N-алкилпроизводные антибиотиков или их фармацевтически приемлемые соли, обладающие противогрибковой активностью, соединение в качестве промежуточного соединения в синтезе n-алкилпроизводных антибиотиков и способ получения n-алкилпроизводных антибиотика и - RU2032693C1
Код документа: RU2032693C1
Чертежи

Описание
Изобретение рассматривает N-алкил производные антибиотического комплекса BU-3608. Эти соединения активны в качестве противогрибковых агентов.
Известны методики ферментации, выделения и очистки антибиотиков BU-3608, BU-3608 B, BU-3608 С. Структура упомянутых
антибиотиков дана формулами Ia-Ie


Ib: BU-3608 B, R1 CH3, R2 H, R3 CH3
Ic: BU-3608 C, R1 CH3, R2 O-ксилозил, R3 H
Id: BU-3608 D, R1 H, R2 O-ксилозил, R3 CH3
Ie: BU-3608 E, R1 H, R2 O-ксилозил, R3 H
BU-3608, однако, имеет очень ограниченную растворимость в воде. Таким образом, целью данного изобретения является получение водорастворимых производных различных компонентов антибиотического комплекса BU-3608.
Изобретение рассматривает соединения, имеющие формулу I:


B-D-ксилозил представляет
собой фрагмент:

Другим аспектом данного изобретения являются бесксилозильные производные BU-3608 C (IIIa), BU-3608 D (IIIб) и BU-3608 E (IIIв), либо их фармакологически приемлемые соли.


