Способ для извлечения органического соединения из растворов - RU2184148C2
Код документа: RU2184148C2
Чертежи






Описание
Изобретение относится к способу, с помощью которого кристаллизующееся органическое соединение может быть извлечено из растворов, содержащих указанное соединение. В частности, настоящее изобретение относится к способу, с помощью которого растворимые в воде органические соединения могут быть кристаллизованы из перенасыщенных водных растворов, имеющих очень высокую вязкость, а затем извлечены из этих растворов.
Двумя принципиальными стадиями кристаллизации являются образование зародышей кристаллов (зародышеобразование) и рост кристаллов. В большинстве промышленных процессов кристаллизация прежде всего основывается на росте кристаллов. Литературные данные в отношении кристаллизации представлены, например, в Mathlouthi, M. and Reiser, P. (ed.), Sucrose, Properties and Applications, Blackie Academic & Professional, Suffolk, Great Britian, 1995, p. 49 и последующие страницы. Это описание объясняет механизм кристаллизации как по отношению к зародышеобразованию, так и к росту кристаллов. В отношении промышленной кристаллизации сахарозы эта публикация утверждает, например, что необходимо избегать концентрирования раствора до зоны зародышеобразования, то есть зоны, в которой легко происходит самопроизвольное образование центров кристаллизации (с. 58); необходимо избегать образования неконтролируемого количества затравочных кристаллов (с. 59); отношение кристаллов к маточной жидкости не может превышать определенное значение (с. 59-60); и кристаллизация должна осуществляться в метастабильной области, не слишком близкой к зоне зародышеобразования и кривой насыщения (с. 60-61 и с. 63-64). Метастабильная область представляет собой область, где самопроизвольное образование кристаллов может осуществляться только тогда, когда присутствуют кристаллы. Подчеркивается, что в этой области не образуются новые кристаллы в отсутствие затравки. Кроме того, в соответствии с этой публикацией (сравни, например, с. 57 и 58), примеси уменьшают скорость роста кристаллов и даже могут полностью заблокировать рост.
При увеличении степени перенасыщения и уменьшении температуры раствора вязкость раствора также увеличивается, таким образом замедляя и в конечном счете полностью блокируя диффузию молекул через жидкий слой, окружающий кристаллы или зародыши кристаллов, к поверхности кристалла, и, следовательно, известные из литературы способы, основанные на росте кристаллов, не являются более возможными. В соответствии с предшествующим уровнем техники высокая вязкость также рассматривается как прямое препятствие для выделения кристаллов из маточной жидкости.
В отношении кристаллизации сахарозы эти проблемы также были обсуждены в указанной выше публикации Mathlouthi, М. and Reiser, P. (ed.), Sucrose, Properties and Applications.
Способы для извлечения сахарозы, используемые в сахарной промышленности, обычно содержат три последовательных стадии кристаллизации. На последней стадии, которая известна как кристаллизация "С", содержание сахарозы в сиропе исходного материала составляет примерно 73-75% по отношению к сухому веществу; этот способ кристаллизации является медленным и трудным, а еще чистота сахарозы (% сахарозы по отношению к сухому веществу) в оттеке, то есть в мелассе, все еще обычно так же высока как примерно 58%. Существует несколько способов, с помощью которых, как предполагается, можно улучшить выход сахарозы, то есть уменьшить степень чистоты сахарозы в мелассе. Эти способы включают способы Квентина (Quentin) и Стеффена (Steffen) и способы фракционирования мелассы типа тех, которые описаны в Патенте Финляндии 77845 (Suomen Sokeri Oy; Heikkilae, Melaja, Millner, Virtanen; соответствует международной опубликованной заявке WO 81/02420). Такие способы фракционирования делают возможным извлечение фракции, обогащенной бетаином, откуда бетаин может быть извлечен для получения из мелассы.
В обычных способах ксилозу можно кристаллизовать, только если степень чистоты ксилозы составляет по крайней мере примерно 70 мас.% по отношению к сухому веществу. В этой связи необходимо сначала очистить содержащий ксилозу раствор, полученный, например, в результате гидролиза полученного из растений материала, до требуемой степени чистоты с помощью различных способов ультрафильтрации, ионного обмена, обесцвечивания, ионной эксклюзии или хроматографических способов разделения, или их сочетаний; после этого для кристаллизации ксилозы используют вспомогательные растворители, уменьшающие растворимость ксилозы.
Указанные выше способы разделения, очистки и кристаллизации ксилозы описаны, например, в патентах США 4631129 (Heikkilae, H.; Suomen Sokeri Oy), 4075406 (Melaja, A. J. & Haemaelaeinen, L. ; Suomen Sokeri Oy), 5084104 (Heikkilae, H. and Hyoky, G.; Cultor, Ltd) и 4168988 (Riehm, Т. & Hofenk, G. ; Institut voor Bewaring en Verwerking van Landbouwprodukten) и в публикациях, упоминаемых в них.
Когда ксилозу получают путем гидролиза ксилана, альтернативой указанным выше способам является очистка ксилана перед его гидролизом до ксилозы для получения раствора ксилозы с достаточной степенью чистоты. Эта процедура также является очень сложной и громоздкой, как ясно из Browning, B.L., Methods of wood Chemistry, II, Interscience publishers, New York, 1967, и Fry, S. C., The growing plant cell wall: chemical and metabolic analysis, Longman Scientific & Technical, England 1988.
В соответствии с German Offenlegungsschrift 1643940 (Eickenmeyer, R. & Scholler, H. ) кристаллическую ксилозу извлекают из гидролизата природных веществ, содержащих пентозан и целлюлозу, путем кристаллизации из сиропа, содержащего по крайней мере примерно 70% ксилозы. Сироп вводят в кристаллизатор при 60-70oС и кристаллическую массу, содержащую 15-33% ксилозы по отношению к количеству ксилозы, подаваемому в кристаллизатор, извлекают из кристаллизатора при 48-52oС. Кристаллы отделяют от этой кристаллической массы путем центрифугирования и маточную жидкость, количество которой составляет 300-100% свежего сиропа, подаваемого в систему, объединяют с исходным материалом гидролизата. Полученную смесь маточной жидкости и гидролизата обрабатывают в катионобменнике и анионобменнике и после последующей обесцвечивающей обработки смесь упаривают для получения сиропа, который будет подаваться в кристаллизатор. В дополнение к громоздким обработкам очистки способ таким образом содержит очень дорогостоящую стадию рециркулирования, в соответствии с этой публикацией малые количества ксилозы, получаемой при одной кристаллизации (малый выход по сравнению с ксилозой, подаваемой в кристаллизатор), обусловлены тем фактом, что когда температура падает ниже примерно 48oС, скорость кристаллизации будет очень малой из-за того факта, что вязкость раствора по существу повышается с падением температуры.
Патент США 3981739 (Dmitrovsky et al.; Amstar Corporation) относится к способу непрерывной кристаллизации cахаров (сахарозы, декстрозы, фруктозы, лактозы, углеводов). Способ включает контролируемый рост кристаллов при двухстадийной кристаллизации с испарением, начинающейся с затравочных кристаллов малого размера. Кристаллы на первой стадии являются по существу больше, чем затравочные кристаллы, а кристаллы увеличенного размера получают на второй стадии.
Патент США 4199373 (Dwivedi et al.; Chimicassa GmbH) относится к способу производства свободно текучих смесей фруктозы и глюкозы, избегая недостатков существующих ранее способов (таких как необходимость в сложных механических устройствах и тщательном контроле, большая стоимость энергии и низкий выход). Процесс представляет собой способ затвердевания; он не включает разделения кристаллов и маточной жидкости. Сильно концентрированный раствор затравляют и оставляют его стоять (давая таким образом возможность осуществиться кристаллизации) при конкретной температуре и относительной влажности, извлекают, сушат и измельчают. Слишком низкая концентрация приводит к тестообразной массе, слишком высокая концентрация приводит к стеклообразной смеси. Главное, чтобы окружающий воздух имел относительную влажность ниже 50% и температуру между 50-90oF (10-32oC).
Другие способы с полным затвердеванием обсуждаются, например, в патентах США 4297146 (Mise et al.; CPC International Inc.), 4595418 (Yoshino; Sanwa Kosan Kabushiki Kaisha) и 4640717 (Shukla et al; Tate & Lyle Public Limited Company).
Патент США 4634472 (Niekamp et al.; A.E. Staley Manufacturing Company) предлагает способ производства обогащенного сиропа фруктозы. В этом способе устанавливают температуру питающего сиропа (концентрация сухого твердого вещества 75-89%), соответствующую кристаллизации глюкозы. Из литературы хорошо известно, что легкая кристаллизация глюкозы, даже при низкой степени чистоты, часто является проблемой, как, например, в случае меда (типичная концентрация твердого вещества 81-85%, примерно 40% глюкозы и примерно 30% фруктозы по отношению к сухому твердому веществу). Также известно (Harold E. Horn, "Dextrose: An Alternative to Sucrose in Panned Confections", The Manufacturing Confectioner for 1977), что кристаллизация глюкозы все больше и больше замедляется при вязкостях 10000-100000 сП (10-100 Па•с). Согласно вычислениям из примера 1 патента США 4634472 вязкость при кристаллизации составляет только примерно 2000 сП, что представляет очень низкую вязкость раствора. Вода не может быть использована в качестве разбавителя в способе по патенту США 4634472 (столбец 5, строки 20-25), поскольку кристаллы будут растворяться.
Патент США 4816079 (Ahrens et al.; Fried Krupp GmbH) относится к способу непрерывной кристаллизации моногидрата декстрозы. Способ представляет собой, в принципе, традиционный способ кристаллизации с охлаждением, основанный на росте кристаллов. Часть питающего сиропа подвергают процессу обработки сдвигом в течение периода 0,01-2 секунды, чтобы инициировать зародышеобразование для получения затравочных кристаллов для процесса.
Соответственно, существует необходимость в экономичном и эффективном способе для достижения высокой степени извлечения кристаллического продукта из содержащего его раствора, особенно из источника, имеющего более низкие уровни кристаллизующегося продукта, чем можно непосредственно обрабатывать для достижения такого же выхода при сравнимых условиях.
Главной целью изобретения является достижение улучшения общего выхода извлекаемых кристаллизующихся органических соединений из содержащих их растворов.
Дополнительной целью является экономное использование в таких способах потоков материалов, содержащих кристаллизующиеся органические соединения с примесями.
Также целью является использование потока оттека или рецикла из промышленных процессов, содержащего кристаллизующиеся органические соединения, в качестве источника для эффективного извлечения таких соединений с хорошим выходом.
