Производные 16-гидрокси-11-(замещенный фенил)-эстра-4,9-диена, соединение, фармацевтическая композиция - RU2187510C2
Код документа: RU2187510C2
Чертежи

Описание
Изобретение относится к некоторым производным 16-гидрокси-11-(замещенный фенил)-эстра-4,9-диена, способам их получения, содержащим их фармацевтическим композициям и к их использованию в лекарственной терапии, в особенности при лечении или профилактике глюкокортикоидзависимых заболеваний.
В патенте США 5089635 описаны некоторые 13-алкил-11β-фенилгонаны, оказывающие антигестагеническое и антиглюкокортикоидное действие. В Европейском патенте 0057115 описаны 19-нор-стероиды и 19-нор-D-гомостероиды, имеющие антиглюкокортикоидную активность.
В настоящее время обнаружен ряд производных 16-гидрокси-11-(замещенный фенил)-эстра-4,9-диена, которые имеют высокоселективное сродство к глюкокортикоидным рецепторам и имеют мощную антиглюкокортикоидную активность in vivo.
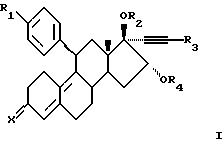
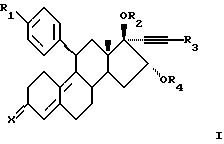
Соответственно, настоящее изобретение относится к соединениям формулы I

где R1 представляет C1-6-алкил, С3-6-циклоалкил, C1-6-алкокси, трифлат, пиридил или фенил, где фенильная группа необязательно замещена одним или несколькими заместителями, выбранными из циано, галогена и C1-4-алкила;
R2 представляет водород, C1-6-алкил, 1-oкco-C1-6-алкил или карбокси-1-оксо-C1-6-алкил;
R3 представляет водород, галоген или C1-6-алкил, необязательно замещенный одним или несколькими заместителями, выбранными из C1-6-алкокси и галогена;
R4 представляет водород, C1-6-алкил, 1-окco-C1-6-алкил или карбокси-1-оксо-C1-6-алкил; и
Х представляет (Н, ОН), О или NOH;
или их фармацевтически приемлемой соли или сольвату.
Настоящее изобретение включает производные 16-гидрокси-11-(замещенный фенил)-эстра-4,9-диена формулы I, в которых:
1. R1
представляет фенил, трифлат или C1-6-алкил, например трет-бутил, изопропил или метил.
2. R2 представляет водород.
3. R3 представляет водород, галоген, например хлор, C1-6-алкил, например метил, этил, пропил или трет-бутил, необязательно замещенный C1-6-алкокси, таким как метокси.
4. R4 представляет водород или метил.
5. Х представляет О.
6. R1, R2, R3, R4 и Х являются таковыми, как определено в пп.1-5 выше, или их фармацевтически приемлемые соли или сольваты.
7. R1 представляет C1-6-алкил, С3-6-циклоалкил, трифлат или фенил; R2 представляет водород, C1-6-алкил, 1-оксo-C1-6-алкил или карбокси-1-оксо-C1-6-алкил; R3 представляет водород, галоген или C1-6 -алкил, необязательно замещенный одним или несколькими заместителями, выбранными из C1-6-алкокси и галогена; R4 представляет водород, C1-6-алкил, 1-oкco-C1-6-алкил или карбокси-1-оксо-C1-6-алкил; и Х представляет (Н, ОН), О или NOH; или его фармацевтически приемлемую соль или сольват.
Дополнительные примеры соединений формулы I выше включают примеры 1-4.
Как используется здесь, термин "алкил" означает алкильную группу с прямой или разветвленной цепью. Такие алкильные группы включают метил, этил, изопропил, н-пропил, н-бутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил и неогексил. Указание на циклоалкил включает циклопропил, циклопентил и циклогексил.
Термин "алкокси" имеет значение, понятное специалистам, и включает прямые и разветвленные цепи. Примеры алкоксигрупп включают метокси и этокси. Предпочтительные алкоксигруппы включают C1-4-алкокси.
Термин "галоген" включает хлор, бром, фтор и иод.
Трифлат означает трифторметансульфонат.
1-Оксо-C1-6-алкил или карбокси-1-оксо-C1-6-алкил группы включают 1-оксометил, 1-оксоэтил, 1-оксопропил, 3-карбокси-1-оксопропил, 3-карбокси-1-оксобутил и 3-карбокси-1-оксопентил.
Предпочтительными примерами R1 являются фенил и C1-6-алкил, например трет-бутил (1,1-диметилэтил), изопропил (1-метилэтил) или метил, наиболее предпочтительно трет-бутил и фенил.
R2 предпочтительно представляет водород.
Предпочтительные примеры R3 включают C1-6-алкил, наиболее предпочтительно метил.
R4 предпочтительно представляет водород или C1-6-алкил, в частности метил.
Х предпочтительно представляет О.
Предпочтительные соединения формулы I включают те, в которых R1 представляет фенил или C1-6-алкил, например трет-бутил, изопропил или метил; R2 представляет водород; R3 представляет C1-6-алкил, наиболее предпочтительно метил; R4 представляет водород или C1-6-алкил, предпочтительно метил; и Х представляет О; или их фармацевтически приемлемые соли или сольваты.
Особо предпочтительные
производные 16-гидрокси-11-(замещенный фенил)-эстра-4,9-диена формулы I включают:
(11β,16α,17β
)-11-(4-трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)-эстра-4,
9-диен-3-он;
(11β,16α,17β)-11-(4-иэопропилфенил)-16,17-дигидрокси-17-(1-пропинил)-эстра-4,9-диен-3-он;
(11β,16α,17β)-11-(4-метилфенил)-16,
17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-он;
(11β,16α,17β)-11-(1,1'-бифенил-4-ил)-16,
17-дигидрокси-17-(1-пропинил)-эстра-4,9-диен-3-он;
(11β,
16α,17β)-16,17-дигидрокси-11-[4-[(трифторметил-сульфонил)окси] фенил]-17-(1-пропинил)эстра-4,9-диен-3-он
или их фармацевтически приемлемый сольват.
