3-амино-6,7-дикислородсодержащие стероиды, фармацевтическая композиция, способы лечения - RU2291873C2
Код документа: RU2291873C2
Чертежи










Описание
Область техники
Настоящее изобретение относится к 3-амино-6,7-дикислородсодержащим стероидам, композициям, включающим эти стероиды, а также к их терапевтическому применению.
Уровень техники
Воспалительная реакция (воспаление)
Воспаление представляет собой важнейшую локализованную реакцию организма-хозяина на вторжение микроорганизма или повреждение ткани, эта реакция связана с клетками иммунной системы. Классические признаки воспаления включают покраснение (эритему), распухание (отек), боль и увеличенное продуцирование тепла (гипертермию) в зоне воспаления. Воспалительная реакция дает возможность организму специфически распознавать и уничтожать вторгшийся микроорганизм и/или восстанавливать повреждение ткани. Многие из острых видоизменений в зоне воспаления являются либо прямым, либо косвенным следствием массивного притока лейкоцитов (например, нейтрофилов, эозинофилов, лимфоцитов, моноцитов), который свойствен этой реакции. Инфильтрация и аккумуляция лейкоцитов в тканях приводит к их активации и последовательному высвобождению медиаторов воспалительного процесса, таких как LTB4, простагландины, фактор некроза опухолей TNF-α, интерлейкины IL-β, IL-8, IL-5, IL-4, гистамин, протеазы и, например, реакционноспособные формы кислорода.
Обычное воспаление представляет собой чрезвычайно регулируемый процесс, который постоянно контролируется на нескольких уровнях для каждого типа клеток, участвующих в этой реакции. Например, экспрессия провоспалительного цитокина TNF-α контролируется на уровне генной экспрессии, трансляции, пост-трансляционной модификации и высвобождения готовой формы из клеточной мембраны. Множество белков, активированных при воспалении, контролируется фактором транскрипции, NF-kB. Сопутствующие воспалению реакции в некоторых случаях регулируются эндогенными противовоспалительными механизмами, например, такими как генерирование интерлейкина IL-10. Характерной особенностью обычной воспалительной реакции является то, что она временна по своей природе и после нее наступает фаза прекращения воспалительных явлений, в которой ткани возвращаются в свое первоначальное состояние. Как полагают, фаза прекращения воспалительных явлений связана с позитивной регуляцией и действием противовоспалительных механизмов, например, с участием интерлейкина IL-10, а также с негативной модуляцией процессов, сопутствующих воспалению.
Воспалительное заболевание
Воспалительное заболевание происходит в том случае, когда воспалительная реакция инициируется тогда, когда в этом нет необходимости, и/или не прекращается нормальным образом, а продолжается и переходит в хроническое воспалительное состояние. Воспалительное заболевание может относиться ко всему организму (например, волчанка) или быть локализованным в конкретных тканях или органах, и таким образом являться ужасным бременем для личности и тяжелым экономическим грузом для общества. Примеры некоторых наиболее распространенных и проблемных заболеваний включают астму, аллергию, ревматоидный артрит, воспалительные заболевания желудочно-кишечного тракта, псориаз, эмфизему, колит, заболевания, связанные с реакцией "трансплантат против хозяина", контактный дерматит и связанные с ишемией нарушения кровообращения. Другие болезненные состояния, такие как иммунологическая недостаточность, как известно, связаны с изменениями регуляции в системе хемокин/цитокин и соответствующих рецепторах, что может видоизменить репликацию вирусов и привести к синдрому приобретенного иммунодефицита - патогенезу при СПИД.
Сообщалось о множестве процессов в тканях, клетках, а также биохимических процессах, которые нарушаются при воспалительных заболеваниях, и это учитывалось при разработке экспериментальных моделей и подходов для того, чтобы свести к минимуму такие болезненные состояния. Такие исследования in vitro и in vivo предоставили возможность для отбора и скрининга соединений, которые с высокой вероятностью являются терапевтически эффективными для соответствующих воспалительных заболеваний. Например, способность соединения ингибировать аллерген-индуцированную аккумуляцию клеток зоны воспаления, таких как эозинофилы и лимфоциты, в промывной жидкости, полученной от сенсибилизированных животных, является показателем противоастматической активности. В частности, эта модельная система полезна для оценки влияния соединений при лечении на поздних стадиях воспалительной реакции и при гиперчувствительности, которая характерна для астмы, в тех случаях, когда обнаруживается воспаление легкого.
Астма и аллергия
Астма и аллергия тесно связаны, убедительным подтверждением чему являются данные клинических исследований, свидетельствующие о том, что имеется определенная корреляция между выраженностью астмы и степенью атопии (аллергии). Чувствительность к аллергенам, как полагают, является наиболее важным фактором риска астмы как у детей, так и у взрослых, приблизительно в 90% случаев астмы проявляется атопия.
Аллергия характеризуется повышением содержания IgE (антител) в сыворотке крови. Для того чтобы инициировать атопию и последующую астматическую или аллергическую реакцию, обычно необходимы повторные воздействия аллергенов, в процессе, называемом сенсибилизацией. Как только B-клетки подвергаются воздействию аллергенов, они начинают продуцировать антитела, которые связываются с поверхностью тучных клеток. Сшивание двух антител с участием антигена запускает серию реакций, приводящих к дегрануляции и высвобождению ряда медиаторов, которые модулируют воспалительную реакцию. Медиаторы, которые высвобождаются или генерируются при астматической или аллергической реакции, включают гистамин, лейкотриены, простагландины, цитокины и триптазы.
Астма характеризуется гиперчувствительностью дыхательных путей, эпизодическими периодами бронхоспазма и хроническим воспалением легких. Обструкция дыхательных путей является обратимой во времени или в результате реакции на лекарственную терапию. Пациенты с нормальной проходимостью дыхательных путей могут быть гиперчувствительными к различным природным раздражителям, например, таким как холодный воздух, физическая нагрузка, химические продукты и аллергены. Наиболее распространенным явлением, вызывающим астматическую реакцию, является непосредственная аллергическая реакция на общеизвестные аллергены, включая пыльцу амброзии, пыльцу трав, различные грибки, плесень, пыль, тараканов и домашних животных. Симптомы данного заболевания включают затрудненность дыхания, хрипы, нехватку воздуха и кашель. Заболеваемость астмой и связанная с этим смертность возрастают по всему миру, удвоившись за последние двадцать лет, несмотря на современные способы лечения.
Реакция дыхательных путей на аллергены является комплексной и состоит из начальной астматической реакции (EAR), пик которой приходится на время через 20-30 мин после воздействия раздражителя, она характеризуется наличием бронхостеноза и обычно прекращается через 1,5-2 часа. Поздняя астматическая реакция (LAR), как правило, происходит через 3-8 часов после первоначального воздействия раздражителя и включает как наличие бронхостеноза, так и развитие воспаления и отека в легочной ткани. Такое воспаление часто становится хроническим, с наличием повреждения эпителия и инфильтрации легких клетками зоны воспаления, такими как эозинофилы и нейтрофилы.
Современные методы лечения астмы
Глюкокортикоиды (стероиды) представляют собой наиболее эффективное средство длительной терапии для лечения астмы. Например, вследствие наличия воспаления дыхательных путей даже при легком течении астмы вдыхание стероидов используют даже на ранних стадиях лекарственной терапии. Хотя стероиды являются эффективными противовоспалительными средствами, они не слишком пригодны для устранения острого астматического приступа. Введение стероидов перорально связано со значительными побочными эффектами, вследствие чего их постоянное применение для устранения острого астматического приступа является минимальным. Комбинированная терапия часто используется в случае перорального введения стероидов, в этом случае комбинированная терапия может быть подразделена следующим образом: противовоспалительные лекарственные средства (например, стероиды для ингаляции или перорального использования), бронходилаторные средства (например, β2-агонисты, ксантины, антихолинергики) и ингибиторы медиаторов (например, антагонисты кромолинов и лейкотриена). Вообще говоря, имеющийся в настоящее время арсенал медикаментозных средств не слишком хорошо помогает пациентам, страдающим астмой, находящимся в состоянии от средней тяжести до тяжелого. Лекарственные средства, которые безопасны, лишь в самой малой степени эффективны, в то время как эффективным лекарственным средствам присуще неприемлемое побочное действие, связанное с необходимостью всестороннего контроля за состоянием пациента. Находящиеся в состоянии разработки продукты должны удовлетворять требованиям в отношении нежелательных побочных эффектов (например, таких как побочный эффект - рвота в случае некоторых ингибиторов фосфодиэстеразы 4), и исключать плохие фармакокинетические характеристики, а также неудовлетворительные параметры метаболизма. Имеется существенная необходимость в терапевтических средствах, которые позволят осуществить безопасное и эффективное лечение воспалительных заболеваний, таких как астма и аллергия. Настоящее изобретение обеспечивает достижение этого и других полезных результатов, как показано в настоящем описании.
Сущность изобретения
Согласно первому аспекту настоящее изобретение относится к соединениям формулы (1) и их фармацевтически приемлемым солям, сольватам, стереоизомерам и пролекарствам, по отдельности или в смеси,

в которой, в каждом случае независимо друг от друга:
R1 и R2 выбирают из водорода, кислорода таким образом, чтобы образовать нитро или оксим, амино, -SO3-R, и органических групп, содержащих 1-30 атомов углерода и необязательно содержащих 1-6 гетероатомов, выбранных из азота, кислорода, фосфора, кремния и серы, и где R2 может представлять прямую связь с атомом под номером 3, или R1 и R2 могут вместе с атомом азота, к которому они оба присоединены, образовывать гетероциклическую структуру, которая может являться частью органической группы, содержащей 1-30 атомов углерода и необязательно содержащей 1-6 гетероатомов, выбранных из азота, кислорода и кремния, и где R1 может представлять цепь из двух или трех атомов, присоединенную к атому под номером 2 таким образом, что -NR1- образует часть конденсированной бициклической структуры, включающей ядро А;
R3 и R4 выбирают из прямых связей с атомами под номерами 6 и 7 соответственно таким образом, чтобы образовались карбонильные группы, из водорода или из защитной группы, при условии, что R3 и/или R4 представляют часть защитной группы гидроксила или карбонила;
номера от 1 до 17 каждый представляют атом углерода, где атомы углерода под номерами 1, 2, 4, 11, 12, 15, 16 и 17 могут быть независимо замещены:
(a) одной из групп: =O, =C(R5)(R5), =C=C(R5)(R5), -C(R5)(R5)(C(R5)(R5))n- и -(O(C(R5)(R5))nO)-, где n изменяется в диапазоне от 1 до приблизительно 6, или
(b) двумя из следующих групп, которые выбирают независимо: -X, -N(R1)(R2), -R5 и -OR6;
и где атомы углерода под номерами 5, 8, 9, 10, 13 и 14 могут быть независимо замещены одной из групп -X, -R5, -N(R1)(R2) или -OR6;
в дополнение к группам -OR3 и -OR4, как показано, каждый из атомов углерода под номерами 6 и 7 может быть независимо замещен одной из групп -X, -N(R1)(R2), -R5 или -OR6;
каждое из ядер A, B, C и D является независимо полностью насыщенным, частично насыщенным или полностью ненасыщенным;
R5 в каждом случае независимо выбирают из H, X и C1-30-органического фрагмента, который необязательно может содержать, по меньшей мере, один гетероатом, выбранный из группы, состоящей из бора, галогена, азота, кислорода, кремния и серы, где две геминальные группы R5 могут вместе образовывать ядро вместе с атомом углерода, к которому они обе присоединены;
R6 представляет H или защитную группу, такую, что -OR6 представляет защищенную гидроксильную группу, где вицинальные группы -OR6 могут вместе образовывать циклическую структуру, которая защищает вицинальные гидроксильные группы, и где геминальные группы -OR6 могут вместе образовывать циклическую структуру, которая защищает карбонильную группу, и
X представляет фторид, хлорид, бромид и иодид.
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, включающей стероидное соединение, как указано выше, фармацевтически приемлемый носитель, наполнитель или разбавитель.
Согласно другому аспекту настоящее изобретение относится к способу лечения воспаления, включающему введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
Согласно другому аспекту настоящее изобретение относится к способу профилактики воспаления, включающему введение субъекту, нуждающемуся в этом, профилактически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения астмы, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения аллергического заболевания, включающего, но не ограничивающегося этим, наличие кожных и глазных показаний, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения хронического обструктивного легочного заболевания, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения аллергического дерматита, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения твердого новообразования, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения СПИД, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения нарушения кровообращения при ишемии, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения сердечной аритмии, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества стероидного соединения, как указано выше.
Эти и имеющие к ним отношение аспекты настоящего изобретения описаны более подробно ниже.
Краткое описание чертежей
На фиг. 1А и 1В приведены схемы синтетических превращений, которые могут быть использованы для превращения 3-аминостероида в 3-азотсодержащий стероид настоящего изобретения.
На фиг. 2А, 2В и 2С приведен набор столбчатых диаграмм, показывающих влияние соединения 89 (реакция на дозировку, 4 дозы ежедневно, перорально) на индуцированную овальбумином аккумуляцию клеток зоны воспаления в легочной промывочной жидкости, полученной от сенсибилизированных крыс линии Brown Norway. На фиг. 2А показана аккумуляция эозинофилов, на фиг. 2В показана аккумуляция нейтрофилов и на фиг. 2С показана аккумуляция лимфоцитов.
На фиг. 3А, 3В и 3С приведен набор столбчатых диаграмм, показывающих влияние соединения 28 (реакция на дозировку, 4 дозы ежедневно, перорально) на индуцированную овальбумином аккумуляцию клеток зоны воспаления в легочной промывочной жидкости, полученной от сенсибилизированных крыс линии Brown Norway. На фиг. 3А показана аккумуляция эозинофилов, на фиг. 3В показана аккумуляция нейтрофилов и на фиг. 3С показана аккумуляция лимфоцитов.
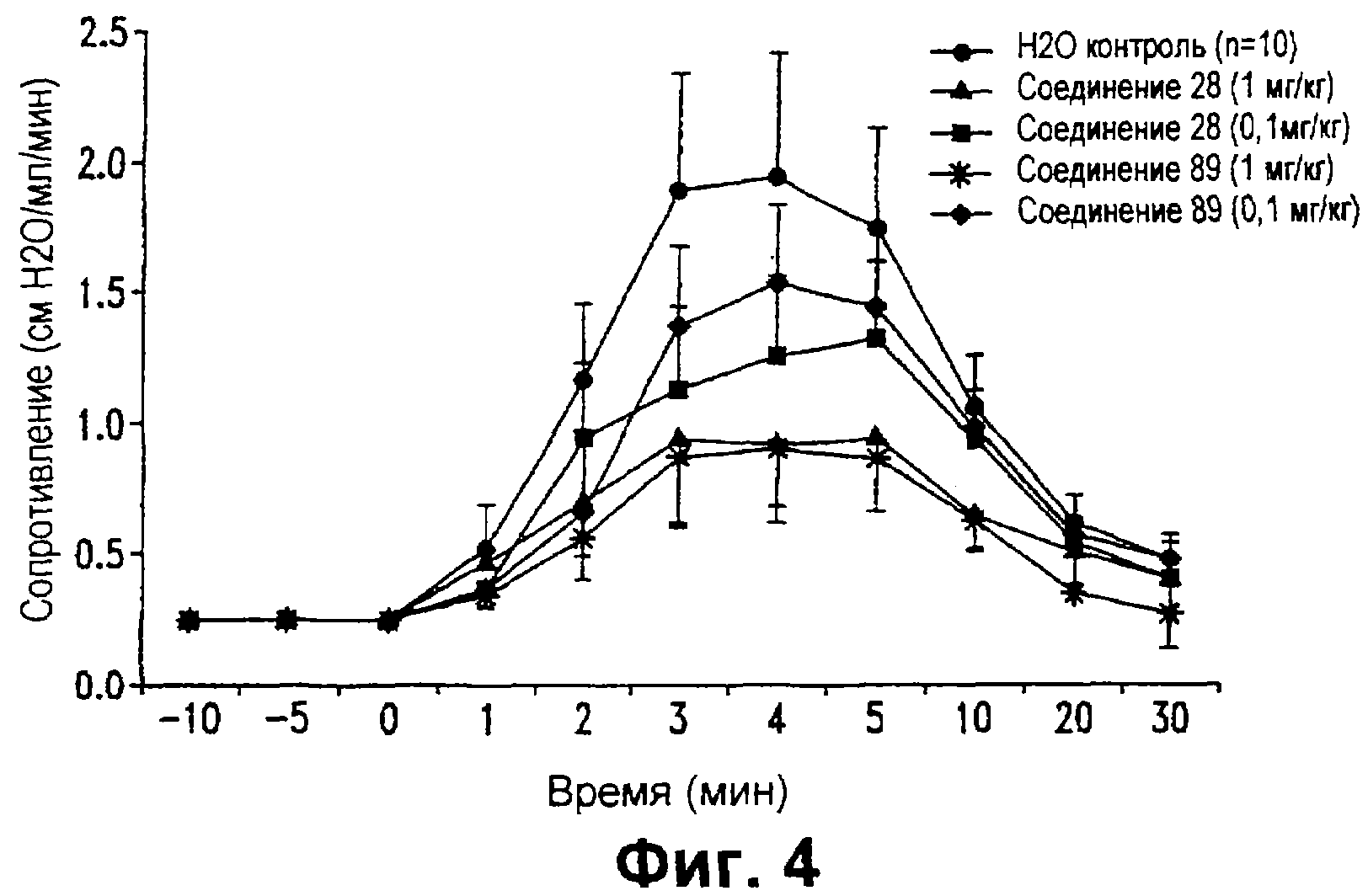
На фиг. 4 представлен график, показывающий влияние тестируемых соединений 28 и 89, при введении перорально один раз в день в течение 4 дней до введения провоцирующей пробы, на индуцированные аллергеном изменения сопротивления легкого у сенсибилизированных морских свинок.
На фиг. 5 представлен график, показывающий влияние тестируемых соединений 28 и 89, при введении перорально один раз в день в течение 4 дней до введения провоцирующей пробы, на индуцированные аллергеном изменения эластичности легкого у сенсибилизированных морских свинок.
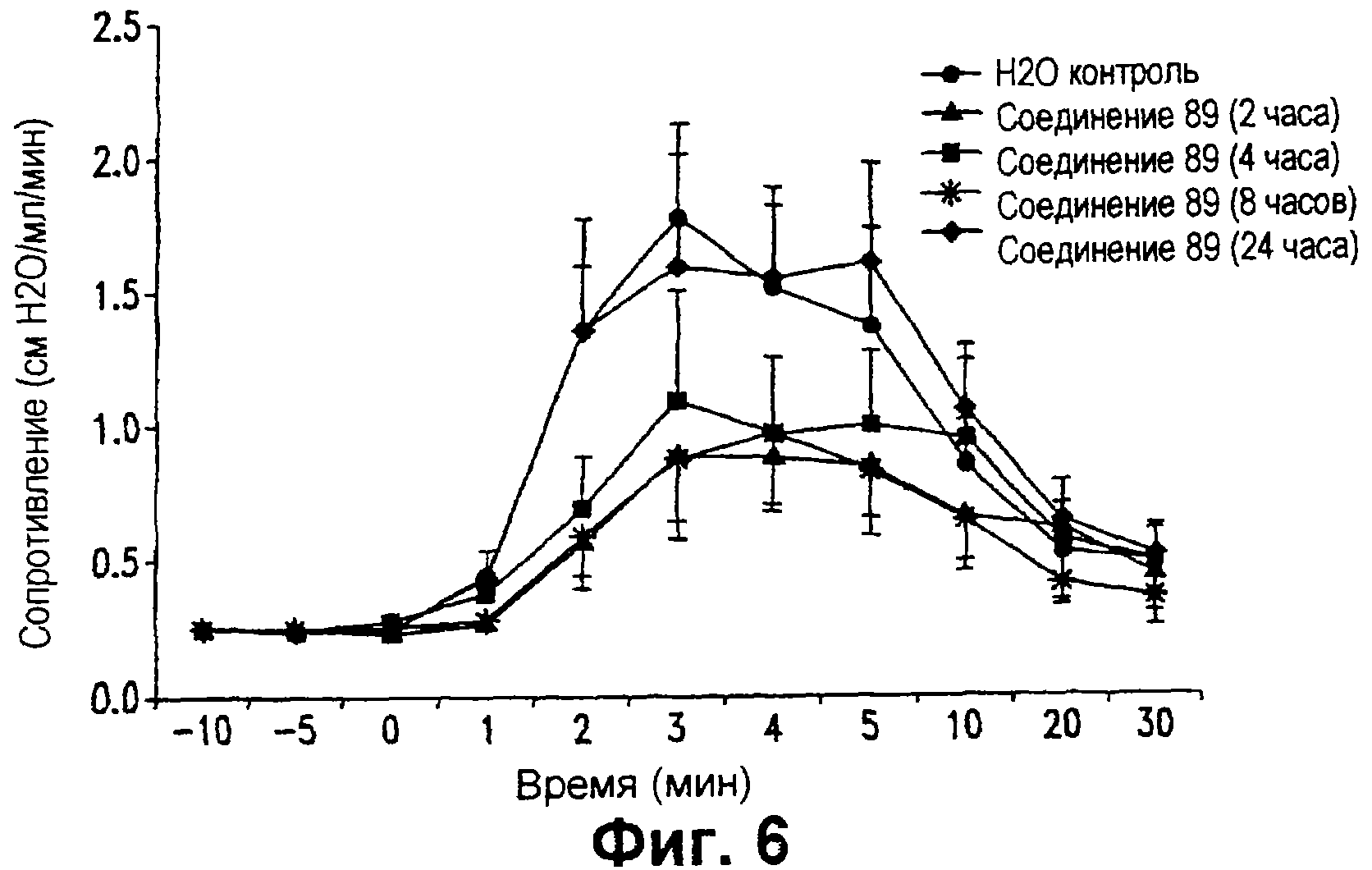
На фиг. 6 представлен график, показывающий продолжительность антибронхоспастической активности тестируемого соединения 89, при введении перорально в количестве 1 мг/кг один раз в день в течение 4 дней до введения провоцирующей пробы, на индуцированные аллергеном изменения сопротивления легких у сенсибилизированных морских свинок.
На фиг. 7 представлен график, показывающий продолжительность антибронхоспастической активности тестируемого соединения 89, при введении перорально в количестве 1 мг/кг один раз в день в течение 4 дней до введения провоцирующей пробы, на индуцированные аллергеном изменения эластичности легких у сенсибилизированных морских свинок.
Подробное описание изобретения
Настоящее изобретение относится к соединениям, композициям и способам, пригодным для лечения и/или профилактики различных болезненных состояний. Например, в соответствии с одним из аспектов настоящее изобретение предоставляет способ лечения и/или профилактики воспалительного заболевания. Способ включает введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы (1) или его фармацевтически приемлемых соли, сольвата, стереоизомера или пролекарства, или эффективного количества композиции, содержащей соединение формулы (1) или его фармацевтически приемлемую соль, сольват, стереоизомер или пролекарство.
Перед тем, как описывать изобретение более подробно, некоторым терминам, которые используются в настоящем описании, будут даны приведенные ниже определения, а также будут раскрыты некоторые обозначения, используемые в настоящем описании.
А. Определения терминов.
При использовании в настоящем описании приведенные ниже термины имеют указанное значение, если не указано иное.
"Алкил" представляет собой моновалентную насыщенную или ненасыщенную, линейную, разветвленную или циклическую, алифатическую (т.е. не ароматическую) углеводородную группу. В соответствии с различными воплощениями изобретения алкильная группа содержит 1-20 атомов углерода, т.е. представляет собой С1-С20 (или С1-С20) группу, или представляет собой С1-С18 группу, С1-С12 группу, С1-С6 группу или С1-С4 группу. Независимо, в различных воплощениях изобретения, алкильная группа содержит ноль разветвлений (т.е. представляет собой линейную цепь), одно разветвление, два разветвления или более чем два разветвления; является насыщенной; является ненасыщенной (где ненасыщенная алкильная группа может содержать одну двойную связь, две двойные связи, более чем две двойные связи и/или одну тройную связь, две тройные связи или более чем две тройные связи); представляет собой или включает циклическую структуру; является ациклической. Примеры алкильных групп включают C1алкильные группы (то есть -CH3 (метил)), C2алкильные группы (то есть -СН2CH3 (этил), -CH=CH2 (этенил) и -C≡CH (этинил)) и C3алкильные группы (то есть -CH2CH2CH3 (н-пропил), -СН(CH3)2 (изо-пропил), -CH=CH-CH3 (1-пропенил), -CC≡ CH3 (1-пропинил), -CH2-CH=CH2 (2-пропенил), -CH2-C≡CH (2-пропинил), -C(CH3)=CH2 (1-метилэтенил) и -CH(CH2)2 (циклопропил)).
"Арил" представляет собой моновалентную ароматическую углеводородную циклическую систему. Эта циклическая система может быть моноциклической или конденсированной полициклической (например, бициклической, трициклической и т.д.). В различных воплощениях моноциклические арильные ядра состоят из C5-C10, или C5-C7, или C5-C6, где эти числа при атомах углерода означают количество атомов углерода, образующих циклическую систему. Предпочтительной арильной группой является С6 циклическая система, то есть фенильное ядро. В различных воплощениях полициклическое ядро представляет собой бициклическую арильную группу, где С8-С12 или С9-С10 являются предпочтительными бициклическими арильными группами. Предпочтительной полициклической арильной группой является нафтильное ядро, которое состоит из 10 атомов углерода.
"Гетероалкил" представляет собой алкильную группу (как определено ранее), в которой, по меньшей мере, один из атомов углерода замещен гетероатомом. Предпочтительными гетероатомами являются азот, кислород, сера и галоген. Гетероатом может, хотя обычно это и не так, иметь такое же количество валентных связей, как и углерод. Соответственно, в том случае, когда углерод заменяется на гетероатом, может потребоваться увеличить или уменьшить количество атомов водорода, связанных с гетероатомом, с тем, чтобы их количество соответствовало количеству валентных связей гетероатома. Например, если углерод (валентность четыре) заменяется на азот (валентность три), то один из атомов водорода, ранее присоединенный к замененному углероду, должен быть удален. Аналогичным образом, если углерод заменяется на галоген (валентность один), то три (то есть все) атомы водорода, ранее присоединенные к замененному углероду, должны быть удалены.
"Гетероарил" является одновалентной ароматической циклической системой, содержащей углерод и, по меньшей мере, один гетероатом в ядре. В различных воплощениях гетероарильная группа может содержать один гетероатом или 1-2 гетероатома, или 1-3 гетероатома, или 1-4 гетероатома в ядре. Гетероарильные ядра могут быть моноциклическими или полициклическими, причем полициклическое ядро может содержать конденсированные, спиро или мостиковые соединение ядер. В одном из воплощений гетероарил выбирают из моноциклических и бициклических ядер. Моноциклические гетероарильные ядра могут содержать приблизительно 5-10 атомов-членов (углерод и гетероатомы), предпочтительно 5-7 и наиболее предпочтительно 5-6 атомов-членов в ядре. Бициклические гетероарильные ядра могут содержать приблизительно 8-12 атомов-членов или 9-10 атомов-членов в ядре. Гетероарильное ядро может быть замещенным или незамещенным. В одном из воплощений гетероарильное ядро является незамещенным. В другом воплощении гетероарильное ядро является замещенным. Примеры гетероарильных групп включают бензофуран, бензотиофен, фуран, имидазол, индол, изотиазол, оксазол, пиперазин, пиразин, пиразол, пиридазин, пиридин, пиримидин, пиррол, хинолин, тиазол и тиофен.
"Гетероатом" представляет собой атом галогена, азота, кислорода, фосфора, кремния или серы. Группы, содержащие более одного гетероатома, могут содержать различные гетероатомы.
"Углеводородные группы" представляют собой химические группы, образованные только водородом и углеродом; "галогенуглеродные группы" представляют собой химические группы, образованные только галогеном и углеродом, и "галогенуглеводородные группы" представляют собой химические группы, образованные только водородом, галогеном и углеродом.
Термины "органические группы" и "органические фрагменты" используются как синонимы и относятся к стабильным структурам, которые состоят из указанного числа атомов указанного типа.
"Фармацевтически приемлемая соль" и "его соли" в случае соединений настоящего изобретения относится к аддитивным солям кислоты и к аддитивным солям основания.
Термин "аддитивная соль кислоты" относится к солям, полученным из соединений согласно настоящему изобретению и неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и им подобных, и/или органических кислот, таких как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота и им подобных кислот.
Термин "аддитивная соль основания" относится к солям, полученным из соединений согласно настоящему изобретению и неорганических оснований, таких как соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и им подобных. Подходящие соли включают соли аммония, калия, натрия, кальция и магния, полученные из фармацевтически приемлемых органических нетоксичных оснований, и включают соли первичных, вторичных и третичных аминов, включая замещенные амины природного происхождения, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаины, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурин, пиперазин, пиперидин, N-этилпиперидин и им подобные.
В тех случаях, когда какая-либо переменная встречается чаще чем один раз в каком-либо компоненте или соединении формулы (1), ее определение в каждом случае является независимым от ее определения в любом другом случае. Допустимы только такие комбинации заместителей и/или переменных, которые приводят к стабильным соединениям. Соединения, пригодные для использования в способах и в композициях согласно настоящему изобретению, также как и соединения настоящего изобретения могут содержать асимметрические центры или находиться в виде рацематов, рацемических смесей и индивидуальных диастереомеров или энантиомеров, и все их изомерные формы входят в объем настоящего изобретения. Рацемат или рацемическая смесь не подразумевает смесь стереоизомеров в соотношении 50:50.
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, включающей соединение формулы (1), как указано выше, в сочетании с фармацевтически приемлемым носителем, наполнителем или разбавителем. Такие композиции могут быть полезны для лечения воспаления или других состояний, как показано в настоящем описании. Данные композиции могут быть также представлены в виде лекарственной формы, которая может быть полезна, например, для лечения воспаления.
Данные композиции полезны, например, в качестве образцов для исследования, удобны для насыпания, пригодны как фармацевтические композиции. Необходимое для испытаний количество соединения согласно настоящему изобретению представляет собой количество, которое легко можно измерить, используя общепринятые методики и процедуры количественного анализа, которые хорошо известны и понятны специалистам в данной области техники. Необходимое для испытаний количество соединения согласно настоящему изобретению будет, как правило, варьироваться от приблизительно 0,001 мас.% до приблизительно 100 мас.% от суммарной массы композиции. Инертные носители включают любое вещество, которое не распадается или другим образом ковалентно не взаимодействует с соединением формулы (1). Примерами подходящих инертных носителей являются вода; водные буферы, такие как буферы, обычно используемые в анализах методом высокоэффективной жидкостной хроматографии (ВЭЖХ); органические растворители, такие как ацетонитрил, этилацетат, гексан и подобные им растворители, и фармацевтически приемлемые носители.
"Фармацевтически приемлемые носители" для терапевтического применения хорошо известны в фармацевтике и описаны, например, в Remingtons Pharmaceutical Sciences, Mack Publishing Co (A.R.Gennaro edit. 1985). Например, могут быть использованы стерильный солевой раствор и забуференный фосфатом солевой раствор с физиологическим значением рН. Консерванты, стабилизаторы, красители и вещества, придающие вкус и запах, могут быть включены в фармацевтическую композицию. Например, в качестве консервантов могут быть добавлены бензоат натрия, сорбиновая кислота и сложные эфиры п-гидроксибензойной кислоты (там же, с.1449). В дополнение, могут быть использованы антиоксиданты и суспендирующие агенты (см. там же).
Стероидные соединения настоящего изобретения содержат, по меньшей мере, четыре ядра, обычно обозначаемых как А, В, С и D, как показано ниже, где ядро А может быть конденсировано с дополнительным ядром:

В. Соединения.
Настоящее изобретение относится к соединениям формулы (1) и их фармацевтически приемлемым солям, сольватам, стереоизомерам и пролекарствам, по отдельности или в смеси,

где, в каждом случае независимо:
R1 и R2 выбирают из водорода, кислорода таким образом, чтобы образовать нитро или оксим, амино, -SO3-R и органических групп, содержащих 1-30 атомов углерода и необязательно содержащих 1-6 гетероатомов, выбранных из азота, кислорода, фосфора, кремния и серы, и где R2 может представлять прямую связь с атомом под номером 3, или R1 и R2 могут вместе с атомом азота, к которому они оба присоединены, образовывать гетероциклическую структуру, которая может являться частью органической группы, содержащей 1-30 атомов углерода и необязательно содержащей 1-6 гетероатомов, выбранных из азота, кислорода и кремния; или где R1 может представлять цепь из двух или трех атомов, присоединенную к атому под номером 2 таким образом, что -NR1- образует часть конденсированной бициклической структуры, включающей ядро А;
R3 и R4 выбирают из прямых связей с атомами под номерами 6 и 7 соответственно таким образом, чтобы образовались карбонильные группы, из водорода или из защитной группы, при условии, что R3 и/или R4 представляют часть защитной группы гидроксила или карбонила;
номера от 1 до 17 каждый представляет атом углерода, где атомы углерода под номерами 1, 2, 4, 11, 12, 15, 16 и 17 могут быть независимо замещены:
(a) одной из групп: =O, =C(R5 )(R5), =C=C(R5)(R5), -C(R5)(R5)(C(R5)(R5))n- и -(O(C(R5)(R5))nO)-, где n изменяется в диапазоне от 1 до приблизительно 6; или
(b) двумя из следующих групп, которые выбирают независимо: -X, -N(R1)(R2), -R5 и -OR6;
и где атомы углерода под номерами 5, 8, 9, 10, 13 и 14 могут быть независимо замещены одной из групп -X, -R5, -N(R1)(R2) или -OR6;
в дополнение к группам -OR3 и -OR4, как показано, каждый из атомов углерода под номерами 6 и 7 может быть независимо замещен одной из групп -X, -N(R1)(R2), -R5 или -OR6;
каждое из ядер A, B, C и D является независимо полностью насыщенным, частично насыщенным или полностью ненасыщенным;
R5 в каждом случае независимо выбирают из H, X и C1-30-органического фрагмента, который необязательно может содержать, по меньшей мере, один гетероатом, выбранный из группы, состоящей из бора, галогена, азота, кислорода, кремния и серы, причем две геминальные группы R5 могут вместе образовывать ядро вместе с атомом углерода, к которому они обе присоединены;
R6 представляет H или защитную группу, такую, что -OR6 представляет защищенную гидроксильную группу, где вицинальные группы -OR6 могут вместе образовывать циклическую структуру, которая защищает вицинальные гидроксильные группы, и где геминальные группы -OR6 могут вместе образовывать циклическую структуру, которая защищает карбонильную группу, и
X представляет фторид, хлорид, бромид и иодид.
В соответствии с одним из аспектов настоящего изобретения R1 и R2выбирают из водорода и органических групп, содержащих 1-30 атомов углерода и необязательно содержащих 1-6 гетероатомов, выбранных из азота, кислорода, фосфора, кремния и серы. Необязательно R2 представляет прямую связь с атомом под номером 3. В соответствии с другим аспектом группы R1, R2 и атом N, к которому они обе присоединены, образуют гетероциклическую структуру, которая может быть частью органической группы, содержащей 1-30 атомов углерода и необязательно содержащей 1-6 гетероатомов, выбранных из азота, кислорода и кремния. В соответствии с другим аспектом R1 может представлять цепь из двух или трех атомов, присоединенную к атому под номером 2 таким образом, что -NR1- образует часть конденсированной бициклической структуры, включающей ядро А, и 2 или 3 атома выбирают из C, N и O таким образом, чтобы образовалась стабильная структура. Необязательно в соответствии с данными и другими аспектами настоящего изобретения органическая группа содержит 1-20 атомов углерода, тогда как в другом необязательном воплощении изобретения органическая группа содержит 1-10 атомов углерода.
Согласно предпочтительному аспекту настоящего изобретения каждый из R1 и R2 представляет водород. Данные стероиды не только обладают необходимой биологической активностью, но также являются удобными соединениями-предшественниками для получения других стероидов согласно настоящему изобретению, в которых R1 и/или R2 не являются водородом.
Например, согласно одному из воплощений изобретение относится к соединению формулы (1), в которой R1 и R2 представляют водород, R3 и R4 выбирают из прямых связей с атомами под номерами 6 и 7 соответственно, таким образом, чтобы образовались карбонильные группы, из атома водорода или защитной группы таким образом, чтобы R3 и/или R4 являлись частью защитной группы гидроксила или карбонила, и в дополнение к приведенным группам -OR3 и -OR4, как показано, каждый из атомов углерода под номерами 6 и 7 замещен атомами водорода в тех возможных случаях, когда -OR3 или -OR4 представляет карбонильную группу; атомы углерода под номерами 1, 2, 4, 11, 12, 15 и 16 каждый замещен двумя атомами водорода; атомы углерода под номерами 5, 8, 9 и 14 каждый замещен одним атомом водорода; атом углерода под номером 10 замещен метилом; атом углерода под номером 13 в том случае, когда он не является частью ненасыщенной связи, замещен метилом; атом углерода под номером 17 замещен (a) одной из групп: =O, =C(R5)(R5), =C=C(R5)(R5), -C(R5)(R5)(C(R5)(R5))n- и -(O(C(R5)(R5))nO)-, где n изменяется в диапазоне от 1 до приблизительно 6; или (b) двумя из следующих групп, которые выбирают независимо: -X, -N(R1)(R2), -R5 и -OR6; каждое из ядер A, B, C и D является независимо полностью насыщенным, частично насыщенным или полностью ненасыщенным; R5 в каждом случае независимо выбирают из H, X и C1-30-органического фрагмента, который необязательно может содержать, по меньшей мере, один гетероатом, выбранный из группы, состоящей из бора, галогена, азота, кислорода, кремния и серы, причем две геминальные группы R5 могут вместе образовывать кольцо вместе с атомом углерода, к которому они обе присоединены; R6 представляет H или защитную группу, такую, что -OR6 представляет защищенную гидроксильную группу, где вицинальные группы -OR6 могут вместе образовывать циклическую структуру, которая защищает вицинальные гидроксильные группы, и где геминальные группы -OR6 могут вместе образовывать циклическую структуру, которая защищает карбонильную группу; и X представляет фторид, хлорид, бромид и иодид.
Согласно другому воплощению изобретение относится к соединению формулы (1), в которой R1 и R2 представляют водород, R3 и R4 выбирают из водорода или защитных групп таким образом, чтобы R3 и/или R4 являлись частью защитной группы гидроксила; атомы углерода под номерами 1, 2, 4, 11, 12, 15 и 16 каждый замещен двумя атомами водорода; атомы углерода под номерами 5, 8, 9 и 14 каждый замещен одним атомом водорода; атом углерода под номером 10 замещен метилом; атом углерода под номером 13 в том случае, когда он не является частью ненасыщенной связи, замещен метилом; атом углерода под номером 17 замещен(a) одной из групп: =C(R5)(R5), =C=C(R5)(R5); или (b) двумя из следующих групп, которые выбирают независимо: -X, -N(R1)(R2) и -R5; каждое из ядер A, B, C и D является независимо полностью насыщенным или частично насыщенным; R5 в каждом случае независимо выбирают из H, X и C1-30-углеводородной, галогенуглеродной и галогенуглеводородной групп; и X представляет фторид, хлорид, бромид и иодид.
Согласно еще одному воплощению изобретение относится к соединению формулы (1), в которой R1 и R2 представляют водород, R3 и R4 выбирают из атома водорода или защитных групп таким образом, чтобы R3 и/или R4 являлись частью защитной группы гидроксила; атомы углерода под номерами 1, 2, 4, 11, 12, 15 и 16 каждый замещен двумя атомами водорода; атомы углерода под номерами 5, 8, 9 и 14 каждый замещен одним атомом водорода; атом углерода под номером 10 замещен метилом; атом углерода под номером 13 в том случае, когда он не является частью ненасыщенной связи, замещен метилом; атом углерода под номером 17 замещен (a) одной из групп: =C(R5)(R5), =C=C(R5)(R5); или (b) двумя группами -R5; каждое из ядер A, B, C и D является независимо полностью насыщенным или частично насыщенным; R5 в каждом случае независимо выбирают из H и C1-30-углеводородной группы.
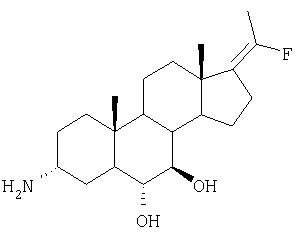
Конкретные соединения настоящего изобретения в которых R1 и R2 представляют водород, включают следующие:





Согласно другому аспекту изобретение относится к стероидам, которые являются 3-азотзамещенными, где 3-азот замещен органической группой. Например, изобретение относится к стероидным соединениям, в которых R1 выбирают из -C(=O)-R7, -C(=O)NH-R7 и -SO2-R7, где R7 выбирают из алкильной, гетероалкильной, арильной и гетероарильной групп. В родственном воплощении R1 представляет водород и R2 представляет -CH2-R7, где R7 выбирают из алкильной, гетероалкильной, арильной и гетероарильной групп. В одном из воплощений R7 выбирают из C1-10-углеводородных групп. В другом воплощении -C(=O)-R7 включает биотин. В другом воплощении R7 выбирают из алкилзамещенного фенила, галогензамещенного фенила, алкоксизамещенного фенила, арилоксизамещенного фенила и нитрозамещенного фенила.
Согласно другому аспекту (R1 )(R2)N- представляет гетероцикл, то есть N, входящий в (R1)(R2)N-, может быть частью гетероциклического ядра. Примеры включают:
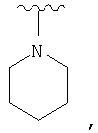



Согласно другому аспекту один или оба из R1 и R2 включают гетероциклическое ядро или карбоциклическое ядро. Предпочтительным гетероциклическим ядром является

и предпочтительным карбоциклическим ядром является фенил, включая замещенный фенил, такой как 3-метилфенил, 4-гидроксифенил и 4-сульфонамидфенил.
Согласно другому аспекту R1 может представлять цепь из двух или трех атомов, присоединенную к атому под номером 2 таким образом, что -NR1 - образует часть конденсированной бициклической структуры, включающей ядро A. Таким образом, настоящее изобретение позволяет получить соединения формулы, приведенной ниже, где Z представляет 2 или 3 атома, выбранных из C, N, и O. Ядро, включающее Z, может быть насыщенным или ненасыщенным.

Примеры таких соединений с конденсированными ядрами включают:

Согласно другому аспекту R1 представляет водород и R2 включает С1-10углеводородную группу.
Согласно другому аспекту R1 представляет водород и R2 представляет гетероалкил. Подходящий гетероалкил включает, без ограничений, С1-10алкил-W-С1-10алкилен, где W выбирают из O и NH; HO-C1-10алкилен- и HO-C1-10алкилен-W-C1-10алкилен-, где W выбирают из O и NH.
Согласно другому аспекту каждый из R1 и R2 независимо выбирают из галогена и органических групп, содержащих 1-20 атомов углерода и необязательно содержащих 1-5 гетероатомов, выбранных из азота, кислорода, кремния и серы.
Согласно другому аспекту каждый из R1 и R2 независимо выбирают из водорода, R8, R9, R10, R11 и R12, где R8 выбирают из С1-10алкила, С1-10гетероалкила, содержащего 1, 2 или 3 гетероатома, С6-10арила и С3-15гетероарила, включающего 1, 2 или 3 гетероатома; R9 выбирают из (R8)r-C1-10алкилена, (R8)r-C1-10 гетероалкилена, включающего 1, 2 или 3 гетероатома, (R8)r-C6-10арилена и (R8)r-C3-15гетероарилена, включающего 1, 2 или 3 гетероатома; R10 выбирают из (R9)r-C1-10алкилена, (R9)r-C1-10гетероалкилена, включающего 1, 2 или 3 гетероатома, (R9)r-C6-10арилена и (R9)r-C3-15гетероарилена, включающего 1, 2 или 3 гетероатома; R11 выбирают из (R10)r-C1-10 алкилена, (R10)r-C1-10гетероалкилена, включающего 1, 2 или 3 гетероатома, (R10)r-C6-10арилена и (R10)r-C3-15гетероарилена, включающего 1, 2 или 3 гетероатома; R12 выбирают из (R11)r-C1-10алкилена, (R11)r-C1-10 гетероалкилена, включающего 1, 2 или 3 гетероатома, (R11)r-C6-10арилена и (R11)r-C3-15гетероарилена, включающего 1, 2 или 3 гетероатома, и r выбирают из 0, 1, 2, 3, 4 и 5, с тем условием, что R1 и R2 могут присоединяться к общему атому таким образом, чтобы образовать с указанным общим атомом кольцо.
Согласно другому аспекту изобретение относится к стероидам, структура которых описана выше, в которой каждый из R1 и R2 независимо выбирают из водорода, R8, R9, R10, R11 и R12, где R8 выбирают из алкила, гетероалкила, арила и гетероарила; R9 выбирают из (R8)r-алкилена, (R8)r-гетероалкилена, (R8)r-арилена и (R8)r-гетероарилена; R10 выбирают из (R9)r -алкилена, (R9)r-гетероалкилена, (R9)r-арилена и (R9)r-гетероарилена; R11 выбирают из (R10)r-алкилена, (R10)r-гетероалкилена, (R10)r-арилена и (R10)r-гетероарилена; R12 выбирают из (R11)r-алкилена, (R11)r-гетероалкилена, (R11)r-арилена и (R11)r-гетероарилена, и r выбирают из 0, 1, 2, 3, 4 и 5, при условии, что R1 и R2 могут присоединяться к общему атому таким образом, чтобы образовать с указанным общим атомом кольцо; R3 и R4 выбирают из атома водорода и защитных групп таким образом, чтобы R3 и/или R4 являлись частью защитной группы гидроксила; атомы углерода под номерами 1, 2, 4, 11, 12, 15 и 16 каждый замещен двумя атомами водорода; атомы углерода под номерами 5, 8, 9 и 14 каждый замещен одним атомом водорода; атом углерода под номером 10 замещен метилом; атом углерода под номером 13 в том случае, когда он не является частью ненасыщенной связи, замещен метилом; атом углерода под номером 17 замещен (a) одной из групп: =C(R5)(R5) и =C=C(R5)(R5), или (b) двумя группами -R5; каждое из ядер A, B, C и D является независимо полностью насыщенным или частично насыщенным; R5 в каждом случае независимо выбирают из H и C1-10углеводородных групп.
Например, R1 и R2 выбирают из атома водорода, CH3-, CH3(CH2)2-, СН3(СН2)4-, СН3СО-, С6 Н5СО-, (СН3)2CHSO2-, C6H5SO2-, C6H5NHCO-, CH3(CH2)2NHCO-, CH3(CH2)2NH(CH2)2-, (CH3)2N(CH2)2-, HOCH2CH2-, HOCH2(CH2)4-, HOCH2CH2NHCH2CH2-, 3-(CH3)C6H4-, 4-(HO)C6H4-, 4-(H2NSO2)C6 H4-, 4-((CH3)CH)C6H4-CH2-, 2-(F)C6H4-CH2-, 3-(CF3)C6H4-CH2-, 2-(CH3O)C6H4-CH2-, 4-(CF3O)C6H4-CH2-, 3-(C6H5O)C6H4-CH2-, 3-(NO2)C6H4-CH2-,

или R1 и R2 могут объединяться вместе с атомом азота, к которому они оба присоединены, и образовывать гетероцикл, выбранный из

Конкретные соединения настоящего изобретения, в которых R1 представляет водород, но R2 не является водородом, включают




Таким образом, в одном из семейств предпочтительных соединений согласно настоящему изобретению R1 представляет водород, но R2 не является водородом.
В стероидных соединениях настоящего изобретения, как показано выше, в соответствии с одним аспектом R3 и R4 представляют водород, т.е. стероид является гидроксизамещенным у каждого из атомов углерода, находящихся под номерами 6 и 7. В соответствующем варианте одна или обе гидроксигруппы у атомов углерода, находящихся под номерами 6 и 7, находятся в защищенной форме, т.е. связаны с группой, являющейся защитной для гидроксигруппы. Такие защитные группы хорошо известны в данной области техники и раскрыты, например, в Greene and Wuts, "Protective Groups in Organic Synthesis", John Wiley & Sons, New York, N.Y. (1999). Подходящая защитная группа представляет собой кетальную группу, вследствие чего настоящее изобретение относится к соединениям следующей структуры:

Как указывалось выше, настоящее изобретение относится к стероидным соединениям, которые включают соединения определенной стереохимии. Одно из таких соединений имеет стереохимию, приведенную в виде следующей структуры для R3О- и R4О-:

Как также указывалось выше, настоящее изобретение относится к солевым формам стероидных соединений согласно настоящему изобретению, предпочтительно к фармацевтически приемлемым солям. Согласно одному из воплощений -N(R1)(R2) находится в солевой форме. Другими словами, -N(R1)(R2) является протонированным таким образом, что N несет положительный заряд. В таком случае стероидное соединение настоящего изобретения представляет собой аддитивную соль кислоты, как определено выше в настоящем описании. В соответствии с предпочтительным аспектом настоящее изобретение относится к гидрохлоридным солям соединения стероидной структуры, приведенной выше. В соответствии с предпочтительным аспектом настоящее изобретение относится к ацетатным солям соединения стероидной структуры, приведенной выше.
Как также указывалось выше, настоящее изобретение относится к пролекарствам конкретных соединений, представленных формулой (1). Согласно одному из аспектов настоящее изобретение направлено на получение пролекарств любого из конкретных соединений, представленных формулой (1). В соответствии с другим аспектом из настоящего изобретения исключены пролекарства конкретных соединений, представленных формулой (1), т.е. один из аспектов настоящего изобретения направлен на получение соединений формулы (1) и фармацевтически приемлемых солей, сольватов, стереоизомеров, но не пролекарств указанных соединений, в отдельности или в смеси.
В стероидных соединениях настоящего изобретения, как указывалось выше, в предпочтительном воплощении, атом под номером 17 замещен группой =C(R5)(R5), и R5 выбирают из водорода, галогена, C1-6алкила, C1-6гидроксиалкила и -CO2-C1-6алкила. В другом предпочтительном воплощении атом 17 замещен C1-6алкилом или C1-6галогеналкилом, или атом под номером 17 замещен -OR6 или =O, где R6 представляет водород.
В стероидных соединениях настоящего изобретения, как указывалось выше, в предпочтительном воплощении, по меньшей мере, один из атомов под номерами 10 и 13 замещен метилом.
В стероидных соединениях настоящего изобретения, как указывалось выше, в предпочтительном воплощении, атомы под номерами от 1 до 16 каждый представляют водород, где атомы углерода под номерами 1, 2, 4, 11, 12, 15 и 16 могут быть независимо замещены: (a) одной из групп: =O, =C(R5)(R5), =C=C(R5)(R5), -C(R5)(R5)(C(R5)(R5))n- и -(O(C(R5)(R5))nO)-, где n изменяется в диапазоне от 1 до приблизительно 6, или (b) двумя из следующих групп, которые выбирают независимо: -X, -N(R1)(R2), -R5 и -OR6, и атом под номером 17 представляет атом углерода, замещенный: (a) одной из групп: =C(R5a)(R5a), =C=C(R5a)(R5a) и -C(R5a)(R5a)(C(R5a)(R5a))n-, где n изменяется в диапазоне от 1 до приблизительно 6, или (b) двумя из следующих групп, которые выбирают независимо: -X, -N(R1)(R2) и -R5a, где R5a в каждом случае независимо выбирают из H, X и C1-30органического фрагмента, который необязательно может содержать, по меньшей мере, один гетероатом, выбранный из группы, состоящей из бора, галогена, азота, кремния и серы; где две геминальные группы R5 могут вместе образовывать кольцо с атомом углерода, к которому они обе присоединены. Необязательно R5a в каждом случае независимо выбирают из C1-30углеводородной, C1-30 галогенуглеродной, C1-30галогенуглеводородной групп, H и X. В альтернативном необязательном воплощении R5a в каждом случае независимо выбирают из C1-10углеводородной, C1-10галогенуглеродной, C1-10галогенуглеводородной групп, H и X. Необязательно каждое из перечисленных воплощений согласно настоящему изобретению предполагает дальнейшее воплощение, в котором R1 и R2 выбирают из водорода, кислорода таким образом, чтобы образовалась нитрогруппа или оксим, аминогруппы, -SO3-R и органических групп, содержащих 1-30 атомов углерода и независимо содержащих 1-6 гетероатомов, выбранных из кислорода, фосфора, кремния и серы, где R2 может быть прямой связью с атомом под номером 3, или R1 и R2 вместе с атомом N, к которому они обе присоединены, могут образовывать гетероциклическую структуру, которая может быть частью органической группы, содержащей 1-30 атомов углерода и необязательно включающей 1-6 гетероатомов, выбранных из кислорода и кремния; или R1 может представлять цепь из двух или трех атомов, присоединенную к атому под номером 2 таким образом, что -NR1- образует часть конденсированной бициклической структуры, включающей ядро А. Необязательно в каждом из перечисленных воплощений настоящим изобретением предлагается дальнейшее воплощение, в котором атомы углерода под номером 1, 2, 4, 11, 12, 15 и 16, в том случае, когда они не являются частью ненасыщенной связи, каждый замещен двумя атомами водорода; атомы углерода под номерами 5, 8, 9 и 14, в том случае, когда они не являются частью ненасыщенной связи, каждый замещен одним атомом водорода; атом углерода под номером 10 замещен метилом и атом углерода под номером 13 в том случае, когда он не является частью ненасыщенной связи, замещен метилом. Необязательно в каждом из перечисленных воплощений настоящим изобретением предлагается дальнейшее воплощение, согласно которому атомы углерода под номерами 1, 2, 4, 11, 12, 15 и 16 каждый замещен двумя атомами водорода; атомы углерода под номерами 5, 8, 9 и 14 каждый замещен одним атомом водорода; атом углерода под номером 10 замещен метилом и атом углерода под номером 13 в том случае, когда он не является частью ненасыщенной связи, замещен метилом.
В стероидных соединениях настоящего изобретения, как показано в данном описании, в соответствии с предпочтительным воплощением каждый из R1 и R2 представляет водород и/или каждый из R3 и R4 представляет водород; и/или атом углерода под номером 17 замещен (a) одной из групп: =C(R5a)(R5a), =C=C(R5a)(R5a) и -C(R5a)(R5a )(C(R5a)(R5a))n-, где n изменяется в диапазоне от 1 до приблизительно 6; или (b) двумя из следующих групп, которые выбирают независимо из -X, -N(R1)(R2) и -R5a, где R5a в каждом случае независимо выбирают из H, X и C1-30органического фрагмента, который необязательно может содержать, по меньшей мере, один гетероатом, выбранный из группы, состоящей из бора, галогена, азота, кремния и серы, причем две геминальные группы R5 могут вместе образовывать кольцо вместе с атомом углерода, к которому они обе присоединены.
В стероидах настоящего изобретения, если не указано иное, каждое из ядер A, B, C и D является независимо полностью насыщенным, частично насыщенным или полностью ненасыщенным. Таким образом, атомы водорода, присоединенные к атомам углерода в положениях 1-17, могут быть опущены таким образом, чтобы сделать возможным ненасыщенность ядер А, B, C и/или D. Например, в том случае, когда указано, что атомы углерода под номерами 5, 8, 9 и 14 замещены одним атомом водорода, и также указано, что каждое из ядер А, B, C и D является независимо полностью насыщенным, частично насыщенным или полностью ненасыщенным, в этом случае какой либо один или более из атомов водорода, присоединенных к атомам углерода под номерами 5, 8, 9 и 14, может быть исключен для того, чтобы сделать возможной ненасыщенность у атома углерода.
Соединения настоящего изобретения предполагается применять в качестве фармацевтических средств. Предпочтительно молекулярная масса соединения согласно настоящему изобретению является относительно невысокой, то есть менее чем приблизительно 5000 г/моль, обычно менее чем приблизительно 4000 г/моль, более типично менее чем приблизительно 3000 г/моль, еще более типично менее чем приблизительно 2000 г/моль и совсем типично менее чем приблизительно 1000 г/моль, где минимальная молекулярная масса соединения согласно настоящему изобретению составляет приблизительно 300 г/моль, и каждый их таких типичных интервалов представляет отдельное воплощение настоящего изобретения.
Стероидные соединения настоящего изобретения включают фармацевтически приемлемые соли, сольваты, стереоизомеры и пролекарства, 3-азот-6,7-дикислородсодержащих стероидов, описанных выше, по отдельности или в смеси друг с другом.
Стероидные соединения настоящего изобретения могут и обычно существуют в виде твердых веществ, включая кристаллические твердые вещества, которые могут кристаллизоваться из общепринятых растворителей, таких как этанол, N,N-диметилформамид, вода и подобные им растворители. Процесс кристаллизации может, в зависимости от условий кристаллизации, приводить к получению различных кристаллических структур. Обычно термодинамически более стабильная полиморфная форма более выгодна для производства стероидного соединения согласно настоящему изобретению в промышленном масштабе и является предпочтительной формой соединения.
Часто процесс кристаллизации приводит к получению сольвата стероидного соединения согласно настоящему изобретению, имеющего структуру, показанную выше. При использовании в настоящем описании термин "сольват" относится к агрегатам, которые включают одно или более 3-азот-6,7-дикислородсодержащее стероидное соединение настоящего изобретения и одну или более молекул растворителя. Растворитель может представлять собой воду, в этом случае сольват может быть гидратом. В качестве альтернативы растворитель может представлять собой органический растворитель. Таким образом, соединения настоящего изобретения могут существовать в виде гидрата, включая моногидрат, дигидрат, полугидрат, полуторагидрат, тригидрат, тетрагидрат и подобные им соединения, а также в виде соответствующих сольватированных форм. Стероидные соединения могут являться истинными сольватами, хотя в других случаях могут просто удерживать случайное количество воды или представлять собой смесь с водой плюс с некоторыми случайными растворителями.
При использовании в настоящем описании термин "фармацевтически приемлемый сольват" относится к сольвату, который сохраняет биологическую эффективность и свойства биологически активного 3-азот-6,7-дикислородсодержащего стероидного соединения согласно настоящему изобретению. Примеры фармацевтически приемлемых сольватов включают, но не ограничиваются этим, сольваты, содержащие воду, изопропанол, этанол, метанол, ДМСО (диметилсульфоксид), этилацетат, уксусную кислоту и этаноламин. Специалистам в данной области техники понятно, что сольватированные формы эквивалентны несольватированным формам и, как предполагается, входят в объем настоящего изобретения. Sykes P.A., Guidebook to Mechanism in Organic Chemistry, 6th Ed (1986, John Wiley & Sons, N.Y.) является примером ссылки, в которой описаны сольваты.
Соединения настоящего изобретения могут существовать в виде отдельных стереоизомеров, рацематов, и/или смесей энантиомеров, и/или диастереомеров. Все такие отдельные стереоизомеры, рацематы и их смеси, как предполагается, входят в объем настоящего изобретения. В соответствии с предпочтительным аспектом соединения настоящего изобретения используют в оптически чистой форме.
"Фармацевтически приемлемое пролекарство", как подразумевается, означает соединение, которое может быть превращено в физиологических условиях или путем сольволиза в биологически активное 3-азот-6,7-дикислородсодержащее стероидное соединение, описанное выше. Таким образом, термин "пролекарство" относится к метаболическому предшественнику стероидного соединения согласно настоящему изобретению, который является фармацевтически приемлемым. Пролекарство может быть неактивным в тот момент, когда его вводят субъекту, но in vivo превращаться в активное 3-азот-6,7-дикислородсодержащее стероидное соединение настоящего изобретения. Пролекарства, как правило, быстро трансформируются in vivo с получением материнского соединения вышеуказанной формулы, например, при гидролизе в крови.
Обсуждение пролекарств приведено у T.Higuchi, V.Stella, "Prodrugs as Novel Delivery Systems", Vol.14, ACS Symposium Series, а также в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, оба эти источника информации включены в настоящее описание в качестве ссылки. Типичное пролекарство представляет собой производное стероидного соединения согласно настоящему изобретению, которое содержит химически или метаболически расщепляемые группы и превращается, в результате сольволиза или гидролиза в физиологических условиях, в соединения согласно настоящему изобретению, которые являются фармацевтически активными in vivo. Пролекарство в производной форме часто имеет преимущества в растворимости, совместимости с тканями или характеризуется замедленным высвобождением в организме млекопитающего (см. Bundgard H., Design of Prodrugs, pp.7-9, 21-24, Elsevier, Amsterdam 1985). Предпочтительное пролекарство представляет собой соединение, замещенное у 3-азота стероида настоящего изобретения, причем заместитель отщепляется in vivo, с получением фармацевтически активного соединения.
Стероиды согласно настоящему изобретению, замещенные азотом у С3 и замещенные кислородом у атомов в положениях 6 и 7, обладают неожиданными свойствами, которые усиливают эффективность таких соединений. Например, стероид настоящего изобретения обладает прекрасной метаболической стабильностью в S9 фракции печени человека. Например, 100% соединений 28, 89, 139 и 143 остаются неизмененными после 15 и даже 40 мин инкубирования с фракциями S9 человека. Неожиданно было обнаружено, что азотные заместители, расположенные у атома С3, значительно снижают глюкоронидацию молекул в плазме. В дополнение, стероиды настоящего изобретения, содержащие азотные заместители, расположенные у атома С3, такие как соединения 28, 89 и 83, хорошо растворимы в водных растворах, проявляя растворимость в воде > 100 мг/мл. Кроме того, эффективность и фармакокинетический профиль стероидов согласно настоящему изобретению, содержащих азотные заместители, расположенные у атома С3, весьма подходят для фармацевтического использования. Для доз <1,0 мг/кг, один раз в день, воспроизводимо показана значительная противовоспалительная активность in vivo на моделях воспаления. В случае крыс соединения 28 и 89 характеризуются средним значением периода полупревращения 7,5 часа и оральной биодоступностью ˜100%, тогда как у обезьян значение периода полупревращения составляет 15 часов, и оральная биодоступность составляет 25-30%. Максимальная концентрация в плазме для обоих видов предсказуема и изменяется по линейному закону.
С. Получение соединений.
Соединения согласно настоящему изобретению могут быть получены способами с проведением стадий, известных специалистам в данной области техники или аналогичных им стадий. Общие методики проведения реакций стероидов могут быть найдены в "Steroid Reactions", C.Djerassi, Ed. Holden Day, San Francisco, Calif., 1963, и в приведенных там ссылках. Общие методики синтеза могут быть найдены в "Comprehensive Organic Transformations", R.C. Larock, VCH Publishers, New York. N.Y. 1989, и в приведенных там ссылках. Дополнительные литературные ссылки, полезные для синтеза соединений настоящего изобретения, являются следующими: T.Reichstein; C.H.Meystre, Helv. Chim. Acta, 1932, 22, 728; H. Westmijze; H.Kleyn; P.Vermeer; L.A. van Dijck, Tet. Lett. 1980, 21, 2665; K.Prezewowsky; R.Wiechert, US Pat. No.3682983; P.Kaspar; H.Witzel, J.Steroid Biochem. 1985, 23, 259; W.G.Dauben; T.Brookhart, J. Am. Chem. Soc. 1981, 103, 237; A.J.Manson et al., J. Med. Chem. 1963, 6, 1; R.O. Clinton et al., J. Am. Chem. Soc. 1961, 83, 1478, и J.A.Zderic et al., Chem. And Ind. 1960, 1625.
В предпочтительном способе в качестве исходных веществ или промежуточных соединений используют С3, С6, C7 и С17 полиоксигенированные стероиды. Методики введения С6 и С7 атомов кислорода в коммерчески доступные исходные вещества описаны в патенте США 6046185. В этом патенте США также раскрыто множество способов, согласно которым замещение с предпочтительной стереохимией может быть осуществлено в положениях С1, С2, С4, С5, С8, С9, С10, С11, С12, С13, С14, С16 и С17. В соответствии с настоящим изобретением атомы кислорода у С6 и С7 могут присутствовать в виде гидроксилов или в виде защищенных гидроксилов. 6- и 7-Гидроксилы могут быть защищены индивидуально или они могут составлять часть ядра. Подходящие защитные группы перечислены в Greene and Wuts, "Protective Groups in Organic Synthesis", John Wiley & Sons, New York, N.Y. (1999).
Как показано на схеме А, кетоны, представляющие собой соединения 2, или аналогичные им соединения могут быть проалкилированы различными алкилирующими группами с получением стероидов настоящего изобретения, содержащих, но, не ограничиваясь указанным, алкильные, циклоалкильные, арильные и гетероарильные заместители. Например, алкилирование 17-кетона, представляющего собой соединение 2, анионом ацетилена приводит к получению 17α-этинил-17β-гидроксильного промежуточного соединения 3. Инверсия стереохимии заместителя у атома С17 может быть осуществлена образованием сначала метансульфоната с последующей обработкой нитратом серебра (I) в тетрагидрофуране (ТГФ) и воде. Дегидратация соединения 3 с использованием POCl3 в 2,4-лутидине дает соединение 4. Фторид тетрабутиламмония в ТГФ удаляет трет-бутилдиметилсилильную (TBS) защитную группу 3-гидроксила с получением соединения 5. Обработка 3α-гидроксильного соединения 5 с использованием ZnN6·2py, трифенилфосфина и диизопропилазодикарбоксилата (DIAD) в толуоле приводит к 3β-азидосоединению 6. ZnN6·2py получают взаимодействием Zn(NO3)2 и NaN3 c последующей обработкой пиридином в соответствии с опубликованной методикой M.C.Viaud, P. Rollin, Synthesis, 1990, 130. Восстановление азида алюмогидридом лития в диэтиловом эфире (Et2O) дает амин 7. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 8.
Схема А

i) KCCH; ii) POCl3, 2,4-лутидин; iii) Bu4NF, ТГФ; iv) ZnN6·2py, Ph3P, DIAD, толуол; v) LiAlH4, Et2O, вода, MeCN.
Как показано на схеме В, стероиды настоящего изобретения имеющие алленовую функциональную группу, могут быть получены из промежуточного соединения, аналогичного соединению 3. Примером является реакция соединения 3 с LiAlH4 и AlCl3 в ТГФ с получением аллена 9. Фторид тетрабутиламмония в ТГФ удаляет защитную группу 3-гидроксила с образованием при этом соединения 10. Обработка 3α-гидроксильного соединения 10 с использованием ZnN6·2py, трифенилфосфина и диизопропилазодикарбоксилата (DIAD) в толуоле приводит к 3β-азидосоединению 11. Восстановление азида 11 алюмогидридом лития в Et2O дает амин 12. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 13.
Схема B

i) LiAlH4, AlCl3, ТГФ; ii) Bu4NF, ТГФ; iii) ZnN6·2py, Ph3P, DIAD, толуол; iv) LiAlH4, Et2O; v) HCl, вода, MeCN.
Как показано на схеме С, стероиды настоящего изобретения, содержащие алкинильную функциональную группу, могут быть получены из алленовых промежуточных соединений. Примером является обработка соединения 9 с использованием н-BuLi в ТГФ, приводящая к получению 17β-этинильного соединения 14. Фторид тетрабутиламмония в ТГФ удаляет защитную группу 3-гидроксила с образованием при этом соединения 15. Обработка 3α-гидроксильного соединения 15 с использованием ZnN6·2py, трифенилфосфина и DIAD в толуоле приводит к 3β-азидосоединению 16. Восстановление азида 16 алюмогидридом лития в Et2O дает амин 17. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 18.
Схема С

i) н-BuLi, ТГФ; ii) Bu4NF, ТГФ; iii) ZnN6·2py, Ph3P, DIAD, толуол; iv) LiAlH4, Et2O; v) HCl, вода, MeCN.
Как показано на схеме D, стероиды настоящего изобретения и содержащие алкенильную функциональную группу, могут быть получены из алкиновых промежуточных соединений. Примером является контролируемое гидрирование соединения 14 с использованием в качестве катализатора Pd-CaCO3 с получением алкена 19. Фторид тетрабутиламмония в ТГФ удаляет защитную группу 3-гидроксила с образованием при этом соединения 20. Обработка 3α-гидроксильного соединения 20 с использованием ZnN6·2py, трифенилфосфина и DIAD в толуоле приводит к 3β-азидосоединению 21. Восстановление азида 21 алюмогидридом лития в Et2O дает амин 22. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 23.
Схема D

i) H2, Pd-CaCO3; ii) Bu4NF, ТГФ; iii) ZnN6·2py, Ph3P, DIAD, толуол; iv) LiAlH4, Et2O; v) HCl, вода, MeCN.
Соединение 2 может быть использовано во множестве реакций получения олефиновых соединений, включая реакцию типа реакции Виттига, с получением соединений настоящего изобретения и содержащих экзоциклическую олефиновую группу у С17. Например, как показано на схеме Е, соединение 2 может быть обработано этилтрифенилфосфонийбромидом и трет-бутоксидом калия (КОtBu) с получением соединения 24, у которого R1=метил и R2=водород. Фторид тетрабутиламмония в ТГФ удаляет защитную группу 3-гидроксила с образованием при этом соединения 25. Обработка 3α-гидроксильного соединения 25 с использованием ZnN6·2py, трифенилфосфина и DIAD в толуоле приводит к 3β-азидосоединению 26. Восстановление азида 26 алюмогидридом лития в Et2O дает амин 27. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 28, у которой R1=метил и R2= водород.
По аналогии с синтезом, показанным на схеме Е, кетоны, такие как соединение 2, могут быть подвергнуты взаимодействию с другими реагентами типа реагентов Виттига, такими как (но не ограничиваясь указанным) метил-, пропил-, бутил-, пентил- или гексилтрифенилфосфонийбромид с получением стероидов настоящего изобретения и аналогичных соединению 28, у которых R2= водород и R1= водород, этил, пропил, бутил или пентил.
Схема E
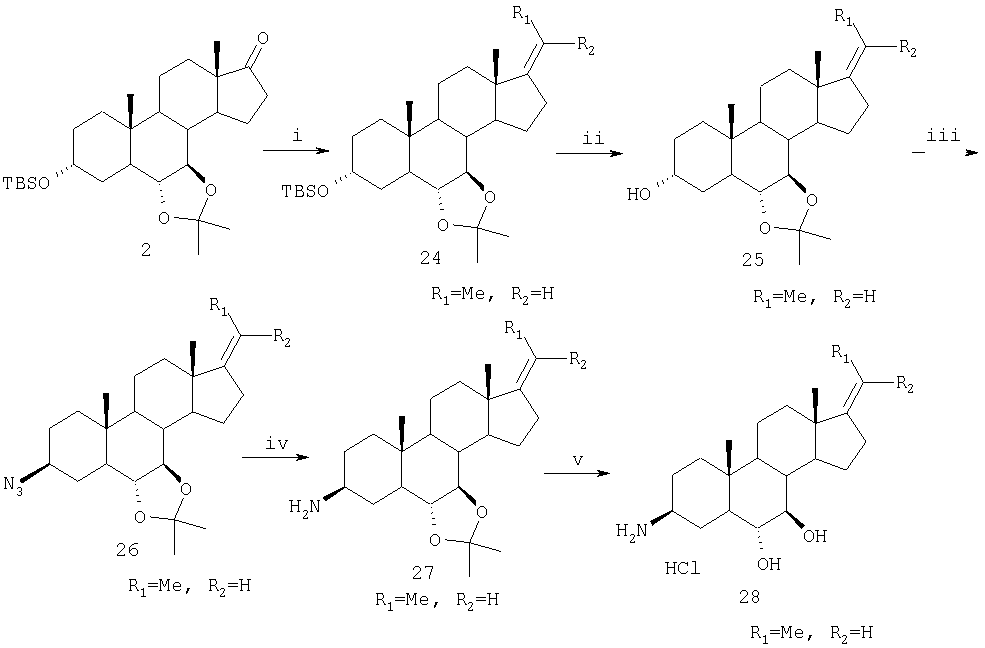
i) EtPPh3Br, KOtBu, толуол; ii) Bu4NF, ТГФ; iii) ZnN6·2py, Ph3P, DIAD, толуол; iv) LiAlH4, Et2O; v) HCl, вода, MeCN.
Стероиды настоящего изобретения могут содержать экзоциклические двойные связи Е и/или Z геометрии. Например, как показано на схеме Е, Z-олефин 24 в циклогексане может быть подвергнут УФ-облучению в присутствии дифенилдисульфида, что приводит к изомеризации в Е-олефин 29. Фторид тетрабутиламмония в ТГФ удаляет защитную группу 3-гидроксила с образованием при этом соединения 30. Обработка 3α-гидроксильного соединения 30 с использованием ZnN6·2py, трифенилфосфина и DIAD в толуоле приводит к образованию 3β-азидосоединения 31. Восстановление азида 31 алюмогидридом лития в Et2O дает амин 32. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 33.
Схема F

i) (PhS)2, hν, циклогексан; ii) Bu4NF, ТГФ; iii) ZnN6·2py, Ph3P, DIAD, толуол; iv) LiAlH4, Et2O; v) HCl, вода, MeCN.
Множество стероидов настоящего изобретения с функционализированными боковыми цепями может быть получено с использованием таких способов, как промотированная кислотами Льюиса конденсация с альдегидами и акцепторами Михаэля. Например, как показано на схеме G, соединение 24 может взаимодействовать с метилпропиолатом в присутствии диэтилалюминийхлорида с образованием при этом соединения 34. Двойные связи могут быть подвергнуты гидрированию с использованием катализатора, например, такого как платина, с получением соединения 35. Фторид тетрабутиламмония в ТГФ удаляет защитную группу 3-гидроксила с образованием при этом соединения 36. Обработка 3α-гидроксильного соединения 36 с использованием ZnN6·2py, трифенилфосфина и DIAD в толуоле приводит к образованию 3β-азидосоединения 37. Восстановление азида 37 алюмогидридом лития в Et2O дает амин 38. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 39.
Схема G

i) HCCCO2Me, Et2AlCl; ii) H2, Pt; iii) Bu4NF, ТГФ; iv) ZnN6 ·2py, Ph3P, DIAD, толуол; v) H2, Pd, EtOAc; vi) 80%-ная уксусная кислота.
Используя 3-аминостероид, такой как любой из стероидов, полученных в соответствии со схемами А-G, может быть синтезировано множество соединений настоящего изобретения, содержащих вторичные и третичные аминогруппы. На фиг. 1А и 1В приведены некоторые направления синтеза, которые могут быть использованы для получения 3-аминосоединений настоящего изобретения. Например, методы восстановительного аминирования могут быть использованы для конденсации первичных (см. фиг. 1А) и вторичных (см. фиг. 1В) аминов с альдегидами (RC(=O)H) и кетонами (RC(=O)R1). Хотя это не показано на фиг. 1А и 1В, соединения, содержащие две альдегидные группы, т.е. диальдегиды общей формулы НС(=O)R-С(=O)H, могут взаимодействовать с 3-аминостероидами с образованием при этом стероидов, содержащих гетероциклические структуры в положении 3. В дополнение (или в качестве альтернативы) методы восстановительного аминирования могут быть использованы для конденсации 3-кетостероидов с вторичными гетероциклическими аминами. При использовании таких подходов настоящее изобретение позволяет получить соединения, в которых R1 и R2 могут вместе с атомом N, к которому они оба присоединены, образовывать гетероциклическую структуру, которая может являться частью органической группы, содержащей 1-30 атомов углерода, и необязательно содержащую 1-6 гетероатомов, выбранных из азота, кислорода и кремния. Коммерческие источники и ссылки на химическую литературу делают доступным для обычного специалиста в данной области техники получение множества альдегидов (включая диальдегиды) и кетонов, которые могут быть использованы для получения стероидных соединений настоящего изобретения. Методы восстановительного аминирования описаны, например, в Synthesis 1975, 135; J. Am. Chem. Soc. 1971, 93, 2897; M. Freifelder in "Catalytic Hydrogenation in Organic Synthesis" J. Wiley & Sons 1978, Ch. 10; Russ. Chem. Rev. 1980, 49, 14, а также в приведенных там ссылках. См. также J. Chem. Soc. Perkin Trans 1 1998, 2527, и Synlett 1999, 1781, а также приведенные там ссылки.
Первичные (см. фиг. 1А) и вторичные (см. фиг. 1 В) амины могут быть подвергнуты конденсации с арильными соединениями (ArX) для получения множества арилзамещенных аминных соединений настоящего изобретения. Коммерческие источники и ссылки на химическую литературу делают доступным для обычного специалиста в данной области техники получение множества арильных соединений, которые могут быть использованы для получения стероидных соединений настоящего изобретения. Примеры методов аминирования арильных соединений могут быть найдены в J. Org. Chem. 2000, 65, 1158, а также в обзоре, опубликованном в Angew. Chem. Int. ed. 1998, 37, 2046, и в приведенных там ссылках.
Методики проведения взаимодействия первичных (см. фиг. 1А) и вторичных (см. фиг. 1 В) аминов с хлорангидридами (RC(=O)Cl) и сульфонилхлоридами (RSO2Cl) с образованием при этом амидных и сульфонамидных производных настоящего изобретения соответственно хорошо известны специалистам в области органической химии, в контексте других аминных соединений, и те же самые методики могут использоваться в отношении аминных производных согласно настоящему изобретению. Коммерческие источники и ссылки на химическую литературу делают доступным для среднего специалиста в данной области техники получение множества хлорангидридов и сульфонилхлоридов, которые могут быть использованы для получения стероидных соединений настоящего изобретения.
Методики проведения взаимодействия первичных (см. фиг. 1А) и вторичных (см. фиг. 1 В) аминов с изоцианатами (RN=С=O) и с изотиоцианатами (RN=С=S) для получения производных мочевины и тиомочевины соответственно хорошо известны специалистам в области органической химии, в контексте других аминных соединений, и те же самые методики могут использоваться в отношении аминных производных согласно настоящему изобретению. Коммерческие источники и ссылки на химическую литературу делают доступным для обычного специалиста в данной области техники получение множества изоцианатов и изотиоцианатов, которые могут быть использованы для получения стероидных соединений настоящего изобретения. Обзорная статья в Russ. Chem. Rev. 1985, 54, 249, и приведенные в ней ссылки, в которых описаны примеры синтеза множества замещенных мочевин и тиомочевин, могут быть использованы при осуществлении изобретения.
Таким образом, при использовании подходящим образом выбранных альдегидов, кетонов, арильных соединений, хлорангидридов, сульфонилхлоридов, изоцианатов и/или изотиоцианатов специалист в данной области техники может получить стероидные соединения, в которых R1 и R2 выбирают из водорода и органических групп, содержащих 1-30 атомов углерода и необязательно содержащих 1-6 гетероатомов, выбранных из азота, кислорода, фосфора, кремния и серы.
Стероиды настоящего изобретения могут содержать конденсированные гетероциклы, например, такие как пиразол, изоксазол и пиримидин (но не ограничиваясь ими). Как показано на схеме Н, соединение 43 представляет собой пример конденсированного пиразола согласно настоящему изобретению, для которого синтез исходного соединения 40 описан в патенте США 6046185. Обработка соединения 40 этилформиатом в пиридине в присутствии NaOMe приводит к получению гидроксиметиленового промежуточного соединения 41. Взаимодействие соединения 41 с гидразингидратом в EtOH приводит к образованию пиразольного производного 42, обработка которого фторидом тетрабутиламмония в ТГФ дает соединение 43.
Схема H

