Способ получения флуметазона, соединение - RU2260596C1
Код документа: RU2260596C1
Описание
Настоящее изобретение относится к способу получения флуметазона, флуметазон 21-ацетата и его 17-карбоксиландростенового аналога, а также к некоторым исходным веществам для осуществления такого способа.
Флуметазон, 6α,9α-дифтор-16α -метилпреднизолон был впервые описан в 1962 году. Несмотря на то что этот кортикостероид обладает повышенной противовоспалительной активностью, он не нашел широкого применения в клинической практике. В настоящее время экономичное получение этого препарата в промышленном масштабе приобретает большую важность, поскольку это вещество также является отличным исходным сырьем для производства новых дифтор-17-карбоксиландростенов, которые становятся все более важными веществами с клинической точки зрения.
Флуметазон и его получение являются предметом ряда патентов, включающих патент US 3499016 (1962) и патент GB 902292 (1970). Новые синтетические технологии, развиваемые с 1970 г., позволяют осуществлять более эффективное получение флуметазона со значительно более высоким выходом по сравнению с выходами, которые были достигнуты в ранних патентах.
Был разработан новый способ получения флуметазона, а также получения 6α,9α-дифтор-11β,17α -дигидрокси-16α-метил-17β-карбоксиандроста-1,4-диен-3-она, известного также под названием «гидроксикислота», представляющего собой превосходный исходный материал для получения флутиказона и других новых противовоспалительных соединений андроста-1,4-диенового ряда. «Гидроксикислота» впервые была описана и заявлена в патенте US 3636010 (приоритет 1968 г.).
В европейском патенте 0610138 В1 (1994) описывается новый синтетический метод получения так называемой «гидроксикислоты». Однако настоящее изобретение обладает значительными, неожиданными преимуществами по сравнению со способом, описанным в указанном патенте:
- реакционная последовательность сокращается на одну стадию посредством исключения стадии десольватации 6α,9α -дифтор-11β,17α-дигидрокси-16α-метил-17β-метоксикарбониландроста-1,4-диен-3-она, дополнительной стадии процесса,
- способ настоящего изобретения исключает применение высокотоксичного реагента, диметилсульфата,
- обеспечивается одновременное деацетилирование и окислительная деградация боковой цепи прегнана, в результате чего непосредственно образуется эквивалентное андростановое производное,
- повышается выход гидроксикислоты высокой чистоты.
В то время как все реакционные стадии настоящего изобретения, за исключением последней стадии, реализованы в прегнановом ряду, что обеспечивает эффективное получение флуметазона, последовательность реакций согласно EP 0610138 В1 обеспечивает преобразование традиционного исходного материала обоих процессов, например, на первой стадии, в соединения андростанового ряда.
В соответствии с настоящим изобретением предусматривается способ получения флуметазона (6α,9α-дифтор-11β,17α,21-тригидрокси-16α-метилпрегна-1,4-диен-3,20-диона), флуметазон 21-ацетата или его 17-карбоксиландростенового аналога формулы:

причем такой способ включает:
(а) реакцию соединения формулы (II)

c хлористым бензоилом с образованием 3-енольного сложного эфира формулы (IIIa)

b) реакцию енолбензоата (IIIa) с электрофильным фторирующим агентом с целью введения фтора в С6-положение с образованием нового соединения формулы (IIIb)

(c) снятие защитной группы с положения С3 в соединении (IIIb) с образованием соединения формулы (IV)
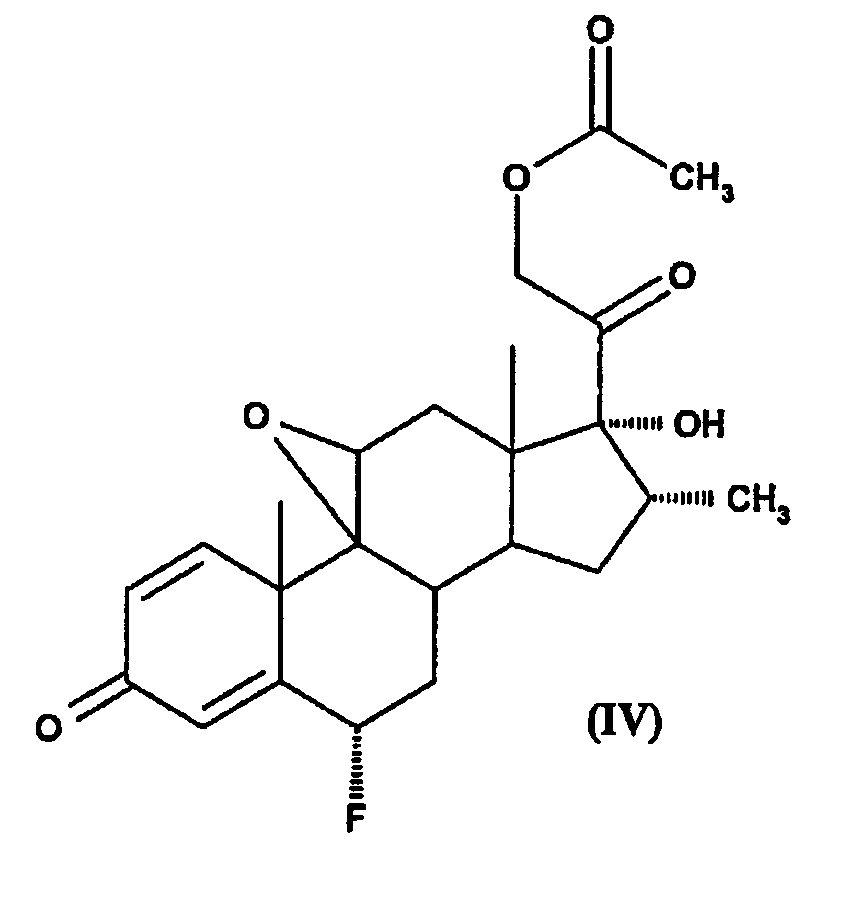
d) фторирование 9,11-эпоксигруппы соединения IV реакцией с фтористоводородной кислотой с образованием флуметазон 21-ацетата; и
е) необязательный гидролиз флуметазон 21-ацетата в присутствии или в отсутствие окислительного агента, с образованием соединения (I) или флуметазона соответственно.
Кроме этого, настоящее изобретение также предусматривает соединения формулы (V)
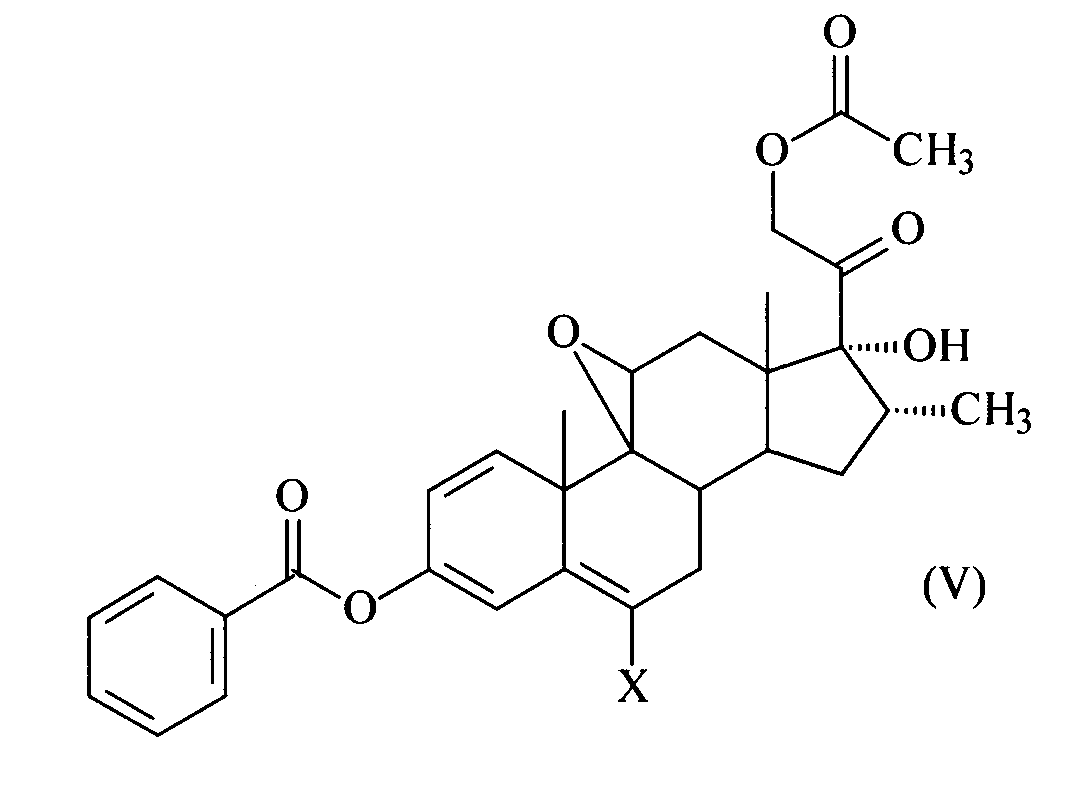
где Х представляет собой водород или фтор.
