Способ получения производных морфолинилантрациклина - RU2563638C2
Код документа: RU2563638C2
Описание
Настоящая заявка, не являющаяся предварительной, поданная в соответствии с §1.53(b) п.37 Свода федеральных правил (CFR), испрашивает, согласно параграфу §119(e) главы 35 Свода законов США (USC), приоритет на основании предварительной заявки на патент США №61/418949, поданной 2 декабря 2010 г., содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
Настоящее изобретение относится к способу получения производных морфолинилантрациклина, характеризующемуся тем, что морфолиновое кольцо соединено через атом кислорода с остатком сахара по положению С-4′.
Указанные производные морфолинилантрациклина, способ их получения, содержащие их фармацевтические композиции и их применение в качестве терапевтических агентов, в частности для лечения рака, описаны и заявлены в международной заявке WO 98/02446.
Морфолинилантрациклины представляют собой полусинтетические аналоги антрациклинов и отличаются выдающейся противоопухолевой активностью (см.: Bioactive Molecules Vol.6, ED. J.W. Lown, Elsevier 1988; Curr Pharm Des. Mar 5(3):217-27, 1999).
Указанные соединения могут быть получены в соответствии с известными химическими способами путем проведения реакции N-оксида производного морфолинилантрациклина с солью железа в присутствии агента комплексообразования железа согласно описанию в международной заявке WO 98/02446, упоминаемой выше.
Конъюгаты антител с морфолинилантрациклинами обладают направленной противоопухолевой активностью (WO 2009/099741; WO 2010/009124).
Авторами настоящего изобретения неожиданно было обнаружено, что указанные производные морфолинилантрациклина могут быть успешно получены с помощью нового способа, обеспечивающего получение целевых продуктов с высоким выходом и чистотой.
Таким образом, первым объектом настоящего изобретения является способ получения производного морфолинилантрациклина формулы (I)
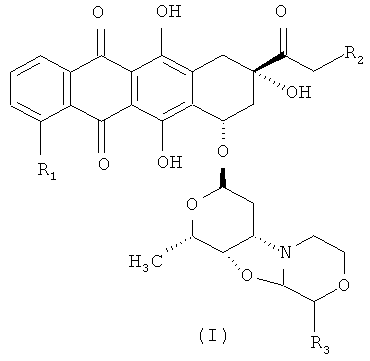
где R1 представляет собой водород, ОН или ОСН3,
R2 представляет собой водород или ОН, и
R3 представляет собой водород или ОС1-С5 алкил, или его фармацевтически приемлемую соль присоединения кислоты;
включающий:
(i) реакцию цианурхлорида с N-оксидным производным антрациклина формулы (II)

где R1, R2 и R3 такие, как описано выше, и
(ii) необязательно, превращение полученного соединения формулы (I) в его фармацевтически приемлемую соль присоединения кислоты.
Конкретными примерами производных морфолинилантрациклина формулы (I) являются перечисленные ниже соединения:
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-доксорубицин (1);
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-идарубицин (2);
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-даунорубицин (3);
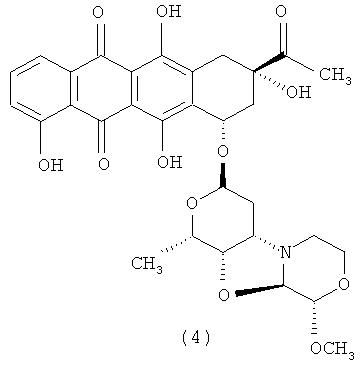
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-карминомицин (4); и
3′-деамино-3′′-4′-ангидро-[2′′(S)-этокси-3′′(R)-гидрокси-4′′-морфолинил]-доксорубицин (5),
или их фармацевтически приемлемой солью присоединения кислоты.
Конкретным примером производного морфолинилантрациклина формулы (I) является 3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-доксорубицин (1). Формула (I) представляет собой метаболит неморубицина, также известный как PNU-159682, (Quintieri et al. (2005) Clinical Cancer Research, 11(4):1608-1617; Beulz-Riche et al. (2001) Fundamental & Clinical Pharmacology, 15(6):373-378; EP 0889898; WO 2004/082689; WO 2004/082579). PNU-159682 формулы (1) более цитотоксичен in vitro, чем неморубицин и доксорубицин, и эффективен в моделях опухолей in vivo. Конъюгаты антител с лекарственными средствами, содержащие PNU-159682 формулы (1), обеспечивают направленный киллинг клеток (WO 2010/009124).

