Способ встраивания нужной днк в геном клетки млекопитающего и векторная система для его осуществления - RU2233334C2
Код документа: RU2233334C2
Чертежи






































































Описание
Область, к которой относится изобретение
Настоящая заявка относится к способу направленной интеграции нужной экзогенной ДНК в конкретный участок генома клетки млекопитающего. Более конкретно, в настоящем изобретении описан новый способ идентификации транскрипционно активного сайта-мишени ("горячей точки") в геноме млекопитающего, и встраивания нужной ДНК в этот сайт посредством гомологичной рекомбинации. Настоящее изобретение также, но необязательно, относится к способности амплификации гена нужной ДНК в этом сайте путем коинтеграции амплифицируемого селектируемого маркера, например, DHFR, в комбинации с экзогенной ДНК. Кроме того, в настоящем изобретении описано конструирование новых векторов, подходящих для осуществления вышеуказанных способов, а также продуцирование указанными способами клеточных линий млекопитающих, которые содержат нужную экзогенную ДНК, интегрированную в целевой "горячей точке".
Предпосылки создания изобретения
Техника экспрессии рекомбинатных белков как в прокариотических, так и в эукариотических микроорганизмах хорошо разработана. С точки зрения продуцирования белков, клетки млекопитающих имеют значительные преимущества по сравнению с бактериями или дрожжами благодаря своей способности к правильной сборке, гликозилированию и пост-транскрипционной модификации рекомбинантно экспрессированных белков. После трансфекции в клетки-хозяева, рекомбинантные экспрессионные конструкции могут быть сохранены в виде внехромосомных элементов, либо они могут быть интегрированы в геном клетки-хозяина. Генерирование стабильно трансфецированных клеточных линий млекопитающих обычно предусматривает встраивание вышеуказанной конструкции, то есть, ДНК-конструкцию, кодирующую нужный ген, вместе с геном резистентности к лекарственному средству (доминантным селективным маркером) вводят в клетку хозяина; а затем эти клетки выращивают в присутствии лекарственного средства, позволяющего проводить отбор клеток, содержащих успешно интегрированную экзогенную ДНК. Во многих случаях, нужный ген присоединяют к селективному маркеру резистентности к лекарственному средству, который может быть затем подвергнут генной амплификации. Для этих целей чаще всего используют ген, кодирующий дигидрофолат-редуктазу (DHFR). Выращивание клеток в присутствии метотрексата, конкурентного ингибитора DHFR, приводит к повышенному продуцированию DHFR посредством амплификации гена DHFR. По мере амплификации также фланкирующих участков ДНК, осуществляемая ко-амплификация гена, присоединенного к DHFR, в трансфецированной клеточной линии может приводить к увеличению продуцирования белка, что будет обеспечивать высокий уровень экспрессии нужного гена.
Хотя этот подход дает хорошие результаты, однако описанная система создает ряд проблем, связанных со случайной природой событий интеграции. Эти проблемы возникают из-за того, что уровни экспрессии в значительной степени подвержены влиянию эффектов локального генетического окружения в генном локусе, причем эти явления были хорошо описаны в литературе, и обычно называются "эффектами положения" (см., например, Al-Shawi et al., Mol. Cell. Biol., 10:1192-1198 (1990); Yoshimura et al., Mol. Cell. Biol., 7:1296-1299 (1987)). Поскольку в своем подавляющем большинстве ДНК млекопитающих находится в транскрипционно неактивном состоянии, то способы случайной интеграции не обеспечивают контроля над транскрипционным состоянием интегрированной ДНК. Поэтому может происходить широкое варьирование уровня экспрессии интегрированных генов в зависимости от сайта интеграции. Так, например, интеграция экзогенной ДНК в неактивные или транскрипционно "молчащие" области генома будет приводить к небольшой экспрессии или вовсе к ее отсутствию. В противоположность этому, интеграция в транскрипционно активный сайт может приводить к высокому уровню экспрессии.
Поэтому, если целью исследования является получение высоких уровней экспрессии гена, достижение которых обычно является желаемым результатом методов генной инженерии, то для выявления такого высоко продуцирующего клона, в основном, необходимо скринировать большое число трансфектантов. Кроме того, в некоторых случаях, случайная интеграция экзогенных ДНК в геном может вызвать разрушение важных клеточных генов, что приводит к изменению фенотипа. Из-за этих факторов, генерирование высокоэкспрессирующих стабильных клеточных линий млекопитающих может стать сложным и трудоемким процессом.
Недавно в нашей лаборатории было описано использование ДНК-векторов, содержащих трансляционно ослабленные доминантные селективные маркеры, для экспрессии гена млекопитающих. (Это было описано в заявке на патент США peг. №08/147696, поданной 3 ноября 1993 и недавно признанной патентоспособной).
Эти векторы содержат трансляционно ослабленный ген неомицин-фосфотрансферазы (nео) в качестве доминантного селектируемого маркера, искусственно сконструированного так, чтобы он содержал интрон, в который был встроен ген DHFR вместе с нужным геном или нужными генами. Было обнаружено, что по сравнению со стандартными экспрессирующими векторами млекопитающих, использование этих векторов в качестве экслрессирующих конструкций значительно снижает общее число продуцируемых колоний, резистентных к лекарственному средству, что тем самым облегчает процедуру скрининга. Кроме того, значительный процент клонов, полученных с использованием этой системы, представляет собой высокоэкспрессирующие клоны. Эти результаты, очевидно, обусловлены модификациями, внесенными в селектируемом маркер nео. Вследствие трансляционного ослабления гена neo, трансфецированные клетки не будут продуцировать белок neo на уровне, достаточном для выживания клеток, устойчивых к лекарственному средству, что, тем самым, приводит к снижению общего числа колоний, резистентных к этому лекарственному средству. Кроме того, более высокий процент выживших клонов будет содержать экспрессирующий вектор, интегрированный в сайты в геноме, где базальные уровни транскрипции являются достаточно высокими, что будет приводить к сверхпродуцированию гена neo, и тем самым позволит избежать повреждения гена neo в этих клетках. В соответствии с этим, нужные гены, присоединенные к гену neo, будут подвергаться аналогичному повышению уровня транскрипции. Те же самые преимущества могут быть достигнуты также в результате создания искусственного интрона в гене neo; и в этом случае выживание клонов зависит от синтеза функционального гена neo, который, в свою очередь, зависит от правильного и эффективного сплайсинга интронов neo. Более того, эти критерии, по всей вероятности, являются удовлетворительными в том случае, если ДНК вектора интегрирована в область, которая уже является в высокой степени транскрипционно активной.
После интеграции вектора в транскрипционно активную область, амплификацию гена осуществляют путем отбора на ген DHFR. С применением этой системы можно получить клоны, отобранные с использованием низких уровней метотрексата (50 нМ), содержащих небольшое количество (< 10) копий вектора, который секретирует высокие уровни белка (> 55 пг/клетка/день). Кроме того, это может быть достигнуто за относительно короткий период времени. Однако такая амплификация осуществляется с переменным успехом. Некоторые транскрипционно активные сайты не могут быть амплифицированы, а поэтому частота и степень амплификации из конкретного сайта является непредсказуемой.
В целом использование этих транскрипционно ослабленных векторов представляет собой значительно усовершенствованный способ по сравнению с другими способами случайной интеграции. Однако, как уже обсуждалось, отсутствие контроля в отношении сайта интеграции создает значительные проблемы.
Одним из способов решения проблем, связанных с случайной интеграцией, является способ целевого переноса гена, в результате чего обеспечивается направленное встраивание экзогенной ДНК в специфический локус в геноме хозяина. Экзогенная ДНК встраивается посредством гомологичной рекомбинации, происходящей между последовательностями ДНК в экспрессирующем векторе и соответствующей гомологичной последовательности в геноме. Однако в дрожжах и других грибковых микроорганизмах, этот тип рекомбинации происходит в природе с высокой частотой, тогда как в высших эукариотических организмах этот тип рекомбинации является исключительно редким событием. Было установлено, что в клетках млекопитающих, частота гомологичной рекомбинации по отношению к негомологичной рекомбинации (случайной интеграции) составляет в пределах от 1/100 до 1/5000 (см., например, Capecchi, Science, 244:1288-1292 (1989); Morrow & Kucherlapati, Curr. Op. Biotech., 4:577-582 (1993)).
В одной из наиболее ранних работ описана гомологичная рекомбинация в клетках млекопитающих, содержащих искусственную систему, созданную в мышиных фибробластах (Thomas et al., Cell, 44:419-428 (1986)). Была создана клеточная линия, содержащая мутированную нефункциональную версию гена neo, интегрированного в геном хозяина, а затем в нее была введена путем целевого переноса вторая нефункциональная копия гена neo, имеющая другую мутацию. Восстановление функционального гена neo может происходить только при направленном переносе гена. Гомологичные рекомбинанты были идентифицированы путем отбора клеток на резистентность к G418, и подтверждена путем анализа геномной ДНК, выделенной из резистентных клонов.
Недавно сообщалось об использовании гомологичной рекомбинации для замены генов тяжелой и легкой цепи иммуноглобулина в эндогенном локусе в антитело-секретирующих клетках (патент США № 5202238, Fell et al., (1993)). Однако этот конкретный способ не имеет широкого применения, поскольку он ограничен продуцированием иммуноглобулинов в клетках, которые эндогенно экспрессируют иммуноглобулины, например, в В-клетках и клетках миеломы. Эта экспрессия также ограничена уровнями однокопийного гена вследствие того, что после гомологичной рекомбинации не происходит ко-амплификации. Кроме того, этот метод осложняется тем фактом, что для продуцирования функционального иммуноглобулина требуются два отдельных события интеграции; одно для гена легкой цепи, а затем одно для гена тяжелой цепи.
Дополнительный пример системы этого типа описан для клеток NS/0, где рекомбинантные иммуноглобулины экспрессируются путем гомологичной рекомбинации в локусе 2А гамма-иммуноглобулина (Hollis et al. Международная патентная заявка # PCT/IB95 (00014) ). Уровни экспрессии, полученные из этого сайта были исключительно высокими - порядка 20 пг/клетку/день от однокопийного интегранта. Однако, как и в примере, указанном выше, экспрессия ограничена этим уровнем вследствие того, что амлифицируемый ген не ко-интегрирован в этой системе. Другие исследователи также сообщали об аберрантном гликозилировании рекомбинантных белков, экспрессированных в клетках NS/0 (см., например, Flesher et al., Biotech. and Bioeng., 48:399-407 (1995)), что тем самым ограничивает применение этого подхода.
