Способ получения улипристала ацетата и его промежуточных продуктов - RU2624007C2
Код документа: RU2624007C2
Описание
Область техники, к которой относится изобретение.
[0001] Настоящее изобретение относится к медицине, в частности, к способу приготовления лекарственного средства, а более конкретно, к способу получения улипристала ацетата с функцией антипрогестерона и антиглюкокортикоидов, а также ключевому промежуточному соединению и способу его получения.
Предпосылки создания изобретения.
[0002] Улипристала ацетат (Соединение I; химическое название: 17α-ацетоксил-11β-(4-N,N-диметиламино-фенил)-19-норпрегна-4,9-диен-3,20-дион) является сильным антипрогестеронным и антиглюкокортикоидным медицинским препаратом. Структурная формула улипристала ацетата выглядит следующим образом:

[0003] Улипристала ацетат был одобрен для продажи в Европе и Америке для использования в течение пяти дней после незащищенного полового акта или после известных или подозреваемых случаев неудачного использования средств контрацепции. Улипристала ацетат является эффективным и безопасным средством экстренной контрацепции.
[0004] Известны следующие способы получения Улипристала ацетата.
[0005] 1. Способ, описанный в патенте США US 4954490 (показан в уравнении I)
[0006] Уравнение I:

[0007] Способ использует 3-метоксил-19-норпрегна-1,3,5(10),17(20)-тетраен в качестве исходного материала, и после реакции присоединения, окисления, восстановления, гидролиза и реакции присоединения-расщепления, 17α-гидроксил-19-норпрегна-4,9-диен-3,20-дион (Соединение V2), получают путем окисления, затем Улипристала ацетат (соединение I) получают проведением в общей сложности десяти реакций, включающих реакцию конденсации этилен гликоля, m-хлороперокси эпоксидирования бензойной кислоты, реагента Гриньяра, кислотного гидролиза и ацетилирования, и с помощью рекристаллизации воды/метанола получается продукт с точкой плавления между 118°С и 121°С. Способ трудно адаптируем к промышленному производству, потому что включает в себя слишком много этапов, трудно получить исходный материал, условия реакции являются сложными, некоторые промежуточные соединения должны быть очищены хроматографией, общий выход составляет только 0,62%, расходы очень высоки, продукт не достаточно стабилен, чтобы быть использованным в медицинских целях.
[0008] 2. Известен еще один способ, описанный в патенте США US 5929262 (как показано в уравнении II).
[0009] Уравнение II:

[0010] Способ использует 3,3-этилендиоксил-17β-циано-19-норпрегна-5(10),9(11)-диен-17α спирт (соединение III) в качестве исходного материала, и 17α-гидрокси, защищенный диметилфталатом хлорметил кремнеуглеводородом, исходный материал - кислоты, гидролизованные после реакции с реагентом DBB/Li при низкой температуре -70°С, затем получают дикетал путем реакции конденсации этиленгликоля, целевой продукт получают путем реакции эпоксидирования, реакции Гриньяра, реакции кислотного гидролиза и реакция ацетилирования, и продукт желтого цвета с точкой плавления 183°С-185°С получают путем кристаллизации изопропанола, этилацетата и этилового эфира. Способ также труднореализуем в промышленном масштабе, поскольку стоимость исходного материала и DBB очень высоки, реакционные условия являются жесткими, необходимы ультранизкая температура и безводная анаэробная реакция, низкий выход (общий выход только 14%), а расходы также очень высоки.
[0011] 3. Третий способ был показан в международной заявке РСТ, публикация WO 2004078709 (как показано в уравнении III), описывается получение целевого продукта с использованием 17α-гидроксил-19-норпрегна-4,9(10)диен-3,17-дион (Соединение V2) при помощи ацетилизации, 3-карбонил конденсации, эпоксидирования и гидролиза. Синтез является простым, но исходный материал получают из соединения VI путем гидролиза в кислой среде, общий выход только 11,8% (в расчете из соединения VI) и фактически процесс получается многостадийным, выход продукта ниже, а затраты выше, таким образом, этот метод не применим к масштабам промышленного производства.
[0012] Уравнение III:

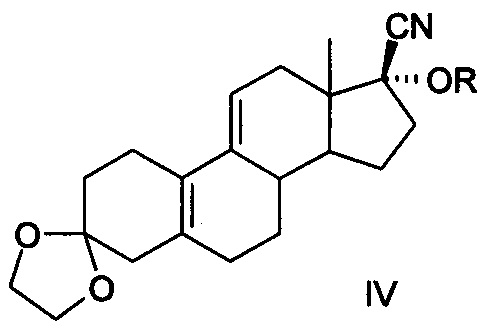
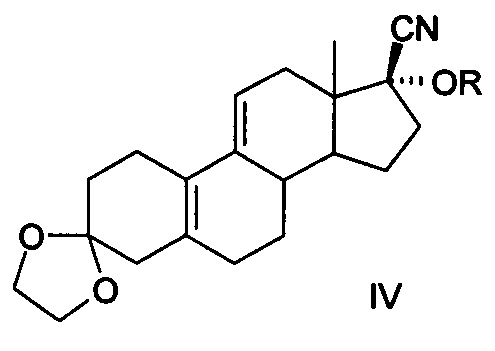
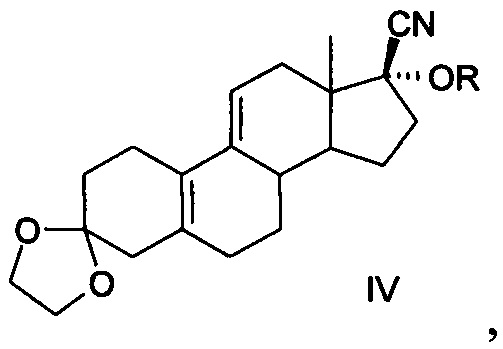
[0013] 4. Четвертый способ был описан в китайской патентной заявке CN 200780021915.9 (как показано в уравнении IV)
[0014] Уравнение IV:

[0015] Известный способ использует 3,3-этилендиоксил-19-норпрегна-5(10),9(11) диен-17-один (3-этилен кеталь для краткости, соединение II) в качестве исходного материала, желаемый продукт получается в общей сложности в результате девяти реакций, включающих реакцию присоединения ацетилена, реакцию с фенилсульфенил хлоридом, гидролиза метоксида натрия, кислотного гидролиза, конденсацию этиленгликоля, эпоксидирование, реакцию Гриньяра, реакцию кислотного гидролиза и реакцию ацетилирования, кристаллы без сольватов получают после кристаллизации изопропанолом и нагревания с помощью этанола и воды в течение 14 ч при 70°С. Реализация способа требует повышенных мер безопасности при использовании ацетилена и фенилсульфенил хлорида с выделением резкого запаха, при этом фенилсульфенил хлорид не стабилен и имеет сложные условия для хранения, а примеси, полученные при разложении, приводят к низкому выходу продукта, кроме того, фенилсульфенил хлорид сильно загрязняет окружающую среду, кроме того при нагревании в течение длительного времени при высокой температуре при реакции кристаллизации получаются новые примеси, общий выход составляет 13,8%-15,8%, расходы высоки, следовательно, и этот метод трудно адаптируем к промышленному производству.
[0016] Среди обычных способов, указанных выше, способы 1, 2 и 4 связаны с получением соединения VI, а исходный материал по способу 3 получен гидролизом из соединения VI.
Сущность заявляемого изобретения в части способа получение улипристала ацетата заключается в том, что он предусматривает следующие этапы:

где R представляет собой гидроксильную защитную группу, выбранную из -СН(СН3)-OR1, где R1 выбран соответственно из C1-С10 алкильного радикала, а пунктирные линии в формуле V представляют собой расположение двойных связей в 5(10), 9(11) или 4(5), 9(10); и включающий стадии:
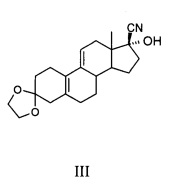
a) использование 3,3-(этилендиоксил)-19-норпрегна-5(10),9(11)-диен-3,17-диона, представленного формулой II в качестве исходного материала, и использованием спиртов, как растворителя реакции, в присутствии реагента циана, чтобы подготовить соединение 3,3-(этилендиокси)-17β-циано-7α-гидроксил-19-норпрегна-5(10),9(11)-диен, представленное формулой III, в слабой кислой среде при температуре от -10°С до комнатной температуры;
b) получение Соединения, представленного формулой IV, с помощью реакции Соединения, представленного формулой III, и гидроксильной защитной группы реагента в кислотной среде в растворителе;
c) взаимодействие Соединения, представленного формулой IV, с реагентом метилирования, и гидролиз Соединения, представленного формулой IV, в кислотной среде после реакции метилирования с получением 5(10),9(11) диен-3,20-диона, или 4(5),9(10)-диенен-3,20-диона, представленного формулой V, или их смеси;
d) взаимодействие Соединения, представленного формулой V, с этиленгликолем путем катализа р-толуолсульфоновой кислоты и триметилортоформиата или триэтилортоформиата при комнатной температуре в дихлорметане с выходом соединения 3,3-(этилен-диокси)-17α-гидрокси-19-норпрегна-5(10),9(11)-диен, представленного формулой VI;
e) эпоксидирование соединения, представленного формулой VI пероксидом водорода с выходом соединения 3,3,20,20-бис(этилендиоксил)-17α-гидроксил-5,10-эпокси-19-норпрегна-9(11)-ен, представленного формулой VII;
f) проведение дополнительной реакции между Соединением, представленным формулой VII, и 4-(N,N-диметил амидоген)фенилмагнийбромида реактива Гриньяра с выходом Соединения 3,3,20,20-бис(этилендиокси)-5α-17α-дигидрокси-11β-[4-(N,N-диметиламино)-фенил-]-19-норпрегна-9(11)-ене, представленного формулой VIII;
g) гидролиз Соединения, представленного формулой VIII в кислотных условиях с получением 17α-гидрокси-11β-[4(N,N-диметиламино)-фенил-]-19-норпрегна-9(11)-диен-3,20 дион, представленного формулой IX; и
h) ацетилирование соединения, представленного формулой IX с уксусной кислотой, хлорной кислотой, уксусным ангидридом при температуре 0-10°С, в присутствии дихлорметана с получением Соединения - Улипристала ацетата, представленного формулой I.
В частных случаях реализации соединение, представленное формулой III, реагирует с реагентом гидроксильной защитной группы с получением Соединения, представленного формулой IV, к которым непосредственно добавляют щелочь для изменения значения рН до нейтрального или щелочного, при этом Соединение, представленное формулой IV, реагирует с метиллитием или метальным реактивом Гриньяра, и полученный продукт гидролизуют в растворителе в кислотных условиях с получением Соединения V.
Сущность изобретения в части соединения заключается в том, что соединение представлено формулой IV,

где R представляет собой гидроксильную защитную группу, выбранную из -СН(СН3)-OR1, где R1 выбран соответственно из C1-С10 алкильного радикала.
В частных случаях реализации соединение по п. 3, а именно:

где R1 определен выше, а обозначенные пунктиром линии представляют R конфигурацию.
В частных случаях реализации соединение представлено

, в котором обозначенные пунктиром линии представляют R конфигурацию.
Сущность изобретения в части способа получения соединения, представленного формулой IV заключается в том, что он включает взаимодействие соединения, представленного формулой III и соединения, содержащего гидроксильную защитную группу СН(СН3)-OR1, где R1 выбран соответственно из C1-С10 алкильного радикала, с получением соединения, представленной формулой IV:

В частных случаях реализации в качестве гидроксильной защитной группы выбран виниловый эфир из следующих:
этилвиниловый эфир, n-пропилвиниловый эфир, n-бутилвиниловый эфир, изобутилвиниловый эфир и метилвиниловый эфир.
В частных случаях реализации способа получения соединения, представленного формулой V,

где пунктирные линии показывают расположение двойных связей: 5(10), 9(11) или 4(5), 9(10); причем способ содержит этапы: взаимодействие соединения, представленного формулой IV с реагентом метилирования, взятым из группы: метиллитий или метальный реактив Гриньяра, а также гидролиз соединения, представленного формулой IV, после реакции метилирования в кислотных условиях, с получением соединения, представленного формулой V:

где R представляет собой гидроксильную защитную группу, выбранную из -СН(СН3)-OR1, где R1 выбран соответственно из С1-С10 алкильного радикала.
Краткое изложение сущности изобретения.
[0017] С учетом вышеописанных проблем, одна из целей изобретения заключается в создании способа получения улипристала ацетата.
[0018] Для достижения вышеуказанной цели, в соответствии с одним из вариантов осуществления настоящего изобретения, предложен способ получения улипристала ацетата и ключевых промежуточных соединений для получения улипристала ацетата.
[0019] Способ согласно изобретению использует 3-этиленкеталь (Соединение II) в качестве исходного материала. 17β-циано группу (Соединение III) получают путем реакции присоединения между 3-этиленкеталем и реагентом циана в растворителе. Группа 17α-гидрокси Соединения III защищена, и получаем Соединение IV. Соединение V получают путем кислотного гидролиза соединения IV, после того как Соединение IV прореагирует с метилом лития или метил-реагентом Гриньяра. 3,3,20,20-бис(этилендиоксил)-17α-гидроксил-19-норпрегна-5(10),9(11) диен (Соединение VI) получают после того, как Соединение V прореагирует с этиленгликолем в присутствии п-толуолсульфоновой кислоты и триметилортоформиата или триэтилортоформиата, затем 3,3,20,20-бис(этилендиоксил)-17α-гидроксил-5α,10α-эпокси-19-норпрегна-9(11)-ене (Соединение VII) получают путем окисления Соединения VI с перекисью водорода. 3,3,20,20-bis(этилендиоксил)-5α-17α-дигидроксил-11β-[4-(N,N-диметиламино)-фенил-]-19-норпрегна-9(11)-ене (Соединение VIII) получают путем реакции Гриньяра - Соединения VII и 4-(N,N-диметиламино)фенилмагнийбромида реагентом Гриньяра. 17α-гидрокси-11β-[4(N,N-диметиламино)фенил-]-19-норпрегна-9(11) диен-3,20-диона (Соединение IX), получают путем гидролиза Соединение VII в кислотной среде. Улипристала ацетат (Соединение I) получают в результате реакции Соединения IX с ацетилирующим реагентом, содержащим безводную уксусную кислоту, хлорную кислоту и уксусный ангидрид.
Способ получения Улипристала ацетата (Соединение I) дополнительно содержит этап рекристаллизации Улипристала ацетата (Соединение I) с этанолом: изопропанол (0,5-1:9). Стадии синтеза следующие:

[0020] R представляет собой гидроксильную защитную группу, выбранную из Сг3(R4)R5, -COR2 или 2-тетрагидропиран; R3, R4 и R5 выбирают, соответственно, из водорода, гидроксила, галогена, OR1, замещенного или незамещенного С1-С10-алкила; R1 и R2 выбраны соответственно из замещенного или незамещенного С1-С10 алкила; заместитель выбран из гидроксила, галогена, нитрогруппы или амидогена.
[0021] Пунктирные линии в формуле V показывают расположение двойных связей на 5(10), 9(11) или 4(5), 9(10).
[0022] Предпочтительно, R представляет собой гидрокси-защитную группу (защитную группу гидроксильной группы), выбранную из СН(СН3)OR1, -COR2 или 2-тетрагидропиран. R1 и R2 выбраны соответственно из С1-С10 алкильного или арильного радикала.
[0023] Также R может быть СН(СН3)OR1, -COR2 или 2-тетрагидропиран, при этом соединения, представленные формулой IV, содержат соединения - изомеры IV1, IV2, IV3 или IV4 или их рацемическую модификацию, подробная формула выглядит следующим образом:
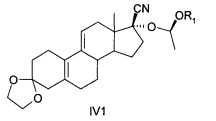
или

или

или

[0024] R1 и R2 имеют указанные выше значения. Пунктирные линии обозначают соответственно R или S конфигурацию или рацемические модификации.
[0025] В предпочтительном варианте R1 и R2 выбраны из представленных ниже соединений:
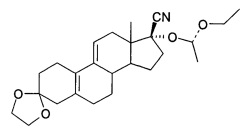
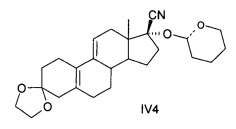
Пунктирные линии обозначают соответственно R или S конфигурацию или рацемические модификации.
[0026] В частности, способ по настоящему изобретению включает в себя этапы, на которых:
[0027] а) Используют 3,3-этилендиоксил-19-норпрегна-5(10),9(11)-диен-17-дион (3-этиленкеталь для краткости) в качестве исходного материала и используют спирты, как реагирующий растворитель. 17β-циано группу (Соединение III) получают путем реакции исходного вещества и реагента циана в кислотной среде при температуре от -10°С до комнатной температуры. Спирты выбирают из метанола, этанола и изопропанола, при этом метанол является предпочтительным. Циан-реагенты выбраны из цианида натрия, цианида калия, ацетонциангидрина, цианистого водорода и т.д., при этом цианид натрия и цианид калия являются предпочтительными. Кислоту предпочтительно выбирают из муравьиной кислоты или безводной уксусной кислоты. Температура реакции предпочтительно между -10°С и 25°С. Мольное отношение исходного материала и циан-реагентов 1:1.1-1.5, и время реакции составляет 2-24 ч.
[0028] b) Соединение IV получают путем реакции Соединения III и реагента гидрокси защитной группы (группы, защищающей гидроксилы) в кислотной среде в растворителе. Растворитель выбирают из галогенированного углеводорода, например, дихлорметана и хлороформа, или эфира, например, ТГФ (тетрагидрофуран), этилового эфира или изопропилового эфира. Растворитель предпочтительно выбирают из дихлорметана, ТГФ или этилового эфира. Реагенты гидрокси-защитной группы выбираются из органосилильных веществ, таких как триметилсилил лития, триметилхлорсилан и хлорхлорметилдиметилсилан или винилового эфира СН2=CHOR1, например, этил-винилового эфира, n-пропил-винилового эфира, n-бутил винилового эфира и метил винилового эфира, или ангидридкарбоновой кислоты, такой как уксусный ангидрид и ангидрид пропановой, или карбоновой кислоты, хлорангидрида и т.д., или 2,3-дигидропирана и т.д., при этом реагенты гидрокси-защитной группы предпочтительно выбирают из этил-винилового эфира или 2,3-дигидропирана. Кислоту выбирают из п-толуолсульфоновой кислоты, количество кислоты составляет

[0029] с) 5(10),9(11)-диен-3,20-дион (Соединение V1) или 4,9(10)-диенен-3,20-дион (Соединение V2), или смешанное соединение V, включающее Соединение V1 и Соединение V2, было получено путем гидролиза соединения IV в кислотной среде после взаимодействия Соединения IV с реагентом метилирования. Реагирующий растворитель выбирают из эфиров, например, диэтилового эфира, изопропилового эфира или ТГФ; или галогенированного углеводорода, например, дихлорметана или хлороформа. Реакционно-способный растворитель предпочтительно выбирают из диэтилового эфира, ТГФ или дихлорметана. Реагент метилирования выбирают из метила лития или метил-реагента Гриньяра, и предпочтительно выбирают из метила лития. Температура реакции от -30°С до температуры дефлегмации и, предпочтительно, комнатная температура; мольное отношение исходного материала и реагента метилирования 1:1.1-5, а предпочтительно 2-3 эквивалента. Время реакции составляет 0.5-24 ч. Растворитель для гидролиза в кислой среде выбран из ацетона или бутанона; метанола или этанола; диэтилового эфира, ТГФ, этиленгликоля или диметилового эфира; уксусного эфира или ментилацетата; дихлорметана или хлороформа. Реагент предпочтительно выбирают из бутанона, метанола, ТГФ или диэтилового эфира.
[0030] Ледяную воду добавляют для охлаждения после реакции, затем добавляют кислоту для гидролизирования или извлекающий реагент, не растворимый в воде, добавляют для извлечения (экстракции). Извлекающий реагент может быть выбран из галогенированных углеводородов, таких как дихлорметан или хлороформ, или сложного эфира - растворителя, такого как уксусный эфир (этилацетат) или метилацетат, или простого эфира, такого как диэтиловый эфир или изопропиловый эфир, или аренового реагента, такого как бензол или толуол. Кислоту непосредственно добавляют для гидролиза после экстракции, или реагент, растворяют в воде, и кислоту добавляют для гидролиза после концентрирования. Кислоту, используемую в гидролизе, выбирают из минеральных кислот, таких как серная кислота, соляная кислота, бисульфат калия или бисульфат натрия, или органических кислот, таких как муравьиная кислота или уксусная кислота. 1-6 N соляная кислота является предпочтительным выбором. Температура гидролиза составляет от -40°С до 100°С, предпочтительно между 25°С и 50°С.
[0031] Кроме того, изобретение раскрывает, что посредством регулирования температуры гидролиза основным продуктом гидролиза является Соединение V1, а затем Соединение V1 медленно превращается в стабильное Соединение V2, или чистое Соединение V2 может быть получено путем кристаллизации с растворителем, таким как метанол, и т.д. На самом деле, соотношение соединений V1 и V2 могут быть изменены путем управления условиями гидролиза, такими как кислотность, температура и время реакции, но соотношение не оказывает влияние на следующей стадии. Например, возьмем этил-виниловый эфир в качестве защитной группы, соединение, представленное формулой IV подвергается гидролизу и превращается в имин после реакции присоединения с метиллитием или метил-реактивом Гриньяра, соединение V (Соединение V представляет собой Соединение V1, Соединение V2 или их смесь) получают после гидролиза имина.