Для терапевтического применения пригодны те соли соединений формулы I, в которых фармацевтически приемлем противоион. Однако соли кислот и оснований, которые фармацевтически неприемлемы, также могут найти применение, например, при получении или очистке фармацевтически приемлемых соединений. Все соли, независимо от того, являются они фармацевтически приемлемыми или нет, включены в предмет настоящего изобретения.
Соли по изобретению включают соли аммония, соли щелочных металлов, такие как соли натрия или калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли с органическими основаниями, такими как дициклогексиламин и N-метил-D-глюкамин, и соли с аминокислотами, такими как аргинин и лизин.
Сольваты по изобретению включают гидраты.
В следующем аспекте изобретение предлагает соединения формулы I и их фармацевтически приемлемые соли или сольваты для применения в терапии, более конкретно при лечении и профилактике зависимых от глюкокортикоида заболеваний или симптомов, таких как синдром Кушинга, диабет, глаукома, расстройства сна, депрессия, беспокойство, атеросклероз, гипертония, ожирение, остеопороз, привыкание к приему наркотиков или лекарств, и при лечении симптомов при лишении привычного вещества, например наркотиков, кокаина и алкоголя. Соединения также находят применение при лечении нейродегенеративных заболеваний, таких как болезнь Альцгеймера, и психических расстройств, таких как шизофрения, мания, гиперактивность, привыкание к веществам, рвота и шизофрениеподобные расстройства.
Соединения формулы I и их фармацевтически приемлемые соли и сольваты и, в особенности, описанные выше предпочтительные соединения, являются полезными при лечении депрессии.
Настоящее изобретение далее включает способ лечения животного, например млекопитающего, включая человека, страдающего или склонного к заболеванию зависимым от глюкокортикоида заболеванием, включая любое из вышеперечисленных заболеваний или симптомов, который включает введение эффективного количества соединения формулы I или его фармацевтически приемлемых соли или сольвата.
В еще одном аспекте настоящее изобретение относится к применению соединения формулы I или его фармацевтически приемлемых соли или сольвата для приготовления лекарства для лечения или профилактики любого из вышеперечисленных заболеваний или симптомов.
Количество соединения формулы I или его фармацевтически приемлемых соли или сольвата, называемого здесь также активным ингредиентом, которое требуется для достижения терапевтического эффекта, будет, конечно, меняться в зависимости от конкретного соединения, способа введения, возраста и состояния пациента и конкретного расстройства или заболевания, подвергающегося лечению.
Подходящая суточная доза для любого из вышеупомянутых расстройств находится в интервале от 0,001 до 50 мг на килограмм веса тела пациента (например, человека) в день, предпочтительно в интервале от 0,01 до 20 мг на килограмм веса тела в день и наиболее предпочтительно в интервале от 0,1 до 10 мг на килограмм веса тела в день. Необходимая доза может быть введена в виде одной, двух, трех, четырех, пяти или более суб-доз, вводимых через соответствующие интервалы времени в течение суток.
Хотя активный ингредиент может вводиться в чистом виде, предпочтительно вводить его в виде фармацевтической композиции. Соответственно, настоящее изобретение предлагает далее фармацевтическую композицию, включающую соединение формулы I или его фармацевтически приемлемые соль или сольват вместе с его фармацевтически приемлемым носителем и, необязательно, другими терапевтическими агентами. Носитель должен быть "приемлемым" с точки зрения совместимости с другими ингредиентами композиции и безвредности для пациента.
Композиции включают носители, подходящие для орального, ректального, назального, топического (включая трансдермальное, защечное и подъязычное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное, внутрикожное и через стекловидное тело) введения. Композиции могут быть приготовлены любыми способами, хорошо известными в фармацевтической практике, например, при использовании таких способов, как описанные в Gennaro et al. . Remington's Pharmaceutical Sciences (18th ed., Mack Publishing company, 1990, см. в особенности Part 8: Pharmaceutical Preparations and their Manufacture). Такие способы включают стадию смешения активного ингредиента с носителем, который состоит из одного или нескольких вспомогательных ингредиентов. Такие вспомогательные ингредиенты включают те добавки, которые обычно применяются в практике, такие как наполнители, связующие, разбавители, разрыхлители, смазывающие, красители, ароматизирующие вещества и увлажняющие агенты.
Композиции, предназначенные для орального введения, могут представлять собой дискретные единицы, такие как пилюли, таблетки или капсулы, каждая из которых содержит определенное количество активного ингредиента; могут быть в виде порошков или гранул, в виде раствора или суспензии. Активный ингредиент может также представлять собой болюс или пасту или может содержаться в жировых шариках.
Композиции для ректального введения могут представлять собой свечи или клизмы.
Композиции для парентерального введения включают водные и неводные стерильные инъекции. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например в запаянных сосудах и ампулах, и могут храниться в высушенном замораживанием (лиофилизированном) виде, требуя только добавления стерильного жидкого носителя, например воды, перед использованием.
Композиции для введения назальной ингаляцией включают мелко измельченные порошки или пудры, которые могут быть получены посредством сжатых аэрозолей с отмеренной дозировкой, распылителей или аппаратов для вдувания.
Настоящее изобретение далее включает следующие способы получения соединения формулы I или его фармацевтически приемлемых соли или сольвата.
Соединения формулы I могут быть получены различными способами, известными обычно в практике органической химии. Исходные материалы либо являются известными и легко доступными из химических источников, либо могут быть получены по известным методикам.
В нижеследующем описании символы R1, R2, R3, R4 и Х имеют значения, приписанные им в формуле I, если не оговорено иное.
В соответствии с первым общим способом (А) соединения формулы I могут быть
получены путем дегидратации и удаления защиты у соединения формулы II:

где Р представляет защищенную кетогруппу и R5 представляет группу R4, как она определена для формулы I, или соответствующим образом защищенную группу R4. Подходящие защитные группы и способы их удаления известны в практике, например из T. W. Green: Protective Groups in Organic Synthesis (Wiley, NY, 1981). Наиболее подходящими защитными группами для защиты кетогрупп являются ацетали, например 1,2-этиленкеталь. Такие группы могут быть удалены, например, кислым гидролизом.
Дегидратацию можно проводить, используя известные способы. Обычно реакцию проводят в присутствии минеральной кислоты, такой как хлористоводородная кислота или серная кислота, в подходящем растворителе, например ацетоне, в интервале температур от -20 до 25oС.
