Способ получения 6α, 9α-дифторированных стероидов, промежуточные соединения - RU2127739C1
Код документа: RU2127739C1
Чертежи





Описание
Предметом настоящего изобретения является новый метод получения 6α, 9α-дифторированных стероидов и новые полученные производные.
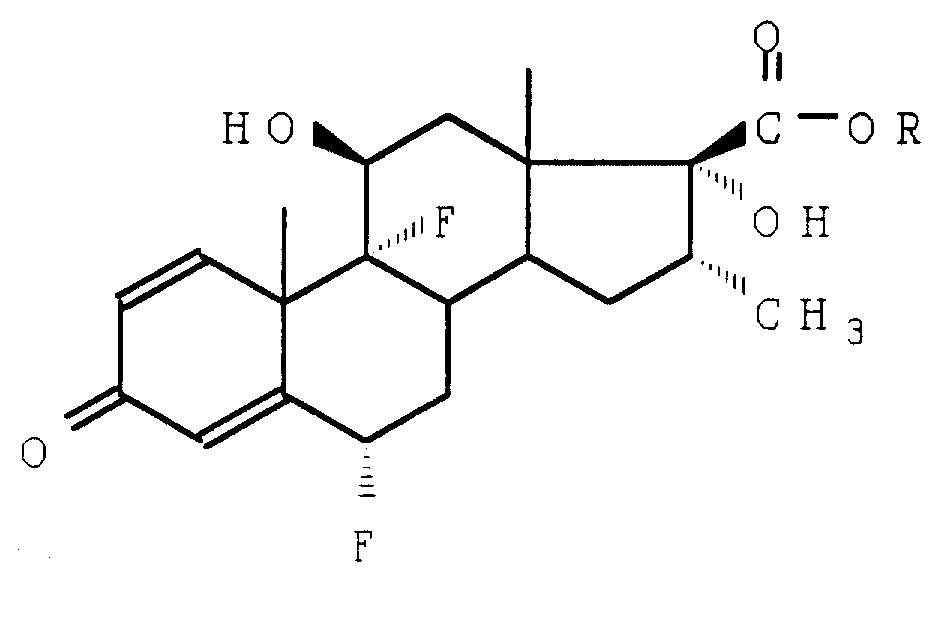
Таким образом, предметом настоящего изобретения является метод получения соединений формулы I

в которой R представляют собой атом водорода или сложноэфирный остаток,
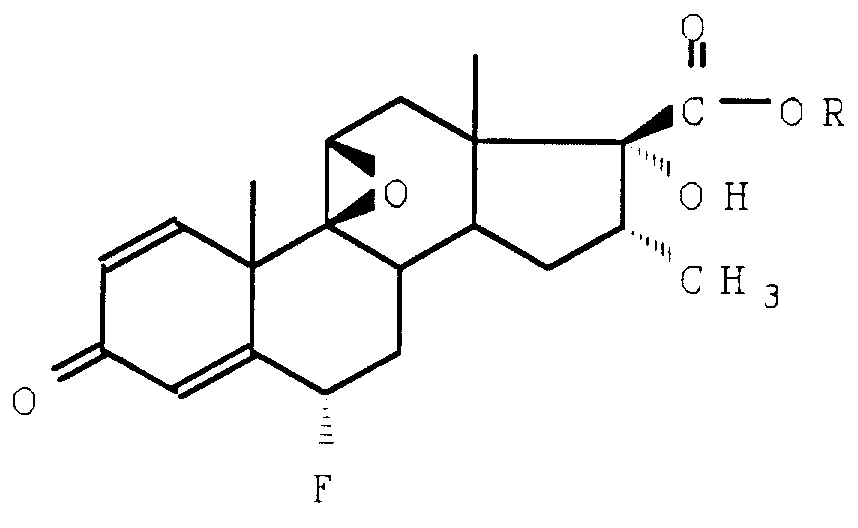
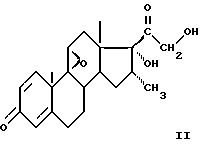
отличающийся тем, что соединение формулы II