[0032] Кроме того, в процедуре, описанной в стадии с), Соединение IV может быть вовлечено в следующую стадию реакции в виде одного изомера или смеси изомеров, предпочтительно в смеси изомеров.
[0033] d) 3,20-дикеталь Соединение VI получают из Соединения V, реагирующего с этиленгликолем в присутствии п-толуолсульфоновой кислоты и триметилортоформиата или триэтилортоформиата при комнатной температуре. В качестве растворителя предпочтительно выбирают дихлорметан, температура реакции между 0°С и комнатной температурой, и время реакции составляет 1-8 ч.
[0034] е) Соединение VII получают путем взаимодействия соединения VI с окислителем в дихлорметане в щелочных условиях и пергалогено-ацетоном при комнатной температуре. Щелочь выбирают из группы: пиридин, дикалийфосфат, калийфосфат, динатрийфосфат, мононатрийфосфат и т.д. Окислитель выбирают из группы: пероксид водорода, m-хлорпероксибензойная кислота, перекись водорода; температура реакции -10-10°С.
[0035] f) Соединение VIII синтезируют из соединения VII и 4-(N,N-диметиламино)фенилмагнийбромида-реактива Гриньяра с помощью реакции Гриньяра в присутствии катализатора хлорида меди. Молярное отношение исходного материала и реагента Гриньяра составляет 1:1.5-5, при температуре реакции от -10-40°С, и время реакции составляет 2-8 ч.
[0036] g) Полученное Соединение VIII подвергают гидролизу разбавленной кислотой в дихлорметане при температуре 0-25°С. Кислоту выбирают из минеральных кислот, таких как соляная кислота, серная кислота, или используют бисульфат натрия, предпочтительно 0,2-4 N HCl растворителе, реакцию при температуре от -10-50°С, и время реакции составляет 1-5 ч.
[0037] h) Соединение IX ацетилируется безводной уксусной кислотой, хлорной кислотой или уксусным ангидридом, предпочтительно хлорной кислотой и уксусным ангидридом в смеси с уксусной кислотой с выходом улипристала ацетата (Соединение I). Температура реакции -40-25°С, предпочтительно от -10-25°С; соотношение уксусной кислоты 1-50% об/об, предпочтительно 10-15% об/об (процентное соотношение объемов), реагент ацетилирования, содержащий уксусную кислоту, хлорную кислоту и уксусный ангидрид, реакцию можно провести при 0-25°С без побочных реакций при добавлении уксусной кислоты. Тем не менее, аналогичную реакцию в примере заявки CN 200780021915.9 необходимо контролировать при температуре от -30 до -20°С.
[0038] i) Неочищенный улипристала ацетат кристаллизуют из этанола и изопропанола, получая улипристала ацетат с чистотой более 99%;
[0039] На стадии b) представлен способ получения первого промежуточного соединения улипристала ацетата, представленного формулой IV.
[0040] На стадии с) представлен способ получения другого промежуточного соединения улипристала ацетата, представленного формулой V.
[0041] В другом аспекте, настоящее изобретение относится к способу получения промежуточного соединения, представленного формулой V. Соединение III реагирует с реагентом гидроксильной защитной группы с получением соединения, представленного формулой IV. Щелочь непосредственно добавляют для изменения значения рН до 7-8 без разделения, а затем Соединение IV реагирует с метиллитием или метил-реактивом Гриньяра. Продукт подвергают гидролизу в растворителе в кислотной среде сразу после реакции или после обработки с получением Соединения V. Реагент гидрокси-защитной группы выбирают из ангидрида кислоты, кислоты или хлорангидрида, винилового эфира, такого как диэтилвиниловый эфир, n-пропил-виниловый эфир, n-бутилвиниловый эфир, изобутилвиниловый эфир и метилвиниловый эфир, или 2,3-дигидропиран и т.д.
[0042] Кроме того, изобретение обеспечивает еще один способ получения улипристала ацетата. Соединение VI постоянно реагирует без разделения на промежуточные соединения для получения улипристала ацетата. Целевой продукт получают путем реакции в одном реакторе со стадии f) до стадии i) с высоким выходом. Способ содержит следующие стадии: концентрацию эпоксида в надлежащем объеме после реакции эпоксидирования, изменение величины рН до 1-2 после реакции с реагентом Гриньяра, и их охлаждение водным раствором хлорида аммония, перемешивание в течение 1-2 ч для гидролиза, а затем экстракцию дихлорметаном, промывку, сушку безводным сульфатом магния, ацетилирование ацетилирующим реагентом сразу после фильтрации для получения целевого продукта, т.е. улипристала ацетата (Соединение I).
[0043] Кроме того, изобретение раскрывает, что промежуточное соединение, представленное формулой IV, является способным взаимодействовать с метиллитием или метил-реактивом Гриньяра. Условия реакции мягкие, защитную группу можно легко удалить после реакции, побочных реакций мало, последующая обработка проста, выход высок, реагенты дешевы, а расходы на низком уровне. Например, когда реагент гидроксильной защитной группы 17α-гидрокси выбирают из этилвинилового эфира или 2,3-дигидропирана. Соединение V получают путем реакции Соединения III и промежуточного Соединения, представленного формулой IV. Выход двухступенчатой реакции составляет 70-75%. Чистота более 98%. Очевидно, промежуточное соединение, представленное формулой IV, является ключевым промежуточным соединением для получения улипристала ацетата, и является важной частью изобретения.
[0044] Выход 3,20-диона (Соединение VI) увеличивается за счет использования промежуточного соединения, представленного формулой IV, для приготовления улипристала ацетата, выход из Соединения II к Соединению VI составляет 68% при низких затратах.
[0045] Способ, предложенный настоящим изобретением, является простым, включая всего восемь стадий, условия реакции мягкие, способность к реакции высокая. Общий выход составляет около 25-27%. Фактически Соединение IV не должно быть отделено и улипристала ацетат получают с высоким выходом посредством реакции в одном реакторе со стадии f) до стадии i). В результате, только выделение трех промежуточных соединений III, V и VI является необходимым. Операция проста и может быть адаптирована к промышленному производству.
[0046] Настоящее изобретение также относится к способу очистки улипристала ацетата. Способ включает в себя следующие этапы: добавление горячего растворителя этанол : изопропанол (0.5-1:9) в неочищенный улипристала ацетат и охлаждение горячего растворителя до 0-25°С для кристаллизации. Растворитель используется в соотношении 5-20 раз к сырому (неочищенному) ацетату улипристала. Продукт имеет чистоту 99%.
[0047] Способ прост и условия реакции мягкие с высоким выходом. Чистота полученных продуктов является высокой. Кроме того, реагенты, участвующие в способе, дешевы и легко получаемы. В результате уровень затрат сравнительно низкий. Способ применим к объемам промышленного производства и имеет высокое значение в промышленном применении.
Подробное описание вариантов осуществления изобретения.
[0048] Настоящее изобретение далее иллюстрируется, но не ограничивается следующими предпочтительными вариантами осуществления.
[0049] Спектрометром ЯМР (ядерного магнитного резонанса) является Varian INOVA-400, ядерный магнитно-резонансный спектрометр для исследований.