Недавно была получена система рекомбинации cre-loxP, происходящая от бактериофага Р1 и использованная для направленного переноса гена в эукариотические клетки. В частности, была описана сайт-специфическая интеграция экзогенной ДНК в геном клеток яичника китайского хомячка (СНО) с использованием рекомбиназы сrе и серия 1ох-содержащих векторов. (Fukushige & Sauer, Proc. Natl. Acad. Sci., USA, 89:7905-7909 (1992) ). Эта система привлекательна тем, что она обеспечивает репродуцируемую экспрессию в одном и том же положении хромосомы. Однако не было сделано каких-либо попыток идентифицировать сайт хромосомы, в котором экспрессия гена является оптимальной, и, как и в предыдущем примере, экспрессия в этой системе ограничена однокопийными уровнями. И в этом случае возникают осложнения, обусловленные тем фактом, что необходимо обеспечивать экспрессию функционального фермента рекомбиназы в клетках млекопитающих.
Сообщалось также об использовании гомологичной рекомбинации между введенной ДНК-последовательностью и ее эндогенным хромосомным локусом для разработки эффективных способов генной манипуляции в клетках млекопитающих, а также в дрожжевых клетках (см., например, Bradley et al., Meth. Enzymol., 223:855-879 (1993); Capecchi, Science, 244:1288-1292 (1989); Rothstein et al., Meth. Enzymol., 194; 281-301 (1993)). До настоящего времени исследования, относящиеся к целевому переносу гена в большинство клеток млекопитающих, были направлены на разрушение гена ("нокаут") или сайт-направленный мутагенез локусов отобранных целевых генов в стволовых клетках мышиных эмбрионов (ES). Создание этих мышиных моделей "нокаута" дало возможность ученым определять конкретные структурно-функциональные выходы и оценивать биологическую ценность мириадов мышиных генов. Эта область исследований также имеет важное значение с точки зрения возможного применения в генной терапии.
Недавно, Celltech (Kent, U.К.) описали векторы, которые были нацелены на транскрипционно активные сайты в клетках N30, не требующих амплификации гена (Peakmann et al., Hum. Antibod. Hybridomas, 5:65-74 (1994)). Однако в этой работе не сообщалось, что уровни секреции иммуноглобулинов в этих неамплифицированных клетках могут превышать 20 пг/клетку/день, а в амплифицированных клетках СНО могут быть получены уровни, которые достигают 100 пг/клетку/день (там же).
Было бы в высокой степени желательно разработать такую систему целевого переноса гена, которая могла бы обеспечивать репродуцируемую интеграцию экзогенной ДНК в предварительно определенный сайт генома, о котором известно, что он является транскрипционно активным. Кроме того, было бы также желательно, чтобы такая система целевого переноса гена могла облегчить ко-амплификацию встроенной ДНК после ее интеграции. Конструирование такой системы позволило бы осуществлять репродуцируемую и высокоэффективную экспрессию любого представляющего интерес клонированного гена в клетке млекопитающего и несомненно представляло бы большой интерес для исследователей.
В этой заявке нами была описана новая экспрессирующая система млекопитающих, полученная на основе гомологичной рекомбинации, происходящей между двумя искусственными субстратами, содержащимися в двух различных векторах. В частности, в этой системе используются два новых экспрессирующих вектора млекопитающих, названных "маркирующим вектором" и "вектором целевого переноса".
В основном маркирующий вектор обеспечивает идентификацию и маркирует сайт генома млекопитающего, являющийся транскрипционно активным, то есть сайт, в котором уровни экспрессии гена являются высокими. Этот сайт может рассматриваться как "горячая точка" в этом геноме. После интеграции этого маркирующего вектора, рассматриваемая экспрессирующая система обеспечивает интеграцию другой ДНК в этом сайте, то есть вектора целевого переноса, посредством гомологичной рекомбинации, происходящей между ДНК-последовательностями, общими для этих двух векторов. Эта система имеет значительные преимущества по сравнению с другими системами гомологичной рекомбинации.
В отличие от большинства других систем гомологичной рекомбинации, используемых в клетках млекопитающих, эта система не дает фонового уровня. Поэтому клетки, которые подвергаются лишь случайной интеграции вектора, не проходят отбор. Таким образом, нужный ген, клонированный в плазмиду для целевого переноса, экспрессируется на высоких уровнях из маркированной "горячей точки". В соответствии с этим, рассматриваемый способ экспрессии гена позволяет в основном или полностью решить проблемы, связанные с системами случайной интеграции, подробно описанными выше. Более того, эта система обеспечивает репродуцируемую и высокоэффективную экспрессию любого рекомбинантного белка в одном и том же транскрипционно активном сайте генома млекопитающего. Кроме того, амплификация гена может быть осуществлена в этом конкретном транскрипционно активном сайте путем включения амплифицируемого доминантного селективного маркера (например, DHFR) как части маркирующего вектора.
Цели настоящего изобретения
Таким образом, целью настоящего изобретения является разработка улучшенного способа целевого переноса нужной ДНК в специфический сайт клетки млекопитающего.
Более конкретной целью настоящего изобретения является разработка нового способа целевого переноса нужной ДНК в специфический сайт клетки млекопитающего посредством гомологичной рекомбинации.
Другой конкретной целью настоящего изобретения является получение новых векторов для обеспечения сайт-специфической интеграции нужной ДНК в клетке млекопитающего.
Другой целью настоящего изобретения является получение новых клеточных линий млекопитающего, содержащих нужную ДНК, интегрированную в заранее определенный сайт, который обеспечивает высокий уровень экспрессии.
Более конкретной целью настоящего изобретения является разработка нового способа достижения сайт-специфической интеграции нужной ДНК в клетках яичника китайского хомячка (СНО).
Другой более конкретной целью настоящего изобретения является разработка нового способа интеграции генов иммуноглобулина или любых других генов в клетках млекопитающих в заранее определенных хромосомных сайтах, которые обеспечивают высокий уровень экспрессии.
Другой конкретной целью настоящего изобретения является получение новых векторов или комбинаций векторов, подходящих для интеграции генов иммуноглобулина в клетки млекопитающих в заранее определенные сайты, которые обеспечивают высокий уровень экспрессии.
Другой целью настоящего изобретения является получение клеточных линий млекопитающих, содержащих гены иммуноглобулина, интегрированных в заранее определенные сайты, которые обеспечивают высокий уровень экспрессии.
Еще более конкретной целью настоящего изобретения является разработка нового способа интеграции генов иммуноглобулина в клетках СНО, которые обеспечивают высокий уровень экспрессии, а также получение новых векторов или комбинаций векторов, подходящих для такой интеграции генов иммуноглобулина в клетках СНО.
Кроме того, конкретной целью настоящего изобретения является получение новых клеточных линий СНО, которые содержат гены иммуноглобулина, интегрированные в заранее определенные сайты, обеспечивающие высокий уровень экспрессии, и которые были амплифицированы путем отбора с использованием метотрексата для секреции еще больших количеств функциональных иммуноглобулинов.
Фиг.1 изображает карту маркирующей плазмиды настоящего изобретения, названной Desmond. Эта плазмида показана в кольцевой форме (1а) и в линеаризованном варианте, используемом для трансфекции (1b).
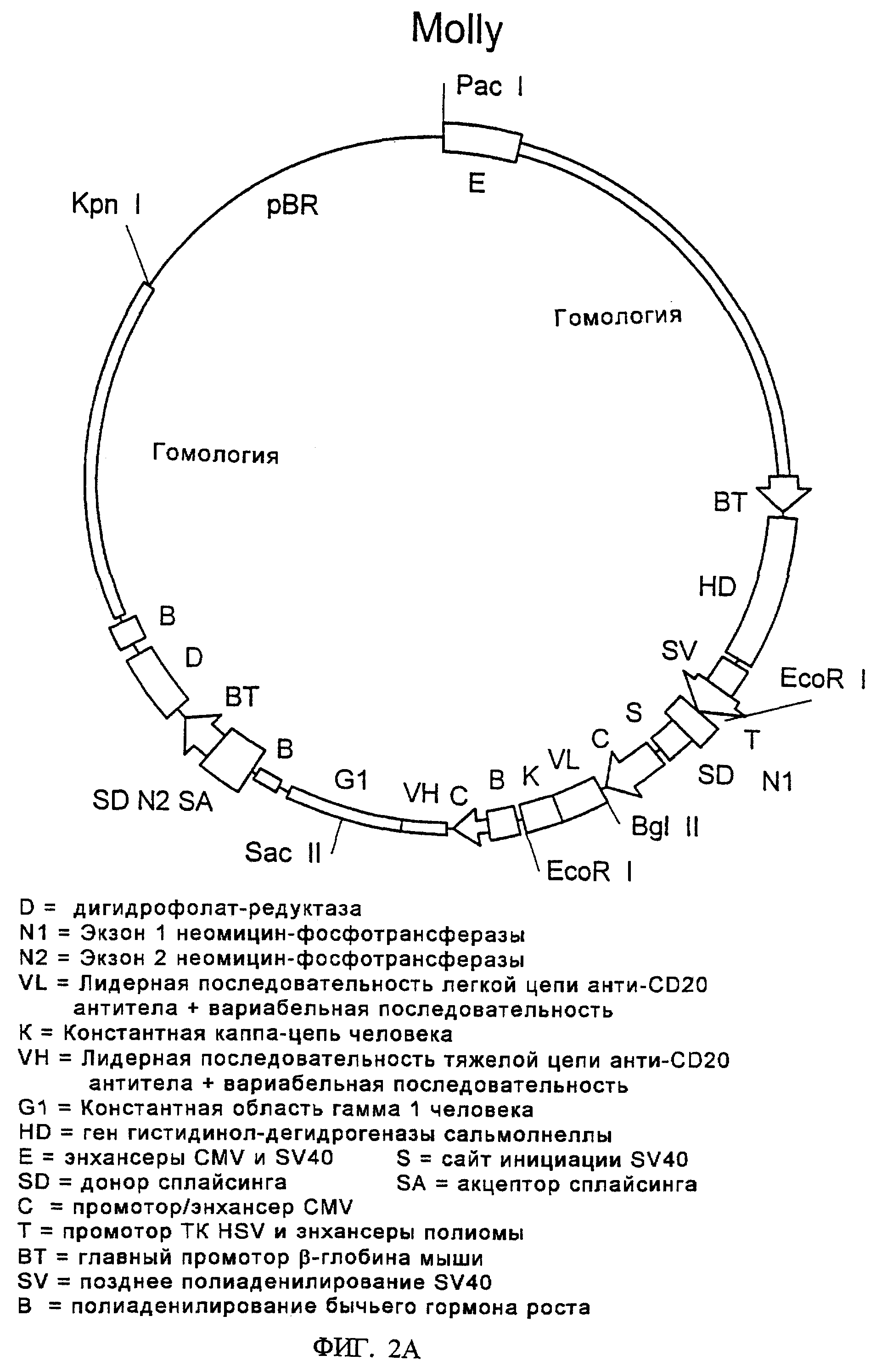
Фиг.2 (а) изображает карту плазмиды целевого переноса, названную "Molly". Показанная здесь плазмида Molly кодирует гены иммуноглобулина против CD20, экспрессия которых описана в Примере 1.
Фиг.2(b) изображает линеаризованную версию Molly после ее гидролиза рестриктазами KpnI и PacI. Эту линеаризованную форму использовали для трансфекции.