подвергают обработке окислительным разрушающим средством для получения соединения формулы III

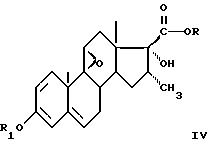
в котором кетоновую функциональную группу в положении 3 защищают в форме енолового эфира или сложного эфира и, при необходимости, кислую функциональную группу в положении 17β в форме сложного эфира для получения соединения формулы IV

в которой R определен, как указано выше; R1 представляет собой эфирный или сложноэфирный еноловый остаток, на который воздействуют электрофильным фторирующим средством для получения соединения формулы V

который подвергают обработке нуклеофильным фторирующим средством для получения искомого соединения формулы (I), который, при необходимости, когда R является сложноэфирным остатком, подвергают омылению для получения соответствующей кислоты.
Под сложноэфирным остатком понимают любой известный специалистам остаток, в частности алкинильный радикал, включающий от 1 до 6 атомов углерода, арильный радикал, включающий от 6 до 10 атомов углерода, или аралкильный радикал, включающий от 7 до 12 атомов углерода.
В тех случаях, когда R представляет собой алкильный радикал, речь идет, например, о метильном, этильном, пропильном, изопропильном, н-бутильном, втор-бутильном, трет-бутильном, пентильном или гексильном.
В тех случаях, когда R представляет собой арильный радикал, речь идет, например, о фенильном радикале или о фенильном радикале, замещенном, в частности, одним или несколькими алкильными радикалами.
В тех случаях, когда R представляет собой аралкильный радикал, речь идет, например, о бензильном или фенетильном радикале.
Под сложноэфирным остатком понимают также силилированный остаток, например триалкилсилильный остаток, такой как триметилсилил, трет-бутилдиметилсилил, или же, например, триарилсилильный остаток, такой как трифенилсилильный остаток, такой как дифенил-трет-бутилсилильный.
Под эфирноеноловым остатком в положении 3 понимают любой известный специалистам остаток, позволяющий заблокировать указанное положение 3 в указанной форме, в частности алкильный радикал, включающий от 1 до 6 атомов углерода, например метильный, этильный или пропильный, бензильный радикал, тетрагидропиранильный радикал, или силилированная функциональная группа, например одна из вышеуказанных групп.
Под сложноэфирноеноловым остатком в положении 3 понимают остаток формулы -COR, в которой R представляет собой алкильный радикал, как определенно выше, или арильный или алкильный радикал, как определенно выше, который может быть замещен одним или несколькими радикалами, такими как нитрильный, галогенный, в частности хлор, или алкильные, включающие от 1 до 4 атомов углерода.
Предметом настоящего изобретения, в частности, является вышеописанный метод, отличающийся тем, что на исходном
этапе защищают
кислую функциональную группу соединения формулы (III), для получения соединения формулы III

