Способ синтеза клофарабина - RU2663584C2
Код документа: RU2663584C2
Чертежи




Описание
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к способу получения клофарабина с высоким выходом и без образования нежелательных α-N9 стереоизомеров.
Предшествующий уровень техники настоящего изобретения
Клофарабин (систематическое название IUPAC - 5-(6-амино-2-хлор-пурин-9-ил)-4-фтор-2-(гидроксил-метил)оксолан-3-ол) представляет собой антиметаболит пуринового нуклеотида, используемый для лечения различных типов лейкозов, в частности острого лимфобластного лейкоза.
На данный момент в уровне техники известны несколько способов получения клофарабина. Пути синтеза, как правило, основываются на реакции сочетания пуриновых производных и арабинофуранозильного фрагмента. В зависимости от способа введения атома фтора такие схемы синтеза могут быть разделены на две группы: (i) способы, при которых атом фтора включается в рибофуранозный фрагмент и вводится в молекулу на стадии сочетания; (ii) способы, при которых атом фтора вводится после стадии сочетания путем химических превращений предварительно полученного нуклеозида.
В патенте США №5034518 раскрывается оригинальная схема синтеза для получения клофарабина, разработанная Montgomery с соавторами. Путь синтеза показан на фиг. 1. Вкратце, 1-бром-2-фтор-сахар 1 соединяют с 2,6-дихлорпурином 2 с последующими стадиями аминирования и снятия защиты. Эта стадия сочетания приводит к образованию смеси α- и β-аномеров продукта 3. После хроматографического разделения можно выделить желаемый β-аномер с выходом всего лишь 32%. Главные недостатки этого способа заключаются в низком общем выходе желаемого продукта (13% исходя из 1), низкой стабильности исходного бромида 1 и трудной очистке продукта 3 реакции сочетания из-за присутствия α-изомера.
В патенте США №6949640 раскрывается модификация вышеупомянутого пути синтеза, предусматривающая (i) применение солей, образованных путем обработки 2-хлор-6-замещенного пурина сильными основаниями (такими как NaH) вместо NH-форм пуринов, и (ii) применение 2-дезокси-2-фтор-3,5-ди-O-бензоил-α-D-арабинофуранозила бромида 4 вместо O-ацетильного производного 1 (этот путь синтеза показан на фиг. 2). Эти модификации позволили повысить общий выход клофарабина до приблизительно 40% исходя из соединения 4. Однако схема синтеза затрудняется низкой стабильностью бромида 4, который трудно получить, и образованием смеси аномеров на стадии сочетания, что требует трудную колоночную хроматографию для отделения желаемого продукта.