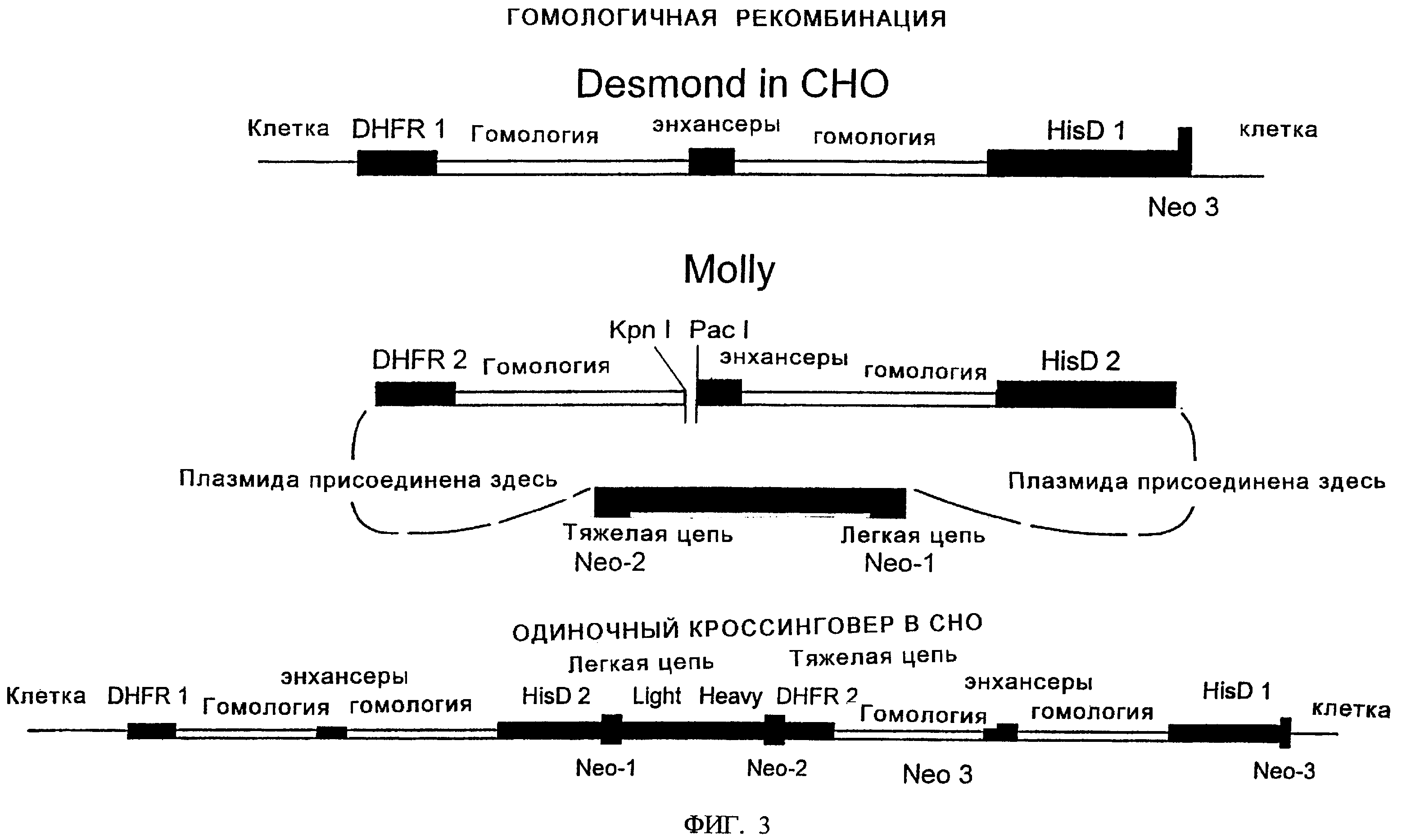
Фиг.3 иллюстрирует возможное сравнение первичных интегрированных в СНО-геном последовательностей Desmond и вставленных последовательностей плазмиды целевого переноса Molly. Представлена также одна из возможных структур последовательноси Molly, интегрированной в последовательность Desmond после гомологичной рекомбинации.
Фиг.4 иллюстрирует Саузерн-анализ однокопийных клонов Desmond. Образцами являлись:
Дорожка 1: маркер размера λHindIII-ДНК
Дорожка 2; клон Desmond 10F3
Дорожка 3: клон Desmond 10C12
Дорожка 4: клон Desmond 15C9
Дорожка 5: клон Desmond 14B5
Дорожка 6: клон Desmond 9В2.
Фиг.5 иллюстрирует Нозерн-анализ однокопийных клонов Desmond. Образцами являлись: Панель А: Нозерн-клоны, зондированные CAD- и DHFR-зондами, как показано на рисунке. Панель В: дубликатные Нозерн-клоны, зондированные CAD- и HisD-зондами, как показано РНК-образцами, загруженными на Панелях А и В, являлись: дорожка 1: клон 9B2; дорожка 2: клон 10C12; дорожка 3: клон 14В12; дорожка 4: клон 15C9; дорожка 5: контрольная РНК из клеток СНО, трансфецированных HisD- и DHFR-содержащей плазмидой; дорожка 6; нетрасфецированные СНО.
Фиг.6 иллюстрирует Саузерн-анализ клонов, полученных в результате гомологичной интеграции Molly в Desmond. Образцами являются: дорожка 1: маркер размера λHindIII-ДНК; дорожка 2: 20F4; дорожка 3: 5F9; дорожка 4: 21С7; дорожка 5: 24G2; дорожка 6: 25Е1; дорожка 7: 28С9; дорожка 8: 29F9; дорожка 9: 39G11; дорожка 10: 42F9; дорожка 11: 50G10; дорожка 12: плазмидная ДНК Molly, линеаризованная ферментом BgIII (верхняя полоса) и разрезанная ферментами BgIII и KpnI (нижняя полоса); дорожка 13: нетрансфецированная Desmond.
Фиг.7А-7С представляют Список последовательностей для Desmond.
Фиг.8A-8I представляют Список последовательностей для плазмиды Molly, содержащей ген иммуноглобулина против CD20.
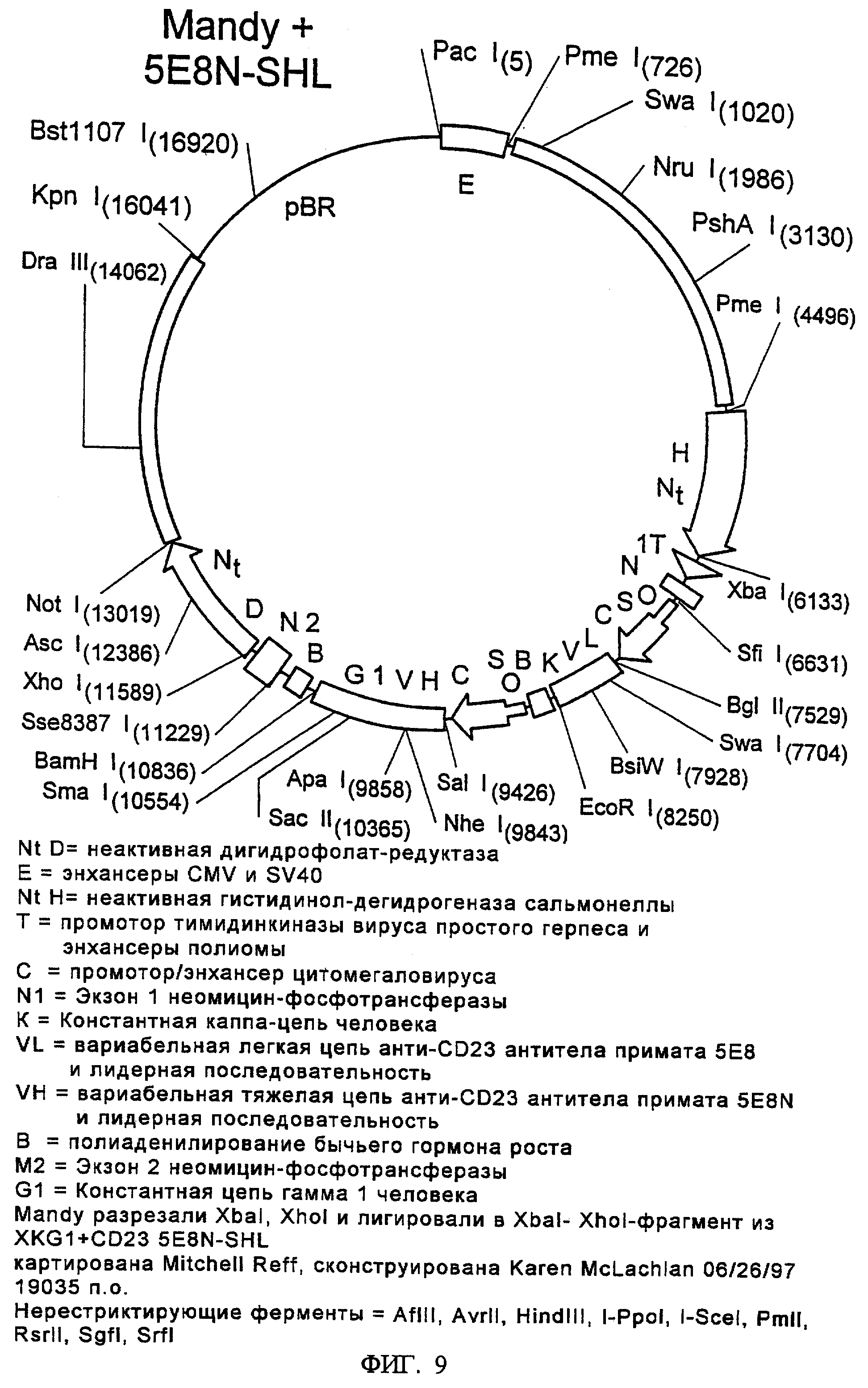
Фиг.9 представляет карту плазмиды целевого переноса, "Mandy", где показаны гены, которые кодируют иммуноглобулин против CD23 и экспрессия которых описана в Примере 5.
Фиг.10А-10N представляет список последовательностей плазмиды "Mandy", содержащей гены иммуноглобулина против CD23, описанные в Примере 5.
Подробное описание изобретения
Настоящее изобретение относится к новому способу интеграции нужной экзогенной ДНК в целевой сайт генома клеток млекопитающих посредством гомологичной рекомбинации. Настоящее изобретение также относится к новым векторам для достижения сайт-специфический интеграции ДНК в целевой сайт генома клеток млекопитающих.
Более конкретно, рассматриваемый способ клонирования обеспечивает сайт-специфическую интеграцию нужной ДНК в клетке млекопитающего путем трансфекции этой клетки "маркерной плазмидой", содержащей уникальную последовательность, которая является чужеродной для генома этих клеток млекопитающих, с последующей трансфекцией "плазмидой целевого переноса", содержащей последовательность, которая обеспечивает гомологичную рекомбинацию с уникальной последовательностью, содержащейся в маркерной плазмиде, и кроме того, включающей нужную ДНК, которую необходимо интегрировать в клетку млекопитающего. В основном, интегрированная ДНК будет кодировать нужный белок, такой как иммуноглобулин или другой гликопротеин, секретируемый клетками млекопитающего.
