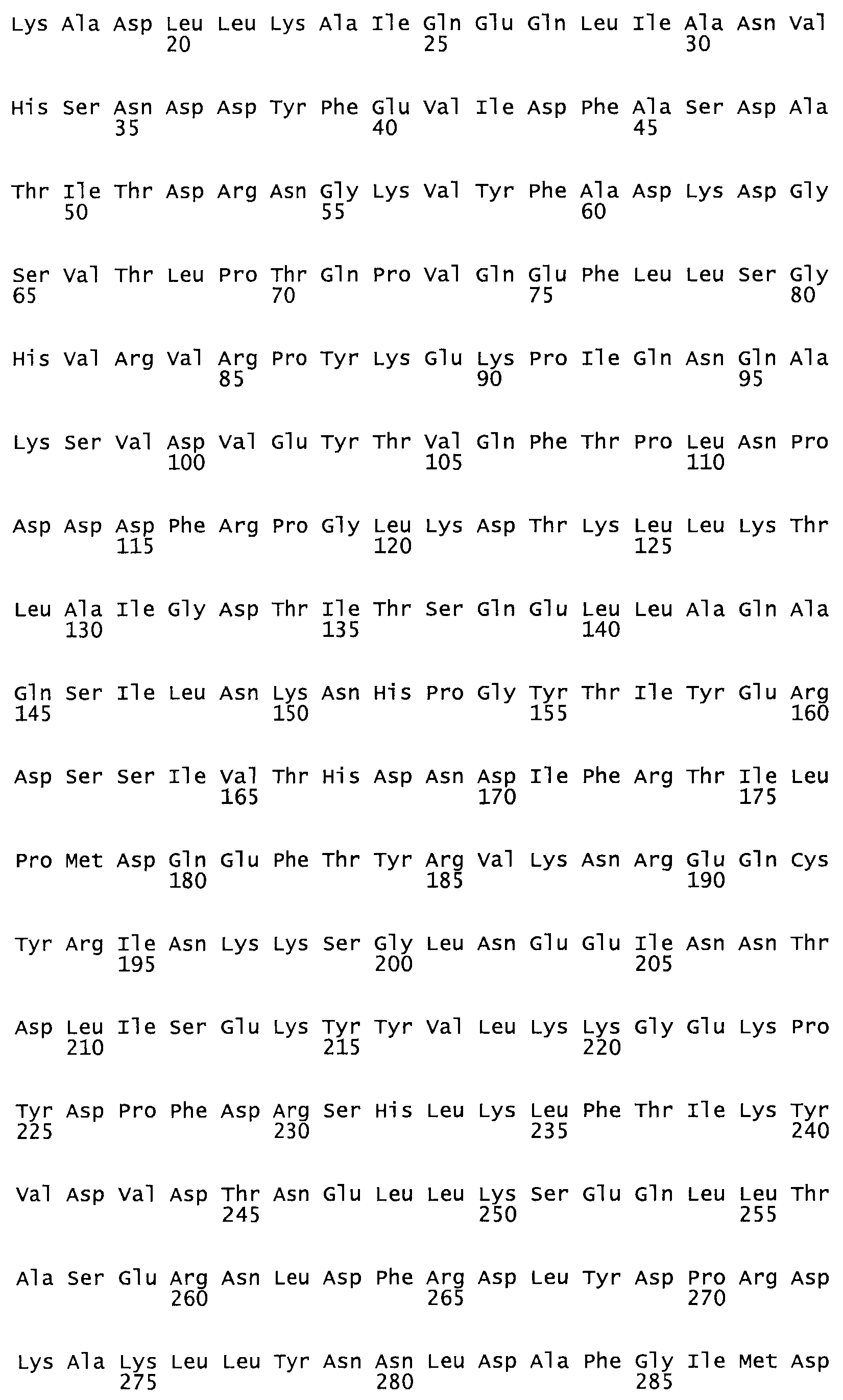


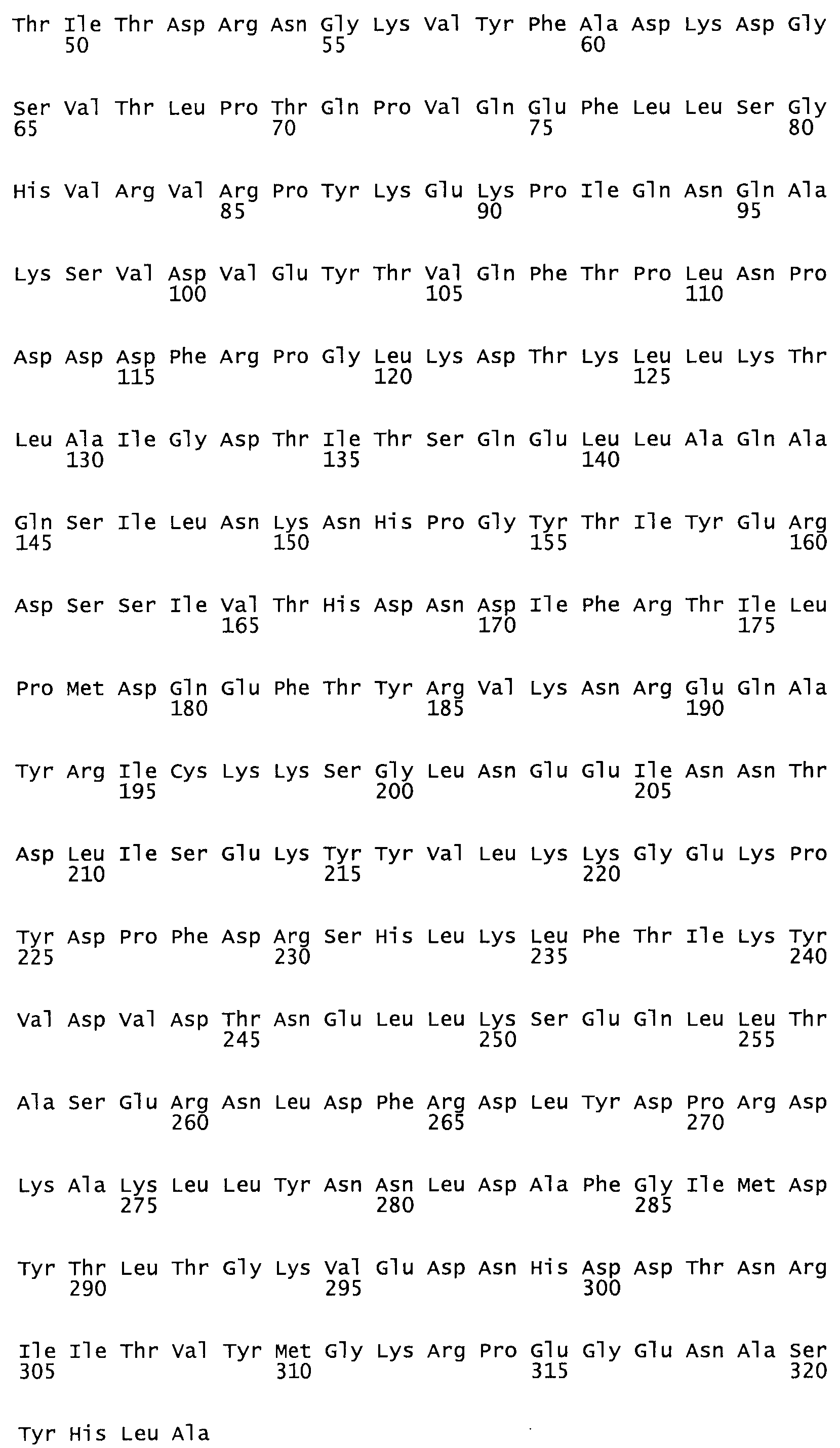
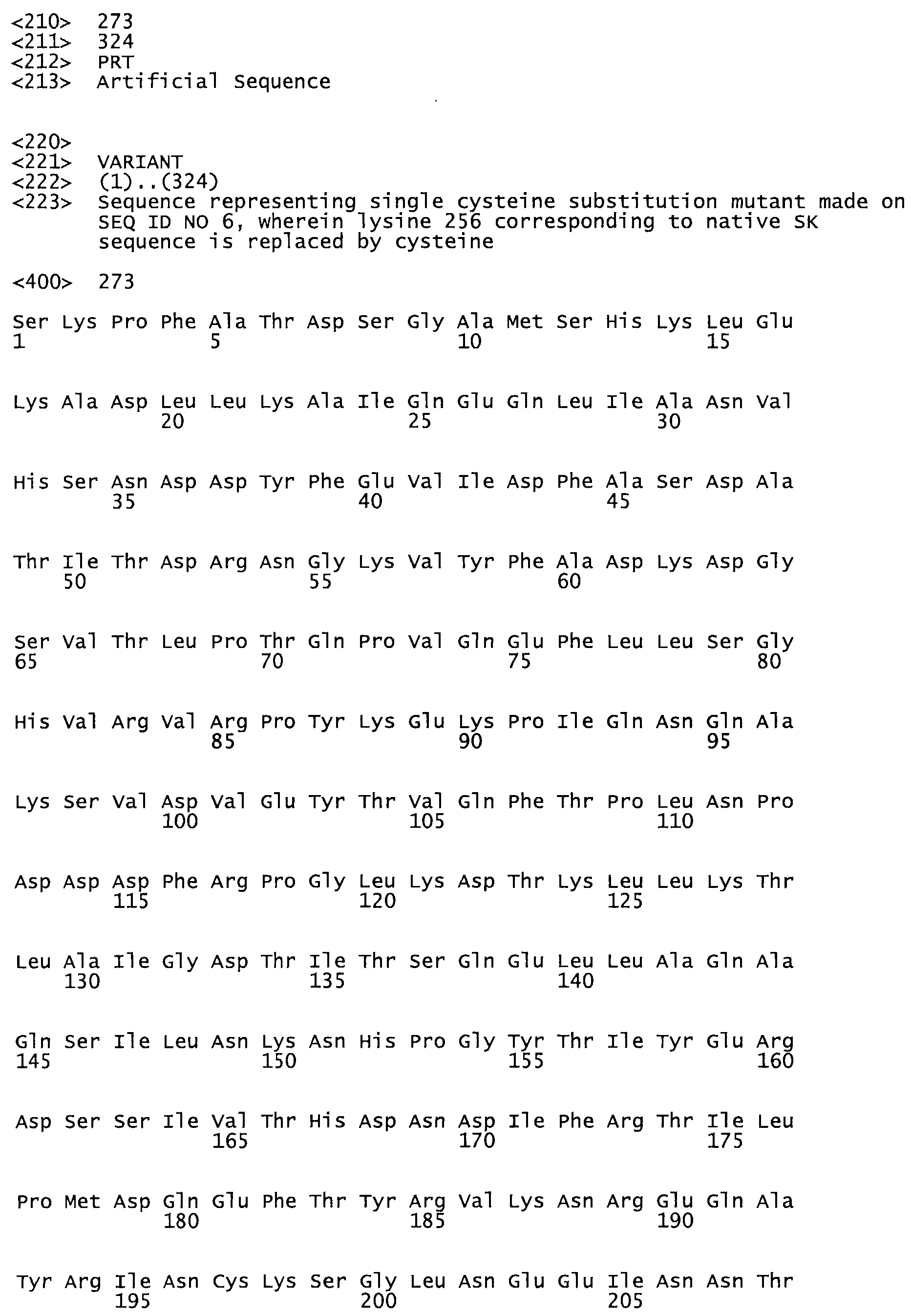

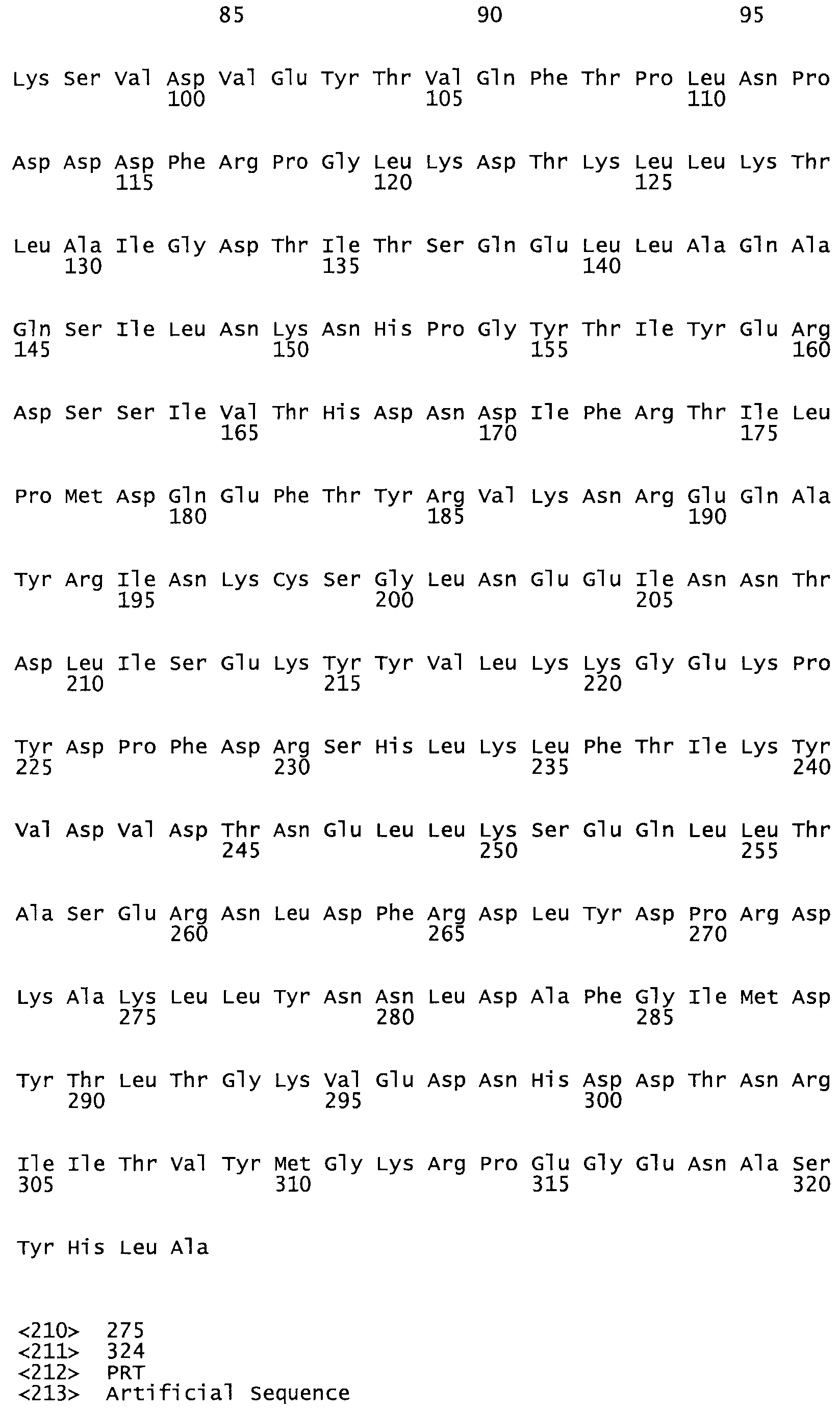
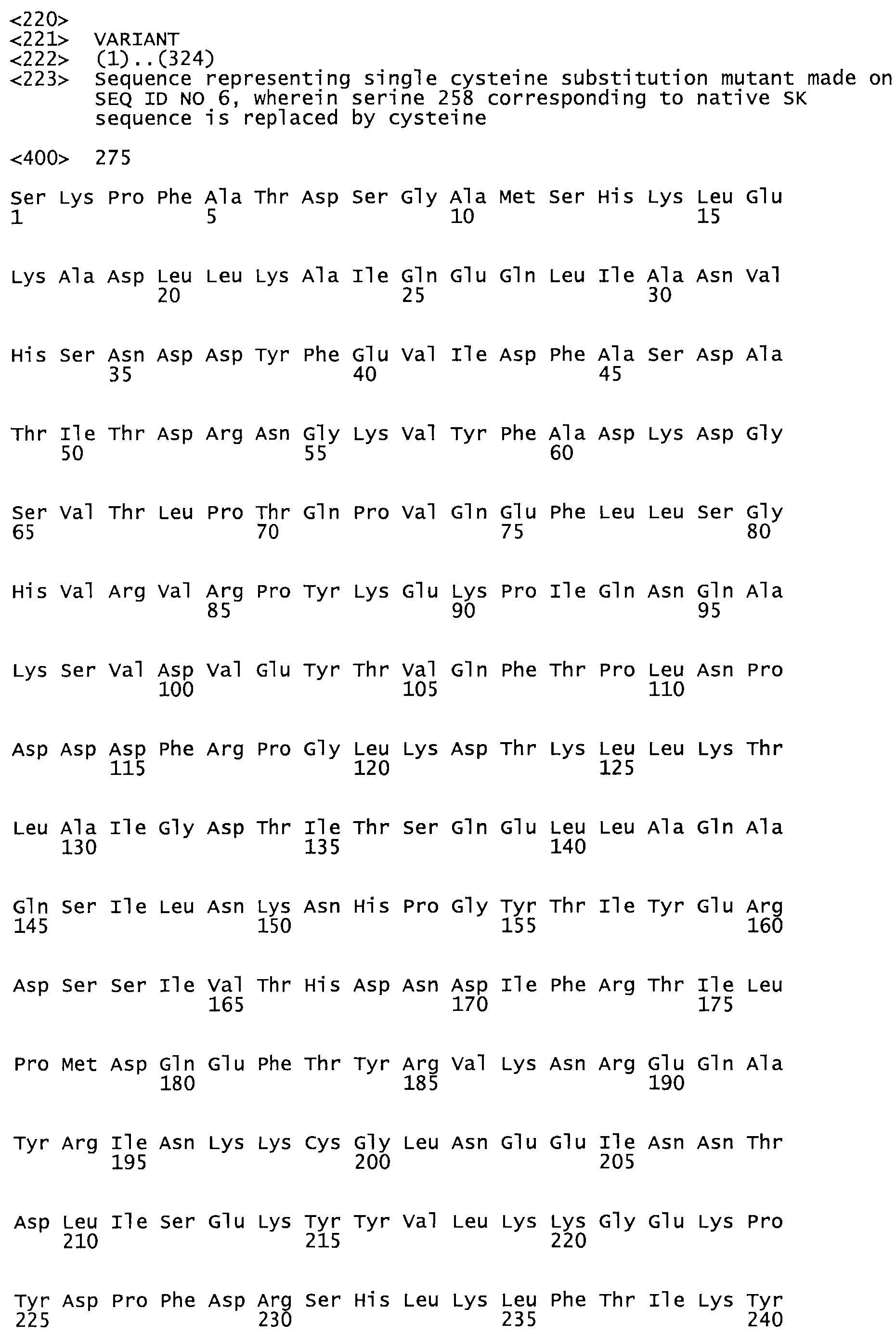

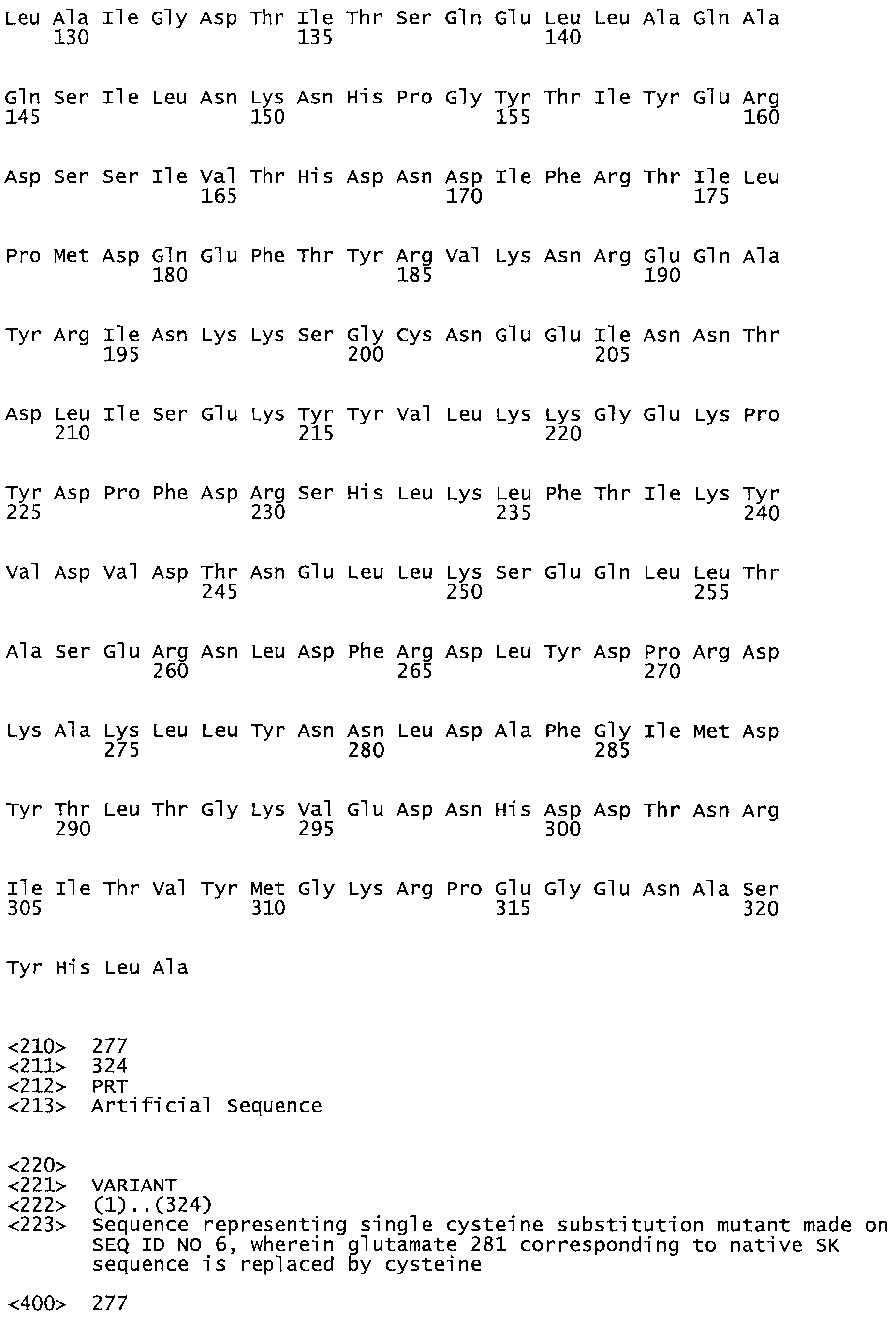
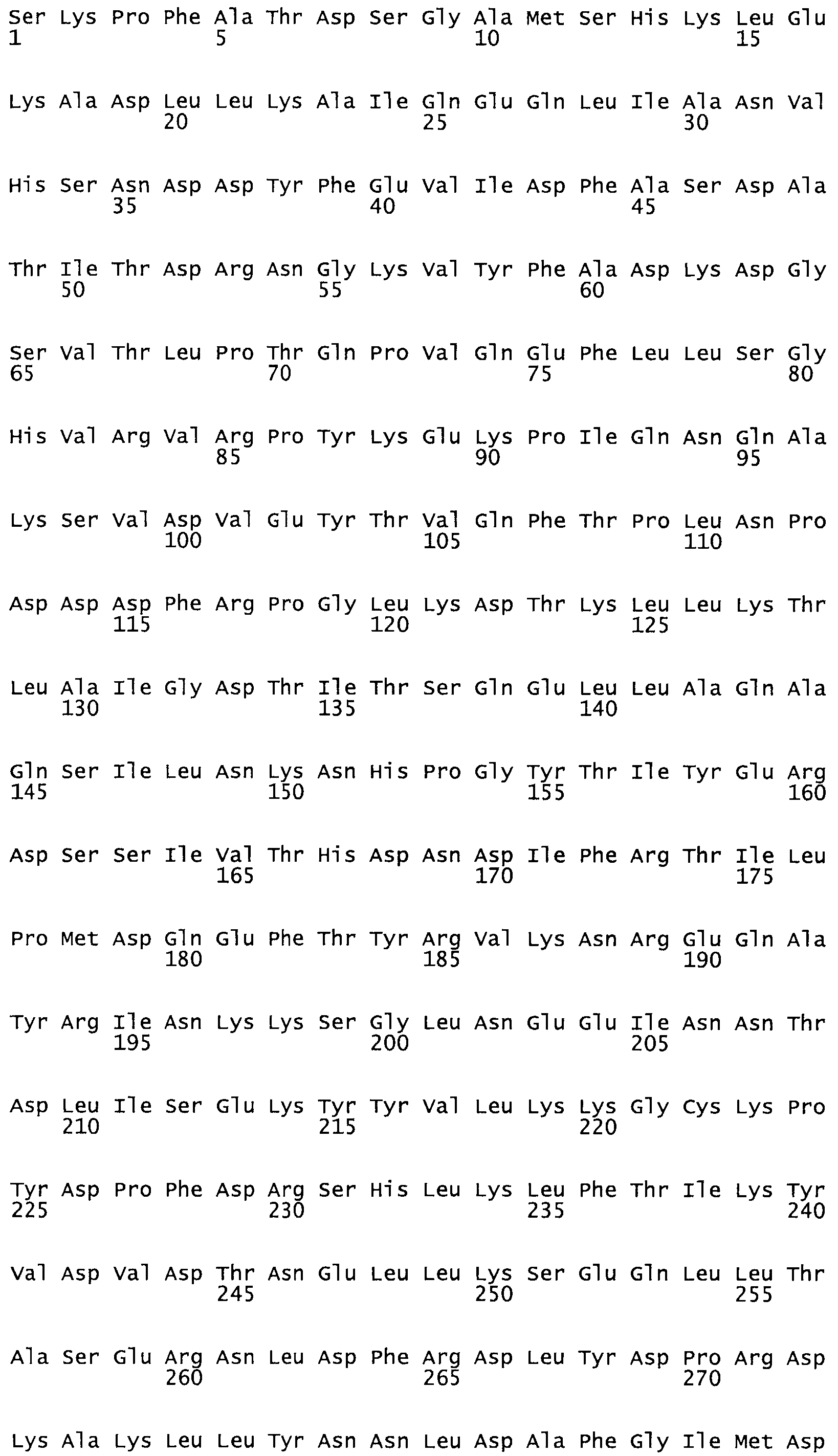





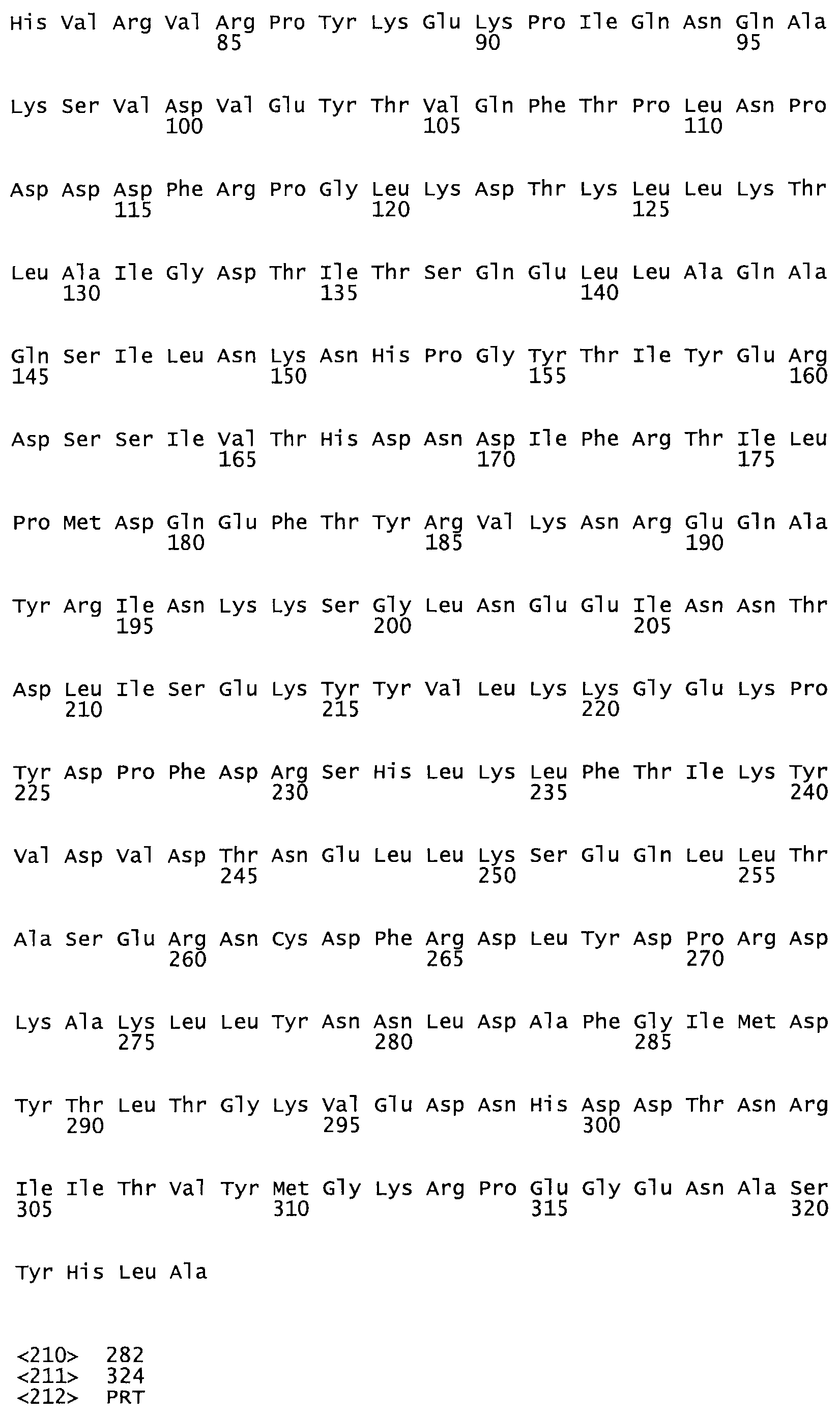
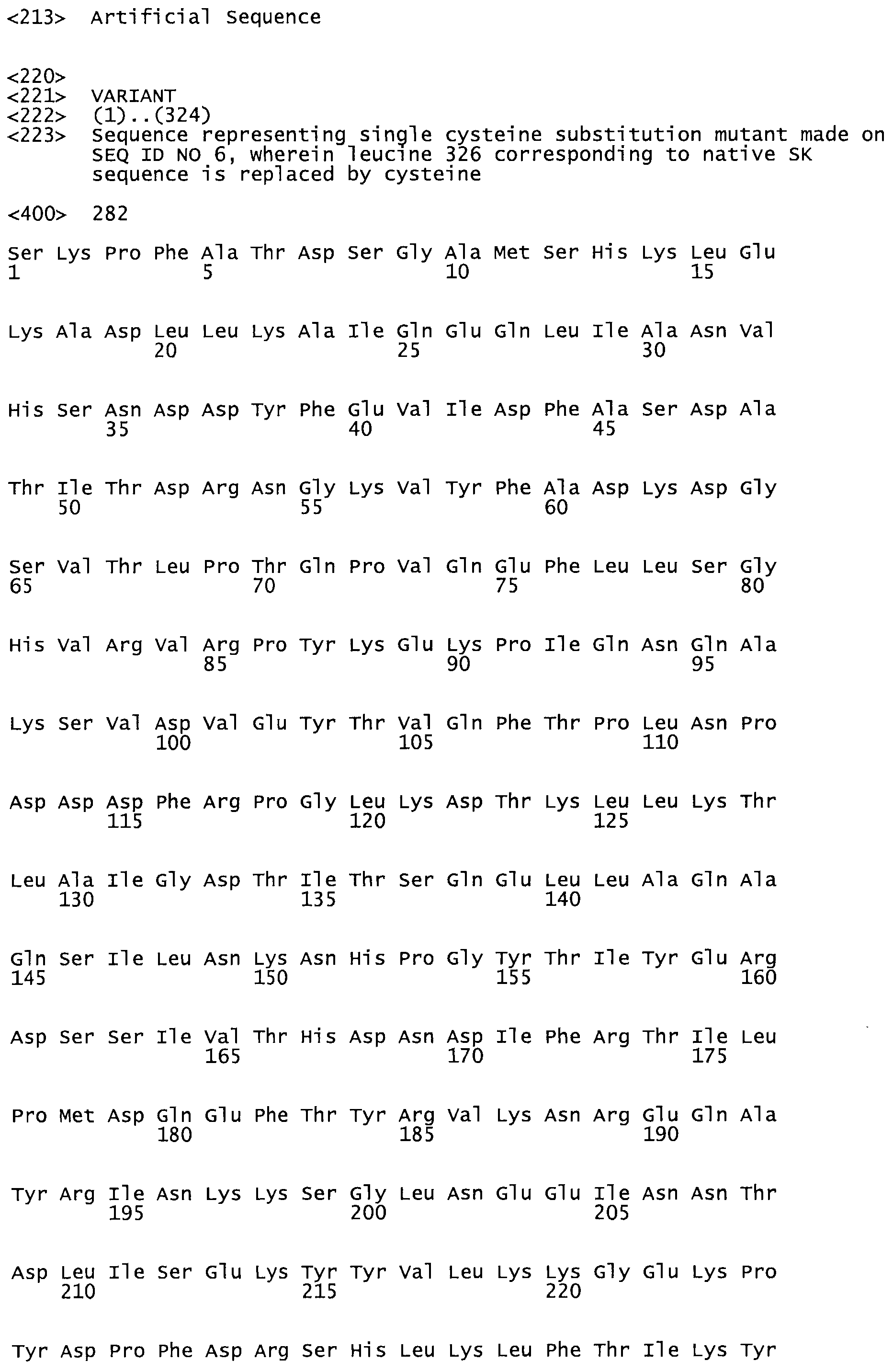

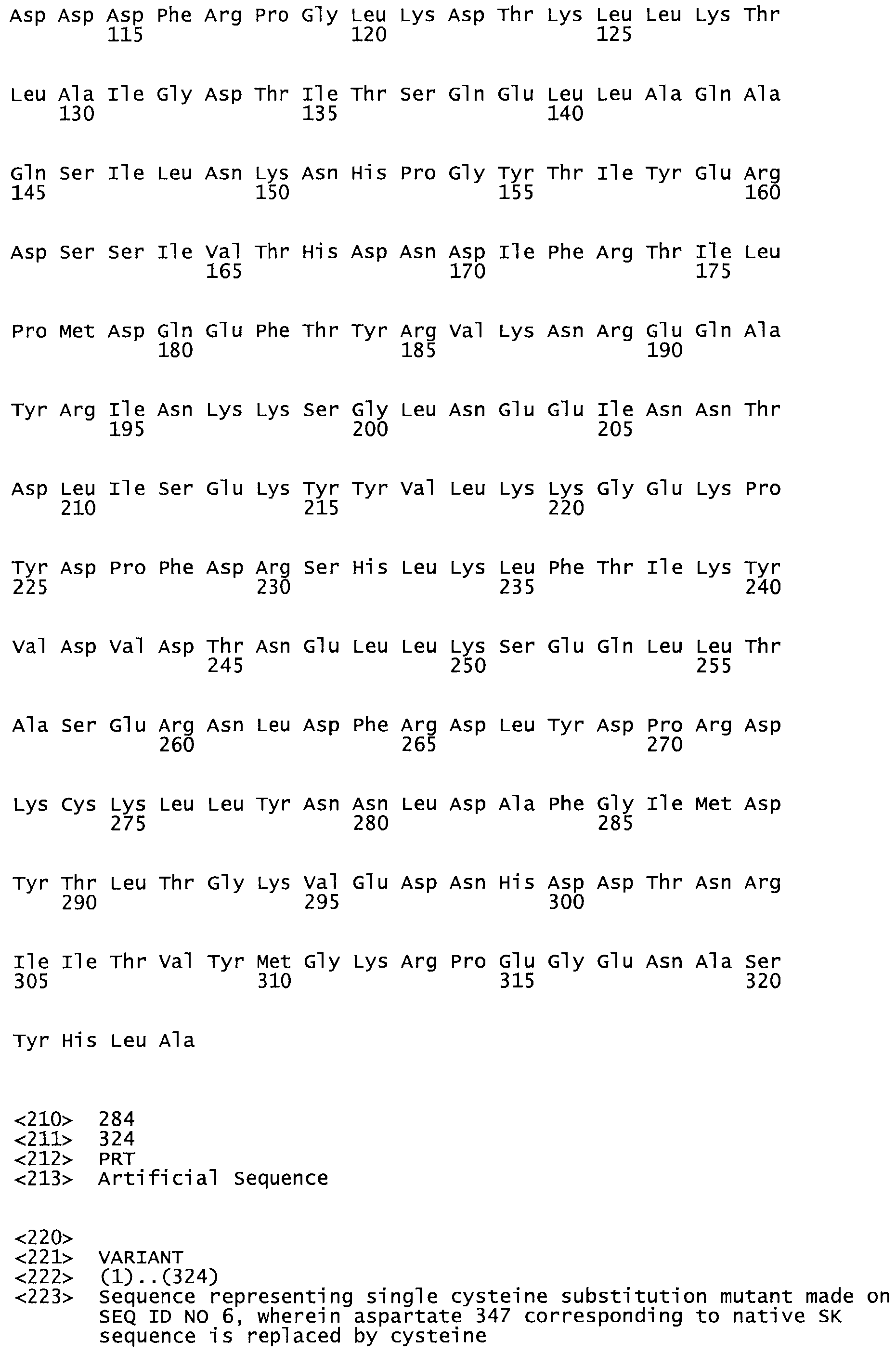
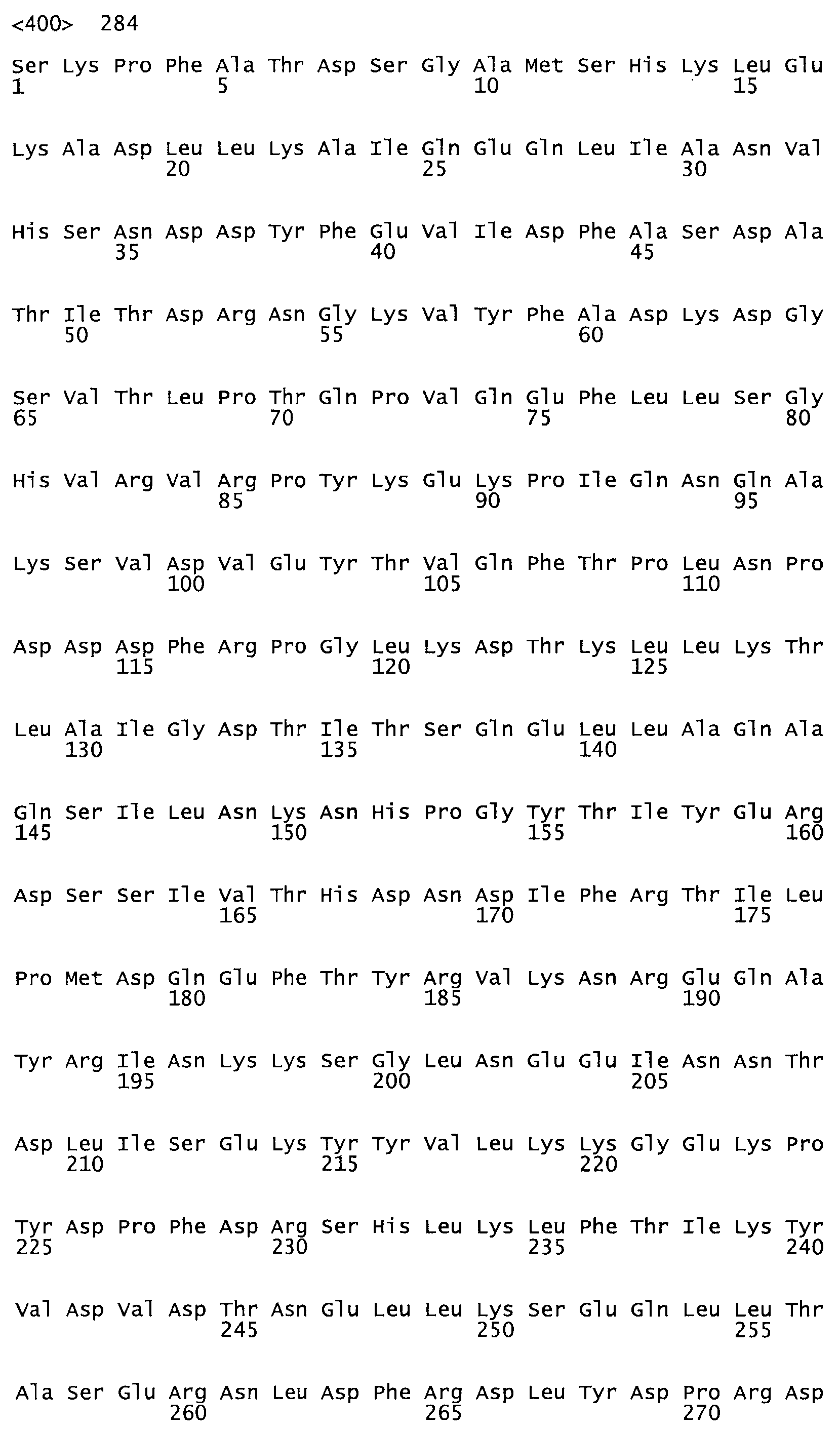


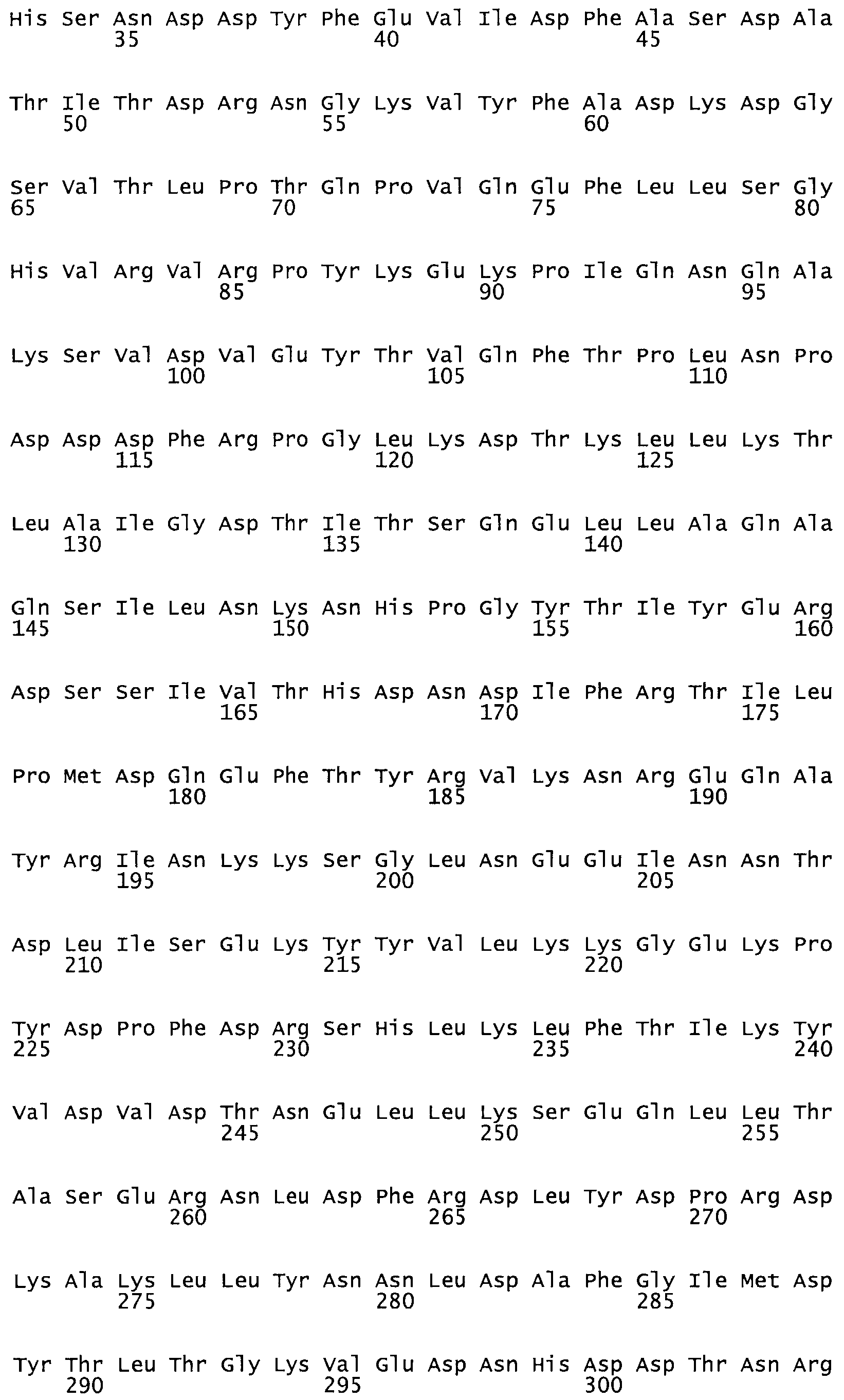


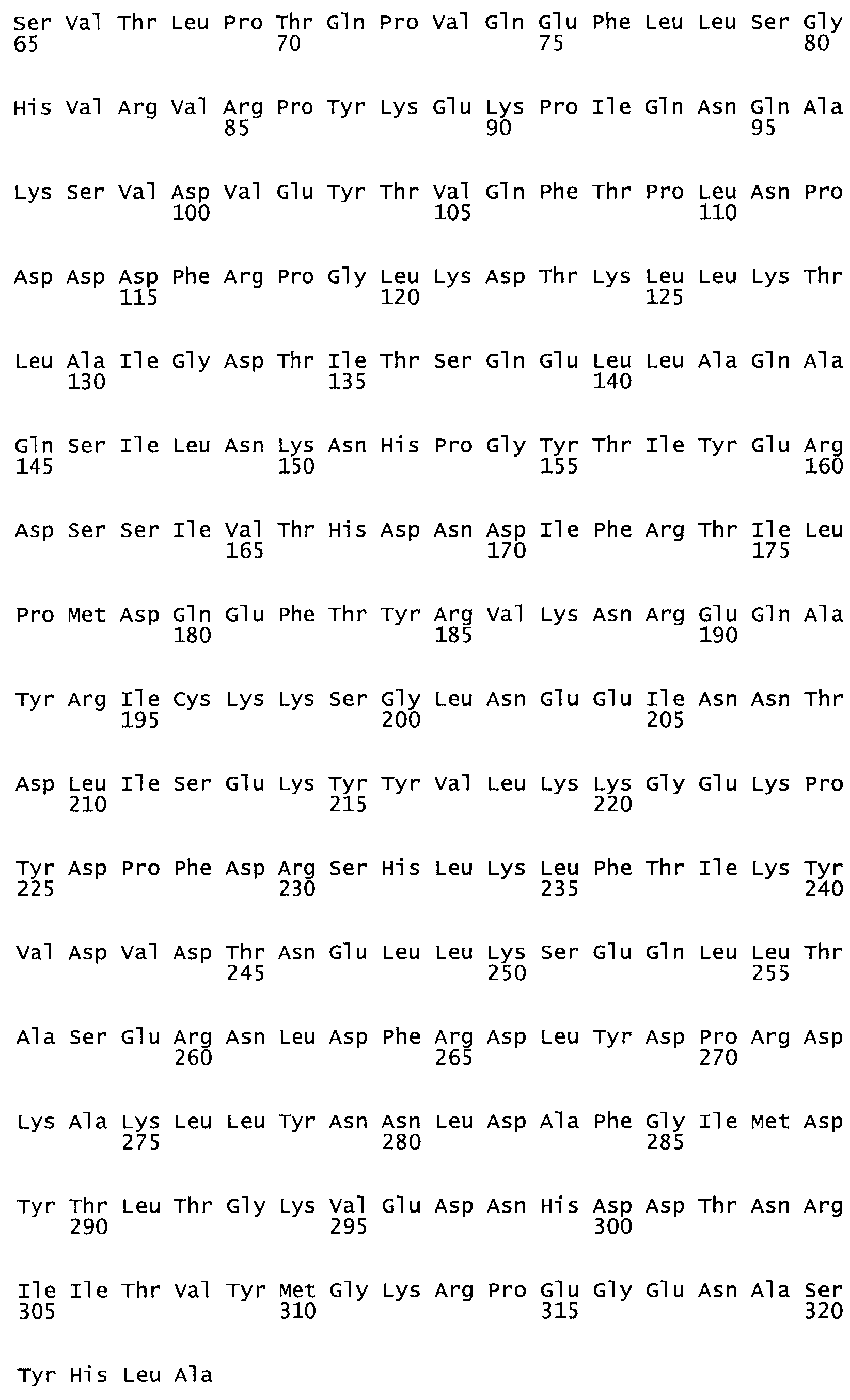


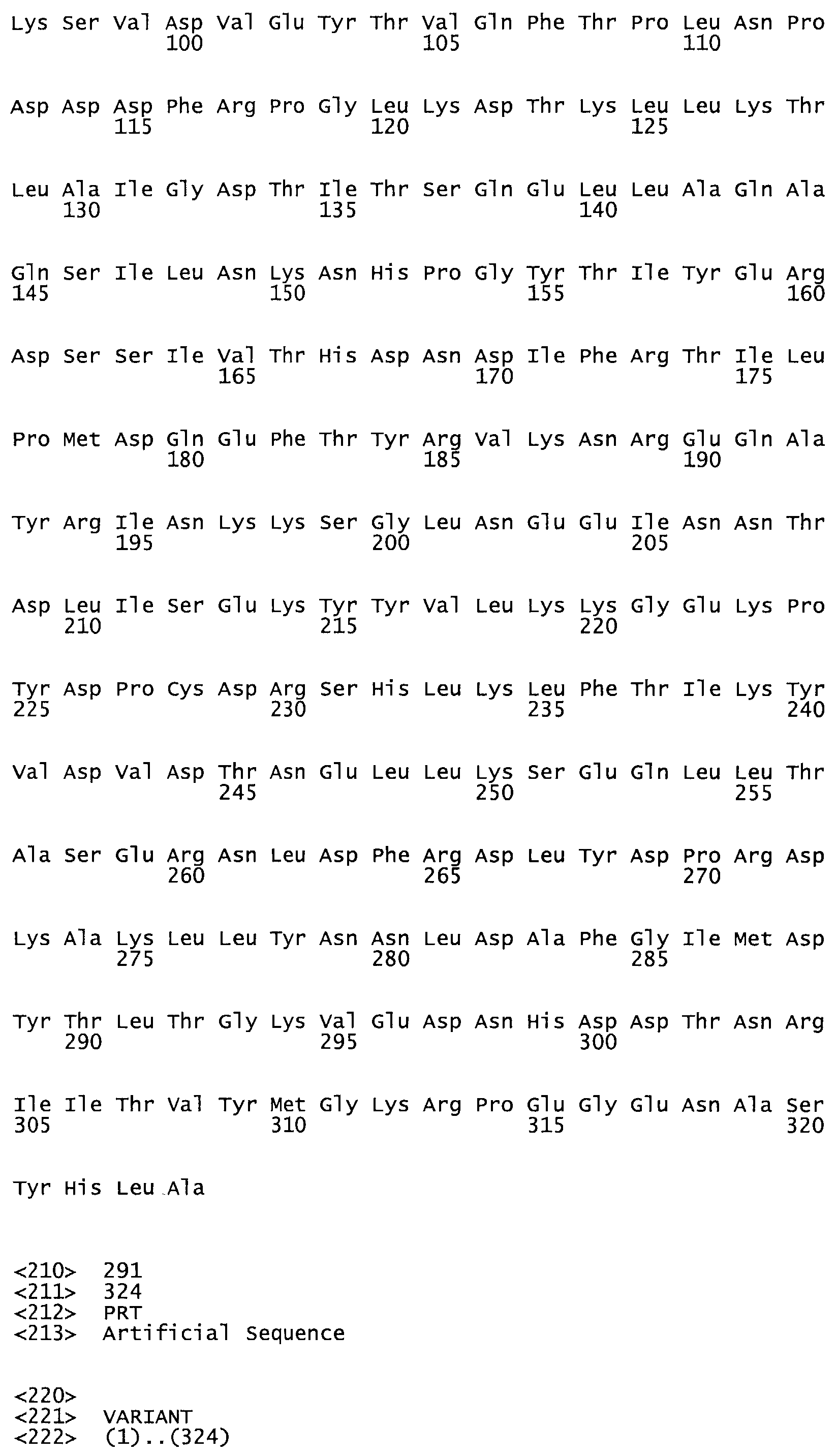
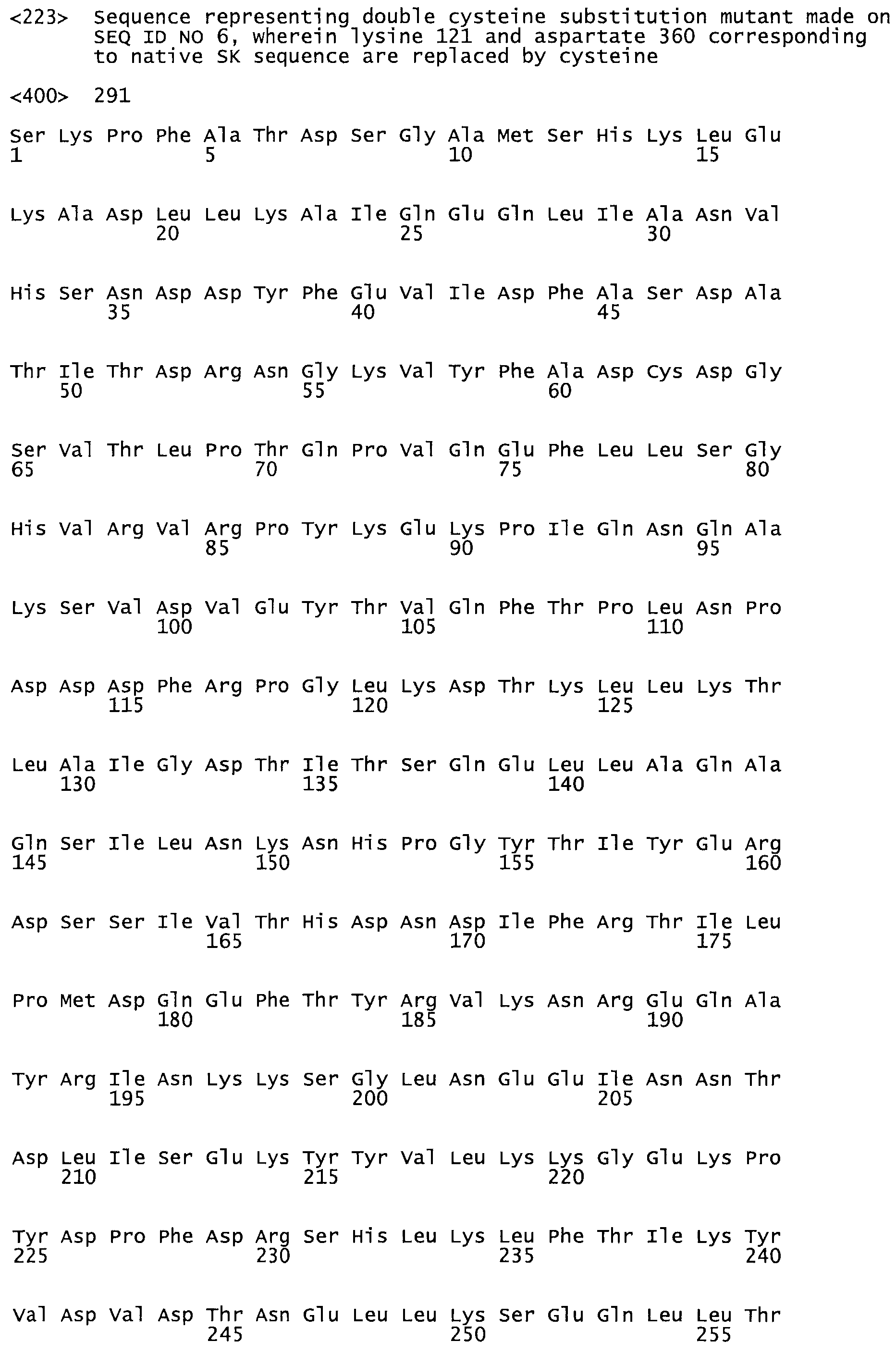


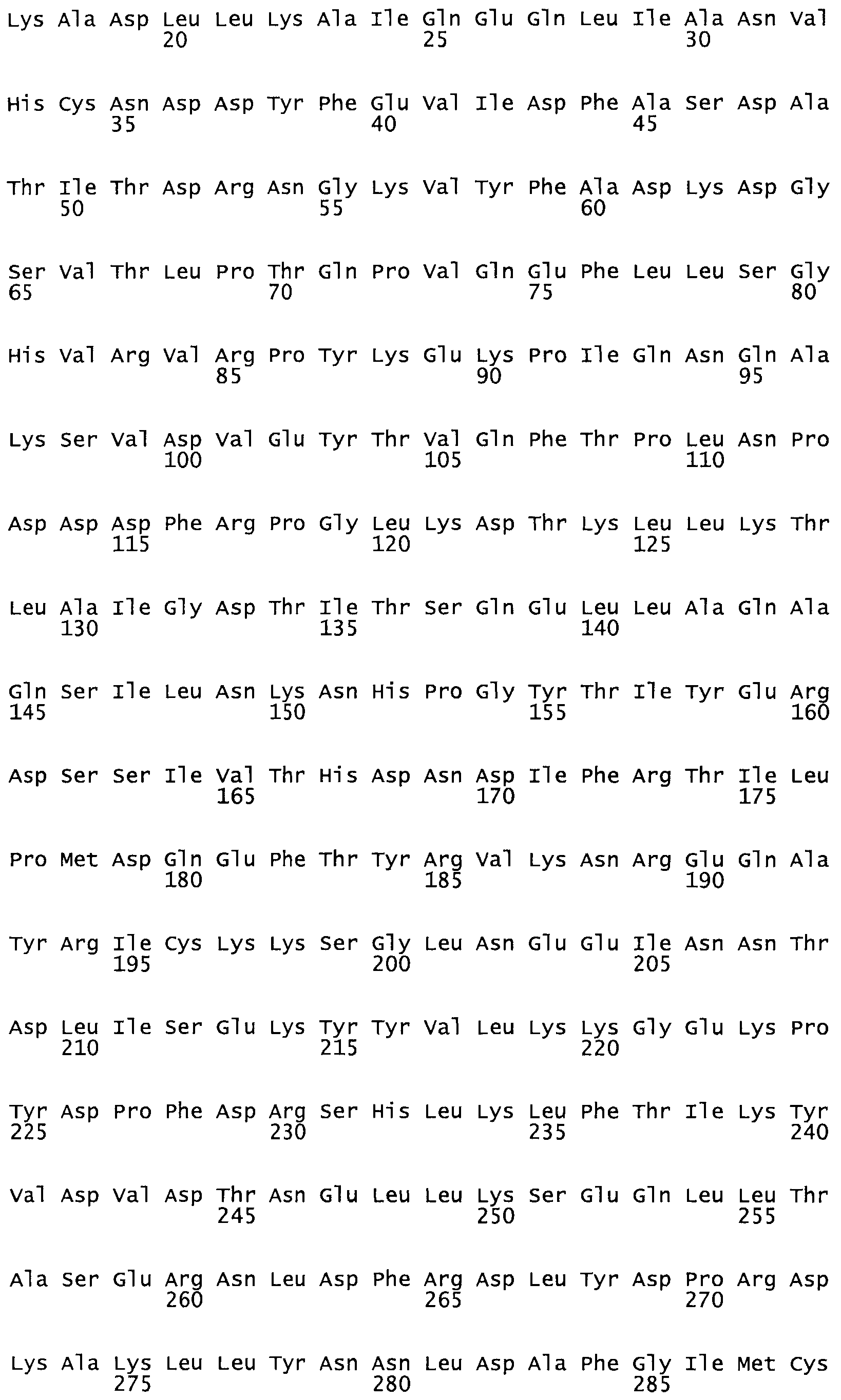


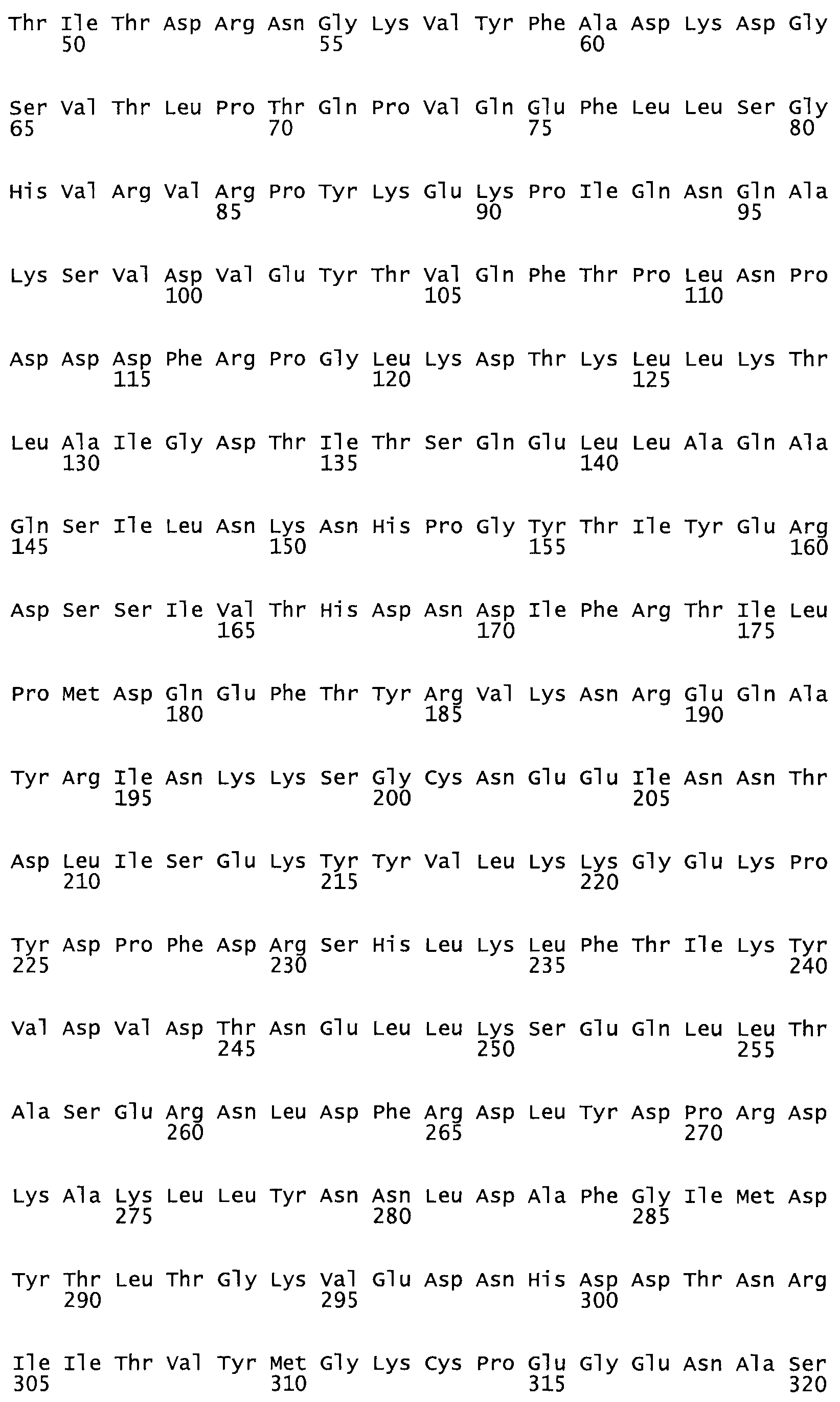


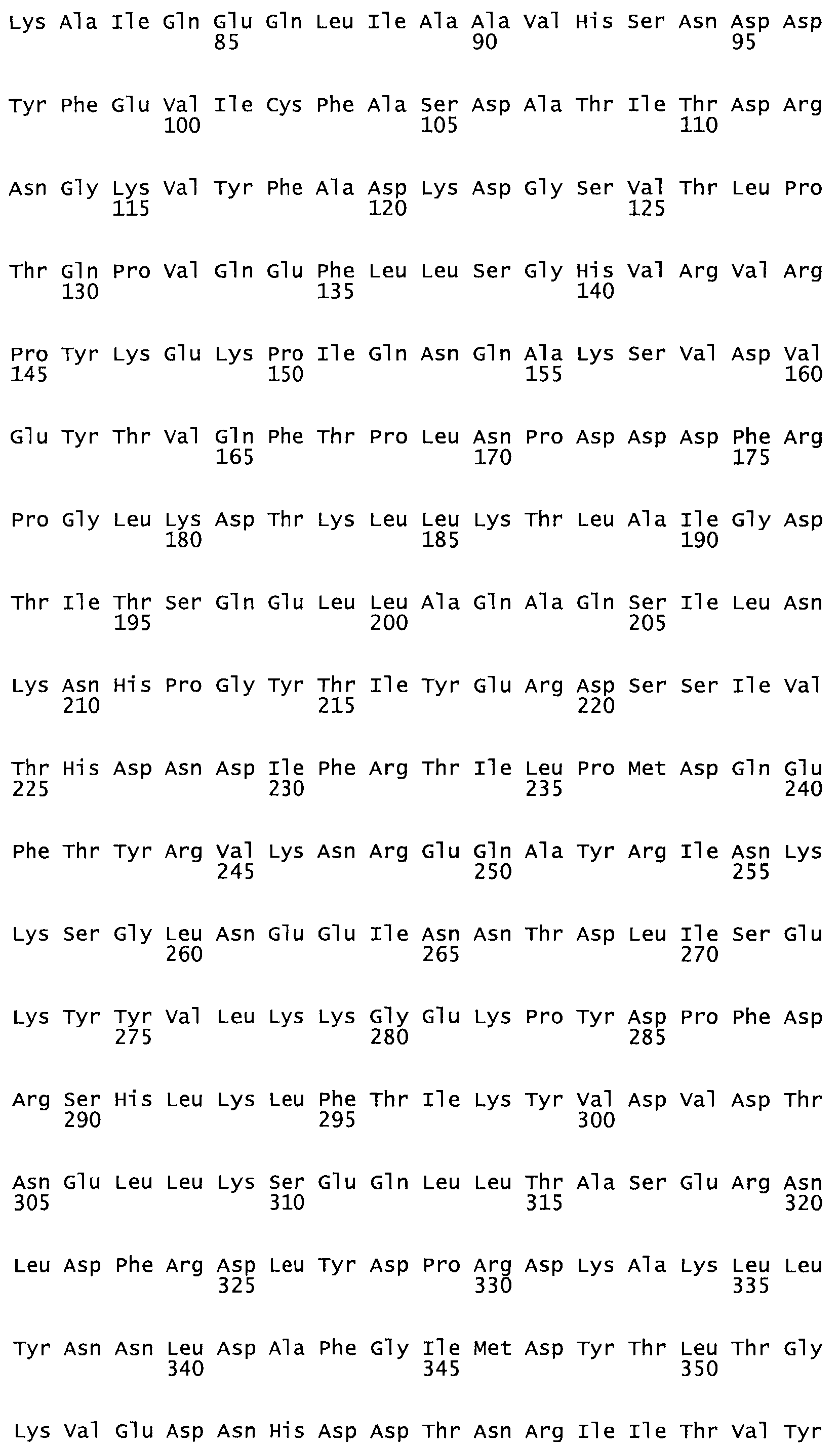




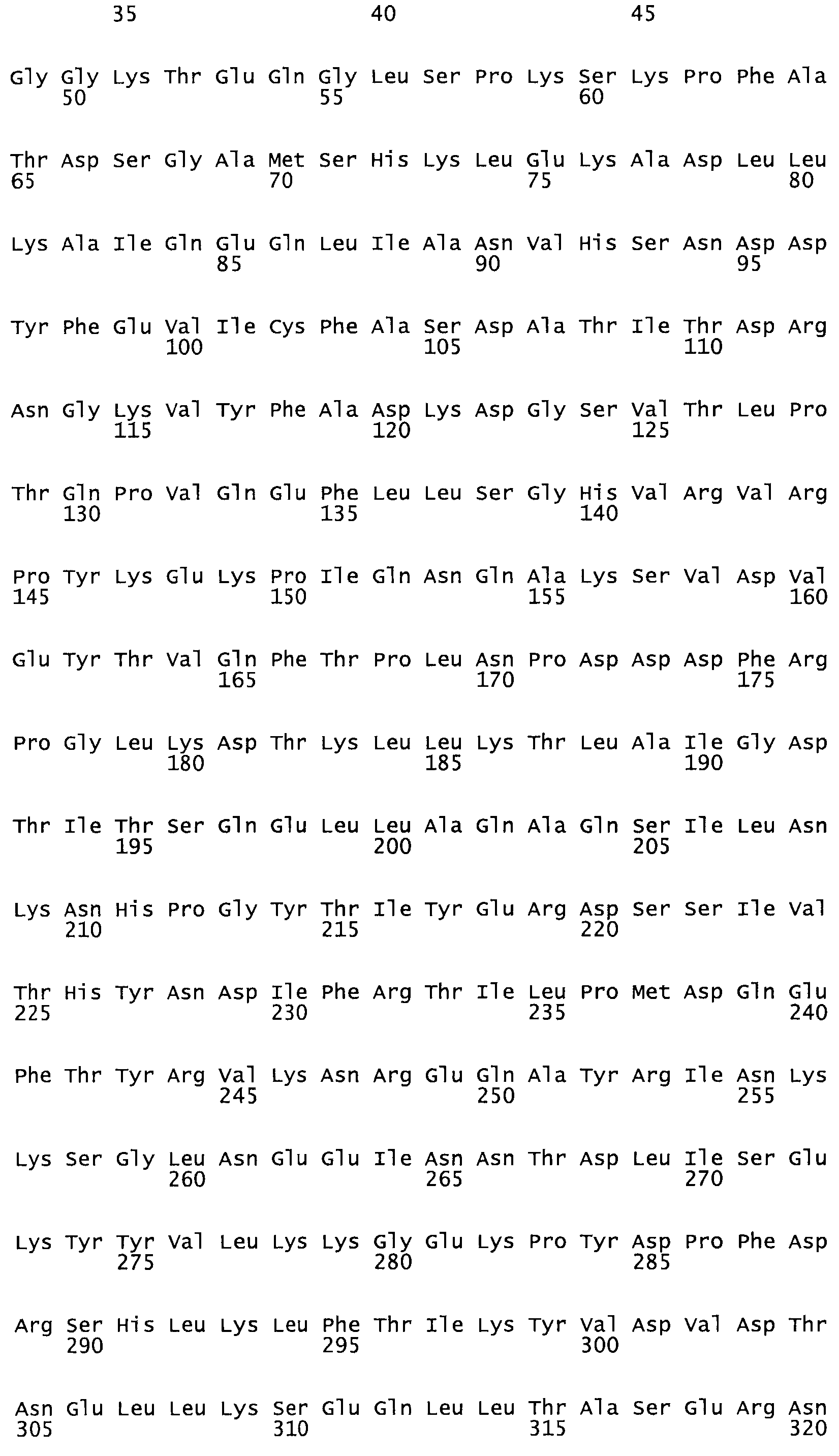

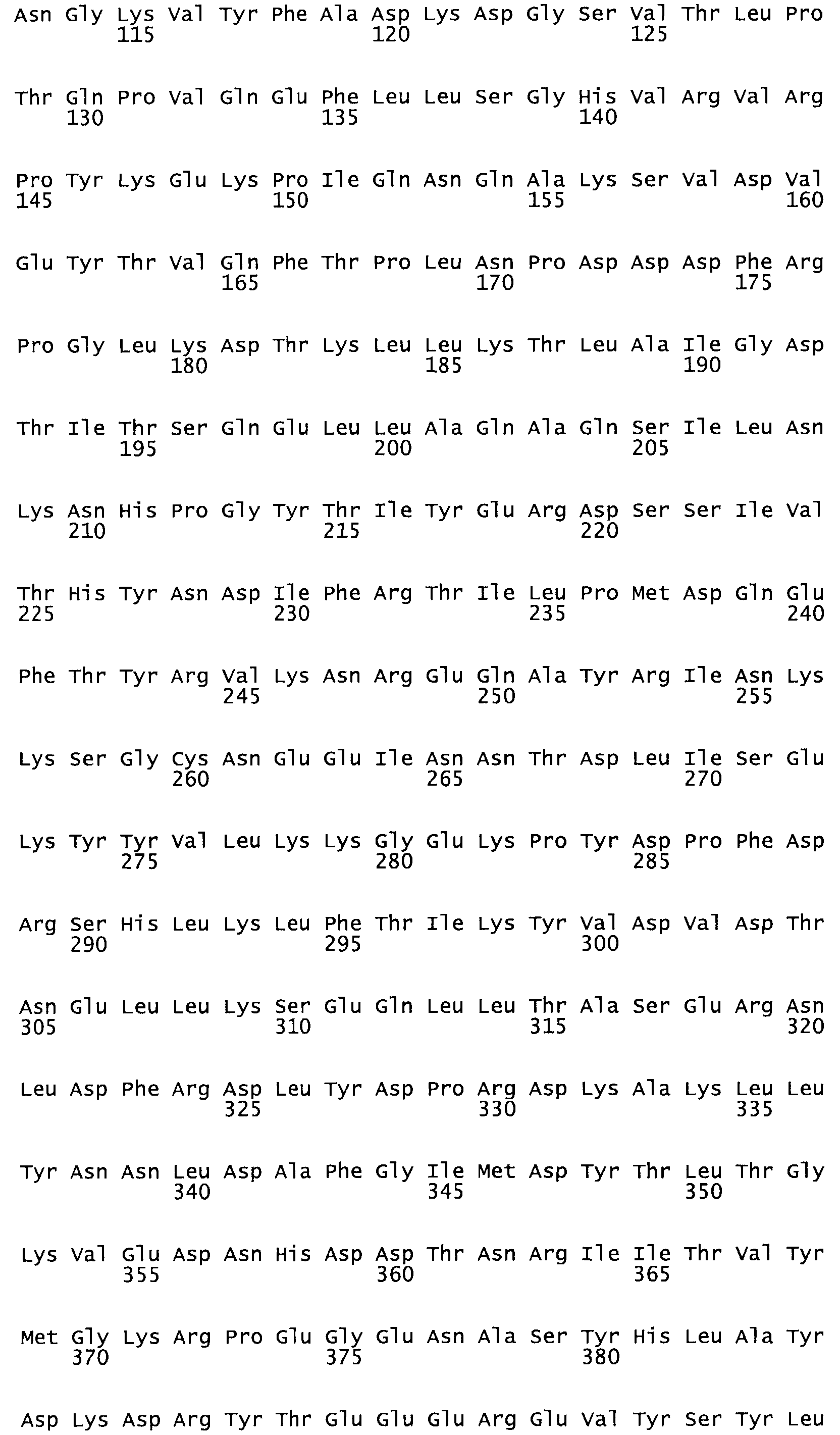

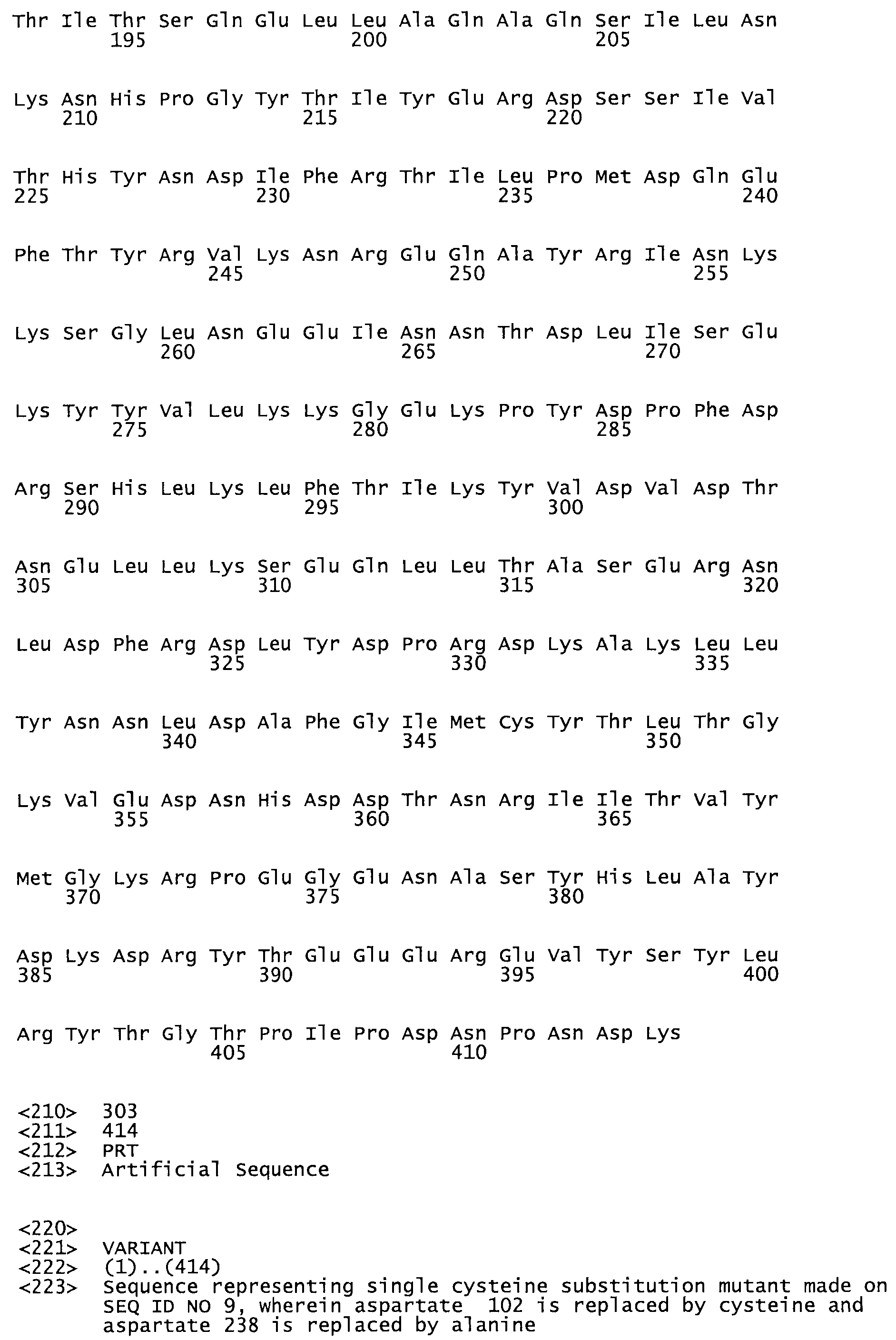
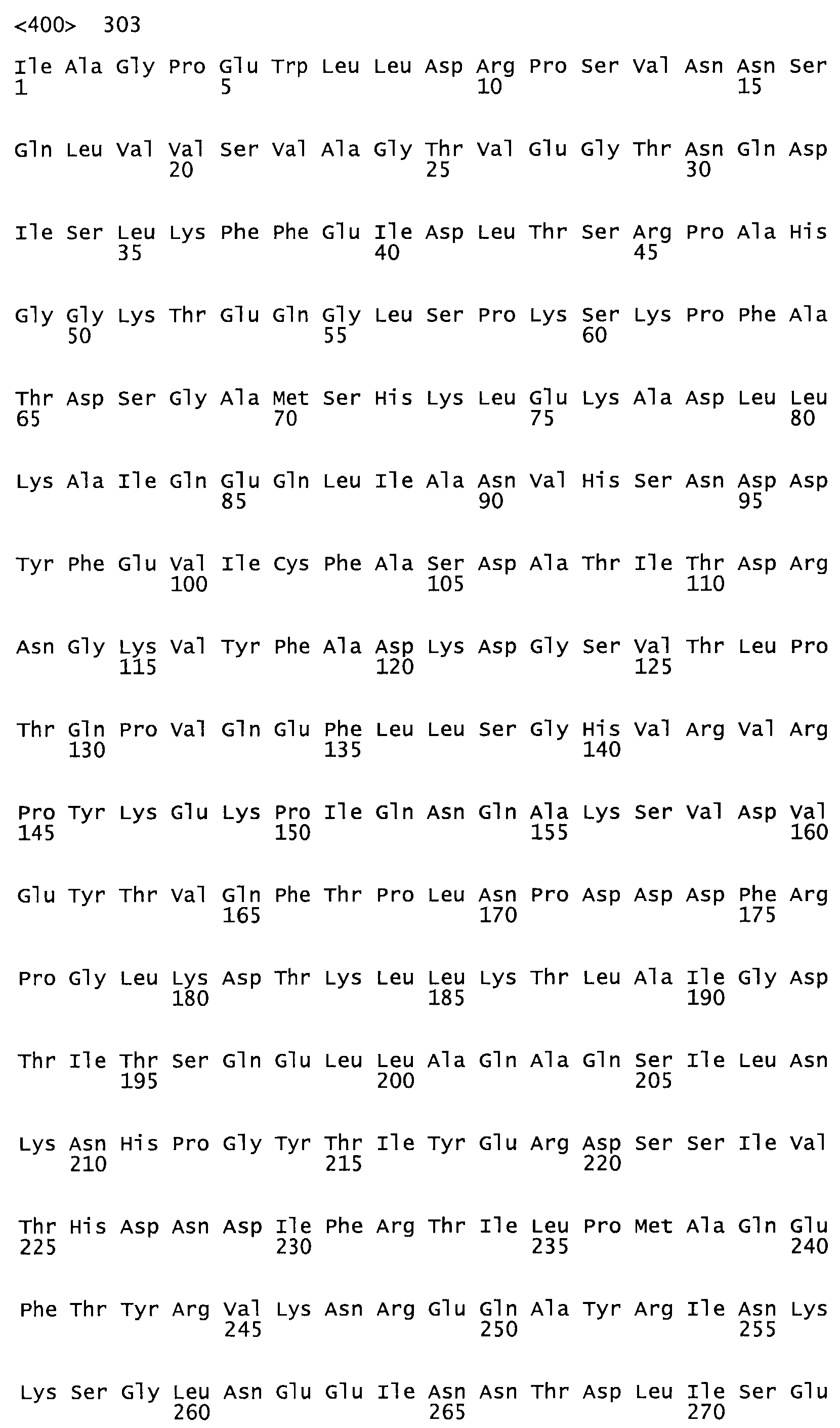

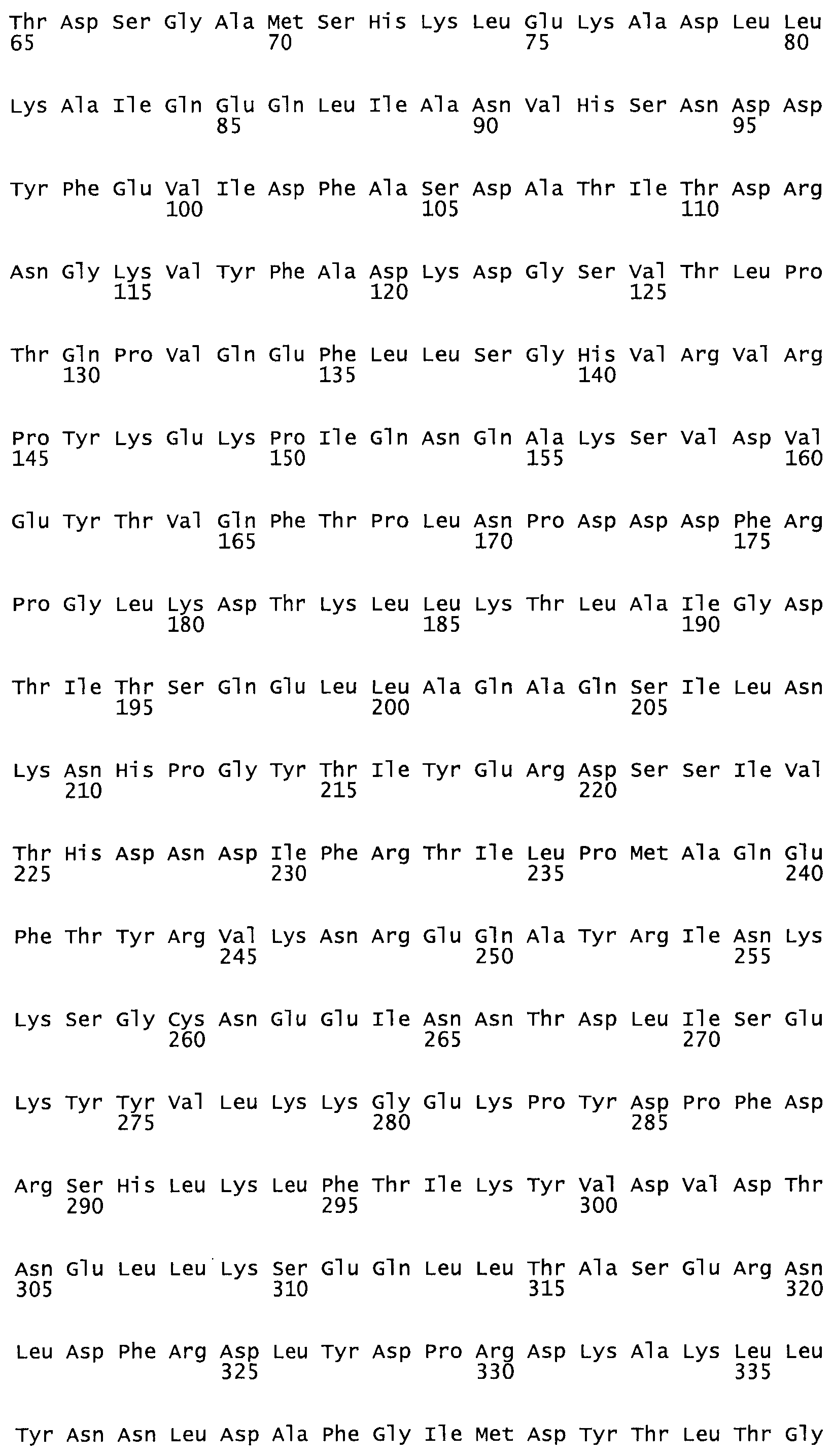

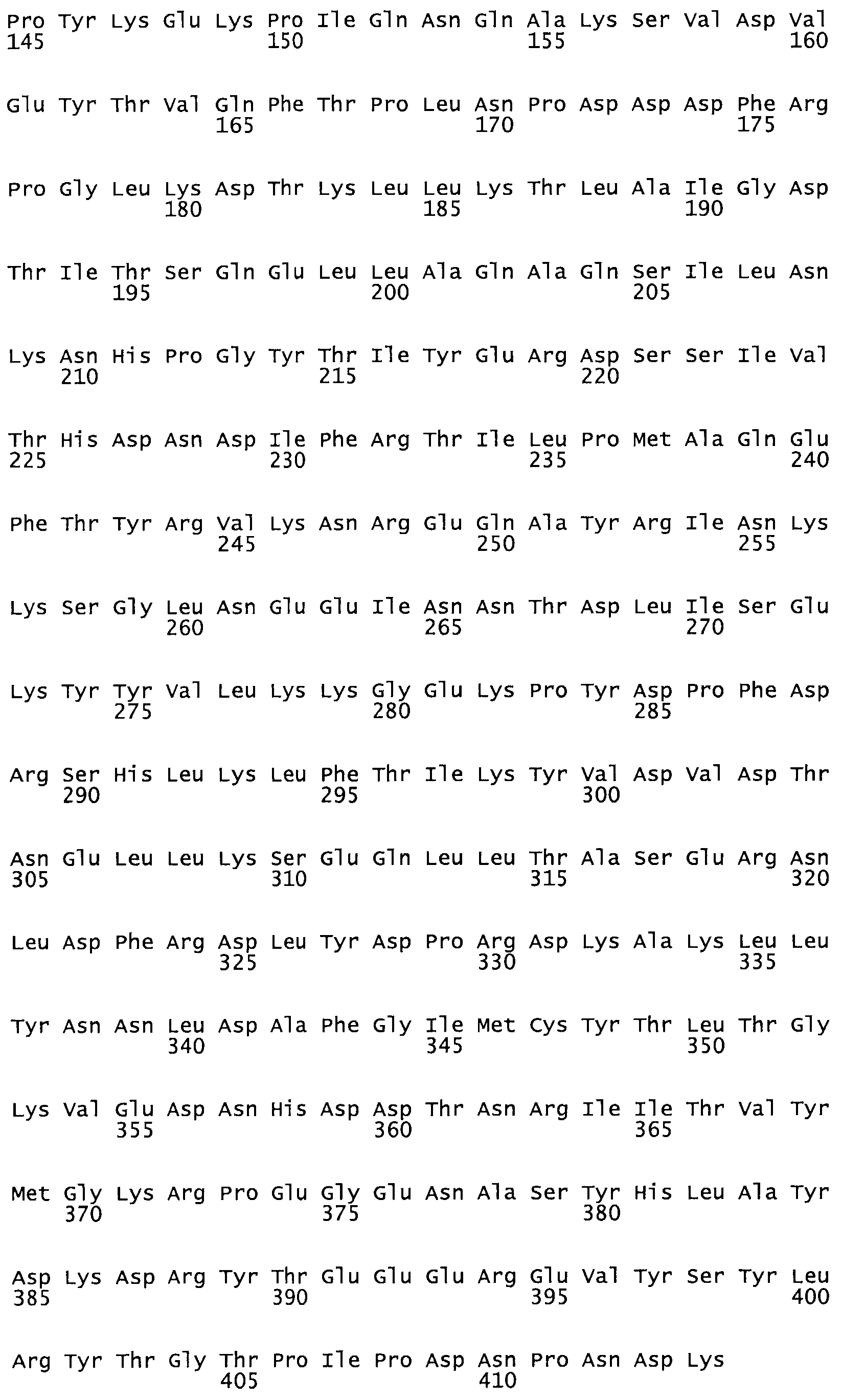

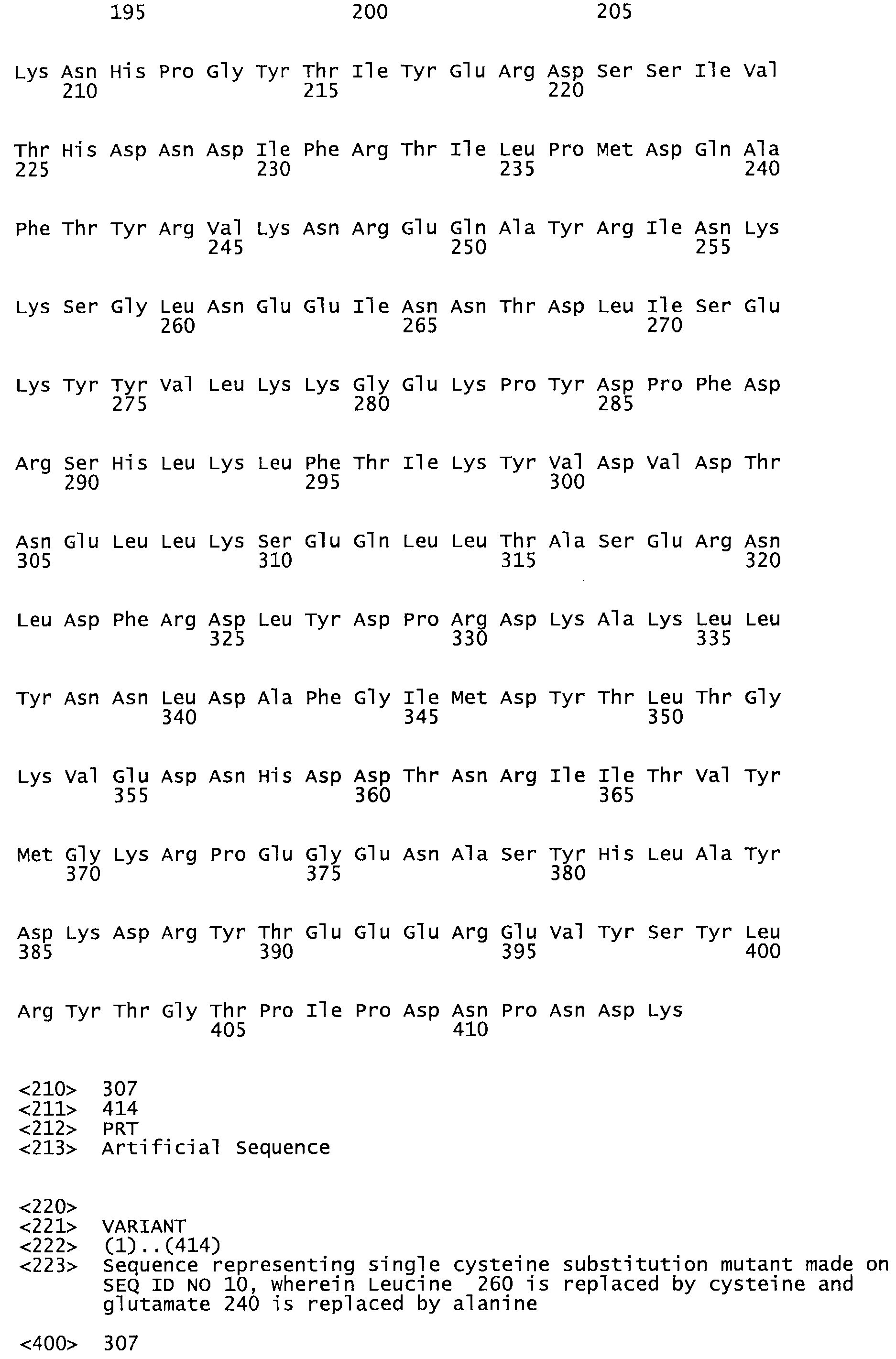
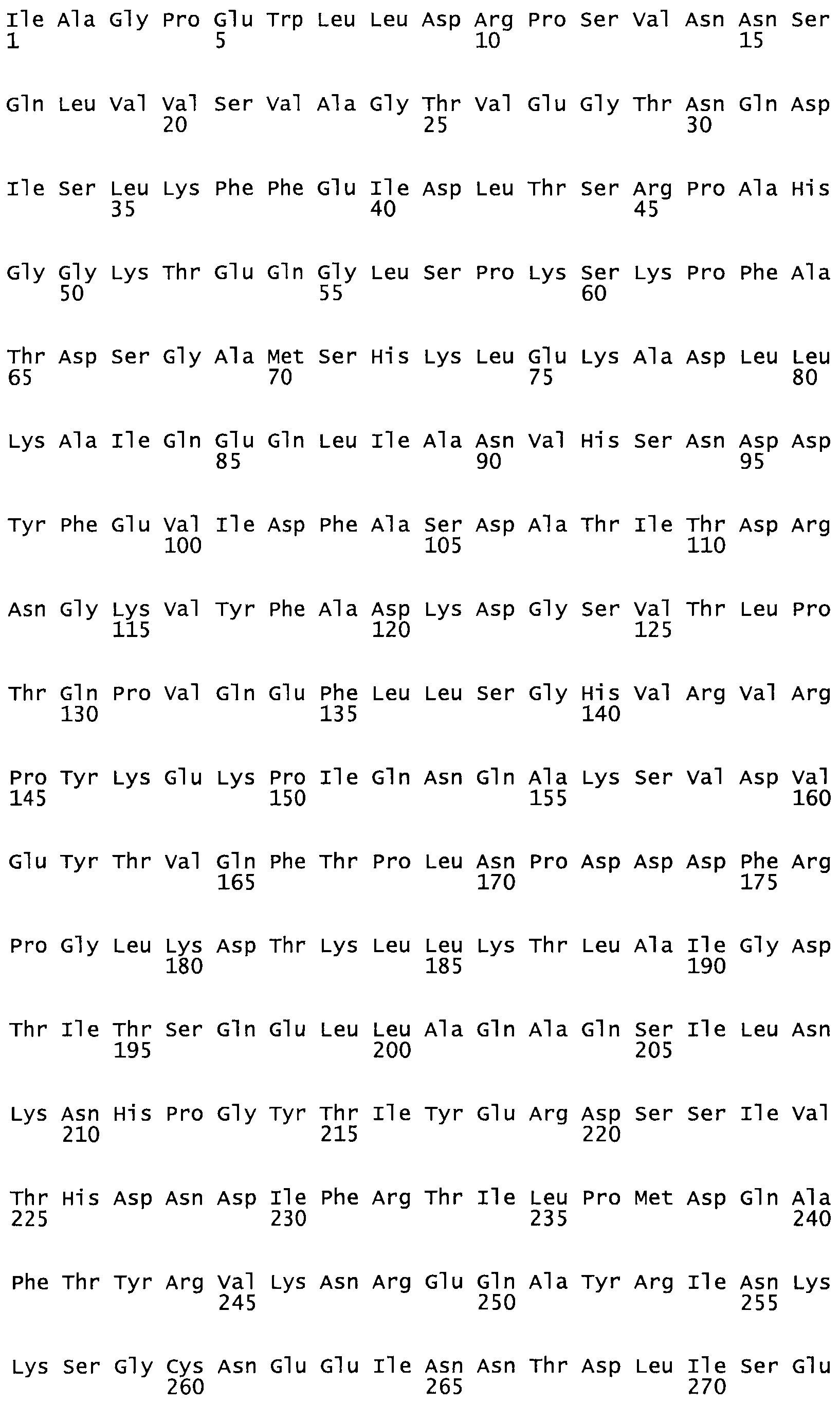



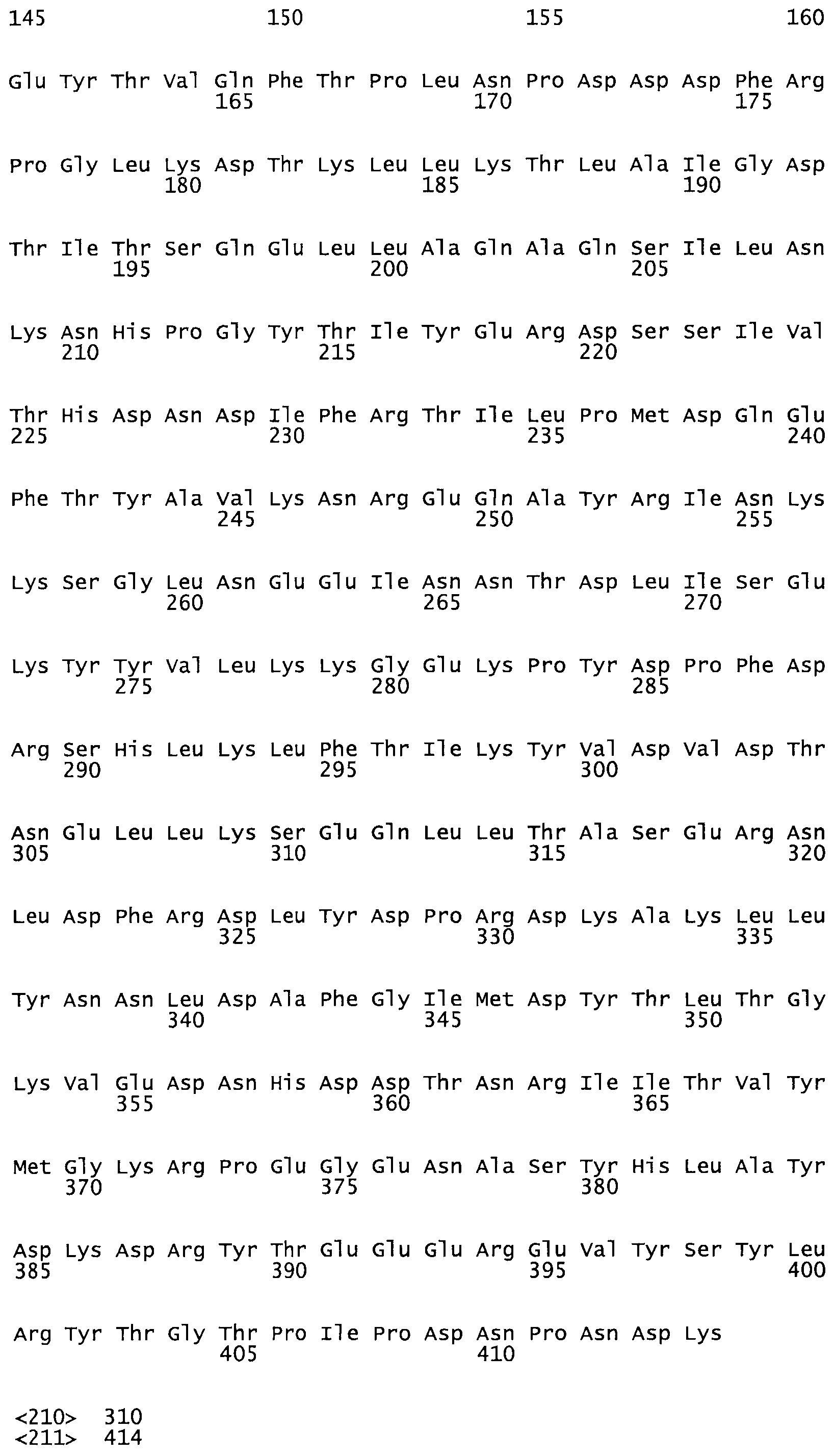
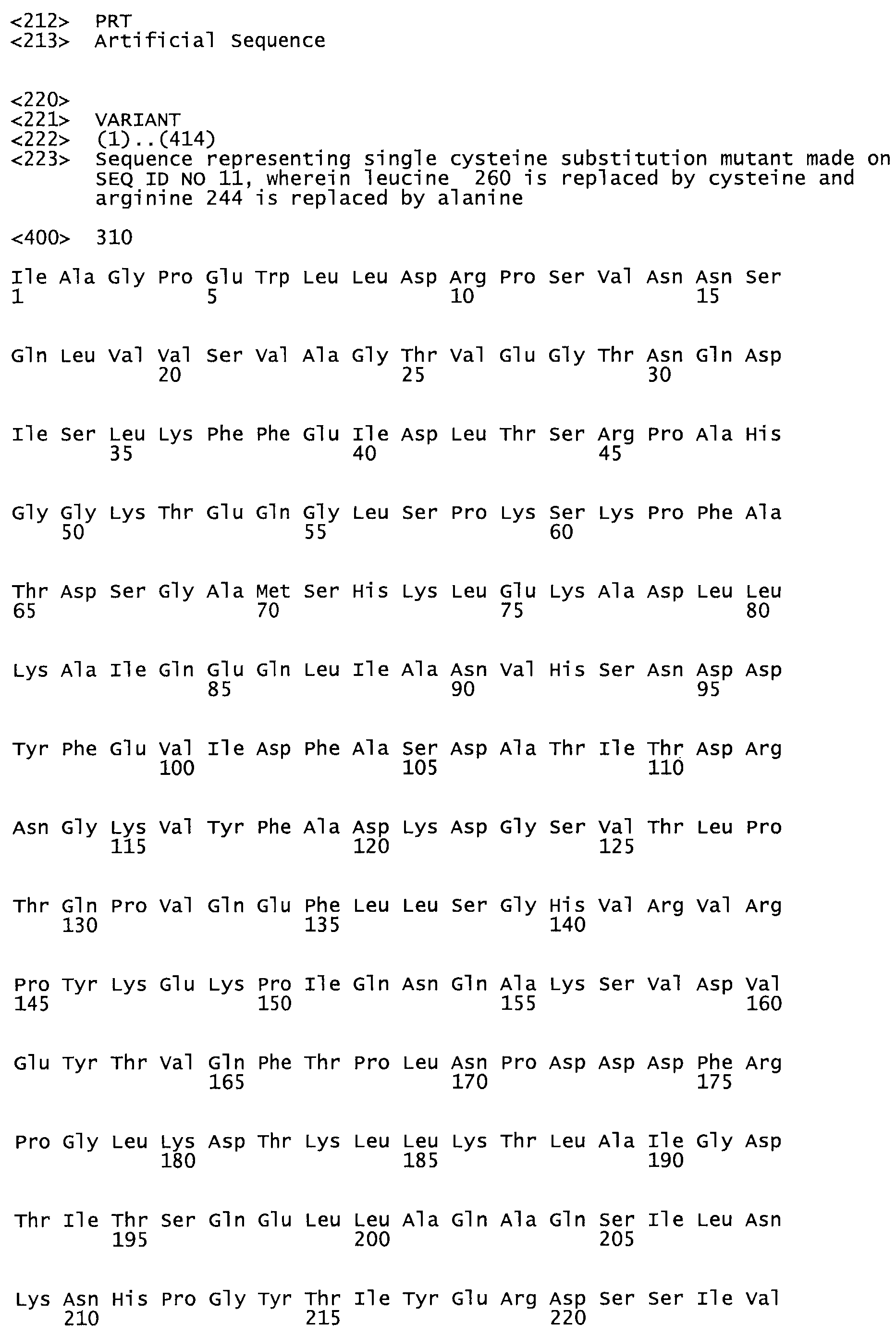

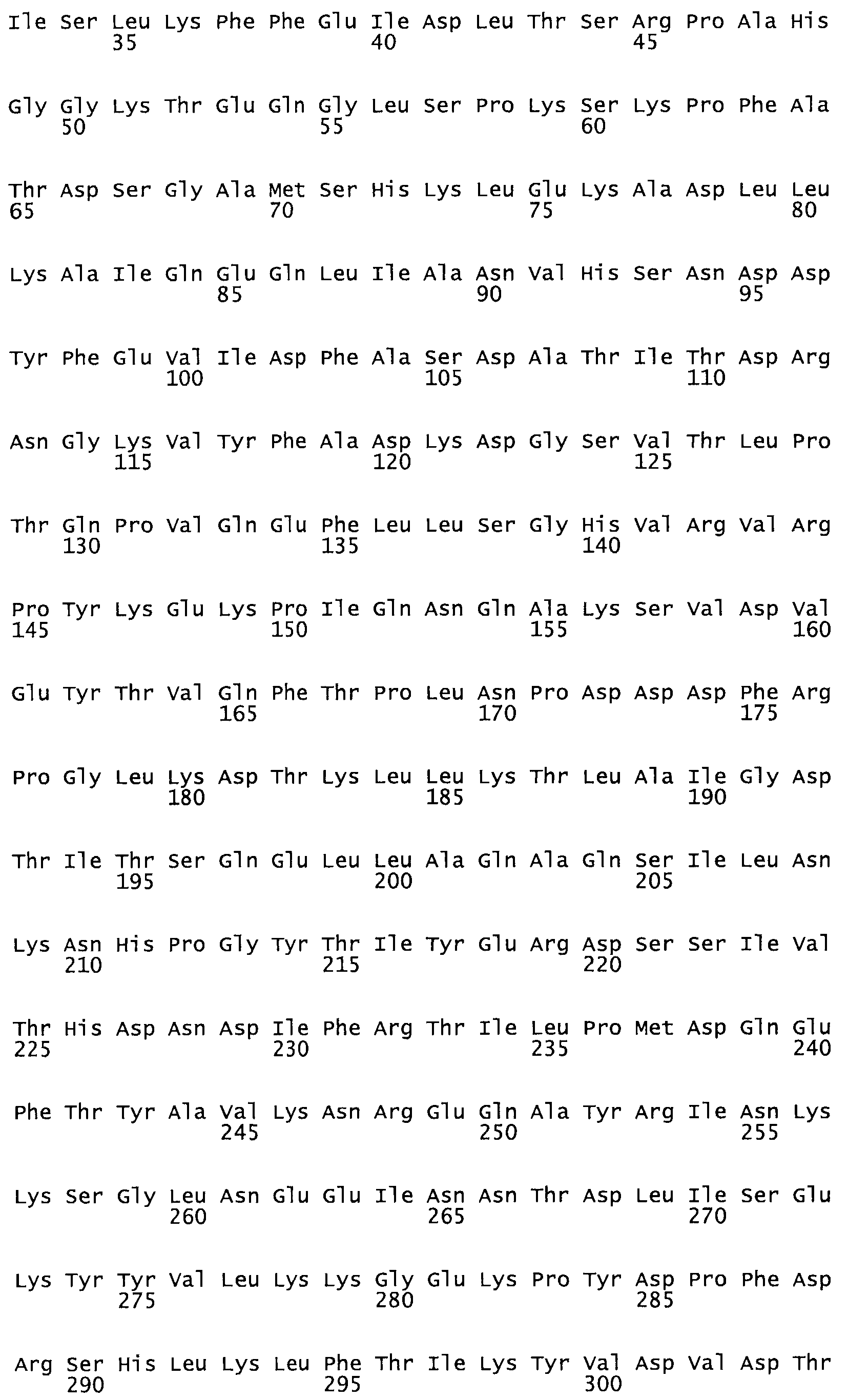

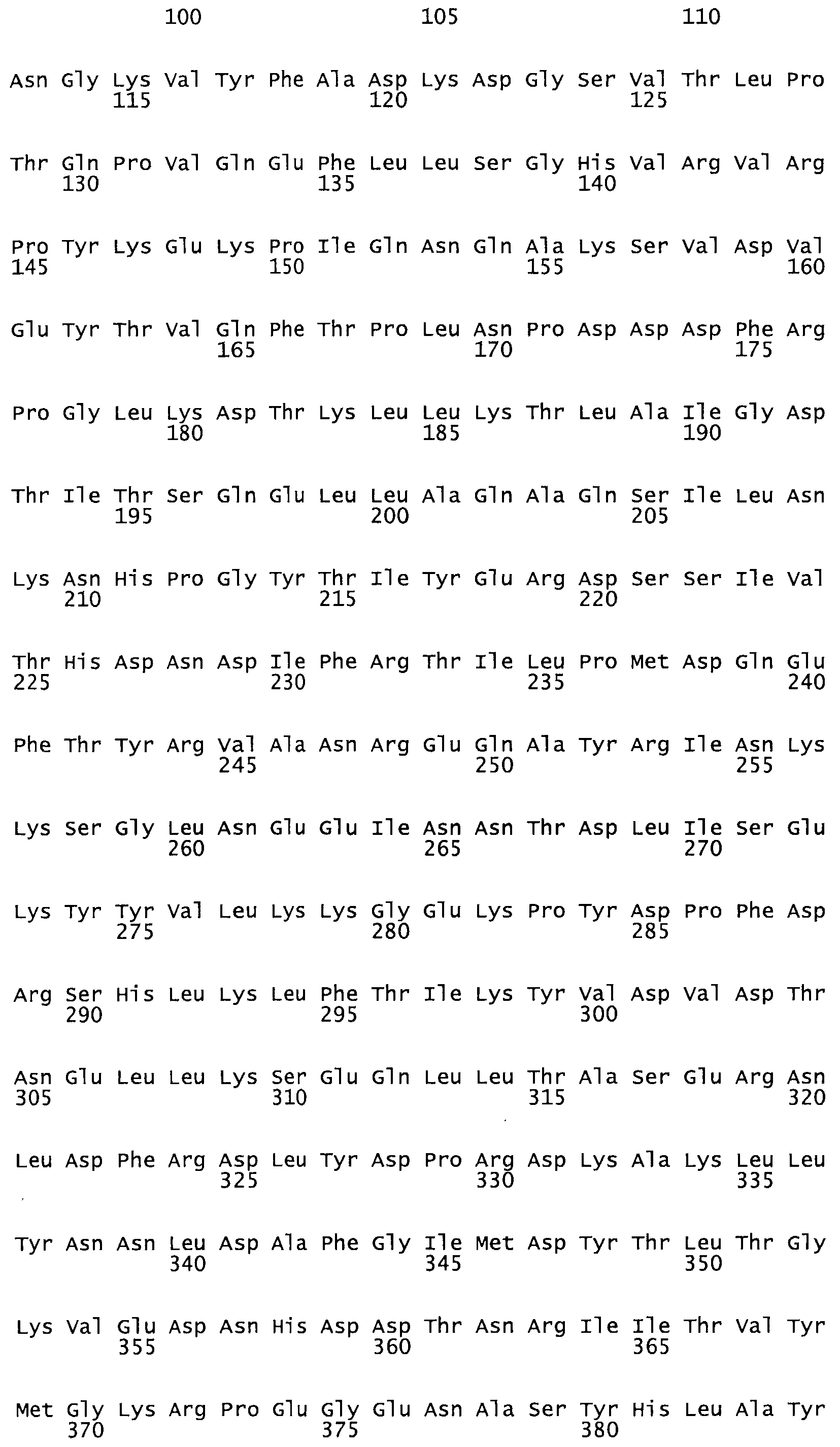


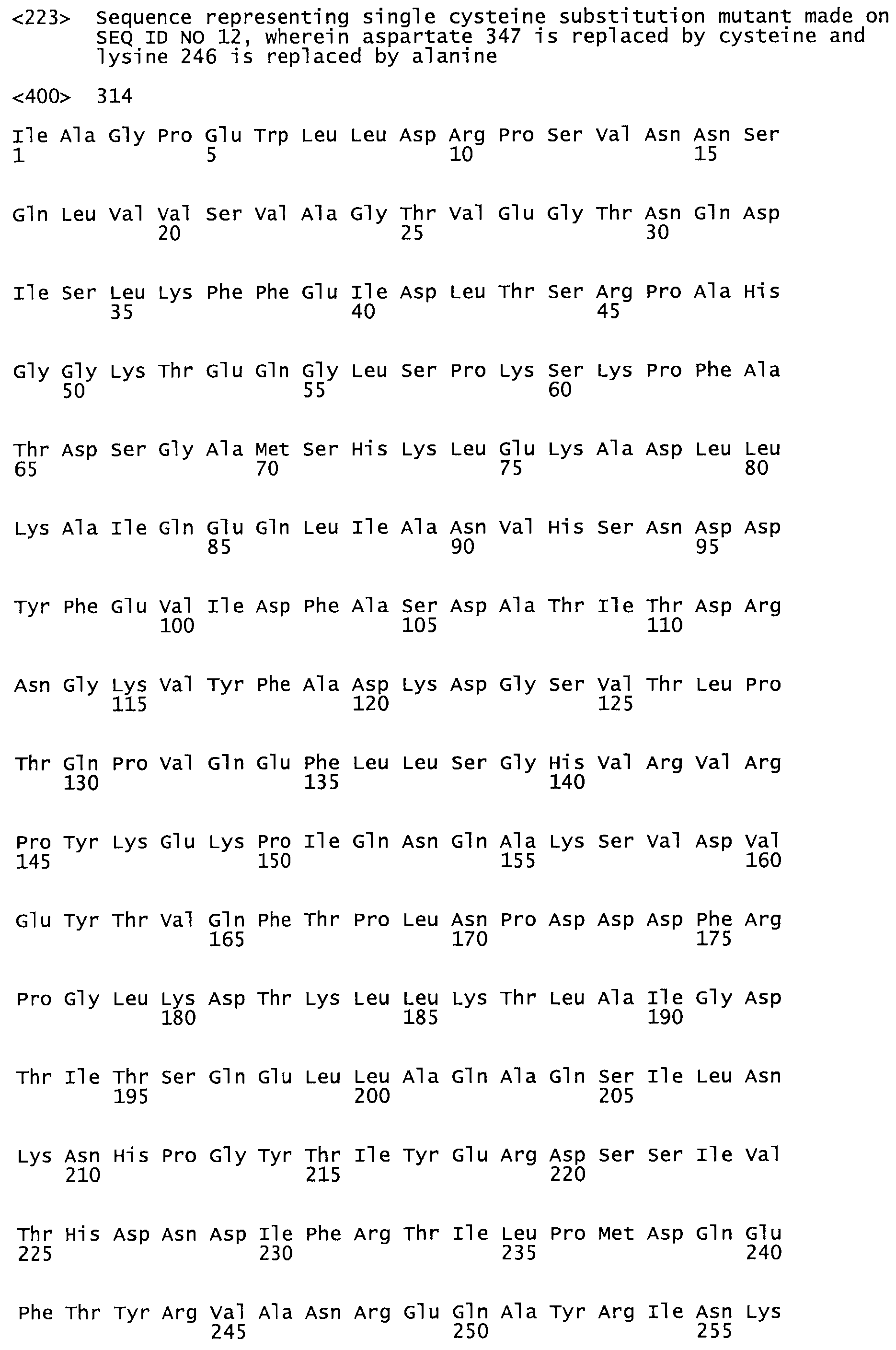



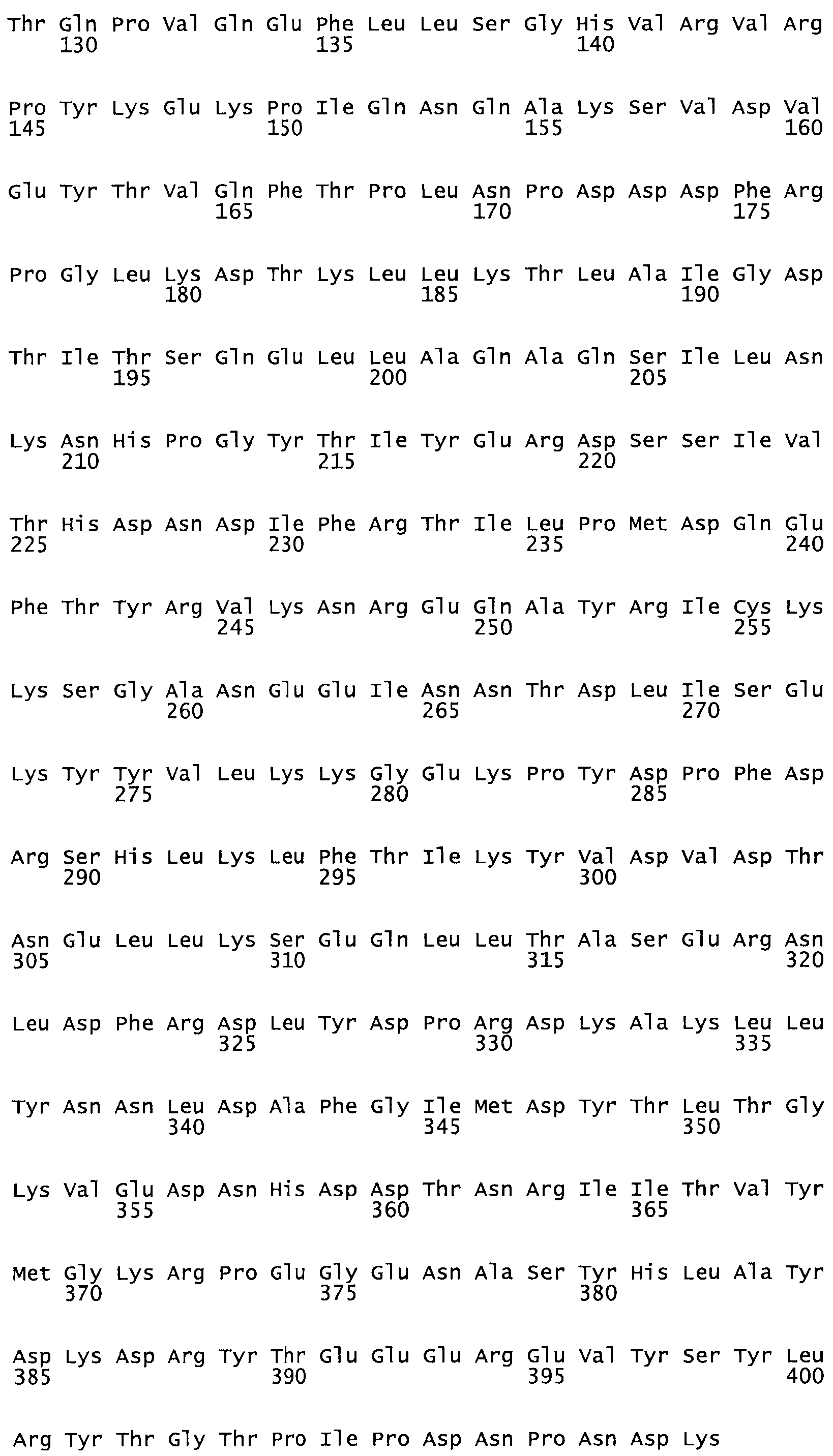


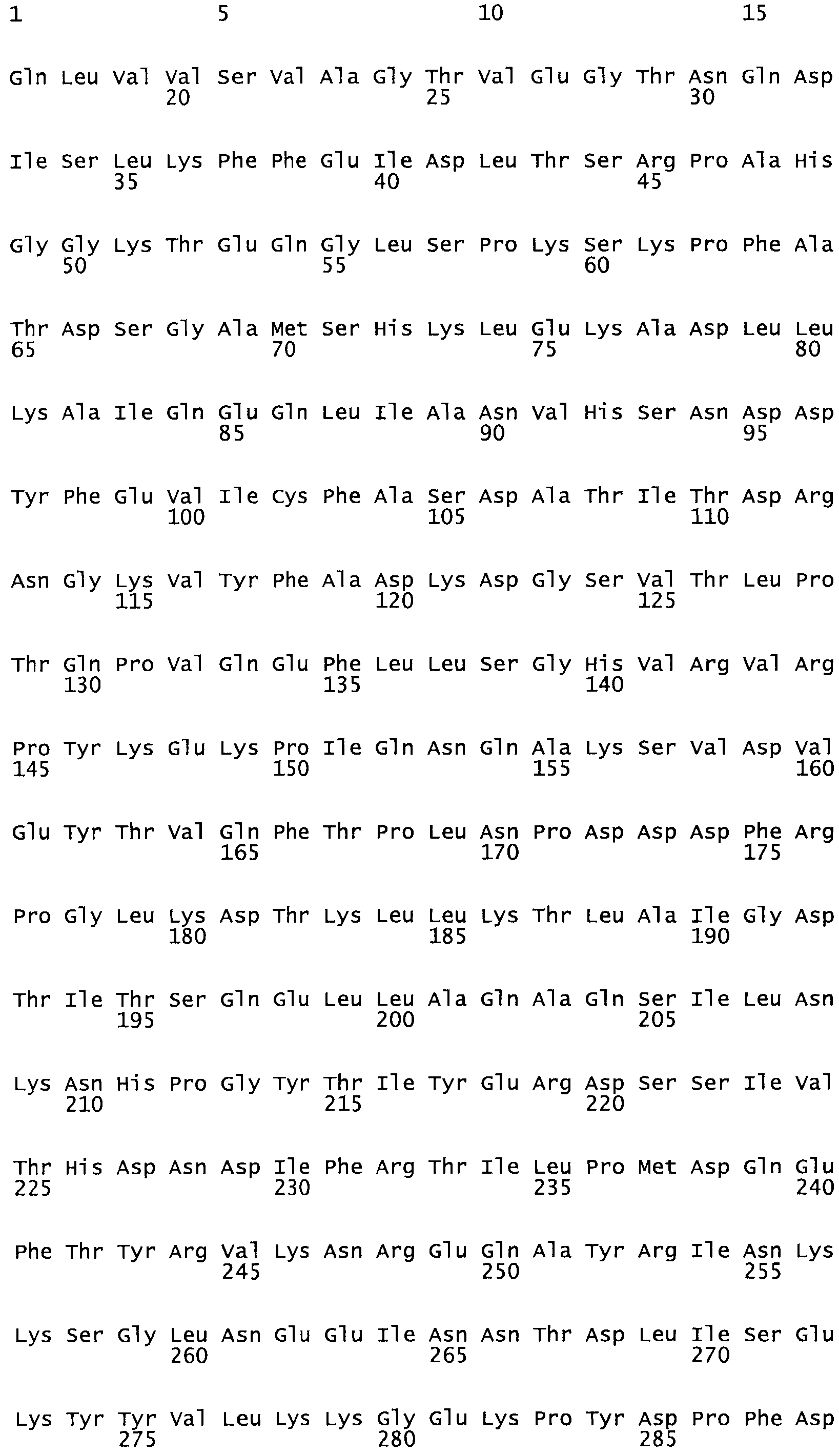

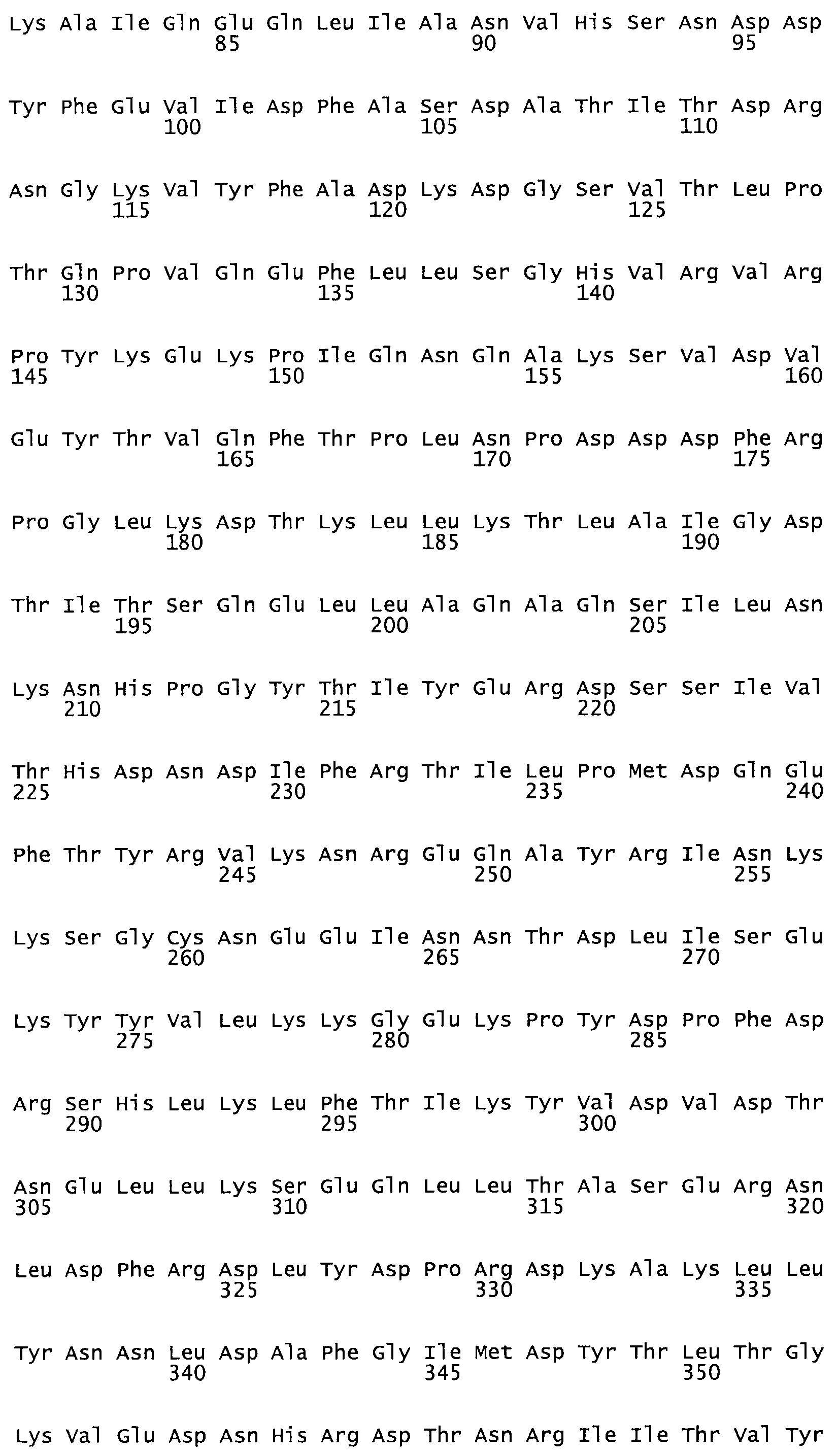

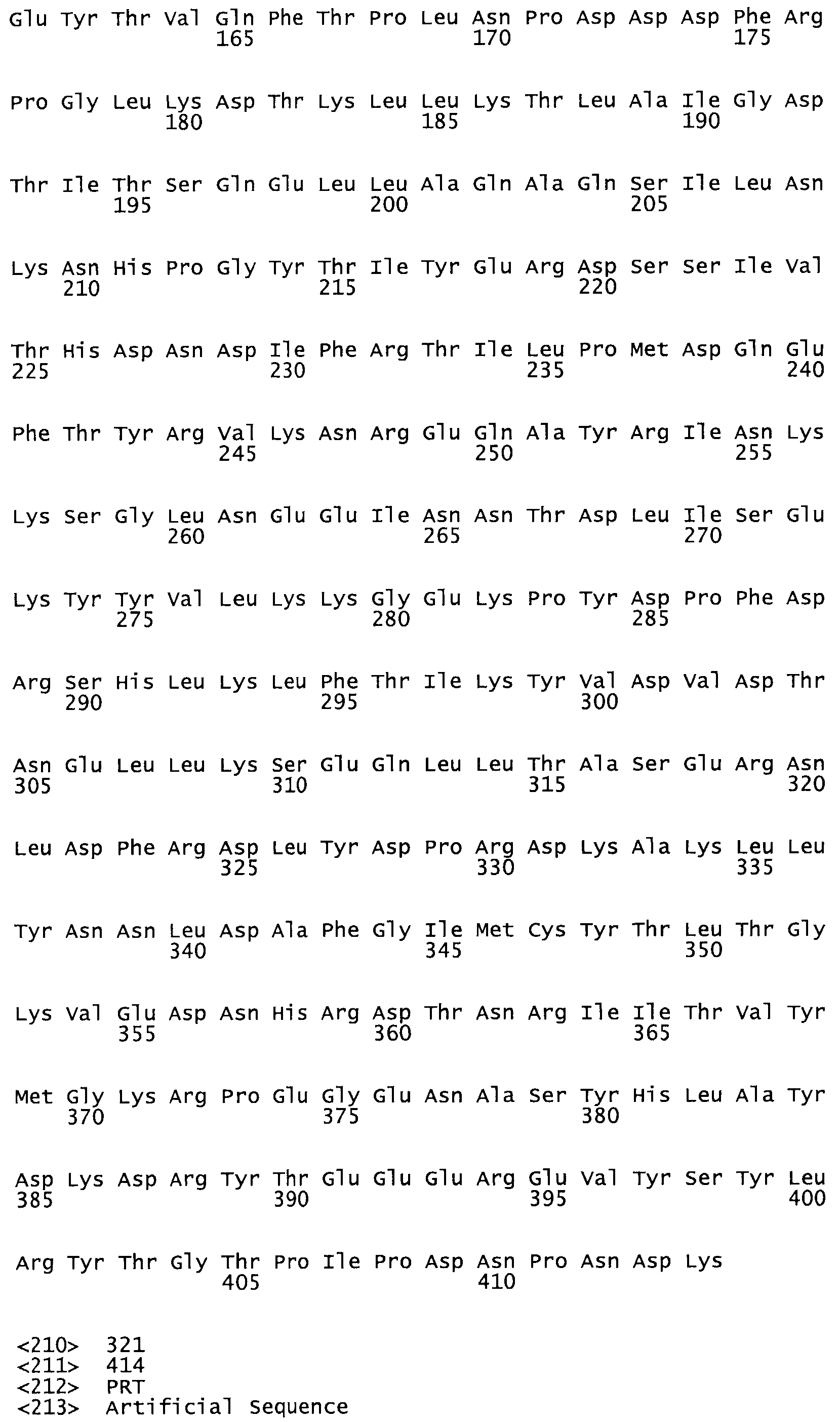
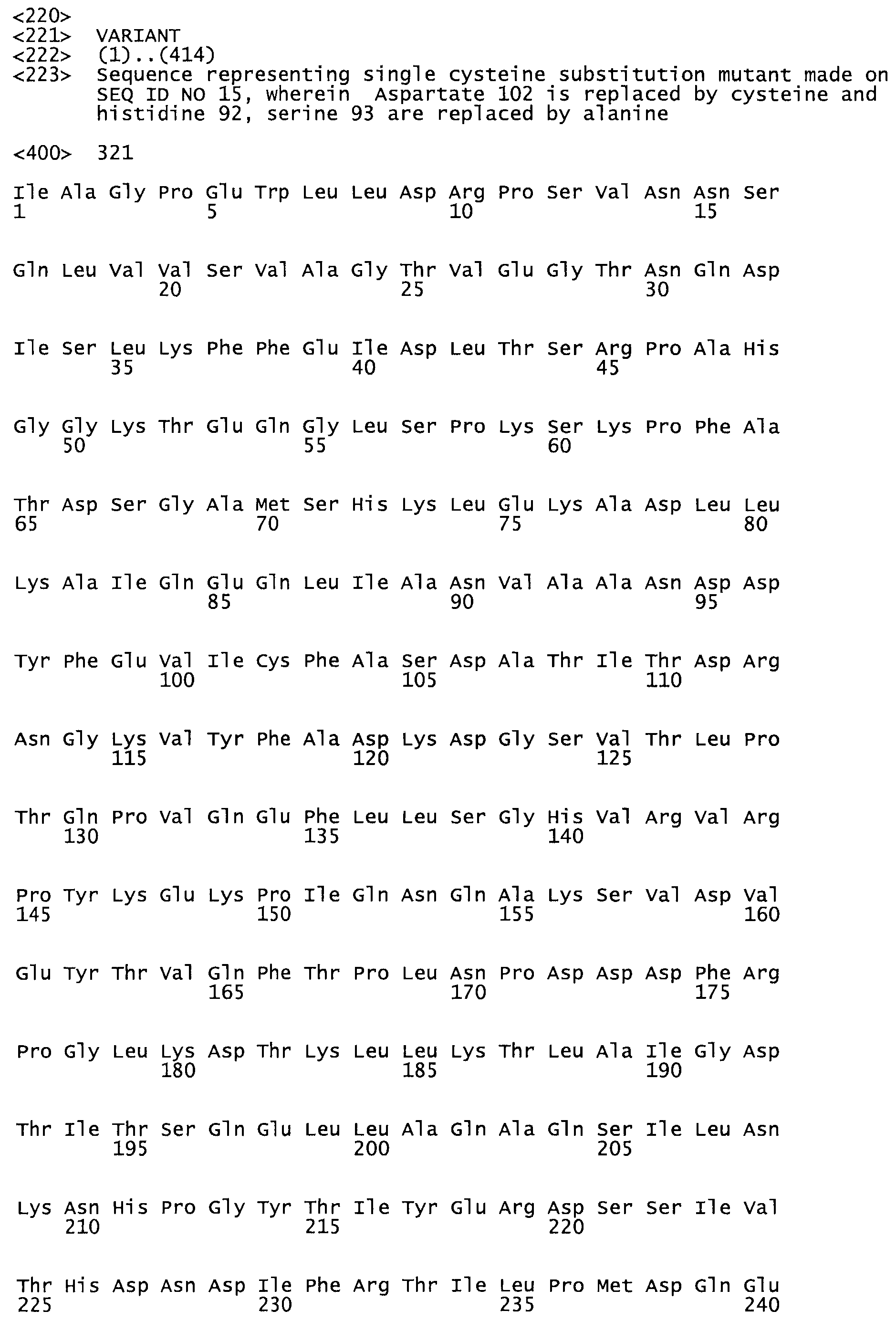