IIIa: R1 CH3, R3 H
IIIв: R1 H, R3 CH3
IIIc: R1 H, R3 H
Антибиотики BU-3608, BI-3608 B, BU-3608 C, BU-3608 и BU-3608 E использованы в качестве исходных материалов для соединений данного изобретения и могут быть получены при культивировании производящего антибиотик вида Actinomodura hibisca N Р157-2 и 0278-4, которые хранятся в Американском собрании культур видов (American Type Culture Collection) (Рокквилл, Мериленд) и имеют вспомогательные номера АТС 53557 и АТСС 53646 соответственно. Мутантная линия, полученная из линии Р157-2 обработкой N-метил-N-нитро-N-нитрозогуанидина (НТГ) производит D и Е компоненты в больших количествах, чем и Р157-2, и 0278-4. Эта мутантная линия, обозначенная как А2660 также хранится в АТСС и имеет вспомогательный номер АТСС 53762.
BU-3608 B является бесксилозильным производным BU-3608, BU-3608 B, бесксилозильные BU-3608, C, D, и Е могут быть получены при нагревании BU-3608, BU-3608 C, D, и Е соответственно с соляной кислотой столько времени, сколько достаточно для отщепления ксилозной группы и установлении рН раствора для осаждения желаемого продукта.
Аминогруппы BU-3608, BU-3608 C, D, E или их соответствующих бесксилозильных производных могут быть алкилированы при помощи восстановительного алкилирования, которое включает первоначальное взаимодействие исходного антибиотика с альдегидом или кетоном с образованием имина, и последующую реакцию этого имина. Конденсация и восстановление могут проводиться в одном реакционном сосуде в одну стадию или в две раздельные стадии. Первичная аминогруппа BU-3608 C, E или их бесксилозильных производных может быть превращена в третичный амин, имеющий две одинаковых алкильных группы при обработке по меньшей мере двумя эквивалентами карбонильного соединения в расчете на антибиотик, за которой следует восстановление, либо в третичный амин, имеющий два различных алкильных заместителя, который может быть получен при использовании строго определенного количества первого карбонильного реагента для превращения первичного амина во вторичный, который затем реагирует с другим карбонильным соединением, давая третичный амин.
Карбонильный реагент может быть альдегидом или кетоном, имеющим от одного до пяти атомов углерода, например, формальдегидом, ацетальдегидом, пропиональдегидом или ацетоном. Восстановление имина может быть проведено с использованием таких восстанавливающих агентов как гидриды металлов например, боргидрид натрия, цианоборгидрид натрия, алюмогидрид лития. Реакцию следует проводить в полярном органическом растворителе, либо их смеси, таком как вода, ацетонитрил низшие спирты и диметилсульфоксид. Температура реакции в частности не ограничивается и может быть от комнатной до примерно 100оС.
В наших экспериментах реакция алкилирования проводимая при комнатной температуре, завершается в пределах 24 ч. Оптимальные реакционные условия будут зависеть конечно, от природы и реакционной способности конкретных используемых реагентов.
Растворимость различных антибиотиков была определена в фосфатном буферном солевом растворе (ФБР) и полученные данные приведены ниже в табл.1. ФБР(-) означает раствор, в каждом литре которого содержится 0,2 г KCl, 0,2 г KH2PO4, 8 г NaCl и 1,15 г Na2HPO4, ФБР(+) дополнительно содержит 100 мг MgCl2.6H2O и 100 мг CaCl2.
Для сравнения, растворимость BU-3608 составляет 16-18 мг/мл и 9-23 мг/мл в растворах ФБР(-) и ФБР(+) соответственно. Таким образом, соединения формулы II демонстрируют повышенную растворимость по сравнению с BU-3608.
Биологические свойства.