В соответствии со вторым общим способом (В) соединения формулы I, где Х представляет (Н,ОН), могут быть получены путем взаимодействия соответствующего соединения формулы I, в котором Х представляет О, с подходящим восстановителем, например, восстановлением боргидридом натрия в присутствии растворителя, такого как метанол, обычно в интервале температур от 0 до 25oС.
Согласно третьему общему способу (С) соединения формулы I, где Х представляет NOH, могут быть получены путем конденсации соединения формулы I, в котором Х представляет О, с подходящим оксимобразующим агентом, например, путем реакции соответствующего 3-кетосоединения с гидроксиламином в присутствии подходящего растворителя, такого как пиридин.
Если необходимо или желательно, вслед
за вышеописанным процессом может быть проведена любая одна или несколько из следующих
дополнительных стадий в любом порядке:
(i) преобразование соединения формулы I в фармацевтически
приемлемую соль или сольват соединения формулы I;
(ii) преобразование
фармацевтически приемлемой соли или сольвата соединения формулы I в соединение формулы I; и
(iii) преобразование
фармацевтически приемлемой соли или сольвата соединения формулы I в другие
фармацевтически приемлемую соль или сольват соединения формулы I.
В подходящем способе получения производных формулы II в качестве исходного продукта используется эстра-5(10), 9(11)-диен-3,17-дион-3-(циклический 1,2-этандиил ацеталь), который может быть получен способами, описанными в ЕР 0683172, или по способу, описанному здесь ниже в примере А. Это соединение сначала преобразуют в 17-силиленовый сложный эфир, используя хорошо известные способы, например взаимодействием с диизопропиламидом лития и триметилсилилхлоридом, после чего сразу осуществляют взаимодействие с трибромидом фенилтриметиламмония в пиридине. Полученное 16α-бромпроизводное затем преобразуют в соответствующий 3-защищенный 16α-гидроксистероид гидроокисью натрия в воде и пиридине по методу, описанному в J.Am.Chem.Sоc., 102, 5402 (1980). Алкинилирование по С17 с последующей (необязательной) защитой 16-гидроксигруппы (например, как TBDMS эфир - см. T.W. Green: Protective Groups in Organic Synthesis, Wiley, NY, 1981) и эпоксидирование 5(10) двойной связи (например, с перекисью водорода, трифторацетофеноном и пиридином в дихлорметане по ЕР 0298020) дает 16-гидрокси(защищенный)-3-кетозащищенный 5α,10α-эпокси-17α-алкинил-17β-гидрокси-эстр-9(11)-ен-3-он. Катализируемая медью реакция Гриньяра с этим эпоксидом в конце концов приводит к соединениям формулы II.
По другому варианту соединения формулы II могут быть легко получены при использовании эстра-5(10),9(11)-диен-3, 17-дион-3-(циклического 1,2-этандиил ацеталя) в качестве исходного вещества, как было описано здесь ранее. Это соединение может быть преобразовано в соответствующий 5α10α-эпоксид, например, с помощью перекиси водорода, трифторацетофенона и пиридина в дихлорметане по методу, описанному в ЕР 0298020. Катализируемая медью реакция Гриньяра дает соответствующий 3-защищенный-11-арил-5-гидрокси-эстр-9,10-ен-17-он. Последующее преобразование в силилпроизводное 17-енола (обработкой ЛДА и триметилсилилхлоридом) с последующим бромированием трибромидом фенилтриметиламмония в пиридине дает соответствующий 16-бромид. Необходимую 16α-гидроксигруппу вводят путем нуклеофильного замещения [гидроокись натрия, пиридин/вода в соответствии с методом, описанным в J.Am.Chem Sоc., 102, 5402 (1980)] . Пропинилирование (пропин, n-BuLi) в конце приводит к желаемому соединению II.
Соединения формулы I могут быть преобразованы в фармацевтически приемлемый 1-оксо-С14-алкил или карбокси-1-оксо-С14-алкил путем взаимодействия с подходящим этерифицирующим агентом, например взаимодействием с соответствующим образом активированной карбоновой кислотой, как, например, с хлорангидридом кислоты, или с активированной дикарбоновой кислотой, как, например, циклический ангидрид, при использовании хорошо известных методов.
Соединения формулы I могут быть преобразованы в их фармацевтически приемлемые соли обычным образом, например путем взаимодействия с соответствующей кислотой.
Настоящее изобретение дополнительно включает все описанные здесь новые промежуточные соединения и, в частности, соединения формулы II.
Наиболее
предпочтительные промежуточные соединения включают:
3-(циклический 1,2-этандиил
ацеталь) 5α,16α,17β-тригидрокси-11β-[4-трет-бутилфенил]-17α
-пропинил-эстр-9-ен-3-она;
3-(циклический 1,2-этандиил ацеталь) 5α,16α,17β
-тригидрокси-11β-[4-трет-бутилфенил]-17α-пентинил-эстр-9-ен-3-она;
3-(циклический 1,2-этандиил ацеталь) 5α,16α,17β-тригидрокси-11β
-[4-трет-бутилфенил]-17α-гексинил-эстр-9-ен-3-она;
3-(циклический 1,2-этандиил ацеталь)
5α,16α,17β-дигидрокси-11β-[4-трет-бутилфенил]-16α
-метокси-17α-пропинил-эстр-9-ен-
3-(циклический 1,2-этандиил
ацеталь)21-хлор-11-(4-трет-бутилфенил)-5α,16α,17β-тригидрокси-19-норпрегн-9-ен-20-ин-3-она;
или их фармацевтически приемлемую соль или сольват.
Следующие примеры предназначены только для иллюстрации и не предназначены для того, чтобы каким-либо образом ограничить предмет изобретения.