В рассматриваемой системе гомологичной рекомбинации, в качестве селективного доминантного маркера используется ген неомицин-фосфотрансферазы. Этот конкретный маркер был использован исходя из следующих ранее опубликованных данных:
(i) продемонстрированной способности этого маркера к направленному переносу и сохранению функции мутированного варианта гена neo (как было указано ранее); и
(ii) нашей разработки трансляционно ослабленных экспрессирующих векторов, в которых ген neo был искусственно создан в виде двух экзонов с нужным геном, встроенным в интрон, где указанные экзоны neo правильно сплайсируются и транслируются in vivo, продуцируя функциональный белок и тем самым сообщая полученной клеточной популяции резистентность к G418. В этом применении, ген neo был разделен на три экзона. Третий экзон neo присутствует на "маркерной" плазмиде и интегрируется в геном клетки хозяина после интеграции маркерной плазмиды в клетки млекопитающего. Экзоны 1 и 2 присутствуют на плазмиде целевого переноса, и разделены интроном, в который был клонирован, по крайней мере, один нужный ген. Гомологичная рекомбинация вектора целевого переноса с интегрированным маркерным вектором приводит к правильному сплайсингу всех трех экзонов гена neo и тем самым к экспрессии функционального белка neo (как было определено путем отбора на резистентность колоний к G418). Перед конструированием рассматриваемой экспрессирующей системы, нами был проведен экспериментальный тест на функциональность такой трижды сплайсированной neo-конструкции в клетках млекопитающих. Результаты этого контрольного эксперимента показали, что все три neo-экзона были правильно сплайсированы, что дало основание предположить о технической реализуемости настоящего изобретения.
Хотя настоящее изобретение проиллюстрировано с использованием гена neo, а более конкретно гена neo с тройным расщеплением, однако общая методика должна быть эффективной с использованием других доминантных селективных маркеров.
Как более подробно обсуждается ниже, настоящее изобретение имеет множество преимуществ по сравнению со стандартными способами экспрессии генов, включая как способ случайной интеграции, так и способ целевого переноса гена. В частности, настоящее изобретение относится к способу, который позволяет осуществить репродуцируемую сайт-специфическую интеграцию нужной ДНК в транскрипционно активный домен клетки млекопитающего. Более того, поскольку рассматриваемый способ позволяет вводить искусственную область "гомологии", которая действует как уникальный субстрат для гомологичной рекомбинации, и встраивать нужную ДНК, то для эффективного осуществления настоящего изобретения не требуется, чтобы эта клетка эндогенно содержала или экспрессировала специфическую ДНК.
Таким образом, этот способ генетически применим ко всем клеткам млекопитающих и может быть использован для экспрессии любого типа рекомбинантного белка.
Использование трижды сплайсированного селективного маркера, например, рассматриваемой трижды сплайсированной neo-конструкции, дает гарантию того, что все продуцированные G418-резистентные колонии будут образовываться в результате событий гомологичной рекомбинации (случайные интегранты не будут продуцировать функциональный ген neo, а следовательно, они не будут выживать при С418-отборе). Таким образом, настоящее изобретение позволяет легко проводить скрининг на нужное событие гомологичной рекомбинации. Кроме того, очевидно, что частота дополнительных случайных интеграций в клетке, которая подвержена событиям гомологичной рекомбинации, является низкой.
Исходя из вышеуказанного, очевидно, что значительное преимущество настоящего изобретения заключается в том, что оно позволяет, в основном, снизить число колоний, которое необходимо скринировать для идентификации высокопродуктивных клонов, то есть, клеточных линий, содержащих нужную ДНК, которая секретирует соответствующий белок на высоких уровнях. В среднем, клоны, содержащие интегрированную нужную ДНК, могут быть идентифицированы путем скрининга от около 5 до 20 колоний (по сравнению с несколькими тысячами, которые должны быть скринированы при использовании стандартной техники случайной интеграции, или несколькими сотнями, которые должны быть скринированы с использованием ранее описанных векторов с интронной инсерцией). Кроме того, поскольку сайт интеграции был предварительно отобран и содержит транскрипционно активный домен, все экзогенные ДНК, экспрессированные в этом сайте, должны продуцировать сравнимые, т.е. высокие уровни нужного белка.
Кроме того, настоящее изобретение является более предпочтительным в том, что оно обеспечивает инсерцию амплифицированного гена после интеграции маркерного вектора. Таким образом, если нужный ген направлен в этот сайт посредством гомологичной рекомбинации, то настоящее изобретение позволяет экспрессировать ген, еще более усиливаемый посредством амплификации генов. В этой связи в литературе сообщалось, что различные геномные сайты имеют различную способность к амплификации генов (Meinkoth et al, Mol. Cell. Biol., 7:1415-1424 (1987)). Следовательно, эта техника является более предпочтительной, поскольку она позволяет помещать нужный ген в специфический сайт, который является как транскрипционно активным, так и легко амплифицируемым. Поэтому, эта техника должна приводить к существенному снижению временных затрат, необходимых для выделения таких высокоэффективных продуцентов.
В частности, стандартные методы конструирования в высокой степени экспрессирующих клеточных линий млекопитающих могут занимать от 6 до 9 месяцев, тогда как настоящее изобретение позволяет выделять эти клоны, в среднем, лишь за 3-6 месяцев. Это обусловлено тем фактом, что для получения удовлетворительных уровней экспрессии генов, выделенные стандартным способом клоны обычно должны подвергаться, по крайней мере, трем циклам амплификации гена, резистентного к лекарственному средству. Поскольку гомологично продуцированные клоны были генерированы из предварительно выбранного сайта, который представляет собой высоко экспрессионный сайт, то для достижения нужного уровня продуцирования требуется проведение меньшего числа циклов амплификации.
Кроме того, настоящее изобретение обеспечивает репродуцируемый отбор высокопродуктивных клонов, в которых вектор интегрируется с низким числом копий, обычно с одной копией. Это является преимуществом, которое позволят повысить стабильность клонов и избежать других возможных нежелательных побочных эффектов, связанных с высоким числом копий. Как описано выше, в рассматриваемой системе гомологичной рекомбинации используется комбинация "маркерной плазмиды" и "плазмиды целевого переноса", которые более подробно описаны ниже.
"Маркерная плазмида", которая используется для мечения и идентификации транскрипционной "горячей точки", содержит, по крайней мере, следующие последовательности:
(i) область ДНК, которая является гетерологичной или уникальной для генома клетки млекопитающего, которая функционирует как источник гомологии, обеспечивая гомологичную рекомбинацию (с ДНК, содержащейся во второй плазмиде целевого переноса). Более конкретно, эта уникальная область ДНК (i), в основном, содержит бактериальную, вирусную, дрожжевую, синтетическую или другую ДНК, которая обычно не присутствует в геноме нормальной клетки млекопитающего и которая, кроме того, не обладает значительной гомологией или идентичностью последовательности по отношению к ДНК, присутствующей в геноме данной клетки млекопитающего. В основном, эта последовательность должна достаточно отличаться от ДНК млекопитающего тем, что не подвергается значительной рекомбинации с геномом клетки-хозяина посредством гомологичной рекомбинации. Так как некоторые другие исследователи отмечали повышение частоты целевой рекомбинации по мере увеличения размера области гомологии (Capecchi, Science, 244:1288-1292 (1989)), то размер такой уникальной ДНК будет, в основном, составлять, по крайней мере, около 2-10 тысяч пар оснований или выше, а более предпочтительно, по крайней мере, около 10 т.п.о.
Верхний предел размера уникальной ДНК, которая служит в качестве сайта для гомологичной рекомбинации с последовательностью во втором целевом векторе, главным образом, обусловлен возможными ограничениями стабильности (если ДНК слишком крупная, то она не может быть легко интегрирована в хромосому, что затрудняет работу с очень крупными ДНК).
(ii) ДНК, включающей фрагмент селективной маркерной ДНК, обычно экзона доминантного селективного маркерного гена. Главным отличительным признаком этой ДНК является то, что она не кодирует функциональный селектируемый маркерный белок, несмотря на то, что она экспрессируется совместно с последовательностью, содержащейся в целевой плазмиде. Обычно целевая плазмида содержит остальные экзоны доминантного селективного маркерного гена (которые не присутствуют в "плазмиде направленного переноса"). В основном функциональный селективный маркер должен продуцироваться только, если происходит гомологичная рекомбинация (приводящая к связыванию и экспрессии этой маркерной ДНК-последовательности(i) вместе с частью (частями) фрагмента селективной маркерной ДНК, которая содержится в целевой плазмиде).
Как было указано, настоящее изобретение иллюстрирует использование гена неомицин-фосфотрансферазы в качестве доминантного селективного маркера, который "расщепляется" на два вектора. Однако подходящими могут быть также и другие селективные маркеры, например ген гистидинол-дегидрогеназы сальмонеллы, ген гигромицин-фосфотрансферазы, ген тимидин-киназы вируса простого герпеса, ген аденозин-дезаминазы, ген глутамин-синтетазы и ген гипоксантин-гуанинфосфорибозил-трансферазы.
(iii) ДНК, кодирующую функциональный селективный маркерный белок, где указанный селективный маркер отличается от селективной маркерной ДНК (ii). Этот селективный маркер обеспечивает успешный отбор клеток млекопитающих, где маркерная плазмида успешно интегрируется в клеточную ДНК. Более предпочтительно, чтобы эта маркерная плазмида содержала два таких доминантных селективных маркерных ДНК, расположенных на противоположных концах вектора. Этот вариант является преимущественным, поскольку в этом случае интегранты могут быть отобраны с использованием различных селективных агентов и, кроме того, могут быть отобраны клетки, которые содержат полный селективный вектор. Помимо этого, один маркер может быть амплифицируемым маркером для облегчения амплификации гена, как обсуждалось ранее. Может быть использован любой из доминантных селективных маркеров, перечисленных в (ii), а также могут быть использованы другие маркеры, хорошо известные специалистам.
Более того, маркерная плазмида может, но необязательно, дополнительно содержать редкий сайт рестрикции эндонуклеазы. Этот вариант является потенциально желательным, поскольку в этом случае может быть облегчено расщепление. Такой редкий рестрикционный сайт, если он присутствует, должен быть расположен почти у середины уникальной области, которая служит в качестве субстрата для гомологичной рекомбинации. Предпочтительно, чтобы такая последовательность составляла, по крайней мере, около 12 нуклеотидов. Сообщалось, что введение двухцепочечного разрыва с помощью аналогичной методики увеличивает частоту гомологичной рекомбинации (Choulika et al., Mol. Cell Biol., 15:1968-1973 (1995)). Однако присутствие такой последовательности не имеет решающего значения.
"Плазмида целевого переноса" включает, по крайней мере, следующие последовательности:
(1) ту же самую уникальную область ДНК, которая присутствует в маркерной плазмиде, или область, имеющую достаточную гомологию или идентичность с этой областью, где указанная ДНК может быть объединена посредством гомологичной рекомбинации с уникальной областью (i) в маркерной плазмиде. Подходящие типы ДНК описаны выше в описании уникальной области ДНК (1) в маркерной плазмиде.