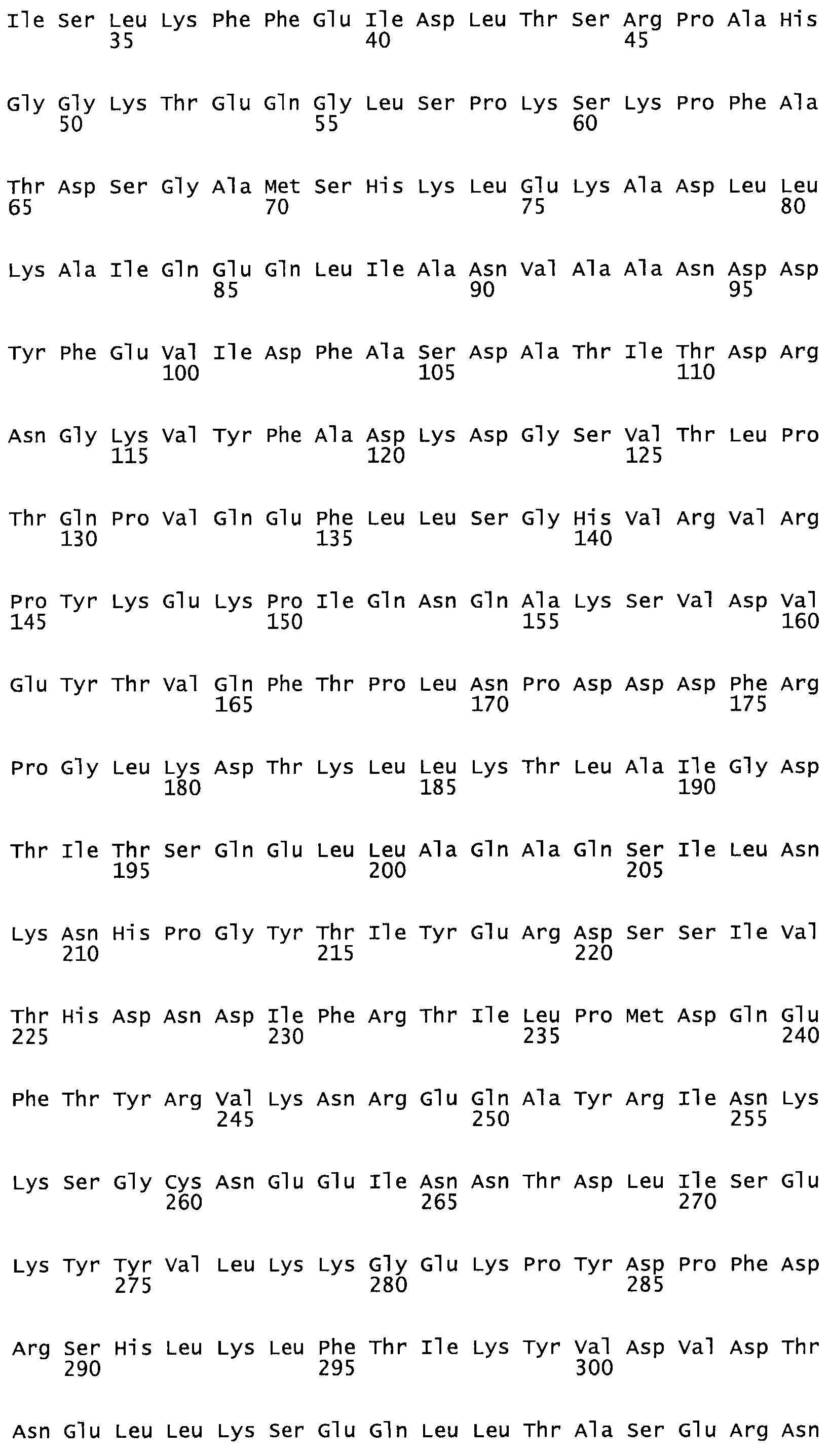

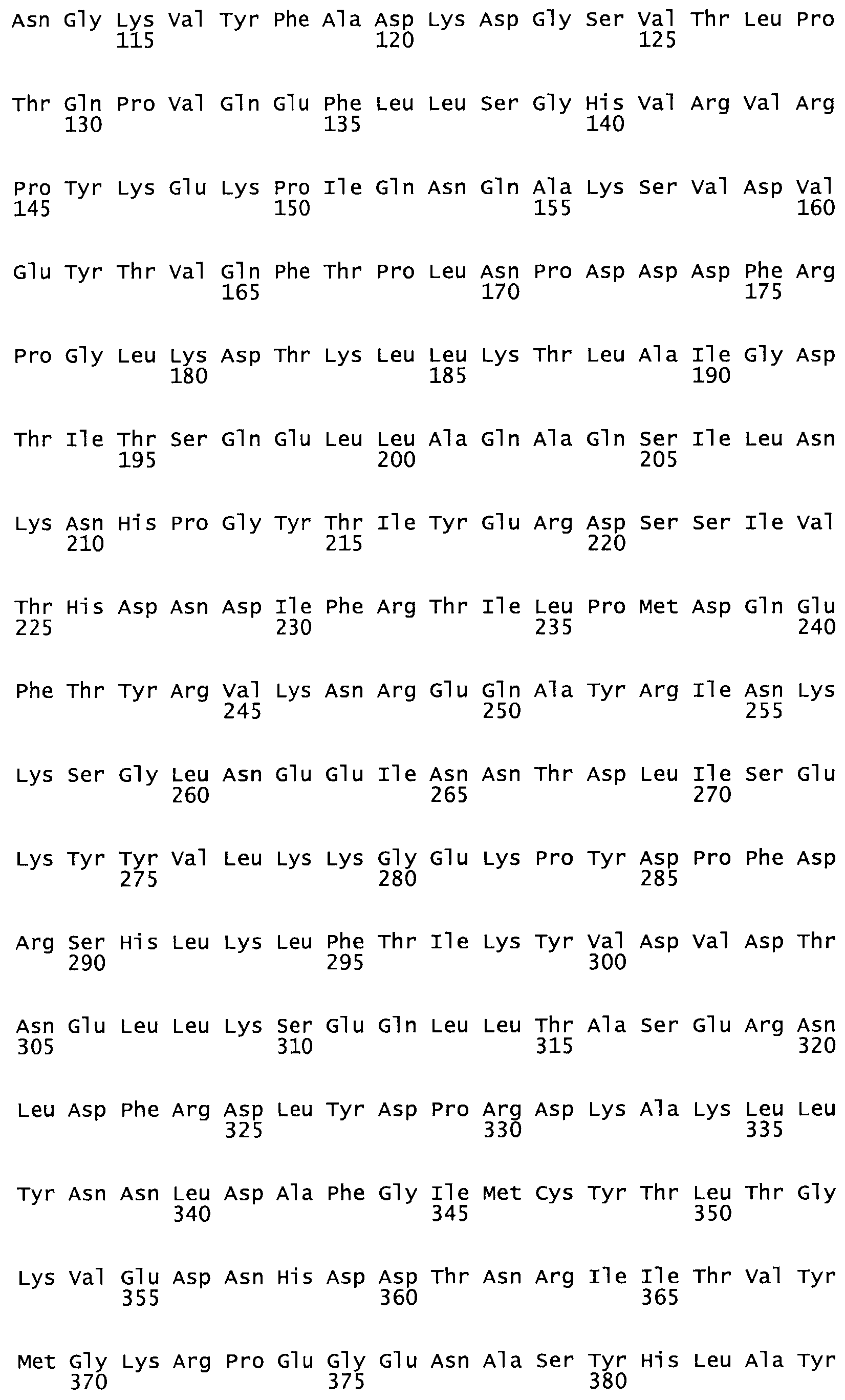

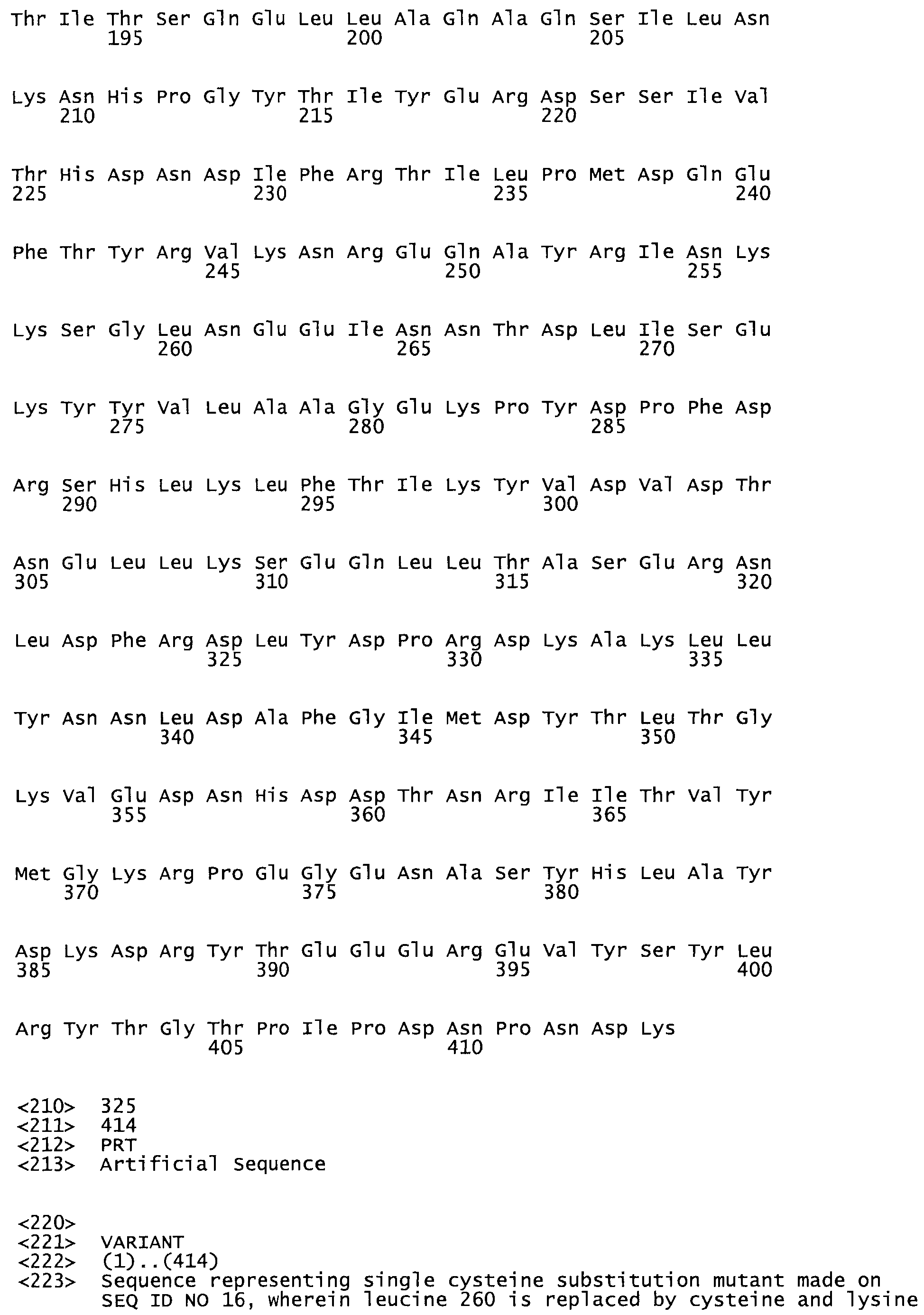
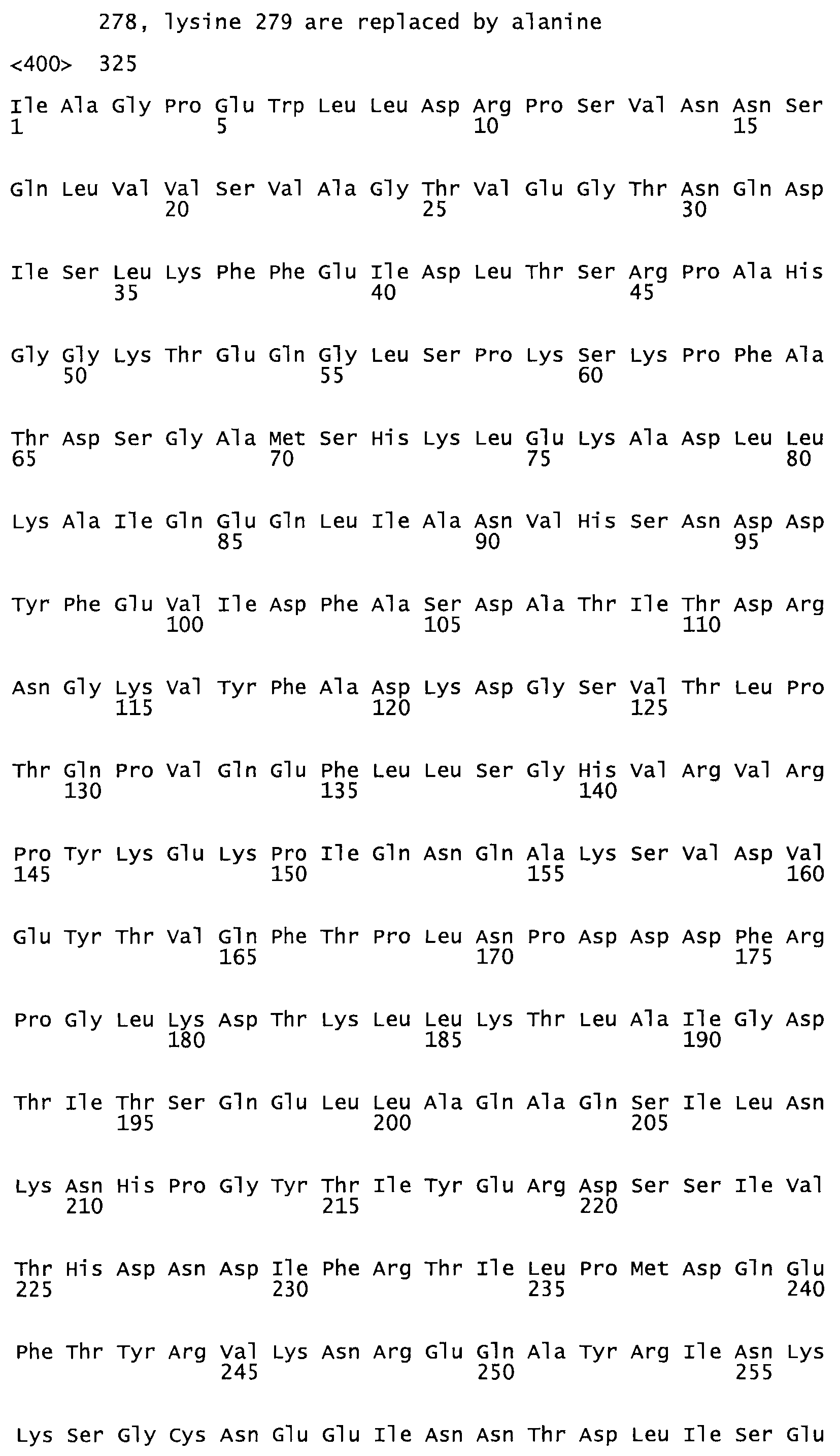

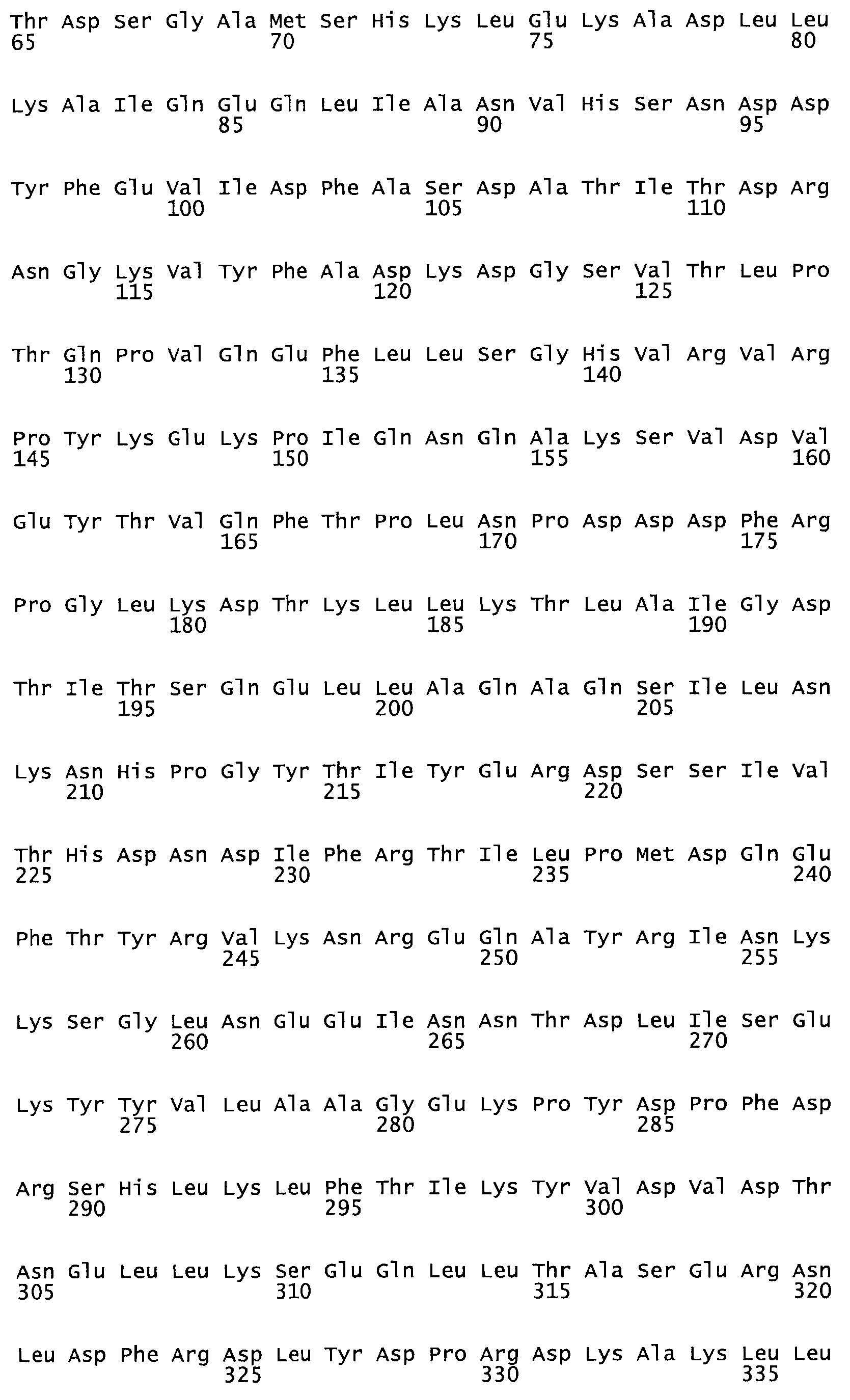


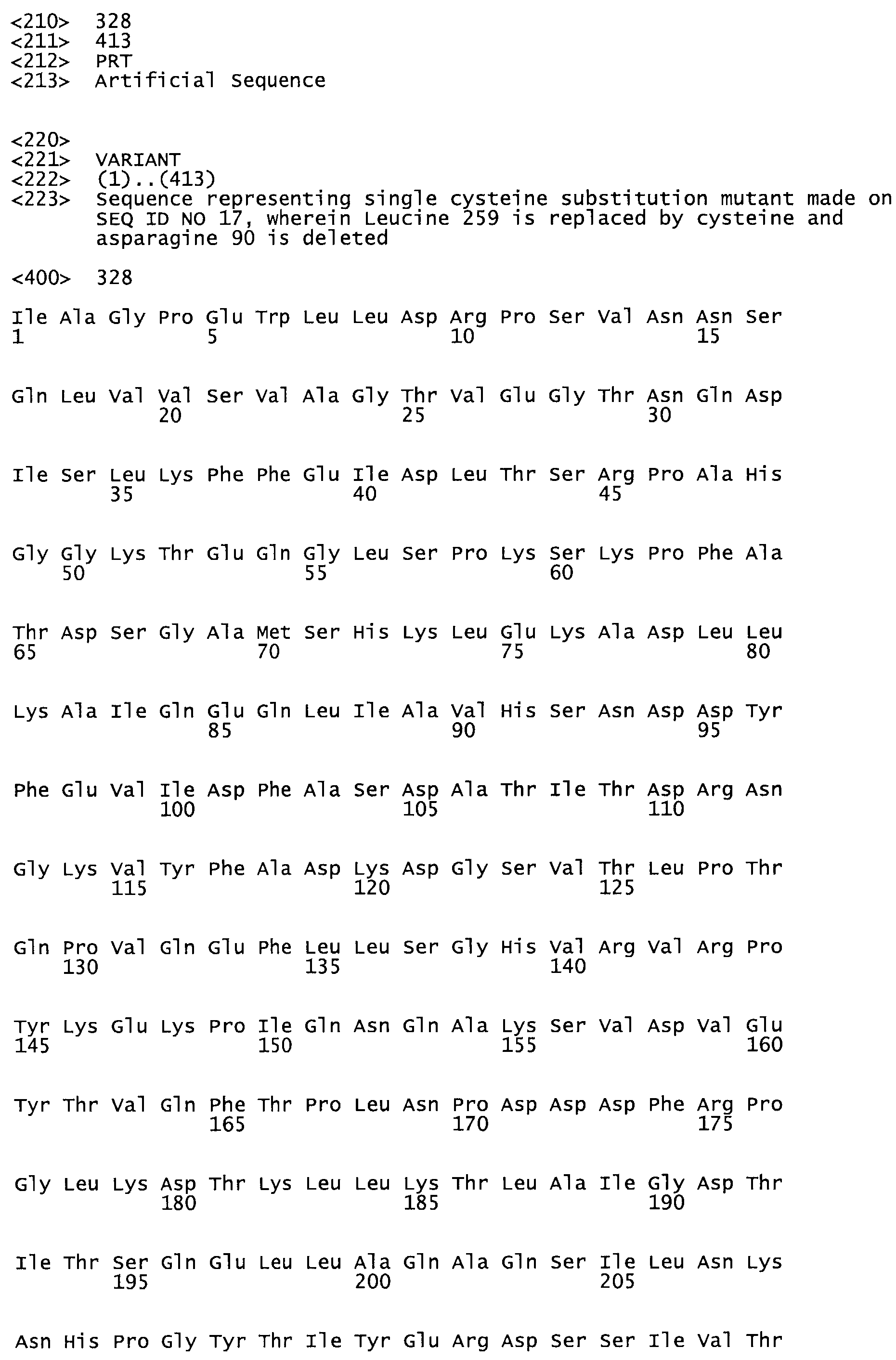

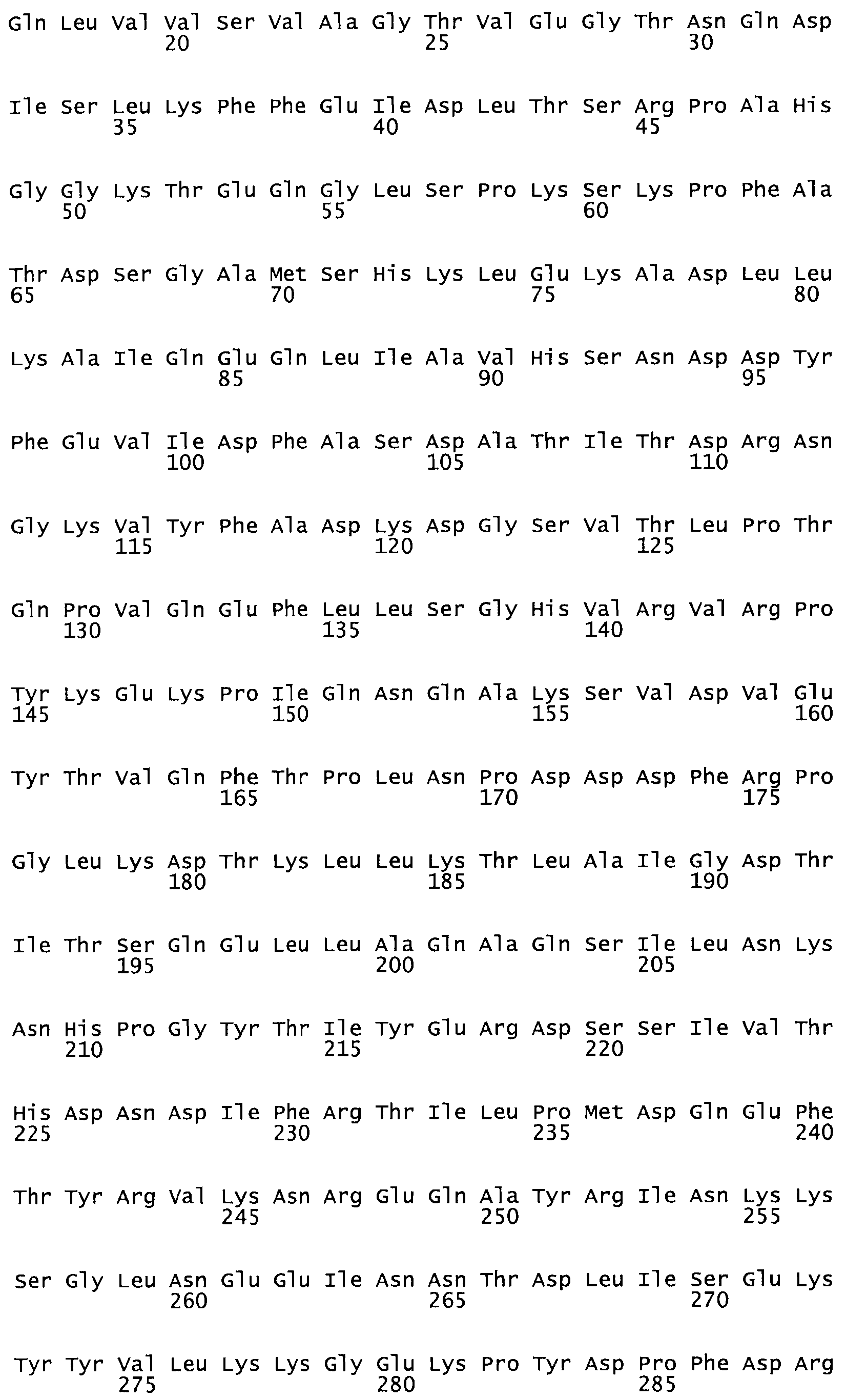

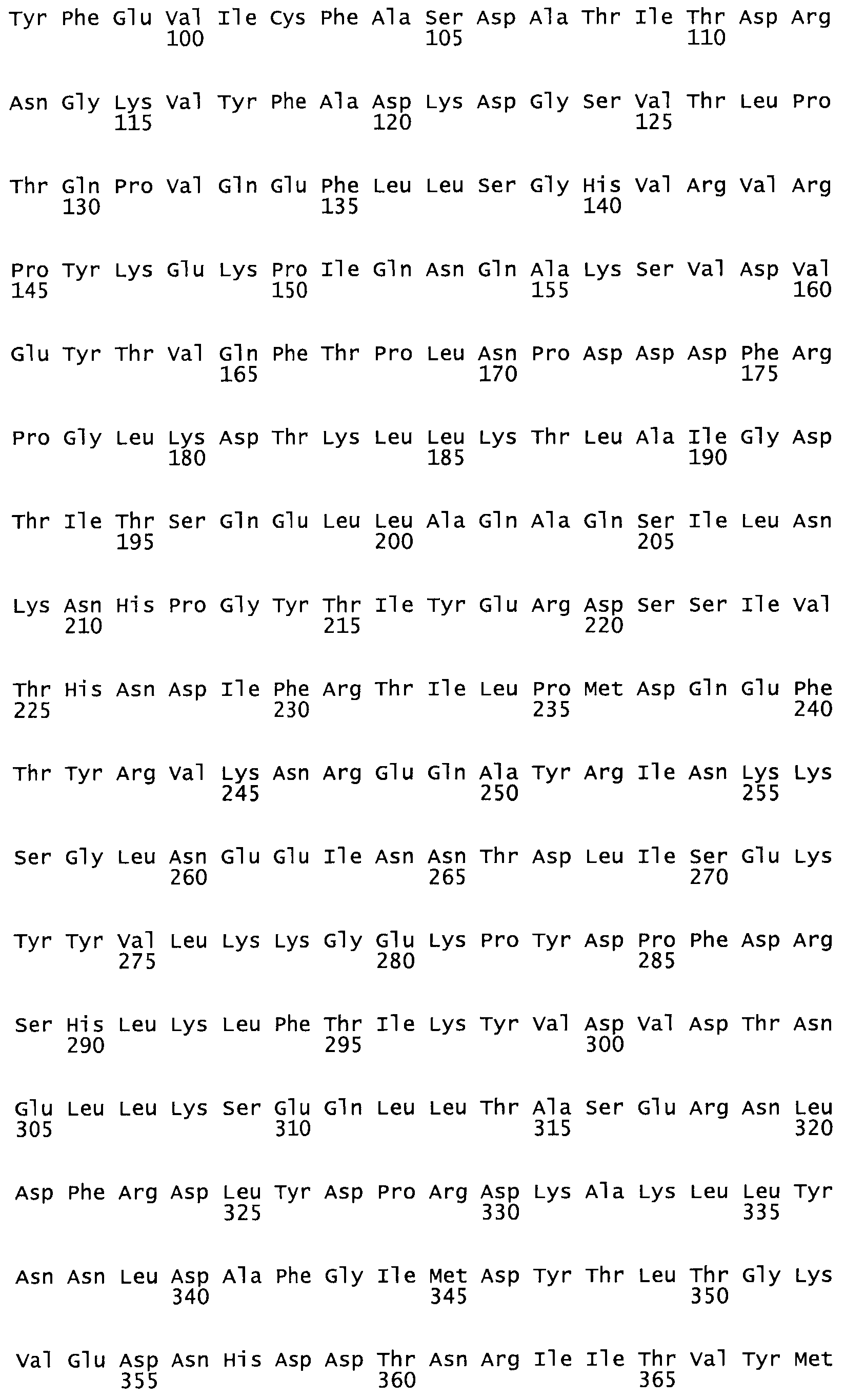

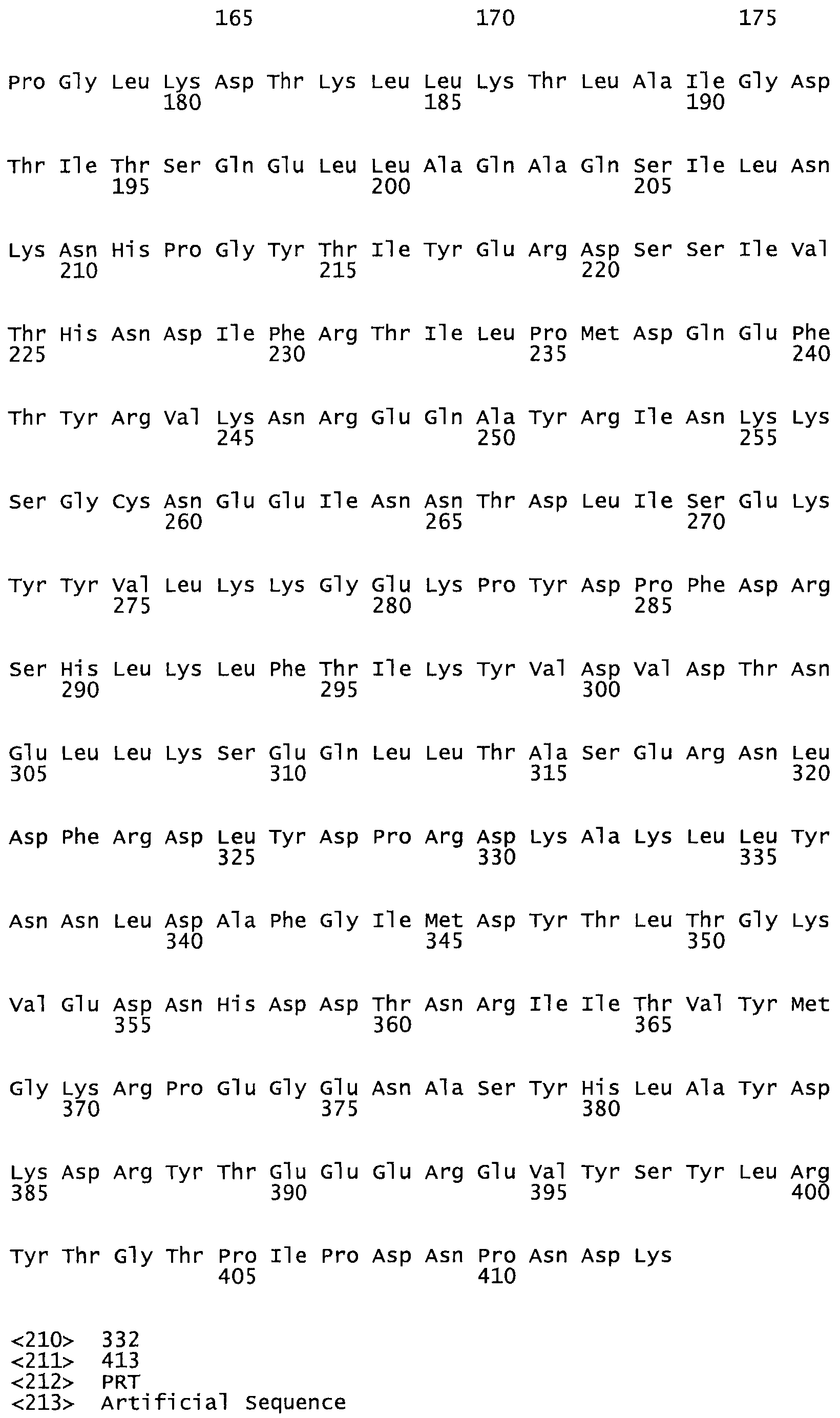


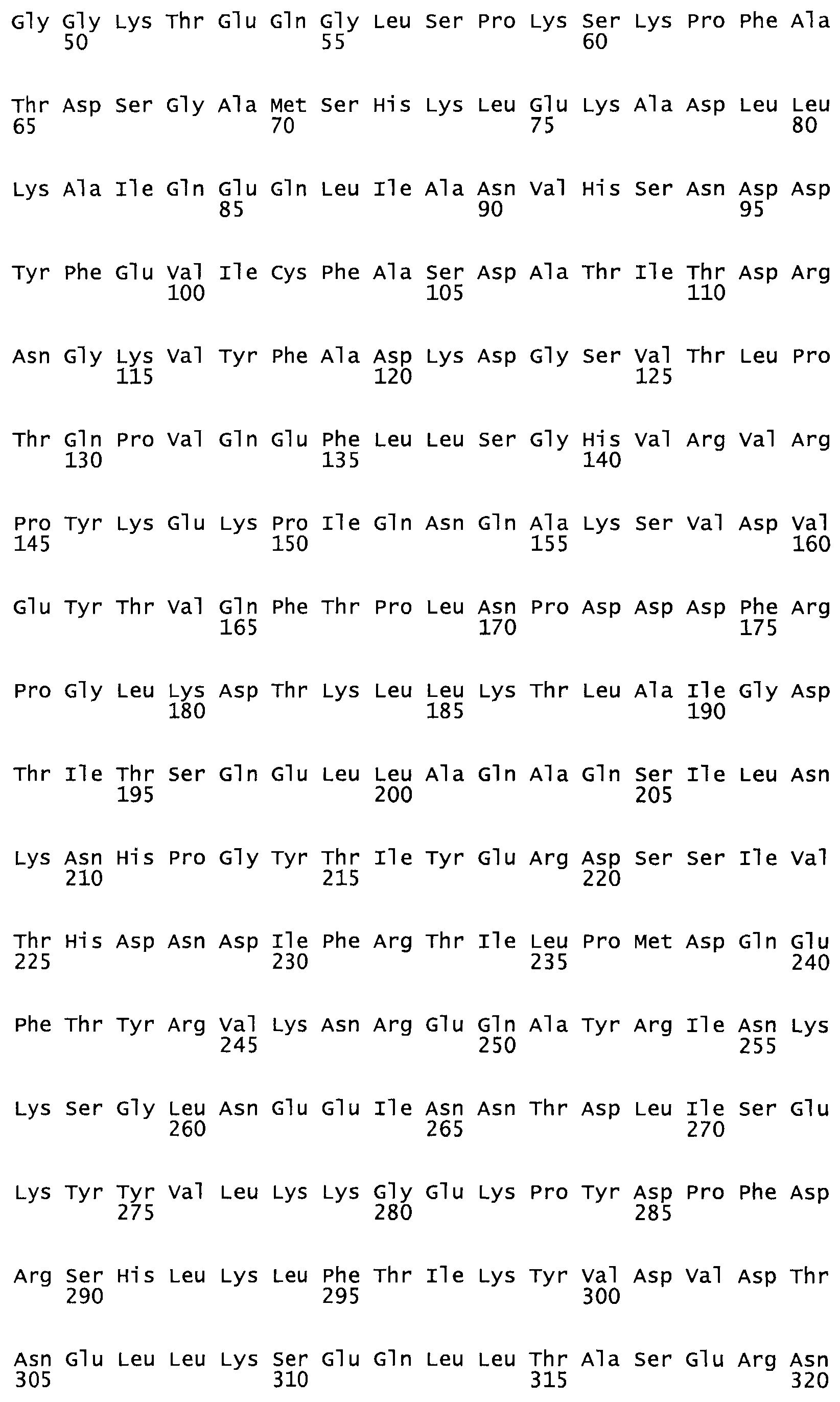

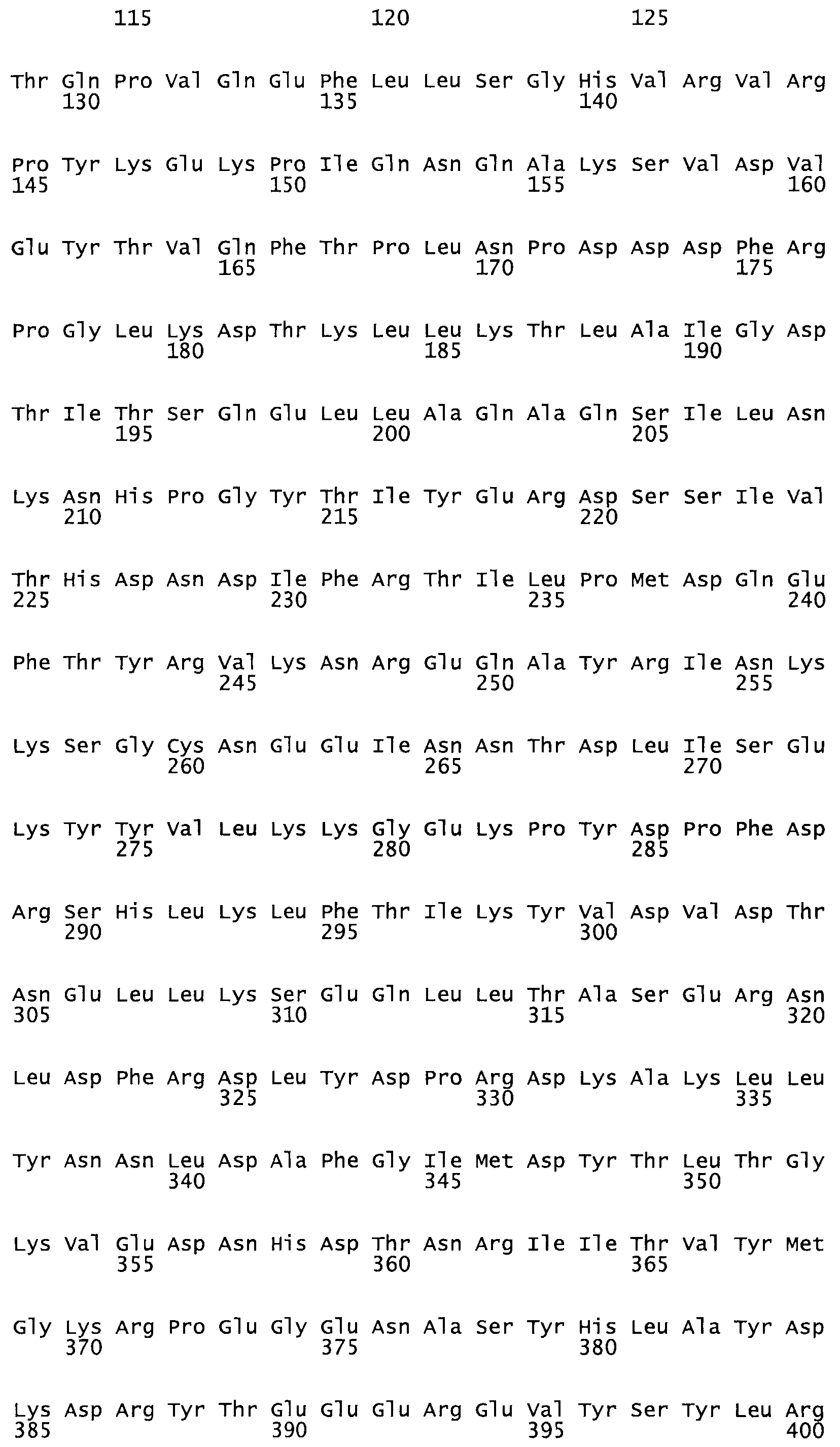

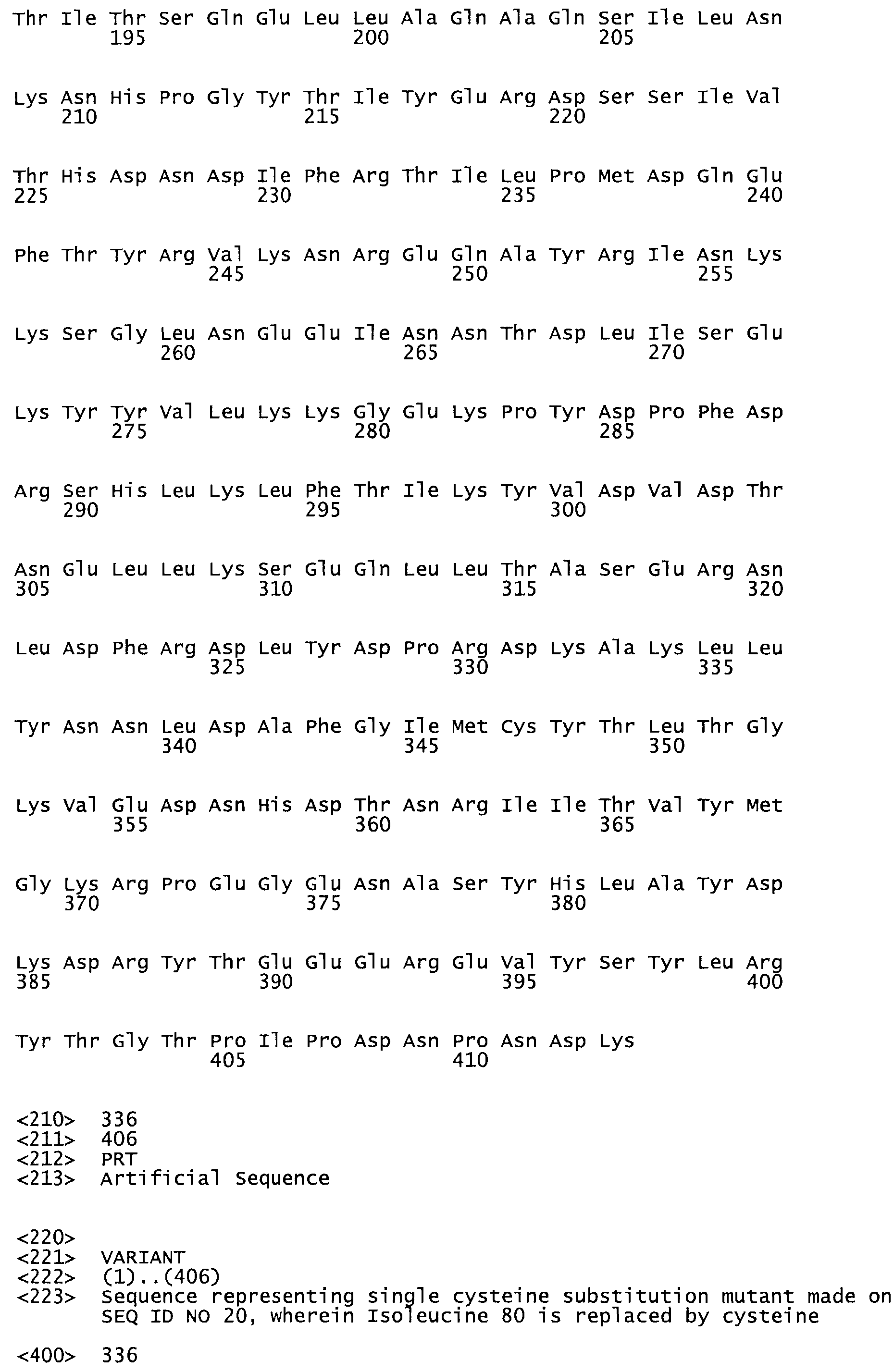


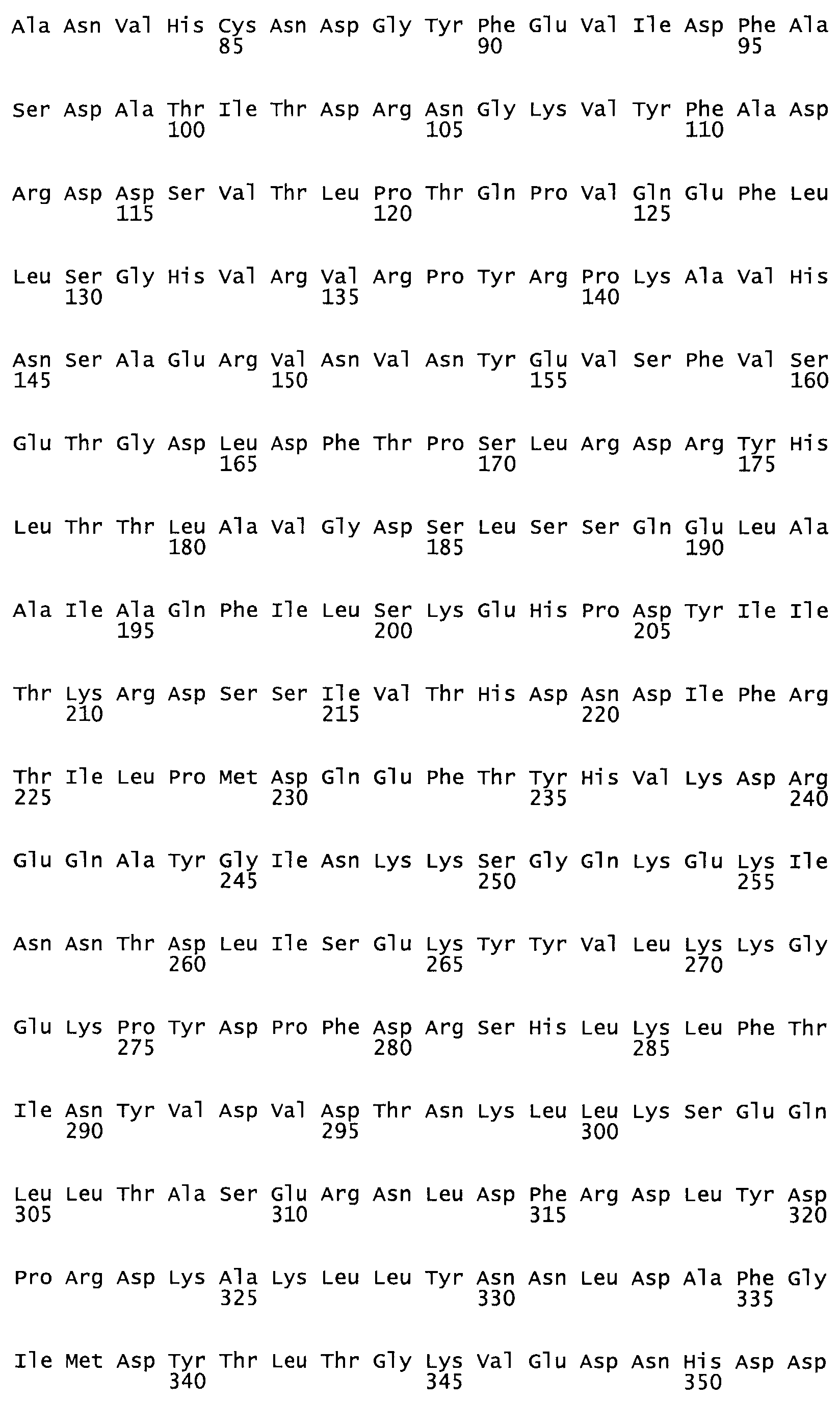

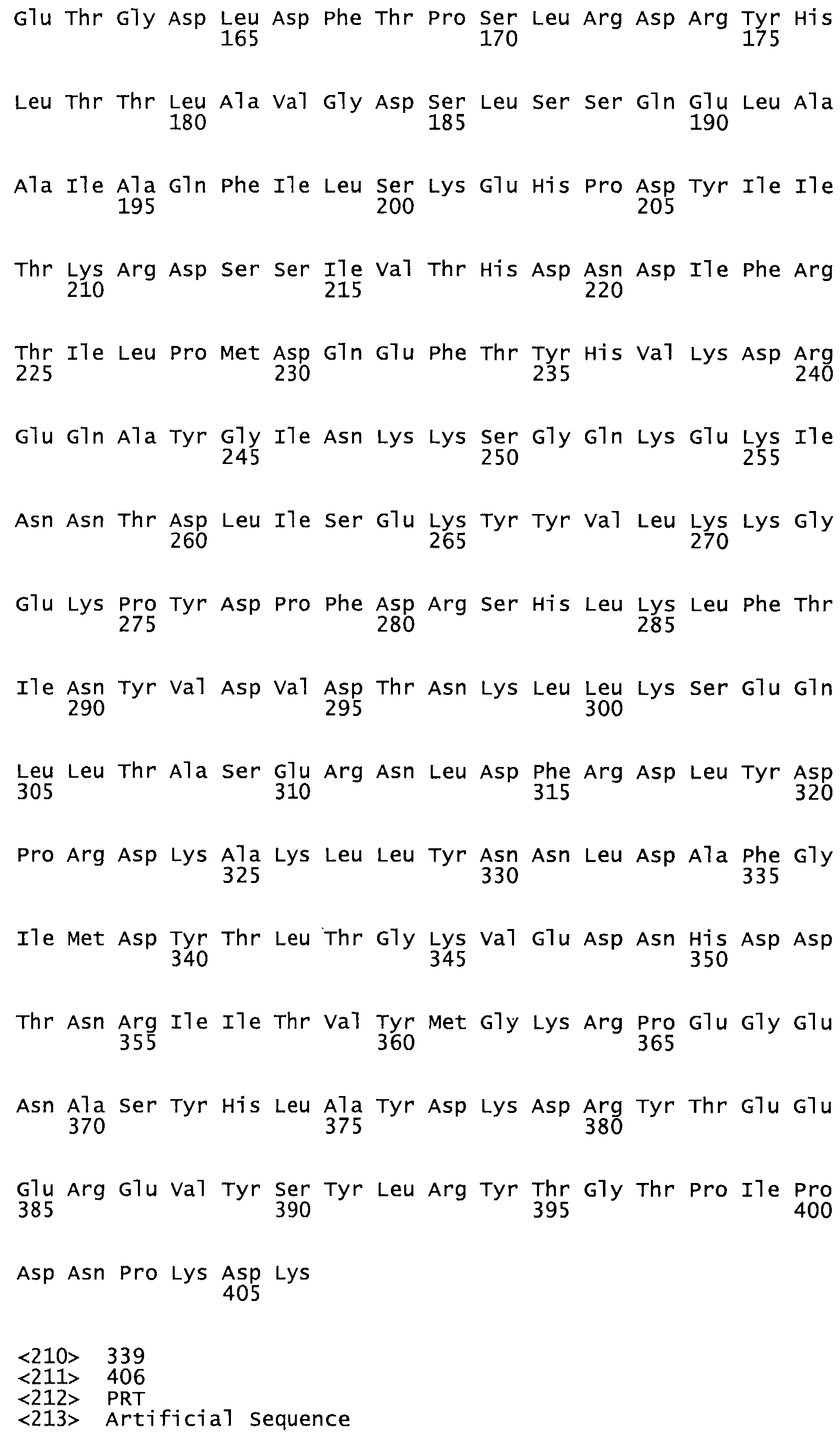
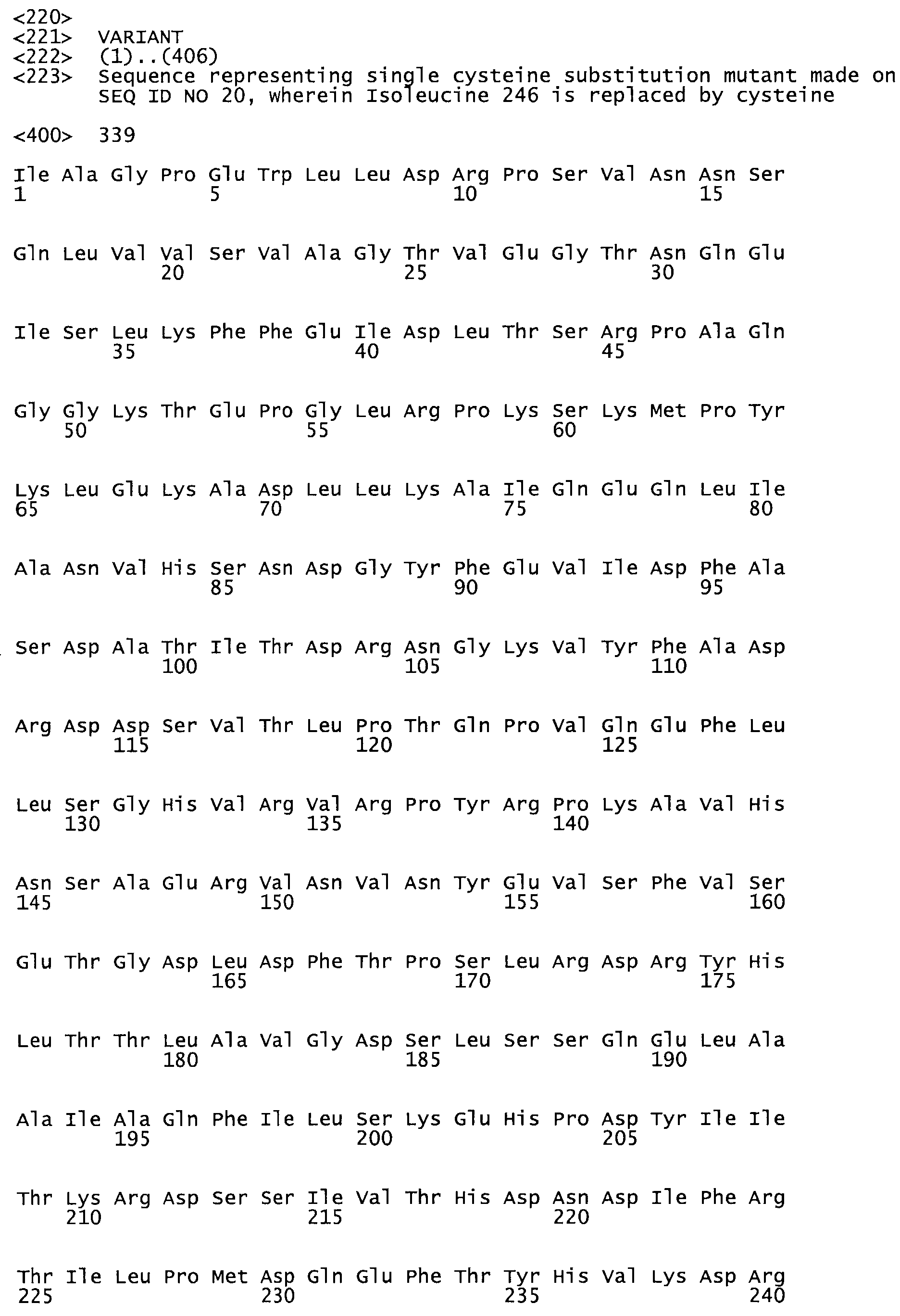

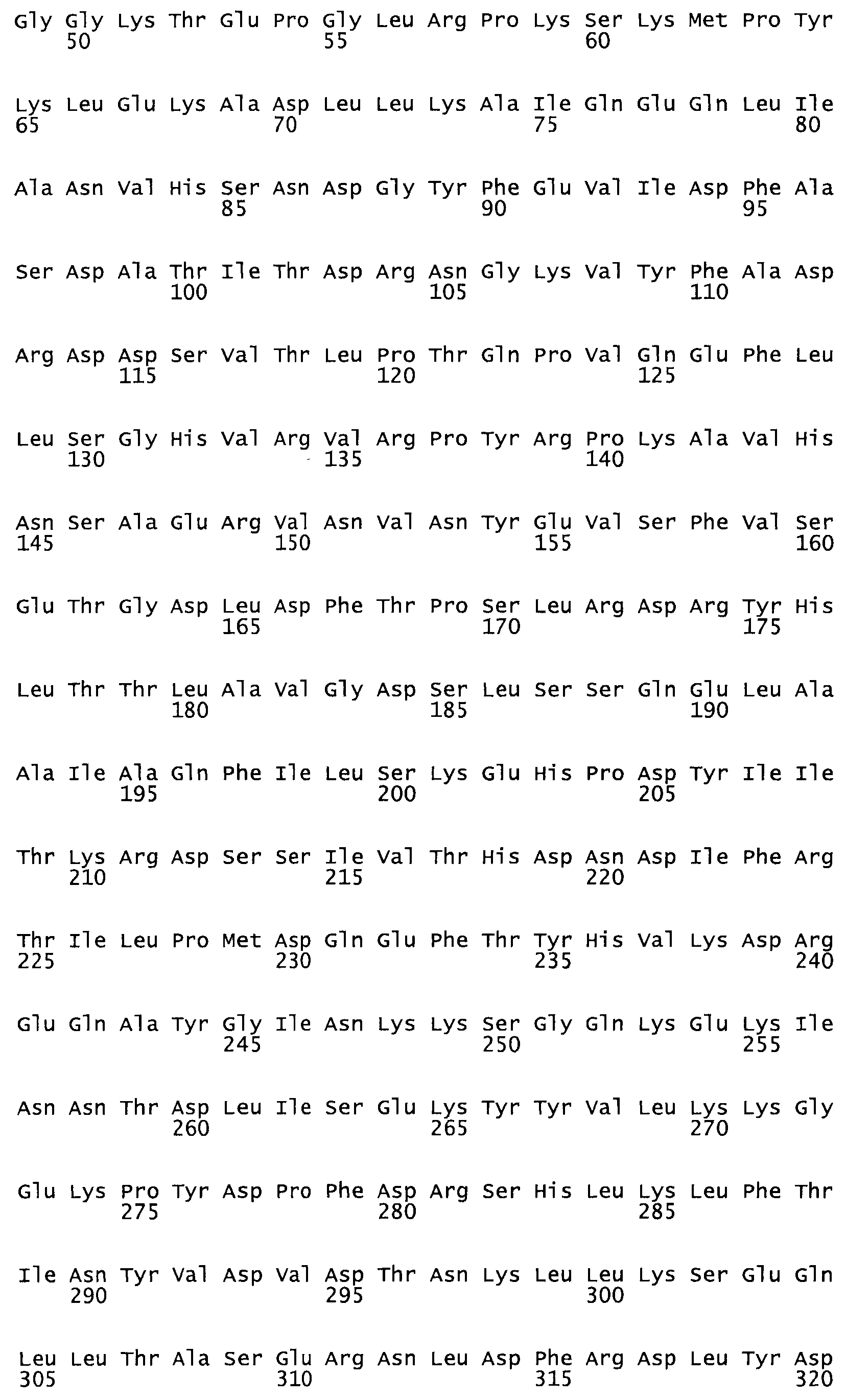

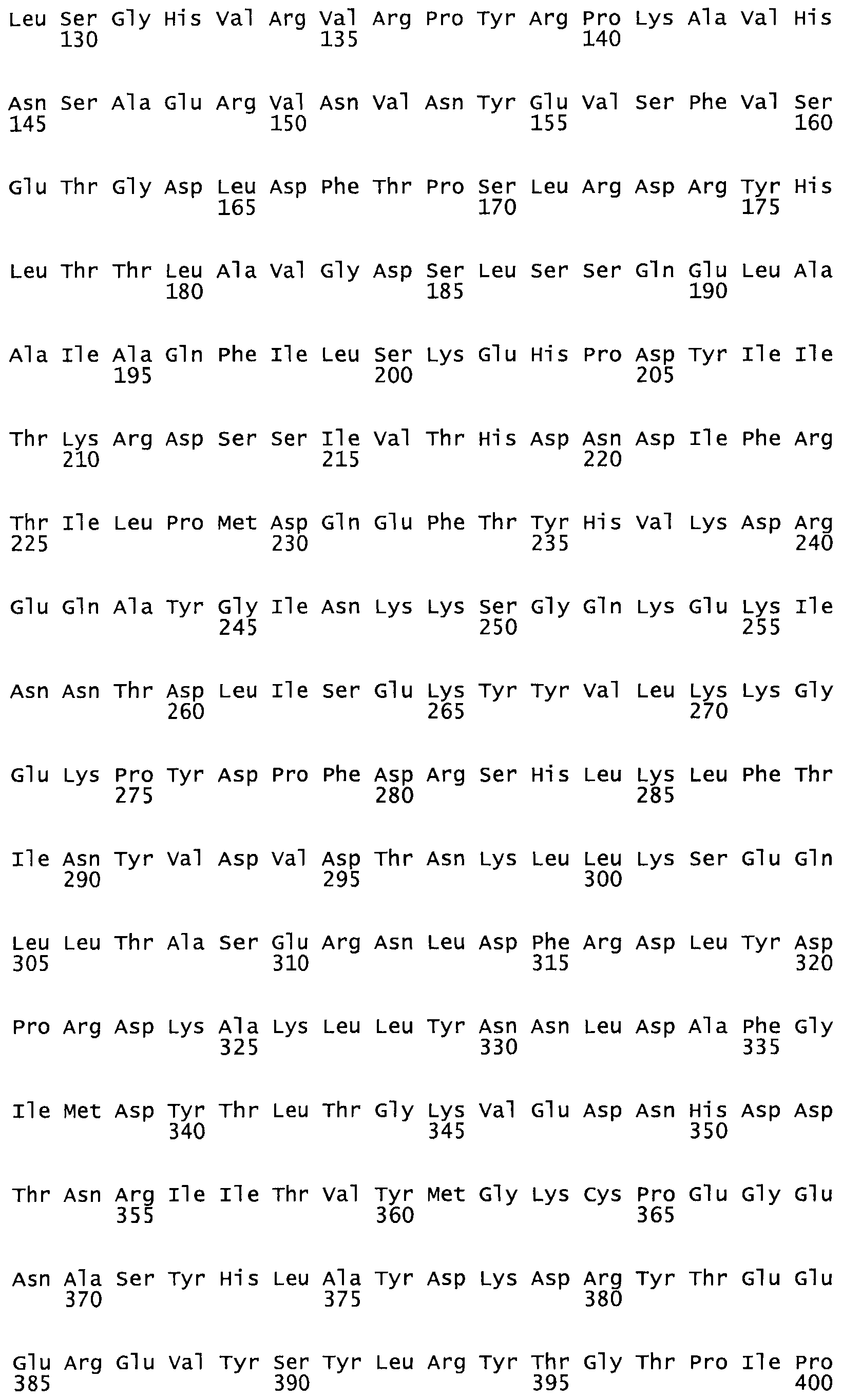

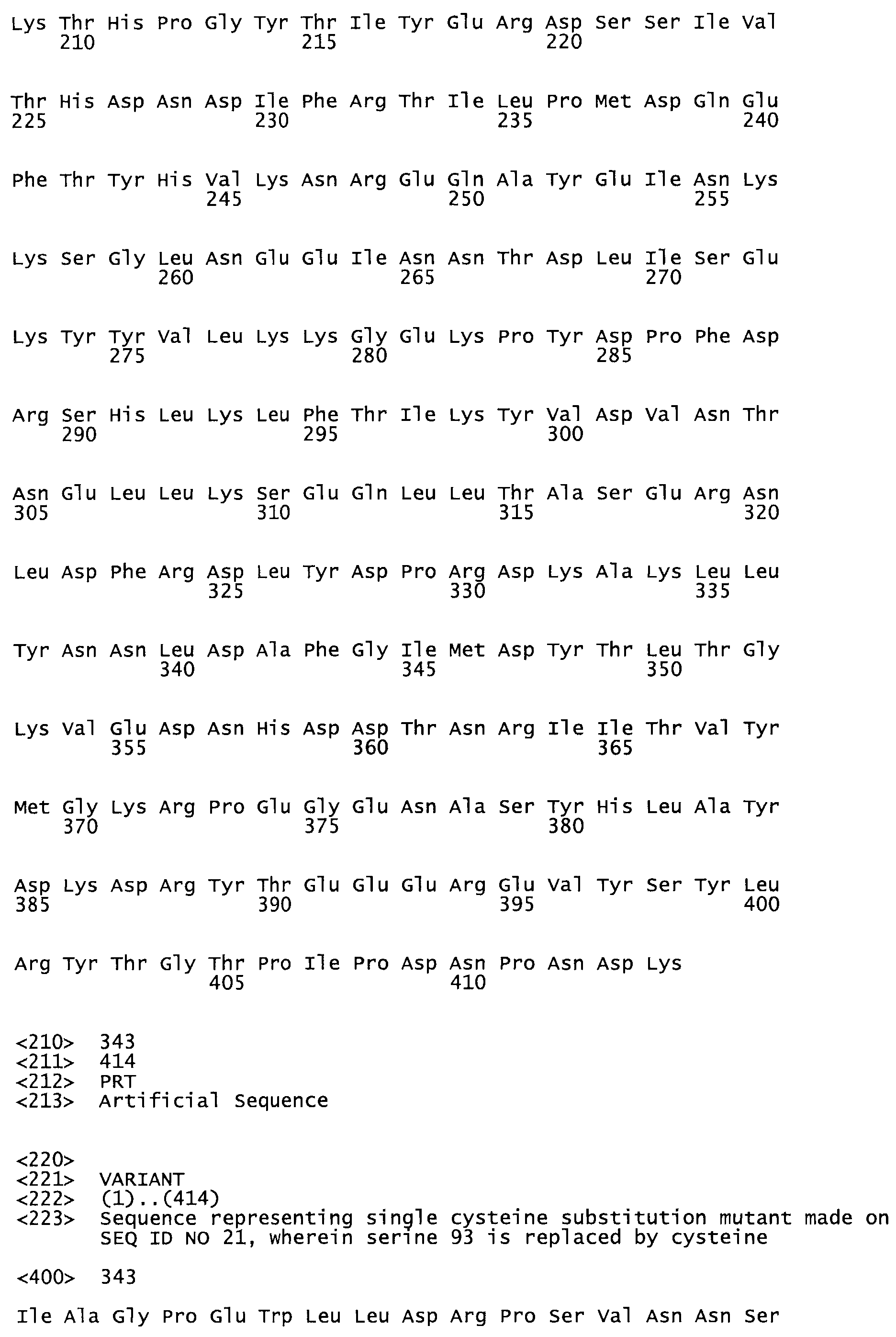
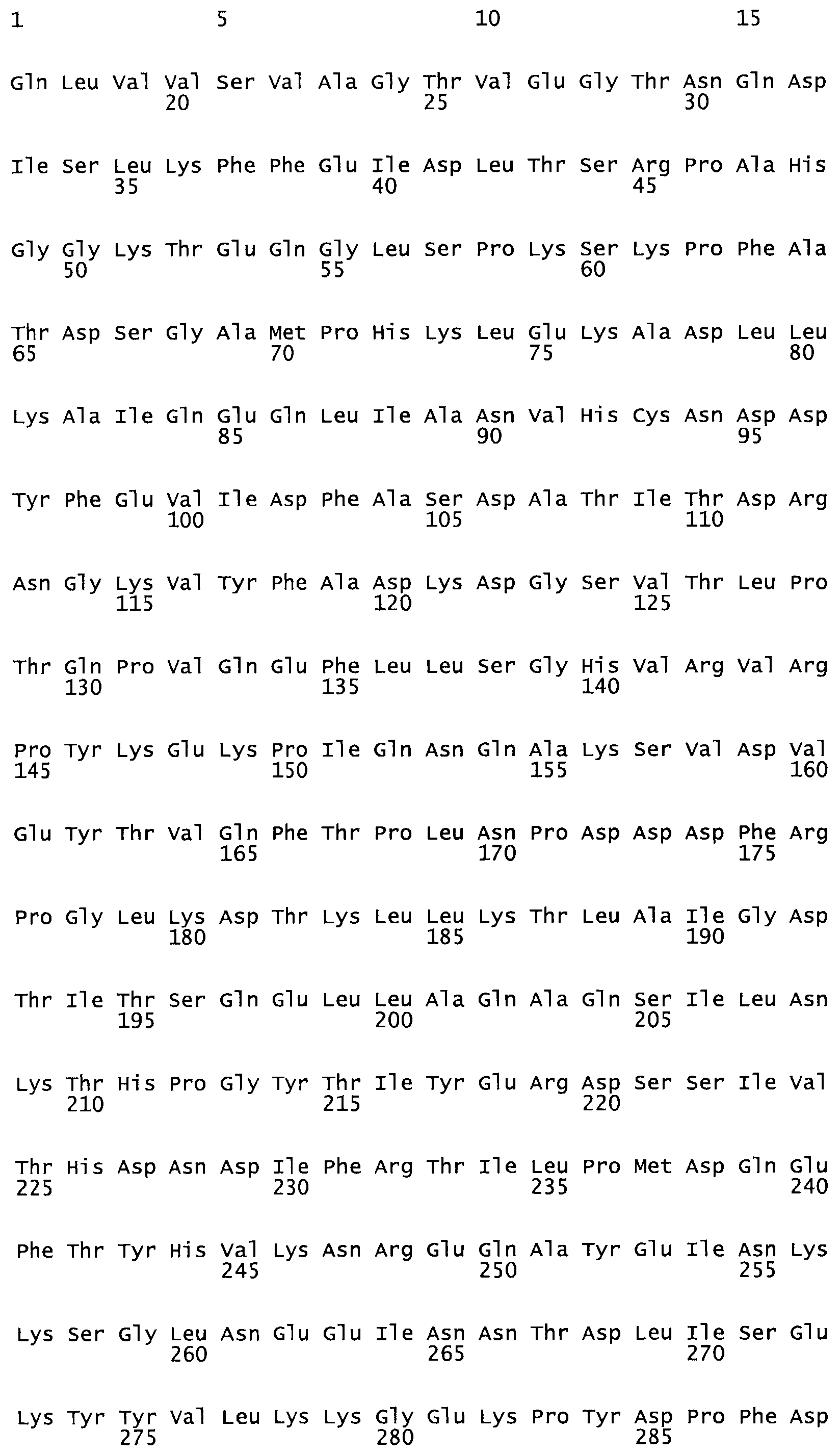

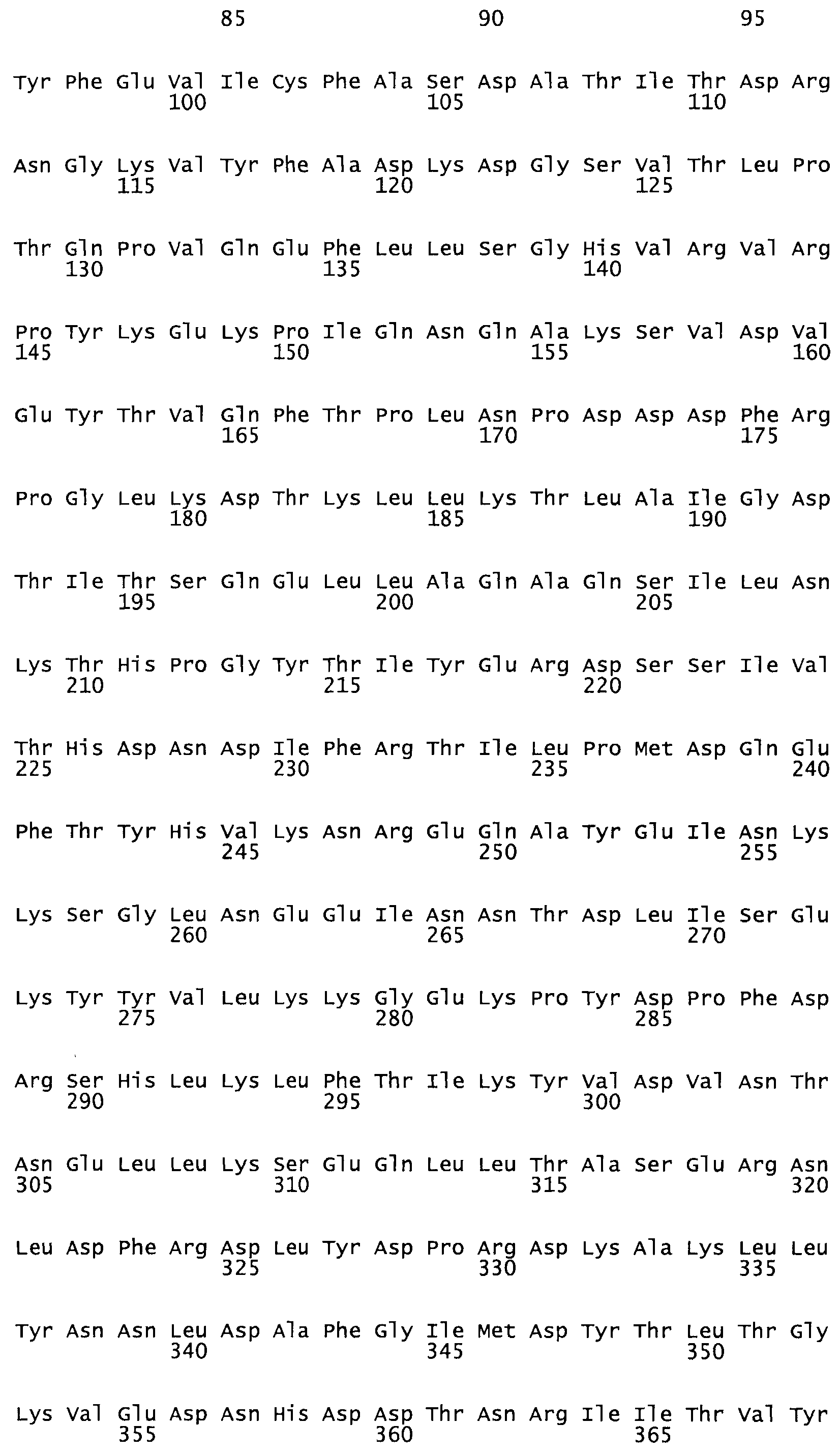

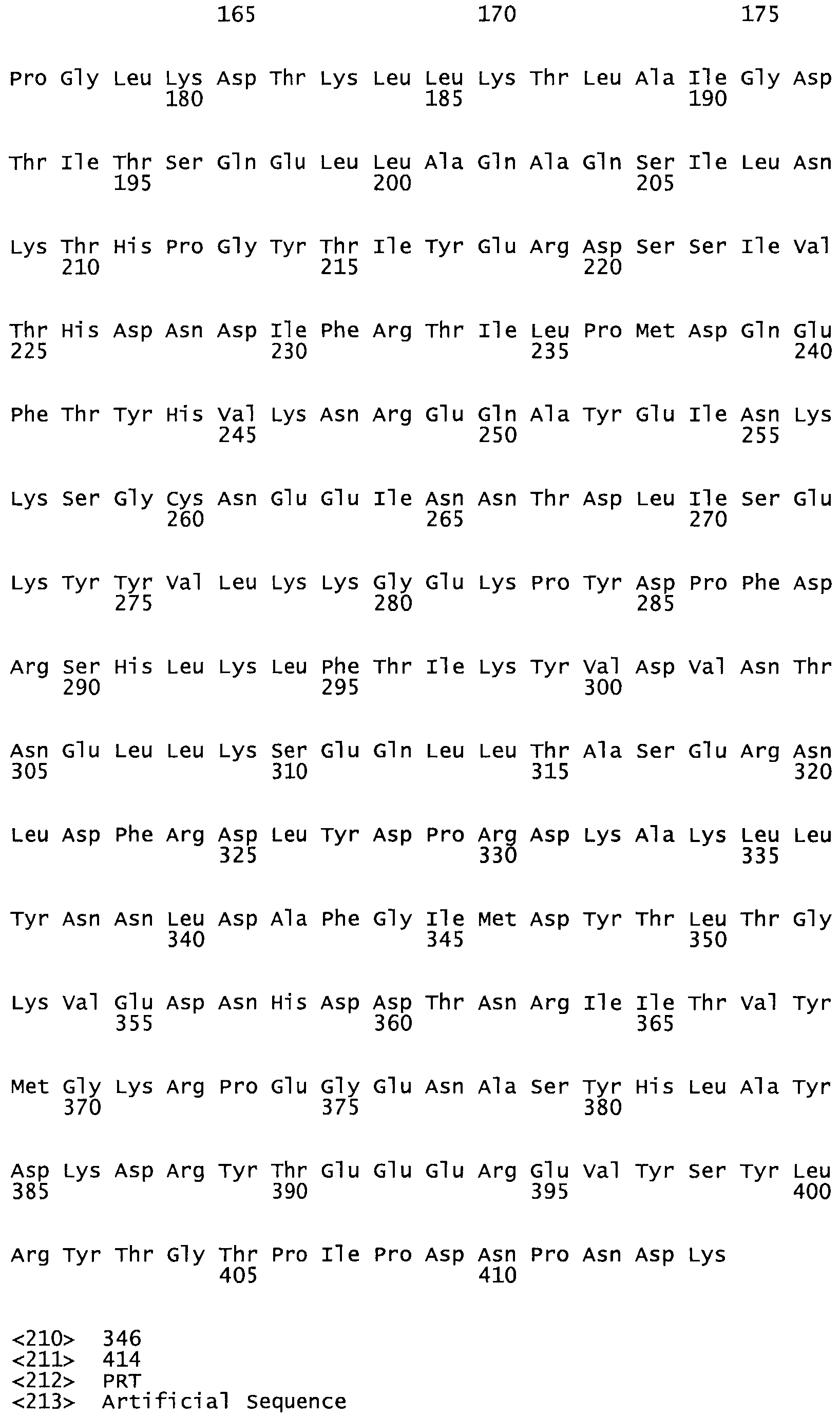
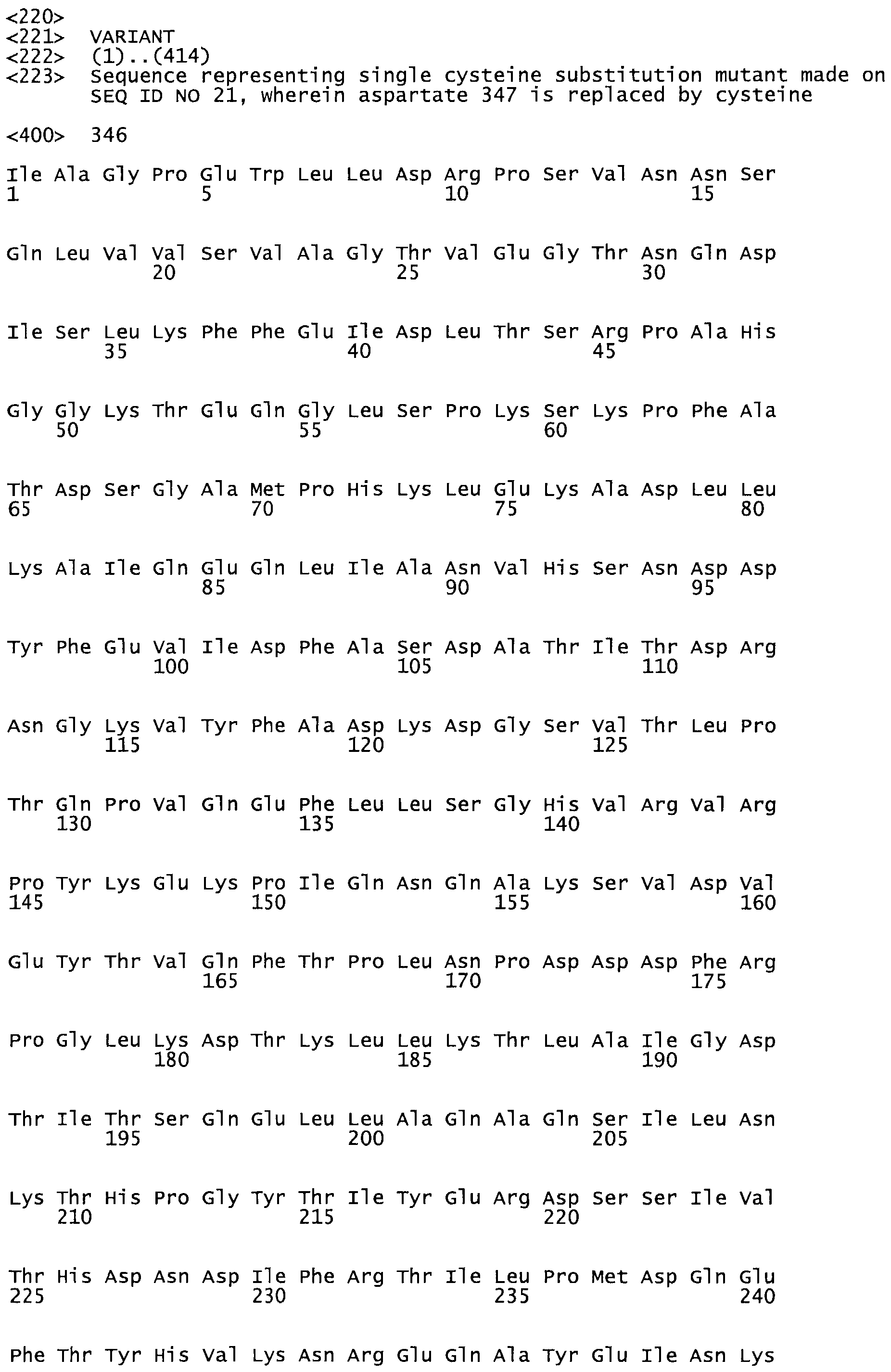

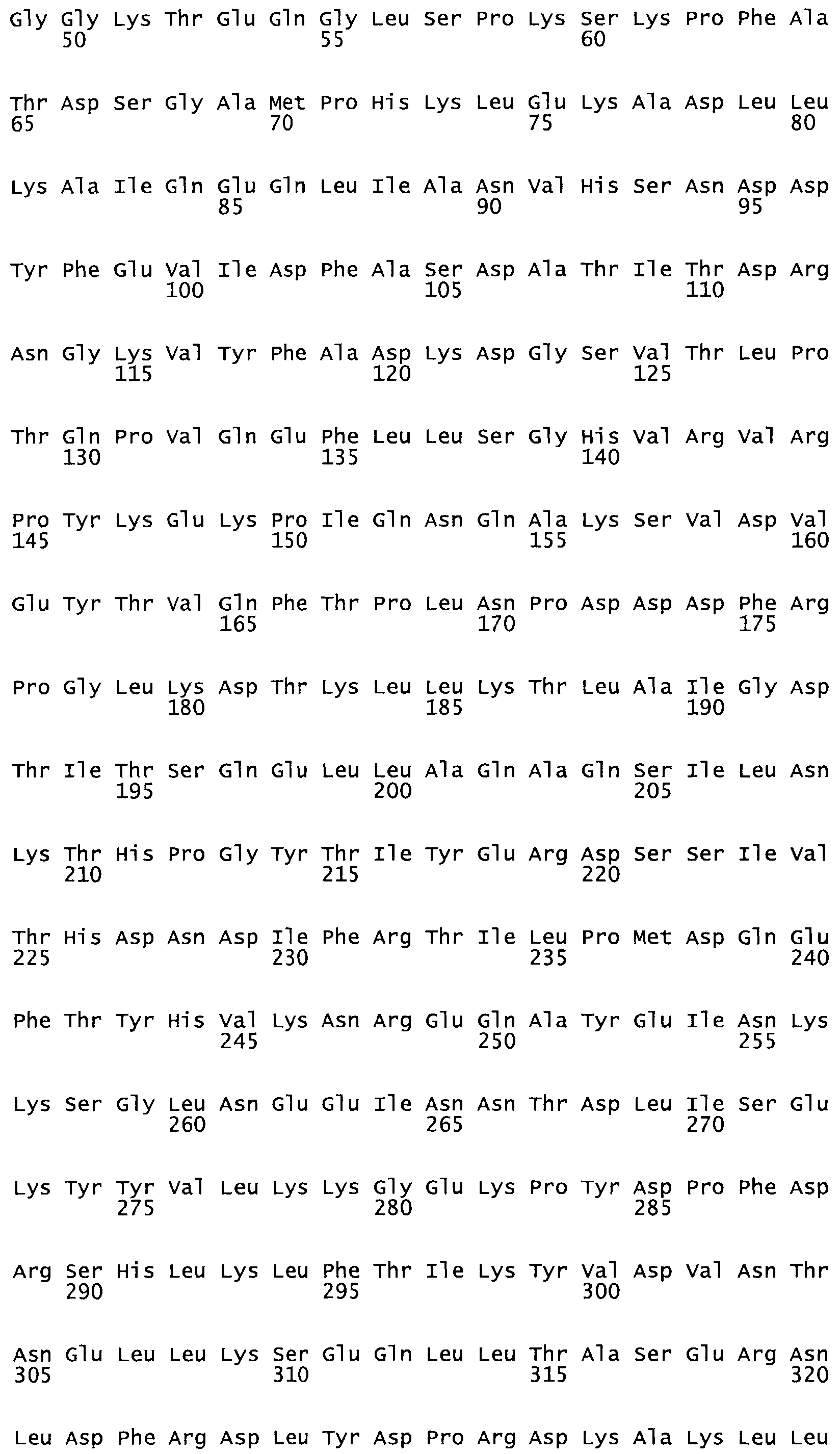

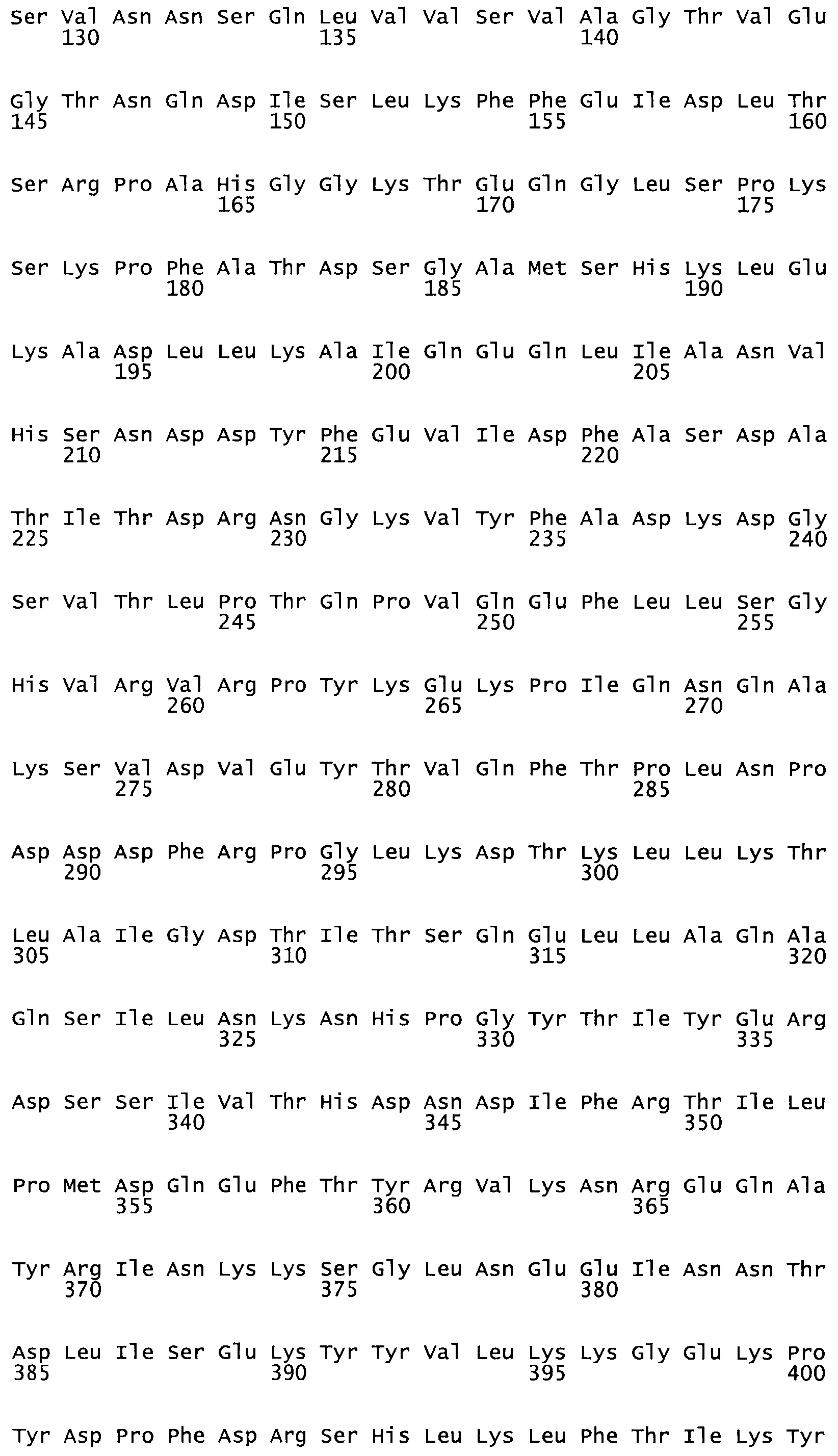



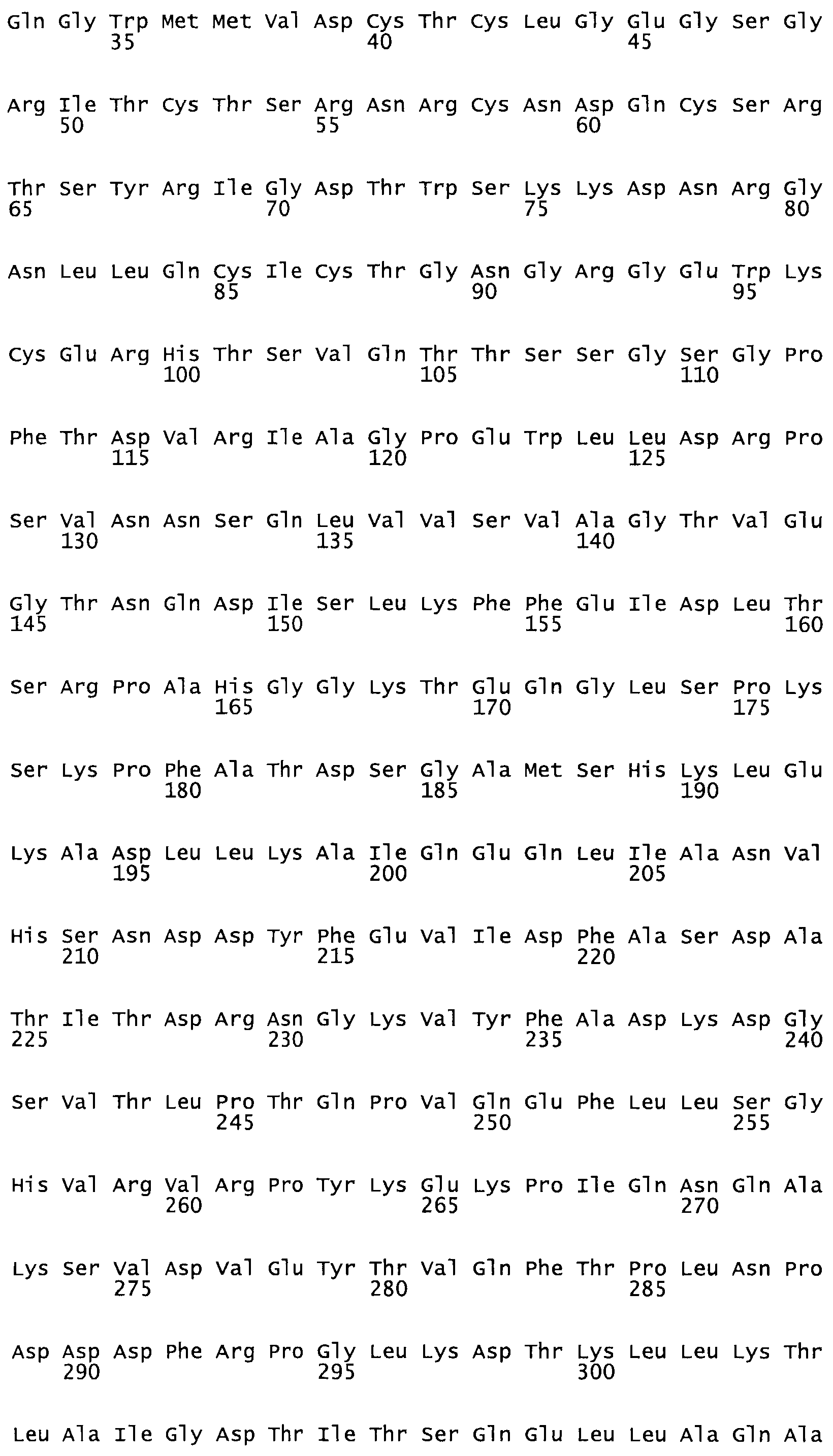
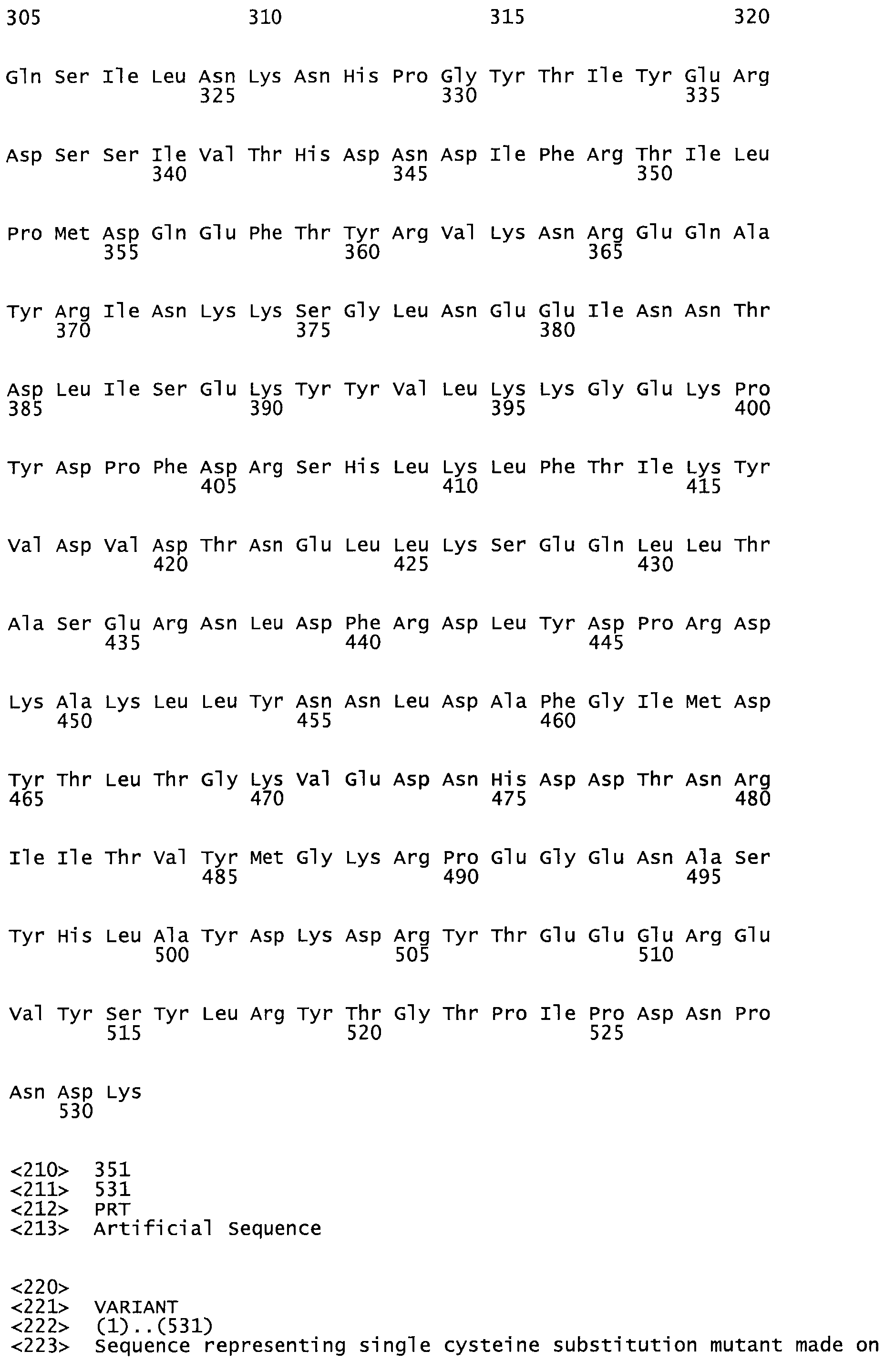
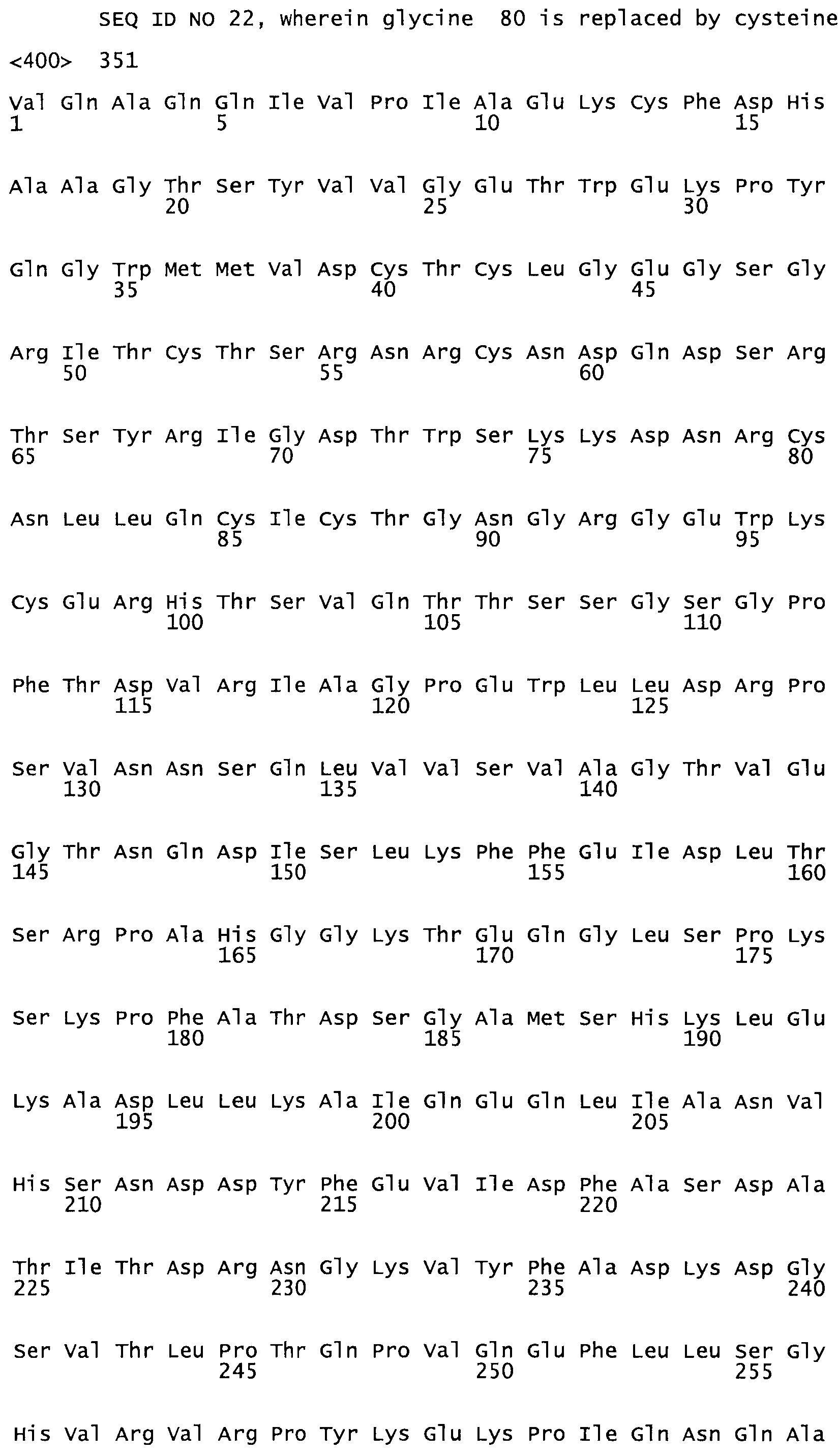
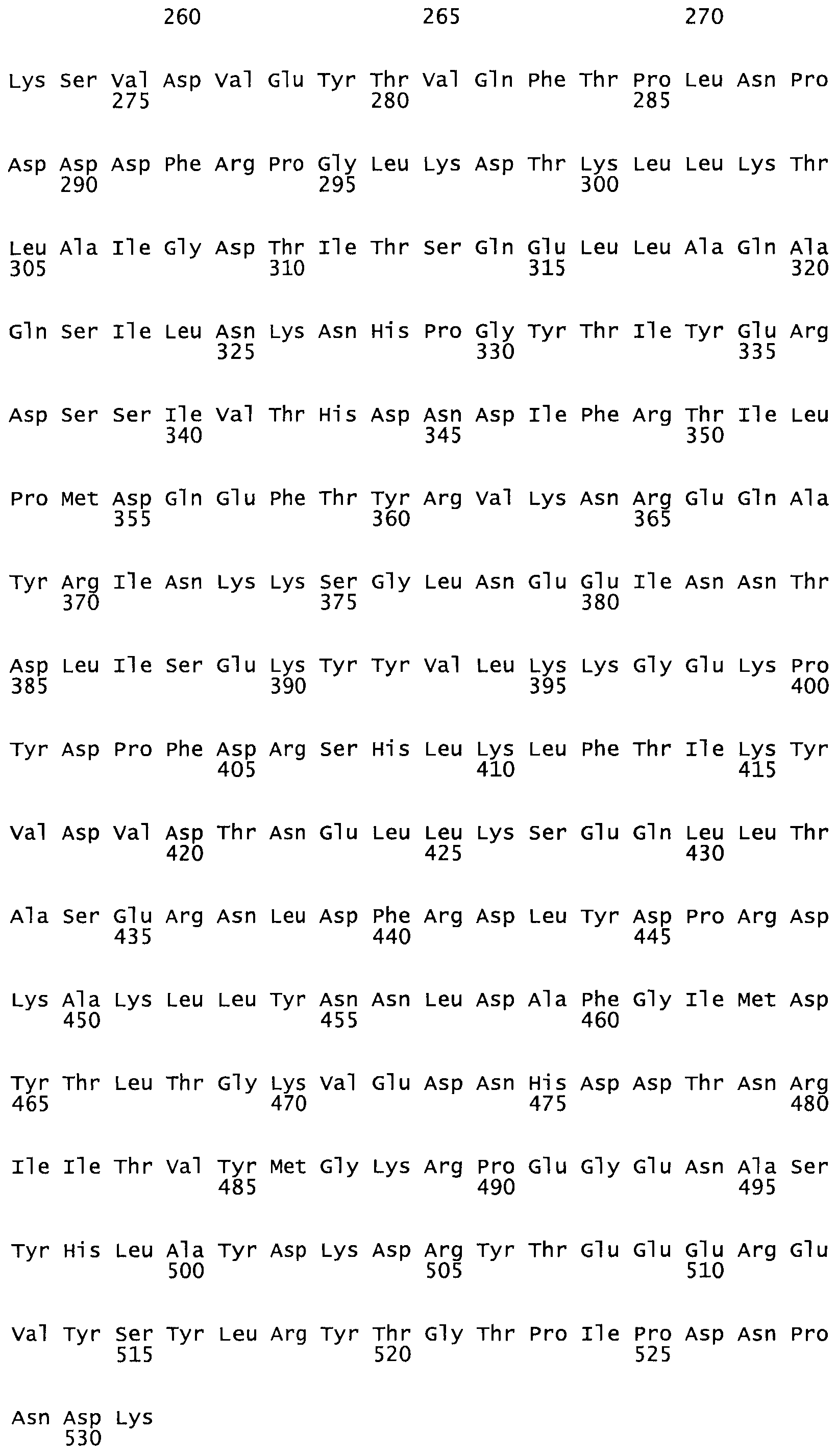
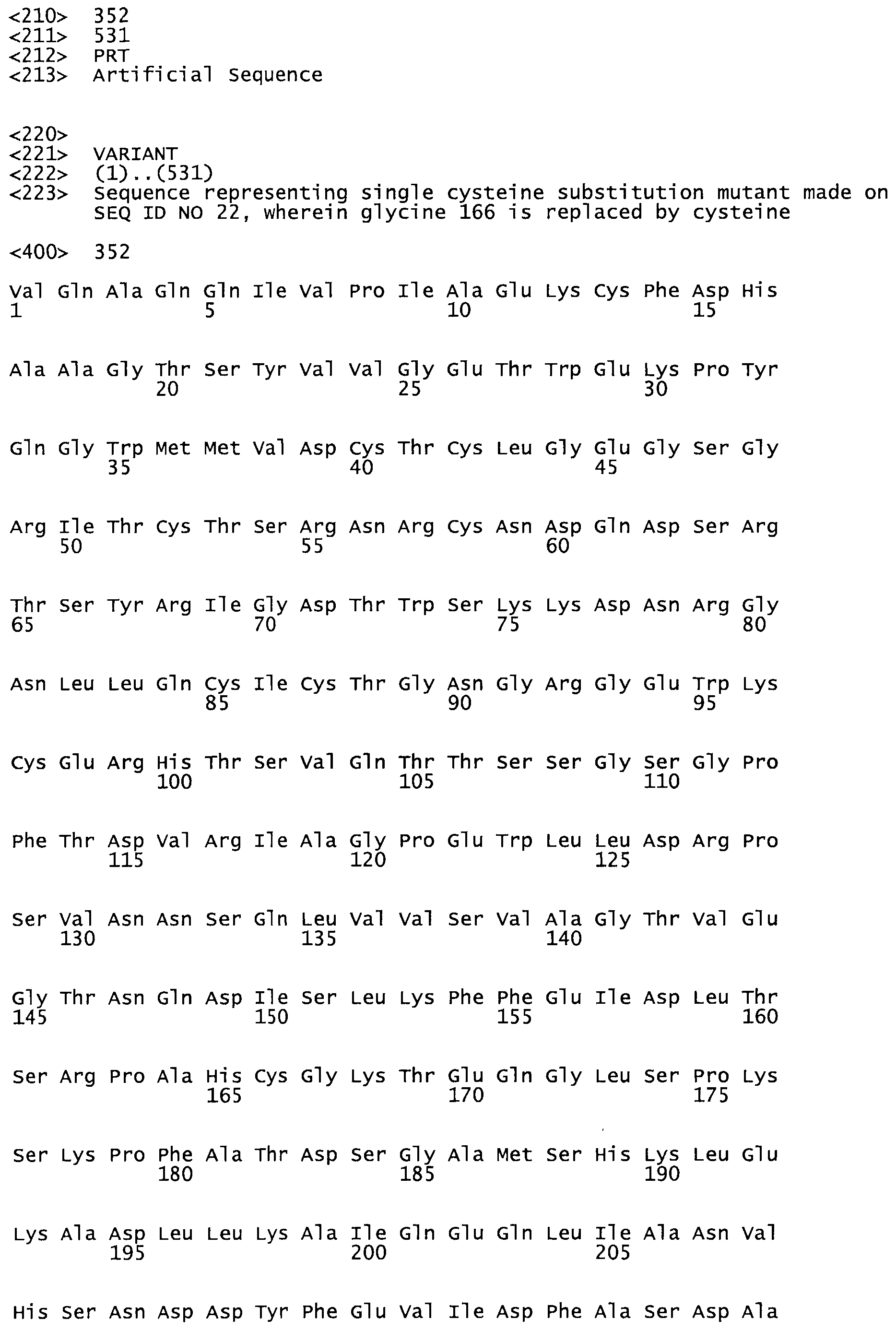
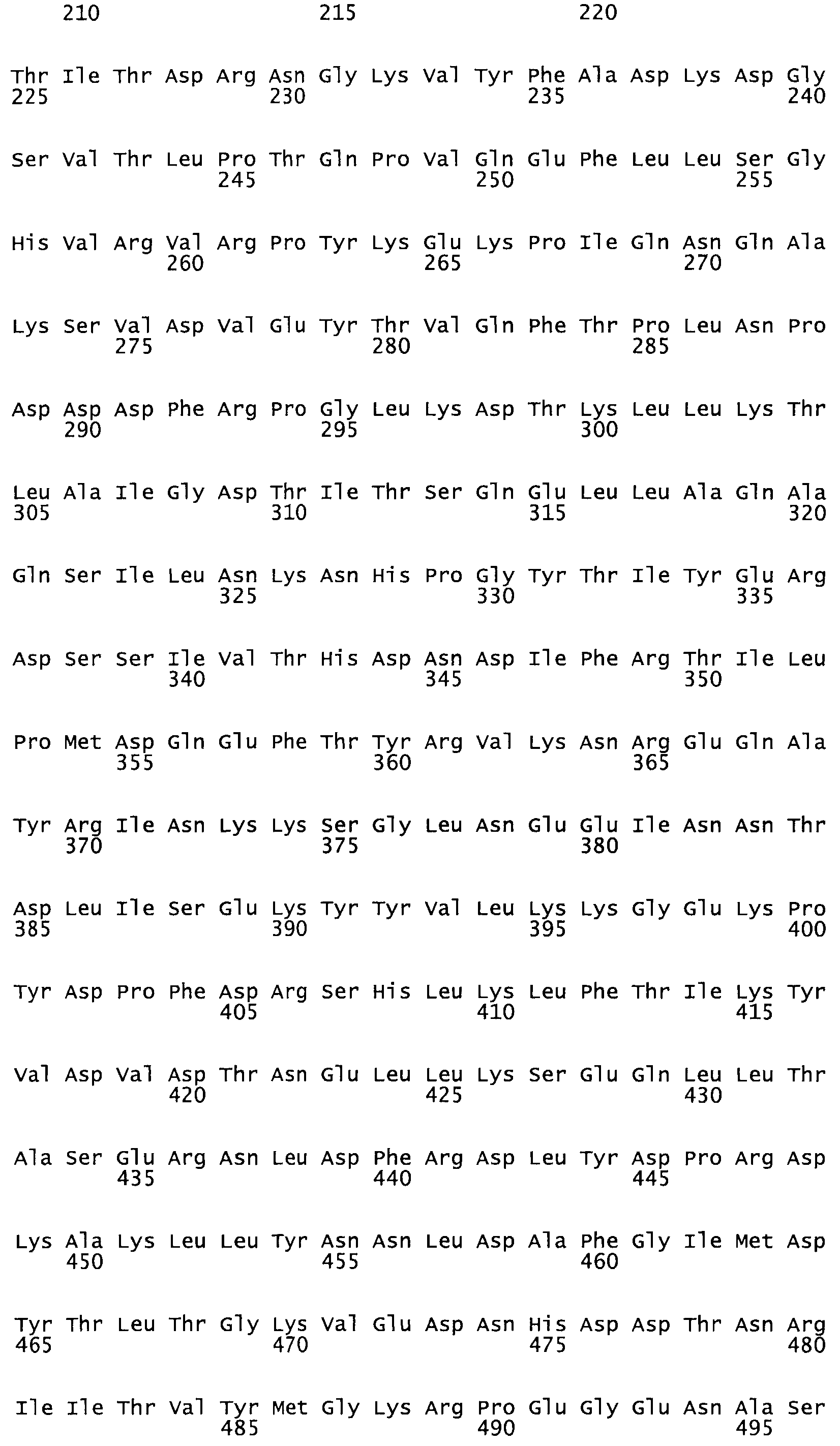