Термин «ОС1-С5 алкил» относится к прямым или разветвленным насыщенным алифатическим гидрокарбильным группам, содержащим от 1 до 5 атомов и связанным с остальной частью молекулы через атом кислорода.
Реакция циклизации согласно Примеру 1 проходит с образованием одного изомера. Указанную реакцию обычно проводят в апротонных растворителях, таких как дихлорметан, хлороформ, ацетон, 1,4-диоксан, диметилформамид, 1,2-дихлорэтан или ацетонитрил, и в присутствии основания, такого как триэтиламин, 4-диметиламинопиридин, карбонат натрия, карбонат цезия или карбонат калия. Указанную реакцию, как правило, проводят при температуре от 0°C до комнатной в течение 5-60 минут. Типовые условия включают ацетонитрил в качестве растворителя и карбонат калия в качестве основания, при комнатной температуре в течение 30 минут.
Исходное N-оксидное соединение формулы (II) может быть получено путем окисления диметилдиоксираном производного морфолинилантрациклина согласно описанию в GB 2296495 А.
Следующие производные морфолинилантрациклина, в общем описанные и заявленные в международной патентной заявке WO 98/02446, а также содержащие их фармацевтические композиции и их применение в качестве терапевтических агентов, в частности при лечении рака, являются новыми:



Подходящие пути введения включают парентеральное введение. Жидкий состав для парентерального введения может быть получен с применением активного соединения и стерильного разбавителя или носителя, который может либо растворять активное соединение, либо обеспечивать его суспензирование. Состав для парентерального введения может быть получен в форме стерильного твердого вещества для предшествующего введению восстановления подходящей основой, такой как физиологический солевой раствор, стерильная вода или другая стерильная основа.
Соединения согласно настоящему изобретению подходят для применения в способах лечения гиперпролиферативных заболеваний, таких как лейкемия, аденокарцинома толстой кишки, и других солидных опухолей и гематологических злокачественных образований.
Пациенту, имеющему гиперпролиферативное заболевание, такое как опухоль, вводят терапевтически эффективное количество для облегчения или улучшения состояния указанного пациента. Может быть введено количество, достаточное для подавления прогрессирования заболевания, например роста опухоли. Назначаемая доза может быть индивидуализирована с помощью модификации известных диапазонов доз для доксорубицина и даунорубицина на основе активности, демонстрируемой настоящим соединением в противоопухолевом тесте in vitro и in vivo. Подходящая дозировка, как правило, находится в диапазоне от 0,01 до 100 мг/м2, в зависимости от природы и тяжести заболевания, лечение которого проводится, и общего состояния пациента.
Биологическая активность: Анализ клеточной пролиферации in vitro
Клетки яичника человека А2780 и рака молочной железы человека MCF7 (1250 клеток/лунка) высевали в белые 384-луночные планшеты на полную среду (RPMI1640 или ЕМЕМ, плюс 10% фетальной бычьей сыворотки) и обрабатывали соединениями, растворенными в 0,1% ДМСО, через 24 ч после высевания. Клетки инкубировали при 37°C и 5% CO2; спустя 72 часа планшеты обрабатывали с применением набора для анализа CellTiter-Glo® (Promega), следуя инструкциям производителя.
CellTiter-Glo® представляет собой гомогенный метод, основанный на количественном определении присутствующего АТФ, индикатора метаболически активных клеток. Количество АТФ определяют с применением системы, основанной на люциферазе и D-люциферине, которые обеспечивают генерацию света. Люминесцентный сигнал пропорционален числу клеток, присутствующих в культуре.
Раствор реагента в количестве 25 мкл на лунку добавляли в каждую лунку; после 5 минут встряхивания микропланшеты считывали с использованием люминометра для определения значений IC50. Люминесцентный сигнал пропорционален числу клеток, присутствующих в культуре.
Следующие примеры иллюстрируют настоящее изобретение, но не ограничивают его объем.
ПРИМЕР 1
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-доксорубицин (1).