i) EtO2CH, NaOMe, пиридин; ii) N2H4, EtOH; iii) Bu4NF, ТГФ.
Как показано на схеме I, промежуточные соединения, такие как соединение 41, могут быть превращены в изоксазолы, примером которых является соединение 44. Обработка гидроксиметиленового промежуточного соединения 41 гидроксидом аммония в пиридине с последующим удалением защиты 6- и 7-гидроксилов с использованием фторида тетрабутиламмония в ТГФ приводит к изоксазолу 44.
Схема I

i) HONH2·HCl, пиридин; ii) Bu4NF, ТГФ.
Как показано на схеме J, промежуточные соединения, такие как соединение 41, могут быть превращены в пиримидины, примером которых является соединение 44а. Обработка гидроксиметиленового промежуточного соединения 41 гидрохлоридом бензамидина и гидроксидом калия в этаноле с последующим удалением защиты 6- и 7-гидроксилов с использованием фторида тетрабутиламмония в ТГФ приводит к пиримидину 44а.
Схема J

i) гидрохлорид бензамидина, KOH, EtOH; ii) BU4NF, ТГФ.
Кроме того, при использовании в качестве промежуточных соединений таких соединений, которые содержат кетозаместители у атома углерода в положении 3 и =СНОН заместители у атома углерода в положении 2, настоящее изобретение позволяет получить множество соединений, в которых R1 может представлять цепь из двух или трех атомов, присоединенную к атому под номером 2 таким образом, что -NR1- образует часть конденсированной бициклической структуры, включающей ядро А.
Взаимодействие 3-кетостероидов с гидроксиламином и пиридином может быть использовано для получения оксимов стероидов настоящего изобретения. Оксим стероида содержит R2 в виде прямой связи с атомом под номером 3, обеспечивая таким образом двойную связь между атомом углерода под номером 13 и N, а R1 представляет ОН. Первичные амины могут быть окислены до нитросоединений с использованием, например, диметилдиоксирана. Таким образом, R1 и R2 могут представлять кислород. Способы получения функциональной нитрогруппы описаны в J. Org. Chem. 1989, 54, 5783. Взаимодействие 3-кетонов с диметилгидразином приводит к N,N-диметилгидразоновым производным стероидов, являющимся предметом настоящего изобретения, в которых R2 представляет прямую связь с атомом под номером 3, а R1 представляет NMe2. Обработка диметилгидразоновых производных стероидов гидразином приводит к получению гидразоновых производных стероидов настоящего изобретения. Описание способов синтеза N,N-диметилгидразонов и гидразонов можно найти в J. Org. Chem. 1966, 31, 677. Первичные амины также могут быть подвергнуты взаимодействию согласно методикам, известным в данной области техники, с сульфоновой/серной кислотами и сложными эфирами с получением сульфаматных производных, т.е. стероидов, в которых атом под номером 3 связан с -N-SO3-R, а R представляет Н или органическую группу, содержащую 1-30 атомов углерода и необязательно содержащую 1-6 гетероатомов, выбранных из азота, кислорода, фосфора, кремния и серы.
D. Фармацевтические композиции.
Настоящее изобретение позволяет получить фармацевтические или ветеринарные композиции (здесь и далее просто указываются как фармацевтическая композиция), содержащие соединение формулы (1), как описано выше, в смеси с фармацевтически приемлемым носителем. Изобретение также позволяет получить композицию, предпочтительно фармацевтическую композицию, содержащую эффективное количество соединения, как описано выше, в сочетании с фармацевтически приемлемым носителем.
Фармацевтические композиции настоящего изобретения могут находиться в любой форме, которая позволяет осуществить введение композиции пациенту. Например, композиция может находиться в виде твердого вещества, жидкого вещества или газообразного вещества (аэрозоля). Типичные способы введения включают, не ограничиваясь ими, пероральное, местное, парентеральное, сублингвальное, ректальное, вагинальное, глазное и интраназальное введение. Термин парентеральное введение при использовании в настоящем описании означает подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или методику вливания. Фармацевтические композиции настоящего изобретения могут быть составлены таким образом, чтобы содержащийся в них активный ингредиент был биодоступным при введении композиции пациенту. Композиции, которые будут вводиться пациенту, имеют форму одной или более единичных дозированных форм, причем, например, таблетка может представлять собой единичную дозированную форму, а контейнер с соединением формулы (1) в аэрозольной форме может заключать множество единичных дозированных форм.
Вещества, используемые для получения фармацевтических композиций, должны быть фармацевтически чистыми и нетоксичными в используемых количествах. Специалисту в данной области техники будет понятно, что оптимальная дозировка активного ингредиента (ингредиентов) в фармацевтической композиции будет зависеть от множества факторов. Определяющие факторы включают, не ограничиваясь этим, вид субъекта (например, человек), конкретную форму активного ингредиента, способ введения и используемую композицию.
Как правило, фармацевтическая композиция включает одно или более активных соединений формулы (1), как описано выше, в смеси с одним или более носителями. Носитель (носители) могут быть такими, что композиции, находятся, например, в форме таблетки или порошка. Носитель (носители) могут быть жидкими, так что композиция, например, будет представлять сироп для перорального введения или жидкость для инъекций. В дополнение, носитель (носители) могут быть газообразными, чтобы, таким образом, получить аэрозольную композицию, пригодную, например, для ингаляционного введения.
В том случае, когда композиция предназначается для перорального введения, она предпочтительно находится либо в твердой, либо в жидкой форме, причем полутвердые, полужидкие формы, суспензии и гелевые формы также входят в число форм, которые рассматривают в настоящем описании как твердые, либо как жидкие формы.
Что касается твердой композиции для перорального введения, то композиция может быть приготовлена в виде порошка, гранул, прессованных таблеток, пилюль, капсул, жевательной резинки, вафель или в виде подобных им форм. Такая твердая композиция будет обычно содержать один или более инертных разбавителей или съедобных носителей. Дополнительно может присутствовать один или более из следующих адъювантов: связующие вещества, например, такие как карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза или желатин; наполнители, такие как крахмал, лактоза или декстрины; средства, способствующие распаду лекарственной формы, такие как альгиновая кислота, альгинат натрия, Примогель, кукурузный крахмал и подобные им вещества; смазывающие вещества, такие как стеарат магния или стеротекс (Sterotex); средства, улучшающие скольжение, такие как коллоидный диоксид кремния; подслащивающие вещества, такие как сахароза или сахарин; вещества, придающие вкус и запах, такие как мятное масло, метилсалицилат и апельсиновый ароматизатор, и окрашивающее средство.
В том случае, когда композиция находится в форме капсулы, например желатиновой капсулы, она может содержать, в дополнение к вышеуказанным типам веществ, жидкий носитель, такой как полиэтиленгликоль, циклодекстрин или жирные (нелетучие) масла.
Композиция может находиться в форме жидкости, например в виде эликсира, сиропа, раствора, эмульсии или суспензии.
Жидкость может быть предназначена для перорального применения или для введения посредством инъекции, в виде двух образцов. В том случае, когда композиция предназначена для перорального введения, предпочтительные композиции содержат, в дополнение к соединениям, являющимся предметом настоящего изобретения, одно или более подслащивающих веществ, консервантов, подкрашивающее вещество/краситель и усилитель вкуса и запаха. В том случае, когда композиция предназначена для введения посредством инъекции, может быть включено одно или более поверхностно-активное вещество, консервант, средство для улучшения смачиваемости, диспергирующий агент, суспендирующий агент, буфер, стабилизатор и изотоническое средство.
Жидкие фармацевтические композиции настоящего изобретения вне зависимости от того, находятся ли они в виде раствора, суспензии или другой подобной формы, могут включать один или более из следующих адъювантов: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический раствор, раствор Рингера, изотонический раствор хлорида натрия, жирные (нелетучие) масла, такие как синтетические моно- и диглицериды, которые могут служить в качестве растворителя или суспендирующей среды, полиэтиленгликоли, глицерин, циклодекстрин, пропиленгликоль или другие растворители; антибактериальные средства, например, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; комплексообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетатные, цитратные или фосфатные буферы, и средства для регулировки изотоничности, такие как хлорид натрия и декстроза. Препарат для парентерального введения может быть заключен в ампулы, одноразовые шприцы или флаконы, содержащие многократное количество разовых доз, выполненные из стекла или пластика. Физиологический раствор является предпочтительным адъювантом. Композиция для инъекций предпочтительно является стерильной.
Жидкая композиция, предназначенная либо для парентерального, либо для перорального введения, должна содержать такое количество соединения формулы (1), чтобы была получена подходящая дозировка. Обычно это количество составляет, по меньшей мере, 0,01% соединения настоящего изобретения в композиции. В том случае, если предполагается пероральное введение, это количество может варьироваться от 0,1% до приблизительно 80% от массы всей композиции. Предпочтительные композиции для перорального введения содержат от приблизительно 4% до приблизительно 50% активного соединения формулы (1). Предпочтительные композиции и препараты в соответствии с настоящим изобретением приготавливают таким образом, чтобы единичная дозированная форма для парентерального введения содержала от 0,01 до 2 мас.% активного соединения.
Фармацевтическая композиция может предназначаться для местного применения, и в этом случае носитель может подходящим образом состоять из растворной, эмульсионной, мазевой или гелевой основы. Основа, например, может включать одно или более из следующего: вазелин, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, и эмульгаторы и стабилизаторы. В фармацевтической композиции для местного применения могут присутствовать загустители. В том случае, если предполагается трансдермальное введение, композиция может включать трансдермальный пластырь или устройство для лекарственного электрофореза. Составы для местного применения могут содержать соединение формулы (1) в концентрации от приблизительно 0,1% до приблизительно 10% мас./об. (единица массы на единицу объема).
Композиции могут быть предназначены для ректального введения, в виде, например, суппозитория, который расплавляется в прямой кишке и высвобождает лекарственное средство. Композиция для ректального введения может содержать масляную основу как подходящий, не вызывающий раздражения наполнитель. Такие основы включают, не ограничиваясь этим, ланолин, масло какао и полиэтиленгликоль.
Композиции могут включать различные вещества, которые модифицируют физическую форму твердой или жидкой разовой дозированной формы. Например, композиция может включать вещества, которые образуют покрывающую оболочку вокруг активных ингредиентов. Вещества, которые образуют покрывающую оболочку, обычно являются инертными и могут быть выбраны, например, из сахарозы, шеллака и других средств, образующих энтеросолюбильную оболочку. В качестве альтернативы активные ингредиенты могут быть помещены в желатиновую капсулу.
Композиции в твердой или жидкой форме могут включать агент, который связывается с активным компонентом (компонентами) и таким образом способствует доставке активных компонентов. Подходящие агенты, которые могут выступать в этом качестве, включают моноклональное или поликлональное антитело, белок или липосому.
Фармацевтическая композиция настоящего изобретения может находиться в виде газообразной дозированной формы, например находиться в виде аэрозоля. Термин "аэрозоль" используется для обозначения разнообразия систем, варьирующих между коллоидными природными системами и системами, заключенными в упаковке под давлением. Доставка может быть осуществлена посредством сжиженного или сжатого газа или подходящей насосной установкой, которая распределяет активные ингредиенты. Аэрозоли соединений настоящего изобретения могут быть доставлены в однофазной, двухфазной или трехфазной системе для того, чтобы осуществить доставку активного(ых) ингредиента(ов). Система доставки аэрозоля включает необходимый контейнер, активаторы, клапаны, субконтейнеры, фиксирующие устройства и тому подобное, которые вместе могут образовывать набор. Предпочтительные аэрозоли могут быть определены специалистом в данной области техники, без излишних экспериментов.
Вне зависимости от формы (твердой, жидкой или газообразной) фармацевтическая композиция настоящего изобретения может содержать одно или более фармацевтических средств, применяемых при лечении воспаления (включая астму, аллергию, ревматоидный артрит, множественный склероз и т.д.), пролиферативные нарушения (новообразования), заболевания, подлежащие лечению посредством регулировки кальция (включая гипертензию, сердечную аритмию и т.п.) и синдром приобретенного иммунного дефицита (СПИД).
Фармацевтические композиции могут быть получены согласно методикам, хорошо известным в области фармацевтики.
Композиции, предназначенные для введения посредством инъекций, могут быть получены при смешении соединения формулы (1) с водой таким образом, чтобы получить раствор. Для облегчения получения гомогенного раствора или суспензии может быть добавлено поверхностно-активное вещество. Поверхностно-активные вещества представляют собой соединения, которые не ковалентно взаимодействуют с соединением формулы (1) таким образом, чтобы облегчить растворение или гомогенизацию суспензии активного соединения в водной системе доставки.
Е. Биологическая активность.
Соединения, раскрытые в настоящем описании и представленные формулой 1, или композиции, включающие одно или более из этих соединений и фармацевтически приемлемый носитель, разбавитель или наполнитель, могут быть использованы при осуществлении способа лечения или профилактики воспалительного состояния или заболевания пациента, причем указанный способ включает введение пациенту, нуждающемуся в этом, соединения или композиции согласно настоящему изобретению, в количестве, которое является эффективным для лечения или профилактики воспалительного состояния или заболевания пациента.
Воспалительное состояние или заболевание может включать респираторное воспаление (например, где респираторное заболевание представляет собой астму, или где респираторное заболевание представляет собой хроническое обструктивное легочное заболевание, или где респираторное заболевание представляет собой эмфизему); воспалительное состояние может быть аутоиммунным состоянием или заболеванием; воспалительное состояние или заболевание может быть красной волчанкой; воспалительное состояние или заболевание может включать острое или хроническое воспаление кости и/или хрящевого отделения суставов; воспалительное состояние или заболевание может представлять собой артрит, выбранный из ревматоидного артрита, подагрического артрита, болезни Стилла; воспалительное состояние или заболевание может представлять собой заболевание центральной нервной системы; состояние или заболевание может быть связано с инфильтрацией лейкоцитов; состояние или заболевание может быть связано с отеком; состояние или заболевание может быть связано с ишемическим нарушением кровообращения; состояние или заболевание может быть связано с повышенным уровнем цитокинов зоны воспаления (например, где цитокин зоны воспаления представляет собой интерлейкин (IL)-4, или где цитокин зоны воспаления представляет собой IL-5, или где цитокин зоны воспаления представляет собой IL-10, или где цитокин зоны воспаления представляет собой IL-13, или где цитокин зоны воспаления представляет собой IL-9, или где цитокин зоны воспаления представляет собой IL-1, или где цитокин зоны воспаления представляет собой IL-2, или где цитокин зоны воспаления представляет собой IL-6, или где цитокин зоны воспаления представляет собой IL-18, или где цитокин зоны воспаления представляет собой IL-3, или где цитокин зоны воспаления представляет собой IL-8, или где цитокин зоны воспаления представляет собой IL-12, или где цитокин зоны воспаления представляет собой фактор некроза опухоли TNF-α, или где цитокин зоны воспаления представляет собой TGF-β, или где цитокин зоны воспаления представляет собой GM-CSF, или где цитокин зоны воспаления представляет собой IFN-γ, или где цитокин зоны воспаления представляет собой LTB4, или где цитокин зоны воспаления представляет собой член семейства цистениллейкотриенов, или где цитокин зоны воспаления представляет собой цитокин, выделяемый неиммунными Т клетками при активации (RANTES), или где цитокин зоны воспаления представляет собой эотоксин 1, 2, или 3, или где цитокин зоны воспаления представляет собой воспалительный белок макрофагов (MIP)-1α, или где цитокин зоны воспаления представляет собой хемоаттрактант моноцитов - белок-1, 2, 3 или 4); состояние или заболевание может быть связано с изменившимся уровнем молекул адгезии при воспалении (например, где молекула адгезии представляет собой молекулу адгезии сосудистого эндотелия (VCAM-1 или 2), где молекула адгезии представляет собой молекулу межклеточной адгезии (ICAM-1 или 2), где молекула адгезии представляет собой антиген-4 самых поздних стадий (VLA-4); где молекула адгезии представляет собой антиген-1, ассоциированный с функцией лейкоцитов (LFA-1); где молекула адгезии представляет собой селектин); воспалительное состояние или заболевание может представлять собой рассеянный (множественный) склероз; воспалительное состояние или заболевание может представлять собой легочный саркадоз; воспалительное состояние или заболевание может представлять собой глазное воспаление или аллергию; воспалительное состояние или заболевание может представлять собой аллергический ринит; воспалительное состояние или заболевание может представлять собой воспалительное заболевание желудочно-кишечного тракта (например, гранулемотозную болезнь - болезнь Крона или неспецифический язвенный колит); воспалительное состояние или заболевание может представлять собой кожное воспалительное заболевание (например, псориаз или дерматит); воспалительное состояние или заболевание может представлять собой реакцию "трансплантат против хозяина"; воспалительное состояние или заболевание может васкулярным (например, васкулит); воспалительное состояние или заболевание может представлять собой атеросклеротическое заболевание.
Кроме того, настоящее изобретение предоставляет способ лечения или профилактики заболевания или состояния пациента, где заболевание или состояние пациента связано с патологическими состояниями, которые включают инфильтрацию лейкоцитов, причем указанный способ включает введение пациенту, нуждающемуся в этом, соединения или композиции согласно настоящему изобретению в количестве, которое является эффективным для лечения или профилактики заболевания или состояния, связанного с патологическими состояниями, которые включают инфильтрацию лейкоцитов.
Кроме того, настоящее изобретение предоставляет способ лечения или профилактики астмы у пациента, включающий введение пациенту, нуждающемуся в этом, соединения или композиции настоящего изобретения в количестве, которое является эффективным для лечения или профилактики астмы.
Кроме того, настоящее изобретение предоставляет способ лечения или профилактики аллергии у пациента, включающий введение пациенту, нуждающемуся в этом, соединения или композиции настоящего изобретения в количестве, которое является эффективным для лечения или профилактики аллергии.
В соответствии со способом согласно настоящему изобретению соединение формулы (1) или композиция, включающая соединение формулы (1) и фармацевтически приемлемый носитель, разбавитель или наполнитель, могут, хотя это и не требуется, приводить к достижению одного или более из следующих желаемых результатов у субъекта, которому введено соединение формулы (1), как определено выше, или композиция, содержащая одно из указанных соединений и фармацевтически приемлемый носитель, разбавитель или наполнитель:
1) ингибирование инфильтрации лейкоцитов (например, нейтрофилов, эозинофилов и т.д.);
2) ингибирование активации лейкоцитов;
3) изменение соотношения лимфоцитов (например, соотношения клеток ТН1 и ТН2);
4) ингибирование хемотаксиса лейкоцитов;
5) ингибирование продуцирования и/или высвобождения TNF-α;
6) ингибирование продуцирования и/или высвобождения хемокина (например, эотаксина и т.д.);
7) ингибирование продуцирования, высвобождения и/или функционирования молекулы адгезии (например, VCAM, VLA-4 и т.д.);
8) ингибирование отека;
9) ингибирование продуцирования и/или высвобождения цитокина - интерлейкина (например, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-9, IL-10, IL-12, IL-13, IL-18);
10) ингибирование высвобождения противовоспалительного медиатора (например, лейкотриенов, триптазы, аденозина и т.п.);
11) ингибирование высвобождения гистамина;
12) ингибирование проявлений астмы;
13) ингибирование проявлений аллергии.
Соединения, представленные в настоящем описании формулой (1) (т.е. соединения формулы (1) или иначе, соединения настоящего изобретения), или композиции, включающие одно или более из данных соединений и фармацевтически приемлемый носитель, разбавитель или наполнитель, могут быть использованы при осуществлении способа лечения или профилактики пролиферативного нарушения у пациента, причем указанный способ включает введение пациенту, нуждающемуся в этом, соединения или композиции согласно настоящему изобретению в количестве, которое является эффективным для лечения или профилактики пролиферативного нарушения у пациента. При использовании в настоящем изобретении термин пролиферативное нарушение включает, без ограничений, все виды лейкемии и твердых новообразований, которые восприимчивы к прохождению процесса дифференцировки или апоптоза с прерыванием их клеточного цикла.
Соединения, представленные в настоящем описании формулой (1) (т.е. соединения формулы (1) или иначе, соединения настоящего изобретения), или композиции, включающие одно или более из данных соединений и фармацевтически приемлемый носитель, разбавитель или наполнитель, могут быть использованы при осуществлении способа лечения или профилактики заболеваний, поддающихся лечению посредством регулировки кальция у пациента, причем указанный способ включает введение пациенту, нуждающемуся в этом, соединения или композиции согласно настоящему изобретению в количестве, которое является эффективным для лечения или профилактики заболевания у пациента. При использовании в настоящем изобретении термин "заболевание, поддающиеся лечению посредством регулировки кальция у пациента" включает, без ограничений, сердечную аритмию, фибрилляцию предсердий, острый коронарный синдром, гипертензию, ишемическое нарушение кровообращения, внезапный приступ, "удар", эпилепсию, демиелинизирующие заболевания, такие как множественный (рассеянный) склероз, боль, эпилептическое состояние, атеросклероз и диабет.
Соединения, представленные в настоящем описании формулой (1) (т.е. соединения формулы (1) или соединения настоящего изобретения), или композиции, включающие одно или более из указанных соединений и фармацевтически приемлемый носитель, разбавитель или наполнитель, могут быть использованы при осуществлении способа лечения или профилактики синдрома приобретенного иммунодефицита (СПИД) у пациента, причем указанный способ включает введение пациенту, нуждающемуся в этом, соединения или композиции согласно настоящему изобретению в количестве, которое является эффективным для лечения или профилактики синдрома приобретенного иммунодефицита у пациента. При использовании в настоящем изобретении термин "синдром приобретенного иммунодефицита" при инфицировании вирусом иммунодефицита человека типа I включает, без ограничений, связанные с ним осложнения, такие как комплекс слабоумия при синдроме приобретенного иммунодефицита и нейросиндромы приобретенного иммунодефицита.
Таким образом, способ согласно настоящему изобретению может быть полезен для лечения воспаления, включая как острое, так и хроническое воспаление, а также пролиферативные нарушения (раковые заболевания, новообразования), заболевания, поддающиеся лечению посредством регулировки кальция у пациента, и СПИД. При использовании в данном описании термин "воспаление" включает, без ограничений, анкилозирующий спондилоартрит, артрит (где этот термин включает более 100 разновидностей ревматических заболеваний), астму, хроническое обструктивное заболевание легких, аллергию, аллергический ринит, болезнь Крона, синдром фибромиалгии, подагру, воспаления мозга (включая множественный склероз, комплекс слабоумия при синдроме приобретенного иммунодефицита, энцефалопатию Лайма, герпетический энцефалит, болезнь Крейтцфельда-Якоба и церебральный токсоплазмоз), эмфизему, воспалительное заболевание желудочно-кишечного тракта, слизистый колит, ишемическое нарушение кровообращения, аллергический дерматит, юношеский эритематозный легочный саркоидоз, болезнь Кавасаки, остеоартрит, воспалительное заболевание почечных лоханок, псориатический артрит (псориаз), ревматоидный артрит, псориаз, трансплантат ткани/органа, реакцию "трансплантат против хозяина", склеродермию, спондилоартропатию, системную красную волчанку, легочный саркоидоз, васкулит, атеросклероз, кардиомиопатию, аутоиммунный миокардит и неспецифический язвенный колит.
Способ настоящего изобретения предполагает введение терапевтически эффективного количества соединения формулы (1), включая его соли, композиции и т.д. При использовании в настоящем описании фактическое количество, обозначаемое термином "терапевтически эффективное количество", будет зависеть от способа введения, типа теплокровного животного, подвергнутого лечению, и физических характеристик конкретного рассматриваемого теплокровного животного. Эти факторы и их взаимодействие при определении такого количества хорошо известны специалисту в области медицины. Это количество и способ введения могут быть заданы таким образом, чтобы достичь оптимальной эффективности, но будут зависеть от таких факторов, как вес, диета, сопутствующее лечение и другие факторы, которые известны специалисту в области медицины.
Эффективное количество соединения или композиции согласно настоящему изобретению будет достаточным для лечения воспаления, пролиферативных заболеваний, заболевания, подлежащего лечению посредством регулировки кальция, или СПИД у теплокровных животных, таких как человек. Способы введения эффективного количества противовоспалительных средств хорошо известны в данной области техники и включают введение посредством ингаляции, пероральные или парентеральные формы. Такие дозированные формы включают, но не ограничиваются ими, парентеральные растворы, таблетки, капсулы, имплантаты замедленного высвобождения и системы трансдермальной доставки, или дозирующие системы лекарственных форм для ингаляции, использующие пульверизаторы для сухого порошка, или дозирующие устройства под давлением, позволяющие ввести множество доз.
Величину дозы и частоту выбирают таким образом, чтобы достичь эффективное содержание средства без вредного воздействия. Такое количество, как правило, варьируется между дозировкой, равной приблизительно от 0,001 до 100 мг/кг/день, и обычно от приблизительно 0,01 до 10 мг/кг/день, при пероральном или внутривенном введении. Дозировка также варьируется приблизительно от 0,0001 до 10 мг/кг/день при интраназальном или ингаляционном введении.
Соединения формулы (1), включая соединения, используемые согласно способам и в композициях, указанных выше, могут быть получены в соответствии со схемами, приведенными в следующих примерах. Следующие примеры предназначены для иллюстрации и не являются ограничивающими объем изобретения.
Если не указано иное, флэш-хроматографию и колоночную хроматографию осуществляют, используя силикагель 60 фирмы Merck (230-400 меш). Флэш-хроматографию можно проводить в соответствии с методикой, приведенной в "Purification of Laboratory Chemicals", 3rd edition, Butterworth-Heinemann Ltd., Oxford (1998), Eds. D.D.Perrin and W.L.F.Armarego, page 23. Колоночная хроматография относится к способу, при котором скорость прохождения потока элюента через материал-наполнитель определяется гравитацией. Во всех случаях флэш-хроматография и радиальная (круговая) хроматография могут использоваться взаимозаменяемо. Радиальную хроматографию осуществляют, используя силикагель на приборе Chromatotron Model#7924T (Harrison Research, Palo Alto, California). Если не указано иное, указанные значения Rf получают методом тонкослойной хроматографии, используя силикагель 60 фирмы Merck (Silica Gel 60 F254, Merck KgaA, 64271, Darmstadt, Germany). Рассол представляет собой насыщенный раствор хлорида натрия.
Также, если не указано иное, химические продукты и реагенты получают от общеизвестных фирм - поставщиков химической продукции, таких как фирмы Aldrich (Milwaukee, WI); EM Industries, Inc (Hawthorne, NY); Fisher Scientific Co. (Hampton, NH) и Lancaster Synthesis, Inc. (Windham, NH). Сульфо-NHS-биотин получают от Pierce (Rockford, IL). Смолу MP-TsOH, смолу PS-DIEA, смолу PS-Trisamine и смолу PS-Benzaldehyde получают от Argonaut Technologies (San Carlos, CA). Газы получают от Praxair (Vancouver, B.C.). Линии клеток, если не указано иное, получают из государственных или коммерческих источников, например из Американской коллекции культур тканей (ATCC, Rockville, MD).
Примеры синтеза
Пример 1
3-Амино-6,7-дигидрокси-17-этилиденстероид
Соединение 49, типичный представитель соединения настоящего изобретения, получают в соответствии со схемой 1. Немало соединений, родственных соединению 49, может быть получено при использовании подобной методологии. Исходное соединение 45 может быть получено согласно методике, описанной в патенте США 6046185. Олефинирование кетона 45 осуществляют с использованием этилтрифенилфосфонийбромида и KOtBu в толуоле. Обработка 3β-гидроксильного соединения 46 с использованием ZnN6·2py, трифенилфосфина и DIAD в толуоле приводит к образованию 3α-азидосоединения 47. Восстановление азида алюмогидридом лития в Et2O дает амин 48. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 49.
Схема 1