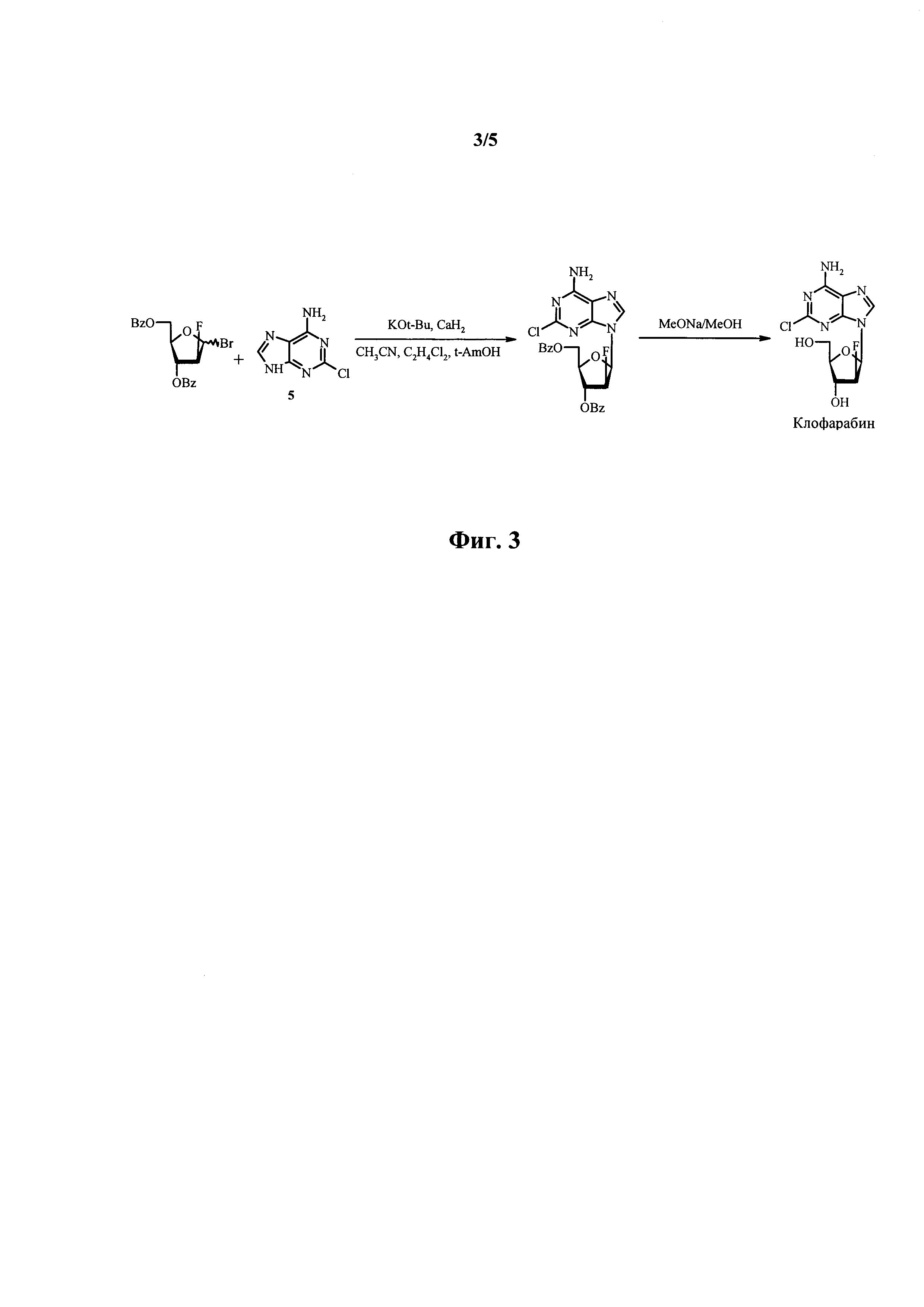
Дополнительная модификация способа Montgomery и соавторов раскрывается в патенте США №6680382 и дополнительно иллюстрируется на фиг. 3. Эта модификация предусматривает применение калиевой соли 2-хлораденина 5, получаемого путем обработки 2-хлораденина с помощью t-BuOK, вместо калиевой соли 2,6-дихлорпурина, применение трехкомпонентной смеси растворителей (например, трет-амилового спирта, CH2Cl2, CH3CN) и добавление CaH2 для полного удаления воды из растворителей. Применение растворителей с низкой полярностью на стадии сочетания позволило достичь стереоселективного протекания реакции (с отношением аномера β к α, доходящим до 15:1). Кроме того, присутствие аминогруппы в исходном материале 2-хлораденине позволяло избежать стадии аминирования продукта реакции сочетания. В результате общий выход очищенного клофарабина из фторированного сахара 4 составляет приблизительно 32%. Однако данный способ по-прежнему обладает двумя недостатками, заключающимися в сложном получении фторированного сахара 4 (получаемого в четыре стадии из 1-O-ацетил-2,3,5-три-O-бензоил-β-D-рибофуранозы) и в его низкой стабильности.
Способ, раскрытый в публикации заявки на выдачу патента США №2012/0010397, относится к вышеупомянутой второй группе способов получения клофарабина. Он показан на фиг. 4. На первой стадии получают 2',3',5'-три-O-бензоил-2-хлораденозин 8 путем соединения сахара 6 и TMS-защищенного 2-хлораденина 7. Дальнейшее дебензоилирование с помощью гидразина гидрата приводит к образованию смеси 3',5'- и

Следовательно, сохраняется потребность в улучшенных способах синтеза клофарабина, которые преодолевают ограничения известных схем синтеза. В частности, необходима схема синтеза, которая обеспечивает высокий выход продукта и не предусматривает образование нежелательных стереоизомеров.
Соответственно, целью настоящего изобретения является обеспечение такого способа получения клофарабина.