На стадии (d) способа образуется флуметазон 21-ацетат формулы
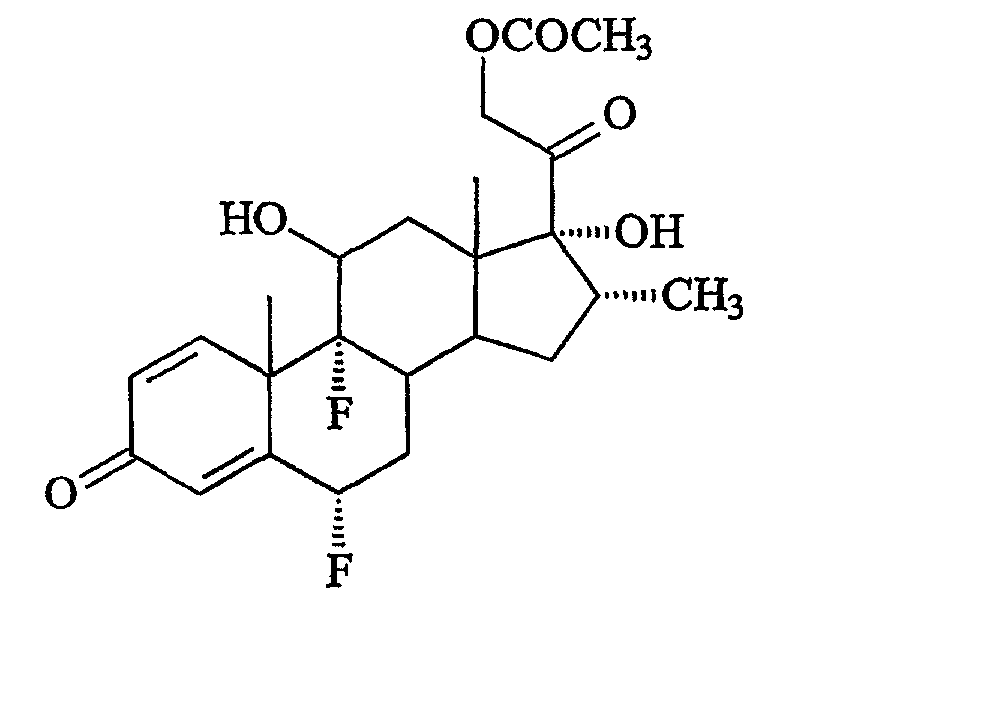
т.е. 6α,9α-дифтор-11β,17α,21-тригидрокси-16α-метилпрегна-1,4-диен-3,20-дион, 21-ацетат. В результате гидролиза этого соединения, предпочтительно с помощью раствора гидроксида калия в метаноле, образуется флуметазон, не содержащий спирта.
С другой стороны, в результате гидролиза ацетата флуметазона, предпочтительно с помощью раствора гидроксида калия в метаноле, и последующего окисления, предпочтительно с помощью раствора пероксида водорода, получают так называемую «гидроксикислоту», отвечающую формуле

Настоящее изобретение обеспечивает прямое превращение ацетата флуметазона в соединение I.
Два новых соединения IIIa и IIIb отвечают общей формуле (V), приведенной в пункте 8.
Соединение I также может быть получено в соответствии с патентом US 3636010 в результате окисления флуметазона, не содержащего спирта.
Исходный материал настоящего изобретения является коммерчески доступным продуктом и широко используется для получения таких кортикостероидов, как дексаметазон и икометазон.