(2) оставшиеся экзоны доминантного селективного маркера, из которых один экзон включен как (ii) в маркерную плазмиду, определенную выше. Основной особенностью этого ДНК-фрагмента является то, что он обеспечивает продуцирование функционального (селективного) маркерного белка только в том случае, если целевая плазмида интегрируется посредством гомологичной рекомбинации (где такая рекомбинация приводит к связыванию этой ДНК с другим фрагментом селективной маркерной ДНК, присутствующей в маркерной плазмиде), а также то, что он способствует встраиванию нужной экзогенной ДНК. В основном эта ДНК будет включать остальные экзоны селективной маркерной ДНК, которые разделены интроном. Так, например, эта ДНК может содержать первые два экзона гена neo, а маркерная плазмида может содержать третий экзон (третий сзади от neo).
(3) Целевая плазмида будет также содержать нужную ДНК, например ДНК, кодирующую нужный полипептид и предпочтительно, встроенную во фрагмент селективной маркерной ДНК, присутствующий в плазмиде. В основном эта ДНК будет встроена в интрон, который находится между экзонами селективной маркерной ДНК. Это способствует тому, что нужная ДНК также интегрируется в том случае, если происходит гомологичная рекомбинация целевой плазмиды и маркерной плазмиды. Этот интрон может быть природным, либо он может быть сконструирован в доминантном селективном маркерном ДНК-фрагменте.
Эта ДНК будет кодировать любой нужный белок, предпочтительно, белок, имеющий фармацевтические или другие желательные свойства. В основном большинство ДНК будут кодировать белок млекопитающего, а в нижеприведенных примерах - иммуноглобулин или иммуноадгезин. Однако настоящее изобретение никоим образом не ограничивается продуцированием иммуноглобулинов.
Как обсуждалось ранее, способ клонирования настоящего изобретения может быть применим к любой клетке млекопитающего, поскольку для его эффективности не требуется присутствия какой-либо специфической последовательности или последовательностей млекопитающего. В основном такими клетками млекопитающего являются клетки, обычно используемые для экспрессии белков, например клетки СНО, миеломные клетки, клетки COS, клетки ВНК, клетки Sp2/0, клетки NIH 3Т3 и HeLa. В примерах, приведенных ниже, используются клетки СНО. Их преимущество заключается в их способности к эффективному росту в подходящей культуральной среде, в их способности к эффективному росту и высокой плотности в культуре, а также в их способности экспрессировать белки млекопитающих, такие как иммуноглобулины, в биологически активной форме.
Кроме того, клетки СНО были выбраны, главным образом, из-за предыдущего использования этих клеток авторами настоящего изобретения для экспрессии иммуноглобулинов в (с использованием трансляционно ослабленных векторов, содержащих доминантный селективный маркер, и описанных ранее). Таким образом, наша лаборатория имеет существенный опыт в использовании таких клеток для экспрессии. Однако, исходя из примеров, приведенных ниже, имеются все основания ожидать, что аналогичные результаты будут получены и с использованием других клеток млекопитающих.
В основном трансформация или трансфекция клеток млекопитающих в соответствии с настоящим изобретением может быть проведена стандартными методами. Поэтому, для лучшего понимания настоящего изобретения, в нижеприведенных примерах описано конструирование иллюстративных векторов и их использование для продуцирования интегрантов.
Пример 1
Конструирование и получение маркерного плазмидного ДНК-вектора и плазмидного ДНК-вектора для целевого переноса
Маркерную плазмиду, обозначенную в данном описании "Desmond", собирали из следующих ДНК-элементов:
(а) Гена мышиной дигидрофолат-редуктазы (DHFR), введенного в транскрипционный кластер, содержащий промотор мышиного бета-глобина с 5'-стороны по отношению к инициирующему сайту DHFR, и сигнал полиаденилирования бычьего гормона роста с 3'-стороны по отношению к стоп-кодону. Транскрипционный кластер DHFR выделяли из ТСАЕ6, экспрессирующего вектора, созданного ранее в этой лаборатории (Newman et al., 1992, Biotechnology, 10:1455-1460).
(b) Гена β-галактозидазы E.coli, коммерчески доступного гена, поставляемого Promega в качестве контрольного вектора pSV-β-галактозидазы, каталог # Е1081.
(c) Бакуловирусной ДНК, коммерчески доступной ДНК, поставляемой от Clontech в виде рВАКРАК8, кат. # 6145-1.
(d) Кластера, включающего промоторные и энхансерные элементы от цитомегаловируса и вируса SV40. Этот кластер был сконструирован с помощью PCR с использованием производного экспрессирующего вектора ТСАЕ8 (Reff et al., Blood, 83:435-445 (1994)). Этот энхансерный кластер был введен в бакуловирусную последовательность, которую сначала модифицировали путем встраивания сайта множественного клонирования.
(e) Гена GUS (глюкуронидазы) E.coli, коммерчески доступного гена, поставляемого Clontech в виде рВ101, кат. # 6017-1.
(f) Гена люциферазы светляка, коммерчески доступного гена, поставляемого Promega в виде pGEM-Luc (каталог # Е1541).
(g) Гена гистидинол-дегидрогеназы S.typhimurlum (HisD). Этот ген был первоначал любезно предоставлен Donahue et al., Gene, 18:47-59 (1982), а затем был введен в транскрипционный кластер, содержащий промотор мышиного бета-глобина с 5'-стороны по отношению к гену, и сигнал полиаденилирования SV40 с 3'-стороны по отношению к гену.
ДНК-элементы, описанные в (а)-(g) были объединены в плазмидный остов, происходящий от pBR, с продуцированием непрерывного ДНК-фрагмента в 7,7 т.п.о., обозначенного в прилагаемых иллюстрациях как "гомологичный" ("homology"). Термин "гомологичный" в данном случае относится к последовательностям ДНК, которые не являются частью генома млекопитающих и используются для стимуляции гомологичной рекомбинации между транфецированными плазмидами, имеющими общие гомологичные ДНК-последовательности.
(h) Гена неомицин-фосфотрансферазы от TN5 (Davis & Smith, Ann. Rev. Micro., 32:469-518 (1978)). Полный ген neo субклонировали в pBluescript SK-(Stratagene catalog # 212205) для облегчения генетической манипуляции. Затем синтетический линкер встраивали в уникальный Pstl-сайт, охватывающий кодоны гена neo для аминокислот 51 и 52. Этот линкер кодировал нужные ДНК-элементы для создания искусственного донорского сайта сплайсинга, интрона и акцепторного сайта сплайсинга в гене neo, что создавало два отдельных экзона, обозначенных в настоящем описании как neo-экзон 1 и 2. Neo-экзон 1 кодировал первые 51 аминокислоты гена neo, а экзон 2 кодировал остальные 203 аминокислоты плюс стоп-кодон белка А. В этом интроне был также создан Notl-сайт клонирования.
Neo-экзон 2 был, кроме того, подразделен на neo-экзоны 2 и 3. Это было достигнуто следующим образом: была сконструирована серия PCR-праймеров для амплификации области ДНК, кодирующей neo-экзон 1, интрон и первые 111 2/3 аминокислот экзона 2. 3'-РСК-праймер приводил к введению нового 5'-сайта сплайсинга, который находился непосредственно за вторым нуклеотидом кодона для аминокислоты 111 в экзоне 2, и тем самым, к генерированию нового меньшего экзона 2. Затем этот ДНК-фрагмент, кодирующий теперь сходный экзон 1, интрон и новый экзон 2, был субклонирован и размножен в векторе, происходящем от pBR. Остальную часть первоначального экзона 2 использовали в качестве матрицы для другого цикла PCR-амплификации, в котором был генерирован "экзон 3" 5'-праймер для этого цикла амплификации вводил новый акцепторный сайт сплайсинга со 5'-стороны нового созданного экзона 3, то есть перед конечным нуклеотидом кодона для аминокислоты 111. Полученные 3 экзона гена neo кодируют следующие элементы: экзон 1 - первые 51 аминокислоты гена neo; экзон 2 - следующие 111 2/3 аминокислот, и экзон 3 - конечные 91 1/3 аминокислот плюс кодон терминации транскрипции гена neo.
Neo-экзон 3 был введен вместе с вышеуказанными ДНК-элементами в маркерную плазмиду "Desmond". Neo-экзоны 1 и 2 были введены в плазмиды целевого переноса "Molly". Notl-сайт клонирования, созданный в интроне между экзонами 1 и 2, использовали в последующих стадиях клонирования для встраивания нужных генов в плазмиду целевого переноса.
Была также сконструирована вторая плазмида целевого переноса "Mandy". Эта плазмида почти идентична плазмиде "Molly" (были изменены некоторые рестрикционные сайты на векторе) за исключением того, что в плазмиде "Molly" были инактивированы первоначально присутствующие гены HisD и DHFR. Эти изменения были сделаны потому, что клеточная линия, содержащая плазмиду Desmond, не может больше культивироваться в присутствии гистидинола, а поэтому, очевидно, нет необходимости включать вторую копию гена HisD. Кроме того, ген DHFR был инактивирован для гарантии того, что в любой полученной клеточной линии будет амплифицирован только один ген DHFR, а именно ген, присутствующий Desmond-маркированном сайте. Плазмида "Mandy" была получена из "Molly" путем следующих модификаций:
(i) Синтетический линкер был введен в середину DHFR-кодирующей области. Этот линкер вводит стоп-кодон и смещает остальную часть DHFR-кодирующей области за пределы рамки считывания, что делает этот ген нефункциональным.
(ii) Часть гена HisD была делегирована и заменена PCR-генерированным фрагментом HisD, в котором отсутствовал промотор и старт-кодон этого гена.
На фиг.1 показано расположение этих ДНК-элементов в маркерной плазмиде "Desmond". На фиг.2 показано расположение этих ДНК-элементов в первой плазмиде целевого переноса, "Molly". На фиг.3 проиллюстрировано возможное расположение различных ДНК-элементов в геноме клеток СНО после целевого переноса и интеграции ДНК Molly в Desmond-маркированные клетки СНО. На фиг.9 показана плазмида целевого переноса "Mandy".
Конструирование маркерной плазмиды и плазмид целевого переноса из вышеуказанных ДНК-элементов осуществляли в соответствии со стандартной техникой клонирования (см., например, Molecular Cloning, A laboratory manual, J. Sambrook et al., 1987, Cold Spring Harbor Laboratory Press, и Current Protocols in Molecular Biology, F. M. Ausubel et al., eds., 1987, John Wiley & Sons). Все плазмиды были размножены и поддерживались в XLI blue E.coli (Stratagene, cat. # 200236). Крупномасштабные плазмидные препараты были получены с использованием системы для очистки ДНК (Promega Wizard Maxiprep DNA Purification System®) в соответствии с инструкциями производителей.
Пример 2
Конструирование маркерной клеточной линии СНО
1. Процедуры культивирования и трансфекции клеток для продуцированной маркерной клеточной линии СНО
Маркерную плазмидную ДНК линеаризовали путем гидролиза в течение ночи при 37°С ферментом Bst1107I. Линеаризованный вектор осаждали этанолом и ресуспендировали в стерильном ТЕ до получения концентрации 1 мг/мол. Линеаризованный вектор вводили в DHFR-клетки яичника китайского хомячка (клетки СНО), клетки DG44 (Urlaub et al., Som. Cell and Mot. Gen., 12:555-566 (1986) путем электропорации следующим образом.