В настоящее время обнаружено, что кристаллизующиеся органические соединения, которые образуют растворы, имеющие высокую вязкость, могут быть извлечены из таких растворов с помощью кристаллизации, когда значение перенасыщения является достаточно высоким и когда кристаллизацию осуществляют главным образом посредством зародышеобразования, то есть таким образом, что рост кристаллов не является важным в процессе. Таким образом, соединения могут быть кристаллизованы даже из сравнительно загрязненных растворов, из которых они ранее не могли бы быть кристаллизованы. В контексте настоящего описания и формулы изобретения высокая вязкость означает вязкость, при которой рост кристаллов по существу заторможен. Конкретно, в настоящем изобретении раствор рассматривается как имеющий высокую вязкость, если его вязкость находится в интервале от примерно 105 сП до примерно 106 сП (100-1000 Па•с).
Таким образом, настоящее изобретение предлагает способ извлечения кристаллизуемого органического соединения из растворов, содержащих указанное соединение, отличающийся тем, что соединение кристаллизуют главным образом путем зародышеобразования из раствора, имеющего высокую вязкость и высокую степень перенасыщенности по отношению к соединению, которое необходимо извлечь, и извлекают образовавшиеся кристаллы.
Кристаллизацию по настоящему изобретению предпочтительно осуществляют вне метастабильной зоны, то есть используя терминологию Mathlouthi, M. and Reiser, P. (ed. ). Sucrose, Properties and Applications, например, в зоне зародышеобразования, которую в соответствии с указанной публикацией следует избегать при кристаллизации, например, сахарозы.
В способе по настоящему изобретению зародышеобразование усиливается путем эффективного перемешивания, давая таким образом возможность зародышеобразованию происходить самопроизвольно. Перемешивание осуществляют настолько энергично, насколько позволяет высокая вязкость, для достижения эффективного непрерывного перемешивания частей кристаллизационной массы в и из областей с более высоким сдвигом, в которых более легко осуществляется зародышеобразование, чтобы максимизировать кристаллизацию во всей массе. Таким образом избегают затвердевания кристаллизационной массы, и зародыши способны приобретать кристаллическую форму и расти до тех пор, пока рост кристаллов не будет блокирован, посредством чего реализуются наивысшие возможные выходы. Для индуцирования зародышеобразования к перенасыщенному раствору могут быть добавлены также затравочные кристаллы. Конечный размер кристаллов обычно ограничен до примерно 10-120 мкм. Усовершенствование в извлечении кристаллизующегося соединения, достигаемое с помощью настоящего изобретения, по существу основано на механизме зародышеобразования в очень вязких растворах при непрерывном перемешивании, с помощью которого максимизируется общий выход кристаллизации.
После начала зародышеобразования высокая вязкость маточной жидкости полученной суспензии (то есть кристаллизационной массы) по существу предотвращает рост кристаллов и выход из перенасыщенного состояния; однако зародышеобразование продолжается благодаря эффективному перемешиванию. Во время первого периода процесса зародышеобразования по настоящему изобретению суспензию охлаждают до достижения и поддержания высокой степени перенасыщенности маточной жидкости. После этого температура кристаллизационной массы и концентрация общих твердых веществ являются практически постоянными. Степень перенасыщения маточной жидкости поддерживают высокой во время всей фазы кристаллизации, то есть систему постоянно поддерживают по существу выше интервала метастабильности.
В настоящем описании и формуле изобретения
перенасыщение раствора означает его очевидную перенасыщенность в отношении органического соединения, которое необходимо извлечь, то есть безразмерное отношение измеренного содержания и растворимости
указанного соединения, которое вычисляют из уравнения:
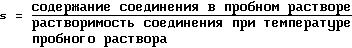
где s представляет собой степень перенасыщения, и единица измерения для содержания и растворимости соединения представляет г чистого соединения/100 г растворителя. Также термины "перенасыщенный" и "перенасыщение" относятся только к насыщению раствора по отношению к соединению, которое необходимо извлечь. Степень чистоты вещества означает его процентное содержание в сухом веществе.
Высокая степень перенасыщения означает перенасыщение, при котором процесс зародышеобразования является доминирующим, а рост кристаллов замедляется. Как правило, в настоящем изобретении раствор рассматривается как имеющий высокую степень перенасыщения, когда s составляет между 1,4 и 4.
Из уровня техники известно, что относительно высокая степень перенасыщения является необходимой для зародышеобразования, и она наиболее эффективно поддерживается при низкой вязкости путем применения сильного перемешивания. Если зародышеобразование как таковое является предпочтительным, тогда очевидными рабочими условиями могут быть относительно низкая вязкость и энергичное перемешивание. В противоположность этому, в настоящем изобретении используют высокие вязкости, тем самым может быть получено превосходное извлечение кристаллизующегося соединения.
В соответствии с предшествующим уровнем техники проблемы, связанные с выделением мелкокристаллического продукта из маточной жидкости при высокой вязкости кристаллизационной массы, также затрудняют промышленное применение кристаллизации, основанной главным образом на зародышеобразовании. Сильное зародышеобразование (самопроизвольное образование кристаллов), следовательно, обычно рассматривается как недостаток традиционных процессов кристаллизации. В соответствии с дополнительным аспектом изобретения мелкокристаллический продукт, полученный при зародышеобразовании, тем не менее может быть выделен из кристаллизационной массы, когда вязкость кристаллизационной массы понижается непосредственно перед извлечением кристаллов или в связи с извлечением кристаллов. Вязкость может быть понижена, например, путем нагрева кристаллической массы и/или ее разбавления либо разбавленным раствором исходного материала, либо растворителем, содержащимся в нем. Также является возможным добавление другого растворителя, в котором кристаллы по существу не растворяются; например, при извлечении сахарозы в качестве такого растворителя может быть использован глицерин.
Конкретным воплощением изобретения является извлечение с помощью фильтрации мелкокристаллической фракции, полученной путем зародышеобразования.
В типичных процессах кристаллизации, известных из предшествующего уровня техники, значительные количества продукта теряются в конечных маточных жидкостях. Настоящее изобретение приводит к значительным улучшениям в извлечении желаемого продукта из маточной жидкости. Извлеченный материал может быть дополнительно очищен с помощью традиционных способов кристаллизации. Типичное улучшение в общем выходе, достигаемое с помощью настоящего изобретения, составляет 5-30% или даже более по сравнению с известными способами, и из водных растворов, где традиционная кристаллизация является неэффективной, могут быть получены общие выходы вплоть до 80%.
Способ по настоящему изобретению является особенно пригодным для извлечения углеводов, которые предпочтительно являются легко кристаллизуемыми, таких как альдозы и альдитолы, например сахара и сахарные спирты, и гидрокси- и аминокислоты и бетаин, из их водных растворов. Термин "водный раствор", как он используется в описании, означает среду, в которой кристаллизуемое соединение изначально растворено, тем самым обеспечивая единую гомогенную непрерывную фазу, содержащую достаточную концентрацию кристаллизуемых соединений, так что, когда раствор концентрируют до его перенасыщенного состояния, легко происходит зародышеобразование. Понятно, что водный раствор может содержать другие смешиваемые с ним вещества либо как примеси в загрузке, либо как вспомогательные вещества для облегчения дальнейшей обработки.
Поскольку способ дает возможность извлечения этих соединений даже из достаточно загрязненных растворов, он является пригодным для использования при извлечении органических соединений из водных растворов, полученных из биомассы. Такие растворы включают мелассу и барду, гидролизаты биомассы или их части, или полученные из них концентраты, такие как варочные растворы в целлюлозной промышленности. Такие водные растворы также включают оттеки (маточные жидкости, из которых выделены кристаллы), полученные в современных промышленных процессах кристаллизации, в которых степень чистоты соединения, которое необходимо извлечь, в растворе исходного материала является сравнительно высокой, и примеси выгружаются (выходят вместе) с оттеками. Кроме того, этот способ является пригодным для извлечения продуктов, получаемых путем ферментации, таких как глюконаты, глютаматы и молочная кислота, из их ферментационных растворов.
Способ по настоящему изобретению является пригодным для извлечения, например, следующих соединений: ксилоза, манноза, ксилит, маннитол, лактоза, лактит, сахароза, глюкоза, фруктоза, мальтоза, мальтит, изомальтоза, изомальтулоза, лактулоза, α-D-глюкопираноцил (1-->6) маннит, α-D-глюкопираноцил (1-->6)сорбит, β-циклодекстрин, итаконовая кислота, лимонная кислота, бетаин, инозит, 1,4-ангидроглюцит.
Способ по настоящему изобретению является особенно преимущественным в случаях, где кристаллизующееся вещество извлекают из растворов путем кристаллизации с помощью известных способов до той степени, которая является технически возможной или экономически оправданной. Другими словами, способ является особенно преимущественным при извлечении кристаллизующегося вещества из растворов, имеющих низкую степень чистоты вещества.
Раствор, из которого органическое соединение извлекают с помощью способа по настоящему изобретению, сначала доводят до состояния значительного перенасыщения для получения зародышеобразования. Обычно это достигается с помощью концентрирования и/или охлаждения. Предпочтительный способ концентрирования представляет собой выпаривание при давлении ниже атмосферного. Раствор может быть концентрирован, например, до содержания сухого вещества 75-98 маc. %; предпочтительное содержание сухого вещества зависит от раствора, который необходимо обработать, и может составлять, например, 82-95 мас.%. Степень перенасыщения максимизируют при условиях, получаемых в пределах осуществимой вязкости.
Для получения кристаллов из перенасыщенного раствора чаще всего используют охлаждение, на время и скорость охлаждения влияют качество и склонность к кристаллизации раствора, который необходимо обработать. На стадии зародышеобразования скорость охлаждения перенасыщенного раствора и приложение рабочей энергии к процессу перемешивания на практике взаимосвязаны с целью предотвращения затвердевания кристаллизационной массы и для ограничения роста кристаллов, чтобы получить кристаллы в целом в пределах не более чем 10-100 мкм, способствуя при этом, например, дальнейшему зародышеобразованию кристаллизационной массы. Как правило, слишком высокая локальная скорость сдвига без эффективного перемешивания может привести к затвердеванию кристаллизационной массы и ее необходимо избегать. При данной вязкости и входной мощности распределение кристаллов по размерам контролируется скоростью охлаждения. Источники с более высокой степенью чистоты могут охлаждаться быстрее, в то время как источники с большим количеством примесей или природных ингибиторов могут требовать более медленной скорости.
Перед началом охлаждения к раствору предпочтительно добавляют тонко измельченные затравочные кристаллы соединения, которое необходимо извлечь; однако кристаллизация может также быть инициирована путем самопроизвольного затравливания. Термин "полная затравка", используемый ниже в связи с затравливанием, является общеизвестным в литературе (смотри "Beet-Sugar Technology", 3rd Edition, edited by R.A. McGinnis (1982) pp. 371-481) и вычисляется из размера затравочных кристаллов, размера кристаллов в желаемом конечном продукте и выхода, при условии, что число кристаллов не изменяется.