Краткое раскрытие настоящего изобретения
В одном аспекте настоящее изобретение относится к способу получения клофарабина, включающему
(a) получение 2-хлораденозина ферментативным трансгликозилированием 2-хлораденина и соединения формулы 1,
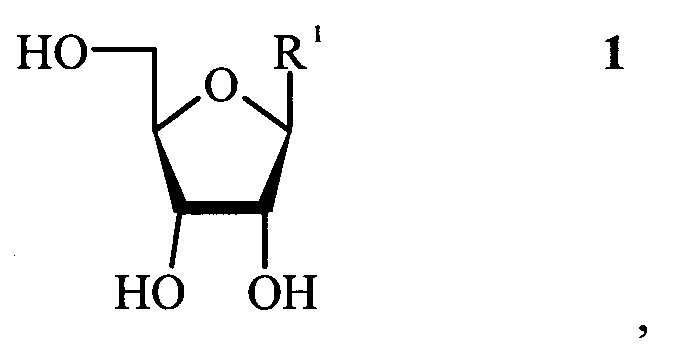
в которой R1 представляет собой пуриновое или пиримидиновое основание формулы 2 или формулы 3, соответственно,
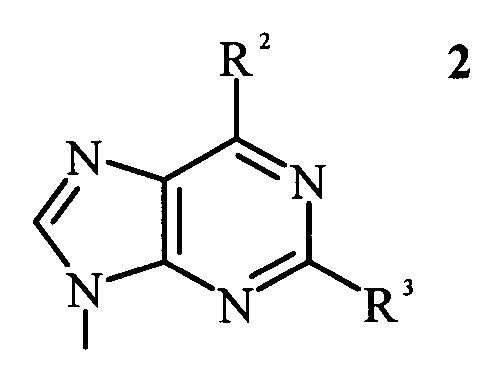

в которой R2 и R3 независимо выбраны из группы, состоящей из -H, -NH2, -OH и -CH3; и R4 выбран из группы, состоящей из -H и -CH3;
(b) частичную защиту гидроксильных групп 2-хлораденозина с получением смеси соединения формулы 4 и соединения формулы 5,


в которых R5 независимо представляет собой защитную группу гидроксила;
(c) изомеризацию соединения формулы 4 в соединение формулы 5;
(d) получение соединения формулы 6 из соединения формулы 5,

в которой OR6 представляет собой уходящую группу;
(e) фторирование соединения формулы 6 до соединения формулы 7,
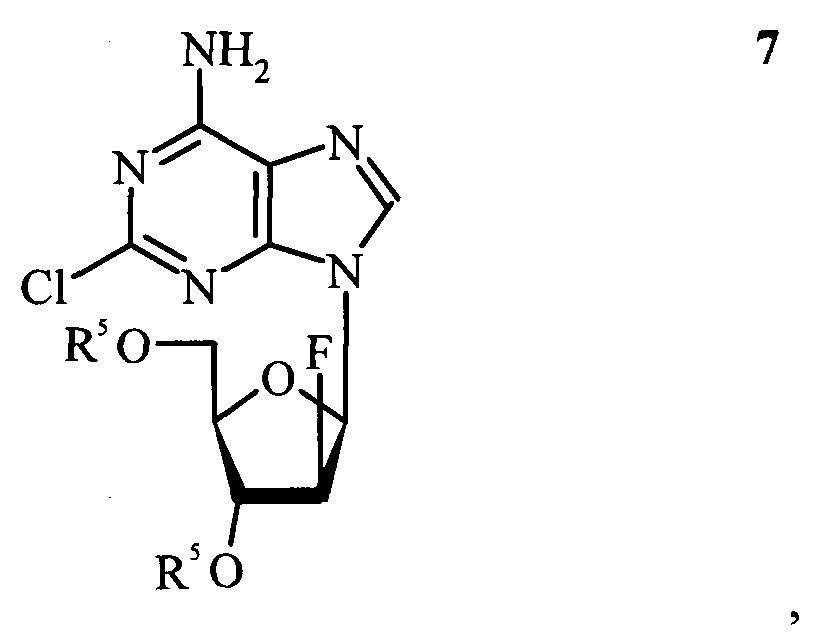
(f) снятие защиты соединения формулы 7 с получением клофарабина.
Согласно предпочтительным вариантам осуществления трансгликозилирование на стадии (a) выполняют с использованием пурин-нуклеозидфосфорилазы или комбинации пурин-нуклеозидфосфорилазы и уридинфосфорилазы.
Согласно следующим предпочтительным вариантам осуществления фторирование на стадии (e) выполняют с использованием фторирующего средства. Предпочтительно фторирующее средство выбрано из группы, состоящей из фтористоводородной кислоты и смеси фтористоводородной кислоты и органического основания Льюиса. Особенно предпочтительным органическим основанием Льюиса (используемым в смеси с фтористоводородной кислотой) является амин.