Клетки с экспоненциальным ростом собирали путем центрифугирования, один раз промывали охлажденным на льду SBC (забуференном сахарозой растворе, 272 мМ сахарозы, 7 мМ фосфата натрия, рН 7,4, 1 мМ хлорида магния), а затем ресуспендировали в SBC до получения концентрации 107 клеток/мл. После 15-минутного инкубирования на льду, 0,4 мл клеточной суспензии смешивали с 40 мкг линеаризованной ДНК в одноразовой кювете для электропорации. Клетки подвергали электрошоку с использованием манипулятора с электроячейками ВТХ (San Diego, СА), имеющего рабочие параметры: напряжение 230 В, электрическая емкость 400 микрофарад и сопротивление 13 Ом. Затем, клетки, подвергнутые электрошоку, смешивали с 20 мл предварительно нагретой среды для культивирования СНО (СНО-S-SFMII, Gibco/BRL, каталог # 31033-012) и засевали в 96-луночные планшеты для культивирования тканей. Через 48 часов после электропорации, в планшеты добавляли селективную среду (в случае трансфекции плазмидой Desmond, селективной средой была среда CHO-S-SFMII без гипоксантина или тимидина, в которую был добавлен 2 мМ гистидинол (Sigma catalog # H6647)). Эти планшеты поддерживали в селективной среде вплоть до 30 дней, или до тех пор, пока в лунках не наблюдался рост клеток. Затем эти клетки удаляли из 96-луночных планшетов, и, наконец, размножали в роллерных 120 мл-колбах, где они все время находились в селективной среде.
Пример 3
Характеризация маркированных клеточных линий СНО
(а) Саузерн-анализ
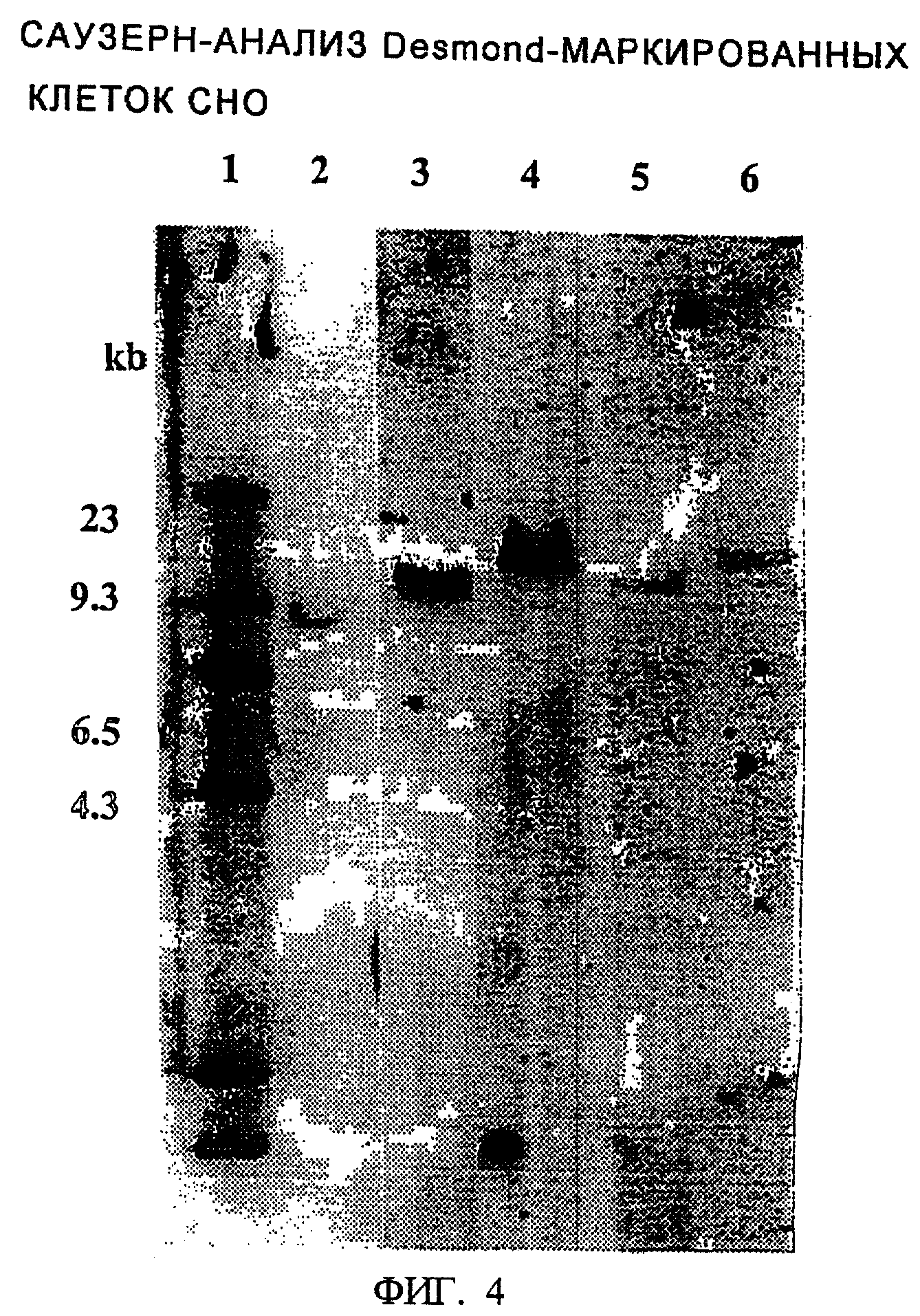
Геномную ДНК выделяли из всех стабильно растущих Desmond-маркированных клеток СНО. ДНК выделяли с использованием набора Invitrogen Easy® DNA kit, в соответствии с инструкциями производителя. Затем геномную ДНК гидролизовали ферментом HindiI II в течение ночи при 37°С, и подвергали Саузерн-анализу с использованием PCR-генерированного меченного дигоксигенином зонда, специфичного для DHFR-гена. Гибридизации и промывки осуществляли с использованием набора для гибридизации Boehringer Mannhiem's DIG easy hyb (catalog # 1603558), промывки DIG Wash и блокирующего буфера Block Buffer Set (catalog # 1585762) в соответствии с инструкциями производителей. Предполагалось, что ДНК-образцы, содержащие одну полосу, гибридизующуюся с DHFR-зондом, представляли собой Desmond-клоны, происходящие от одной клетки, которая имела одну интегрированную копию плазмиды. Эти клоны были оставлены для последующих анализов. Из всех 45 HisD резистентных выделенных клеточных линий, только 5 представляли собой однокопийные интегранты. На фиг.4 показан Саузерн-блот, содержащий все 5 этих однокопийных Desmond-клонов. Обозначения клонов даны в надписях.
(b) Нозерн-анализ
Полную РНК выделяли из всех однокопийных Desmond-клонов с использованием реагента TRIzol (Gibco/BRL, cat # 15596026) в соответствии с инструкциями производителей. 10-20 мкг РНК от каждого клона анализировали на дубликатных формальдегидных гелях. Полученные блоты зондировали с использованием PCR-генерированных меченных дигоксегинином ДНК-зондов для (i) DHFR-транскрипта, (ii) HisD-транскрипта, и (iii) CAD-транскрипта. CAD представляет собой трехфункциональный белок, участвующий в биосинтезе уридина (Wahl et al., J. Biol. Chem., 254, 17:8679-8689 (1979)), и одинаково экспрессируется в клетках всех типов. В данном случае, он использовался в качестве внутреннего контроля для облегчения количественной оценки загрузки РНК. Гибридизации и промывки осуществляли с использованием вышеупомянутых реагентов Boehringer Mannhiem. Результаты Нозерн-анализов показаны на Фиг.5. Однокопийным Desmond-клоном, обнаруживающим самые высокие уровни как HisD-, так и DHFR-транскрипта, был клон 15С9, показанный на дорожке 4 на обеих панелях рисунка. Этот клон был назван "маркированной клеточной линией" и использован для последующих экспериментов по направленного переноса генов в СНО, примеры которых представлены в следующих главах.
Пример 4
Экспрессия антитела против CD20 в Desmond-маркированных клетках СНО
С2В8, химерное антитело, которое распознает антиген CD20 на поверхности В-клеток, было ранее клонировано и экспрессировано в нашей лаборатории. (Reff et al., Blood, 83:434-45 (1994)). ДНК-фрагмент в 4,1 т.п.о., содержащий гены легкой и тяжелой цепи С2В8, вместе с необходимыми регуляторными элементами (эукариотическим промотором и сигналами полиаденилирования) встраивали в искусственный интрон, созданный между экзонами 1 и 2 гена neo, присутствующего в векторе клонирования, происходящего от pBR. Этот вновь генерированный ДНК-фрагмент в 5 т.п.о. (содержащий neo-экзон 1, С2В8 и neo-экзон 2) вырезали и использовали для сборки плазмиды целевого переноса Molly. Другие ДНК-элементы, использованные для конструирования плазмиды Molly, были идентичными элементам, использованным для конструирования маркерной плазмиды Desmond, идентифицированной выше. Полная карта плазмиды Molly показана на фиг.2.
Вектор целевого переноса Molly был линеаризован, а затем трансфецирован путем гидролиза ферментами KpnI и PacI, осажден этанолом и ресуспендирован в стерильном ТЕ до концентрации 1,5 мг/мл. Линеаризованную плазмиду вводили в экспоненциально растущие Desmond-маркированные клетки, в основном, так, как описано выше, за исключением того, что в каждой стадии электропорации использовали 80 мкг ДНК. Через сорок восемь часов после электропорации, в 96-луночные планшеты добавляли селективную среду - CHO-SSFMII, в которую было добавлено 400 мкг/мл генетицина (Geneticin) (Gibco/BRL catalog # 10131-019). Планшеты поддерживали в селективной среде вплоть до 30 дней, либо до тех пор, пока в некоторых лунках не стал наблюдаться рост клеток. Предполагается, что такой рост является результатом клональной экспансии одиночной С418-резистентной клетки. Супернатанты от всех G418-резистентных лунок анализировали на продуцирование С2В8 с помощью стандартной ELISA-техники, и все продуктивные клоны были, в конечном счете, размножены в роллерных 120 мл-колбах, а затем проанализированы.
Характеризация антитело-секретирующих клеток-мишеней
В этом эксперименте было проведено всего 50 электропорации с использованием плазмиды целевого переноса Molly, каждая из которых была внесена в отдельные 96-луночные планшеты. Всего было получено 10 жизнеспособных клонов, секретирующих антитело против CD20, и эти клоны были размножены в роллерных 120 мл-колбах. Из всех клонов выделяли геномную ДНК и осуществляли Саузерн-анализы для того, чтобы определить, представляют ли эти клоны лишь события гомологичной рекомбинации, или в этих же самых клетках происходят, кроме того, события случайной интеграции. Способы выделения ДНК и Саузерн-гибридизации описаны в предыдущем разделе. Геномную ДНК гидролизовали EcoRI и зондировали PCR-генерированным меченным дигоксигенином зондом, специфичным для сегмента константной области тяжелой цепи CD20. Результаты этого Саузерн-анализа представлены на фиг.6. Как видно из фиг.6 восемь из 10 клонов обнаруживали одну полосу, гибридизующуюся с СD20-зондом, что указывало на то, что в этих клетках происходят лишь события гомологичной рекомбинации. Два из десяти клонов, 24G2 и 28С9, обнаруживали присутствие дополнительной полосы (полос), что указывало на то, что в этом геноме, кроме того, имеет место случайная интеграция.