Раствор, приведенный до состояния перенасыщения, необходимого для зародышеобразования, и суспензия, образованная с помощью такого раствора, и кристаллы, содержащиеся в нем, также будут в дальнейшем определяться как кристаллизационная масса.
Способ по настоящему изобретению является особенно преимущественным и представительно описан при извлечении ксилозы из растворов, имеющих сравнительно низкое содержание ксилозы по отношению к сухому веществу, то есть примерно 30-50 маc. % по отношению к растворенному сухому веществу. В этом случае могут быть значительно сокращены или вообще исключены процессы разделения, включенные в известные в уровне техники способы, а также может быть исключено использование вспомогательных растворителей, тем самым делая способ по существу более дешевым, чем известные из уровня техники способы, и ксилоза может быть извлечена в форме кристаллического продукта из растворов ксилозы, которые сложно очищать, например, с помощью хроматографического разделения, которое, следовательно, не дает такие степени чистоты ксилозы, которые требуются в способах кристаллизации предшествующего уровня техники. В частности, целью настоящего изобретения является такой способ извлечения ксилозы из продуктов гидролиза биомассы, которые могут также быть содержащими ксилозу фракциями побочных продуктов, получаемыми в деревоперерабатывающей промышленности, такими как сульфитный варочный раствор или его часть, или полученный из него концентрат, например концентрат, хроматографически полученный из сульфитного варочного раствора, или предгидролизатная часть варочного раствора, или его постгидролизат или его ультрафильтрационное растворенное вещество.
В случае, когда раствор, который необходимо обработать, является водным раствором ксилозы (степень чистоты ксилозы примерно 30-50%), в соответствии с предпочтительным воплощением настоящего изобретения количество затравочных кристаллов, которое необходимо использовать, является высоким, по крайней мере, 10-кратным по сравнению с полной затравкой. Таким образом, перенасыщение во время кристаллизации составляет 1,4-3,0, предпочтительно 1,5-2,5. Размер получаемых кристаллов (длина кристаллов) обычно составляет 10-100 мкм.
Предпочтительным путем осуществления кристаллизации ксилозы в соответствии с настоящим изобретением является охлаждение затравленной кристаллизационной массы при относительно высокой скорости, за время примерно 10-50 часов или менее, до значения перенасыщения, необходимого для зародышеобразования. Здесь температура кристаллизационной массы составляет обычно 20-50oС в зависимости от содержания сухого вещества кристаллизационной массы, и вязкость кристаллизационной массы находится в интервале 100-600 Па•с.
Суспензию перемешивают до тех пор, пока не будет достигнута достаточная степень кристаллизации (выход, уменьшение степени чистоты ксилозы маточной жидкости). Например, кристаллизационный сосуд, снабженный перемешивающими лопастями длиной 1,3-1,7 м (от вала до верхушки) с областями высокого сдвига обычно используют при скорости вращения сначала 3-6 об/мин, а во время периода с высокой вязкостью - 0,5-3 об/мин. Скорость сдвига по отношению к эффективности перемешивания контролируют, чтобы избежать затвердевания кристаллизационной массы при поддержании зародышеобразования. Как правило, мощность, приложенная к мешалке, составляет между примерно 100 Вт/м3 и примерно 800 Вт/м3. Такой предел обеспечивает эффективное перемешивание, посредством чего материал с зародышами переносится во внутреннюю часть кристаллизационной массы. Период осаждения 1-4 дня или даже меньше может уменьшить (преобразовать в кристаллический продукт) уровень ксилозы в маточной жидкости до примерно 20% или менее.
После этого перенасыщение кристаллизационной массы уменьшают путем увеличения температуры и/или разбавления кристаллизационной массы водой или содержащим ксилозу раствором без значительного растворения кристаллов до тех пор, пока вязкость кристаллизационной массы не уменьшится до степени, достаточной для эффективного выделения закристаллизованного материала. Типичная вязкость кристаллизационной массы составляет после понижения вязкости 5-100 Па•с. Кристаллы могут быть выделены путем фильтрации, декантации, центрифугирования и тому подобное, предпочтительно путем фильтрации. Маточная жидкость (то есть оттек), отделенная таким образом, может иметь содержание ксилозы, пониженное до очень низких значений (настолько низких как 16% по отношению к сухому веществу). Степень чистоты ксилозы из полученной кристаллической фракции составляет обычно 60-90% по отношению к сухому веществу, в зависимости от степени чистоты ксилозы из кристаллизационной массы и осуществления процесса, и указанная фракция легко может быть очищена, если это необходимо, например, с помощью обычных методик кристаллизации. Степень чистоты кристаллической фракции, полученной с помощью способа по настоящему изобретению, может быть повышена путем замещения некоторого количества маточной жидкости растворителем или воздухом.
Невозможно кристаллизовать ксилозу из растворов, имеющих степень чистоты менее чем примерно 70%, с помощью известных из уровня техники методов без того, чтобы подвергнуть растворы громоздким обработкам для очистки. Новый способ, разработанный в настоящее время, дает возможность достижения кристаллизации при степенях чистоты ксилозы вплоть до 30% по отношению к сухому веществу.
В соответствии с другим предпочтительным воплощением настоящего изобретения сахароза может быть кристаллизована из ее водных растворов, таких как из мелассы, полученной в сахарной промышленности. В этом случае небольшое количество глицерина (или какого-либо другого органического растворителя, который может присутствовать в мелассе) может быть добавлено к сырой мелассе перед ее подачей в процесс кристаллизации.
Полученный таким образом раствор выпаривают при пониженном давлении до содержания сухого вещества (СВ) примерно 90-95 г/100 г, и полученную кристаллизационную массу при примерно 80-90oС переносят в кристаллизатор.
Кристаллизационную массу затравливают измельченной сахарозой (средний размер кристаллов от 5 до 10 мкм) при 70-90oС. Затравочные кристаллы используют в количестве, которое составляет вплоть до 100-кратного, по сравнению со случаем, где кристаллизация основывается главным образом на росте кристаллов. Количество затравочных кристаллов не является очень важным, поскольку во время эффективного перемешивания путем зародышеобразования образуется множество новых кристаллов.
Кристаллизацию проводят в кристаллизаторе в течение примерно 10 дней. Кристаллизационную массу охлаждают до примерно 50oС за 2-3 дня и перемешивают при этой температуре в течение примерно 7 дней перед приготовлением для фильтрации. Самое высокое значение вязкости кристаллизационной массы составляет менее 800 Па•с, и она уменьшается при осуществлении кристаллизации.
Перед фильтрованием вязкость кристаллизационной массы понижают путем увеличения температуры на 5-15oС и/или необязательного добавления глицерина и/или воды в количестве вплоть до примерно 10 мас.%. Размер полученных кристаллов обычно составляет примерно 10-50 мкм.
Кристаллическую фракцию предпочтительно извлекают путем фильтрования под давлением. Эффективный выход сахарозы, получаемый после фильтрования при проводимых таким образом экспериментах, составляет примерно 30% сахарозы, содержащейся в исходной мелассе, имеющей степень чистоты сахарозы 40-60%, основанной на содержании сухого вещества (СВ). Выход может быть улучшен путем дальнейшей оптимизации условий процесса.
Фильтрование для извлечения кристаллического продукта может быть удобным образом осуществлено с фильтром, работающим под давлением, например фильтром Larox с 10-20 пластинами, с использованием волокнистой ткани умеренной пористости, разделения при 2-16 бар и при времени прессования 0,5-1,0 часа.
Конкретные воплощения способа настоящего изобретения будут проиллюстрированы более подробно посредством последующих примеров, которые не предназначены для ограничения объема изобретения.
В некоторых примерах концентрация кристаллизующегося соединения повышается путем добавления чистого соединения для демонстрации работоспособности изобретения при различных степенях чистоты.
Содержания сухого вещества определяют с помощью метода титрования по Карлу Фишеру (СВ) или с помощью рефрактометрического метода (РСВ).
Углеводы анализируют с помощью жидкостной хроматографии (ВЭЖХ) с использованием колонок, в которых ионообменная смола находится в Na+ и Pb2+ формах, или с помощью ИЭДЖХ (то есть ВЭЖХ с использованием импульсного электрохимического детектора). Цвет определяют с помощью адаптированного метода ICUMSA [смотри Sugar Analysis; Official and Tentative Methods Recommended by the International Commission for Uniform Methods of Sugar Analysis (ICUMSA), ed. Schneider, F. , ICUMSA, Peterborough, England., 1979, pp. 125-128] при рН 5 (пример кристаллизации ксилозы) и при рН 7 (другие примеры), и путем проведения измерения из отфильтрованного раствора (0,45 мкм) при 420 нм.
Пример 1
Кристаллизация ксилозы
150 л фракции ксилозы, полученной из сульфитного варочного раствора на основе магния из букового дерева с помощью хроматографического разделения (по
существу в соответствии с первой стадией способа, описанного в патенте США 4631129), содержащей примерно 105 кг сухого вещества и имеющей степень чистоты ксилозы 39,3%, выпаривают при давлении ниже
атмосферного при примерно 60oС до объема примерно 80 л. Эту фракцию ксилозы затравливают при 58oС 25 г измельченной ксилозы при степени перенасыщения 2,24, и кристаллизационную
массу переносят в 100-литровый кристаллизатор.
Кристаллизационную массу подвергают линейному охлаждению от 58oС до примерно 20oС при одновременном перемешивании (вязкость 190 Па•с, измеренная вискозиметром Brookfield, модель RVDV-1+) через примерно 25 часов, в течение этого времени перенасыщение понижают сначала до 1,66 через 3,7 часа, затем увеличивают до 1,93 (время от затравливания 20,9 час, температура 30,7oС), а после этого снова постепенно понижают (при 20oС степень перенасыщения составляет примерно 1,70). Кристаллизационную массу дополнительно перемешивать при примерно 20oС. Фильтр, работающий под давлением, типа Larox PF 0,1 Н2 используют для выделения кристаллической фракции из кристаллизационной массы. Образцы (примерно 20-200 г) отбирают из кристаллизационной массы при различных временах для отделения маточной жидкости и перемешивание остатка кристаллизационной массы продолжают. Перед фильтрованием кристаллизационной массы ее температуру повышают до примерно 30oС, чтобы уменьшить вязкость.