Для того чтобы ввести фтор в положении С6 в результате электрофильного фторирования, сначала необходимо активировать положение С6. Для этой цели 3-кетогруппу подвергают енолизации с помощью хлорангидрида карбоновой кислоты с образованием остатка енольного сложного эфира формулы -COR, в которой R представляет собой арильную или аралкильную группу. Предпочтительным веществом для енолизации является хлористый бензоил, при взаимодействии с которым образуется вещество формулы IIIa в присутствии такого третичного амина, как пиридин. Предпочтительным растворителем является N,N'-диметилацетамид, и реакцию предпочтительно проводить при температуре 80-85°С, в результате чего получают Δ3, 5-енолбензоат. После этого соединение IIIa реагирует с электрофильным фторирующим агентом с получением соответствующего С6-фтор производного. Предпочтительный фторирующий агент представляет собой 1-(хлорметил)-4-фтор-1,4-диазонийбицикло[2.2.2]октан-бис(тетрафторборат), Selectfluor®. Для проведения фторирования в положении С6 предпочтительным растворителем является ацетонитрил в присутствии воды при предпочтительной температуре -5°С+2°С. После фторирования в положении С6 3-енольный сложный эфир может быть легко превращен в 3-кето-1,4-диеновую систему с образованием соединения IV. Элиминирование енольного сложного эфира предпочтительно осуществлять с использованием водного раствора метабисульфита натрия в аммиаке.
На следующей стадии известным per se способом при температуре ниже 25°С проводят реакцию между 9,11-эпоксигруппой соединения IV и концентрированным водным раствором фтористоводородной кислоты или раствором фтористого водорода в N,N'-диметилформамиде. После того как соединение IV практически полностью прореагировало, реакционную смесь переливают в смесь льда с аммиаком, что достаточно для нейтрализации фтористоводородной кислоты и одновременного осаждения с высоким выходом флуметазон 21-ацетата высокой чистоты. Полученный продукт может быть перекристаллизован, например, из метанола. Далее, полученный 21-ацетат может быть подвергнут гидролизу любыми известными способами с образованием флуметазона, не содержащего спирта. Один из предпочтительных способов осуществляют в дегазированном растворе гидроксида калия в метаноле при температуре в интервале -15°С - -5°С. Через 1 час методом ВЭЖХ устанавливают окончание реакции, которая считается завершенной, если количество исходного соединения составляет менее 1%.
В соответствии с известным способом, с целью проведения окислительной деградации, флуметазон суспендируют в тетрагидрофуране и каплями добавляют раствор окисляющего агента. Вначале происходит растворение субстрата с последующим его осаждением. Окисление проводят предпочтительно при 20°С с использованием, например, периодной кислоты. Через 1 час перемешивания завершение реакции определяют методом ВЭЖХ. Если количество непрореагировавшего флуметазона составляет менее 0,3%, реакцию считают завершенной. После этого реакционную смесь, содержащую соединение I, осаждают добавлением реакционной смеси водного раствора метабисульфита натрия и льда.
В соответствии с настоящим изобретением флуметазон 21-ацетат может быть одновременно подвергнут деацетилированию и окислению раствором гидроксида калия в метаноле и водным раствором пероксида водорода с образованием, после завершения реакции, желаемой гидроксикислоты, соединения I, подкислением реакционной смеси разбавленным раствором хлористоводородной кислоты до рН 2. Такую реакцию проводят при 10°С+2°С при перемешивании до ее завершения.
Совокупный стехиометрический выход в способе, описанном в EP 0610138 B1, в расчете на неперекристаллизованное соединение I составляет 48,9% от 9,11β-эпокси-17α, 21-дигидроксипрегна-1,4-диен-3,20-диона. В соответствии с настоящим изобретением, согласно данным, приведенным в примерах 1b, 2 и 4, совокупный стехиометрический выход составляет 62,4% при использовании 21-ацетата в качестве исходного вещества. Для проведения обоснованного сравнения выходов исходный материал EP 0610138 В1 вначале ацетилировали с выходом 110% масс/масс и совокупный стехиометрический выход рассчитывали на основании этого значения и значений, полученных в примерах 1b, 2 и 4, получая величину в 61,7%.
Следующие ниже примеры представлены для иллюстрации настоящего изобретения и не ограничивают его область.
ПРИМЕР 1
9,11β-эпокси-6α-фтор-17α,21-дигидрокси-16а-метилпрегна-1,4-диен-3,20-дион, 21-ацетат (формула V)
а) В инертной атмосфере 50 г 9,11β-эпокси-17α,21-дигидрокси-16α-метилпрегна-1,4-диен-3,20-дион, 21-ацетата растворяли в 25 мл N, N'-диметилацетамида (DMA). Добавляли 65 мл пиридина и реакционную смесь нагревали при перемешивании до температуры между 80 и 85°С.