Для всех десяти клонов были оценены уровни экспрессии антитела против CD20, и данные этих оценок представлены в таблице 1.
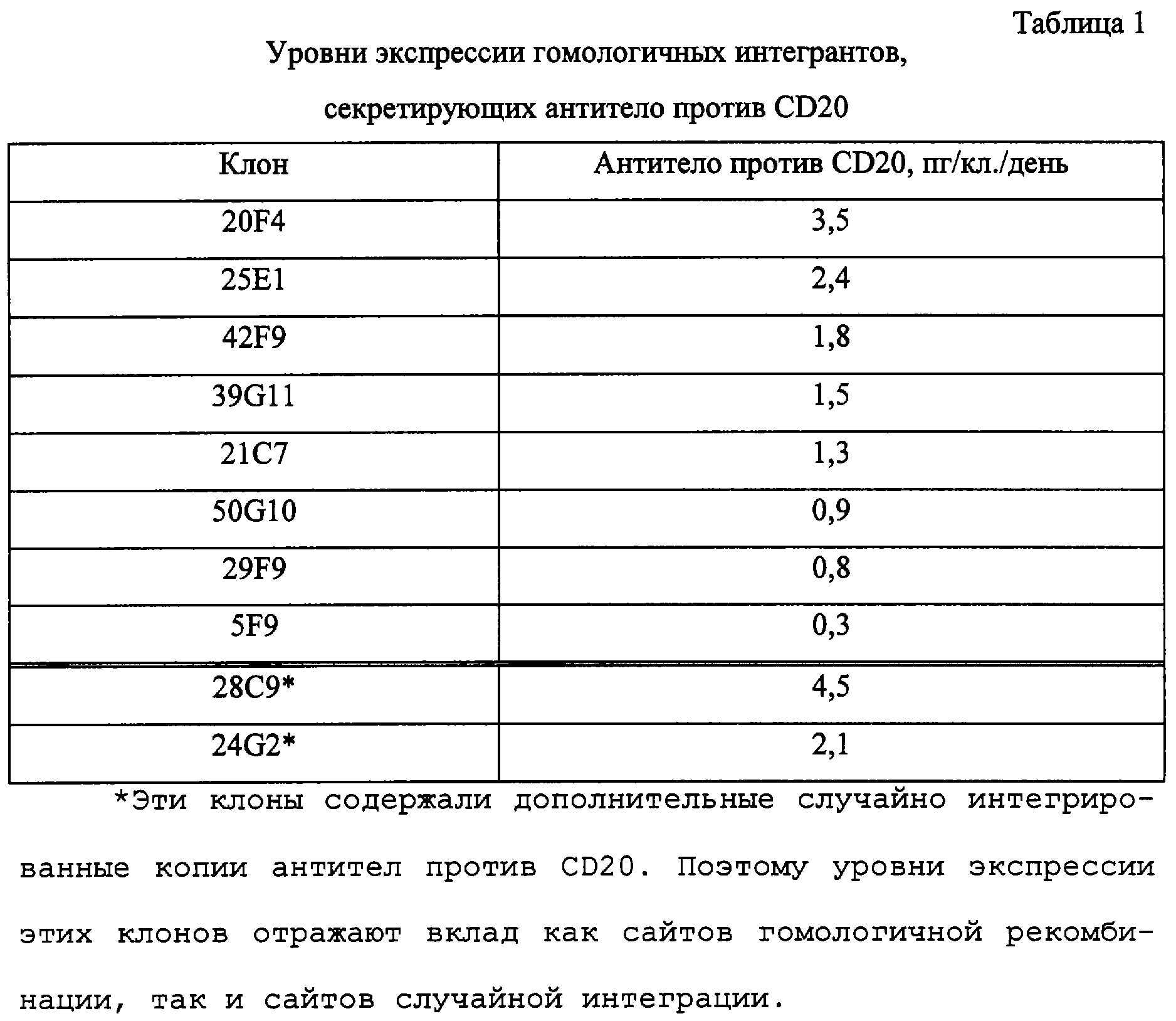
Уровни экспрессии, секретированные отдельными клонами, выражены в пикограммах на клетку в день (пг/кл./день) и представляют собой уровни, полученные из трех ELISA-анализов, проведенных на образцах, взятых из роллерных 120 мл-колб.
Как видно из этих данных, между клонами с наибольшей экспрессией и клонами с наименьшей экспрессией, секреция антитела варьируется приблизительно в десять раз. Этот результат оказался несколько неожиданным, так как мы предполагали получить аналогичные уровни экспрессии для всех клонов, исходя из того факта, что все анти-СD20-гены были интегрированы в тот же самый Desmond-маркированный сайт. Тем не менее, этот наблюдаемый диапазон экспрессии является чрезвычайно малым по сравнению с тем, что наблюдалось с использованием любого традиционного метода случайной интеграции или с использованием нашей трансляционно ослабленной векторной системы.
Клон 20F4, продуцирующий наиболее высокий уровень однокопийного интегранта, был отобран для дальнейшего исследования. В таблице 2 представлены результаты ELISA и данные по культивированию клеток, полученные из семидневных стадий продуцирования этого клона.

Клон 20F4 засевали при концентрации 2×10 мл в роллерной 120 мл-колбе на день 0. В последующие шесть дней проводили подсчет клеток, при этом подсчет проводили в дубликатах и из этой колбы брали 1 мл образцов супернатанта и анализировали с помощью ELISA на секретированные антитела против CD20.
Исходя из данных ELISA, этот клон секретировал в среднем 3-5 пг антитела на одну клетку в день. Этот уровень был аналогичен уровню, полученному от других высокоэкспрессирующих однокопийных клонов, полученных в нашей лаборатории с использованием ранее сконструированных трансляционно ослабленных векторов для случайной интеграции. Этот результат свидетельствует о том, что:
(1) сайт, присутствующий в геноме клеток СНО, маркированных Desmond-маркирующим вектором, является в высокой степени транскрипционно активным, а поэтому он представляет собой наиболее подходящий сайт, из которого экспрессируются рекомбинантные белки; и
(2) направленный перенос генов посредством гомологичной рекомбинации может быть осуществлен с использованием векторов настоящего изобретения и происходит с частотой, достаточно высокой для того, чтобы сделать эту систему осуществимой и желаемой альтернативой методам случайной интеграции.
Для дополнительной иллюстрации эффективности этой системы, нами было также продемонстрировано, что этот сайт может быть амплифицирован, что может приводить даже к более высоким уровням экспрессии гена и секреции белка. Амплификацию осуществляли путем посева 20Г4-клеток в серийных разведениях, начиная с плотности 2, 5×104 клеток/мл, в 96-луночные планшеты для культивирования тканей, и культивирования этих клеток в средах (CHO-SSFMII), в которые было добавлено 5, 10, 15 или 20 нМ метотрексата. Антителосекретирующие клоны скринировали с использованием стандартной ELISA-техники, и наиболее высокопродуцирующие клоны были размножены, а затем проанализированы. Результаты этого эксперимента по амплификации представлены в таблице 3.

Амплификацию с использованием метотрексата 20F4 проводили, как описано в тексте, с использованием концентраций метотрексата, указанных в таблице. 3 Супернатанты от всех выживших 96-луночных колоний анализировали с помощью ELISA, и диапазон уровней экспрессии антител против CD20, продуцированных этими клонами, указан в столбце 3. Исходя из этих результатов, наиболее высокопродуцирующие клоны размножали в роллерных 120 мл-колбах и супернатанты от спин-культур подвергали нескольким анализам ELISA для определения уровней экспрессии (пг/клетку/день), приведенных в столбце 5.
Полученные данные ясно указывают на то, что этот сайт может быть амплифицирован в присутствии метотрексата. Было обнаружено, что клоны от 10 и 15 нМ-амплификации продуцируют уровни порядка 15-20 пг/клетку/день.
Клон 15 нМ, обозначенный 20F4-15A5, был отобран в качестве наиболее высокоэкспрессирующей клеточной линии. Этот клон был получен из 96-луночного планшета, в котором рост наблюдался только в 22 лунках, а поэтому можно допустить, что он происходит от одной клетки. Клон 15 нМ, обозначенный 20F4-15A5, был отобран в качестве наиболее высокоэкспрессирующей клеточной линии. Этот клон был получен из 96-луночного планшета, в котором рост наблюдался только в 22 лунках, а поэтому можно допустить, что он происходит от одной клетки. Затем клон подвергали дополнительному циклу амплификации с использованием метотрексата. Как описано выше, культуру в серийных разведениях высевали в 96-луночные планшеты, и культивировали в среде CHO-SS-FMII, в которую было добавлено 200, 300 или 400 нМ метотрексата. Выжившие клоны были скринированы с помощью ELISA, и несколько высокопродуцирующих клонов размножали с получением спин-культур, а затем анализировали. Результаты этого второго эксперимента по амплификации представлены в таблице 4.

Амплификацию с использованием метотрексата 20F4-15A5 проводили, как описано в тексте, с использованием концентраций метотрексата, указанных в таблице. 4 Клоны в лунках с наиболее высокой степенью продуцирования, номера которых указаны в столбце 4, размножали в роллерных 120 мл-колбах. Уровни экспрессии от клеточных линий, полученных из этих лунок, выражены в пг/клетку в день и приведены в столбце 5.
Наиболее высоко продуцирующие клоны были получены в результате амплификации с использованием 250 нМ метотрексата. 250 нМ-Клон, 20F4-15A5-250A6, был получен из 96-луночного планшета, в котором рост обнаруживали только клетки, а поэтому можно предполагать, что он происходит от одной клетки. Данные таблиц 3 и 4, взятые вместе, со всей очевидностью показывают, что два цикла амплификации с использованием метотрексата вполне достаточно для достижения уровней экспрессии 60 пг/кл/день, которые приближаются к максимальному уровню секреции иммуноглобулина в клетках млекопитающих (Reff, M.E., Curr. Opin. Biotech., 4:573-576 (1993)). Возможность достичь этого уровня экспрессии только за две стадии амплификации повышает ценность этой системы гомологичной рекомбинации. Обычно в методах случайной интеграции для достижения такого уровня экспрессии требуется более чем две стадии амплификации, и они обычно менее эффективны с точки зрения простоты амплификации. Таким образом, система гомологичной рекомбинации дает более эффективный и требующий меньшего времени способ достижения высокого уровня экспрессии генов в клетках млекопитающих.