Через 74,3 часа после затравливания вязкость
образца кристаллизационной массы составляет 66 Па•с при примерно 30oС. Образец кристаллизационной массы фильтруют с помощью указанного выше фильтра, работающего под давлением, Larox,
сначала используя давление фильтрования 13 бар в течение 15 минут, а затем давление фильтрования 14,5 бар в течение пяти минут. Полученная кристаллическая лепешка имеет толщину примерно 2,5 см. Выход
сухого вещества в кристаллизационной массе перед фильтрованием составляет 20,2%, а выход ксилозы - 50,4%. Результаты анализов представлены в таблице 1, в которой обозначения и сокращения имеют
следующие значения:
Начальн. = образец кристаллизационной массы перед началом охлаждения;
рН 5% = рН, определенный для образца, разбавленного водой до РСВ 5%;
Проводим. =
проводимость, определенная для образца, разбавленного до РСВ 5%;
Зола = содержание золы, вычисленное по проводимости с помощью использования сахарозного коэффициента для сульфатной золы;
Фильтр. = кристаллизационная масса, подаваемая на фильтр.
Проведенные тесты показывают, что выход и степень чистоты ксилозы подвержены влиянию времени перемешивания кристаллизационной массы в зоне зародышеобразования (в данном случае, в интервале температур примерно 20-30oС). Степень чистоты ксилозы отфильтрованной кристаллической фракции составляет в лучшем случае 83,8% (время после затравливания составляет 76,2 час; вязкость кристаллизационной массы составляет 66 Па•с при 29,8oС; фильтрование при 14,5 бар в течение пяти минут), степень чистоты ксилозы фильтрата, то есть отека, составляет самое меньшее 18,1% (время после затравливания 220 час; вязкость кристаллизационной массы 59 Па•с при 29,2oС; фильтрование при 13-14 бар в течение 15 минут). Выход ксилозы в кристаллы из кристаллизационной массы составляет самое большее 63,2% (время после затравливания 49,3 час).
Пример 2
Кристаллизация ксилозы
Если не утверждается иного, процедура подобна процедуре примера 1. Содержащий ксилозу раствор, который должен быть обработан (20,5 кг), получают путем объединения
фракции ксилозы, полученной из сульфитного варочного раствора из бука на основе магния с помощью хроматографического разделения, и водного раствора кристаллической лепешки, полученной из
предварительных исследований кристаллизации зародышеобразованием. Раствор имеет содержание сухого вещества (СВ) 62,7% и степень чистоты ксилозы 53,0%.
Раствор выпаривают до содержания сухого вещества (СВ) 89,7%. 13,4 кг полученной кристаллизационной массы переносят в 10-литровый кристаллизатор. Осуществляют затравливание при 65oС 5 г измельченной ксилозы (размер кристаллов 50 мкм) при перенасыщении 1,96 и линейное охлаждение от 65oС до примерно 20oС через примерно 17 часов. В течение этого времени степень перенасыщения уменьшается до 1, 71, и она остается в пределах 1,70-1,76, когда кристаллизационную массу перемешивают в зоне зародышеобразования (при температуре 20-22oС). Через 21,5 часа после затравливания (вязкость 183 Па•с при 22oС) кристаллизационную массу нагревают до 32oС и фильтруют с помощью фильтра, работающего под давлением (15 минут, давление фильтрования 13,5 бар).
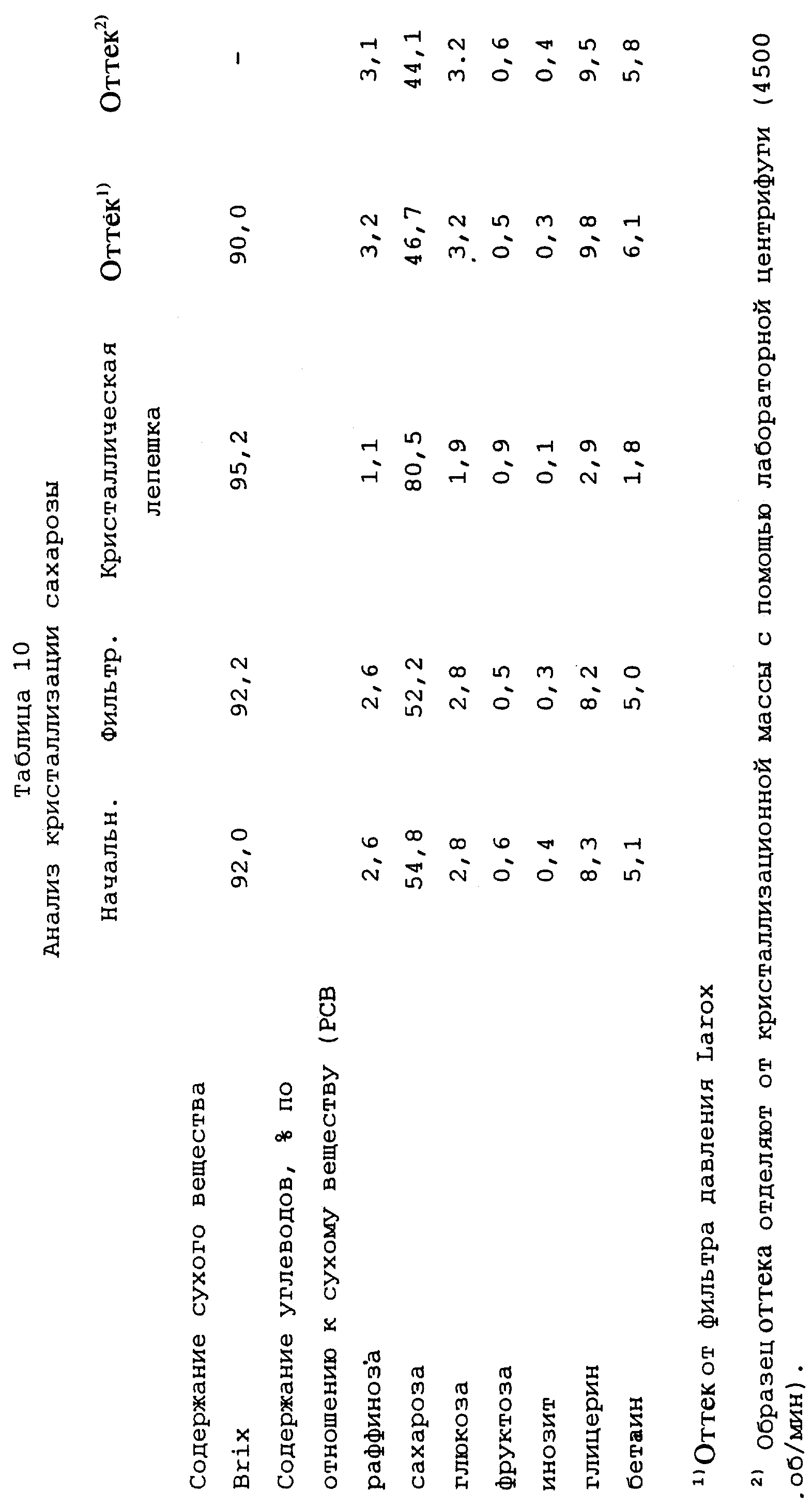
Выход сухого вещества в кристаллы из кристаллизационной массы перед фильтрованием составляет 38,1%, а выход ксилозы - 72,1%. Результаты анализа представлены в таблице 2, в которой определения и сокращения имеют те же самые значения, что и в примере 1.
Пример 3
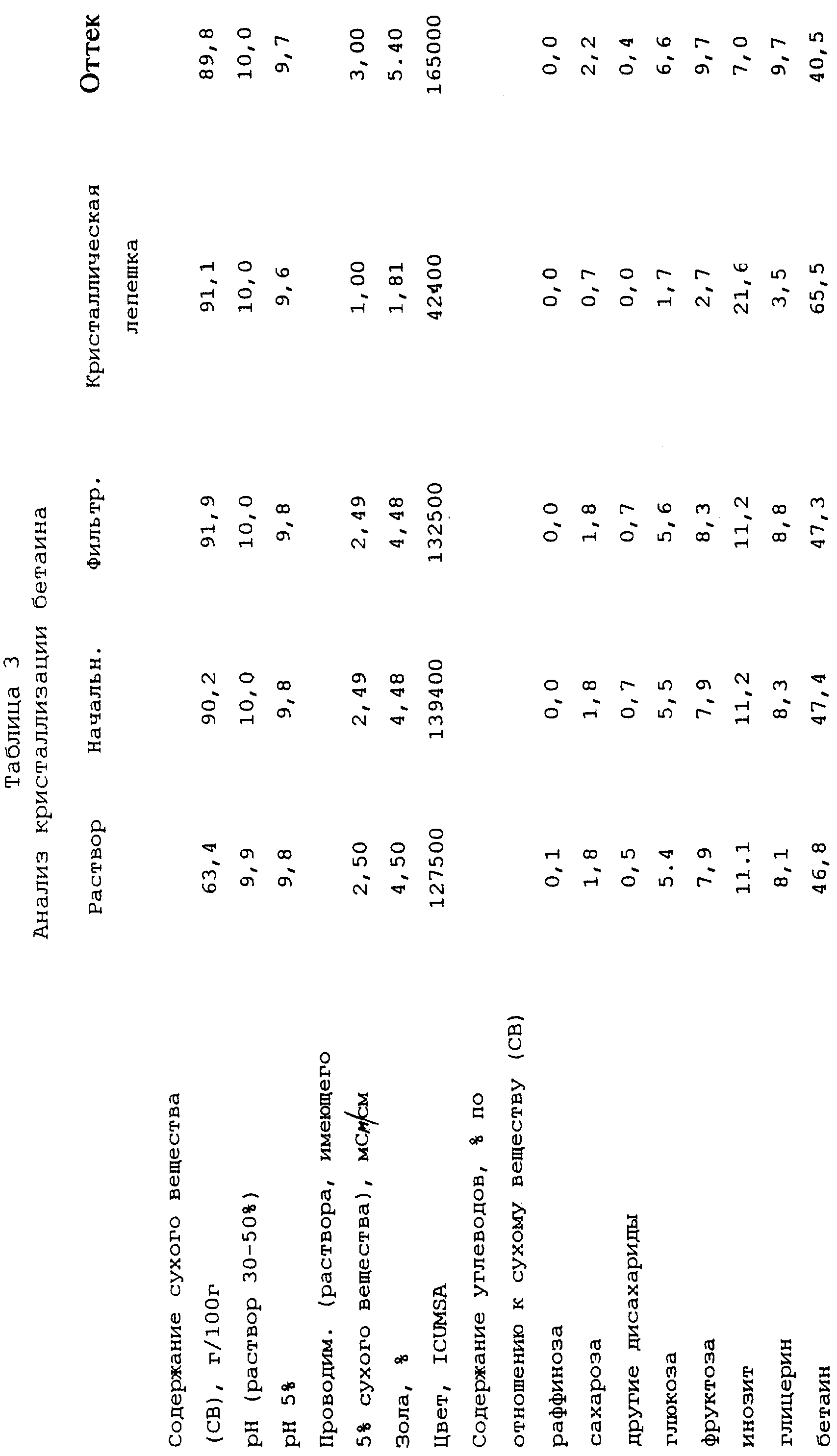
Кристаллизация бетаина
Раствор, который необходимо обработать, представляет собой
оттек, полученный путем кристаллизации бетаина из фракции бетаина от хроматографического разделения мелассы (указанный выше патент Финляндии 77845; Международная заявка WO 81/02420). Содержание сухого
вещества (СВ) этого раствора составляет 63,4 г/100 г, и результаты его анализа представлены в таблице 3.