i) CH3CH2PPh3Br, KOtBu, толуол; ii) ZnN6 ·2py, Ph3P, DIAD, толуол; iii) LiAlH4, Et2O; iv) HCl, ТГФ, вода.
Синтез соединения 46
Раствор, содержащий КОtBu (0,24 г, 2,0 ммоль), EtPPh3Br (0,75 г, 2,0 ммоль) и толуол (2,5 мл), перемешивают при комнатной температуре в атмосфере аргона. Через 1 час темно-красный раствор охлаждают на льду и добавляют кетон 45 (184 г, 0,508 ммоль), полученному раствору дают нагреться до комнатной температуры. После перемешивания в течение ночи реакционную смесь гасят добавлением 10 мл воды, разбавляют 60 мл этилацетата (EtOAc), разделяют и промывают 2×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки колоночной хроматографией с элюированием смесью EtOAc/гексан в соотношении 1:1 получают 171 мг (90%) соединения 46 в виде бесцветной пленки.
Синтез соединения 47
DIAD (0,44 мл, 2,14 ммоль) добавляют по каплям в течение 10 минут к находящемуся при комнатной температуре раствору, содержащему 3β-гидроксисоединение 46 (400 мг, 1,07 ммоль), ZnN6·2py (246 мг, 0,80 ммоль), Ph3Р (560 мг, 2,14 ммоль) и толуол (10,7 мл) в атмосфере аргона. Через 4 часа реакционную смесь загружают на колонку, заполненную силикагелем в смеси 10% этилацетат/гексан, и элюируют смесью 20% этилацетат/гексан с получением 422 мг (99%) соединения 47 в виде белого твердого вещества.
Синтез соединения 48
Алюмогидрид лития (42 мг, 1,04 ммоль) добавляют к охлаждаемому льдом раствору азида 47 (415 мг, 1,04 ммоль) в 5,2 мл Et2O в атмосфере аргона. Реакционной смеси дают нагреться до комнатной температуры. После 2 часов охлаждения на льду раствор разбавляют 25 мл диэтилового эфира и медленно гасят добавлением 2 мл насыщенного раствора Na2SO4. Через 10 минут образуется белый осадок, раствор разбавляют 50 мл этилацетата, промывают 3×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество очищают, используя колонку с силикагелем, предварительно заполненную в смеси 1% Et3N/CH2Cl2 и промытую смесью 5% MeOH/CH2Cl2. Неочищенное вещество загружают в CH2 Cl2, элюируют смесью 5% MeOH/CH2Cl2 и затем смесью в соотношении 95:5:2 CH2Cl2:МеОН:Et3N, получая белую пену, которая, по данным1Н ЯМР-спектроскопии, содержит следы Et3N. Вещество помещают в 50 мл гексана, промывают 2×20 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 322 мг (83%) соединения 48 в виде белой пены.
Синтез соединения 49
Раствор, содержащий 3α-аминосоединение 48 (317 мг, 0,850 ммоль), 4 М HCl в диоксане (255 мкл, 1,02 ммоль), ТГФ (13,6 мл) и воду (3,4 мл), перемешивают при комнатной температуре в течение ночи. Раствор концентрируют досуха, растирают с ацетоном порциями 3×10 мл, выпаривая ацетон после каждого растирания. После концентрирования получают 301 мг (96%) соединения 49 в виде белого твердого вещества. Жидкостная хроматография/масс-спектроскопия (ЖХ/МС, LC/MS - прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 334,16; С21Н36NO2.
Пример 2
3-Амино-6,7-дигидрокси-17-метилиденстероид
Дополнительный пример алкенов, родственных соединению 49, приведен на схеме 2. Олефинирование кетона 45 с использованием метилтрифенилфосфонийбромида и КОtBu в тетрагидрофуране приводит к 17-метилиденовому соединению 50. Обработка с использованием ZnN6·2py, PPh3 и DIAD в толуоле приводит к образованию 3α-азидосоединения 51. Восстановление алюмогидридом лития в ТГФ приводит к образованию 3α-азидосоединения 52. Обработка 80%-ной уксусной кислотой приводит к удалению защитной ацетонидной группы и образованию аммонийноацетатной соли 53. Альтернативно, соединение 52 обрабатывают хлористоводородной кислотой в ацетонитриле и водой, получая гидрохлоридную соль 54.
Схема 2

i) CH3PPh3Br, KOtBu, ТГФ; ii) ZnN6·2py, Ph3P, DIAD, толуол; iii) LiAlH4, Et2O; iv)80%-ная уксусная кислота или HCl, вода, MeCN.
Синтез соединения 50
Раствор, содержащий КОtBu (2,0 г, 16,9 ммоль), MePPh3Br (6,0 г, 16,8 ммоль) и 27 мл ТГФ, перемешивают при комнатной температуре в атмосфере аргона. Через 1 час к желтому раствору добавляют кетон 45 (2,00 г, 5,52 ммоль), полученный раствор кипятят с обратным холодильником в течение 1 часа. Реакционную смесь гасят добавлением 50 мл насыщенного раствора хлорида натрия, разбавляют 100 мл этилацетата (EtOAc), разделяют и промывают 25 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки колоночной хроматографией с элюированием смесью EtOAc/гексан в соотношении 1:1 получают 1,90 г (95%) соединения 50 в виде белого твердого вещества.
Синтез соединения 51
DIAD (0,85 мл, 4,10 ммоль) добавляют по каплям в течение 15 минут к находящемуся при комнатной температуре раствору, содержащему 3β-гидроксильное соединение 50 (739 мг, 2,05 ммоль), ZnN6·2py (473 мг, 1,54 ммоль), Ph3Р (1,075 мг, 4,10 ммоль) и толуол (20 мл), в атмосфере аргона. Через 4 часа реакционную смесь загружают на колонку, заполненную силикагелем в смеси 10% этилацетат/гексан, и элюируют смесью 20% этилацетат/гексан с получением 743 мг (94%) соединения 51 в виде белой пены.
Синтез соединения 52
Алюмогидрид лития (77 мг, 1,93 ммоль) добавляют к охлаждаемому льдом раствору 3α-азида 51 в 5 мл ТГФ и 5 мл диэтилового эфира в атмосфере аргона. Реакционной смеси дают нагреться до комнатной температуры. Через 4 часа раствор охлаждают на льду, разбавляют 25 мл диэтилового эфира и медленного гасят добавлением 5 мл насыщенного раствора Na2SO4. Через 10 минут образуется белый осадок, раствор разбавляют 50 мл этилацетата, промывают 3×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество очищают на колонке с силикагелем, предварительно заполненной смесью 1% Et3N/CH2Cl2 и промытой смесью 5% MeOH/CH2Cl2. Неочищенное вещество загружают в CH2 Cl2, элюируют смесью 5% MeOH/CH2Cl2 и затем смесью в соотношении 95:5:2 CH2Cl2:МеОН:Et3N, получая 636 мг (92%) соединения 52 в виде белого твердого вещества.
Синтез соединения 53
Раствор, содержащий 3α-амин 52 (287 мг, 0,799 ммоль) и 10 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 1 часа. Реакционную смесь концентрируют, получая белую пену. Добавляют ацетон (10 мл) и раствор подвергают воздействию ультразвука для того, чтобы растворить вещество, а затем упаривают. Добавляют еще 10 мл ацетона, подвергают воздействию ультразвука и упаривают, получая 301 мг (99%) соединения 53 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 320,19; С20Н34NO2.
Синтез соединения 54
Раствор 4 М HCl в диоксане добавляют к раствору амина 52 в 1 мл ацетонитрила и 50 мкл воды. Полученное резиноподобное твердое вещество разбавляют 2 мл ацетонитрила и энергично перемешивают до тех пор, пока не образуется твердое вещество. Твердое вещество отфильтровывают и сушат, получая 50 мг (63%) соединения 54. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 320,19; С20Н34NO2.
Пример 3
3-Амино-6,7-дигидрокси-17-метилиденстероид (альтернативный синтез)
Промежуточное соединение 52 также синтезируют, используя альтернативный подход, показанный на схеме 3. Азидирование спирта 45 с использованием ZnN6·2py, трифенилфосфина и DIAD в толуоле приводит к образованию 3α-азидосоединения 55. Гидрирование азида с использованием в качестве катализатора палладия (Pd) на углероде дает амин 56. Олефинирование соединения 56 с использованием метилтрифенилфосфонийбромида и КОtBu в тетрагидрофуране приводит к 17-метилиденовому соединению 52.
Схема 3

i) ZnN6·2py, Ph3P, DIAD, толуол; ii) H2, Pd, EtOAc; iii) CH3PPh3Br, KOtBu, ТГФ.
Синтез соединения 55
DIAD (2,4 мл, 11,6 ммоль) добавляют по каплям в течение 20 минут к находящемуся при комнатной температуре раствору, содержащему 3β-гидроксильное соединение 45 (2,108 г, 5,81 ммоль), ZnN6·2py (1,34 г, 4,36 ммоль), Ph3Р (3,05 г, 11,6 ммоль) и толуол (58 мл), в атмосфере аргона. После взаимодействия в течение ночи реакционную смесь загружают на колонку, заполненную силикагелем, и элюируют смесью 20% этилацетат/гексан с получением 1,14 г (50%) соединения 55 в виде белой пены.
Синтез соединения 56
Раствор, содержащий азид 55 (1,10 г, 2,84 ммоль), 10%-ный Pd на углероде (60 мг, 0,057 моль) и 28 мл этилацетата, перемешивают при комнатной температуре в течение ночи в атмосфере водорода. Раствор фильтруют через целит, элюируя этилацетатом. После очистки методом радиальной хроматографии с элюированием смесью CH2Cl2 :МеОН:Et3N в соотношении 95:5:2 получают 892 мг (81%) соединения 56 в виде белого твердого вещества.
Синтез соединения 52
Раствор, содержащий КОt Bu (175 мг, 1,48 ммоль), MePPh3Br (528 мг, 1,48 ммоль) и 3 мл ТГФ, перемешивают при комнатной температуре в атмосфере аргона. Через 1 час к желтому раствору добавляют кетон 56 (100 мг, 0, 277 ммоль) и полученный раствор оставляют при перемешивании при комнатной температуре в течение ночи. Реакционную смесь гасят добавлением 5 мл воды, разбавляют 50 мл этилацетата (EtOAc), разделяют и промывают 10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки радиальной хроматографией с элюированием смесью CH2Cl2 :МеОН:Et3N в соотношении 95:5:2 получают 96 мг (97%) соединения 52 в виде белого твердого вещества.
Пример 4
3-Амино-6,7-дигидрокси-17-фторэтилиденстероид
Галогенированные аналоги, родственные соединению 49, могут быть получены с использованием для олефинирования галогенированных реагентов. На схеме 4 показан синтез 20-фторсодержащего аналога соединения 64. Гидроксильную группу в соединении 45 защищают посредством обработки трет-бутилдиметилсилилхлоридом и имидазолом в диметилформамиде (ДМФ). Олефинирование кетона 57 с использованием аниона триэтил-2-фторо-1-фосфоноацетата приводит к получению смеси соединения 58 и его геометрического изомера. Соединения возможно разделить хроматографией на силикагеле. Восстановление алюмогидридом лития сложноэфирной группы в среде Et2O дает аллиловый спирт 59. Обработка комплексом триоксида серы и пиридина в ТГФ с последующим добавлением алюмогидрида лития позволяет получить дегидроксилированное соединение 60. Тетрабутиламмонийфторид в ТГФ удаляет защитные группы 3-гидроксильного соединения с получением соединения 61. Азидирование с использованием ZnN6·2py, PPh3 и DIAD в толуоле приводит к образованию 3α-азидосоединения 62. Восстановление азида алюмогидридом лития в ТГФ дает 3α-амин 63. Обработка с использованием HCl в ТГФ и воды приводит к удалению защиты 6- и 7-гидроксильных групп и образованию аммонийнохлоридной соли 64.
Схема 4

i) TBSCl, имидазол, ДМФ; ii) (EtO)2P(O)CHFCO2Et, LiN(TMS)2, ТГФ; iii) LiAlH4, Et2O; iv) SO3Py, ТГФ, LiAlH4; v) Bu4NF, ТГФ; vi) ZnN6·2Py, Ph3P, DIAD, толуол; vii) LiAlH4, Et2O; viii) HCl, ТГФ, вода.
Синтез соединения 57
Раствор, содержащий кетон 45 (4,73 г, 13,1 ммоль), TBSCl (3,01 г, 19,6 ммоль), имидазол (2,67 г, 39,2 ммоль) и ДМФ (52 мл), перемешивают при комнатной температуре в течение ночи. Белую суспензию разбавляют 250 мл EtOAc, промывают 2×50 мл воды и 50 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 5,97 г (96%) соединения 57 в виде белого твердого вещества.
Синтез соединения 58
Бис(триметилсилил)амид лития (10,0 мл 1,0 М раствора в ТГФ, 10,0 ммоль) добавляют к находящемуся при комнатной температуре раствору (EtO)2P(O)CHFCO2Et (2,65 г, 10,5 ммоль) в ТГФ (22 мл) в атмосфере аргона. Через 1 час добавляют раствор кетона 57 (2,50 г, 5,25 ммоль) в ТГФ (20 мл) и полученный раствор кипятят с обратным холодильником в течение 4,5 часа, после чего перемешивают при комнатной температуре в течение ночи. Реакционную смесь гасят добавлением 1,5 мл насыщенного раствора NaHCO3 и затем частично концентрируют для того, чтобы удалить большую часть ТГФ. Остаток разбавляют 200 мл EtOAc, промывают 3×20 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество очищают колоночной хроматографией, элюируя смесью сначала 2,5%, а затем 5% EtOAc/гексан, получая 1,50 г (50%) соединения 58 в виде белого твердого вещества.
Синтез соединения 59
Алюмогидрид лития (106 мг, 2,66 ммоль) добавляют к охлаждаемому льдом раствору сложного эфира 58 (1,50 г, 2,66 ммоль) в Et2O (13 мл) в атмосфере аргона. Раствору дают нагреться до комнатной температуры. Через 3 часа раствор охлаждают на льду и медленно добавляют 20 мл насыщенного раствора Na2SO4. Через 10 минут раствор разбавляют 150 мл EtOAc, промывают водой и насыщенным раствором хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 1,44 г (выход количественный) соединения формулы 59 в виде белой пены.
Синтез соединения 60
Комплекс триоксида серы и пиридина (69,5 мг, 0,428 ммоль) добавляют к охлаждаемому льдом раствору аллилового спирта 59 (149 мг, 0,285 ммоль) в ТГФ (2,8 мл) в атмосфере аргона. Через 6 часов добавляют алюмогидрид лития (68 мг, 1,71 моль), после чего раствору дают нагреться до комнатной температуры в течение ночи. Раствор охлаждают на льду и медленно добавляют 5 мл насыщенного раствора Na2SO4. Через 10 минут раствор разбавляют 75 мл EtOAc, промывают водой и насыщенным раствором хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 109 мг (76%) соединения формулы 60 в виде белого твердого вещества.
Синтез соединения 61
Раствор, содержащий соединение 60 (410 мг, 0,810 ммоль), Bu4NF (0,89 мл 1,0 М раствора в ТГФ, 0,89 ммоль) и ТГФ (5 мл), кипятят с обратным холодильником в атмосфере аргона. Через 1,5 часа раствор охлаждают до комнатной температуры, разбавляют 75 мл EtOAc, промывают водой (20 мл) и насыщенным раствором хлорида натрия (2×20 мл), сушат над MgSO4, фильтруют и концентрируют. Остаток фильтруют через силикагель, элюируя EtOAc, и получают 318 мг (100%) соединения формулы 61 в виде белого твердого вещества.
Синтез соединения 62
DIAD (0,33 мл, 1,59 ммоль) добавляют по каплям в течение 10 минут к находящемуся при комнатной температуре раствору 3β -спирта 61 (312 мг, 0,796 ммоль), ZnN6·2py (183 мг, 0,597 ммоль), Ph3Р (417 мг, 1,59 ммоль) и толуола (8,0 мл) в атмосфере аргона. Через 3 часа реакционную смесь загружают на колонку, заполненную силикагелем в смеси 10% EtOAc/гексан, и элюируют смесью 20% EtOAc/гексан, получая 322 мг (97%) соединения 62 в виде кристаллического вещества.
Синтез соединения 63
Алюмогидрид лития (29 мг, 0,75 ммоль) добавляют к охлаждаемому льдом раствору азида 62 (314 мг, 0,753 ммоль) в Et2O (7,5 мл) в атмосфере аргона. Раствору дают нагреться до комнатной температуры при перемешивании в течение ночи. Раствор охлаждают на льду и медленно гасят добавлением 10 мл насыщенного раствора Na2SO4. Через 10 минут образуется белый осадок, раствор разбавляют 75 мл этилацетата (EtOAc), промывают 20 мл воды, 2×20 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество очищают, используя колонку с силикагелем, предварительно заполненную смесью 1% Et3N/CH2Cl2 и промытую смесью 5% MeOH/CH2Cl2. Неочищенное вещество загружают в CH2Cl2, элюируют смесью 5% MeOH/CH2Cl2 и затем смесью CH2Cl2:МеОН:Et3N в соотношении 95:5:2, получая белое твердое вещество. По данным анализа методом1Н ЯМР-спектроскопии вещество содержит следы Et3N, поэтому вещество помещают в 75 мл СН2Cl2 и промывают водой (2×25 мл), сушат над MgSO4, фильтруют и концентрируют, получая 137 мг (47%) соединения 63 в виде бесцветной пленки.
Синтез соединения 64
Раствор, содержащий 3α-аминосоединение 63 (137 мг, 0,35 ммоль), 4 М раствор HCl в диоксане (105 мкл, 0,42 ммоль), ТГФ (5,6 мл) и воду (1,4 мл), перемешивают при комнатной температуре в течение ночи. Раствор концентрируют, остаток дважды помещают в 3 мл метанола и концентрируют, получая 130 мг (96%) соединения 64 в виде не совсем белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 352,14; С21Н35FNO2.
Пример 5
3-Амино-6, 7-дигидрокси-17-карбометоксиэтилиденстероид
Олефинирование соединений, родственных соединению 57, также можно проводить для того, чтобы синтезировать 21-карбоалкоксизамещенные аналоги. На схеме 5 показан пример синтеза 21-карбометоксизамещенного соединения 69. Олефинирование кетона 57 с использованием аниона триметил-2-фосфоноацетата дает смесь соединения 65 и его геометрического изомера. Указанные соединения можно разделить хроматографией на силикагеле. Тетрабутиламмонийфторид в ТГФ удаляет защитные группы 3-гидроксильного соединения с образованием соединения 66. Введение азидной группы с использованием ZnN6·2py, PPh3 и DIAD в толуоле приводит к образованию 3α-азидосоединения 67. Гидрирование азида с использованием Pd на углероде дает 3α-амин 68. Обработка с использованием 80%-ной уксусной кислоты приводит к удалению защиты 6- и 7-гидроксильных групп и образованию аммонийноацетатной соли 69.
Схема 5

i) (MeO)2P(O)CHFCO2Me, LiN(TMS)2, ТГФ; ii) Bu4NF, ТГФ; iii) ZnN6·2py, Ph3P, DIAD, толуол; iv) H2, Pd, EtOAc; v) 80%-ная уксусная кислота.
Синтез соединения 65
Бис(триметилсилил)амид лития (2,00 мл 1,0 М раствора в ТГФ, 2,00 ммоль) добавляют к находящемуся при комнатной температуре раствору (МеО)2P(O)CH2CO2Ме (390 мг, 2,10 ммоль) в ТГФ (22 мл) в атмосфере аргона. Через 3 часа добавляют раствор кетона 57 (509 мг, 1,07 ммоль) в ТГФ (2 мл) и полученный раствор кипятят с обратным холодильником в течение 3 дней. Реакционную смесь гасят добавлением 5 мл воды, разбавляют 75 мл EtOAc, промывают 2×15 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество очищают колоночной хроматографией, элюируя смесью 5% EtOAc/гексан, получая 307 мг (54%) соединения 65 в виде бесцветной пленки. Также выделяют 119 мг (21%) Z-изомера.
Синтез соединения 66
Раствор, содержащий соединение 65 (296 мг, 0,550 ммоль), Bu4NF (0,61 мл 1,0 М раствора в ТГФ, 0,61 ммоль) и ТГФ (3 мл), кипятят с обратным холодильником в атмосфере аргона. Через 1 час раствор охлаждают до комнатной температуры, разбавляют 20 мл EtOAc, промывают 10 мл воды и 2×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 230 мг (100%) соединения 66 в виде белого твердого вещества.
Синтез соединения 67
DIAD (252 мкл, 1,28 ммоль) добавляют по каплям к находящемуся при комнатной температуре раствору, содержащему 3β-спирт 66 (230 мг, 0,55 ммоль), ZnN6·2py (147 мг, 0,48 ммоль), Ph3Р (335 мг, 1,28 ммоль) и толуол (6,4 мл), в атмосфере аргона. Через 2 часа реакционную смесь очищают радиальной хроматографией, элюируя смесью 15% EtOAc/гексан, и получая 203 мг (84%) соединения 67.
Синтез соединения 68
Раствор, содержащий азид 67 (203 мг, 0,45 ммоль), 10%-ный Pd на углероде (48 мг, 0,045 ммоль) и 4,5 мл EtOAc, перемешивают при комнатной температуре в атмосфере водорода в течение 3 дней. Раствор фильтруют через целит, используя в качестве элюента EtOAc и MeOH, и получая 165 мг (88%) соединения 68.
Синтез соединения 69
Раствор, содержащий амин 68 (165 мг, 0,40 ммоль) и 2 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 1 часа. Реакционную смесь разбавляют 10 мл толуола, после чего концентрируют для того, чтобы удалить остаточную уксусную кислоту. Растирание остатка с 10 мл циклогексанона с последующей фильтрацией и сушкой позволяет получить 117 мг (67%) соединения 69 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 378,17; С22Н36NO4.
Пример 6
Соль уксусной кислоты 3α-амино-6α,7β-дигидроксиандростан-17-она
Аналогичную методику можно использовать для получения соединений с различными функциональностями у атома С17. Например, 17-кетозамещенное соединение получают обработкой соединения 56 80%-ной уксусной кислотой с получением соли уксусной кислоты 3α-амино-6α,7β-дигидроксиандростан-17-она (70) (см. таблицу 2).
Синтез соединения 70
Раствор, содержащий кетон 56 (67 мг, 0,16 ммоль) и 1 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 1 часа. Реакционную смесь разбавляют 10 мл толуола, после чего концентрируют с получением 63 мг (100%) соединения 70. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 322,18; С19Н32NO3.
Пример 7
3-Амино-6,7-дигидрокси-17-гидроксистероид
17-Гидроксилзамещенные аналоги могут быть получены из кетонов, родственных соединению 56, таким образом, как показано на схеме 6. Карбонильную группу в соединении 56 восстанавливают с помощью NaBH4 в метаноле, получая исключительно 17β-гидроксильный изомер 71. Обработка 80%-ной уксусной кислотой приводит к удалению ацетонидной защитной группы и образованию аммонийноацетатной соли 72.
Схема 6

