Соединение, способы его получения, фармацевтическая композиция соединения - RU2277100C2
Код документа: RU2277100C2
Описание
Данное изобретение относится к органическим соединениям, их получению и к их использованию в качестве фармацевтических препаратов.
Согласно одному из аспектов настоящее изобретение обеспечивает получение соединений формулы,

где R означает одновалентную циклическую органическую группу, содержащую от 3 до 15 атомов в циклической системе.
Термины, используемые в спецификации, имеют следующие значения:
"(С1-С4)алкил" означает (С1-С4)алкил с прямой или разветвленной цепью, который может означать метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил или трет-бутил;
"(С1-С4)алкиламиногруппа" означает аминогруппу, замещенную (С1 -С4)алкилом, как определено выше;
"ди((С1-С4)алкил)аминогруппа" означает аминогруппу, дважды замещенную (С1-С4)-алкилом, как определено выше;
"(С1-С4)алкилсульфонил" означает сульфонил, замещенный (С1-С4)алкилом, как определено выше;
"галоид(С1-С4)алкил" означает (С1-С4)алкил, как определено выше, замещенный одним или несколькими, предпочтительно одним, двумя или тремя атомами галоида, предпочтительно атомами фтора или хлора;
"гидрокси(С1-С4)алкил" означает (С1-С4)алкил, установленный выше, замещенный одной или несколькими, предпочтительно одной, двумя или тремя гидроксильными группами;
"(С1-С4)алкоксигруппа" означает (С1-С4)алкоксигруппу с прямой или разветвленной цепью, которая может быть метоксигруппой, этоксигруппой, н-пропоксигруппой, изопропоксигруппой, н-бутоксигруппой, изобутоксигруппой или трет-бутоксигруппой;
"(С1-С4 )алкоксикарбонил" означает карбонил, замещенный (C1-С4)алкоксигруппой, как установлено выше, и может быть метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, втор-бутокси- или трет-бутоксикарбонилом;
"(С1-С4)алкилтиогруппа" означает (С1-С4)алкилтиогруппу с прямой или разветвленной цепью и может быть метилтио-, этилтио-, н-пропилтио-, изопропилтио-, н-бутилтио-, изобутилтио-, втор-бутилтио- или трет-бутилтиогруппой;
"(С1-С4)ацил" означает карбонил, замещенный (С1-С4)алкилом, как установлено выше;
"(С1-С4)ацилоксигруппа" означает карбонилоксигруппу, замещенную (С1-С4 )алкилом, как установлено выше;
"(С1-С4)ациламиногруппа" означает аминогруппу, замещенную формилом или (С1-С4)-ацилом, как установлено выше.
R может быть карбоциклической группой или гетероциклической группой с одним или несколькими кольцевыми гетероатомами, выбранными из азота, кислорода и серы. В одном из вариантов осуществления изобретения R является циклоалифатической группой, содержащей от 3 до 8 атомов углерода, например, (С3-С8)циклоалкилом, как, например, циклопропил, метилциклопропил, циклобутил, метилциклобутил, циклопентил, циклогексил, метилциклогексил, диметилциклогексил или циклогептил, предпочтительно (С3-С6)циклоалкилом.
В другом варианте осуществления изобретения R является, как минимум, частично насыщенной гетероциклической группой, содержащей от 5 до 10 атомов в кольце, из которых один или несколько являются кольцевыми гетероатомами, выбранными из азота, кислорода и серы, предпочтительно группой, содержащей от 5 до 7 кольцевых атомов, из которых один или два являются гетероатомами, выбранными из азота и кислорода, в частности пятичленной гетероциклической группой с одним кольцевым гетероатомом, как, например, тетрагидрофуриловая или оксотетрагидрофуриловая группа.
Еще в другом варианте осуществления изобретения R является карбоциклической или гетероциклической ароматической группой, содержащей от 5 до 15 атомов в кольце. Например, R может быть такой ароматической группой, где кольцевая система является незамещенной или замещенной одним или несколькими заместителями, выбранными из галогена, циангруппы, (С1-С4)алкила, галоид(С1-С4 )алкила, (С1-С4)алкоксигруппы, (С1-С4)алкилтиогруппы, гидроксила, (С1-С4)ацила, (С1-С4)ацилоксигруппы, аминогруппы, (С1-С4)алкиламиногруппы, ди((С1-С4)алкил)аминогруппы, (С1-С4)ациламиногруппы, ацил((С1-С4 )алкил)аминогруппы, (С1-С4)алкилсульфонил((С1-С4)алкил)аминогруппы, (С1-С4)алкоксикарбонила, или 5-членным гетероциклическим остатком, обычно N-гетероциклическим остатком с одним или двумя атомами азота. Одним предпочтительным классом таких ароматических групп является фенил или нафтил, необязательно замещенный одним или несколькими, предпочтительно одним, двумя или тремя заместителями, выбранными из циангруппы, (С1-С4)алкила, галоид(С1-С4)алкила, (С1-С4 )алкоксигруппы, галогена, гидроксила, (С1-С4)ацилоксигруппы, аминогруппы, (С1-С4)алкиламиногруппы, ди((С1-С4)алкил)аминогруппы, (С1-С4)ациламиногруппы, (С1-С4)ацил((С1-С4)алкил)аминогруппы, (С1-С4)-алкилсульфонил((С1-С4 )-алкил)аминогруппы или (С1-С4)алкоксикарбонила, особенно предпочтительны подобные ароматические группы, включающие фенил, цианфенил, толил, диметилфенил, этилфенил, (трифторметил)фенил, диметоксифенил, диэтоксифенил, гидроксифенил, (метиламино)фенил, (метансульфонилметиламино)фенил и (метоксикарбонил)фенил.
Другим предпочтительным классом таких ароматических групп является гетероциклическая ароматическая группа с 6-членным гетероциклическим кольцом с одним, двумя или тремя гетероатомами в кольце, предпочтительно с азотом, гетероциклическое кольцо является незамещенным или замещено одним или несколькими, предпочтительно одним, двумя или тремя заместителями, выбранными из галогена, циангруппы, гидроксила, (С1-С4 )ацилоксигруппы, аминогруппы, (С1-С4)-алкиламиногруппы, ди((С1-С4)алкил)аминогруппы, (С1-С4)алкила, гидрокси(С1-С4)-алкила, галоид(С1-С4)алкила, (С1-С4)алкоксигруппы или (С1-С4)алкилтиогруппы, и гетероциклическое кольцо необязательно сконденсировано с бензольным кольцом. Предпочтительные подобные гетероциклические ароматические группы включают те, в которых гетероциклическая группа содержит один или два атома азота в кольце, в частности пиридиновое, пиримидиновое, пиразиновое или пиридазиновое кольцо. Особо предпочтительными гетероциклическими ароматическими группами являются пиридильная, пиримидильная и пиразинильная группы, необязательно замещенные одним или двумя заместителями, выбранными из галогена (в особенности, хлора) или (С1-С4)алкила (в частности, метила или н-бутила).
Другим предпочтительным классом таких ароматических групп является гетероциклическая ароматическая группа, имеющая 5-членное гетероциклическое кольцо с одним, двумя или тремя кольцевыми гетероатомами, выбранными из азота, кислорода и серы, при этом гетероциклическое кольцо является незамещенным или оно замещено одним или двумя заместителями, выбранными из галогена, (С1-С4 )алкила, галоид(С1-С4)алкила, (С1-С4)алкоксигруппы, (С1-С4)алкилтиогруппы, циангруппы или гидрокси(С1-С4)алкила, и гетероциклическое кольцо необязательно сконденсировано с бензольным кольцом. Такие предпочтительные гетероциклические ароматические группы включают те, в которых гетероциклическое кольцо содержит в кольце один атом азота, кислорода или серы или один атом кислорода и один или два атома азота в кольце, или один атом серы и один или два атома азота в кольце, в частности кольцо пиррола, фурана, тиофена, оксазола, изоксазола, имидазола, пиразола, фуразана, тиазола или тиадиазола. Особенно предпочтительными гетероциклическими ароматическими группами являются пирролильная, фурильная и тиенильная группы, необязательно замещенные одним или двумя заместителями, выбранными из галогена (в частности, хлора или брома), (С1-С4)алкила (в частности, метила или этила), галоид-(С1-С4)алкила (в частности, трифторметила), (С1-С4)алкоксигруппы (в частности, метоксигруппы), (С1-С4)алкилтиогруппы (в частности, метилтиогруппы), циангруппы или гидрокси(С1-С4)алкила (в частности, гидроксиметила), изоксазолильная, имидазолильная, пиразолильная, тиазолильная или тиадиазолильная группы, необязательно замещенные одной или двумя (С1-С4)алкильными группами, и бензофурильная, бензотиенильная и бензофуразанильная группы.
В соединениях формулы I указанная метильная группа в положении 16 кортикостероидной циклической системы может иметь α- или β-конформацию. Предпочтительны 16-α-метилпроизводные.
Соединения формулы I, в которых R содержит основную группу, способны образовывать кислотно-аддитивные соли, в особенности фармацевтически приемлемые кислотно-аддитивные соли. Фармацевтически приемлемые кислотно-аддитивные соли соединения формулы I включают соли неорганических кислот, например галоидводородных кислот, как, например, фтористоводородная кислота, соляная кислота, бромистоводородная кислота или иодистоводородная кислота, азотной кислоты, серной кислоты, фосфорной кислоты, и органических кислот, например алифатических монокарбоновых кислот, как, например, муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота и масляная кислота, алифатических оксикислот, как, например, молочная кислота, лимонная кислота, винная кислота или яблочная кислота, дикарбоновых кислот, как, например, малеиновая кислота или янтарная кислота, ароматических карбоновых кислот, как, например, бензойная кислота, п-хлорбензойная кислота, дифенилуксусная кислота или трифенилуксусная кислота, ароматических гидроксикислот, как, например, о-гидроксибензойная кислота, п-гидроксибензойная кислота, 1-гидроксинафталин-2-карбоновая кислота или 3-гидроксинафталин-2-карбоновая кислота, и сульфокислот, как, например, метансульфокислота или бензолсульфокислота. Эти соли могут быть получены из соединений формулы I с помощью известных методик образования солей.
Особенно предпочтительные соединения формулы I включают те, которые описаны ниже в примерах, в частности соединения из примеров 3, 11, 14, 17, 19, 26, 34, 37, 39, 51, 60, 67, 72, 73, 90, 99 и 101.
Согласно другому аспекту настоящее изобретение представляет способ получения соединений формулы I, который включает
(А) превращение карбоновой кислоты формулы,