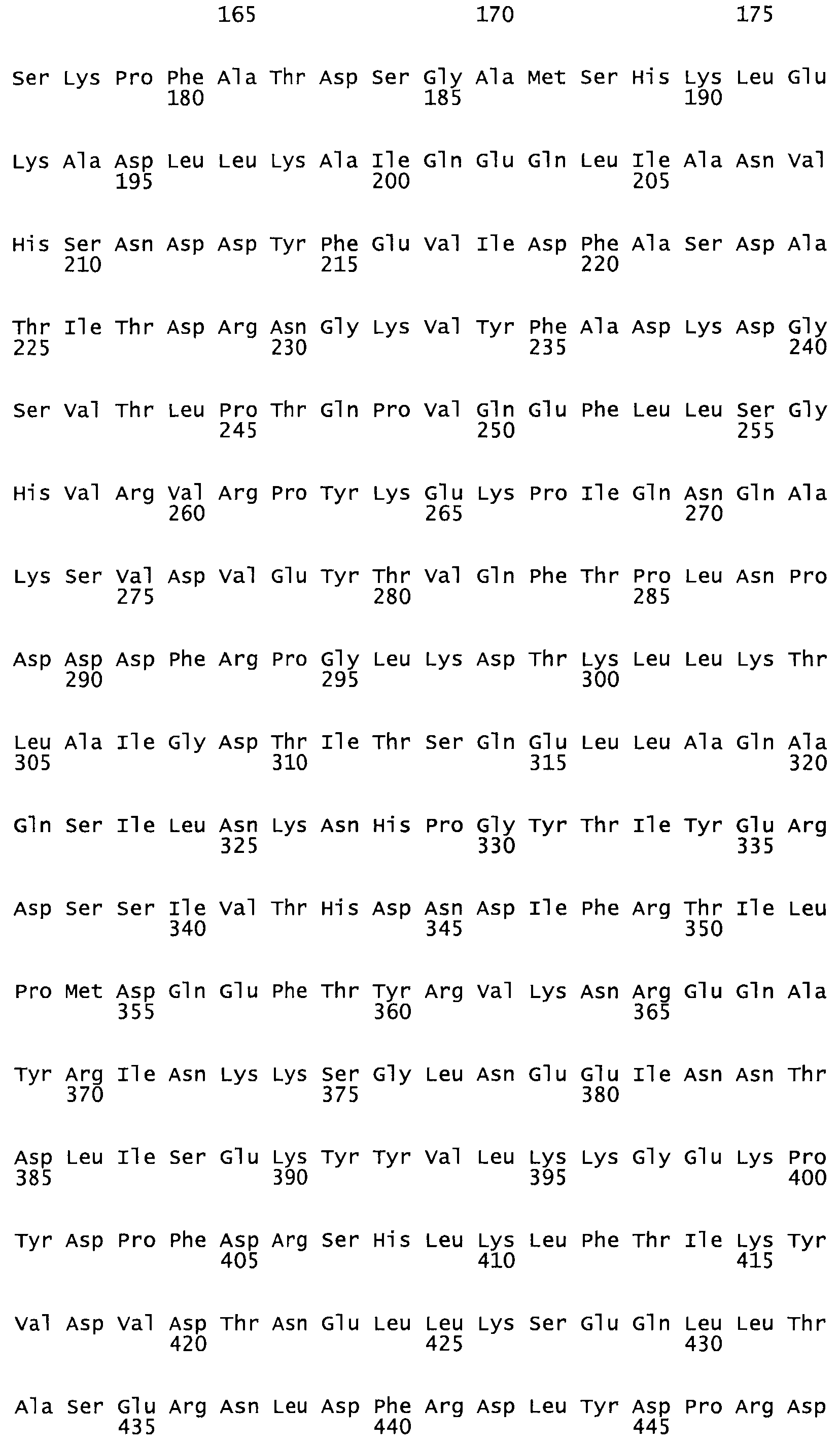

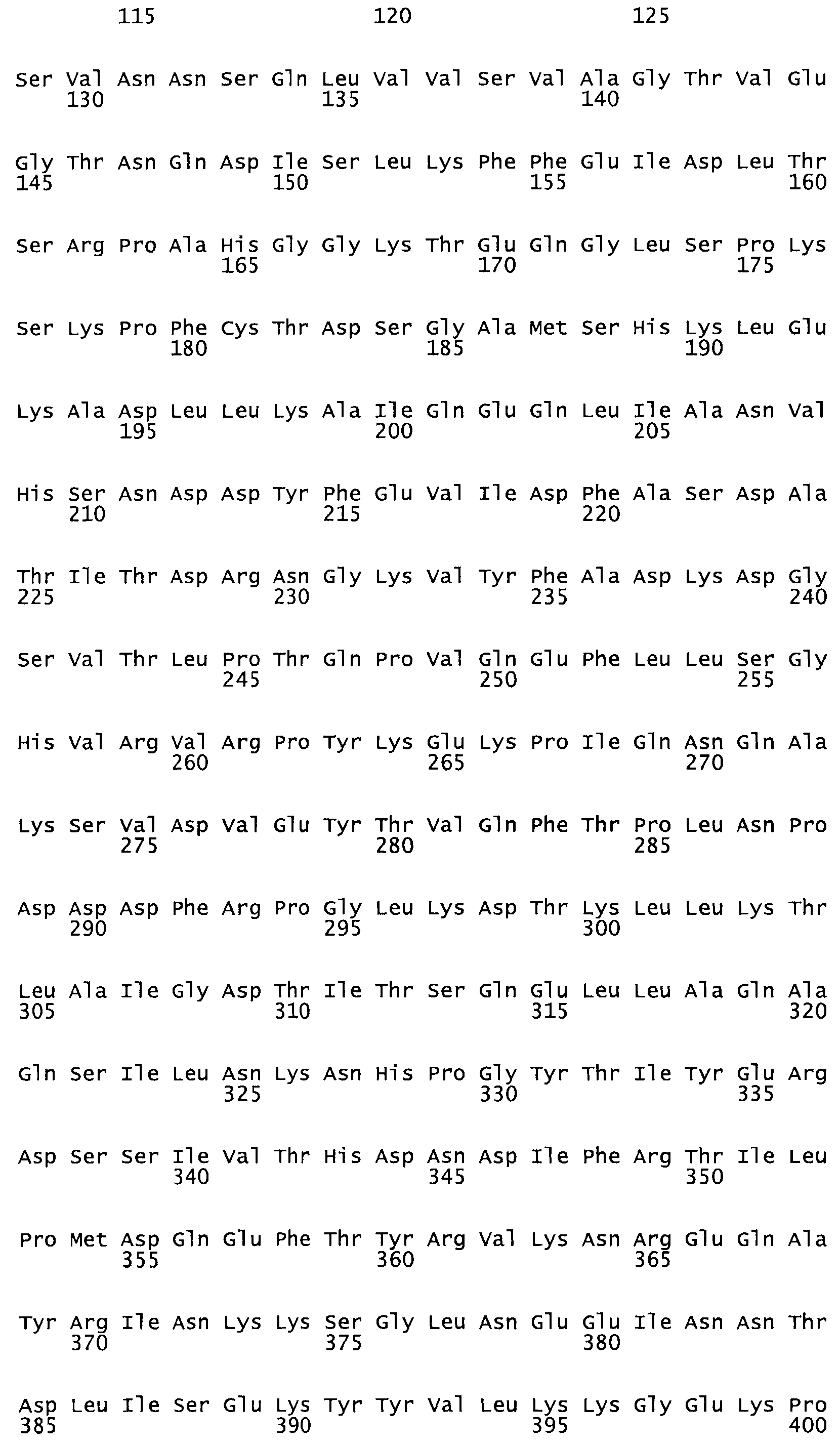

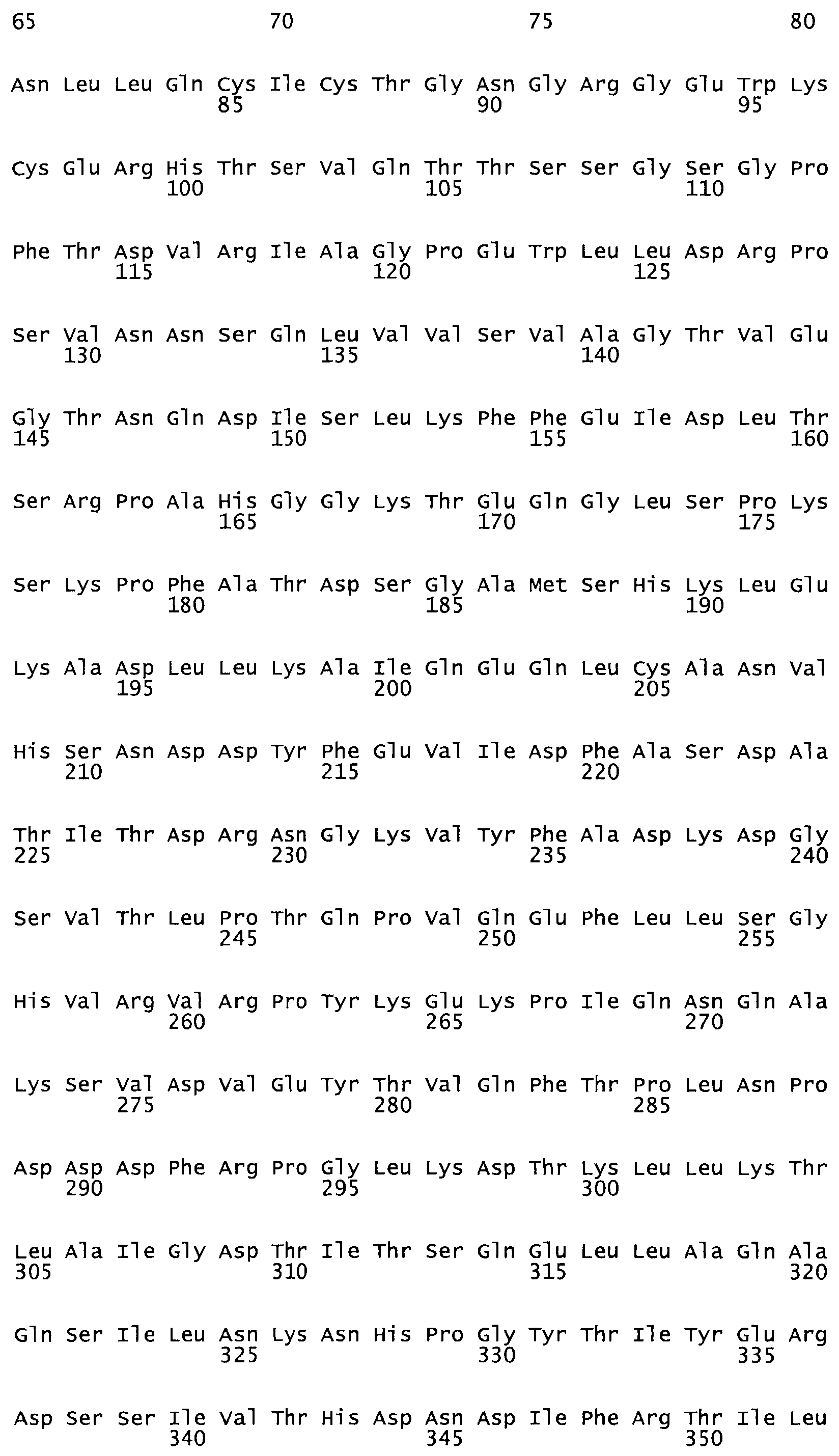

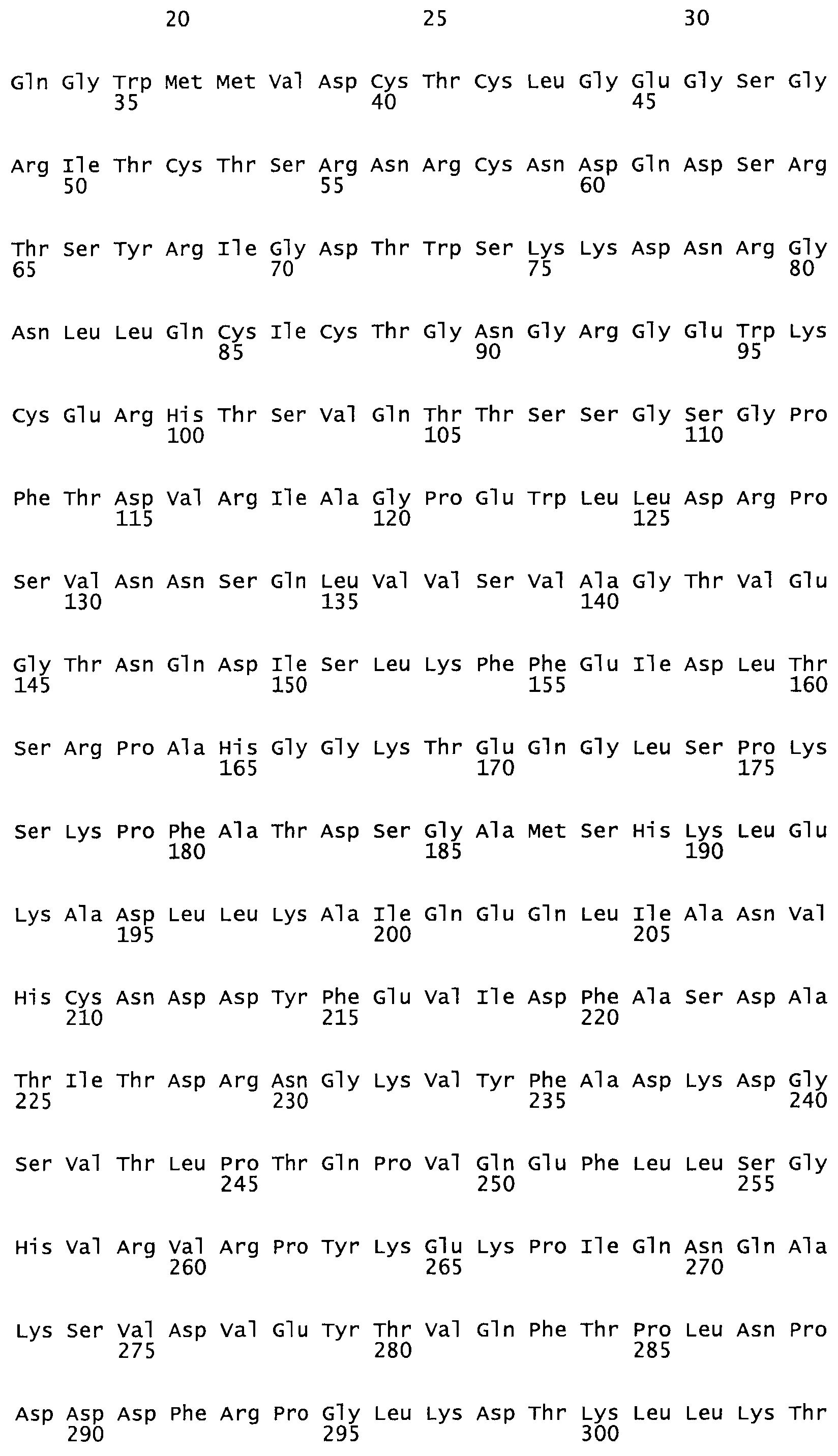
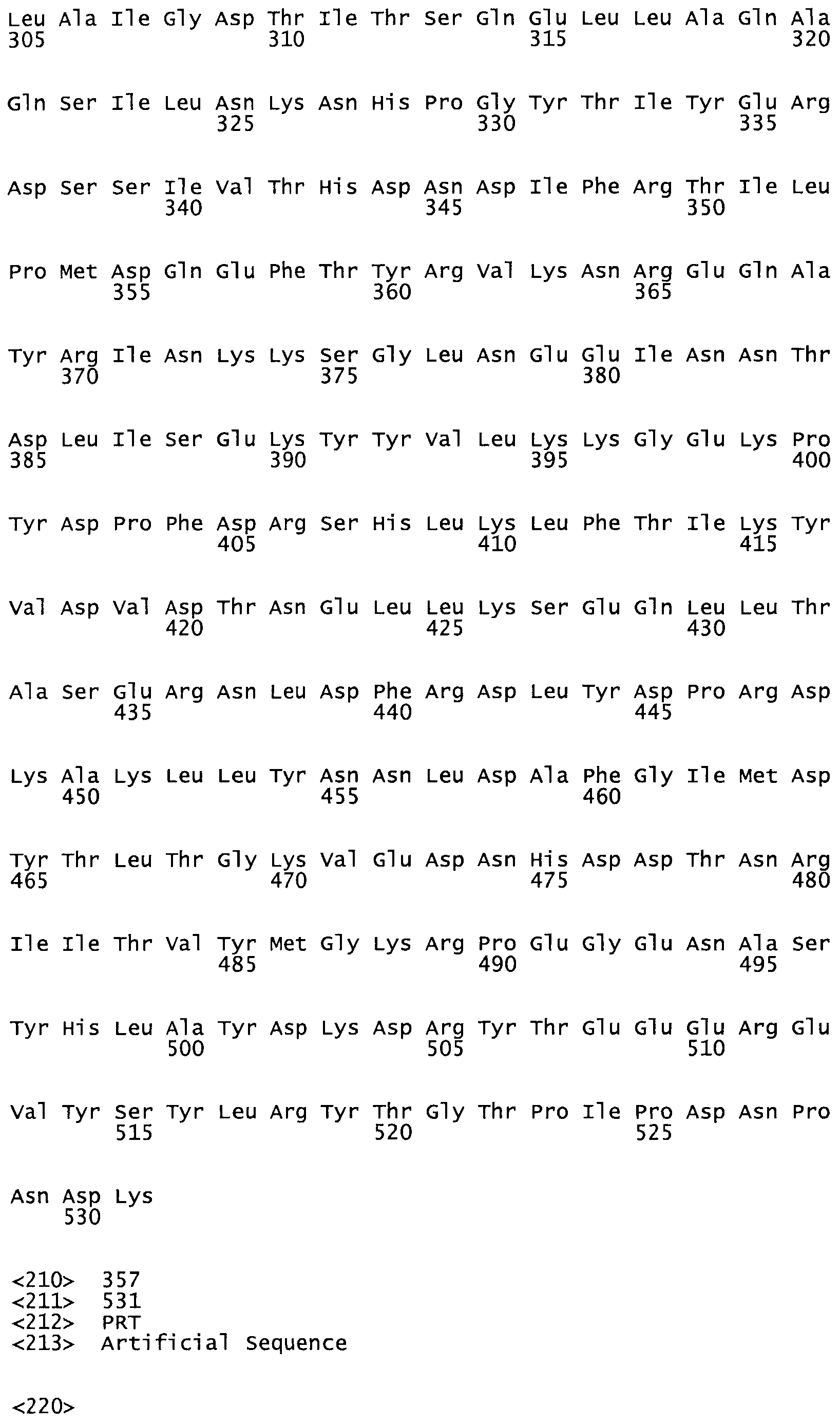



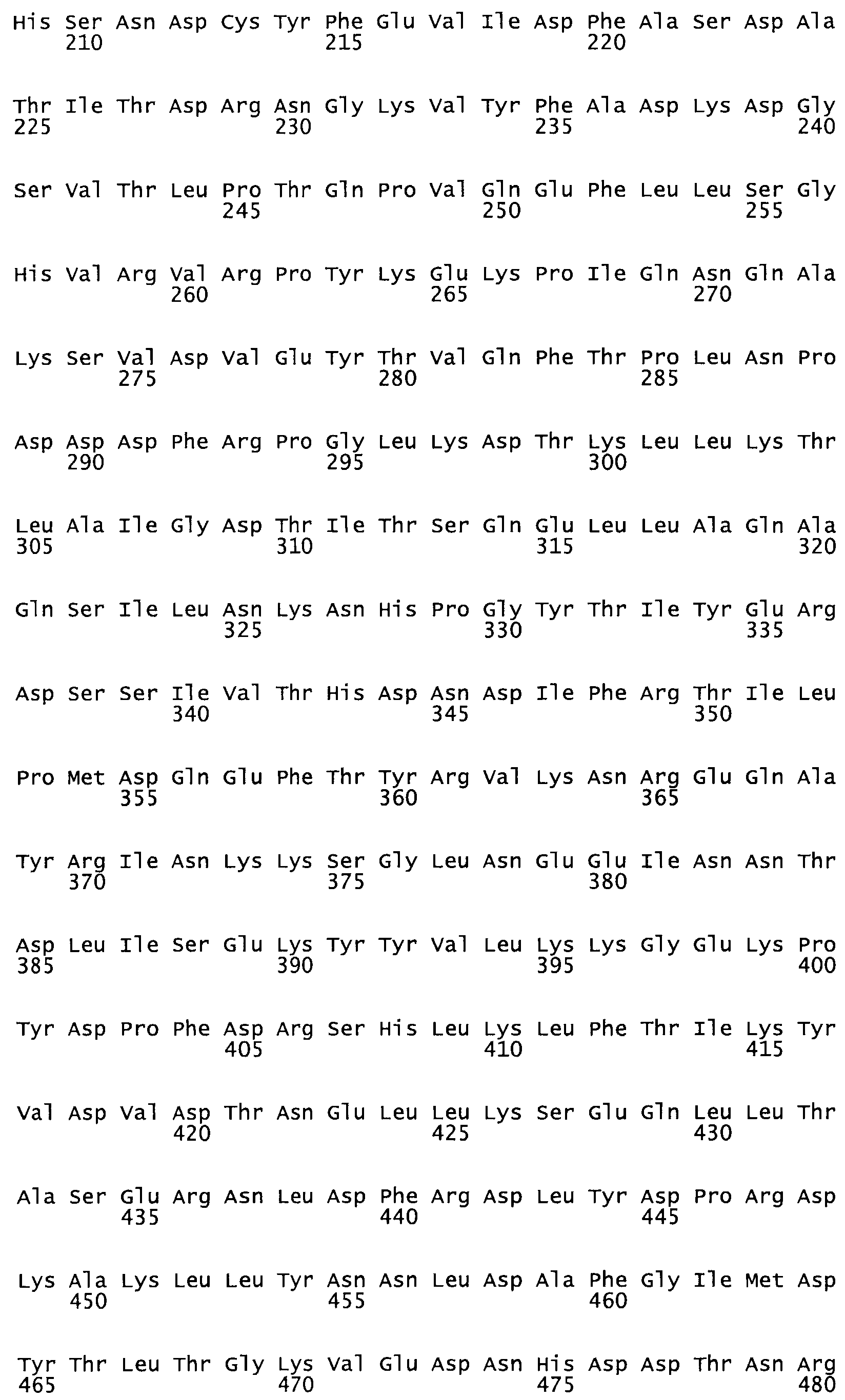

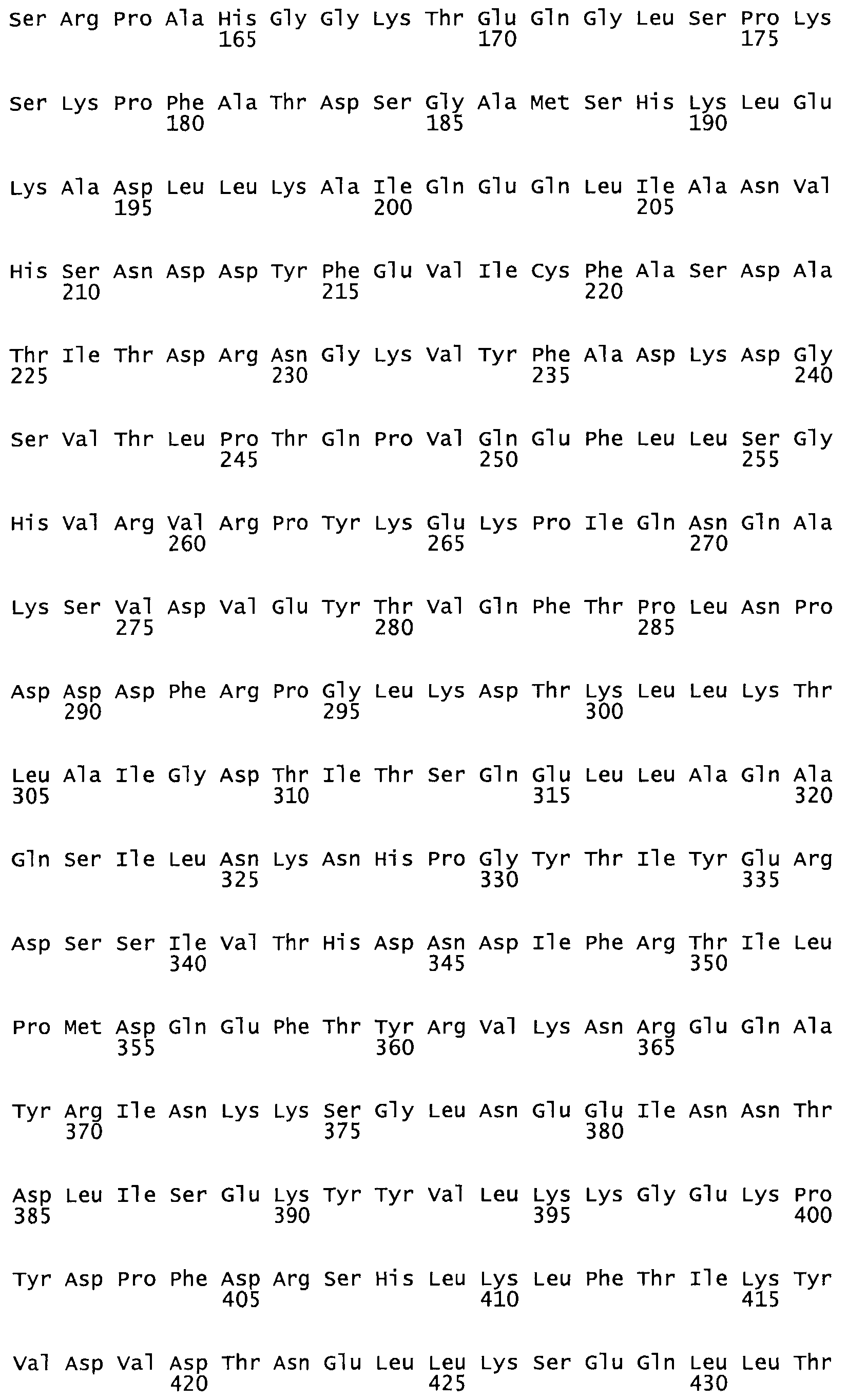

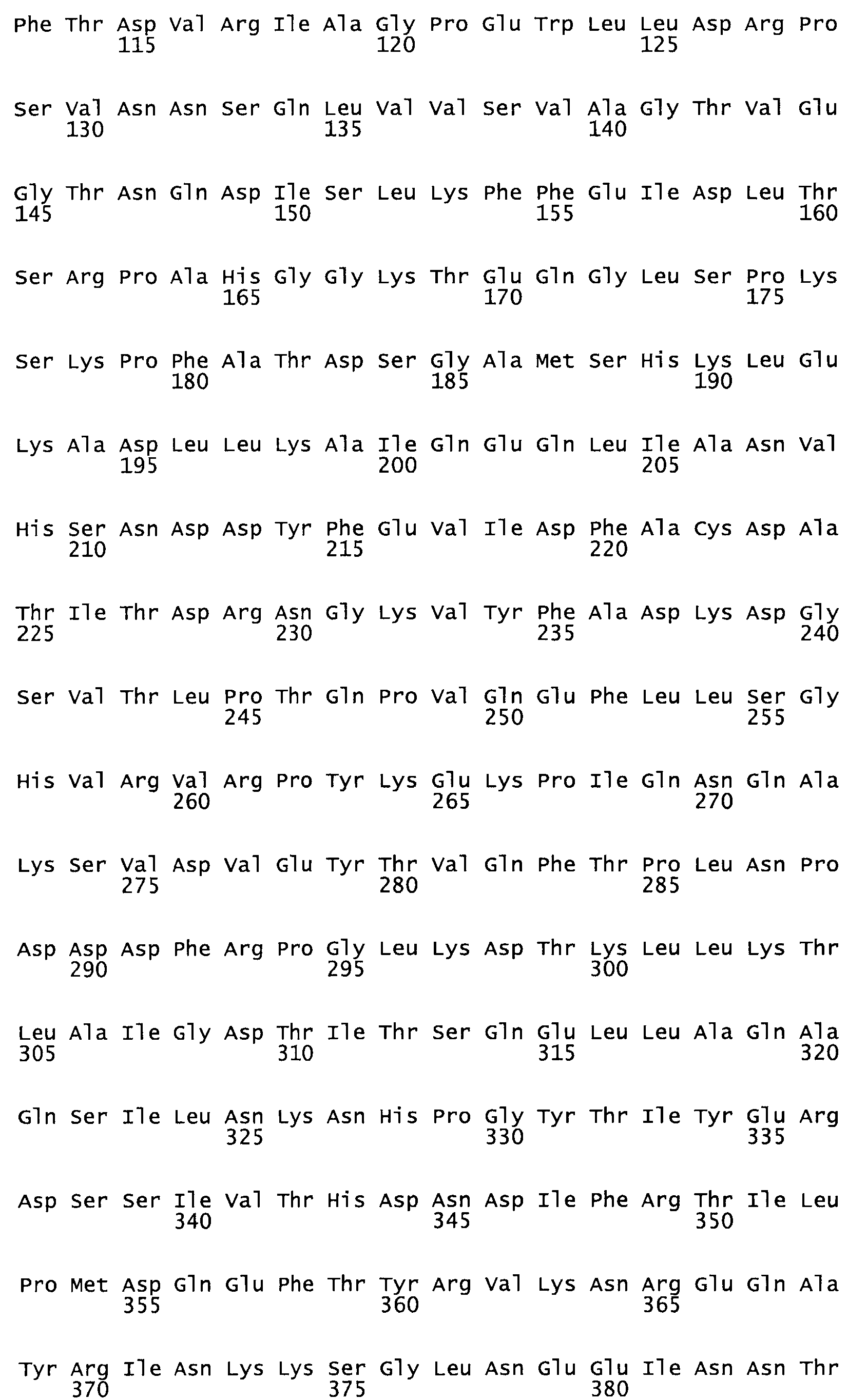

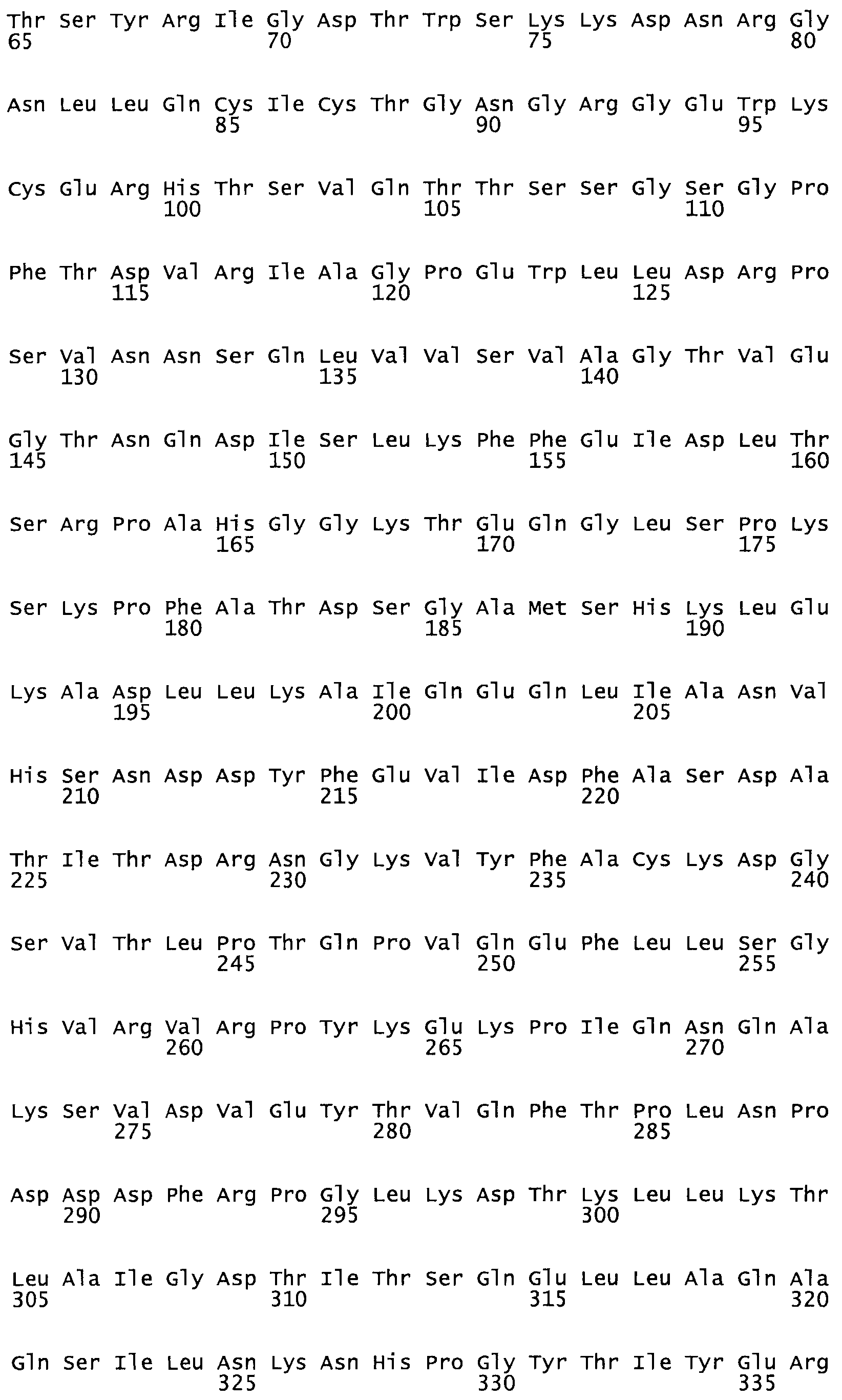




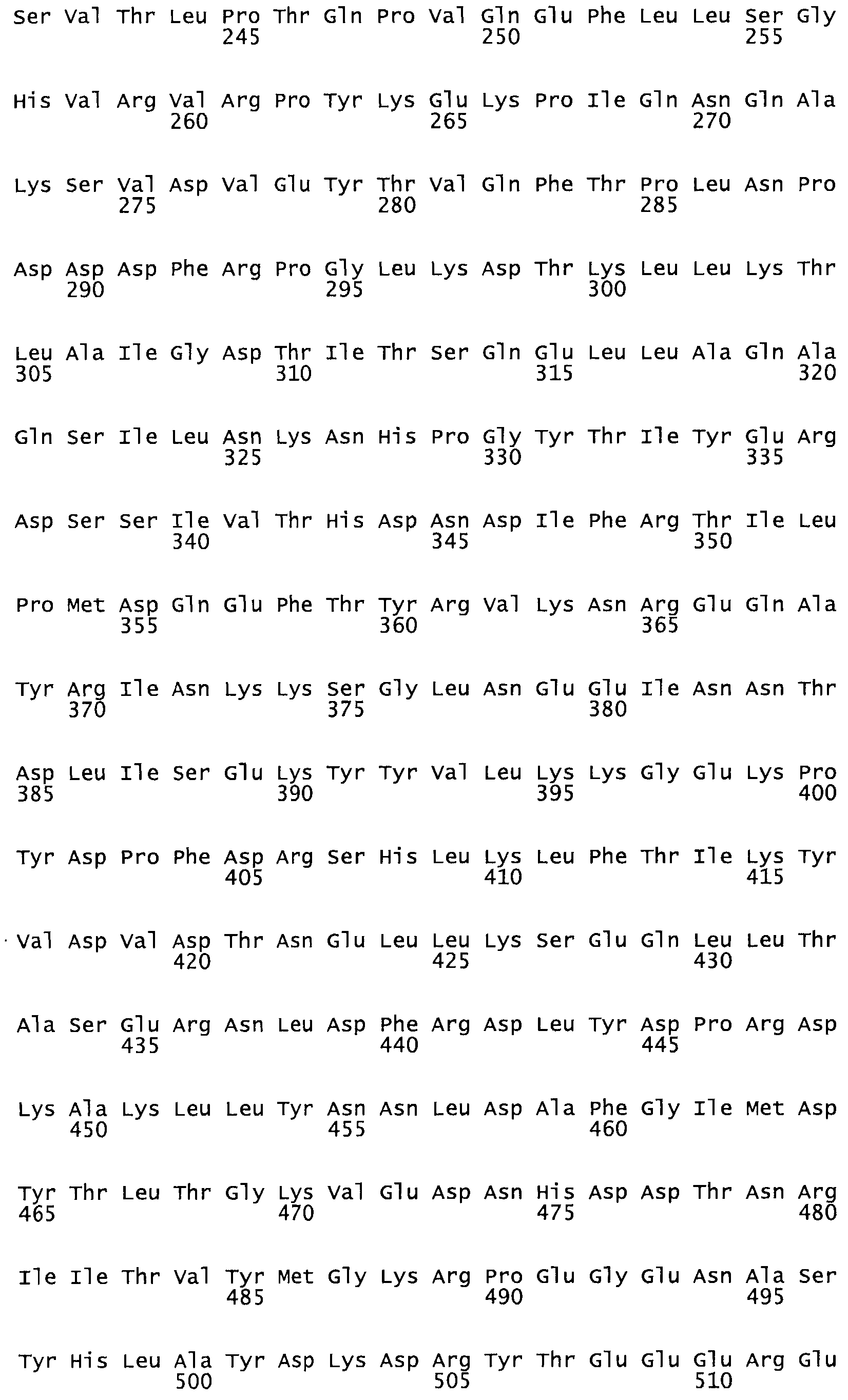

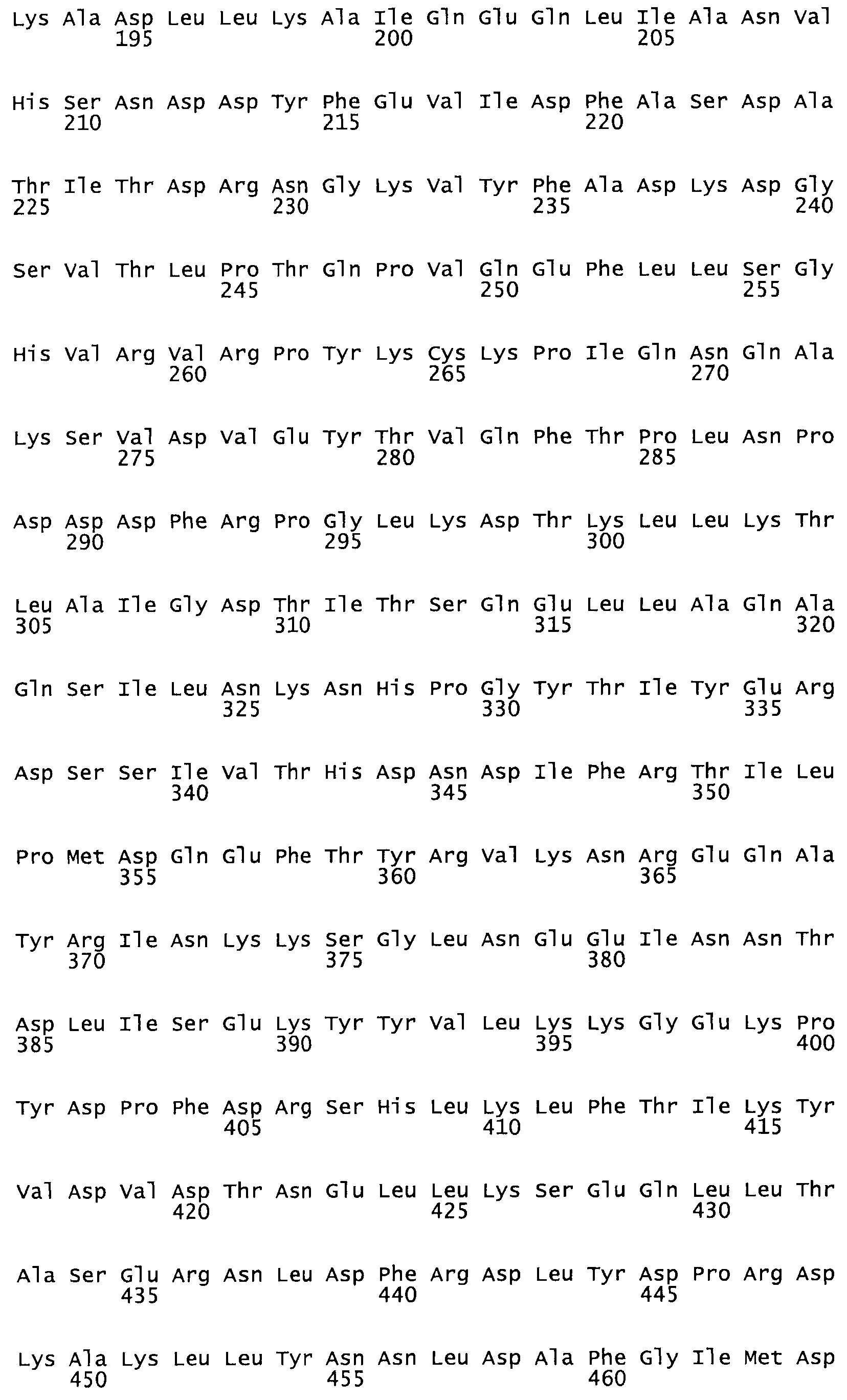

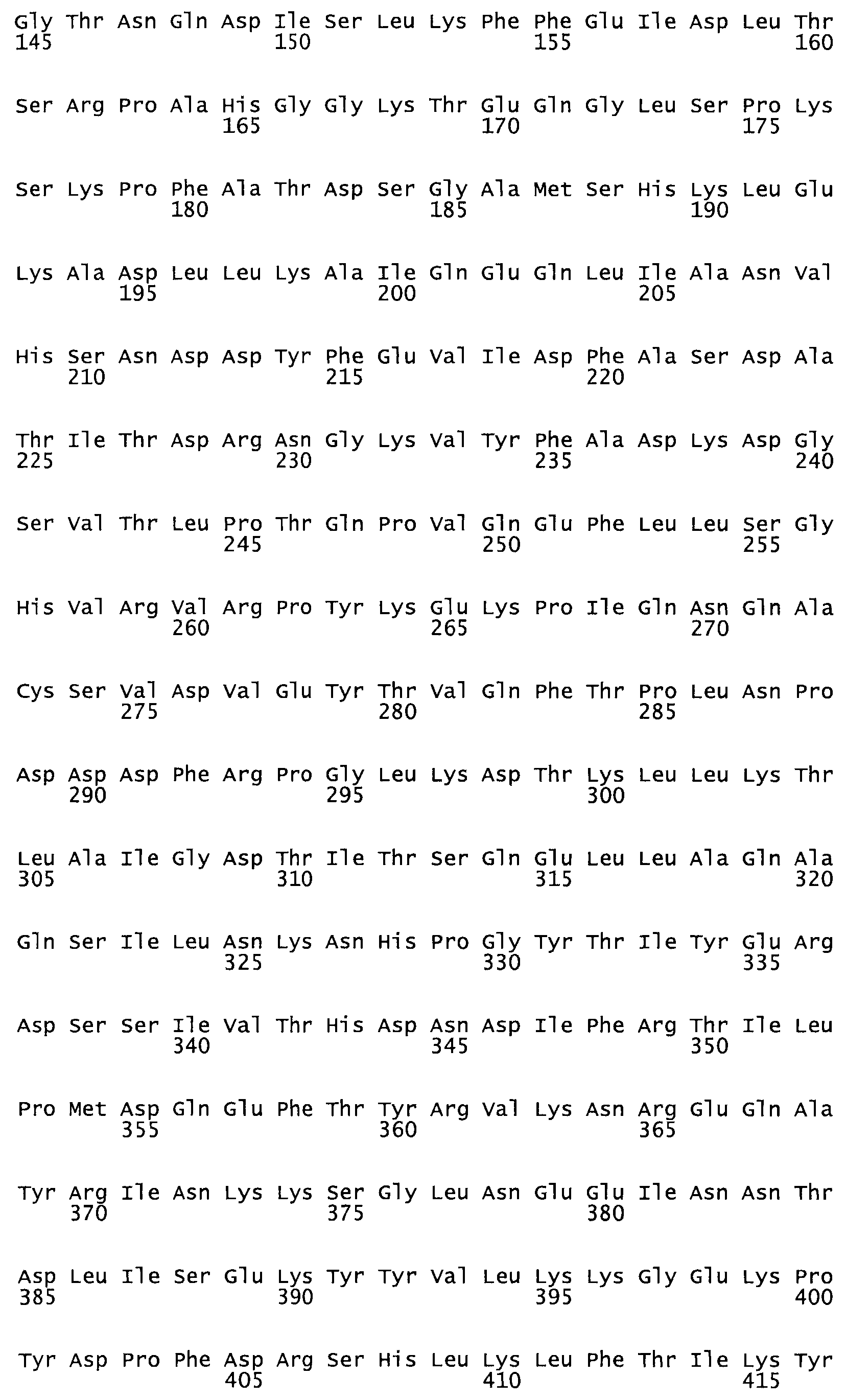

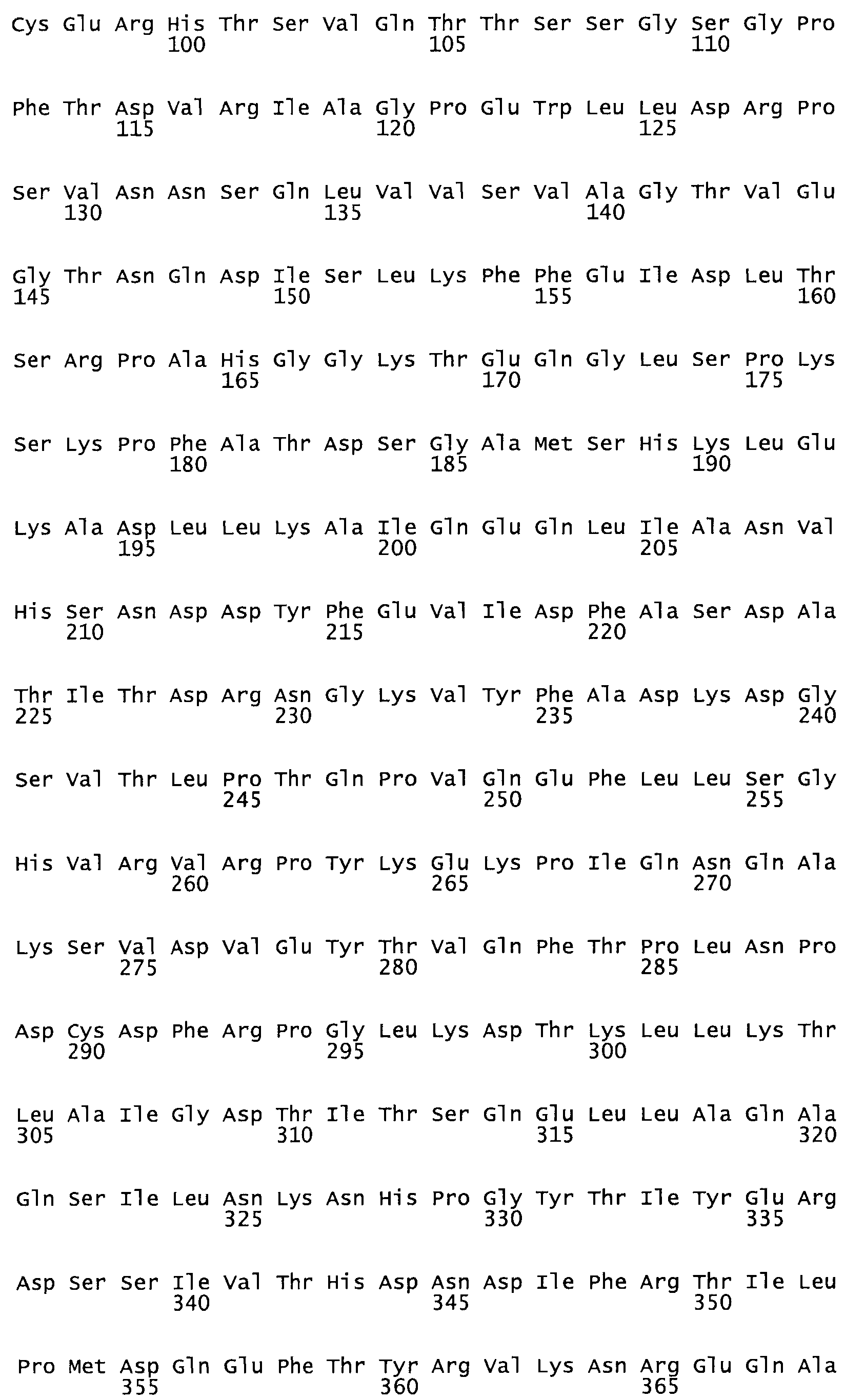

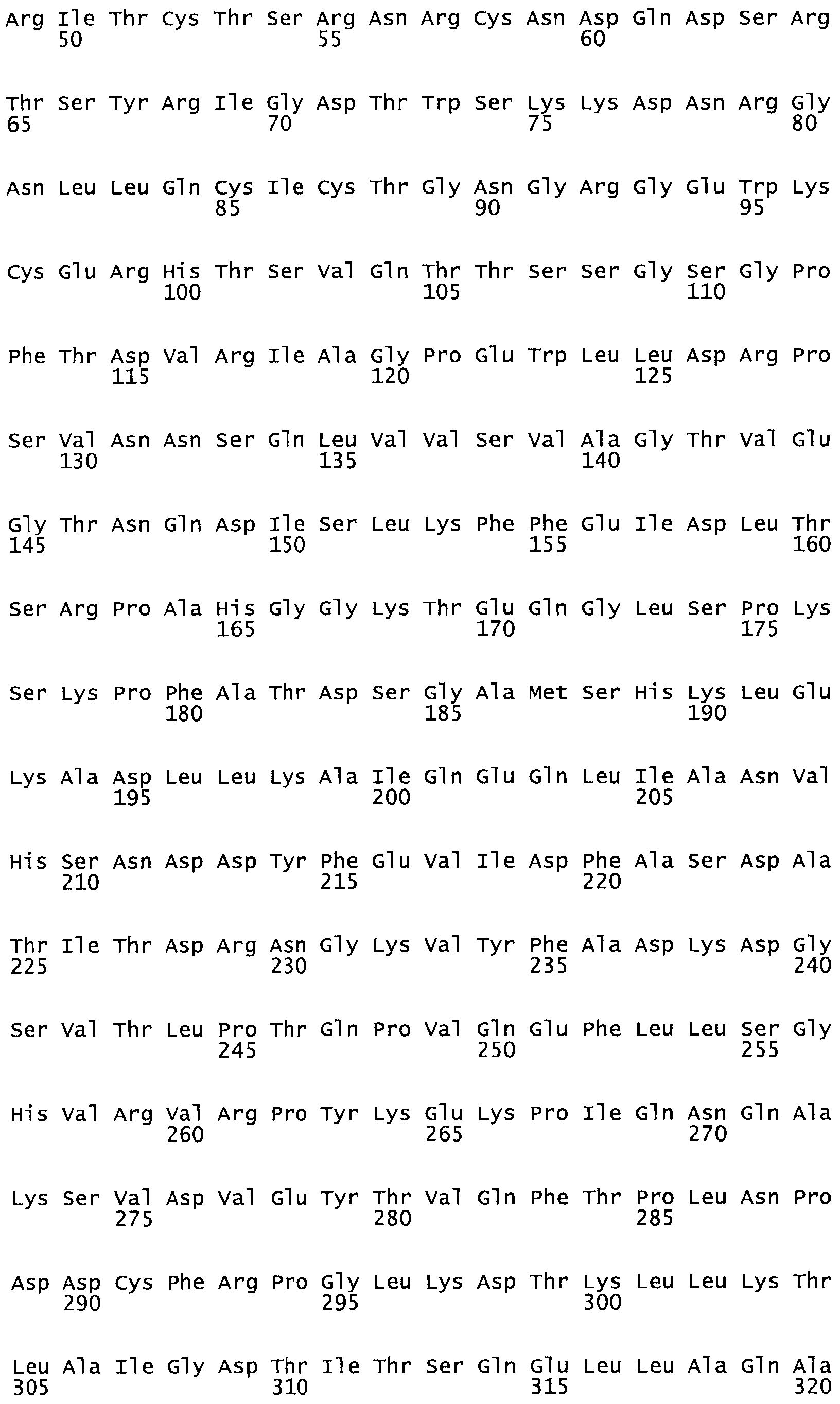
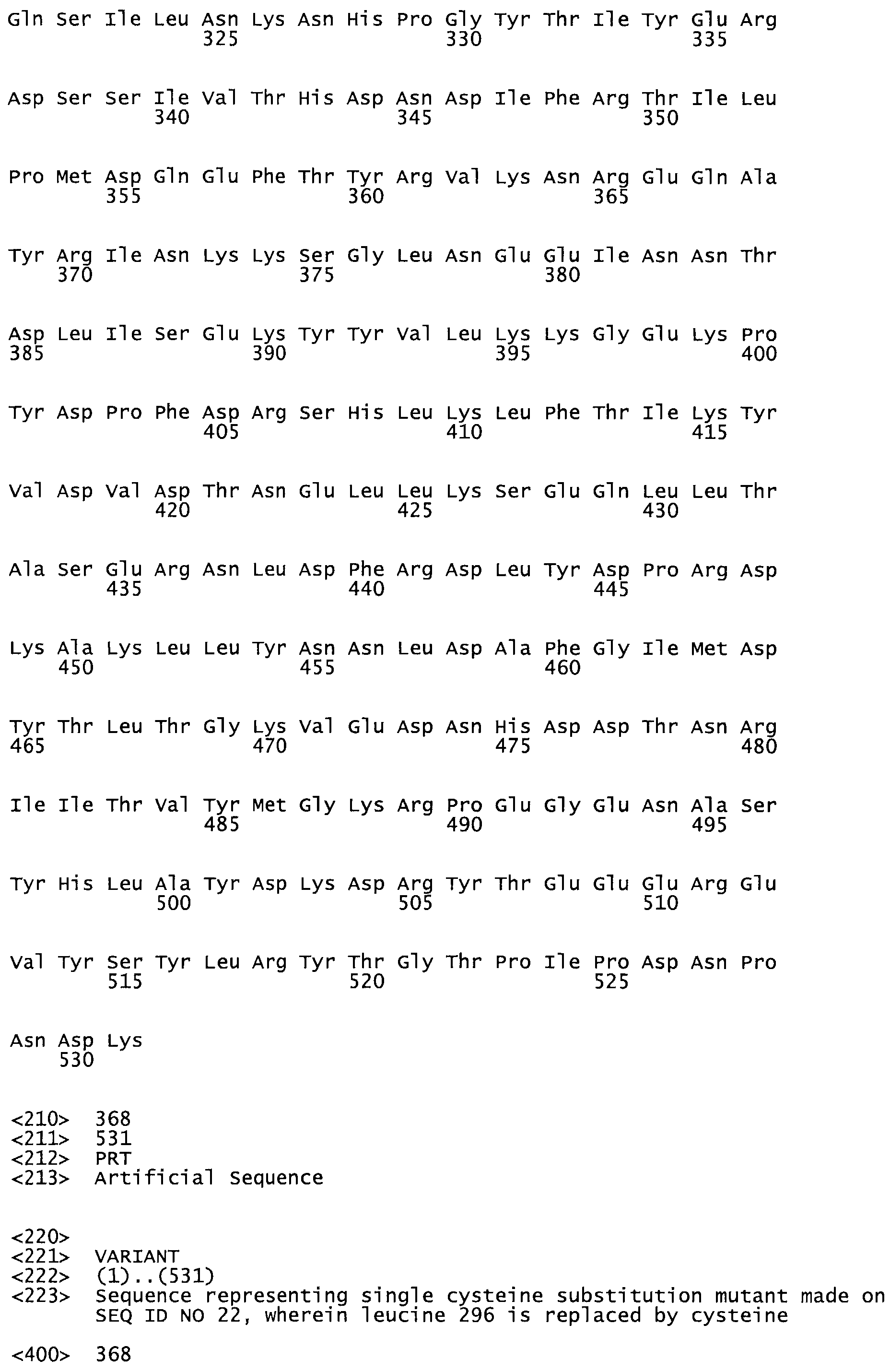
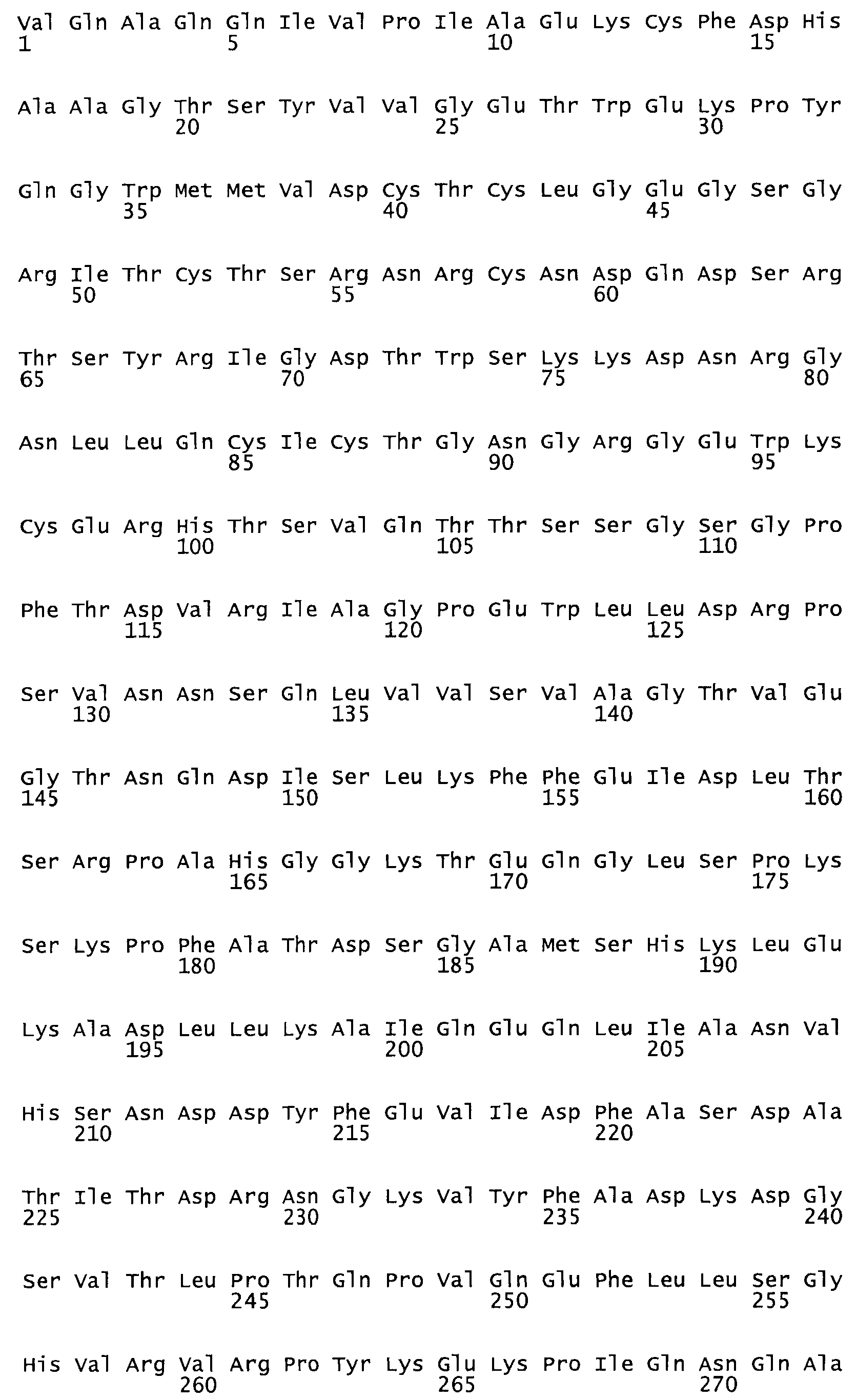
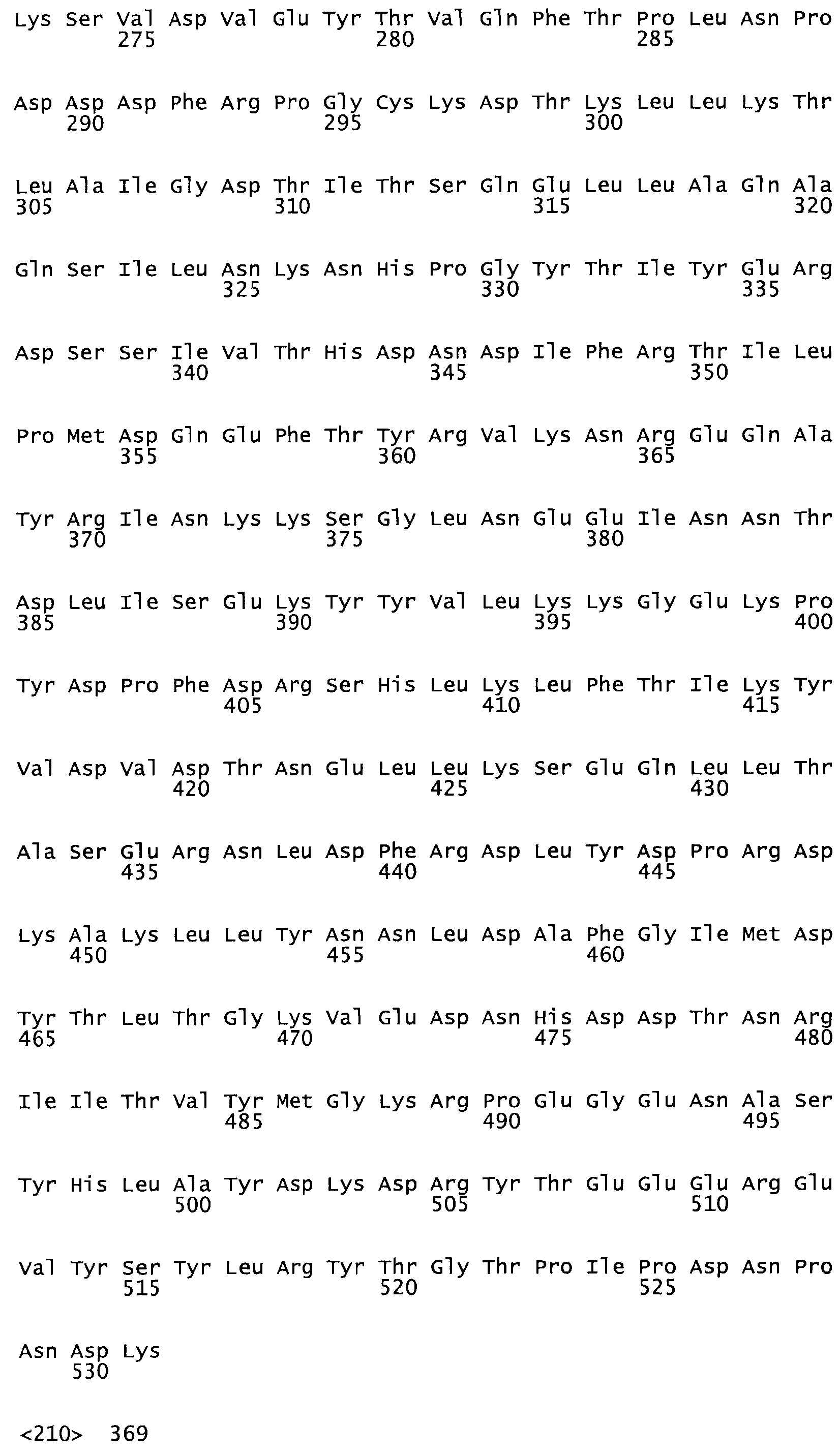
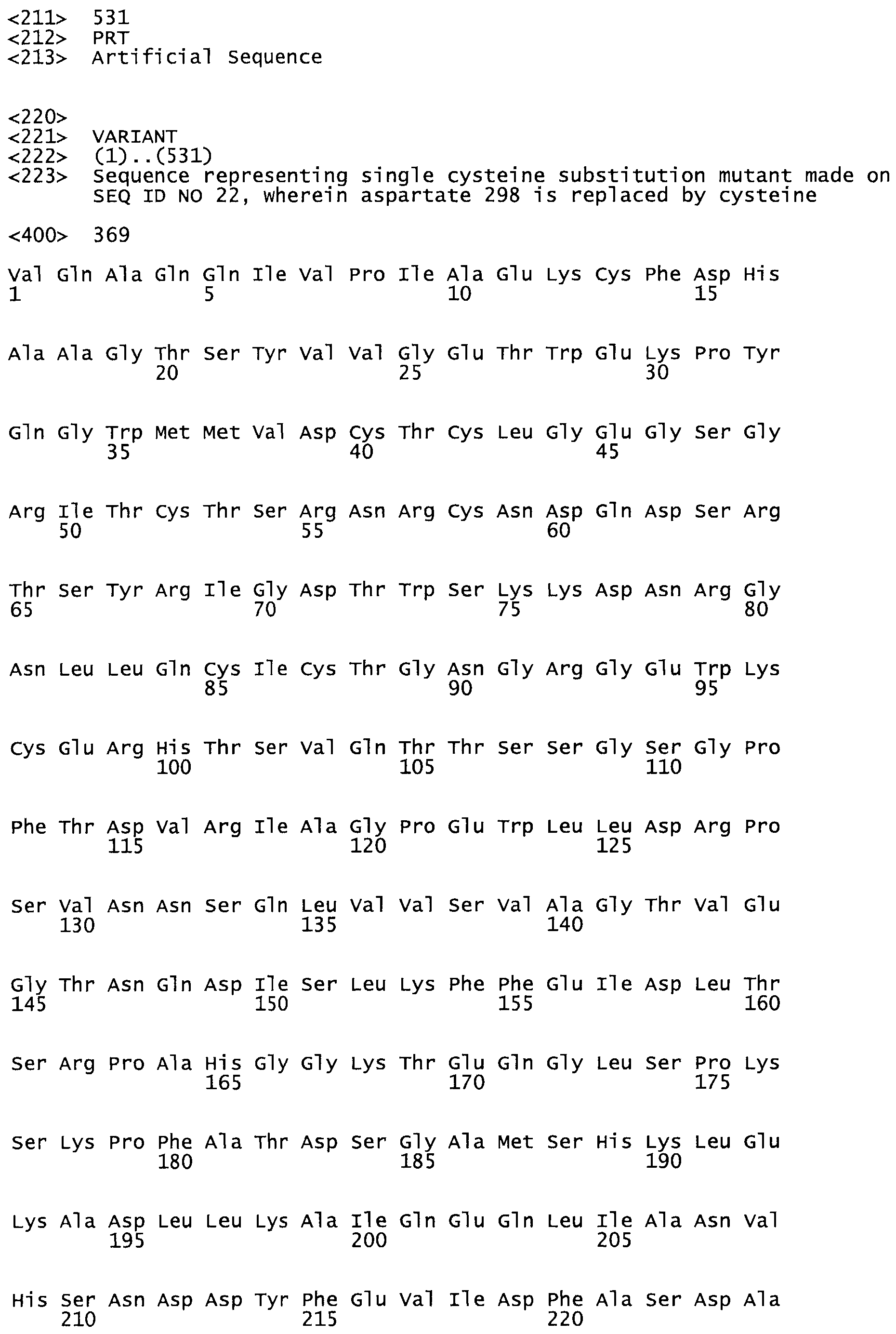


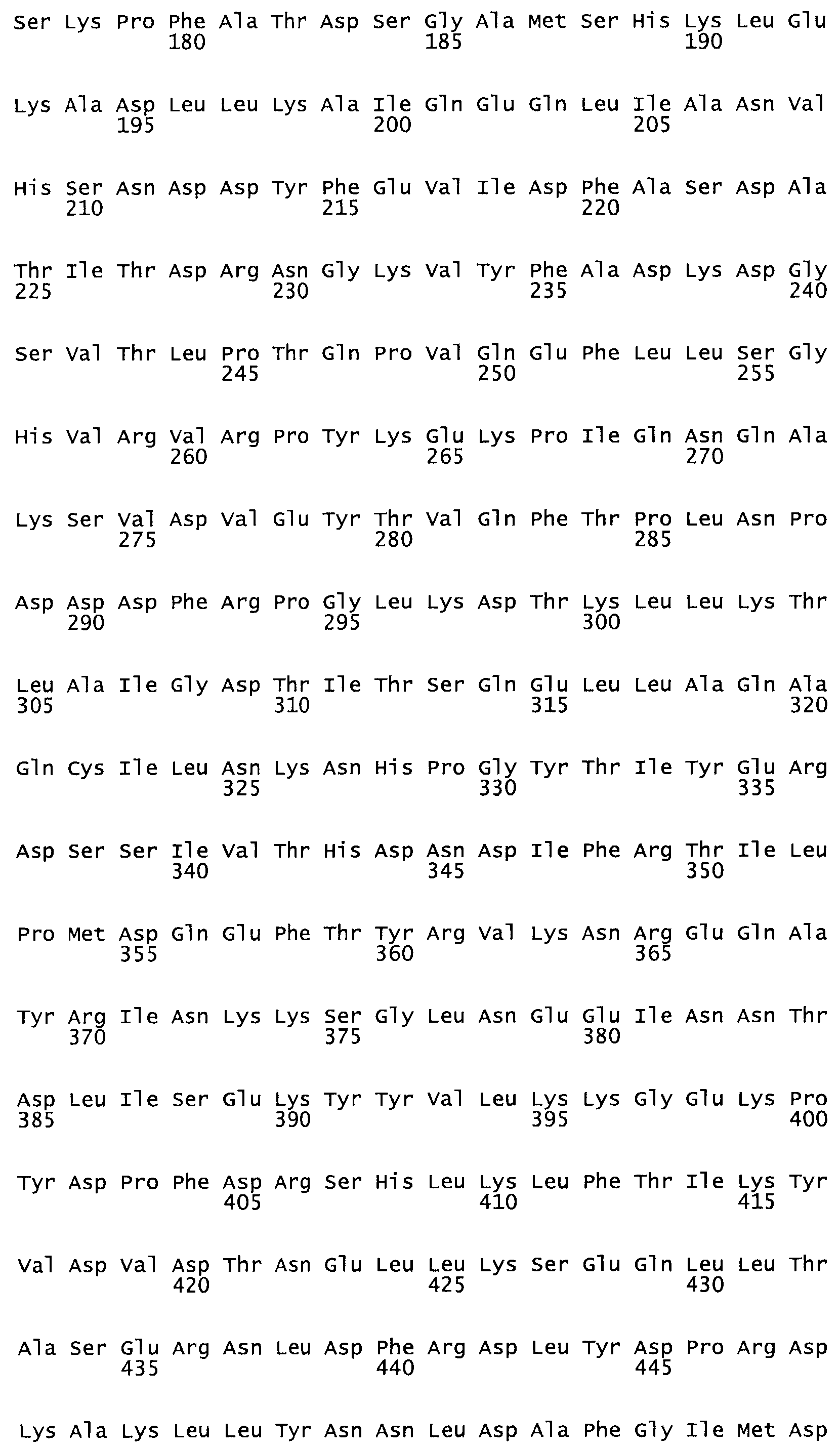






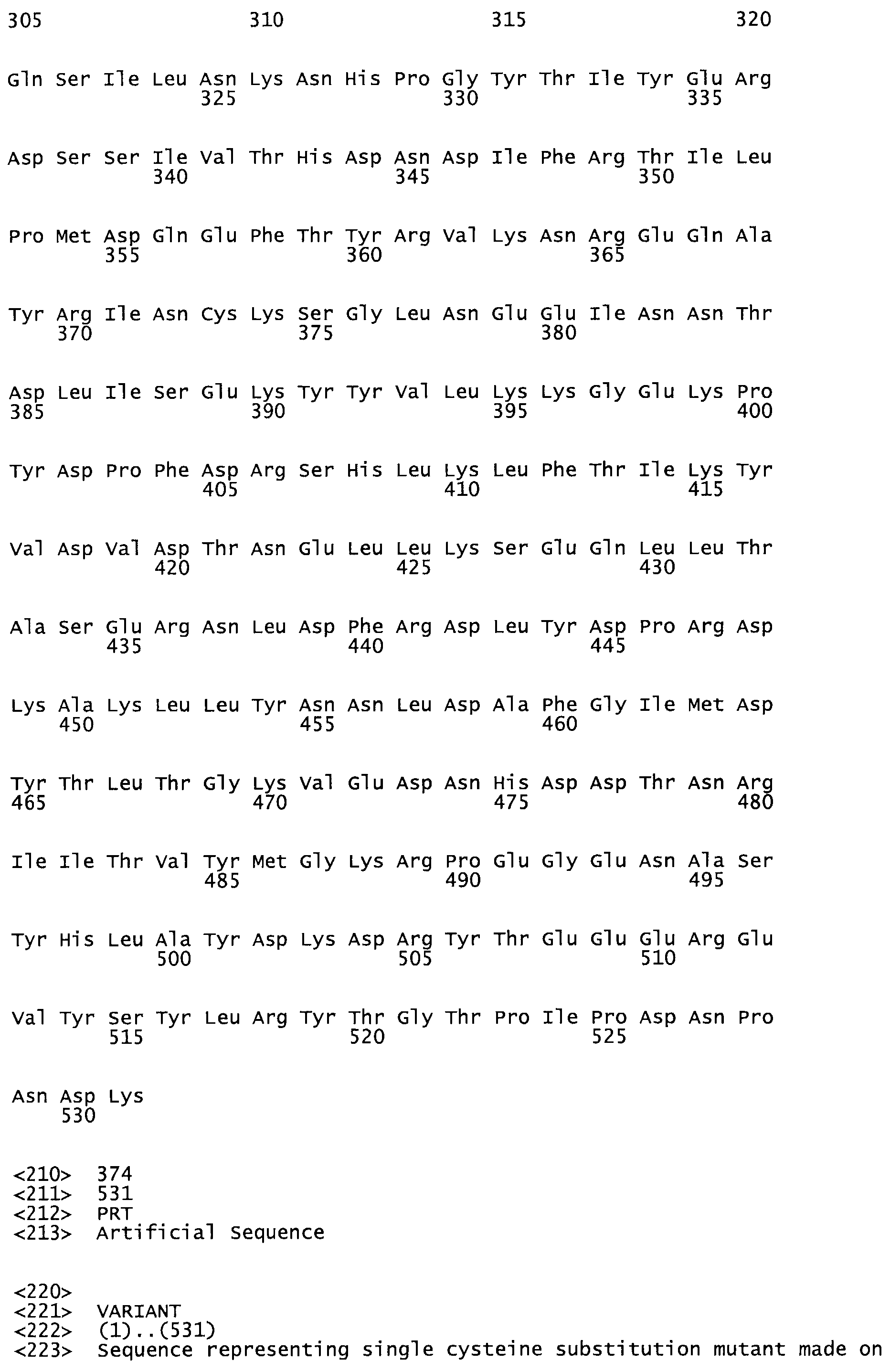

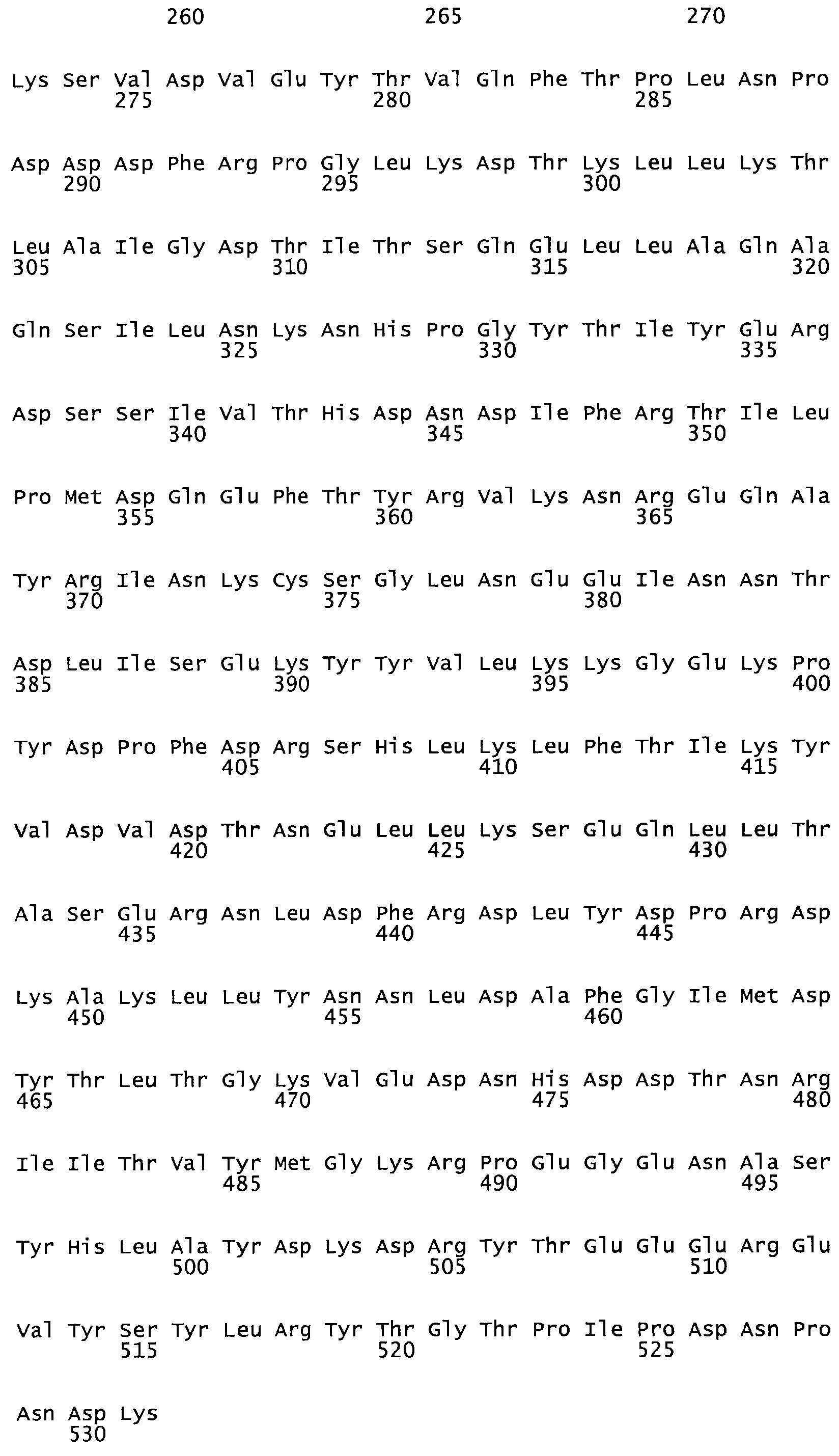

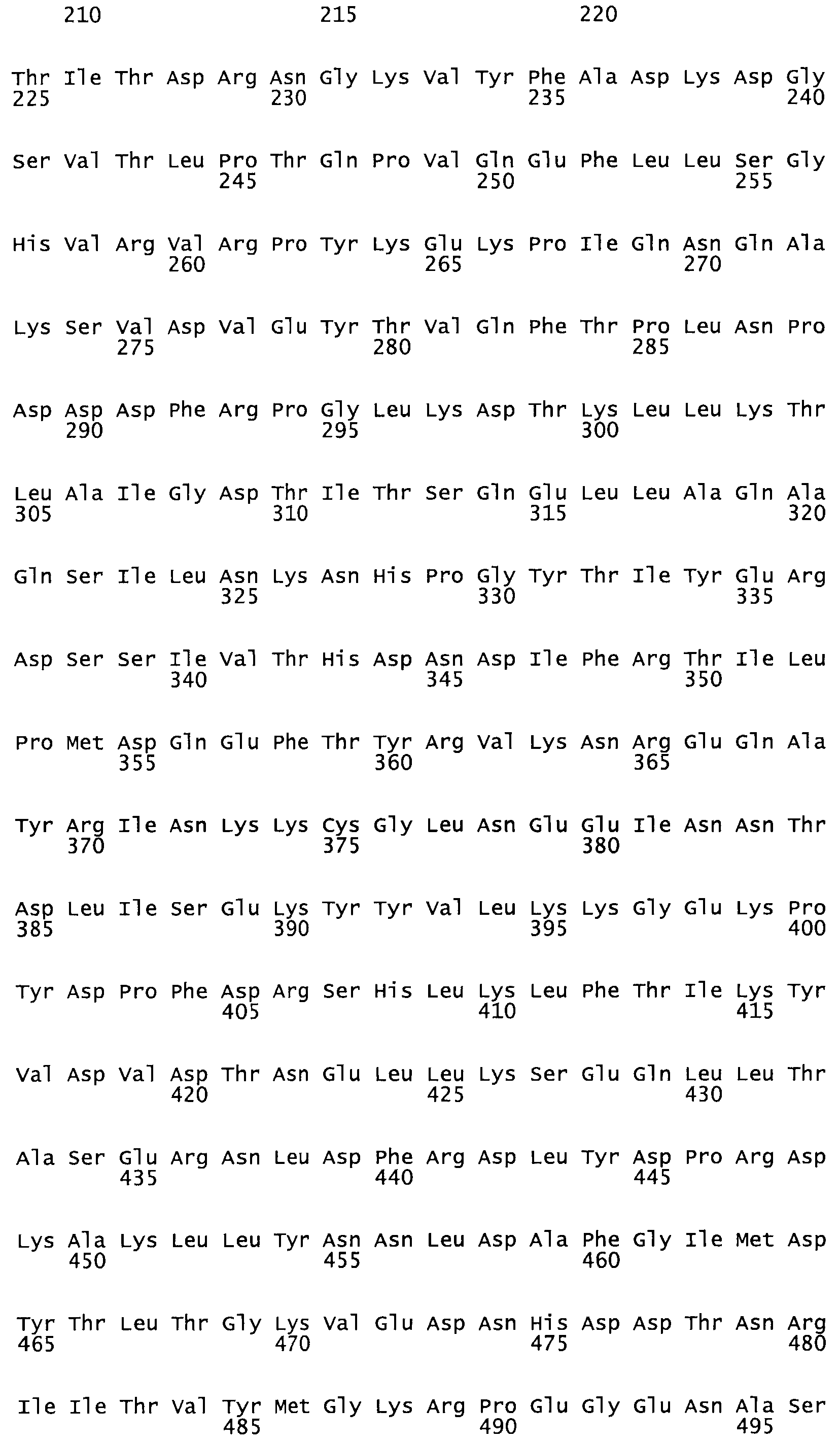

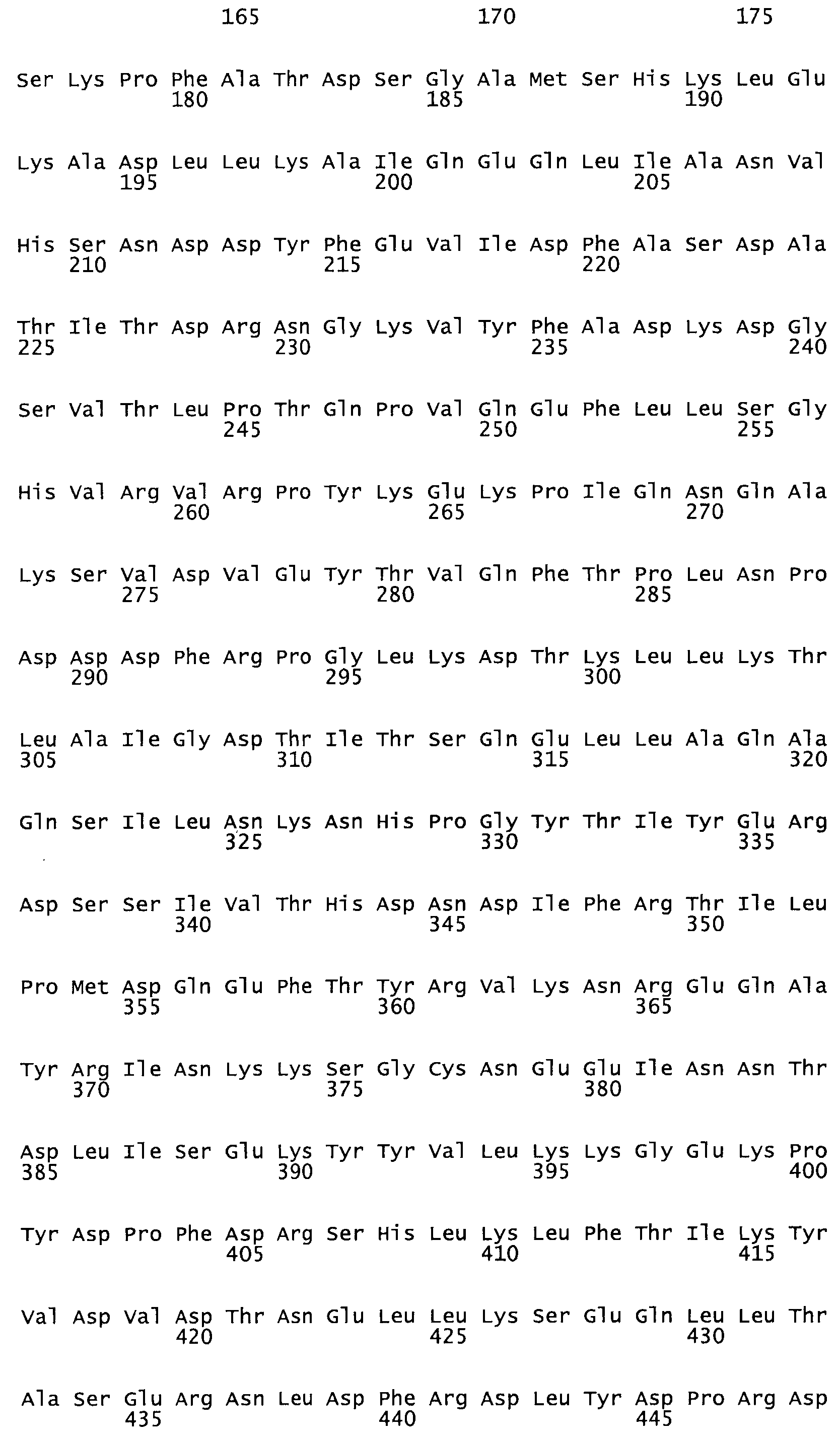

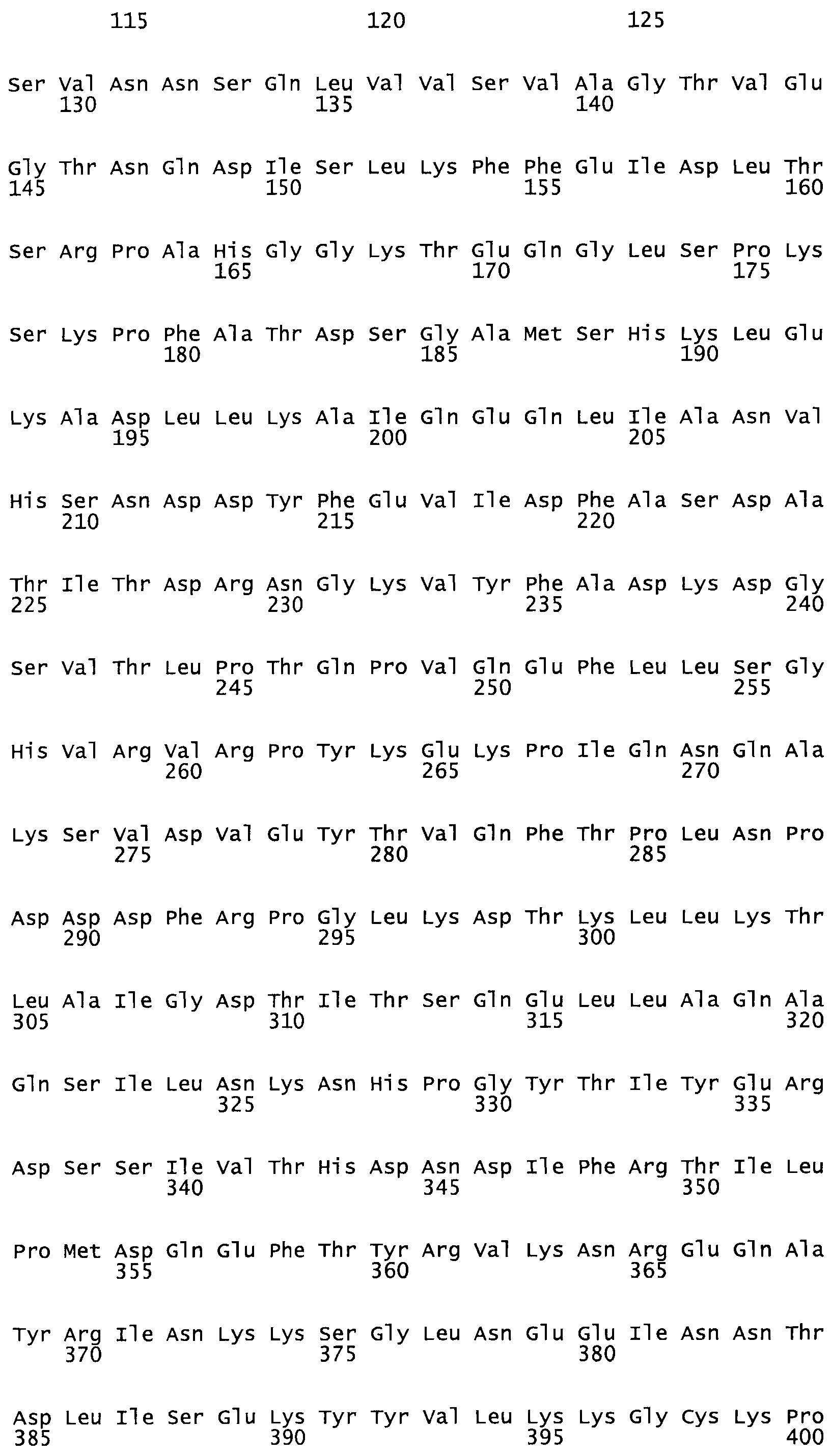

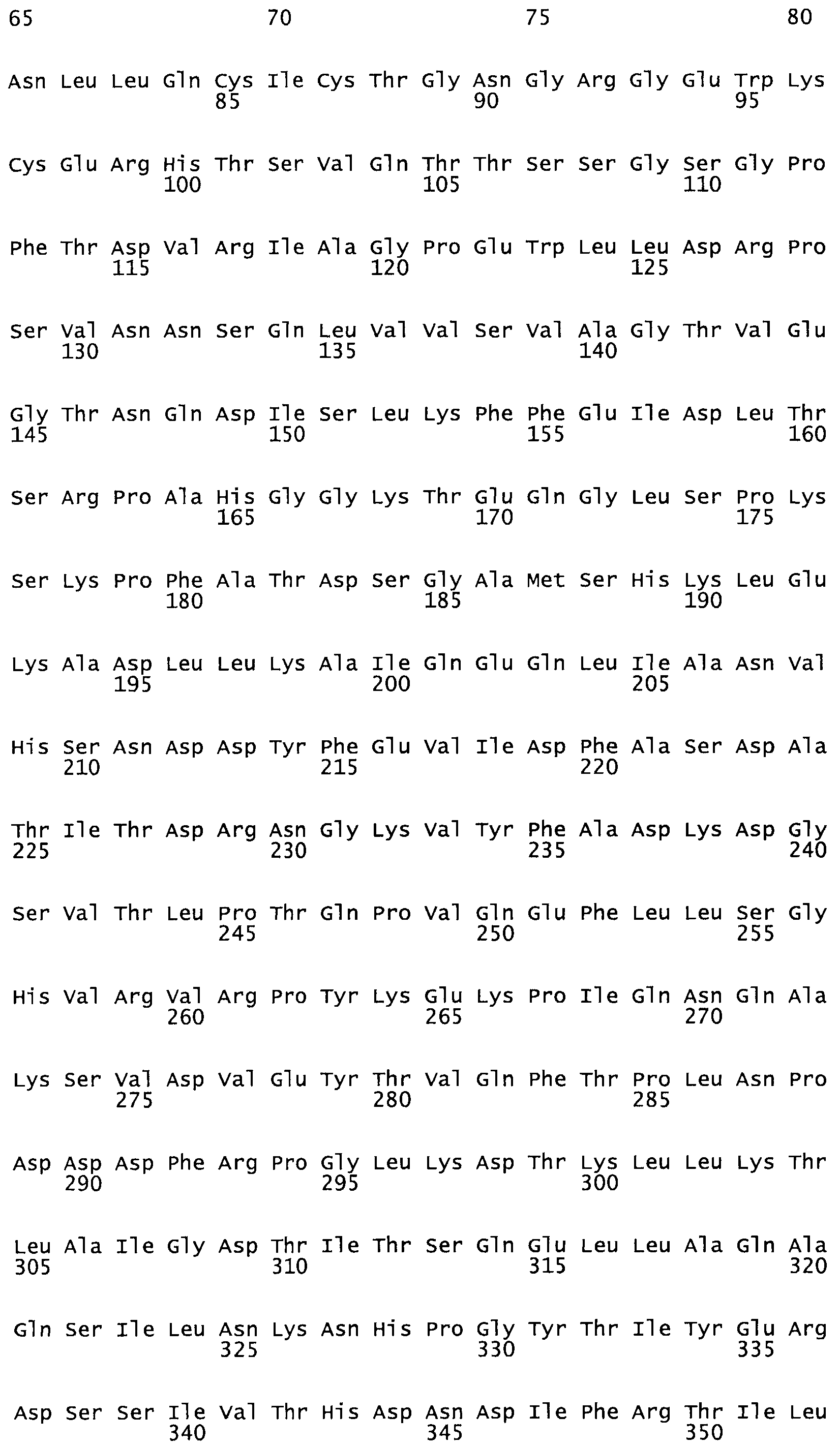


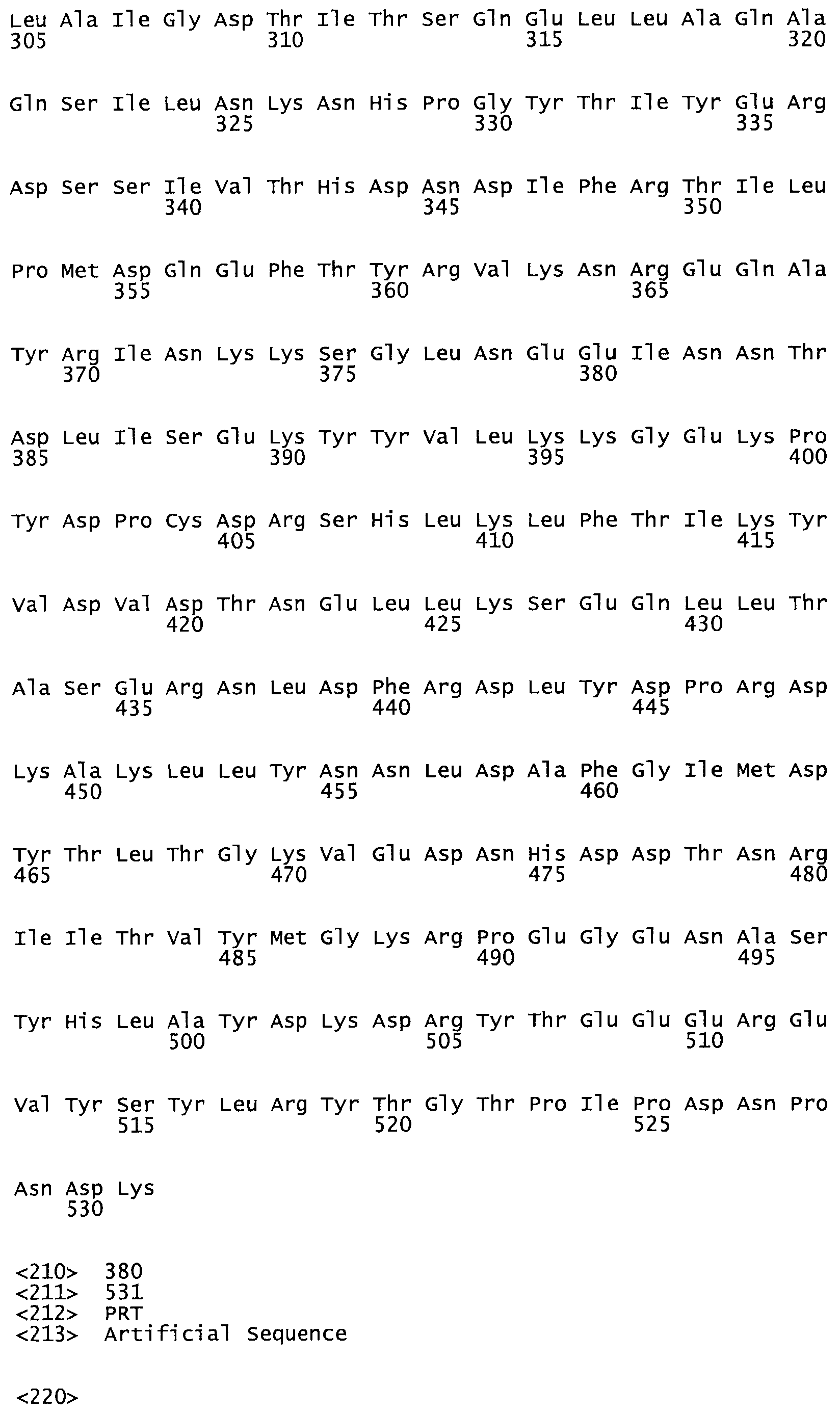

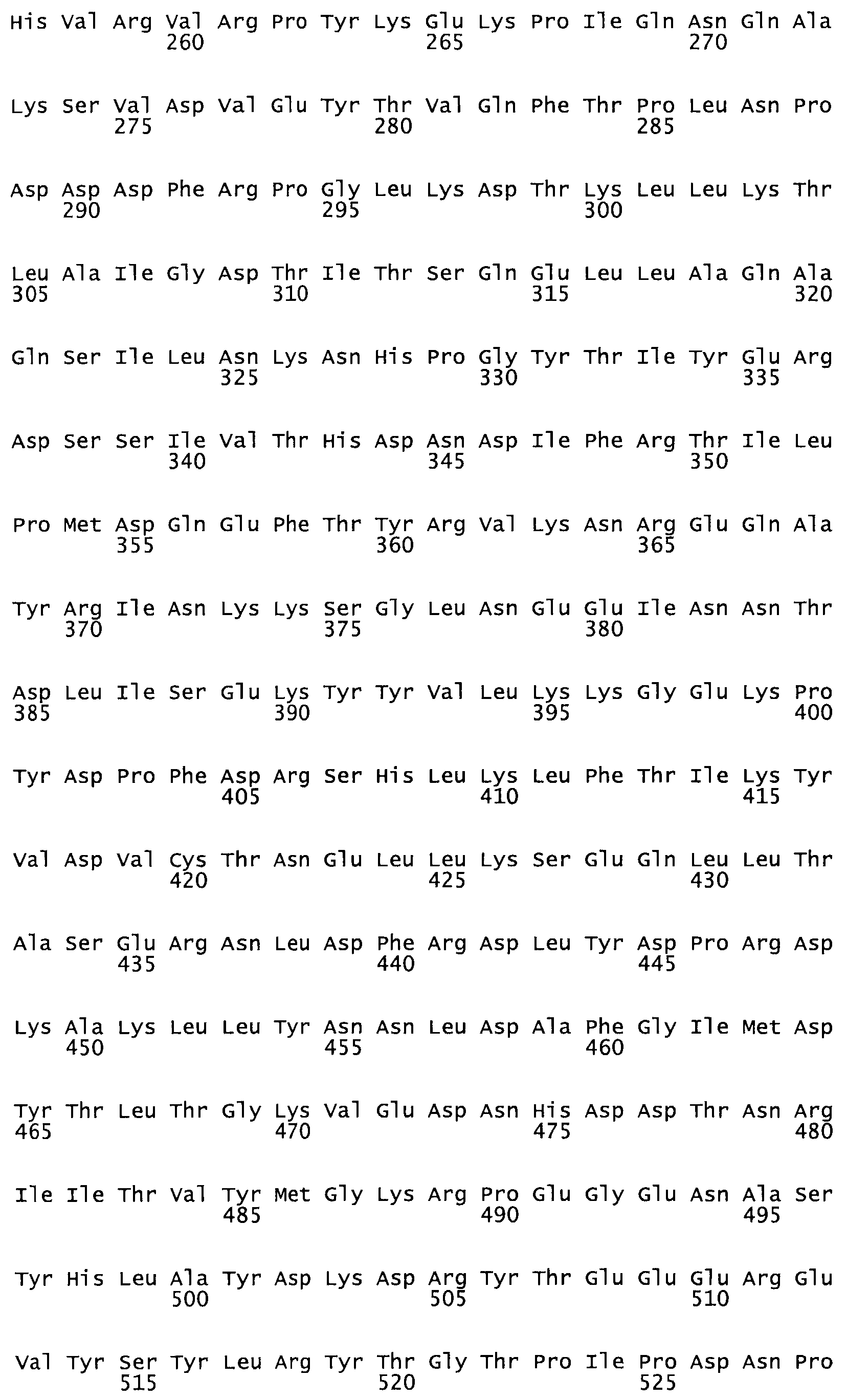



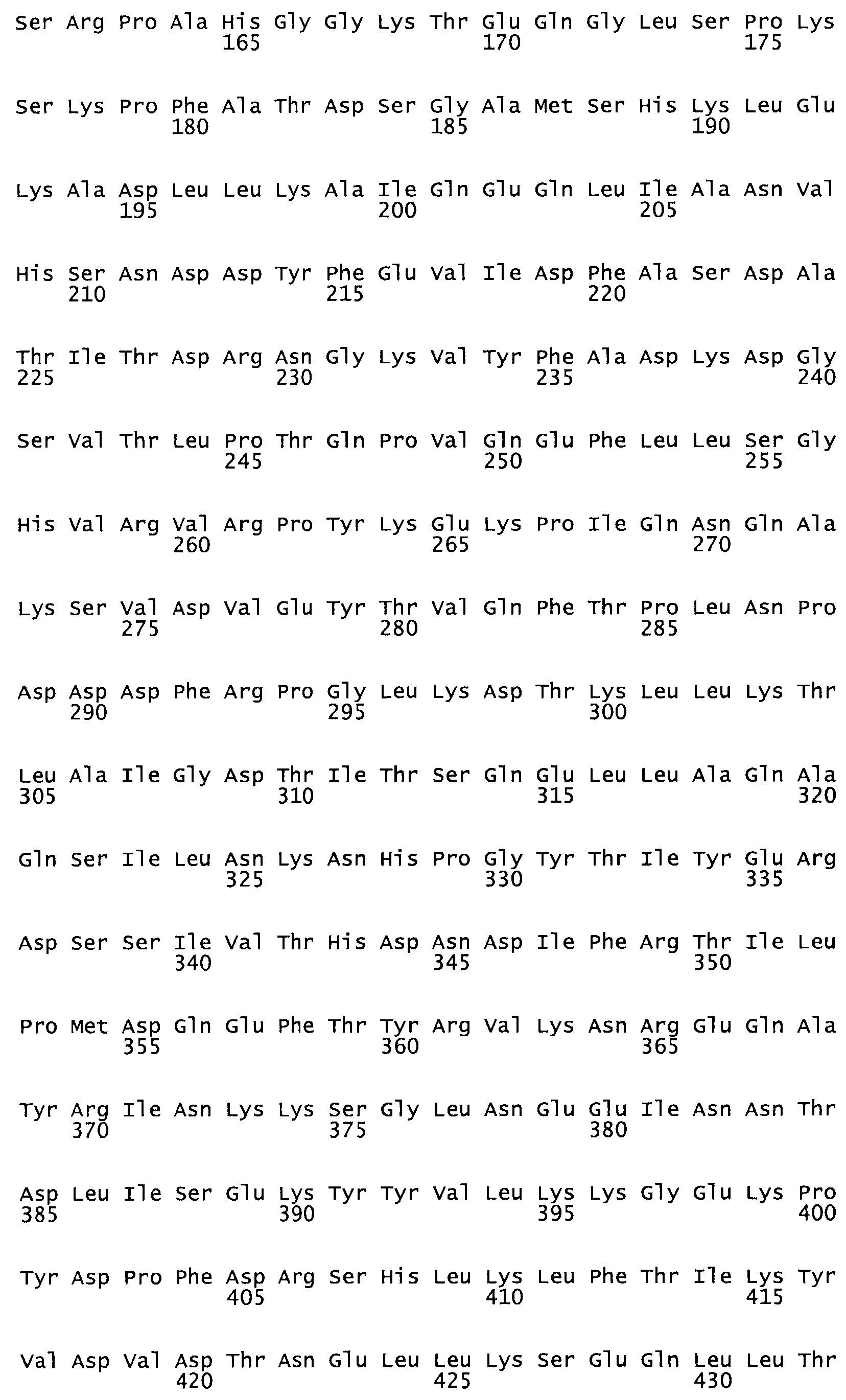

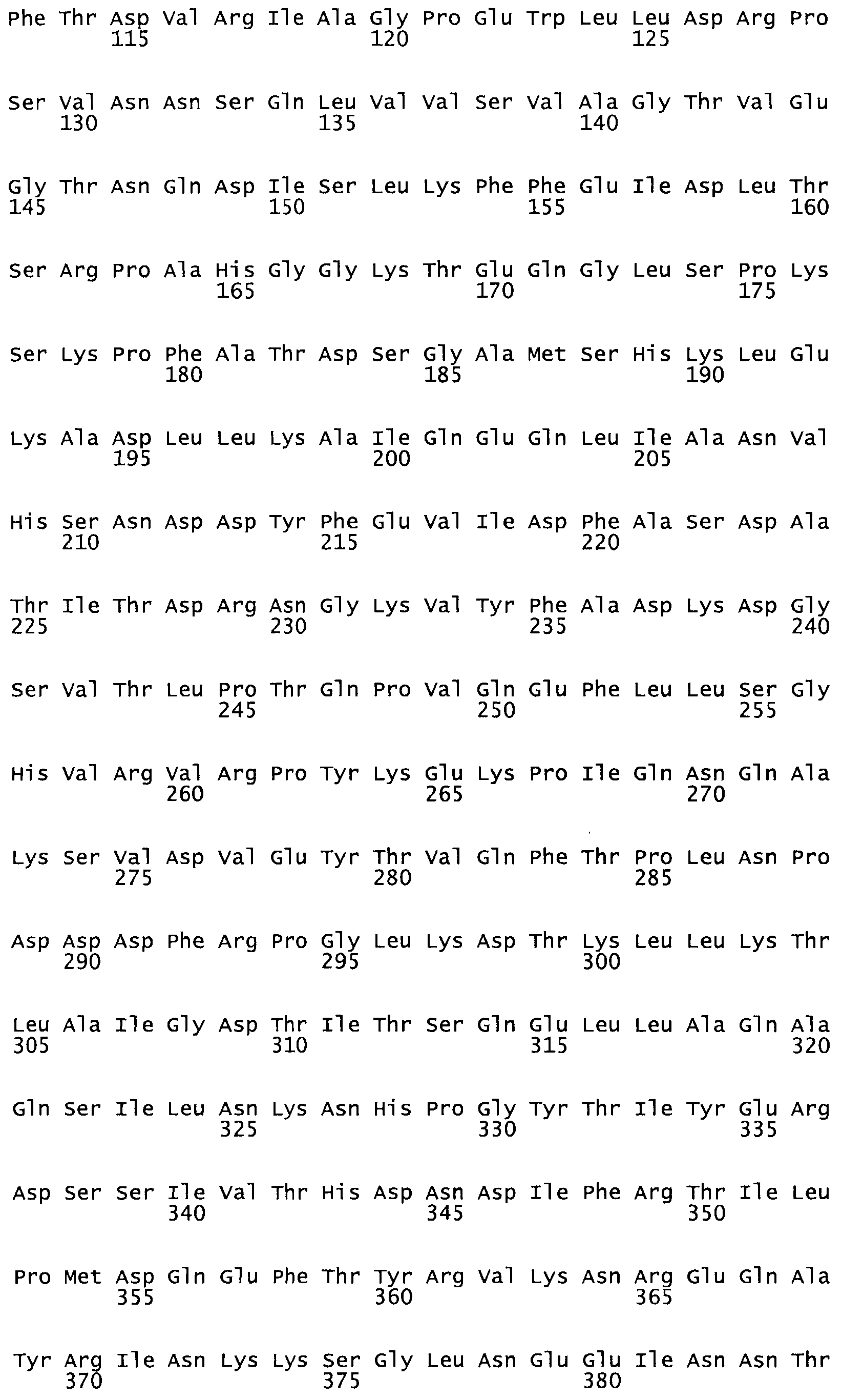

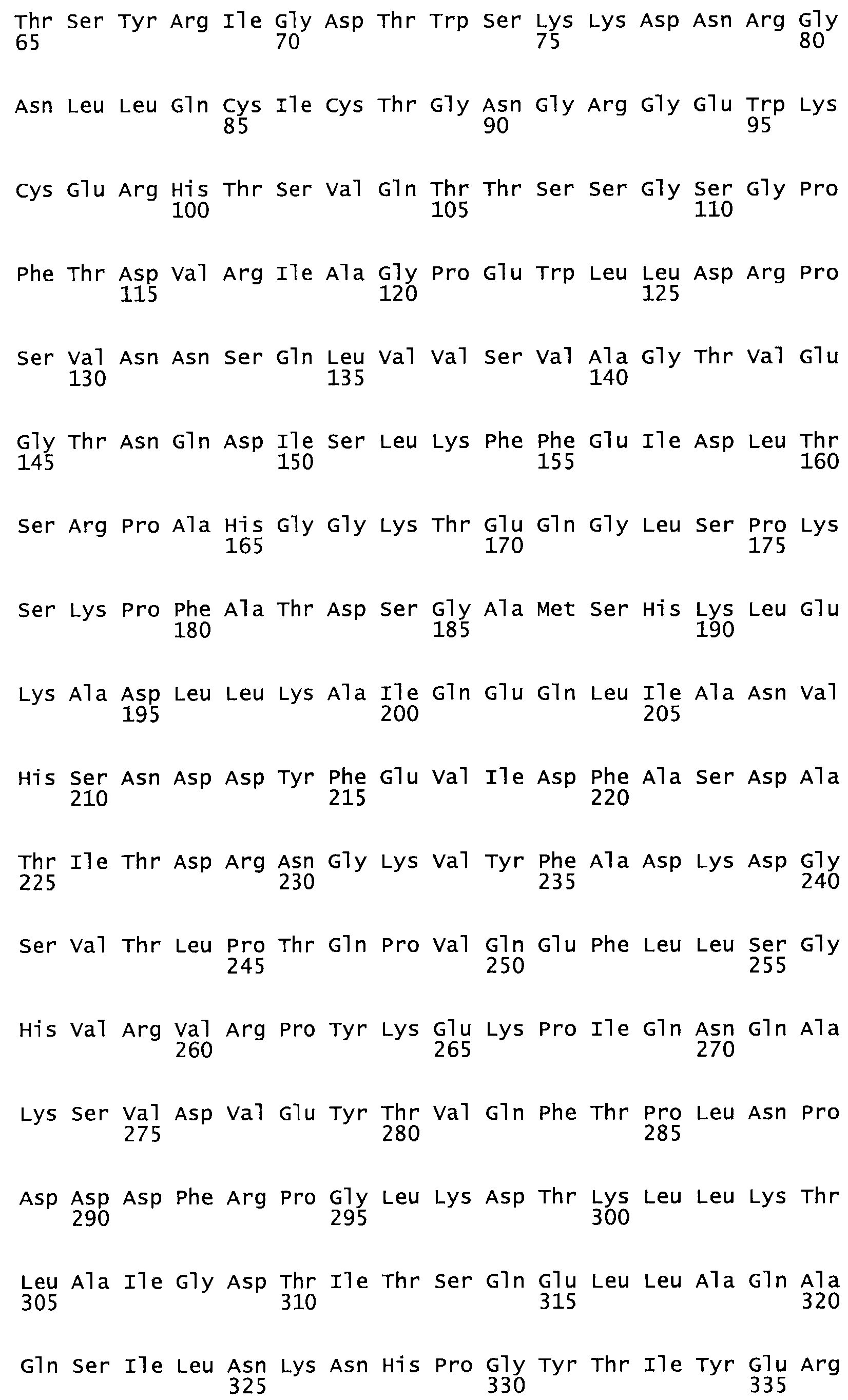

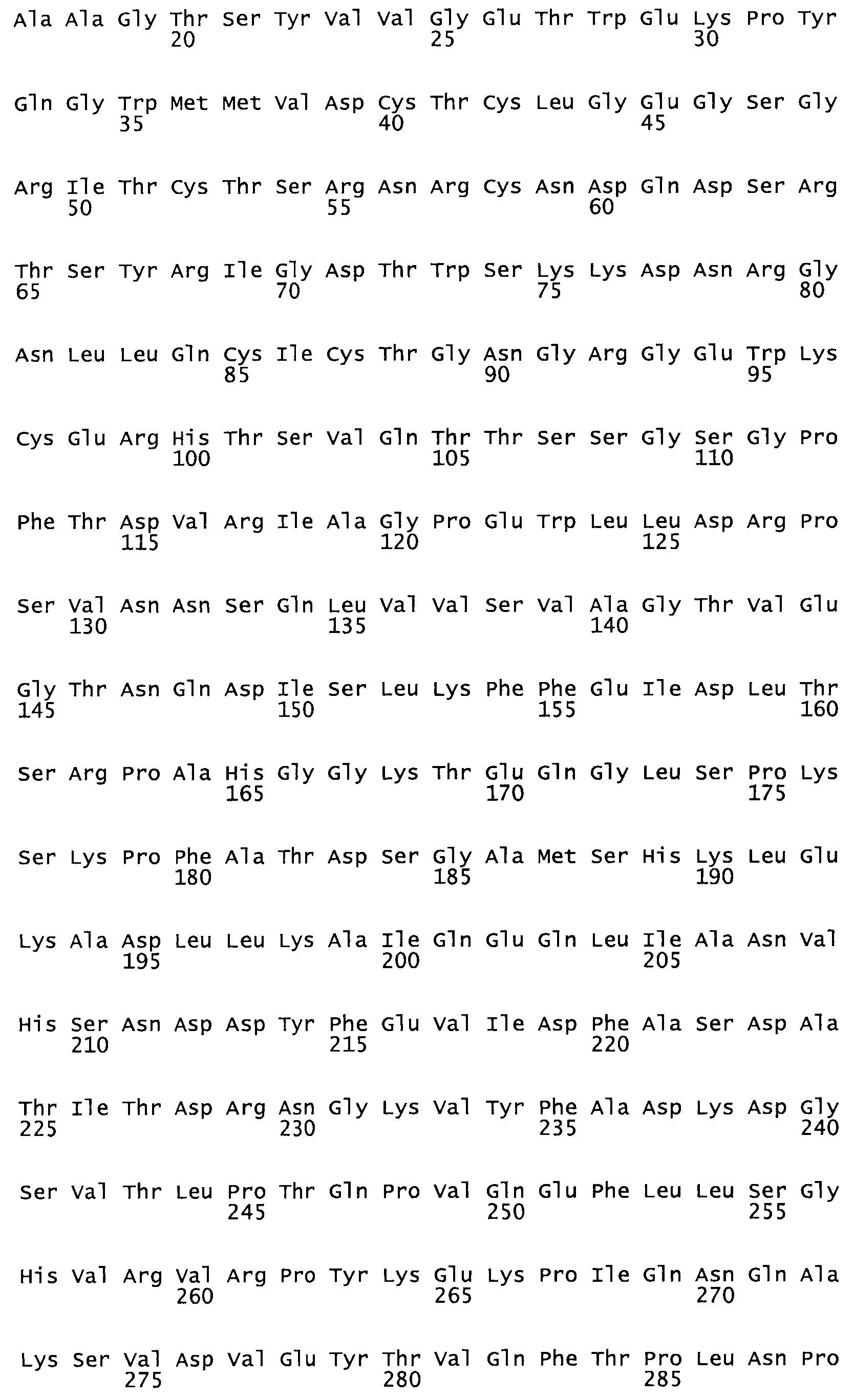
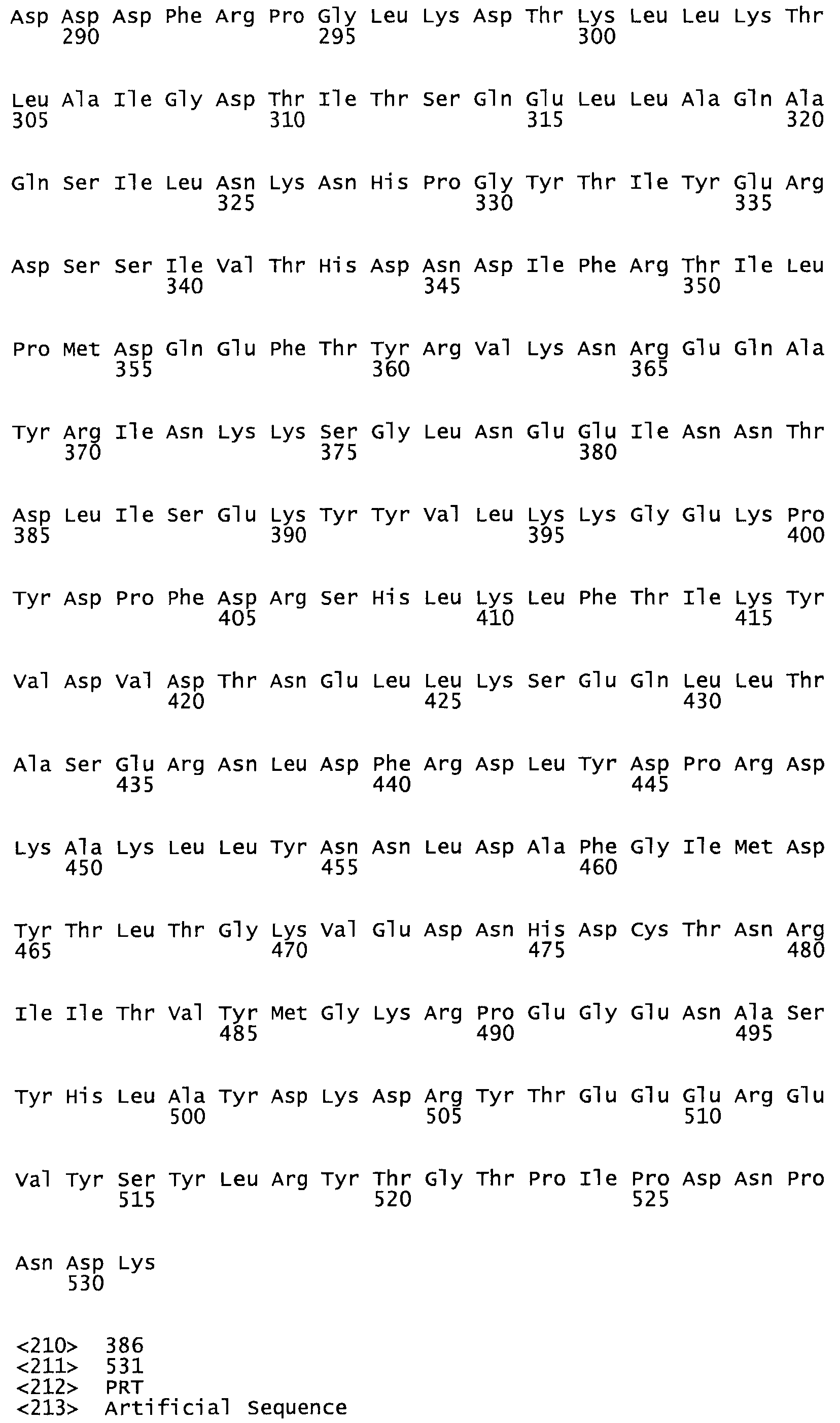
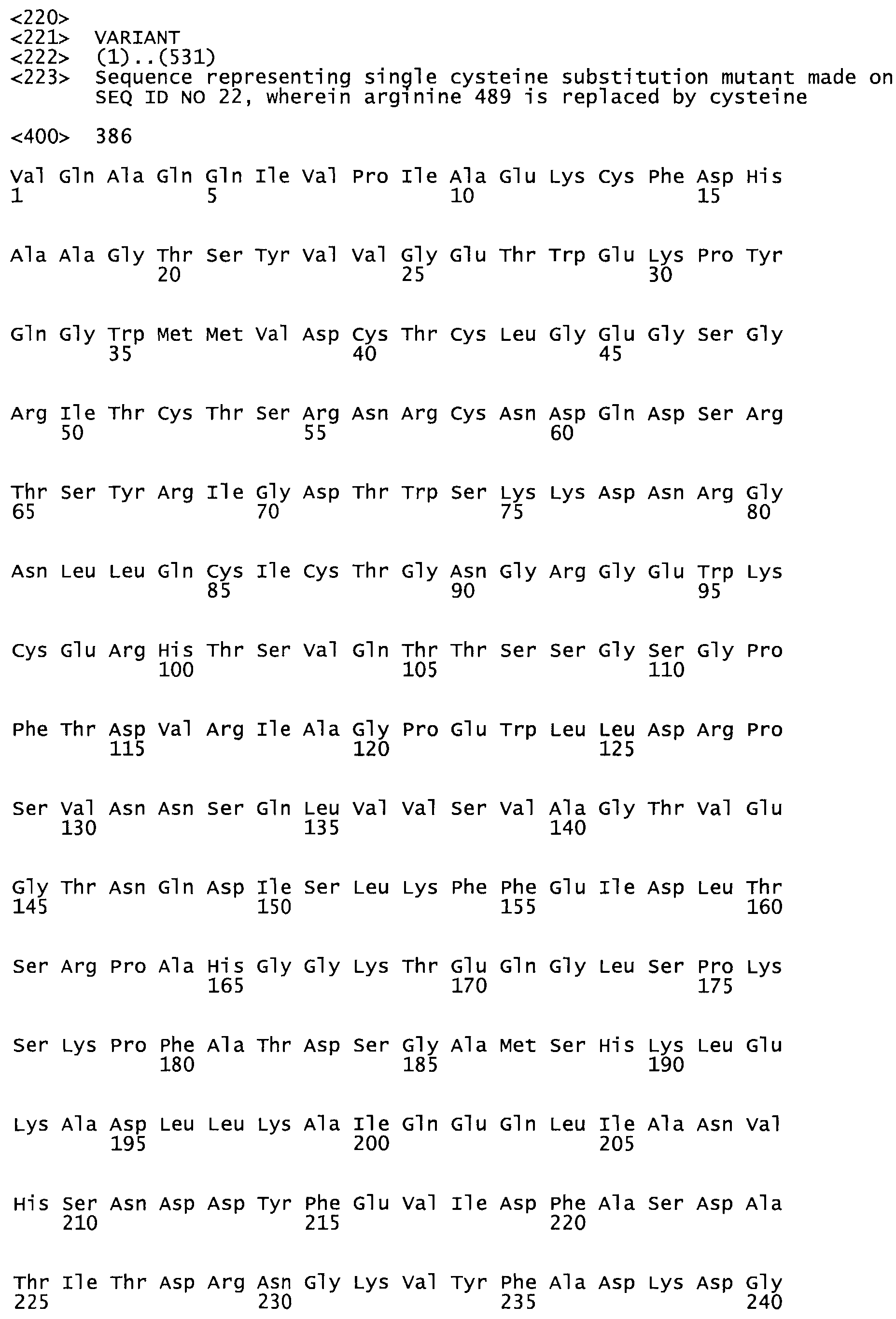
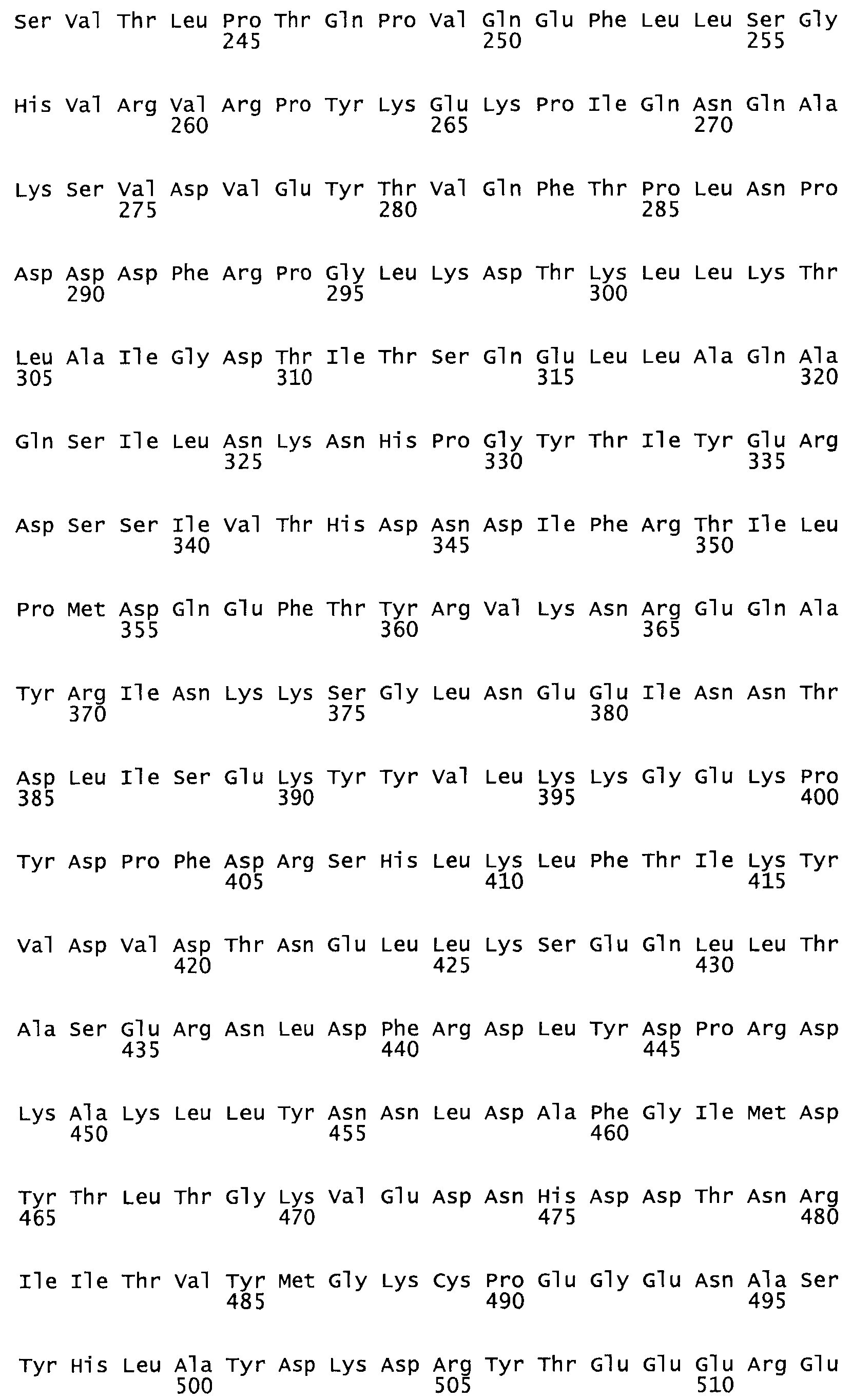