в которой R' представляет собой сложноэфирный остаток, в котором затем защищают кетоновую функциональную группу в положении 3 в форме енолового эфира или сложного эфира для получения соединения формулы (IV), как определено выше, в которой R имеет значение, указанное выше для R', после чего синтез продолжают, как указано выше.
Защита кислой функциональной группы в положении 17 может обеспечиваться одним или другим из вышеуказанных сложноэфирных остатков, алкильным остатком, при этом предпочтение отдается метильному и этильному радикалу.
Защита кетоновой функциональной группы в положении 3 может обеспечиваться одним или другим из вышеуказанных эфирных или сложноэфирных остатков, при этом предпочтение отдается сложноэфирному остатку. Можно, в частности, назвать бензоильный остаток, который может быть замещен одним или несколькими радикалами, такими как нитрильный, хлорный или метильный, или ацетильным, пропильным, бутирильным остатком или же валерильным остатком.
Предметом настоящего изобретения, в частности, также являются вышеописанный метод, отличающийся тем, что в рамках одной и той же операции выполняют защиту кетоновой функциональной группы в положении 3 и кислой функциональной группы в положении 17β, соответственно в форме енолового эфира и сложного эфира, для получения соединения формулы (IV), как определено выше, в которой R и R1 представляют собой одну и ту же защитную группу, после чего синтез продолжают, как указано выше.
В этом случае защита в положении 3 и 17 производится, в первую очередь, соответственно в форме силилированного эфира или сложного эфира, как указано выше.
Предметом настоящего изобретения, в частности, также является вышеописанный метод, отличающийся тем, что защите подвергают только кетоновую функциональную группу в положении 3 в форме енолового эфира или сложного эфира для получения соединения формулы (IV), как определено выше, в которой R представляет собой атом водорода, после чего синтез продолжают, как указано выше.
В этом случае защита в положении 3 производится, в первую очередь, в форме енолового сложного эфира, в частности одного из перечисленных выше.
Окислительным разрушающим средством, используемым в методе, являющемся предметом настоящего изобретения, может быть, например, иодная кислота, тетрауксуснокислый свинец, перманганат калия, перекись водорода, каталитическая иодная кислота, используемая в присутствии перекиси водорода, щелочные персульфаты, такие как оксон® (тройная соль 2KHSO5-KHSO4-K2SO4) или моноперсульфат калия. Предпочтение отдается иодной кислоте.
Различные вышеуказанные блокировки в положении 3 и 17, то есть выбор соответствующих реактивов и их использование, вполне доступны специалистам. Блокировка в форме сложного эфира в положении 17 может осуществляться, например, за счет воздействия спиртом в кислой среде или в присутствии дициклогексилкарбодиимида и диметиламинопиридина, или же, в случае сложного метилового эфира, за счет воздействия диазометаном, диметилсульфатом или метилкарбонатом.
Может оказаться целесообразным выполнять операции на уровне гетерогенных фаз в присутствии катализатора переноса фазы, которым может быть, в частности, четвертичная аммонийная соль, например бромид тетрабутиламмония, хлорид триэтилбензиламмония или хлорид трикаприлметиламмония, или же фосфонийная соль. Используемым органическим растворителем может быть хлорированный растворитель, например трихлорметан, метиленхлорид или дихлорметан, ароматический растворитель, такой как, например, толуол, ксилен или бензоил, или алифатический или циклоалифатический растворитель, например гексан или циклогексан.
Блокировка в форме енолового сложного эфира в положении 3 может осуществляться, например, за
счет
воздействия соответствующим кислотным хлорангидридом, причем операции выполняются в присутствии азотосодержащего основания или за счет переэтерификации с использованием енолового сложного эфира,
например ацетата, типа