где R является таким, как указано выше, или ее функционального производного, образующего сложный эфир, в сложный метиловый эфир этой кислоты; или
(Б) присоединение хлористого водорода к соединению формулы,

где R является таким, как указано выше.
Вариант способа (А) может быть осуществлен с применением известных методик превращения карбоновой кислоты или ее образующего сложный эфир функционального производного, например ее галоидангидрида, в соответствующий сложный метиловый эфир. Обычно карбоновая кислота подвергается реакции с метиловым эфиром сильной кислоты, предпочтительно с диметилсульфатом, в присутствии апротонного органического основания, как, например, 1, 8-диазабицикло[5.4.0]ундец-7-ен (ДБУ). Реакцию обычно проводят в инертном органическом растворителе, например в амиде, как, например, диметилформамид, в простом эфире, как, например, тетрагидрофуран, или в их смеси. Подходящей является температура реакции от температуры окружающей среды до 100°С.
Вариант способа (Б) может быть осуществлен при использовании известных методик присоединения хлористого водорода, например при пропускании газообразного хлористого водорода в раствор соединения формулы III в инертном органическом растворителе, например в углеводороде, как, например, толуол. Подходящей является температура реакции от температуры окружающей среды до 60°С.
Соединения формулы II являются новыми и могут быть получены при соответствующем ацилировании подходящего 17-гидроксипроизводного, т.е. соединения формулы


Ацилирование может быть проведено при использовании известных методик, например при реакции соединения формулы IV с галоидангидридом формулы RCOX, где R является таким, как указано выше, и Х означает галоген, например бром или предпочтительно хлор. Реакцию обычно проводят в присутствии основания, предпочтительно третичного органического основания, как, например, пиридин. Подходящей является температура от температуры окружающей среды до 50°С. Ацилирование может быть также осуществлено при реакции соединения формулы IV с карбоновой кислотой формулы RCO2H в присутствии активирующего средства, как, например, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N', N'-тетраметилурония (HATU), и основания, предпочтительно третичного органического основания, как, например, N,N-диизопропилэтиламин. Реакция может быть проведена в диполярном апротонном растворителе, как, например, N,N-диметилформамид (ДМФА), или в хлоруглеводородном растворителе, как, например, дихлорметан (ДХМ). Подходящей является температура реакции от температуры окружающей среды до 60° С. Соединения формулы IV могут быть получены при присоединении хлористого водорода к соответствующему 9,11-эпоксипроизводному формулы V,

например, используя известные методики присоединения хлористого водорода, такие, как описанные выше. Соединение формулы V, где метильная группа в положении 16 имеет α-конформацию, может быть получено, как описано Aigbirhio и др., J. Labelled Compd. Radiopharm. (1997), 39 (7), 567-584. Соединение формулы V, где метильная группа в положении 16 имеет β-конформацию, может быть получено, как описано в патенте US №4607028.
Соединения формулы III являются новыми и могут быть получены путем превращения соответствующей 17-карбоновой кислоты формулы