Противогрибковое действие представленных соединений данного изобретения было определено как in vitro так и in vivo. Минимальная ингибирующая концентрация (МИК) против различных грибков, была определена методом последовательных разбавлений с использованием агара декстрозы Saborand. Примерно 0,003 мл грибковой суспензии, содержащей 106 клеток на мл было нанесено на поверхность агарового слоя, содержащего испытуемый антибиотик. Значения МИК, записанные после инкубации культур в течение 44 ч при 28оС представлены ниже в табл.2.
Действие соединений данного изобретения in vivo было испытано против Candila albicans A9540 на мышах.
Исследуемые организмы содержались 18 ч при 28оС в среде ДГП (дрожжевой экстракт, глюкоза, пептон, K2HРO4, MgSO4) и затем суспендировались в солевом растворе. Самцы мышей ICP, весом 20-24 г инфицировались внутривенно примерно 10-кратной летальной дозой испытуемого препарата.
Антибиотик в различных дозах вводили внутривенно по группам из 5 мышей в каждой сразу же после инфицирования. Доза, которая защищает 50% животных от инфекции (РД50 мг/кг) было вычислено из доли выживших животных на 20 день после грибковой инфекции. Все контрольные животные умерли с 7 по 15 день после инфекции. РД50 для соединений примеров 5, 7, 8 составляет 14 мг/кг (токсическая), 11 мг/кг и 3,5 мг/кг соответственно.
Таким образом другой аспект данного изобретения представляет собой метод лечения грибковых инфекций, который включает в себя введение в организм, инфицированный грибком эффективного количества противогрибкового соединения данного изобретения. Для лечения грибковых инфекций у животных и человека, антибиотики данного изобретения могут быть введены любым принятым путем, которые включают, но не ограничиваются ими, внутривенный, внутримышечный, оральный, носовой, и для поверхностный инфекций местный. Рецептуры для парентерального введения включают стерильные водные и неводные растворы, суспензии или эмульсии. Они могут также производиться в форме стерильных твердых смесей, которые могут быть растворены в стерильной воде, физиологическом растворе, либо некоторых других стерильных средах непосредственно перед введением. Оральные рецептуры могут быть в форме таблеток, желатиновых капсул, порошков, облаток, сиропов и т.п. Для местного применения соединение может быть включено в лосьоны, мази, гели, кремы, притирания, настойки и т. п. Дозировочные единицы могут быть получены с использованием общеизвестных в фармакологии методов.
При лечении грибковых инфекций чувствительных к антибиотикам данного изобретения, предпочтительный путь введения и применяемая доза будет на усмотрении лечащих врачей, и будут различаться в зависимости от конкретного организма, его чувствительности к антибиотикам, рода и пути инфекции, а также таких характеристик пациента, как возраст, вес тела, скорость выделения, сопутствующие медикаменты и общее физическое состояние.
Следующие примеры являются иллюстративными без ограничения области изобретения.
П р и м е р 1. Ферментация Actinomadura hibisca штамма Р157-2
а. Агаровый носитель Actinomadura hibisca штамма Р157-2 проращивают на агаровом носителе, состоящем
из: 0,5% растворимого крахмала (Ничиден Кагаку Ко), 0,5% глюкозы, 0,1% экстракта из мяса рыбы (Микуни Кагаку), 0,1% дрожжевого экстракта (Ориентл Уэст Ко), 0,2% N фактора
(Шеффилд), 0,1% CaCO3, 0,2% NaCl, 1,6% агара. Культуру инкубируют при 28оС в течение 7 дней.
б. Рассада культуры. Порцию выросшей культуры переносят в колбу Эрленмейера на 500 мл, содержащую питательную среду следующего состава: 1% глюкозы, 2% растворимого крахмала (Ничиден Кагаку Ко), 0,5% N-амина А (Шеффилд), 0,5% дрожжевого экстракта (Ориентл Уэст), 0,1% CaCO3. рН среды устанавливают 7,2 до стерилизации. Рассаду культуры инкубируют при 28оС 4 дня на роторном смесителе при 200 об/мин.