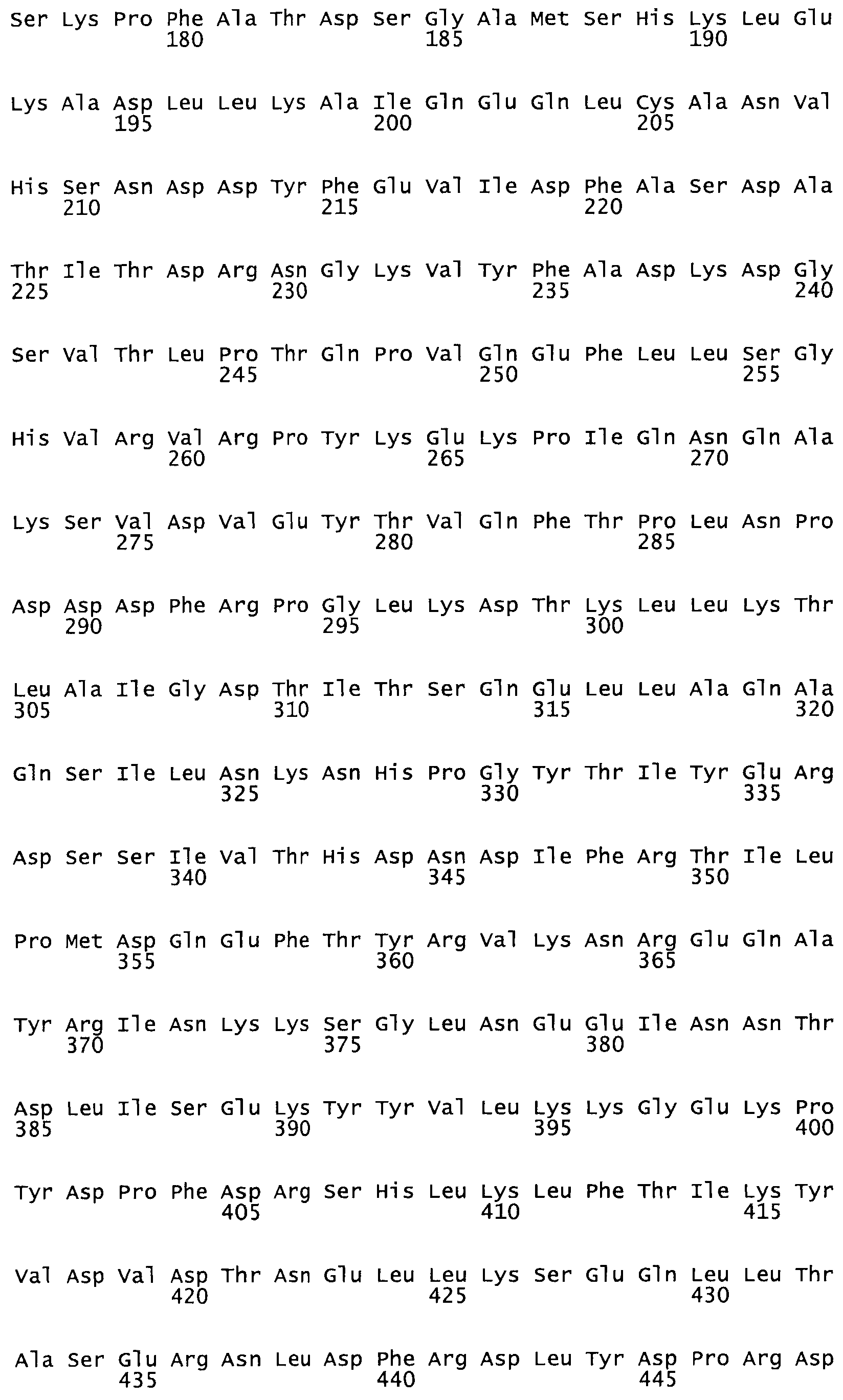

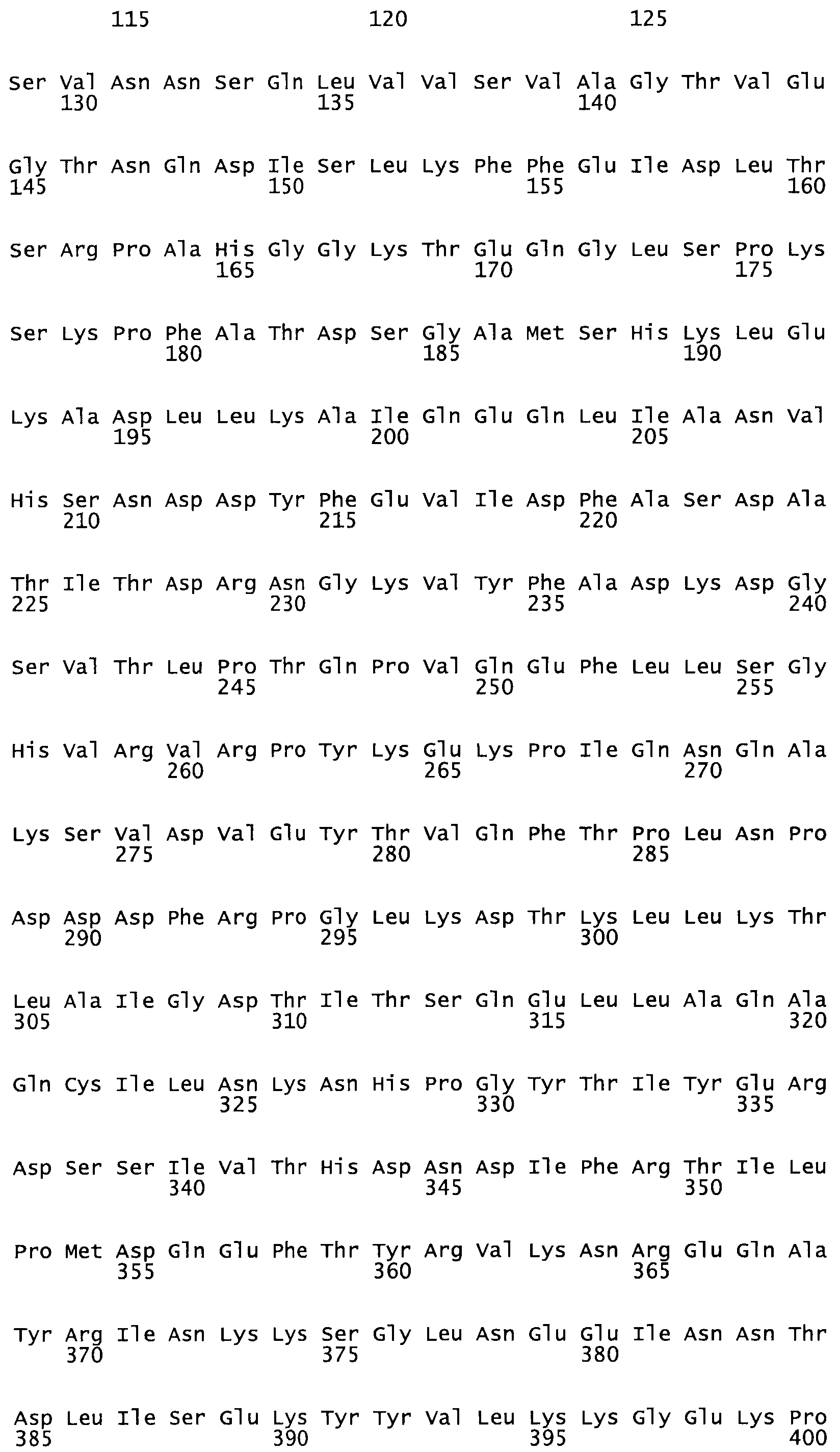

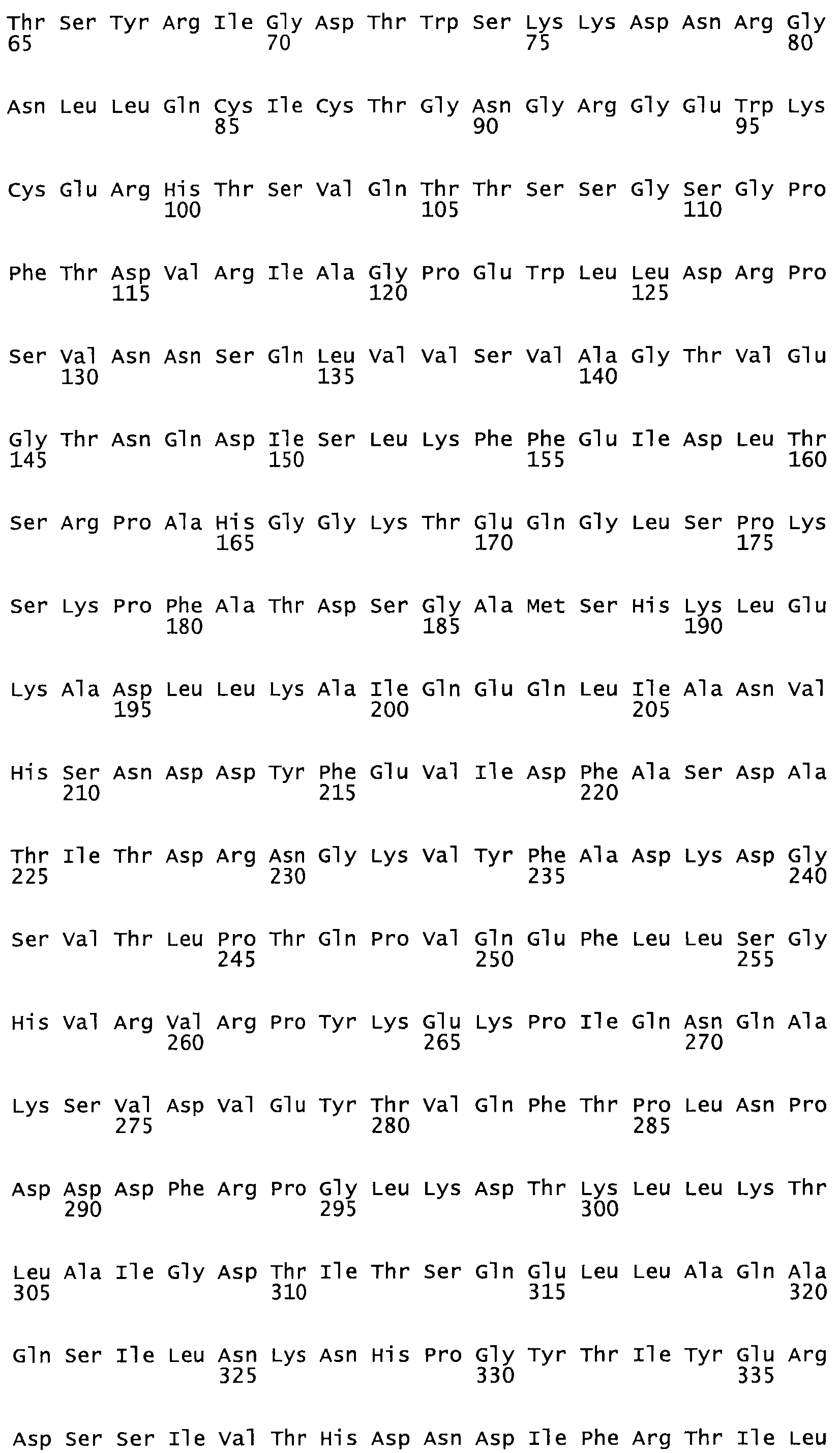

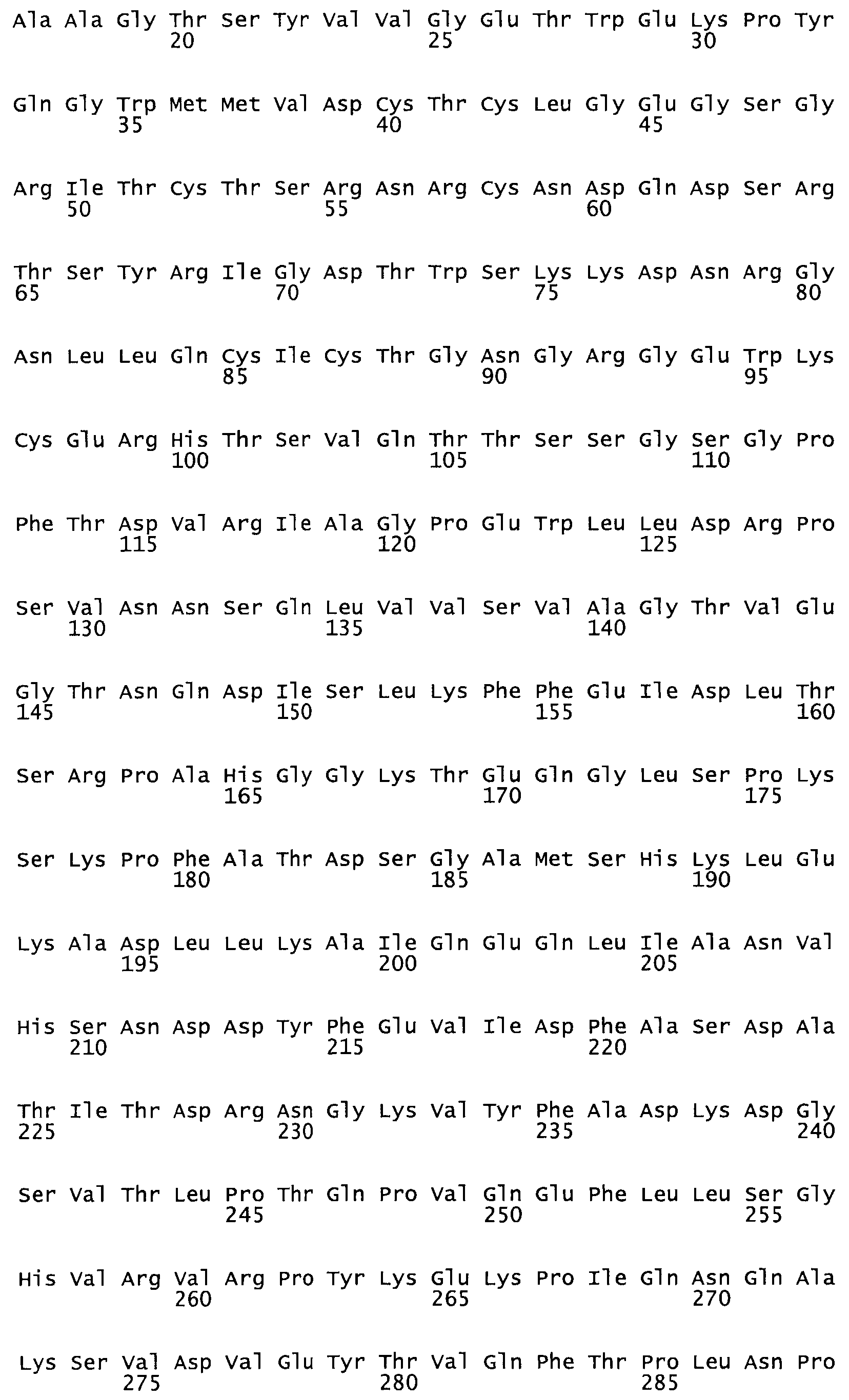
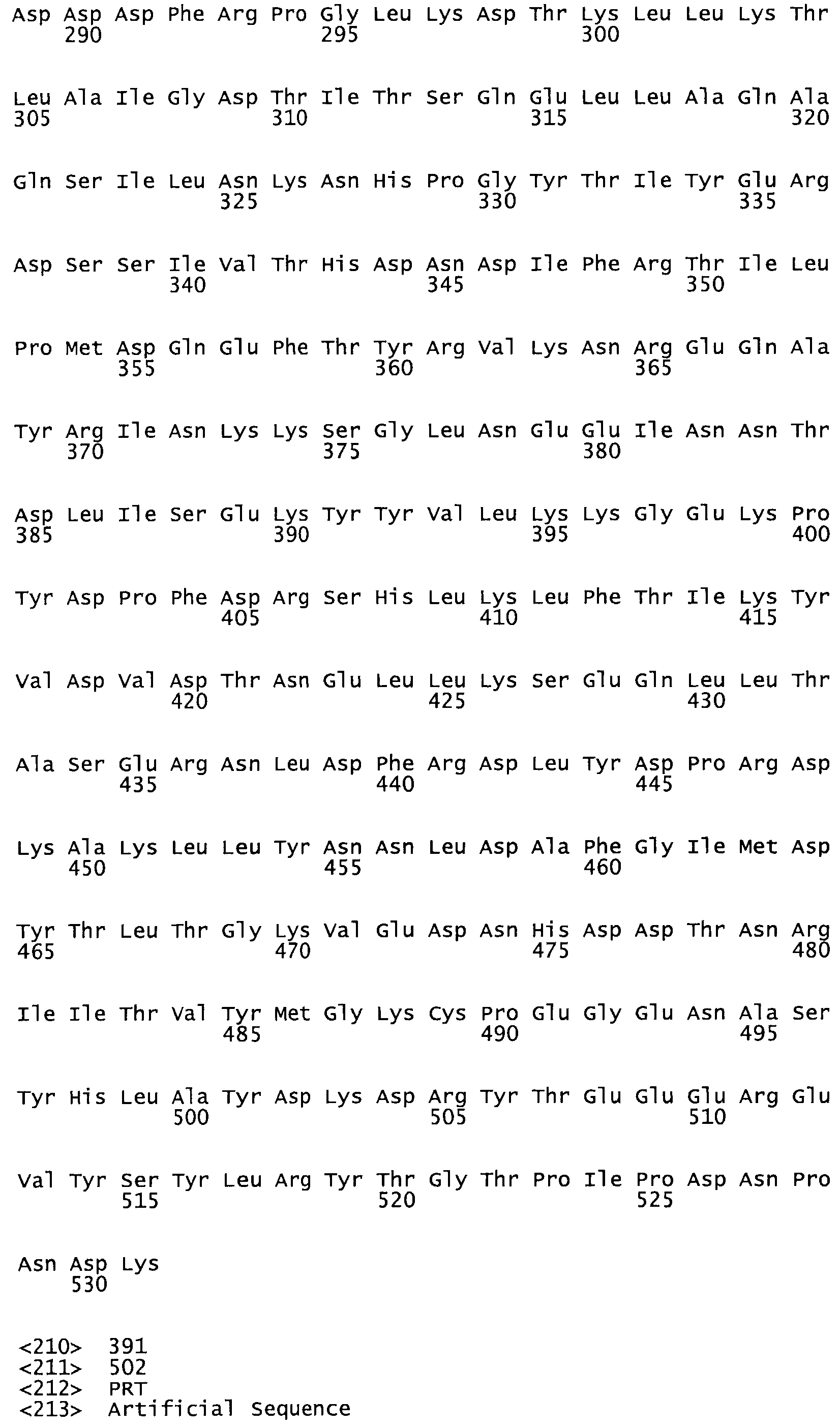

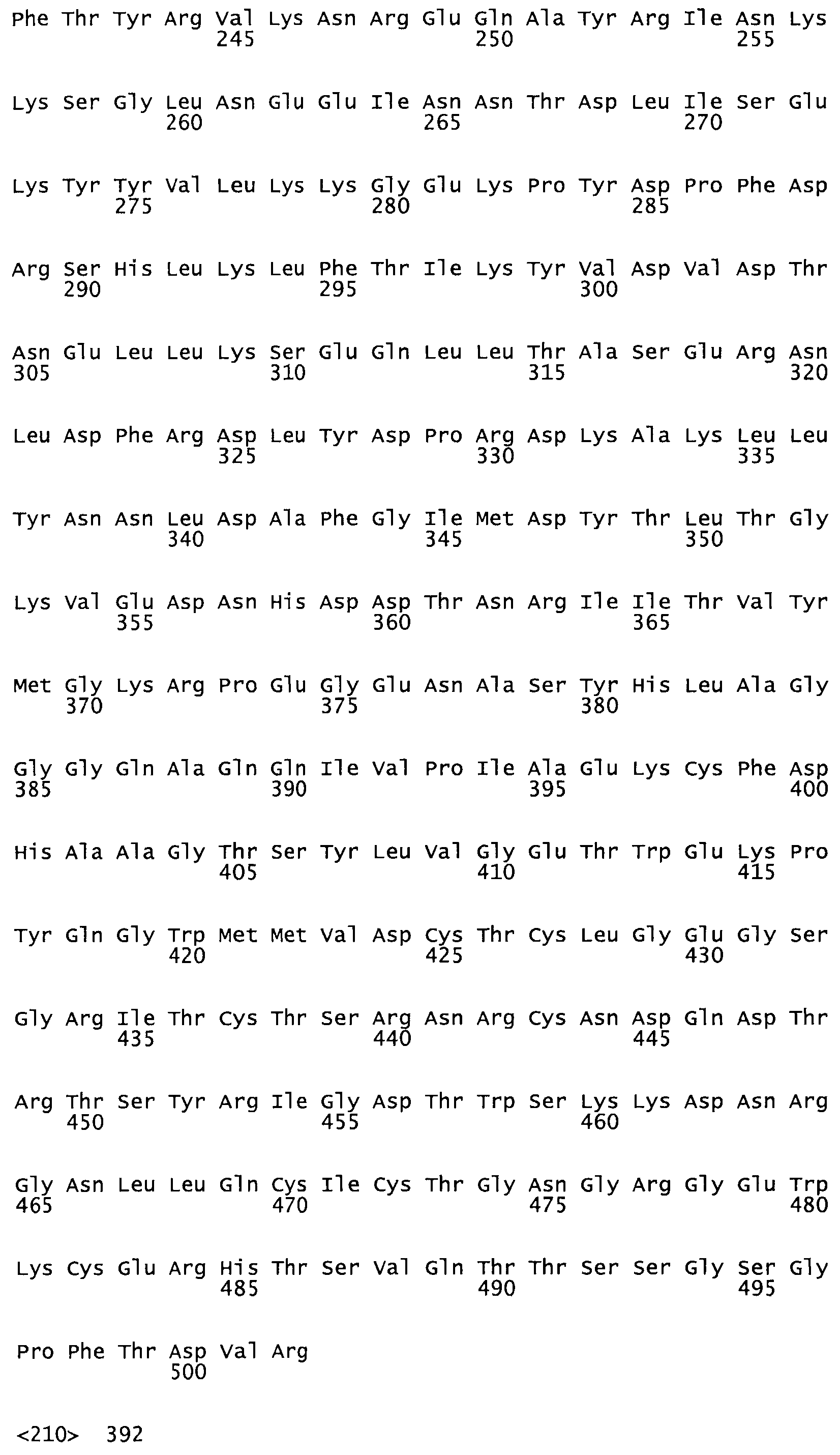

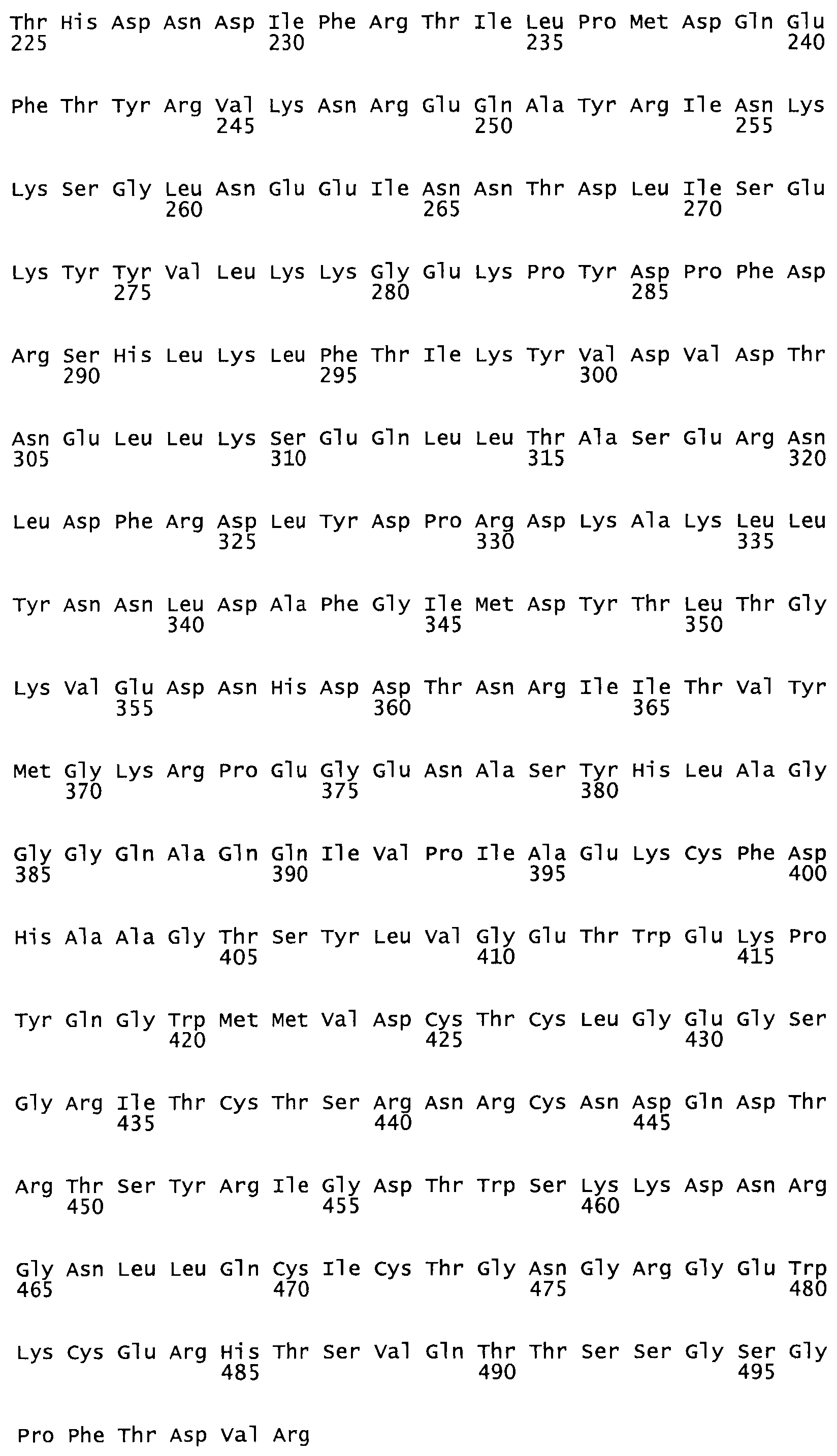


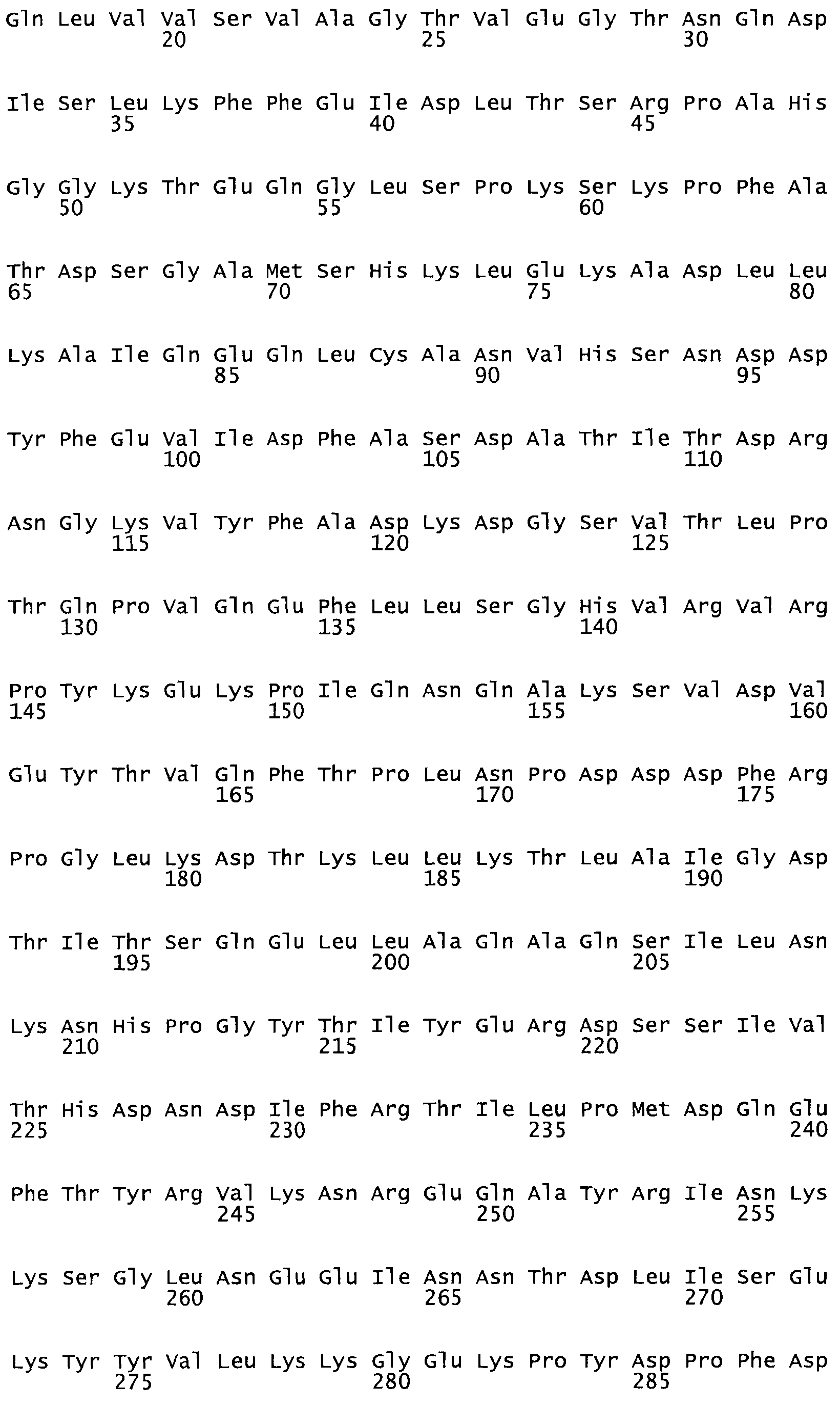
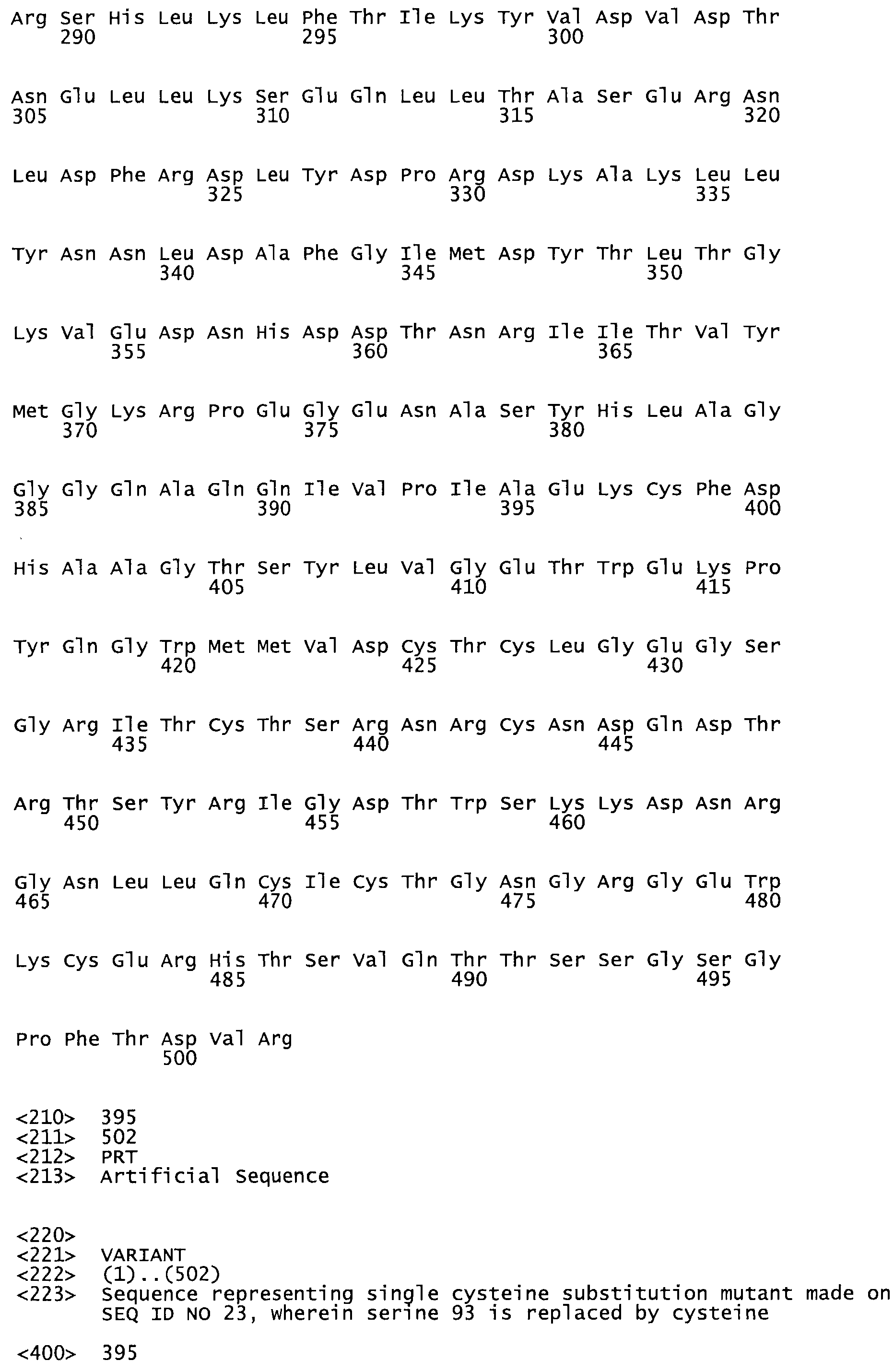
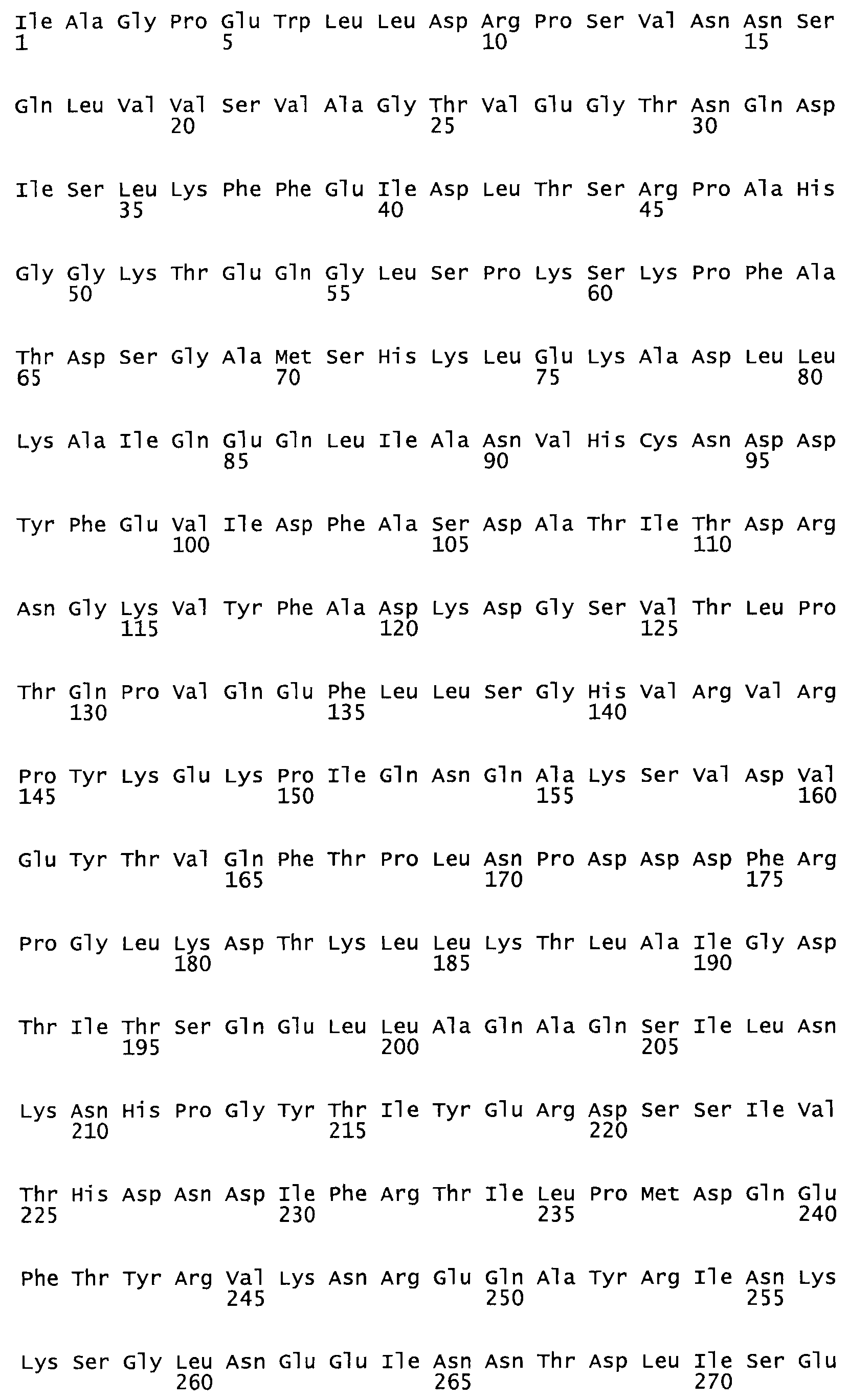

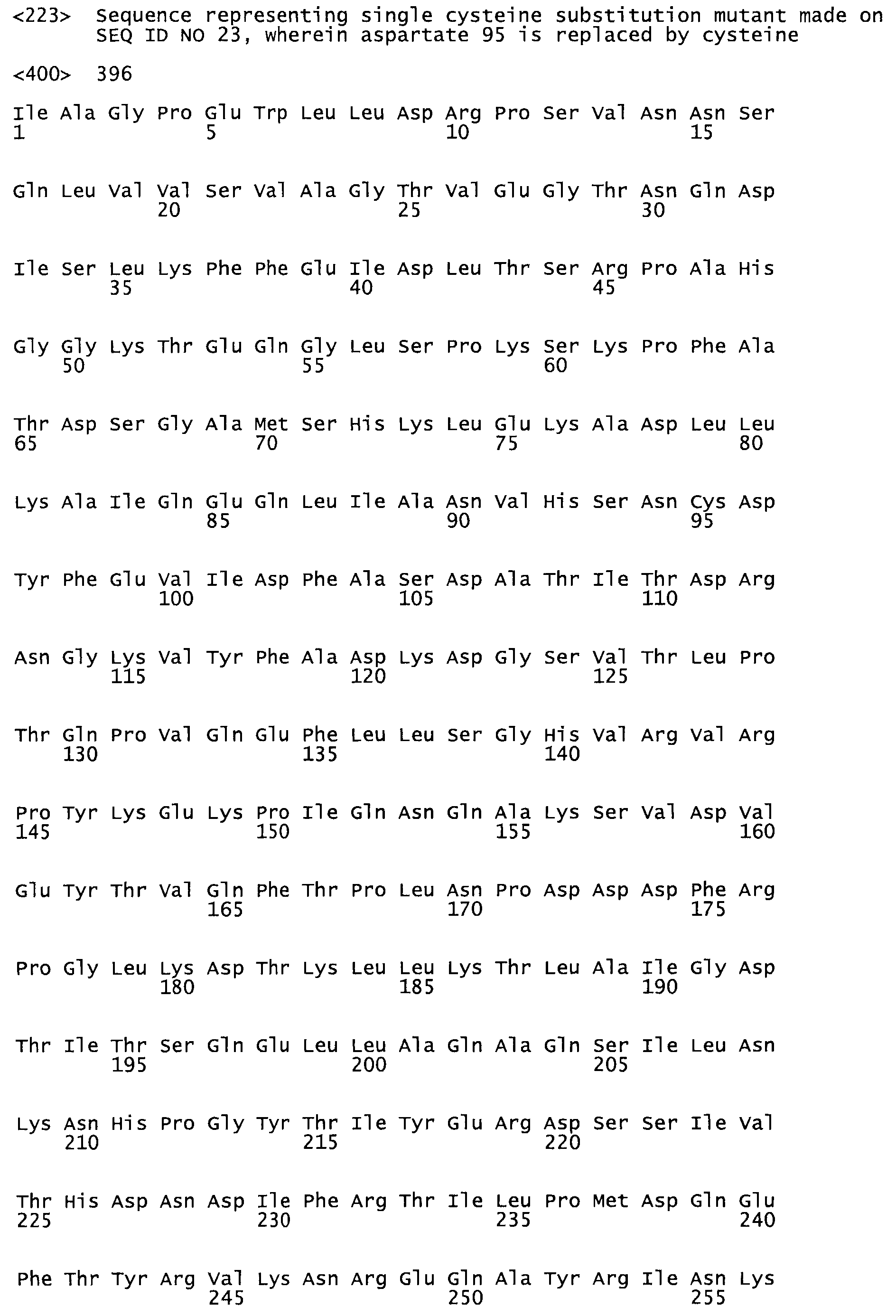

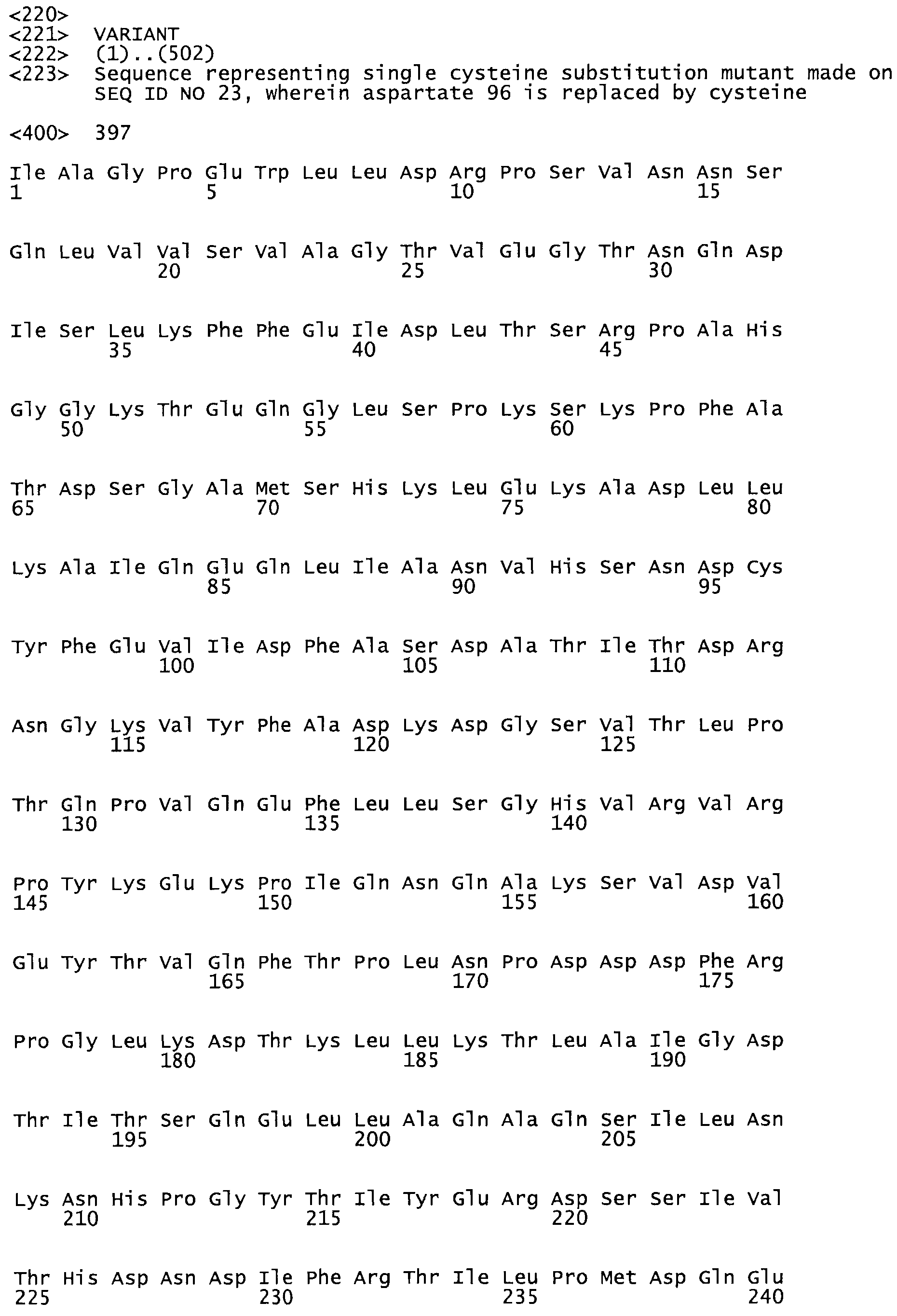

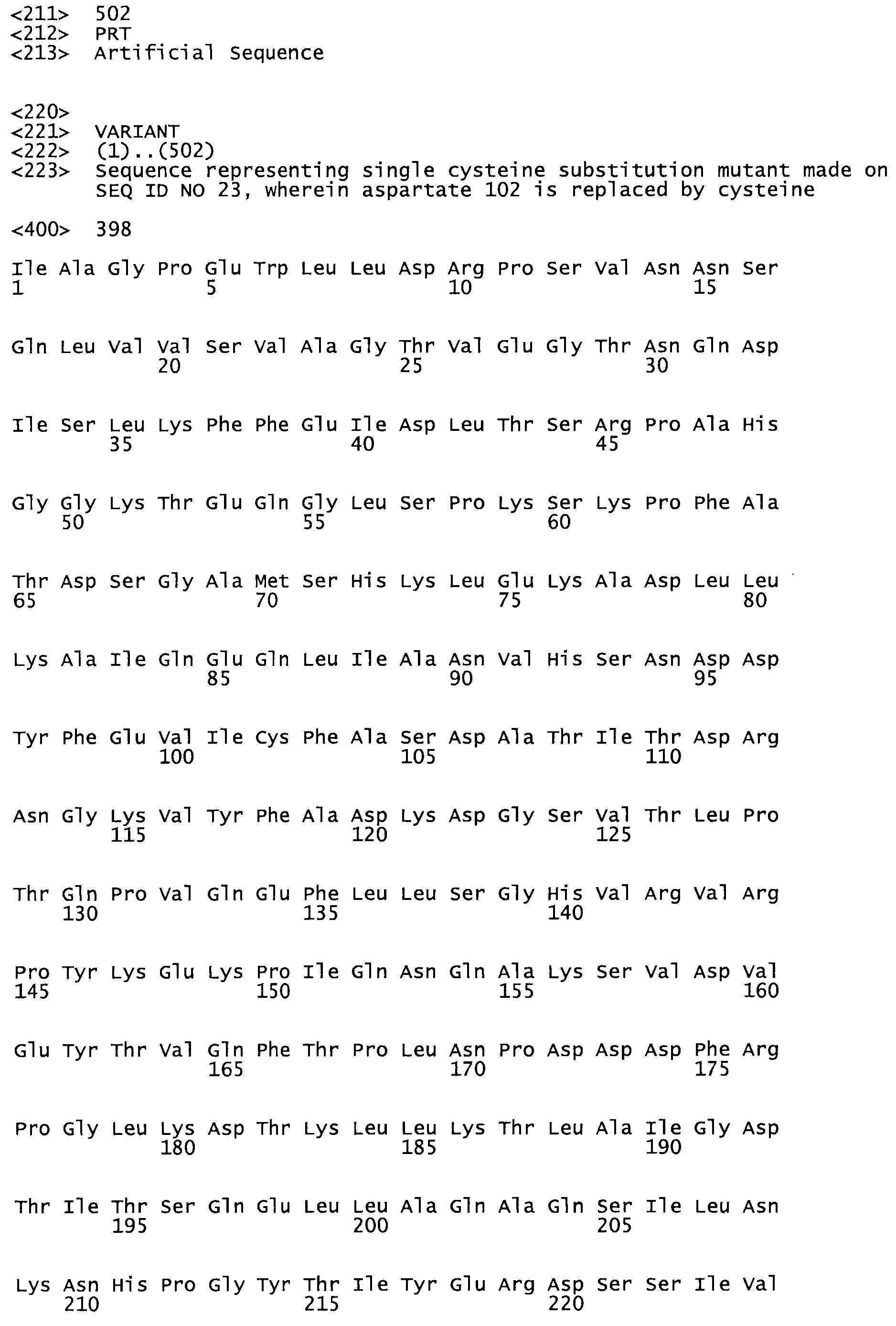


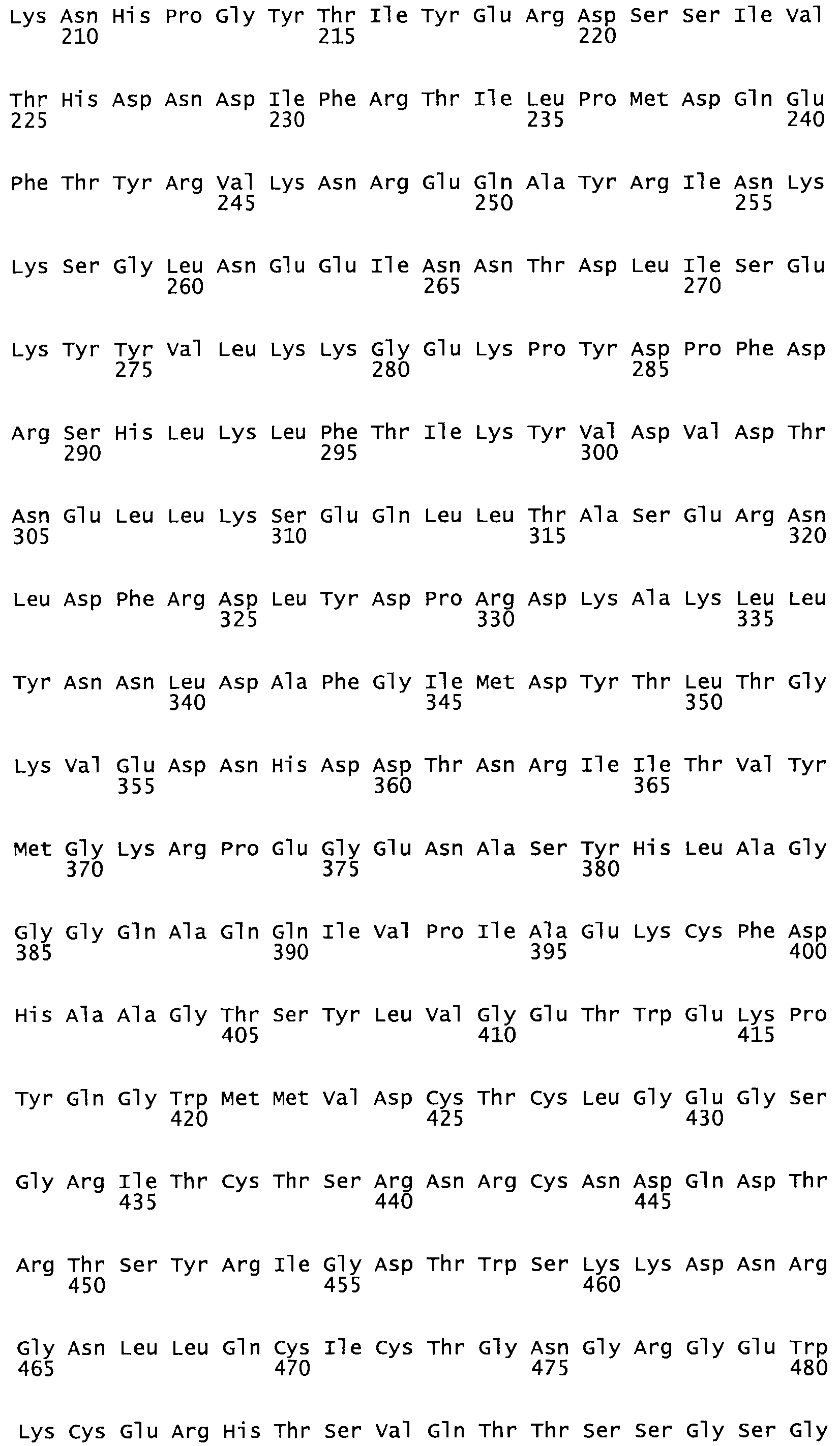





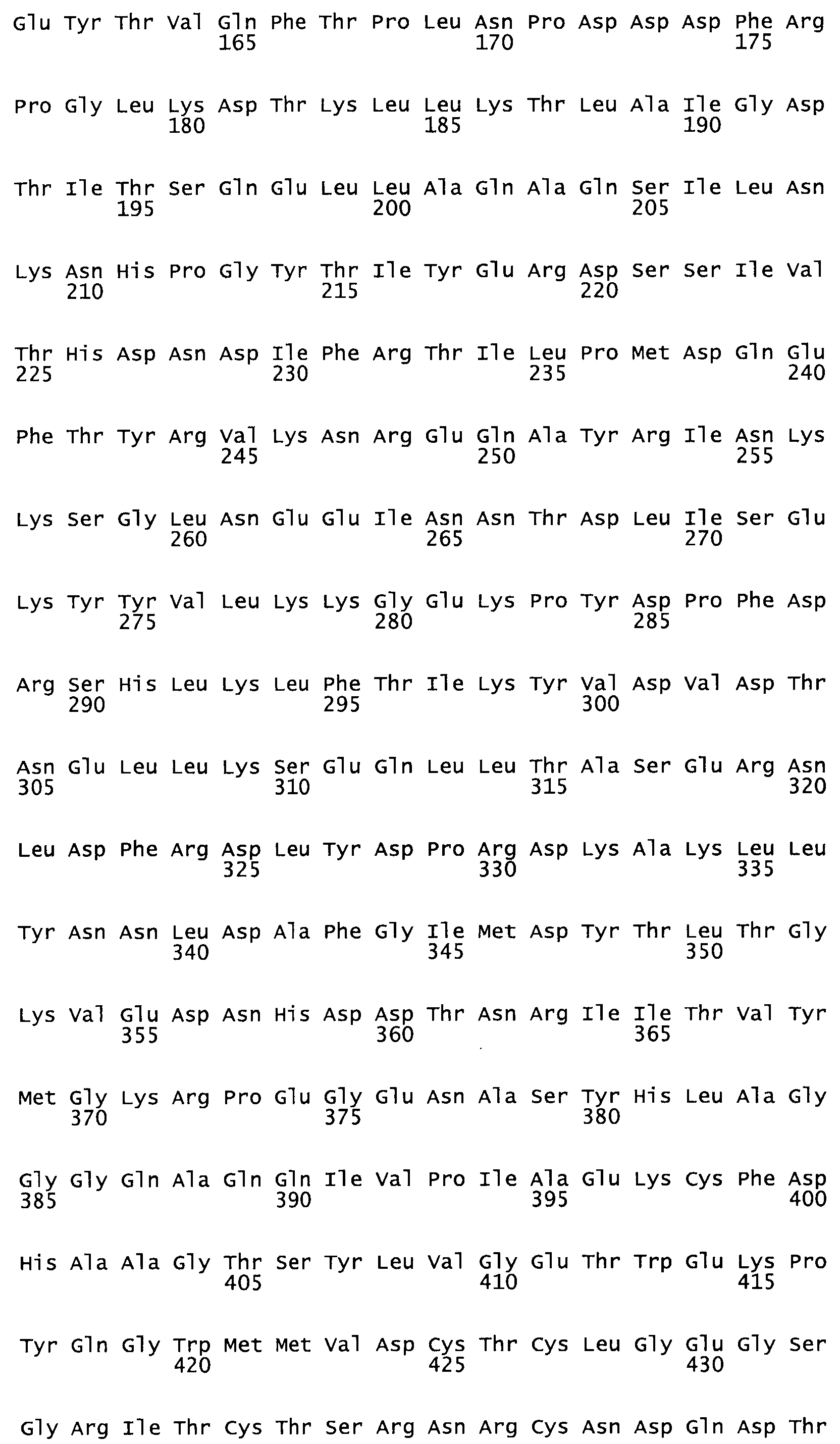

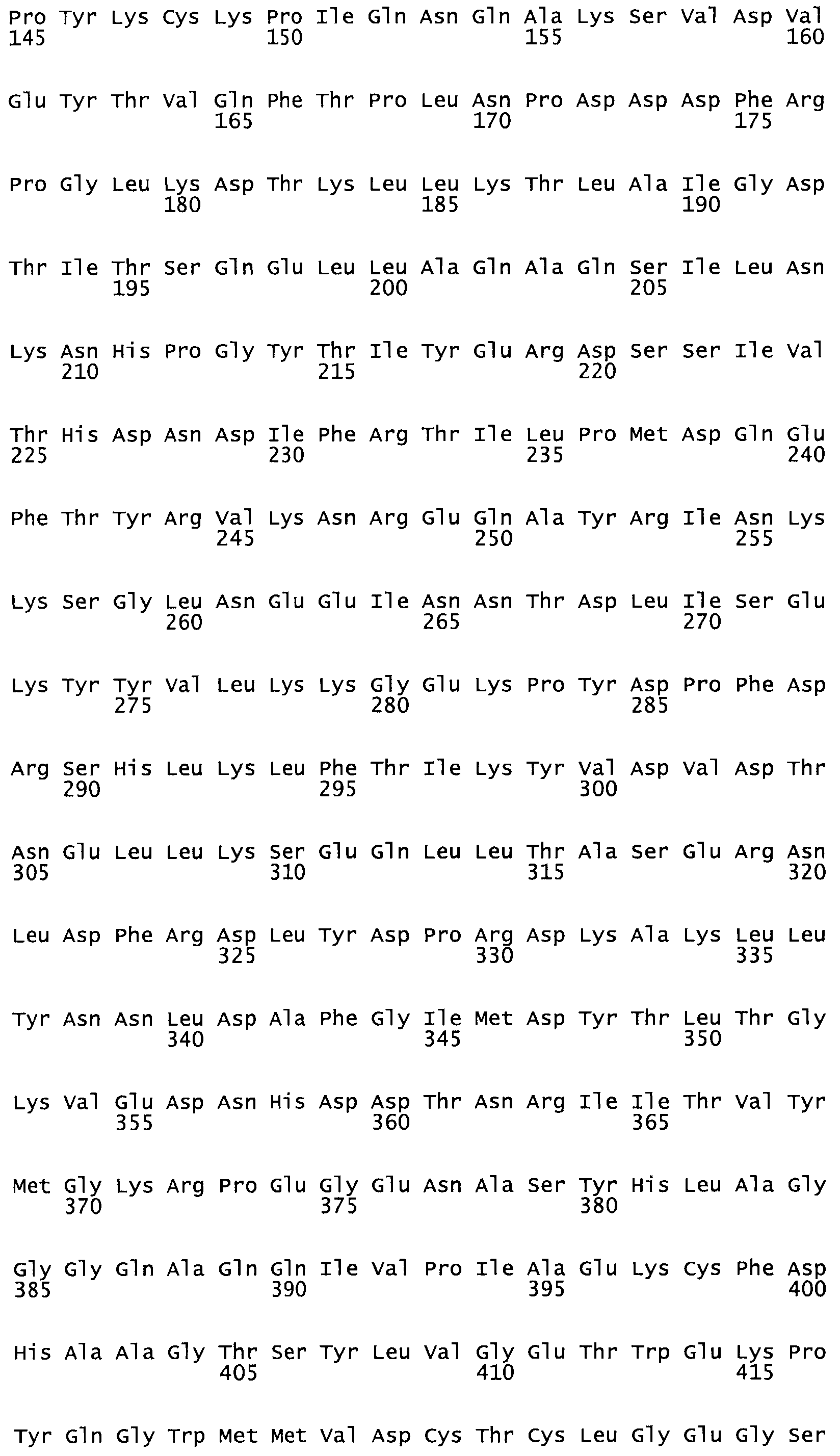

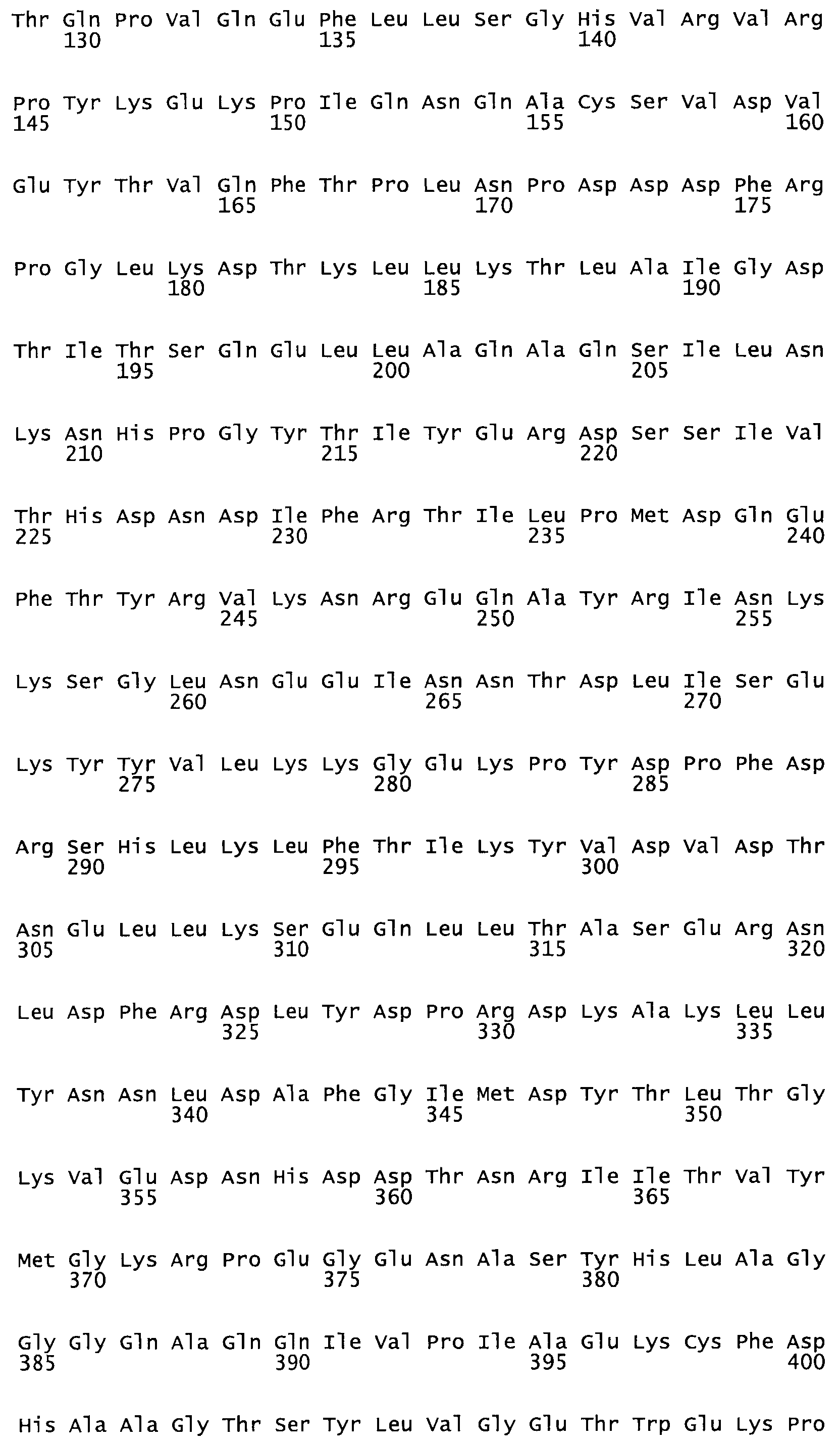
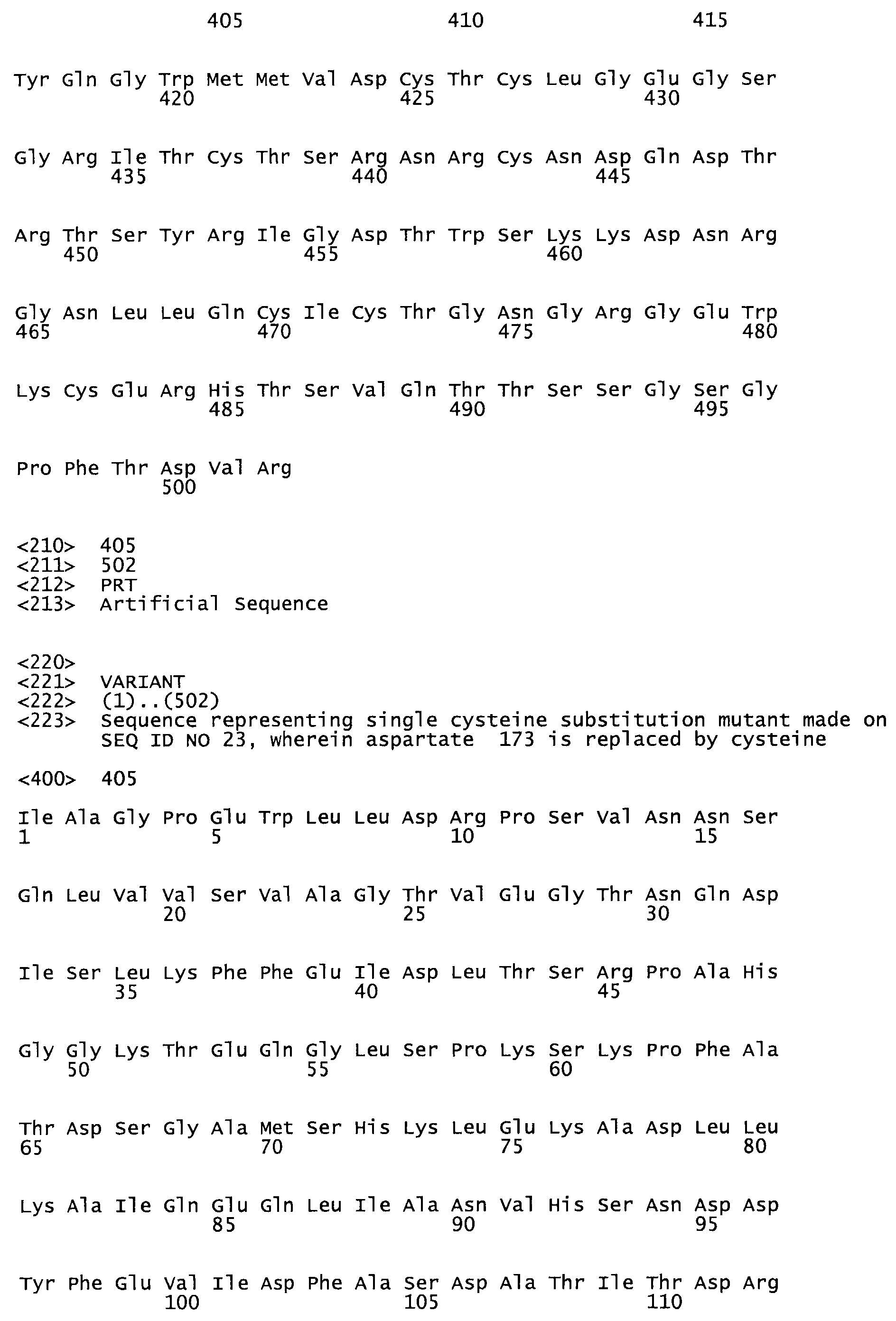


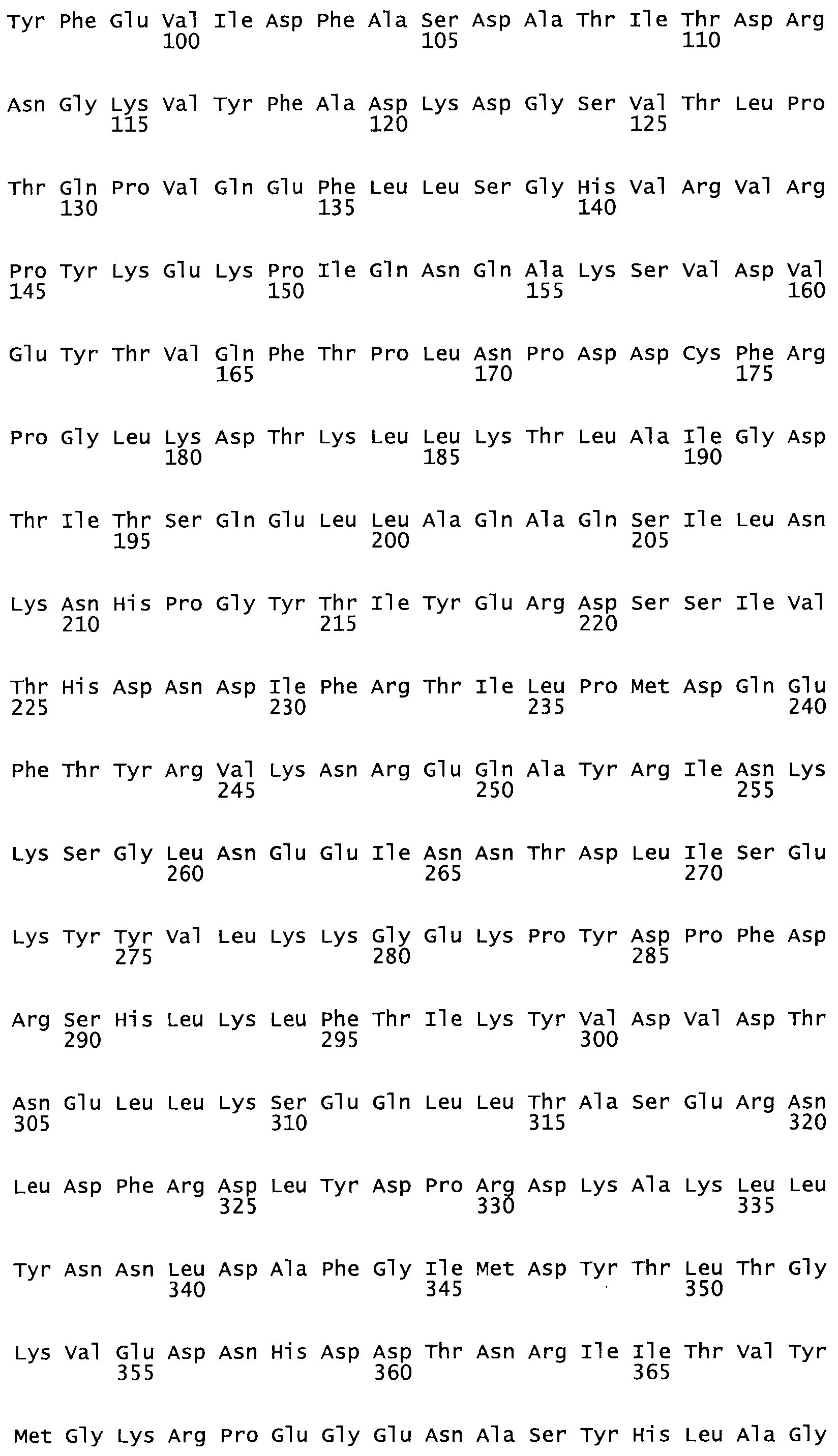

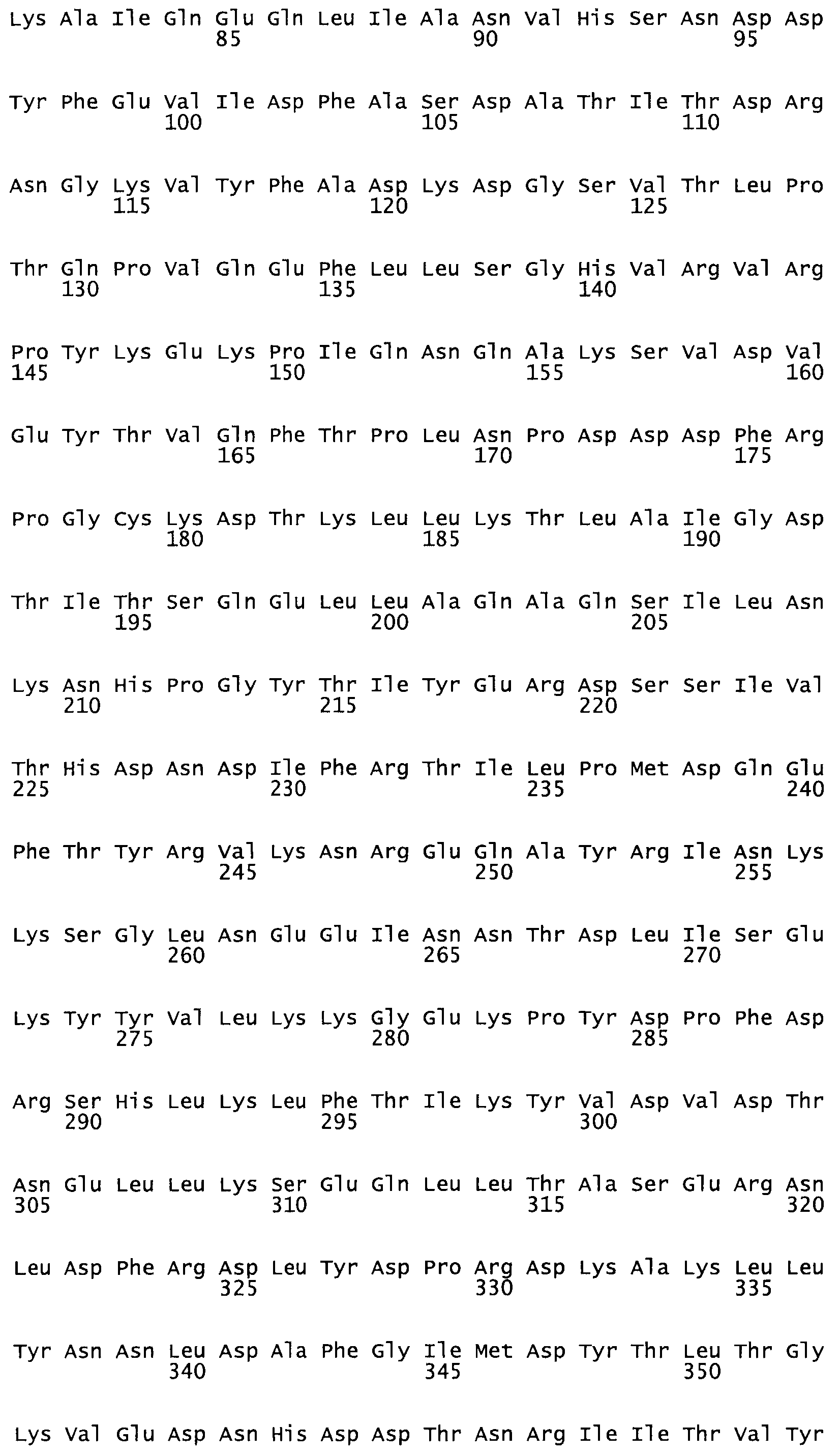

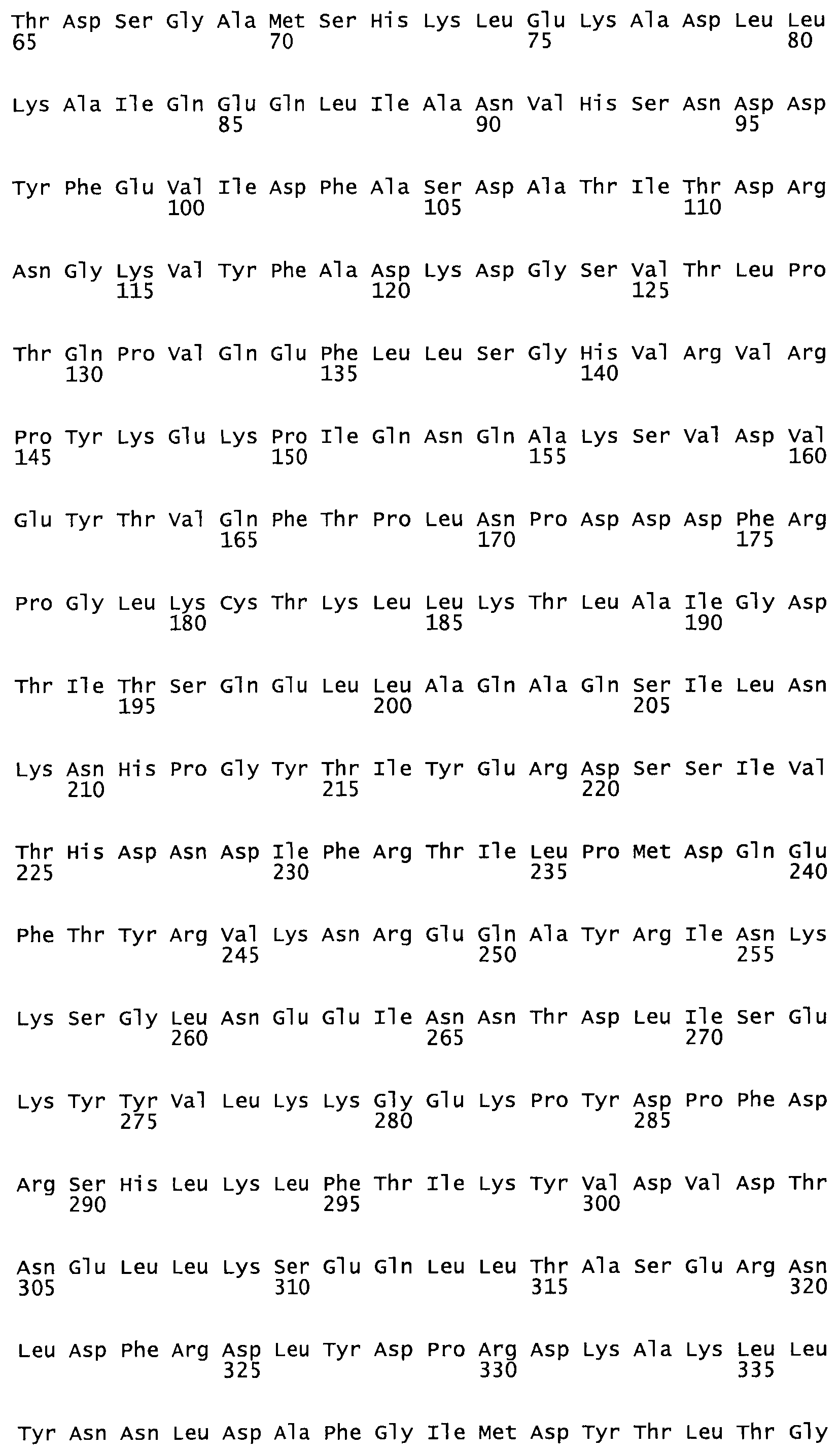

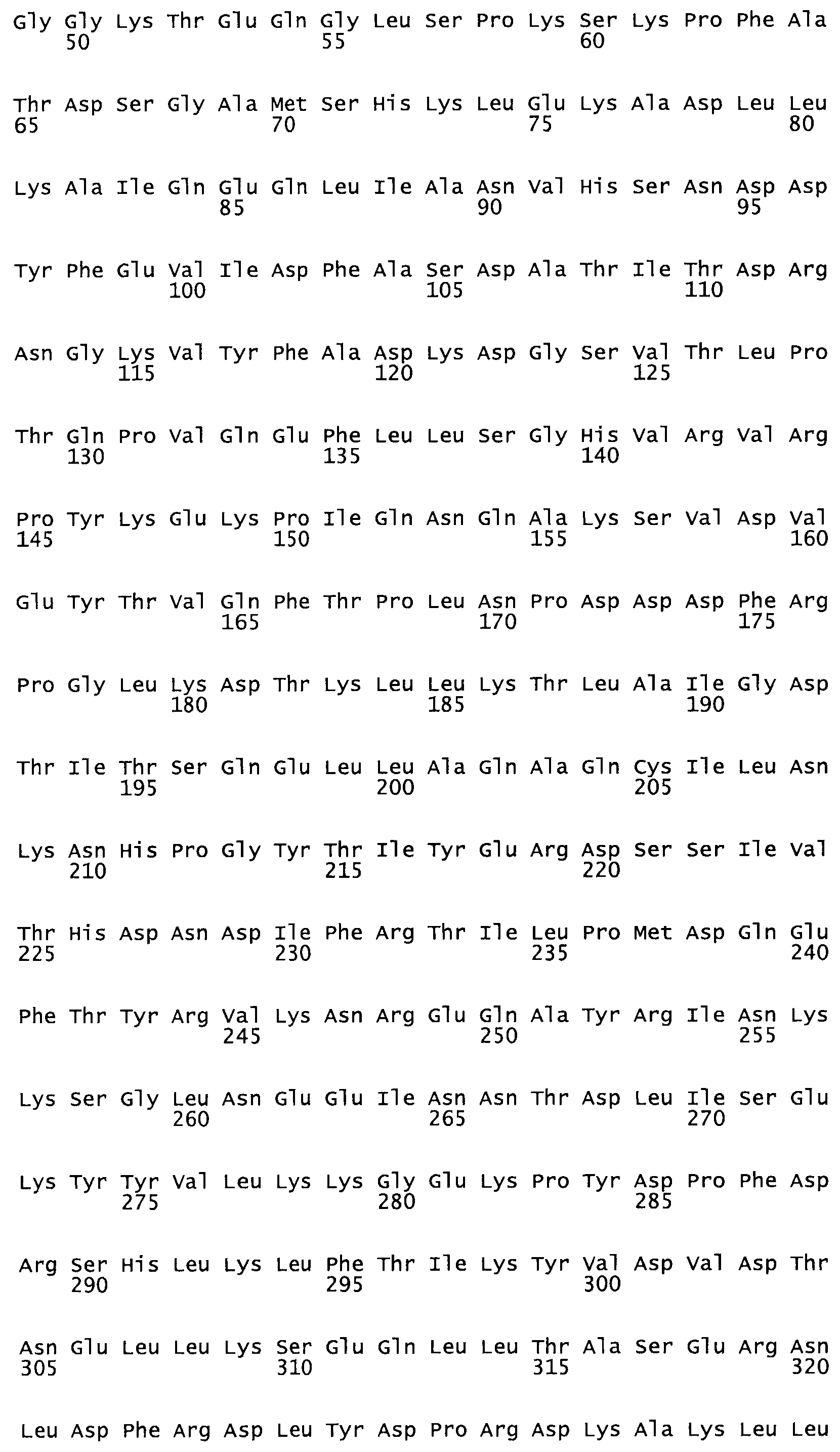

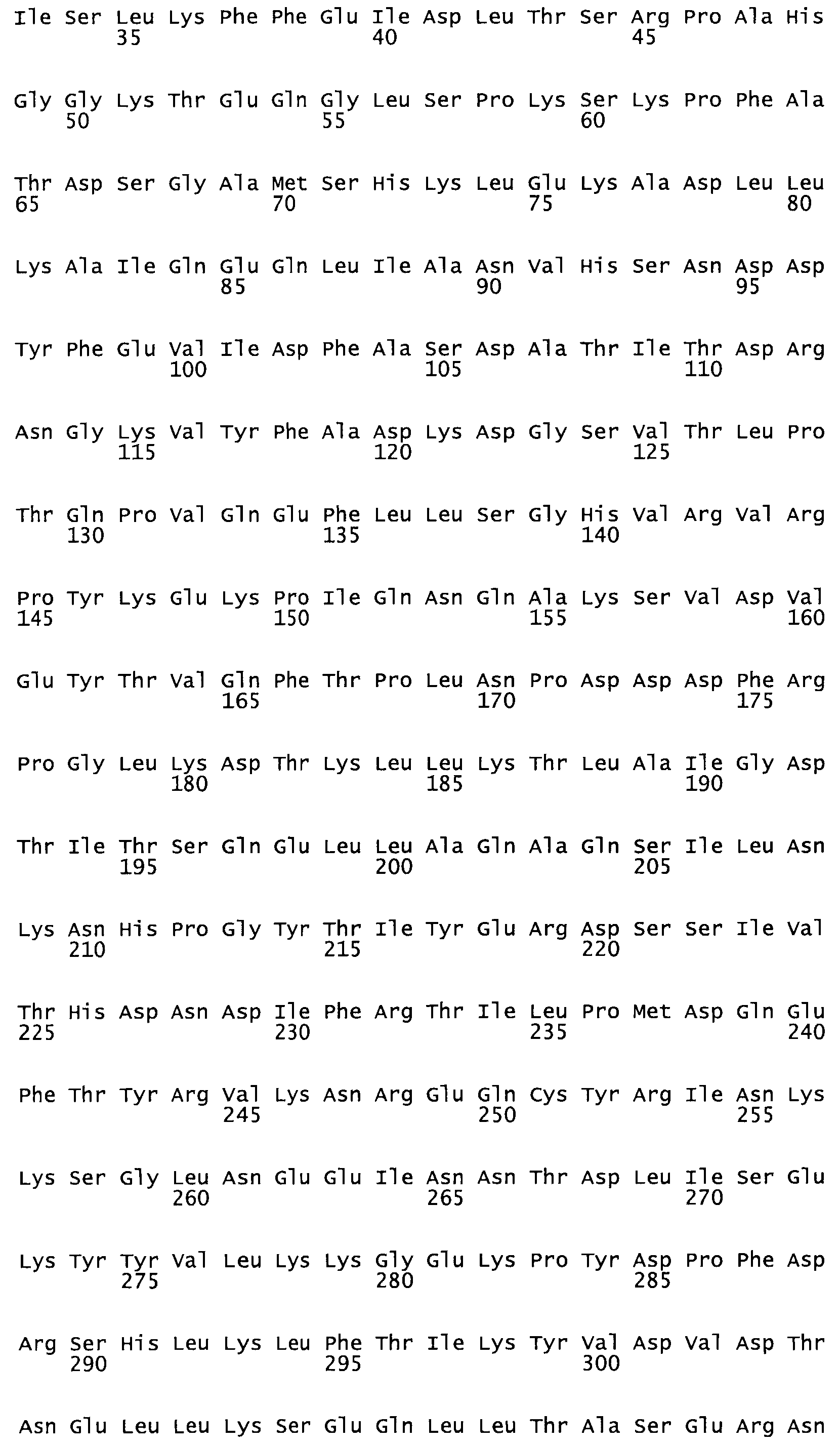



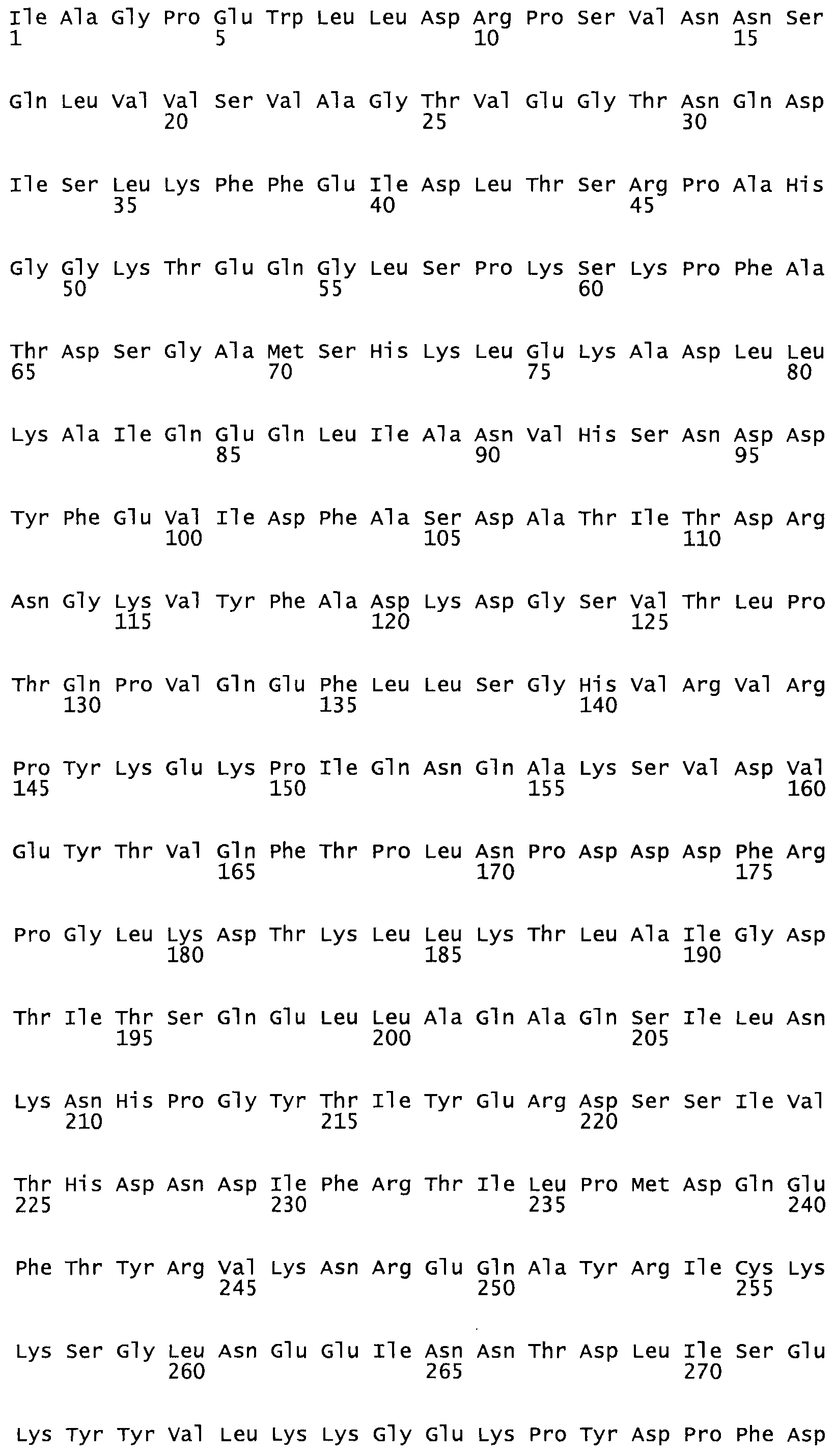

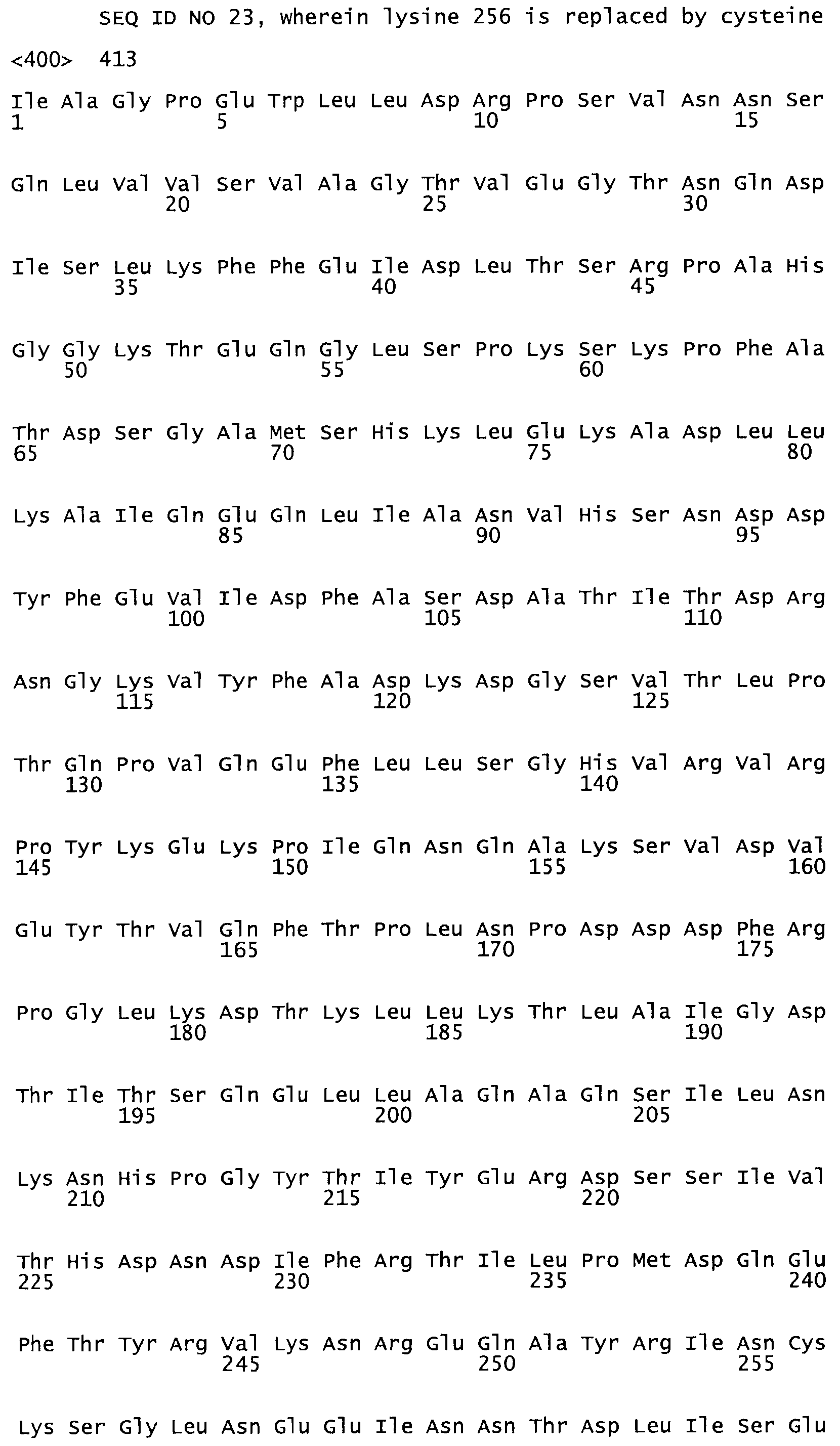


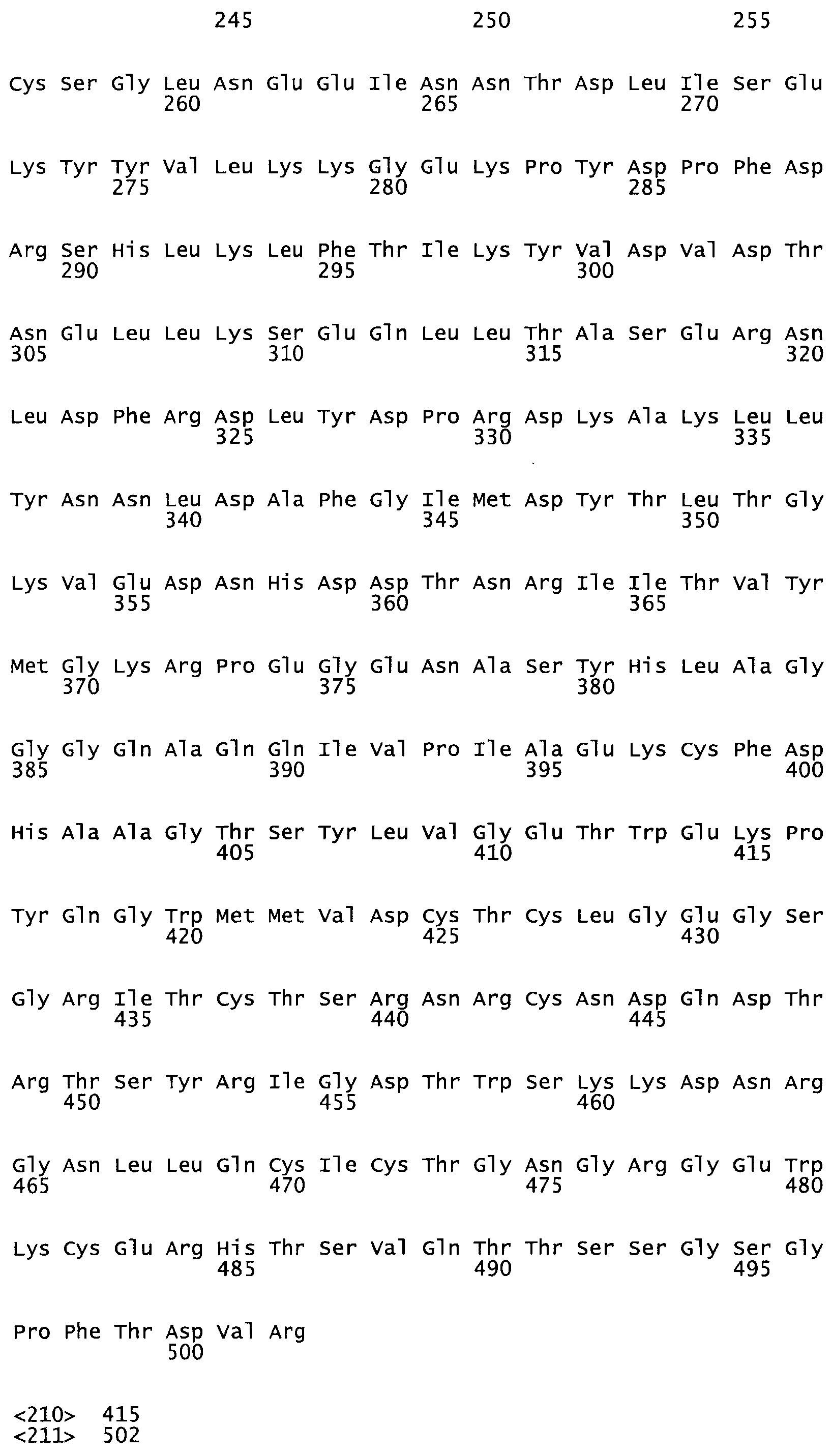
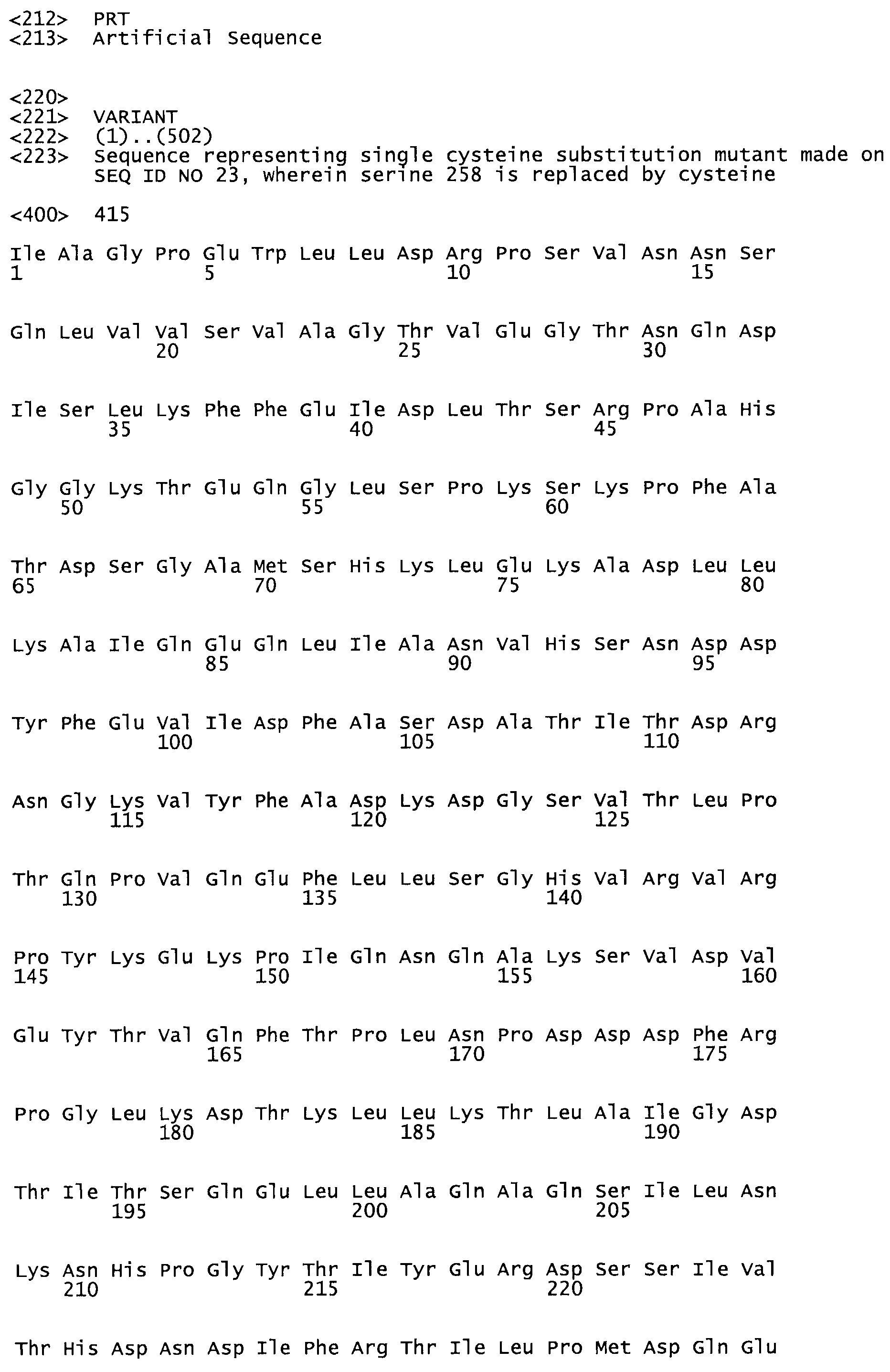

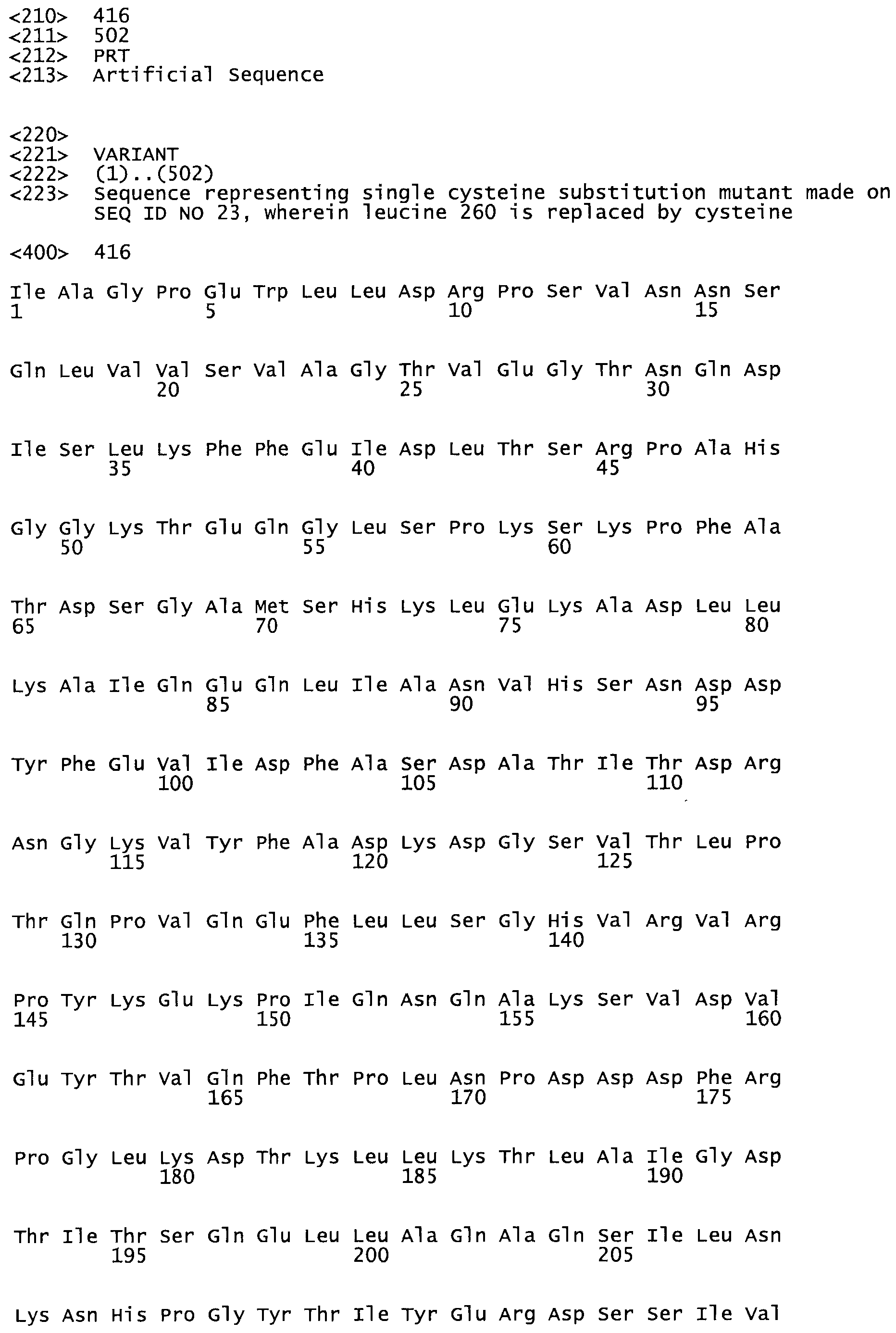
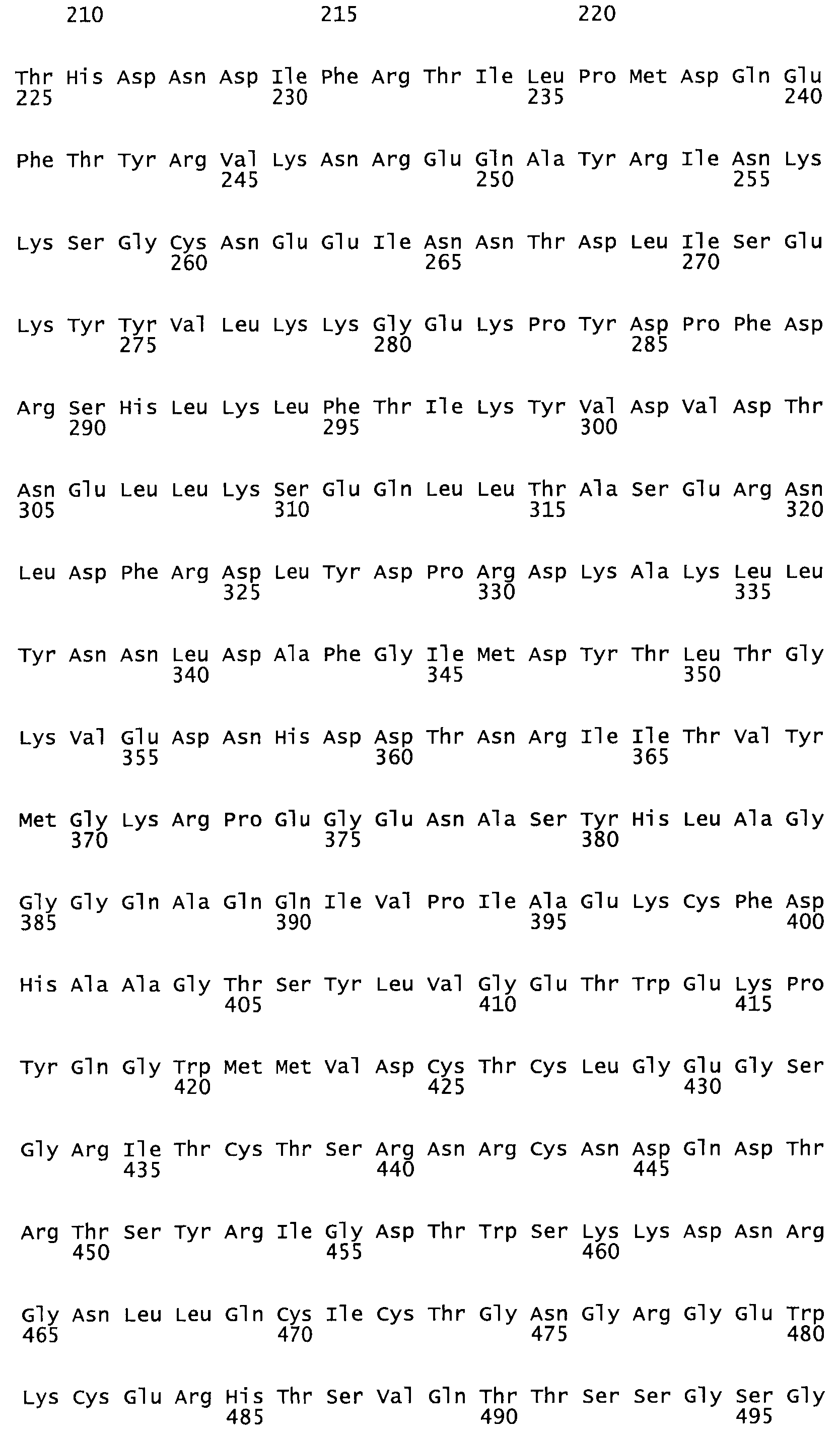