Согласно следующим предпочтительным вариантам осуществления заместитель R1 представляет собой пиримидиновое основание, являющееся уридином; и/или заместитель R5 представляет собой защитную группу гидроксила, являющуюся бензоилом; и/или заместитель OR6 представляет собой уходящую группу, являющуюся трифторметансульфонатом.
Краткое описание графических материалов
На фиг. 1 показана схема синтеза для получения клофарабина, раскрытая в патенте США №5034518.
На фиг. 2 показана схема синтеза для получения клофарабина, раскрытая в патенте США №6949640.
На фиг. 3 показана схема синтеза для получения клофарабина, раскрытая в патенте США №6680382.
На фиг. 4 показана схема синтеза для получения клофарабина, раскрытая в публикации заявки на выдачу патента США №2012/0010397.
На фиг. 5 показана схема синтеза для получения клофарабина в соответствии с настоящим изобретением.
Подробное раскрытие настоящего изобретения
Настоящее изобретение основывается на неожиданном открытии того, что, если начинается с 2-хлораденозина, выполнение каскада реакций, включающего стадии ферментативного трансгликозилирования, бензоилирования, изомеризации, образования сульфоната сложного эфира (т.е. сульфонилирования), фторирования и снятия защиты, приводит к получению клофарабина с высоким выходом без сопутствующего образования нежелательных стереоизомеров, таким образом, преодолевая главные недостатки известных способов и обеспечивая более эффективную и менее трудоемкую схему синтеза.
Далее настоящее изобретение будет описываться в отношении конкретных вариантов осуществления и в отношении конкретных графических материалов, но следует учитывать, что настоящее изобретение ограничивается не ими, а только прилагаемой формулой изобретения. Описываемые графические материалы являются исключительно схематическими и иллюстративными и считаются неограничивающими.
Используемый в настоящем описании и формуле изобретения термин «включающий» («содержащий») не исключает другие элементы или стадии. Для целей настоящего изобретения термин «состоящий из» считается предпочтительным вариантом термина «содержащий». Если далее в настоящем документе группа определяется как содержащая по меньшей мере некоторое число вариантов осуществления, то это также следует понимать как раскрытие группы, которая предпочтительно состоит только из этих вариантов осуществления.
Использование существительного в единственном числе, предусматривает множественное число этого существительного, если специально не указано иное.
В случае указания числовых значений в контексте настоящего изобретения специалисту в данной области будет понятно, что технический эффект рассматриваемого признака обеспечивают в диапазоне точности, который, как правило, охватывает отклонение числового значения в пределах ±10% и предпочтительно ±5%.
Кроме того, термины первые, второй, третий, (a), (b), (c) и подобные в настоящем описании и в формуле изобретения используют для различения подобных элементов и необязательно для описания последовательного или хронологического порядка. Следует понимать, что термины, используемые таким образом, являются взаимозаменяемыми при соответствующих обстоятельствах, и что варианты осуществления настоящего изобретения, описываемые в настоящем документе, могут работать в последовательностях, отличных от описываемых или иллюстрируемых в настоящем документе.
Следующие определения терминов будут приведены далее в контексте использования этих терминов. Следующие термины или определения представлены исключительно в целях объяснения настоящего изобретения. Эти определения не должны быть истолкованы в объеме, меньшем, чем известный рядовому специалисту в данной области.