12,3 кг этого раствора выпаривают при давлении ниже атмосферного в роторном испарителе при температуре, превышающей 80oС, до содержания сухого вещества (СВ) 90,2 г/100 г (результаты анализов представлены в таблице 3). Программа линейного охлаждения полученного таким образом концентрированного раствора в 6-литровом кристаллизаторе начинается с 95oС, перенасыщение раствора затем составляет 1,74. В течение полной кристаллизации кристаллизационную массу энергично перемешивают. Через 6,2 часа температура составляет 76,5oС, степень перенасыщения составляет 3,18, и никакой кристаллизации не происходит. В этот момент добавляют 0,6 г измельченного моногидрата бетаина и начинают зародышеобразование. Образцы (примерно 20-200 г) отбирают из кристаллизационной массы в различные моменты времени для отделения маточной жидкости и продолжают перемешивание кристаллизационной массы. Охлаждение продолжают линейно до 30oС (время от затравливания моногидратом бетаина - 31,1 час), степень перенасыщения затем составляет 2, 43. Кристаллизационную массу перемешивают при этой температуре в течение 3,8 часа, после чего температуру повышают до 35oС за 0,8 часа (после этого вязкость составляет 113 Па•с), а дополнительно до 37oС за 0,9 часа. В этот момент вязкость составляет 84 Па•с, и кристаллическую фракцию отделяют от кристаллизационной массы с помощью фильтра, работающего под давлением, Larox из примера 1, используя давление фильтрования 14-15 бар, в течение 30 минут. Получают сухую кристаллическую лепешку, имеющую толщину 8 мм.
Результаты анализов представлены в таблице 3, в которой определения и сокращения имеют те же самые значения, как и в предыдущих примерах, за исключением того, что цвет измеряют при рН 7. Кроме того, термин "Раствор", использованный в первом столбце, относится к раствору сырого материала перед выпариванием.
Выход бетаина в кристаллическую фракцию составляет 37,7% бетаина, содержащегося в исходном растворе, а выход инозита в кристаллическую фракцию составляет 55,5% инозита, содержащегося в растворе.
Проведенные исследования показали, что выход и степень чистоты бетаина и инозита подвержены влиянию времени перемешивания кристаллизационной массы в зоне зародышеобразования. Степень чистоты объединенных бетаина и инозита отфильтрованной кристаллической фракции составляет в самом лучшем случае 87,1% (время от затравливания составляет 37 час). Степень чистоты бетаина фильтрата, то есть оттека, отделенного от кристаллической массы, составляет самое меньшее 33,3%, и чистота инозита составляет 7,0 (время от затравливания 31 час).
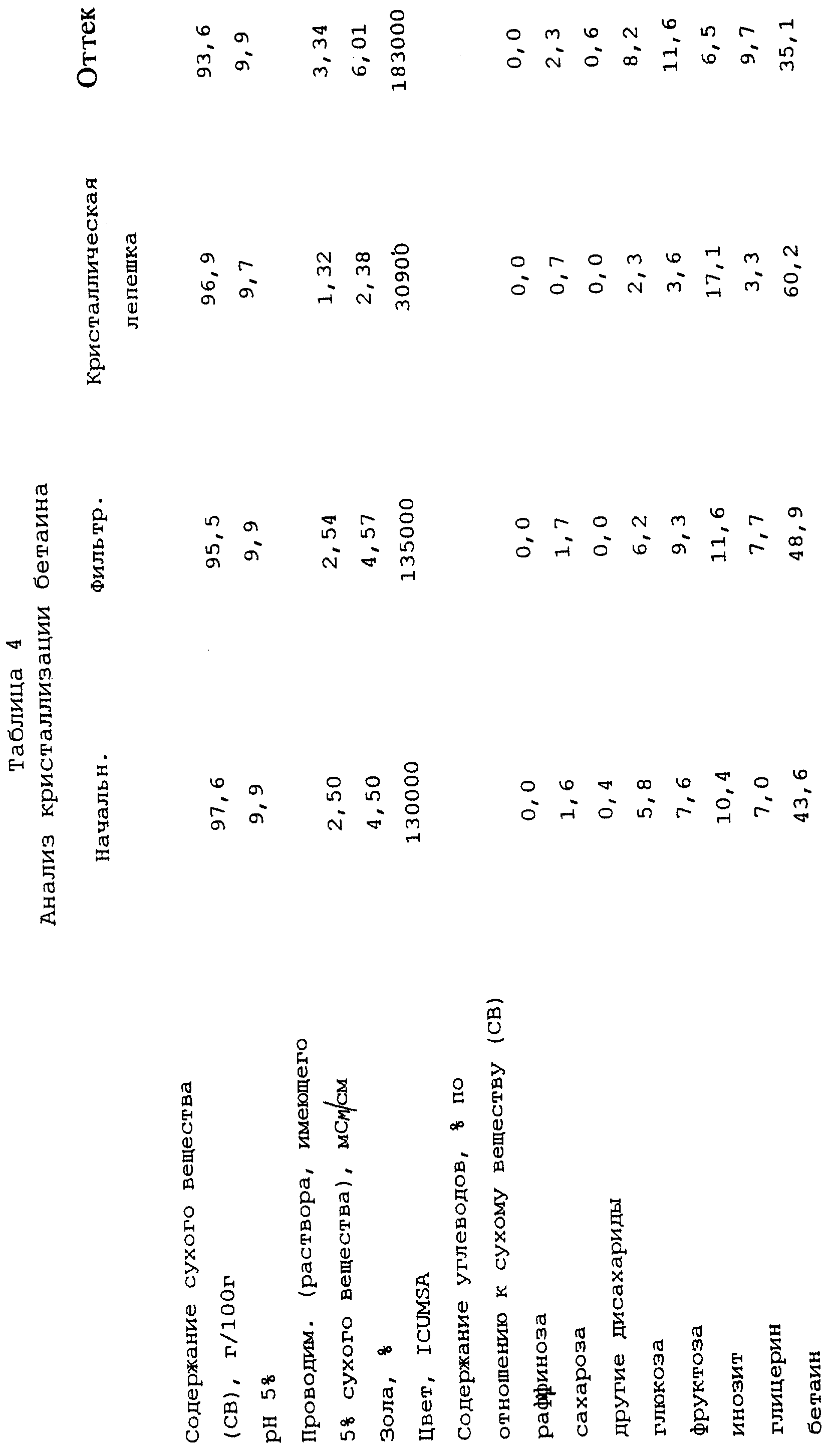
Пример 4
Кристаллизация бетаина
Раствор, который необходимо обработать, такой же, как в примере 3. 13,6 кг
данного раствора выпаривают при давлении ниже атмосферного в роторном испарителе при температуре несколько ниже 80oС до содержания сухого вещества (СВ) 97,6 г/100 г. В этой ситуации
затравливание самопроизвольное, степень перенасыщения составляет 3,69. 7 кг кристаллизационной массы переносят в 6-литровый кристаллизатор, нагревают до 95oС и добавляют 150 мл воды при
примерно 90oС. Полученную таким образом кристаллизационную массу линейно охлаждают путем энергичного перемешивания в течение 10 часов от 95oС до 70oС.
Кристаллизационную массу перемешивают при этой температуре в течение примерно девяти часов (в течение ночи), после чего ее охлаждают в течение примерно пяти часов до постоянной температуры 36oС, при которой ее перемешивают в течение примерно 62 часов. Вязкость кристаллизационной массы составляет 15,6 Па•с при 70oС, 55 Па•с при 45oС и, после
перемешивания в течение 90 часов (от затравливания) - 347 Па•с при 36oС. После указанного выше времени перемешивания температуру кристаллизационной массы сначала повышают до 48oС (вязкость 75 Па•с), а затем кристаллическую фракцию отделяют от кристаллизационной массы при 45oС (вязкость 116 Па•с, степень перенасыщения 17,87) с помощью фильтра,
работающего под давлением, Larox из примера 1, используя давление фильтрования 14,5 бар в течение 30 минут. Получают по существу кристаллическую сухую лепешку, имеющую толщину 8 мм.
Результаты анализа представлены в таблице 4, в которой определения и сокращения соответствуют определениям и сокращениям из примера 3.
Выход бетаина в кристаллическую фракцию составляет 47,0% бетаина, содержащегося в исходном растворе, а выход инозита составляет 60,5% инозита, содержащегося в растворе.
Степень чистоты объединенных бетаина и инозита отфильтрованной кристаллической фракции составляет в самом лучшем случае 77,3%, а степень чистоты бетаина в оттеке составляет самое меньшее 30,9%, и степень чистоты инозита составляет самое меньшее 6, 5%.
Пример 5
Кристаллизация ксилита
Раствор, который необходимо обработать, представляет собой оттек, полученный от кристаллизации ксилита. Его выпаривают с помощью
роторного испарителя при давлении 40 мбар при 70oС до содержания сухого вещества (РСВ определяют с помощью таблиц-диаграмм для ксилита) 93,8 г/100 г.
12,3 кг полученной кристаллизационной массы переносят в 10-литровый кристаллизатор, нагревают до 50oС (s=l,5), затравливают 10 г измельченного ксилита и охлаждают до 25oС в течение 10 час. Примерно через три часа после того, как достигается температура 25oС, кристаллизационная масса имеет вязкость 61,5 Па•с (s=3,9). Кристаллизационную массу перемешивают при этой температуре в течение в целом 8 часов, после чего температуру дополнительно понижают (температура охлаждающей воды - 15oС). Примерно через 3 часа кристаллизационная масса имеет температуру 16o С (s=4,9). Кристаллизационную массу перемешивают при этой температуре бани в течение 18 часов, после чего вязкость составляет 250 Па•с (s=3,0), когда кристаллизационная масса имеет температуру 18oС.
После этого температуру кристаллизационной массы повышают до 25oС в течение примерно трех часов (после этого вязкость составляет 81,5 Па•с (s= 2,1)), а далее - до 28oС в течение примерно двух часов.