в ее метиловый эфир. Это превращение может быть осуществлено с применением описанных выше методик для варианта способа (А). Соединения формулы VI могут быть получены при соответствующем ацилировании соответствующего 17-гидроксипроизводного формулы V; такое ацилирование может быть осуществлено с использованием известных методик, например, описанных выше для ацилирования соединений формулы IV.
Соединения формулы I полезны в качестве фармацевтических препаратов. Соответственно, изобретение также предоставляет соединение формулы I для применения в качестве фармацевтического препарата. Соединения формулы I обладают важными фармакологическими свойствами. Например, они проявляют высокую противовоспалительную активность, которая может быть продемонстрирована при ингибировании ими синтеза фактора некроза опухоли (α-TNF) и выделения в линии клеток макрофага человека и при их подавлении воспалительных состояний, в частности, в дыхательных путях, например, ингибирование активации эозинофилов, на моделях животных, например воспаление дыхательных путей на моделях мышей и крыс, например, как описано Scarka и др., J. Immunol. Methods (1997) 202: 49-57; Renzi и др., Am. Rev. Respir. Dis. (1993) 148: 932-939; Tsuyuki и др., J. Clin. Invest. (1995) 96: 2924-2931; и Cernadas и др. (1999) Am. Respir. Cell Mol. Biol. 20: 1-8.
Соединения формулы I обнаруживают в терапевтически эффективных дозах на удивление слабые общие побочные действия. Соединения формулы I характеризуются длительной продолжительностью действия, обеспечиваемой введением один раз в день.
Ингибирование синтеза α-TNF и выделения из линии клеток U937 макрофага человека соединениями формулы I может быть продемонстрировано и измерено в исследовании, описанном Sajjadi и др., J. Immunol. 1996; 156: 3435-3442. Соединения из примеров 3, 11, 14, 17, 19, 26, 34, 37, 39, 51, 60, 67, 72, 73, 90, 99 и 101 имели в данном исследовании значения IC50 (нМ) (IC50 - концентрация, вызывающая 50%-ное ингибирование) 0,035, 0,025, 0,100, 0,05, 0,046, 0,024, 0,10, 0,102, 0,101, 0,048, 0,048, 0,048, 0,102, 0,159, 0,076, 0,106 и 0,208 соответственно.
Противовоспалительная активность in vivo может быть определена при подавлении легочной эозинофилии у крыс, применяя модификацию метода Szarka и др. в цитируемой статье. Самцов крыс линии Brown Norway (примерно 200 г) сенсибилизировали в день 1 с помощью внутрибрюшинной инъекции 0,5 мл смеси овальбумина (0,02 мг/мл) и гидроокиси алюминия (20 мг/мл), а затем бесклеточной коклюшевой адсорбат-вакциной (0,2 мл в разведении 1:4 с помощью 0,9% физиологического раствора). Процедуру повторяют в день 15 и в день 21. В день 28 вводят внутритрахеально исследуемое соединение в виде сухой смеси с порошком лактозы при анестезии изофлюраном. Через 24 часа сенсибилизированных введенной дозой крыс подвергают воздействию аэрозоля из овальбумина (5 мг/мл) в течение 60 минут и затем еще через 24 часа их умерщвляют. Легкие удаляют и после промывания раствором Хенкса (сбалансированный солевой раствор, 100 мл; этилендиаминтетрауксусная кислота /ЭДТК/ 100 мМ, 100 мл; N-2-гидроксиэтилпиперазин-N'-2-этан-сульфокислота /HEPES/ 1M, 10 мл, вода 1000 мл) количественно оценивают число эозинофилов в выделенном растворе, непосредственно используя аппарат Corbas Helios 5 Diff (фирма Hoffman-LaRoche).
Соединения из примеров 14, 17, 26, 34, 37, 39, 51, 60, 73, 99 и 101 понижают в данном анализе количества эозинофилов по сравнению с контролем при использовании наполнителя на 65, 71, 63, 90, 61, 76, 69, 67, 43, 48 и 40% соответственно. Соединения из примеров 14, 26, 34 и 99 вводили в дозе 3 мг/кг, а остальные в дозе 1 мг/кг.
Общие побочные действия могут быть оценены по уменьшению массы вилочковой железы при длительном введении крысам. Самцов крыс линии Sprague-Dawley (массой около 250 г) обрабатывают один раз в день в течение 4 дней исследуемым соединением, вводимым в дозе 1 мг/кг либо перорально в виде суспензии с гидроксипропилцеллюлозой или внутритрахеально при анестезии изофлюраном в виде сухой смеси с порошком лактозы. В день 5 крыс умерщвляют, проводят аутопсию и определяют массу тимуса. В данном исследовании соединения из примеров 17, 26, 34, 37, 73, 99 и 101 при пероральном введении вызывают уменьшение массы тимуса по сравнению с используемым в качестве контроля наполнителем на 20, 2, 0, 19, 9, 0 и 2% соответственно. В данном исследовании соединения из примеров 17, 26, 73 и 99 при внутритрахеальном введении вызывают уменьшение массы тимуса по сравнению с используемым в качестве контроля наполнителем на 78, 55, 31 и 70% соответственно.
Учитывая их противовоспалительную активность, соединения формулы I полезны при лечении воспалительных состояний, в частности воспалительных или обструктивных заболеваний дыхательных путей. Лечение согласно изобретению может быть симптоматическим или профилактическим.
Воспалительные или обструктивные заболевания дыхательных путей, к которым применимо настоящее изобретение, включают астму любого типа или происхождения, включая врожденную (неаллергическую) астму и вызываемую внешними условиями (аллергическую) астму, слабую астму, астму средней тяжести, тяжелую астму, бронхиальную астму, вызванную физической нагрузкой астму, профессиональную астму и астму, индуцированную бактериальной инфекцией. Под лечением астмы следует понимать лечение, воздействующее на субъектов, например, возраста 4 или 5 лет, проявляющих симптомы свистящего дыхания и диагностируемых или которым можно поставить диагноз "страдающие астмой дети младшего возраста", это установленная категория больных, вызывающая особое беспокойство медиков, сейчас часто идентифицируется как начальные астматики или астматики на ранней стадии болезни. (Для удобства на это особое астматическое состояние ссылаются как на "астматический синдром у детей".)
Свидетельством эффективности профилактики в лечении астмы является уменьшенная частота или тяжесть симптоматических приступов, например острого астматического или бронхостенозного приступа, улучшение функционирования легких или улучшенная повышенная реактивность дыхательных путей. Об эффективности профилактики далее могут свидетельствовать сниженная потребность в другой симптоматической терапии, то есть, терапии с целью ограничения или раннего купирования симптоматического приступа, когда он возникает, например терапии противовоспалительной (например, кортикостероидом) или бронхорасширяющей. Польза профилактики при астме может, в частности, быть очевидной для субъектов, склонных к "утренней глубокой пальпации". "Утренняя глубокая пальпация" является общепризнанным астматическим синдромом, обычным для значительного процента астматиков и характеризующимся астматическим приступом, например, между 4 и 6 часами утра, то есть, в такое время, которое, по существу, отдалено от любой ранее примененной симптоматической терапии астмы.
Другие противовоспалительные или обструктивные заболевания дыхательных путей или состояния, к которым применимо данное изобретение, включают острое легочное повреждение (ALI), респираторный дистресс-синдром взрослых (ARDS), хроническое обструктивное легочное заболевание, заболевание дыхательных путей или заболевание легких (COPD, COAD или COLD), включая хронический бронхит или связанную с ним одышку, эмфизему, а также обострение повышенной реактивности дыхательных путей, являющееся результатом лечения другими лекарствами, в особенности, лечения другими вдыхаемыми препаратами. Изобретение также применимо к лечению бронхита любого вида или происхождения, включая, например, острый, арахноидальный, катаральный, крупозный, хронический или бронхит при туберкулезе. Другие воспалительные или обструктивные заболевания дыхательных путей, к которым применимо настоящее изобретение, включают пневмокониоз (воспалительное, обычно связанное с профессией заболевание легких, часто сопровождаемое обструкцией дыхательных путей, как хроническое, так и острое, и вызываемое неоднократным вдыханием всякой пыли) любого типа или происхождения, включающий, например, алюминоз, антракоз, асбестоз, халикоз, птилоз, сидероз, силикоз, табакоз и биссиноз.