В одном аспекте настоящее изобретение относится к способу получения клофарабина, включающему
(a) получение 2-хлораденозина ферментативным трансгликозилированием 2-хлораденина и соединения формулы 1,

в которой R1 представляет собой пуриновое или пиримидиновое основание формулы 2 или формулы 3, соответственно,
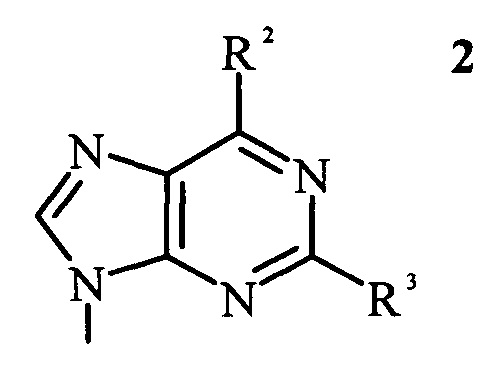
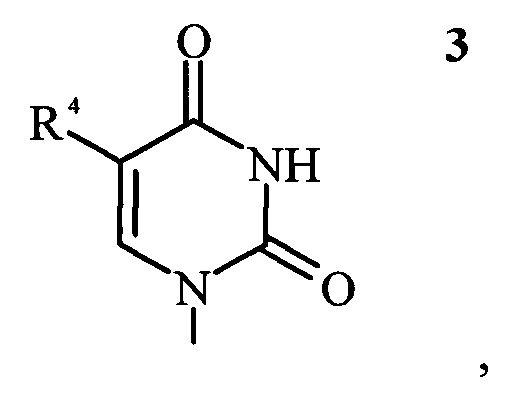
в которых R2 и R3 независимо выбраны из группы, состоящей из -H, -NH2, -OH и -CH3; и R4 выбран из группы, состоящей из -H и -CH3;
(b) частичную защиту гидроксильных групп 2-хлораденозина с получением смеси соединения формулы 4 и соединения формулы 5,


в которых R5 независимо представляет собой защитную группу гидроксила;
(c) изомеризацию соединения формулы 4 в соединение формулы 5;
(d) получение соединения формулы 6 из соединения формулы 5,

в которой OR6 представляет собой уходящую группу;
(e) фторирование соединения формулы 6 до соединения формулы 7,

(f) снятие защиты соединение формулы 7 с получением клофарабина.
Способ в соответствии с настоящим изобретением схематически приведен на фиг. 5. Далее нумерация соединений соответствует номенклатуре, используемой на фиг. 5.
2-Хлораденозин (III) получают путем реакции ферментативного трансгликозилирования между 2-хлораденином (II) и любым нуклеозидом, который содержит рибозу, предпочтительно уридином (I) (поскольку он легко растворяется и легко доступен), с использованием ферментов, таких как предпочтительно пурин-нуклеозидфосфорилаза (PNP) и уридинфосфорилаза (UP). Эта реакция является обратимой. Для смещения равновесия в сторону продукта (III) используют избыток уридина от 1,5 до 5 эквивалентов, предпочтительно 3,3. Реакция может быть проведена при температуре от 40 до 70°C, предпочтительно при температуре от 58 до 61°C.
Из-за стереоспецифичности ферментативного трансгликозилирования единственным продуктом гликозилирования является β-аномер (III). 2-Хлораденозин может быть очищен от исходных реагентов (I), (II) и урацила (побочного продукта), например, с использованием препаративной хроматографии низкого давления. Как правило, выбирают условия, обеспечивающие удерживание 2-хлораденозина сорбентом, тогда как большая часть примесей не удерживается в колонке. Это позволяет упростить процесс очистки путем включения только стадий «внесения» и «промывания».
Затем полученный 2-хлораденозин предпочтительно бензоилируют бензоилхлоридом в присутствии пиридина. В этой реакции бензоилхлорид может быть использован в количестве, варьирующем от 1,8 до 2,5 эквивалента, предпочтительно 2,3 эквивалента, исходя из триольного соединения формулы (III). Реакция может быть проведена при температуре от -10 до 20°C, предпочтительно при температуре от 0 до 5°C.
В качестве побочного продукта может образовываться до 35% 2',3',5'-три-O-бензоил-2-хлораденозина (IV). Однако присутствие этого побочного продукта не влияет на последующую стадию изомеризации, а также его легко можно отделить при кристаллизации 3',5'-ди-O-бензоильного производного. Таким образом, почти все количество исходного соединения 2-хлораденозина в конечном итоге приводит к образованию 3',5'-ди-O-бензоильного производного.
Далее выполняют изомеризацию 2',5'-ди-О-бензоильного изомера (V) до 3',5'-ди-O-бензоильного изомера (VI). Перегруппировка предпочтительной бензоильной группы из положения 2' в 3' в нуклеозидах известна в уровне техники (см. inter alia Maruyama, T. et al. (1999) Chem. Pharm. Bull. 47, 966-970). Предпочтительно реакцию изомеризации выполняют путем кипячения в метаноле в течение 40-45 часов при энергичном перемешивании. Полученные в результате кристаллы после горячей фильтрации реакционной смеси характеризуются чистотой >98% (по HPLC), в то время как почти все примеси остаются в остаточной жидкости, из которой после повторной обработки может быть выделен 2-хлораденозин.