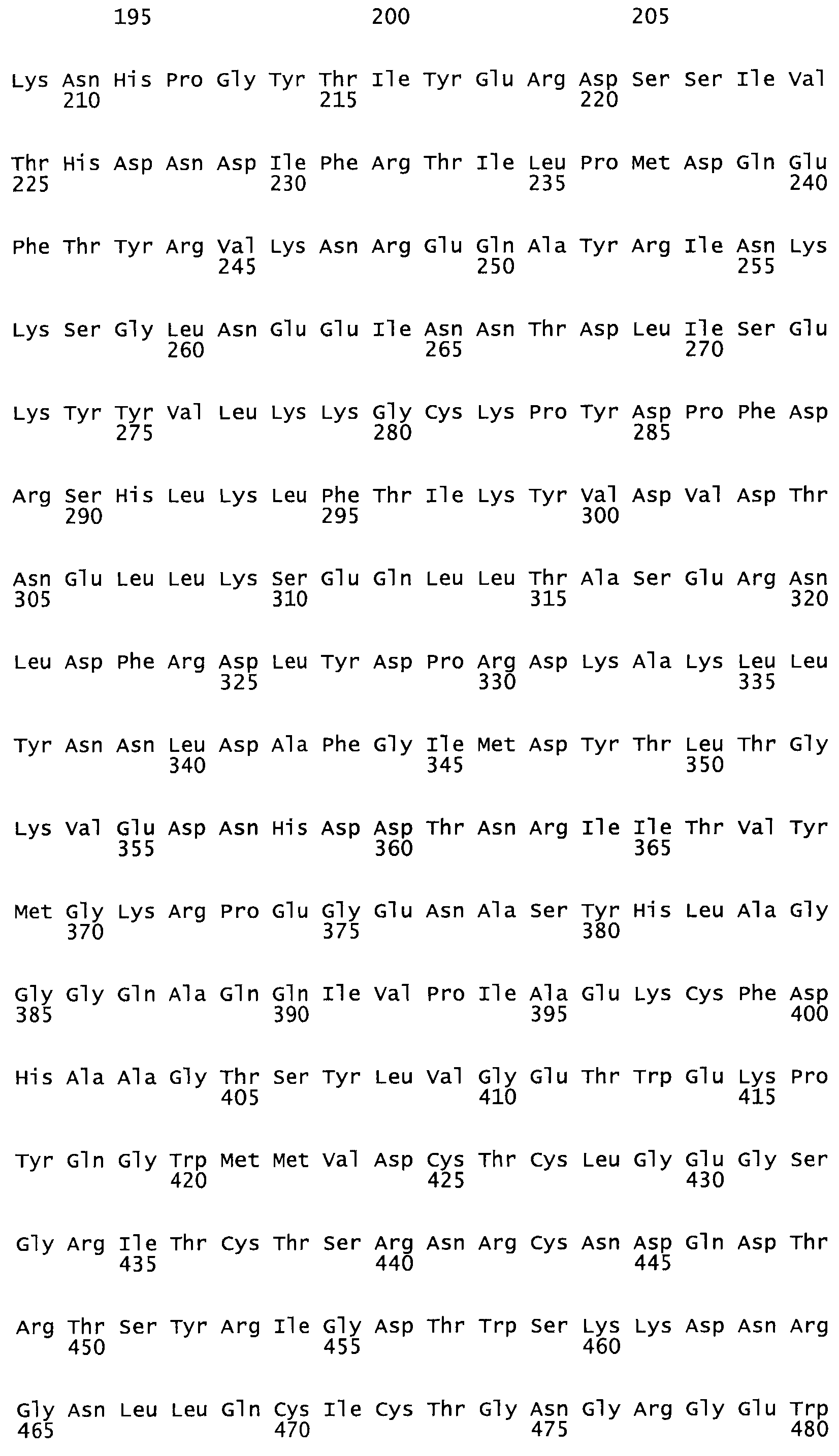

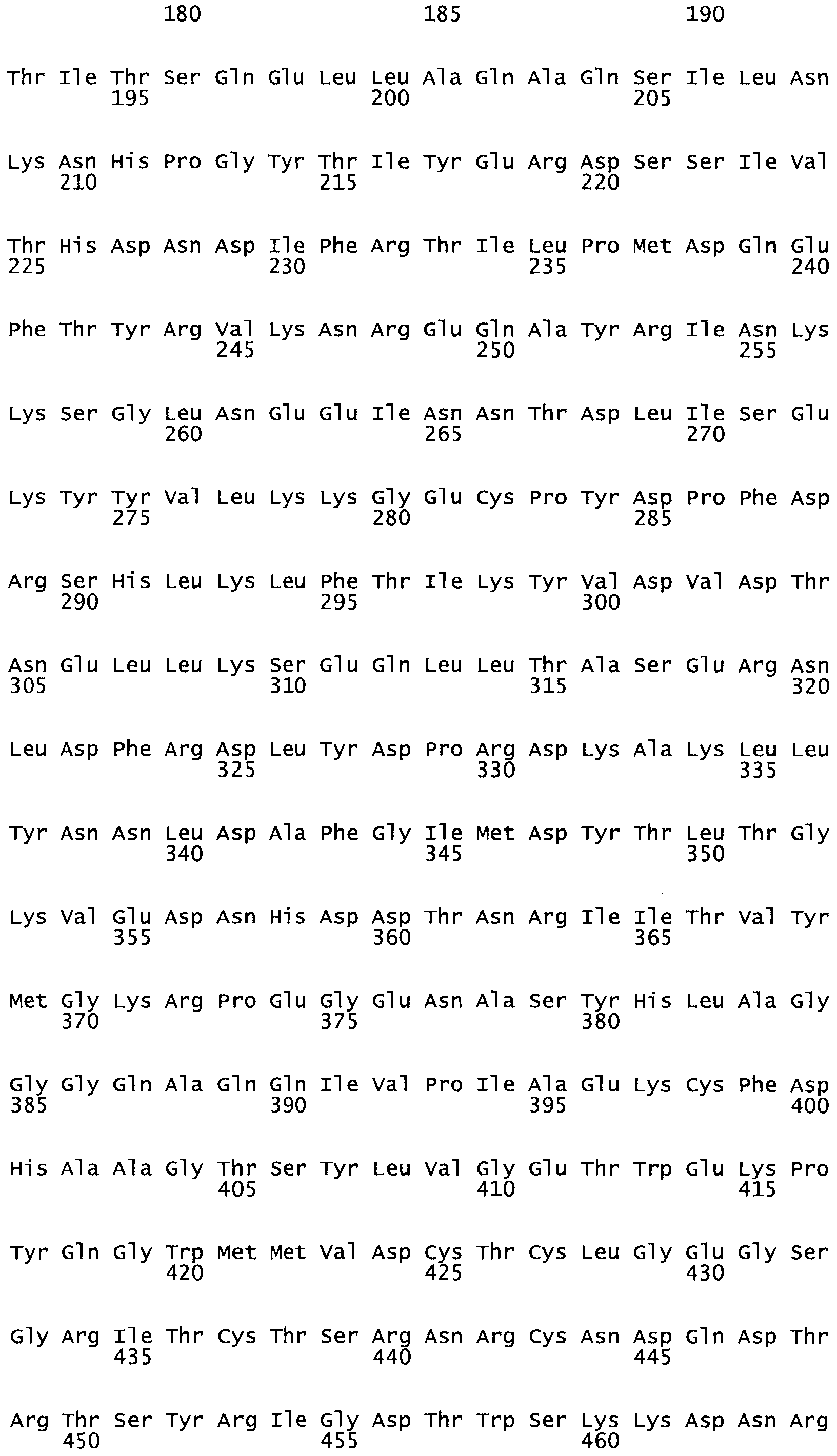

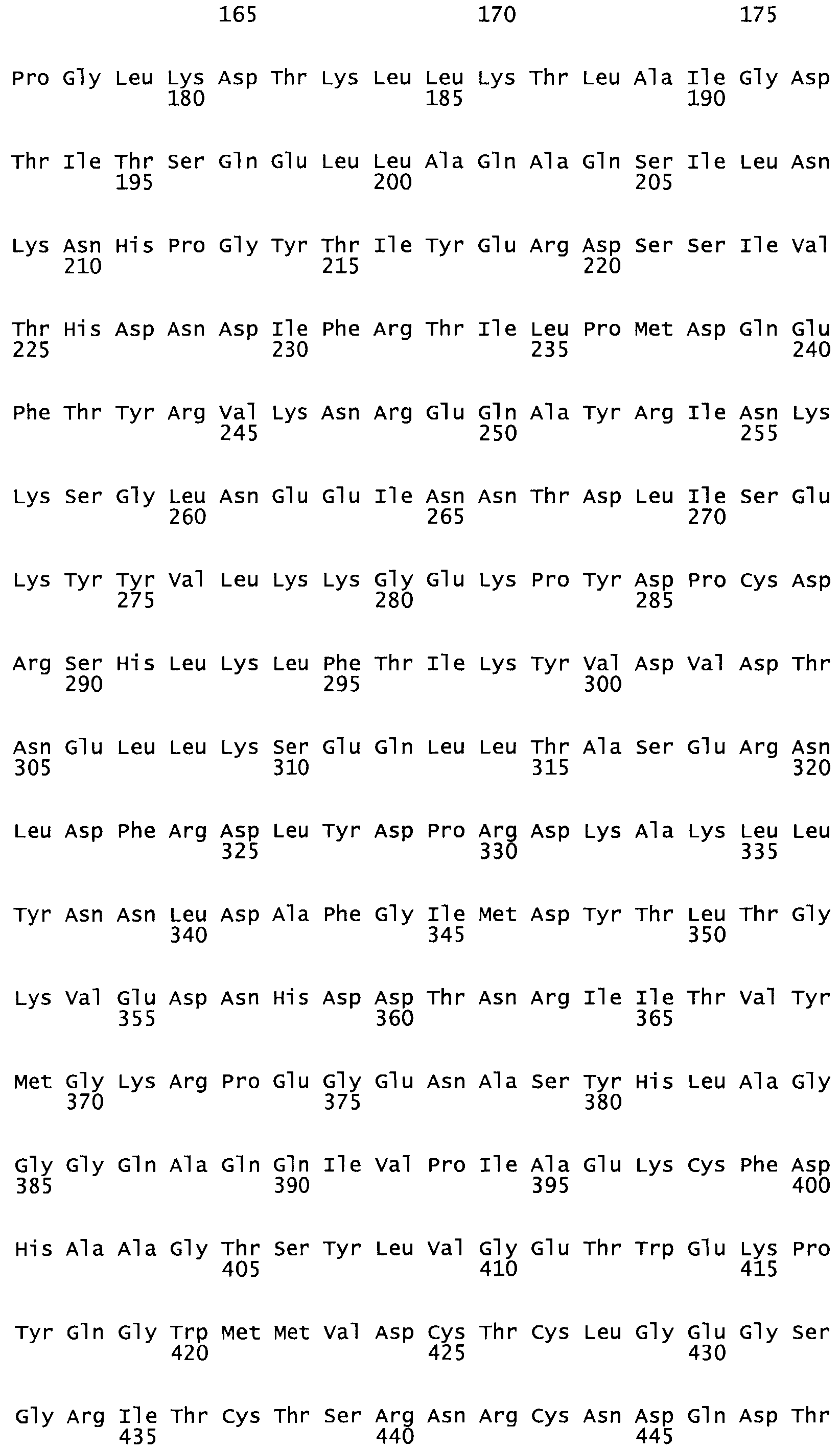

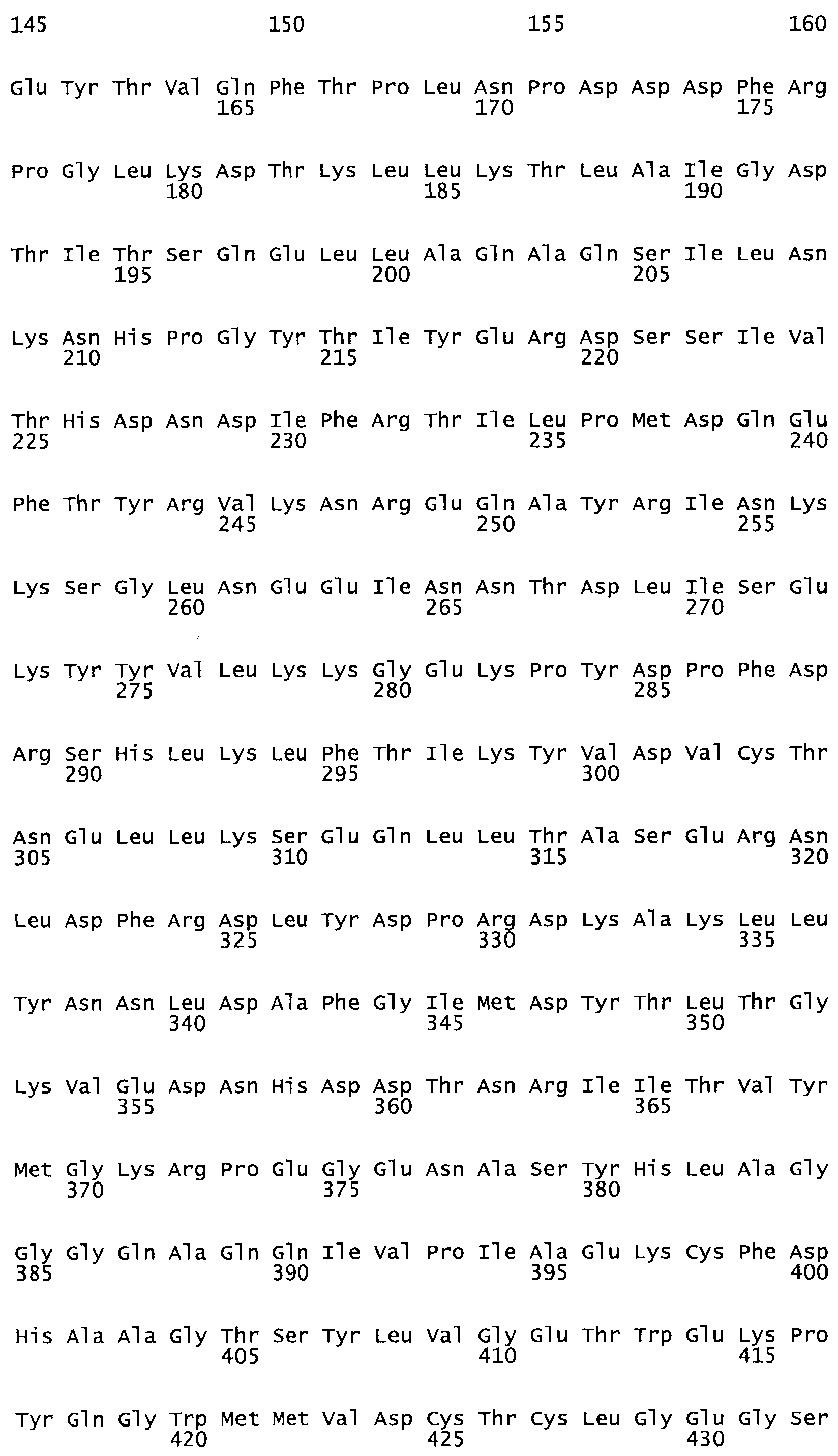

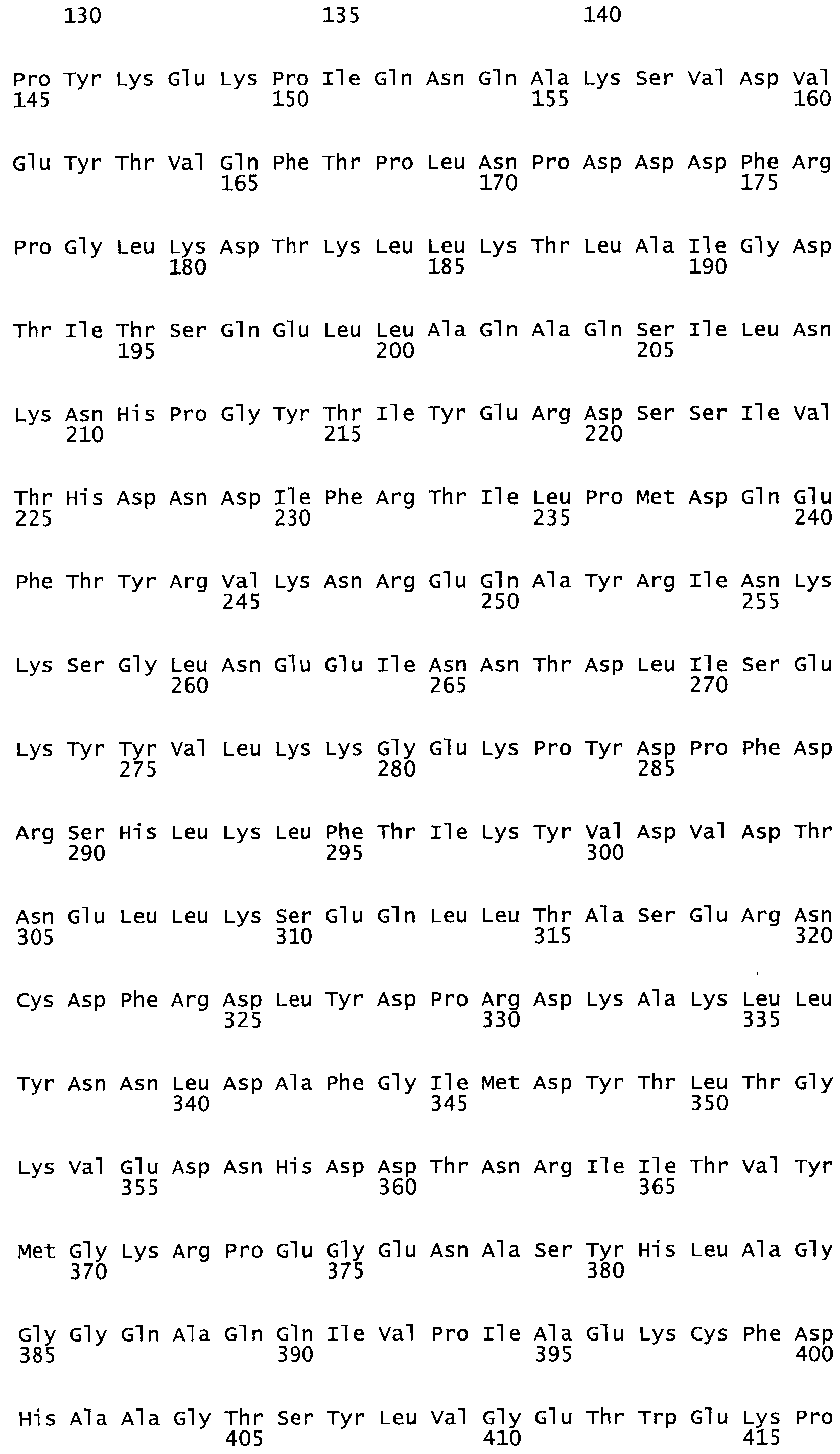

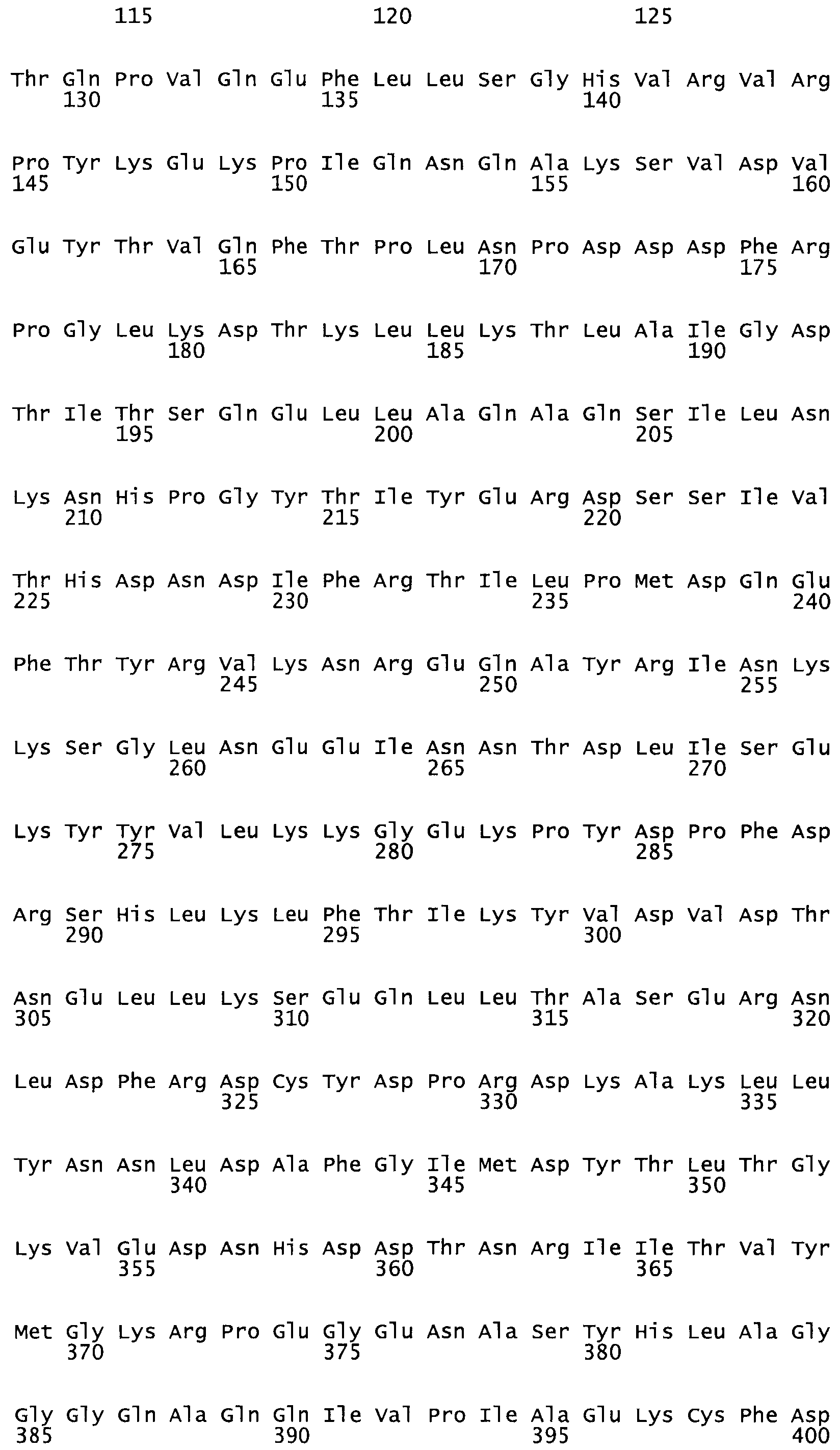

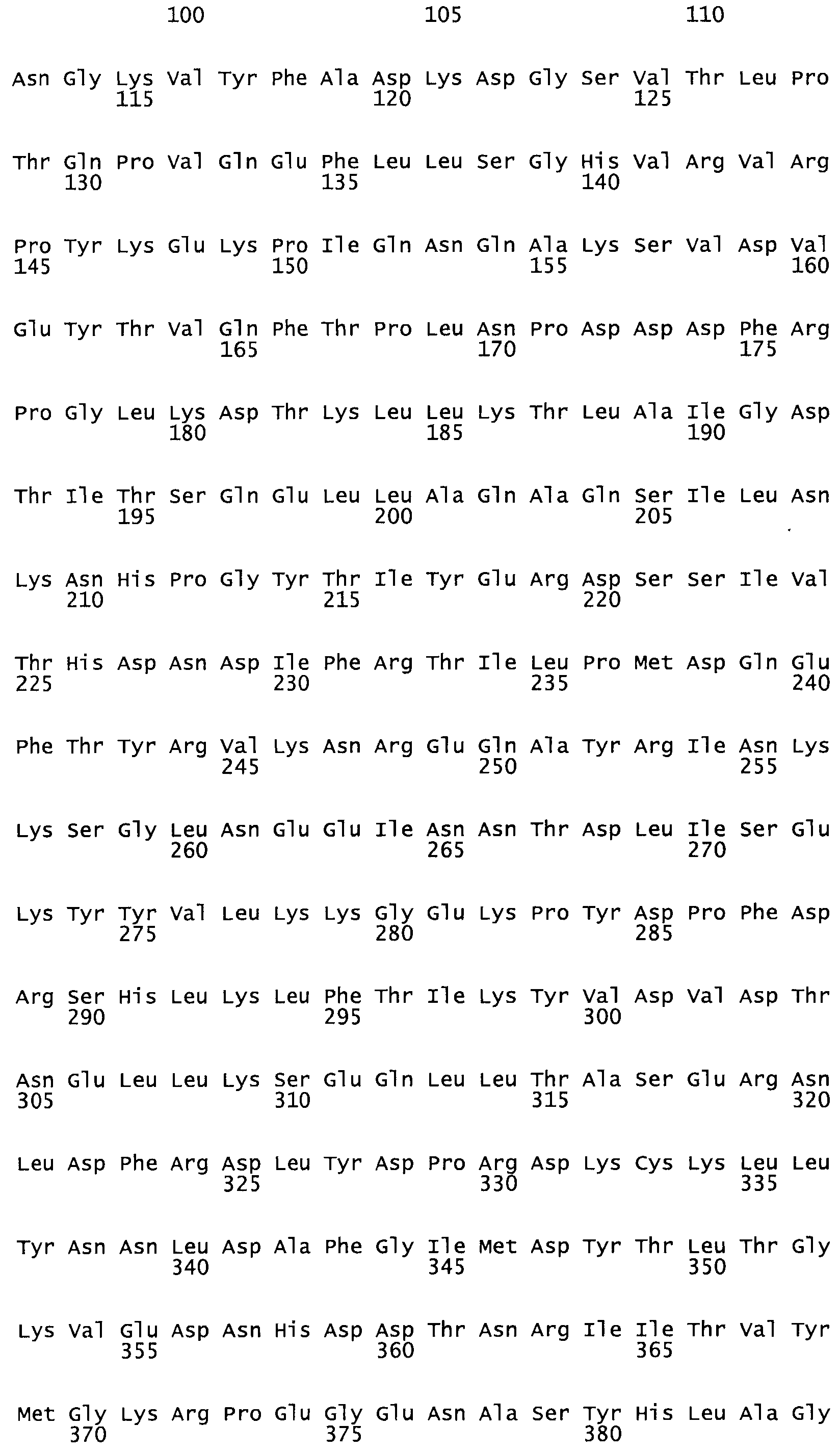

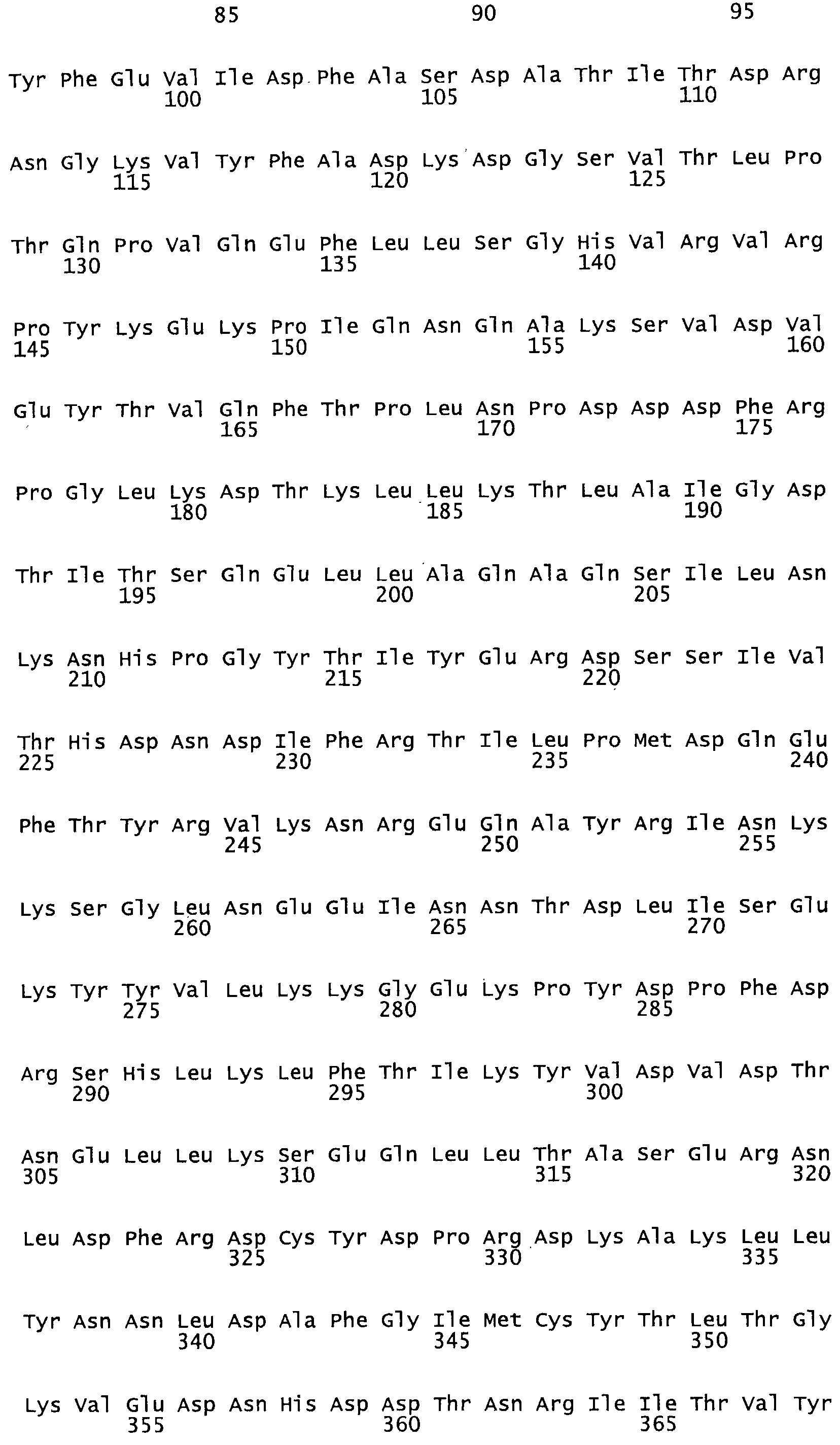

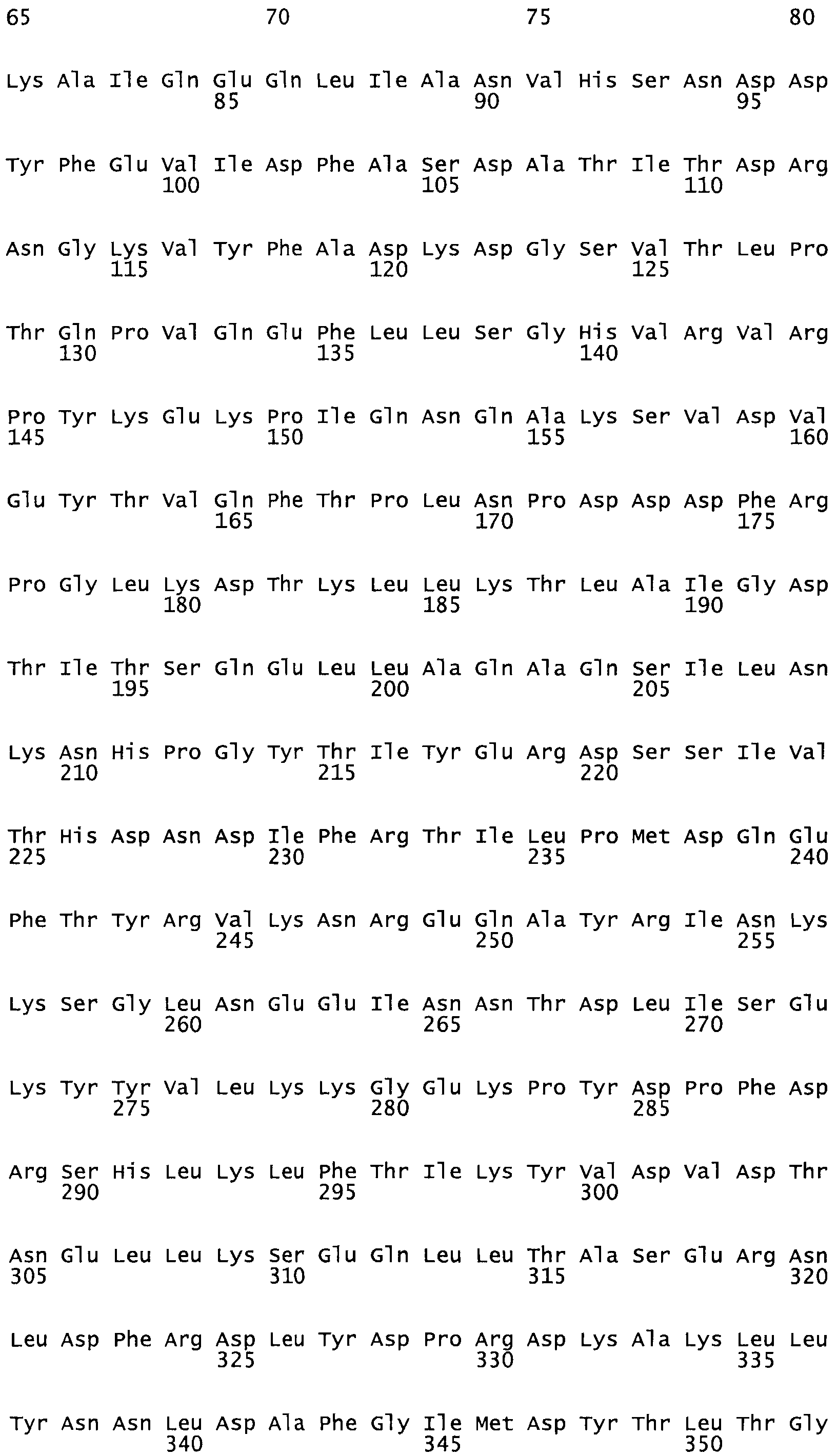

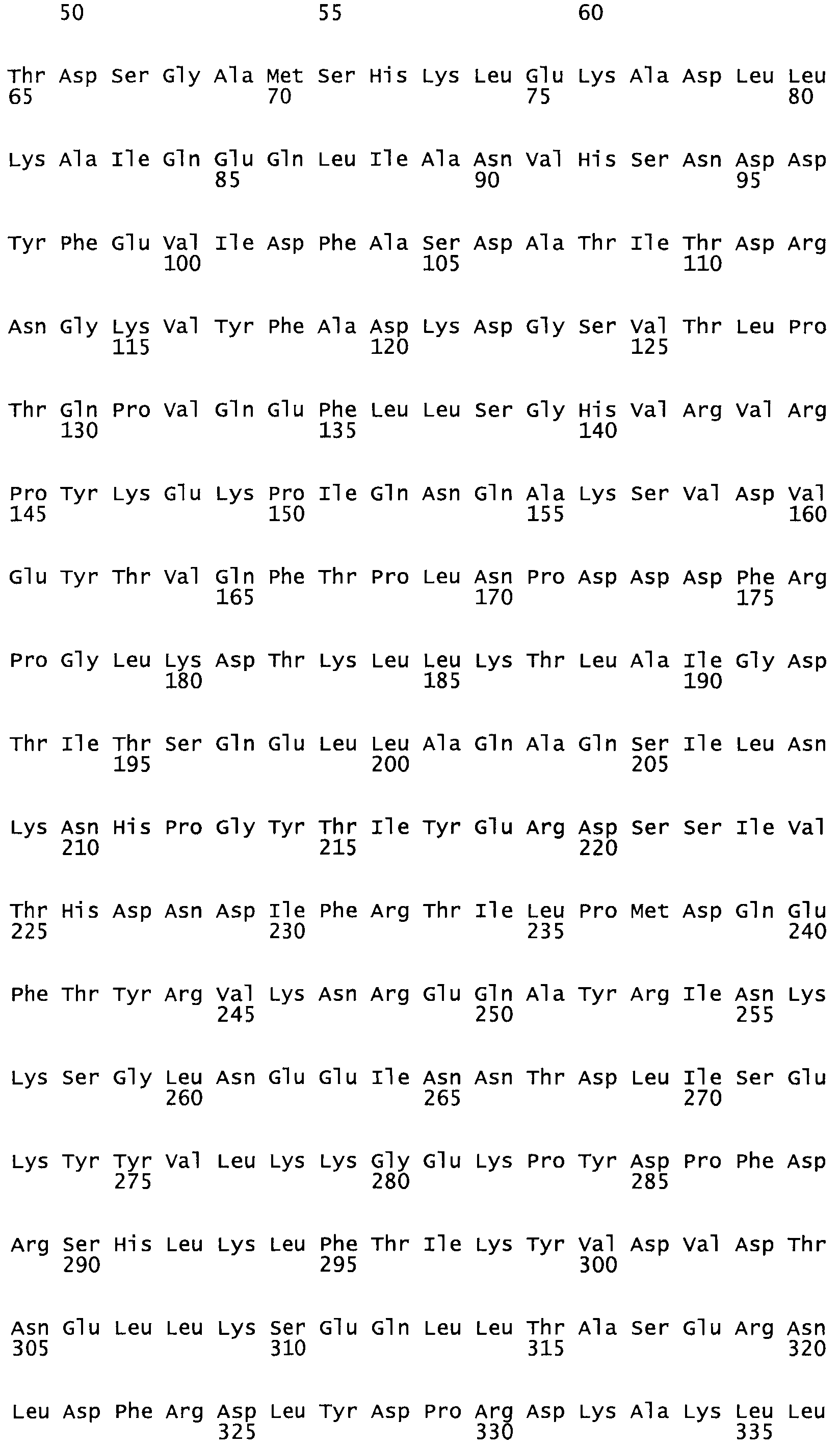

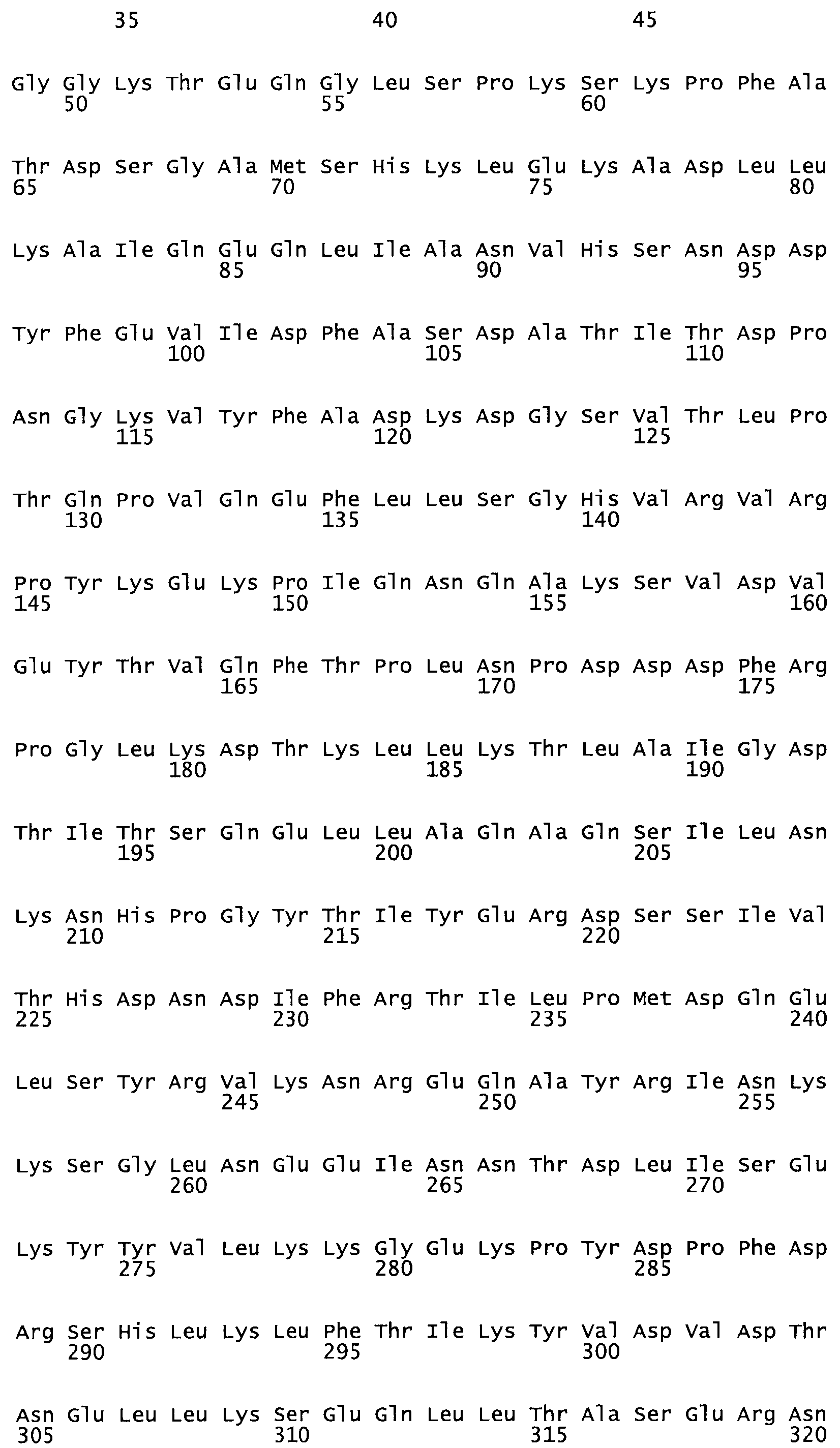

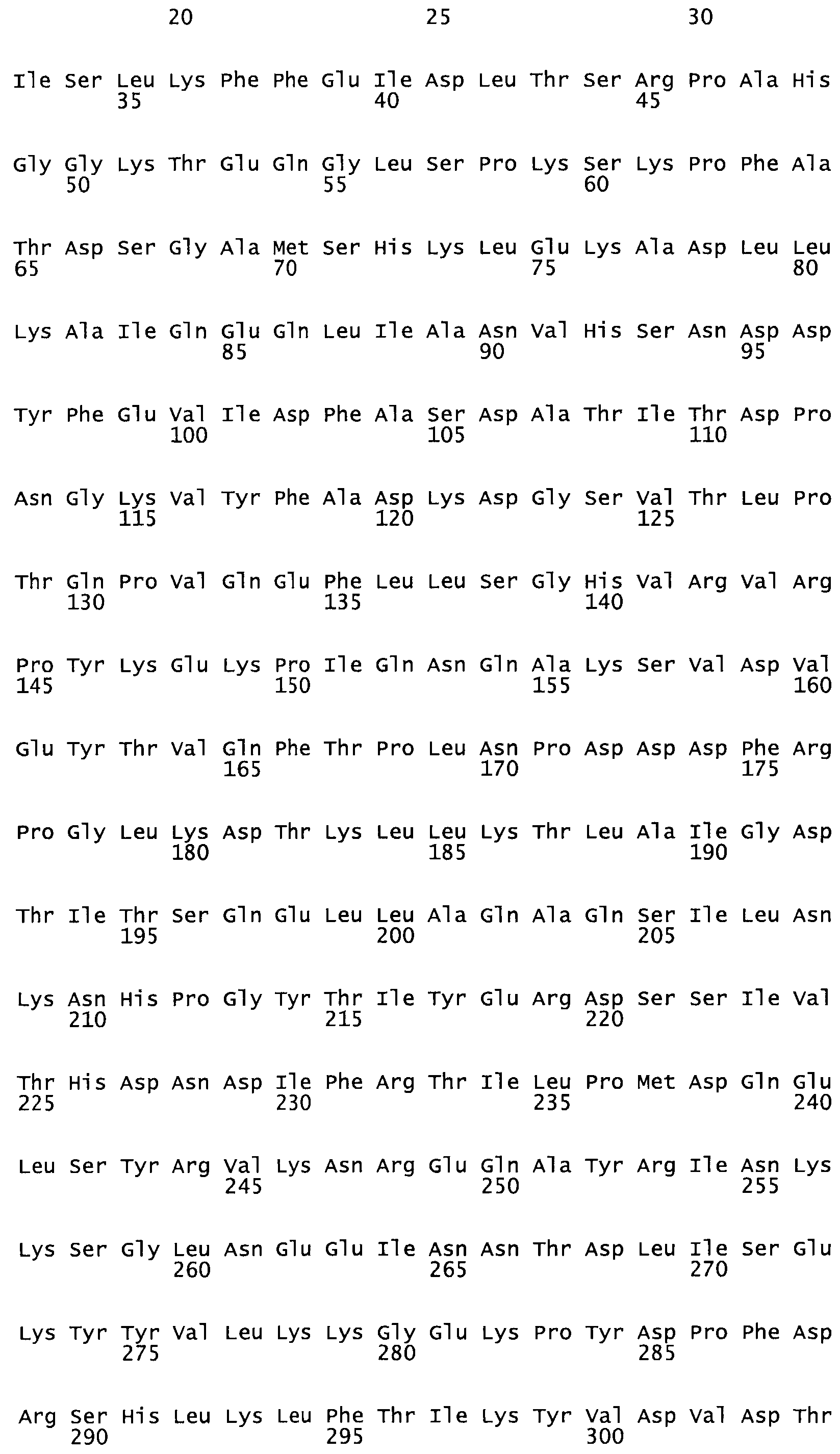


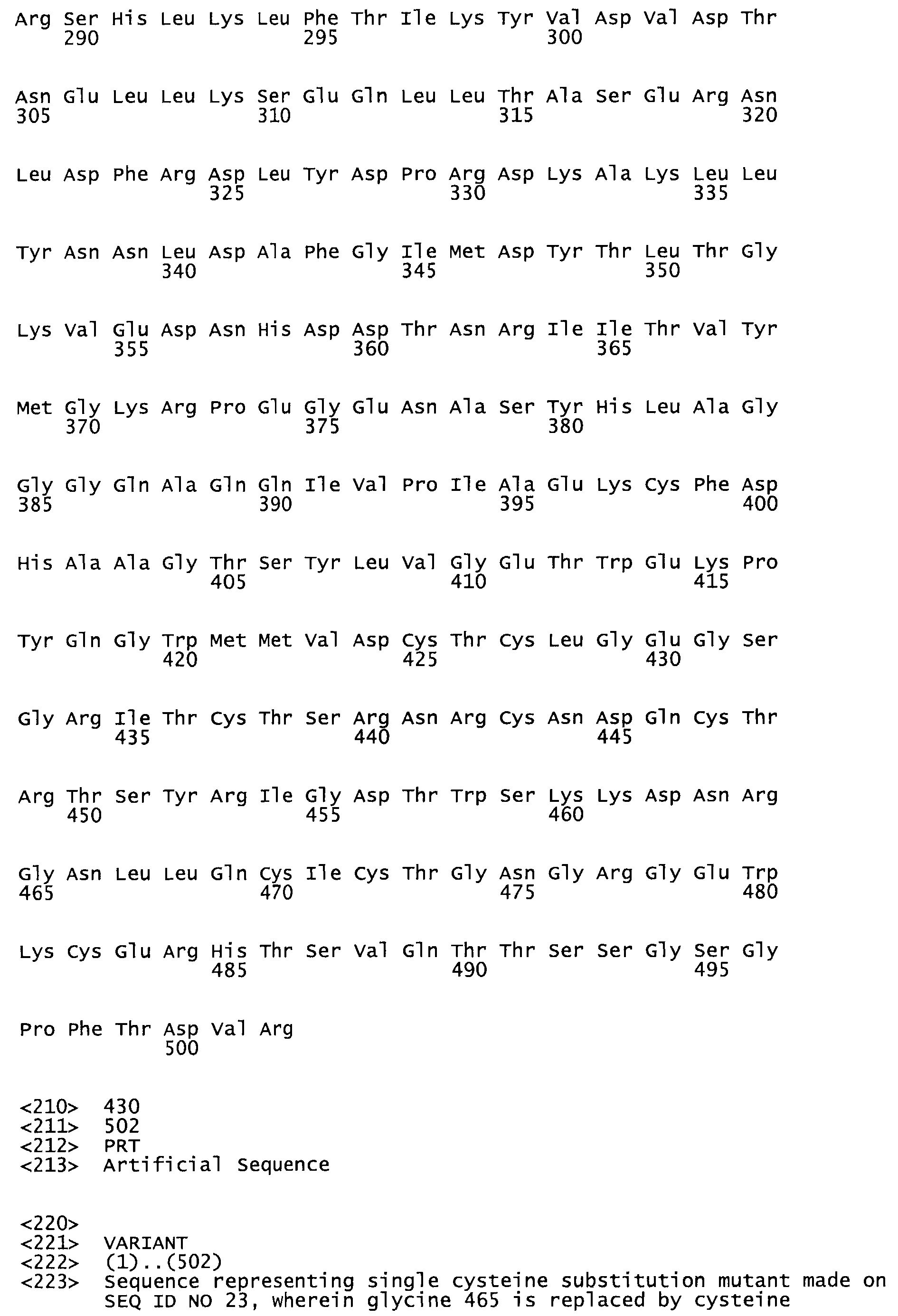
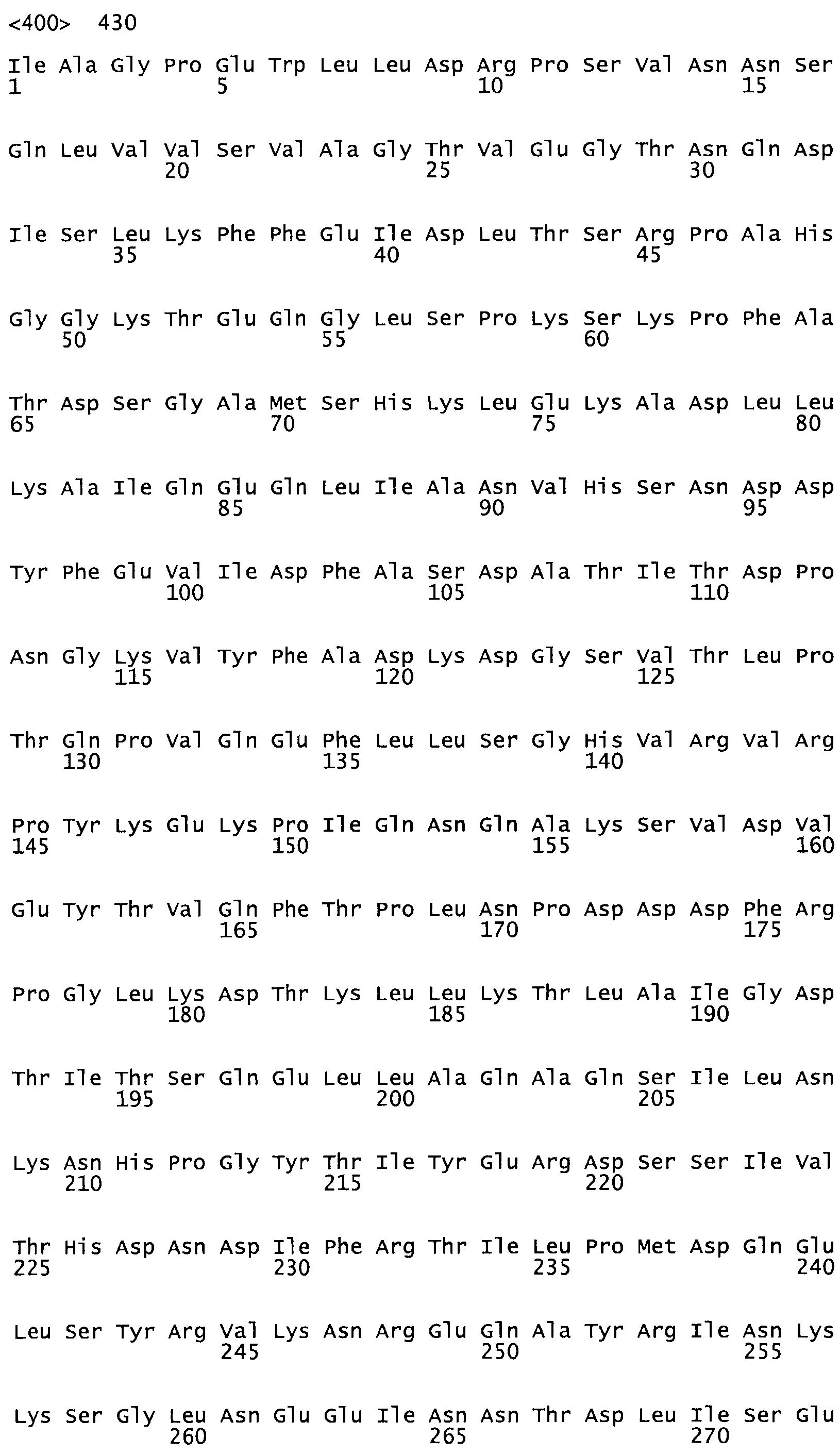


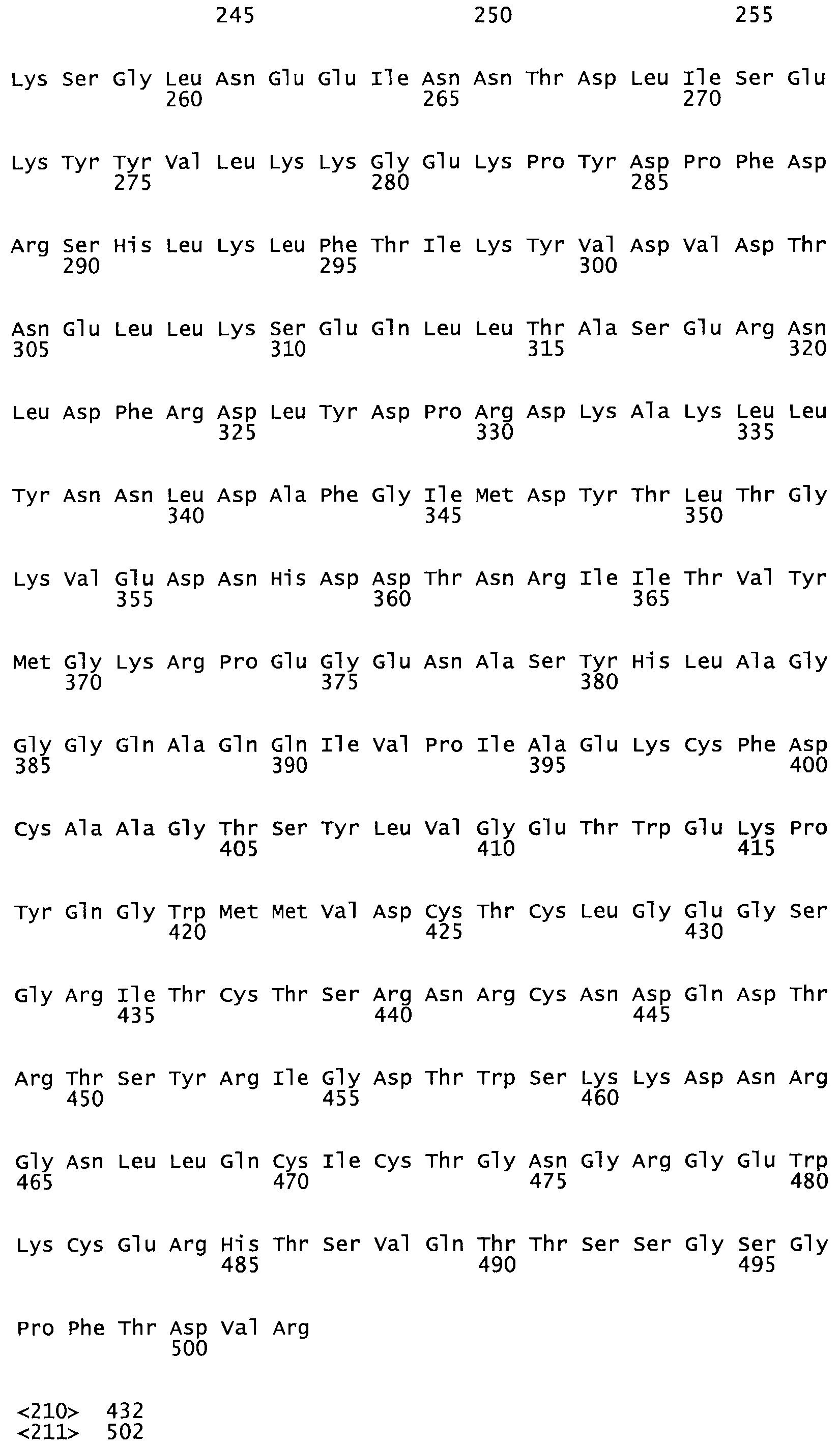
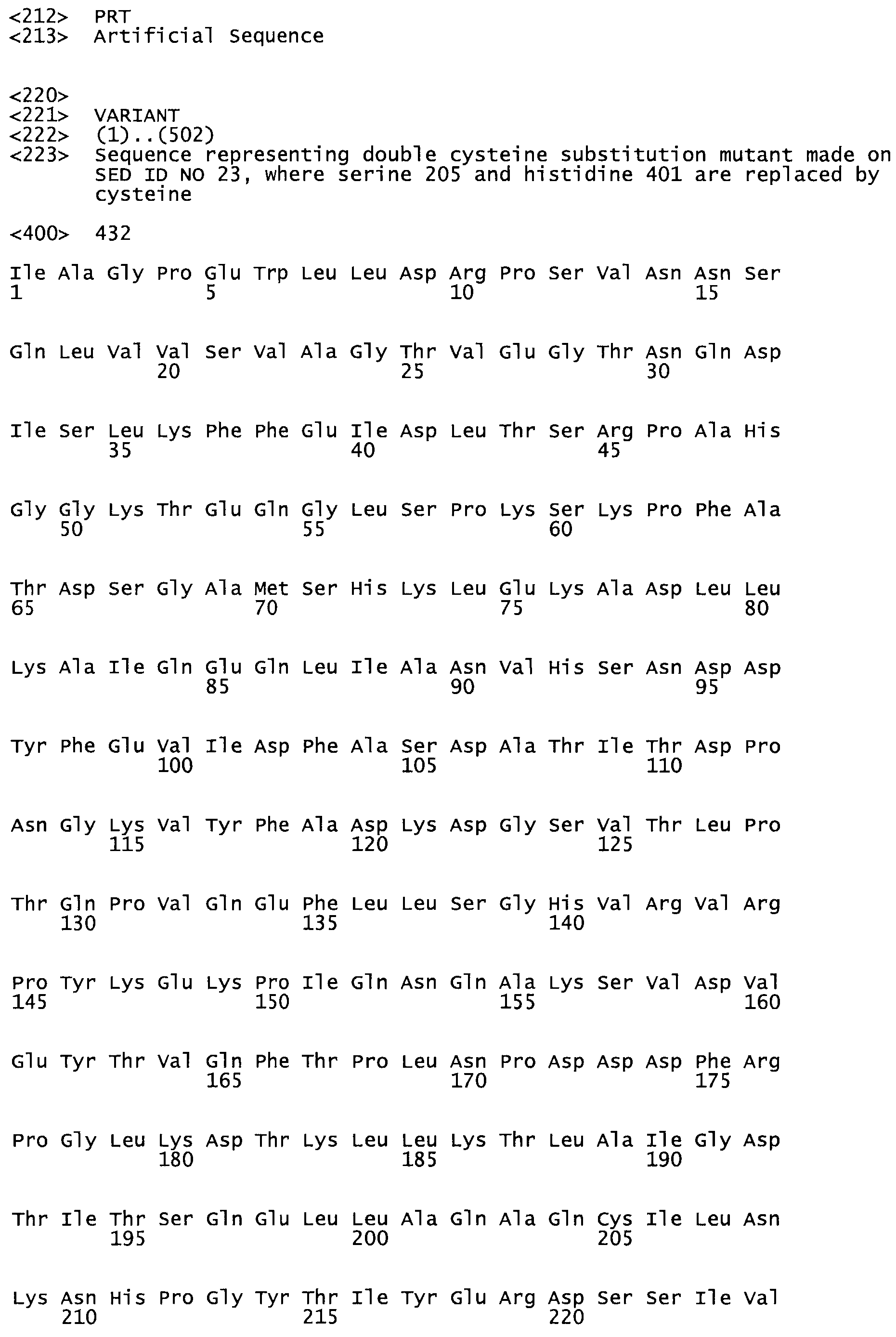
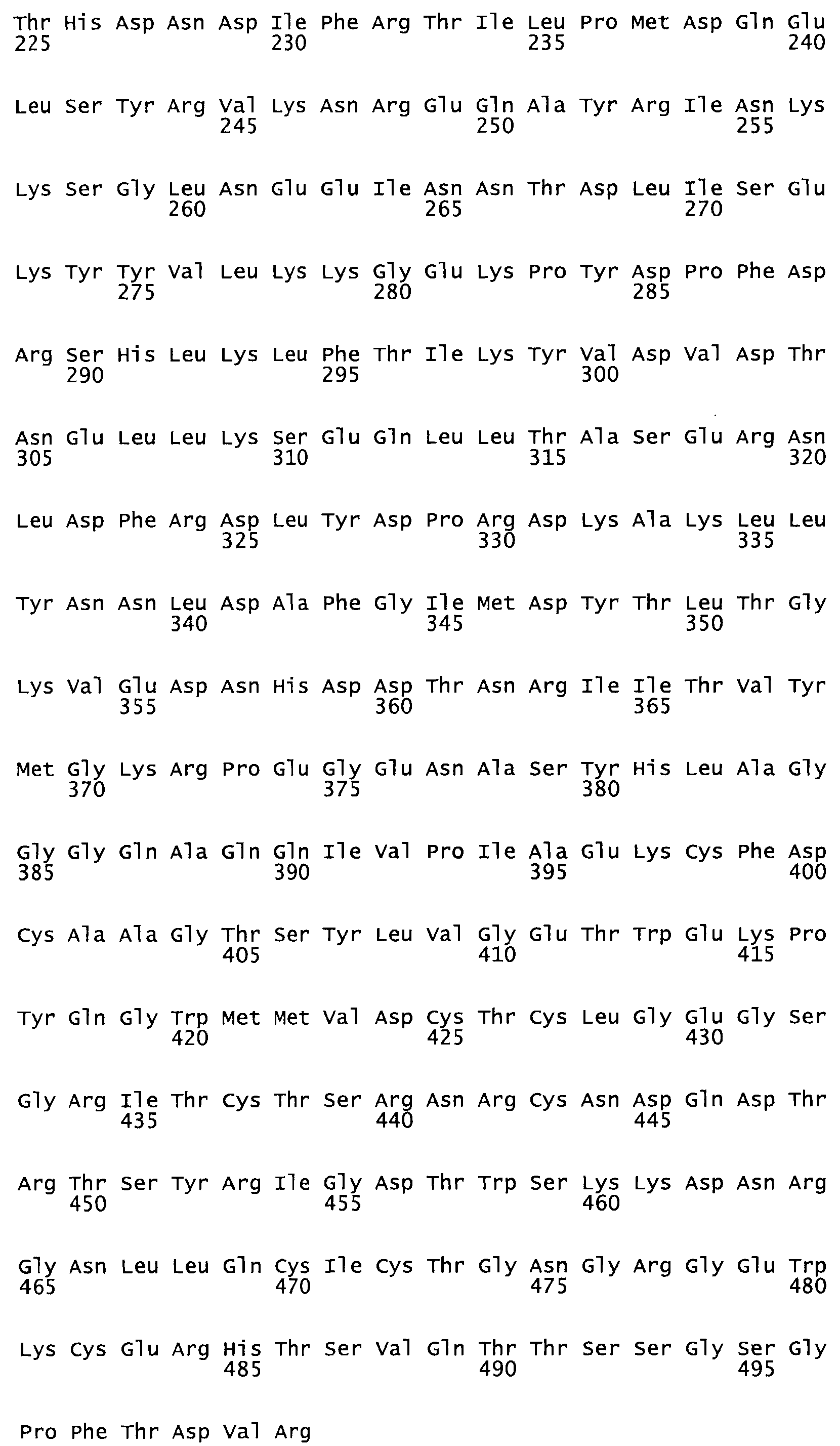

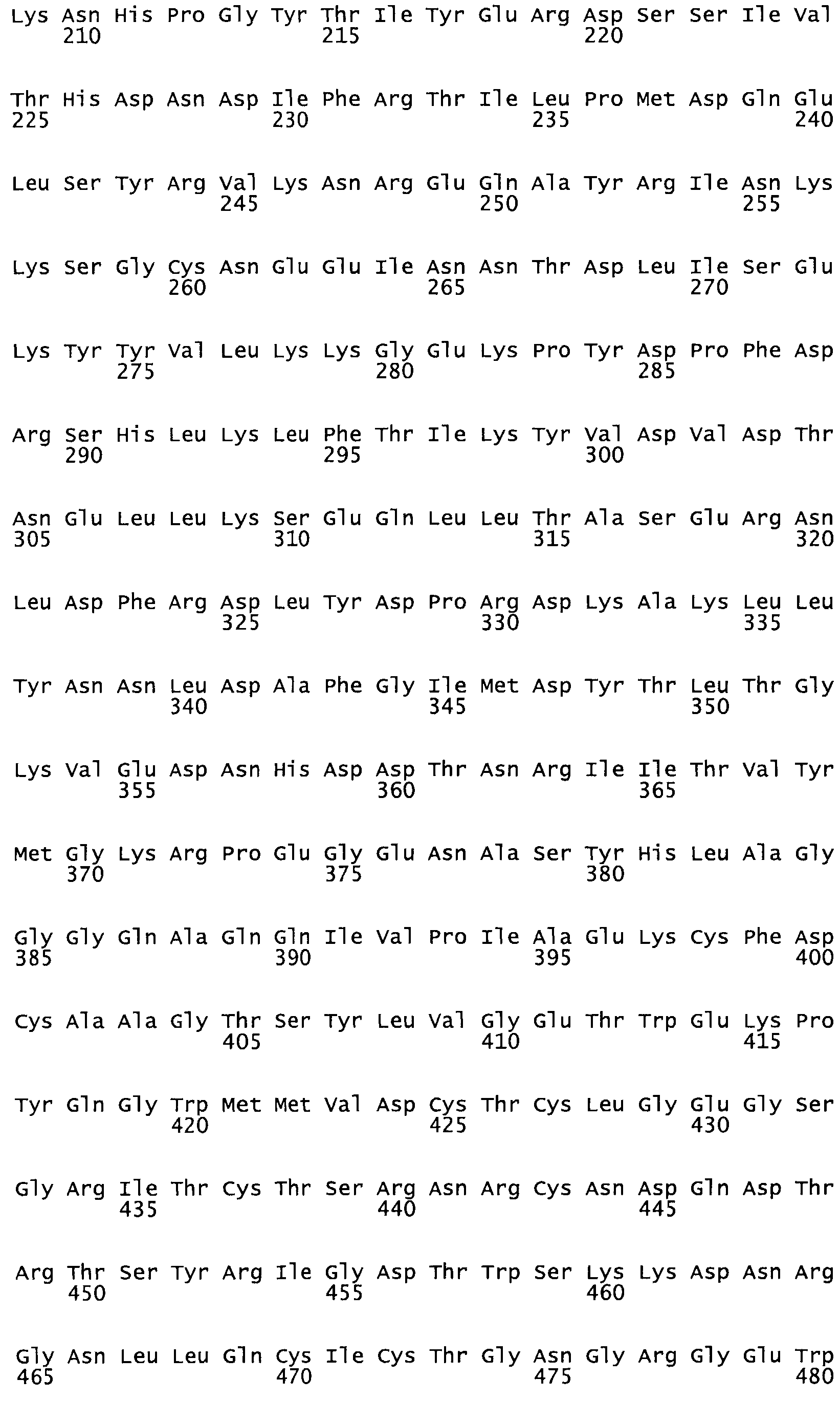

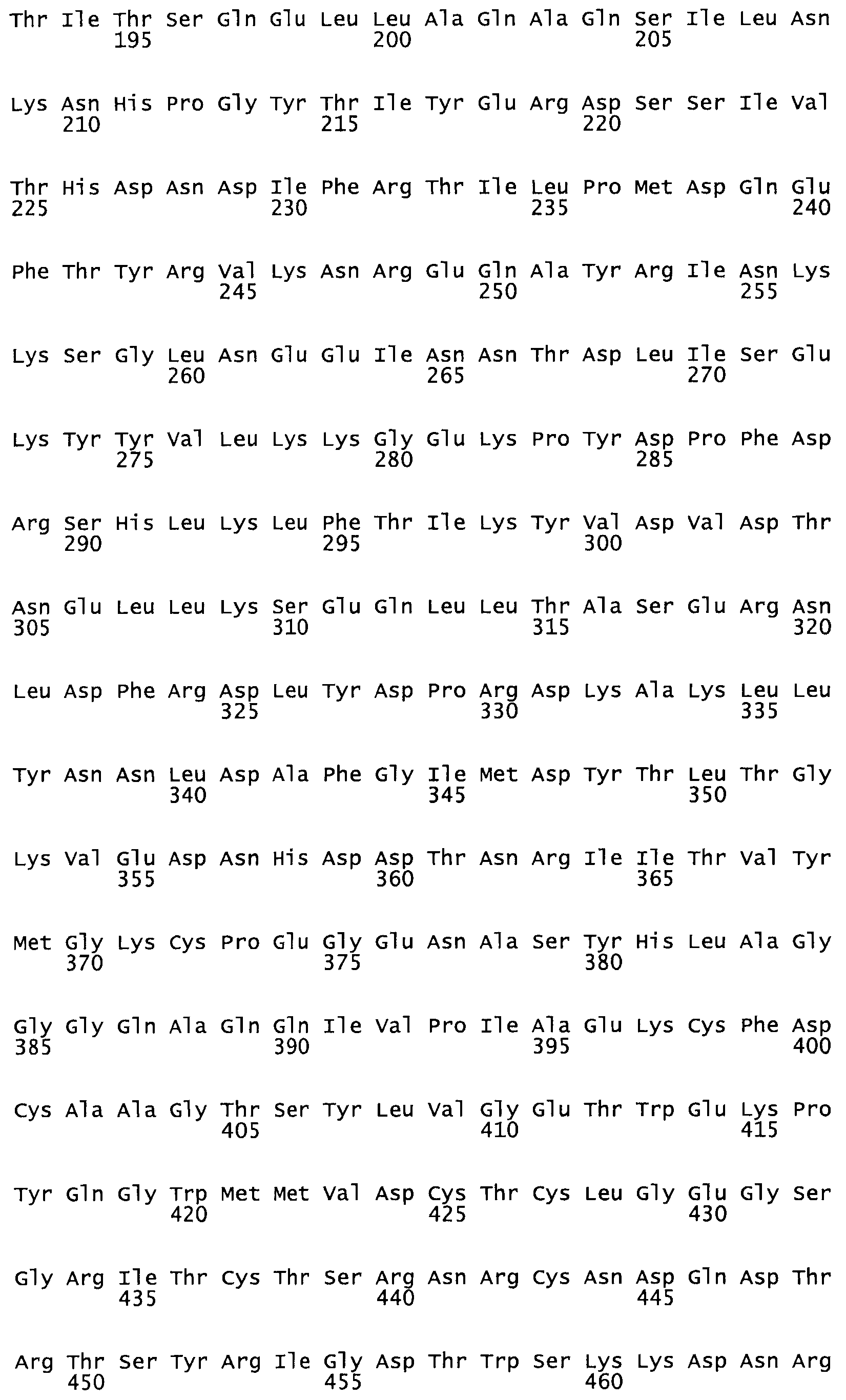



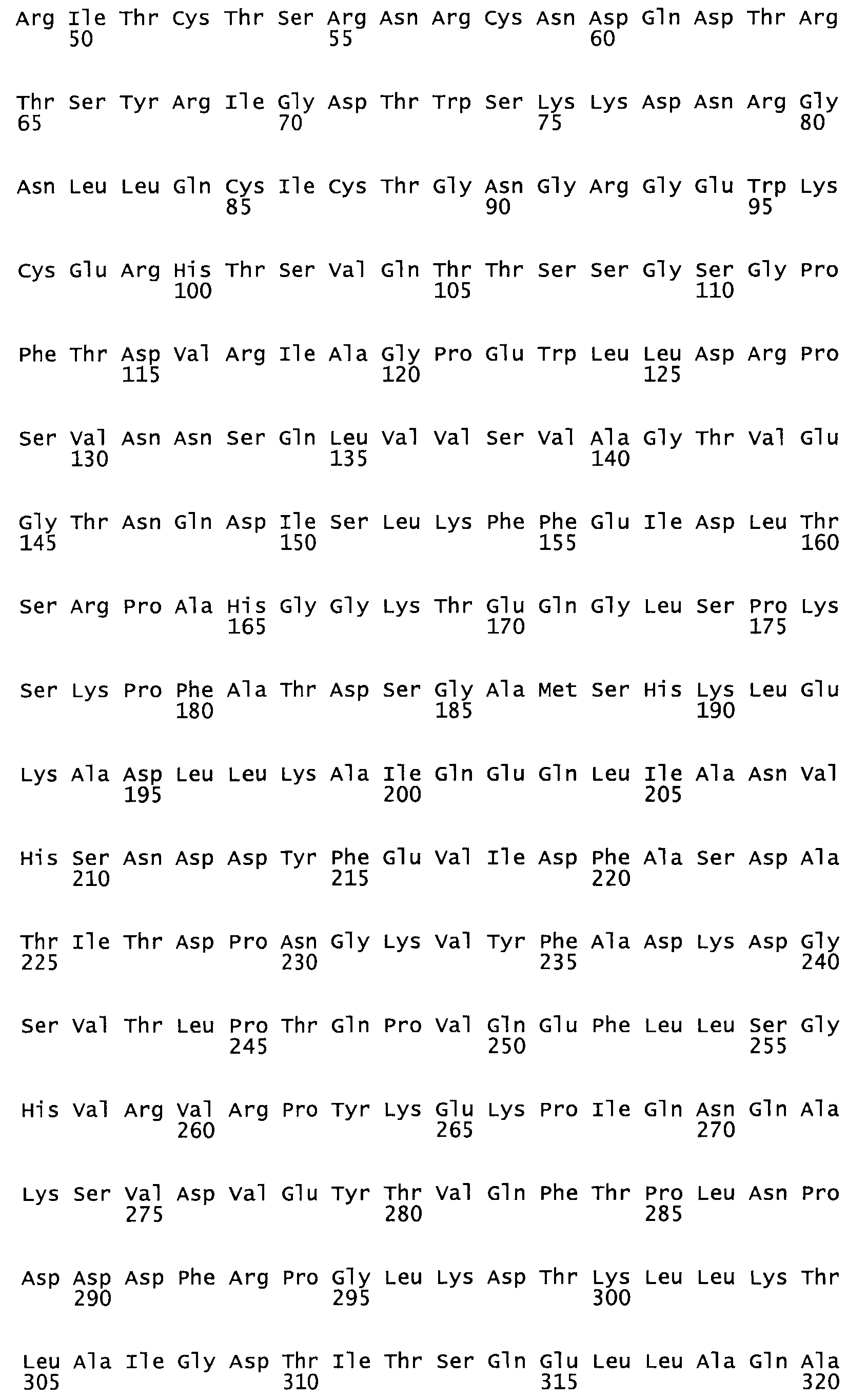
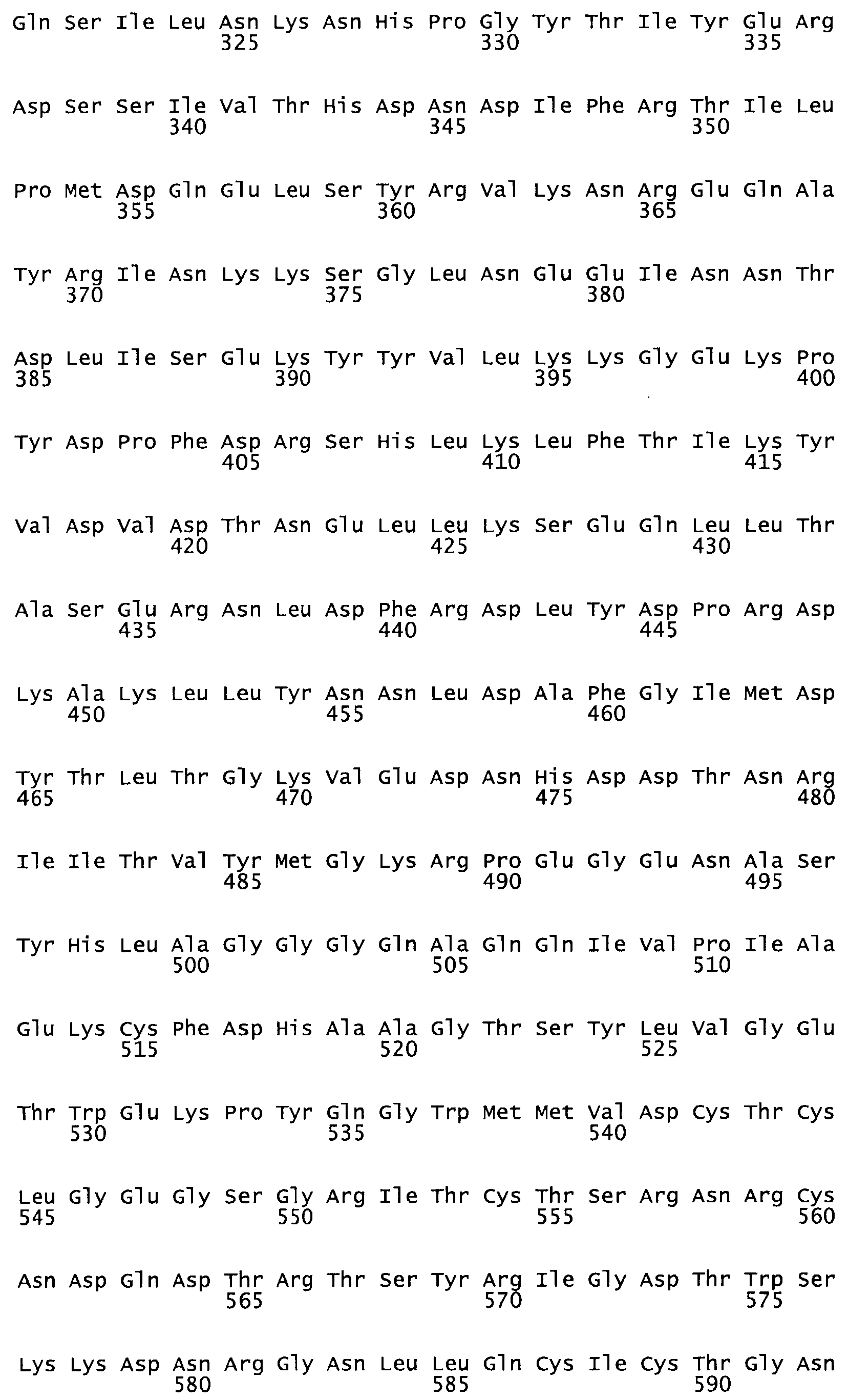



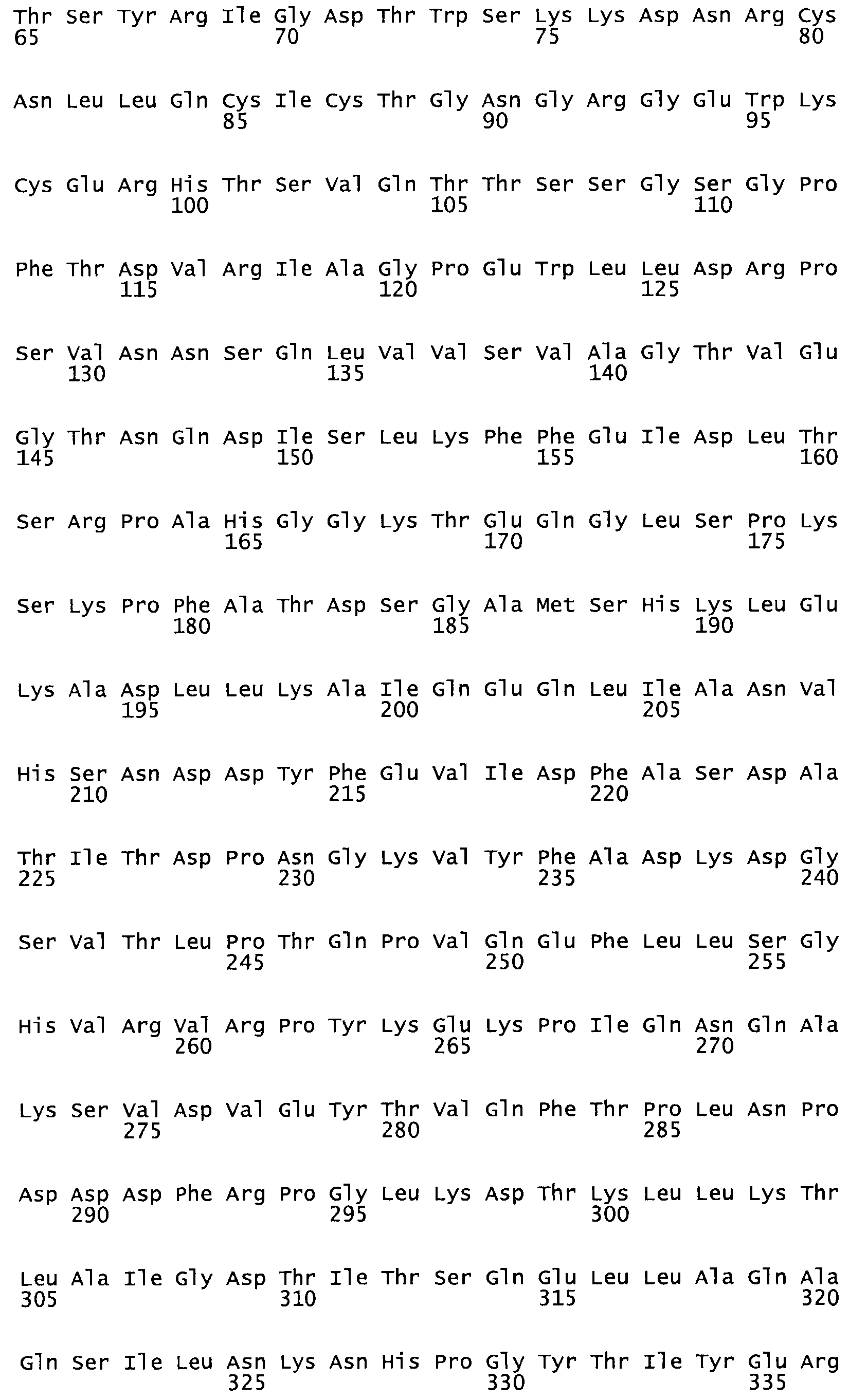
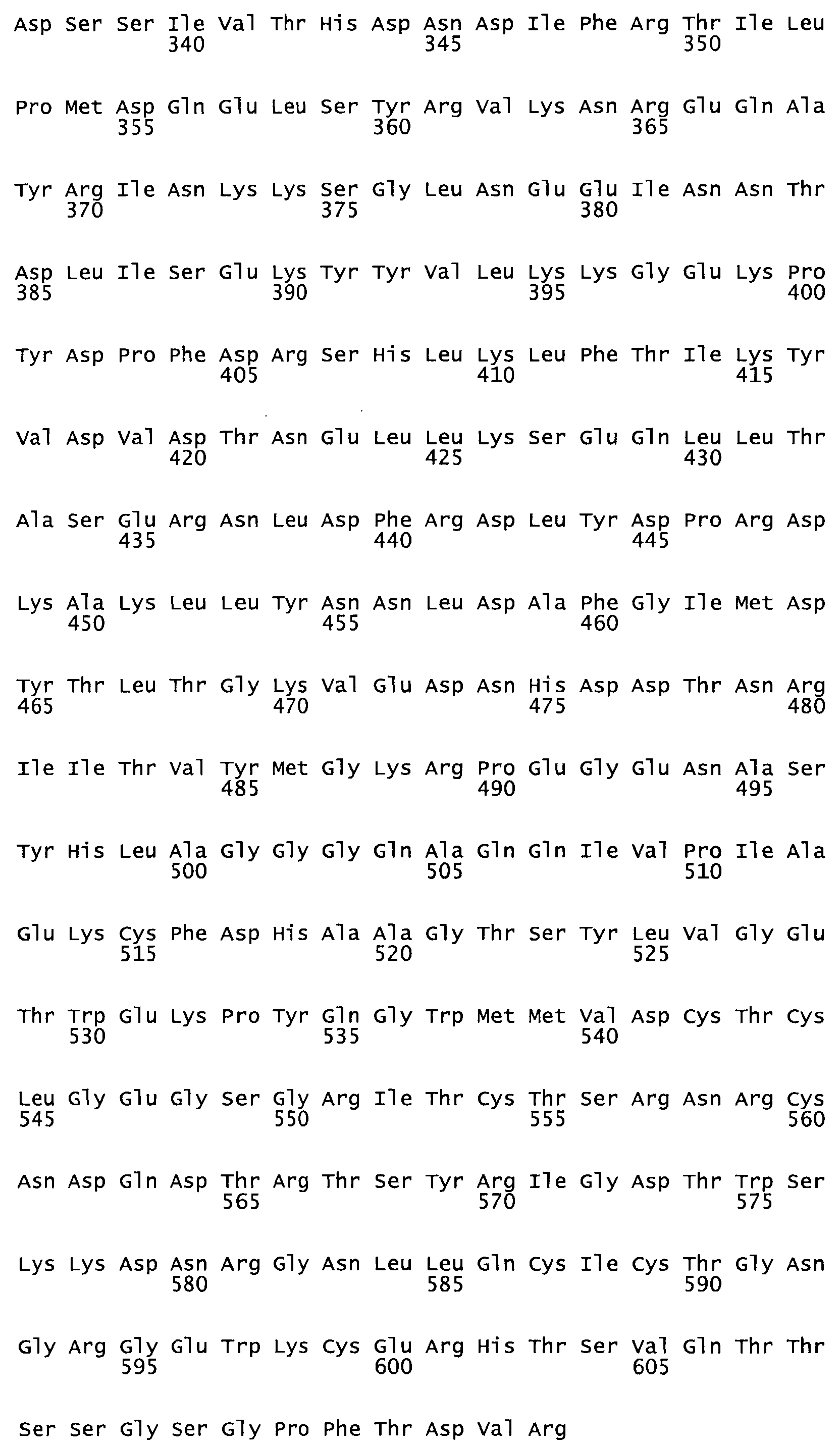

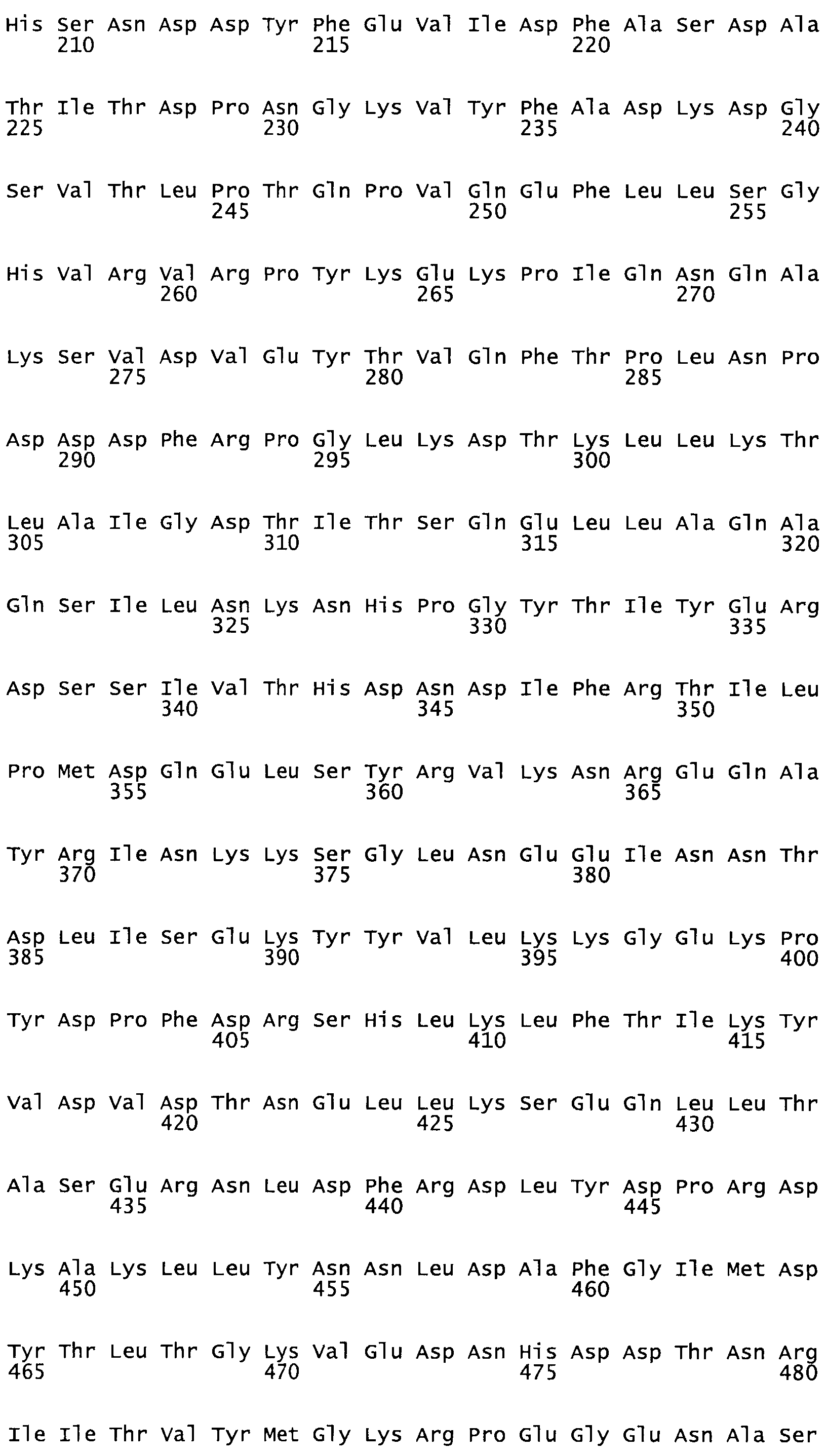

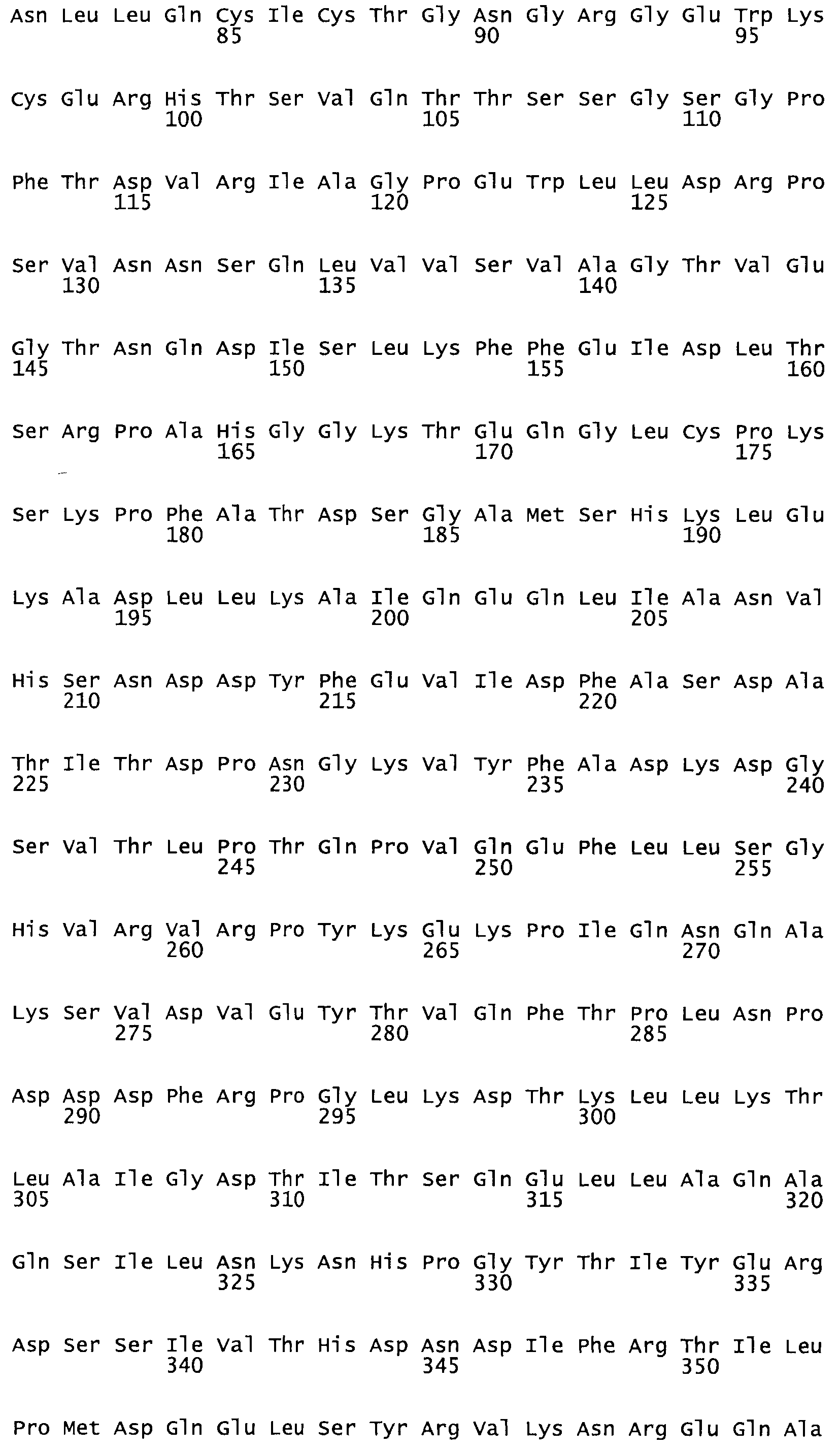
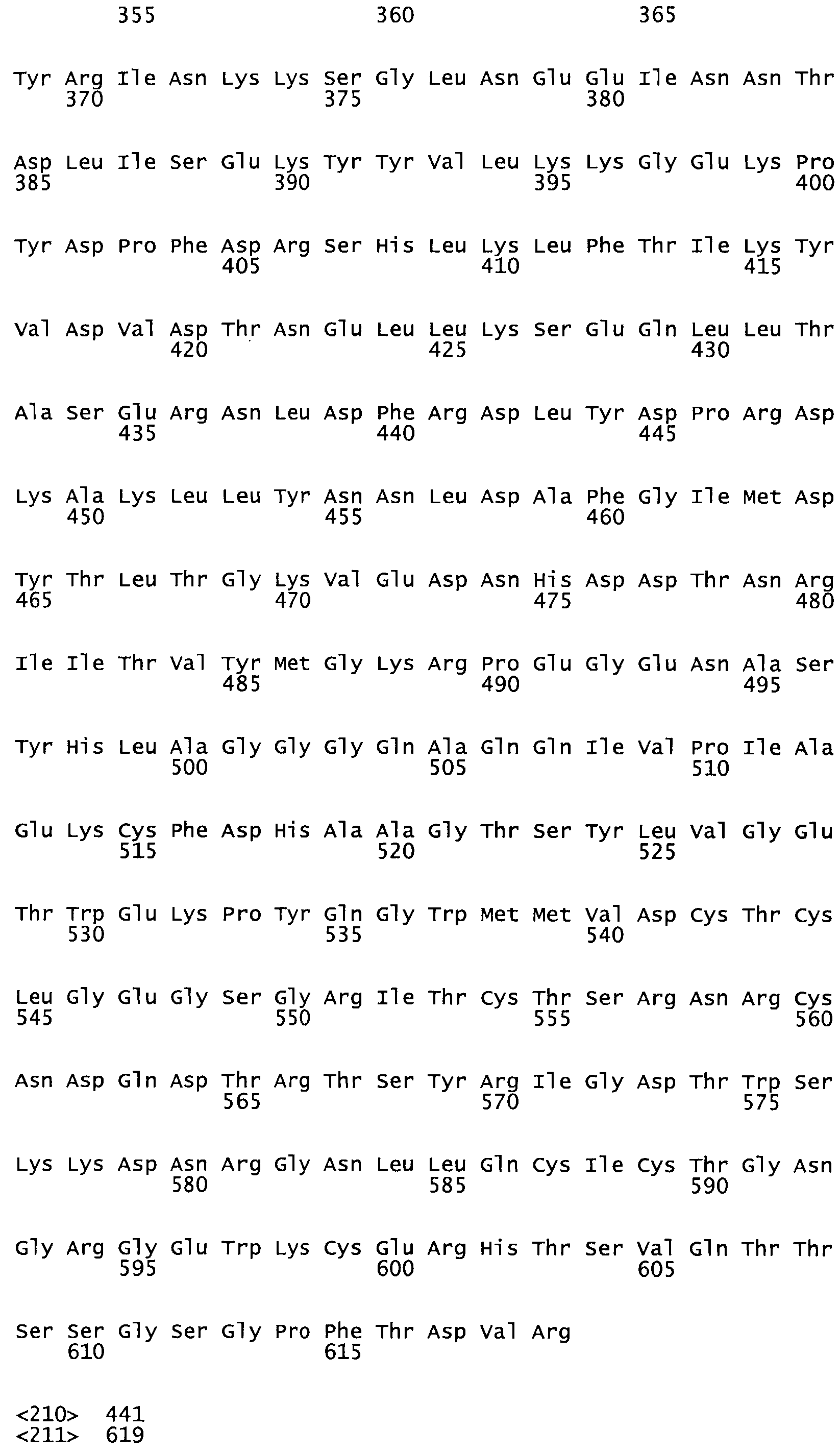

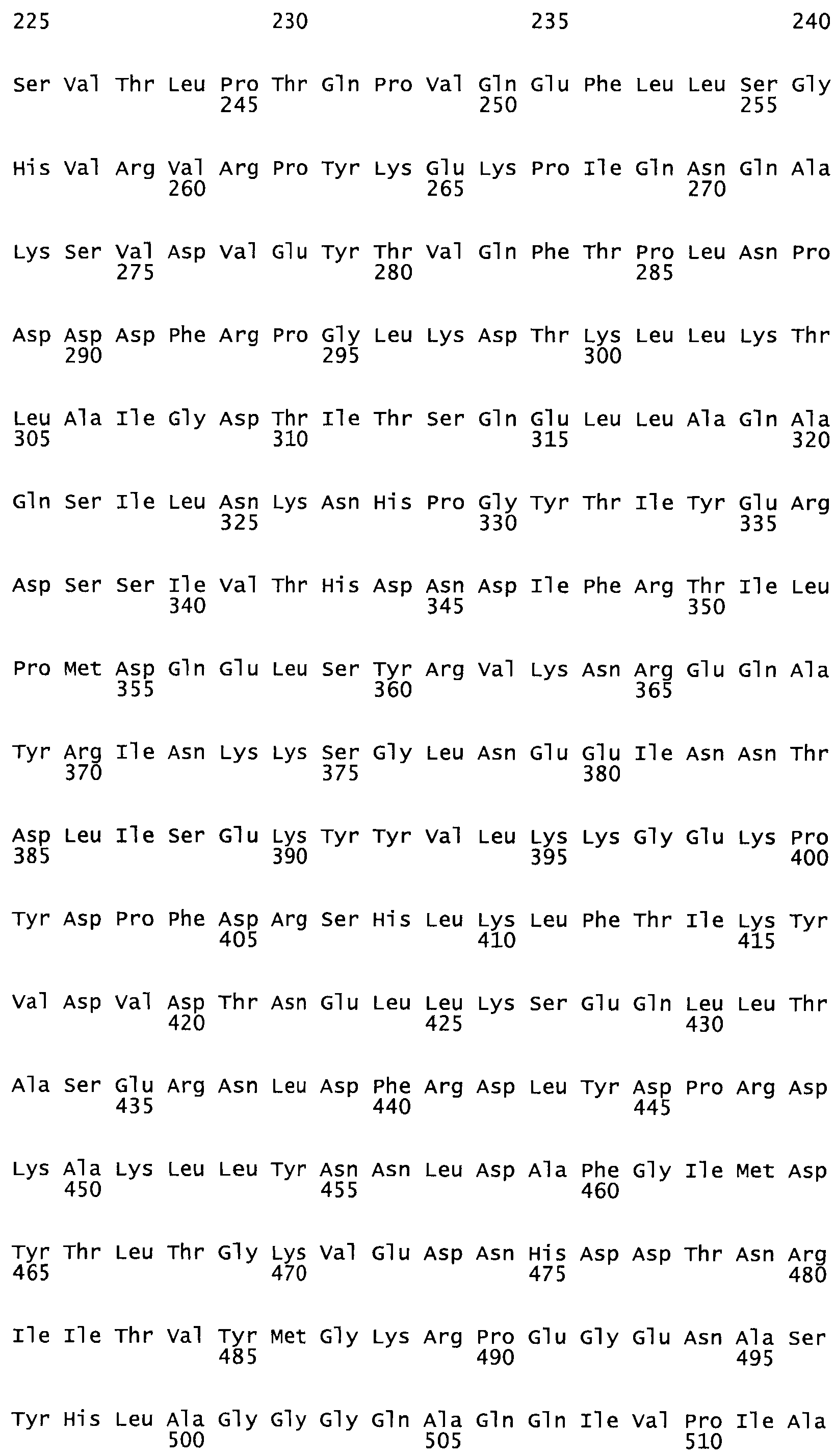


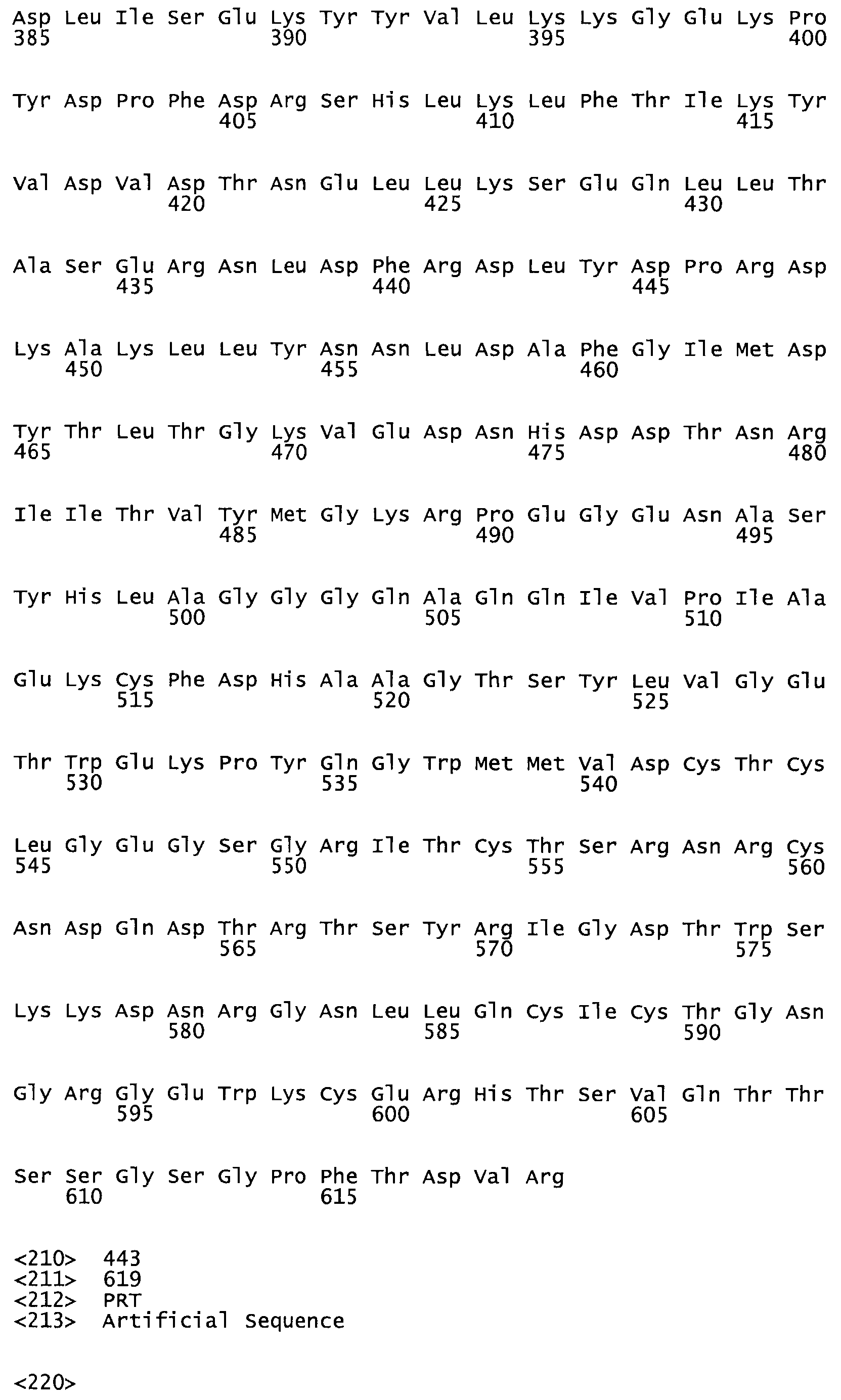
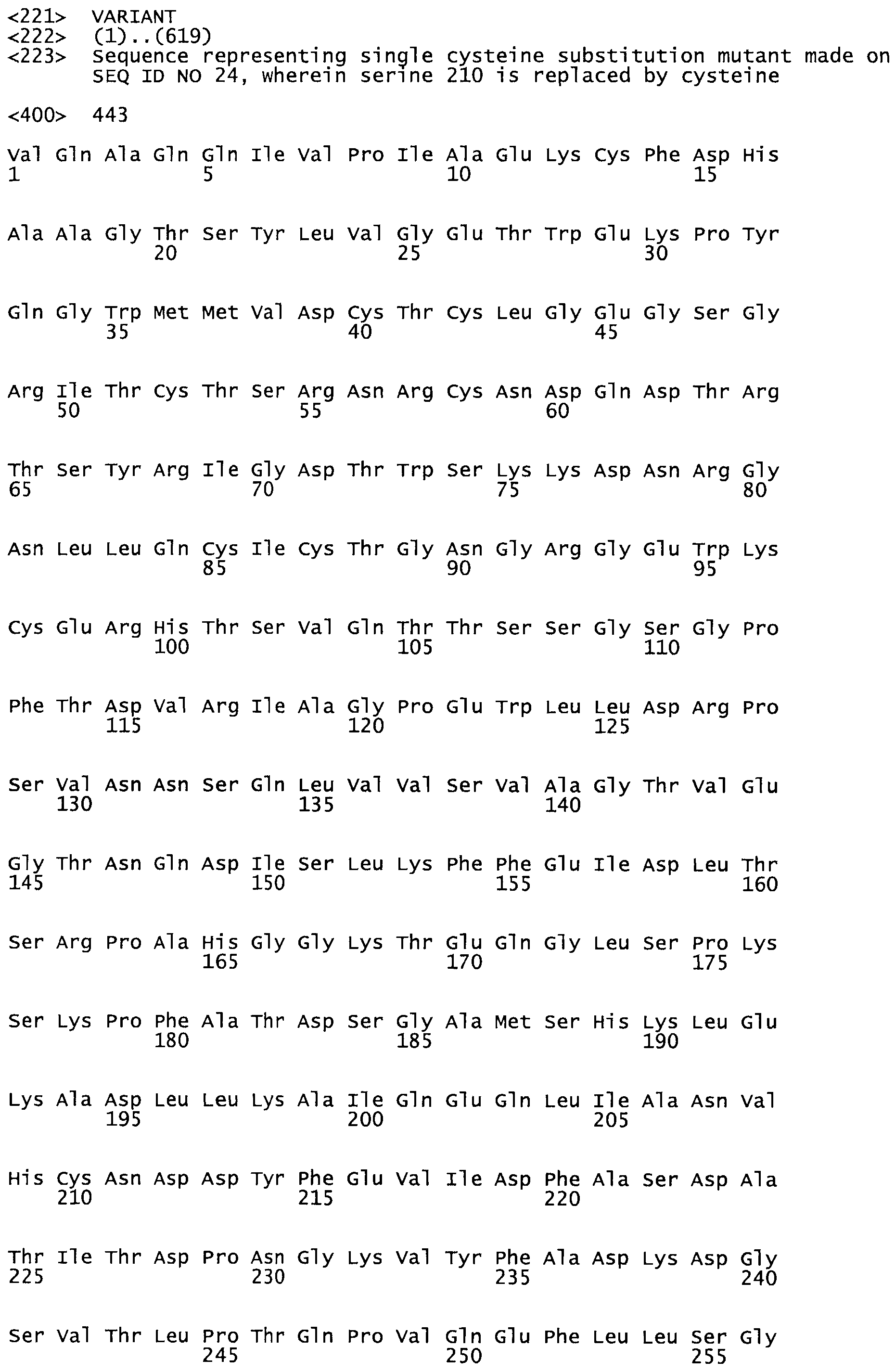
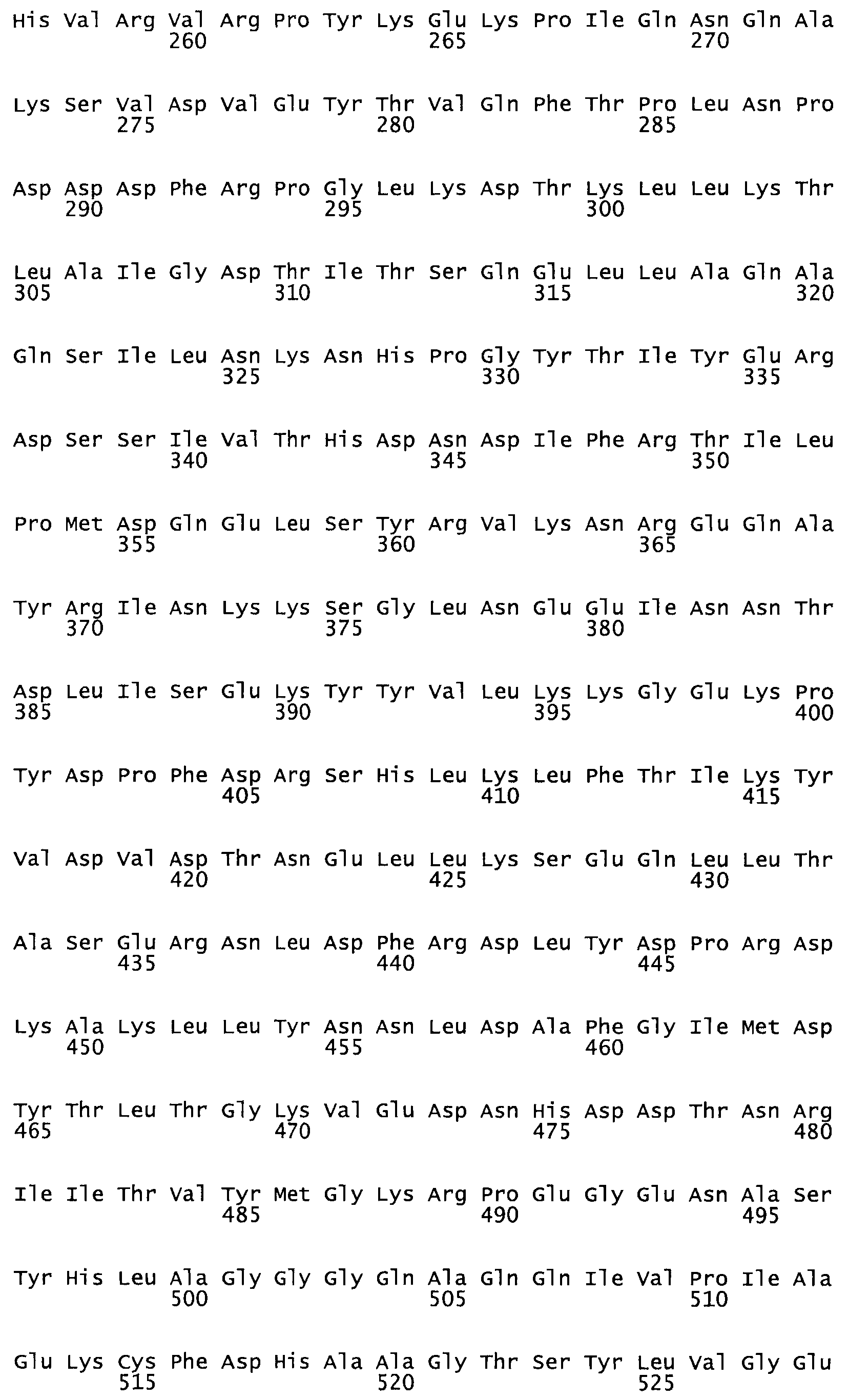

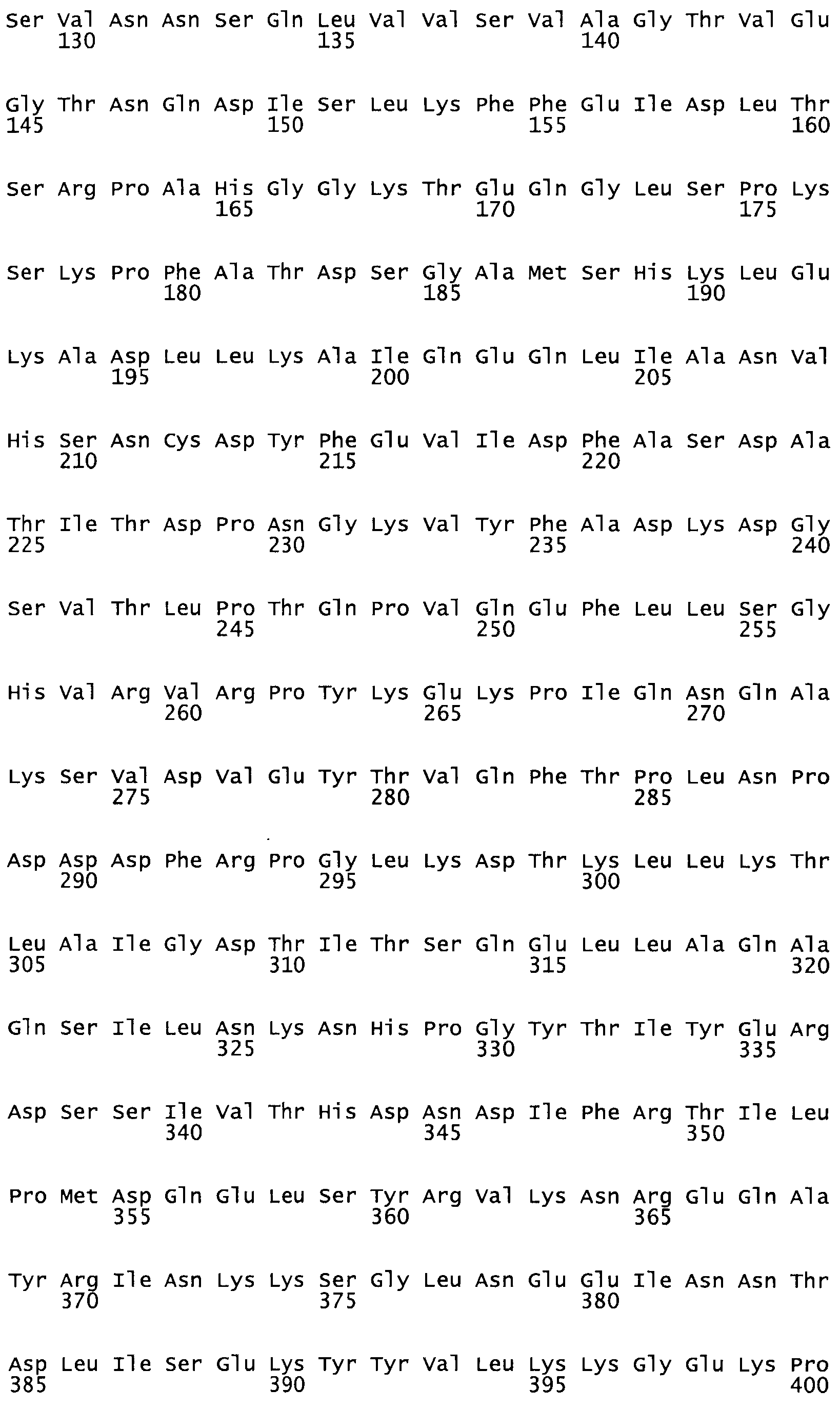
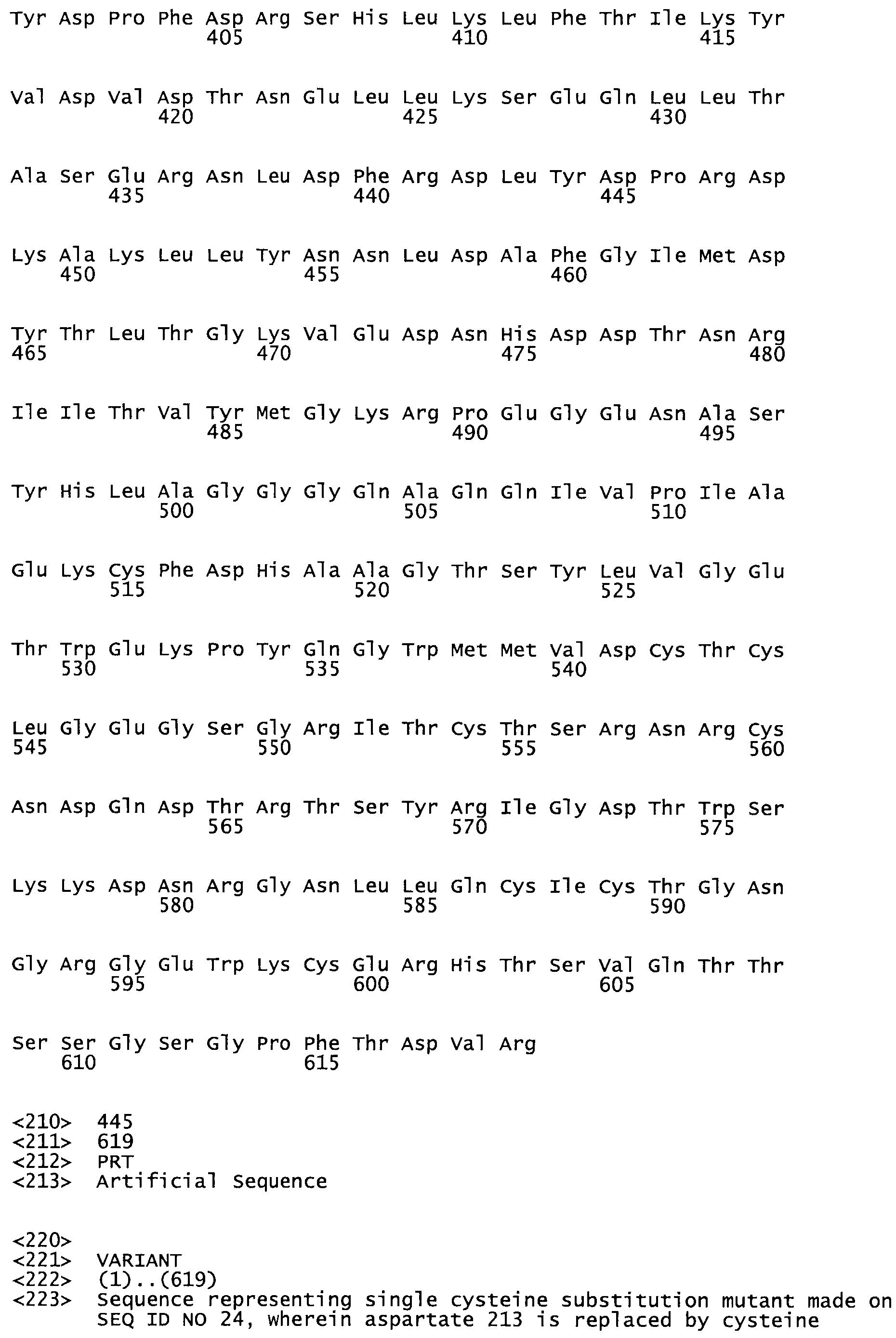
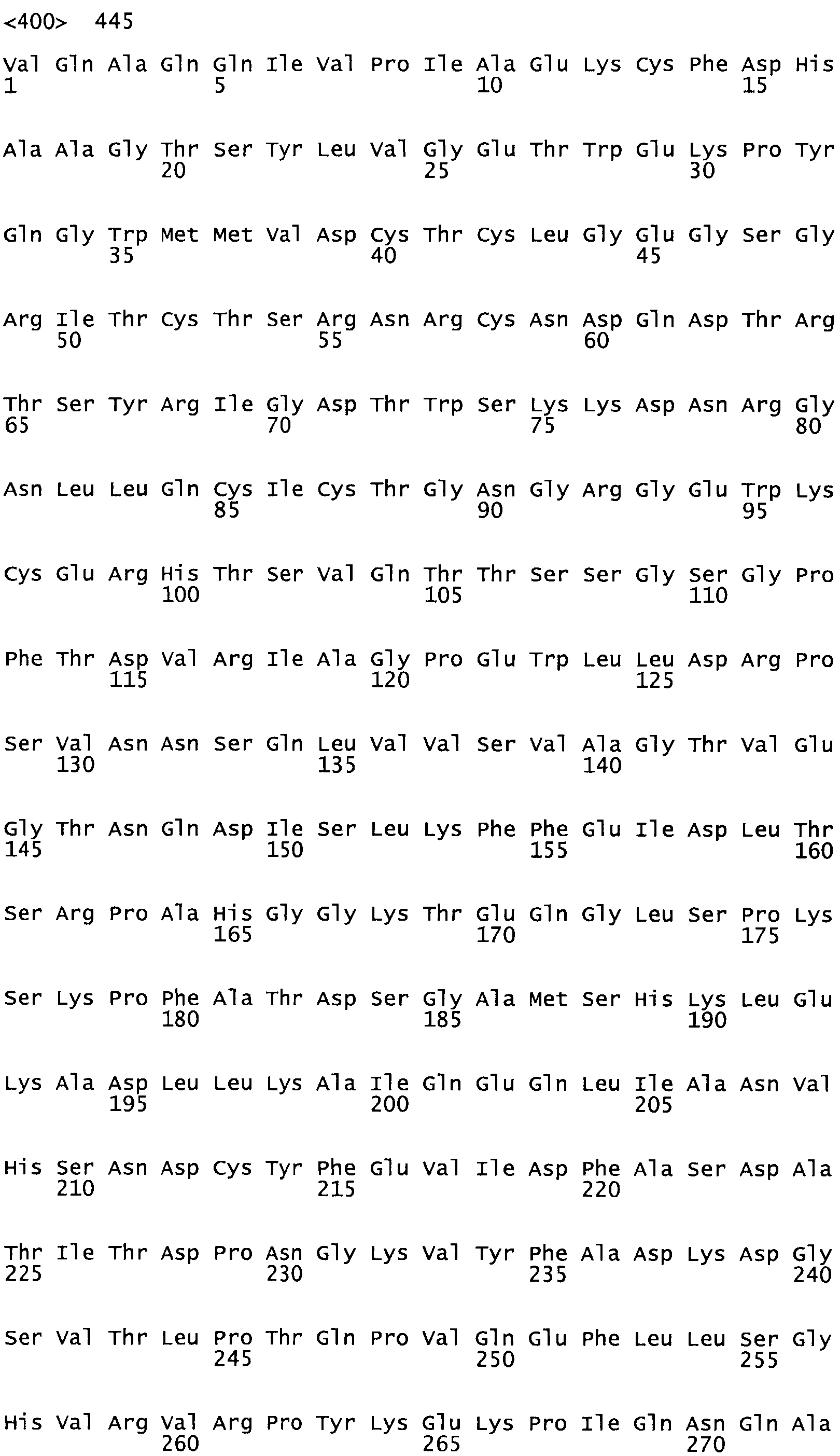
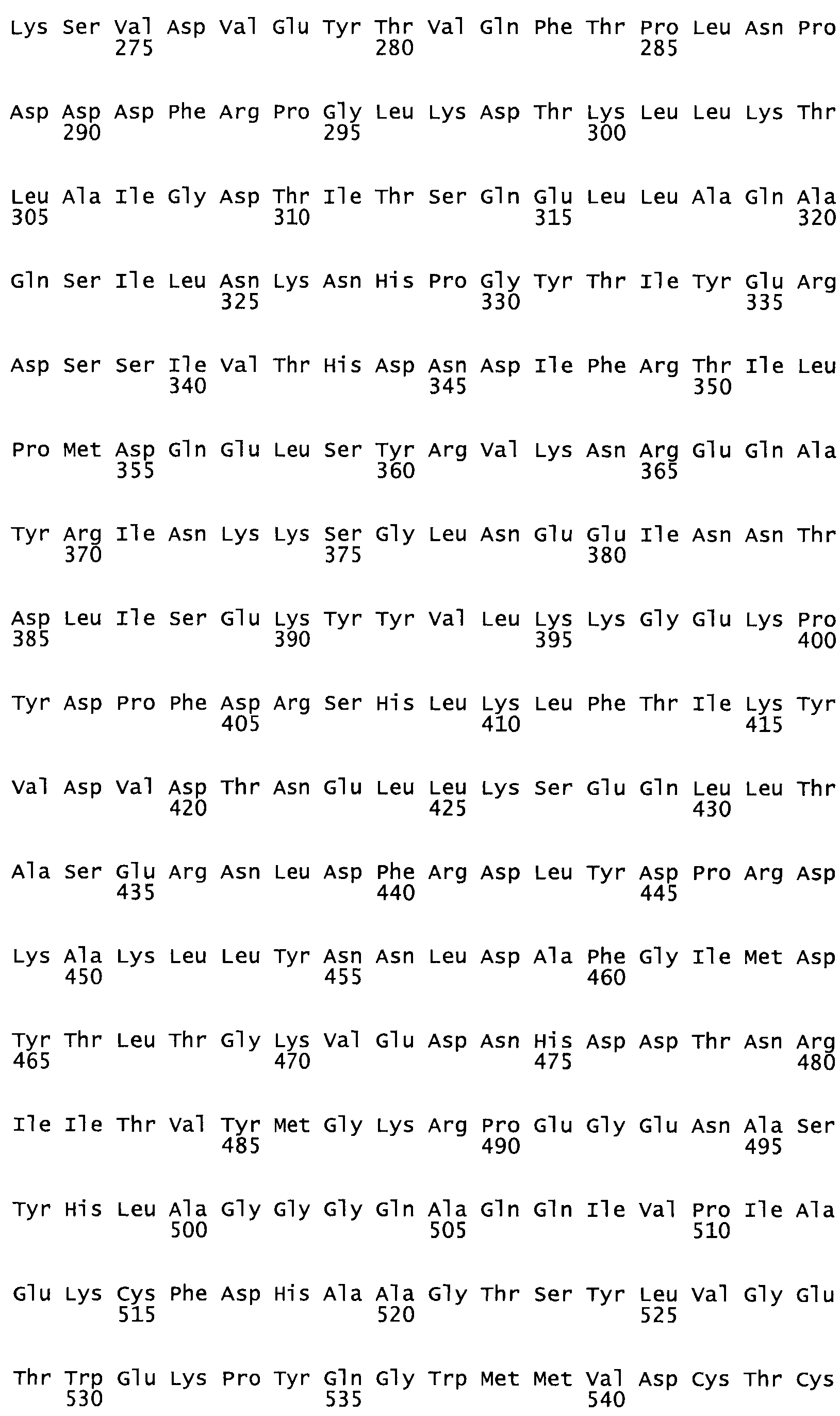