Пример А. 3,3-этиленкеталь эстра-5(10),9(11)-диен-3,
17-диона
Смесь 500 мл циклогексана, 183 мл триэтилортоформиата, 92 мл этиленгликоля и 0,9 г
п-толуолсульфокислоты перемешивают в течение 30 минут при комнатной температуре, после чего
нагревают до кипения с обратным холодильником. Образовавшийся этанол отгоняют в смеси с циклогексаном, но
объем поддерживают постоянным путем добавления циклогексана. Спустя 4,5 часа отгоняют остаток
циклогексана и 1 экв. остатка добавляют в качестве улавливателя воды в атмосфере азота к 1 г эстра-4,
9-диен-3,17-диона, 0,1 экв. хлористого водорода в диоксане и 1,5 экв. этиленгликоля в 15 мл
диметоксиэтана при -10oС. Спустя 75 мин реакционную смесь выливают в насыщенный водный раствор
гидрокарбоната натрия. Кристаллическую массу отфильтровывают, промывают водой и сушат в
вакууме, после чего получают 1,1 г 3,3-этиленкеталя эстра-5(10), 9(11)-диен-3,17-диона. После кристаллизации из
этанола получают 1 г продукта, имеющего чистоту выше 97%.
Пример 1.
(11β,16α,17β)-11-(4-Трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)-эстра-4,9-диен-3-он
а) 16-Бром-эстра-5(10),9(11)-диен-3,17-дион-3-(циклический 1,
2-этандиилацеталь)
Раствор 192 ммоль ЛДА (полученного добавлением 120 мл 1,6М раствора н-BuLi к раствору 34 мл
диизопропиламина в 340 мл сухого ТГФ) добавляют по каплям к холодному (-30oС) раствору 50 г (159 ммоль) эстра-5(10),9(11)-диен-3,17-дион-3-(циклического 1,2-этандиилацеталя) в 480 мл
сухого ТГФ. Перемешивание продолжают в течение 15 мин при -30oС. Затем
добавляют по каплям 60 мл (473 ммоль) триметилсилилхлорида и раствору дают нагреться до 0oС за примерно 1
час. К этому раствору енольного эфира медленно добавляют трибромид
фенилтриметиламмония (60 г, 160 ммоль), растворенного в 60 мл пиридина и, спустя один час, реакционную смесь нагревают до комнатной
температуры. ТСХ показывает полное исчезновение исходного материала
и образование более липофильного продукта (толуол/этилацетат; 85/15). Процесс завершают выливанием реакционной смеси в холодный
раствор хлористого аммония с последующей экстракцией дихлорметаном.
Сушка над сульфатом магния, фильтрация и выпаривание растворителей дают полутвердую массу. Кристаллизация из этанола дает 41,8 г
целевого бромида (преимущественно альфа) в виде беловатых кристаллов
(т.пл. 166,8-167,8oС).
b) 16α-Гидрокси-эстра-5(10), 9(11)-диен-3,17-дион-3-(циклический 1,
2-этандиилацеталь)
40 г (101 ммоль) продукта, полученного на стадии
а), растворяют в 840 мл сухого пиридина. При перемешивании добавляют 240 мл воды и затем 120 мл 1 н. раствора NaOH.
Поддерживают температуру ниже 25oС. После перемешивания в течение 30 мин
при комнатной температуре ТСХ показывает полную конверсию. Смесь выливают в насыщенный раствор хлористого аммония.
Экстракция дихлорметаном, сушка сульфатом магния и выпаривание растворителей дают
сырое гидроксисоединение в виде масла. Колоночная хроматография (силикагель, гептан/-этилацетат, 8/2) дает 18,8 г
чистого 16α-гидрокси-эстра-5(10),9(11)-диен-3,17-дион-3-(циклический 1,
2-этандиилацеталя) в виде пены. Образец для анализа, перекристаллизованный из диэтилового эфира, дает белые кристаллы;
т.пл. 188,4-190,6oС.
c) 16α,17β
-Дигидрокси-17α-пропинил-эстра-5(10),9(11)-диен-3,17-дион-3-(циклический 1,2-этандиилацеталь)
В
трехгорлую колбу, оборудованную трубкой для ввода газа и капельной воронкой, содержащую
130 мл сухого ТГФ при -70oС, добавляют 106 мл 1,6 н. раствора н-BuLi в гексане. Раствор становится
желтым. Через раствор барботируют газообразный пропин до исчезновения желтого окрашивания.
Образуется белая суспензия и перемешивание продолжают в течение 15 мин при -70oС. Затем к
холодному раствору пропин-аниона по каплям добавляют раствор 18 г (54,3 ммоль) полученного перед
этим продукта в 150 мл сухого ТГФ. После добавления раствору дают медленно нагреться до -20o
С. После 2 час перемешивания при этой температуре ТСХ показывает полное преобразовние исходного
продукта. Операцию завершают, выливая смесь в насыщенный раствор хлористого аммония с последующей
экстракцией дихлорметаном. Сушка над сульфатом магния и выпаривание растворителей дает 19,8 г сырого
продукта. Очистка с использованием короткой колонки (силикагель, гептан/этилацетат 1/1) дает 15,9
г чистого целевого соединения. Кристаллизация образца для анализа из диэтилового эфира дает белые
кристаллы, т.пл. 71oС.
d) 16-ТБДМС эфир 16α,17β
-дигидрокси-17α-пропинил-эстра-5(10),9(11)-диен-3,17-дион-3-(циклического 1,2-этандиилацетадя)
15,9 г (42,9 ммоль) продукта, полученного на стадии с), растворяют в 60 мл сухого ДМФА.
К этому раствору добавляют 15 г имидазола и после этого 15 г (120 ммоль) трет-бутилдиметилсилилхлорида. После
перемешивания в течение 3 час при 40oС анализ ТСХ показывает количественное
преобразование исходного соединения в один липофильный продукт. Смесь гасят раствором хлористого аммония, после
чего экстрагируют дихлорметаном. Сушка над сульфатом магния с последующим выпариванием
растворителей дает 35 г сырого силильного соединения, которое используют как таковое на следующей стадии.