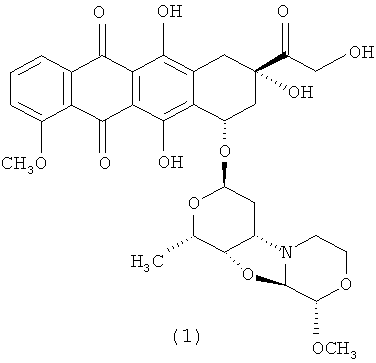
К раствору 3′-деамино-3′[2(S)-метокси-4-морфолинил]-доксорубицин N-оксида (полученному согласно описанию в GB 296495 А) (50,0 мг; 0,076 ммоль) в 12,5 мл сухого ацетонитрила, добавляли карбонат калия в виде порошка (31,5 мг; 0,228 ммоль) и цианурхлорид (2,4,6-Трихлор-1,3,5-триазин, рег. № CAS 108-77-0; 28,0 мг; 0,152 ммоль). Реакционную смесь интенсивно перемешивали в темноте в течение 20 минут, пока стартовый материал не становился неразличим (ТСХ-анализ, EtOH:CH2Cl2=1:9). Затем к реакционной смеси добавляли раствор 3-амино-1,2-пропандиола (42,0 мг, 0,46 ммоль) в воде (1 мл) и водную фазу экстрагировали дихлорметаном (4×30 мл). Объединенные органические фазы высушивали над безводным сульфатом натрия, фильтровали и выпаривали под вакуумом. Неочищенный продукт очищали посредством колоночной флэш-хроматографии (EtOH:CH2Cl2=0,2:9,8) на силикагеле (230-400 меш) с получением 24,4 мг 3′-деамино-3”-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]доксорубицина (1) в виде твердого вещества красного цвета (выход=50%).1Н ЯМР (500 МГц; АЦЕТОНИТРИЛ-d3) δ ч./млн. 1,29 (d, J=6,41 Гц; 3Н) 1,68 (dt, J=15,02; 5,86 Гц; 1Н) 1,89 (dt, J=15,02; 5,50 Гц; 1Н) 2,07-2,13 (m, 1Н) 2,46 (dt, J=14,66; 2,02 Гц; 1Н) 2,69-2,75 (m, 1Н) 2,76-2,81 (m, 1Н) 2,95 (d, J=18,50 Гц; 1Н) 3,08 (t, J=5,50 Гц; 1Н) 3,14 (dd, J=18,59; 1,92 Гц; 1Н) 3,37 (s, 3Н) 3,41-3,47 (m, 1Н) 3,52-3,58 (m, 1Н) 3,73 (ddd, J=11,50; 8,11; 2,93 Гц; 1Н) 4,01 (s, 3Н) 4,02-4,08 (m, 2Н) 4,25 (d, J=2,93 Гц; 1Н) 4,53 (d, J=2,93 Гц; 1Н) 4,61 (s, 1Н) 4,63-4,75 (m, 2Н) 5,22 (dd, J=3,94; 2,11 Гц; 1Н) 5,36 (t, J=5,59 Гц; 1Н) 7,54 (d, J=8,06 Гц; 1Н) 7,84 (t, J=8,06 Гц; 1Н) 7,96 (dd, J=7,69; 0,73 Гц; 1Н). МС (ИЭР): 642 [М+Н]+. Время удерживания=4,88.
В соответствии с методикой, применяемой для получения 3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]доксорубицина (1), но с использованием подходящих замещенных производных, получали следующие соединения:
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-идарубицин (2).

1Н ЯМР (АЦЕТОНИТРИЛ-d3) δ: 8,29-8,34 (m, 2Н); 7,86-7,95 (m, 2Н); 5;35 (t, J=5,6 Гц; 1H); 5,19 (dd, J=4,1; 2,1 Гц; 1Н); 4,54 (s, 1H); 4,54 (s, 1Н); 4,26 (d, J=2,9 Гц; 1 H); 4,09 (dd, J=6,6; 1,7 Гц; 1Н); 4,03 (dd, J=7,1; 1,8 Гц; 1H); 3,74 (ddd, J=11,5; 8,2; 3,0 Гц; 1Н); 3,51-3,58 (m, 1Н); 3,44 (q, J=6,0 Гц; 1Н); 3,37 (s, 3Н); 3,06-3,11 (m, 1Н); 2,91-2,98 (m, 1Н); 2,67-2,81 (m, 2Н); 2,44 (dt, J=14,8; 2,1 Гц; 1Н); 2,35 (s, 3Н); 2,06 (dd, J=14,6; 4,4 Гц; 1Н); 1,85-1,91 (m, 1Н); 1,71 (dt, J=15,0; 5,9 Гц; 1Н); 1,29 (d, J=6,6 Гц; 3Н). МС расчета.: 596,2127; МС экспериментальн.: 596,2117. МС (ИЭР): 596 [М+Н]+. Время удерживания=6,32 мин.