Последующее сульфонилирование соединения формулы VI выполняют путем обработки с помощью Tf2O с использованием пиридина в качестве основания. В этой реакции трифторметансульфоновый ангидрид может быть использован в количестве, варьирующем от 1,3 до 2,0 эквивалента, предпочтительно 1,6 эквивалента, исходя из соединения формулы (VI). Растворителем может быть органический растворитель, такой как ацетонитрил, тетрагидрофуран, дихлорметан, хлороформ, предпочтительно дихлорметан. Реакция может быть проведена при температуре от -20 до 30°C, предпочтительно при температуре от -10 до 0°C.
На следующей стадии может быть получено фторпроизводное (VIII) путем обработки трифлата (VII) фторирующими средствами, такими как NEt3*3HF, TBAF или TBAF*(t-BuOH)4, предпочтительно NEt3*3HF, например, с использованием диизопропилэтиламина в качестве основания. Растворителем может быть, например, этилацетат, тетрахлорметан, толуол, ацетонитрил, предпочтительно толуол. Реакция может быть проведена при температуре от 0 до 100°C, предпочтительно при температуре от 35 до 40°C. Полученный защищенный клофарабин может быть кристаллизован из смеси этилацетата и метанола. При вышеупомянутых условиях выход продукта реакции после кристаллизации составляет приблизительно 60%.
В конечном итоге, с защищенного клофарабина снимают защиту при мягких условиях. Такие мягкие условия (30 минут при 30°C) позволяют выполнять реакцию без каких-либо побочных реакций. Образованный метилбензоат отделяют путем экстракции метиленхлоридом, а клофарабин, содержащийся в водной фазе, очищают с использованием препаративной хроматографии низкого давления с последующей кристаллизацией. Полученный клофарабин характеризуется высокой чистотой без выявляемых количеств примесей. Общий выход клофарабина составляет приблизительно 30-40%.
Настоящее изобретение далее описывается с помощью графических материалов и следующих примеров, которые приводятся исключительно в целях иллюстрации конкретных вариантов осуществления настоящего изобретения и не должны считаться как ограничивающие каким-либо образом заявляемый объект изобретения.
Примеры
Пример 1а. Получение 2-хлораденозина (III) из 2-хлораденина и уридина
400 г уридина и 150 г KH2PO4 растворяли при перемешивании в смеси воды (52 л) и DMSO (1,8 л) при 58-61°C. В полученный раствор добавляли первую порцию (0,75 л) раствора, полученного из 2-хлораденина (85 г), воды (7 л) и KОН (120 г). Регулировали pH полученной в результате смеси до 7,1-7,2 водным раствором KOH. Добавляли растворы уридинфосфорилазы и пурин-нуклеозидфосфорилазы при перемешивании при 58-61°C. Оставшуюся вторую часть раствора 2-хлораденина постепенно добавляли в реакционную смесь при перемешивании в течение 3-часового периода при 58-61°C с поддерживанием pH в диапазоне 7,1-7,2 водным раствором HCl. Затем реакционную смесь перемешивали в течение 1 часа при 58-61°C и добавляли NaOH для доведения pH до 11.
Полученный в результате раствор, содержащий 2-хлораденозин, очищали посредством препаративной хроматографии с последующим выделением продукта, включающим кристаллизацию в воде, с получением 120 г названного соединения. Как правило, чистота 2-хлораденозина составляла >99%, а выход исходя из 2-хлораденина составлял >85%.
Пример 1b. Получение 2-хлораденозина (III) из 2-хлораденина и гуанозина 229 г гуанозина и 75 г KH2PO4 растворяли при перемешивании в смеси воды (52 л) и DMSO (1,8 л) при 58-61°C. В полученный раствор добавляли первую порцию (0,75 л) раствора, полученного из 2-хлораденина (42 г), воды (7 л) и КОН (60 г). Регулировали pH полученной в результате смеси до 7,1-7,2 водным раствором KOH. В реакционную смесь добавляли раствор пурин-нуклеозидфосфорилазы при перемешивании при 58-61°C. Оставшуюся вторую часть раствора 2-хлораденина постепенно добавляли в реакционную смесь при перемешивании в течение 3-часового периода при 58-61°C с поддерживанием pH в диапазоне 7,1-7,2 водным раствором HCl. Затем реакционную смесь перемешивали в течение 1 часа при 58-61°C и добавляли NaOH для доведения pH до 11.