Что же касается блокировки в форме енолового эфира, то она может осуществляться за счет воздействия алкильным галогенидом в щелочной среде, или же за счет дигидропирана, алкильного ортоформиата или спирта в кислой среде.
Блокировки в положении 17 в форме сложного эфира и в положении 3 в форме енолового эфира могут также осуществляться в результате одной и той же операции, в частности в случае блокировки в форме силилированного сложного эфира или эфира за счет воздействия соответствующим галогенидом. Следует отметить, что в подобном случае исключительная неустойчивость силилированного сложного эфира вызывает возврат к кислоте при гидролизе, следующем сразу после фторирования в положении 6.
Фторирующим средством, используемым в методе, являющемся предметом настоящего изобретения, является, как указано выше, электрофильное фторирующее средство.
Можно, в частности, назвать фторид перхлорида, фторид трифторметансульфонила и его производные, N-фторпиридинпиридингептафтордиборат, гипофторид ацетила или трифторацетила, N-фторпиридиний, N-фторсульфонамиды или N-фторсульфонимиды, например N-фторбензолсульфонимид или, в первую очередь, селектфтор® или N-фтор- N-хлорметилтриэтилендиамин-бис-тетрафторборат. Операции выполняются в среде растворения, например в среде тетрагидрофурана, ацетона, метиленхлорида, толуола и, в частности, в случае фторирования с использованием селектфтора®, в среде полярного растворителя, протонного или апротонного, такого как, например, диметилформамид, метанол или, в первую очередь, ацетонитрил, преимущественно в присутствии воды. Реакция выполняется либо при комнатной, либо при более низкой температуре. Операции могут выполняться в присутствии катализатора переноса фазы, в частности одного из перечисленных выше и при необходимости, в присутствии сорастворителя, в частности хлорированного растворителя, такого как вышеперечисленные. После гидролиза электрофильное фторирование в положении 6 вызывает разблокировку кетона в положении 3 и возврат к системе двойных связей Δ 1, 4.
Нуклеофильным фторирующим средством, которым воздействуют на соединение формулы (V), может быть, например, фтористоводородная кислота, в частности водная фтористоводородная кислота или комплексное соединение фтористоводородной кислоты с тетрагидрофураном или, в первую очередь, с диметилформамидом. Операции выполняются, например, при температуре от -10 до +25oC в присутствии или, что лучше, при отсутствии сорастворителя.
Возможное конечное омыление производится с использованием методов, хорошо известных специалистам. Можно назвать гидролиз или алкоголиз в присутствии основания, такого как, например, гидроокись щелочного или щелочноземельного металла, или в присутствии соответствующего азотированного основания.
Предметом настоящего изобретения также являются, в качестве новых промышленных соединений и, в частности, в
качестве промежуточных продуктов, необходимых для внедрения вышеуказанного метода, соединения формулы VI