[0050] Для исследований монокристаллов используется рентгеновский монокристальный автоматический дифрактометр Bruker SMART АРЕХ-II; Для проведения испытаний требуются: излучение CuKa, графитовый монохроматор, диаметр единственной камеры ф=0.50 мм, расстояние между кристаллом и CCD (ПЗС) детектором D=60.3 мм, напряжение трубки 40 кВ, ток рентгеновской трубки 30 мА; режим сканирования: ф/ω сканирование.
Пример 1. Получение 3,3-(этилен-диокси)-17β-циано-17α-гидрокси-19-норпрегна-5(10),9(11) диена (Соединение III)
[0051] 3-этилен кеталь (2,0 кг, 6,37 моль), метанол (12 л), цианид натрия (343 г, 7,0 моль) и уксусную кислоту (440 мл) добавляли в реакционный объем, а затем перемешивали в течение ночи при комнатной температуре. Добавляли ледяную воду и перемешивали реакционную смесь в течение 30 мин. Выпавший в осадок кристаллический продукт отфильтровывали, осадок на фильтре промывали три раза водой, а затем сушили с получением 2,06 кг белого порошка, точка плавления: 176-178°С (с разложением), выход составил 95%, чистота по ВЭЖХ (Высокоэффективная жидкостная хроматография) была выше 98%; MS: 342 (М+1).
[0052] Конфигурация Соединения III иллюстрируется в соответствии с проведенными испытаниями монокристалла:
Пример 2. Получение 17α-[(±)1-(1-этокси)этил]окси-17β-циано-3,3-(этилен-диокси)-19-норпрегна-5(10),9(11)-диена
[0053] К суспензии, содержащей Соединение III (2.0 кг, 5.87 моль), полученное в примере 1, ТГФ (14 л) и п-толуолсульфоновую кислоту (12.0 г, 70 ммоль), добавляли при комнатной температуре в присутствии ледяной воды метил-виниловый эфир (668 мл, 7.04 моль). Реакционную смесь перемешивали в течение 4 ч при той же температуре. Добавляли триэтиламин (15 мл), воду и перемешивали в течение 10 мин. Водную фазу экстрагировали дихлорметаном, и органическую фазу объединяли и промывали водой, сушили над безводным сульфатом магния и концентрировали в вакууме с получением 2.43 кг желтоватого или бесцветного масла. Выход был определен количественно (выход составил 100%).
[0054] Масло отображалось в виде двух соединений с помощью ТСХ (тонкослойной хроматограммы) этилацетат : петролейный эфир = 1:5. Твердое вещество осаждали путем замораживания масла. Продукты были кристаллизованы из смеси этилацетат : петролейный эфир (1:2) с получением продуктов с высокой полярностью, ВЭЖХ>90%. Небольшое количество продуктов были разделено хроматографией (300-400 ячеек) с получением соединения IV1 (с низкой полярностью) и соединение (IV2 с высокой полярностью).
[0055] Соединение IV1 было желтоватым маслом: MS: 315 (М+1), 651 (2М+Na); 1Н ЯМР (CDCl3): 0,96 (д, 3Н)), 1,20 (т, 3Н), 1.24-1.34 (м), 1,37 (д, 3Н) 1.44-1.50 (м, 1H), 1,52 (с, 1Н), 1,75-1,97 (м, 8Н), 2,14-2,22 (м), 2,28 (с, 2Н), 2,50-2,70 (м), 3.46-3.60 (м), 4,0 (с, 4Н), 5,10 (д, 1Н), 5,60 (д, 1H);
[0056] Соединение IV2 было белым кристаллом: точка плавления: 131-134°С; MS: 315 (М+1), 651 (2М+Na); 1Н ЯМР (CDCl3): 0,97 (д, 3Н)), 1,26 (т, 3Н), 1,33 (д, 3Н), 1.45-1.49 (м), 1,52 (с, 1Н), 1.77-1.97 (м, 8Н), 2.13-2.28 (м, 6Н), 2,52 (д, 1Н), 2,77 (д, 1Н), 3.55-3.74 (м, 2Н), 3,98 (с, 4Н), 5,02 (д, 1H), 5,59 (д, 1Н).
Пример 3. Получение 17α-[(±)1-(1-этокси)этил]окси-17β-циано-3,3-(этилен-диокси)-19-норпрегна-5(10),9(11)-диена
[0057] К суспензии, содержащей Соединение III (50,0 г, 0,147 моль), полученное в Примере 1, дихлорметан (500 мл) и п-толуолсульфоновую кислоту (0,3 г, 1,74 ммоль), добавляли при комнатной температуре в присутствии ледяной воды метил-виниловый эфир (17 мл, 0,18 моль). Реакционную смесь перемешивали в течение 4 часов. Было получено 60,7 г желтоватого масла 17α-[(±)1-(1-этоксильных)этил]-17β-циано-3,3-этилендиоксил-19-норпрегна-5(10),9(11)-диена. Выход был зафиксирован количественно, и масло было идентифицировано в виде двух соединений методом тонкослойной хроматографии. Результат идентифицирования структуры соединения был таким же, как и результат в Примере 2.
Пример 4. Получение 17α-[(±)1-(1-н-пропил окси)этил]окси-17β-циано-3,3-(этилен-диокси)-19-норпрегна-5(10),9(11)-диена
[0058] N-пропил-виниловый эфир был использован в качестве исходного материала, а желтоватое масло было получено как в Примере 3. Выход был определен количественно; масло отображалось в виде двух соединений с помощью ТСХ; МС: 324 (М-ОСН(ОС3Н7)СН3), 368 (М-ОС3Н7), 450 (М+Na), 877 (2М+Na).
Пример 5. Получение 17α-[(±)1-(1-н-бутил-окси)этил]окси-17β-циано-3,3-(этилен-диокси)-19-норпрегна-5(10),9(11)-диена
[0059] N-бутил-виниловый эфир был использован в качестве исходного материала. Желтоватое масло было получено как в Примере 3. Выход был определен количественно; масло отображалось в виде двух соединений с помощью ТСХ; МС: 324 (М-ОСН(ОС4Н9)СН3), 368 (М-ОС4Н9), 464 (М+Na), 905 (2М+Na).
Пример 6. Получение 17α-[(±)1-(1-изобутокси)этил]окси-17β-циано-3,3-(этилендиокси)-19-норпрегна-5(10),9(11)-диена
[0060] Изобутилвиниловый эфир был использован в качестве исходного материала. Желтоватое масло было получено как в Примере 3. Выход был определен количественно; масло отображалось в виде двух соединений с помощью ТСХ; МС: 324 (М-ОСН(ОС4Н9)СН3), 368 (М-ОС4Н9), 464 (М+Na), 905 (2М+Na).
Пример 7. Получение 17α-[(±)1-(1-тетрагидропиран)этил]окси-17β-циано-3,3-(этилен-диокси)-19-норпрегна-5(10),9(11)-диена
[0061] Дигидропиран был использован в качестве исходного материала. Желтоватое масло было получено как в Примере 3. Выход был определен количественно; масло отображалось в виде двух соединений с помощью ТСХ; МС: 426 (М+1), 873 (2М+Na).
Пример 8. Получение Соединения V
[0062] 17α-[(±)1-(1-этокси)этил]окси-17β-циано-3,3-(этилен-диокси)-5(10),9(11)-диен (12,0 г, 29 ммол) и безводный ТГФ (120 мл) добавляли в реакционную колбу и охлаждали ледяной водой. 1,0 М метиллитий 2-метил-тетрагидрофуран (58 мл, 58 ммоль) добавляли при 0-10°С, затем перемешивали при 0-10°С в течение 4 часов. Добавляли 50 мл воды и перемешивали в течение 10 мин. Органическую фазу отделяли и водную фазу экстрагировали этилацетатом. Органическую фазу объединяли и концентрировали в вакууме. Добавляли 50 мл метанола и 2N HCl и перемешивали в течение 2 ч при 25°С. Реакционную смесь выливали в ледяную воду. Органическая фаза отделялась. Далее происходило экстрагирование водной фазы этилацетатом, объединение органической фазы, сушка, фильтрация, и концентрирование в вакууме, получали 6,6 г желтоватого порошка, точка плавления 184-188°С. Выход составил 73%, и порошок был показан в виде двух соединений с помощью ТСХ (этилацетат : петролейный эфир = 1:2).
[0063] Смесь этилацетат : петролейный эфир = 1:5 была использована в качестве элюанта, и продукты были разделены хроматографией с получением Соединения V1 (с низкой полярностью) и Соединения V2 (с высокой полярностью). Образец для анализа кристаллизовали этилацетатом, где этилацетата было в 5 раз больше использовавшихся продуктов.
[0064] Соединение V1 было желтоватым порошком, MP: 196-200°С; Масс-спектр: 15 (М+1); 1Н-ЯМР (CDCl3): 0,71 (с, 3Н), 1.28-1.48 (м, 2Н), 1,64 (м, 1Н),1,75-1,81 (дд, 1Н), 1.89-2.02 (м, 5Н), 2.22-2.24 (ш, 1Н), 2,27 (с, 3Н), 2.45-2.53 (м, 3Н), 2.67-2.80 (м, 3Н), 2,80 (с, 1Н), 2,86 (шс, 2Н), 5,62 (д, 1Н).
[0065] Абсолютная конфигурация Соединения V1 иллюстрируется в соответствии с исследованиями монокристалла:

[0066] Соединение V2 было желтоватым порошком, MP: 197-202°С; МС: 315 (М+1); 1Н-ЯМР (CDCl3): 0,87 (с, 3Н), 1.34-1.46 (м, 2Н), 1.48-1.53 (м, 1Н), 1.59-1.66 (м, 1Н), 1,83-1,96 (м, 4Н), 2,10 (дт, 1Н), 2.26-2.30 (с, 3Н, м, 1Н), 2.2-2.56 (м, 5Н), 2,73 (дт, 1Н), 2.81-2.91 (м, 3Н), 5,67 (с, 1Н).
[0067] Абсолютная конфигурация соединения V2 иллюстрируется в соответствии с исследованиями монокристалла:

Пример 9. Получение Соединения V.
[0068] 17α-[(±)1-(1-этокси)этил]окси-17β-циано-3,3-этилендиокси-5(10),9(11)-диен (10,0 г, 24,2 ммоль) и безводный ТГФ (100 мл) добавляли к реакционной колбе и охлаждали ледяной водой. 1,0 М бромистого лития 2-метил тетрагидрофурана (48,4 мл, 48,4 ммоль) добавляли при 0-10°С, затем перемешивали в течение 4 часов. Добавляли 10 мл 4 N HCl и перемешивали в течение 30 мин. Органическую фазу отделяли и водную фазу экстрагировали этилацетатом. Далее происходило объединение органической фазы, сушка, концентрация в вакууме и кристаллизация из смеси этилацетат : петролейный эфир, чтобы получить 5,4 г желтоватого порошка, точка плавления 185-188°С, выход 71%.
[0069] Опционально использовали 1,6 М метиллитий этиловый эфир и получали аналогичный результат.
Пример 10. Получение Соединения V.
[0070] В соответствии со способом, описанным в Примере 9 с использованием для реакции безводного этилового эфира и 1,6 М метиллитий этилового эфира. Был получен аналогичный результат, и выход составил 74%.
Пример 11. Получение Соединения V.
[0071] Использование 17α-[(±)1-(1-этокси)этил]окси-17β-циано-3,3-этилендиолксил-5(10),9(11)-диен (4,0 г, 9,68 ммоль) в качестве исходного материала и заменой метиллития на заново полученный метиловый эфир-реактив Гриньяра. Смесь перемешивали в течение 2 часов. 2,1 г желтоватого порошка был получено с помощью реакций, описанных в Примере 9, с выходом 68%.
Пример 12. Получение Соединения V.
[0072] Использовали 17α-[(±)1-(1-н-пропил окси)этил]окси-17β-циано-3,3-этилендиолксил-19-норпрегна-5(10),9(11)-диена (8,0 г, 18,7 ммоль) в качестве исходного вещества. 4,0 г желтоватого порошка было получено с помощью реакций, описанных в Примере 9, и выход составил 70%.
Пример 13. Получение Соединения V.
[0073] Использовали 17α-[(±)1-(1-изобутил окси)этил]окси-17β-циано-3,3-этилендиолксил-19-норпрегна-5(10),9(11) диена (10,0 г, 22,7 ммоль) в качестве исходного вещества. 4,7 г желтоватого порошка было получено с помощью реакций, описанных в Примере 9, и выход составил 67%.
Пример 14. Получение Соединения V.
[0074] Использовали соединение (21,3 г, 50,1 ммоль), полученное в Примере 7, в качестве исходного вещества. 11,2 г желтоватого порошка было получено с помощью реакций, описанных в Примере 9, и выход составил 72%.
Пример 15. Получение соединения V2.
[0075] 1,6 М метиллитий этиловый эфир (380 мл, 0,61 моль) добавляли к смеси, содержащей 17α-[(±)1-(1-этокси)этил]окси-17β-циано-3,3-(этилен диокси)-5(10),9(11)-диен (50,0 г, 0,121 ммоль) и этиловый эфир (500 мл) при 0-10°С. Реакционную смесь перемешивали в течение 4 ч и выливали в ледяную воду. Органическую фазу отделяли. Производили экстрагирование водной фазы этилацетатом до тех пор, пока продукт не был получен. Органическую фазу объединяли, промывали один раз водой и сушили, затем концентрировали в вакууме с получением 58 г продукта. Затем добавляли 20% ацетон и 80 мл 4 N HCl и перемешивали в течение 8 ч при 25°С, затем реакционную смесь выливали в 5 раз больший объем ледяной воды. Водную фазу экстрагировали с помощью дихлорметанового эфира. Органическую фазу объединяли и промывали один раз насыщенным раствором бикарбоната натрия и один раз водой, сушили и концентрировали в вакууме. После этого добавляли 50 мл этилацетата и охлаждали, затем перемешивали в течение 30 мин и выпавший кристаллический осадок отфильтровывали. Получили 27,4 г желтоватого твердого вещества, точка плавления 195-199°С, выход 72%; твердое вещество отображается с помощью ТСХ как соединение V2, ВЭЖХ 98%.
Пример 16. Получение Соединения V.
[0076] К раствору, содержащему Соединение III (1,5 кг, 4,40 моль), ТГФ (7,5 л), п-толуолсульфоновую кислоту (5 г), добавляли этил-виниловый эфир (632 мл, 6,6 моль) при комнатной температуре в присутствии из ледяной воды. Реакционную смесь перемешивали в течение 3 ч при комнатной температуре, а затем охлаждали до 0°С. Изменяли рН смеси до нейтральной реакции с триэтиламином. 1,6 М метиллитий этиловый эфир (8,0 л, 12,8 моль) добавляли при 0-10°С. Реакционную смесь перемешивали в течение 8 ч при той же температуре. Затем добавляли 300 мл 2 N HCl и перемешивали в течение 4 ч при 25°С. Органическую фазу отделяли. Водную фазу экстрагировали шесть раз уксусным эфиром, 600 мл * 6. Органическую фазу объединяли, промывали водой и сушили, концентрировали в вакууме до постоянного веса, нагревали и растворяли, используя в 5 раз больший объем уксусного эфира, а затем охлаждали. Осажденный кристаллический продукт отфильтровывали, получали 1036 г желтоватого порошка, точка плавления: 183-187°С, выход 75%. ВЭЖХ показало, что около 15% продукта составляет V1 и около 83% продукта составляет V2.
Пример 17. Получение 3,3,20,20-бис(этилен-диокси)-17α-гидрокси-19-норпрегна-5(10),9(11) диен (Соединение VI)