i) NaBH4, MeOH; ii) 80% уксусная кислота.
Синтез соединения 71
Охлаждаемый льдом раствор кетона 56 (100 мг, 0,28 ммоль), NaBH4 (16 мг, 0,41 ммоль) и 1,4 мл МеОН подвергают взаимодействию в течение 2,5 часов. Реакционную смесь гасят добавлением 1 мл воды и концентрируют для того, чтобы удалить большую часть МеОН. Остаток разбавляют 40 мл СН2Cl2, промывают 2×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 90 мг (90%) соединения 71.
Синтез соединения 72
Раствор, содержащий амин 71 (90 мг, 0,25 ммоль) и 2 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 2 часов. Реакционную смесь разбавляют 10 мл толуола, после чего концентрируют для того, чтобы удалить остаточную уксусную кислоту. Остаток растворяют в 1 мл МеОН и 5 мл гексана, концентрируют и сушат, получая 86 мг (90%) соединения 72 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 324,19; С19Н34NO3.
Пример 8
Соли 3α-амино-6, 7-дигидрокси-17-метилиденстероида
6- и 7-Гидроксильные группы могут быть защищены с использованием разнообразных защитных групп. Подходящие защитные группы перечислены в Greene and Wuts, "Protective Groups in Organic Synthesis", John Wiley & Sons, New York, N.Y. (1999). На схеме 7 показаны примеры аналогов, которые могут быть синтезированы с использованием 6- и 7-гидроксильных групп, защищенных как простой метиловый эфир. Исходное соединение 73 описано в патенте США 6046185. Генерирование дианиона соединения 73 с использованием NaH в диметилформамиде с последующим алкилированием метилиодидом позволяет получить соединение 74. Обработка 80%-ной уксусной кислотой приводит к удалению защитных групп как циклических кетальных, так и трет-бутилдиметилсилильных простых эфирных защитных групп. Олефинирование кетона 75 с использованием метилтрифенилфосфонийбромида и КОtBu в ТГФ приводит к 17-метилиденовому соединению 76. Азидирование с использованием ZnN6·2py, PPh3 и DIAD в толуоле дает 3α-азидосоединение 77. Восстановление алюмогидридом лития в ТГФ приводит к 3α-амину 78. Обработка с использованием HCl в ТГФ и воды приводит к образованию аммонийнохлоридной соли 79. После обработки соединения 78 уксусной кислотой получают аммонийноацетатную соль 80.
Схема 7

i) NaH, MeI, ДМФ; ii) 80% уксусная кислота; iii) CH3 PPh3Br, KOtBu, ТГФ; iv) ZnN6·2Py, Ph3P, DIAD, толуол; v) LiAlH4, Et2O; vi) HCl, Et2O, MeOH или уксусная кислота.
Синтез соединения 74
К раствору диола 73 (1,49 г, 3,10 ммоль) в 15 мл ДМФ в атмосфере азота добавляют гидрид натрия (0,50 г, 12,4 ммоль). Через 2 часа раствор охлаждают на льду и добавляют по каплям в течение 30 секунд MeI (1,93 мл, 30,9 ммоль). Реакционной смеси дают нагреться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь разбавляют 100 мл Et2O, промывают 10 мл воды и 2×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 1,73 г неочищенного соединения 74 в виде бледно-желтого масла.
Синтез соединения 75
Раствор, содержащий неочищенное соединение 74 (1,73 г, 3,10 ммоль) и 15 мл 80%-ной уксусной кислоты, перемешивают в течение 4 часов при комнатной температуре. Раствор концентрируют, остаток помещают в 50 мл EtOAc, промывают 2×20 мл насыщенного раствора NaHCO3и 2×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 1,18 г неочищенного соединения 75 в виде белой пены.
Синтез соединения 76
Раствор, содержащий КОtBu (1,09 г, 9,20 ммоль), MePPh3Br (3,30 г, 9,20 ммоль) и 15 мл ТГФ, перемешивают при комнатной температуре в атмосфере азота. Через 2 часа к желтому раствору добавляют неочищенный кетон 75 (1,17 мг, 3,08 ммоль) и полученный раствор оставляют при перемешивании при комнатной температуре в течение ночи. Реакционную смесь гасят добавлением 2 мл воды, разбавляют 100 мл этилацетата (EtOAc), промывают 3×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки колоночной хроматографией с элюированием смесью 80% EtOAc/гексан получают 890 мг загрязненного примесями соединения 76 в виде белого твердого вещества.
Синтез соединения 77
DIAD (1,05 мл, 5,08 ммоль) добавляют по каплям в течение 10 минут к находящемуся при комнатной температуре раствору 3β-спирта 76 (885 мг, 2,54 ммоль), ZnN6·2Py (585 мг, 1,90 ммоль), Ph3Р (1,33 г, 5,08 ммоль) и толуола (25 мл) в атмосфере аргона. Через 11 часов реакционную смесь очищают колоночной хроматографией, элюируя смесью 15% EtOAc/гексан и получая 594 мг (63%) соединения 77 в виде кристаллического твердого вещества.
Синтез соединения 78
Алюмогидрид лития (0,79 мл 1 М раствора в Et2O, 0,79 ммоль) добавляют к охлаждаемому льдом раствору азида 77 (588 мг, 1,57 ммоль) в 15,7 мл Et2O в атмосфере аргона. Через 10 мин раствору дают нагреться до комнатной температуры, продолжая перемешивание в течение ночи. Через 1 час охлаждения на льду раствор медленно гасят добавлением 10 мл насыщенного раствора Na2SO4. Через 10 минут образуется белый осадок, и жидкость декантируют. Остаток промывают 2×25 мл EtOAc и промывочные жидкости объединяют с ранее декантированным эфирным раствором. Раствор промывают 3×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки колоночной хроматографией с элюированием смесью CH2Cl2:МеОН:Et3N в соотношении 95:5:2 получают 434 мг (79%) соединения 78 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 348,20; С22Н38NO2.
Синтез соединения 79
Хлористый водород (0,26 мл 1,0 М раствора в Et2O, 0,26 ммоль) добавляют к раствору амина 78 (60 мг, 0,17 ммоль) в 2 мл Et2O. Полученное гелеобразное вещество растворяют в 5 мл метанола и концентрируют. Остаток растворяют в 1 мл метанола, разбавляют 5 мл циклогексана и концентрируют, получая 66 мг (100%) соединения 79 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 348,20; С22Н38NO2.
Синтез соединения 80
Раствор, содержащий амин 78 (61 мг, 0,17 ммоль) и 1 мл уксусной кислоты, оставляют стоять при комнатной температуре в течение 30 минут. Раствор разбавляют 5 мл толуола и концентрируют.Остаток помещают в 5 мл гексана, концентрируют, затем полученный остаток сушат в течение 2 часов, используя прибор Абдерхальдена для высушивания с орошением ацетоном, и получают 71 мг (100%) соединения 80 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 348,20; С22Н38NO2.
Пример 9
Соли 3β-амино-6,7-дигидрокси-17-метилиденстероида
Стереохимия у атома С3 может быть изменена с получением производных 3β-аммонийных солей разнообразных соединений, родственных соединению 49. Стереохимия у атома С3 может быть подвергнута инверсии в 3 стадии синтеза, как показано на схеме 8 для синтеза соединений 28 и 83. 3β-Гидроксильное соединение 46 превращают в 3β-мезилат 81, используя метансульфонилхлорид и пиридин. Нагревание соединения 81 и обработка ацетатом цезия в ДМФ при 100°С приводит к образованию 3α-ацетатного соединения 82. Последовательность превращений для инверсии завершается метанолизом ацетатной группы в соединении 82 с использованием метоксида натрия и получением 3α-гидроксильного соединения 25. Обработка соединения 25 ZnN6·2py, трифенилфосфином и DIAD в толуоле приводит к получению 3β -азидосоединения 26. Восстановление азида алюмогидридом лития в Et2O приводит к образованию 3β-аминосоединения 27. Обработка с использованием HCl в ТГФ и воды приводит к удалению ацетонидной группы и образованию аммонийнохлоридной соли 28. Подобным образом в результате обработки соединения 27 80%-ной уксусной кислотой, удаляют ацетонидную группу и получают аммонийноацетатную соль 83. При использовании методов, показанных на схеме 8, соединение 50 превращают в соединение 89, а соединение 61 превращают в соединение 95 (см. таблицу 1). Соединения 26, 27, 87, 88, 89, 93, 94 и 95 представляют собой примеры соединений настоящего изобретения и имеющих 3β стереохимию.
Схема 8
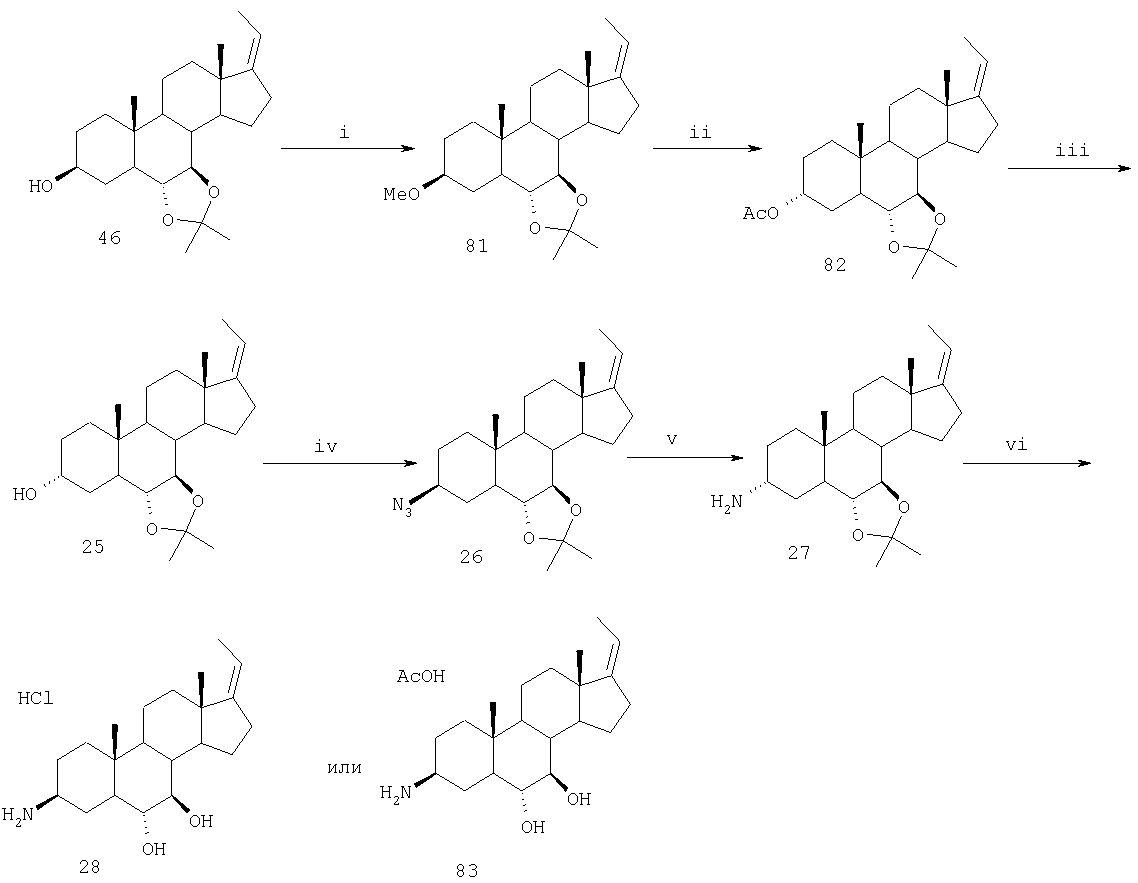
i) MsCl, пиридин; ii) CsOAc, ДМФ, 100°C; iii) NaOMe, MeOH; iv) ZnN6·2py, Ph3P, DIAD, толуол; v) LiAlH4, Et2O; vi) 4 М HCl в диоксане, ТГФ, вода или 80%-ная уксусная кислота.
Синтез соединения 81
Метансульфонилхлорид (1,2 мл, 16 ммоль) добавляют к охлажденному льдом раствору 3β-гидроксильного соединения 46 (3,0 г, 8,0 ммоль) в пиридине (20 мл) в атмосфере аргона. Через 4 часа раствор охлаждают льдом и добавляют 20 мл насыщенного раствора NaHCO3. Через 15 минут раствор разбавляют 150 мл EtOAc и промывают 3×25 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 3,6 г (100%) соединения 81 в виде не совсем белой пены.
Синтез соединения 82
Раствор, содержащий мезилат 81 (3,6 г, 8,0 ммоль), ацетат цезия (4,6 г, 24 ммоль) и 40 мл ДМФ, нагревают при 100°С в течение 24 часов. Раствор разбавляют 100 мл воды, экстрагируют 2×100 мл Et2O, промывают 2×50 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая приблизительно 3 г неочищенного соединения 82.
Синтез соединения 25
Раствор, содержащий Na (398 мг, 17,3 ммоль) в МеОН (21,5 мл), добавляют к 3α-ацетату 82 (1,8 г, 4,3 ммоль) в ТГФ (10 мл). Через 2 часа добавляют 20 мл воды и полученный раствор разбавляют 100 мл EtOAc, последовательно промывают насыщенным раствором NaHCO3, водой и насыщенным раствором хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 1,58 г (98%) неочищенного соединения 25 в виде желтого вспененного вещества.
Синтез соединения 26
DIAD (1,70 мл, 8,24 ммоль) добавляют по каплям в течение 10 минут к находящемуся при комнатной температуре раствору 3α-спирта 25 (1,54 г, 4,12 ммоль), ZnN6·2py (0,94 г, 3,99 ммоль), Ph3Р (2,16 г, 8,24 ммоль) и толуола (44 мл) в атмосфере аргона. После перемешивания в течение ночи реакционную смесь загружают на колонку, заполненную силикагелем в смеси 10% EtOAc/гексан, и элюируют смесью 10% EtOAc/гексан с получением 0,89 г (61%) соединения 26 в виде белого твердого вещества.
Синтез соединения 27
Алюмогидрид лития (146 мг, 3,66 ммоль) добавляют к охлажденному льдом раствору азида 26 (1,46 г, 3,66 ммоль) в 18,3 мл Et2O в атмосфере аргона. Реакционной смеси дают нагреться до комнатной температуры. Через 1,5 час раствор охлаждают на льду, разбавляют 25 мл Et2O и медленно гасят добавлением 20 мл насыщенного раствора Na2SO4. Через 10 минут образуется белый осадок, раствор разбавляют 50 мл EtOAc, промывают 3×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 1,31 г (96%) соединения 27 в виде белой пены.
Синтез соединения 28
Раствор, содержащий 3β-аминосоединение 27 (227 мг, 0,609 ммоль), 4 М HCl в диоксане (183 мкл, 0,73 ммоль), ТГФ (9,7 мл) и воду (2,4 мл), перемешивают при комнатной температуре в течение ночи. После упаривания ТГФ и воды остаток помещают в 5 мл метанола, затем концентрируют, растирают с 5 мл ацетона, в результате чего получают 224 мг (100%) соединения 28 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 334,10; С21Н36NO2.
Синтез соединения 83
Раствор, содержащий амин 27 (412 мг, 1,10 ммоль) и 5 мл 80%-ной уксусной кислоты, перемешивают при комнатной температуре в течение 4 часов. Реакционную смесь разбавляют 5 мл толуола, после чего концентрируют.Остаток во второй раз растворяют в 5 мл толуола и концентрируют, для того чтобы удалить остаточную уксусную кислоту. Полученный остаток дважды растирают с 10 мл СН2Cl2, после чего концентрируют, получая 430 мг (99%) соединения 83 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 334,19; С21Н36NO2 .
Синтез соединения 84
Метансульфонилхлорид (0,33 мл, 4,2 ммоль) добавляют к охлаждаемому льдом раствору 3β-гидроксильного соединения 50 (754 мг, 2,09 ммоль) в пиридине (5,3 мл) в атмосфере аргона. Через 4 часа раствор охлаждают на льду и добавляют 5 мл насыщенного раствора NaHCO3. Через 15 минут раствор разбавляют 60 мл этилацетата и промывают 3 раза насыщенным раствором хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 860 мг (94%) соединения 84 в виде не совсем белого твердого вещества.
Синтез соединения 85
Раствор, содержащий мезилат 84 (860 мг, 1,96 ммоль), ацетат цезия (1,13 г, 5,88 ммоль) и 10 мл ДМФ, нагревают при 95°С в течение 32 часов. Раствор разбавляют 50 мл воды, экстрагируют 2×100 мл Et2O, промывают 2×30 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки колоночной хроматографией с элюированием смесью 5%, а затем 8% EtOAc/гексан, получают 558 мг (71%) соединения 85 в виде белого твердого вещества.
Синтез соединения 86
Раствор, содержащий Na (128 мг, 5,56 ммоль) в МеОН (7 мл), добавляют к 3α-ацетату 85 (558 мг, 1,38 ммоль). Через 2 часа добавляют 5 мл насыщенного раствора NaHCO3 и полученный раствор разбавляют 100 мл EtOAc. Раствор промывают водой (2×20 мл) и насыщенным раствором хлорида натрия (2×20 мл), сушат над MgSO4, фильтруют и концентрируют, получая 491 мг (99%) соединения 86 в виде белого твердого вещества.
Синтез соединения 87
DIAD (0,57 мл, 2,74 ммоль) добавляют по каплям в течение 15 минут к находящемуся при комнатной температуре раствору 3α-гидроксисоединения 86 (493 мг, 1,37 ммоль), ZnN6·2py (315 мг, 1,03 ммоль), Ph3Р (718 мг, 2,74 ммоль) и толуола (13,7 мл) в атмосфере аргона. Через 3,5 часа реакционную смесь загружают в колонку, заполненную силикагелем в смеси 10% EtOAc/гексан, и элюируют смесью 10% EtOAc/гексан с получением 390 мг (74%) соединения 87 в виде вязкого масла.
Синтез соединения 88
Алюмогидрид лития (40 мг, 1,01 ммоль) добавляют к охлаждаемому льдом раствору азидосоединения 87 (390 мг, 1,01 ммоль) в 5 мл диэтилового эфира в атмосфере аргона. Реакционной смеси дают нагреться до комнатной температуры. Через 2 часа раствор охлаждают на льду, разбавляют 25 мл диэтилового эфира и медленно гасят добавлением 2 мл насыщенного раствора Na2SO4. Через 10 минут образуется белый осадок, после чего раствор разбавляют 40 мл этилацетата, промывают 3×15 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество очищают колоночной хроматографией, используя колонку с силикагелем, предварительно заполненную в смеси 1% Et3 N/CH2Cl2 и промытую смесью 5% MeOH/CH2Cl2. Неочищенное вещество загружают в CH2Cl2, элюируют смесью 5% MeOH/CH2Cl2 и затем смесью CH2Cl2:МеОН:Et3N в соотношении 95:5:2, получая 277 мг (76%) соединения 88 в виде белого твердого вещества.
Синтез соединения 89
Раствор, содержащий аминосоединение 88 (270 мг, 0,752 ммоль) и 10 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 1 часа. Реакционную смесь концентрируют, получая белую пену. Добавляют еще 10 мл ацетона, подвергают воздействию ультразвука и упаривают с получением 285 г (100%) соединения 89 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 320,26; С20Н34NO2.
Синтез соединения 90
Метансульфонилхлорид (0,20 мл, 2,56 ммоль) добавляют к охлаждаемому льдом раствору 3β-гидроксильного соединения 61 (501 мг, 1,28 ммоль) в пиридине (3,2 мл) в атмосфере аргона. Через 4 часа раствор охлаждают на льду и добавляют 5 мл насыщенного раствора NaHCO3. Через 15 минут раствор разбавляют 50 мл EtOAc и промывают 3 раза насыщенным раствором хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 590 мг (98%) соединения 90 в виде белой пены.
Синтез соединения 91
Раствор, содержащий мезилат 90 (590 мг, 1,25 ммоль), ацетат цезия (722 мг, 3,76 ммоль) и 6,2 мл ДМФ, нагревают при 100°С в течение 24 часов. Раствор разбавляют 50 мл воды, экстрагируют с использованием 2×50 мл Et2O, промывают 2×30 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки колоночной хроматографией с элюированием смесью 8% EtOAc/гексан, получают 297 мг (55%) соединения 91 в виде белого твердого вещества.
Синтез соединения 92
Раствор, содержащий Na (63 мг, 2,7 ммоль) в МеОН (3,4 мл), добавляют к 3α-ацетату 91 (297 мг, 0,684 ммоль) в ТГФ (1 мл). Раствор перемешивают в течение ночи. Добавляют 5 мл воды и 80 мл EtOAc, промывают дважды водой и дважды насыщенным водным раствором хлорида натрия, сушат над MgSO4, фильтруют и концентрируют, получая 251 мг (94%) соединения 92 в виде белого твердого вещества.
Синтез соединения 93
DIAD (0,26 мл, 1, 24 ммоль) добавляют по каплям в течение 10 минут к находящемуся при комнатной температуре раствору, содержащему 3α-спирт 92 (243 мг, 0,620 ммоль), ZnN6·2py (143 мг, 0,465 ммоль), Ph3Р (325 мг, 1,24 ммоль) и толуол (6,2 мл), в атмосфере аргона. Через 4 часа реакционную смесь загружают в колонку, заполненную силикагелем в смеси 10% EtOAc/гексан, и элюируют смесью 20% EtOAc/гексан с получением 209 мг неочищенного соединения 93 в виде желтого масла.
Синтез соединения 94
Алюмогидрид лития (20 мг, 0,50 ммоль) добавляют к охлаждаемому льдом раствору неочищенного азида 93 (209 мг, 0,50 ммоль) в 5 мл Et2O в атмосфере аргона. Реакционной смеси дают нагреться до комнатной температуры. Через 4 часа раствор охлаждают на льду, разбавляют 25 мл диэтилового эфира (Et2O) и медленно гасят добавлением 2 мл насыщенного раствора Na2SO4. Через 10 минут образуется белый осадок, после чего раствор разбавляют 50 мл этилацетата (EtOAc), промывают 3×10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество хроматографируют, используя колонку с силикагелем, предварительно заполненную смесью 1% Et3N/CH2Cl2 и промытую смесью 5% MeOH/CH2Cl2. Неочищенное вещество загружают в CH2Cl2, элюируют смесью 5% MeOH/CH2Cl2 и затем смесью CH2Cl2:МеОН:Et3N в соотношении 95:5:2, получая 97 мг неочищенного соединения 94 в виде белого твердого вещества.
Синтез соединения 95
Раствор, содержащий 3β-аминосоединение 94 (97 мг, 0,25 ммоль), 4 М HCl в диоксане (74 мкл, 0,30 ммоль), ТГФ (4 мл) и воду (1 мл), перемешивают при комнатной температуре. Через 4 часа раствор концентрируют, остаток помещают в 5 мл метанола и концентрируют. Полученный остаток дважды растирают с 5 мл ацетона и концентрируют. Белое твердое вещество растворяют в приблизительно 0,5 мл воды и медленно добавляют ацетон (5 мл), до тех пор, пока не появятся кристаллы. Кристаллы отфильтровывают, промывают ацетоном и сушат, получая 66 мг соединения 95 в виде бесцветных тонких игл. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 352,09; С21Н35FNO2.
Пример 10
3-Амино-6,7-дигидрокси-17-алкилстероид
Любые соединения, содержащие 17(20)-алкенильную функциональную группу, могут содержать двойную связь, которая подвергается гидрированию с использованием Н2 в присутствии катализатора, такого как 10% Pd на углероде. Например, соединение 96 получают из соединения 28, как показано на схеме 9. Аналогичным образом, соединение 97 получают из соединения 49, используя ту же самую методику, которая показана на схеме 9 (см. таблицу 2).
Схема 9