в которой R имеет вышеуказанное значение, X представляет собой атом водорода или фтора, а Y, связанный с пунктирными линиями, представляет собой систему 3-кето Δ4 или X представляет собой атом водорода, а Y, связанный с пунктирными линиями, представляет собой систему 3-OR1 Δ3,5, в которой R1 определен, как указано выше.
Соединение формулы (II) описано в патенте США US 3 947 409.
Описание соединений формулы (I) приводится, например, во французском патенте 2 026 919. Сложные эфиры обладают, в частности, противовоспалительными свойствами. Приводимые далее примеры иллюстрируют изобретение, вместе с тем не ограничивая его.
Пример 1: 6α, 9α- дифтор 11β- метил
17α- дигидрокси 16α метил 17β- метоксикарбониландроста-1,4-диен-3-он
Этап A: 9,11β- эпокси 16α- метил 17α- гидрокси 17β- карбоксиандроста 1,
4-диен-3-он
Для начала смешивают 200 г 9,11β- эпокси 16α- метил 17α,21- дигидрокси 20-кетопрегна-1,4-диен-3-она и 800 см3 метанола, после чего при температуре
не выше + 40oC и при взбалтывании добавляют за 20 мин 128,5 г ортоиодной кислоты. Взвесь выдерживают в течение 1 ч при взбалтывании в среде инертного газа, при температуре от +23 до
+25oC, после чего за 5 мин вливают в смесь 1000 см3 воды, 2000 г льда и 200 г метабисульфита натрия. Затем смесь выдерживают в течение 30 мин при взбалтывании при температуре от
0 до +
10oC, фильтруют и промывают водой. После сушки получают 192 г искомого продукта, который без дополнительной обработки используют на следующем этапе.
Инфракрасный
спектр:
(CHCl3)
Поглощение 3600 см-1: OH; 1706 см-1: C = 0 кисл.; 1662, 1623, 1607 см-1: кето -3 Δ 1,4.
Спектр ЯМР: (CDCl3 - 300
МГц - млн-1)
0,97 (d): 16-CH3; 1,09 (s): 18 - CH3; 1,45 (s): 19-CH3; 3,21 (s): H11; 3,94: OH; 6,19 (s): H4; 6,
23 (dd); 6,64
(d): H1.
Этап Б: 9,11β- эпокси 16α- метил 17α- гидрокси 17β- метоксикарбониландроста 1,4-диен-3-он
Для начала смешивают
192 г продукта,
полученного на Этапе A, 800 см3 метиленхлорида и 4 г бромида тетрабутиламмония. Затем в среде инертного газа при температуре + 18o/+22oC примерно за 5
мин. вводят 400
см3 2 н. едкого натра, а затем 45,8 см3 диметилсульфата. После этого смесь выдерживают при взбалтывании в течение 1 ч 30 мин., а затем отстаивают и повторно
экстрагируют водную
фазу с использованием метиленхлорида. Затем соединенные органические фазы промывают водой, концентрируют примерно до 400 см3, после чего продолжают дистиллирование,
заменяя метиленхлорид
простым изопропиловым эфиром. Затем смеси дают остыть за 1 ч при комнатной температуре, продолжая взбалтывать, после чего выдерживают в течение еще 1 ч в указанных условиях,
центрифугируют и промывают
кристаллы простым изопропиловым эфиром и высушивают их. В результате получают 185,4 г искомого продукта.
Инфракрасный спектр: (CHCl3)
Поглощение 3600 и 3540 см-1: OH; 1743, 1713 и 1438 см-1: CO2Me; 1662, 1624 и 1608 см-1: кето-3 Δ 1,4.
Спектр ЯМР: (CDCl3 - 300
МГц - млн-1)
0,93 (d): 16 - CH3; 0,98 (s): 18 - CH3; 1,44 (s): 19 - CH3; 2,97 (s): OH; 3,21 (t): H11; 3,77 (s): CO2CH3; 6,15 (s): H4; 6,
19 (dd): H2; 6,61 (d): H1.
Химический состав (C22H28O5: 372,5)
% расчетный: С 70,9;
Н 7,6
% полученный: С 71,0;
Н 7,8
Этап В: 3-бензоилокси 9,11β- эпокси 16α- метил 17α- гидрокси 17β- метоксикарбониландроста-1,3,5-триен
При
температуре + 20o/+22oC в среде инертного газа смешивают 30 г продукта, полученного на Этапе Б, 75 мг гидрохинона и 42 см3 пиридина, нагревают до температуры 70oC и добавляют 13 см3
бензоилхлорида. Затем выдерживают в течение 6 ч при +70oC, после чего температуре дают опуститься до +40oC. Затем добавляют 30 см3 метанола, выдерживают 30 мин. при
взбалтывании при температуре +40oC и дают температуре опуститься до уровня комнатной. После этого раствор вливают в смесь 300 см3 воды
и 44 см3 хлористоводородной
кислоты при 22oBé, добавляют 270 см3 метанола и выдерживают при взбалтывании в течение 1 ч. Затем кристаллы центрифугируют,
промывают водой и высушивают. В результате
получают 36,95 г искомого продукта, который можно очистить, растворив его в двух объемах метиленхлорида, добавив 5 объемов метанола и выполнив
дистиллирование метиленхлорида. После возвращения
температуры при взбалтывании до уровня комнатной, а затем за 1 ч до температуры 0oC, выделяют 24,97 г искомого сухого продукта.
Инфракрасный спектр: (CHCl3)
Поглощение 3600 и 3540 см-1: OH; 1730 см-1: C=0; 1438 см-1: OCH3; 1657, 1620, 1603 и 1585 см-1: C=C ароматич.
Спектр ЯМР:
(CDCl3 - 300 МГц - млн-1)
0,94 (d, J= 7): 16 - CH3; 0,98 (s): 18 - CH3; 1,28 (s): 19 - CH3; 3,11 (s): H - 11; 3,78 (s): CO2
CH3; 5,49 (d, J = 10): H - 1; 5,80 (dd): H - 2; 5,8: H - 6; 5,93 (s): H - 4; 7,48: H мета; 7,61 (tt): H пара; 8,08: H орто.
Химический состав (C29H32
O6: 476,6)
% расчетный: С 73,1; Н 6,8
% полученный: С 72,9; Н 6,9
Этап Г: 6α- фтор 9,11β
- эпокси 16α- метил 17α- гидрокси 17β
- метоксикарбониландроста-1,4-диен-3-он
При температуре +20oC в среде инертного газа смешивают 20 г продукта, полученного
на Этапе В, и 100 см3 ацетонитрила, после чего
добавляют 2 см3 воды. Затем взвесь охлаждают до температуры -1o/+1oC и медленно добавляют 17,4 N-фтор
N-хлорметилтриэтилен диамин-бис-тетрафторбората. После окончания
вышеуказанного добавления выдерживают 1 ч при взбалтывании при температуре -1o/+1oC и вливают взвесь в раствор
400 см3 воды и 10 см3 20-процентного
нашатырного спирта. Затем при взбалтывании вводят 0,4 г метабисульфита натрия и продолжают взбалтывание в течение 30 мин при комнатной
температуре. После этого при необходимости добавляют достаточное
количество 20-процентного нашатырного спирта для получения pH 8, после чего центрифугируют, промывают кристаллы водой и высушивают. В
результате получают 16,34 г искомого продукта.
Спектр ЯМР: (CDCl3 - 300 МГц - млн-1)
0,93 (d):