е)16-ТБДМС-эфир5α,10α-эпокси-17α-пропинил-17β
-гидрокси-эстр-9(11)-ен-3-он-3-(циклического 1,2-этандиилацеталя)
35 г сырого продукта, полученного на
стадии d) (максимальное количество чистого вещества 20,8 г, 42,9 ммоль), растворяют в
300 мл дихлорметана; последовательно добавляют 2 мл пиридина, 5,3 мл трифторацетофенона и 70 мл 30% перекиси
водорода и полученную двухфазную систему интенсивно перемешивают при температуре
окружающей среды в течение 48 часов. Смесь выливают на воду, органический слой отделяют и дважды промывают насыщенным
раствором тиосульфата натрия. Сушка над безводным сульфатом магния, фильтрация и
выпаривание дают полутвердый остаток. Очистка колоночной хроматографией дает 16,4 г целевого α-эпоксида в виде
аморфного вещества.
f) 16-ТБДМС-эфир 3-(циклического 1,
2-этандиилацеталя) 5α,16α-17β-дигидрокси-11β-[4-трет-бутилфенил]-17α
-пропинил-эстр-9-ен-3-она
330 мг CuCl добавляют при температуре 0-5oС к
раствору бромида 4-трет-бутилфенилмагния (приготовленного из 0,83 г (35 ммоль) Мg и 6,0 мл (34,5 ммоль)
4-бром-трет-бутилбензола в 50 мл сухого ТГФ). После перемешивания в течение 30 мин при 0-5o С добавляют по каплям 2,5 г (5 ммоль) ранее полученного эпоксида, растворенного в 30 мл сухого
ТГФ, поддерживая температуру ниже 10oС. Перемешивание продолжают в течение одного
часа при температуре окружающей среды. Операцию завершают, выливая смесь в насыщенный раствор хлористого
аммония и экстрагируя этилацетатом (2х). Объединенные органические слои промывают насыщенным
солевым раствором, сушат над безводным сульфатом магния, фильтруют и концентрируют. Колоночная
хроматография (силикагель, гептан/этилацетат 7/3) дает 2,7 г чистого 11-замещенного соединения в виде
белого аморфного вещества.
g) (11β,16α,17β
)-11-(4-Трет-бутилфенил) -16,17-дигидрокси-17-(1-пропинил)-эстра-4,9-диен-3-он
2,7 г (4,26 ммоль) соединения,
полученного на стадии f), растворяют в 50 мл ацетона. При комнатной температуре
добавляют 3 мл 6 н. Н2SO4 и смесь перемешивают в течение двух часов. Затем холодный раствор
выливают в насыщенный раствор бикарбоната натрия и смесь экстрагируют этилацетатом
(2х). Объединенные органические слои промывают насыщенным солевым раствором, сушат над безводным сульфатом магния,
фильтруют и концентрируют. Хроматография (дихлорметан/ацетон 8/2) дает 1,6 г целевого
(11β,16α,17β)-11-(4-трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)-эстра-4,9-диен-3-она в
виде белого твердого вещества. Кристаллизация из диэтилового эфира дает 1,2 г белых
кристаллов, т.пл. 251,6-253,8oС.
Альтернативная методика
а) 5α,10α
-Эпокси-эстра-9(11)-диен-3,17-дион-3-(циклический 1,2-этандиилацеталь)
150
г эстра-5(10),9(11)-диен-3,17-дион-3-(циклического 1,2-этандиилацеталя) (478 ммоль) растворяют в 2,2 л дихлорметана,
последовательно добавляют 14,4 мл пиридина, 48 мл трифторацетофенона и 666 мл 30%
перекиси водорода и полученную двухфазную систему интенсивно перемешивают при температуре окружающей среды в течение
48 часов. Смесь выливают на воду, органический слой отделяют и дважды промывают
насыщенным раствором тиосульфата натрия. Сушка над безводным сульфатом магния, фильтрация и выпаривание дают
полутвердый остаток. Кристаллизация из смеси эфир/гептан дает 80 г целевого α
-эпоксида в виде белого кристаллического вещества (т.пл. 153oС).
b) 5α
-Гидрокси-11-(4-трет-бутилфенил)-эстр-9-ен-3,17-дион-3-(циклический 1,2-этандиилацеталь)
900 мг CuCl добавляют при температуре
0-5oС к раствору 20 г (60 ммоль) ранее полученного
эпоксида, растворенного в 30 мл сухого ТГФ. Смесь перемешивают в течение 20 мин и медленно
добавляют по каплям раствор бромида 4-трет-бутил-фенилмагния (приготовленного из 5 г (200 ммоль) Мд и 32,8
мл (200 ммоль) 4-бром-трет-бутилбензола в 150 мл сухого ТГФ), поддерживая температуру ниже
10oС. Перемешивание продолжают в течение одного часа при температуре окружающей среды. Операцию
завершают, выливая смесь в насыщенный раствор хлористого аммония и экстрагируя этилацетатом
(2х). Объединенные органические слои промывают насыщенным солевым раствором, сушат над безводным сульфатом
магния, фильтруют и концентрируют. Колоночная хроматография (силикагель, гептан/этилацетат
7/3) дает 28 г целевого 5α-гидрокси-11β-(4-трет-бутил-фенил)-эстр-9-ен-3-он-3-(циклического 1,
2-этандиилацеталя).
с) 16α-Бром-5α-гидрокси-11β
- [4-трет-бутилфенил} эстра-9-ен-3-он-3-(циклический 1,2-этандиилацеталь)
Раствор 181 ммоль ЛДА
(полученного добавлением 113 мл 1,6М раствора н-BuLi к раствору 25,6 мл диизопропиламина в 400
мл сухого ТГФ) добавляют по каплям к холодному (-60oС) раствору 28 г (60 ммоль) 5α
-гидрокси-11β-(4-трет-бутил-фенил)-эстр-9-ен-3-он-3-(циклического 1,2-этандиилацеталя) в 700
мл сухого ТГФ. Перемешивание продолжают в течение 30 мин при -50oС. Затем добавляют по
каплям 38,3 мл триметилсилилхлорида и раствор перемешивают при -45oС в течение 1 часа.