[0077] К раствору, содержащему Соединение V (1035 г, 3,30 моль), дихлорметан (11 л), этиленгликоль (1000 мл, 17,9 моль) и триметилортоформиат (1400 мл, 8,4 моль), добавляли п-толуолсульфоновую кислоту (30 г, 0,15 моль) при 25°С. Исходный материал полностью прореагировал после реакции в течение 5 ч при комнатной температуре. Реакционную смесь выливали в насыщенный раствор бикарбоната натрия (5 кг) и перемешивали в течение 30 мин. Водную фазу дважды экстрагировали 2 л * 2. Органическую фазу объединяли и промывали водой, затем сушили с безводным сульфатом магния. 10 мл пиридина и полученную смесь концентрировали в вакууме при 40°С до тех пор, до исчезновения дихлорметана. Добавляли 600 мл метанола, охлаждали до 0-10°С и перемешивали в течение 30 мин. Выпавший кристаллический осадок отфильтровали и сушили с получением 1192 г Соединения VI с выходом 90%.
Пример 18. Получение 3,3,20,20-бис(этилен-диокси)-17α-гидрокси-5α,10α-эпокси-19-норпрегна-9(11)-ена (Соединение VII)
[0078] Раствор, содержащий Соединение VI (1190 г, 2,96 моль), дихлорметан (12 л), пиридин (20 мл) и гексафторацетон тригидрат (270 мл, 1,93 моль) охлаждали до 0-5°С и добавляли к нему 50% Н2О2 (970 мл, 20 моль). Полученную смесь перемешивали в течение 3-4 ч при температуре -5-5°С до тех пор, пока исходные вещества полностью прореагируют. Органическую фазу отделяли. Водную фазу дважды экстрагировали с дихлорметаном. Органическую фазу объединяли и промывали один раз 10% водным раствором тиосульфата (500 мл), промывали водой (500 мл * 2), и сушили с безводным сульфатом магния. Органическую фазу концентрировали в вакууме до постоянного веса (1237 г, теоретически) 1310 г было получено, в котором 5α,10α-эпокси : 5β,10β-эпокси = 8:2, (продукт был обнаружен с помощью ВЭЖХ), который был использован при следующей стадии без очистки.
Пример 19. Получение 3,3,20,20-бис(этилендиокси)-5α-17α-дигидрокси-11β-[4-(N,N-диметиламино)фенил]-19-норпрегна-9(11)-ена (Соединение VIII).
[0079] К смеси, содержащей Mg (165 г, 6,87 моль), 1,2-дихлорметан (2 мл) и ТГФ (200 мл), были по каплям медленно добавлены 4-бром-N,N-диметиланилин (1380 г, 6,9 моль) и ТГФ (3000 мл) при температуре между 40-50°С. Смесь перемешивали при 40-50°С в течение 3 ч с получением реактива Гриньяра, который охлаждали до 25°С. Добавляли хлорид меди (43 г, 0,44 моль) и охлаждали ледяной водой. Добавляли раствор эпоксида (содержит около 2,3 моль 5α,10α-эпокси), полученного в Примере 20, в дихлорметане (4 л), медленно по каплям при температуре от 10 до 20°С, затем перемешивали в течение 2 часов. Реакционную смесь выливали в 3000 мл насыщенного льдом NH4Cl и перемешивали в течение 10 мин. Органическую фазу отделяли. Водную фазу экстрагировали с помощью дихлорметана, 2000 мл * 5. Органическую фазу объединяли и промывали три раза водой, затем сушили с безводным сульфатом магния. Органическую фазу концентрировали в вакууме до тех пор, пока органическая фаза не достигла состояния пены. Добавляли 800 мл этилацетата и нагревали при 70°С в течение 10 мин, затем охлаждали до 10-20°С и перемешивали в течение 30 мин. Выпавший в осадок кристаллический продукт отфильтровывали, дважды промывали этилацетатом и сушили с получением 957 г светлого порошка. Выход двухступенчатой реакции составил 60%, точка плавления: 230-234°С, ВЭЖХ>95%.
Пример 20: Получение 17α-гидрокси-4-11β-[(N,N-диметиламино)фенил-]-19-норпрегна-4,9(10)диен-3,20-диона (Соединение IX).
[0080] К раствору 2 N HCl (4000 мл), добавляли Соединение VIII (950 г, 1,76 моль), полученное в Примере 21. Смесь перемешивали при 25°С в течение 2 ч до тех пор, пока исходные вещества полностью прореагируют (по данным ТСХ). Реакционную смесь экстрагировали пять раз дихлорметаном 3000 мл * 2, 1000 мл * 3, соответственно. Органическую фазу объединяли и промывали один раз насыщенным раствором бикарбоната натрия и один раз водой, затем сушили с безводным сульфатом магния, фильтровали и концентрировали до примерно 3000 мл для дальнейшего использования.
Пример 21. Получение Улипристала ацетата (I).
[0081] К раствору, содержащему Соединение IX, полученное в Примере 22, и дихлорметан, добавляли уксусную кислоту (200 мл, 3,50 моль). Смесь охлаждали до -10°С и добавляли 70% хлорную кислоту (237 мл, 3,92 моль). Затем добавляли медленно по каплям уксусный ангидрид (1400 мл, 14,9 ммоль) при 0-10°С. После перемешивания в течение 1-2 ч, добавляли 3 кг ледяной воды. Органическую фазу отделяли и водную фазу экстрагировали шесть раз дихлорметаном, 500 мл * 6. Объединенную органическую фазу промывали один раз в 800 мл насыщенного раствора бикарбоната натрия и один раз в 800 мл воды, затем сушили над безводным сульфатом магния, отфильтровывали и фильтрат концентрировали в вакууме до достижения постоянного веса. Добавляли 800 мл изопропанола и перемешивали в течение 30 мин. Выпавший кристаллический осадок отфильтровывали и промывали изопропанолом, затем высушивали при 60°С с получением 790 г желтоватого твердого вещества, которое растворяли в 8000 мл раствора изопропанол : этанол (95:5) при нагревании и обесцвечивали 1% активированным углем. Смесь фильтровали, охлаждали до 10°С, и перемешивали в течение 1 часа. Выпавший кристаллический осадок отфильтровывали и промывали раствором изопропанол : этанол (95:5), и сушили при 60°С с получением 586 г желтоватого твердого вещества, точка плавления: 151-153°С. Структура улипристала ацетата была подтверждена, и выход составил около 70%, ВЭЖХ>99%.
Пример 22. Получение Улипристала ацетата (I)