Что касается их противовоспалительной активности, в особенности, в связи с ингибированием активации эозинофилов, соединения формулы I также пригодны для лечения нарушений, связанных с эозинофилами, например эозинофилия, в частности, связанные с эозинофилами нарушения дыхательных путей (например, включающие вызванную болезнью инфильтрацию эозинофилов легочных тканей), включая гиперэозинофилию, так как она влияет на дыхательные пути и/или легкие, а также, например, связанные с эозинофилами нарушения дыхательных путей, вызываемые или сопутствующие синдрому Леффлера, эозинофильную пневмонию, паразитарную (в частности, относящуюся к многоклеточным) инвазию (включая тропическую эозинофилию), бронхолегочный аспергиллез, полиартритные узловатости (включая синдром Чарга-Штраусса), эозинофильную гранулему и связанные с эозинофилами нарушения, наносящие вред дыхательным путям, вызванные взаимодействиями с лекарствами.
Соединения формулы I также полезны при лечении воспалительных состояний кожи, например псориаза, контактного дерматита, атопического дерматита, гнездной алопеции, многообразных эритем, сходного с герпесом дерматита, склеродермии, витилиго, васкулита повышенной чувствительности, крапивницы, пузырчатого пемфигоида, красной волчанки, обыкновенной пузырчатки, приобретенного буллезного эпидермолиза и других воспалительных состояний кожи.
Соединения формулы I могут быть также использованы для лечения других заболеваний или состояний, в частности заболеваний или состояний, включающих воспалительный компонент, например для лечения заболеваний и состояний глаз, как, например, конъюнктивит, кератоконъюнктивит сухой и весенний конъюнктивит, заболеваний, влияющих на нос, включая аллергический ринит, заболеваний суставов, как, например, ревматоидный артрит, и воспалительного заболевания кишечника, как, например, язвенный колит, и болезни Крона.
Соединения формулы I также полезны в качестве сотерапевтических средств для применения в сочетании с другими лекарственными веществами для лечения заболеваний дыхательных путей, особенно с бронхорасширяющими или противовоспалительными лекарственными препаратами, в особенности при лечении обструктивных или воспалительных заболеваний дыхательных путей, таких как указанные выше, например, в качестве средств, потенциирующих терапевтическую активность таких лекарственных препаратов, или в качестве средств для снижения требуемой дозы или возможных побочных действий таких лекарственных препаратов. Соединение формулы I может быть смешано с другим лекарственным препаратом в установленной фармацевтической композиции или оно может быть применено отдельно, до, одновременно с или после другого лекарственного препарата. Эти другие лекарственные препараты включают антихолинергические или антимускариновые средства, в частности бромид ипратропия, бромид окситропия и бромид тиотропия, антагонисты LTB4 (лейкотриен B4), как, например, описанные в патенте US №5451700, антагонисты LTD4 (лейкотриен D4), как, например, монтелюкаст и зафирлюкаст, агонисты допаминового рецептора, как, например, каберголин, бромокриптин, ропинирол и 4-гидрокси-7-[2-[[2-[[3-(2-фенилэтокси)пропил]сульфонил]этил]амино]этил]-2(3Н)-бензотиазолон и их фармацевтически приемлемые соли (гидрохлорид, являющийся Viozan®-AstraZeneca), ингибиторы PDE4, например, арифло®(фирма GlaxoSmith Kline), рофлюмиласт (фирма Byk Gulden), V-11294A (фирма Napp), BAY19-8004 (фирма Bayer), SCH-351591 (фирма Schering-Plough) и PD 189659 (фирма Parke-Davis), и β2-агонисты адренергических рецепторов, как, например, сальбутамол, тербуталин, сальметерол и, в частности, формотерол, и их фармацевтически приемлемые соли, и соединения (в свободной форме или в виде солей или сольватов) формулы I из опубликованной международной заявки на патент WO 00/75114 по договору о патентной кооперации (РСТ), на которую здесь ссылаются, предпочтительно соединения из приведенных там примеров, в частности соединение формулы
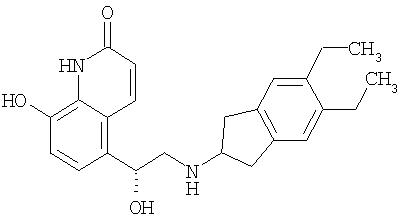
в свободной форме или в виде фармацевтически приемлемой соли или сольвата.
Комбинации соединений формулы I и β2-агонистов, ингибиторов PDE4 или антагонистов LTD4 может быть использована, например, при лечении COPD или, в частности, астмы. Комбинация средств по изобретению и антихолинергических или антимускариновых средств, ингибиторов PDE4, антагонистов LTB4 или допаминовых агонистов может быть использована, например, при лечении астмы или, в особенности, COPD.
В соответствии с изложенным выше изобретение также обеспечивает способ лечения воспалительного состояния, в особенности, воспалительного или обструктивного заболевания дыхательных путей, который включает введение субъекту, в частности человеку, в случае необходимости эффективного количества соединения формулы I, как описано выше. В другом аспекте изобретение обеспечивает применение соединения формулы I, описанного выше, для приготовления лекарства для лечения воспалительного состояния, в особенности, воспалительного или обструктивного заболевания дыхательных путей.
Соединение формулы I может быть введено любым подходящим способом, например, перорально, в виде таблетки или капсулы; парентерально, например, внутривенно; путем ингаляции, например при лечении воспалительного или обструктивного заболевания дыхательных путей; внутриназально, например при лечении аллергического ринита; местно на кожу, например при лечении атопического дерматита; или через прямую кишку, например при лечении воспалительного заболевания кишечника.
Согласно еще одному аспекту изобретение также предоставляет фармацевтическую композицию, включающую в качестве активного ингредиента соединение формулы I, необязательно вместе с фармацевтически приемлемым для этого разбавителем или носителем. Композиция может содержать сотерапевтическое средство, как, например, бронхорасширяющее или противовоспалительное лекарственное средство, как описано выше. Такие композиции могут быть приготовлены с использованием обычных разбавителей или наполнителей и методик, известных в производстве галеновых препаратов. Таким образом, пероральные лекарственные формы могут включать таблетки и капсулы. Готовые формы для местного применения могут быть в виде кремов, мазей, гелей или трансдермальных систем доставки лекарственного вещества, например пластырей. Композиции для ингаляции могут включать аэрозоль или другие тонко измельченные готовые лекарственные формы или готовые формы в виде сухого порошка.
Изобретение включает (А) соединение формулы I в форме для вдыхания, например в виде аэрозоля или другой мелкоизмельченной композиции или в виде вдыхаемых твердых частиц, например частиц микронных размеров, (Б) вдыхаемое лекарственное средство, включающее соединение формулы I в ингаляционной форме; (В) фармацевтический продукт, включающий соединение формулы I в ингаляционной форме вместе с приспособлением для ингаляции; и (Г) приспособление для ингаляции, содержащее соединение формулы I в форме для вдыхания.
Дозировки соединений формулы I, применяемые в практике настоящего изобретения, будут, конечно, варьироваться в зависимости, например, от конкретного, подвергающегося лечению состояния, от желаемого эффекта и от способа введения. Обычно подходящие ежедневные дозы для введения путем ингаляции составляют от 0,005 до 10 мг, в то время как подходящие дневные дозы для перорального введения составляют от 0,05 до 100 мг.
Изобретение иллюстрируется следующими примерами.
Примеры 1-101
Соединения формулы I и способы их получения представлены в следующей таблице 1, где Et означает этил и n-Bu означает н-бутил, способы описаны далее. В примерах №34 и 45 метильная группа в положении 16 имеет β-конформацию; во всех других примерах она имеет α-конформацию.