После охлаждения реакционной смеси до -60oС по каплям добавляют 27,7 г (72,4 ммоль)
трибромида фенилтриметиламмония, растворенного в 100 мл пиридина. После 2 часов перемешивания при -60oС ТСХ показывает полное исчезновение исходного материала и образование более
липофильного продукта (гептан/-этилацетат; 6/4). Операцию завершают выливанием реакционной смеси в раствор
хлористого аммония с последующей экстракцией этилацетатом. Сушка над сульфатом магния,
фильтрация и выпаривание растворителей дают полутвердую массу. Кристаллизация из гептана с последующей
кристаллизацией из этанола дает 20 г целевого бромида в виде белых кристаллов (т.пл. 164-165oС).
d) 5α,16α-дигидрокси-11β-[4-трет-бутилфенил]
эстр-9-ен-3,17-дион-3-(циклический 1,2-этандиилацеталь)
12,5 г (23,0 ммоль) продукта, полученного
на стадии а), суспендируют в 400 мл 75% пиридина в воде. Добавляют 27,5 мл 1М NaOH. После
перемешивания в течение 30 мин при комнатной температуре исходное вещество растворяется и ТСХ показывает
полное превращение. Реакционную смесь выливают в раствор хлористого аммония и экстрагируют
этилацетатом. Сушка над сульфатом магния, выпаривание растворителей и совместное испарение с толуолом дают
сырое гидроксисоединение. Кристаллизация из диизопропилового эфира дает 9,0 г целевого
соединения (т.пл. 180-182oС).
e) 5α,16α,17α
-тригидрокси-11β-[4-трет-бутилфенил] -17-(1-пропинил)-эстр-9-ен-3-он-3-(циклический 1,
2-этандиилацетадь)
В трехгорлую колбу, оборудованную трубкой для ввода газа и капельной воронкой,
содержащую 130 мл сухого ТГФ, при температуре -70oС добавляют 95 мл 1,6 н.
раствора н-BuLi в гексане. Раствор становится желтым. Через раствор барботируют газообразный пропин до
исчезновения желтого окрашивания. Образуется белая суспензия и перемешивание продолжают в течение
15 мин при -70oС. Затем к холодному раствору пропинаниона по каплям добавляют раствор 18 г
(38 ммоль) полученного перед этим продукта в 150 мл сухого ТГФ. После добавления раствору дают
медленно нагреться до -20oС. После 2 час перемешивания при этой температуре ТСХ показывает
полное превращение исходного продукта. Операцию завершают, выливая смесь в насыщенный раствор
хлористого аммония с последующей экстракцией дихлорметаном. Сушка над сульфатом магния и выпаривание
растворителей дает 19,8 г сырого продукта. Очистка с использованием короткой колонки (силикагель,
гептан/этилацетат 1/1) дает 18,5 г чистого целевого соединения в виде белой пены.
f)
(11β,16α,17β)-11-(4-Трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)-эстра-4,
9-диен-3-он
2,7 г (4,26 ммоль) соединения, полученного на стадии е), растворяют в 50 мл
ацетона. При комнатной температуре добавляют 3 мл 6 н. H2SO4 и смесь перемешивают
в течение двух часов. Затем холодный раствор выливают в насыщенный раствор бикарбоната натрия и
смесь экстрагируют этилацетатом (2х). Объединенные органические слои промывают насыщенным солевым
раствором, сушат над безводным сульфатом магния, фильтруют и концентрируют. Хроматография
(дихлорметан/ацетон 8/2) дает 1,6 г целевого (11β,16α,17β)-11-(4-трет-бутилфенил)-16,
17-дигидрокси-17-(1-пропинил)-эстра-4,9-диен-3-она в виде белого твердого вещества.
Кристаллизация из диэтилового эфира дает 1,2 г белых кристаллов, т.пл. 251,6-253,8oС.
При
замене 4-бром-трет-бутилбензола на 4-бромизопропилбензол, 4-бромтолуол и
4-бромбифенил получают следующие продукты:
1А. (11β,16α,17β)-11-(4-изопропилфенил)-16,
17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-он; т.пл. 153,4-154,9o
С.
1В. (11β,16α,17β)-11-(4-метилфенил)-16,17-дигидрокси-17-(1-пропинил)эстра-4, 9-диен-3-он; т.пл. 212,2-213,8oС.
1С. (11β, 16α,17β)-11-(1,1'-бифенил-4-ил)-16,17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-он; т.пл. 254,8-256,2oС.
1D. 3E- и 3Z-оксим (11β,16α,17β
)-11-(4-трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-она:
Получение: 200 мг (0,44 ммоль)
(11β,16α,17β)-11-(4-трет-бутилфенил)-16,
17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-она (полученного, как описано в 1f) растворяют в 1 мл пиридина. Добавляют 70 мг (0,88 ммоль)
гидроксиламингидрохлорида и смесь перемешивают при
температуре кипения с обратным холодильником в течение 40 минут. Смесь выливают в воду, нейтрализуют разбавленной хлористоводородной кислотой и
экстрагируют этилацетатом. Органический слой промывают
насыщенным солевым раствором, сушат над сульфатом магния, фильтруют и упаривают до сухого остатка. Сырой оксим подвергают разделению ВЭЖХ
(ацетонитрил/вода 40/60-->60/40), получая 39 мг
3г-оксима (11β,16α,17β)-11-(4-трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-она ([α]20D=+30o (с=0,2, диоксан))
и
75 мг 3Е-оксима (11β,16α,17β)-11-(4-трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил) эстра-4,
9-диен-3-она([α]20D=+60o
(с=0,6, диоксан)).
Пример 2. (11β,16α,17β)-11-(4-трет-бутилфенил)-16,17-дигидрокси-19,21,
27-тринорхолэста-4,9-диен-20(22)-ин-3-он
a) 3-(циклический 1,
2-этандиилацеталь) 5α,16α,17β-тригидрокси-11β-[4-трет-бутилфенил]-17α
-гексинил-эстр-9-ен-3-она
1-Гексин (2,88 г, 4 мл, 35 ммоль) растворяют в 75 мл сухого
ТГФ; после охлаждения раствора до -20oС добавляют по каплям 20 мл 1,6М раствора н-BuLi и смесь
перемешивают при -20oС в течение 15 мин. Затем добавляют по каплям раствор 2,4 г
3-(циклического 1,2-этандиилацеталя) 5α,16α,17β-тригидрокси-11β
-[4-трет-бутилфенил] -эстр-9-ен-3,17 диона в 50 мл ТГФ и перемешивание продолжают в течение 1 ч при -20oС.