Полученный в результате раствор, содержащий 2-хлораденозин, очищали посредством обращенно-фазовой колоночной хроматографии низкого давления с последующим выделением продукта, включающим кристаллизацию в воде, с получением 53 г названного соединения. Как правило, чистота 2-хлораденозина составляла >99%, а выход исходя из 2-хлораденина составлял >70%.
Пример 2. Бензоилирование 2-хлораденозина
Раствор 2-хлораденозина (750 г) в пиридине (7,5 л) охлаждали до -5-0°C. Затем в реакционную смесь медленно добавляли раствор бензоилхлорида (720 г) в ацетонитриле (1440 мл) при перемешивании и охлаждении. При этом внутренняя температура реакционной смеси не должна была превышать 5°C. Смесь выдерживали в течение 30 минут при тех же условиях. После этого растворители выпаривали при пониженном давлении и при температуре 60°C. Остаток растворяли в CH2Cl2 и последовательно промывали 1 M водным раствором H2SO4, насыщенным водным раствором NaHCO3 и водой. Органическую фазу выпаривали при пониженном давлении с получением смеси 2-хлор-9-(2',5'-ди-O-бензоил-β-D-рибофуранозил)-аденина и 2-хлор-9-(3',5'-ди-O-бензоил-β-D-рибофуранозил)-аденина (оба в сумме составляли приблизительно 65% по HPLC), а также 2',3',5'-три-O-бензоил-2-хлораденозина (приблизительно 30% по HPLC).
Пример 3. Изомеризация 2-хлор-9-(2',5'-ди-O-бензоил-β-D-рибофуранозил)-аденина (V) до 2-хлор-9-(3',5'-ди-O-бензоил-β-D-рибофуранозил)-аденина (VI)
Смесь 2-хлор-9-(2',5'-ди-O-бензоил-β-D-рибофуранозил)-аденина и 2',3',5'-три-O-бензоил-2-хлораденозина, полученную в примере 2, добавляли в 25 л MeOH. Полученную в результате смесь нагревали с обратным холодильником в течение 40-45 часов при энергичном перемешивании. После этого смесь подвергали горячему фильтрованию в вакууме, а отфильтрованный остаток промывали с помощью MeOH. Фильтрат, содержащий 2',3',5'-три-O-бензоил-2-хлораденозин и 2-хлор-9-(2',5'-ди-O-бензоил-β-D-рибофуранозил)-аденин, направляли на повторную обработку для получения 2-хлораденозина путем гидролиза. Твердое вещество сушили в вакууме при 50°C с получением 880 г 2-хлор-9-(3',5'-ди-O-бензоил-β-D-рибофуранозил)-аденина с чистотой по HPLC, составляющей >98%.
Пример 4. Получение 2-хлор-9-(3',5'-ди-O-бензоил-2'-O-трифторметилсульфонил-β-D-рибофуранозил)-аденина (VII)
2-Хлор-9-(3',5'-ди-O-бензоил-β-D-рибофуранозил)-аденин (880 г) суспендировали в смеси пиридина (0,8 л) и CH2Cl2 (8 л). В атмосфере N2 раствор трифторметансульфонового ангидрида (455 мл) в CH2Cl2 (1500 мл) медленно добавляли в реакционную смесь при перемешивании и охлаждали при -10-0°C. Реакционную смесь выдерживали в течение 30 минут при тех же условиях. Затем добавляли насыщенный водный раствор NaHCO3 при перемешивании для доведения pH полученной в результате смеси до 7. Органическую фазу отделяли, промывали водой и выпаривали при пониженном давлении с получением 1000 г (выход 95%) названного продукта, характеризующегося чистотой по HPLC, составляющей >95%.
Пример 5. Получение 2-хлор-9-(3',5'-ди-O-бензоил-2'-дезокси-2'-фтор-β-D-арабино-фуранозил)-аденина (VIII)
2-Хлор-9-(3',5'-ди-O-бензоил-2'-O-трифторметилсульфонил-β-D-рибофуранозил)-аденин, полученный в примере 4, суспендировали в 5 л толуола. Постепенно добавляли N,N-диизопропилэтиламин (350 мл) и триэтиламинтригидрофторид (1,2 л) при перемешивании. Реакционную смесь перемешивали в течение 48 часов при 35-40°C и выпаривали при пониженном давлении. Остаток обрабатывали этилацетатом и фильтровали. К фильтрату добавляли насыщенный водный раствор NaHCO3 при перемешивании до доведения pH полученной в результате смеси до 7. Органическую фазу отделяли, промывали водой и выпаривали при пониженном давлении. Остаток растворяли в метаноле (1,5 л) при перемешивании и при 60°C. Полученный раствор выдержали при 2-6°C в течение 2-3 часов до полного осаждения. Образованный осадок фильтровали, промывали метанолом и сушили с поучением 540 г (выход 60%) названного продукта, характеризующегося чистотой по HPLC, составляющей >95%.