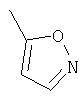

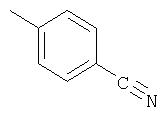






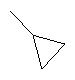






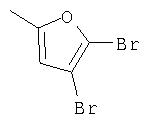




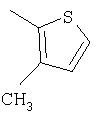





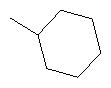
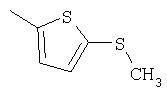


















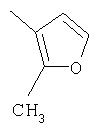




























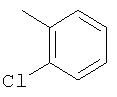







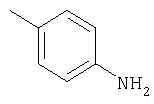





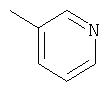





Способ А
Стадия 1
Растворяют (6S,9S,10S,11S, 13S,16R,17R)-9,11-эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]-фенантрен-17-карбоновую кислоту (50 г) в диоксане (500 мл). Через раствор в течение 15 минут пропускают путем барботирования HCl-газ и реакционную смесь перемешивают при комнатной температуре. Через 4 часа образовавшийся в результате осадок собирают при фильтрации и промывают метанолом. Неочищенный продукт кипятят в метаноле и фильтруют горячим. Фильтрат упаривают и получают (6S,9R,10S,11S,12S,13S,16R,17R)9-хлор-6-фтор-11,17-дигидрокси-10,13,16-триметил-3-оксо-6, 7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]-фенантрен-17-карбоновую кислоту. Избранные сигналы1Н-ЯМР-спектров (d6-ДМСО) δ 6,10 (1H,d), 6,30 (1H, dd), 7,25 (1H, d).
Стадия 2
Продукт со стадии 1 (250 мг) растворяют в пиридине (1,5 мл) и прибавляют к хлорангидриду 4-метил-1,2,3-тиадиазол-5-карбоновой кислоты (108 мг). Реакционную смесь перемешивают при комнатной температуре 2 часа, затем прибавляют по каплям к 6 н. соляной кислоте. Полученный в результате осадок собирают при фильтрации и сушат, получают (6S,9R,10S, 11S,13S,16R,17R)-17-карбокси-9-хлор-6-фтор-11-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]фенантрен-17-иловый эфир 4-метил-[1,2, 3]тиадиазол-5-карбоновой кислоты. Время удерживания при ВЭЖХ 0,849 минуты; условия ВЭЖХ: колонна высокого разрешения Zorbax, А=0,1% трифторуксусная кислота (ТФК) в воде, В=0,1% ТФК в ацетонитриле, градиент от 30 до 95% В в А в 1 минуту при 4 мл/мин, 50°С.
Стадия 3
Продукт со стадии 2 (326 мг) растворяют в диметилформамиде (ДМФА, 0,5 мл) и тетрагидрофуране (ТГФ, 1 мл). Прибавляют 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, 101 мг), а затем диметилсульфат (84 мг). Реакционную смесь перемешивают при комнатной температуре 2 часа, затем распределяют между водой и дихлорметаном (ДХМ). Органический слой сушат над сульфатом магния и упаривают. Соединение очищают с помощью флэш-хроматографии на силикагеле, элюируя гексаном-этилацетатом (1:1), получают (6S,9R,10S, 11S,13S,16R,17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]фенантрен-17-иловый эфир 4-метил-[1,2, 3]тиадиазол-5-карбоновой кислоты.
Способ Б1
Стадия 1
(6S,9S,10S,11S,13S,16R,17R)-9,11-Эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12, 13,14,15,16,17-додекагидро-3Н-циклопента[а]фенантрен-17-карбоновую кислоту (5 г) растворяют в пиридине (15 мл) и охлаждают до 0°С. Прибавляют хлорангидрид фуран-2-карбоновой кислоты (1,82 г) и реакционную смесь перемешивают при комнатной температуре. Через 2 часа реакционную смесь по каплям добавляют к интенсивно перемешиваемому раствору 6М соляной кислоты. Прибавляют ДХМ, фазы разделяют и органическую фазу промывают водой и соляным раствором. После высушивания над сульфатом магния и упаривания получают (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-карбокси-10,13, 16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]фенантрен-17-иловый эфир фуран-2-карбоновой кислоты. Избранные сигналы1Н-ЯМР (CDCl3) δ 6,30 (1H, dd), 6,5 (2Н, m), 6,55 (1H, d), 7,20-7,65 (2H, m).
Стадия 2
Продукт со стадии 1 (6,20 г) растворяют в этилацетате (100 мл). Последовательно прибавляют ДБУ (2,20 г) и диметилсульфат (1,83 г). Реакционную смесь перемешивают при комнатной температуре 2 часа, затем распределяют между этилацетатом и водой. Органический слой промывают водой и соляным раствором, сушат над сульфатом магния и упаривают. Кристаллизация из метанола приводит к (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3Н-циклопента[а]фенантрен-17-иловому эфиру фуран-2-карбоновой кислоты. Избранные сигналы1Н-ЯМР (CDCl3): δ 3,75 (3Н, s), 6,30 (1H, dd), 6,45 (1H, d), 6,55 (2H, m), 7,15 (1H, d), 7,60 (1H, d).
Стадия 3
Продукт со стадии 2 (4,5 г) растворяют в толуоле (150 мл). Через раствор пропускают путем барботирования HCl-газ в течение 15 минут и реакционную смесь перемешивают при комнатной температуре 6 часов. Растворитель выпаривают и технический продукт кристаллизуют из изопропилового спирта, получают (6S,9R,10S,11S,13S,16R, 17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]-фенантрен-17-иловый эфир фуран-2-карбоновой кислоты.
Способ Б2
Стадия 1
(6S,9S,10S,11S,13S,16R,17R)-9,11-Эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3Н-циклопента[а]фенантрен-17-карбоновую кислоту (20 г) растворяют в пиридине (50 мл) и охлаждают до 0°С. Прибавляют хлорангидрид 3-метилтиофен-2-карбоновой кислоты (9,39 г) и реакционную смесь перемешивают при комнатной температуре. Через 2 часа реакционную смесь прибавляют по каплям к интенсивно перемешиваемому раствору 6М соляной кислоты. Прибавляют ДХМ, фазы разделяют и органическую фазу промывают водой и соляным раствором. После высушивания над сульфатом магния и упаривания подвергают флэш-хроматографии на силикагеле, элюируя ДХМ-метанолом (25:1), получают (6S,9S, 10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-карбокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]фенантрен-17-иловый эфир 3-метилтиофен-2-карбоновой кислоты. Избранные сигналы1Н-ЯМР (CDCl3): δ 0,90 (3Н, d), 0,95 (3Н, s), 1,40 (3Н, s), 2,40 (3Н, s).