Операцию завершают выливанием смеси в насыщенный раствор хлористого аммония с последующей экстракцией этилацетатом (2х), промывкой объединенных органических слоев насыщенным солевым раствором, сушкой над сульфатом магния и выпариванием растворителей. Это дает сырой продукт в виде масла. Растирание с диизопропиловым эфиром дает 800 мг чистого 3-(циклического 1, 2-этандиилацеталя) 5α,16α,17β-тригидрокси-11β -[4-трет-бутилфенил]-17α-гексинил-эстр-9-ен-3-она в виде белых кристаллов; т.пл. 182-183oС.
b) (11β,16α,17β)-11-(4-трет-бутилфенил)-16,
17-дигидрокси-19,21,27-три-норхолэста-4,9-диен-20(22)-ин-3-он
3-(Циклический 1,2-этандиилацеталь)5α,16α,
17β-тригидрокси-11β-[4-трет-бутилфенил]-17α
-гексинил-эстр-9-ен-3-она, полученный на предшествующей стадии, растворяют в 50 мл ацетона; добавляют 1 мл 6 н. Н2SO4 и продолжают перемешивание в течение 30 мин. ТСХ
(гептан/этилацетат 1/1) показывает полное превращение исходного вещества в два липофильных продукта. Операцию завершают добавлением
насыщенного раствора NаНСО3 с последующей экстракцией
этилацетатом (2х), промывкой объединенных органических слоев насыщенным солевым раствором и сушкой над сульфатом магния. Выпаривание
растворителей дает сырое соединение в виде масла. Колоночная
хроматография (гептан/этилацетат 8/2-->6/4) дает 500 мг целевого (11β,16α,17β)-11-(4-трет-бутилфенил)-16,
17-дигидрокси-19,21,27-три-норхолэста-4,9-диен-20(22)-ин-3-она в
виде аморфного белого вещества; [α]20D=34,2 (с=0,5, диоксан).
Следующие продукты были получены согласно примерам 2а и 2b при использовании, соответственно, 1-пентина, 3-метокси-пропина и ацетилена.
2А. (11β,16α,17β)-11-(4-трет-бутилфенил)-16, 17-дигидрокси-17-(1-пентинил) эстра-4,9-диен-3-он; [α ]20D=37,8 (с=1, диоксан).
2В. (11β,16α,17β)-11-(4-трет-бутилфенил)-16, 17-дигидрокси-17-(3-метокси-1-пропинил) эстра-4,9-диен-3-он; т.пл. 171,0-171,6oC.
2С. (11β,16α,17β)-11-(4-трет-бутилфенил)-16, 17-дигидрокси-19-норпрегна-4,9-диен-20-ин-3-он; [α]20D=46,9 (с=1, диоксан).
2D. (11β,16α,17β)-16, 17-дигидрокси-11-(4-[(трифторметил-сульфонил)оксил] фенил)-17-(1-пропинил)эстра-4,9-диен-3-он; [α]20D=8,9 (с=1 диоксан).
Пример 3. (11β,
16α,17β)-11-(4-Трет-бутилфенил)-17-гидрокси-16-метокси-17-(1-пропинил)эстра-4,9-диен-3-он
a) 3-(Циклический 1,2-этандиилацеталь) 5α-гидрокси,11β
-[4-трет-бутилфенил]-16α-метокси-эстр-9-ен-3,17-диона
2,0 г (4,16 ммоль) 3-(Циклического 1,2-этандиилацеталя)
5α,16α,17β-тригидрокси-11β
-[4-трет-бутилфенил]-эстр-9-ен-3,17-диона растворяют в 150 мл сухого дихлорметана. К этому раствору добавляют 4,5 г 2,
6-ди-трет-бутил-4-метилпиридина (15,75 ммоль) и 1,3 г
триметилоксоний-тетрафторбората (8,3 ммоль) и раствор перемешивают при комнатной температуре. Спустя три часа анализ ТСХ (гептан/этилацетат/этанол
10/10/1) показывает полное превращение исходного
вещества. Операцию завершают добавлением насыщенного раствора бикарбоната натрия с последующей экстракцией дихлорметаном. Выпаривание растворителей
дает 2,6 г сырого продукта, достаточно чистого для
того, чтобы быть использованным на следующей стадии.
b) 5α,17β-дигидрокси-11β-[4-трет-бутилфенил] -16α
-метокси-17α
-пропинил-эстр-9-ен-3-он-3-(циклический 1,2-этандиилацеталь)
Вещество, полученное на предшествующей стадии, растворяют в 10 мл сухого ТГФ и добавляют по каплям к раствору
пропиниллития
(полученному добавлением газообразного пропина к раствору 20 мл 1,3М н-BuLi до исчезновения желтого окрашивания и перехода в белую суспензию) при -60oС. Раствору дают
нагреться до -20oС и продолжают перемешивание в течение 1 ч. Операцию завершают, выливая смесь в насыщенный раствор хлористого аммония с последующей экстракцией дихлорметаном. Осушка
сульфатом магния и
выпаривание растворителей дают сырой продукт, который очищают колоночной хроматографией (гептан/этилацетат 1/1).
Выход: 1,15 г целевого 5α,17β -дигидрокси-11β -[4-трет-бутилфенил] -16α-метокси-17α-пропинил-эстр-9-ен-3-он-3-(циклического 1,2-этандиилацеталя) в виде аморфного белого вещества.
с) (11β,
16α,17β
)-11-(4-трет-бутилфенил)-17-гидрокси-16-метокси-17-(1-пропинил)-эстра-4,9-диен-3-он
Вещество, полученное в предыдущей реакции (1,15 г), растворяют в 20 мл ацетона,
добавляют 2 мл 2 н. НС1 и
смесь перемешивают в течение 2 ч при температуре окружающей среды. Нейтрализация раствором бикарбоната натрия и экстракция этилацетатом после промывки насыщенным солевым
раствором и сушки над
сульфатом магния дают сырой продукт. Колоночная хроматография (гептан/этилацетат 1/1) дает 700 мг чистого целевого соединения в виде аморфного вещества;
[α]20D=50,1 (c=0,5, диоксан).
Пример 4. (11β,16α,17β)-21-хлор-11-(4-трет-бутилфенил)-16,17-дигидрокси-19-норпрегна-4,9-диен-20-ин-3-он
a) 3-(циклический
1,2-этандиилацеталь) 21-хлор-11-(4-трет-бутидфенил)-5α,16α,17β-тригидрокги-19-норпрег-9-ен-20-ин-3-она
Метиллитий (11 мл 2,2М раствора в
диэтиловом эфире) добавляют по
каплям при 0oС к раствору 1,2 г транс-1,2-дихлорэтена (12 ммоль) в 5 мл сухого диэтилового эфира. Перемешивание продолжают при комнатной температуре в течение
1,5 час. Затем добавляют по
каплям раствор 1,4 г (3 ммоль) 3-(циклического1,2-этандиилацеталя)5α,16α,17β-тригидрокси-11β-[4-трет-бутилфенил] -эстр-9-ен-3,17 диона в 20 мл
сухого толуола и продолжают
перемешивание в течение 1 ч при температуре окружающей среды. Операцию завершают добавлением насыщенного раствора хлористого аммония с последующей экстракцией этилацетатом.