в. Ферментация в колбе. 5 мл микрофлоры переносят из рассады культуры в колбу Эрленмейера на 500 мл, которая содержит 100 мл среды следующего состава: 3% глюкозы, 3% соевой муки (Никко Сейю Ко ), 0,5% "Фармамедиа" (Трейдерс Протеин) 0,1% дрожжевого экстракта (Ориентл Уэст), 0,3% CaCO3.
Ферментацию проводят при 28оС в течение 5-6 дней на роторном смесителе. Производство антибиотика при ферментации контролируют методом разбавления с использованием Candida Albicans A9540 как индикатор в бульоне декстрозы. Также используется наблюдение в УФ при 500 нм в 0,01 н NaOH-MeOH растворе. Производство антибиотика достигает максимума 650 мкг/мл на пятый день.
г. Ферментация в резервуаре, 3 л рассады культуры прививают на 120 л стерильной среды, содержащейся в 200 мл ферментаторе. Состав среды такой же, как использовался при ферментации в колбе. Ферментатор выдерживают при 28оС при перемешивании со скоростью 250 об/мин и аэрации со скоростью 120 л/мин. После 96 ч ферментации достигается содержание антибиотика 500 мкг/мл и рН бульона 7,9.
П р и м е р 2. Выделение и очистка антибиотика.
Методика выделения и очистки различных компонентов антибиотического комплекса.
Созревший бульон (рН 7,8) центрифугируют, всплывший слой подкисляют до рН 2,60 6 н. HCl для осаждения бис/неактивных соединений. После того, как осадок удален, рН фильтрата устанавливают 5,0 при помощи 6 н. NaOH и раствор перемешивают 30 мин при комнатной температуре. Полученный темно-красный осадок отфильтровывают и сушат в вакууме. Этот осадок затем растворяют в смеси 3: 1: 4 н-бутанол-метанол-1% NaCl и перемешивают смесь 30 мин. Нижний водный слой отделяют, промывают еще раз свежим верхним слоем, подкисляют до рН 2,0 6 н. HCl и затем экстрагируют н-бутанолом. Экстракт промывают водой, упаривают в вакууме и лиофилизируют получая полуочищенный гидрохлорид BU-3608. Раствор твердого продукта в н-бутаноле встряхивают со щелочной водой (рН 9,0). Водный слой подкисляют до рН 2,0 и промывают этилацетатом. Экстракция н-бутанолом с последующим упариванием растворителя дает более чистый образец BU-3608. HCl. Этот продукт затем подвергают хроматографии с обращенной фазой на силикагеле (ODS-60, 350/250 меш, Ямамура Кемикал Лэб, колонка 4,5 х 90 см). Образец растворяют в воде и наносят на колонку, которая находится в равновесии при помощи смеси ацетонитрил-0,15% KH2PO4 (рН 3,5) 17:83 (объемн).
Эту колонку последовательно промывают 5 л каждой из смесей ацетонитрил-0,15% KH2PO4 с соотношением компо- нентов 17:83, 18:82, 19:81, 20:80 и затем той же смесью растворителей с соотношением 22:78. Элюат собирают во фракции по 100 мл за которыми следят при помощи микротеста с С. albicans A9540 и ТСХ (силикагель, метилацетат-н-пропанол-28% водный аммиак 45:105: 60). Фракции, содержащие в основном одно соединение объединяют и очищают далее, получая BU-3608.
В хроматографии с обращенной фазой на силикагеле, описанной выше, фракции элюирующие до и после гомогенного BU-3608 объединяют. Объединенный элюат раскисляют, используя смолу НР-20, чтобы получить продукты, содержащие BU-3608 B и С. Продукт содержащий BU-3608 B растворяют в воде и наносят на колонку ODS-60 (8,0 х 90 см) и промывают смесью ацетонитрил-0,15% KH2PO4 22: 78 (рН 3,5). Фракции собирают и анализируют при помощи HPLC. Фракции, содержащие BU-3608 B, объединяют, упаривают в вакууме и, раскисляют при помощи хроматографии на смоле НР-20, получая загрязненный BU-3608, чистый BU-3608, и BU-3608 B, BU-3608 и затем очищают при помощи препаративной HPLC на колонке Microsorb Short One C18 (4,6 мм/D. 100 мм, 3 мм, Рэйнин Инструмент Ко), элюируя смесью ацетонитрил-0,15% KH2PO4 21:79 (рН 3,5). Для текущего анализа элюата используется HPLC.