Стадия 2
Продукт со стадии 1 (11,1 г) растворяют в этилацетате (200 мл). Последовательно прибавляют ДБУ (4,05 г) и диметилсульфат (3,36 г) и реакционную смесь перемешивают при комнатной температуре 2 часа, затем распределяют между этилацетатом и водой. Органический слой промывают водой и соляным раствором, сушат над сульфатом магния и упаривают. Кристаллизация из метанола приводит к (6S,9S,10S,11S,13S,16R,17R)-9, 11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]-фенантрен-17-иловому эфиру 3-метилтиофен-2-карбоновой кислоты. Избранные сигналы1Н-ЯМР (CDCl3): δ 0,90 (3Н, d), 0,92 (3Н, s), 1,40 (3Н, s), 2,40 (3Н, s), 3,65 (3Н, s).
Стадия 3
Продукт со стадии 2 (16 г) растворяют в толуоле (250 мл). Через раствор в течение 15 минут пропускают путем барботирования HCl-газ и реакционную смесь перемешивают при комнатной температуре 16 часов. Растворитель выпаривают и технический продукт кристаллизуют из ацетонитрила и второй раз из изопропанола, получают (6S,9R,10S,11S,13S,16R,17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11, 12,13,14,15,16,17-додекагидро-3Н-циклопента[а]фенантрен-17-иловый эфир 3-метилтиофен-2-карбоновой кислоты.
Способ В
Стадия 1
(6S,9S,10S,11S,13S,16S,17R)-9, 11-Эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновую кислоту (1 г) растворяют в пиридине (5 мл). Прибавляют хлорангидрид циклопропилкарбоновой кислоты (330 мг) и реакционную смесь перемешивают при комнатной температуре. Через 2 часа раствор прибавляют по каплям к интенсивно перемешиваемому раствору 6М соляной кислоты. Образующийся в результате осадок собирают при фильтрации и сушат, получают (6S,9S,10S,11S,13S,16S,17R)-9,11-эпокси-17-циклопропанкарбонилокси-6-фтор-10,13,16-триметил-3-оксо-6,7,8,9, 10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновую кислоту. Избранные сигналы1H-ЯМР (CDCl3): δ 6,25 (1H, dd), 6,45 (1H, d), 6,55 (1H, d).
Стадия 2
Продукт со стадии 1 (1,1 г) растворяют в этилацетате (25 мл). Прибавляют ДБУ (450 мг), а затем диметилсульфат (370 мг). Реакционную смесь перемешивают при комнатной температуре 2 часа и затем распределяют между этилацетатом и водой. Органический слой промывают водой и соляным раствором, сушат над сульфатом магния и упаривают. Технический продукт кристаллизуют из метанола, получают метиловый эфир (6S,9S,10S,11S,13S,16S,17R)-9,11-эпокси-17-циклопропан-карбонилокси-6-фтор-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновой кислоты. Избранные сигналы1Н-ЯМР (CDCl3): δ 3,65 (3H, s), 6,25 (1H, dd), 6,45 (1H, d), 6,55 (1H, d).
Стадия 3
Продукт со стадии 2 (500 мг) растворяют в толуоле (20 мл). Через раствор барботирует HCl-газ в течение 5 минут и реакционную смесь перемешивают при комнатной температуре 18 часов. Растворитель выпаривают и технический продукт кристаллизуют из изопропанола-метанола, получают метиловый эфир (6S,9R,10S,11S,13S,16S,17R)-9-хлор-17-циклопропанкарбонилокси-6-фтор-11-гидрокси-10, 13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]-фенантрен-17-карбоновой кислоты.
Способ Г
Стадия 1
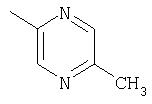
Прибавляют N, N-диизопропилэтиламин (2,3 мл) к охлажденному (0°С) раствору 5-метилпиразин-2-карбоновой кислоты (736 мг) в ДМФА (7 мл), а затем прибавляют гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N', N'-тетраметилурония (HATU, 2,26 г). Суспензию перемешивают при комнатной температуре 10 минут, затем прибавляют раствор (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-гидрокси-10,13, 16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновой кислоты (2 г) в ДМФА (7 мл). Через 2 часа реакционную смесь прибавляют по каплям к раствору 1М соляной кислоты. Продукт собирают при фильтрации, несколько раз промывают водой и сушат, получают (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-карбокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12, 13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 5-метилпиразин-2-карбоновой кислоты. Избранные сигналы1Н-ЯМР (CDCl3): δ 0,95 (3H, d), 1,10 (3H, s), 1, 40 (3H, s), 2,65 (3H, s).
Стадия 2
Продукт со стадии 1 (2,50 г) растворяют в ДМФА (11 мл) и охлаждают до 0°С. Прибавляют ДБУ (1,68 г), а затем через 10 минут диметилсульфат (953 мг). Реакционную смесь перемешивают при комнатной температуре 2 часа, выливают в воду и экстрагируют этилацетатом. Объединенные органические фазы промывают водой и соляным раствором, затем сушат над сульфатом натрия. После упаривания получают (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 5-метилпиразин-2-карбоновой кислоты. МС, наблюдаемая масса (М+Н)+ 511.
Стадия 3
Продукт со стадии 2 (1,92 г) растворяют в толуоле (100 мл). Через раствор путем барботирования пропускают HCl-газ в течение 90 минут и реакционную смесь перемешивают при комнатной температуре 16 часов. Растворитель выпаривают, технический продукт растирают с горячим этанолом и сушат, получают (6S,9R,10S,11S,13S,16R,17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14, 15,16,17-додекагидро-3H-циклопента[а]-фенантрен-17-иловый эфир 5-метилпиразин-2-карбоновой кислоты.
Способ Д
Стадия 1