Химический состав (C22H27FO5: 390,45)
% расчетный: С 67,67; Н 6,97; F 4,87
%
полученный: С 67,9; Н 6,9; F 4,7
Этап Д: 6α,9α
-дифтор-11β,17α-дигидрокси-16α- метил -17β- метоксикарбониландроста-1,4-диент-3-он
В среде
инертного газа смешивают 180 см3 комплексного соединения
фтористоводородной кислоты и диметилформамида и 18 г продукта, полученного, как описано на Этапе Г. Затем взбалтывают в течение 3 ч
при температуре 22oC ± 3oC и вливают
раствор в смесь при 0o/2oC, включающую 1,8 л воды и 9 см3 нашатырного спирта при 22oBe.
Поддерживая температуру смеси ниже + 10oC, добавляют за
30 мин. 290 см3 нашатырного спирта при 22oBe, то есть количество, необходимое для поддерживания pH 4,5 ±
0,5.
Затем взбалтывают в течение 1 ч, давая температуре подняться, после чего оставляют в состоянии покоя на 1 ч. Кристаллы центрифугируют, промывают водой с нейтральным pH и высушивают. Таким образом получают 18,86 г сырого продукта, который поглощают в примерно 7 объемах трихлорметана. Затем доводят до температуры кипения и дистиллируют примерно 2/3 трихлорметана, после чего медленно охлаждают до температуры 0o/+5oC и выдерживают в течение 1 ч при этой температуре. Затем центрифугируют и высушивают кристаллы. Таким образом получают 18,3 г целевого продукта, который обязательно следует подвергнуть десольватации. Для этого кристаллы вводят в 10 объемов воды и, взбалтывая, доводят за 30 мин. до температуры 90o/95oC, дистиллируя трихлорметан. После этого охлаждают, центрифугируют кристаллы, промывают водой и высушивают. В результате получают 15,8 г продукта (tпл 227oC).
α= + 60o ± 1o (c = 1% ДМФ). Расчетн.% F: 9,25, полученный % F: от 9 до 9,2.
Комплексное соединение фтористоводородной
кислоты и диметилформамида, используемое на начальном этапе, получают следующим образом:
При температуре +19o/+21oC в среде инертного газа взбалтывают в течение 10 мин.
210 см3 диметилформамида. Затем медленно вводят конденсированную фтористоводородную кислоту, охлажденную до
температуры -15o/-20oC, дав затем температуре подняться
примерно до +45oC, после чего это повышение температуры ограничивают +50o/+60oC с
помощью внешней ванны с температурой -15o/-20oC. Таким образом
в сумме добавляют за 1 ч 15 мин. 250 г фтористоводородной кислоты. Затем раствор выдерживают в течение нескольких
минут в состоянии взбалтывания в среде инертного газа, перед вводом стероида, который
производится в вышеуказанных условиях.
Пример 2: 6α,9α- дифтор -11β,
17α- дигидрокси -16α- метил -17β- карбоксиандроста-1,4-диен-3-он
В среде инертного газа смешивают 7,9 г продукта, полученного в Примере 1, 75 см3 метанола и 4
см3 воды и выдерживают при взбалтывании при комнатной температуре в течение 10 мин.
Затем добавляют за 5 мин 2,5 г едкого кали в 20 см3 воды, после чего медленно доводят до
температуры кипения. После выдержки в течение 3 ч 30 мин при температуре кипения смесь охлаждают до
+50oC и добавляют уксусную кислоту до получения pH = 6, то есть примерно 3 см3
. После этого раствор концентрируют примерно до 40 см3 и охлаждают до температуры +20oC, после чего добавляют воды. Кристаллы центрифугируют, промывают смесью метанола и воды, а
затем водой и высушивают.
В результате получают 7,3 г целевого продукта, который очищают путем сгущения в горячем состоянии в метаноле и рекристаллизации с использованием ацетона и обработкой активированным углем.
α= + 65,5 (с = 1% ДМФ).
Химический состав (C21H26F2O5)
%
расчетный: С 63,79; Н 6,63; F 9,61
% полученный: С 63,7; Н 6,6; F 9,3
Спектр ЯМР: (CDCl3 - 300 МГц - млн-1)
1,02 (d): CH3; 1,26 (s): 18
- CH3; 1,58 (s): 19 - CH3; 4,40 (d, m): H - 11; 5,40 (d, m): H - 6; 6,33 (d): H - 2; 7,18
(d): H - 1; 6,43 (s): H - 4.
Инфракрасный спектр: (вазелиновое масло)
Поглощение 3559 - 3541 см-1: OH; 1698 - 1661 см-1: C = 0 и C = 0 сопряж. ;
1615-1603 см-1: C = C.
Пример 3: 6α- фтор -9,11β- эпокси
-16α- метил -17α- гидрокси -17β- метокси-карбониландроста-1,4-диен-3-он