Фракции содержащие BU-3608 C собирают и упаривают в вакууме. Полученный водный раствор раскисляют с помощью хроматографии на смоле НР-20, получая почти чистый BU-3608 С и почти чистый BU-3608.
В процессе хроматографии с обращением фаз на силикагеле, описанном выше, фракции элюирующиеся ранее BU-3608 C отдельно собирают и объединяют. Объединенные бледно-оранжевые фракции раскисляют с использованием хроматографии на Diaion HP-20. Полученный продукт относительно обогащен D и Е компонентами, однако еще содержит большое количество С компонента. Объединенные продукты наносят на колонку с обращенной фазой (ODS-60, силикагель, 8,0 х 90 см) и элюируют смесью ацетонитрил-0,15% KH2PO4 21:79 (рН 3,5). Элюат анализируют при помощи HPLC с использованием вышеописанной колонки в смеси тех же растворителей 7: 17 (рН 3,5) в качестве подвижной фазы при скорости течения 1,2 л/мин и УФ-детектировании (254 нм). BU-3608 E элюируется первым за ним следует BU-3608 D. Фракции, содержащие BU-3608 E объединяют, концентрируют в вакууме и раскисляют при помощи НР-20, получая почти однородный BU-3608 Е HCl. При установлении рН водного раствора BU-3608 Е HCl равным 5,0 0,1 н. NaOH чистый BU-3608 Е осаждается в виде цвиттер-иона. Аналогично в виде цвиттер-иона получается и BU-3608 D.
П р и м е р 3. Получение
бесксилозильного BU-3608 Е (IIIс)
Раствор гидрохлорида BU-3608 E (97 мг) в 2 н HCl (12 мл) нагревают при 115оС 70 мин в запаянной ампуле. рН полученного раствора устанавливают
равным 5,5 прибавлением 1 н NaOH, и затем центрифугируют.
Полученный таким образом твердый продукт промывают изопропанолом и ацетоном получая 87 мг бесксилозильного BU-3608 Е.
Т.пл. 205-209оС (с разл.).
УФ 0,01 н NaOH λмакс, нм (ε): 235,2 (23600), 319,2 (11100), 498,4 (10800)
П р и м е р 4. Получение
бесксилозильного BU-3608 С (IIIa)
Используя методику примера 3 из гидрохлорида BU-3608 С получают названное соединение.
Т.пл. 208-215оС (разл.)
П р и м е р
5. Получение N-метил BU-3608 B (II, R2 H, R1 R3 R4 CH3)
Смесь BU-3608 (540 мг) в 2 н HCl (60 мл) нагревают при 115оС 70 мин в
запаянной ампуле и затем охлаждают. Полученный твердый продукт отделяют центрифугированием (3000 об/мин) и суспендируют в воде и устанавливают рН 11,7 6 н. NaOH. Раствор (60 мл) прибавляют к ацетону
(300 мл и отделяют полученный твердый BU-3608 B. Продукт растворяют в воде (20 мл) и добавляют 1 н HCl до установления рН 8,3 и затем разбавляют 20 мл ацетонитрила. Водный HCHO (37% 0,8 мл) и NaBH3CN (120 мл) последовательно добавляют к раствору при комнатной температуре и раствор перемешивают 15 ч. Растворитель удаляют в вакууме и водный остаток по каплям при перемешивании прибавляют к
ацетону. Полученный осадок промывают ацетоном и сушат получая 440 мг N-метил BU-3608 B в виде натриевой соли. Часть этой соли растворяют в воде и подкисляют 1 н HCl до рН 6. Полученный осадок
промывают водой и лиофилизируют, получая 33 мг цвиттер-ионной формы.
Т.пл. 211-215оС (разл.)
УФ 0,01 н NaOH-MeOH λмакс, нм (ε): 240,8
(29700), 319,2 (13400), 499,2 (13100)
П р и м е р 6. Получение П,П-диметил бесксилозильного BU-3608 Е (II, R1 R2 H R3 R4 CH3).
К раствору бесксилозильного BU-3608 E (52,9 мг) в воде (5 мл) приливают MeCN (5 мл). Водный HCHO (0,2 мл) и NaBH3CN (30 мг) последовательно прибавляют к раствору при комнатной
температуре и полученный раствор перемешивают 14 часов. Растворитель удаляют в вакууме и водный остаток (рН 11,3) при перемешивании прибавляют к ацетону. Полученный осадок растворяют в воде и
устанавливают рН 5,5, получая твердый продукт, который последовательно промывают водой, изопропанолом, ацетоном и сушат получая 28,7 мг N,N-диметил бесксилозильного BU-3608 Е
Т.пл.
205-208оС
УФ 0,01 н NaOH λмакс, нм (ε): 232,8 (34000), 319,2 (15700), 497,6 (14900)