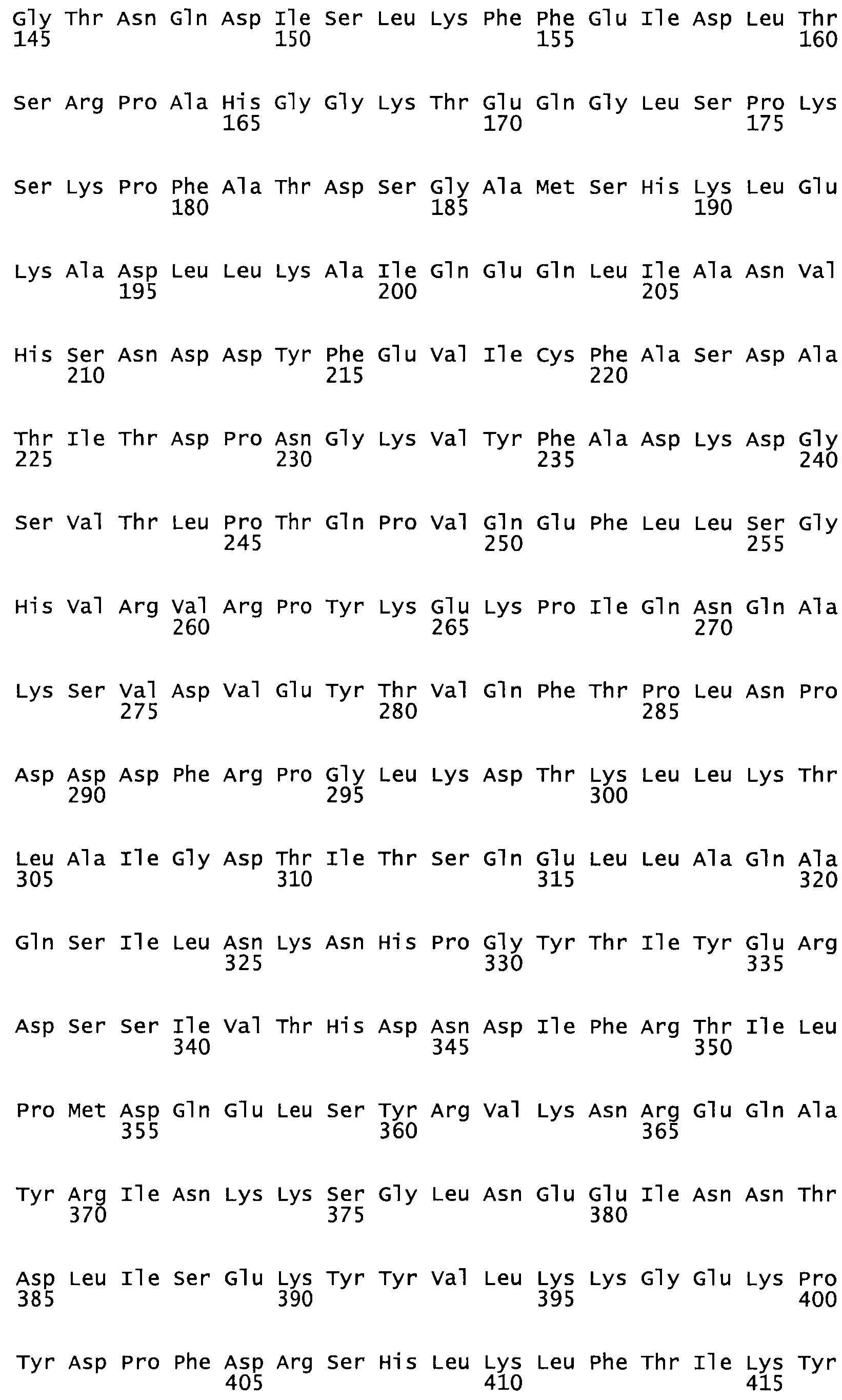

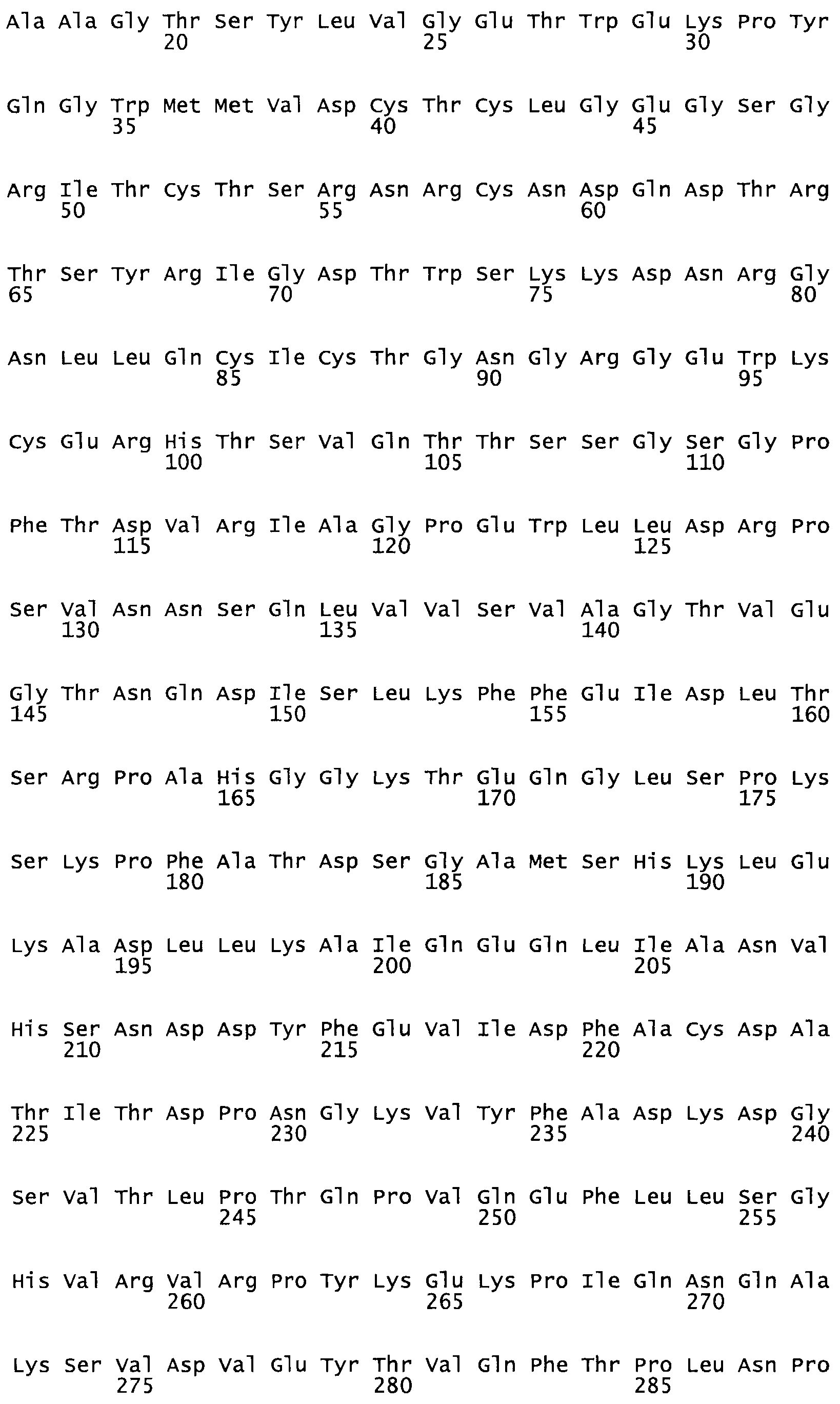
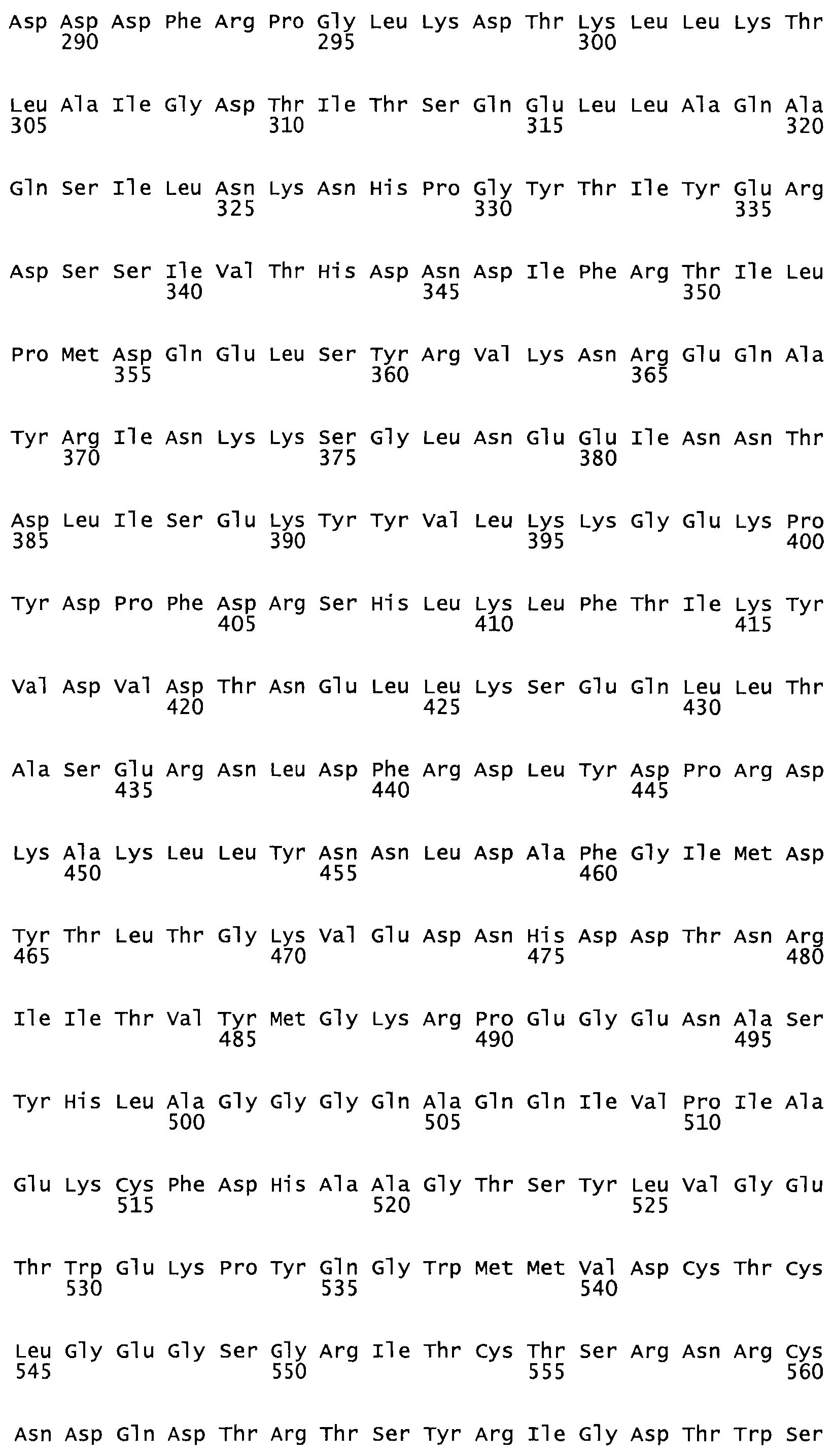

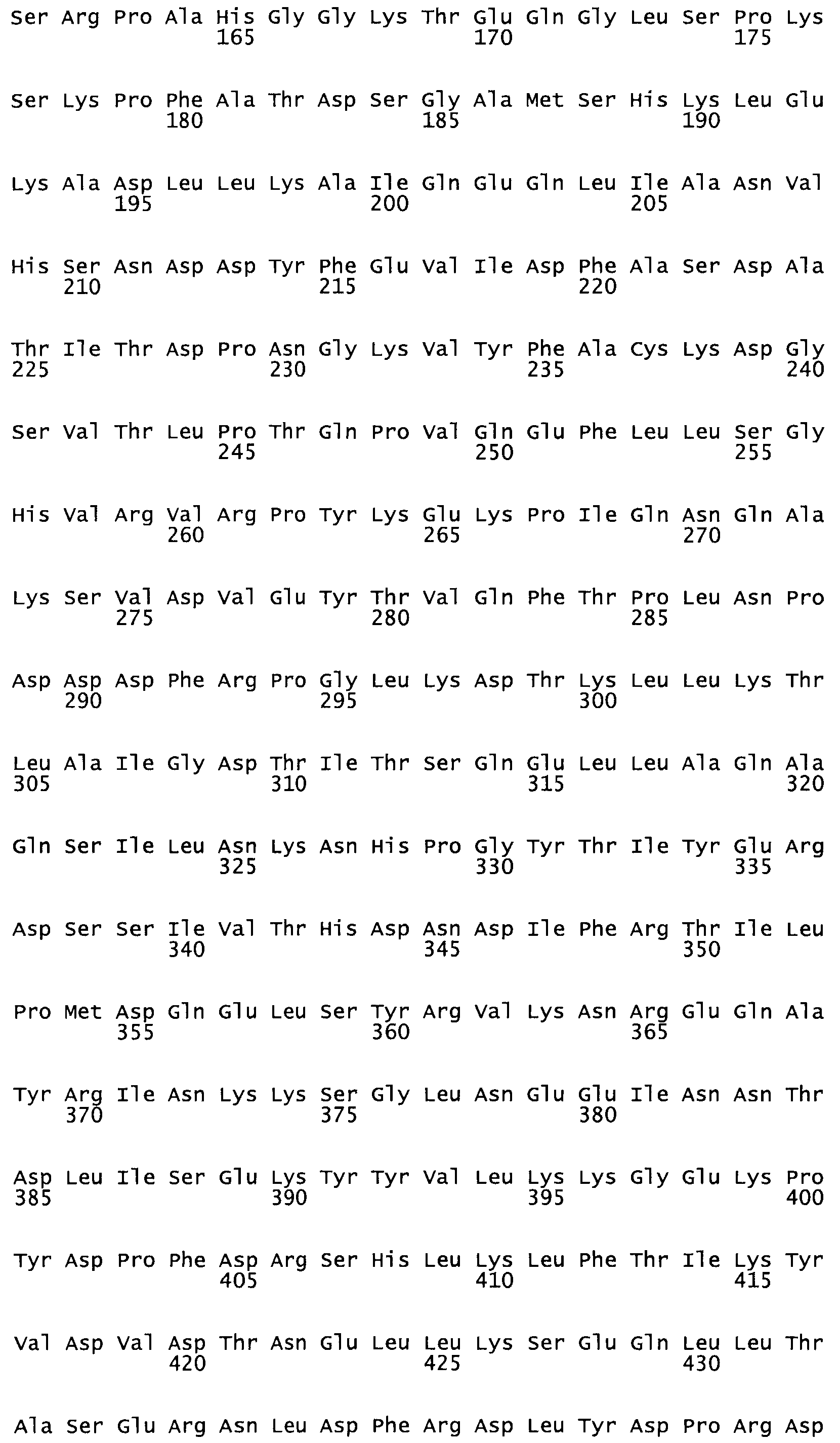

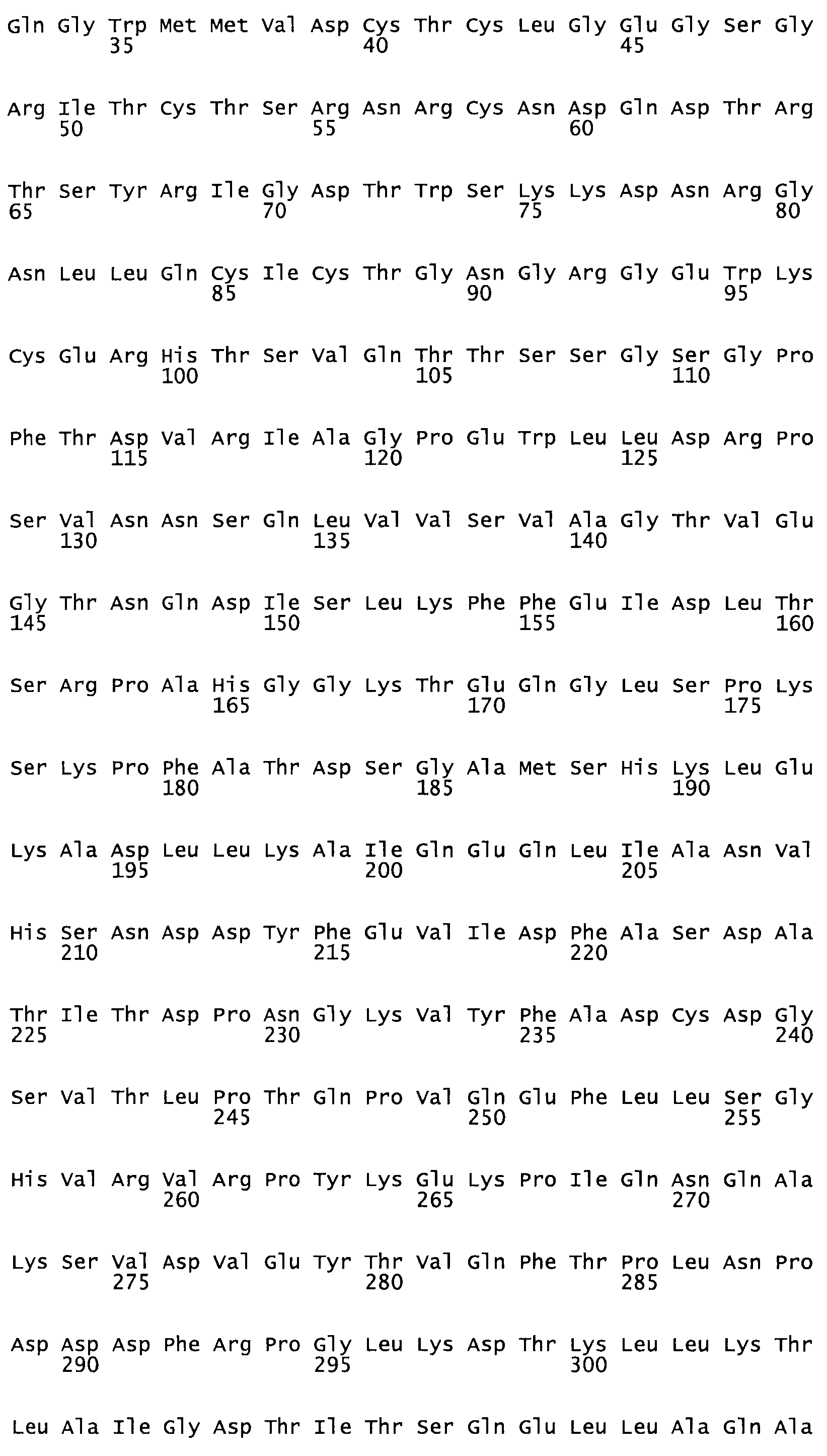
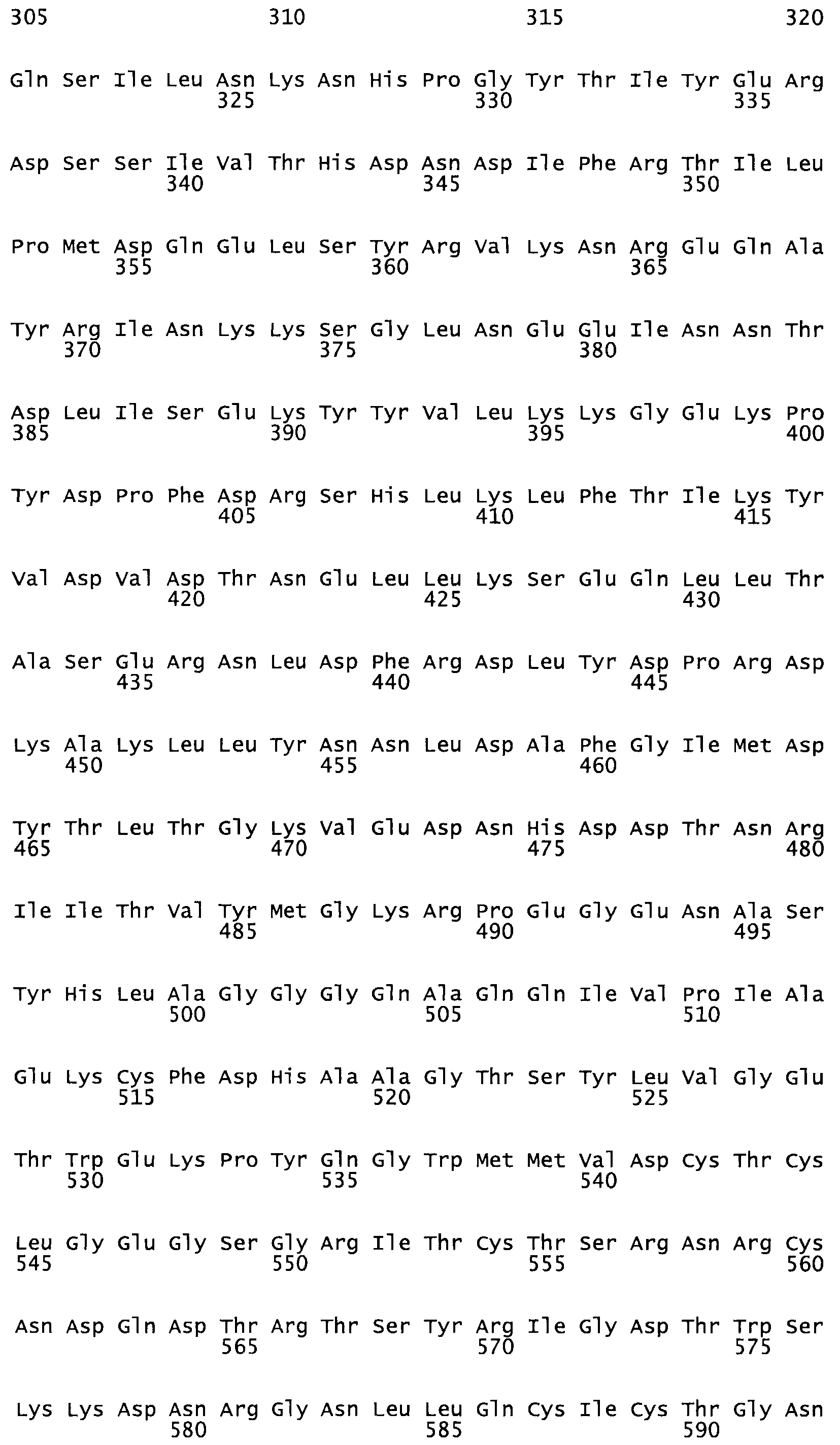

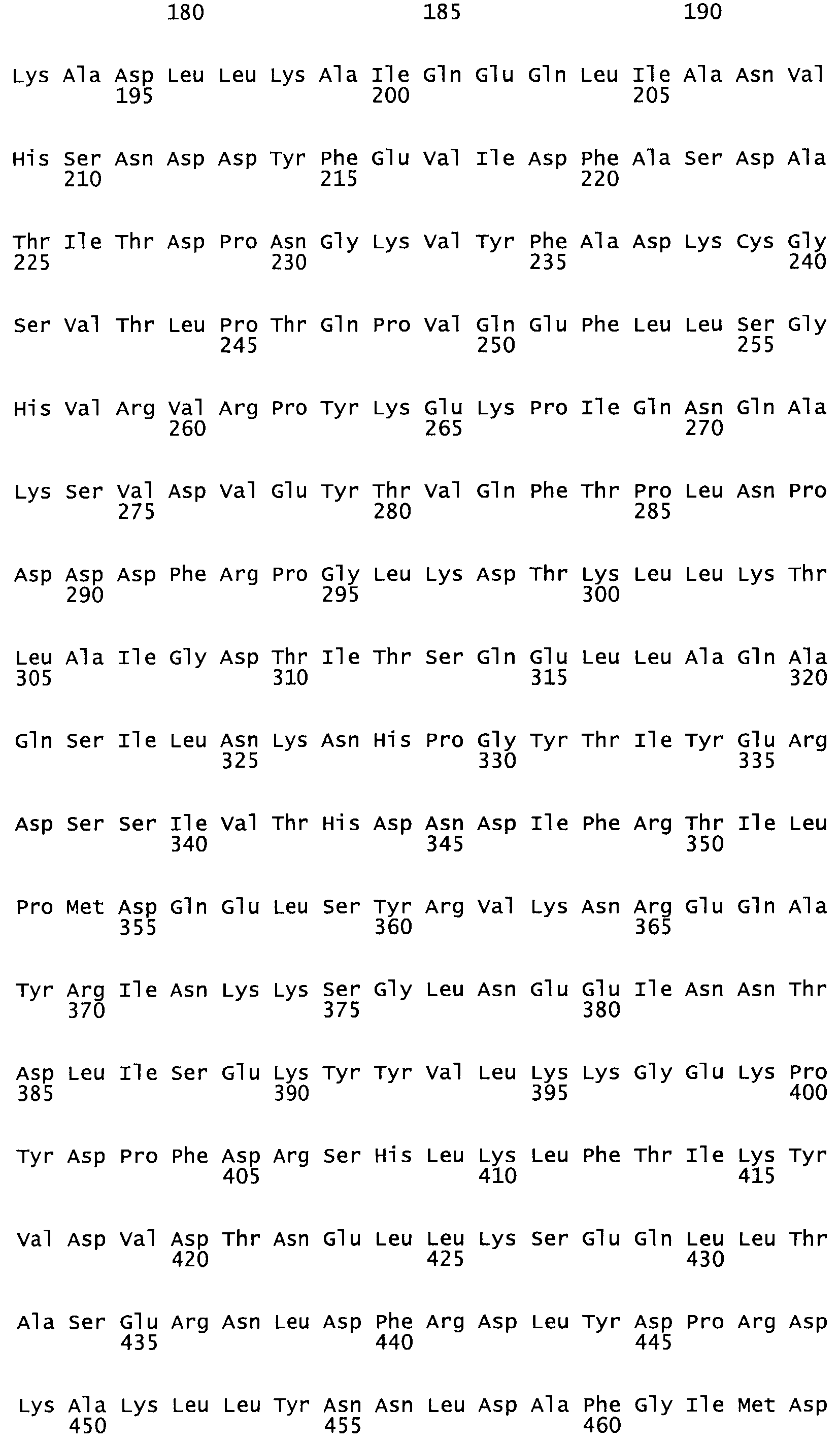

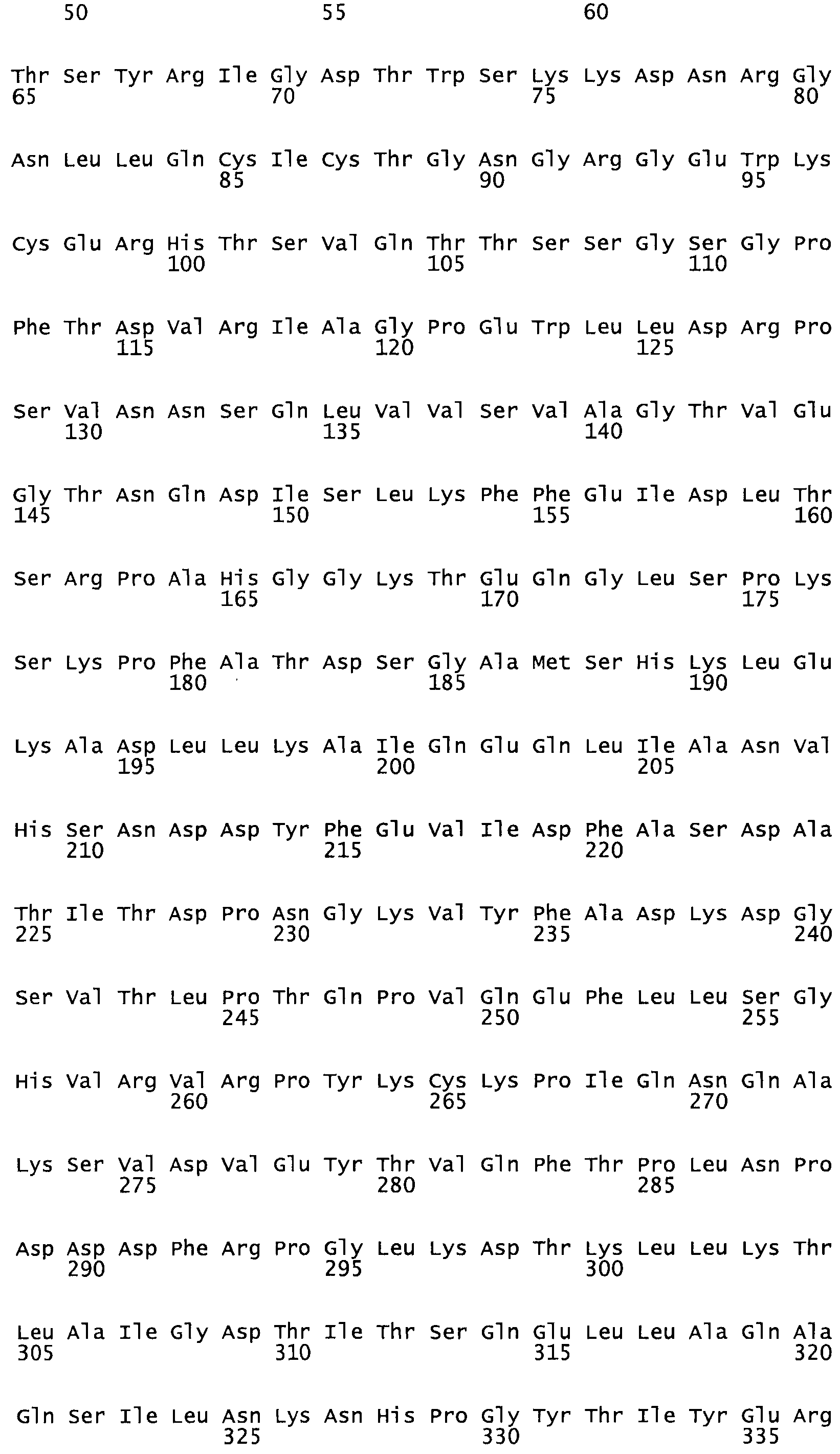





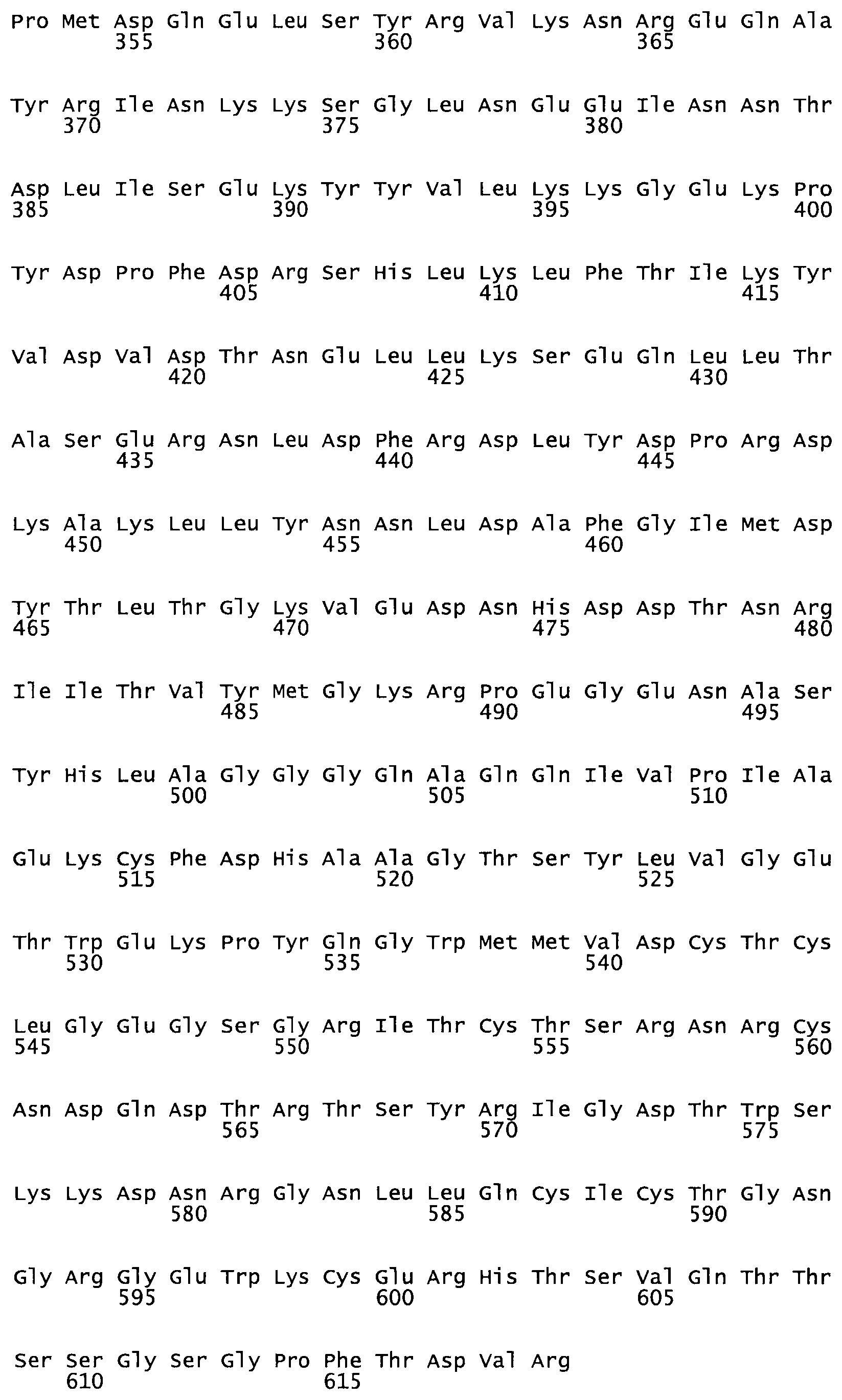

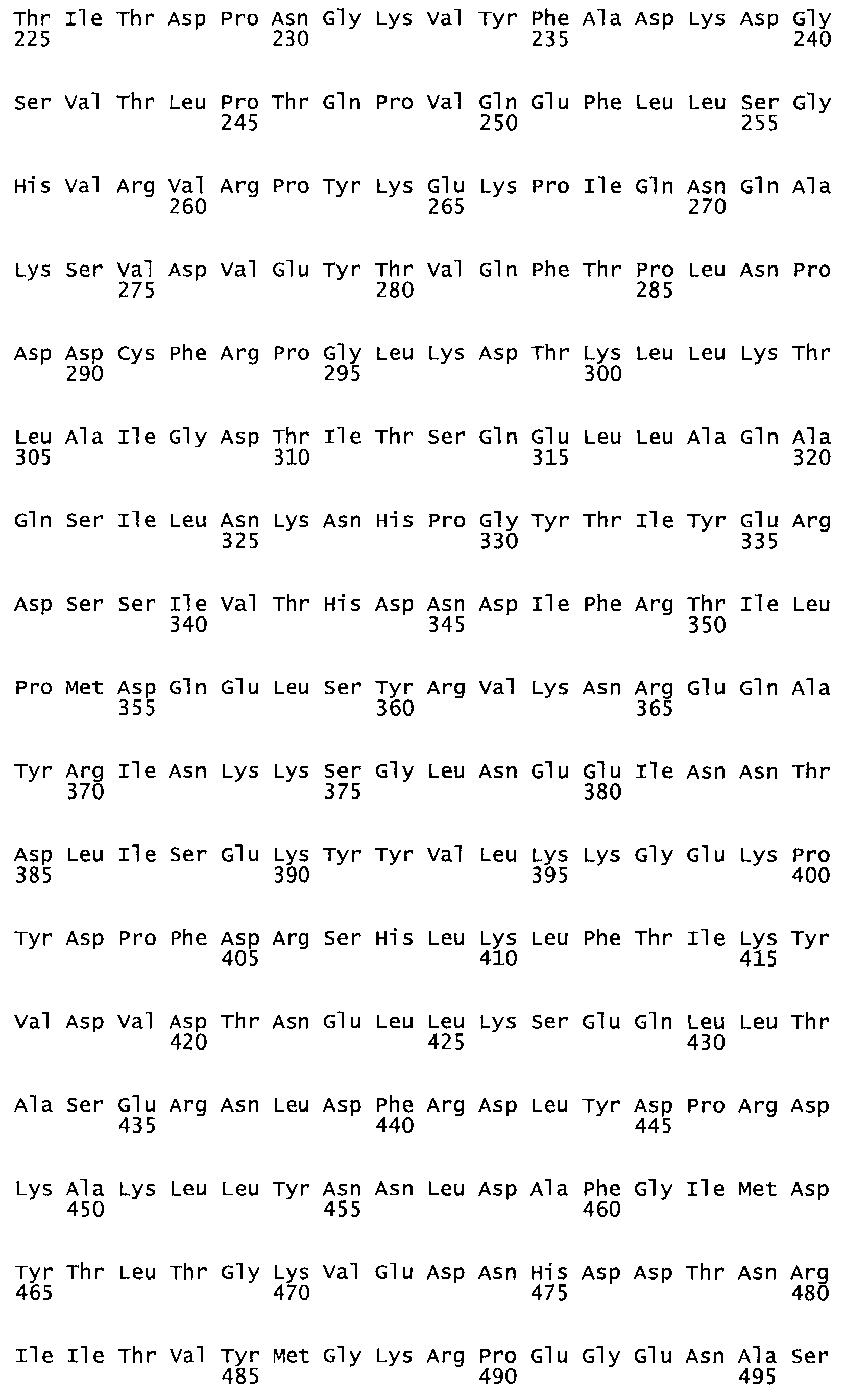


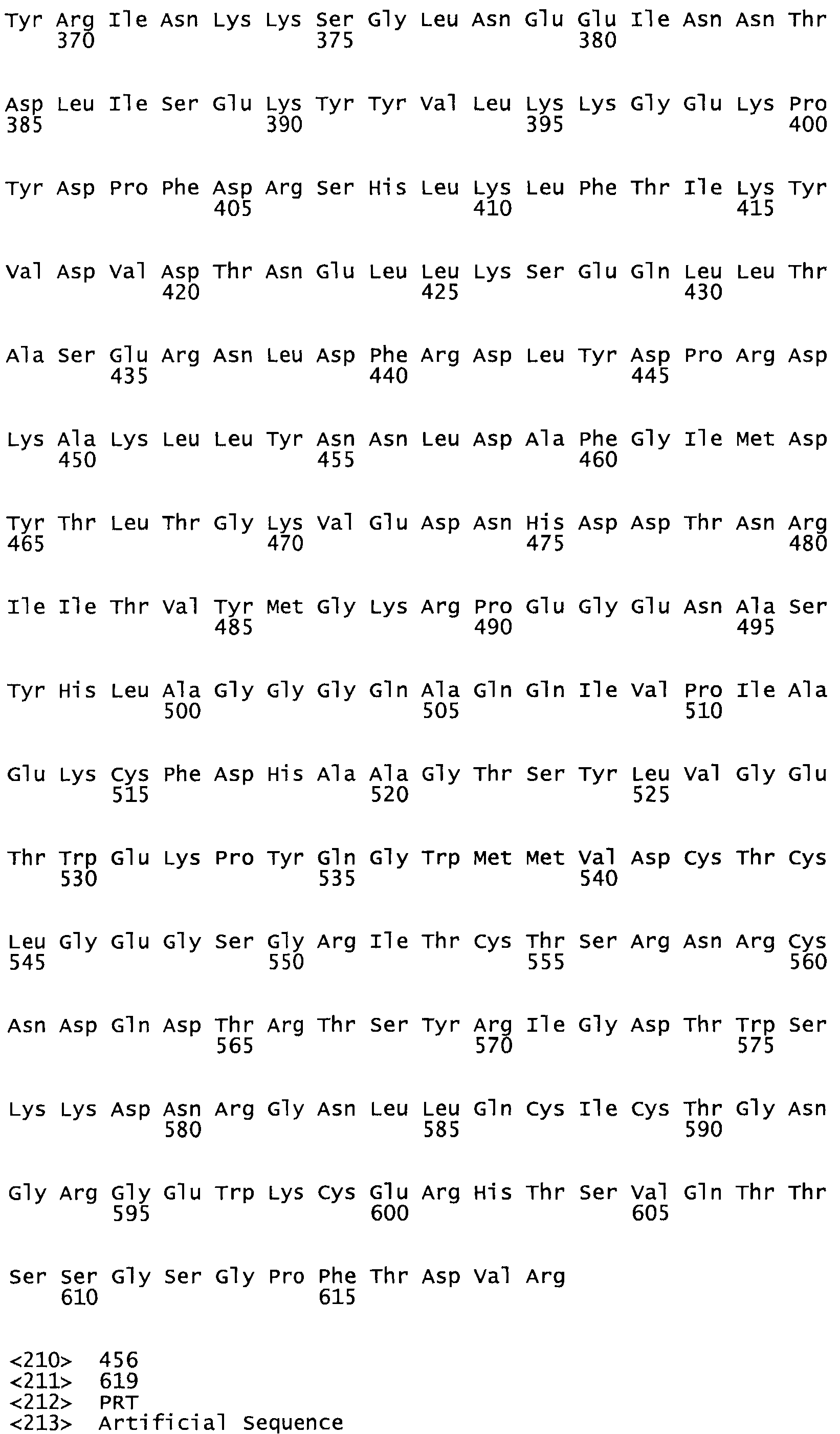
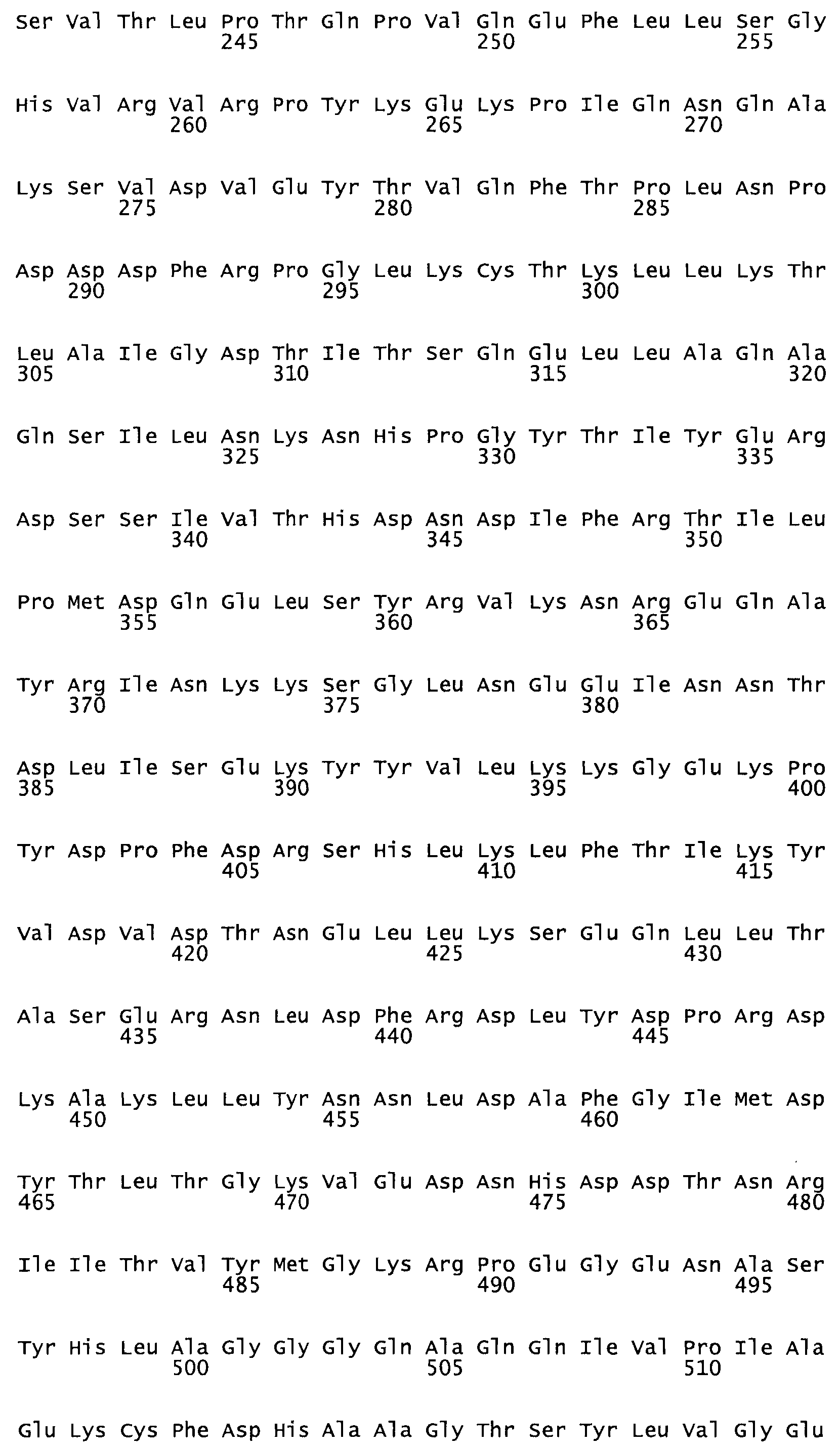

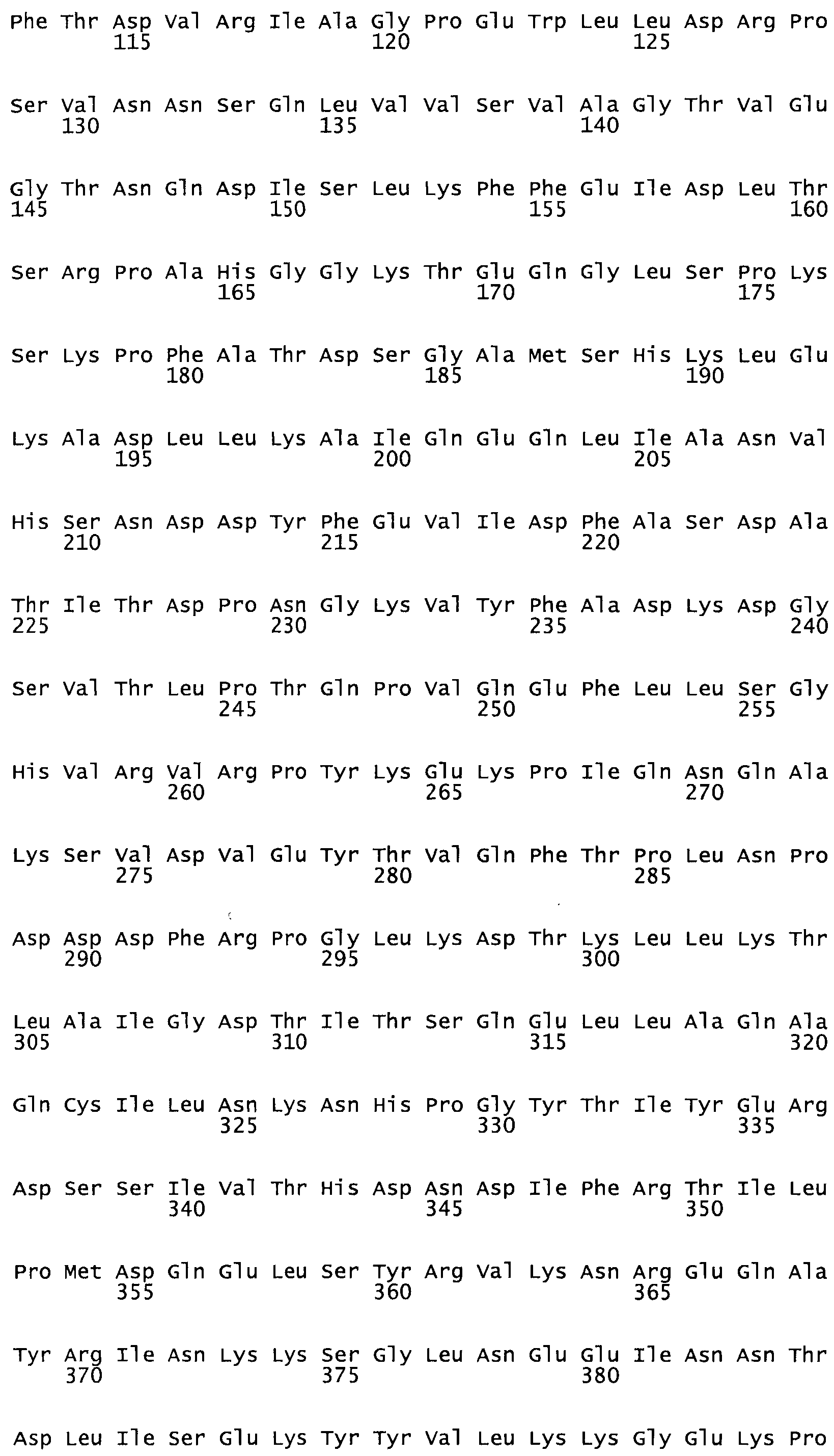
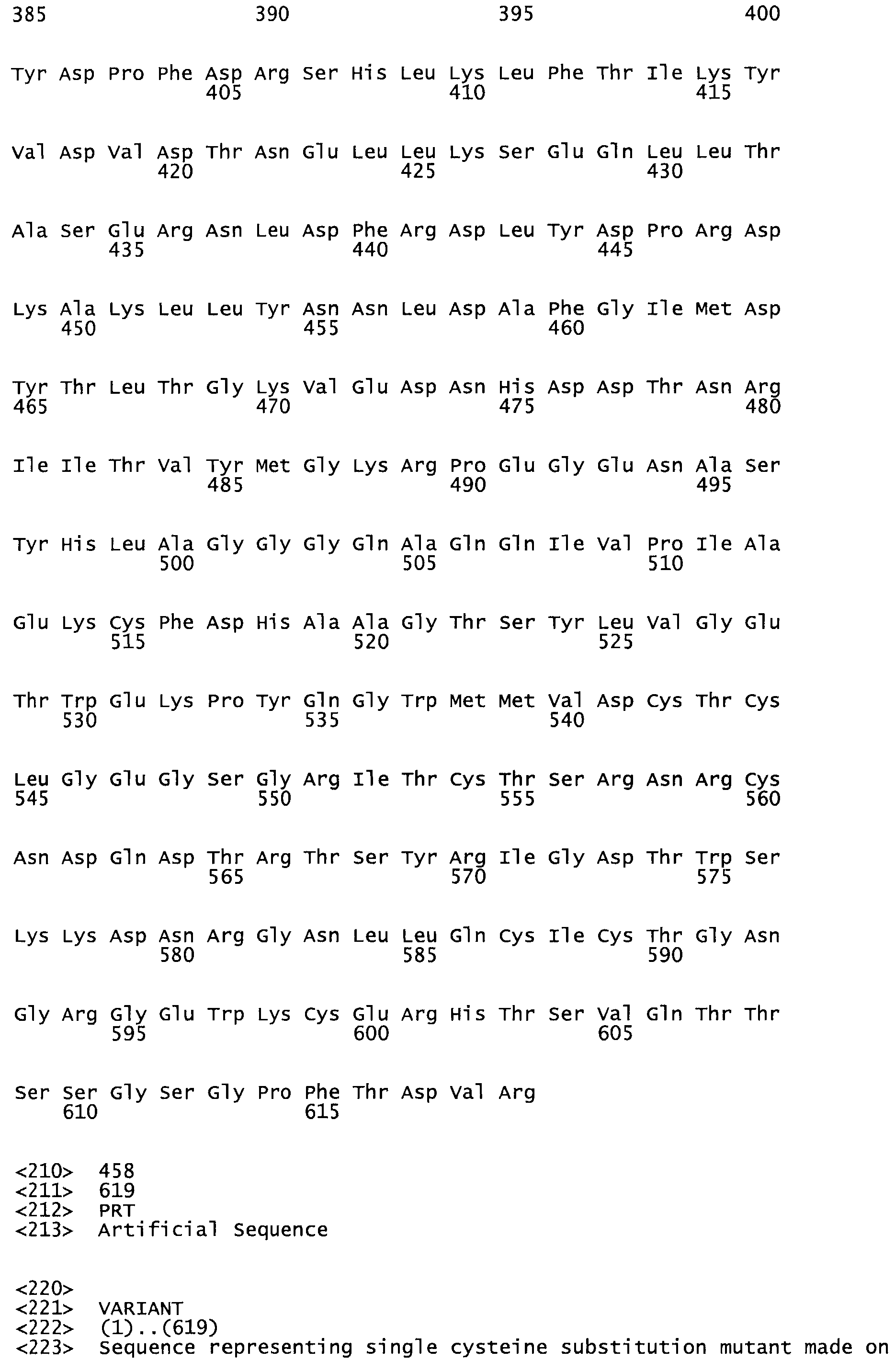

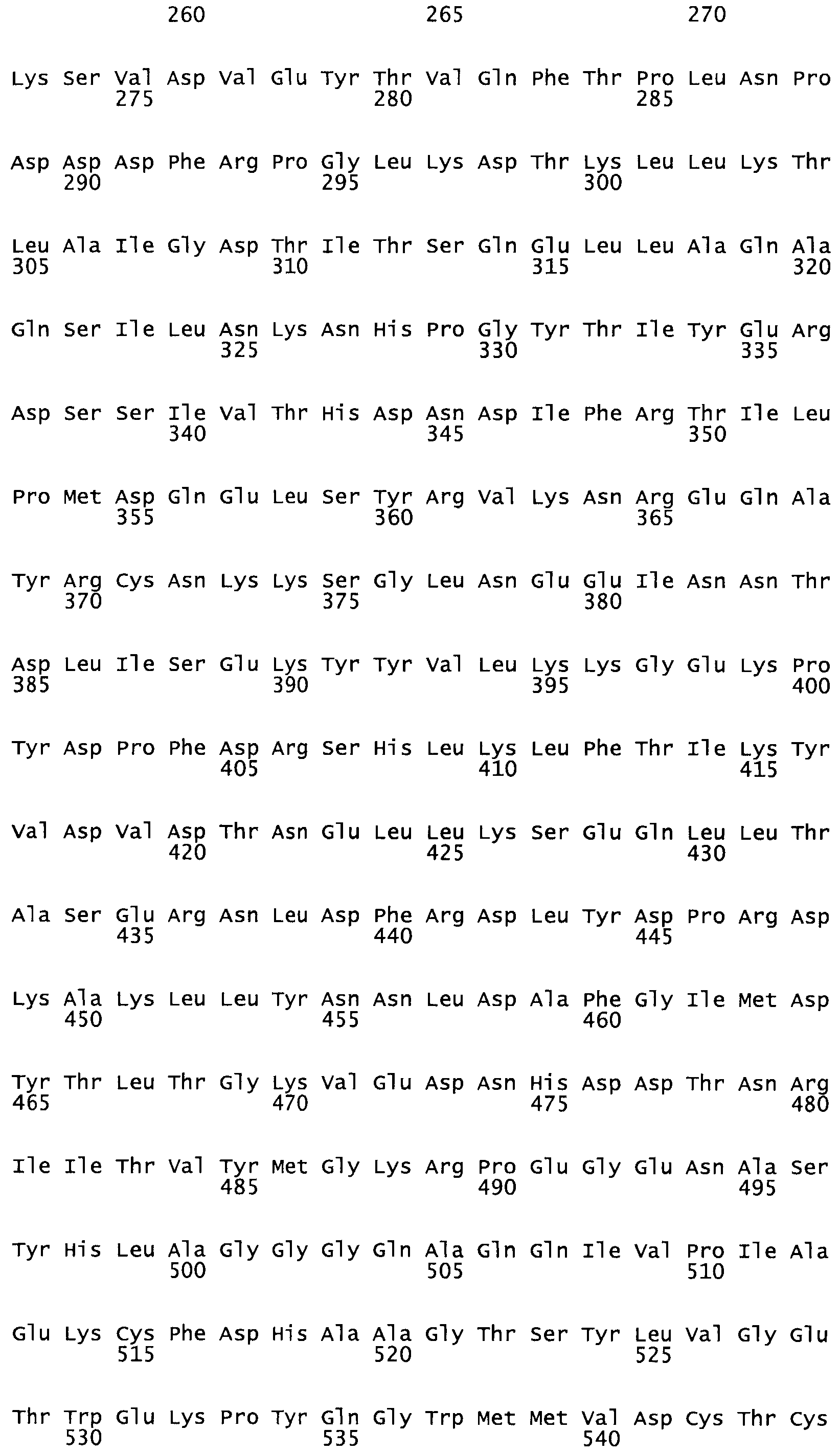

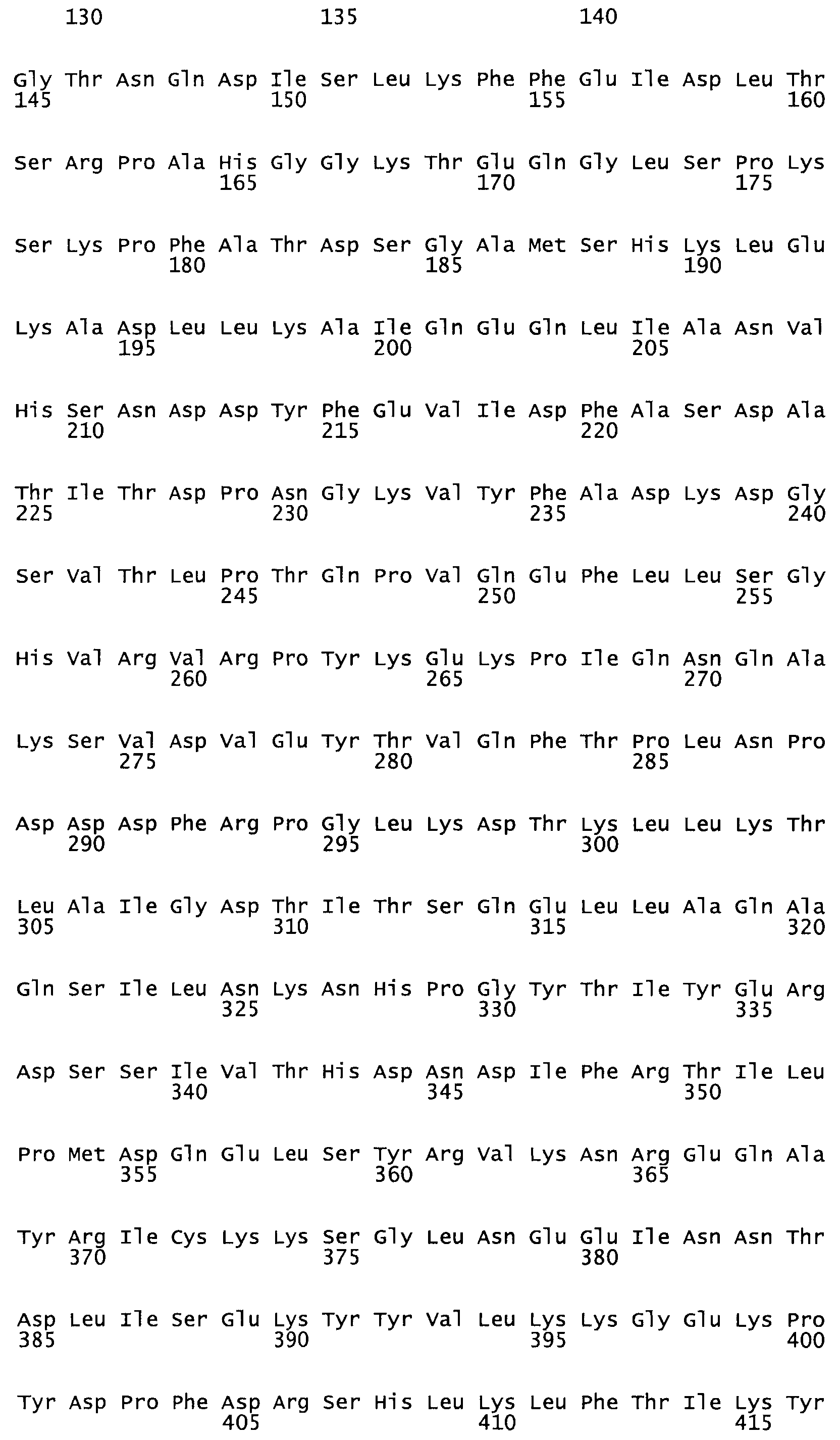


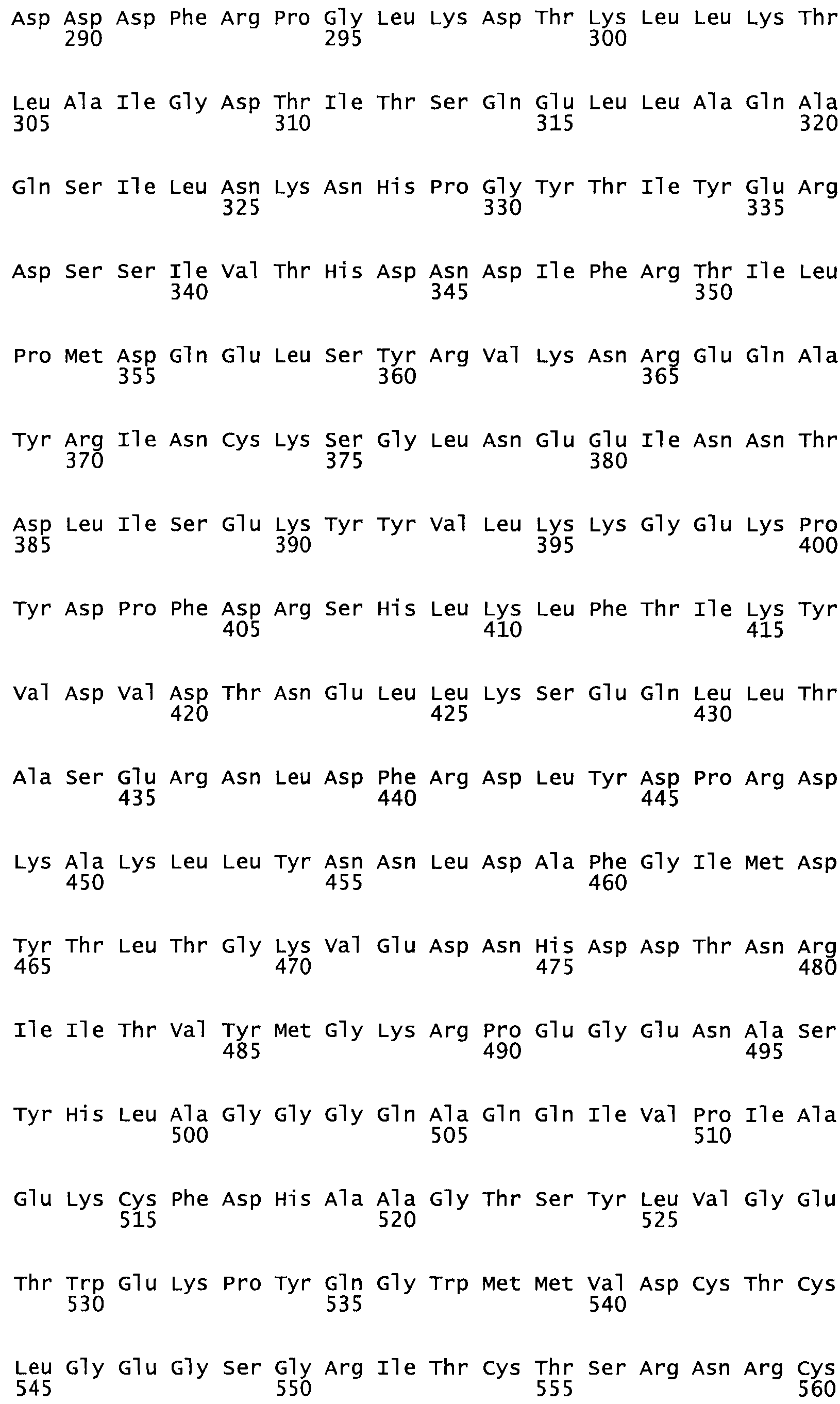

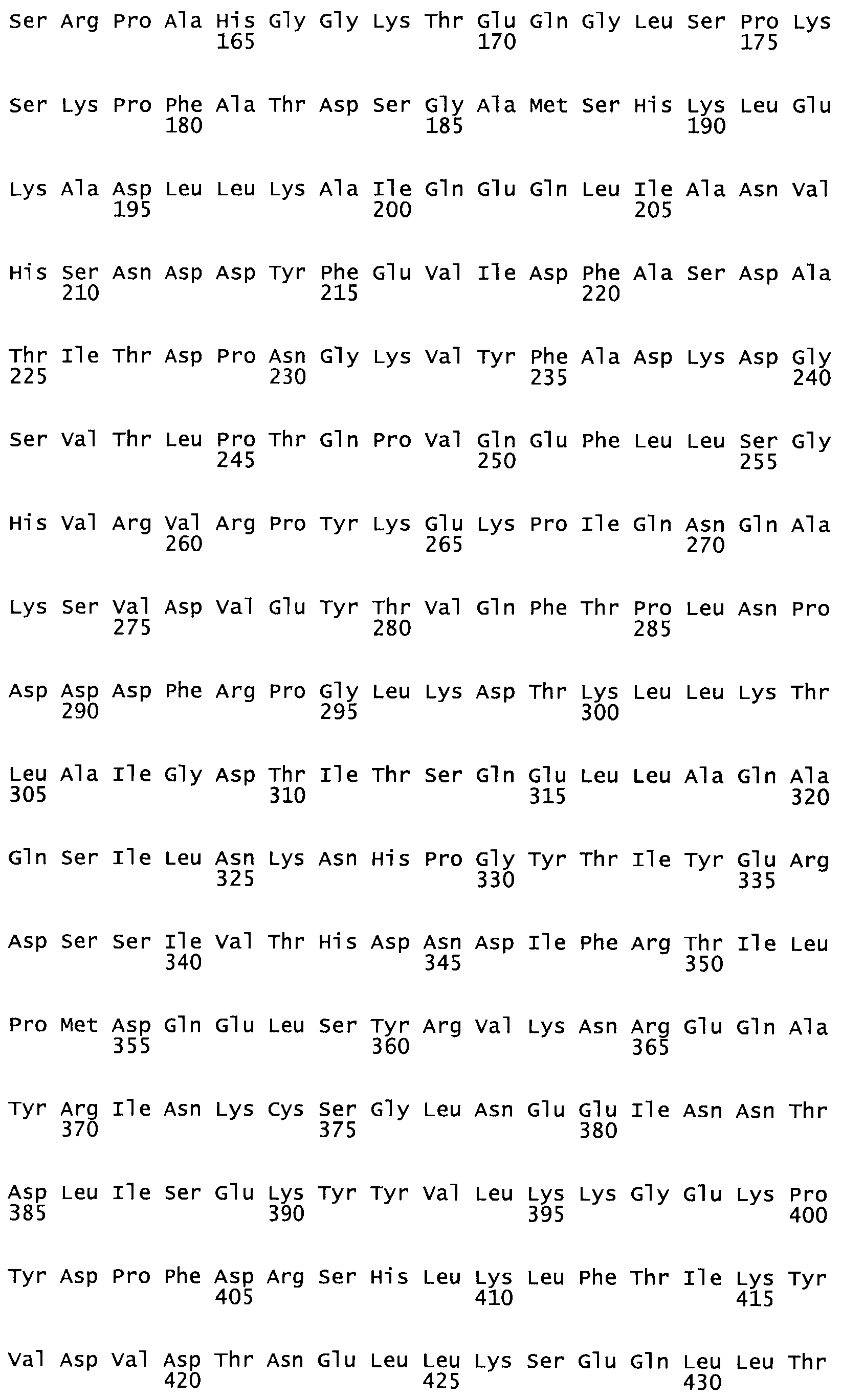
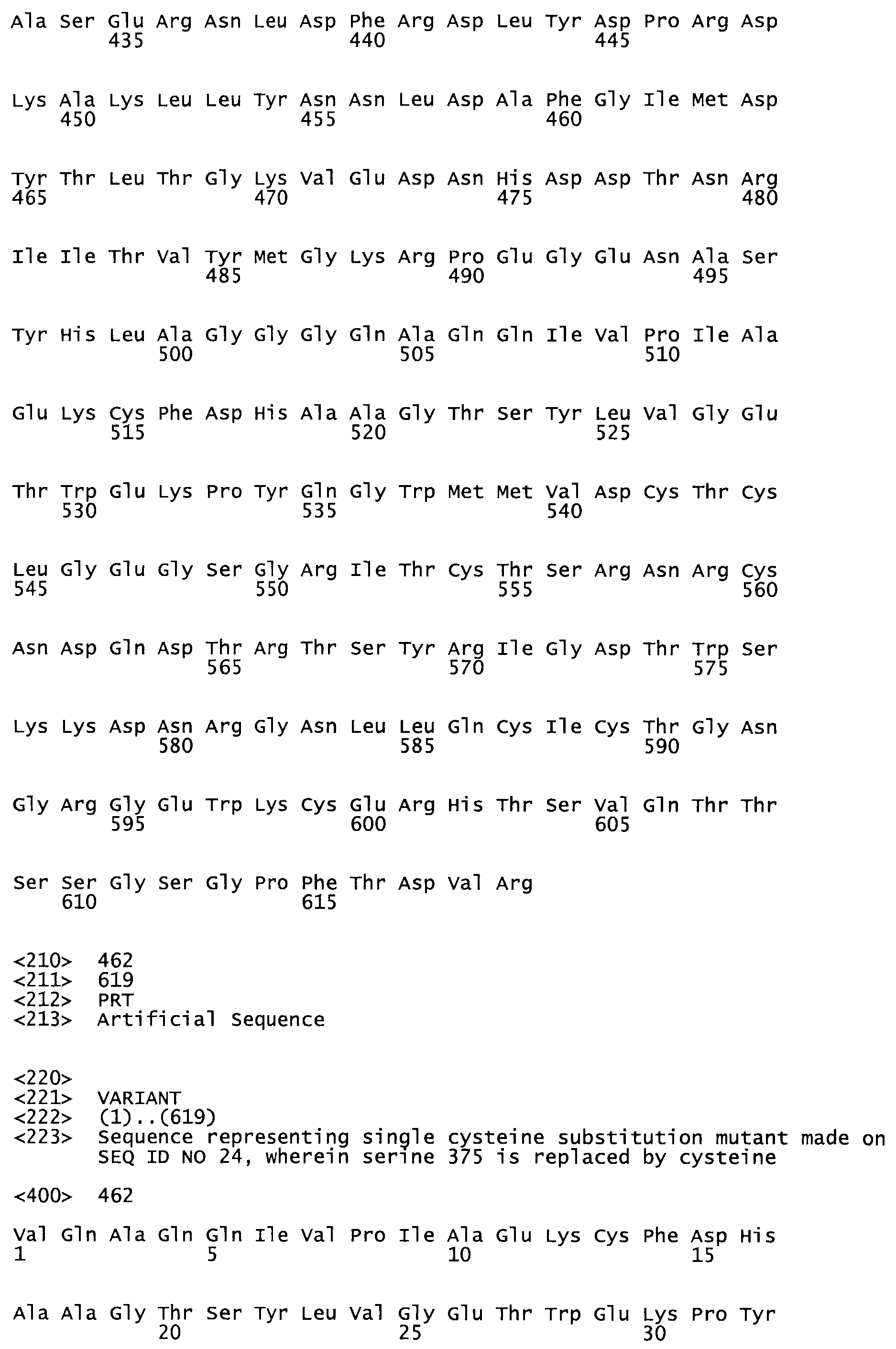
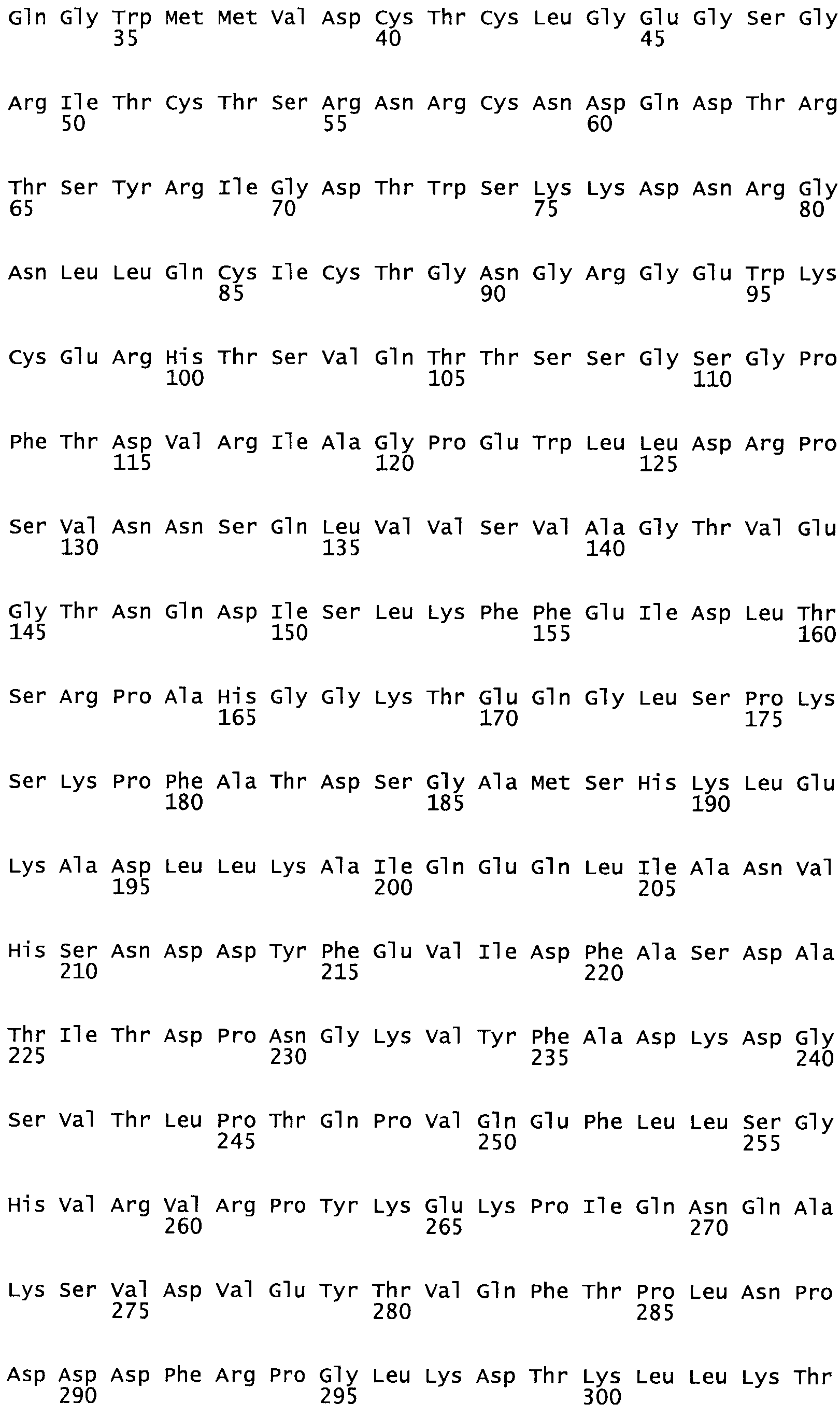
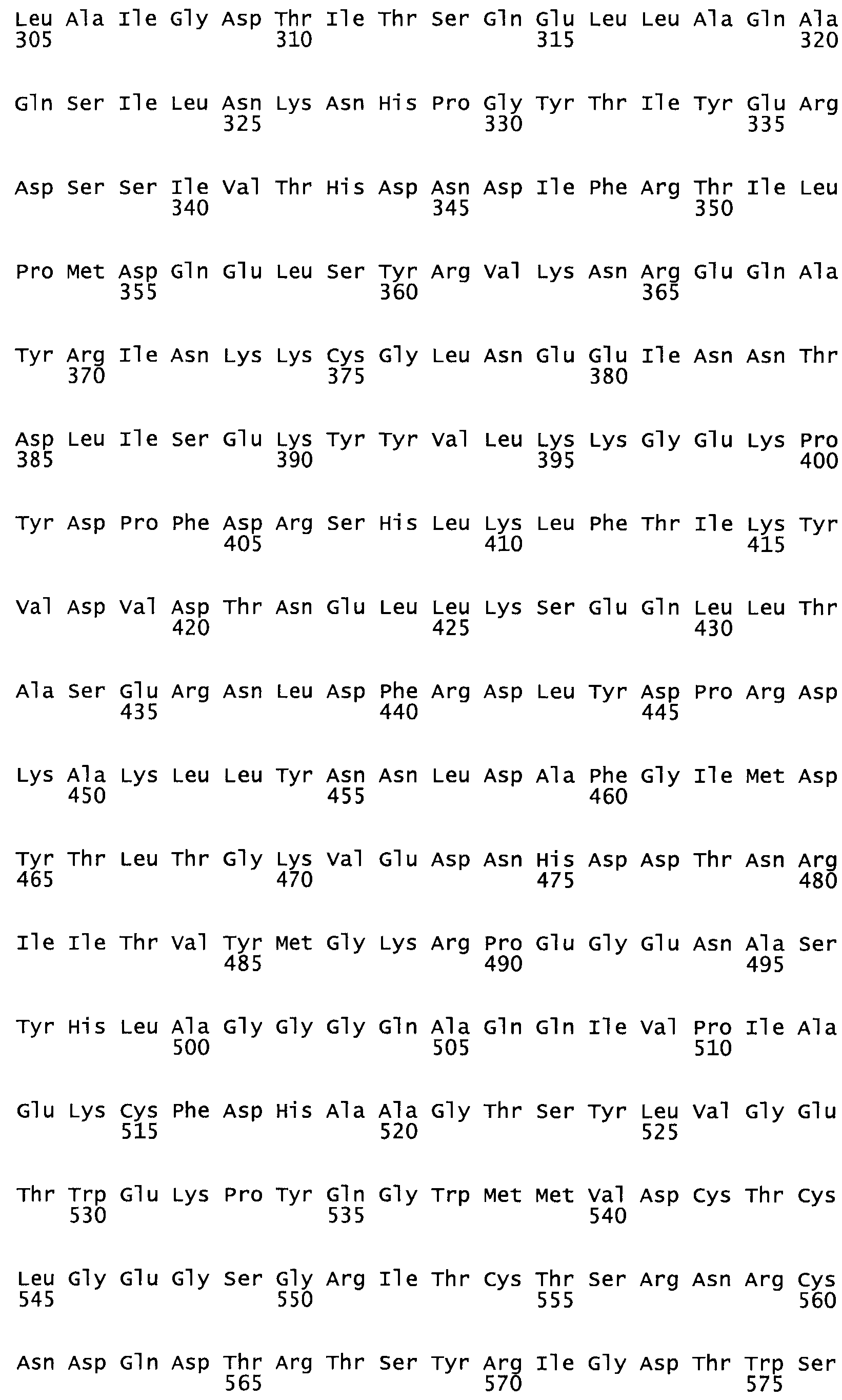

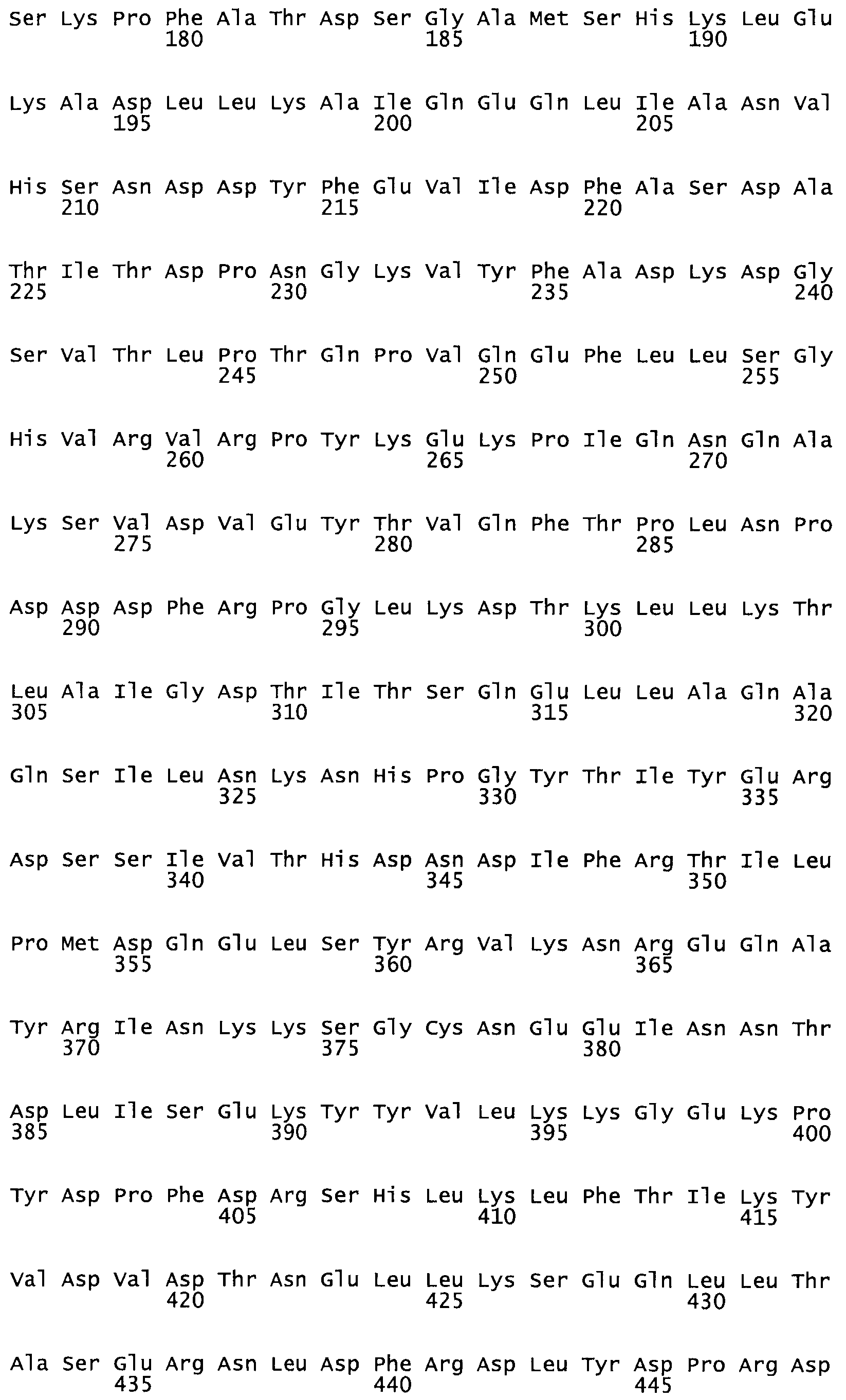

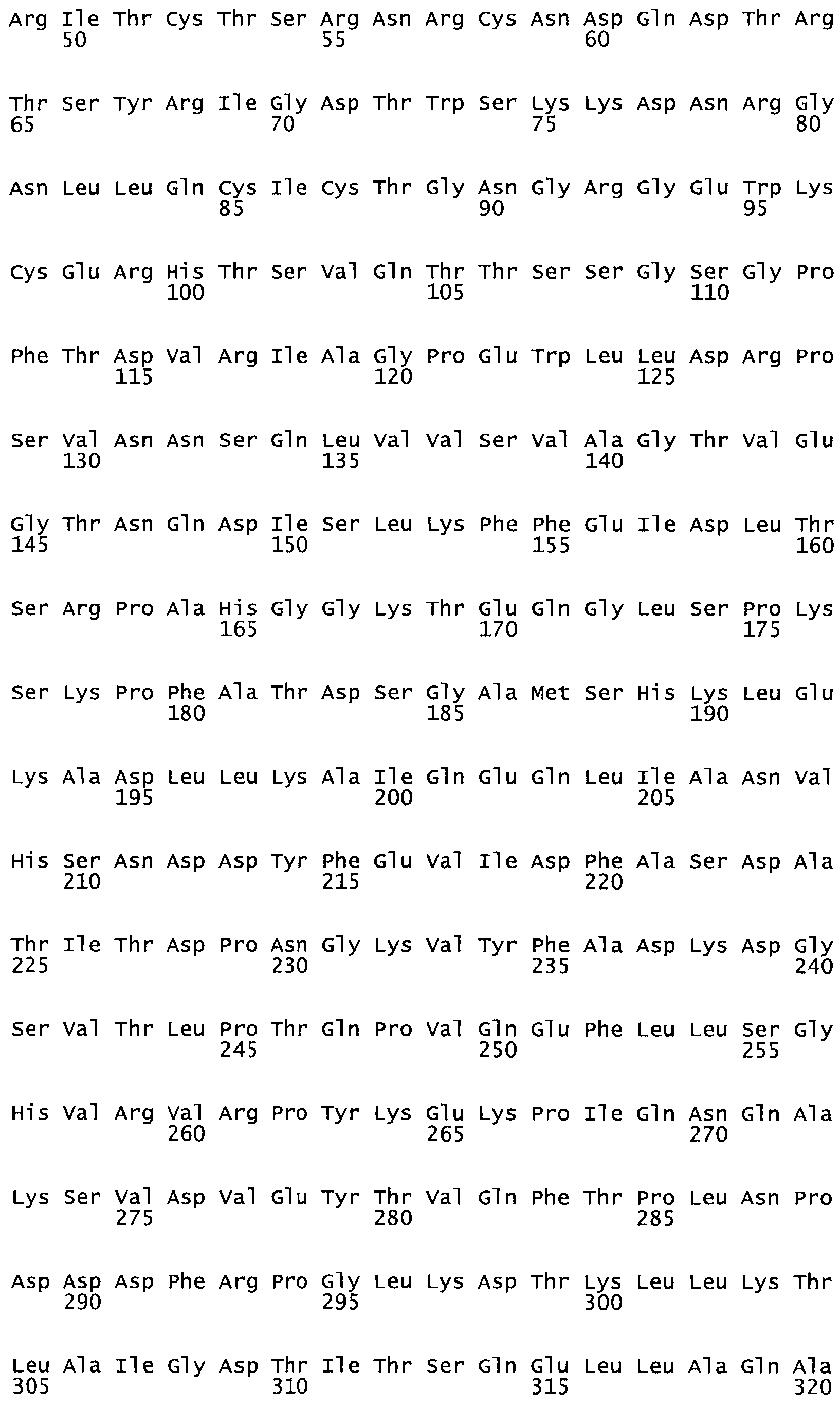


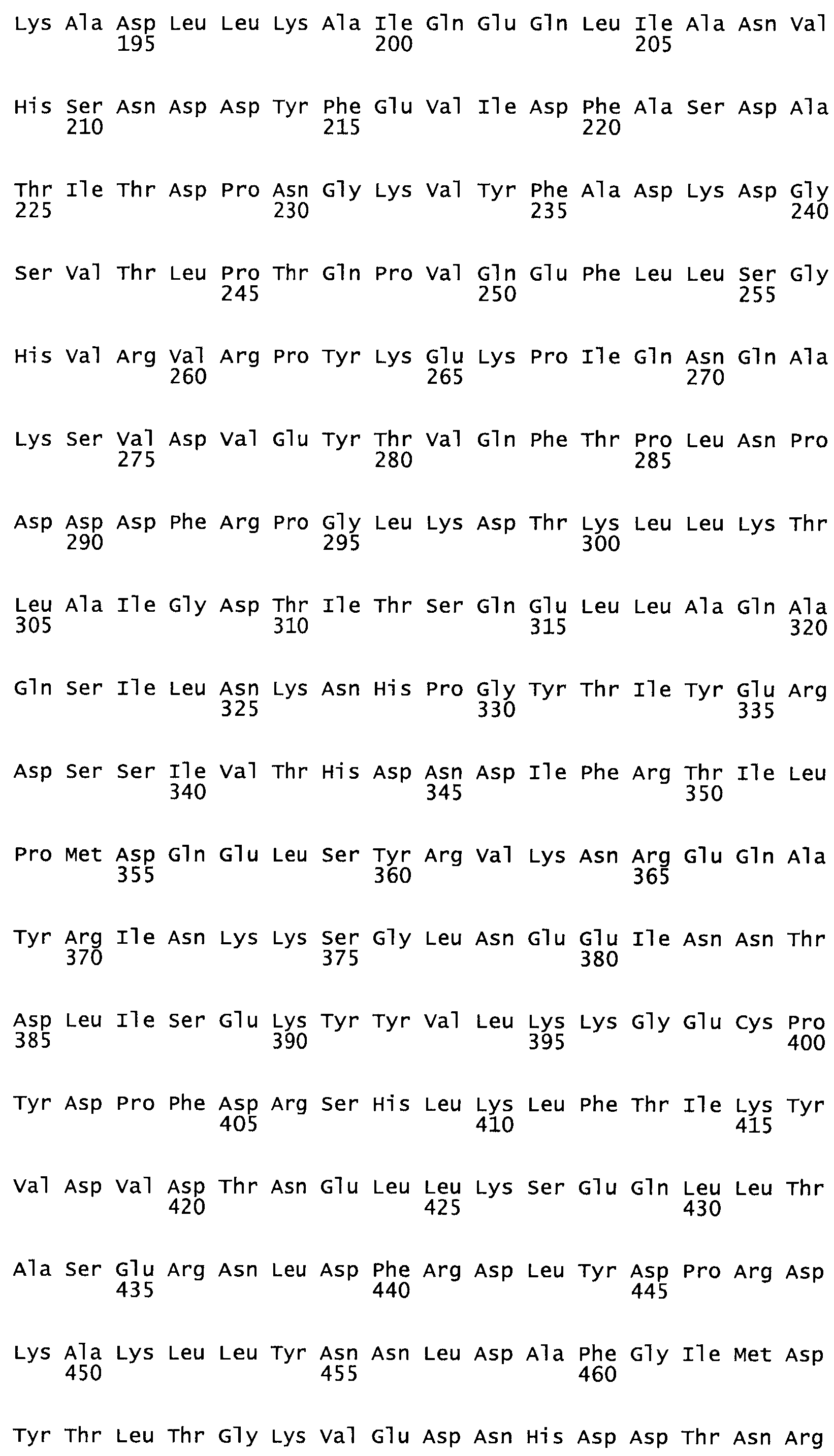

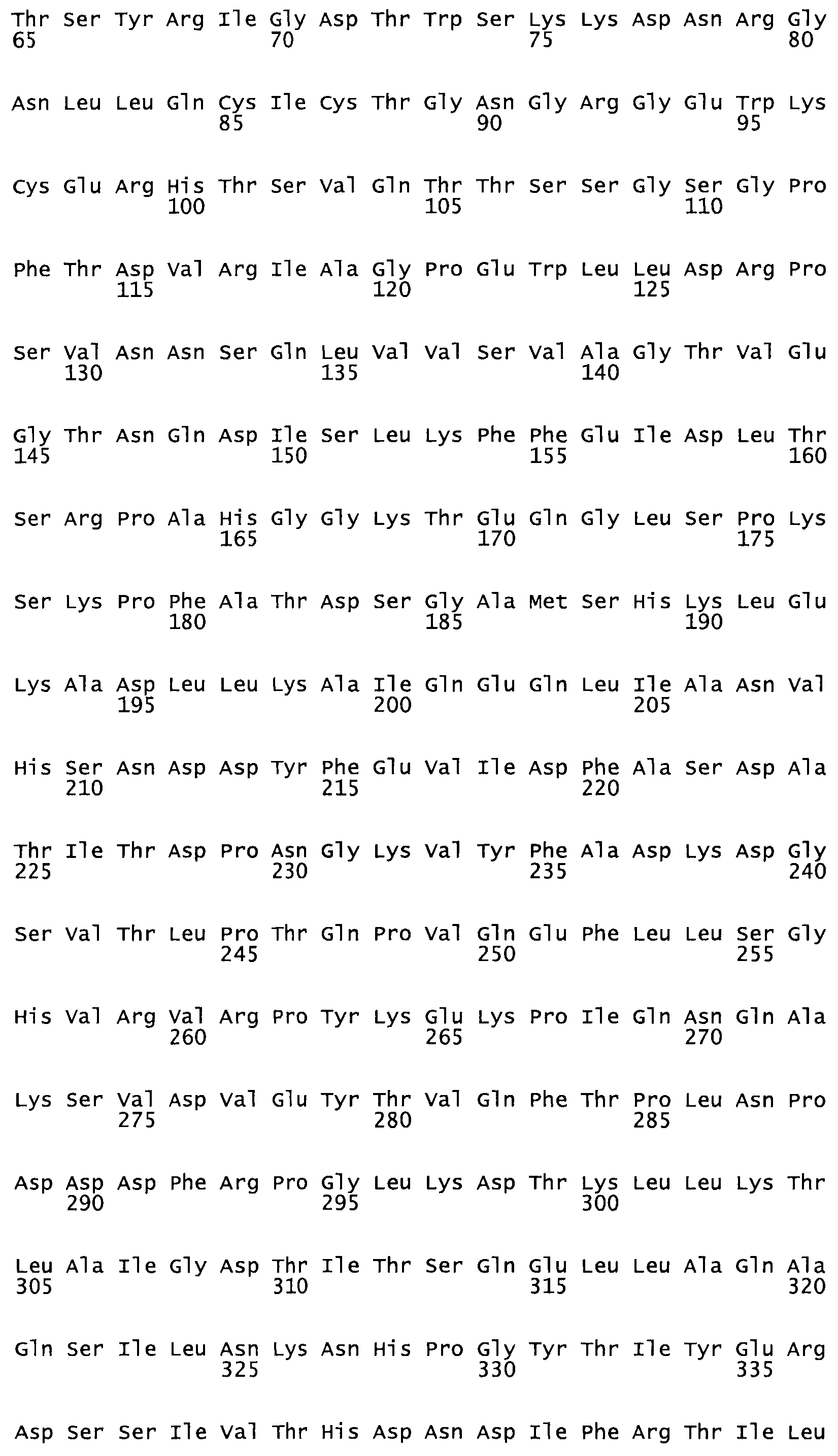

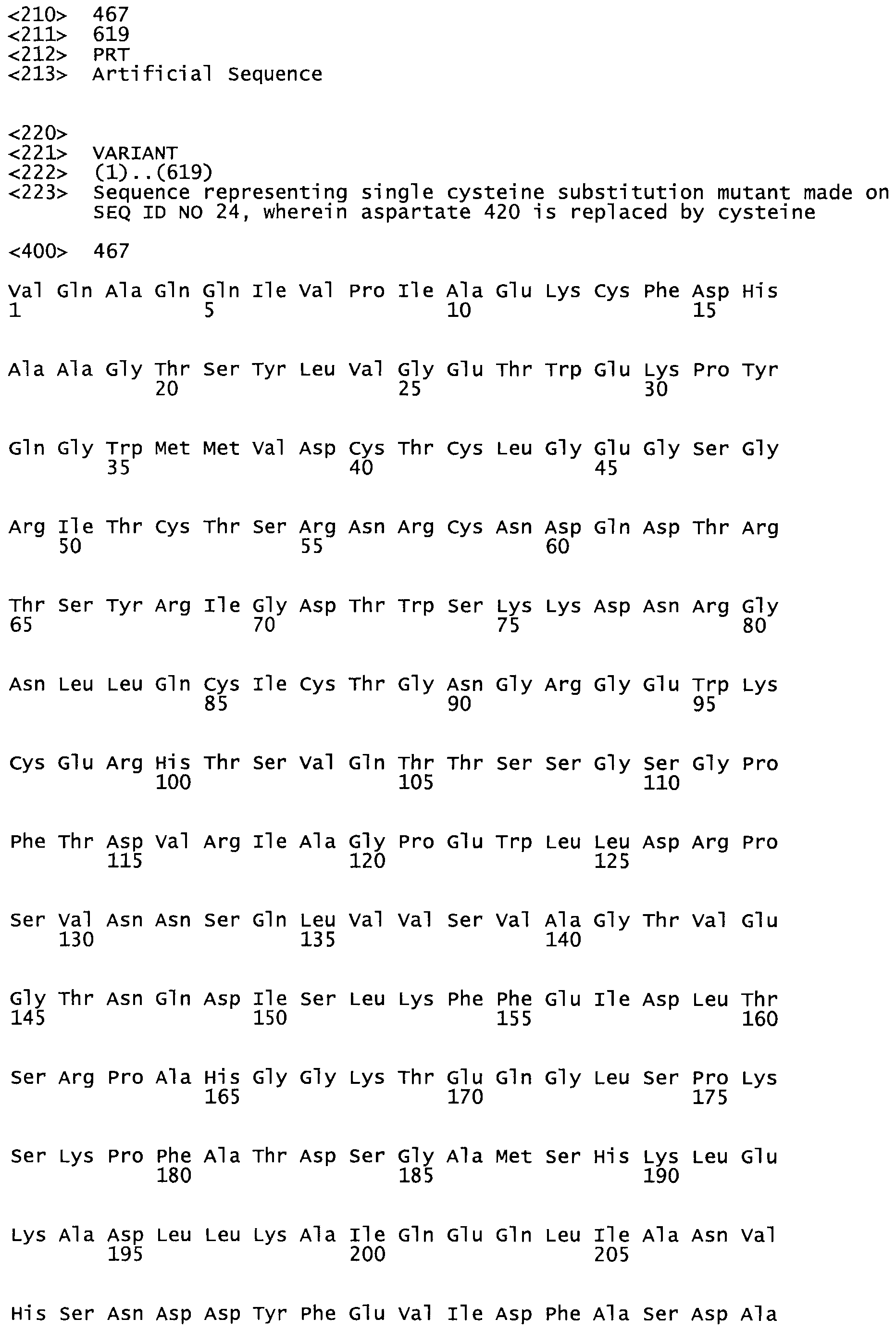



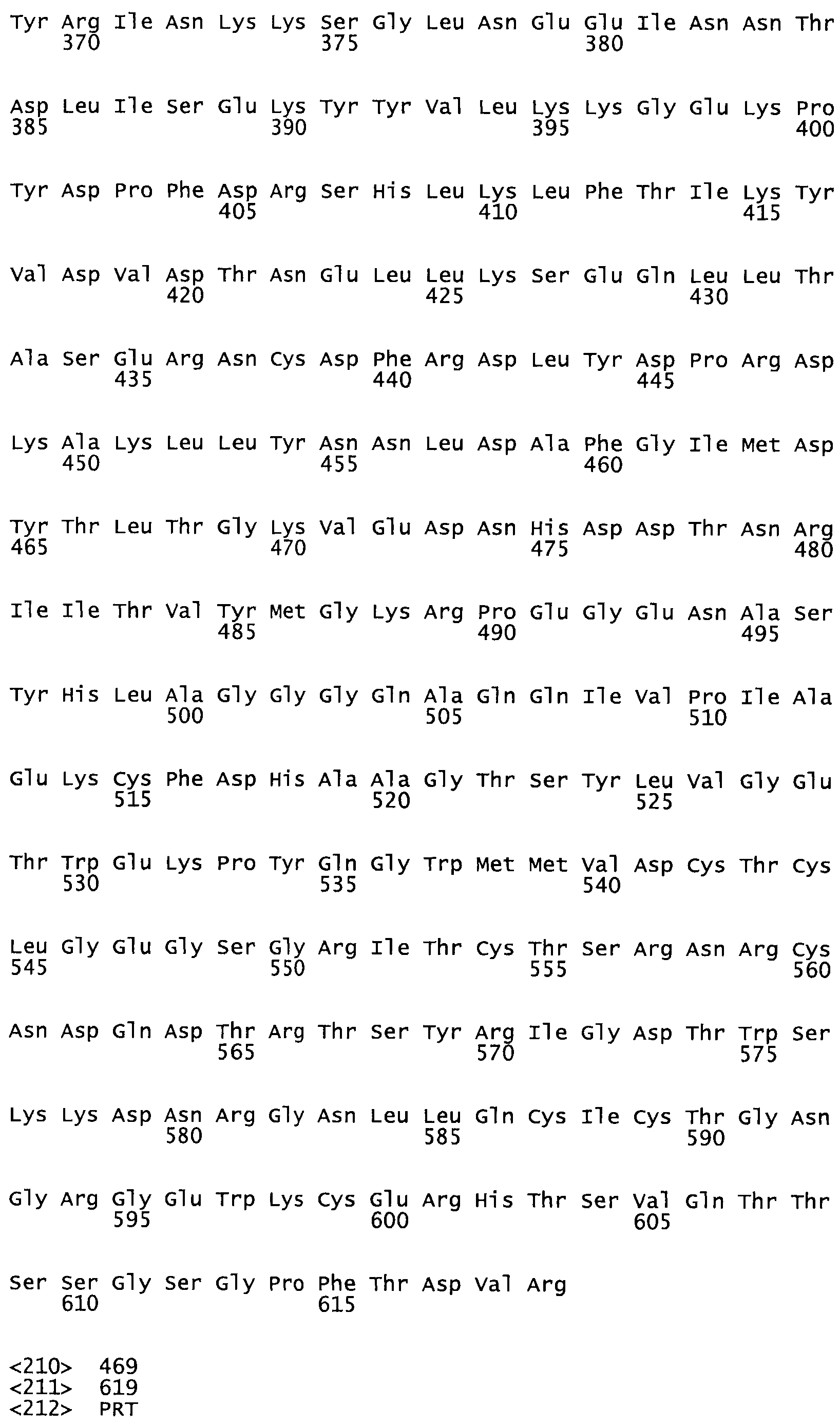
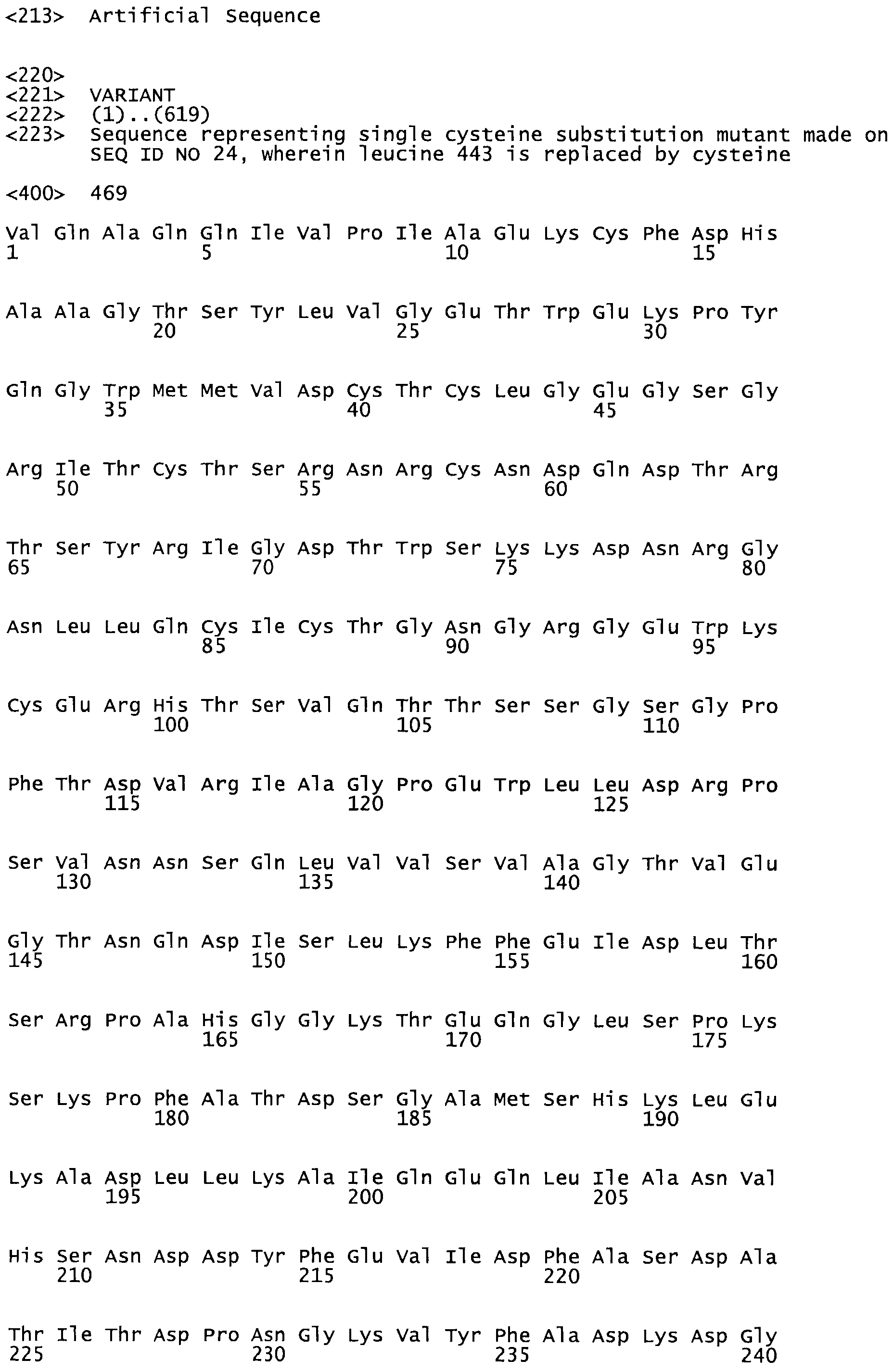
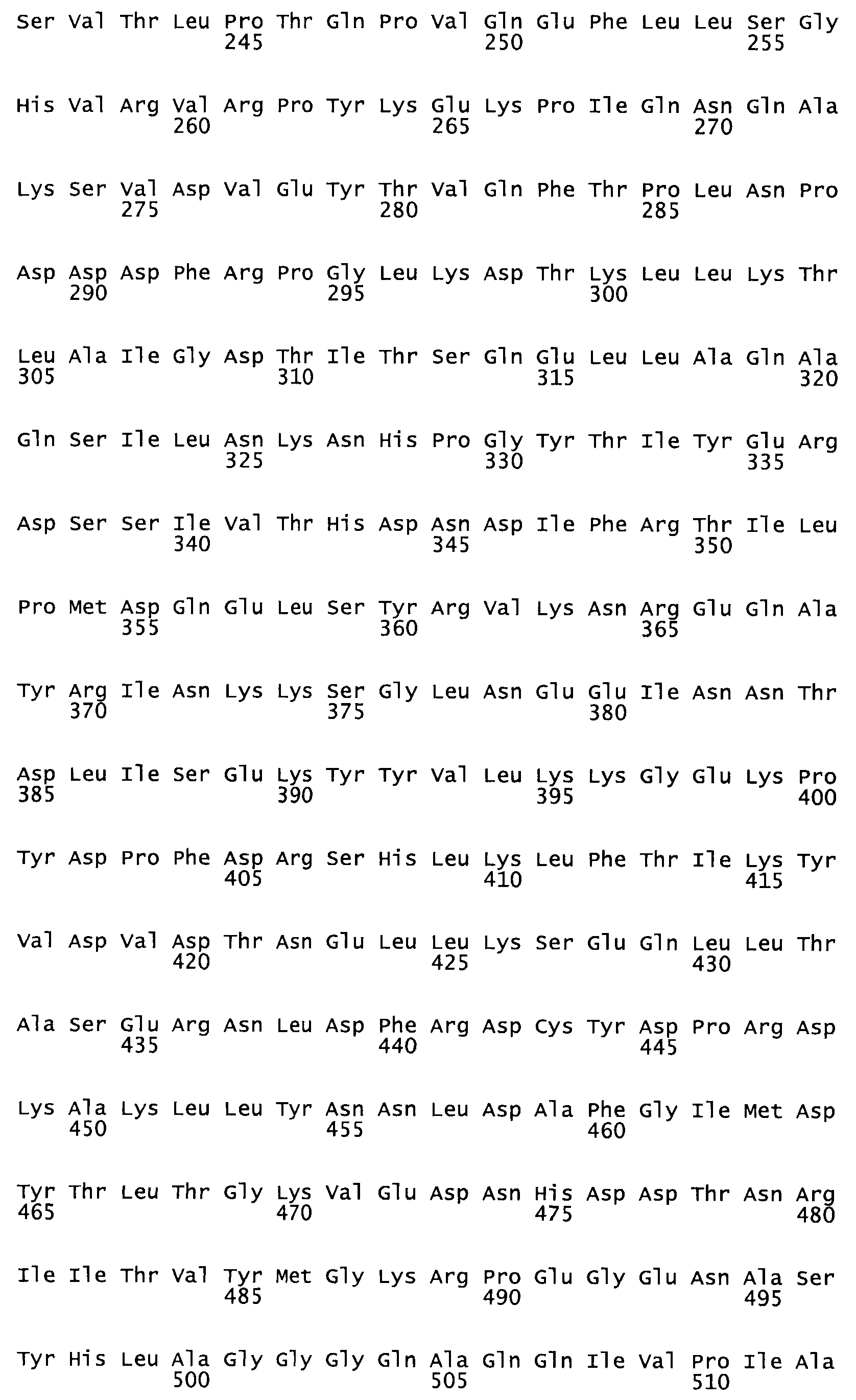


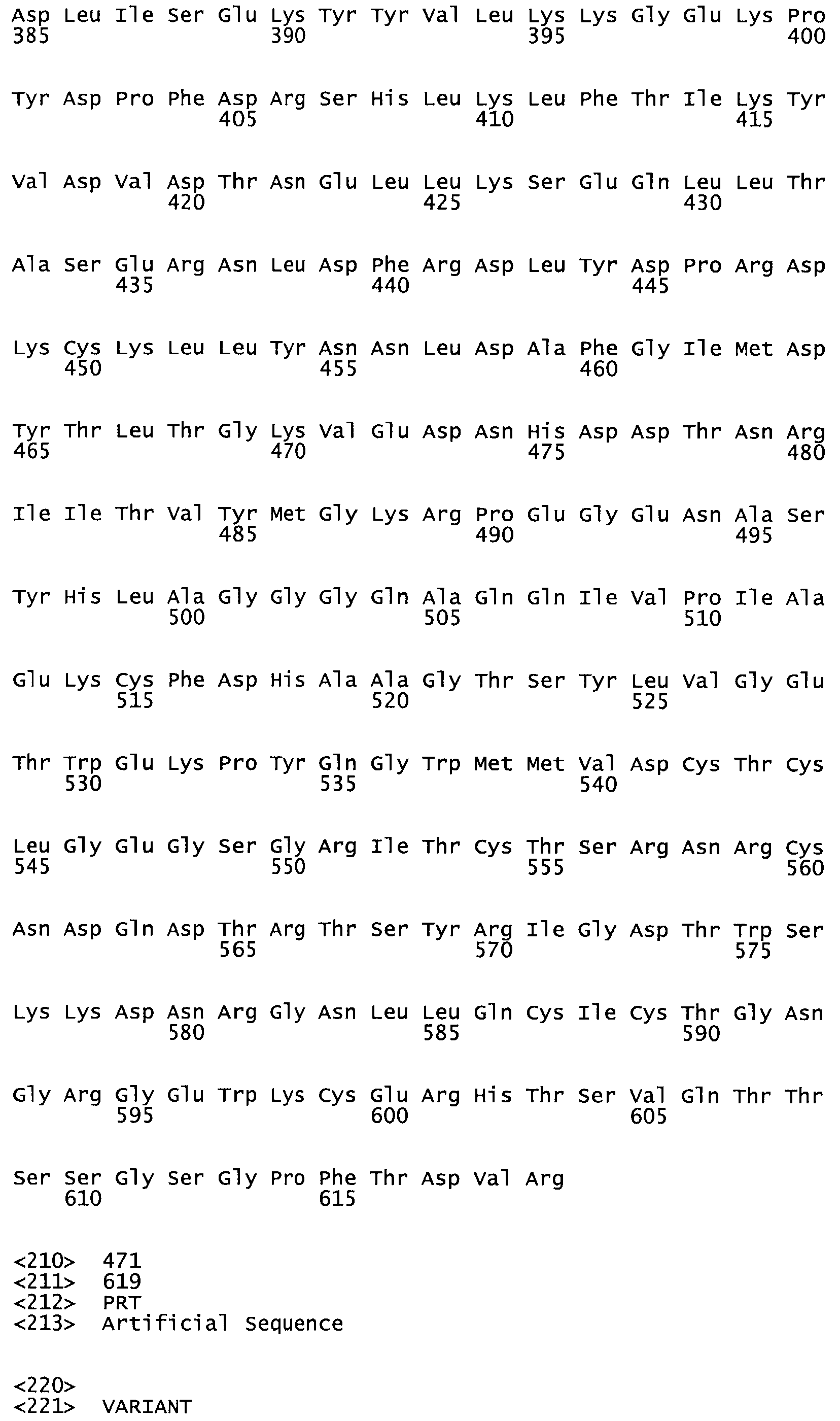
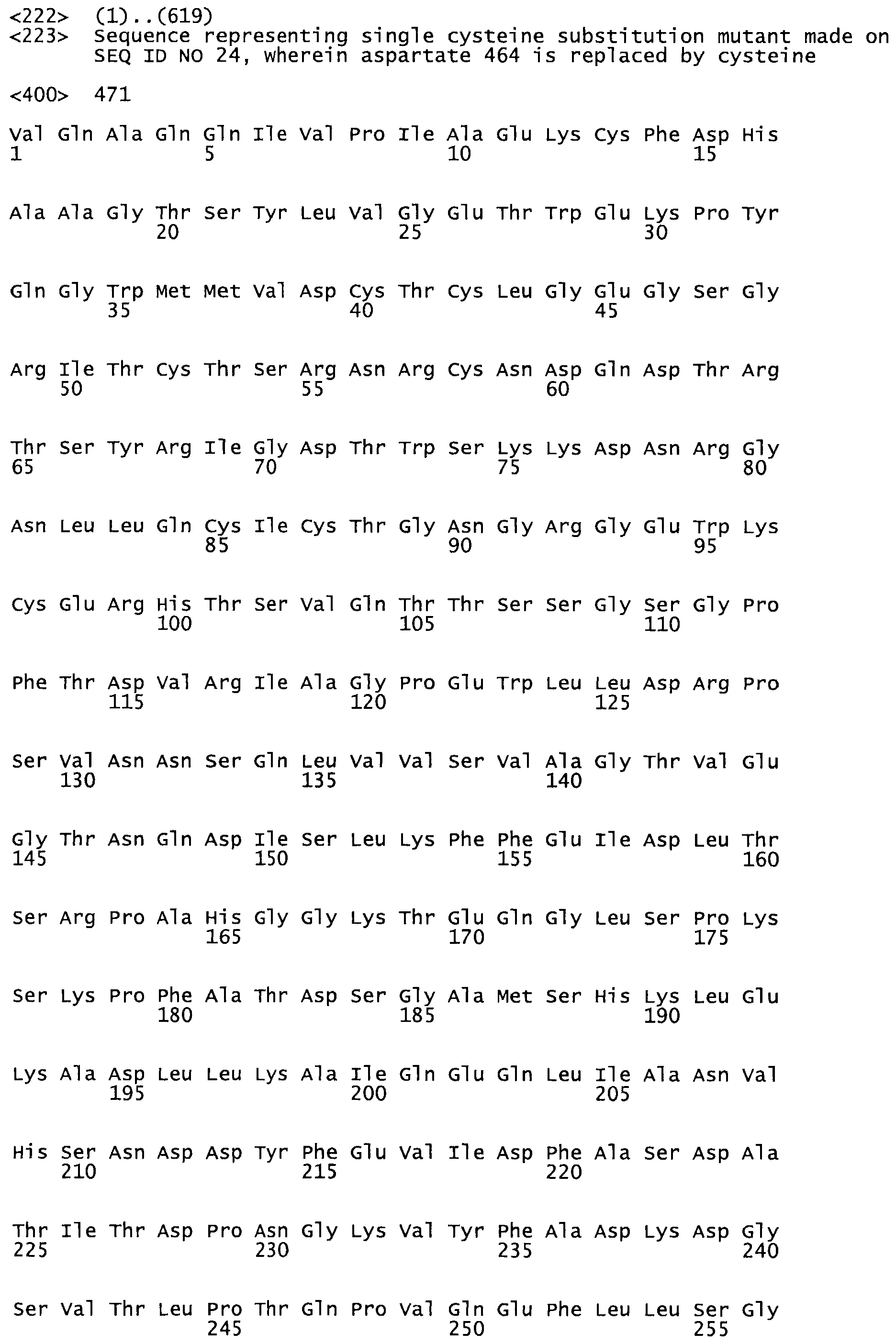