В этот момент кристаллизационная масса имеет вязкость 59,5 Па•с (s=2,0), и кристаллическую фракцию отделяют от кристаллизационной массы с помощью фильтра, работающего под давлением, Larox из предыдущих примеров, используя давление фильтрования 12 бар в течение 15 минут. Давление убирают перед формированием подходящей кристаллической лепешки.
Результаты анализа представлены в таблице 5, в которой определения и сокращения имеют такие же значения, как и в примере 3.
Выход ксилита в кристаллическую фракцию составляет 67% ксилита, содержащегося в исходном растворе.
Пример 6
Кристаллизация сахарозы
Раствор, который необходимо обработать,
представляет собой мелассу, полученную с завода для переработки сахарной свеклы. Раствор выпаривают в роторном испарителе до содержания сухого вещества (РСВ определяют с помощью таблиц-диаграмм для
сахарозы) 90,3 г/100 г.
14,5 кг полученной кристаллизационной массы переносят в 10-литровый кристаллизатор, нагревают до температуры 62oС и затравливают 10 г измельченной сахарозы, и охлаждают при одновременном энергичном перемешивании до 40oС в течение 40 часов. Примерно через 25 часов после того, как достигается температура 40oС, кристаллизационная масса имеет вязкость в 550 Па•с. Температуру кристаллизационной массы повышают до 53oС в течение примерно пяти часов, после чего вязкость составляет 111 Па• с и кристаллическую фракцию отделяют с помощью фильтра, работающего под давлением, Larox, используя давление фильтрования 12 бар, в течение 15 минут. Давление убирают перед формированием по существу сухой кристаллической фракции.
Результаты анализа представлены в таблице 6, в которой определения и сокращения имеют такие же значения, как в примере 3.
Пример 7
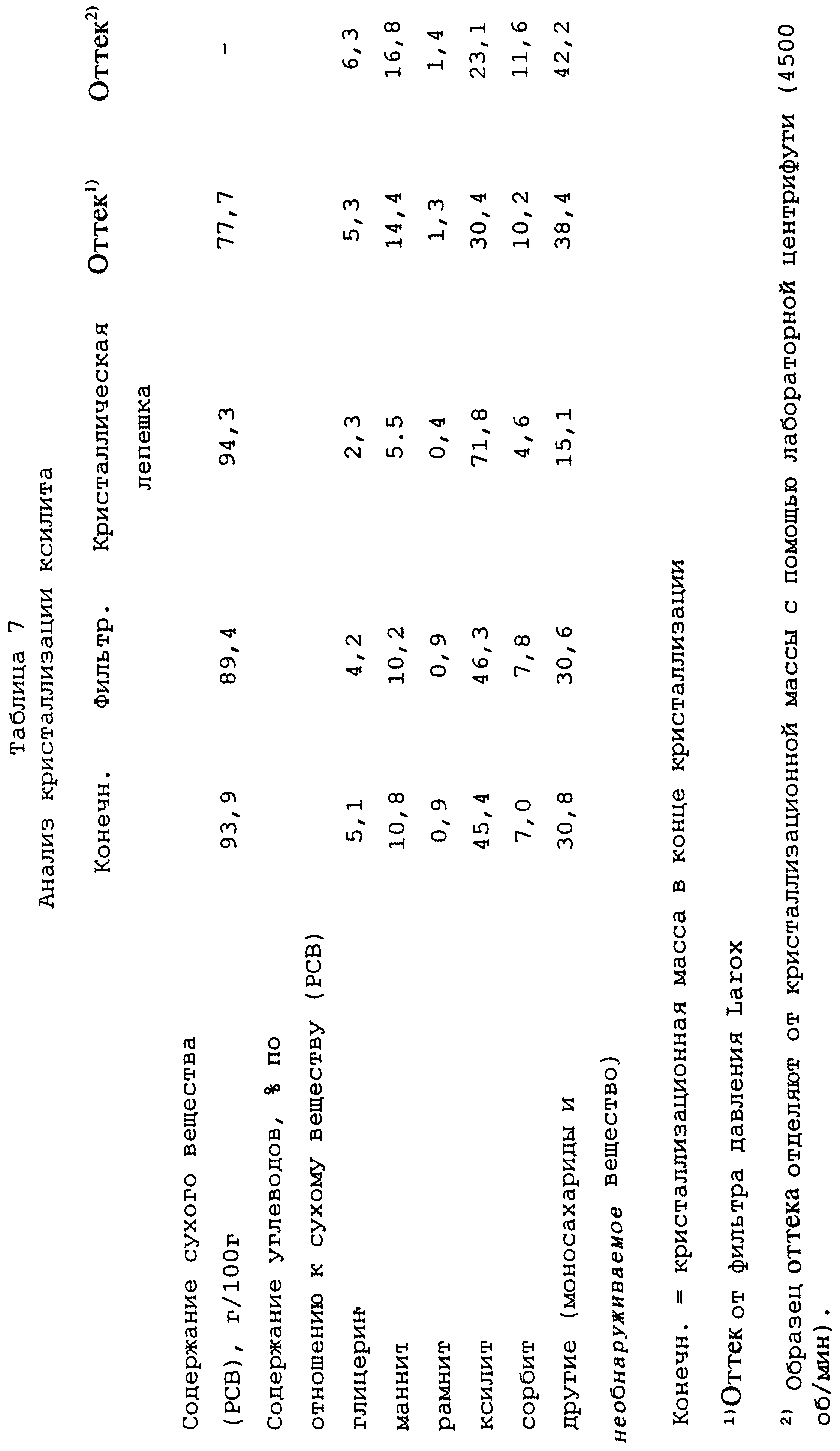
Кристаллизация ксилита
Исходным материалом является оттек, полученный от предыдущих кристаллизаций ксилита. Его фильтруют с помощью лабораторного вакуумного фильтра. Степень чистоты
ксилита по отношению к сухому веществу (РСВ) в полученном растворе увеличивают до примерно 46% путем добавления чистого кристаллического ксилита. Раствор выпаривают с помощью лабораторного вакуумного
испарителя при температуре бани 60-70oС в течение 6 часов до содержания сухого вещества (РСВ) 94,1 г/100 г.
13,58 кг (объем 10 л) полученной таким образом кристаллизационной массы переносят в 10-литровый кристаллизатор с температурой бани 50oС и перемешивают в течение двадцати минут. В это время кристаллизационная масса имеет температуру 51oС, и степень перенасыщения составляет 1,7. Затем кристаллизационную массу затравливают 10 г измельченного ксилита и подвергают линейному охлаждению от 50oС до 23oС (температура охлаждающей воды) в течение 15 часов. В конце этого периода охлаждения кристаллизационная масса имеет температуру 24oС, вязкость в 110 Па•с и степень перенасыщения 3,2.
Кристаллизационную массу дополнительно перемешивают при этой температуре в течение примерно 2 часов, после чего ее температуру понижают до 20oС в течение примерно 3 часов (после этого вязкость составляет 200 Па•с, степень перенасыщения 3,5), и дополнительно до 16oС в течение примерно 3 часов. После этого кристаллизационная масса имеет вязкость 345 Па• с. Перемешивание кристаллизационной массы примерно при этой температуре (охлаждающая вода при постоянной температуре 15oС) продолжают в течение 42 часов. Образец, отобранный через 17 часов перемешивания при этой температуре, имеет вязкость 400 Па•с и степень перенасыщения 4,0. В конце этого периода перемешивания вязкость кристаллизационной массы составляет 407 Па•с.
После этого температуру кристаллизационной массы повышают за полчаса до 20oС (после этого вязкость составляет 256 Па•с),
и далее - в течение трех часов до 23oС. В этот момент кристаллизационная масса имеет вязкость 198 Па•с. В этот момент времени отбирают образец кристаллизационной массы, и от нее с помощью лабораторной центрифуги отделяют
образец оттека.
Затем кристаллизационную массу удаляют из кристаллизатора, добавляют воду в количестве 5 маc.% от кристаллизационной массы для понижения вязкости и кристаллическую фракцию отделяют с помощью фильтра, работающего под давлением, Larox из предыдущих примеров, используя давление фильтрования 15 бар в течение 30 минут.
Результаты анализа представлены в таблице 7, в которой определения и сокращения имеют такие же значения, как в примере 5.
Выход ксилита в кристаллическую лепешку при фильтровании с помощью фильтра Larox составляет 57% от ксилита, содержащегося в исходном растворе.
Пример 8
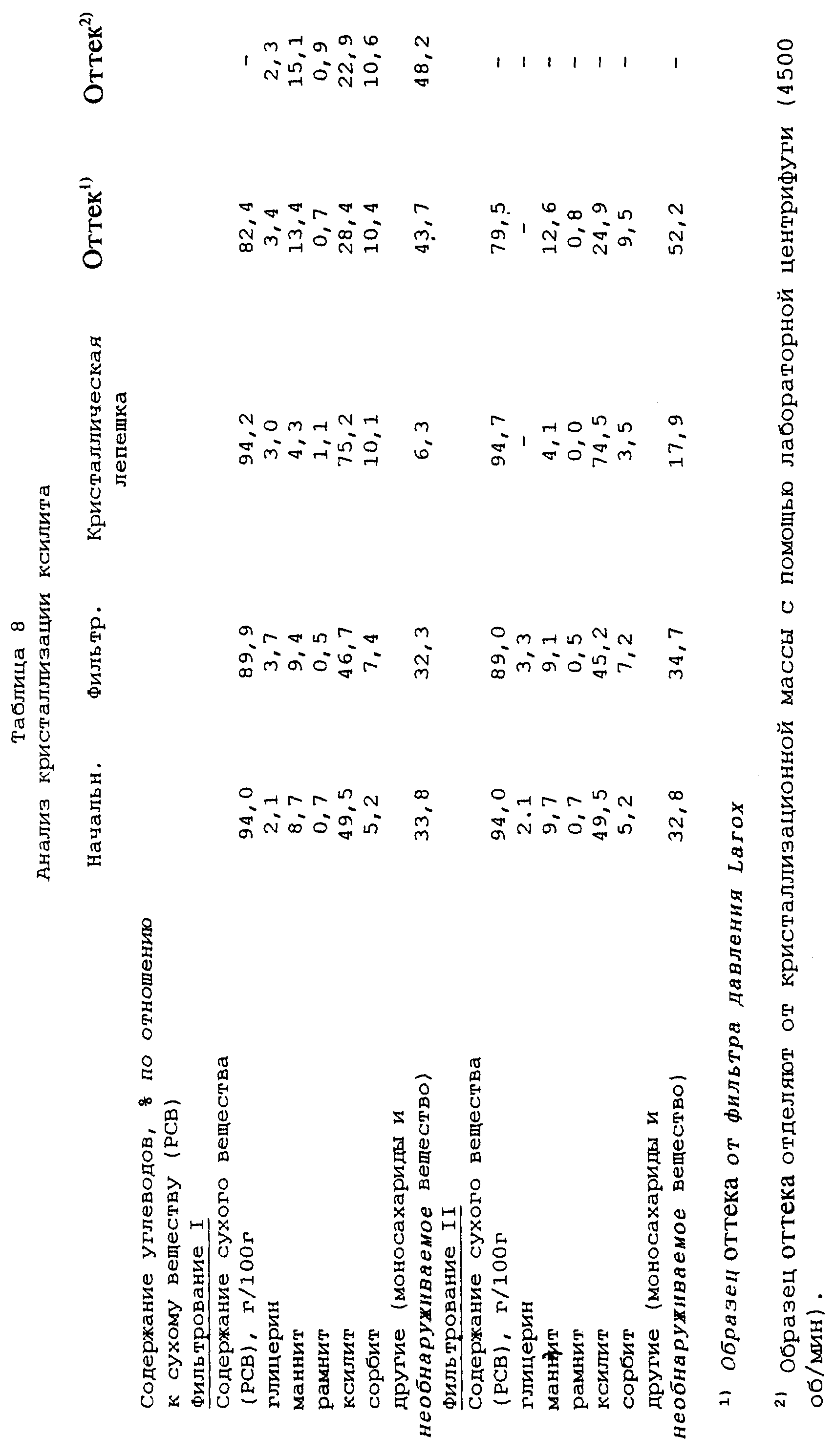
Кристаллизация ксилита
Используют такой же исходный материал, как и в примере 7. Степень чистоты ксилита в
отфильтрованном растворе увеличивают до примерно 47% добавлением чистого кристаллического ксилита. Раствор выпаривают с помощью роторного испарителя при температуре бани 70oС до содержания
сухого вещества (РСВ) 94,4 г/100 г.