После этого при защите системы от попадания света добавляли 33 мл хлористого бензоила. Перемешивание продолжали в течение двух-трех часов при указанной температуре. После завершения реакции смесь охлаждали до 40°С. Затем добавляли 75 мл метанола и перемешивание продолжали в течение 30 минут при 40°С, после чего реакционную смесь охлаждали до 20-25°С. Далее, реакционную смесь добавляли в 1000 мл воды, содержащей 57,5 мл хлористоводородной кислоты и 100 мл дихлорметана. Фазы разделяли и водную фазу дополнительно экстрагировали 100 мл дихлорметана. Органические фазы объединяли и промывали водой и водным раствором гидроксида натрия. Полученный таким образом дихлорметановый раствор досуха выпаривали в вакууме с образованием масла, представляющего собой 21 ацетат 3-бензоилокси-9,11β-эпокси-17α, 21-дигидрокси-16α-метилпрегна-1,3,5-триен-20-она (формула IIIa), который обрабатывали 150 мл ацетонитрила, охлажденного до -5 - 0°С. Затем раствор енолбензоата добавляли к суспензии из 44,5 г 1-хлорметил-4-фтор-1,4-диазонийбицикло[2.2.2]октан-бис(тетрафторбората), Selectfluor®, в 175 мл ацетонитрила, содержащего 5 мл воды.
После завершения реакции фторирования в положении С6 (формула IIIb) реакционную смесь переливали в раствор, содержащий 100 мл воды, 1,2 г метабисульфита натрия, 5 мл 25% аммиака и 200 мл дихлорметана. рН раствора доводили до значения в интервале 7-8 и смесь перемешивали в течение 30 минут, после чего фазы разделяли и органическую фазу промывали раствором аммиака (12,5%). Далее, органическую фазу выпаривали досуха в вакууме и добавляли метанол. Целевое соединение кристаллизовали, и его затем отфильтровывали, и сушили при 40-45°С с получением 40 г целевого продукта с чистотой 90%, рассчитанной по площади пиков, определенных ВЭЖХ.
b) Повторяли методику примера 1а, однако образовавшийся 3-енолбензоат не экстрагировали и не изолировали. К реакционной смеси, полученной в примере 1а, медленно и непосредственно добавляли 44,5 г кристаллов Selectfluor®, проводя добавление четырьмя порциями. При содержании исходного материала в количестве менее 1% реакционную смесь переливали в 100 мл воды, содержащей 1,2 г метабисульфита натрия. Затем рН доводили до значения в интервале 7-7,5, раствор перемешивали в течение 30 минут и полученный осадок отфильтровывали и промывали. Далее полученный таким образом продукт промывали путем его суспендирования в 150 мл метанола при перемешивании. После перемешивания в течение 30 минут продукт отфильтровывали и сушили при температуре 40-45°С с получением 46 г 21-ацетата 9,11β-эпокси-6α-фтор-17α,21-дигидрокси-1,4-диен-3,20-диона с чистотой 91,6%.
ПРИМЕР 2
21-ацетат 6α,9α -дифтор-11β,17α,21-тригидрокси-16α-метилпрегна-1,4-диен-3,20-диона (21-ацетат флуметазона).
36 г соединения, полученного в примере 1, растворяли в инертной атмосфере в 360 мл комплекса из фтористого водорода и N,N'-диметилформамида (˜64% масс/масс) при температуре 20±3°С. После перемешивания в течение трех часов при этой температуре перемешиваемый продукт переливали в смесь из 3000 мл воды, 1000 мл льда и 800 мл аммиака (25%), поддерживая температуру в ходе всей операции осаждения на значении ниже 25°С. рН доводили до значения в интервале 4,5-5 с помощью раствора аммония и перемешивание продолжали еще в течение одного часа. Затем осадок отфильтровывали и промывали водой до нейтрального значения рН. После высушивания соединение растворяли в смеси 333 мл дихлорметана и 148 мл метанола. Полученный раствор концентрировали до объема 89 мл с кристаллизацией целевого продукта. После фильтрации кристаллы сушили при температуре 40-45°С с получением 29,2 г 6α,9α-дифтор-11β,17α,21-тригидрокси-16α-метилпрегна-1,4-диен-3,20-дион, 21-ацетата с чистотой 95%, определенной по площади пика, определенной методом ВЭЖХ.