i) Н2, Pd на углероде, метанол.
Синтез соединения 96
Раствор, содержащий олефин 28 (52 мг, 0,14 ммоль), 10% Pd на углероде (15 мг, 0,014 ммоль) и метанол (3 мл), перемешивают при комнатной температуре в течение ночи. Раствор фильтруют через целит, элюируя 50 мл метанола, и концентрируют, получая 50 мг (96%) соединения 96 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 336,24; С21Н38NO2.
Синтез соединения 97
Раствор, содержащий олефин 49 (844 мг, 2,28 ммоль), 10% Pd на углероде (243 мг, 0,228 ммоль) и метанол (11 мл), перемешивают при комнатной температуре в течение ночи. Раствор фильтруют через целит, элюируя 50 мл метанола, затем концентрируют. Остаток растирают с 10 мл ацетона, фильтруют и сушат, получая 801 мг (94%) соединения 97 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 336,21; С21Н38NO2.
Пример 11
3-втор-Амино-6, 7-дигидрокси-11-метилиденстероид
Любые амины, родственные соединению 52, могут быть подвергнуты конденсации с альдегидом или кетоном для того, чтобы получить вторичные или третичные амины. Взаимодействие соединения 52 с раствором 4-изопропилбензальдегида и изопропоксидом титана в ТГФ с последующим восстановлением борогидридом натрия приводит к получению соединения 99. Обработка 80%-ной уксусной кислотой приводит к удалению ацетонидных групп и образованию аммонийноацетатной соли соединения 100. Соединения примеров 101-107 были синтезированы с использованием методов, показанных на схеме 10 (см. таблицу 6).
Схема 10
i) 4-изопропилбензальдегид, Ti(OiPr)4, ТГФ; NaBH4, MeOH; ii) 80%-ная уксусная кислота.
Синтез соединения 99
Изопропоксид титана (IV) (120 мкл, 0,42 ммоль) добавляют к находящемуся при комнатной температуре раствору, содержащему амин 52 (100 мг, 0,28 ммоль), 4-изопропилбензальдегид (46 мкл, 0,31 ммоль) и 1,4 мл ТГФ в атмосфере азота. Через 12 часов добавляют раствор NaBH4 (29 мг, 0,78 ммоль) в 1 мл EtOH и взаимодействие проводят в течение еще 8 час. Реакционную смесь гасят добавлением 3 мл насыщенного раствора хлорида натрия, разбавляют 30 мл EtOAc, разделяют, промывают насыщенным раствором хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки радиальной хроматографией получают 50 мг (36%) соединения 99.
Синтез соединения 100
Раствор, содержащий амин 99 (50 мг, 0,10 ммоль) и 1 мл 80%-ной уксусной кислоты, перемешивают при 40°С в течение 3 часов. Реакционную смесь дважды обрабатывают порциями толуола по 5 мл, после чего концентрируют, а затем по одному разу обрабатывают ацетоном и гексаном, получая 25 мг (51%) соединения 100. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 452,27; С30Н46NO2.
Синтез соединения 101
Используя методику, описанную для синтеза соединения 99, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с 2-фторбензальдегидом (32 мкл, 0,32 ммоль), получая 43 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 1 мл 80%-ной уксусной кислоты, при 40°С в течение 3 часов. Реакционную смесь разбавляют 5 мл толуола и концентрируют. Полученный остаток растворяют в 1 мл ацетона, разбавляют 5 мл гексана и концентрируют, получая 49 мг (37%) соединения 101 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 428,22; С27Н39FNO2.
Синтез соединения 102
Используя методику, описанную для синтеза соединения 99, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с 3-(трифторметил)бензальдегидом (41 мкл, 0,31 ммоль), получая 61 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 1 мл 80%-ной уксусной кислоты, при 40°С в течение 3 часов. Реакционную смесь разбавляют 5 мл толуола и концентрируют. Полученный остаток растворяют в 1 мл ацетона, разбавляют 5 мл гексана и концентрируют, получая 64 мг (45%) соединения 102 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 478,18; С28Н39F3NO2.
Синтез соединения 103
Используя методику, описанную для синтеза соединения 99, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с о-анисовым альдегидом (42 мг, 0,31 ммоль), получая 30 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 1 мл 80%-ной уксусной кислоты, при 40°С в течение 3 часов. Реакционную смесь разбавляют 5 мл толуола и концентрируют. Полученный остаток растворяют в 1 мл ацетона, разбавляют 5 мл гексана и концентрируют, получая 18 мг (14%) соединения 103. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 440,23; С28Н42NO3.
Синтез соединения 104
Используя методику, описанную для синтеза соединения 99, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с 4-(трифторметокси)бензальдегидом (44 мкл, 0,31 ммоль), получая 86 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 1,5 мл 80%-ной уксусной кислоты, при 40°С в течение 3 часов. Реакционную смесь разбавляют 5 мл толуола и концентрируют. Полученный остаток растворяют в 1 мл ацетона, разбавляют 5 мл гексана и концентрируют, получая 84 мг (57%) соединения 104 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 494,15; С28Н39F3NO3.
Синтез соединения 105
Используя методику, описанную для синтеза соединения 99, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с 3-феноксибензальдегидом (60 мг, 0,32 ммоль), получая 73 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают 1 мл 80%-ной уксусной кислоты, при 40°С в течение 3 часов. Реакционную смесь разбавляют 5 мл толуола и концентрируют. Полученный остаток растворяют в 1 мл ацетона, разбавляют 5 мл гексана и концентрируют, получая 87 мг (58%) соединения 105 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 502,20; С33Н44NO3.
Синтез соединения 106
Используя методику, описанную для синтеза соединения 99, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с 3-нитробензальдегидом (46 мг, 0,31 ммоль), получая 18 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 1 мл 80%-ной уксусной кислоты, при 40°С в течение 3 часов. Реакционную смесь разбавляют 5 мл толуола и концентрируют. Полученный остаток растворяют в 1 мл ацетона, разбавляют 5 мл гексана и концентрируют, получая 18 мг (14%) соединения 106 в виде не совсем белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 455,20; С27 Н39N2O4.
Синтез соединения 107
Используя методику, описанную для синтеза соединения 99, амин 52 (200 мг, 0,55 ммоль) подвергают взаимодействию с 3-пиридилкарбоксальдегидом (82 мкл, 0,61 ммоль), получая 100 мг аминного промежуточного соединения. Суспензию, содержащую аминное промежуточное соединение, 4 М HCl в диоксане (65 мкл, 0,26 моль), 110 мкл воды и 2,2 мл ацетонитрила, перемешивают при комнатной температуре в течение 1 часа. Раствор фильтруют и сушат твердое вещество, получая 77 мг (30%) соединения 107 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 411,21; С26Н39N2O2.
Пример 12
3-Циклоамино-6,7-дигидрокси-17-этилиденстероид
Любой кетон, родственный соединению 108, может быть подвергнут конденсации с амином с использованием способов, показанных на схеме 11. Синтез исходного соединения 108 описан в патенте США 6046185. Взаимодействие соединения 108 с пиперидином и цианоборогидридом натрия в метаноле дает соединение 109 в виде смеси изомеров у атома С3. Обработка 80%-ной уксусной кислотой приводит к удалению защитных ацетонидных групп и образованию аммонийноацетатной соли 110. Соединение 111 согласно примеру синтезируют с использованием способов, показанных на схеме 11, за тем исключением, что вместо уксусной кислоты используют хлористоводородную кислоту (см. таблицу 5). 3-Циклоаминная группа является группой, присоединенной в положении 3, где атом углерода в положении 3 присоединен непосредственно к атому азота, а данный атом азота представляет собой часть гетероциклического кольца.
Схема 11

i) пиперидин, Ti(OiPR)4, ТГФ; NaBH4, MeOH; ii) 80%-ная уксусная кислота.
Синтез соединения 109
Раствор, содержащий кетон 108 (200 мг, 0,54 ммоль), пиперидин (266 мкл, 2,68 ммоль), 100 мг молекулярных сит, 3Å, NaBH3CN (24 мг, 0,38 ммоль) и 5,4 мл МеОН, перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь разбавляют 20 мл воды и экстрагируют 2×20 мл CH2Cl2. Объединенные экстракты промывают 10 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. Неочищенное вещество очищают радиальной хроматографией, элюируя 20% МеОН/CH2Cl2 и получая 112 мг (47%) соединения формулы 109 в виде белого твердого вещества.
Синтез соединения 110
Раствор, содержащий амины 109 (102 мг, 0,23 ммоль) и 5 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 1 часа. Раствор концентрируют, остаток помещают в 2 мл MeOH, разбавляют 15 мл толуола и концентрируют. Остаток растирают с 5 мл ацетона, фильтруют и сушат с получением 44 мг (42%) соединения 110 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 402,31; С26Н44NO2.
Синтез соединения 111
Используя методику, описанную для синтеза соединения 109, кетон 108 (200 мг, 0,54 ммоль) подвергают взаимодействию с морфолином (234 мкл, 2,68 ммоль), получая 56 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 5 мл 80%-ной уксусной кислоты, при 40°С в течение 1 часа. Раствор концентрируют, разбавляют 5 мл MeOH и концентрируют. С помощью1H и13C ЯМР-исследования обнаруживают, что ацетонидная группа была удалена, но соли образовалось немного либо вообще не образовалось. Вещество обрабатывают 4 М раствором HCl в диоксане (32 мкл, 0,13 ммоль) и 2 мл ацетона с получением белого осадка. Суспензию разбавляют 2 мл ацетона, фильтруют и сушат с получением 48 мг (20%) соединения 111 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 404,20; С25Н42NO3.
Пример 13
Превращение 3-оксостероида в 3-втор-аминостероид
Любые кетоны, родственные соединению 108, могут быть подвергнуты конденсации с амином с использованием методологии, показанной на схеме 12. Метиламин добавляют к раствору, содержащему соединение 108 и изопропоксид титана в ТГФ, с последующим восстановлением борогидридом натрия. Раствор фильтруют и элюируют через MP-TsOH-смолу, получая соединение 112 в виде смеси изомеров у С3. Обработка с использованием HCl в ацетонитриле и воды приводит к образованию аммонийнохлоридной соли 113. Соединения 114-129 согласно примеру были синтезированы с использованием методик, показанных на схеме 12, за тем исключением, что вместо хлористоводородной кислоты используют уксусную кислоту в случае примеров, в соответствии с которыми получают аммонийноацетатные соли (см. таблицу 5).
Схема 12

i) гидрохлорид метиламина, Ti(OiPR)4, ТГФ; NaBH4, MeOH; ii) HCl, вода, ацетонитрил.
Синтез соединения 112
Изопропоксид титана (IV) (270 мкл, 0,92 ммоль) добавляют в атмосфере азота к находящемуся при комнатной температуре раствору, содержащему кетон 108 (250 мг, 0,67 ммоль), гидрохлорид метиламина (41 мг, 0,61 ммоль) и 1,5 мл ТГФ. Через 12 часов добавляют раствор NaBH4 (65 мг, 1,7 ммоль) в 2,3 мл EtOH и взаимодействие проводят в течение еще 10 час. Реакционную смесь гасят добавлением 0,5 мл воды и отфильтровывают для того, чтобы удалить белый осадок. Раствор загружают на колонку, содержащую 600 мг MP-TsOH-смолы и элюируют 3 мл МеОН, затем 4 мл 2 М NH3 в МеОН. Фракцию в NH3 /МеОН концентрируют, получая 76 мг (32%) соединения 112.
Синтез соединения 113
Суспензию, содержащую соединение 112 (76 мг, 0,21 ммоль), 4 М HCl в диоксане (75 мкл, 0, 30 моль), 50 мкл воды и 1 мл ацетонитрила, перемешивают при комнатной температуре в течение 1 часа. Раствор фильтруют и сушат твердое вещество, получая 38 мг (13%) соединения 113 в виде серого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 348,19; С22Н38NO2.
Синтез соединения 114
Используя методику, описанную для синтеза соединения 112, кетон 108 (200 мг, 0,53 ммоль) подвергают взаимодействию с гидрохлоридом пропиламина (47 мг, 0,49 ммоль), получая 72 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, получая 34 мг (17%) соединения 114. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 376,22; С24Н42NO2.
Синтез соединения 115
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с амиламином (70 мкл, 0,61 ммоль), получая 82 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, получая 75 мг (28%) соединения 115 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 404,28; С26Н46NO2.
Синтез соединения 116
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с циклопентиламином (60 мкл, 0,61 ммоль), получая 99 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 200 мкл уксусной кислоты в течение 1 часа, и дважды разбавляют, а затем концентрируют из порций толуола объемом 1 мл. Остаток растирают с 1 мл циклогексана, фильтруют и сушат с получением 98 мг (35%) соединения 116. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 402,27; С26Н44NO2.
Синтез соединения 117
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с циклогексиламином (70 мкл, 0,61 ммоль), получая 120 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 0,5 мл уксусной кислоты в течение 1 часа, и дважды разбавляют, а затем концентрируют из порций толуола объемом 1 мл. Полученный остаток растирают с 1 мл циклогексана, фильтруют и сушат с получением 101 мг (32%) соединения 117. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 416,25; С27Н46NO2.
Синтез соединения 118
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с пирролидином (51 мкл, 0,61 ммоль), получая 70 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 50 мл уксусной кислоты в течение 1 часа, и добавляют 1 мл циклогексана, получая твердое вещество, которое фильтруют и сушат с получением 65 мг (22%) соединения 118. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 388,28; С25Н42NO2 .
Синтез соединения 119
Используя методику, описанную для синтеза соединения 112, кетон 108 (200 мг, 0,53 ммоль) подвергают взаимодействию с N-пропилэтилендиамином (60 мкл, 0,49 ммоль), получая 99 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, получая 47 мг (20%) соединения 119. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 419,32; С26Н47N2O2.
Синтез соединения 120
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с N,N-диметилэтилендиамином (65 мкл, 0,61 ммоль), получая 93 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, получая 77 мг (29%) соединения 120 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 405,28; С25Н45N2O2.
Синтез соединения 121
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с пиперазином (52 мг, 0,61 ммоль), получая 33 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 200 мкл уксусной кислоты в течение 1 часа, и дважды разбавляют, а затем концентрируют из порций толуола объемом 1 мл. Полученный остаток растирают с 1 мл циклогексана, фильтруют и сушат с получением 39 мг (14%) соединения 121. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 403,23; С25Н43N2O2.
Синтез соединения 122
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с этаноламином (33 мкл, 0,61 ммоль), получая 136 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора в HCl, получая 124 мг (50%) соединения 122 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 378,19; С23Н40NO3.
Синтез соединения 123
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с 5-амино-1-пентанолом (63 мг, 0,61 ммоль), получая 129 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора в HCl, получая 65 мг (24%) соединения 123 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 420, 25; С26Н46NO3.
Синтез соединения 124
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с 2-(2-аминоэтиламино)этанолом (62 мкл, 0,61 ммоль), получая 90 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, получая 79 мг (28%) соединения 124 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 421,24; С25Н45N2O3.
Синтез соединения 125
Используя методику, описанную для синтеза соединения 112, кетон 108 (200 мг, 0,53 ммоль) подвергают взаимодействию с м-толуидином (52 мкл, 0,49 ммоль), получая 95 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, с получением 43 мг (19%) соединения 125. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 424,23; С28Н42NO2 .
Синтез соединения 126
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с 4-аминофенолом (67 мг, 0, 61 ммоль), получая 138 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, с получением 41 мг (14%) соединения 126. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 426,18; С27Н40NO3.
Синтез соединения 127
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с сульфаниламидом (105 мг, 0,61 ммоль), получая, после очистки радиальной хроматографией, 24 мг аминного промежуточного соединения. Аминное промежуточное соединение обрабатывают, используя 75 мкл раствора HCl, с получением 23 мг (7%) соединения 127. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 489,17; С27Н41N2O4S.
Синтез соединения 128
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с 3-аминометилпиридином (62 мкл, 0,61 ммоль), получая 108 мг аминного промежуточного соединения. Аминное промежуточное соединение подвергают взаимодействию с 1 мл 80%-ной уксусной кислоты при 40°С в течение 1 часа. Реакционную смесь концентрируют и дважды разбавляют, а затем концентрируют из 1 мл толуола. Полученный остаток растирают с 1 мл циклогексана, фильтруют и сушат с получением 117 мг (41%) соединения 128. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 425,24; С27Н41N2O2.
Синтез соединения 129
Используя методику, описанную для синтеза соединения 112, кетон 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с гистамином (68 мг, 0,61 ммоль), получая 120 мг аминного промежуточного соединения. Аминное промежуточное соединение подвергают взаимодействию с 1 мл 80%-ной уксусной кислоты при 40°С в течение 1 часа. Реакционную смесь концентрируют и дважды разбавляют, а затем концентрируют из 1 мл толуола. Полученный остаток дважды растирают с 1 мл циклогексана, фильтруют и сушат с получением 128 мг (38%) соединения 129. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 428,23; С26Н42N3O2.
Пример 14
Превращение 3-аминостероида в 3-ациламиностероид
Амидные и сульфонамидные аналоги могут быть получены из какого-либо амина, родственного соединению 52. На схеме 13 показан синтез амида 131. Ацетилирование амина 52 в CH2Cl2, c использованием хлорангидрида уксусной кислоты и диэтиламина, связанного со смолой, приводит к получению амида 130. Обработка 80%-ной уксусной кислотой приводит к удалению ацетонидных групп с получением дигидроксиамида 131.
Схема 13

i) хлорангидрид уксусной кислоты, PS-DIEA, CH2Cl2; PS-Trisamine; ii) 80%-ная уксусная кислота.
Синтез соединения 130
Раствор, содержащий амин 52 (100 мг, 0,28 ммоль), хлорангидрид уксусной кислоты (50 мкл, 0,70 ммоль), 440 мг смолы PS-DIEA и 2,4 мл CH2Cl2, перемешивают при комнатной температуре в течение 16 часов. Смолу отфильтровывают и фильтрат инкубируют в течение 2 часов с 260 мг смолы PS-Trisamine. Смолу отфильтровывают и фильтрат концентрируют. После очистки радиальной хроматографией получают 69 мг (62%) соединения 130.
Синтез соединения 131
Раствор, содержащий амид 130 (69 мг, 0,17 ммоль) и 1 мл 80% уксусной кислоты, нагревают при 40°С в течение 1 часа. Реакционную смесь дважды разбавляют и концентрируют из 5 мл толуола, затем один раз из 5 мл МеОН и один раз из 1 мл ацетона и 5 мл гексана с получением 62 мг (62%) соединения 131 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 384,16; С22Н35NNaO3, 362,20; С22Н36NO3, 344,18; С22Н34NO2.
Синтез соединения 132
Используя методику, описанную для синтеза соединения 130, амин 52 (88 мг, 0,24 ммоль) подвергают взаимодействию с бензоилхлоридом (65 мкл, 0,56 ммоль), получая 64 мг амидного промежуточного соединения. Раствор, содержащий амидное промежуточное соединение и 2 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 1 часа. Реакционную смесь дважды разбавляют и концентрируют из 5 мл толуола, затем один раз из 5 мл МеОН и один раз из 1 мл ацетона и 5 мл гексана с получением 55 мг (55%) соединения 132 в виде белого твердого вещества (см. таблицу 3). ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 446,18; С27Н37NNaO3, 424,29; С27Н38NO3, 406,19; С27Н36NO2.
Синтез соединения 133
Используя методику, описанную для синтеза соединения 130, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с изопропилсульфонилхлоридом (63 мкл, 0,56 ммоль), получая 38 мг сульфонамидного промежуточного соединения. Раствор, содержащий сульфонамидное промежуточное соединение и 1,5 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 1 часа. Реакционную смесь дважды разбавляют и концентрируют из 5 мл толуола, затем один раз из 5 мл МеОН и один раз из 1 мл ацетона и 5 мл гексана с получением 35 мг (29%) соединения 133 в виде не совсем белого твердого вещества (см. таблицу 3). ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 426,14; С23Н40NO4S.
Синтез соединения 134
Используя методику, описанную для синтеза соединения 130, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с бензолсульфонилхлоридом (90 мкл, 0,70 ммоль), получая 105 мг сульфонамидного промежуточного соединения. Раствор, содержащий сульфонамидное промежуточное соединение и 2 мл 80%-ной уксусной кислоты, нагревают при 40°С в течение 5 часов. Реакционную смесь дважды разбавляют и концентрируют из 5 мл толуола, затем один раз из 5 мл МеОН и один раз из 1 мл ацетона и 5 мл гексана с получением 83 мг (65%) соединения 134 в виде белого твердого вещества (см. таблицу 3). ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 482,11; С26Н37NNaO4S, 477,17; С26Н41N2O4S, 460,15; С26Н38NO4S.
Пример 15
3-Ациламинобиотин-6,7-гидрокси-17-этилиденстероид
На схеме 14 показан синтез амида 135. Взаимодействие амина 83 с триметиламином и водорастворимой формой сложного биотинового эфира N-гидроксисукцинимида в метаноле и воде приводит к получению биотинилированного амидного аналога 135.
Схема 14

i) сульфо-NHS-биотин, Et3N, MeOH, вода.
Синтез соединения 135
Раствор, содержащий соединение 83 (97 мг, 0,25 ммоль), Et3N (104 мкл, 0,75 ммоль), сульфо-NHS-биотин (120 мг, 0,27 ммоль), 2,5 мл MeOH и 2,5 мл воды, перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют и очищают, используя колоночную хроматографию с обращенной фазой и элюируя смесью 5% воды/МеОН, с получением 89 мг (64%) соединения 135 в виде не совсем белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 560,30; С31Н50N3O4S.
Пример 16
3-Мочевина-6,7-гидрокси-17-метилиденстероид
Любые амины, родственные соединению 52, могут быть подвергнуты взаимодействию с изоцианатами или изотиоцианатами с получением соединений, содержащих мочевинные или тиомочевинные функциональные группы. Соединения 136, 137 и 138 представляют собой примеры мочевин, которые были синтезированы с использованием способов, показанных на схеме 15 (см. таблицу 3).
Схема 15

i) фенилизоцианат, PS-Trisamine, CH2Cl2; ii) 80%-ная уксусная кислота.
Синтез соединения 136
Раствор, содержащий амин 52 (100 мг, 0,28 ммоль), фенилизоцианат (76 мкл, 0,70 ммоль) и 2,4 мл CH2Cl2, перемешивают при комнатной температуре в течение 16 часов. Раствор инкубируют в течение 2 часов с 260 мг смолы PS-Trisamine. Смолу отфильтровывают и фильтрат концентрируют. После очистки радиальной хроматографией получают 95 мг (71%) соединения 136.
Синтез соединения 137
Раствор, содержащий мочевину 136 (95 мг, 0,20 ммоль) и 2 мл 80% уксусной кислоты, нагревают при 80 С в течение 2 часов. Реакционную смесь разбавляют и концентрируют из 5 мл толуола, затем из 5 мл МеОН и после этого из 5 мл гексана. После очистки радиальной хроматографией с элюированием смесью CH2Cl2:МеОН:Et3N в соотношении 95:5:2 получают 40 мг (33%) соединения 137. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 461,18; С27Н38N2NaO3, 439,22; С27Н39 N2O3, 421,25; С27Н37N2O2.
Синтез соединения 138
Используя методику, описанную для синтеза соединения 137, амин 52 (100 мг, 0,28 ммоль) подвергают взаимодействию с пропилизоцианатом (52 мкл, 0,56 ммоль), получая 72 мг мочевинного промежуточного соединения. Раствор, содержащий мочевинное промежуточное соединение и 2 мл 80%-ной уксусной кислоты, нагревают при 80°С в течение 2 часа. Реакционную смесь дважды разбавляют и концентрируют из 5 мл толуола, затем один раз из 5 мл МеОН и один раз из 5 мл гексана с получением 51 мг (45%) соединения 138 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 427, 21; С24Н40N2NaO3, 405,25; С24Н41N2O3.
Пример 17
3-Мочевина-6, 7-гидрокси-17-диметилненасыщенный стероид
Любые соединения, родственные соединениям 88 или 89, могут претерпевать перегруппировку с использованием методики, приведенной на схеме 16. Обработка соединения 88 нагретым до 50°С раствором хлористоводородной кислоты в метаноле и воды приводит к удалению ацетонидных защитных групп, способствуя миграции 18-метильной группы к атому С17 и образованию аммонийнохлоридной соли 139. Обработка соединения 89 в тех же самых условиях приводит к соединению 139. Соединения в соответствии с примерами 140-148 были синтезированы способом, приведенным на схеме 16 (см. таблицу 4).
Схема 16