Промывка органических
слоев насыщенным солевым раствором, сушка над сульфатом магния и выпаривание растворителей дает 2 г сырого продукта. Колоночная хроматография (гептан/этилацетат 1/1) дает 1,2 г
целевого
3-(циклического 1,2-этандиилацеталя) 21-хлор-11-(4-трет-бутилфенил)-5α,16α,17β-тригидрокси-19-норпрег-9-ен-20-ин-3-она в виде аморфного белого вещества, достаточно
чистого для
использования на следующей стадии.
b) (11β,16α,17β)-21-хлор-11-(4-трет-бутилфенил)-16,17-дигидрокси-19-норпрегна-4,9-диен-20-ин-3-он
Полученное на
предыдущей стадии вещество по способу, описанному в примере 3с, дает после колоночной хроматографии 460 мг целевого (11β,16α,17β)-21-хлор-11-(4-трет-бутилфенил)-16,
17-дигидрокси-19-норпрегна-4,9-диен-20-ин-3-она, который может быть перекристаллизован из диэтилового эфира; т.пл. 202,2-202,7oС (разлагается).
Пример 5. Сродство
связывания
рецепторов глюкокортикоида (ГР) и рецепторов прогестерона (ПР)
В таблице представлены данные по сродству с рецепторами соединений по изобретению для рецепторов глюкокортикоида
(ГР) по
сравнению с рецепторами прогестерона (ПР).
Глюкокортикоидное сродство соединений измеряли для рецепторов глюкокортикоида, присутствующих в неповрежденных клетках множественной миеломы человека, и сравнивали со сродством дексаметазона (согласно методике, описанной H. J. Kloosterboer et al. , J. Steroid Biochem., Vol.31, 567-571 (1988)). Прогестероновое сродство соединений определяли для цитоплазменных рецепторов прогестерона, присутствующих в клетках рака груди человека, и сравнивали со сродством (16α)-16-этил-21-гидрокси-19-норпрегн-4-ен-3,20-диона (согласно методике, описанной E.W. Bergink et al., J.Steroid Biochem., Vol.19, 1563-1570 (1983)).
Соединения по настоящему изобретению сравнивали с 11β-(4-ацетилфенил)-16α, 17β -дигидрокси-17α-(1-пропинил)эстра-4,9-диен-3-оном (соединение "11 п-ацетил"), описанным в патенте США 5089635 и RU (38)486. Соединения по настоящему изобретению показали высокие значения ГРцит, тогда как нежелательная ПРцит-активность была низкой.
ПРИМЕРЫ 6,7 фармацевтической композиции в виде капсул
Ингредиент - мг
Активное соединение
(11β,16α,
17β)-11-(4-трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-он - 1
Гидроксипропилцеллюлоза (связующее) - 9,45
Кукурузный крахмал
(разрыхлитель) - 157,50
Стеарат магния (смазывающее) - 1,58
Лактоза (растворитель) - До общей массы в 315,0
Ингредиент - мг
Активное соединение (11β,
16α,17β
)-11-(4-трет-бутилфенил)-16,17-дигидрокси-17-(1-пропинил)эстра-4,9-диен-3-он - 50
Гидроксипропилцеллюлоза - 9,45
Кукурузный крахмал - 157,50
Стеарат
магния - 1,58
Лактоза - До общей массы в 315,0о
Реферат
Изобретение относится к производным 16-гидрокси-11-(замещенный фенил)-эстра-4,9-диена, соответствующим формуле I, где R1 - C1-6 - алкил, трифлат или фенил, где фенильная группа необязательно замещена одним или несколькими заместителями, выбранными из циано, галогена и С1-4-алкила, R2 - водород, или карбокси -1-оксо-С1-6-алкил; R3 - водород, галоген или С1-6- алкил, необязательно замещенный одним или несколькими С1-6-алкокси, R4 - водород, или С1-6-алкил, и Х, О или NOH; или их фармацевтически приемлемой соли или сольвату; описаны способы их получения и содержащая их фармацевтическая композиция, предназначенная для использования в медицинской терапии, в особенности при лечении или профилактике глюкокортикоидзависимых заболеваний или симптомов. 3 с. и 6 з.п.ф-лы, 1 табл.
Формула
где R1 представляет собой С1-6-алкил, трифлат или фенил, где фенильная группа необязательно замещена одним или несколькими заместителями, выбранными из циано, галогено и С1-4-алкила;
R2 представляет собой водород или карбокси-1-оксо-С1-6 -алкил;
R3 представляет собой водород, галоген или С1-6-алкил, необязательно замещенный одним или несколькими С1-6-алкокси;
R4 представляет собой водород или С1-6-алкил;
Х представляет собой О или NOH,
или его фармацевтически приемлемая соль или сольват.
(11β,16α,17β )-11-(4-трет-бутилфенил)-16, 17-дигидрокси-17-(1-пропинил) эстра-4,9-диен-3-она;
(11β,16α,17β)-11-(4-изопропилфенил)-16,17-дигидрокси-17-(1-пропинил) эстра-4,9-диен-3-она;
(11β, 16α,17β)-11-(4-метилфенил)-16,17-дигидрокси-17-(1-пропинил) эстра-4,9-диен-3-она;
(11β,16α,17β)-11-(1,1'-бифенил-4-ил)-16, 17-дигидрокси-17-(1-пропинил) эстра-4,9-диен-3-она;
(11β,16α,17β)-16,17-дигидрокси-11-[4-(трифторметилсульфонил)окси] фенил] -17-(1-пропинил) эстра-4,9-диен-3-она;
или его фармацевтически приемлемые соль или сольват.

где R1, R2 и R3 являются таковыми, как определено в п.1;
R5 представляет собой группу R4, как она определена в п.1, или соответствующим образом защищенную группу R4;
Р представляет собой защищенную кетогруппу.
Комментарии