В атмосфере инертного газа при температуре 20oC смешивают 10 г продукта, полученного на
этапе A предыдущего примера, и 50 см3 ацетонитрила, затем добавляют 1 см3
воды. Охлаждают суспензию до -9, -11oC, затем медленно добавляют 5,08 г N-фтор-2,
6-дихлор-пиридиний-тетрафторбората. После окончания подачи реагентов при перемешивании поддерживают
температуру -9, -11oC в течение 3 ч 45 мин, затем эмульсию вливают в раствор из 200
см3 и 0,2 г метасульфита натрия. Продолжают перемешивание, добавляя достаточное количество
20%-го гидроксида аммония для доведения pH до 7 - 7,5, т.е. 2,3 см3. Обезвоживают и
промывают кристаллы водой, затем сушат их. Получают 8,18 г сырого продукта, который очищают
перекристаллизацией в ацетоне с 10% воды. Получают 5,1 г искомого продукта, подобного тому, который получают
на этапе Г заявки.
Пример 4. 6α- фтор -9,11β- эпокси -16α- метил -17α- гидрокси -17β- метокси-карбониландроста-1,4-диен-3-он.
Этап А: 3-(4-нитробензоилокси) -9,11β- эпокси -16α- метил -17α - гидрокси -17β- метоксикарбониландроста-1,3,5-триен.
Действуют также, как на этапе B заявки, используя 4-нитро-бензоилхлорид вместо бензоилхлорида, и получают целевой продукт.
Этап Б: 6α- фтор -9,11β- эпокси -16α- метил -17α- гидрокси -17β - метоксикарбониландроста-1,4-диен-3-он.
В атмосфере инертного газа при 20oC смешивают 100 г продукта, полученного на этапе А, и 500 см3 ацетонитрила, затем добавляют 10 см3 воды. Суспензию охлаждают до -12, -10oC, затем медленно добавляют 68 г 1-гидрокси-4-фтор-1,4-диазонийбицикло-(2,2,2-октан бис тетрофторбората). После окончания подачи реагентов выдерживают при перемешивании при температуре -10, -12oC в течение 2 ч 40 мин, затем суспензию вливают на раствор из 1850 см3 воды и 3 г метабисульфита натрия. Затем при перемешивании подают при температуре +22oC 60 см3 20%-го гидроксида аммония. Затем продолжают перемешивание в течение 30 мин при температуре окружающей среды. Добавляют при необходимости достаточное количество 20%-го гидроксида аммония для доведения pH до 7,5, затем обезвоживают и промывают кристаллы водой, затем сушат их. Получают 73,85 г искомого продукта, подобного тому, который получают на этапе Г заявки.
Этот продукт затем преобразуется в 6α,9α- дифторпроизводное согласно способу, описанному в заявке.
Реферат
Объект изобретения - новый способ получения 6α, 9α-дифторированных стероидов формулы I, где R - атом водорода или сложноэфирный остаток, отличается тем, что соединение формулы II подвергают обработке окислительным разрушающим средством для получения соединения формулы III, в котором кетоновую функциональную группу в положении 3 защищают в форме енолового эфира или сложного эфира и, при необходимости, кислую функциональную группу в положении 17β в форме сложного эфира для получения соединения формулы IV, где значение R как указано выше, а R1 эфирный или сложноэфирный еноловый остаток, на который воздействуют электрофильным фторирующим средством для получения соединения формулы V, который подвергают обработке нуклеофильным фторирующим средством для получения соединения формулы I и который, при необходимости, когда R сложноэфирный остаток, подвергают омылению для получения соответствующей кислоты. Преимущество предложенного способа заключается в том, что защите подвергают только кетоновую функциональную группу в положении 3 в форме енолового эфира или сложного эфира для получения соединения формулы IV. Другим объектом изобретения являются новые соединения формулы VI, где R имеет значение, указанное в формуле I, X представляет атом водорода или фтора, а Y, связанный с пунктирными линиями, представляет собой систему 3-кетоΔ4 или Х - атом водорода, а Y, связанный с пунктирными линиями, представляет собой систему 3-OR1 Δ 3,5, в которой R1 определен, как указано в формуле I, которые могут быть использованы как промежуточные соединения при получении стероидных препаратов. 2 с. и 7 з.п.ф-лы.