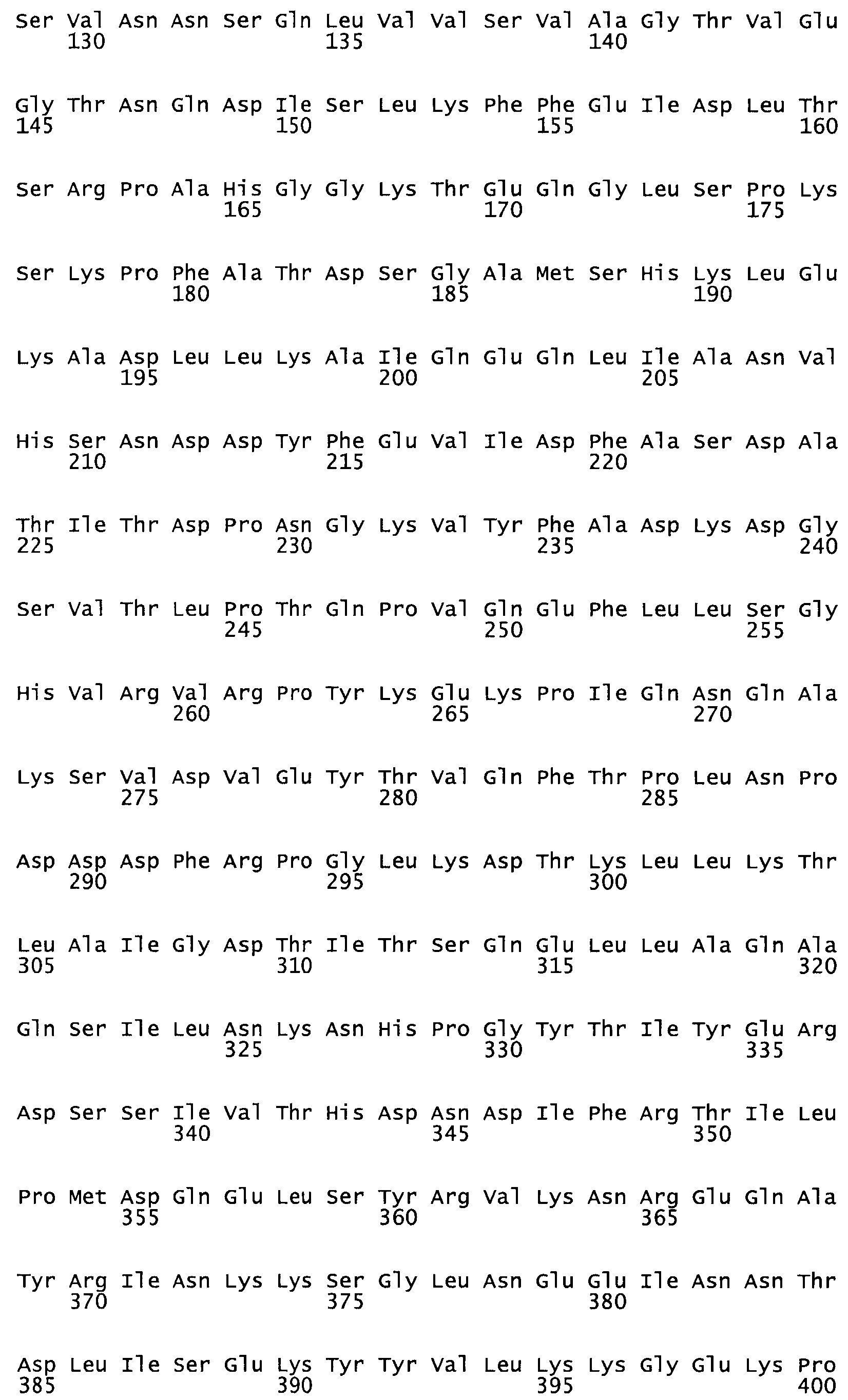
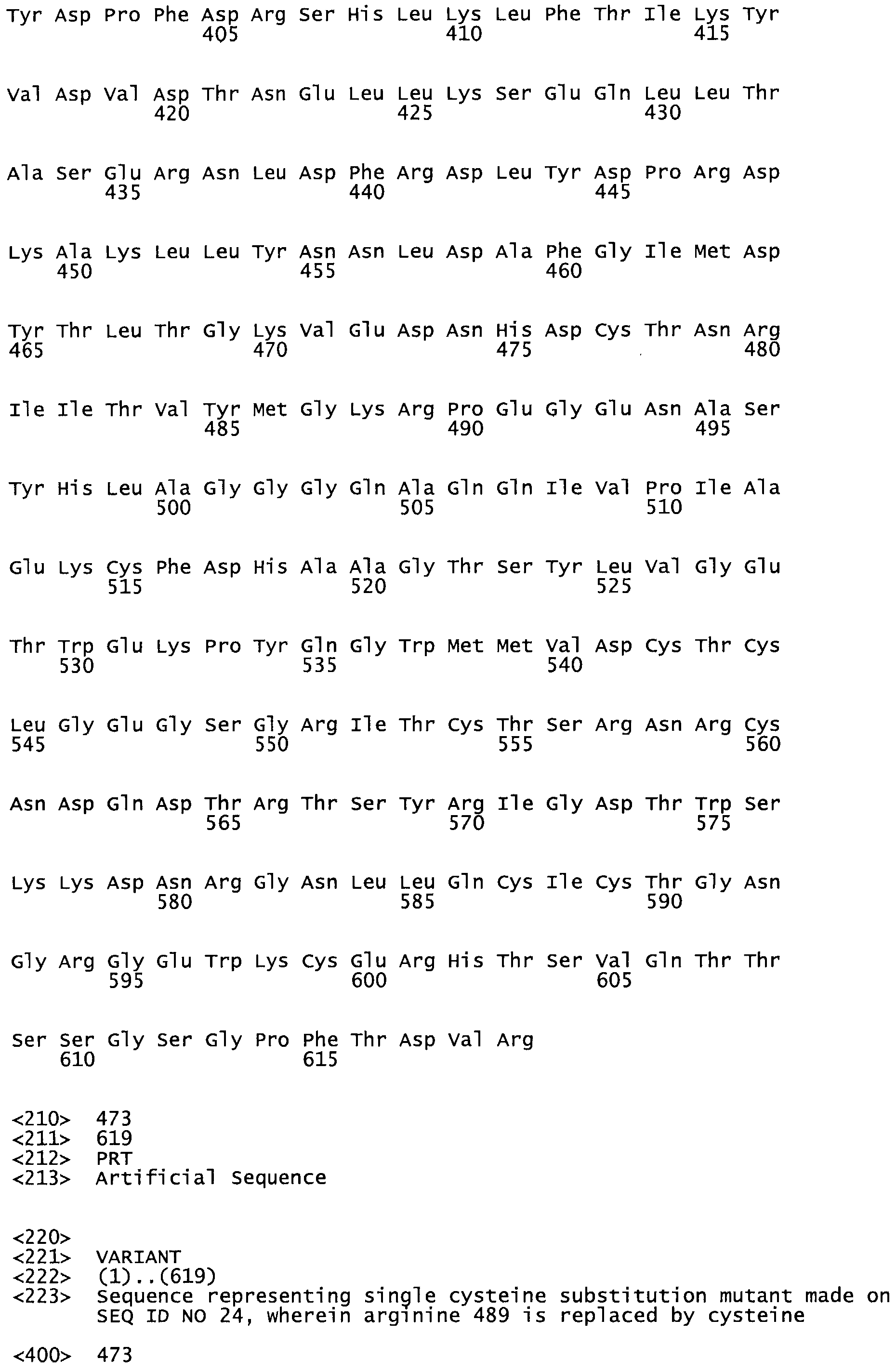
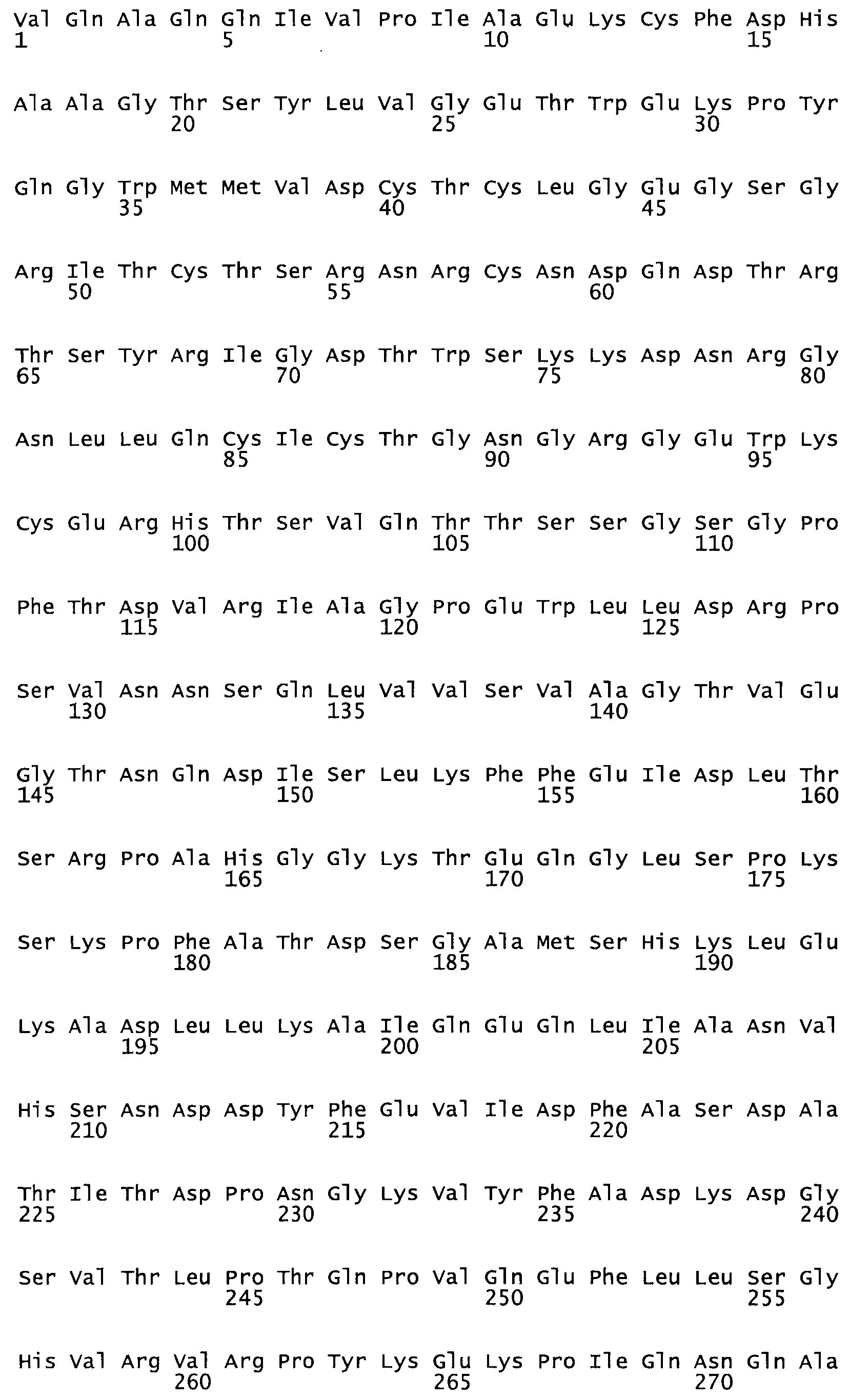
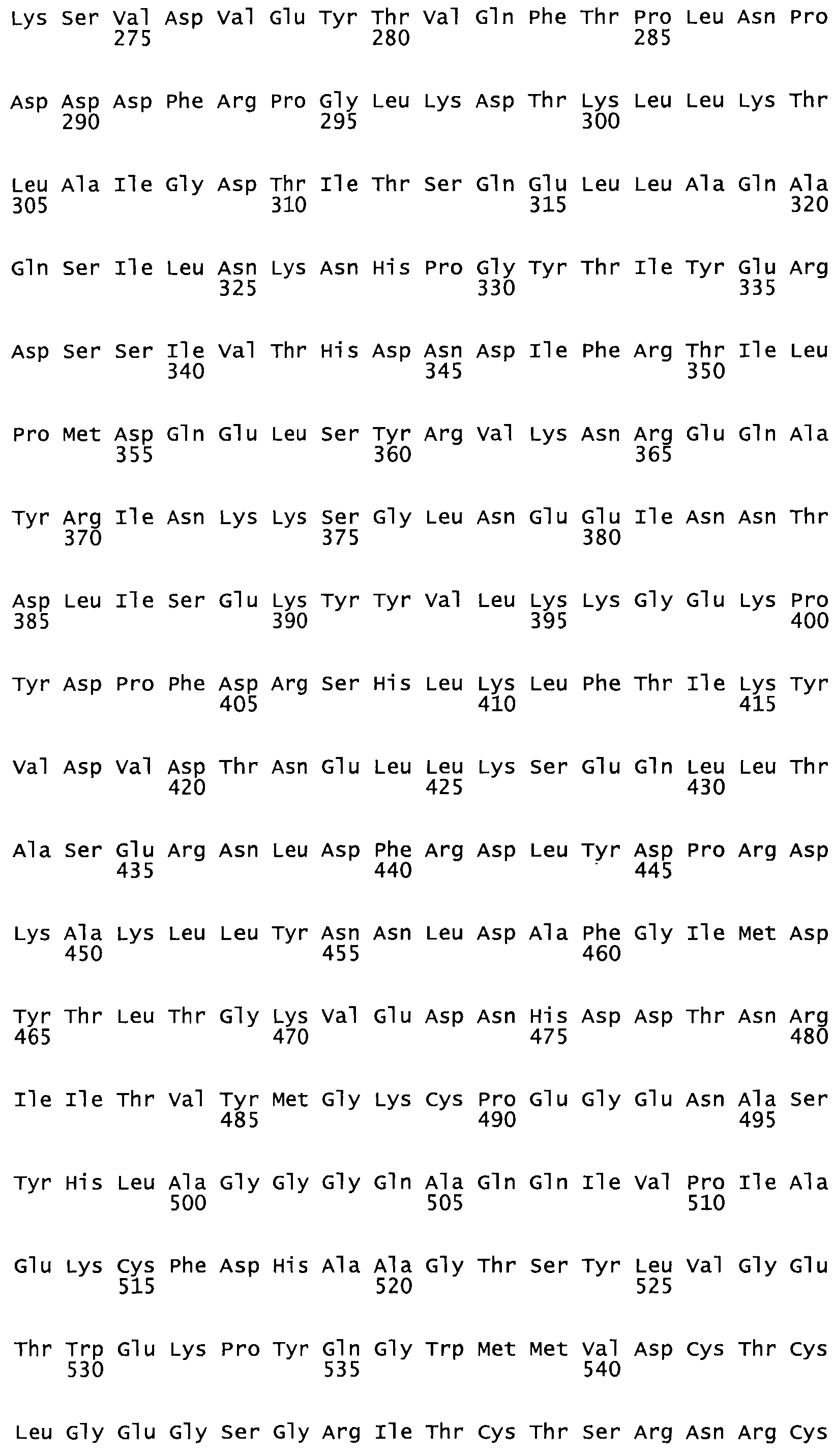


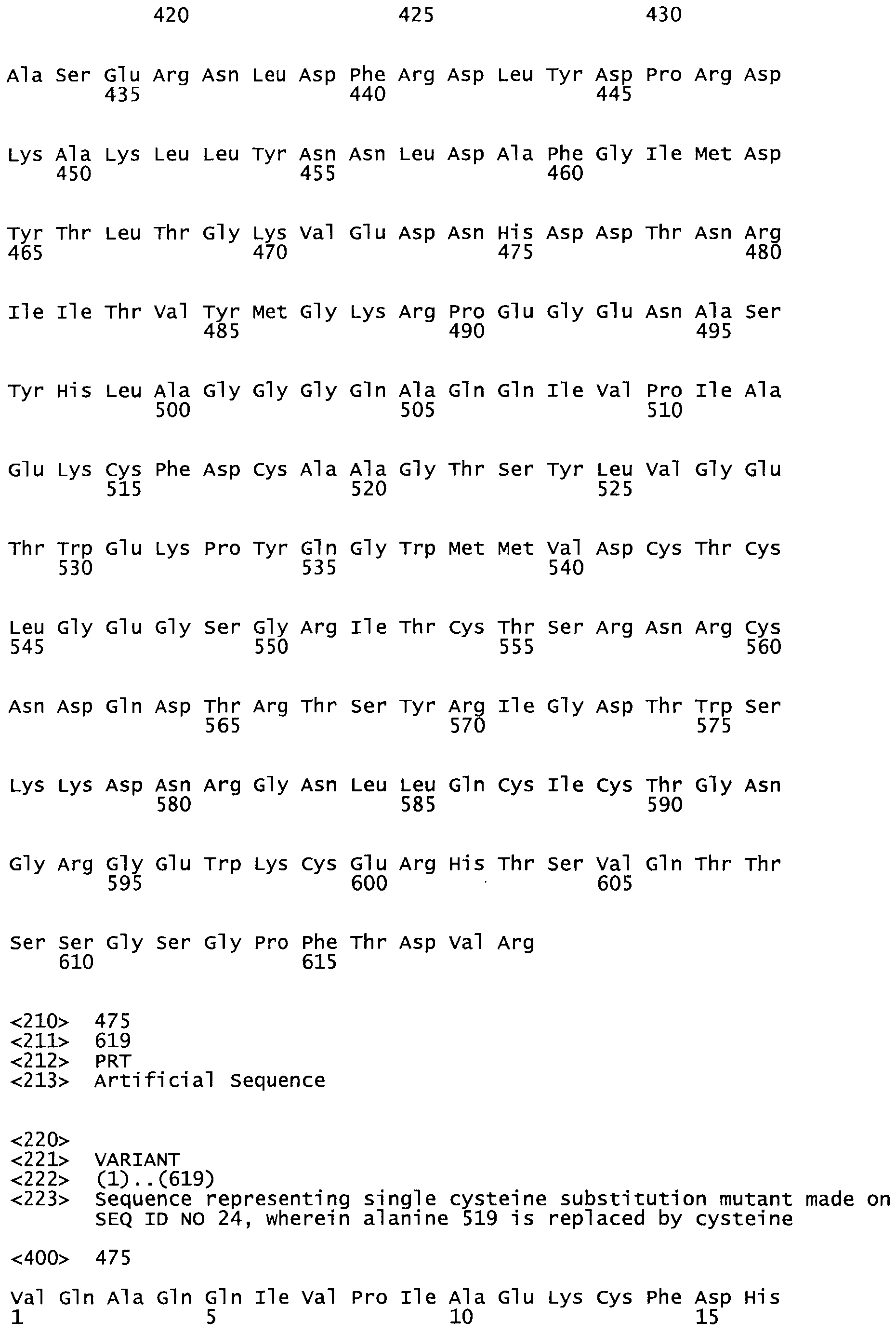

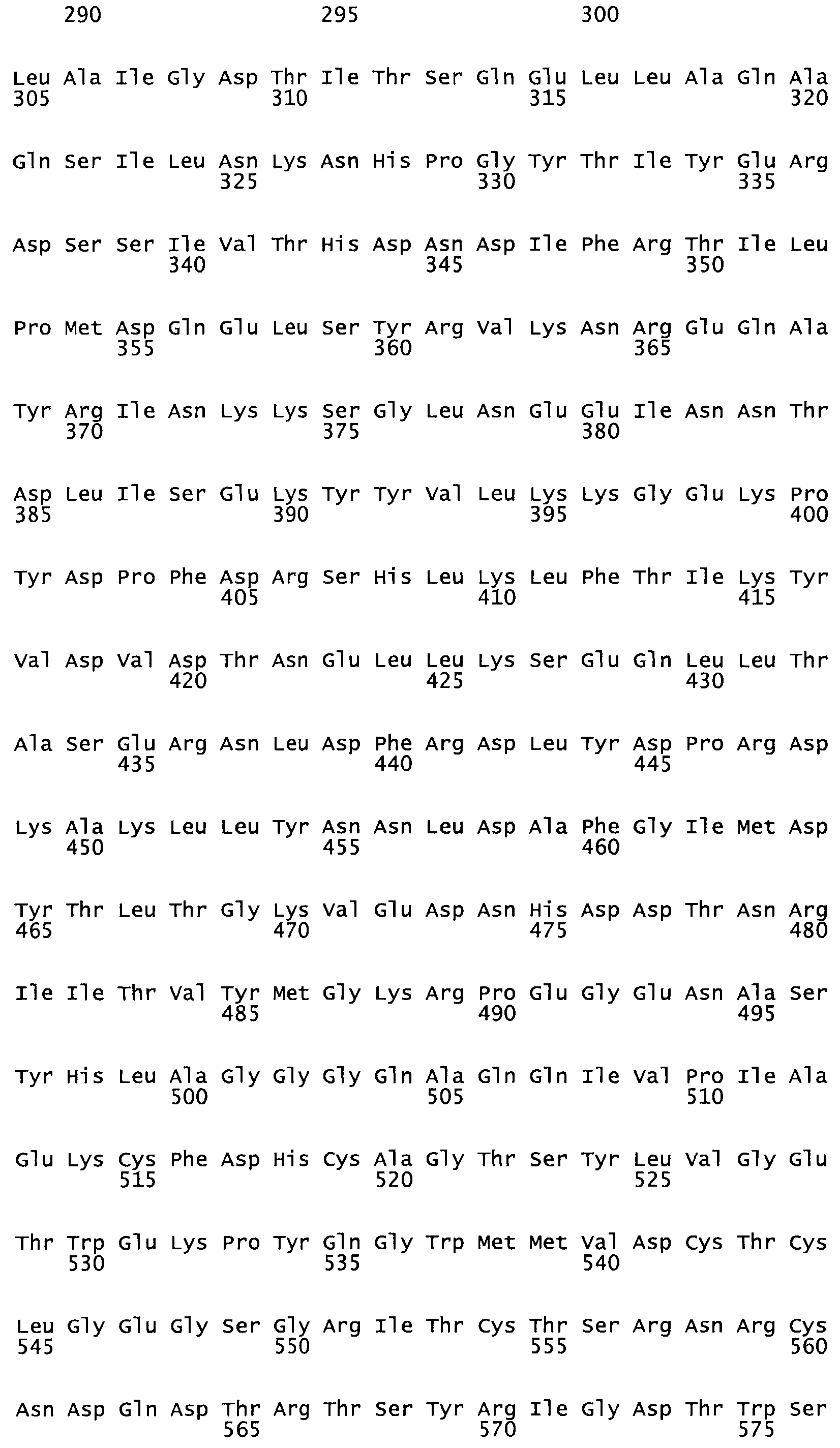

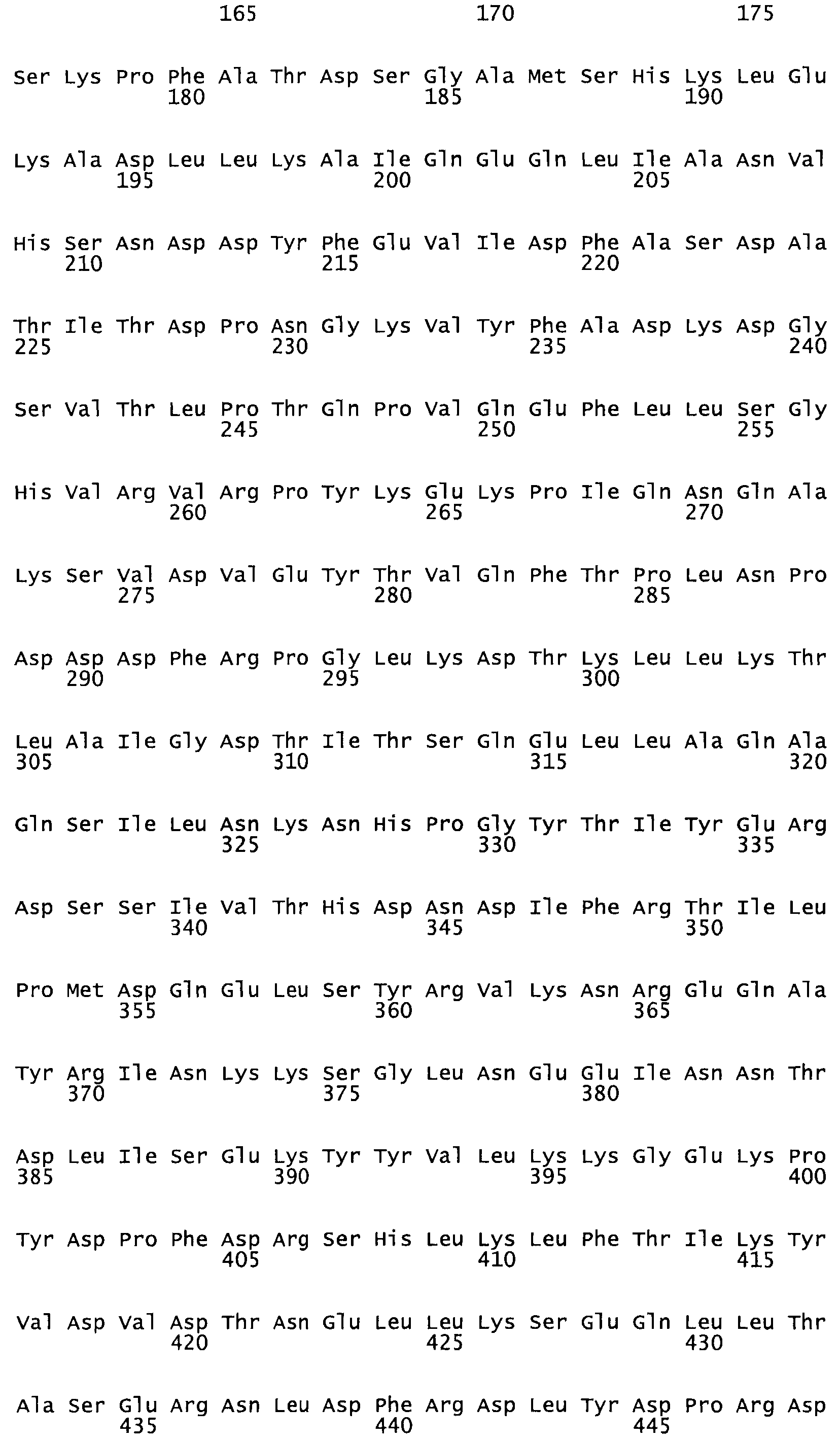

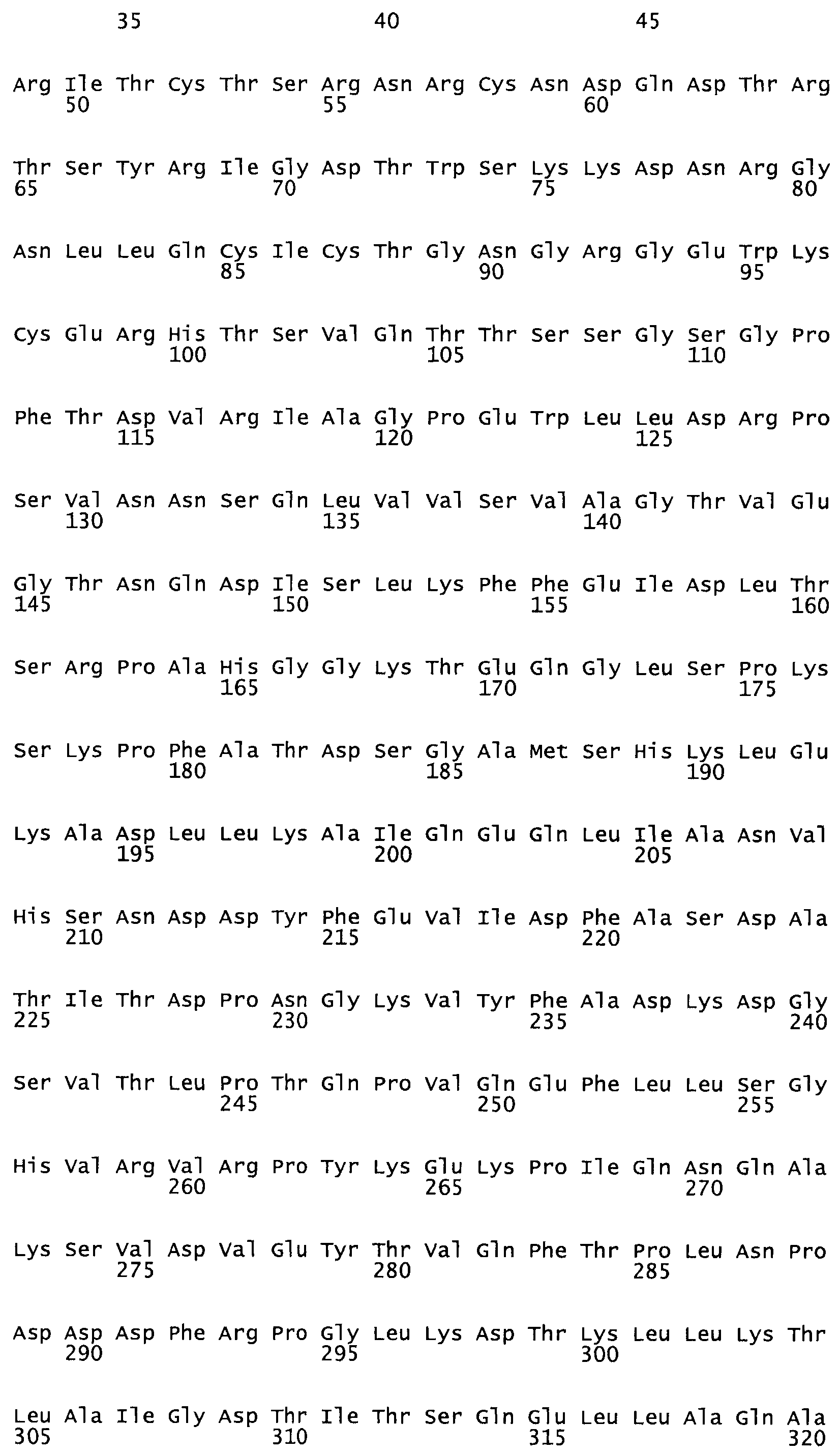
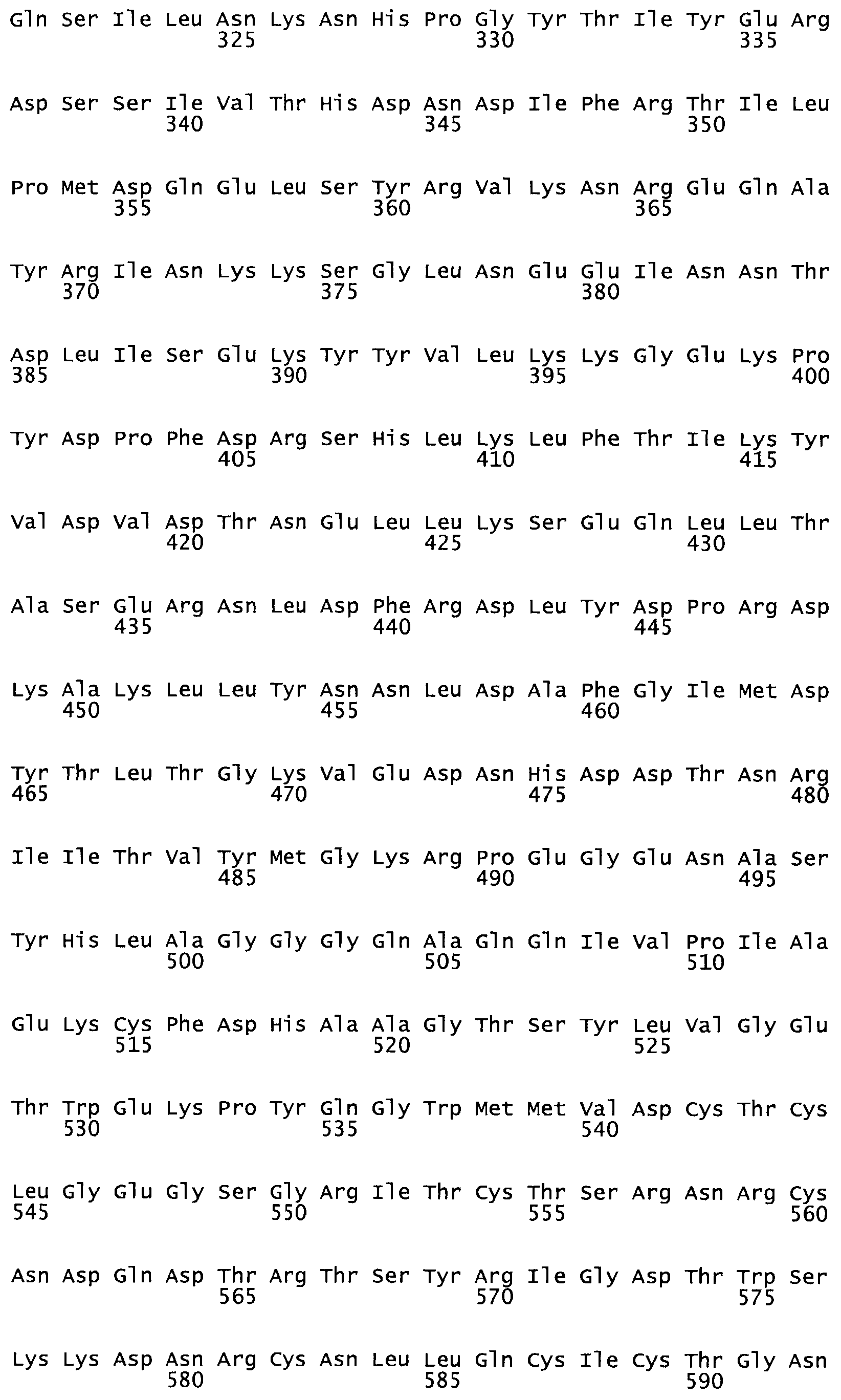

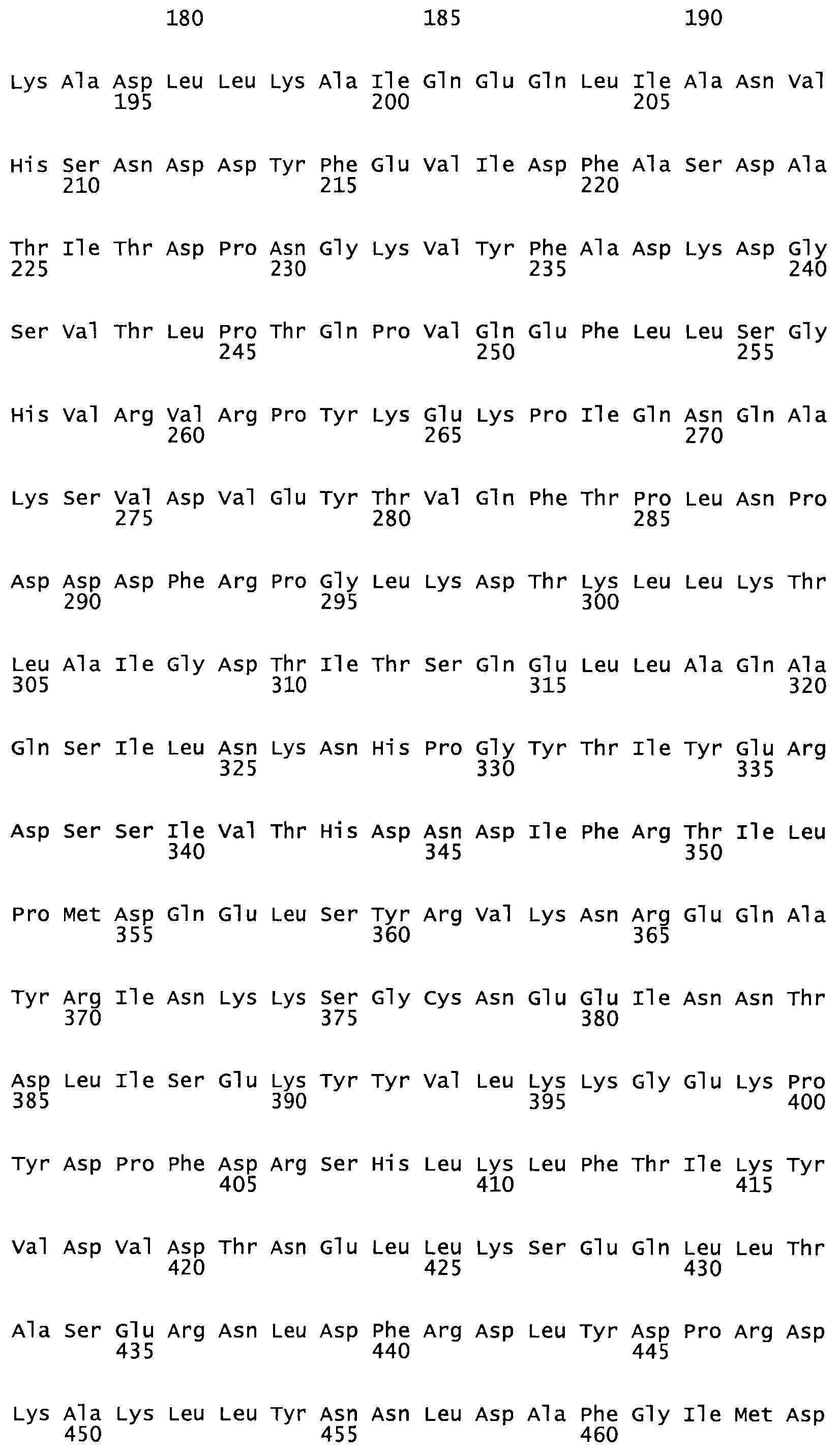


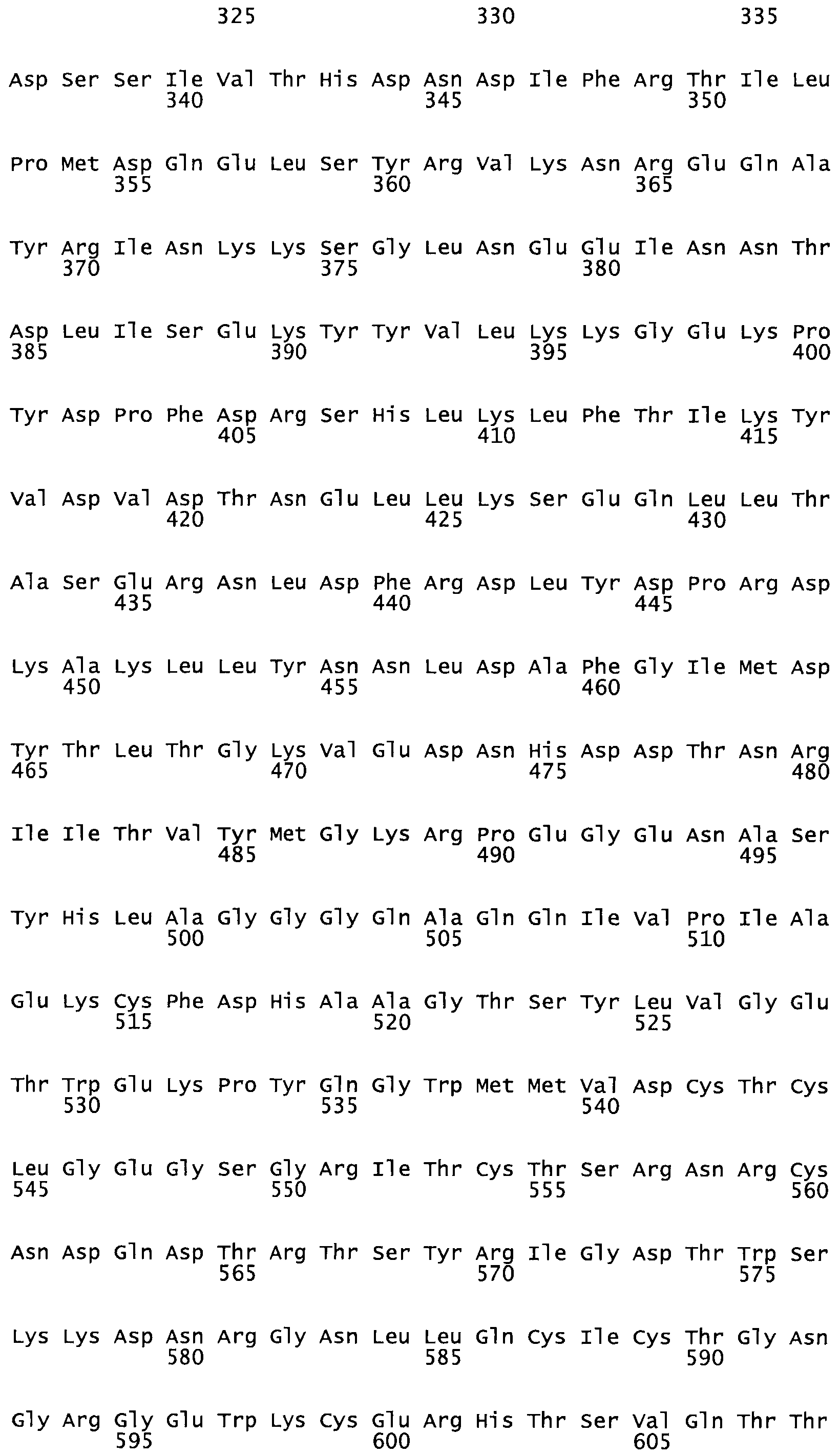



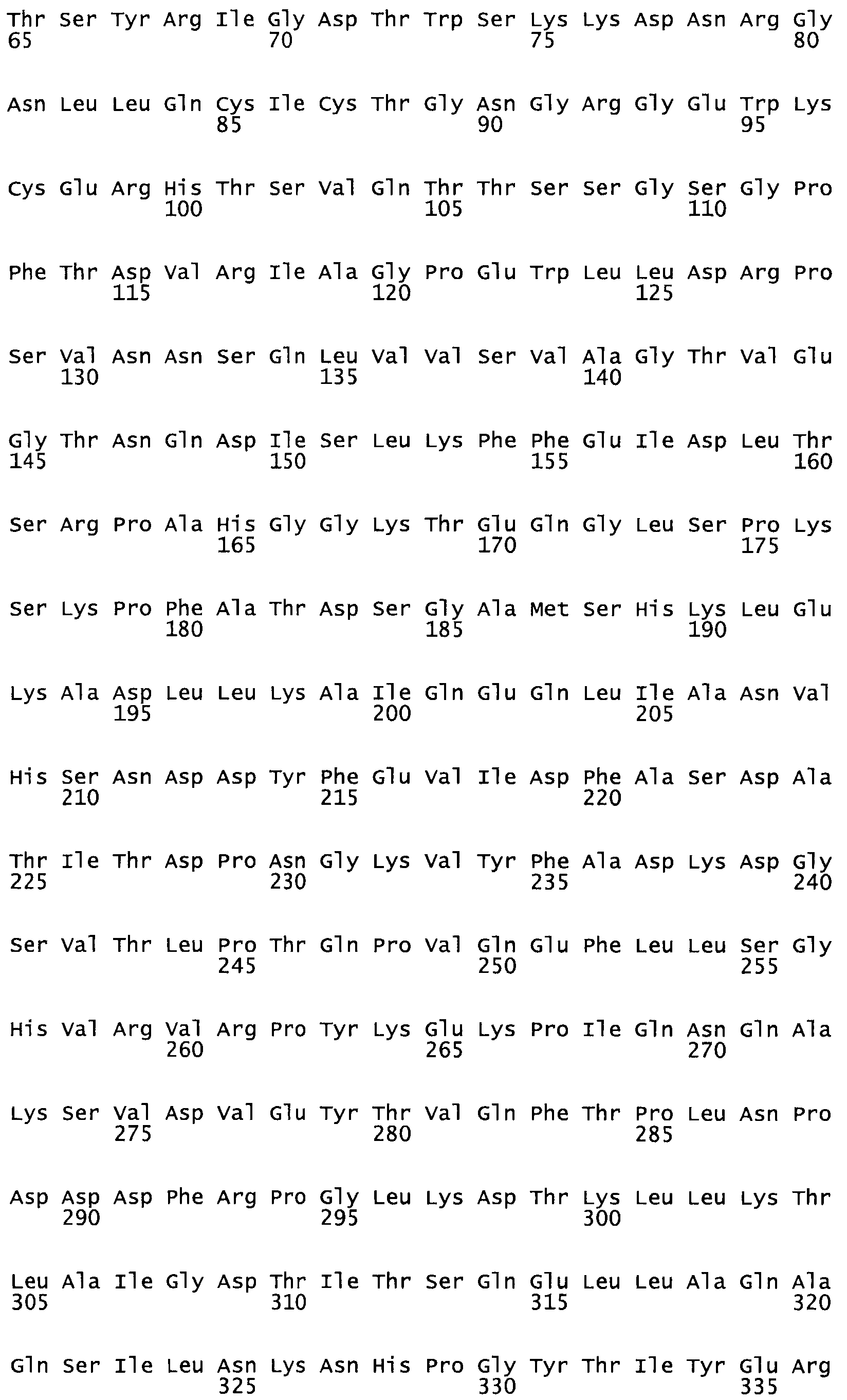
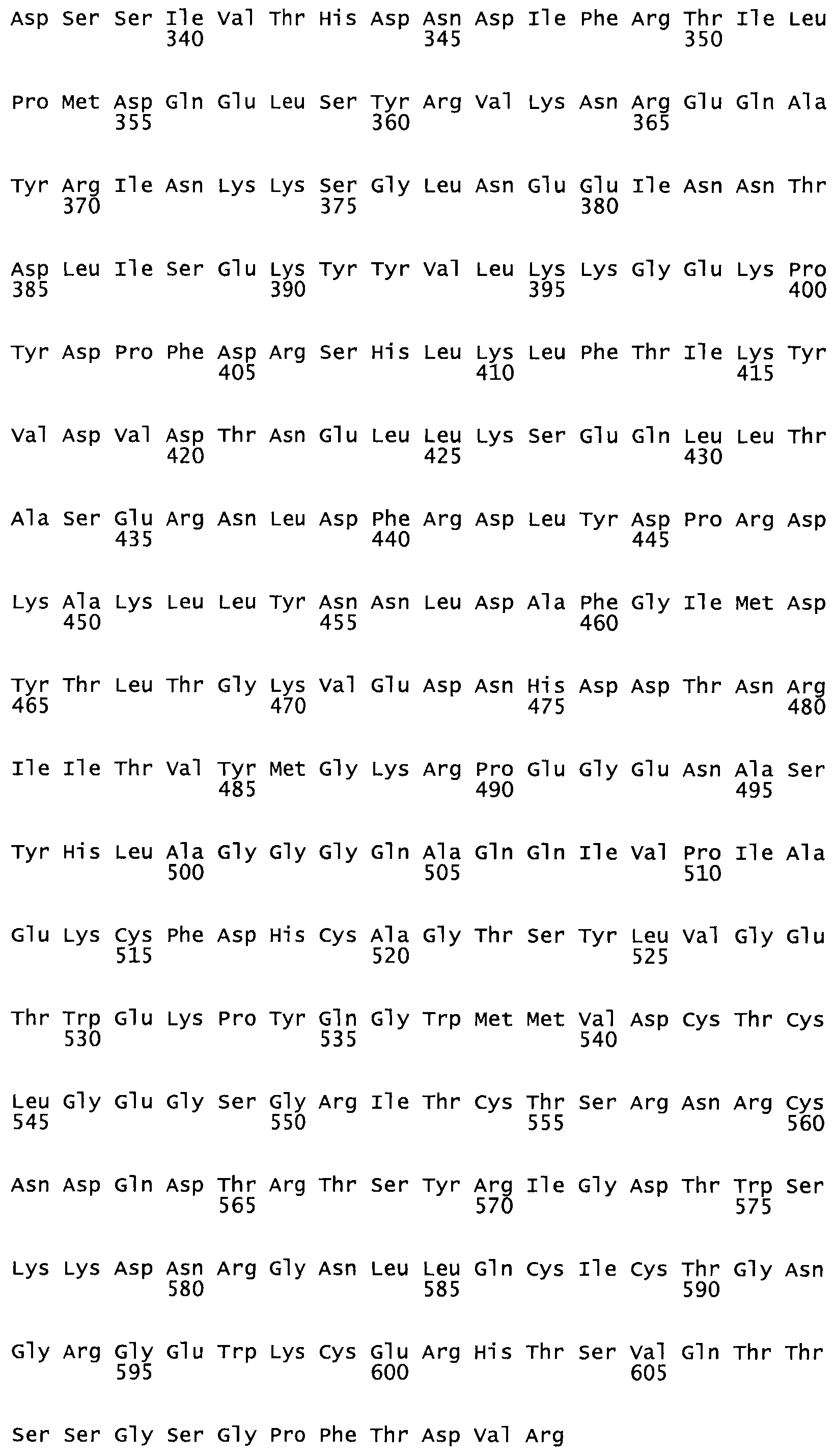

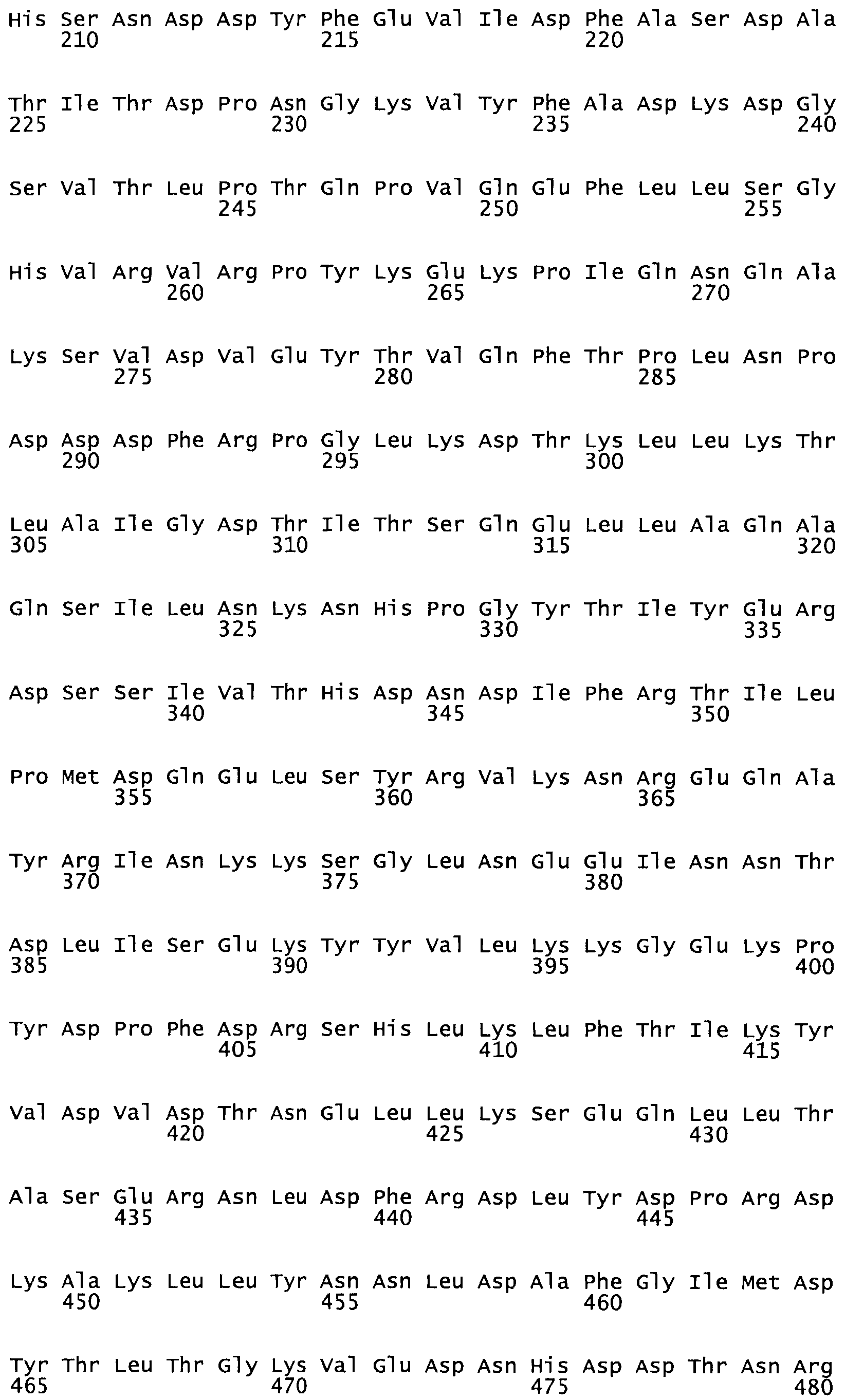

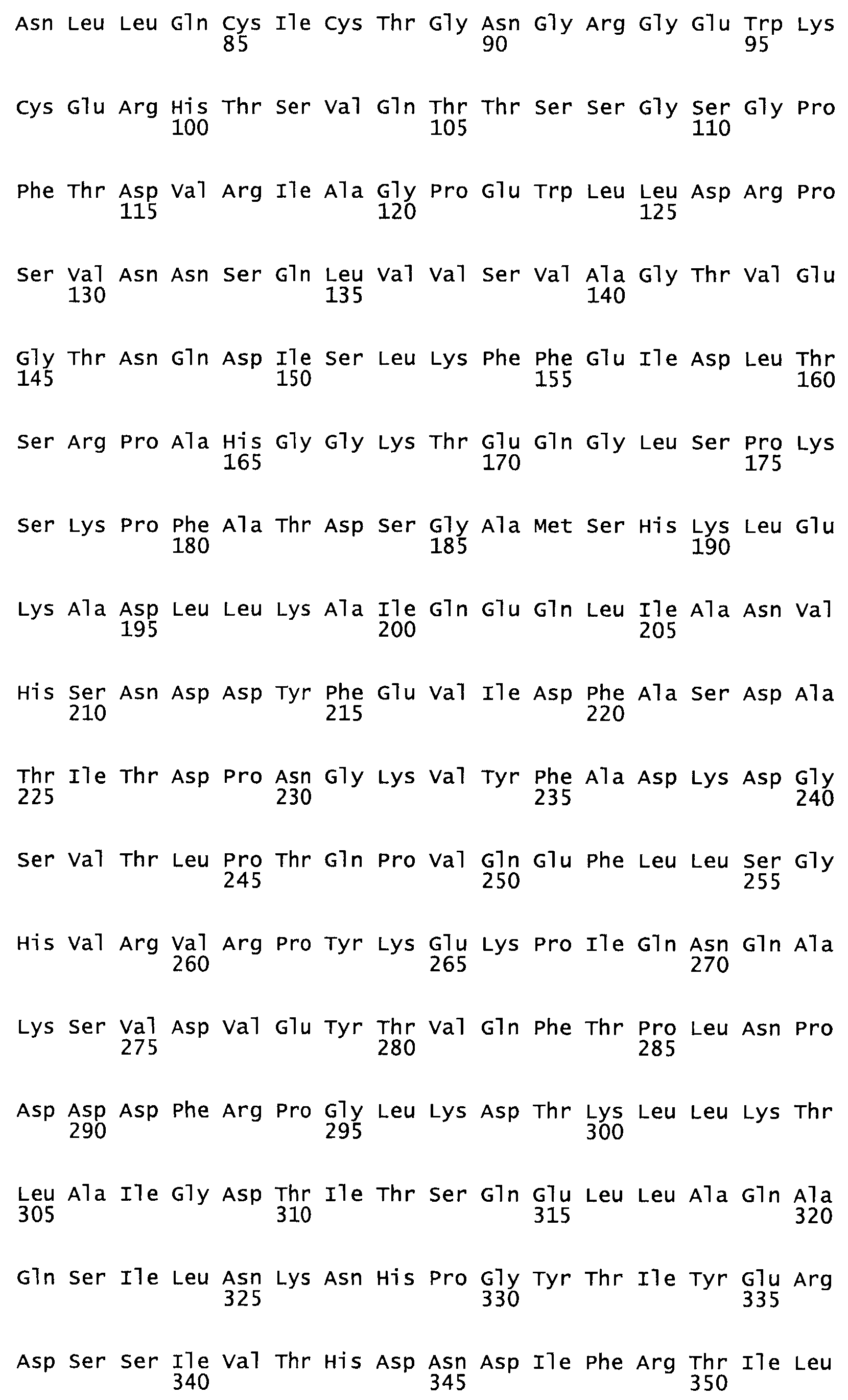
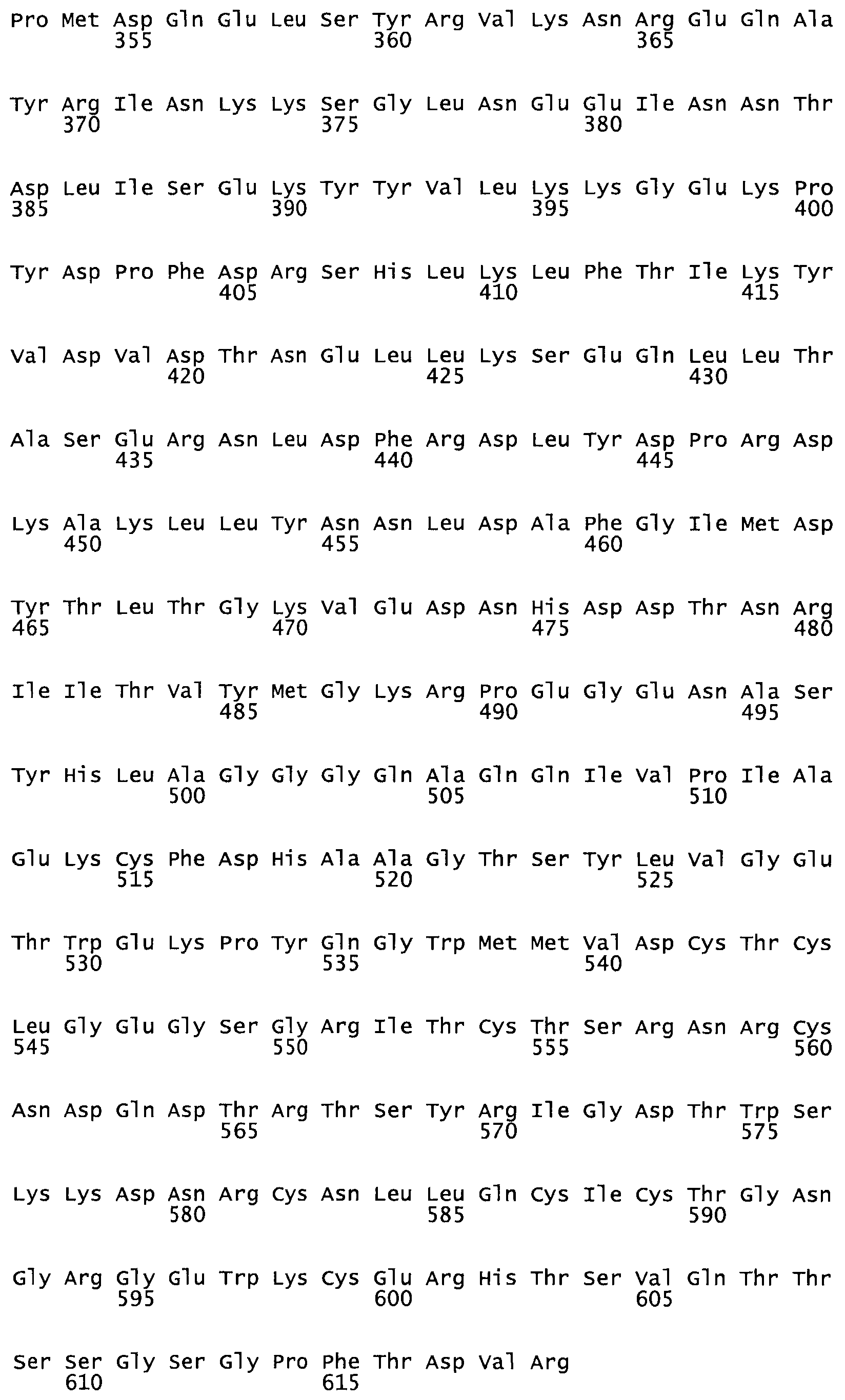

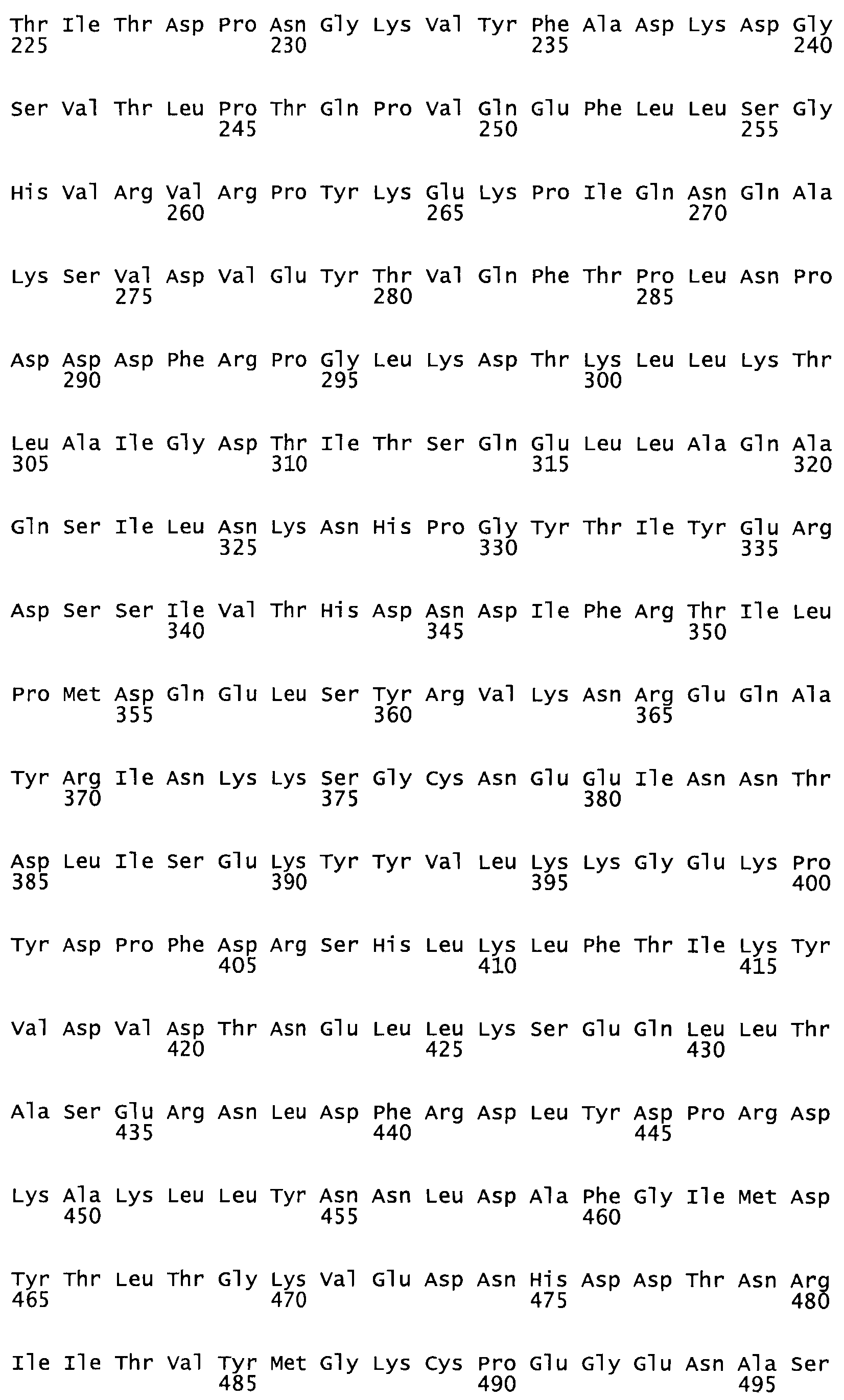

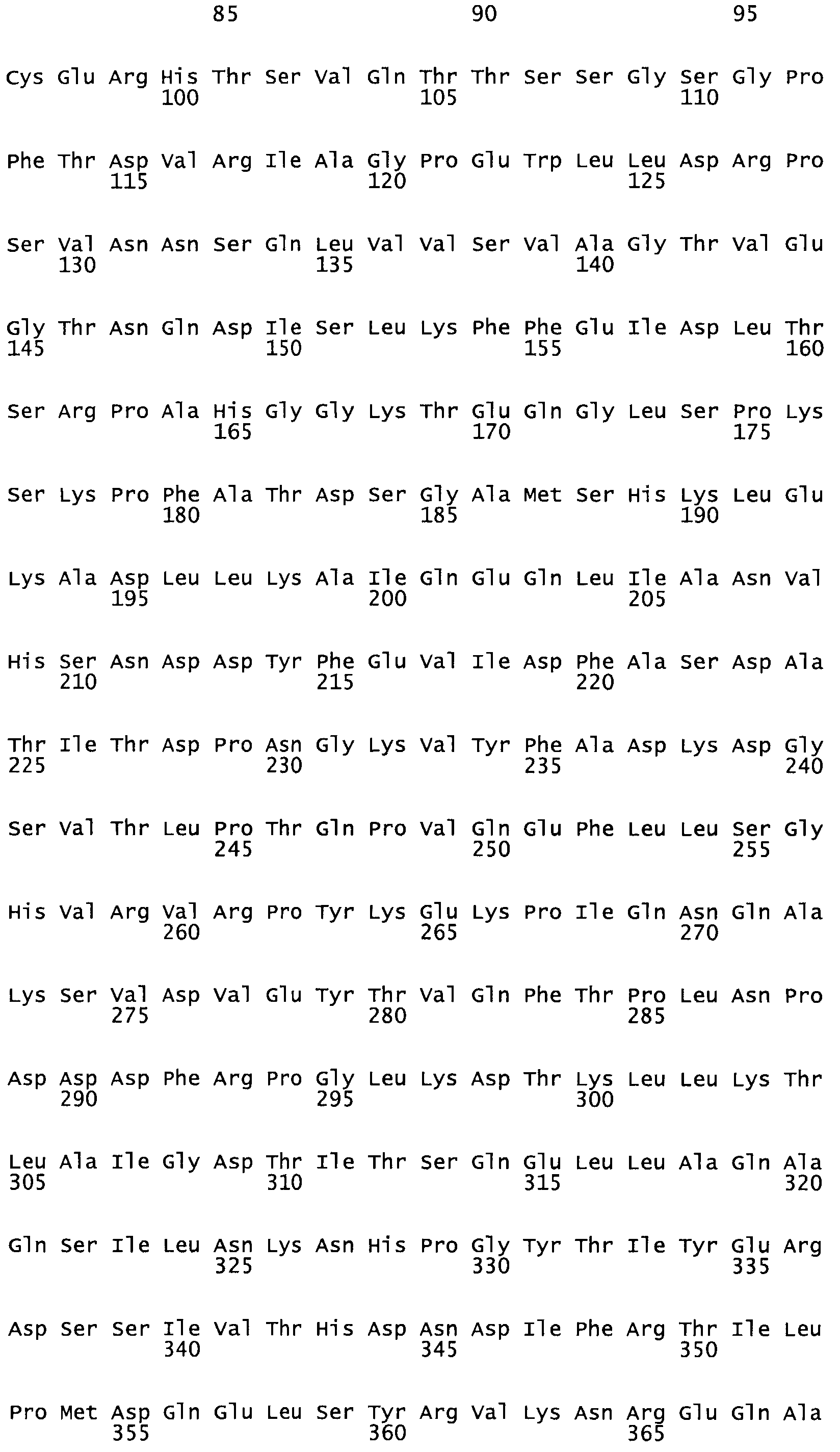
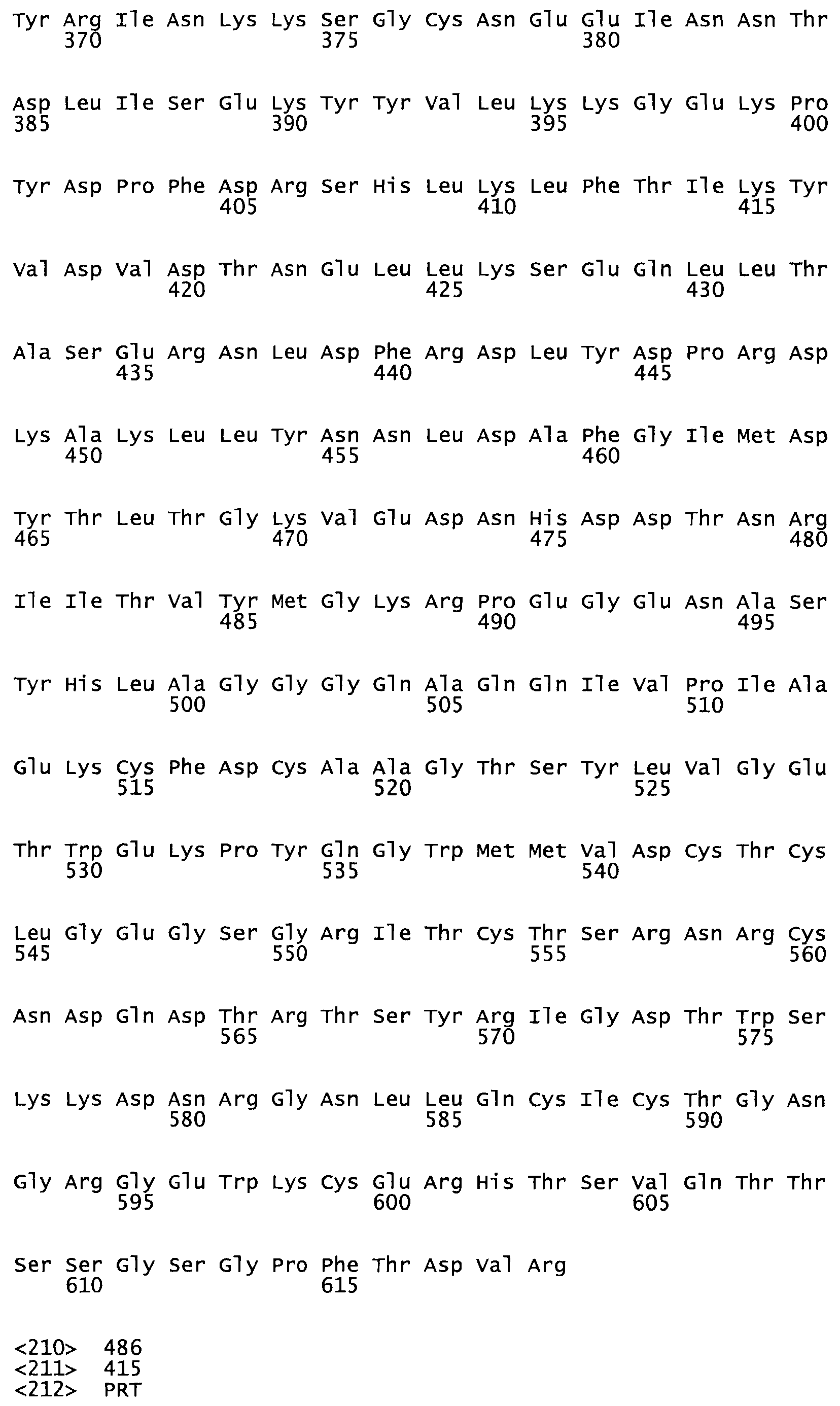
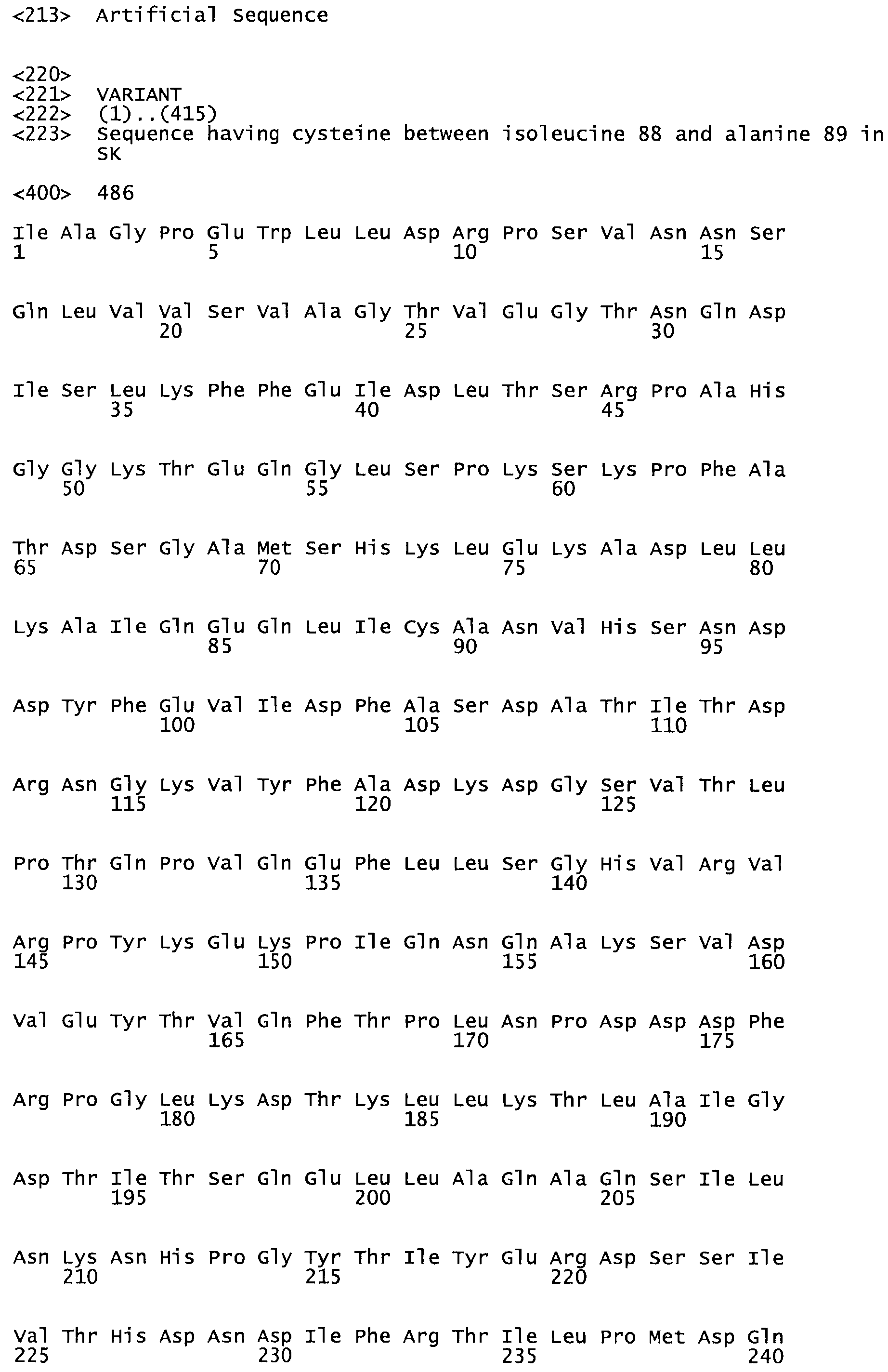

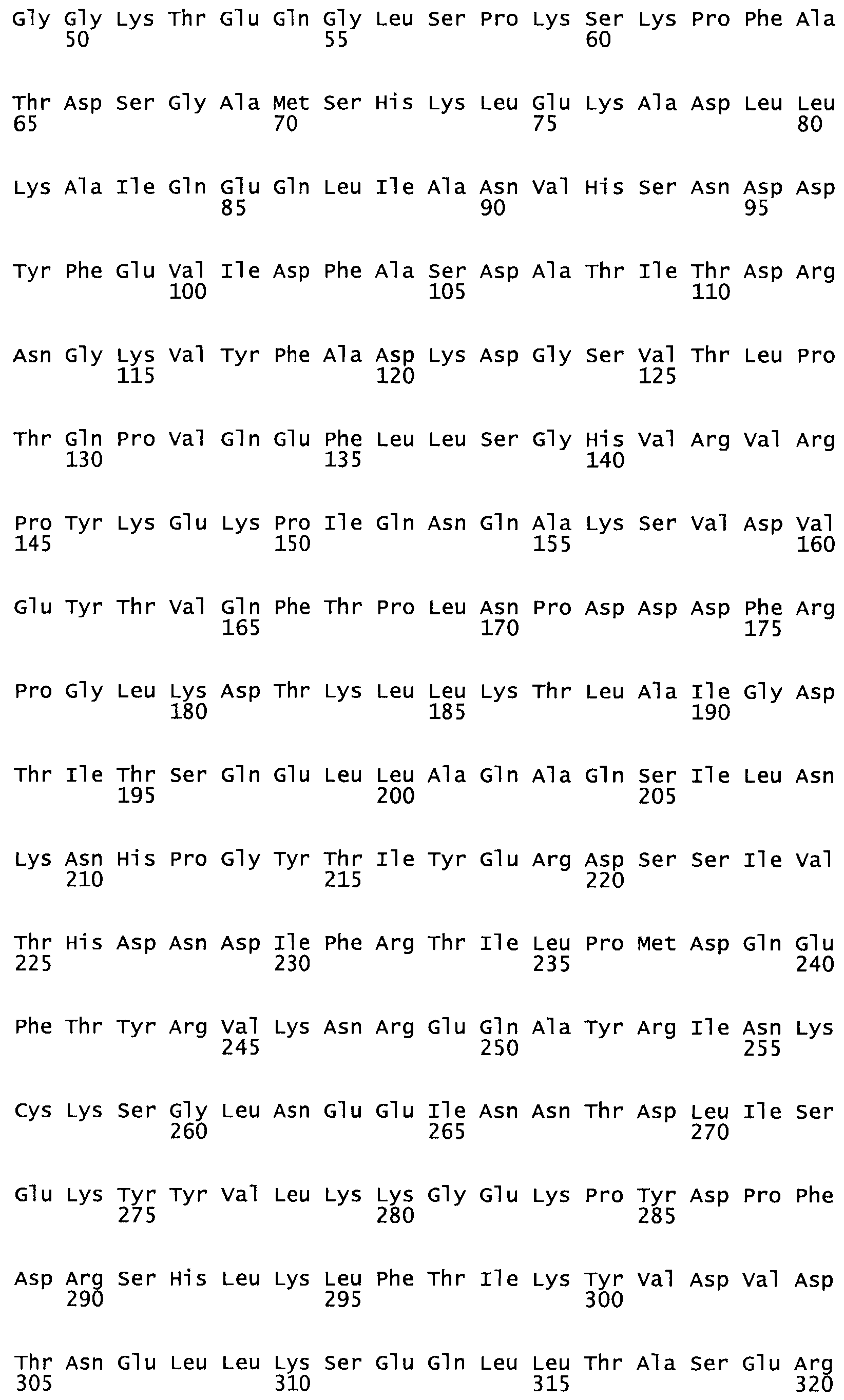

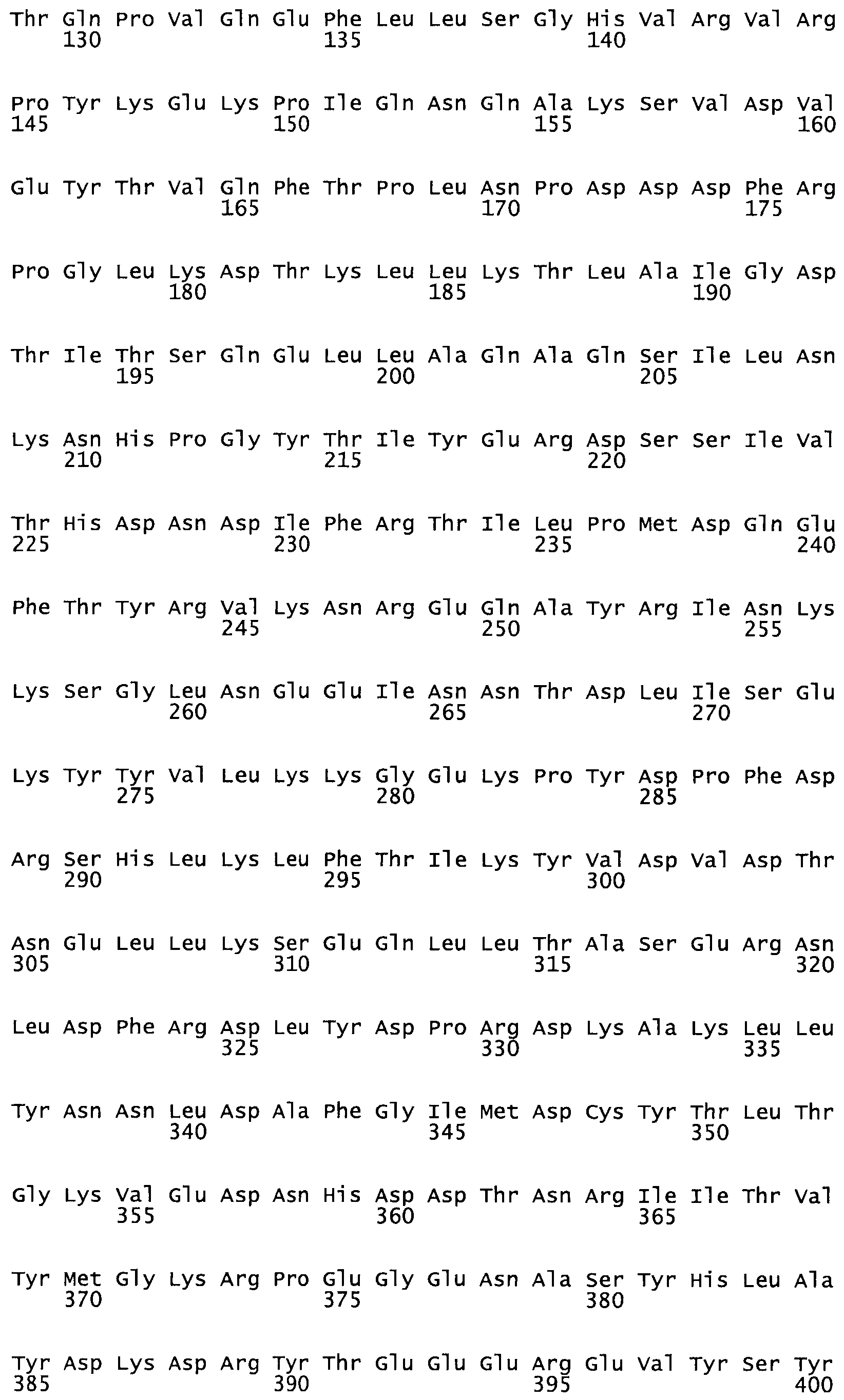

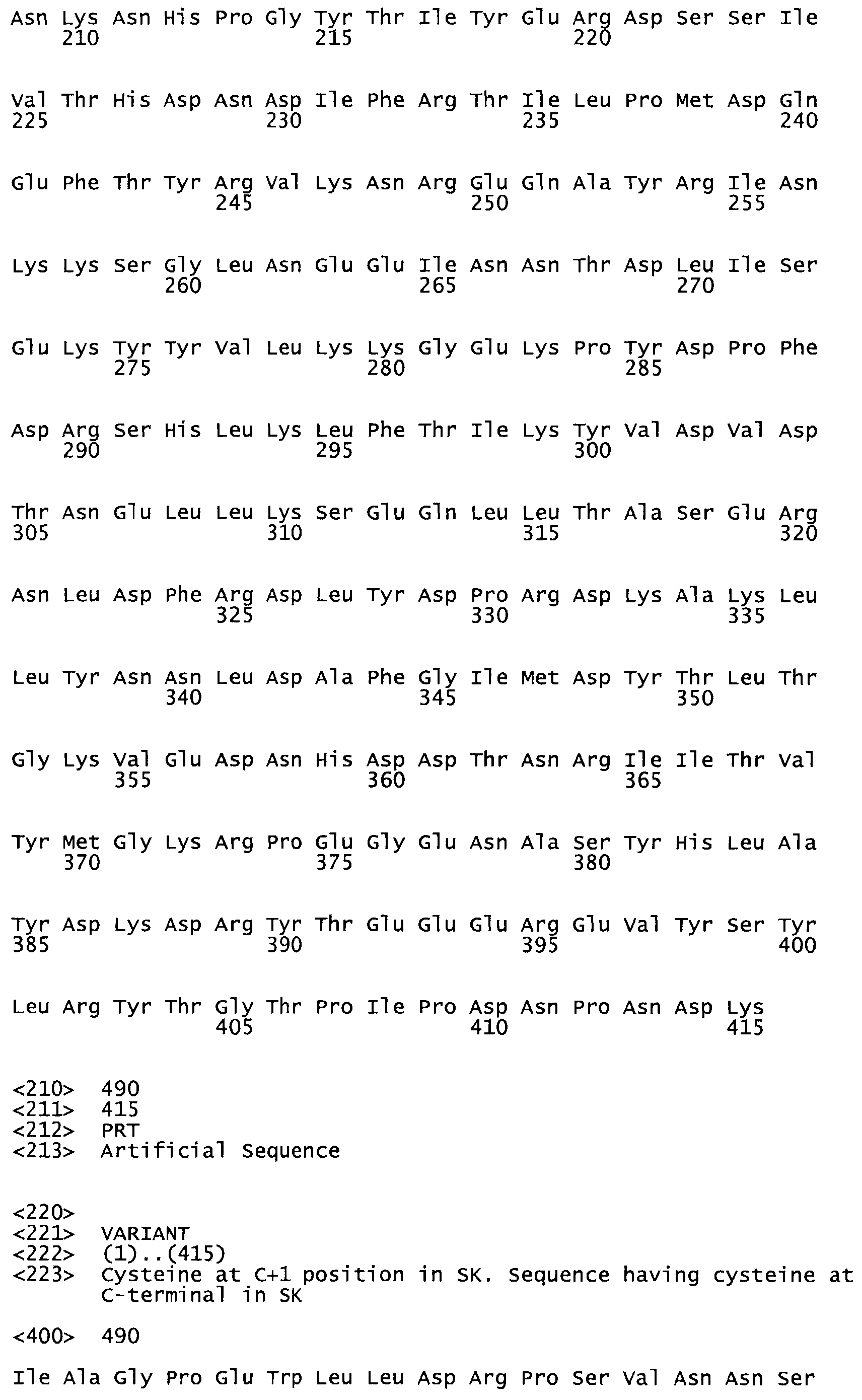
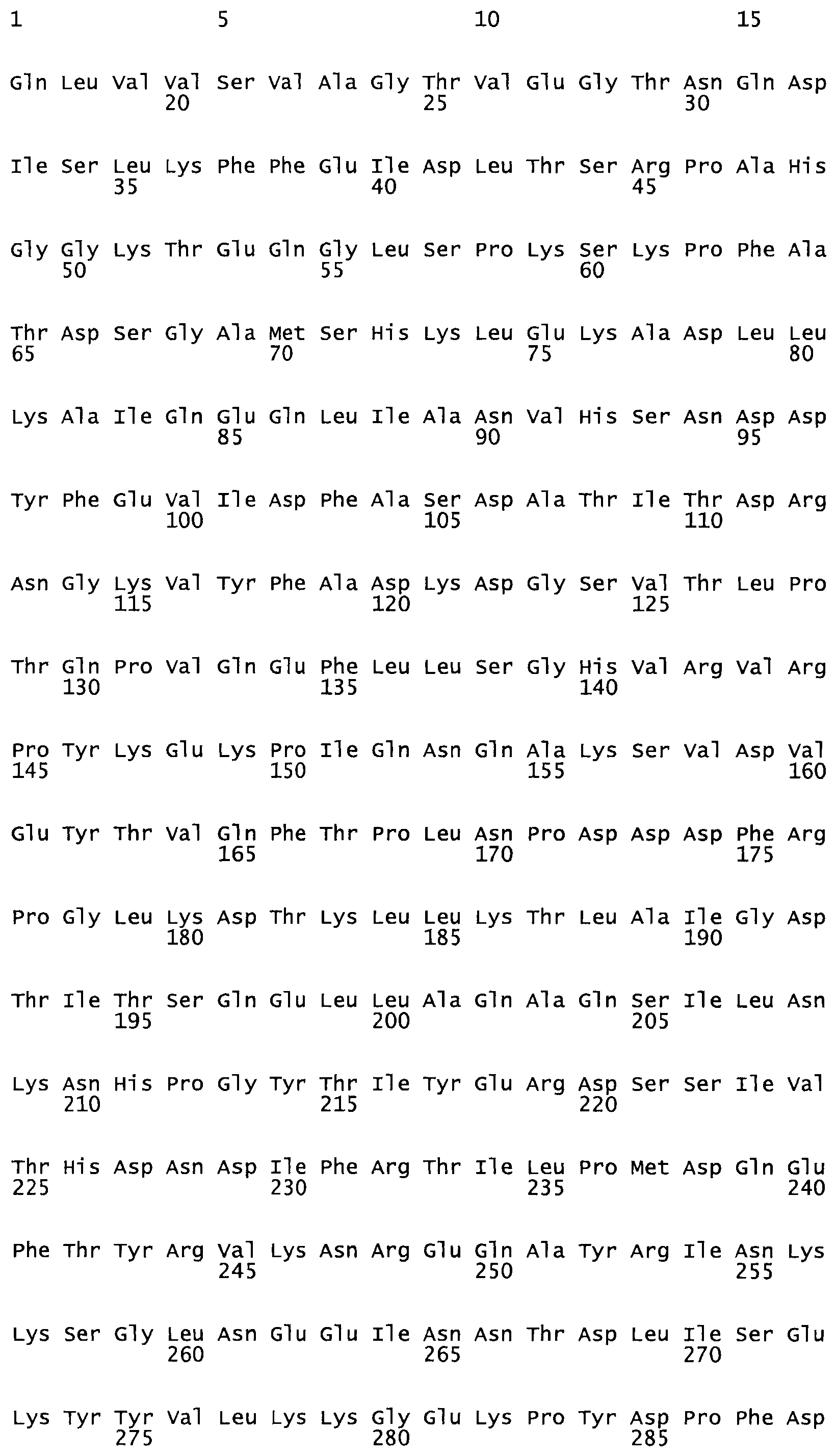