13,52 кг полученной таким образом кристаллизационной массы переносят в 10-литровый кристаллизатор. Как и в примере 7 кристаллизационную массу эффективно перемешивают во время всей процедуры. Кристаллизационную массу затравливают при температуре 56oС (s=l,4) с помощью 10 г измельченного ксилита и подвергают линейному охлаждению. Примерно через 26 часов кристаллизационная масса достигает температуры 20,5oС. Кристаллизационную массу перемешивают при этой температуре в течение 42 часов, после этого времени степень перенасыщения составляет 3,6. Вязкость кристаллизационной массы в конце этого периода перемешивания составляет 280 Па•с.
После этого температуру кристаллизационной массы повышают примерно в течение двух часов до 25oС (после этого вязкость составляет 176 Па•с, s=3,1) и перемешивание при этой температуре продолжают в течение одного часа. В этот момент времени отбирают образец кристаллизационной массы и с помощью лабораторной центрифуги отделяют от нее образец оттека.
Часть кристаллизационной массы затем удаляют из кристаллизатора и к ней добавляют воду в количестве 5 мас.% для понижения вязкости, которая после добавления воды составляет 28 Па•с. Кристаллическую фракцию отделяют от этой части с помощью фильтра, работающего под давлением, Larox из предыдущих примеров, используя давление фильтрования 16 бар в течение 1 час 15 мин.
К остатку кристаллизационной массы в кристаллизаторе добавляют этанол в количестве 5 мас.% кристаллизационной массы, которую перемешивают при 25oС в течение примерно получаса. Затем кристаллизационную массу удаляют из кристаллизатора и фильтруют таким же путем, как описано выше для первой части кристаллизационной массы.
Результаты анализа представлены в таблице 8, в которой определения и сокращения имеют такие же значения как в примере 5, "Фильтрование I" относится к фильтрованию с помощью фильтра Larox с добавлением воды, а "Фильтрование II" - к фильтрованию с помощью фильтра Larox с добавлением этанола.
Выход ксилита в кристаллическую лепешку от первого фильтрования (с добавлением воды) составляет 68%, а от второго фильтрования (с добавлением этанола) - 74% от ксилита, содержащегося в исходном растворе.
Пример 9
Кристаллизация сахарозы
Исходный материал, который необходимо обработать, представляет собой мелассу, полученную с завода по обработке сахарной свеклы. Мелассу
фильтруют и фильтрат выпаривают при пониженном давлении до содержания сухого вещества Вх 93,0.
100 л полученной кристаллизационной массы переносят в 100-литровый кристаллизатор, затравливают 100 г измельченной сахарозы при 78,5oС и охлаждают при одновременном эффективном перемешивании до 50oС в течение примерно 60 часов. Вязкость кристаллизационной массы после этого составляет примерно 800 Па•с, и перемешивание продолжают, поддерживая температуру по существу неизменной. Примерно через 170 часов после того, как достигается температура 50oС, кристаллизационная масса имеет вязкость примерно 670 Па•с. Через 172 часа при примерно 50oС температура кристаллизационной массы повышается до примерно 60oС в течение примерно пяти часов, и после примерно 24 часов при этой температуре кристаллизационная масса имеет вязкость примерно 280 Па•с. Примерно через 60 часов после достижения температуры 60oС кристаллизационная масса имеет вязкость 241 Па•с.
Вязкость дополнительно понижают добавлением воды (2 мас.%) и кристаллическую фракцию отделяют с помощью фильтра, работающего под давлением, Larox, используя давление фильтрования 16 бар в течение 60 минут. Температуру подачи на фильтр Larox (фильтр.) быстро повышают до 69oС непосредственно перед фильтрованием.
Результаты анализа представлены в таблице 9, в которой определения и сокращения имеют такие же значения, как и в предыдущих примерах, если не указано иного.
Пример 10
Кристаллизация сахарозы
Исходный материал, который необходимо обработать, представляет собой такую же мелассу, как в примере 9, и ее сначала обрабатывают, как
описано в примере 9, за исключением того, что перед выпариванием к отфильтрованному раствору добавляют некоторое количество фракции глицерина. Фракцию глицерина, полученную при хроматографическом
фракционировании барды, используют для добавления глицерина, и добавленное количество глицерина составляет 10% от сухого вещества, содержащегося в отфильтрованном растворе.
100-литровый кристаллизатор загружают полученной таким образом кристаллизационной массой (Вх 92,0) и кристаллизационную массу затравливают 100 г измельченной сахарозы при 76oС. Кристаллизационную массу охлаждают при одновременном энергичном перемешивании до примерно 50oС в течение примерно 60 часов. После этого вязкость кристаллизационной массы составляет примерно 210 Па•с, и перемешивание продолжают при этой температуре в течение 11 часов. После этого температуру понижают до 46,5oС, в результате чего вязкость сначала увеличивается до примерно 280 Па•с и постепенно уменьшается до примерно 220 Па•с в течение 145 часов при этой температуре.
Затем температуру постепенно повышают до 53oС (вязкость 120 Па•с) и в течение примерно 30 часов при этой температуре кристаллическую фракцию отделяют с помощью фильтра, работающего под давлением, Larox, используя давление фильтрования 16,2 бар в течение 65 минут.
Результаты анализа представлены в таблице 10, в которой определения и сокращения имеют такие же значения, как и в предыдущих примерах.
Выход сахарозы в кристаллическую лепешку во время фильтрования с помощью фильтра Larox составляет 35% от сахарозы в исходном питающем сиропе.
Пример 11
Кристаллизация сахарозы
Исходный материал, который необходимо обработать, представляет собой мелассу, полученную с завода по обработке сахарного тростника. Мелассу выпаривают при пониженном давлении до содержания сухого
вещества СВ 88,1 (определяют по методу Карла Фишера).
12,3 кг полученной кристаллизационной массы переносят в 10-литровый кристаллизатор, затравливают 10 г измельченной сахарозы при примерно 75oС и охлаждают при одновременном эффективном перемешивании до 50oС в течение примерно 60 часов. Вязкость кристаллизационной массы после этого составляет примерно 860 Па•с, и перемешивание продолжают, поддерживая температуру по существу неизменной. Через одиннадцать дней после того, как достигается температура 50oС, кристаллизационная масса имеет вязкость примерно 800 Па•с, первый образец оттека отделяют от кристаллизационной массы с помощью лабораторной центрифуги и перемешивают с массой 50 мл воды для понижения вязкости.
Через четыре дня после добавления воды кристаллизационная масса имеет вязкость примерно 510 Па•с и температуру 50oС, отделяют от кристаллизационной массы с помощью лабораторной центрифуги второй образец оттека и смешивают с массой 200 мл воды для понижения вязкости.
Через четыре дня после добавления 200 мл воды вязкость дополнительно понижают путем повышения температуры кристаллизационной массы до примерно 60oC в течение примерно пяти часов. Примерно через час при этой температуре кристаллизационная масса имеет вязкость примерно 75 Па•с, и кристаллическую фракцию отделяют с помощью фильтра, работающего под давлением, Larox, используя давление фильтрования 16 бар в течение 60 минут. Скорость фильтрования - медленная. Ткань фильтра представляет собой Tamfelt 71-2209-L1 с размером пор примерно 17 микрометров.
Результаты анализа представлены в таблице 11, в которой определения и сокращения имеют такие же значения, как и в предыдущих примерах, если не указано иного.