ПРИМЕР 3
6α,9α-дифтор-11β,17α,21-дигидрокси-16α-метилпрегна-1,4-диен-3,20-дион (Флуметазон)1,4 г гидроксида калия растворяли в 140 мл дегазированного метанола в инертной атмосфере и полученный раствор охлаждали до 0 - -5°С. Далее, при перемешивании раствор добавляли к суспензии 28 г соединения, полученного в предыдущем примере, в 700 мл дегазированного метанола. Полученную смесь перемешивали 1-2 часа при температуре -10°С+2°С. После завершения реакции, согласно данным ВЭЖХ, в систему добавляли уксусную кислоту до рН 7. Затем объем системы уменьшали в вакууме до ˜224 мл. После этого проводили охлаждение до 10°С и добавляли 140 мл холодной воды. После перемешивания в течение часа при температуре между 5 и 10°С полученное вещество отфильтровывали, промывали водой и сушили при 45°С с образованием 22,4 г целевого соединения с чистотой 96% согласно определению по площади пика методом ВЭЖХ.
ПРИМЕР 4
6α,9α-дифтор-11β,17α-дигидрокси-16α -метил-17β-карбоксиандроста-1,4-диен-3-он (формула I, «гидроксикислота»)
В инертной атмосфере 2 г гидроксида калия растворяли в смеси из 100 мл метанола и 100 мл воды. После этого добавляли 10 мл водного раствора пероксида водорода (130 объемов) и реакционную смесь охлаждали до 10°С+2°С, к которой добавляли 5 г 21-ацетата флуметазона. Реакционную смесь перемешивали в течение ночи при указанной температуре и после завершения реакции хлористоводородной кислотой доводили рН до значения 2. Полученный осадок отфильтровывали, промывали водой до нейтрального значения рН и сушили при 45°С. Выход целевого соединения составил 80% масс/масс, что соответствует стехиометрическому выходу 91,3%.
ПРИМЕР 5
6α, 9α-дифтор-11β,17α-дигидрокси-16α-метил-17β-карбоксиандроста-1,4-диен-3-он
22 г флуметазона, не содержащего спирта, полученного в примере 3, в инертной атмосфере суспендировали в 110 мл тетрагидрофурана и охлаждали до 20°С+2°С. При перемешивании медленно добавляли 17,6 г периодной кислоты в 70 мл воды. После перемешивания при той же температуре завершение реакции определяли методом ВЭЖХ. Определили, что реакция обычно завершается за два часа. После этого реакционную смесь переливали в раствор из 33 г метабисульфита натрия в 770 мл воды и 330 мл льда. Осажденный продукт реакции фильтровали и промывали водой до нейтрального значения рН, после чего сушили при 40-45°С с получением 21 г целевого соединения с чистотой 96% согласно определению по площади пика методом ВЭЖХ. После перекристаллизации образовавшегося соединения в этаноле получали 6α,9α-дифтор-11β,17α-дигидрокси-16α -метил-17β-карбоксиандроста-1,4-диен-3-он высокой чистоты, обладающий следующими аналитическими значениями:
- оптическое вращение = +64,4° (с=1% ДМФ);
- KF - 0,09%;
- чистота, определенная методом ВЭЖХ по площади пика = 99,2%.
- Продукт характеризуется основными пиками поглощения в инфракрасной области при длинах волн 1698 см-1, 1660 см-1, 1614 см-1и 1603 см-1.
Реферат
Описывается улучшенный способ получения флуметазона (6α,9α-дифтор-11β,17α,21-тригидрокси-16α-метилпрегна-1,4-диен-3,20-диона), флуметазон 21-ацетата или его 17-карбоксил-андростенового аналога формулы I, действием хлористого бензоила в среде пиридина или его смеси с N,N'-диметилацетамидом на соединение II с получением 3-енольного сложного эфира IIIa, с последующим его взаимодействием с 1-(хлорметил)-4-фтор-1,4-диазоний-бицикло[2.2.2]октан-бис(тетрафторборатом) в среде ацетонитрила и воды с получением IIIв, после чего в положении С3 у IIIв снимают защитную группу в среде водного метабисульфита и аммиака с получением IV: после фторирования 9,11-эпоксигруппы в IV с помощью HF получают 21-ацетат флуметазона, после чего проводят необязательный гидролиз с помощью КОН в СН3ОН в присутствии или в отсутствие Н2O2 с получением I или флуметазона соответственно. 2 н. и 1 з.п. ф-лы.





Формула






Комментарии