Формула

в которой R - атом водорода или сложноэфирный остаток,
отличающийся тем, что соединение общей формулы II

подвергают обработке окислительным разрушающим средством для получения соединения общей формулы III

в котором 3-кетогруппу защищают в форме сложного енолового эфира или эфира, и при необходимости, карбоксильную группу в положении 17β защищают в форме сложного эфира для получения соединения общей формулы IV

в которой R имеет вышеуказанные значения;
R1 - эфирный или сложноэфирный еноловый остаток,
после чего воздействуют электрофильным фторирующим средством для получения соединения общей формулы V

в которой R имеет вышеуказанные значения,
и подвергают обработке нуклеофильным фторирующим средством для получения целевого соединения общей формулы I, которое, при необходимости, когда R - сложноэфирный остаток, подвергают омылению для получения соответствующей кислоты.

в которой R1 - сложноэфирный остаток, и в котором затем защищают 3-кетогруппу в форме енолового сложного эфира или эфира для получения соединения общей формулы IV по п.1, в которой R имеет значение, указанное для R1, после чего синтез продолжают как указано выше.


в которой R имеет значение, указанное в п.1;
Х - атом водорода или фтора;
У, связанный с пунктирными линиями, представляет собой систему 3-кето Δ4 или Х - атом водорода;
У, связанный с пунктирными линиями, представляет собой систему 3-OR1 Δ3,5, в которой R1 имеет значения, как в п.1.
Комментарии