Пример 5
Экспрессия антитела против CD23 человека в Desmond-маркированных клетках СНО
CD23 представляет собой низкоаффинный рецептор IgE, который опосредует связывание IgE с В- и Т-лимфоцитами (Sutton, B.J. & Gould, H.J., Nature, 366:421-428 (1993)). Моноклональное антитело против CD23 человека 5Е8 представляет собой моноклональное антитело гамма-1, которое было недавно клонировано и экспрессировано в нашей лаборатории. Это антитело описано в совместной переуступленной заявке peг. №08/803085, поданной 20 февраля 1997.
Гены тяжелой и легкой цепи антитела 5Е8 были клонированы в экспрессирующий вектор млекопитающего N5KG1, полученный из вектора NEOSPLA (Barnett et al., Antibody Expression and Endineering, H.Y. Yang & T. Imanaka, eds., p. 27-40 (1995)), а затем в эти гены были введены две модификации. Недавно мы наблюдали несколько более высокую секрецию легких цепей иммуноглобулина по сравнению с тяжелыми цепями в других экспрессирующих конструкциях, полученных в лаборатории (Reff et al., неопубликованные наблюдения). В попытке компенсировать этот дефицит, мы модифицировали ген тяжелой цепи 5Е8, путем добавления более сильного промотора/энхансера непосредственно выше от сайта инициации. В последующих стадиях, 2,9 т.п.о. - ДНК-фрагмент, содержащий модифицированные гены легкой и тяжелой цепи 5Е8, выделяли из вектора N5KG1 и встраивали в вектор целевой доставки Mandy. Получение 5Е8-содержащего вектора Molly и электропорацию в клетки СНО, содержащие Desmond 15C9, осуществляли, в основном, как описано в предыдущем разделе.
В соответствии с ранее описанной схемой, одна модификация была осуществлена в культуральной среде используемого типа. Desmond-маркированные клетки СНО культивировали в среде CD-CHO, не содержащей белка (Gibco-BRL, catalog # AS21206), в которую было добавлено 3 мг/л рекомбинантного инсулина (3 мг/мл маточного раствора, Gibco-BRL, catalog # AS22057), и 8 мМ L-глутамина (200 мг/мл маточного раствора, Gibco-BRL, catalog # 25030-81). Затем трансфецированные клетки отбирали в вышеуказанной среде, в которую было добавлено 400 мкг/мл генетицина. В этом эксперименте было осуществлено 20 электропораций, и полученные клетки засевали в 96-луночные планшеты для культивирования тканей. Клетки росли и секретировали антитело против CD23 всего в 68 лунках, все из которых, предположительно, представляли клоны, происходящие от одной С418-клетки. Двенадцать из этих клонов размножали в роллерных 120 мл-колбах для последующего анализа. Мы предполагаем, что повышенное число клонов, выделенное в этом эксперименте (68 по сравнению с 10 для антитела против CD20, описанного в Примере 4), обусловлено более высокой эффективностью клонирования и уровнем выживания клеток, культивированных в среде CD-CHO по сравнению с клетками, культивированными в среде CHO-SS-FMII. Уровни экспрессии для этих клонов, проанализированных в спин-культуре, составляли в пределах 0,5-3 пг/кл./день, что почти соответствует уровням, наблюдаемым для клонов, секретирующих антитело против CD20. Для увеличения уровней экспрессии клона, продуцирующего наиболее высокие уровни антитела против CD23 и обозначенного 4Н12, его подвергали амплификации с использованием метотрексата. Эту амплификацию осуществляли способом, аналогичным способу, использованному для анти-CD20-клони, описанного в Примере 4. Экспоненциально растущие 4Н12-клетки в серийных разведениях засевали в 96-луночные планшеты для культивирования тканей и культивировали в среде CD-CHO, в которую было добавлено 3 мг/л инсулина, 8 мМ глутамина и 30, 35 или 40 нМ метотрексата. Результаты этого эксперимента по амплификации представлены в таблице 5.

Наиболее высокоэкспрессирующим полученным клоном был 30 нМ клон, выделенный из планшета, в котором рост обнаруживали 22 лунки. Этот клон, обозначенный 4H12-30G5, репродуцируемо секретировал 18-22 пг антитела на клетку в день. Этот же самый интервал уровней экспрессии наблюдался для первой амплификации анти-С020 клона 20F4 (клон 20F4-15AS, который продуцировал 15-18 пг/кл./день, как описано в Примере 4). Эти данные служат для дополнительного подтверждения наблюдения того факта, что амплификация в этом маркированном сайте в СНО является репродуцируемой и эффективной. Вторая амплификация этой 30 нМ-клеточной линии проводится в настоящее время. Ожидается, что уровни насыщения экспрессии могут быть достигнуты для антитела против CD23 лишь за две стадии амплификации, как и в случае антитела против CD20.
Пример 6
Экспрессия иммуноадгезина в Desmond-маркированных клетках СНО
CTLA-4, член суперсемейства 1д, обнаружен на поверхности Т-лимфоцитов, и, очевидно, играет определенную роль в антиген-специфической активации Т-клеток (Dariavach et al., Eur. J. Iinmunol., 18:1901-1905 (1988); и Linsley et al., J. Exp. Med., 174:561-569 (1991)). Для дальнейшего исследования конкретной роли молекулы CTLA-4 в пути активации, был сконструирован растворимый слитый белок, содержащий внеклеточный домен CTLA-4, присоединенный к усеченной форме константной области IgGI человека (Linsley et al., там же). Недавно нами была осуществлена экспрессия этого слитого белка CTLA-4-Ig в экспрессирующем векторе BLECH1 млекопитающего, производном плазмиды NEOSPA (Barnett et al., in Antibody Expression and Engineering, H.Y. Yang & T. Imanaka, eds., p. 27-40 (1995)). 800 п.о. - фрагмент, кодирующий CTLA-4-Ig, был выделен из этого вектора, и вставлен между SacII- и BgIII-сайтами в Molly.
Получение CTLA-4-Ig-Molly и электропорацию в клетках СНО Desmond-клона 15С9 осуществляли как описано в предыдущем примере, относящемся к анти-СD20-клону. Было осуществлено двадцать процедур электропорации, а затем материал высевали в 96-луночные культуральные планшеты, как описано ранее. Из 96-луночных планшетов были выделены восемнадцать CTLA-4-экспрессирующих лунок и помещены непосредственно в роллерные 120 мл-колбы. Для определения того, сколько гомологичных клонов содержат дополнительные случайные интегранты, был проведен Саузерн-анализ геномной ДНК, выделенной из этих клонов. Геномную ДНК гидролизовали ферментом BglII и зондировали PCR-генерированным и меченным дигоксигенином зондом для константной области IgGI человека. Результаты этого анализа указывали на то, что 85% CTLA-4-клонов являлись только гомологичными интегрантами; остальные 15% содержали один дополнительный случайный интегрант. Этот результат подтверждает факты, выявленные в результате экспрессии анти-СD20-клона и обсуждаемые выше, где 80% клонов являлись только гомологичными интегрантами. Следовательно, исходя из этого можно сделать вывод, что эта система экспрессии позволяет репродуцируемо получать только целевые гомологичные интегранты, по крайней мере, в 80% от всех продуцируемых клонов.
Уровни экспрессии для гомологичных CTLA-4-Ig-клонов составляли в пределах от 8-12 пг/кл./день. Этот диапазон несколько выше, чем диапазон, указанный для клонов антитела против CD20 и антитела против CD23, обсуждаемых выше. Однако ранее мы наблюдали, что экспрессия этой молекулы с использованием интронной инсерционной векторной системы также дает значительно более высокие уровни экспрессии, чем те, которые были получены для иммуноглобулинов. В настоящее время мы не можем дать объяснения этому наблюдению.
Пример 7
Направленное введение антитела против CD20 в другую Desmond-маркированную клеточную линию СНО
Как было описано в предыдущем разделе, получили 5 однокопийных Desmond-маркированных клеточных линий СНО (см. фиг.4 и 5). Для иллюстрации того, что успех нашей стратегии целевого переноса не обусловлен некоторым уникальным свойством Desmond-клона 15С9 и не ограничен только этим клоном, мы ввели анти-СD20-плазмиду Molly в Desmond-клон 9В2 (дорожка 6 на фиг.4, дорожка 1 на фиг.5). Получение ДНК Molly и электропорацию в Desmond-клон 9В2 осуществляли точно так же, как описано в предыдущем примере, относящемся к антителу против CD20. В этом эксперименте мы получили один гомологичный интегрант. Этот клон был размножен в роллерной 120-мл колбе, где он продуцировал в среднем 1,2 пг/кл/день антитела против CD20. Этот уровень экспрессии значительно меньше, чем тот, который мы наблюдали с использованием плазмиды Molly, введенной путем целевого переноса в Desmond-клон 15С9. Однако этот результат можно было предвидеть, если учесть проведенный нами Нозерн-анализ Desmond-клонов. Как можно видеть из фиг.5, уровни мРНК от клона 9В2 были значительно ниже, чем уровни от 15С9, что указывало на то, что сайт в этом клоне не является транскрипционно активным, как в клоне 15С9. Поэтому этот эксперимент не только демонстрирует репродуцируемость этой системы, - предположительно, любой маркированный сайт Desmond может быть введен путем целевого переноса с использованием Molly, - но и также подтверждает данные Нозерн-анализа, указывающие на то, что сайт в Desmond-клоне 15С9 является наиболее транскрипционно активным.
Исходя из вышеуказанного следует отметить, что хотя конкретные варианты настоящего изобретения были описаны здесь в целях иллюстрации, однако в настоящее изобретение могут быть внесены различные модификации, не выходящие за рамки объема изобретения. В соответствии с этим настоящее изобретение не ограничено прилагаемыми пунктами формулой изобретения.
Реферат
Изобретение относится к области генной инженерии и может быть использовано в биотехнологической промышленности. Предложен способ интеграции интересующей ДНК в сайт генома клетки млекопитающего, характеризующийся высокой транскрипционной активностью. Указанный сайт предварительно маркируют путем введения в клетку специально сконструированной плазмиды (“маркерной”), включающей а) фрагмент гетерологичной по отношению к геному клетки ДНК, которая после интеграции в геном дает уникальный сайт для гомологичной рекомбинации; б) фрагмент ДНК, кодирующий часть первого селективного маркера и в) по крайней мере одну маркерную последовательность ДНК, обеспечивающую возможность отбора клеток, в которых успешно прошла интеграция “маркерной” плазмиды. Отобранные на первой стадии клетки затем трансформируют второй (“целевой”) плазмидой, включающей (а) фрагмент ДНК, обладающий достаточной для осуществления рекомбинации гомологией с уникальным сайтом а) “маркерной” плазмиды; б) фрагмент ДНК, кодирующий вторую часть первого селективного маркера, совместная экспрессия которого с элементом (б) “маркерной” плазмиды обеспечивает синтез полного маркерного белка и в) ДНК, которую предполагается встроить в геном. Путем скрининга на экспрессию первого селективного маркера отбирают клетки, в которых ДНК “целевой” плазмиды и соответственно “нужная” ДНК, включилась в состав геномной ДНК. Предложен набор для осуществления способа, содержащий по крайней мере “маркерную” и “целевую” плазмиды. Применение изобретения обеспечивает высокий уровень экспрессии при получении любых рекомбинантных белков. 2 с. и 39 з.п. ф-лы, 10 ил., 5 табл.

Комментарии