Пример 6. Получение 2-хлор-9-(2'-дезокси-2'-фтор-β-D-арабинофуранозил)-аденина (т.е. клофарабина)
2-Хлор-9-(3',5'-ди-O-бензоил-2'-дезокси-2'-фтор-β-D-арабинофуранозил)-аденин, полученный в примере 5, суспендировали в 1,5 л метанола. Добавляли 30 мл 10% метанольного раствора MeONa и реакционную смесь перемешивали в течение 30 минут при 30°C. После этого в реакционную смесь добавляли воду (1,5 л) и CH2Cl2 (1,5 л). После перемешивания в течение 10 минут водную фазу отделяли и выпаривали при пониженном давлении до объема приблизительно 1,5 л. Полученный в результате раствор, содержащий клофарабин, очищали посредством препаративной колоночной хроматографии низкого давления с последующим выделением продукта, включающим кристаллизацию в смеси воды и ацетона, с получением 260 г названного соединения. Как правило, чистота клофарабина составляла >99,9%, а общий выход (после 6 стадий) составлял 32%.
Настоящее изобретение, иллюстративно описываемое в настоящем документе, может быть соответствующим образом осуществлено при отсутствии какого-либо элемента или элементов, ограничения или ограничений, специально не раскрытых в настоящем документе. Так, например, термины «содержащий», «включающий», «имеющий» и т.д. следует понимать расширительно и без ограничения. Кроме того, термины и выражения, используемые в настоящем документе, были использованы в качестве терминов описания, а не ограничения, и такие термины и выражения не предназначены для применения с целью исключения каких-либо эквивалентов показанных и описанных признаков или их частей, но следует отметить, что возможны различные модификации, входящие в объем заявляемого настоящего изобретения. Таким образом, следует понимать, что, хотя настоящее изобретение было конкретно раскрыто с помощью вариантов осуществления и необязательных признаков, специалистами в данной области могут быть использованы модификации и вариации настоящего изобретения, предусмотренные в нем, и что такие модификации и вариации попадают в объем настоящего изобретения.
Настоящее изобретение было описано в настоящем документе в широком смысле и в общих чертах. Каждые из более узких объектов и субгенерических групп, попадающих в общее раскрытие, также составляют часть настоящего изобретения. Это касается общего описания настоящего изобретения с условием или отрицательным ограничением, исключающим какой-либо заявляемый объект из родового понятия, независимо от того, указан или нет исключенный материал конкретно в настоящем документе.
Другие варианты осуществления попадают в объем следующей формулы изобретения. Кроме того, поскольку признаки или аспекты настоящего изобретения описаны в терминах групп Маркуша, специалистам в данной области должно быть понятно, что настоящее изобретение тем самым также описывается при помощи любого отдельного представителя или подгруппы представителей группы Маркуша.
Реферат
Изобретение относится к биотехнологии и фармацевтике, а именно к способу получения клофарабина. Получают 2-хлораденозин ферментативным трансгликозилированием 2-хлораденина и соединения формулы 1,в которой Rпредставляет собой пуриновое или пиримидиновое основание. Проводят частичную защиту гидроксильных групп 2-хлораденозина с получением смеси соединения формулы 4 и соединения формулы 5,в которых Rнезависимо представляет собой защитную группу гидроксила. Изомеризуют соединение формулы 4 до соединения формулы 5. Получают соединение формулы 6в которой ORпредставляет собой уходящую группу. Фторируют соединение формулы 6 до соединения формулы 7и снимают защиту соединения формулы 7 с получением клофарабина. Изобретение позволяет получать противораковый нуклеозид клофарабин с высоким выходом и без образования нежелательных α-N9 стереоизомеров. 7 з.п. ф-лы, 5 ил., 5 пр.
Формула




Документы, цитированные в отчёте о поиске
Способ получения производных 2'-дезокси-2'-фторрибонуклеозидов или их фармацевтически приемлемых солей
Комментарии