Прибавляют N,N-диизопропилэтиламин (0,508 мл) к охлажденному (0°С) раствору 5-ацетоксифуранкарбоновой кислоты (258 мг) в ДМФА (1 мл), а затем прибавляют HATU (556 мг). Суспензию перемешивают при комнатной температуре 10 минут, затем прибавляют раствор (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновой кислоты (500 мг) в ДМФА (1 мл). Через 2 часа реакционную смесь медленно прибавляют к раствору 0,2 М соляной кислоты и экстрагируют ДХМ. Органическую фазу промывают водой и соляным раствором, затем сушат над сульфатом магния и упаривают, получают (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-карбокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 5-ацетоксифуран-2-карбоновой кислоты. ТСХ, Rf 0,5 (проявление ДХМ-метанолом 10:1).
Стадия 2
Продукт со стадии 1 (721 мг) растворяют в этилацетате (25 мл). Прибавляют ДБУ (242 мг), затем диметилсульфат (201 мг) и реакционную смесь перемешивают при комнатной температуре 2 часа, затем разбавляют 0,2 М соляной кислотой. Органическую фазу промывают водой и соляным раствором, затем сушат над сульфатом магния и упаривают. Очистка с помощью флэш-хроматографии на силикагеле при элюировании гексаном-этилацетатом (2:1) приводит к (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-иловому эфиру 5-ацетоксифуран-2-карбоновой кислоты. Избранные сигналы ЯМР (CDCl3): δ 0,96 (3H, d), 0,98 (3H, s), 1,40 (3H, s), 2,18 (3H, s), 2,76 (3H, s).
Стадия 3
Продукт со стадии 2 (555 мг) растворяют в метаноле (10 мл) и прибавляют 2М раствор гидроокиси натрия в метаноле (1 мл). Через 1 час растворитель выпаривают и остаток распределяют между водой и этилацетатом. Органическую фазу сушат над сульфатом магния и упаривают. Очистка с помощью флэш-хроматографии при элюировании гексаном-этилацетатом (1:1) приводит к (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-иловому эфиру 5-гидроксифуран-2-карбоновой кислоты. ТСХ, Rf0,25 (проявление гексаном-этилацетатом 1:1).
Стадия 4
Продукт со стадии 3 (250 мг) растворяют в толуоле (10 мл) и диоксане (10 мл). Через раствор пропускают путем барботирования HCl-газ в течение 10 минут и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Растворитель выпаривают и технический продукт очищают с помощью флэш-хроматографии на колонке с силикагелем, элюируя гексаном-этилацетатом (1:1), затем растирают с эфиром, получают (6S,9R,10S,11S,13S,16R,17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 5-гидроксифуран-2-карбоновой кислоты.
Способ Е
Стадия 1
(6S,9S,10S,11S,13S,16R,17R)-9,11-Эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10, 11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновую кислоту превращают при обработке, аналогичной таковой на стадии 1 способа Г, в (6S,9S,10S,11S,13S,16R,17R)-9, 11-эпокси-6-фтор-17-карбокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]-фенантрен-17-иловый эфир 3-ацетоксибензойной кислоты. Время удерживания при ВЭЖХ 0, 762 минуты, условия такие, как по способу А.
Стадия 2
Продукт со стадии 1 превращают при обработке, аналогичной таковой на стадии 2 способа Г, в (6S,9S,10S,11S,13S,16R, 17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 3-ацетоксибензойной кислоты. Наблюдаемая масса 552 (М+).
Стадия 3
Продукт со стадии 2 превращают при обработке, аналогичной таковой на стадии 3 способа Д, в (6S,9S,10S,11S,13S,16R,17R)-9, 11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 3-гидроксибензойной кислоты. Наблюдаемая масса 510 (М+).
Стадия 4
Продукт со стадии 3 превращают при обработке, аналогичной таковой на стадии 4 способа Д, в (6S,9R,10S,11S,13S,16R, 17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 3-гидроксибензойной кислоты.
Способ Ж
Стадия 1
(6S,9S,10S,11S,13S,16R,17R)-9,11-Эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновую кислоту превращают при обработке, аналогичной таковой на стадии 1 способа Г, в (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-карбокси-10,13, 16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]-фенантрен-17-иловый эфир 4-метиламинобензойной кислоты. Время удерживания при ВЭЖХ 0,728 минуты, условия: колонка высокого разрешения Мах RP, А=0,05% ТФК в воде, В=0,105% ТФК в ацетонитриле, градиент от 30 до 95% В в А в 1 минуту при скорости 4 мл/мин, 50°С.
Стадия 2
Продукт со стадии 1 превращают при обработке, аналогичной таковой на стадии 2 способа Г, в (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15, 16,17-додекагидро-3H-циклопента[а]-фенантрен-17-иловый эфир 4-метиламинобензойной кислоты. Наблюдаемая масса 523 (М+).
Стадия 3
Продукт со стадии 2 (42 мг) растворяют в дихлорметане (1 мл) и прибавляют ДБУ (66 мг), а затем через 5 минут хлорангидрид метансульфокислоты (114 мг). Реакционную смесь кипятят с обратным холодильником в течение ночи, после чего растворитель выпаривают и остаток растворяют в ДМФА. Раствор прибавляют по каплям к 1М соляной кислоте и образовавшийся в результате твердый осадок собирают при фильтрации и сушат, получают (6S,9S,10S, 11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 4-(метансульфонилметиламино)бензойной кислоты. Наблюдаемая масса 601 (М+).
Стадия 4
Продукт со стадии 3 (35 мг) растворяют в толуоле (10 мл) и через раствор пропускают путем барботирования HCl-газ в течение 5 минут перед тем, как смесь перемешивают при комнатной температуре 16 часов. Полученный в результате твердый остаток собирают при фильтрации и сушат, получают (6S,9R,10S,11S,13S,16R,17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 4-(метансульфонилметиламино)бензойной кислоты.
Способ З
Стадия 1