П р и м е р 7. Получение N,N-диметил BU-3608 Е (II, R1 H R2 D-ксилозил R3 R4 CH3)
К раствору BU-3608 Е (485 мг) в смеси воды (40 мл) и MeCN (40 мл) при рН 8,0 при комнатной температуре
последовательно прибавляют водный HCHO (37% 1,6 мл) и NaBH3CN (240 мг). Раствор перемешивают при комнатной температуре 15 ч и растворитель удаляют в вакууме. Остаток растворяют в воде и
устанавливают рН 11,0 и по каплям прибавляют к перемешиваемому ацетону (300 мл). Полученный осадок выделяют и растворяют в воде и подкисляют до рН 2,0 6 н HCl. Раствор раскисляют при пропускании через
НР-20. рН раствора, содержащего продукт, устанавливают 5,5 для его осаждения, осадок отделяют промывают водой и ацетоном и сушат, получая 364 мг N,N-диметил BU-3608 Е. Т.пл. 214-218оС
(разл.)
УФ 0,01 н NaOH λмакс, нм (ε ): 233,6 (32900), 319,2 (15500), 497,6 (15100)
Вычислено для C40H44N2O18.1,
5H2O
С 55,36 H 5,46 N 3,23
Найдено: C 55,26; H 5,45 N 3,19
П р и м е р 8. Получение N-метил BU-3608 (II, R1 R3 R4 CH3, R2 D-ксилозил)
К перемешиваемому раствору BU-3608 натриевой соли (550 мг) в 50% водном MeCN (55 мл) прибавляют HCHO (37% 0,75 мл) и NaBH3CN (150 мг) и смесь перемешивают 18 ч
при комнатной температуре. После концентрирования водный концентрат разбавляют (200 мл) подкисляют до рН 3,0 и подвергают колоночной хроматографии на НР-20 (300 мл). После промывания водой и
последующего элюирования 60% водным ацетоном элюат красного цвета концентрируют в вакууме и устанавливают рН 5,5 для осаждения N-метил-BU-3608, который отфильтровывают (425 мг).
Т.пл.
190-195оС
ИК (KBr) см-: 3400, 1605, 1450, 1295,
УФ 50% MeOH λмакс, нм ( ε): 220 (33700), 278 (26800), 488 (11400)
Масс-спектр:
855 (М + Н)+
П р и м е р 9. Получение N-пропил BU-3608 (II, R1 R3 CH3 R2 D-ксилозил R4CH2CH2CH3)
Следуя общей методике примера 8 и используя натриевую соль BU-3608 (200 мг), пропионовый альдегид (1 мл) и NaBH3CN (200 мг) получают N-пропил BU-3608 (127 мг)
Т.пл. 187-192оС
ИК (KBr) см-: 3380, 1605, 1450, 1295, 1255
УФ 50% MeOH λмакс, нм ( ε ): 223 (33000), 278 (27500), 488 (11700)
Масс-спектр: 833 (М + Н)+
П р и м е р 10. Получение четвертичного аммониевого производного BU-3608 (II, Y II(CH3)3.Cl)
Натриевую соль BU-3608
обрабатывают метилиодидом (1,5 мл) и бикарбонатом калия (200 мг), в ДМСО (5 мл) и метаноле (20 мл) при комнатной температуре 43 ч. Смесь концентрируют, разбавляют 0,5 н NaOH (20 мл) и выдерживают 30
мин при 70оС. Устанавливают рН раствора 3,0 и подвергают хроматографии на НР-20 (150 мл). Элюат, содержащий названное соединение упаривают и получают грязный продукт четвертичное аммониевое
производное (144 мг). Этот продукт подвергают хроматографии на силикагеле с обращенной фазой (2,0 х 45 см) с элюентом MeCN-0,15% KH2PO4 20:80 (рН 3,0). Элюат анализируют при
помощи HPLC и фракции, содержащие чисто четвертичное основание объединяют и раскисляют при помощи хроматографии на НР-20 (150 мл) получая чистое названное вещество (71 мг).
Т.пл. 205-210оС.
ИК (KBr) см-: 3400, 1620, 1600, 1440, 1255
УФ 50% MeOH λмакс, нм ( ε): 276 (22300), 498 (9800)
Масс-спектр:
869 (М+) Молекулярная формула C42H49N2O18Cl
П р и м е р 11. Получение бесксилозильного BU-3608 D (IIв)
Методику, описанную в
примере 3 повторяют, используя BU-3608 D HCl, и получают названное соединение.
Т.пл. 205-211оС.
Реферат
Использование: в химии антибиотиков, в частности в синтезе N -алкилпроизводных антибиотиков или их фармацевтически приемлемых солей, а также промежуточных соединений для их синтеза. Сущность изобретения: продукт: новые N -алкилпроизводные антибиотиков общей ф-лы I, где R1 - водород или метил (в этом случае образуется D-аланил); R2 - водород или β - D-ксилозил; Y-группа -