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-даунорубицин (3)
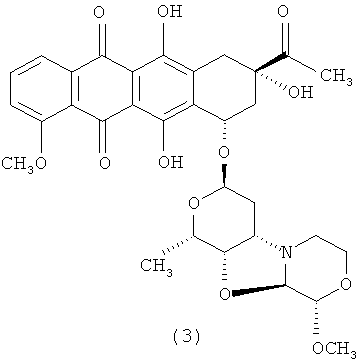
1Н ЯМР (АЦЕТОНИТРИЛ-d3) δ: 7,94-7,99 (m, 1Н); 7,84 (t, J=8,1 Гц; 1Н); 7,54 (d, J=8,5 Гц; 1Н); 5,35 (t, J=5,5 Гц; 1Н); 5,19 (m, 1Н); 4,55 (s, 1H); 4,54 (d, J=2,9 Гц; 1H); 4,26 (d, J=2,7 Гц; 1Н); 4,09 (dd, J=6,6; 1,7 Гц; 1H); 3,97-4,05 (m, 4Н); 3,74 (, 1H); 3,54 (m, 1Н); 3,44 (q, J=6,1 Гц; 1Н); 3,37 (s, 3Н); 3,02-3,10 (m, 1Н); 2,88-3,01 (m, 1H); 2,64-2,86 (m, 2Н); 2,43 (dt, J=14,8; 2,1 Гц; 1Н); 2,34 (s, 3Н); 2,05 (dd, J=14,7; 4,3 Гц; 1Н); 1,88 (dt, J=15,1; 5,7 Гц; 1Н); 1,70 (dt, J=15,1; 5,8 Гц; 1H); 1,29 (d, J=6,6 Гц; 3Н).
МС расчета.: 626,2232; МС экспериментальн.: 626,2208. МС (ИЭР): 626 [М+Н]+. Время удерживания=5,66 мин.
3′-деамино-3′′-4′-ангидро-[2′′(S)-метокси-3′′(R)-гидрокси-4′′-морфолинил]-карминомицин (4).
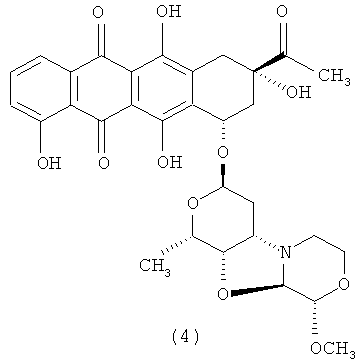
1Н ЯМР (АЦЕТОНИТРИЛ-d3) δ: 7,81-7,91 (m, 1H); 7,84 (m, 1Н); 7,35 (dd, J=8,3; 1,1 Гц; 1H); 5,24-540 (m, 1Н); 5,19 (m, 1Н); 4,54 (d, J=2,9 Гц; 1Н); 4,53 (s, 1H); 4,26 (d, J=2,9 Гц; 1Н); 4,06-4,14 (m, 1Н); 4,04 (dd, J=7,1; 1,8 Гц; 1Н); 3,74 (m, 1Н); 3,55 (m, 1Н); 3,45 (m, 1Н); 3,37 (s, 3Н); 3,07-3,11 (m, 1H); 2,94-2,98 (m, 1Н); 2,69-2,80 (m, 2Н); 2,42-2,46 (m, 1Н); 2,35 (s, 3Н); 1,99-2,11 (m, 1H); 1,85-1,92 (m, 1Н); 1,66-1,75 (m, 1Н); 1,29 (d, J=6,56 Гц; 2Н). МС расчета.: 612,2076; МС экспериментальн.: 612,2054. МС (ИЭР): 612 [М+Н]+. Время удерживания=6,28 мин.
3′-деамино-3′′-4′-ангидро-[2′′(S)-этокси-3′′(R)-гидрокси-4′′-морфолинил]-доксорубицин (5).

1Н ЯМР (АЦЕТОНИТРИЛ-d3) δ: 7,96 (d, J=7,6 Гц; 1Н); 7,83 (t, J=8,1 Гц; 1H); 7,53 (d, J=8,4 Гц; 1Н); 5,36 (t, J=5,6 Гц; 1Н); 5,21 (ушир. синглет, 1 H); 4,69 (t, J=5,4 Гц; 2Н); 4,63 (d, J=2,4 Гц; 1H); 4,62 (s, 1Н); 4,24 (s, 1Н); 4,04-4,04 (m, 2 Н); 4,00 (s, 3Н); 3,70-3,82 (m, 2Н); 3,37-3,60 (m, 3Н); 3,13 (d, J=18,8 Гц; 1Н); 3,08 (t, J=5,3 Гц; 1Н); 2,94 (d, J=18,6 Гц; 1Н); 2,66-2,83 (m, 2Н); 2,46 (d, J=14,9 Гц; 1Н); 2,07-2,12 (m, 1Н); 1,86-1,92 (m, 1Н); 1,63-1,77 (m, 1Н); 1,29 (d, J=6,4 Гц; 3Н); 1,20 (t, J=7,1 Гц; 3Н). МС расчета.: 656,2338; МС экспериментальн.: 656,2325 МС (ИЭР): 656 [М+Н]+. Время удерживания=5,22 мин.