Гидрид натрия (60% дисперсия в минеральном масле, 241 мг) прибавляют к суспензии терефталевой кислоты (1 г) в ДМФА (5 мл), а затем через 5 минут прибавляют 2-(триметилсилил)этоксиметилхлорид (0,998 г). Через 2 часа реакционную смесь прибавляют по каплям к воде и полученный в результате твердый осадок отфильтровывают и сушат. Очистка с помощью флэш-хроматографии на силикагеле с элюированием этилацетатом приводит к моно(2-триметилсилилэтоксиметил)овому эфиру терефталевой кислоты. Время удерживания при ВЭЖХ 0,879 минуты, условия такие же, как для способа Ж.
Стадия 2
(6S,9S,10S,11S,13S,16R,17R)-9,11-Эпокси-6-фтор-17-гидрокси-10,13, 16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновую кислоту превращают при обработке, аналогичной таковой на стадии 1 способа Г, в (6S,9S,10S,11S,13S, 16R,17R)-9,11-эпокси-6-фтор-17-карбокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3Н-циклопента[а]-фенантрен-17-иловый эфир моно(2-триметилсилилэтоксиметил)ового эфира терефталевой кислоты. Время удерживания при ВЭЖХ 1,046 минуты, условия такие же, как для способа Ж.
Стадия 3
Продукт со стадии 2 превращают при обработке, аналогичной таковой на стадии 2 способа Г, в (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир моно(2-триметилсилилэтоксиметил)ового эфира терефталевой кислоты. Время удерживания при ВЭЖХ 1,055 минуты, условия такие же, как для способа Ж.
Стадия 4
Продукт со стадии 3 (320 мг) растворяют в толуоле (10 мл) и через раствор пропускают путем барботирования HCl-газ в течение 5 минут, а затем перемешивают 16 часов при комнатной температуре. Образовавшееся в результате твердое вещество отфильтровывают, растирают с ДХМ и сушат, получают моно[(6S,9R,10S,11S,13S,16R, 17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-ил]овый эфир терефталевой кислоты. Наблюдаемая масса 575,1 (М+).
Стадия 5
Продукт со стадии 4 (24 мг) растворяют в ДМФА (0,5 мл). Прибавляют ДБУ (8 мг), затем диметилсульфид (7 мг) и реакционную смесь перемешивают 1 час при комнатной температуре. Реакционную смесь прибавляют по каплям к 1М соляной кислоте и образовавшийся в результате твердый продукт отфильтровывают и сушат, получают 1-метил-4-[(6S, 9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-ил]овый терефталевой кислоты.
Способ И
Стадия 1
(6S,9S,10S,11S,13S,16R,17R)-9,11-Эпокси-6-фтор-17-гидрокси-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16, 17-додекагидро-3H-циклопента[а]фенантрен-17-карбоновую кислоту превращают при обработке, аналогичной таковой на стадии 1 способа Г, в (6S,9S,10S,11S,13S,16R,17R)-9,11-эпокси-6-фтор-17-карбокси-10,13, 16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]-фенантрен-17-иловый эфир 4-(трет-бутоксикарбониламино)бензойной кислоты. Время удерживания при ВЭЖХ 0,873 минуты, условия такие же, как для способа Ж.
Стадия 2
Продукт со стадии 1 превращают при обработке, аналогичной таковой на стадии 2 способа Г, в (6S,9S,10S,11S,13S,16R,17R)-9, 11-эпокси-6-фтор-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 4-(трет-бутоксикарбониламино)бензойной кислоты. Наблюдаемая масса 609,7 (М+).
Стадия 3
Продукт со стадии 2 (510 мг) растворяют в 150 мл толуола и через раствор пропускают путем барботирования HCl-газ в течение 5 минут, затем перемешивают 48 часов при комнатной температуре. Растворитель выпаривают и технический продукт кристаллизуют из этилацетата-циклогексана, получают (6S,9R,10S,11S,13S,16R, 17R)-9-хлор-6-фтор-11-гидрокси-17-метоксикарбонил-10,13,16-триметил-3-оксо-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[а]фенантрен-17-иловый эфир 4-аминобензойной кислоты.
Дополнительные примеры
Указанные в прилагаемых составах соединения А, В и С являются соединениями из примеров 17, 26 и 99 соответственно.
Пример 102
Аэрозольная композиция, годная для высвобождения под давлением из ингалятора необходимой дозы. Она получена смешением пропеллентов HFA 227 (1,1,1,2,3,3,3-гептафторпропана) и HFA 134a (1,1,1,2-тетрафторэтана), охлажденных до -55° С, при перемешивании с абсолютным этанолом, и затем добавлением микронизированного соединения А. Полученная суспензия интенсивно гомогенизируется перед ее введением в металлический контейнер ингаляционного устройства.
Пример 103
Аэрозольная композиция для высвобождения необходимой дозы из ингалятора, полученная так же, как и в примере 102.
Пример 104
Аэрозольная композиция для высвобождения необходимой дозы из ингалятора, полученная так же, как и в примере 102.
Пример 105
Сухой порошок, пригодный для высвобождения из резервуара мультидозового ингалятора, описанного в WO 97/20589. Получен путем смешения вышеуказанных ингредиентов, где соединение А состоит в основном из частиц диаметром 1-5 мкм, в пневматической мельнице, а частицы моногидрата лактозы имеют диаметр менее 212 мкм.
Пример 106
Сухой порошок, пригодный для высвобождения из резервуара мультидозового ингалятора, описанного в WO 97/20589. Получен, как описано в примере 105, но с использованием соединения В вместо соединения А.
Пример 107
Сухой порошок, пригодный для высвобождения из резервуара мультидозового ингалятора, описанного в WO 97/20589. Получен, как описано в примере 105, но с использованием соединения С.
Реферат
Изобретение относится к соединениям формулы (I),

где R - (С3-С6)циклоалкил, возможно замещенный С1-С4алкилом, или бензофуранил, нафтил, бензотиофенил, бензооксадиазолил; или фенил, возможно замещенный одним или несколькими заместителями. Описаны также способы получения соединения I. Указанные соединения полезны в качестве фармацевтических препаратов, обладающих противовоспалительными свойствами. 5 н. и 7 з.п. ф-лы, 1 табл.
Формула



Комментарии