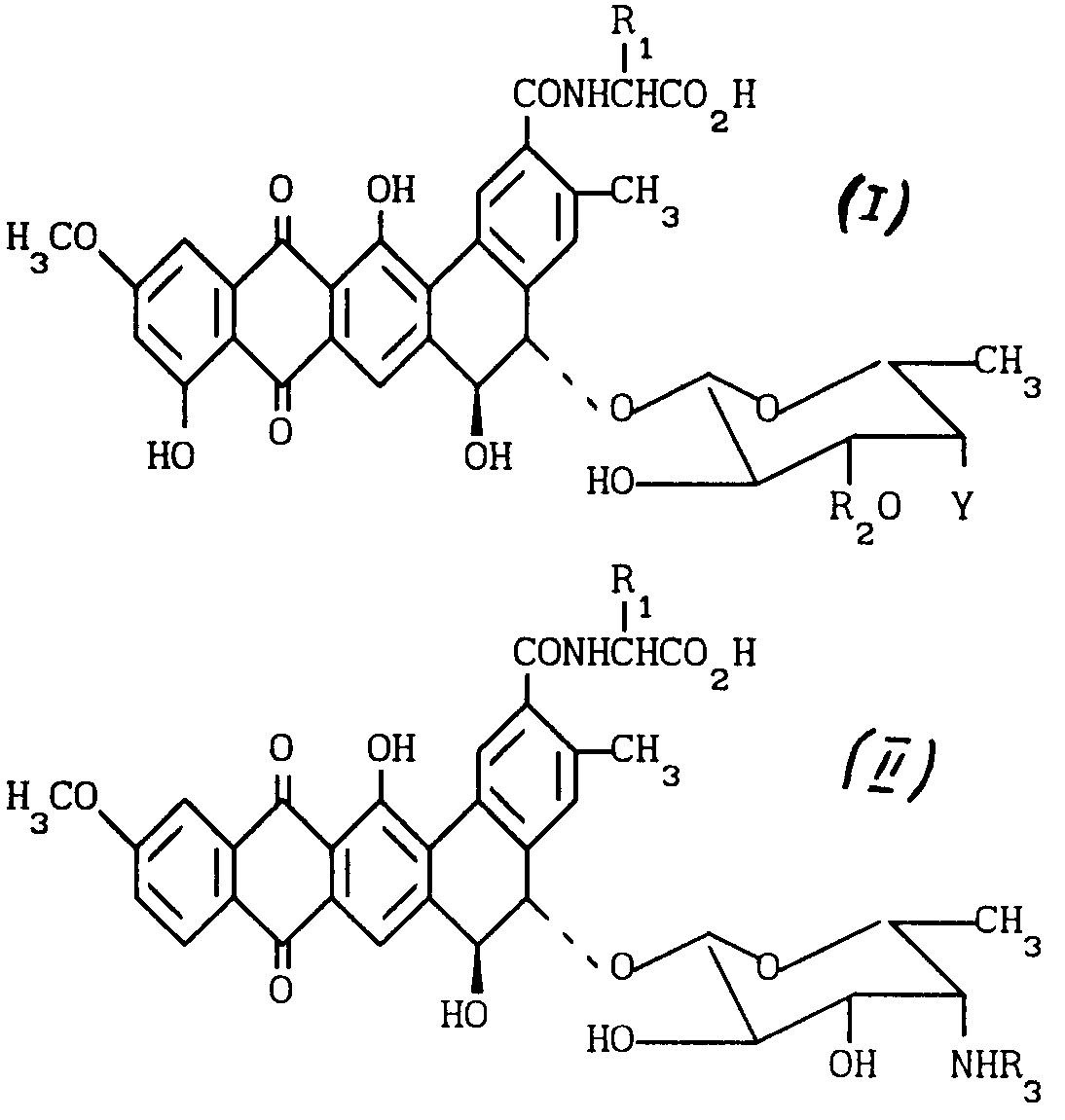
Формула

где R1 водород или R1 -метил и получающийся в результате аланил является D-аланилом;
R2 водород или β-D- ксилозил;
Y NR3R4 или

X- анион,
или их фармацевтически приемлемые соли, обладающие противограбковой активностью.
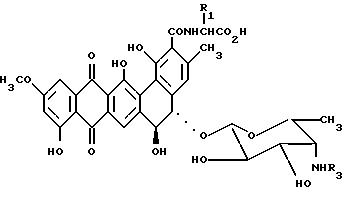
где R1 водород и R3 водород или метил или R1 - метил и получающийся аланил представляет D-аланил и R3 водород,
или его фармацевтически приемлемая соль в качестве промежуточного соединения в синтезе N-алкилпроизводных антибиотиков общей формулы

где R1 водород или метилрадикал, причем в последнем случае получающийся аланил означает D-аланил;
R2 водород;
Y группа общей формулы NR3R4 или

или их фармацевтически приемлемых солей.

где R1 водород или метилрадикал, причем в последнем случае получающийся аланил означает D-аланил;
R2 водород или β-D= -ксилозил;
Y группа формулы NR3 R4 или

X анион,
или их фармацевтически приемлемых солей, отличающийся тем, что соединение общей формулы

где R1 и R3 имеют указанные значения,
или его соль подвергают взаимодействию при рН реакционной среды 8,0 8,5 с альдегидом, имеющим 1 5 атомов углерода, с образованием амина с последующей обработкой последнего восстанавливающим агентом и выделением целевого продукта в свободном состоянии или в виде фармацевтически приемлемой соли.
Комментарии