[0082] Раствор, содержащий 3,20-дикеталь (Соединение VI, 100 г, 0,25 моль), дихлорметан (1 л), пиридин (5 мл) и тригидрат гексафторацетон (20 мл, 143 ммоль) охлаждали до -10-0°С. Медленно добавляли 50% Н2О2 (70 мл, 1,44 моль). Смесь перемешивали при -5-5°С в течение 3-4 ч до тех пор, пока исходные вещества полностью прореагируют. Органическую фазу отделяли. Водную фазу дважды экстрагировали дихлорметаном. Объединенную органическую фазу промывали 10% раствором тиосульфата натрия (10 мл). Органическую фазу промывали водой (50 мл * 2), затем сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме до объема 200 мл.
[0083] К смеси Mg (9,6 г, 0,4 моль), 1,2-дихлорметан (1 мл) и ТГФ (50 мл), медленно по каплям добавили раствор 4-бром-N,N-диметиланилин (71 г, 0,35 моль) и ТГФ (200 мл). Полученную смесь перемешивали при 50-60°С в течение 3 ч с получением реактива Гриньяра, который охлаждали до 25°С. Был добавлен хлорид меди (3 г, 30 ммоль), затем перемешивали при 25°С в течение 30 мин. Смесь охлаждали, и добавляли медленно по каплям раствор эпоксида в дихлорметане, поддерживая температуру 10-20°С, и перемешивали в течение 2 ч при той же температуре. Реакционную смесь выливали в 500 мл насыщенным льдом NH4Cl и перемешивали в течение 10 мин. Органическую фазу отделяли. Водную фазу экстрагировали с помощью дихлорметана 5 раз (400 мл * 5). Объединенную органическую фазу промывали 1000 мл насыщенного водного раствора 2 N HCl, а затем перемешивали в течение 2 ч при 25°С, чтобы отделить органическую фазу. Водную фазу экстрагировали с помощью дихлорметана 3 раза (200 мл * 3). Органическую фазу объединяли, затем промывали один раз насыщенным раствором бикарбоната натрия и один раз водой, сушили над безводным сульфатом магния и фильтровали. Фильтрат выливали в реакционный сосуд, и добавляли ледяную уксусную кислоту (18 мл, 315 ммоль), затем охлаждали до -10-0°С. Добавляли 70% хлорной кислоты (24 мл, 295 ммоль), перемешивали, потом добавляли уксусный ангидрид (140 мл, 1,49 моль) при -10-0°С. После перемешивания в течение 30 мин при той же температуре, водную фазу экстрагировали дихлорметаном 3 раза (200 мл * 3). Объединенную органическую фазу промывали один раз насыщенным раствором бикарбоната натрия и один раз водой, затем органическую фазу сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали до достижения постоянного веса 110 г, перекристаллизовывали с 10-кратным реагентом этанол : изопропанол (95:5), получали 61,2 г желтоватых кристаллов, точка плавления: 145-148°С. Выход составил 52%, ВЭЖХ>97%. Продукты снова перекристаллизовывали с реагентом этанол : изопропанол (95:5) и сушили при 60°С с получением 46,0 г желтоватых кристаллов. Структура улипристала ацетата была подтверждена структурным исследованием, и выход составил 75%, ВЭЖХ>99%.
Способ прост и условия реакции мягкие, с высоким выходом. Чистота полученных продуктов является высокой. Кроме того, реагенты, участвующие в способе, дешевы и легко получаемы. В результате уровень затрат сравнительно низкий. Способ применим к объемам промышленного производства и имеет высокое значение в промышленном применении.
Реферат
Изобретение относится к способу получения улипристала ацетата, содержащему следующие этапы:где R представляет собой гидроксильную защитную группу, выбранную из -СН(СН)-OR, где Rвыбран из С-Салкильного радикала, а пунктирные линии в формуле V представляют собой расположение двойных связей в 5(10), 9(11) или 4(5), 9(10); и включающему стадии: a) использование 3,3-(этилендиоксил)-19-норпрегна-5(10), 9(11)-диен-3,17-диона формулы II в качестве исходного материала и использование спиртов как растворителя реакции в присутствии реагента циана, чтобы подготовить соединение 3,3-(этилендиокси)-17β-циано-7α-гидроксил-19-норпрегна-5(10), 9(11)-диен формулы III в слабой кислой среде при температуре от -10°С до комнатной температуры; b) получение соединения формулы IV с помощью реакции соединения формулы III и гидроксильной защитной группы реагента в кислотной среде в растворителе; c) взаимодействие соединения формулы IV с реагентом метилирования и гидролиз соединения формулы IV в кислотной среде после реакции метилирования с получением 5(10), 9(11)-диен-3,20-диона, или 4(5), 9(10)-диенен-3,20-диона формулы V, или их смеси; d) взаимодействие соединения формулы V с этиленгликолем путем катализа р-толуолсульфоновой кислоты и триметилортоформиата или триэтилортоформиата при комнатной температуре в дихлорметане с выходом соединения 3,3-(этилен-диокси)-17α-гидрокси-19-норпрегна-5(10), 9(11)-диен формулы VI; e) эпоксидирование соединения формулы VI пероксидом водорода с выходом соединения 3,3,20,20-бис(этилендиоксил)-17α-гидроксил-5,10-эпокси-19-норпрегна-9(11)-ен формулы VII; f) проведение дополнительной реакции между соединением формулы VII и 4-(N,N-диметил амидоген) фенилмагнийбромидом реактива Гриньяра с выходом соединения 3,3,20,20-бис(этилендиокси)-5α-17α-дигидрокси-11β-[4-(N,N-диметиламино)-фенил-]-19-норпрегна-9(11)-ен формулы VIII; g) гидролиз соединения формулы VIII в кислотных условиях с получением 17α-гидрокси-11β-[4(N,N-диметиламино)-фенил-]-19-норпрегна-9(11)-диен-3,20-диона формулы IX и h) ацетилирование соединения формулы IX с уксусной кислотой, хлорной кислотой, уксусным ангидридом при температуре 0-10°С, в присутствии дихлорметана с получением соединения улипристала ацетата формулы I. Изобретение также относится к соединению формулы IV, к способу получения соединения формулы IV, к способу получения соединения формулы V. Технический результат: предложен новый способ получения улипристала ацетата с высоким выходом и высокой чистотой конечного продукта, применимый в промышленном производстве. 4 н. и 4 з.п. ф-лы, 22 пр.
Формула




Комментарии