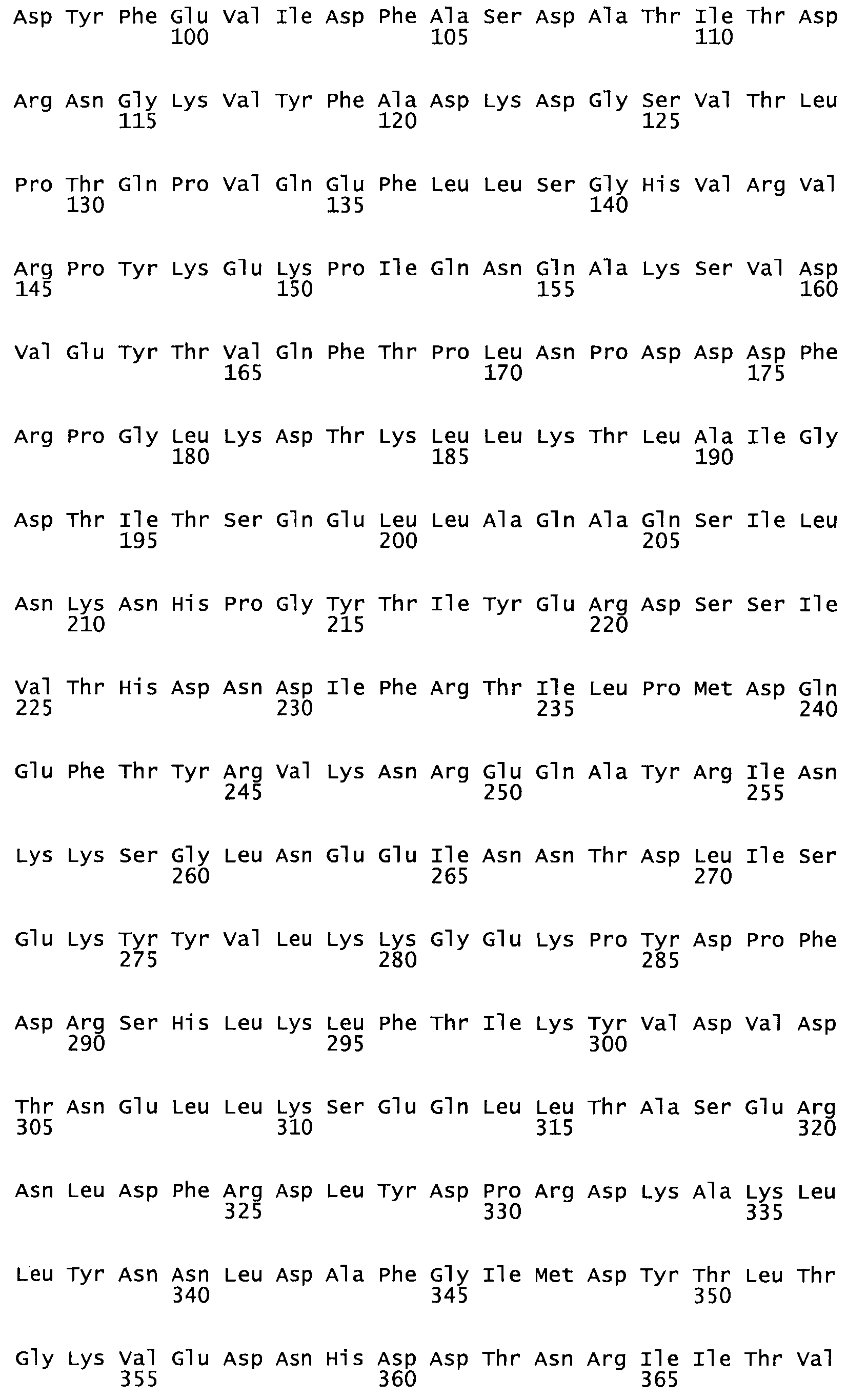


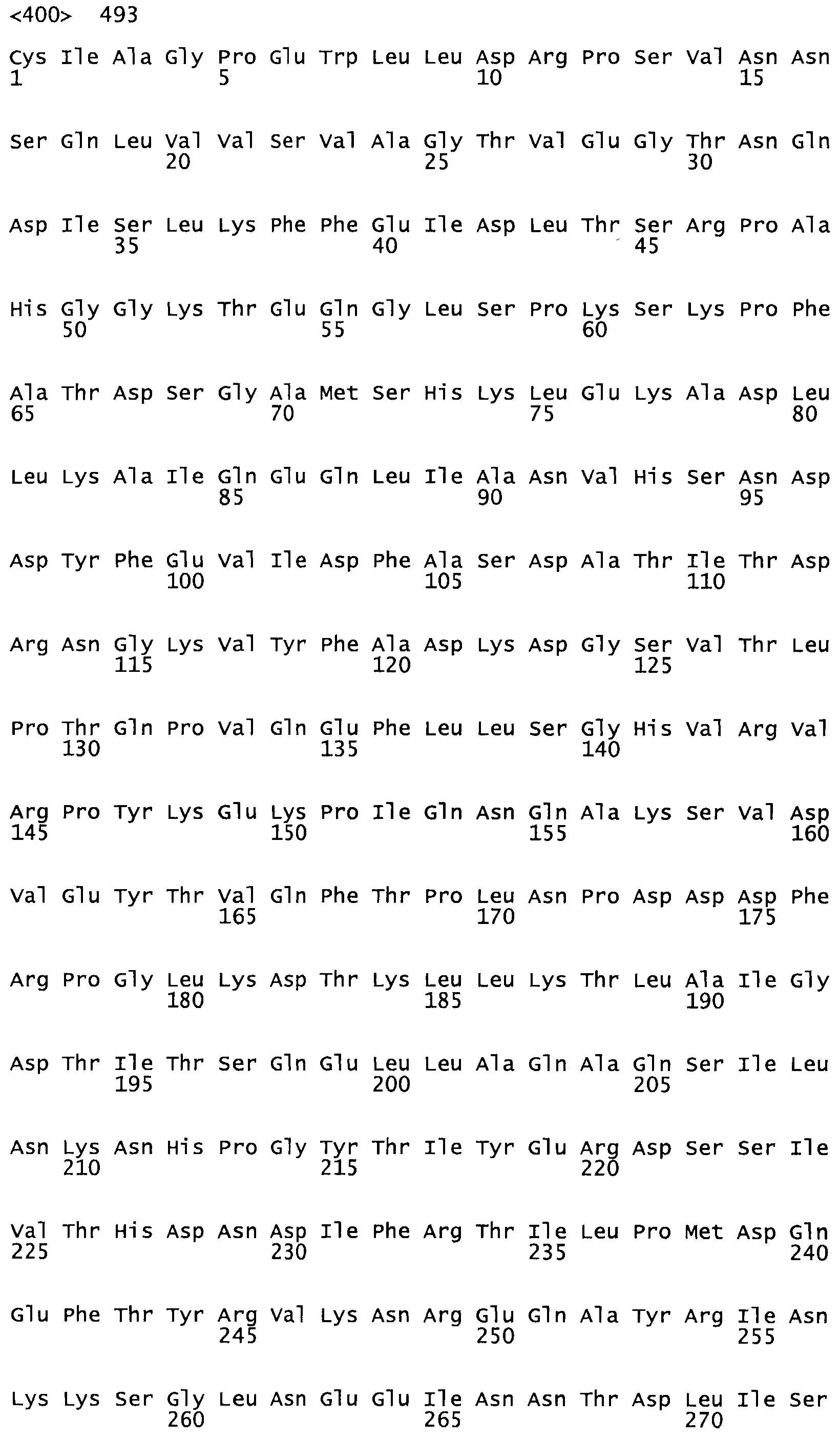

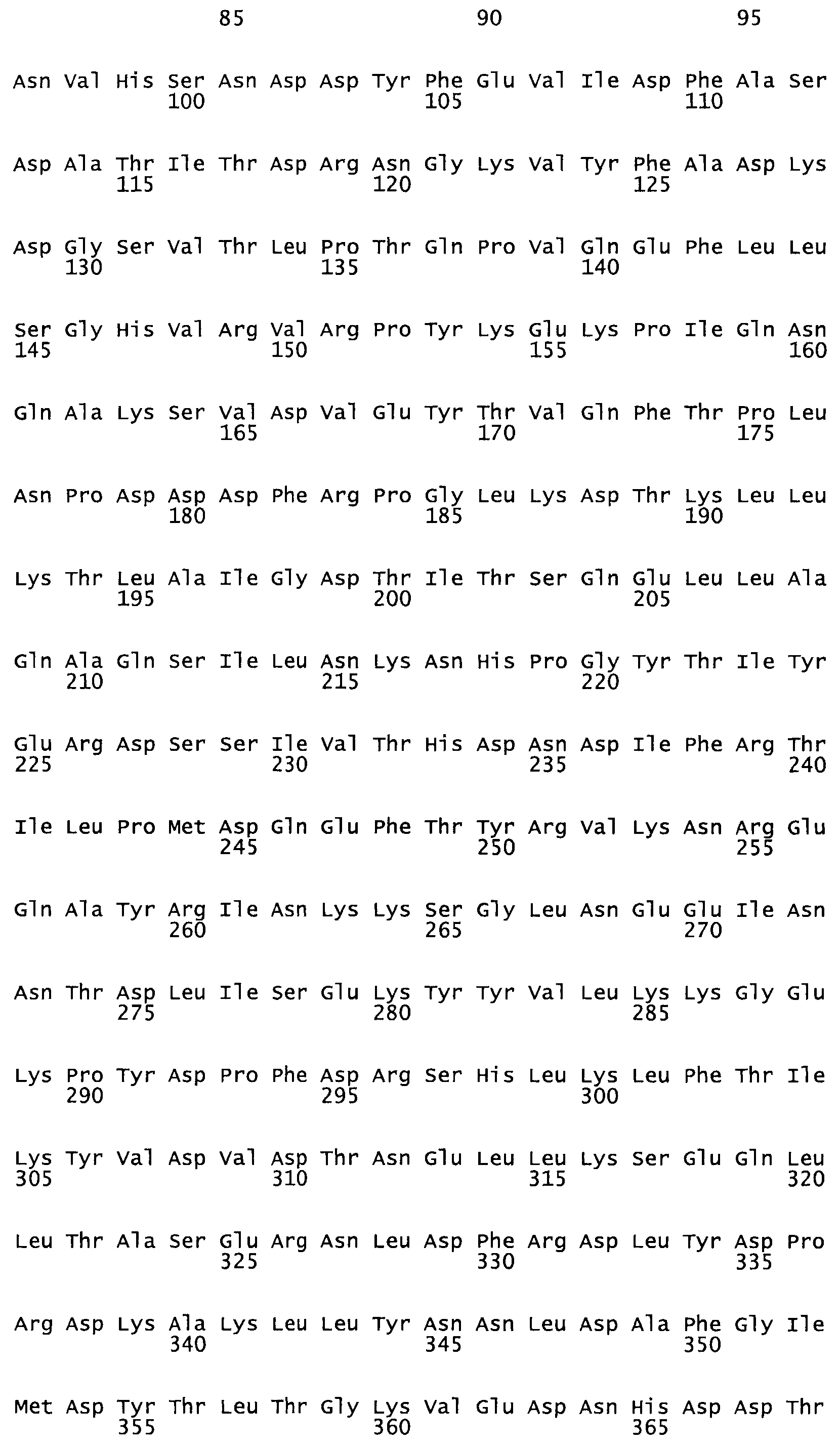

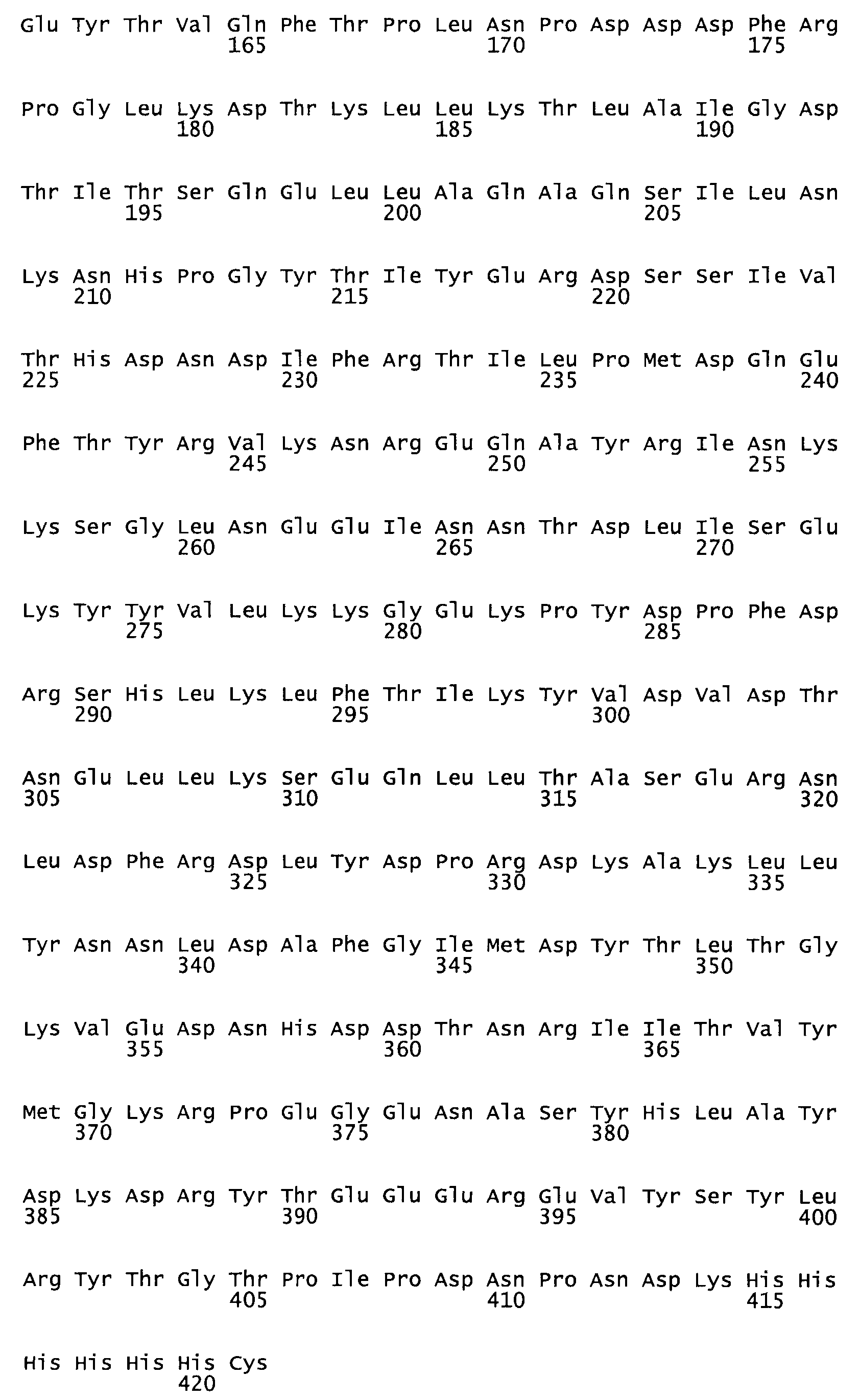
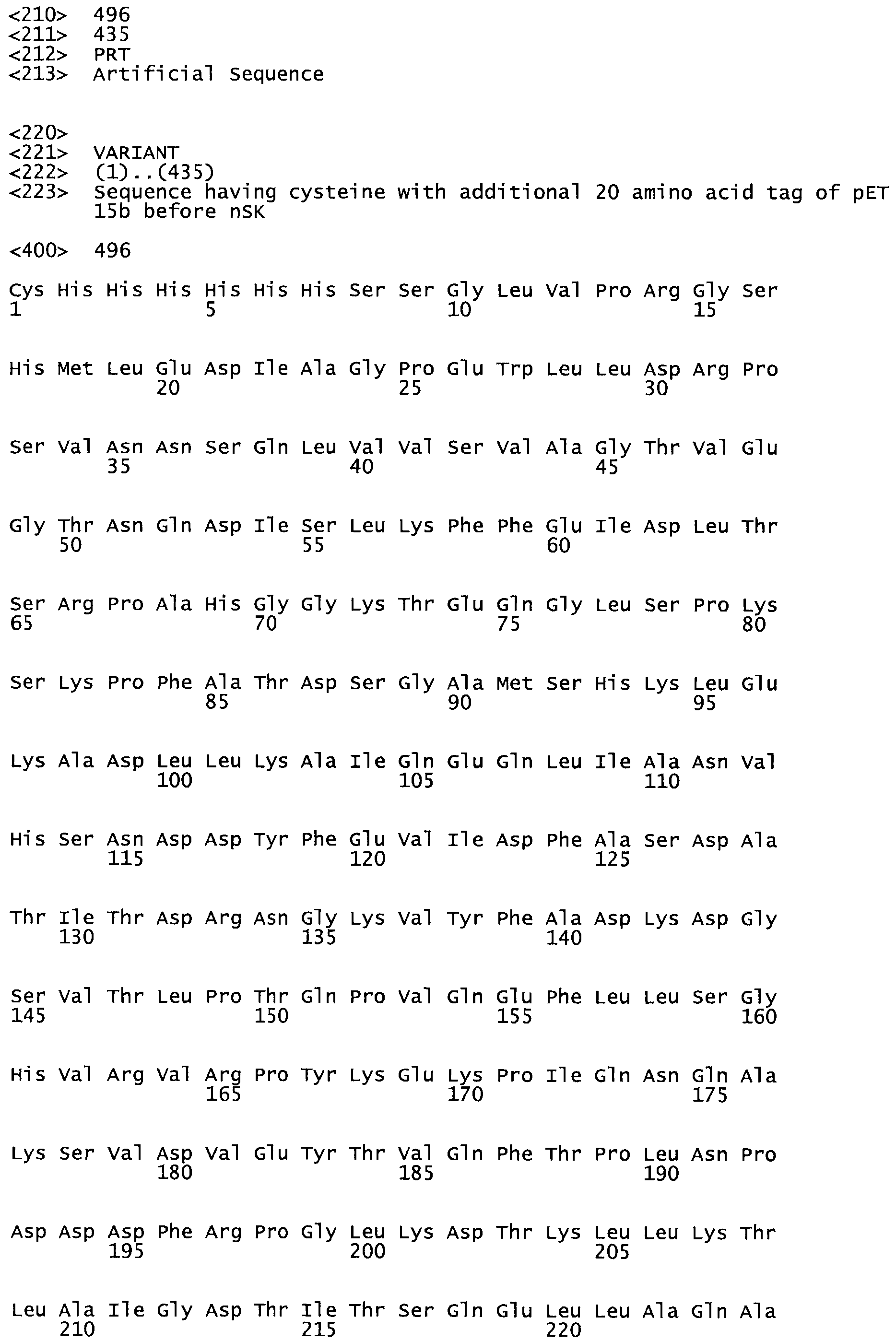
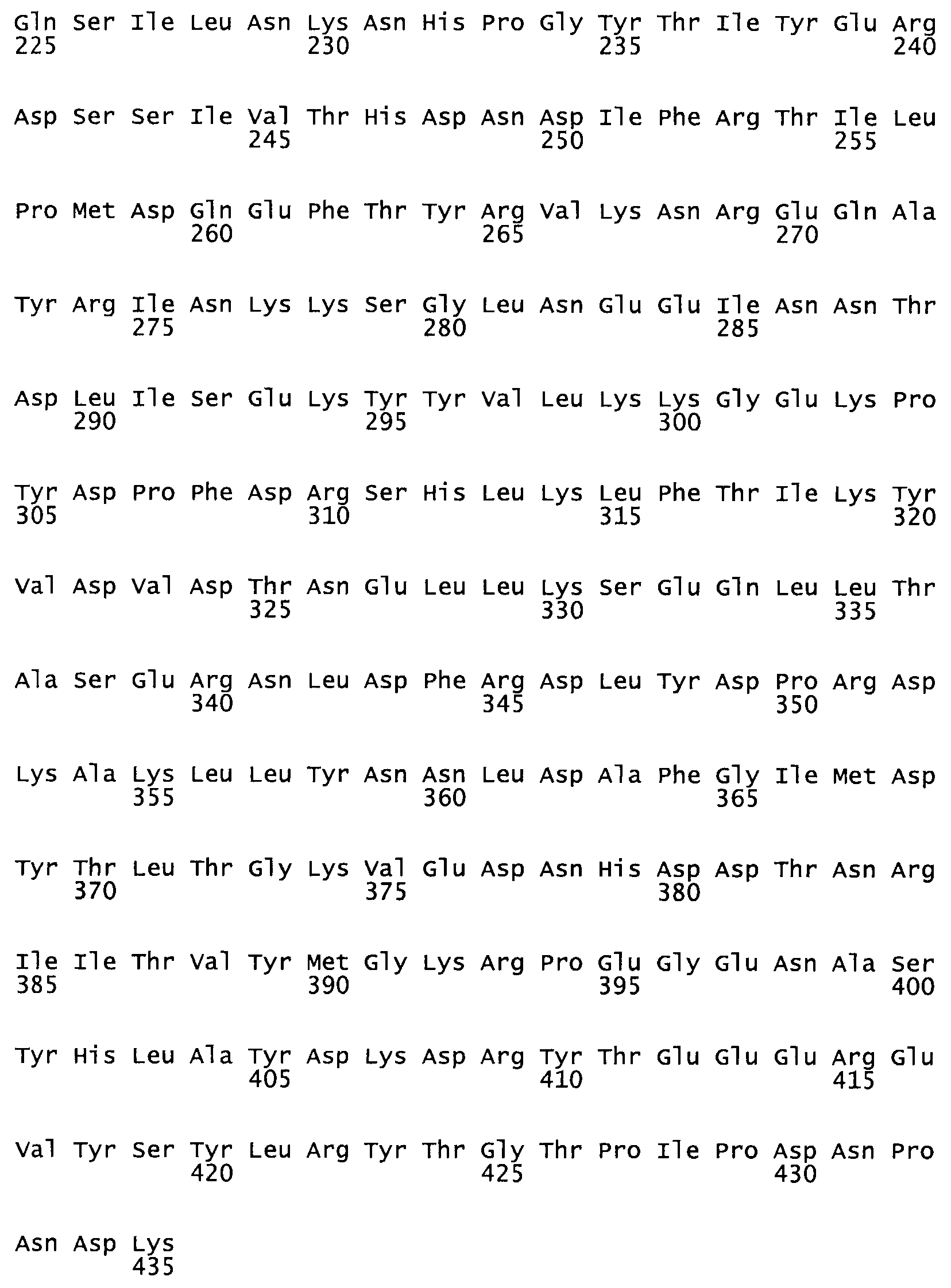
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к мутантам стрептокиназы и их ковалентно модифицированным формам. Настоящее изобретение использует стрептокиназу и ее родственные варианты, полученные заменой, присоединением, удалением или созданием конструкций за счет слияния доменов, модифицированные ПЭГ гомогенно, сайт-специфично и определенным образом, что позволяет использовать их в качестве улучшенных белковых терапевтических средств.
Настоящее изобретение относится к ковалентному присоединению ПЭГ к цистеиновым вариантам стрептокиназы, ее мутеинам, видовым вариантом или продуктам слияния с фибрином, при использовании реагентов на основе производных ПЭГ, реагирующих с тиольными группами. Могут быть также использованы различные значения pKa альфа-аминогрупп для проведения специфического конъюгирования ПЭГ при кислых рН с целью получения монопэгилированных вариантов стрептокиназы или ее мутеинов.
Настоящее изобретение относится также к идентификации различных цистеиновых вариантов стрептокиназы или ее мутантов, в том числе родственных ковалентных вариантов на основе структурной и функциональной информации (Wang et al., 1998). Структурное сравнение с другим однодоменным активатором плазминогена, стафилокиназой (SAK), указывает на значительное сходство с альфа-доменом СК, несмотря на то что оба белка не обладают значительной гомологией последовательностей на аминокислотном уровне (Rabijns et al., 1997). Это указывает на то, что активаторы плазминогена из различных источников сохраняют одинаковый тип структурной укладки, даже если они значительно различаются по своей полипептидной последовательности. Можно ожидать, что эволюционное ограничение удерживает структурную целостность, поскольку активаторы бактериального плазминогена являются белковыми кофакторами, следовательно, они используют множество точек контакта, необходимых для конформационной активации зимогена. Такое структурное подобие еще больше увеличивает масштаб настоящего изобретения, поскольку методы, используемые в данном случае, могут быть применены к активаторам бактериального плазминогена других типов или видов, которые участвуют в подобной активации плазминогена. Поэтому «правила», приводимые здесь для получения биологически активных цистеиновых вариантах стрептокиназы, будут использованы для получения биологически активных цистеиновых вариантов различных других форм стрептокиназы. Конъюгирование этих цистеиновых вариантов с производными ПЭГ, способными реагировать с цистеином, приводит к тем же преимуществам, которые получают при использовании производных в настоящем исследовании. Принципы определения сайта, в котором находится цистеин, главным образом основываются на идее выбора остатков, попадающих либо в петлю, либо в спираль, либо в пограничные области между структурированными и гибкими участками. Для определения поверхностной доступности использована программа DSSP. Программа DSSP (Kabsch et al., 1983) определяет вторичную структуру, геометрические свойства и доступность белков по отношению к растворителям, задаваемую атомными координатами в формате Protein Data Bank. DSSP устанавливает доступность каждого остатка в квадратных ангстремах. Поверхностная доступность аминокислотных остатков стрептокиназы была расшифрована из данных по кристаллической структуре высокого разрешения для стрептокиназы в комплексе с микроплазмином (Wang et al., 1998, PDB ID 1BML). Для областей, которые были пропущены в этой структуре (175-181 и 252-262) при определении поверхностной доступности используют кристаллическую структуру изолированных бета-доменов (Wang et al., 1999, PDB ID 1c4p).
Цистеиновые варианты стрептокиназы, ее мутеины, производные видов и ее ковалентно модифицированные формы далее химически модифицируют присоединением реагентов на сульфгидрильные группы с последующим эмпирическим тестированием на существенное сохранение биологической активности наряду с приобретением новых свойств, таких как пониженная иммуногенность или реакция с анти-СК антителами, пониженная чувствительность к протеолизу, увеличенное выживание in vivo и т.д. Более конкретно, настоящее изобретение относится к получению сконструированных вариантов стрептокиназы с целью применения для использования в фармацевтических композициях для лечения сосудистых заболеваний.
Уровень техники
Образование тромба (кровяного сгустка) в кровеносной системе может вызвать закупорку сосудов, приводящую к фатальным последствиям. Образование сгустка и его растворение представляет собой строго контролируемый процесс гомеостаза. Любое отклонение от нормального свертывания крови приводит к различным клиническим патологическим состояниям, таким как инсульт, эмболия сосудов легких, тромбоз глубоких вен и острый инфаркт миокарда. Патофизиологические состояния, развивающиеся в результате нарушения процесса свертывания крови, требуют незамедленного клинического вмешательства. Наиболее часто практикуемое медицинское вмешательство состоит во введении тромболитических агентов (Collen et al., 1988; Collen, 1990; Francis and Mafder, 1991). Наиболее часто используемые тромболитические агенты включают в себя стрептокиназу СК (SK), урокиназу (UK) и тканевой активатор плазминогена (ТРА). Ранее были проведены многочисленные фармакоэкономические оценки использования различных тромболитиков для коррекции острого инфаркта миокарда (Mucklow, 1995; Gillis and Goa, 1996). Banerjee et al., 2004, рассмотрел клиническое использование стрептокиназы и ее применение в качестве лучшего выбора препарата для лечения. Что касается клинической эффективности, то как стрептокиназа, так и ТРА в равной степени являются хорошими препаратами, но в связи с тем, что стрептокиназа на несколько порядков дешевле и несколько лучше в отношении периода полураспада in vivo, она во всем мире является наиболее предпочтительным тромболитиком (Sherry and Marder, 1991, Wu et al., 1998). Кроме того, использование ТРА несколько более вероятно вызывает инсульт, наиболее частый побочный эффект для обоих лекарств. Однако, стрептокиназа, будучи бактериальным белком, является по природе антигенной и может вызывать клинические осложнения, например, аллергическую реакцию или кровотечение. Кроме того, время полужизни циркуляции стрептокиназы (15-30 мин) недостаточно для эффективного тромболиза (Wu et al., 1998).
Несмотря на все это в последнее время тромболитическая терапия с помощью фибринолитических агентов, например, стрептокиназы (СК), тканевого активатора плазминогена (ТРА) или урокиназы (UK) полностью изменила клиническое применение различных заболеваний органов кровообращения, например, тромбоза глубоких вен, эмболии сосудов легких и острого инфаркта миокарда. Эти агенты проявляют свои фибринолитические свойства за счет активации плазминогена ПГ (PG) при циркуляции путем расщепления лабильных пептидных связей между остатками 561 и 562 в ПГ. В результате неактивный зимоген переходит в активную форму, сериновую протеиназу, плазмин ПН (PN), который затем циркулирует в системе и действует на фибрин, разрушая последний с образованием растворимых продуктов распада. Здесь следует упомянуть, что сам PN не способен активировать ПГ до ПН; эта реакция катализируется высоко специфическими протеазами, подобными ТРА, комплексом СК-плазминоген и UK, все они обладают необычайно высокой специфичностью по отношению к белковым субстратам, а именно способностью расщеплять лабильную пептидную связь в ПГ высоко специфическим образом. Однако, в отличие от UK и ТРА, СК не обладает собственной протеолитической активностью и активирует ПГ в ПН "косвенно", т.е. вначале образуя высокоафинный эквимолярный комплекс с ПГ, известный как активаторный комплекс (рассмотренный Castellino, F.J., 1981). Затем активаторный комплекс действует как протеаза, которая расщепляет другие молекулы субстрата ПГ до ПН.
Несмотря на огромные преимущества терапевтического использования стрептокиназы и других бактериальных тромболитиков имеется несколько недостатков, ограничивающих применимость этих полипептидных лекарств. Эти недостатки включают в себя их подверженность деградации протеолитическими ферментами, малое время полужизни циркуляции, малый срок хранения, быстрое выведение почками и их способность генерировать нейтрализующие антитела. Эти недостатки также иногда присущи многим другим полипептидным лекарствам, по своей природе отличающимся от полипептидов человека. Этот аспект в общем виде рассмотрен Roberts et al.; 2002. Были сделаны различные попытки устранить эти недостатки полипептидных лекарств, например, изменением аминокислотных последовательностей для снижения протеолиза или антигенности, присоединением полипептидов к доменам глобулина или альбумина для увеличения времени полужизни (Osborn et al., 2002).
Эти способы приносят мало пользы для решения проблемы и сопровождаются дополнительными затратами. Основным прорывом в этой области является пэгилирование белков, обеспечивающее единственное решение множества проблем. ПЭГ (полиэтиленгликоль) образуется в результате полимеризации нескольких повторяющихся субъединиц этиленгликоля, приводящей к линейным или разветвленным полимерам ПЭГ заранее запланированных молекулярных масс. Будучи ковалентно конъюгированными с ПЭГ, белки или полипептиды проявляют улучшенные фармакокинетические или фармакодинамические свойства, такие как увеличение растворимости в воде, уменьшение почечного клиренса и часто существенное уменьшение иммунореактивности (Moreadith et al., 2003, Doherty et al., 2005, Basu et al., 2006). Конъюгирование с ПЭГ также делает молекулу менее подверженной протеолизу. Уменьшение взаимодействия с рецепторами или взаимодействия с поверхностными белками клеток, обусловленное присоединением ПЭГ, также способствует подавлению неблагоприятных иммунологических эффектов. Пэгилированные лекарства также более стабильны в широком интервале рН и изменения температуры (Monfardini et al. 1995). Применение ПЭГ одобрено FDA для терапевтических средств и показано, что он практически не обладает токсичностью и выводится из организма в неизменном виде либо через почки, либо вместе с калом. Полезные свойства конъюгирования с ПЭГ могут быть потенциально сообщены СК для того, чтобы сделать его более эффективным и безопасным тромболитиком. Попытка пэгилирования СК при использовании относительно неспецифической реакции химической модификации описана в литературе (Rajagopalan et al., 1985). Терапевтические применения таких модификаций строго ограничены высокой аномальной способностью активации плазминогена. Кроме того, слабо контролируется природа модификации, которая может быть гетерогенной. Причиной такой гетерогенности являются химические превращения, используемые для модификации ПЭГ, не являющиеся адресной модификацией специфического сайта. Поэтому такую стратегию модификации не используют для получения усовершенствованных тромболитиков на основе СК.
Термин стрептокиназа, используемый везде обобщенно в тексте, относится к вариантам стрептокиназы, любому из ее функциональных фрагментов, функциональным мутеинам, изолятам из различных образцов и продуктов конденсации, полученных присоединением олиго- или полипептидов природного или синтетического происхождения.
Известно, что различные функциональные группы, находящиеся в молекуле белка, могут быть использованы для введения ПЭГ. Наиболее часто используемыми способами является дериватизация лизиновых остатков или цистеиновых остатков в белках. Альфа-аминогруппа в N-концевом фрагменте также может быть использована для единичного гомогенного конъюгирования ПЭГ в белках (Baker et al., 2006). Однако, использование цистеиновых остатков для присоединения групп ПЭГ является особенно предпочтительным, т.к. потенциально SH-группы могут быть использованы в качестве мишени сайт-специфическим образом, особенно если белок содержит или может быть устроен таким образом, чтобы он содержал очень ограниченное число цистеиновых остатков. Не будет преувеличением считать, что конъюгирование с ПЭГ становится видом искусства, когда белок лишен любых цистеинов, т.к. он составляет виртуальный чистый холст для присоединения, введения цистеина или замены цистеином с целью сайт-специфического «раскрашивания» ПЭГ, или декорации, белков. Поскольку потенциально присоединение цистеинов к не содержащему цистеина субстрату может оказывать неблагоприятное действие на функцию белка. Поэтому выбор сайтов для получения цистеиновых вариантов требует тщательного планирования и исполнения. В противоположность, скажем, пэгилированию, основанному на модификации лизина, хотя химизм этой реакции и хорошо известен, гетерогенность является существенным недостатком. В случае СК большое число лизиновых остатков равномерно распределено вдоль полипептида и поэтому ограничивают возможность гомогенного сайт-специфического ПЭГ конъюгирования. Интереснее, что в молекуле стрептокиназы нет природного цистеина (Malke et al., 1985), благодаря чему становится возможным получение различных вариантов стрептокиназы. Кроме того, в нативно свернутых ковалентных производных СК, образующихся при слиянии с фибринсвязывающими доменами (ссылка US Patent 7163817). Это дает возможность получения различных вариантов специфичной к тромбам стрептокиназы, содержащей свободный цистеин, в отсутствие действительных факторов, мешающих нормальному сворачиванию белка, богатого цистеином (все цистеиновые остатки участвуют в образовании дисульфидной связи). Вводимый свободный цистеин(ы) может взаимодействовать с различными реагентами на тиольные группы, в том числе с ПЭГ, с образованием цистеиновых аддуктов этих белков.
Стрептокиназа (СК) является видовым обозначением секреторного белка, образованным множеством гемолитических стрептококков, способных индуцировать лизис плазменных сгустков (Tillet and Gamer, 1933). Поэтому она может легко и экономично быть получена из ее основного источника или при использовании рДНК технологии из подходящих гетерологических хозяев, СК очень рентабельна и поэтому является основным тромболитическим лекарственным соединением, особенно для мировых рынков, чутко реагирующих на цену. Показано, что СК очень эффективна при клиническом лечении острого инфаркта миокарда, сопровождающегося тромбозом коронарных артерий (1SIS-3, 1992), и выступает в роли тромболитического агента в течение более трех десятилетий. Однако, она имеет ряд недостатков. Известно, что плазмин, образующийся при активации плазминогена, опосредованной стрептокиназой, расщепляет стрептокиназу вскоре после ее введения (Rajagopalan et al., 1985, Wu et al., 1998). Это ограничивает время полужизни стрептокиназы in vivo приблизительно до 15 мин (Wu et al., 1998). Хотя стрептокиназа остается в кровотоке значительно дольше, чем выбранное другое тромболитическое лекарственное средство, ТРА (со временем полужизни, равным менее 5 мин; Ross, 1999; Ouriel, 2002), все же этого недостаточно для эффективной терапии (Wu et al., 1998). Из-за обнаруженных недостатков, связанных с быстрым выведением in vivo имеющихся активаторов плазминогена, предприняты попытки получать усовершенствованные рекомбинантные производные этих соединений (Nicolini et al., 1992, Adams et al., 1991, Lijnen et al., 1991; Marder, 1993, and Wu et al., 1998). Несмотря на свои внутренние проблемы стрептокиназа остается предпочтительным лекарственным средством, особенно в развивающихся странах, из-за ее относительно низкой стоимости (например, приблизительно US $ 50 или меньше на курс лечения по сравнению почти с US $ 1500 для ТРА).
Впервые показано, что стрептокиназа вызывает лизис кровяных сгустков (Tillet and Gamer (1933)). Однако, позже было показано, что фибринолитическая активность СК обусловлена ее способностью активировать плазминоген человека ПГЧ (HPG), описанная Castellino, 1979. Стрептокиназа главным образом секретируется β-гемолитической группой А, С и G стрептококки. СК представляет собой активатор ПГ человека, хотя сама она не является протеазой, скорее она связывается с ПГ/ПН человека и привлекает другие молекулы HPG в качестве субстрата и превращает их в продукт, ПН. Последний циркулирует в кровотоке. Плазмин, будучи неспецифической протеазой, обобщенной и промежуточной генерацией ПН, после инъекции СК приводит к широкомасштабному разрушению различных факторов крови, что приводит к риску кровотечения, а также к растворению ЕСМ и базальной мембраны (ВМ) и к увеличению бактериальной инвазивности во вторичные центры зараженности в теле хозяина (Esmon and Mather, 1998; Uihteenmaki et al., 2001). Таким образом, имеется острая необходимость минимизировать побочные эффекты конструированием усовершенствованных аналогов СК.
В настоящее время СК интенсивно используют в качестве тромболитического лекарственного средства во всем мире, т.к. она является эффективным растворителем фибриновых сгустков, однако она имеет собственные ограничения. СК является белком, продуцируемым из гемолитической стрептококки, его использование в организме человека индуцирует иммуногенность (McGrath and Patterson, 1984; McGrath et al., 1985; Schweitzer et al., 1991). Известно, что высокие титры анти-СК иммуноглобулинов (Ig), генерируемые после первого введения СК, остаются у пациентов в течение периода от нескольких месяцев до нескольких лет (Lee et al., 1993). Таким образом, анти-СК антитела существенно ограничивают ее применение в качестве пролонгированной терапии либо из-за нейтрализации СК при введении (Spottal and Kaiser, 1974; Jalihal and Morris, 1990), либо из-за того, что она вызывает ряд аллергических реакций (McGrath and Patterson, 1984; McGrath et al., 1985).
Как уже упоминалось выше, использованию стрептокиназы в тромболитической терапии препятствует относительно короткое время полужизни (несколько минут) этого белка in vivo (что в действительности наблюдается для всех используемых в настоящее время тромболитических лекарственных средств), помимо ее иммуногенности. Показано, что чужеродные белки при введении в кровоток позвоночных часто быстро выводятся через почки. Эта ситуация становится еще более острой в случае стрептокиназы, когда постепенно увеличивающиеся дозы белка (для того, чтобы превысить быструю нейтрализацию, обусловленную антителом) могут значительно увеличить вероятность аллергического ответа/ов, что делает повторное введение в значительной мере неэффективным и опасным. Обычно попытки решить эти проблемы в литературе хорошо подтверждены документально, где показано, что для получения усовершенствованных лекарственных веществ целесообразны различные физические и химические модификации, например, смотри: Mateo, С. et al., 2000, Lyczak, J.В. & Morrison, S.L. 1994, Syed, S. et. al.; 1997, Allen, Т.М. 1997. Наиболее перспективной из них в настоящее время является модификация терапевтических белков ковалентным присоединением полиалкиленоксидных полимеров, например, полиэтиленгликолей (ПЭГ). ПЭГ представляет собой неантигенный, инертный полимер и известно, что он увеличивает время полужизни циркуляции белков в организме (Abuchowski et al., 1984; Hershfield, 1987; Meyers et al., 1991). Это обеспечивает продолжительное действие лекарственных веществ при их использовании. Считают, что конъюгирование ПЭГ с белками увеличивает их общий размер и поэтому уменьшает быстрое почечное очищение. Кроме того, присоединение ПЭГ также придает белку или полипептиду большую растворимость в воде и увеличивает их стабильность в условиях in vivo наряду со значительным уменьшением иммуногенности и увеличением стабильности in vivo (Katre et al., 1987; Kcitre, 1990). U.S. Pat. №. 4,179,337 раскрывает применение ПЭГ или полопропиленгликоля, присоединенных к белкам, для получения физиологически активной неиммуногенной водорастворимой полипептидной композиции.
Несмотря на то что химизм конъюгирования ПЭГ является, в основном, универсальным, ключевое расположение полимеров ПЭГ в терапевтическом белке имеет первостепенное значение для достижения успешных результатов. Доступность информации о трехмерной структуре с функциональными активными областями, выявленными благодаря различным исследованиям в растворе, помогает в разработке плана введения мутаций, чтобы сохранить функциональность неизменной.
Полная аминокислотная последовательность СК определена последовательным деградационным анализом по Эдману фрагментов СК, полученных бромциановым и ферментативным способами (Jackson and Tang, 1982). Результаты показали, что молекула с молекулярной массой, Mr, равной 47408 Да, содержит 415 аминокислот в единичной полипептидной цепочечной аминокислотной последовательности.
Нуклеотидная последовательность из S.equisimilis H46A (штамм прототипа для получения СК, которую чаще всего используют терапевтически для лечения людей) была секвенирована Malke с сотрудниками в 1985. Был также изучен транскрипционный контроль этого гена и описан функциональный анализ его комплексного промотора (Grafe et al., 1996). Следовательно, имеется значительная информация для эффективного использования этого гена, для того чтобы с уверенностью получать стрептокиназу в относительно непатогенных микробах.
Цель изобретения
Основной целью настоящего изобретения является обеспечение мутантов стрептокиназы, обладающих повышенной эффективностью благодаря более продолжительному действию и пониженной иммунореактивности.
Другой целью настоящего изобретения является селективная модификация стрептокиназы для улучшения ее фармакокинетических свойств и придания ей свойств терапевтически эффективного тромболитика и увеличения протеолитической стабильности и увеличения времени полужизни in vivo.
Еще одной целью настоящего изобретения является селективная модификация стрептокиназы с тем, чтобы сохранить желаемые биологические свойства, присущие немодифицированной молекуле.
Еще одной целью настоящего изобретения является обеспечение производного СК, которое обеспечивает продолжительный и более эффективный фибринолиз в определенном сайте при пониженной иммунореактивности.
Еще одной целью настоящего изобретения является обеспечение способа получения пэгилированных цистеиновых вариантов стрептокиназы или ее активных мутеинов или молекул гибридных молекул активаторов плазминогена в чистой или биологически активной форме.
Раскрытие изобретения
Следовательно, настоящее изобретение обеспечивает мутанты стрептокиназы, ее функциональные фрагменты или ковалентно модифицированные формы. Производные содержат полипептиды, относящиеся к СК, где один или несколько остатков цистеина замещены одной или несколькими заменимыми аминокислотами белков предпочтительно, производные содержат остаток цистеина, замещенный аминокислотой, выбранной из аминокислот, входящих в области петли, концов альфа-спиралей и даже в области, образующие вторичную структуру, или в области, где остатки цистеина присоединены к N-концу или С-концу молекулы белка, при наличии или отсутствии аминокислотных достроек.
Настоящее изобретение включает в себя общие способы селекции, получения и применения стрептокиназ, проявляющих повышенную протеолитическую стабильность, увеличенное время полужизни из плазмы и пониженную иммуногенность. Производные имеют модифицированные аминокислотные последовательности, но фактически сохраняют свою биологическую активность. Настоящее изобретение также описывает цистеиновые варианты стрептокиназы, ковалентно связанные с одной или несколькими молекулами полиэтиленгликоля (ПЭГ) различных молекулярных масс, например, 5, 10, 20 и 40 кДа. Один из вариантов осуществления настоящего изобретения касается фармацевтических композиций вариантов пэгилированной стрептокиназы вместе с подходящими инертными наполнителями, стабилизаторами и носителями, известными в данной области техники, для эффективного распределения в организме при лечении различных заболеваний кровеносной системы.
Осуществление изобретения
Настоящее изобретение основано на экспериментальных результатах, показывающих, что ковалентное присоединение одной или нескольких молекул ПЭГ к стратегически замещенным или добавленным остаткам цистеина в молекуле стрептокиназы приводит к биологически активной пэгилированной стрептокиназе с повышенной протеолитической стабильностью и увеличением времени полужизни и меньшей иммунореактивностью по сравнению со нативной стрептокиназой. Локализация и число молекул при ПЭГ конъюгировании могут быть специально подобраны при помощи цистеиновых вариантов, сконструированных таким образом, чтобы они не могли ингибировать каталитическую функцию (активацию плазминогена) в стрептокиназе и ее активных вариантов, в том числе в стрептокиназах, специфичных к тромбам (в данном контексте можно сослаться на U.S. Patent 7163817, в котором описано устройство и конструкция вариантов стрептокиназ, специфичных к тромбам, с повышенной фибринолитической активностью благодаря введению фибринсвязывающих доменов по любому или обоим концам молекулы белка СК). Замена, присоединение или внедрение одного или нескольких молекул цистеина в стрептокиназу и полипептиды стрептокиназы, специфичной к тромбам, создает условия для нужной локализации при присоединении ПЭГ различных молекулярных масс к полипептидам при условии, что замена и/или присоединение проводят "стратегически" полностью, избегая потери функциональных свойств, и приводит к получению новых полезных свойств, отсутствующих в немодифицированном нативном белке. Выбор места расположения ПЭГ проектируют, исходя из поверхностной доступности выбранного сайта и его структурно-функциональной значимости.
Пэгилированные стрептокиназы настоящего изобретения обладают большей применимостью в качестве терапевтических средств, а также большими преимуществами при использовании по сравнению с нативной молекулой(ами), поскольку сохраняя нативную или почти нативную биологическую активность, они имеют увеличенный латентный период по сравнению с первыми, которые быстро распадаются in vitro в присутствии плазмина (плазминогена), а также после введения, в результате чего они очень быстро выводятся из кровотока. Напротив, пэгилированные стрептокиназы проявляют значительно более высокую протеолитическую стабильность, являются иммунореактивными и выводятся из кровотока in vivo только после заметно увеличенного периода времени по сравнению с нативными (непэгилированными) белками.
Следовательно, пэгилированные стрептокиназы настоящего изобретения эффективны для лечения субъектов с нарушением кровообращения, например, с венозными или артериальными тромбозами, инфарктом миокарда и т.д., имея то преимущество, что пэгилированные стрептокиназы настоящего изобретения создают потенциал для увеличения эффективности благодаря увеличению срока действия и уменьшению иммунореактивности, что дает возможность повторного введения благодаря минимальной антигенности. Число, размер и локализация групп(ы) ПЭГ могут быть использованы таким образом, чтобы реконструировать стрептокиназы для реализации различных времен полужизни, что позволяет использовать их соответственно требованиям конкретного заболевания/клинического синдрома, или определенного больного.
Настоящее изобретение включает в себя селективную модификацию стрептокиназ для фармацевтического применения как для того, чтобы усилить их фармакокинетические свойства, так и для того, чтобы обеспечить терапевтически эффективные тромболитики. Это изобретение также включает в себя мутанты стрептокиназы, ее природные или искусственные варианты, которые сохраняют желаемые биологические свойства нативной немодифицированной молекулы. Все производные настоящего изобретения могут быть получены экспрессированием последовательности рекомбинантной ДНК, кодирующей нужное производное в клетках хозяина, например, в прокариотических клетках хозяина, например, E.coli или в эукариотических клетках хозяина, например, дрожжевых клетках или клетках млекопитающих, при использовании обычно используемых материалов, известных в данной области техники. Информация по последовательности ДНК для закодированной стрептокиназы из различных препаратов может быть получена из опубликованных данных. Изучен полиморфизм гена стрептокиназы и объяснена его роль в патогенезе (Malke Н, 1993). Также проведено молекулярное эпидемиологическое исследование для определения распределения гена стрептокиназы в группе А стрептококковых штаммов различных М типов и других препаратов стрептококка. Большинство изученных штаммов в этом исследовании проявило положительную стрептокиназную активность по данным оверлейного теста казеин-плазминоген. Суммарные результаты этих исследований показывают, что существует значительная гетерогенность среди стрептокиназ, полученных из различных стрептококковых препаратов (Huang et al.; 1989). Возможно использование любых доступных вариантов стрептокиназы, которые способны активировать плазминоген для мутагенеза цистеина и последующих модификаций реакционноспособными сульфгидрильными реагентами.
Новые последовательности ДНК, кодирующие мутанты и видовые производные могут быть подобным образом клонированы и экспрессированы, как и в случае природных форм. Затем стрептокиназы, полученные экспрессией в генетически сконструированных клетках хозяина, могут быть очищены и при желании переведены в фармацевтические композиции обычными способами.
В качестве предпочтительного аспекта настоящего изобретения стрептокиназы, экспрессированные рекомбинантным путем, реагируют с целевыми реакционноспособными агентами на тиольную группу в условиях, позволяющих осуществить присоединение остатка, реагирующего с тиольной группой, к сульфгидрильной группе введенных цистеиновых остатков в стрептокиназах.
Термин «реагирующий с тиольной группой» определяется здесь как любое соединение, имеющее реакционноспособную группу или способное активироваться с образованием реакционноспособной группы, способной присоединяться к сульфгидрильной группе (-SH) цистеинового остатка. К числу таких соединений относятся полимеры, например, полипропиленгликоль и ПЭГ, полимеры на основе углеводов и полимерные аминокислоты и производные биотина. Соединение, подлежащее конъюгированию, можно активировать сульфгидрильными остатками, например, сульфгидрильной группой, тиолом, трифлатом, трезилатом, азиридином или оксираном или предпочтительно иодацетамидом или малеимидом. Конъюгирующая группа может иметь различные молекулярные массы, предпочтительно, от 5000 до 40000 для ПЭГ. Одним из важнейших признаков настоящего изобретения является придание позиционной селективности пэгилированию или другим типам присоединения при сохранении функциональных свойств белка.
Таким образом, настоящее изобретение обеспечивает мутантный полипептид стрептокиназы с последовательностью аминокислот, выбранных из группы, состоящей из SEQ ID NO: 1-24, где по меньшей мере один цистеиновый остаток заменен или внедрен. В таблице 34 представлены остатки, соответствующие остаткам SEQ ID NO: 1, которые, возможно, нетолерантны к мутациям или замене.
В одном из вариантов настоящего изобретения полученный мутант стрептокиназы является функциональным фрагментом, имеющим SEQ ID NO: 2-6.
В одном из вариантов настоящего изобретения полученные мутанты стрептокиназы являются мутеинами стрептокиназы, имеющими SEQ ID NO: 7-19.
В одном из вариантов настоящего изобретения полученный мутант стрептокиназы представляет собой видовые варианты стрептокиназы, имеющие SEQ ID NO: 20-21.
В одном из вариантов настоящего изобретения для видовых вариантов стрептокиназы гомология аминокислотной последовательности составляет 75%-100% относительно нативной стрептокиназы, имеющей SEQ ID NO: 1.
В другом варианте настоящего изобретения, по меньшей мере, один цистеиновый остаток, замещенный по меньшей мере одной аминокислотой, расположенной по меньшей мере, в одной области стрептокиназы, выбранной из группы, состоящей из: петли 48-64, петли 88-97, области 103-106 или 119-124 или спиралеобразующей области 196-207, или петлеобразующей области 170-181, или петлеобразующей области 254-264, или суперспиральной области 318-347, или области 360-372 в SEQ ID NO: 1 или ее мутеинов или их функциональных фрагментов, где указанное производное обладает биологической активностью, измеренной стандартным тестом.
* В другом варианте настоящего изобретения полипептид мутанта стрептокиназы содержит от одной до трех цистеиновых замен, где цистеиновая замена представляет собой кислоту, локализованную, по меньшей мере, в одной области, соответствующей нативной последовательности аминокислотных остатков стрептокиназы (SEQ ID NO: 1), в области, выбранной из группы, состоящей из петли аминокислотных остатков 48-64, петли аминокислотных остатков 88-97, области аминокислотных остатков 102-106, области аминокислотных остатков 119-124, спиралеобразующей области аминокислотных остатков 196-207, петлеобразующей области аминокислотных остатков 170-181, петлеобразующей области аминокислотных остатков 254-264, суперспиральной области аминокислотных остатков 318-347 и области аминокислотных остатков 360-372, где мутант может активировать плазминоген.
Так, как используется здесь, термин «соответствующий» используют для обозначения пронумерованных положений в эталонном белке, например, в стрептокиназе (СК) дикого типа или SEQ ID NO: 1, и этих положений в запрашиваемом белке (например, мутантной СК), которые выравнивают с положениями в эталонном белке. Таким образом, когда аминокислотная последовательность испытуемой СК, т.е. SEQ ID NO: 2, 3, 4, 5 и т.д., выровнена с аминокислотной последовательностью эталонного белка, т.е. SEQ ID NO: 1, аминокислоты в последовательности испытуемой СК, которая «соответствует» определенным пронумерованным положениям последовательности эталонной СК, является таковой, которая согласовывается с этими положениями в последовательности эталонной СК, но необязательно в этих точных обозначенных цифрами положениях последовательности эталонной СК. Например, мутант Gly34Cys в SEQ ID NO: 4 должен "соответствовать" мутанту Gly49Cys в SEQ ID NO: 1.
** В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, дополнительно содержащий фибринсвязывающий домен, присоединяют к С-концу, N-концу или к обоим концам.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в предыдущем варианте, где фибринсвязывающий домен содержит по меньшей мере одну замену цистеином.
*** В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в предыдущем варианте, где фибринсвязывающий домен присоединен к мутанту стрептокиназы при помощи гибкого связывающего олигопептида.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, где мутант стрептокиназы дополнительно содержит по меньшей мере одну дополнительную аминокислотную замену, причем аминокислотная замена соответствует аминокислотной замене, выбранной из группы, состоящей из Asn90Ala, HisI07Ala, Serl08Ala, Asp227Tyr, Asp238Ala, Glu240Ala, Arg244Ala, Lys246Ala, Leu260Ala, Lys278Ala, Lys279Ala и Asp359Arg в SEQ ID NO: 1.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, где мутант стрептокиназы содержит делецию, причем делеция соответствует аминокислотной делеции, выбранной из группы, состоящей из Asn90, Asp227 и Asp359.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, где мутант дополнительно содержит аминокислотное удлинение у N и/или С-конца.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, где мутант стрептокиназы содержит по меньшей мере одну цистеиновую мутацию в положении, соответствующем G49, S57, А64, 188, S93, D95, D96, DI02, S105, D120, К121, D122, Е148, К156, D173, D174, L179, DI81, S205, А251, 1254, N255, К256, К257, S258, L260, Е281, К282, F287, D303, L32I, L326, А333, D347, D360 или R372 в SEQ ID NO: 1.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, где мутант стрептокиназы содержит по меньшей мере две цистеиновых мутации в положении, соответствующем G49; S57, А64, 188, S93, D95, D96, 0102, S105, D120, К121, D122, Е148, К156, D173, D174, L179, D181, S205, А251, 1254, N255, К256, К257, S258, 1260, Е281, К282, F287, D303, L321, L326, А333, 0347, D360 or R372 SEQ ID NO: 1.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, где мутант стрептокиназы содержит три цистеиновых мутации в положении, соответствующем G49, S57, А64, 188, S93, D95, D96, D102, S105, D120, К121, D122, Е148, К156, L173, D174, L179, D181, S205, А251, 1254, N255, К256, К257, S258, L260, Е281, К282, F287, D303, L321, L326, А333, D347, D360 или R372 в SEQ ID NO: 1.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, где мутант способен лечить заболевание или нарушение, выбранное из группы, состоящей из инфаркта миокарда, сосудистых тромбозов, эмболии легких, инсульта, сосудистых осложнений, ангины, транзиторной ишемической атаки, тромбоза глубоких вен, тромболитической реокклюзии после процедуры коронарной ангиопластики, периферического сосудистого тромбоза, хирургии сердца, хирургии сосудов, сердечной недостаточности, синдрома Х и заболеваний, при которых происходит сужение по меньшей мере одной коронарной артерии.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце *, дополнительно содержащий реакционноспособную группу, взаимодействующую с цистеином, замещенный по меньшей мере в одном из цистеиновых мутантов.
** В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в предыдущем варианте, где реакционноспособной группой, взаимодействующей с цистеином, является полиэтиленгликоль (ПЭГ).
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в предыдущем варианте, где ПЭГ представляет собой линейный или разветвленный полимер с молекулярной массой в диапазоне от 5000 дальтон до 40000 дальтон.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в предыдущем варианте, где мутант обладает повышенной протеолитической стабильностью по сравнению непэгилированным мутантом стрептокиназы.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце **, где мутант обладает пониженной антигенностью и пониженной иммуногенностью in vivo по сравнению непэгилированным мутантом стрептокиназы.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце **, где мутант обладает замедленным почечным клиренсом и увеличенным временем полужизни in vivo по сравнению непэгилированным мутантом стрептокиназы.
*** Другой вариант осуществления относится к слитому полипептиду, причем слитый полипептид содержит домен стрептокиназы, содержащему от одной до трех цистеиновых замен, где цистеиновая замена находится в фибринсвязывающем домене или по меньшей мере в одной области, соответствующей нативной аминокислотной последовательности стрептокиназы (SEQ ID NO: 1), причем эта область выбрана из группы, состоящей из петли аминокислотных остатков 48-64, петли аминокислотных остатков 88-97, области аминокислотных остатков 102-106, области аминокислотных остатков 119-124, спиралеобразующей области аминокислотных остатков 196-207, петлеобразующей области аминокислотных остатков, 170-181, петлеобразующей области аминокислотных остатков 254-264, суперспиральной области аминокислотных остатков 318-347 и области аминокислотных остатков 360-372 в SEQ ID NO: 1, где мутант может активировать плазминоген и связывать фибрин.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в предыдущем варианте, где фибринсвязывающий домен присоединен к N-концу домена стрептокиназы, С-концу домена стрептокиназы или к обоим концам домена стрептокиназы.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в предыдущем варианте, где фибринсвязывающий домен присоединен к N-концу домена стрептокиназы и слитый полипептид содержит по меньшей мере одну цистеиновую мутацию, выбранную из группы, состоящей из Н16, А17, D62, G80, GI66, SI57, AI8I, 1205, S210, D212, D213, D219, D222, D237, К238, D239, Е265, К.273, D290, D29I, L296, D298, S322, 1371, N372, К373, К374, S375, L377, Е398, К399, F404, D420, L438, L443, А450, D464, D477 и R489 в SEQ ID NO: 22.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в предыдущем варианте, где фибринсвязывающий домен присоединен к N-концу домена стрептокиназы и слитый полипептид содержит по меньшей мере одну цистеиновую мутацию, выбранную из группы, состоящей из G49, S57, А64, I88, S93, D95, D96, D102, S105, D120, К121, D122, Е148, К156, D173, D174, L179, D181, S205, А251, I254, N255, К256, К257, S258, L260, Е281, К282, F287, D303, L321, L326, А333, D347, D360, R372, Н401, А402, D447 и G465 в SEQ ID NO: 23.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в абзаце ***, где фибринсвязывающий домен присоединен как к N-концу, так и С-концу домена стрептокиназы и слитый полипептид содержит по меньшей мере одну цистеиновую мутацию, выбранную из группы, состоящей из Н16, А17, D62, G80, G166, S157, AI8I, 1205, S21O, D212, D213, D219, D222, D237, К.238, D239, Е265, К.273, D290, D291, L296, D298, S322, 1371, N372, К373, К374, S375, L377, Е398, К399, F404, D420, L438, L443, А450, D464, D477, R489, Н518, А519, D564 и G582 в SEQ ID NO: 24.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в абзаце ***, где мутант способен лечить заболевание или нарушение, выбранное из группы, состоящей из инфаркта миокарда, сосудистых тромбозов, эмболии легких, инсульта, сосудистых осложнений, ангины, транзиторной ишемической атаки, тромбоза глубоких вен, тромболитической реокклюзии после процедуры коронарной ангиопластики, периферического сосудистого тромбоза, хирургии сердца, хирургии сосудов, сердечной недостаточности, синдрома Х и заболеваний, при которых происходит сужение по меньшей мере одной коронарной артерии.
**** В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в абзаце ***, дополнительно содержащий реакционноспособную в отношении цистеина группу, замещенную по меньшей мере в одном из цистеиновых мутантов.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в предыдущем варианте, где реакционноспособной в отношении цистеина группой является полиэтиленгликоль (ПЭГ).
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в предыдущем варианте, где ПЭГ представляет собой линейный или разветвленный полимер с молекулярной массой в диапазоне от 5000 дальтон до 40000 дальтон.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в абзаце ****, где мутант обладает повышенной протеолитической стабильностью по сравнению непэгилированным мутантом стрептокиназы.
В другом варианте осуществления настоящего изобретения используют мутант, упомянутый в абзаце ****, где мутант обладает пониженной антигенностью и пониженной иммуногенностью in vivo по сравнению непэгилированным мутантом стрептокиназы.
В другом варианте осуществления настоящего изобретения используют мутант, который упомянут в абзаце ****, где мутант обладает замедленным почечным клиренсом и увеличенным временем полужизни in vivo по сравнению непэгилированным мутантом стрептокиназы.
В еще одном варианте осуществления настоящего изобретения SEQ ID NO: 22-24 представляют собой ковалентно модифицированный гибридный полипептид, содержащий по меньшей мере один функциональный фрагмент стрептокиназы (СК) и фибринсвязывающие домены 4 и 5, фибринсвязывающие домены (FBDs) 1 и 2 фибринонектина человека.
В еще одном варианте осуществления настоящего изобретения функциональный фрагмент СК и указанные фибринсвязывающие домены связаны гибким соединительным олигопептидом.
В еще одном варианте осуществления настоящего изобретения описанный выше мутант содержит аминокислотное удлинение у N и/или С-конца.
В еще одном варианте осуществления настоящего изобретения по меньшей мере аминокислота, выбранная из группы, состоящей из: G49, S57, А64, I88, S93, D95, D96, D102, S105, D120, К121, D122, Е148, К156, D173, D174, L179, D181, S205, А251, 1254, N255, К256, К257, S258, L260, Е281, К282, F287, D303, L321, L326, А333, D347, D360, R372 заменена остатком цистеина, где указанное производное обладает биологической активностью, измеренной стандартными тестами.
В еще одном варианте осуществления настоящего изобретения по меньшей мере аминокислота, выбранная из группы, состоящей из: Н16, А17, D62, G80, G166, S157, А181, I205, S210, D212, D213, D219, D222, D237, К238, D239, Е265, К273, D290, D291, L296, D298, S322, 1371, N372, К373, К374, S375, L377, Е398, К399, F404, 0420, L438, L443, А450, D464, D477, R489 в SEQ ID NO: 22, заменена остатком цистеина, где указанное производное обладает биологической активностью, измеренной стандартными тестами.
В еще одном варианте осуществления настоящего изобретения по меньшей мере аминокислота, выбранная из группы, состоящей из: G49, S57, А64, 188, S93, 095, D96, D102, S105, D120, К121, D122, Е148, К156, D173, D174, L179, D181, S205, А251, 1254, N255, К256, К257, S258, L260, Е281, К282, F287, D303, L321, L326, А333, D347, D360, R372, Н401, А402, D447, G465 в SEQ ID NO: 23, заменена остатком цистеина, где указанное производное обладает биологической активностью, измеренной стандартными тестами.
В еще одном варианте осуществления настоящего изобретения по меньшей мере аминокислота, выбранная из группы, состоящей из: Н16, А17, D62, G80, G166, S157, А181, 1205, S210, D212, D213, D219, D222, D237, К238, D239, Е265, К273, D290, D291, L296, D298, S322, 1371, N372, К373, К374, S375, L377, Е398, К399, F404, D420, L438, L443, А450, D464, D477, R489, Н518, А519, D564, G582 в SEQ ID NO: 24, заменена остатком цистеина, где указанное производное обладает биологической активностью, измеренной стандартными тестами.
В еще одном варианте осуществления настоящего изобретения замененный цистеиновый остаток модифицирован реакционноспособной в отношении цистеина группой.
В еще одном варианте осуществления настоящего изобретения цистеиновый остаток модифицирован полиэтиленгликолем.
В еще одном варианте осуществления настоящего изобретения указанная выше молекула ПЭГ представляет собой линейный или разветвленный полимер с молекулярной массой в диапазоне от 5000 дальтон до 40000 дальтон.
В еще одном варианте осуществления настоящего изобретения описанное выше производное обладает повышенной протеолитической стабильностью по сравнению с исходными немодифицированными аналогами.
В еще одном варианте осуществления настоящего изобретения описанное выше производное обладает пониженной антигенностью и пониженной иммуногенностью in vivo по сравнению с исходными немодифицированными аналогами.
В еще одном варианте осуществления настоящего изобретения описанное выше производное обладает замедленным почечным клиренсом, что увеличивает время полужизни in vivo по сравнению исходными немодифицированными аналогами.
В еще одном варианте осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере один из цистеиновых вариантов, необязательно вместе с фармацевтически приемлемым(и) инертным(и) наполнителем(ями).
В еще одном варианте осуществления настоящего изобретения фармацевтическая композиция применима для лечения заболеваний или нарушений, выбранных из группы, состоящей из инфаркта миокарда, сосудистых тромбозов, эмболии легких, инсульта, сосудистых осложнений, ангины, транзиторной ишемической атаки, тромбоза глубоких вен, тромболитической реокклюзии после процедуры коронарной ангиопластики, периферического сосудистого тромбоза, хирургии сердца, хирургии сосудов, сердечной недостаточности, синдрома Х и заболеваний, при которых происходит сужение по меньшей мере одной коронарной артерии.
Особенно настоящее изобретение характеризуется пэгилированными цистеиновыми вариантами стрептокиназы или ее мутеинов или гибридных молекул активатора плазминогена, содержащих полипептидную связь между стрептокиназой (СК), или модифицированными формами СК, или их соответствующими частями, способными к активации плазминогена (ПГ), с фибринсвязывающими участками фибринонектина человека, выбранными из фибринсвязывающих доменов фибринонектина человека (например, пары доменов 4 и 5, или доменов 1 и 2, или их модифицированных форм), вследствие того, что активаторы гибридного плазминогена обладают способностью независимо связываться с фибрином и поэтому становятся специфичным к тромбам благодаря их усиленному сродству к веществу кровяного тромба, т.е. к фибрину (U.S. Patent №. 7163817).
Он обеспечивает моно-, или би-, или мультипэгилированные цистеиновые варианты стрептокиназы или ее процессированных форм, которые не только активны в отношении способности к активации ПГ, но и проявляют новые и неожиданные функциональные свойства. Например, бипэгилированный цистеиновый вариант СК, в котором дополнительные молекулы цистеина находятся в двух концевых частях полипептида, т.е. у N- и С-концов, проявляет неожиданное свойство в отношении его способности к активации плазминогена человека, состоящее в том, что оно обладает значительно более низкой скоростью активации плазминогена (ПГ) по сравнению с немодифицированной СК, но становится полностью способным активировать плазминоген так же, как и немодифицированная СК, через несколько минут после окончания лаг-периода, когда проводят испытание активации ПГ in vitro. Неспособность самоактивироваться незамедленно (как в случае нативной немодифицированной СК, которая активирует ПГ практически при контакте) обусловлена плазминзависимым типом действия. Напротив, для активации нативной СК не нужен никакой плазмин, и она активирует практически как только образует комплекс с ПГ. Таким образом, после инъекции в тело, такое производное СК путешествует по кровеносной системе, все еще находясь в неактивном или частично активном состоянии. Однако оно предпочтительно активируется в непосредственной близости от тромба, в момент его контакта с тромбом, который, как известно, насыщен плазмином, в то время как общий кровоток не насыщен (свободный плазмин быстро инактивируется при «открытой» циркуляции благодаря присутствию плазминспецифических серпинов [ингибиторов сериновых протеиназ], например, альфа-2-антиплазмина и альфа-2-макроглобулина), что таким образом устраняет или значительно уменьшает системную активацию ПГ, происходящую синхронно с введением природной СК, которая сейчас же активирует ПГ при введении, сопровождающемся побочными эффектами, например, кровотечением и крупномасштабным разрушением различных белковых компонентов сердечно-сосудистой системы. Это свойство, т.е. плазминзависимая активация вместе с увеличенным временем полужизни и низкой иммуногенностью и антигенностью, должно приводить не только к общему пониженному образованию свободного плазмина в общем кровотоке, но также и к способности для тромболитика быть повторно введенным при различных заболеваниях кровеносной системы в относительно низких дозах, что позволяет избежать нежелательных иммунных реакций. Суммарным результатом является продолжительный и более эффективный фибринолиз в мишени, принимающей на себя значительно более низкие терапевтически эффективные дозы тромболитического агента при минимизировании побочных эффектов, например, снижении иммунореактивности и уменьшении отрицательных последствий геморрагических осложнений, часто наблюдаемых для нормальной СК.
Настоящее изобретение обеспечивает пэгилированные цистеиновые варианты стрептокиназы или ее мутеинов или гибридных молекул активатора плазминогена, содержащих полипептидную связь между стрептокиназой (СК), или модифицированными формами СК, или ее соответствующими частями, обладающие биологической активностью in vitro, сравнимой с биологической активностью нативной стрептокиназы, определенной тестом на активацию плазминогена, причем активность уменьшается, если в некоторых случаях она хорошо компенсируется увеличением времени полужизни производного и/или уменьшением скорости клиренса.
Настоящее изобретение обеспечивает пэгилированные цистеиновые варианты стрептокиназы, обладающих способностью активировать плазминоген только после протекания лаг-периода со временем более 5 минут после того, как соответствующий плазминоген животного или человека будет подвергнут действию активатора плазминогена.
Настоящее изобретение обеспечивает прокариотические или эукариотические клетки, трансформированные или трансфицированные экспрессирующими векторами, в которых для экспрессирования стрептокиназы, ее мутеинов или ковалентно модифицированных форм клонируют гены, способные экспрессировать цистеиновые варианты стрептокиназы или ее мутеинов или гибридные молекулы активатора плазминогена. Для эффективной экспрессии последовательности ДНК, кодирующие стрептокиназу, ее мутеины и ковалентно модифицированные формы оптимизируют с учетом кодонового предпочтения в отношении используемых для экспрессии хозяев бактериальной или дрожжевой природы.
Настоящее изобретение детализирует способ получения пэгилированных цистеиновых вариантов стрептокиназы или ее активных мутеинов или гибридных молекул активаторов плазминогена в чистой или биологически активной форме для клинических и научных применений.
Настоящее изобретение учитывает то, что при пэгилировании этих цистеиновых вариантов используют матричный полинуклеотид, в котором использованный для экспрессии СК полинуклеотид, кодирующий СК, модифицируют мутагенезом при помощи известных используемых для ДНК биохимических или химических синтетических способов или их комбинации, чтобы активаторная активность плазминогена сохранялась.
Настоящее изобретение учитывает то, что цистеиновые варианты СК или ее процессированные формы, которые подвергают пэгилированию, но также сохраняющие дополнительные фибринсвязывающие домены, соединены полипептидными связями, так, что образующиеся химерные/слитые полипептиды наряду с проявлением способности к активации плазминогена также обладают фибринсвязывающими свойствами. Связывание между фибринсвязывающими доменами может происходить напрямую или также при помощи коротких линкерных пептидных областей, состоящих из участков аминокислотной последовательности, которая не является конформационно жесткой, а является гибкой, например, преимущественно состоящей из Gly, Ser, Asn, GIn и подобных аминокислот.
Цистеиновые варианты СК или ее мутеины или ковалентно модифицированные формы экспрессируют в Е.coli, используя стандартные плазмиды под контролем сильных промоторов, например, tac, trc, T7 РНК полимеразы и т.п., которые также обладают другими хорошо известными свойствами, необходимыми для того, чтобы вызвать высокий уровень экспрессии введенной рамки считывания, которая кодирует СК или ее мутеины или ковалентно модифицированные аналоги СК.
Цистеиновые варианты СК или ее мутеины или видовые варианты или ковалентно модифицированные формы экспрессируют в дрожжевых экспрессирующих системах, используя стандартные плазмиды, где N-концевой сигнальный пептид оптимизируют для эффективной экстраклеточной секреции зрелого полипептида. Информационная последовательность для этих сигнальных пептидов может быть получена по секреторным белкам дрожжевых экспрессирующих систем. Дополнительно такая информация может быть также получена по другим рекомбинантным белкам, которые гипер-секретированы и содержат оптимизированную сигнальную последовательность.
Настоящее изобретение обеспечивает способ, в котором грубые клеточные лизаты, получены при использовании либо химического, механического, либо ферментативного способов, исходя из клеток, содержащих единичные, двойные или тройные цистеиновые варианты СК, или химерные полипептиды СК, подвергают окислению на воздухе или каталитическому окислению тиол-дисульфидным реагентом или тиол-дисульфидному окислительному рефолдингу, катализируемому ферментом, для сворачивания в их биологически активные конформации, содержащие нативные пары цистеинов (в ковалентно модифицированных формах СК), освобождая дополнительные цистеин(ы) от участия в химической модификации сульфгидрильных групп.
Настоящее изобретение обеспечивает способ, в котором грубые клеточные лизаты, полученные при использовании либо химического, механического, либо ферментативного способов, исходя из клеток, содержащих единичные, двойные или тройные или множественные цистеиновые варианты ковалентно модифицированных форм СК, подвергают окислению или рефолдингу при использовании смеси восстановленного и окисленного глутатиона или других подобных реагентов, которые применяют для таких реакций окислительного фолдинга посредством тиол-дисульфидного обмена, например, цистеина и цистина, при соответствующем редокс-потенциале, что позволяет ковалентно модифицированным формам СК сворачиваться в их биологически активные конформации, освобождая дополнительные цистеин(ы) от участия в химической модификации сульфгидрильных групп.
Цистеиновые варианты СК, ее мутеины или ковалентно модифицированные формы экспрессируют в эукариотические организмы, например, животные или растительные клетки, используя стандартные генетические способы либо в качестве включенных генетических единиц в основной геном или в качестве автономных генетических элементов, хорошо известных в данной области для получения высокого уровня экспрессии встроенной открытой рамки считывания, которая кодирует СК или ее мутеины или ковалентно модифицированные формы.
Настоящее изобретение обеспечивает способ, в котором пэгилированный цистеиновый вариант СК или химерный активирующий плазминоген белок СК может быть использован для тромболитической терапии или профилактики различных сосудистых тромбозов. Активатор может быть составлен в композиции в соответствии с обычными процедурами в виде фармацевтической композиции(й), приспособленной для введения человеку, и может включать в себя, но не ограничиваться стабилизаторами, например, сывороточным альбумином человека, маннитолом и т.д., солюбилизирующими агентами или анестезирующими агентами, например, лигнокаином и т.п., а также другими агентами или их комбинациями для того, чтобы стабилизировать и/или облегчать введение производных in vivo.
Настоящее изобретение обеспечивает фармацевтическую композицию, содержащую пэгилированный цистеиновый вариант СК или гибридный активатор плазминогена и стабилизаторы, которая включает в себя, но не ограничивается сывороточным альбумином человека, маннитолом и т.д. и солюбилизирующими агентами, анестезирующими агентами и т.д.
Настоящее изобретение будет описано более детально в следующих примерах, что, однако, не должно ограничивать область действия изобретения. Для специалистов в данной области техники очевидна возможность использования в пределах компетенции настоящего изобретения других производных, комбинаций и усовершенствований. Таким образом, вероятно, подобная работа или ее тщательные повторения должны приводить к подобным или улучшенным свойствам даже у других вариантов стрептокиназы, которые не раскрыты в настоящем изобретении, и относиться к другим изолятам человеческого и отличного от человеческого происхождения.
Общие способы, использованные в примерах
Обычно используют молекулярные способы и методики, хорошо известные в области молекулярной биологии и науки о белках. Они легко доступны из различных стандартных источников, например, учебников и сборника протоколов, имеющих отношение к этой области техники, например, Sambrook et al., Molecular Cloning: A Laboratory Manual (II.sup.nd edition), Cold Sparing Harbor Press, New York, 1989; McPherson, M.J., Quirke, P., and Taylor, G.R., [Ed.] ПЦР: A Practical Approach, IRL Press, Oxford, 1991, Current Protocols in Protein Science, published by John Wiley & Sons, Inc. Для иммунологических экспериментов ссылаются на учебник и сборник протоколов из Iinmu№chemical Protocols, Hudson L, Hay FC (1989) 3rd ed. Blackwell Scientific. Однако, это не ограничивает подробное разъяснение в контексте специальных экспериментов, описывающих настоящее изобретение, особенно, когда проводят модификации известных процедур, указанных в примерах, если это необходимо.
Реагенты
Клонирование гена СК проводят в экспрессирующем векторе на основе промотора Т7 РНК полимеразы, pET-23d и трансформируют в штамм Escherichia coli BL21 (DE3, полученного от Novagen Inc. (Madison, WI). Термостабильная ДНК-полимераза (Pfu), рестриктазы, Т4 ДНК-лигаза и другие ферменты, модифицирующие ДНК, получены от New England Biolabs (Beverly, MA). Олигонуклеотидные праймеры предоставлены одной из компонент: Biobasic, Inc., Canada, Integrated DNA technologies, US или Sigma-Aldrich, US. Очистка ДНК и экстракция ПЦР-амплифицированных продуктов из агарозного геля проведена при использовании наборов, доступных от Qiagen GmbH (Germany). Автоматическое секвенирование ДНК с использованием флуоресцентных красителей проводят на 16-капиллярном секвенаторе ДНК Applied·Biosystems 3130×1 Genetic Analyzer. Glu-плазминоген либо получают от Roche Diagnostics GmbH (Penzberg, Germany), либо выделяют из плазмы человека аффинной хроматографией (Deutsch and Mertz, 1970). N-концевая аминокислотная последовательность определена при помощи Applied Biosystems Sequencer, Model 476A b 491 clc. Urokinases, EACA. Цианоборгидрид натрия, L-лизин предоставлены Sigma Chemical Co., St. Louis, USA. Фенилагароза 6XL и ДЭАЭ-Сефароза (быстрый поток) покупают в Pharmacia Biotech, Uppsala, Sweden, в то время, как while, гранулы Ni-NTA в Qiagen. Все остальные реагенты имеют высшую степень химической чистоты.
Наслаивание казеина-плазминогена для определения активности СК: Активность различных производных СК определяют наслаиванием казеина и плазминогена человека в мягком агаре. Оригинальный способ Malke and Ferretti, 1984; был модифицирован, когда пятно очищенной СК (0,5 микрограмм) наносили непосредственно на отмеченное углубление на чашках с LB-Amp агаром. Затем чашки инкубируют при 37°С в течение 10 минут; после этого казеин-HPG-агарозу наносят, осторожно выливая смесь растворов А и В на верхнюю часть чашек, содержащую пятна. Раствор А получают нагреванием 1 г обезжиренного молока в 15 мл 50 мМ Трис-HCl буфера (рН 7,5), после чего его выдерживают при 37°С в водяной бане до последующего использования. Раствор В получают нагреванием 0,38 г агарозы в 15 мл 50 мМ Трис-HCl буфера (рН 7,5) при 50°С. После охлаждения раствора до 37°С добавляют 3 мкл Тритона Х-100 (0,04% об./об.) и 200 мкг HPG. Затем пластинку инкубируют при 37°С и наблюдают за образованием зоны просвета (образования гало), обусловленной гидролизом казеина с последующей активацией HPG.
Для определения протеолитической стабильности каждое пэгилированное производное и нативную СК инкубируют с протеолитическими ферментами, например, трипсином иди плазмином. Концентрации протеаз, используемых в реакции, варьируют в диапазоне 500-10,000 раз от концентрации белков. Реакции проводят при встряхивании при 25°С в течение двух-четырех часов. То же самое количество трипсинизированного белка как для теста, так и для контроля капают на чашки с LB-Amp агаром и измеряют остаточную активность после наслаивания на чашки казеин-плазмигогена.
Анализ белков методом электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-PAGE): SDS-PAGE проводят по существу согласно Laemmli, U.К., 1970, с небольшими модификациями, если нужно. Вкратце, образцы белка получают смешиванием равных объемов образца и буфера с двукратно увеличенной концентрацией (0,1 М Tris-HCI, pH 6,8; 6% SDS; 30% глицерина; 15% бета-меркаптоэтанола и 0,01% красителя бромфенолового синего). Для проведения невосстанавливающего электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия в образец буфера не включают бета-меркаптоэтанол. Перед нанесением на гель образцы нагревают в кипящей водяной бане в течение 5 минут. Неоднородная система геля обычно содержит 5% (концентрация акриламида) при разделении белков и 10% при разложении геля. Электрофорез проводят, используя буфер Laemmli при величине постоянного тока, равного вначале 15 мА, до тех пор, пока образцы сконцентрируются, а затем при 30 мА до завершения процесса. После завершения электрофореза, гель погружают в 0,1% раствор Кумаси Голубого R250 в системе метанол : уксусная кислота : вода (4:1:5) при осторожном встряхивании и затем обесцвечивают в обесцвечивающем растворе (в 20% метаноле и 10% ледяной уксусной кислоте) до тех пор, пока фон не станет прозрачным.
Пэгилированные белки могут быть дополнительно окрашены иодом по стандартному способу, развитому Kurfiirst (Kurfiirst М.М., 1992), согласно которому специфически окрашиваются молекулы ПЭГ. Для окрашивания очищенных производных ПЭГ иодом сразу же после завершения электрофореза гель вымачивают в 5%-ном растворе глутарового альдегида (Merck) в течение 15 минут для фиксации. После этого гель окрашивают для определения ПЭГ следующим образом. Во-первых гель помещают в раствор 20 мл хлорной кислоты (0,I М) в течение 15 минут, а затем в 5 мл 5%-ного раствора хлорида бария и добавляют 2 мл 0,1 М раствора иода (Merck, Titrisol 9910). Через несколько минут появляется полоса окрашенного пэгилированного белка. Для двойного окрашивания при проведении электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия гели, окрашенные иодом, далее окрашивают Кумаси Голубым R250, используя протокол Laemmli, описанный выше.
Кинетические анализы (Shi et al., 1994, Wu et al., 1987, Wohl et al., 1980) используют для определения активации HPG модифицированной ПЭГ и немодифицированной СК или ее ковалентных производных, особенно когда нужно определить кинетические константы. ПЭГ-модифицированную, либо немодифицированную СК или ее ковалентно модифицированные формы в различных концентрациях (10 нМ - 200 нМ) добавляют к конечному объему в 100 микролитров мультилуночном планшете, содержащем 1-2 мкМ HPG в буфере для количественного определения (50 мМ Tris-HCI буфер, рН 7,5, содержащем 0,5-1 мМ хромогенного субстрата и 0,1 М NaCl). Используемый хромогенный субстрат (S-2251, Roche Diagnostics GmbH, Germany) является плазминспецифическим и дает при расщеплении желтый окрашенный продукт, содержание которого может быть определено при 405 нм.
Аликвоты белка добавляют после добавления в лунку всех других компонентов и первое спектрофотометрическое поглощение принимают за ноль. Затем определяют изменение в поглощении при 405 нм как функцию времени в перестраиваемом ридере для микропланшетов Versa-Max от Molecular Devices USA. Соответствующие разбавления S.equisimilis стрептокиназы полученные при помощи WHO, Hertfordshire, U.K., используют в качестве стандартного образца для калибровки в международных единицах для неизвестных препаратов.
Тест для определения стационарных кинетических констант активаторной HPG активности для ПЭГ-модифицированной и немодифицированной СК и ее ковалентно модифицированных форм.
Для определения кинетических параметров активации HPG в буфер для количественного определения, содержащий различные концентрации HPG (варьирующие в диапазоне от 0,035 до 2,0 микромолей), добавляют фиксированные количества ПЭГ-модифицированной и немодифицированной СК и ее ковалентно модифицированных форм (0,05-0,1 наномоль) в многолуночном планшете, как описано выше. Затем спектрофотометрически измеряют изменение в поглощении при 405 нм в течение 10-40 мин при 25°С. Затем стандартными способами рассчитывают кинетические параметры активации HPG из графика зависимости обратных величин Михаэлиса-Ментон (Wohl et al., 1980).
Различные пэгилированные СК или ее мутеины и нативную СК, меченную радиоактивным иодом, Йод125, (I125) получают в Perkin Elmer Singapore Pte Ltd., при использовании способа с участием иодогена (1,3,4,6-тетрахлор-3α-6α-дифенилглюколурила) (Fraker & Speck, 1978). Согласно этому способу, использованному Fraker and Speck, иодоген растворяют в хлороформе и наносят на стенки трубки из боросиликатного стекла испарением растворителя при помощи струи газообразного азота. Для иодирования раствор белка в фосфатном буфере (PBS) добавляют в трубку, покрытую иодогеном, и смешивают с I125. Приблизительно через 10-30 мин, белок, меченный радиоактивным иодом, отделяют от свободного радиоактивного иода обессоливанием на колонке с Сефадексом G25 (тонкий) (Amersham Biosciences).
Генетические конструкции
Конструирование стрептокиназы
Дизайн и конструирование вектора рЕТ, содержащего ген СК (PET-23d-CK), описаны Nihalani et al.; (1998). Они включают в себя клонирование гена СК Streptococcus equisimilis H46A в pBR 322 (Pratap et al., 1996) с последующим субклонированием в рЕТ-23d, экспрессирующий вектор, содержащий высокоэффективный сайт связывания рибосом из фага Т7 главного капсидного белка (Studier and Moffatt, 1986), и дополнительную модификацию 5' конца гена для минимизации способности к образованию вторичной структуры. Вектор занимает смежное положение внутри рамки инициаторного кодона для Met в начале открытой рамки считывания, кодирующей СК, для того, чтобы экспрессировать белок в качестве Met-CK. За подробностями можно сослаться на Sahni et al.; 2007 (US Patent №. 7163817). СК мутеины могут быть также сконструированы при использовании обновленной матрицы, как в случае СК, для того, чтобы получить высокую внутриклеточную экспрессирующую способность. Эта схема получения укороченных производных СК другим способом подробно объяснена Nihalani et al., 1998. Помимо секвенирования гена аутентичность СК и ее укороченных производных также установлена газофазным секвенированием N-концевых аминокислот белков на белковом секвенаторе Applied Biosystems-Perkin Elmer модели 476А или 491 clc.
Конструкция ковалентно модифицированных форм СК получением гибридных полинуклеотидов ДНК между ДНК, кодирующей СК, и фибринсвязывающими доменами фибринонектина человека и ее клонированием и экспрессией в Е.coli подробно объясняют в US Patent №7,163,817. Вкратце фибринсвязывающие домены присоединяют к стрептокиназе по N-концу или С-концу или как N-, так и по С-концу для получения различных ковалентно модифицированных форм стрептокиназы.
Примеры
Пример 1
Выбор остатков или областей белка для получения цистеиновых вариантов стрептокиназы
Выбор остатка для замены или делеции является ключевым моментом для сохранения функциональности модифицированных полипептидов. Поэтому план цистеинового мутагенеза требует как структурной информации, имеющейся в кристаллической структуре, так и функциональных представлений, полученных из данных по изучению растворов. Подробные структурные и функциональные исследования за многие годы собраны в тома информации о роли различных областей стрептокиназы в активации плазминогена. Для принятия решения относительно остатков или областей, в которых природные аминокислоты могут быть предпочтительно замещены цистеином, нами была использована информация, содержащаяся в трехмерной структуре СК или ее отдельных доменов наряду с их функциональной важностью. Выбор остатков для цистеинового мутагенеза частично основывается на поверхностной доступности остатков. Место введения цистеина также ограничено гибкими областями стрептокиназы. Для определения поверхностного выступа используют программу DSSP. Код DSSP часто используют для описания вторичной структуры белка с единичным буквенным кодом. DSSP расшифровывается как "Словарь Вторичной Структуры Белка". Программа DSSP (Kabsch and Sander, 1983) определяет вторичную структуру, геометрические параметры и доступность белков по отношению к растворителям, заданные атомные координаты в формате Банка Белковых Данных. DSSP определяет доступность каждого остатка в квадратных ангстремах. Однократное выполнение программы DSSP на данном PDB файле приводит к сокращенному DSSP выходному формату информации. Можно получить величину поверхностной доступности под заголовком Acc в выходном формате DSSP. Для определения поверхностной доступности используют разрешенную кристаллическую структуру стрептокиназы (Wang et al., 1998, PDB ID 1BML) в комплексе с микроплазмином. Для определения поверхностной доступности областей, пропущенных в этой структуре (175-181 и 252-262), используют кристаллическую структуру выделенного бета-домена (Wang et al., 1999, PDB ID 1c4p). Некоторые из петель, пропущенных в кристаллической структуре, наращивали в экспериментально полученной структуре и в ней определяли наиболее предпочтительную конформацию при использовании молекулярных моделей. Остатки, которые необязательно обнаруживают в структуре, но определяются как высоко доступные, поскольку они расположены в гибкой области, также выбраны для цистеинового мутагенеза. В таблице 1 представлены величины поверхностной доступности для различных цистеиновых вариантов СК (SEQ:ID №: 1), которые замещены цистеином. Этот перечень, однако, не ограничивает масштаб цистеиновой замены для других природных аминокислот в СК. Величины доступности, рассчитанные по программе DSSP, выбраны непосредственно в качестве меры поверхностной доступности. Программа DSSP включает в себя многие поверхностно доступные остатки. Однако при выборе остатков для цистеиновой замены проведен тщательный отбор. Этот эксперимент включает в себя мутации, равномерно распределенные вдоль трех различных доменов, а именно альфа-, бета- и гамма-стрептокиназы. Кроме того, выбирают мутации, которые попадают во вторичные структурные области. Также выбор включает в себя цистеиновую замену нескольких остатков, которые обладают особенно низкой поверхностной доступностью, только для того, чтобы проиллюстрировать тот факт, что по существу все без исключения остатки стрептокиназы могут быть замещены цистеином и успешно модифицированы реагентами, взаимодействующими с тиольной группой.
Несмотря на разнообразие нуклеотидной и полипептидной последовательностей, существует значительное структурное подобие среди различных бактериальных активаторов плазминогена. Однодоменная стафилокиназа обладает структурной гомологией с альфа-доменом стрептокиназы. Также двухдоменный бычий активатор плазминогена, полученный из Streptococcus uberis, характеризуется структурным подобием с альфа- и бета-доменами стрептокиназы. Эволюционная сохраняемость трехмерной структуры белка среди различных бактериальных активаторов плазминогена дает возможность планировать цистеиновые модификации других вариантов стрептокиназы, выделенных из различных бактериальных видов.
Пример 2
Генетическое конструирование цистеиновых вариантов стрептокиназ
Все генетические конструкции для экспрессирования стрептокиназ обычно конструируют при использовании обычных подходов, известных в данной области техники. Описаны способы манипуляции с ДНК для включения мутаций, например, в Протоколах ПЦР: A Guide to Methods and Applications, edited by Innis, M.A. et at, 1990, Academic Press Inc., San Diego, Calif. and ПЦР Protocols: Current Methods and Applications edited by B.A.White, 1993., Hurriana Press, Inc., Totowa, N.J., USA. Бактериальные и дрожжевые экспрессионные кассеты получают внедрением молекулы ДНК, кодирующей нужные стрептокиназы в соответствующий вектор (или внедрением исходной матричной ДНК в вектор и мутагенезом в ней последовательности по желанию), с последующей трансформацией клеток-хозяев при помощи экспрессионной кассеты при использовании обычных способов, известных в данной области техники. Специфические мутации вводят в желаемые конструкции при использовании различных процедур, например, техники ПЦР-мутагенеза (Innis et al., 1990), набора для мутагенеза, например, такого, который продает Stratagene ("Quick-Change Mutagenesis" kit, San Diego, Calif.) или Promega (Gene Editor Kit, Madison Wis.). Обычно, олигонуклеотиды конструируют для того, чтобы включать нуклеотидные изменения в кодирующую последовательность стрептокиназы, что приводит к замене, делеции или добавлению нужного остатка на природный остаток. Также конструируют мутагенные праймеры для добавления цистеина в начало молекулы зрелого белка, т.е. в положение, ближайшее к N-концевой аминокислоте или следующей за последней аминокислотой в зрелом белке, т.е. после С-концевой аминокислоты стрептокиназы и ее усеченных конструктов. Подобную стратегию используют для внедрения цистеиновых остатков между двумя любыми выбранными аминокислотами стрептокиназы или любых из ее форм, представленных SEQ ID NO:, приведенных в таблице 2. Используя стандартные способы, генерируют соответствующие цистеинсодержащие мутанты на различные формы СК. Затем проводят скрининг трансформированных клонов для различных мутантов и закрепляют результат автоматическим секвенированием ДНК при использовании флуоресцентных красителей на 16 капиллярном секвенаторе ДНК Applied Biosystems 3130×1 16 Genetic Analyzer.
В таблице 2 перечислены различные полипептидные конструкции, экспрессирующие следующие вещества: стрептокиназу; ее мутеины; видовые производные; или ковалентно модифицированные формы. Последовательность нативного полноразмерного полипептида стрептокиназы определяется SEQ ID NO: 1. Укороченная форма СК, где остатки С-конца 31 удалены, отражена в SEQ ID NO: 2. Укороченная форма СК, где остатки N-конца 15 удалены, отражена в SEQ ID NO: 3. Полипептид, содержащий делецию как со стороны N-концевых остатков, так и со стороны С-концевых 31 остатков СК представлен в SEQ ID NO: 4. Функциональный фрагмент СК, в котором удалены остатки N-конца 49, отражен в SEQ ID NO: 5. Конструкция стрептокиназы, в которой удалены остатки как N-конца 59, так и С-конца 31, представлена SEQ ID NO: 6. Полноразмерный полипептид СК (остатки 1-414), который содержит замену аланина на аспарагин 90 в альфа-домене, представлена SEQ ID NO: 7. Бета-домен мутантного полноразмерного полипептида СК, в котором аланин замещен тирозином представлен в SEQ ID NO: 8. Аналогично, SEQ ID NO: 9 представляет бета-домен мутанта СК, где остаток аспартата в положении 238 замещен аланином. SEQ ID NO: 10 определяет бета-домен мутанта стрептокиназы, в котором глутамат в положении 240 замещен аланином. SEQ ID NO: 11 и SEQ ID NO: 12 представляют мутации аргинина на аланин и лизина на аланин в 244 и 246 остатках, соответственно, для полноразмерной СК. Мутант петли 250 бета-домена СК, в котором остаток лейцина в 260 положении замещен аланином представлен SEQ ID NO: 13. Гамма-домен мутанта СК, в котором остаток аспартата в 359 положении замещен аргинином, представлен SEQ ID NO: 14. Двойной мутант СК, в котором как гистидин 92, так и серин 93 замещены на аланин, представлен SEQ ID NO: 15. Другой двойной мутант СК, в котором два последовательных остатка лизина в 278 и 279 положении замещены на аланин, представлен SEQ ID NO: 16. Мутанты СК, в которых аспарагин в 90 положении в альфа-домене, аспартат в 22 положении в бета-домене или аспартат в 359 положении гамма-домена удалены, представлены SEQ ID NO: 17, SEQ ID NO: 18 и SEQ ID NO: 19, соответственно. Зрелые и активные формы СК могут быть получены из производных видов и подвидов вида Streptococcus. Чтобы оценить возможности реализации цистеинового мутагенеза и последующего пэгилирования для всех различных форм СК, были также выбраны производные СК, полученной из вида Streptococcus, а именно, pyogenes и dysgalactiae. Видовые производные СК, полученные из Streptococcus pyogenes, представлены SEQ ID NO: 20 и один, полученный из Streptococcus dysgalactiae, представлен SEQ ID NO: 21. Ковалентно модифицированная форма СК, в которой фибринсвязывающие домены находятся на N-конце СК, определена SEQ ID NO: 22. Продукт присоединения С-концевого фибринсвязывающего домена СК представлен SEQ ID NO: 23. Гибридный полипептид, содержащий фибринсвязывающий домен как на N-, так и на С-конце СК, представлен SEQ ID NO: 24. Генетические конструкции слитых с фибриновым доменом форм СК детально рассмотрены в US Patent №. 7163817. Различные полипептиды, включающие нативную полноразмерную СК, ее мутеины, видовые варианты и ковалентно модифицированные формы дополнительно используют для получения цистеиновых вариантов.
Цистеиновые варианты, полученные из различных форм СК, упомянутые в таблице 2, представлены уникальными SEQ ID NO:. В таблицах 3-28 перечислены отдельные производные с их уникальными SEQ ID NO:.
Таблица 3: варианты, сконструированные на основе нативной полноразмерной СК (SEQ ID NO: 1)
Таблица 4: варианты, сконструированные на основе укороченной СК 1-383 (SEQ ID NO: 2)
Таблица 5: варианты, сконструированные на основе укороченной СК 16-414 (SEQ ID NO: 3)
Таблица 6: варианты, сконструированные на основе укороченной СК 16-383 (SEQ ID NO: 4)
Таблица 7: варианты, сконструированные на основе укороченной СК 50-414 (SEQ ID NO: 5)
Таблица 8: варианты, сконструированные на основе укороченной СК 60-383 (SEQ ID NO: 6)
Таблица 9: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 7)
Таблица 10: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 8)
Таблица 11: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 9)
Таблица 12: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 10)
Таблица 13: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 11)
Таблица 14: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 12)
Таблица 15: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 13)
Таблица 16: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 14)
Таблица 17: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 15)
Таблица 18: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 16)
Таблица 19: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 17)
Таблица 20: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 18)
Таблица 21: варианты, сконструированные на основе полипептида мутантной СК (SEQ ID NO: 19)
Таблица 22: цистеиновые варианты Streptococcus pyogenes MGASI0270 (SEQ ID NO: 20)
Таблица 23: цистеиновые варианты Streptococcus dysgalactiae subsp. equisimilis (SEQ ID NO: 21)
Таблица 24: цистеиновые варианты СК, содержащие присоединенный к N-концу фибринсвязывающий домен (SEQ ID NO: 22)
Таблица 25: цистеиновые варианты СК, содержащие присоединенный к С-концу фибринсвязывающий домен (SEQ ID NO: 23)
Таблица 26: цистеиновые варианты СК, содержащие фибринсвязывающие домены, присоединенные как к N-, так и С-концу (SEQ ID NO: 24)
Таблица 27: мутанты СК, полученные инсерцией цистеина
Таблица 28: варианты СК, в которых цистеин находится у N- или С-конца, что сопровождается или не сопровождается удлинением пептида.
Эти примеры показывают, что можно получить цистеиновые варианты практически всех форм СК, например, нативной полноразмерной, укороченной, удлиненной по N- или С-концу или слитый с другой полипептидной последовательностью. Мы также получили цистеиновые варианты, полученные заменой, инсерцией или делецией мутантов СК. Это доказывает применимость настоящего изобретения к любой форме СК для цистеинового мутагенеза и последующей модификации агентами, взаимодействующими с тиольной группой.
Конструкции, полученные в примере 2, используют во всех дальнейших экспериментах, проводимых для того, чтобы успешно осуществить настоящее изобретение. Однако следует понимать, что перечень цистеиновых вариантов стрептокиназы является просто типичным, а не эксклюзивным. Конструкция и синтез альтернативных и дополнительных цистеиновых вариантов стрептокиназы согласно настоящему изобретению могут быть легко выполнимы специалистами в данной области. Синтез таких производных может быть успешно проведен обычными способами и методами.
Пример 3
Сверхэкспрессия и очистка биологически активных стрептокиназ
Все вещества, нативный белок стрептокиназа, нСК (nCK), ее мутант и их дальнейшие цистеиновые мутанты, которые нужно очищать, выращивают из одной колонии, делают посев штрихом на чашке с LB-Amp из их маточных растворов BL21 (DE3) в глицерине. Первичные культуры получают, инокулируя pET-23d-CK или варианты СК в 10 мл среды LB, содержащей 100 мкг/мл ампицилина (среда LB-Amp) и инкубируют в течение 8-16 часов при 30-37°С при встряхивании (180-280 об/мин). Этот преинокулят используют для засева 500 мл среды LB-Amp при концентрации 2-10% об./об. и дают расти при 30-37°С, при скорости 180-280 об/мин до O.D600 нм (оптическая плотность, измеренная при 600 нанометрах), равной 0,5-1,0. На этой стадии его индуцируют IPTG (конечная концентрация, равная 0,5-1,0 миллимоль) (Chaudhary et al., 1999; Dhar et al., 2002) и далее выращивают при 40°С в течение 6-12 часов при встряхивании. Затем клетки собирают центрифугированием при 6000-7000 g в течение 10 мин. Осадок затем дважды промывают ледяным буфером (конечная концентрация 100-150 мМ NaCl, 10-50 мМ Tris-HCl, pH 8,0 и 1-5 мМ ЭДТА) и подвергают обработке ультразвуком (Heat System, New York) при 4°С, при длительности звуковых импульсов, равной 30 сек, перемежающейся с равными периодами покоя. Затем клеточный лизат центрифугируют при высоких скоростях оборота (10000-14000 g) в течение 15 мин. SDS-PAGE анализ показывает, что более 90% нужного белка уходит в тельца включения (IB). Затем IB солюбилизируют в 8 М мочевине при комнатной температуре в течение 45 мин при постоянном осторожном встряхивании. Белок в супернатанте сворачивается после 10-36-кратного разбавления (Sundram et al., 2003) в буфере для нанесения (0,4 М NaCl в 20 мМ РВ). Затем образец хроматографируют на гранулах фенилагарозы 6XL и элюируют водой. Таким образом полученный белок затем подвергают дополнительной очистке анионообменной хроматографией на колонке с ДЭАЭ-Сефарозой® (GE-Amersham Biosciences). Фракции белка после HIC объединяют и добавляют Трис-HCl буфер при рН 7,5 до конечной концентрации, равной 20 мМ Трис-HCl, после чего ее наносят на колонку с ДЭАЭ-Сефарозой (Fast Flow), предварительно уравновешенную 20 мМ Трис-HCl (рН 7,5). После промывания буфером, содержащим 20 мМ Трис-HCl (рН 7,5) связанный белок элюируют при использовании линейного градиента соли (0-0,5 М NaCl) в 20-25 мМ Трис-HCl. Элюированные белки СК обычно имеют более 95% чистоты, что показано SDS-PAGE. Количество белка в каждой фракции измеряют по методу Брэдфорд определения белков (Bradford., 1976) и подтверждают измерением поглощения при 280 нМ. Все хроматографические стадии проводят при 4°С. Фракции, содержащие белок, анализируют вместе со стандартной СК и эталонами молекулярной массы SDS-PAGE. Нужные фракции консервативно объединяют для получения гомогенного препарата СК или мутантов СК.
Сверхэкспрессия и очистка различных ковалентно модифицированных конструкций, образованных СК и фибринсвязывающими доменами (FBDs) в Е.coli, и их рефолдинг in vitro описаны в US Patent №. 7163817, где экспрессированные белки подвергают рефолдингу in vitro и очищают колоночной хроматографией. Вкратце, солюбилизированные тельца включения разбавляют до конечной концентрации белка, равной 1 мг/мл, используя дистиллированную воду, вместе с добавлением следующих дополнительных компонентов (приведены конечные концентрации в разбавленной смеси): Трис-HCl, рН 8,0, 50 мМ; NaCl 100-150 мМ; ЭДТА 1-5 мМ; смесь восстановленного и окисленного глутатиона 1,25 мМ : 0,5 мМ. Ренатурированную фракцию отделяют и очищают на колонке, наполненной фибрин-сефарозными гранулами. Для детального описания рефолдинга, очистки и свойств ренатурированного белка, пожалуйста, обращайтесь к Sahni et al.; 2007 (US Patent №. 7,163,817).
Пример 4
Ковалентное конъюгирование цистеиновых вариантов стрептокиназы с полиэтиленгликолем
Тиольные группы цистеиновых вариантов стрептокиназы селективно пэгилируют при использовании малеимид-активированного линейного метоксиПЭГ различной длины, например, 5 кДа, 20 кДа и 40 кДа (JenKem Technology, USA). Для реакции пэгилирования полипептид, подлежащий пэгилированию выдерживают в 50-100 мМ Трис-HCl буфере при рН 8,0, содержащем 100-150 мМ NaCl. К нему добавляют 5 молярный избыток ПЭГ-реагента. Молярный избыток рассчитывают, исходя из числа свободных тиольных групп, взаимодействующих с ПЭГ-реагентом, а не только из молярности белка. Реакционную смесь оставляют при перемешивании при комнатной температуре в течение 1,5-4 часов, и затем реакцию останавливают добавлением 1 мМ DTT. Пэгилированный белок из свободного ПЭГ и непрореагировавшей СК очищают анионообменной хроматографией на колонке с ДЭАЭ сефарозой (GE-Amersham Biosciences). Реакционную смесь разбавляют в 10-15 раз 25 мМ буфере фосфата натрия при рН, равном 7,4, после чего ее помещают на колонку, набитую ДЭАЭ-сефарозой (Fast Flow), предварительно прокалиброванную этим же буфером. После промывания буфером, содержащим 25 мМ фосфат натрия, связанный белок элюируют при использовании линейного градиента соли (0-0,5 М NaCl) в 25 мМ фосфата натрия. В качестве альтернативы, если некоторые из пэгилированных производных не удается чисто отделить от непрореагировавшего ПЭГ ионообменной хроматографией, эти реакционные смеси подвергают гель-хроматографии на Сефадексе 75 (Amersham Biosciences), используя буфер при нейтральном рН и конечную концентрацию NaCl, равную 100-150 мМ. Кроме того, в некоторых случаях, когда очищенные цистеинилированные образцы белка, которые содержат димеры, связанные дисульфидными связями, вначале восстанавливают добавлением 10 мМ дитиотреитолом. Образцы, обработанные дитиотреитолом, обессоливают на колонке, набитой гранулами Сефадекса G-25 (тонкого) и сейчас же используют ля конъюгирования с ПЭГ. Гомодимерные формы СК, связанные дисульфидными связями, также могут быть разделены с использованием гельпроникающей хроматографии на Сефадексе G-75 и могут быть использованы вместо мономерной стрептокиназы для терапевтических целей, благодаря их большим размерам и замедленному клиренсу.
Присоединение ПЭГ во всех случаях подтверждено SDS PAGE. Гель-электрофорез показывает, что >95% пэгилированного белка находится во фракциях, которые получены после удаления непрореагировавшего белка и свободного ПЭГ-реагента.
Пример 5
Наслаивание казеина-плазминогена для быстрого определения способности к активации плазминогена
Активность различных цистеиновых вариантов СК и химерных производных СК определяют наслаиванием казеина и HPG в мягком агаре. Первоначальный способ Malke and Ferretti, 1984 модифицирован таким образом, что наносят пробу очищенной СК (0,1-0,5 микрограмм) непосредственно на отмеченную выемку на чашках с LB-Amp-агаром. Затем пластинки инкубируют при 37°С в течение 10 минут; после этого осторожно наслаивают смесь казеин-HPG агарозу осторожным выливанием смеси растворов А и В на поверхность чашки, содержащей пятна. Раствор А получают нагреванием 1 г обезжиренного молока в 15 мл 50 мМ Трис-HCl буфера (рН 7,5), после чего его выдерживают при 37°С на водяной бане до следующего использования. Раствор В получают нагреванием 0,38 г агарозы в 15 мл 50 mM-HCl буфера (рН 7,5) при 50°С. После выдерживания раствора при 37°С добавляют 3 мкл Тритона Х-100 (0,04% об./об.) и 100-200 мкг HPG и осторожно перемешивают без вспенивания. Затем чашку инкубируют при 37°С в течение 1-4 часов и наблюдают за образованием зон просветления (образование гало), обусловленных гидролизом казеина, с последующей активацией HPG. Следует принимать во внимание площадь зоны лизиса (гало), окружающей лунку в агарозной среде, для сравнения способности к активации плазминогена стрептокиназой и ее ковалентно модифицированными производными. Все цистеиновые варианты стрептокиназы сохраняют значительную способность к активации плазминогена, как показано тестом на казеин-плазминогеновый оверлей. Цистеиновые варианты стрептокиназы далее используют для конъюгирования с образцами ПЭГ различных молекулярных масс в диапазоне от 5000 Да до 40000 Да, реагирующих с тиольными группами, и их активность определяют при использовании теста с наслаиванием казеина-HPG с контролем активности конъюгированного ПЭГ в отношении потери его способности к активации плазминогена. Все стрептокиназы проявляют значительную способность к активации плазминогена, как показано казеинолитическим тестом, и терапевтически применимы. Однако, желательная комбинация подходящего времени полужизни, что требуется для клинической практики, а также пониженная иммунореактивность, делает некоторые производные более применимыми по сравнению с другими. Сравнительно более низкая активность в некоторых случаях может быть хорошо компенсирована увеличением протеолитической стабильности и времени полужизни ПЭГ-белковых аддуктов in vivo.
Альтернативно способность к активации плазминогена была также определена, как объяснено ранее. Таблица 29 представляет активаторную активность HPG и кинетические константы для нескольких типичных представителей пэгилированных производных СК. Таблица 30 обобщает данные по диапазону активности различных пэгилированных производных ковалентно модифицированных стрептокиназ, содержащих присоединенный фибриновый домен.
Измерение активности укороченных форм СК (50-414 и 60-383) и их пэгилированных производных требует добавления синтетических пептидов 1-49 для проявления оптимальной амидолитической и плазминогенактиваторной способности. В литературе было показано, что усеченные производные СК, лишенные N-концевого пептида, соответствующего области 1-59, являются плохими активаторами плазминогена и проявляют повышенную активность при добавлении в реакционную смесь либо пептида СК 1-59, либо фибрина для проявления оптимальной амидолитической способности и способности к активации плазминогена (Nihalani et al., 1998 and Sazonova et al., 2004). Для активации плазминогена пэгилированные производные СК, не содержащие пептидов 1-49 или более коротких N-концевых пептидов, в результате проявляют зависимость от присутствия фибрина; их действия ограничены тромбом, благодаря чему они становятся тромбспецифическими.
Пример 6
Протеолитическая стабильность пэгилированных цистеиновых вариантов стрептокиназы
Для определения протеолитической стабильности 50 микрограмм каждого пэгилированного производного и нативную СК в качестве контроля инкубируют с 50 микролитрами 50 мМ Трис-HCl буфера и 100 мМ NaCl. К этой смеси добавляют трипсин до получения конечного соотношения пэгилированный белок:трипсин, равного 1000:1 (мас./мас.). Реакцию проводят при встряхивании при 25°С в течение двух-четырех часов. Равные количества трипсинизированного белка наносят на чашки с LB-Amp-агаром и измеряют остаточную активность так же, как указано в примере 5 для определения активности СК. Равные аликвоты наносят также из контрольных реакций, в которых в реакционную смесь добавляют только трипсин или только СК или в реакционную смесь добавляют гибриды СК. В этом случае для каждой трипсинизированной пэгилированной стрептокиназы или ее ковалентно модифицированных производных также определяют зону лизиса при наслаивании в агарозе. Площадь зоны лизиса дает прямое измерение остаточной способности к активации плазминогена для защищенной стрептокиназы или ее вариантов. Такой тест показывает значительную защиту для различных изучаемых производных СК, когда они конъюгированы с одним, двумя или тремя остатками ПЭГ и инкубированы с трипсином или плазмином. Обнаружено многократное увеличение протеолитической стабильности при присоединении более одной группы ПЭГ к производным СК.
Альтернативно измерения остаточной активности также подтверждаются для трипсинизированной пэгилированной стрептокиназы или ее ковалентно модифицированных производных измерением их способности к активации плазминогена при использовании спектрофотометрического одностадийного теста, описанного ранее при использовании хромогенного субстрата. Трипсинизированная СК и пэгилированные производные СК в различных концентрациях (1-10 нМ) добавляют до конечного объема, равного 100 микролитрам в мультилуночном планшете, содержащем 1-2 мкМ HPG в буфере для проведения испытаний (50 мМ Трис-HCl буфер, рН 7,5, содержащий 0,5-1 мМ хромогенный субстрат и 0,1 М NaCl). Аликвоты белка добавляют после добавления в лунку всех остальных компонентов. Затем измеряют изменение поглощения при 405 нм как функцию времени в настраиваемом планшет-ридере модели Versa-Max от Molecular Devices Inc., USA. Результаты, полученные этим способом, согласуются с результатами, полученными казеинолитическим тестом. Показано, что при инкубации с трипсином во всех пэгилированных формах стрептокиназы и ее ковалентно модифицированных форм сохраняется значительная функциональная активность. Непэгилированные стрептокиназы, попадающие в подобные условия, проявляют едва детектируемую активность и склонны к перевариванию трипсином.
Образцы, подвергнутые трипсинизации, также изучают в восстанавливающем SDS-PAGE для того, чтобы физически наблюдать протеолитическую стабильность и генерирование усеченных фрагментов в результате протеолиза. Это дает качественную оценку защищенного белка, подвергнутого трипсинизации. С этой целью из реакционной смеси отбирают аликвоты в различные интервалы времени и ингибируют реакцию добавлением 20-молярного избытка соевого ингибитора трипсина (GE-Amersham Biosciences) для того, чтобы подавить дальнейшую триптическую активность. Образцы собирают в различные моменты времени (5-180 мин), подвергают электрофорезу на 10% SBS-PAGE и анализируют на содержание защищенного интактного белка. Результаты, полученные в этом эксперименте, также подтверждают правоту выводов, полученных при нашем функциональном изучении трипсинизированных белков. Большая остаточная способность к активации плазминогена в условиях эксперимента также отражается в большей степени защиты белка при рассмотрении физической целостности на SDS-PAGE.
Пример 7
Удлиненные по N- и С-концам варианты СК и их ПЭГ-модифицированные формы.
Чтобы установить, что произвольное удлинение несколькими аминокислотами как с N-, так и с С-конца молекулы стрептокиназы или ее вариантов неизбежно приводит к тому же результату, который был получен с их неудлиненными аналогами; СК или ее цистеиновые варианты модифицируют либо по N-концу, либо по С-концу с небольшим аминокислотным удлинением.
N-концевые удлиненные формы получают при использовании двух различных стратегий, приводящих к полипептидам двух различных размеров, а именно, одного с 6 аминокислотным удлинением и другого с 20 аминокислотным удлинением. Используя стратегию перекрывания удлинений кодированием 18 нуклеотидного удлинения 6 гистидиновых остатков помещают перед N-концевой аминокислотой зрелой стрептокиназы. Продуктом такой модификации является удлиненный со стороны N-конца белок, дополнительно содержащий шесть аминокислот. Для включения удлинений в 20 аминокислот кассеты, кодирующие СК или ее варианты, переносят из рЕТ 23d в рЕТ 15b (Cat. №. 69661-3, Novagen, Inc. US). Помещение кассеты в рЕТ 15b дает N-концевое удлинение на 20 аминокислот, включающее удлинение из шести гистидиновых остатков и сайт расщепления тромбином. Отщепление N-концевого удлинения от полипептида может быть проведено тромбином. Это приводит к удлинению с гистидиновым маркером и к получению процессированного полипептида с аминокислотной последовательностью, свойственной только СК. SEQ ID NO: 496 демонстрирует аминокислотную последовательность СК, полученную клонированием в рЕТ 15b.
Для получения С-концевого удлиненного продукта удлинение из шести гистидиновых остатков добавляют сразу после последней аминокислоты СК или ее производных использованием стратегии перекрывания удлинений. Это приводит к введению шести дополнительных остатков у С-конца. Образцы очищают использованием либо металл-афинной хроматографии, либо применением способов очистки, описанных в примере 2 для получения гомогенно очищенного продукта. Далее такие очищенные N- или С-концевые удлиненные производные СК или ее производных модифицируют ПЭГ, используя химические реакции, описанные в примере 4. Биохимическое исследование функциональной активности и протеолитической стабильности дает результат, аналогичный тому, который получен для соответствующих удлиненных аналогов. Это является серьезным основанием для того, чтобы сделать заключение, что другие N- или С-концевые удлиненные производные СК или ее производные приведут к аналогичным результатам. Специалисты в данной области могут думать о многочисленных возможностях удлинения как со стороны N-, так и С-конца СК или ее производных для получения функциональных форм стрептокиназы, которые могут быть использованы для замены цистеином, инсерции или добавления цистеина и их последующей модификации по тиольным группам ПЭГ или другими реагентами на сульфгидрильную группу.
Пример 8
Приобретение новых функциональных свойств производными СК, в которых группы ПЭГ присоединены по двум концам, а именно, к N- и С-концу СК или ее укороченных вариантов.
Показано, что присоединение групп ПЭГ одновременно как к N-, так и к С-концу молекулы стрептокиназы и любому из ее изучаемых укороченных функциональных делает ее активность зависящей от присутствия плазмина. Такие новые функциональные свойства могут быть определены, когда мы наблюдаем, что на профиле активации плазминогена бипэгилированное производное СК обнаруживает лаг-период, равный нескольким минутам. К нашему удивлению, когда была изучена активация плазминогена бипэгилированным производным СК после того, как оно образовало комплекс с плазмином, оно продемонстрировало нормальную (т.е. подобную нативной СК) кинетику. Неспособность к незамедленной самоактивации (как в случае нативной, немодифицированной СК, которая активирует PG практически при контакте) обусловлена зависящим от плазмина характером ее действия. Напротив, нативная СК не требует для активации никакого плазмина, а активируется практически как только образует комплекс с PG. В экспериментах, где образцы отбирают из реакционной смеси в различные моменты времени на кинетических кривых активации плазминогена, т.е. на ранней стадии (во время лаг-периода), в быстрой фазе активации и т.п. и анализируют при помощи SDS-PAGE для того, чтобы отслеживать тип продуктов, оказалось, что активация тесно связана с образованием усеченных фрагментов, в которых пептидные сегменты, содержащие ПЭГ на концах полипептида, отщепляются под действием плазмина. Подобные результаты получают масс-спектроскопическим анализом. Эти результаты четко устанавливают корреляцию между активацией плазминогена ПЭГ-модифицированной СК и удалением групп ПЭГ, указывая на опосредованный плазмином механизм активации, согласно которому? как только удаляются объемистые группы ПЭГ, у фрагментов СК появляется возможность взаимодействовать с плазминогеном, так же, как и у нСК, и быстро ее активировать. Поэтому понятно, что присутствие групп ПЭГ на концах, помимо придания стабилизирующего влияния на протеолиз, иммунореактивность и т.д., ожидаемых независимо от положения, также приводит к неожиданному возникновению плазмин-зависимого "переключения", которое оказывает сильное благотворное действие на тромболитическую терапию.
Функциональное свойство зависимости от плазмина, т.е. встроенный "плазминовый переключатель"? обнаружено и у других бипэгилированных цистеиновых вариантов укороченной СК, где дополнительные цистеины находятся в двух крайних положениях полипептида, а именно, у N- и С-концов, которые проявляют неожиданную способность в отношении параметров активации плазминогена человека, когда она протекает с заметно более низкой начальной скоростью активации плазминогена (PG) по сравнению с немодифицированной СК, но приобретает полную способность активировать плазминоген так же, как немодифицированная СК после начального лаг-периода, длящегося в течение нескольких минут, когда определяют активацию ПЭГ in vitro. В Таблице 4 представлены стационарные кинетические параметры активации HPG стрептокиназой и двумя различными бипэгилированными производными СК. NC 1-414 относится к производному СК, в котором цистеин вводят как перед встречающейся в природе N-концевой аминокислотой, так и после С-концевой аминокислоты для получения двойного цистеинового мутанта СК. NC 1-383 относится к укороченному варианту СК, в котором один цистеин вводят как перед встречающейся в природе N-концевой аминокислотой, так и после трех последовательных остатков глицина, которые расположены за 383 аминокислотой. Данные показывают, что оба бипэгилированных производных характеризуются выраженным лаг-периодом прежде, чем они станут полностью функциональными. Кинетические параметры, рассчитанные по линейным участкам кинетических кривых после завершения лаг-периода, показывают, что сразу после того, как бипэгилированные производные полностью активируются после завершения начального лаг-периода, приобретают значительную активность в терминах способности к активации PG по сравнению с СК. Аналогичные результаты, полученные с двумя бипэгилированными производными СК, в которых ПЭГ присоединяют по концам, показывают, что проявление этого нового функционального свойства зависит от положения и почти не зависит от присутствия двойной модификации ПЭГ в той же самой молекуле. Таким образом, зависимость от плазмина сообщается молекуле, когда один или два конца при N- и С-концах, в любом функциональном фрагменте СК пэгилированы.
Кроме того, для специалистов в данной области очевидно, что такое функциональное свойство, которое может быть также сообщено молекуле при любой модификации внутри и вблизи двух концов (например, в подходящей боковой цепи лизина) также может привести к появлению зависящего от плазмина лаг-периода при активации плазминогена благодаря протеолитическому процессингу стрептокиназы, при котором такие "блокирующие" группы, которыми могут быть остатки ПЭГ, другие белковые домены, интактные белки, например, альбумин и т.д., удаляются при протеолизе, что позволяет оставшемуся полипептиду стать функционально активным в отношении активации плазминогена. Таким образом, можно ожидать подобных эффектов, когда группы ПЭГ присоединяют к концам, используя любую доступную химическую реакцию. Один из таких химических подходов использует различие в величинах рКа между альфа-аминогруппой у N-конца и обычно участвующей в конъюгировании с ПЭГ реакционноспособной альфа-аминогруппы. Аналогичные свойства у различных укороченных комбинаций концевых групп указывают на то, что как только две группы ПЭГ входят в любое положение внутри или вблизи двух концевых групп, молекула может приобретать зависимость от плазмина.
Таким образом, после введения в организм такое производное СК начинает свое движение по сосудистой системе, находясь еще в неактивном или частично активном состоянии. Однако оно начинает предпочтительно активироваться в непосредственной близости от тромба, в момент контакта с тромбом, который как известно, обогащен плазмином, в то время как общий кровоток нет (свободный плазмин быстро инактивируется в «открытом» кровотоке благодаря присутствию плазминспецифических серпинов (ингибиторов сериновых протеиназ), например, альфа-2-антиплазмина и альфа-2-макроглобулина), что таким образом устраняет или значительно уменьшает системную активацию ПГ, происходящую синхронно с введением природной СК, которая незамедленно активирует PG при введении, сопровождающемся побочными эффектами, например, кровотечением и крупномасштабным разрушением различных белковых компонентов сердечно-сосудистой системы. Это свойство, т.е. плазминзависимая активация вместе с увеличенным временем полужизни и низкой иммуногенностью и антигенностью должно приводить не только к общему пониженному образованию свободного плазмина в общем кровотоке, но также и к способности для тромболитика быть повторно введенным при различных заболеваниях кровеносной системы в относительно низких дозах, что позволяет избежать нежелательных иммунных реакций. Суммарным результатом является продолжительный и более эффективный фибринолиз в мишени, принимающей на себя значительно более низкие терапевтически эффективные дозы тромболитического агента при минимизировании побочных эффектов.
Дальнейшее улучшение этого свойства состоит в придании бипэгилированной молекуле зависимости от фибрина введением групп ПЭГ в два экстремальных положения производных СК 50-414 или СК 60-383. Такой результат возможен, поскольку делеция "каталитического переключателя" (остатки СК 1-59) меняет конформацию альфа-домена СК и превращает такие усеченные фрагменты в фибринзависимый активатор плазминогена, как показано Reed et al., 1999 и Sazonova et al., 2004. Ожидаемым результатом конструирования таких бипэгилированных производных является зависимость от плазмина и улучшенное соотношение фибриновая зависимость/сродство. Эти два свойства, т.е. плазминовая зависимость и соотношение фибриновое сродство/селективность делает молекулу активатором PG, который направлен исключительно против фибриновых тромбов. Такие молекулы могут эффективно устранить проблему системной активации PG и позволить осуществить строгую активацию PG только в непосредственной близости от фибринового тромба.
Пример 9
Фармакокинетический анализ пэгилированных цистеиновых вариантов стрептокиназы
Все белки, которые используют для введения животным, тщательно обрабатывают для удаления эндотоксина пропусканием через колонку, содержащую гель Агароза-Polymyxin В (BioRad Inc., Palo Alto, CA, USA). Различные производные монопэгилированной СК, представляющие собой включение цистеина в каждый домен, и нативную СК (в качестве контроля) радиоактивно метят с помощью125I при использовании иодогена (Fraker & Speck, 1978) и отделяют от свободного радиоактивного иода пропусканием через обессоливающую колонку с Сефадексом G25. CDI мышей (23-25 г) анестезируют 3% изофлуораном и индуцируют слабое расширение кровеносных сосудов, подставляя хвост под флуоресцентную лампу на 100 ватт. Затем мыши вводят приблизительно 7 микрограмм меченного радиоактивным йодом белка в стерильном солевом растворе через хвостовую вену и собирают все пробы крови приблизительно по 50 мкл в течение времени, необходимого для забора крови из хвоста или из ретроорбитальной полости, и хранят в гепаринизированных эппендорфовских пробирках. Образцы обрабатывают для получения плазмы и определяют активность125I в сцинтилляционном счетчике Perkin-Elmer. После определения активности125I к каждой аликвоте плазмы добавляют равный объем 20% ТХУ для определения количества125I, оставшегося связанным с интактным белком. Образцы быстро перемешивают на вортексе и помещают на лед на 15 мин. Аликвоты центрифугируют приблизительно при 3000 g в течение 10 мин, и супернатант, содержащий свободную метку или метку, связанную с фрагментированным белком, отсасывают из каждой пробы с помощью аспиратора. В образующемся после осаждения ТХУ осадке определяют активность125I. Обычно обрабатывают дублирующие образцы и значения усредняют.
Остаточная радиоактивность различных образцов плазмы, полученных осаждением кислотой, после введения пэгилированных цистеиновых вариантов стрептокиназы используют для определения времени полужизни в опытах in vivo. Результаты определения времени полужизни показывают различные степени удерживания in vivo различных производных ПЭГ, когда они конъюгированы с одним, или двумя, или тремя остатками ПЭГ. Эти результаты показывают, что при введении более одной группы ПЭГ получающееся в результате время полужизни также увеличивается. Время полужизни для нескольких выбранных пэгилированных производных СК суммировано в таблице 32, которая показывает, что в среднем происходит 5-120-кратное увеличение времени полужизни для различных моно-, би- и трипэгилированных цистеиновых вариантов стрептокиназы. Показано, что времена удерживания пэгилированных цистеиновых вариантов СК зависят от положения присоединенного ПЭГ. Пэгилированные производные в альфа-петле 88-97 характеризуются максимальным увеличением (~15-20-кратное) времени удерживания in vivo, в то время как пэгилированные производные СК в бета-домене характеризуются промежуточным увеличением (~10-12-кратным) времени удерживания in vivo. Пэгилированные производные в гамма-домене характеризуются приблизительно 4-5-кратным увеличением времени полужизни in vivo. В целом, все пэгилированные стрептокиназы характеризуются многократным увеличением времени полужизни по сравнению с нативной СК (12-15 минут). Это означает, что пэгилированные цистеиновые варианты характеризуются значительным увеличением времени полужизни in vivo, и свидетельствует об увеличенном замедлении действия пэгилированной СК. Аналогичные результаты получены с другими цистеинилированными вариантами СК после пэгилирования. Хорошо известно, что увеличенное замедление действия пэгилированного производного обусловлено остатком ПЭГ и не зависит от положения полимера ПЭГ в молекуле СК. Таким образом, присоединение остатка ПЭГ через цистеиновый остаток приводит к пэгилированной СК или гибридному производному СК, обладающими свойством увеличенного замедления действия, обуславливающего возможность меньшего введения пэгилированного соединения при поддержании высокого уровня соединения в крови в течение продолжительного времени.
Аддитивность в элиминировании времени полужизни при присоединении двух групп ПЭГ показывает, что можно манипулировать по желанию или обеспечивать в соответствии с требованиями элиминирование времени полужизни конъюгированием с группами ПЭГ в нужном количестве, положении и также варьированием длины полимерной молекулы ПЭГ.
Пример 10
Иммунореактивность пэгилированных цистеиновых вариантов стрептокиназ
Реактивность нСК и пэгилированных цистеиновых вариантов СК и ее ковалентно модифицированных форм в отношении поликлональной антисыворотки СК, индуцируемая в крысах, изучена способом на основе метода твердофазного иммуноферментного анализа (ELISA). Процедура ELISA состоит в следующем.
1. СК и пэгилированные варианты СК вначале растворяют в 0,2 М бикарбонатном буфере, рН 9,2, для получения 100 микролитров раствора, содержащего от 0,75 микрограмм до 1,5 микрограмм белка, и раствор добавляют в каждую лунку микротитровального планшета (Nunc 96-Well Microplates, Cole-Parmer USA).
2. Планшет, сенсибилизированный антигеном, покрывают парафином и инкубируют в холодной комнате в течение ночи во влажном боксе, содержащем влажное бумажное полотенце или при комнатной температуре и влажности в течение двух часов при легком встряхивании.
3. Планшет опорожняют и незанятые места блокируют 200 мкл блокирующего буфера, содержащего 5-10% обезжиренного молока в фосфатно-солевом буфере (PBS) в течение одного часа при комнатной температуре.
4. Планшет опорожняют и промывают четыре раза промывным буфером, полученным из PBS.
5. Раствор первичного антитела вначале разбавляют PBS для получения фактора разбавления, равного 50000. В каждую лунку добавляют 100 мкл разбавленного раствора антитела. Затем планшет инкубируют при комнатной температуре в течение 45-60 минут при легком встряхивании.
6. Вновь опорожняют планшет и промывают четыре раза промывным буфером.
7. Меченное ферментом пероксидазой хрена антитело против антигена растворяют соответственно в PBS. По 100 мкл такого раствора добавляют в каждую лунку и инкубируют при комнатной температуре в течение 1 часа.
8. Вновь опорожняют планшет и промывают шесть раз 1 X PBS.
9. В каждую лунку добавляют по 100 мкл ТМВ (жидкий субстрат, тетраметилбензидин, Sigma-Aldrich, USA) и пластинку оставляют на 10 минут при комнатной температуре.
Для остановки реакции в каждую лунку добавляют по 50 мкл 1 N серной кислоты и развитие окраски фиксируют спектрофотометрически при 450 нм.
Значения поглощения при 450 нм в ELISA, полученные для немодифицированной нСК и различных пэгилированных вариантов СК, используют для оценки относительных степеней их иммунореактивности по отношению к поликлональной антисыворотке СК, индуцируемой в крысах. Исследование с использованием ELISA показывают, что конъюгирование с одной группы ПЭГ молекулярной массы 20 кДа в одном из трех доменов СК уменьшает ее реактивность в отношении содержимого лунки менее, чем на 20% против поликлональной антисыворотки СК. Конъюгирование с двумя группами ПЭГ молекулярной массы 20 кДа, т.е. одной с N-, а другой с C-концом СК уменьшает ее реактивность в отношении содержимого лунки менее, чем на 20% против поликлональной антисыворотки. Конъюгирование с двумя группами ПЭГ молекулярной массы 20 кДа, т.е. с одной в каждом домене СК доводит реактивность почти до детектируемого уровня. Поэтому ясно, что конъюгирование молекул(ы) ПЭГ с различными областями СК значительно уменьшает их реакционную способность против поликлональной сыворотки. Тесты in vitro, демонстрирующие пониженную реактивность антитела, доказывают, что сниженная индукция иммунного ответа происходит, как только пэгилированный белок вводят живому животного.
В таблице 33 перечислены данные по иммунореактивности в процентах, оставшейся в пэгилированных вариантах СК, если реактивность немодифицированной СК дикого типа принять за 100%. Таким образом, настоящее изобретение раскрывает пэгилированные варианты стрептокиназы со значительно пониженной иммунореактивностью, но с интактной тромболитической активностью.
Пример 11.
Способность к активации плазминогена у различных ПЭГилированных мутантных полипептидов стрептокиназы.
В таблицах 35 и 36, находящихся ниже, приведены данные по способности к активации плазминогена у различных вариантов SEQ ID NO:4 (16-383), которые содержат замену определенной аминокислоты на цистеин. Анализ был осуществлен по методике, раскрытой в описании настоящей заявки.
Обнаружили, что в профиле активации ПЭГилированные по цистеину варианты SEQ ID NO:4 имеют примерно 10-минутный лаг-период. Данный феномен также наблюдался для мутантов, полученных на основе SEQ ID NO:491 (см. пример 8). Однако примечательно, что когда исследовали способность к активации плазминогена у этих вариантов, после того как они образовали комплекс с эквимолярным количеством плазмина, их профиль активации был таким же, как и у природной стрептокиназы, т.е. без выраженного отставания в активации плазминогена.
Полученные результаты, также обсуждаемые в примере 8 настоящей заявки, указывают на зависимый от плазмина механизм действия. Очевидно, что опосредованная плазмином активация плазминогена представляет собой значительное преимущество в тромболитической терапии, поскольку тромбы обогащены плазмином, в то время как в свободном кровотоке, как правило, нет свободного активного плазмина. Таким образом, активация плазминогена, которая зависит от плазмина, является эффективным тромбоспецифичным механизмом действия, присущим вариантам стрептокиназы по изобретению.
Преимущества изобретения
Преимущества настоящего изобретения состоят в раскрытии дизайна цистеиновых вариантов стрептокиназы, ее мутеинов, видовых вариантов и ковалентно модифицированных форм. Сайтспецифическое конъюгирование ПЭГ с цистеиновыми вариантами, раскрываемое в этом изобретении, сообщает молекуле стрептокиназы различные полезные терапевтические свойства, например повышенную протеолитическую стабильность, улучшенное время полужизни in vivo и меньшую иммунореактивность. Конкретнее, настоящее изобретение относится к получению сконструированных вариантов стрептокиназы для применения в фармацевтических композициях для лечения нарушений кровообращения.
В кинетике активации плазминогена лаг-периода не наблюдалось
ССЫЛКИ
Abuchowski, A., Kazo, G.M., Verhoest, С.R., van Es, Т., Kafkewitz, D., Nucci, M.L. Viau, A.T. and Davis, F.F. Cancer Biochem. Biophys. 1984; 7: 175-186.
Adams D.S, Griffin L.A, Nachajko W.R, Reddy V.B, Wei C.M. A synthetic DNA encoding a modified human urokinase resistant to inhibition by serum plasminogen activator inhibitor. J. Biol. Chem. 1991; 266 (13): 8476-8482.
Alien, T.M. Liposomes: opportunities in drug development. Drugs 1997; 54: Suppl. 4, 8-14.
Baker D.P, Lin E.Y, Lin K, Pellegrini M, Petter R.C, Chen L.L, Arduini R.M, Brickelmaier M, Wen D, Hess D.M, Chen L, Grant D, Whitty A, Gill A, Lindner O.J, Pepinsky R.B. N-terminally PEGylated human interferon-beta-la with. improved pharmacokinetic properties and in vivo efficacy in a melanoma angiogenesis model. Bioconjug Chem. 2006; 17 (1): 179-188.
Banerjee A., Chisti Y., Banerjee U.C. Streptokinase-a clinically useful thrombolytic agent. Biotechnol. Adv. 2004; 22 (4): 287-307.
Basu A., Yang K., Wang M., Liu S., Chintala R., Palm Т., Zhao H., Peng P., Wu O., Zhang Z., Hua J., Hsieh M.C., Zhou J., Petti G., Li X., Janjua A., Mendez M., Liu J., Longley C., Zhang Z.; Mehlig M., Borowski V., Viswanathan M., Filpula D. Structure-function engineering of interferon-beta-lb for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective monoPEGylation. Bioconjug Chem. 2006; 17 (3): 618-630.
Castellino F.J., A unique enzyme-protein substrate modifier reaction plasmin/streptokinase interaction. Trends Biochem. Sci. 1979, 4: 1-5.
Castelino, F.J. Recent advances in the chemistry of the fibrinolytic system. Chem. Rev. 1981; 81: 431-446.
Chaudhary A., Vasudha S., Rajagopal K., Komath S.S., Garg N., Yadav M., Mande S.C., Sahni G. Function of the central domain of streptokinase in. substrate plasminogen docking and processing revealed by site-directed mutagenesis. Protein Sci 1999; 8: 2791-2805.
COLLEN D, STUMP D.C., GOLD H.K., THROMBOLYTIC THERAPY. ANN. REV. MED., 1988, - 39: 405-423.
COLLEN D. CORONARY THROMBOLYSIS: STREPTOKINASE OR RECOMBINANT TISSUE-TYPE PLASMNOGEN ACTIVATOR? ANN INTERN MED. 1990; 112 (7): 529-538.
Deutsch D.G, Mertz E.T. Plasminogen: purification from human plasma by affinity chromatography. Science. 1970; 170 (962): 1095-1096.
Dhar J, Pande A.H, Sundram V, Nanda J.S., Mande S.C., Sahni G. Involvement of a nine-residue loop of streptokinase in the generation of macromolecular substrate specificity by the activator complex through interaction with substrate kringle domains. J. BioI. Chem. 2002; 277, 13257-13267.
Doherty D.H., Rosendahl M.S., Smith D.J., Hughes J.M., Chlipala E.A., Cox G.N. Site-specific PEGylation of engineered cysteine analogues of recombinant human granulocyte-macrophage colony-stimulating factor. Bioconjug Chem. 2005; 16 (5): 1291-1298.
Esmon С.Т., Mather T. Switching serine protease specificity. Nat Struct Biol. 1998; 5 (11): 933-937.
Fraker P.J., Speck J, C., Jr. Protein and cell membrane iodinations with a sparingly soluble chloroamide, 1,3,4,6-tetrachloro-3a,6a-diphrenylglycoluril. Biochem. Biophys. Res. Comm. 1978; 80 (4): 849-857.
Francis C.W., Marder V.J. Fibmolytic therapy for venous thrombosis. Prog. Cardiovasc. Dis. 1991; 34 (3); 193-204.
Grafe S., Ellinger Т., Maike H. Structural dissection and functional analysis of the complex promoter of the streptokinase gene from Streptococcus equisimilis H46A.
Med Microbiol Immunol. 1996; 185 (1): 11-17.
Hershfield M.S., Buckley R.H., Greenberg M.L., Melton A.L., Schiff R., Hatem C., Kurtzberg J., Markert M.L., Kobayashi R.H., Kobayashi A.L. Treatment of adenosine deaminase deficiency with. polyethylene glycol-modified adenosine deaminase. N. Engl. J. Med. 1987; 316 (10): 589-596;
Huang I.Т., Malke H., Ferretti J.J. Heterogeneity of the streptokinase gene in group A streptococci. Infect Immun. 1989; 57 (2): 502-506.
ISIS-3: a randomised comparison of streptokinase vs tissue plasmnogen activator vs anistreplase and of aspirin plus heparin vs aspirin alone among 41 299 cases of suspected acute myocardial infarction. ISIS-3 (Third International Study of Infarct Survival) Collaborative Group. Lancet. 1992; 339 (8796): 753-770.
Innis M.A., Gelfand D.A., Sninsky J.J., White T.J. (1990); ПЦР. protocols. Academic Press, Inc, San Diego
Jackson K.W., Tang J. Complete aminoacid sequence of streptokinase and its homology with serine proteases. Biochemistry. 1982; 21 (26): 6620-6625.
Jalihal S., Morris G.K. Antistreptokinase titres after intravenous streptokinase. Lancet. 1990; 335 (8688): 534.
Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogenbonded and geometrical features. Biopolymers. 1983; 22 (12): 2577-2637.
Katre N.V, Knauf M.J, Laird W.J. Chemical modification of recombinant interleukin 2 by polyethylene glycol increases its potency in the murine Meth A sarcoma model. Proc. Natl. Acad. Sci. USA. 1987; 84 (6): 1487-1491.
Katre N.V. Immunogenicity of recombinant IL-2 modified by covalent attachment of polyethylene glycol. J Immunol. 1990; 144 (1): 209-213.
Kurflirst M.M. Detection and molecular weight determination of polyethylene glycol-modified hirudin by staining after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem. 1992; 200 (2): 244-248.
Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227 (5259): 680-685.
Lahteenmaki K., Kuusela P., Korhonen T.K. Bacterial plasminogen activators and receptors. FEMS Microbiol. Rev. 2001; 25 (5): 531-552. Review.
Lee H.S, Cross S, Davidson R, Reid T, Jennings K. Raised levels of antistreptokinase antibody and neutralization titres from 4 days to 54 months after administration of streptokinase or anistreplase. Eur. Heart J. 1993; 14 (1): 84-89.
Lijnen H.R, Stassen J.M, Vanlinthout I., Fukao H., Okada K., Matsuo O., Collen D. Comparative fibrinolytic properties of staphylokinase and streptokinase in animal models of ve.№us thrombosis. Thromb. Haemost. 1991; 66 (4): 468-473.
Lyczak, J.B. & Morrison, S.L. Biological and pharmacokinetic properties of a novel immunoglobulin-CD4 fusion protein. Arch. Virol. 1994; 139, 189-196.
Malke H., Ferretti J.J. Streptokinase: cloning, expression, and excretion by Escherichia coli. Proc. Natl. Acad. Sci. USA. 1984; 81 (11): 3557-3561.
Malke H., Ferretti J.J. Expression in Escherichia coli of streptococcal plasmid-determined erythromycin resistance directed by the cat gene promoter of pACYC 184. J Basic Microbiol. 1985; 25 (6): 393-400.
Malke H. Polymorphism of the streptokinase gene: implications for the pathogenesis of poststreptococcal glomerulonephritis. Zentralbl. Bakteriol. 1993; 278 (2-3): 246-257.
Marder VJ. Recombinant streptokinase: opportunity for an improved agent. Blood Coagul. Fibrinolysis. 1993; 4 (6): 1039-1040.
Mateo С., Lombardero J., Moreno E., Morales A., Bombino G., Coloma J., Wims L., Morrison S.L., Perez R. Removal of amphipathic epitopes from genetically-engineered antibodies: production of modified immmunoglobulins with reduced immunogenicity. Hybridoma. 2000; 19, 436-471.
McGrath K.G., Patterson R. Anaphylactic reactivity to streptokinase. JAMA. 1984; 252 (10): 1314-1317.
McGrath K, Patterson R. Immunology of streptokinase in human subjects. Clin. Exp. Immunol 1985; 62 (2): 421-426.
Meyers F.J, Paradise C., Scudder S.A, Goodman G., Konrad M. A phase I study including pharmacokinetics of polyethylene glycol conjugated interleukin-2. Clin. Phannacol Ther. 1991; 49 (3): 307-313.
Monfardini, C. et al. A branched monomethoxypolyethylene glycol for protein modification. Bioconjug. Chem. 1995; 6: 62-69.
Moreadith R.W., Collen D. Clinical development of PEGylated recombinant staphylokinase (PEG-Sak) for bolus thrombolytic treatment of patients with acute myocardial infarction. Adv. Drug Deliv Rev. 2003; 55 (10): 1337-45.
Nihalani D., Kumar R., Rajagopal K., Sahni G. Role of the amino-terminal region of streptokinase in the generation of a fully functional plasminogen activator complex probed with synthetic peptides. Protein Sci 1998; 7, 637-648.
Nicolini F.A, Nichols W.W., Saldeen T.G., Khan S., Mehta J, L. Adjunctive therapy with low molecular weight heparin with recombinant tissue-type plasminogen activator causes sustained reflow in canine coronary thrombosis. Am. Heart J. 1992; 124 (2): 280-288.
Osbom B.L., Olsen H.S., Nardelli В., Murray J.H., Zhou J.X., Garcia A., Moody G., Zaritskaya L.S., Sung C. Pharmacokinetic and pharmacodynamic studies of a human serum albumin-interferonalpha fusion protein in cynomolgus monkeys. J. Phannacol Exp. Ther. 2002; 303 (2): 540-548.
Ouriel K. Comparison of safety and efficacy of the various thrombolytic agents. Rev. Cardiovasc. Med. 2002; 3 Supp. 12: S 17-24. Review.
Pratap J., Kaur J., RajaMohan G., Singh D., Dikshit K.L. Role of N-terminal domain of streptokinase in protein transport. Biochem. Biophys. Res. Commun. 1996; 227, 303-310.
Rajagopalan S., Gonias S.L., Pizzo S.V. A nonantigenic covalent streptokinase-polyethylene glycol complex with plasminogen activator function. J Clin Invest. 1985; 75 (2): 413-9.
Rabijns, A., Hendrik, L., De Bondt, H.L. and De Ranter, C. Three dimensional structure of staphylokinase, a plasminogen activator with therapeutic potential. Nat. Struct. Biol. 1997; 4, 357-360.
Reed G.L., Houng A.K., Liu L., Parhami-Seren В., Matsueda L. H., Wang S., Hedstrom L.A catalytic switch and the conversion of streptokinase to a fibrin-targeted plasminogen activator. Proc. Natl. Acad. Sci. USA. 1999; 96 (16): 8879-83.
Roberts, M.J., Bentley, M.D. & Harris, J.M. Chemistry for peptide and protein PEGylation. Adv. Drug Delivery Rev. 2002; 54, 459-476.
Ross A.M. New plasminogen activators: a clinical review. Clin. Cardiol. 1999; 22 (3): 165-171. Review.
Sazo№va I.Y., Robinson B.R., Gladysheva I.P., Castellino F.J., Reed G.L. Alpha-domain deletion converts streptokinase into a fibrin-dependent plasminogen activator through mechanisms akin to staphylokinase and tissue plasminogen activator. J. Biol. Chem. 2004; 279 (24): 24994-5001.
Schweitzer D.H., van der Wall E.E., Bosker H.A., Scheffer E., Macfarlane I.D. Serum-like illness as a complication after streptokinase therapy for acute myocardial infarction. Cardiology, 1991; 78 (1): 68-71.
Sherry S., Marder V.J.. Thrombosis, fibrinolysis, and thrombolytic therapy: a perspective. Prog Cardiovasc. Dis. 1991; 34 (2): 89-100.
Shi G.Y., Chang. B.I., Chen S.M., Wu D.H., Wu H.L. Function of streptokinase fragments in plasminogen activation. Biochem J. 1994; 304, 235-241.
Spotti F., Kaiser R. Rapid detection and quantitation of precipitating streptokinase-antibodies. Thromb Diath. Haemorrh. 1974; 32 (2-3): 608-616.
Studier F.W., Moffatt B.A. Use of bacteriophage T7 RNA polymerase to direct selective high level expression of cloned genes. J. Mol. Biol. 1986; 189, 113-130.
Studier F.W., Rosenberg A.H., Dunn J.J., Dubendorff J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990; 185: 60-89.
Sundram V., Nanda J.S., Rajagopal K, Dhar J., Chaudhary A., Sahni G. Domain truncation studies reveal that the streptokinase-plasmin activator complex utilizes long range protein-protein interactions with macromolecular substrate to maximize catalytic turnover. J. Biol. Chem. 2003; 278 (33): 30569-30577.
Syed, S. et al. Potent antithrombin activity and delayed clearance from the circulation characterize recombinant hirudin genetically fused to albumin. Blood, 1997; 89 (9): 3243-3252.
Tillet W.S., Garner R.L. The fibrinolytic activity of hemolytic streptococci. J. Exp. Med. 1933, 68: 485-488.
Wang X., Lin X., Loy J.A., Tang J., Zhang X.C. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science. 1998; 281 (5383): 1662-1665.
Wang X., Tang J., Hunter В., Zhang X.C. Crystal structure of streptokinase beta-domain. FEBS Lett. 1999; 459 (1): 85-89.
Wohl R.C., Summaria L., Robbins K.C. Kinetics of activation of human plasminogen by different activator species at pH 7.4 and 37 degrees C. J. Biol. Chem. 1980; 255, 2005-2013.
Wu H.L., Shi G.Y., Bender M.L. Preparation and purification of microplasmin. Proc. Nat. Acad. Sci. USA. 1987; 84 (23): 8292-8295.
Wu X.С., Ye R., Duan Y., Wong S.L. Engineering of plasmin-resistant forms of streptokinase and their production in Bacillus subtilis: streptokinase with longer functional half-life. Appi Environ. Microbiol. 1998; 64 (3):824-829.
Реферат
Изобретение относится к биохимии, в частности, к новым мутантам стрептокиназы. Предложены как мутантные полипептиды стрептокиназы, так и слитые белки, обладающие активностью стрептокиназы. Заявленные стрептокиназы входят в состав фармацевтических композиций, пригодных для лечения заболеваний кровообращения, в частности тромбозов. Изобретение позволяет получить более эффективные мутантные полипептиды со стрептокиназной активностью при лечении заболеваний кровообращения. 15 н. и 5 з.п. ф-лы, 37 табл., 10 пр.
Комментарии