Аналитический метод ВЭЖХ/МС
Оборудование для ВЭЖХ состояло из системы Waters 2795 Alliance НТ®, оснащенной детектором 2996 Waters PDA, и одноквадрупольного масс-спектрометра Micromass ZQ, оснащенного источником ионов для электрораспыления (ИЭР). Управление оборудованием, регистрация и обработка данных осуществлялись с помощью программного обеспечения Empower и MassLynx 4.0. ВЭЖХ проводили при 30°C и скорости потока 1,0 мл/мин с применением колонки Waters X Terra MS С 18-3,5 мкМ (4,6×50 мм). Подвижная фаза А представляла собой 5 мМ рН=5,2 ацетат-аммонийный буфер с ацетонитрилом (95:5); подвижная фаза В представляла собой Н2О/ацетонитрил (5:95); градиент составлял от 10 до 90% В за 8 минут, затем быстро повышался до 100% В за 1,0 минут. Масс-спектрометр работал в режиме определения как положительных, так и отрицательных ионов, капиллярное напряжение устанавливали на уровне 3,5 кВ (ЭР+) и 28 В (ЭР-); температура источника составляла 120°C; конус 14 В (ЭР+) и 2,8 кВ (ЭР-); устанавливали режим полного сканирования, диапазон масс от 100 до 1000 m/z.
Реферат
Изобретение относится к противоопухолевым соединениям: 3'-деамино-3”-4'-ангидро-[2”(S)-метокси-3”(R)-гидрокси-4”-морфолинил]-идарубицину, 3'-деамино-3”-4'-ангидро-[2”(S)-метокси-3”(R)-гидрокси-4”-морфолинил]-даунорубицину, 3'-деамино-3”-4'-ангидро-[2”(S)-метокси-3”(R)-гидрокси-4”-морфолинил]-карминомицину и 3'-деамино-3”-4'-ангидро-[2”(S)-этокси-3”(R)-гидрокси-4”-морфолинил]-доксорубицину, их фармацевтически приемлемым солям присоединения кислоты и способу получения соединения формулы (1) или его фармацевтически приемлемых солей присоединения кислотывзаимодействием цианурхлорида с N-оксидным производным формулы (II), где Rявляется Н, ОН или ОСН; Rявляется Н или ОН, Rявляется Н или С-С-алкокси. Предложены новые эффективные противоопухолевые средства и способ их получения, позволяющий с хорошим выходом и чистотой получить соединение формулы (I). 2 н. и 5 з.п. ф-лы, 1 пр., 1 табл.
Формула
где R1 представляет собой водород, ОН или ОСН3;
R2 представляет собой водород или ОН; и
R3 представляет собой водород, или ОС1-С5 алкил, или их фармацевтически приемлемую соль присоединения кислоты;
включающий проведение реакции цианурхлорида с N-оксидным производным антрациклина формулы (II)

где R1, R2 и R3 такие, как описано выше;
с получением тем самым производного морфолинилантрациклина формулы (I).
3'-деамино-3”-4'-ангидро-[2”(S)-метокси-3”(R)-гидрокси-4”-морфолинил]-идарубицина (2);
3'-деамино-3”-4'-ангидро-[2”(S)-метокси-3”(R)-гидрокси-4”-морфолинил]-даунорубицина (3);
3'-деамино-3”-4'-ангидро-[2”(S)-метокси-3”(R)-гидрокси-4”-морфолинил]-карминомицина (4); и
3'-деамино-3”-4'-ангидро-[2”(S)-этокси-3”(R)-гидрокси-4”-морфолинил]-доксорубицина (5),
или их фармацевтически приемлемой соли присоединения кислоты.

где R1 представляет собой водород, ОН или ОСН3;
R2 представляет собой водород, или ОН; и
R3 представляет собой водород или OC1-C5 алкил.
Документы, цитированные в отчёте о поиске
Антрациклиновые гликозиды и их фармацевтически приемлемые кислотно-аддитивные соли, способ их получения, дииодопромежуточное соединение и способ его получения
Комментарии