i) HCl, вода, метанол, 50°C.
Синтез соединения 139
Раствор, содержащий соединение 88 (300 мг, 0,834 ммоль), 4 капли концентрированной HCl, 2 мл метанола и 2 мл воды, нагревают при 50°С в течение 72 часов. Реакционную смесь концентрируют и остаток дважды разбавляют и концентрируют из 5 мл метанола. Остаток обрабатывают, используя 2 мл метанола, разбавляют 15 мл ацетона и концентрируют. Полученный остаток растирают с 5 мл ацетона, фильтруют и сушат, получая 286 мг (96%) соединения 139 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 320,20; С20Н34NO2.
Синтез соединения 139
Используя методику, описанную для синтеза соединения 139 из соединения 88, подвергают взаимодействию соединение 89, получая 145 мг (77%) соединения 139 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 320,20; С20Н34NO2.
Синтез соединения 140
Используя методику, описанную для синтеза соединения 99, амин 88 (200 мг, 0,55 ммоль) подвергают взаимодействию с м-толуиловым альдегидом (90 мкл, 0,61 ммоль). После очистки радиальной хроматографией с элюированием смесью 5% МеОН/EtOAc получают 127 мг аминного промежуточного соединения. Раствор, содержащий аминное промежуточное соединение, 4 капли концентрированной HCl, 1 мл MeOH и 1 мл воды, нагревают при 50°С в течение 20 часов. Реакционную смесь трижды разбавляют, а затем концентрируют из 5 мл MeOH и один раз из 5 мл ацетона, получая 74 мг (30%) соединения 140 в виде не совсем белой пены. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 424,24; С28Н42NO2.
Синтез соединения 141
Используя методику, описанную для синтеза соединения 99, амин 88 (200 мг, 0,55 ммоль) подвергают взаимодействию с 3,4-дифторбензальдегидом (67 мкл, 0,61 ммоль). После очистки с использованием радиальной хроматографии, элюируя смесью 30% EtOAc/гексан получают 88 мг аминного промежуточного соединения. Раствор, содержащий аминное промежуточное соединение, 4 капли концентрированной HCl, 1 мл MeOH и 1 мл воды, нагревают при 50°С в течение 20 часов. Реакционную смесь трижды разбавляют, а затем концентрируют из 5 мл MeOH и один раз из 5 мл ацетона, получая 73 мг (28%) соединения 141 в виде не совсем белой пены. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 466,42; С27 Н38F2NO2.
Синтез соединения 142
Используя методику, описанную для синтеза соединения 59, амин 49 (200 мг, 0,55 ммоль) подвергают взаимодействию с 3,4-диметоксибензальдегидом (70 мкл, 0,61 ммоль). После очистки с использованием радиальной хроматографии, элюируя смесью 40% EtOAc/гексан, получают 67 мг аминного промежуточного соединения. Раствор, содержащий промежуточный амин, 4 капли концентрированной HCl, 1 мл MeOH и 1 мл воды, нагревают при 50°С в течение 20 часов. Реакционную смесь концентрируют и остаток трижды разбавляют, а затем концентрируют из 5 мл MeOH и один раз из 5 мл ацетона, получая 43 мг (16%) соединения 142 в виде желтого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 470,27; С29Н44NO4.
Синтез соединения 143
Раствор, содержащий соединение 28 (200 мг, 0,540 ммоль), 4 капли концентрированной HCl и 3 мл воды, нагревают при 50°С в течение 72 часов. Реакционную смесь концентрируют и остаток трижды разбавляют, а затем концентрируют из 5 мл метанола. Полученный остаток обрабатывают 3 мл метанола, разбавляют 20 мл ацетона и концентрируют. Полученный остаток растирают с 10 мл ацетона, фильтруют и сушат, получая 179 мг (90%) соединения 143 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 334,20; С21Н36NO2 .
Синтез соединения 144
Изопропоксид титана (IV) (270 мкл, 0,92 ммоль) добавляют к находящемуся при комнатной температуре раствору, содержащему кетон 108 (250 мг, 0,67 ммоль), анилин (56 мкл, 0,61 ммоль) и 1,5 мл ТГФ в атмосфере аргона. Через 12 часов добавляют раствор NaBH4 (65 мг, 1,7 ммоль) в 2,3 мл EtOH и взаимодействие проводят в течение еще 8 час. Реакционную смесь гасят добавлением 0,5 мл воды и фильтруют для того, чтобы удалить белый осадок. Раствор загружают на колонку, содержащую 600 мг MP-TsOH-смолы и элюируют 9 мл МеОН, затем 9 мл 2 М NH3 в МеОН. Фракцию в NH3/МеОН концентрируют, остаток помещают в 4 мл ТГФ, обрабатывают, используя 500 мг смолы, содержащей PS-бензальдегид, и отфильтровывают, для того, чтобы удалить остаточный анилин. Раствор концентрируют, остаток помещают в 2 мл смеси в соотношении 9:1 ТГФ и воды и 100 мкл концентрированной HCl. После перемешивания при комнатной температуре в течение ночи реакционную смесь концентрируют. Остаток растирают с 1 мл циклогексана, фильтруют и сушат, получая 62 мг соединения 144 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 410,03; С27Н40NO2.
Синтез соединения 145
Используя методику, описанную для синтеза соединения 144, соединение 108 (250 мг, 0,67 ммоль) подвергают взаимодействию с 3-(трифторметил)анилином. Промежуточный продукт очищают с использованием радиальной хроматографии, после чего подвергают взаимодействию со 100 мкл концентрированной HCl в 2 мл смеси ТГФ и воды в соотношении 9:1. После перемешивания при комнатной температуре в течение ночи реакционную смесь концентрируют, полученный остаток растирают с 1 мл циклогексана, фильтруют и сушат, получая 23 мг соединения 145 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 477,94; С28Н39F3NO2.
Синтез соединения 146
Изопропоксид титана (IV) (216 мкл, 0,73 ммоль) добавляют к находящемуся при комнатной температуре раствору, содержащему кетон 108 (200 мг, 0,54 ммоль), бензиламин (53 мкл, 0,49 ммоль) и 1,2 мл ТГФ в атмосфере аргона. Через 12 часов добавляют раствор NaBH4 (52 мг, 1,4 ммоль) в 1,7 мл EtOH и взаимодействие проводят в течение еще 6 час. Реакционную смесь гасят добавлением 1 мл воды и отфильтровывают для того, чтобы удалить белый осадок. Раствор разбавляют 70 мл CH2Cl2, промывают 10 мл воды и 20 мл насыщенного раствора хлорида натрия, сушат над MgSO4, фильтруют и концентрируют. После очистки радиальной хроматографией, элюируя последовательно 20% EtOAc/гексан, EtOAc и затем смесью CH2Cl2/МеОН/Et3N в соотношении 95:5:2, получают 127 мг 3α-аминного промежуточного соединения и 26 мг 3β-аминного промежуточного соединения. Раствор, содержащий 127 мг 3α-аминного промежуточного соединения, 1 мл смеси ТГФ и воды в соотношении 9:1 и 0,1 мл концентрированной HCl, перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, остаток помещают в 5 мл MeOH и концентрируют. Полученный остаток растирают с 1 мл циклогексана, фильтруют и сушат, получая 118 мг (95%) соединения 146 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 424,20; С28Н42NO2.
Синтез соединения 147
Раствор, содержащий 26 мг 3β-аминного производного, синтез которого был описан при синтезе соединения 146, 1 мл смеси ТГФ и воды в соотношении 9:1 и 0,1 мл концентрированной HCl, перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, остаток помещают в 5 мл MeOH и концентрируют. Полученный остаток растирают с 1 мл циклогексана, фильтруют и сушат, получая 26 мг (100%) соединения 147 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 424,21; С28Н42NO2.
Синтез соединения 148
Раствор соединения 78 (63 мг, 0,18 ммоль), 4 капли концентрированной HCl, 1 мл метанола и 1 мл воды нагревают при 50°С в течение 48 часов. Реакционную смесь концентрируют, полученный остаток дважды помещают в 5 мл метанола и концентрируют. Остаток помещают в 2 мл гексана, концентрируют и сушат в течение 2 часов, используя прибор Абдерхальдена для высушивания, с орошением ацетоном, получая 69 мг (100%) соединения 148 в виде белого твердого вещества. ЖХ/МС (прямое введение, электрораспыление +ve, 10 мМ NH4OAc в смеси воды с MeCN в соотношении 4:1): 348,20; С22Н38NO2.











Примеры применения
Пример А
Влияние некоторых соединений на индуцированное аллергеном воспаление легких
Способность соединения ингибировать индуцированную аллергеном аккумуляцию клеток зоны воспаления, таких как эозинофилы и нейтрофилы в промывочной жидкости, полученной от сенсибилизированных животных, является признаком того, что соединение обладает антиастматической активностью. В частности, эта модельная система пригодна для оценки влияния тестируемых соединений для лечения на поздних стадиях воспалительной астматической реакции, когда воспаление легких и бронхостеноз являются явными. Тест проводили следующим образом.
Крыс-самцов линии Brown Norway сенсибилизировали к овальбумину одинарной интраперитонеальной инъекцией 1 мг овальбумина, адсорбированого на 100 мг Al(OH)3 в 1 мл стерильного солевого раствора (крысы - группа контроля с солевым раствором - получали только стерильный солевой раствор) в первый день (день 1) и учитывали сенсибилизацию до дня 21. Тестируемые соединения вводили перорально ежедневно, в течение трех дней до введения провоцирующей пробы (дни 19, 20, 21) и один день после введения провоцирующей пробы (день 22), а третью дозу давали за 2 часа перед введением провоцирующей пробы, четвертую дневную дозу давали через 24 часа после введения провоцирующей пробы (объем = 300 мкл/доза). В качестве провоцирующей пробы для введения крысам использовали 0,5% овальбумин в солевом растворе, генерируемый с использованием распылителя Девилбиса в течение 60 минут в день 21.
Через 48 часов после введения провоцирующей пробы животных умерщвляли с использованием избыточной дозы вводимого перитонеально пентобарбитола, после чего легкие промывали 2×7 мл холодного забуференного фосфатом солевого раствора. Полученную в результате промывки жидкость помещали на лед. Бронхоальвеолярную промывочную жидкость центрифугировали, супернатант удаляли. Осадок ресупендировали в забуференном фосфатом солевом растворе при 4°С. Цитоцентрифугированием готовили препараты и окрашивали их для дифференцировки и подсчета видов клеток.
Данные, показывающие защитное воздействие различных тестируемых соединений на индуцированное аллергеном воспаление легких, приведены в таблицах 7 и 8. Влияние дозы на вызываемую реакцию для некоторых соединений показано в таблице 9. Тестируемые соединения вводили в 300 мкл кукурузного масла (таблицы 7 и 8) или в воде (таблица 9), которые использовали в качестве носителя. Контрольные животные получали 300 мкл кукурузного масла или только воду, т.е. им не давали лекарственного средства. Данные, приведенные в таблицах 7, 8 или 9, представляют процент ингибирования аккумуляции лейкоцитов по отношению к ингибированию у контрольных животных. Отрицательные значения в таблицах 7, 8 или 9 свидетельствуют об обострении эффекта по сравнению с контрольными животными.
Пример В
Влияние соединения 83 на индуцированный раздражающим средством отек уха у мышей
Некоторое количество мышей однозначно идентифицировали нанесением метки на хвост с помощью маркера для нанесения несмываемых меток. Мышам перорально вводили 15 мг/кг тестируемого соединения в 100 мкл 45% β-циклодекстрина в солевом растворе. Мышей непродолжительно подвергали анестезии с использованием 2% галотана и наносили на внутреннюю и внешнюю стороны левого уха мыши 2 мкг форбол-12-миристата-13-ацетата в 25 мкл ацетона. На правое ухо мыши точно таким же образом наносили ацетон, что служило контролем с носителем. С контрольными животными осуществляли ту же процедуру, но без тестируемого соединения. Через 3 часа мышей умерщвляли посредством смещения шейного отдела позвоночника, иссекали из каждого уха стандартную пробу для биопсии и взвешивали с точностью до 1/10-й миллиграмма. Полученные данные анализировали, определяя разность между данными для левого и правого уха и вычисляя % ингибирования отека ((среднее значение Rx/среднее значение с раздражающим средством) × 100)-100.
Соединения настоящего изобретения проявляют защитное действие в отношении индуцированного раздражающим средством отека уха у мышей. Например, соединение 83 ингибирует индуцированный раздражающим средством отек уха у мышей на 38% по сравнению с контрольными животными.
Пример С
Влияние соединений на высвобождение гексозаминидазы из тучных клеток крыс линии (RBL-2H3)
Антиаллергенное воздействие соединения настоящего изобретения оценивали, измеряя индуцированную антигеном секрецию гексозаминидазы из пассивно сенсибилизированной линии тучных клеток крыс. Способность тестируемых соединений ингибировать высвобождение содержимого тучных клеток, например, гистамина и гексозаминидазы является признаком антиаллергенной и/или антигистаминной активности соединения. Гексозаминидаза высвобождается из тучных клеток вместе с гистамином и другими медиаторами при провокационном введении антигена. Тест осуществляют следующим образом.
Клетки линии RBL-2H3 выращивают в культуре и пассивно сенсибилизируют в течение 1 часа при 37°С к динитрофенолу (DNP), используя антитела человека к DNP (IgE). Клетки инкубируют с тестируемым соединением в течение 30 минут при 37°С и стимулируют, используя 0,5 мкг/мл DNP-HSA (антиген) в течение 30 минут. Отбирают аликвоту супернатанта и используют для измерения количества гексозаминидазы, высвобождаемой при провокационном введении антигена. Количество гексозаминидазы, присутствующей в супернатанте, определяют колориметрически посредством контроля за ферментативным метаболизмом п-нитрофенил-N-ацетил-β-D-глюкозаминида (p-NAG) в течение 60 мин при 405 нм. Воздействие каждого из тестируемых соединений определяют как процентное отношение индуцированной антигеном реакции (минус фоновое высвобождение) к реакции в присутствии только ДМСО. Эти значения используют для определения степени ингибирования индуцированного антигеном высвобождения гексозаминидазы из клеток.
Соединения настоящего изобретения проявляют способность ингибировать индуцированное антигеном высвобождение гексозаминидазы в ответ на стимуляцию антигеном. Соединения тестировали в концентрации 0,3, 3, 10 и 30 мкМ и определяли величину IC50. Эти данные приведены в таблицах 1-6. Например, соединение 119 в концентрации 6,1 мкМ ингибирует высвобождение гексозаминидазы как реакции на стимуляцию антигеном на 50%.
Пример D
Метаболическая стабильность некоторых соединений во фракциях S9 человека
Терапевтическая эффективность тестируемых соединений часто может быть непосредственно связана с его метаболической стабильностью in vivo. Большинство известных имеющихся на рынке лекарственных средств подвергается метаболизму под действием группы ферментов, известных как ферменты P450. Фракция S9 печени человека содержит все ферменты Р450, а также цитоплазматические ферменты, которые могут участвовать в метаболизме новых химических веществ. Исследование метаболизма in vitro с использованием фракции S9 человека является общепринятым исследованием метаболической стабильности новых соединений. Тест осуществляют следующим образом.
Реагенты размораживают на льду и смешивают для приготовления эталонной смеси следующим образом: фосфат калия (100 мМ, рН 7), G6P (0,25 мМ), G6PDH (2 Ед/мл), NADPH (1 мМ), UDP (0,25 мМ), APSS (0,25 мМ) и фракция S9 (2 мг/мл). Эталонную смесь распределяют по 498 мкл в каждую пробирку для микроцентрифугирования. Тестируемое соединение распределяют в количестве 2 мкл 2,5 мМ раствора (конечная концентрация 10 мкМ) в центр крышки соответствующей пробирки и крышки осторожно закрывают таким образом, чтобы не допустить смешения. Синхронное смешение тестируемых соединений и эталонной смеси достигается посредством десятикратного перевертывания пробирок, которые затем инкубируют при 37°С и встряхивают при 150 об/мин в течение подходящего времени инкубирования. В то время как осуществляется инкубирование, все исходные образцы ("момент 0") по отдельности смешивают, переворачивая три раза, с незамедлительным добавлением 500 мкл охлажденного на льду ацетонитрила и переворачиванием три раза, для того чтобы остановить реакцию. Сразу же после истечения каждых 15 минут и 30 минут инкубацию завершают, в каждую пробирку добавляют охлажденный на льду ацетонитрил и содержимое пробирок перемешивают, переворачивая их три раза. Все пробирки с образцами инкубируют при -80°С в течение минимум 15 минут, размораживают и повторно перемешивают, переворачивая. Аликвоты 650 мкл переносят в хроматографически чистые пробирки фильтровальной системы с микроцентрифугированием (нейлоновая мембрана 0,2 мкм, С616805) и центрифугируют при 13000 об/мин в течение 48 секунд. Фильтраты образцов хранят при -80°С.
Фильтраты образцов анализируют на приборе для LC/MS и рассчитывают процент вещества, оставшегося после инкубирования в течение 15 и 30 минут, относительно содержания этого вещества при инкубации в "момент 0".
Данные, характеризующие метаболическую стабильность, приведены в таблице 10 как процент вещества, оставшегося после инкубирования в течение 15 и 30 минут с фракциями печени человека S9.
Пример E
Растворимость некоторых соединений в физиологически совместимых композициях
Соединения настоящего изобретения обладают хорошей растворимостью в воде. Например, соединение 83 растворимо в воде в концентрации 225 мг/мл. Соединение 83, замещенное гидроксильной группой у С3, имеет растворимость в воде менее чем 60 мкг/мл. Соединения 28 и 89 характеризуются растворимостью при комнатной температуре, равной ˜30 мг/мл, которая может быть значительно повышена при нагревании. Эти неожиданно обнаруженные свойства свидетельствует о том, что 3-аминосоединения могут быть легко введены в состав фармацевтической композиции.
Пример F
Влияние некоторых тестируемых соединений на индуцированный антигеном перенос кальция
Повышение концентрации кальция в цитоплазме является обычным и решающим явлением, которое сопутствует активации многих видов рецепторов поверхности клетки. Повышение внутриклеточного содержания кальция, которое наблюдается после агонистической активации гидролиза инозитлипидов, является следствием высвобождения кальция из эндоплазматического ретикулума и переноса кальция через клеточную мембрану. Повышение концентрации кальция в цитоплазме связано с множеством важных реакций клетки в воспалительном процессе, включая адгезию, подвижность, экспрессию гена, пролиферацию и дегрануляцию. Изменения внутриклеточной концентрации кальция могут воздействовать как на быстрые, так и длительные реакции клетки. Методика исследования, предназначенная для оценки воздействия тестируемых соединений на перенос кальция, состоит в следующем.
Клетки Jurcat clone E6.1, выращенные в RPMI среде, поддерживаемой 10% FBS и 2 мМ L-глутамина, переносят в конические пробирки емкостью 50 мл и центрифугируют в течение 5 минут при 900 об/мин для формирования осадка клеток. Полученные супернатанты отбрасывают, а каждый осадок промывают в 10 мл HBSS. Собирают суспензию клеток и центрифугируют в течение 5 минут при 900 об/мин для формирования осадка клеток. Полученный супернатант отбрасывают, а осадок ресуспендируют в HBSS при 1×107 клеток/мл. Суспензию клеток переносят в 20 мм чашки Петри и инкубируют при 37,5°C, 5% СО2 в течение 20 минут.
Один объем Fura 2АМ смешивают с одним объемом детергента Плюроник F127 (Pluronic F127). В клетку вводят метку, используя по 4 мкл раствора для введения метки на каждый миллилитр суспензии клеток. Чашку Петри заворачивают в алюминиевую фольгу для того, чтобы защитить от воздействия света, и помещают в аппарат для встряхивания планшетов на 30 мин при комнатной температуре.
Последующие стадии проводят в ламинарном боксе при выключенном флуоресцентном освещении. Чашки Петри вынимают из аппарата для встряхивания, меченую клеточную суспензию переносят в конические пробирки емкостью 15 мл и дважды промывают HBSS, как описано выше. Осадок клеток ресуспендируют в 12 мл HBSS и оставляют завернутым в фольгу в течение 30 минут при комнатной температуре. Меченую клеточную суспензию в виде аликвот (100 мкл/лунка) переносят в 96-луночный белый непрозрачный планшет для культур тканей фирмы Дайнекс (Dynex). В соответствующие лунки добавляют по 50 мкл каждого тестируемого образца и затем вместе инкубируют в течение 10 минут при 37°С в аппарате для прочтения планшетов Wallac 1920 VictorTM(тестируемые образцы приготавливают в HBSS при 20 мМ, конечная концентрация составляет 20 мкМ). Выбранные соединения тестируют в отношении влияния дозы на соответствующую ей реакцию. Тестируемые образцы и активатор (анти-CD3 mAb c конечной концентрацией 4 мкг/мл, PharMingen) добавляют вручную (50 мкл) таким образом, что минимальное время для получения первого результата после стимуляции составляло приблизительно 30 секунд. Реакцию на перенос кальция в ответ на анти-CD3 mAb измеряют, проводя исследование в конечной точке. Полностью планшет прочитывают за 100 секунд при скорости 1 лунка в секунду.
Планшет прочитывают до добавления анти-CD3 для того, чтобы проконтролировать неспецифическое воздействие образцы/лекарственные средства. Флуоресцентное излучение измеряют при 510 нм при возбуждении, меняющемся от 340 до 380 каждую секунду с использованием фильтрующей пары возбуждение/излучение. Данные для этих парных длин волн представляют в виде соотношения, соответствующего 2 (двум) длинам волн возбуждения. Это соотношение зависит от красителя, находящегося внутри клетки, и концентрации клеток, позволяя провести реальное сравнение данных различных экспериментов. Данные, демонстрирующие влияние некоторых соединений на перенос кальция в подвергнутых воздействию аллергена клетках Jurkat clone E6.1, приведены в таблицах 1-6. Например, соединение 116 ингибирует кальций на 60,9% в концентрации 20 мкМ. Значение IC50 для соединения 119 является меньшим или равным 10 мкМ, в том случае, когда исследуют влияние дозы на ответную реакцию. Это свидетельствует о том, что существенное воздействие на перенос кальция могло бы оказаться полезным в случае некоторые болезненных патологий, при которых кальций является важным вторым посредником или эффекторной молекулой, включая, но не ограничиваясь этим, ишемию нарушения кровообращения, такие как "удар" или инфаркт миокарда, воспалительные заболевания, такие как астма или аллергия, нервные или мышечные расстройства, такие как болезнь Паркинсона или эпилепсия, сердечная аритмия или гипертензия.
Пример G
Влияние некоторых соединений на индуцированные аллергеном изменения функционирования легких
При астме начальная реакция на введение аллергена характеризуется происходящей непосредственно после этого бронхоконстрикцией, которая достигает пика через 20-30 мин после воздействия раздражителя и которая обычно снимается через приблизительно 2 часа. Противовоспалительные средства, как правило, не являются активными бронходилаторными средствами и не очень эффективны для контроля острой астматической бронхоконстрикции. Это приводит к необходимости проведения комбинированной терапии для лечения как бронхоконстрикции, так и воспаления.
Морских свинок линии Cam-Hatley сенсибилизировали к овальбумину (ОА) в группах по 5-6 особей, подвергая воздействию распыленного в виде аэрозоля раствора 1% ОА в солевом растворе в течение 15 мин два дня подряд, используя распылитель DeVilbiss в сочетании с дополнительной однократной интрадермальной инъекцией 3 мкг ОА в солевом растворе в первый день (день 1). Как было обнаружено, пик сенсибилизации животных к антигену наблюдается через 14 дней после первоначального воздействия. На 14-й день (день 14) животных сначала анестезировали, используя кетамин (50 мг/кг внутрибрюшинно) и ксилазин (100 мг/кг внутрибрюшинно), после чего поддерживали, используя 1% галотан, вводимый с помощью конического наконечника в нос. Левую каротидную артерию каннюлируют, используя систему трубок РЕ90, содержащую 200 Ед/мл гепарина в солевом растворе. Осуществляют трахеостомию и в пищевод вводят заполненную жидкостью канюлю (РЕ 160) приблизительно на 7 см. Животное размещают в плетизмографе (приборе для регистрации изменений объема отдельного органа) и трахею присоединяют к зафиксированной в корпусе аппарата трахейной трубке из нержавеющей стали. Каротидную канюлю присоединяют к датчику давления для мониторинга давления крови и частоты сердечных сокращений. У морской свинки вызывают паралич, используя панкуроний бромид (0,8 мг/кг), и вентилируют воздухом с частотой 60 Гц и подаваемым объемом 3 мл, используя небольшой аппарат Харварда для искусственной вентиляции легких животных.
Данные были получены в течение 20-секундных периодов при частоте дискретизации 100 Гц на подсоединенном к компьютеру физиологическом регистрирующем устройстве с использованием физиологического программного обеспечения DIREC и были проанализированы с использованием программного обеспечения ANADAT. Значения сопротивления и динамической податливости легких были получены из данных сигналов, характеризующих объем, пластичность и давление в легких в соответствии с методом Фон Нигуарда и Вирца (Von Neeguard & Wirz, 1927) с использованием для анализа изоволюметрической многоточечной регрессионной модели (Ludwig, Robatto et al., 1991) и вычисления абсолютных изменений в сопротивлении легких (RL, см Н2О/мл/сек) или податливости легких (CDYN, мл/см Н2О). Сигналы, характеризующие объем и давление, калибровали перед каждым комплексом экспериментов в соответствии с общепринятой методикой.
Некоторые измерения, родственные функционированию легких, проводили через 5-10 минут, для уверенности в устойчивости базисной линии, после чего животному вводили провоцирующую пробу с ОА (2% в солевом растворе), вводимого в 6 (шести) объемах воздуха, обмениваемого за одно дыхание, в виде распыленного аэрозоля при скорости потока 5 л/мин. Функционирование легких и сосудистой системы подвергали непрерывному мониторингу в течение всего эксперимента, хотя данные собирали в определенных временных точках, после введения антигена (10 сек, 1, 2, 3, 4, 5, 10, 20 и 30 мин).
Тестируемые соединения вводили при легкой анестезии галотаном при введении через зонд (0,1-1,0 мг/кг/день, каждый день) в 300 мл полиэтиленгликоля-200 в течение 4 дней до провоцирующей пробы, причем последнюю дозу вводили за 2 часа до введения антигена провоцирующей пробы.
Защитное воздействие выбранных тестируемых соединений на индуцированную аллергеном бронхоконстрикцию показано на фиг. 4 и 5. Продолжительность активности соединения 89 показана на фиг. 6 и 7. Данные представлены в виде среднего значения ± стандартное отклонение от среднего значения. Ингибирование бронхоконстрикции с помощью тестируемых соединений могло бы быть полезным при некоторых заболеваниях, при которых проявляется сильное сжатие гладкой мускулатуры в ответ на введение аллергена, таких как астма и аллергия.
Все публикации и патентные заявки, упомянутые в настоящем описании, приводятся в качестве ссылки в равной степени, как если бы каждая отдельная публикация и патентная заявка конкретно и по отдельности была включена в качестве ссылки. Например, книга в Comprehensive Organic Transformations, A Guide to Functional Group Preparations, Second Edition, Richard C. Larock, John Wiley and Sons, Inc., 1999, и приведенные в ней ссылки по отдельности включаются в настоящее описание в качестве ссылок во всей своей полноте.
Исходя из изложенного выше следует принять во внимание то, что хотя в целях иллюстрации в данном описании приведены некоторые конкретные воплощения настоящего изобретения, могут быть выполнены различные модификации, не отклоняясь от сущности и объема настоящего изобретения. Следовательно, изобретение ограничивается только приведенной формулой изобретения.
Реферат
Описываются соединения формулы (1), их фармацевтически приемлемые соли, сольваты, стереоизомеры, где в каждом случае независимо R1 и R2 - водород, возможно замещенный алкил, арил, гетероалкил, где гетероатом - атом азота, гетероарил, где гетероатом - азот, кислород или сера, или R1 и R2вместе с атомом азота, к которому они оба присоединены, образуют гетероциклическую структуру, являющуюся частью органической группы, содержащей 6-12 атомов С и необязательно содержащей 1-6 гетероатомов, выбранных из азота и кислорода; R3 и R4 - водород или защитная группа, при условии, что R3 и/или R4 - часть защитной группы гидроксила, № от 1 до 17 - атомы углерода, где С под №1, 2, 4, 11, 12, 15, 16 могут быть замещены двумя из групп R5, атом C17 может быть замещен одной из групп =C(R5)(R5), =C=C(R5)(R5) или двумя из групп - R5 и OR6, атомы С под №5, 8, 9, 10, 13, 14 могут быть замещены группой R5, где R5 - Н, C1-6 алкил, C1-6 гидроксиалкил, C1-6 галогеналкил, R6 - Н, защитная группа, такая, что OR6 - защищенная ОН-группа, группы OR6 могут образовывать циклическую защитную структуру для вицинальных ОН-групп. Соединения могут входить в состав фармацевтической композиции и полезны для лечения и/или профилактики различных состояний, включая воспаление, астму, аллергическое заболевание, хроническое обструктивное легочное заболевание, аллергический дерматит, твердые новообразования, ишемию и сердечную аритмию. 12 н. и 42 з.п. ф-лы, 10 табл., 7 ил.

Формула












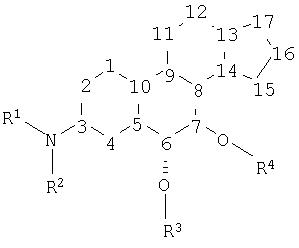








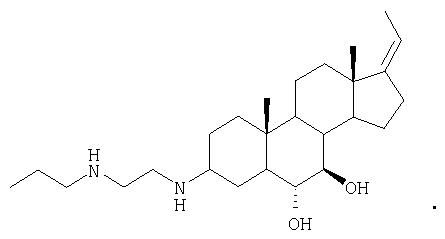

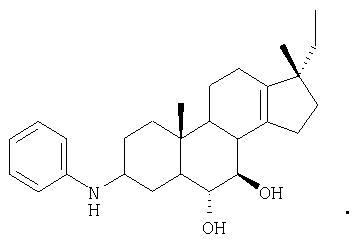

Комментарии