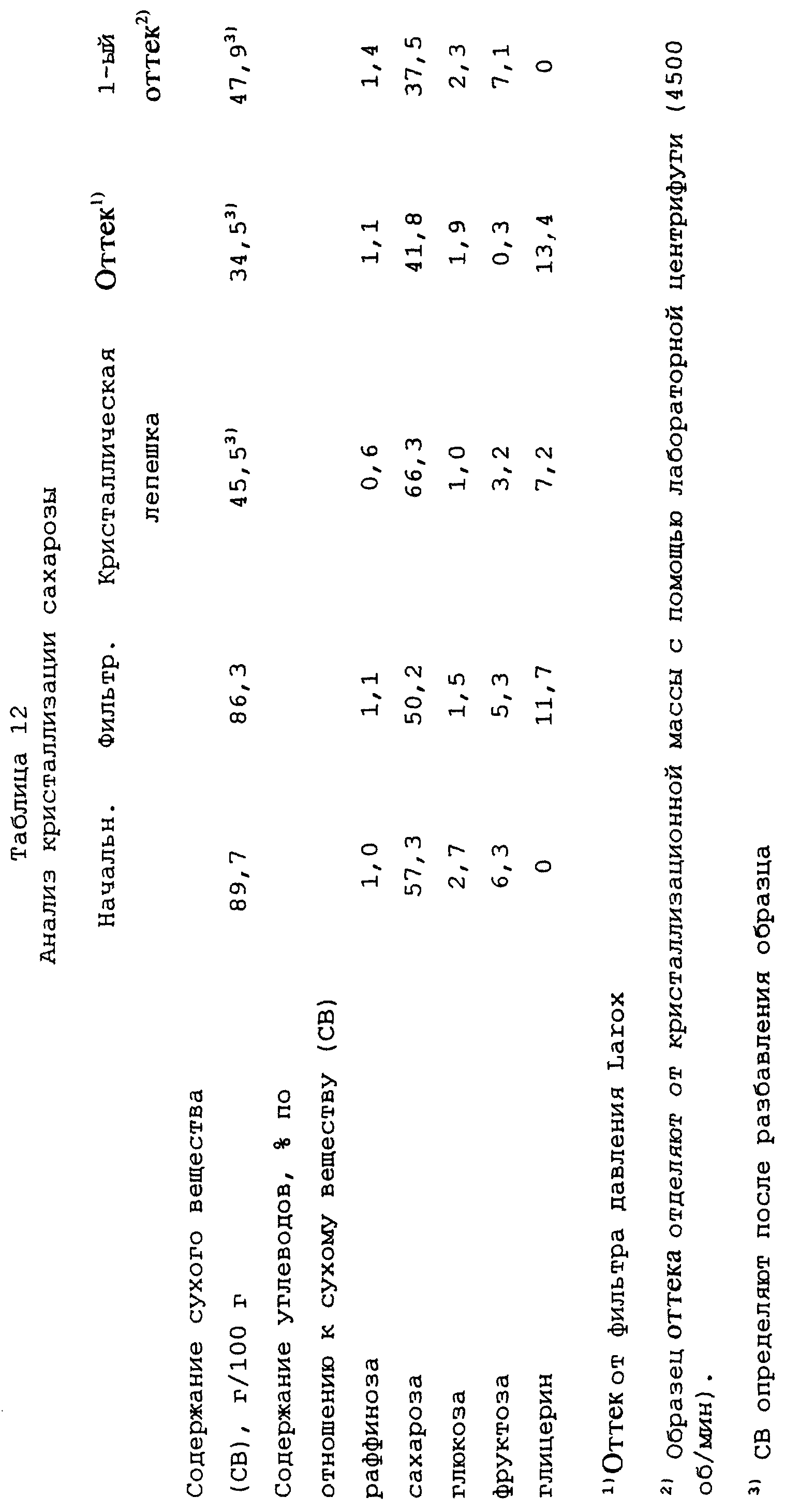
Пример 12
Кристаллизация сахарозы
Исходный материал, который необходимо обработать,
представляет собой такую же мелассу, как в примере 11, за исключением того, что перед выпариванием добавляют некоторое количество сахарозы для повышения степени чистоты питающего сиропа до примерно
58%/СВ. Сироп выпаривают при пониженном давлении до содержания сухого вещества СВ 89,7.
8,4 кг полученной кристаллизационной массы переносят в 6-литровый кристаллизатор, затравливают 8 г измельченной сахарозы при примерно 75oС и охлаждают при одновременном эффективном перемешивании до 50oС в течение примерно 60 часов. Вязкость кристаллизационной массы после этого составляет примерно 900 Па•с, и с массой смешивают 60 мл воды для понижения вязкости, и перемешивание продолжают, поддерживая температуру по существу неизменной. Через восемь дней после того, как достигается температура 50oС, кристаллизационная масса имеет вязкость примерно 720 Па•с, с помощью лабораторной центрифуги отделяют от кристаллизационной массы первый образец оттека и перемешивают 20 мл воды с массой.
Через четыре дня после добавления воды кристаллизационная масса имеет вязкость примерно 610 Па•с и температуру 50o С, и смешивают 1 кг 63% раствора глицерин/вода с массой для понижения вязкости.
Через пять дней после добавления глицерина вязкость кристаллизационной массы составляет 17 Па•с, а температура - 50oС. Через один день перемешивания при этой температуре кристаллическую фракцию отделяют с помощью фильтра, работающего под давлением, Larox, используя давление фильтрования 16 бар в течение 60 минут. Ткань фильтра такая же, как в примере 11.
Результаты анализа представлены в таблице 12, в которой определения и сокращения имеют такие же значения, как и в предыдущих примерах, если не указано иного.
Выход сахарозы в кристаллическую лепешку во время фильтрования с помощью фильтра Larox составляет примерно 45% от сахарозы питающего сиропа.
Выходы сухого вещества (выраженные в мас.%/мас.), приведенные выше в примерах, вычисляют, используя следующую формулу:
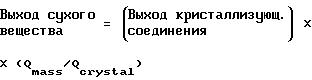
где Qmass представляет собой степень чистоты кристаллизационной массы, и Qcrystal представляет собой степень чистоты кристаллической лепешки.
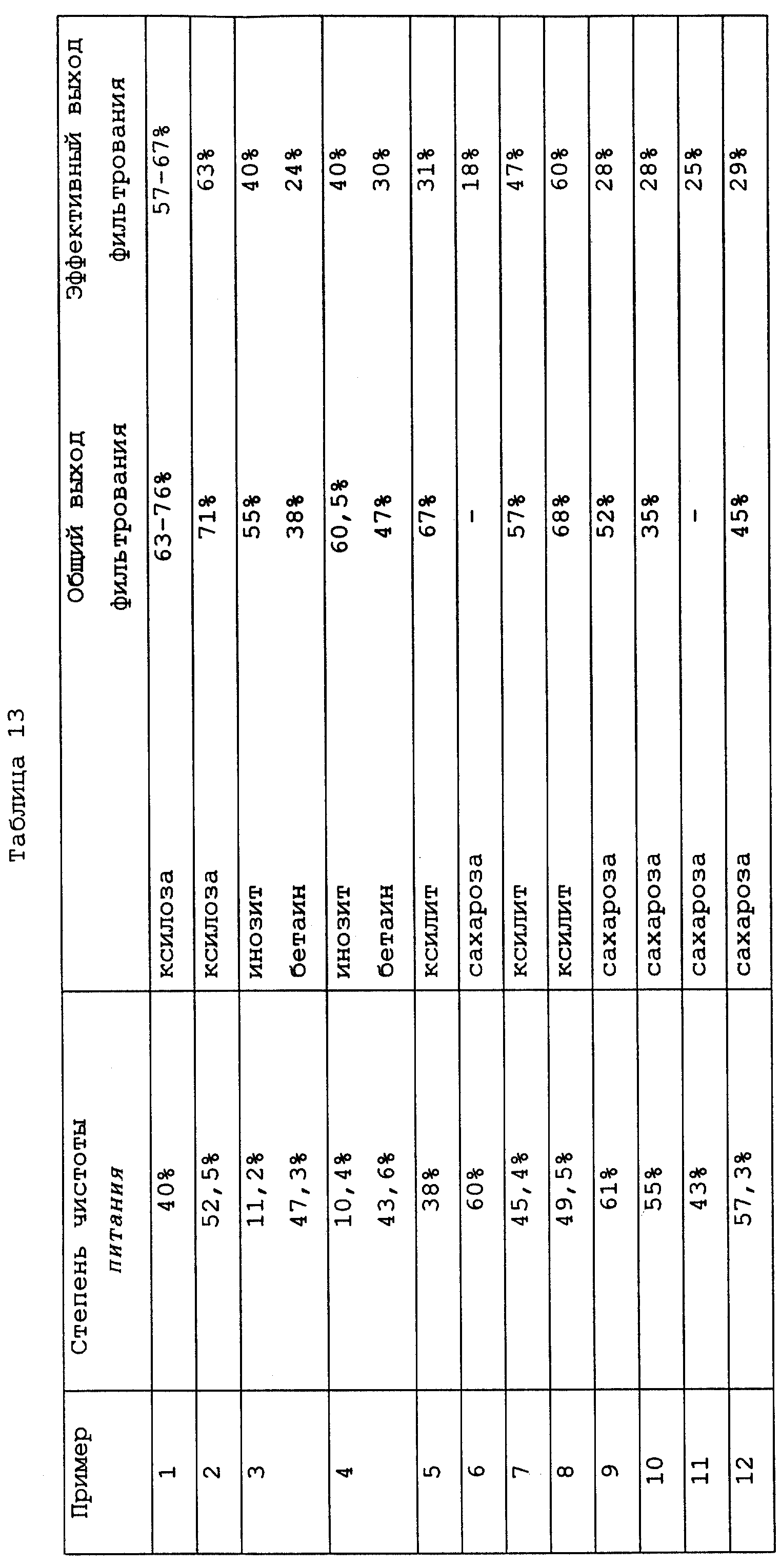
Выходы, полученные в примерах 1-12, а также степень чистоты питающего продукта в каждом случае, то есть концентрация вещества, которое необходимо извлечь, в подаче по отношению к сухому веществу, представлены в таблице 13.
В таблице 13 общие (то есть истинные) выходы
фильтрования вычисляют из чистоты
кристаллизационной массы, оттека при фильтровании и кристаллической лепешки, используя следующую формулу:
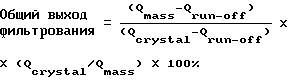
где Qmass и Qcrystal являются такими, как определено выше, а Qrun-off представляет собой степень чистоты оттека при фильтровании.
Например, выходы ксилозы в примере 1, используя данные из таблицы 1, вычисляют следующим образом:
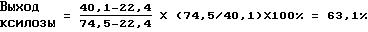
Эффективные выходы фильтрования вычисляют для 100% чистоты кристаллической лепешки. Они говорят о том, сколько чистого соединения может быть очищено из кристаллической лепешки с низкой степенью чистоты.
Реферат
Изобретение относится к технологии кристаллизации органических соединений из содержащих их растворов. Способ извлечения предусматривает приготовление вязкого перенасыщенного исходного раствора, имеющего вязкость 100-1000 Па•с и степень перенасыщения менее чем 4. В этом растворе образуют зародыши кристаллов, размер которых примерно составляет 10-120 мкм. Полученные кристаллы отделяют от межкристального раствора. Исходный раствор перед кристаллизацией выпаривают до достижения перенасыщенного состояния и высокую вязкость раствора достигают путем охлаждения. Извлекаемое органическое соединение представляет собой ксилозу, маннозу, ксилит, маннит, лактозу, лактит, сахарозу, глюкозу, фруктозу, мальтозу, мальтит, изомальтозу, изомальтулозу, лактозу, α-D-глюкопираноцил (1-->6) маннит и др. Образование зародышей кристаллов осуществляют путем эффективного перемешивания исходного раствора для их самопроизвольного возникновения или путем введения затравочных кристаллов извлекаемого соединения. Изобретение обеспечивает повышение выхода извлекаемого органического соединения. 33 з.п. ф-лы, 13 табл.
Формула
28.04.1995 по пп. 1-34;
01.03.1995 по пп. 19-22;
01.03.1995 по п. 26;
01.03.1995 по пп. 31-34.
Комментарии